
Organo ufficiale della Società Infantili (SIMRI) · Per informazioni:Biomedia srl- tel.02/45498282...
65
Transcript of Organo ufficiale della Società Infantili (SIMRI) · Per informazioni:Biomedia srl- tel.02/45498282...



Organo ufficiale della Società Italiana per le Malattie RespiratorieInfantili (SIMRI)
Volume 13, n. 51 - Settembre 2013Spedizione in A.P. - 45%art. 2 comma 20/blegge 662/96 - N. 1047 del 12/07/2002 - PisaReg. Trib. PI n. 12 del 3 giugno 2002
Direttore scientificoRenato Cutrera (Roma)Codirettori scientificiFrancesca Santamaria (Napoli)Luigi Terracciano (Milano)Segreteria scientificaFrancesco Paolo Rossi (Roma)Comitato editorialeEugenio Baraldi (Padova)Angelo Barbato (Padova)Filippo Bernardi (Bologna)Alfredo Boccaccino (Benevento)Attilio L. Boner (Verona)Fabio Cardinale (Bari)Fernando Maria de Benedictis (Ancona)Fulvio Esposito (Napoli)Ahmad Kantar (Bergamo)Mario La Rosa (Catania)Fabio Midulla (Roma)Giorgio L. Piacentini (Verona)Giovanni A. Rossi (Genova)Marcello Verini (Chieti)Gruppo Allergologiacoord. Gianluigi Marseglia (Pavia)Gruppo Disturbi respiratori nel sonnocoord. Luigi Nespoli (Varese)Gruppo Educazionecoord. Stefania La Grutta (Palermo)Gruppo Endoscopia bronchiale e delle Urgenze respiratoriecoord. Fabio Midulla (Roma)Gruppo Fisiopatologia respiratoriacoord. Marcello Verini (Chieti)Gruppo Riabilitazione respiratoriacoord. Giancarlo Tancredi (Roma)Gruppo Infettivologiacoord. Mario Canciani (Udine)Direttore responsabileEugenio Baraldi (Padova)© Copyright 2013 by Primula MultimediaFinito di stampare nel mese di ottobre 2013EditorePrimula Multimedia S.r.L.Via G. Ravizza, 22/b56121 Pisa - Loc. OspedalettoTel. 050 9656242; fax 050 3163810e-mail: [email protected] Massimo PiccioneRealizzazione EditorialePrimula Multimedia S.r.L.StampaLitografia VARO - San Giuliano Terme (PI)
Editoriale3
View point
Manifestazioni respiratorie nelle patologie neuromuscolari
5Respiratory manifestationsin neuromuscular diseasesM.B. Chiarini Testa, V. Caldarelli, M.C. Graziani, M.G. Paglietti,A. Schiavino, R. Cutrera
La bronchiolite obliterante12
Bronchiolitis obliteransS. Cazzato, L. Bertelli, E. di Palmo, A. Pession
La gestione dell’aspergillosi polmonare in età pediatrica
20Management of pulmonary aspergillosisin childrenV. Terlizzi, F. Improta, V. Raia
Le deformità toraciche29
Chest wall deformitiesM. Torre, V. Jasonni
La chirurgia toracoscopica video-assistita nella gestionedell’empiema pleurico
39Video Assisted Thoracic Surgeryin the management of parapneumonicpleural empyemaA. Porreca
Trapianto polmonare in età pediatrica46
Pediatric lung transplantationF. Parisi, S. Alfieri, M.A. Castelluzzo
R U B R I C A : e a n c o r a . . .
