
Scuola di Specializzazione in Medicina Interna
22
Direttore: Prof. Paolo Martelletti Scuola di Specializzazione in Medicina Interna Esame di Specializzazione Medicina Interna IV anno Medico Specializzando: Dr. Federico Marini
Transcript of Scuola di Specializzazione in Medicina Interna

Direttore: Prof. Paolo Martelletti
Scuola di Specializzazione in Medicina Interna
Esame di Specializzazione Medicina InternaIV anno
Medico Specializzando:Dr. Federico Marini

Un caso di shock settico senza focolaio infettivo
Ma che stai a di?
…è impossibile, ma che non le leggi le linee guida?
Questo è proprio andato…

24/07/17Case report Pagina 3
Presentazione paziente
• Sesso: Uomo
• Età: 44 anni
• Anamnesi familiare: Madre con
storia di linfoma mediastinico NH trattato con CHRT attualmente in
abs, 3 figlie (12, 10 e 7 anni) di cui 2 affette da artrite idiopatica
giovanile.
• Anamnesi fisiologica: sviluppo psicofisico regolare; alvo
tendenzialmente diarroico; ex fumatore.

24/07/17Case report Pagina 4
Presentazione paziente
• Anamnesi patologica remota: cirrosi criptogenetica (diagnosi nel
2012); malattia infiammatoria cronica intestinale (dal 2014) non
meglio specificata; recente ricovero per polmonite (Marzo 2016)
complicato da stato settico, risolto con terapia antibiotica; da allora
episodi febbrili ricorrenti.
• Terapia medica domiciliare:Asacol e Budesonide.

24/07/17Case report Pagina 5
Presentazione paziente
• Anamnesi patologica prossima: a Giugno 2016 accesso presso il PS di Bracciano per dispnea. Durante il ricovero episodio di edema polmonare acuto per cui veniva trasferito
presso l’unità di terapia intensiva del nostro Ospedale dove veniva sottoposto a intubazione oro-tracheale. Dopo il miglioramento del quadro acuto veniva trasferito
nell’unità di Medicina d’Urgenza di questo ospedale (al fine di proseguire terapia antibiotica), per poi giungere alla nostra osservazione in buone condizioni cliniche con
diagnosi di shock settico a partenza polmonare.
• Esame obiettivo: fegato di dimensioni aumentate con margine smusso; milza di volume aumentato di consistenza parenchimatosa, presenza di multipli linfonodi laterocervicali,
ascellari e inguinali. Cuore: ndr. Torace: ndr.

24/07/17Case report Pagina 6
Esami ematochimici rilevanti all’ingresso
• Hb 9.5 g/dl, PLT 43.000, GB 3.220 N 1094
• PCR 0,4 mg/dl
• PCT 0,01 ng/ml
• Trigliceridi 380 mg/l (4,8 mmol/L)
• Fibrinogeno 3 g/L
• Ferritina 2300 ng/ml
• positività della sierologia per EBV e
CMV (CMV IgM 19.4 U/ml, CMV IgG > 180 U/ml; EBV VCA IgG >
750 U/ml, IgM < 10 U/ml)
• Emocolture: in corso

24/07/17 Pagina 7
Episodi di shock settico
Dopo circa 3 giorni dal ricovero, a tre giorni dalla sospensione della terapia impostata in terapia intensiva e continuata nel nostro reparto con Merrem 1g x 3 e Targosid 400 mg x 2, il paziente si presentava con un quadro clinico caratterizzato da confusione mentale, dispnea ingravescente (SpO2 88% aa, FR 30), ipotensione (PA 80/40), sudorazione calda, tachicardia (130 bpm ritmico) e febbre (TC 39,7°). Venivano eseguiti:
• Esami ematochimici: PCR 24,3 mg/dl; PCT 104 ng/ml; Hb 9,7 g/dl, GB 5800/mm3 (N 87%); BNP 34 ng/ml.
• EGA in AA: pH 7,48; pO2 54; pCO2 31; HCO3- 24; SpO2 86%; Lat 4,5• RX torace: quadro di edema polmonare acuto in fase interstiziale• TC total body: CRANIO: ndr; TORACE: ispessimento umido dell’interstizio in assenza di focolai
broncopneumonici in atto; ADDOME: fegato in fase cirrotica con aumento delle dimensioni della vena porta e splenomegalia, come per quadro di ipertensione portale, assenza di varici esofagee.
• Trattamento: Flebocortid 1 g ev; Noradrenalina in infusione a 5 gamma; reimpostata terapia antibiotica; ossigenoterapia.
Nonostante la negatività di reperti infettivi di rilievo, nell’ipotesi forte di shock settico, si reimpostava terapia antibiotica in attesa delle emocolture effettuate all’ingresso.Tali episodi si susseguivano a distanza di circa 7-10 giorni l’uno dall’altro ed erano tutti preceduti da sintomatologia diarroica risultando del tutto sconnessi dalla terapia antibiotica impostata.