La discinesia ciliare primitiva:esperienza di un percorso diagnosticoin ambulatorio
52Primary ciliary dyskinesia:experience of an ambulatorydiagnostic work-upR. Padoan, S. Timpano, D. Zaniboni, E. Spinelli, M.M. De Santi,M. Berlucchi, A. Plebani
Conferenze e meeting59
Conferences and meetings
Articoli del prossimo numero63
Forthcoming articles
INDICE SUMMARY

Il sottoscritto, CHIEDE AL PRESIDENTE della Società Italiana per le Malattie Respiratorie Infantili di essereammesso quale socio ordinario. Pertanto, riporta i seguenti dati personali:
DATI PERSONALI
Cognome Nome
Luogo e data di nascita
Domicilio (via/piazza)
CAP Città Prov. Regione
Sede di lavoro Reparto
Indirizzo
Recapiti telefonici: Casa Studio Fax
Ospedale Cellulare e-mail
Laurea in Medicina e Chirurgia - Anno di laurea
Specializzazioni
Altri titoli
CATEGORIA
Universitario Ospedaliero Pediatra di libera scelta
QUALIFICA UNIVERSITARIA
Professore Ordinario Professore Associato Ricercatore Altro
QUALIFICA OSPEDALIERA
Dirigente di 2º Livello Dirigente di 1º Livello Altro
Con la presente autorizzo la Società Italiana per le Malattie Respiratorie Infantili al trattamento dei miei dati personali ai sensi del D.L. del 30giugno 2003 n. 196.
Data Firma del Richiedente
Soci presentatori (cognome e nome) Firma
1)
2)
Compilare in stampatello e spedire insieme con la copia dell’avvenuto versamento (quota sociale di euro 30,00. Specializzandi euro 10,00) a:
Biomedia srl - Segreteria Amministrativa SIP - Via Libero Temolo 4, 20126 Milanoc/c postale N. 67412643 intestato a: Società Italiana di Pediatria
È obbligatoria l’iscrizione anche alla SIP (quota sociale di euro 90,00), può essere fatto un unico versamento indicando chiaramente nella causale per quali società affiliate viene effettuato il versamento.
Per informazioni: Biomedia srl - tel. 02/45498282 - fax 02/45498199 e-mail: [email protected]
Domanda di ammissione per nuovi Soci

Questo fascicolo è stato dedicato a “Le complicanze respiratorie di difficilegestione”, in quanto argomento utile sia al pediatra di famiglia che al pneumologopediatra. Le malattie respiratorie in età pediatrica rappresentano la causa più comu-ne di visita ambulatoriale da parte del pediatra curante e sono tra le prime cause diricovero ospedaliero. Abbiamo chiesto ad alcuni esperti di aggiornarci sulle malattierespiratorie e sulle loro complicanze medico-chirurgiche.Infezioni respiratorie, quali la bronchiolite, la polmonite e l’empiema pleurico pos-
sono complicare patologie croniche note, come le malattie neuromuscolari, portan-do progressivamente ad insufficienza respiratoria, talvolta con esiti infausti.L’aspergillosi polmonare può essere motivo di complicanza nel bambino, soprat-
tutto se non immunocompetente, con rischio di sviluppo di un’infezione invasiva cheinteressa primariamente il polmone, ma con possibilità di disseminazione anche alsistema nervoso centrale.La bronchiolite obliterante, che rappresenta un disordine cronico secondario a
un danno severo a carico delle basse vie aeree, può anch’essa raramente progredi-re verso l’insufficienza respiratoria con esito fatale o richiedere la necessità di inter-venti radicali, come ad esempio il trapianto polmonare.D’altra parte anche la gestione ambulatoriale è fondamentale nel precoce rico-
noscimento di condizioni patologiche, quali ad esempio le malformazioni polmonario le patologie croniche, come la discinesia ciliare primitiva, prevenendo in questomodo importanti ripercussioni respiratorie e sottolineando l’importanza di una cor-retta conoscenza e gestione delle problematiche respiratorie sia in ambito ambula-toriale che ospedaliero.Con l’augurio che anche questa volta “Pneumologia Pediatrica” sia vicina sia ai
pediatri che ai bambini!
Francesca [email protected]
EditorialeView point
Pneumologia Pediatrica 2013; 51: 3 3

La Rivista pubblica contributi redatti in forma di edito-riali, articoli d’aggiornamento, articoli originali, articolioriginali brevi, casi clinici, lettere al Direttore, recensioni(da libri, lavori, congressi), relativi a problemi pneumolo-gici e allergologici del bambino.I contributi devono essere inediti, non sottoposti con-temporaneamente ad altra Rivista, ed il loro contenutoconforme alla legislazione vigente in materia di etica dellaricerca.Gli Autori sono gli unici responsabili delle affermazionicontenute nell’articolo e sono tenuti a dichiarare di averottenuto il consenso informato per la sperimentazionee per la riproduzione delle immagini.La redazione accoglie solo i testi conformi alle normeeditoriali generali e specifiche per le singole rubriche.La loro accettazione è subordinata alla revisione criticadi esperti, all’esecuzione di eventuali modifiche richiesteed al parere conclusivo del Direttore.
NORME GENERALITesto: in lingua italiana o inglese, materialmente digita-to, con ampio margine, con interlinea doppia, massimo25 righe per pagina, con numerazione delle pagine apartire dalla prima, e corredato di:1) titolo del lavoro in italiano, in inglese;2) parola chiave in italiano, in inglese;3) riassunto in italiano, (la somma delle battute, spaziinclusi, non deve superare le 1.500);4) titolo e didascalie delle tabelle e delle figure.Si prega di allegare al manoscritto anche il testo digita-le, purché scritto con programma Microsoft Word ver-sione 4 e succ. (per Mac OS e Win).Nella prima pagina devono comparire: il titolo (conci-so); i nomi degli Autori e l’istituto o Ente di appartenen-za; la rubrica cui si intende destinare il lavoro (decisioneche è comunque subordinata al giudizio del Direttore);il nome, l’indirizzo e l’e-mail dell’Autore cui sono destina-te la corrispondenza e le bozze.Il manoscritto va preparato secondo le norme inter-nazionali (Vancouver system) per garantire la unifor-mità di presentazione (BMJ 1991; 302: 338-341). Èdunque indispensabile dopo una introduzione, descri-vere i materiali e i metodi, indagine statistica utilizzata,risultati, e discussione con una conclusione finale. Glistessi punti vanno riportati nel riassunto.Nelle ultime pagine compariranno la bibliografia, le dida-scalie di tabelle e figure.Tabelle devono essere materialmente digitate in nume-ro contenuto (evitando di presentare lo stesso dato in piùforme), e numerate progressivamente.Figure vanno fornite su supporto digitale in uno deiseguenti formati: tif, jpg e con una risoluzione adeguataalla riproduzione in stampa (300 dpi); oppure immaginigenerate da applicazioni per grafica vettoriale(Macromedia Freehand, Adobe Illustrator per Macintosh).Sono riproducibili, benché con bassa resa qualitativa,anche documenti generati da Power Point. Al contrario,
non sono utilizzabili in alcun modo le immagini inserite indocumenti Word o generate da Corel Draw.La redazione si riserva di rifiutare il materiale ritenutotecnicamente non idoneo.Bibliografia: va limitata alle voci essenziali identificatenel testo con numeri arabi ed elencate al termine delmanoscritto nell’ordine in cui sono state citate. Se gliautori sono fino a tre si riportano tutti, se sono quattroo più si riportano solo i primi tre seguiti da “et al.”.Esempi di corretta citazione bibliografica per:articoli e riviste:Zonana J, Sarfarazi M, Thomas NST, et al. Improved defini-tion of carrier status in X-linked hypohydrotic ectodermaldysplasia by use of restriction fragment lenght polymor-phism-based linkage analysis. J Pediatr 1989; 114: 392-395.libri:Smith DW. Recognizable patterns of human malforma-tion.Third Edition. Philadelphia: WB Saunders Co. 1982.capitoli di libri o atti di Congressi:Krmpotic-Nemanic J, Kostovis I, Rudan P. Aging changes ofthe form and infrastructure of the external nose and its impor-tance in rhinoplasty. In: Conly J, Dickinson JT, (eds). “Plasticand reconstructive surgery of the face and neck”. NewYork, NY: Grune and Stratton 1972: 84-95.
Ringraziamenti, indicazioni di grants o borse di studio,vanno citati al termine della bibliografia.Le note, contraddistinte da asterischi o simboli equiva-lenti, compariranno nel testo a piè di pagina.Termini matematici, formule, abbreviazioni, unità e misu-re devono conformarsi agli standard riportati in Scienze1954; 120: 1078.I farmaci vanno indicati col nome chimico.
Per la corrispondenza scientifica:
Dott. Renato CutreraDirettore UOC BroncopneumologiaDipartimento di Medicina PediatricaOspedale Pediatrico Bambino Gesù - IRCCS Piazza Sant'Onofrio, 400165 [email protected]
RICHIESTA ESTRATTIL’Editore si impegna a fornire agli Autori che ne faccia-no richiesta un pdf del proprio Articolo.