24/07/17 Pagina 8
Quick SOFA e SOFA score

24/07/17 Pagina 9
Quick SOFA e SOFA score

24/07/17 Pagina 10
Quick SOFA e SOFA score

24/07/17 Pagina 11
Ulteriori indagini diagnostiche effettuate:
• Indagini sul trigger gastroenterologico:– EGDS: ndr– RSCS: iperemia della mucosa fino a 5 centimetri dal margine anale
• All’esame istologico: infiltrato infiammatorio aspecifico. Negativa la colorazione rosso congo. Negativa per colite microscopica. Negativa la ricerca di EBV.
• Indagini immunologiche/infettivologiche:– Tipizzazione linfocitaria: bassi livelli di NK all’immunofenotipo (CD16 2.5%) con ridotta
capacità di degranulazione e ridotta proliferazione delle cellule T dopo stimolazione con anti-CD3.
– Dosaggio immunoglobuline: IgG 1024 mg/dl, IgA 216 mg/dl, IgM 366 mg/dl– Sierologia per Leishmania, HIV, virus e batteri pneumotropi, Rickettsie e Brucella, risultava
negativa

24/07/17 Pagina 12
Difetto immunologico?
Il paziente aveva recentemente eseguito, nell’ambito di studi genetici familiari, indagini genetiche con identificazione della variante genomica c.664C >T del gene XIAP. La mutazione di tale gene espone al rischio di crisi di linfoistiocitosi emofagocitica. Tale sindrome clinica poco si differenzia dallo shock settico in quanto condivide lo storm citochinico come base etiopatologica, se ne differenzia solo per l’agente scatenante.

24/07/17 Pagina 13
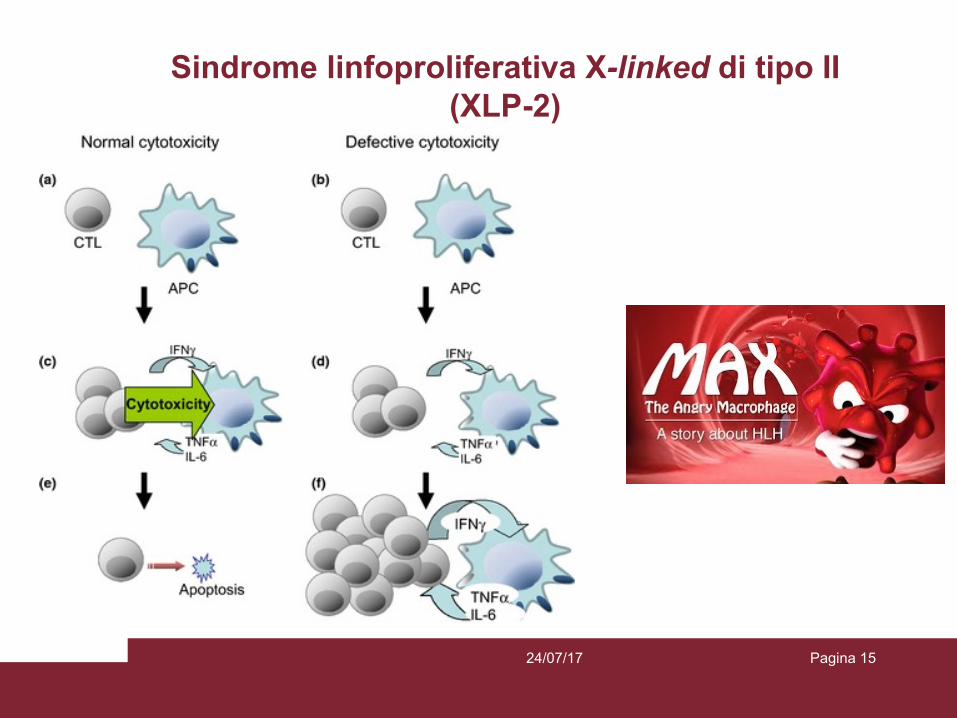
Sindrome linfoproliferativa X-linked di tipo II (XLP-2)
La sindrome linfoproliferativa X-linked di tipo II (XLP-2), identificata per la prima volta nel 2006, è una rara immunodeficienza primitiva, secondaria a mutazioni del gene XIAP (X- Linked inhibitor of apoptosis), implicato nell’inibizione dell’apoptosi cellulare. Dalla prima identificazione sono stati descritti in letteratura poco più di 70 casi e circa 50 differenti tipologie di mutazioni. Si manifesta prevalentemente in età pediatrica con uno spettro clinico tanto più grave quanto più l’insorgenza è precoce. Le manifestazioni cliniche più frequenti sono rappresentate dalla linfoistiocitosi emofagocitica (HLH, presente in più del 50% dei pazienti e spesso secondaria ad infezione da EBV, CMV o HHV6), insieme alla ricorrente splenomegalia (che si associa talvolta a febbre e citopenie) e alle malattie infiammatorie croniche intestinali (IBD). L’HLH, soprattutto nei casi ad insorgenza precoce, presenta spesso un decorso grave e talvolta fatale con un quadro di mononucleosi fulminante. Le IBD associate a XLP-2 frequentemente risultano resistenti alle terapie, con un outcome negativo in circa 10% dei pazienti. Il fenotipo clinico delle XLP-2 può essere infine più raramente caratterizzato da altre manifestazioni quali artriti, ascessi cutanei, eritema nodoso, uveiti e nefriti. L’ipogammaglobulinemia è stata riportata in circa 16% dei pazienti affetti da XLP-2, ma, a differenza delle forme di XLP-1, spesso risulta transitoria. Sono generalmente presenti un’elevata suscettibilità all’apoptosi in vitro ed un ridotto numero di cellule T NK. L’unico trattamento curativo è rappresentato dal trapianto di cellule staminali ematopoietiche, gravato tuttavia da un’elevata mortalità. L’utilizzo di terapia endovenosa con immunoglobuline è stato aneddoticamente descritto nei pazienti con XLP-2 associata ad ipogammaglobulinemia.