ABBONAMENTIPneumologia Pediatrica è trimestrale. Viene inviata gratui-tamente a tutti i soci della Società Italiana per le MalattieRespiratorie Infantili; i prezzi di abbonamento annuo peri non soci sono i seguenti:Italia ed Estero: h 72,00; singolo fascicolo: h 20,00.Le richieste di abbonamento e ogni altra corrisponden-za relativa agli abbonamenti vanno indirizzate a:
Primula Multimedia S.r.L.Via G. Ravizza, 22/b56121 Pisa - Loc. Ospedaletto
Informazioni per gli autoricomprese le norme per la preparazione dei manoscritti

Le malattie neuromuscolari
Le malattie neuromuscolari (MNM), sono ungruppo di malattie rare che globalmente presen-tano una prevalenza di circa una su tremila. Lamaggior parte di queste condizioni sono geneti-che e diventano clinicamente evidenti durantel’infanzia.Il movimento muscolare volontario è controllatodai motoneuroni corticali (primo motoneurone)che, dalla corteccia, decorrono lungo le cornaanteriori del midollo spinale, raggiungono i nervimotori periferici (secondo motoneurone) e, tra-mite la placca neuromuscolare, il muscolo. Le lesio-ni del primo motoneurone determinano, principal-mente, una paralisi spastica mentre le malattie cheinteressano il secondo motoneurone, la placcaneuromuscolare e la muscolatura striata compor-tano riduzione della forza e atrofia muscolare(Tabella 1).
Le MNM dell’infanzia sono forme congenite spes-so evolutive che interessano il secondo motoneu-rone, la placca neuromuscolare e il muscolo.La patologia degenerativa del motoneurone spina-le è detta amiotrofia spinale (SMA) e comprendediversi fenotipi, distinti clinicamente in base all’in-tensità e alla precocità della compromissionemuscolare:• SMA I (malattia di Werdnig Hoffmann);• SMA II;• SMA III (malattia di Kugelberg-Welander);• SMA IV (forma lieve dell’adulto);• SMARD (SMA con paralisi diaframmatica).Le SMA sono malattie congenite ereditate inmaniera autosomica recessiva.Il difetto genetico, identificato nel 1995 con la sco-perta del gene Survival Motor Neuron (SMN), ècausa del 98,6% di queste forme. Il gene si trova
5Pneumologia Pediatrica 2013; 51: 5-11
Maria Beatrice Chiarini Testa, Valeria Caldarelli, Maria Cecilia Graziani, Maria Giovanna Paglietti,Alessandra Schiavino, Renato Cutrera
UOC Broncopneumologia, Ospedale Pediatrico “Bambino Gesù”, Roma
Manifestazioni respiratorienelle patologie neuromuscolari
Respiratory manifestationsin neuromuscular diseases
Parole chiave: malattie neuromuscolari, tosse inefficace, mechanical in-exsufflator, ipoventilazione, ventilazione meccanica noninvasiva
Keywords: neuromuscular diseases, non-efficient cough, mechanical in-exsufflator, hypoventilation, non-invasive mechanical ventilation
Riassunto. Le malattie neuromuscolari dell’infanzia sono forme su base genetica caratterizzate dalla debolezza progressiva einarrestabile della muscolatura volontaria. L’insufficienza muscolare coinvolge anche i muscoli del torace alterando la meccani-ca respiratoria e producendo ostruzione da parte delle secrezioni bronchiali con formazione di atelettasie, polmoniti e stati diinsufficienza ventilatoria acuta e/o cronica. Al momento non sono disponibili terapie risolutive per queste patologie. La fisiote-rapia respiratoria volta alla disostruzione bronchiale, la ventilazione non invasiva a pressione di supporto positiva a lungo ter-mine sono ormai utilizzate largamente nei pazienti pediatrici affetti da malattie neuromuscolari cambiando la storia naturale diqueste patologie e migliorando la sopravvivenza e la qualità di vita di questi pazienti.
Accettato per la pubblicazione il 9 agosto 2013.
Corrispondenza: Maria Beatrice Chiarini Testa, Unità Operativa Complessa Broncopneumologia,Ospedale Pediatrico “Bambino Gesù”
Piazza S. Onofrio 4, 00165 Romae-mail: [email protected]

Chiarini Testa, et al. 6
sul braccio lungo del cromosoma 5 e codifica peruna proteina che mantiene in vita le cellule delmotoneurone spinale.La quantità di proteina è variabile da soggetto asoggetto e i livelli più elevati di SMN 2 sono staticorrelati con le forme più lievi di SMA. Inoltre, laproteina SMN è ubiquitaria e molti degli effetti attri-buiti all’insufficienza neuromuscolare potrebbero
dipendere anche dall’effetto diretto della proteinasu altri apparati esterni al sistema nervoso.L’incidenza dell’SMA va da uno su seimila a uno sudiecimila nati vivi e la frequenza dello stato di por-tatore è di 1 : 40-1 : 60, mentre la prevalenza diqueste forme è di circa uno su trecento. Dopo lafibrosi cistica, l’SMA sono, per frequenza, la secon-da forma trasmissibile in maniera recessiva. L’SMAdi tipo I è la più grave e comprende circa il 50% ditutte le SMA [1-3].La SMARD è caratterizzata da una neuropatiaperiferica, causata dalla mutazione del gene dellaproteina legante le immunoglobuline (IGHBMP2),comporta la degenerazione dei neuroni dellecorna anteriori del midollo spinale producendoipotonia del tronco e del bacino con paralisi dia-frammatica.La distrofia di Duchenne è una distrofia muscolaregeneralizzata che interessa il muscolo striato, ha undecorso relativamente rapido e attivo. Essa colpi-sce quasi esclusivamente il sesso maschile durantei primi anni di vita, e l’incidenza è stata calcolata auno su 3.500 maschi e costituisce il 50% di tutte leforme distrofiche.Le altre forme di miopatie congenite sono in granparte legate a mutazioni dei geni codificanti per leproteine del tessuto muscolare e, a volte, anchedella cute. Le malattie del muscolo striato hannocaratteristiche istologiche e istochimiche peculiari,possono associarsi a dismorfismi e malformazionicongenite (piede torto, artrogripposi, etc.) o mani-festarsi nell’ambito di sindromi polimalformative(malattia di Fukuyama, Eye-brain disease, etc.) [4].Le MNM interessanti i nervi periferici come la sin-drome di Guillain-Barré possono dipendere dal-l’aggressione autoimmune della mielina nel nervoperiferico, mentre le mutazioni dei geni codificantiper le proteine della placca neuromuscolaredeterminano la miastenia Gravis, che riconosceanche un’eziologia autoimmune da autoanticorpicontro i recettori dell’acetilcolina [5].In questa sede saranno prese in considerazionesolo le malattie che portano ad un deficit perma-nente, escludendo quindi alcune MNM acutecome la sindrome di Guillan-Barré, il botulismo, lapoliomielite o altre malattie infettive. La possibilitàdi diagnosi precoce, le cure intensive e le tecnichedi nutrizione e di ventilazione artificiali hanno per-messo di prolungare la sopravvivenza di questimalati e di migliorare le loro condizioni di vitarispetto al passato [6].
Tabella 1 Patologie neuro-muscolari dell’infanzia. Modificatada [5].
Patologie del midollo spinale
- Lesione del midollo cervicale post-traumatica,
- Siringomielia,
- Mielite trasversa.
Patologie del motoneurone
- Atrofie Muscolari Spinali (SMA I, II, III),
- Poliomielite,
- Sindrome post-polio.
Neuromiopatie periferiche
- Lesione del nervo frenico post intervento cardiochirurgico,
- Sindrome di Charcot-Marie-Tooth,
- Sindrome di Guillain-Barrè.
Patologie della giunzione neuro-muscolare
- Miastenia.
Patologie del muscolo
• Distrofie muscolari (DM)
- DM di Duchenne,
- DM di Becker,
- DM dei cingoli,
- DM fascio-scapolo-omerale,
- DM di Emery-Dreyfuss,
- DM Congenite (CDM)
CDM merosino-negativa,
CDM con sindrome del rachide rigido,
CDM di Ullrich,
- Distrofia miotonica congenita
• Miopatie congenite
- Miopatia nemalinica,
- Miopatia centronucleare,
- Miopatia miotubulare.