24/07/17Titolo Presentazione Pagina 14
Sindrome linfoproliferativa X-linked di tipo II (XLP-2)
24/07/17 Pagina 15
Sindrome linfoproliferativa X-linked di tipo II (XLP-2)
24/07/17Titolo Presentazione Pagina 16
Emofagocitosi Linfoistiocitaria

24/07/17Titolo Presentazione Pagina 17
Emofagocitosi Linfoistiocitaria

24/07/17 Pagina 18
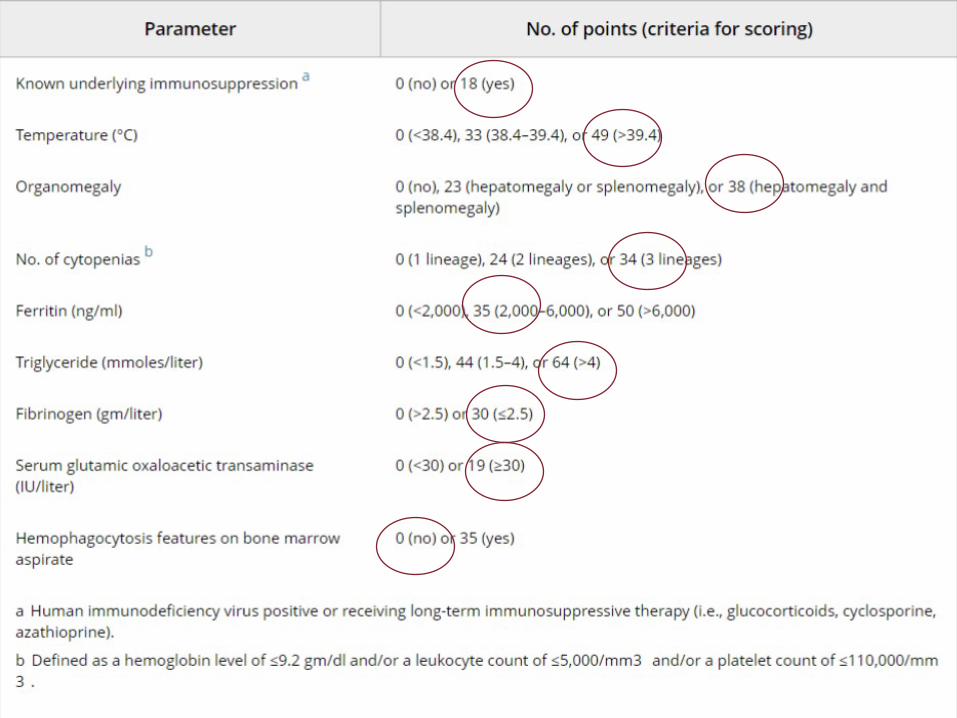
Diagnosi istologica di emofagocitosi
“Among clinicians who are not highly familiar with the syndrome, the presence of hemophagocytosis features on bone marrow aspirate or on biopsy of any other tissue is often thought to be the gold standard for the diagnosis. However, there is no consensus among cytologists regarding which cytologic features are necessary and sufficient for the diagnosis, and in the published literature, cytologic criteria used to define hemophagocytosis are rarely reported” *
* Francois B, Trimoreau F, Vignon P, Fixe P, Praloran V, Gastinne H. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med 1997;103:114–20.
CrossRef | PubMed | CAS | Web of Science® Times Cited: 101* Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia
with hemophagocytosis in critically ill medical patients: a postmortem clinicopathologic analysis. Crit Care Med 2004;32:1316–21.CrossRef | PubMed | Web of Science® Times Cited: 39* Suster S, Hilsenbeck S, Rywlin AM. Reactive histiocytic hyperplasia with hemophagocytosis in hematopoietic organs: a reevaluation of the
benign hemophagocytic proliferations. Hum Pathol 1988;19:705–12.CrossRef | PubMed | CAS | Web of Science® Times Cited: 46