• Miopatie metaboliche
- Miopatie mitocondriali.
• Dermatomiosite giovanile.

Manifestazioni respiratorie nelle patologie neuromuscolari 7
Le complicanze respiratoriedelle malattie neuromuscolari
Le MNM non sono malattie primitive dei polmo-ni, che sono sani, ma dipendono dalla debolezzamuscolare ingravescente che coinvolge anche l’ap-parato respiratorio alterandone profondamentemeccanica e fisiologia. L’interessamento della fun-zione respiratoria è lento, progressivo e inevitabilee rappresenta la principale causa di morbilità emortalità.Dal punto di vista funzionale i test respiratorimostrano un pattern restrittivo con capacità vita-le polmonare totale e capacità funzionale residuadiminuite e con un rapporto conservato tra volu-me espiratorio forzato nel primo secondo (ForcedExpiratory Volume in the 1st second, FEV1) / e capa-cità vitale forzata (Forced Vital Capacity, FVC) (indi-ce di Tiffenau). La cifoscoliosi associata aggrava lamalattia polmonare restrittiva, determina la forma-zione di atelettasie e l’interessamento cardiaco(cuore polmonare cronico) [7].
Tosse inefficace e ritenzionedelle secrezioni bronchiali
In condizioni normali, le secrezioni presenti nellevie aeree vengono trasportate dal movimentodelle ciglia vibratili alla velocità di 20 mm al minu-to a livello della laringe dove vengono espettorateo deglutite (80%) per consentire la lubrificazionedella trachea, priva di ghiandole mucose.La tosse è un altro meccanismo che intervienenella rimozione di materiale che impegna lo spaziodelle vie aeree e che il sistema muco-ciliare non èstato in grado di espellere [5].La tosse dipende dalla compressione e dall’espira-zione forzata, a glottide chiusa, dell’aria inspirata al60-90% della capacità polmonare totale. La debo-lezza muscolare ne determina l’inefficienza ridu-cendo il volume dell’inspirazione, la chiusura dellaglottide e il flusso di aria espirata.Nei pazienti affetti da patologie neuromuscolari lesecrezioni a livello delle vie aeree tendono adaumentare perché continuamente prodotte acausa dello stato infiammatorio cronico e difficil-mente eliminate per la tosse, inefficace a causadella debolezza muscolare.Ne consegue un accumulo di secrezioni a livellodelle vie aeree con rischio elevato di atelettasie epolmoniti per sovrainfezioni batteriche di alterazio-ne del rapporto rapporto ventilazione /perfusione
e conseguenti stati di ipossiemia e/o ipercapnia.Mentre l’aumento di secrezioni tracheo-bronchialidovuto allo stato infiammatorio cronico si correg-ge trattando il paziente con terapia farmacologica,per il problema della tosse inefficace si fa ricorso avari metodi svolti tradizionalmente da un fisiotera-pista respiratorio [5].Oltre alle tecniche manuali di drenaggio, non sem-pre realizzabili in età pediatrica in quanto richie-dono tempo e collaborazione, esistono tecnichedi assistenza manuale della tosse eseguibili quandola funzione muscolare bulbare del paziente è suffi-ciente per mantenere la glottide chiusa [8].Queste consistono in manovre che incrementanoil volume polmonare inspiratorio fino alla capaci-tà massima di insufflazione (Maximum InsufflationCapacity, MIC) attraverso l’insufflazione di aria(aria-stacking) con l’aiuto di un ventilatore o di unpallone ambu (Auxiliary Manual Breathing Unit)oppure in manovre che incrementano il volumeespiratorio attraverso compressioni toraco-addominali [5].Nei pazienti molto deboli – o con incapacità bul-bare a mantenere volumi polmonari idonei, non ingrado di eseguire manovre di tosse manuale oaria-stacking o dove questi metodi non sono effi-caci – deve essere presa in considerazione la tec-nica di insufflazione / essufflazione meccanica(Mechanical Insufflation/Exsufflation, MI-E) attraver-so l’utilizzo di dispositivi meccanici. Ormai da alcu-ni anni si utilizza un apparecchio denominatoMechanical In-Exsufflator noto anche come “mac-china della tosse”, strumento già sperimentato eproposto negli anni Novanta da John R. Bach, cheha poi ottenuto un consenso mondiale ed è statocitato nelle Linee Guida del 2012 come presidioindispensabile alla gestione dei pazienti con inca-pacità di tossire [5].Questo strumento riduce sicuramente il rischio diaccumulo di secrezioni nell’albero bronchiale, aiu-tando i pazienti nella rimozione naturale dellesecrezioni. La sua azione si esplica applicando gra-dualmente alle vie aeree una pressione positivaseguita rapidamente da una pressione negativa. Ilflusso d’aria simula il processo naturale della tosse,permettendo quindi alla persona con debolezzamuscolare di drenare l’albero bronchiale.Le tecniche di disostruzione delle vie tracheo-bron-chiali nei pazienti neuromuscolari devono esseresempre effettuate durante un episodio di riacutiz-zazione infettiva o quando i livelli di saturazione

arteriosa in ossigeno in aria ambiente scendono aldi sotto del 92%. Può essere indicata l’ospedalizza-zione se con tutte le tecniche in uso il valore dellasaturazione di ossigeno rimane sempre basso.
Patologia da inalazione cronica
La patologia polmonare da inalazione è molto fre-quente nei bambini affetti da ipotonia muscolare. Ipazienti possono presentare problemi alla degluti-zione per la disfunzione bulbare, rallentamentodello svuotamento gastrico e reflussi gastroesofa-gei. Il materiale aspirato include saliva e materialealimentare oppure contenuto gastrico.La prevalenza di reflusso gastroesofageo nei bam-bini con MNM non è nota, sebbene l’esperienzaclinica suggerisca che si tratti di un problemacomune che può manifestarsi molto precocemen-te i particolare nei pazienti affetti da SMA di tipo 1e nelle forme gravi di miopatia nemalinica [5].L’inalazione comporta uno stato infiammatoriodelle vie aeree con ostruzione dei bronchi edaggravamento della patologia restrittiva. Inoltre leinalazioni croniche causano nel tempo bronchiec-tasie e fibrosi polmonare.In tutti i pazienti è indicata la valutazione dellamassa corporea, l’esecuzione di un deglutidogram-ma e il follow-up nutrizionale e per cercare un’e-ventuale incompetenza orofaringea. In caso dinecessità bisogna ricorrere a presidi nutrizionaliaggiuntivi e a vie alternative per l’alimentazione(come la gastrostomia endoscopica percutanea,Percutaneous Endoscopic Gastr, PEG o la digiuno-stomia endoscopica percutanea, PercutaneousEndoscopic Jejunostomy, PEJ) [5].
Ipoventilazione cronica
La riduzione progressiva e simmetrica della forzamuscolare e la diminuzione della compliance dellaparete toracica e del polmone aumentano il cari-co del lavoro respiratorio. Quando sopravvieneuno squilibrio tra carico elastico polmonare eforza muscolare, i muscoli respiratori vannoincontro a fatica e diventa difficile generare flussie pressioni respiratori normali, si riducono così ladurata della fase inspiratoria, il volume correntee il volume polmonare totale (Total LungCapacity, TLC).La respirazione diventa superficiale, diminuisco-no gli scambi gassosi, compaiono apnee e si svi-luppa un quadro di ipoventilazione alveolare con
conseguente ipercapnia e/o ipossiemia dapprimanel sonno e poi anche durante la fase di veglia.L’insufficienza ventilatoria cronica che ne derivarappresenta la più frequente causa di mortalità.
Ipoventilazione notturna e i disturbi respiratori nel sonno
Durante il sonno si assiste a modificazioni fisiologi-che dell’apparato cardiovascolare, del tonomuscolare e del “drive” respiratorio centrale. Lafrequenza cardiaca, la frequenza respiratoria ed iltono muscolare si riducono in tutte le fasi delsonno. Nel sonno REM (Rapid Eye Movement) l’i-potonia è più marcata e possono manifestarsieventi di natura ostruttiva delle prime vie aeree(apnee ed ipopnee). Nei pazienti con MNM ilsonno rappresenta, quindi, un momento partico-larmente critico a causa dell’ipotono già presenteper la patologia di base.