24/07/17 Pagina 19
Riassunto caratteristiche XLP tipo 2
- Frequenti episodi di HLH molto spesso causati da infezioni di EBV
- Malattia infiammatoria cronica intestinale- Epatopatia rapidamente progressiva verso la
cirrosi- Difetto immunologico di degranulazione NK- Immunodeficienza ipogamma/disgamma

24/07/17 Pagina 20
Terapia
Unica terapia curativa è il trapianto di midollo osseo, gravato da una elevata mortalità (circa 15%) per l’eccessiva GVHD e la necessità di trattamento condizionante aggressivo.
In alternativa si posso utilizzare i corticosteroidi (qualora le manifestazioni infettive risultassero minori rispetto agli episodi di HLH)
In letteratura sono stati descritti 7 casi in cui il trattamento è stato effettuato con immunoglobuline endovena (IVIg) a scopo immunosostitutivo (i pazienti presentavano severa ipogamma)
Il nostro è il primo caso di trattamento con terapia immunomodulante. A tale scopo abbiamo eseguito il vaccino antipneumococcico 18 valente al paziente per valutare la risposta selettività delle immunoglobuline. È risultato che il paziente rispondeva a soli 6 degli epitopi su 18, per cui è stato inserito tra i pazienti con immunodeficienza funzionale così da poter eseguire il trattamento con IVIg.
Il trattamento immunomodulante va ad agire sulla tempesta citochinica provocando un whashout citochinico che impedisce ai macrofagi di attivarsi e quindi riduce gli effetti della manifestazione clinica più gravosa di questa malattia. Al tempo stesso gli anticorpi introdotti funzionano anche da opsonizzanti batterici, riuscendo a contrastare le manifestazioni cliniche da immunodeficienza.
Da Febbraio-’17 il paziente ha manifestato un solo episodio di febbrile (37,5°C) da lui stesso attribuito ad una faringotonsillite (probabilmente virale), autorisoltasi.

24/07/17 Pagina 21
Conclusioni
1. La sindrome emofagocitica ha un’incidenza stimata di circa 1 caso su 1 milione contro i 300 casi su 100 mila della Sepsi, di questi si stima che circa il 5-10% si manifesti come shock settico (per cui si ipotizza un’incidenza di Shock settico di circa 15-30 casi su 100.000).
2. Il rapporto di incidenza tra le due condizioni è di circa 150-300 a 1, il che significa che ogni 150-300 diagnosi di shock settico ci troviamo di fronte ad un’attivazione macrofagica anomala.
3. La diagnosi va sospettata in tutti quei pazienti con shock settico che presentano diverse emocolture negative.
24/07/17Grazie ziotti Pagina 22


