Ossigenoterapia
L’insufficienza respiratoria può essere secondaria apatologia del parenchima polmonare o dalle vieaeree; in tal caso può essere consigliata una sup-plementazione di ossigeno. Nei casi di ipoventilazione la somministrazione diossigeno comporta IL rischio di arresto respira-torio per l’inefficienza del drive respiratorio daipercapnia.
La ventilazione non invasiva a pressione di supporto positiva a lungo termine
La ventilazione non invasiva a pressione di sup-porto positiva a lungo termine (NoninvasivePositive Pressure Ventilation, NIPPV) è ormai utiliz-zata largamente nei pazienti con patologia neuro-muscolare cambiando la storia naturale di questemalattie in ambito pediatrico [9].Gli obiettivi dell’NIPPV in questo gruppo dipazienti sono i seguenti:• miglioramento degli scambi gassosi;• aumento della sopravvivenza;• riduzione delle infezioni;• riduzione della degenza in ospedale;• riduzione del numero delle ospedalizzazioni [10].Tale metodica di ventilazione ha mostrato dei van-taggi sia nel controllo di problematiche acute chenella gestione domiciliare dei pazienti per MNMnel lungo termine [11].
Chiarini Testa, et al. 8

9Manifestazioni respiratorie nelle patologie neuromuscolari
In Letteratura, i criteri per l’inizio dell’NIPPV nonsono univoci. In caso di ipoventilazione alveolarecon ipercapnia diurna e/o notturna [10] il ricorsoa questo tipo di ventilazione è improrogabile.L’NIPPV favorisce, anche in assenza d’ipoventila-zione, il risparmio energetico riducendo il lavororespiratorio, il rischio di atelettasie e la ricorrenzadi infezioni.La ventilazione non invasiva (Non InvasiveVentilation, NIV) è efficace anche nella patologiaacuta, sia nell’assistenza in casi di lieve entità sianel promuovere l’estubazione precoce deipazienti [10].L’NIPPV viene utilizzata anche in assenza d’ipo-ventilazione conclamata a scopo preventivo comeuna forma di “insufflatore” di aria a fini fisioterapi-ci. Il suo uso comporta la riduzione del rischio diatelettasie, migliora la compliance polmonare epreviene le deformità toraciche con conseguentemigliore sviluppo polmonare [12].L’indicazione principale dell’NIPPV resta comun-que la correzione dell’ipoventilazione alveolarenel sonno. Diversi studi hanno dimostrato che ilricorso all’NIPPV notturna riduce anche l’ipercap-nia diurna. Il miglioramento degli scambi gassosiriduce il numero di riacutizzazioni respiratorie e leconseguenti ospedalizzazioni riducendo quindi lamorbidità legata a queste patologie [13].Una volta raggiunta una buona complianceall’NIPPV, questa dovrebbe essere verificata conuno studio di monitoraggio notturno (polisonno-grafico, esame poligrafico, monitoraggio della pul-sossimetria e capnometria notturna) per verifica-re l’adeguatezza del setting ventilatorio che puòvariare da paziente a paziente e da ventilatore aventilatore [14-15].Quando l’NIV è iniziata precocemente, come nelcaso di quasi tutte l’MNM, è possibile la deforma-zione delle strutture ossee facciali con ipoplasiaossea determinata dalla pressione prolungataesercitata dall’interfaccia. Nei bambini si determi-na spesso un’alterazione delle proporzioni delviso che determina difficoltà di alimentazione.Utilizzare quindi interfacce adeguate e soprattut-to avere l’accortezza di alternarle per variare lasuperficie di appoggio è una delle sfide dell’NIPPVe del progresso delle aziende produttrici dimaschere per NIPPV. Generalmente le mascherenasali o le olive nasali sono tra le interfacce piùconfortevoli e quindi più utilizzate nei pazientipediatrici con MNM. Esistono inoltre maschere
naso-buccali e full-face ma che nell’MNM nonpossono essere utilizzate per il rischio d’inalazio-ne durante eventuali vomiti nei pazienti che nonpossono liberarsi autonomamente dell’interfaccia.Non esistono maschere adeguate per ognipatologia, ma esiste la maschera migliore perogni paziente. Recentemente sono state resedisponibili in Letteratura indicazioni per la scel-ta delle maschere in base all’età e alla patologiadel bambino [16].Da un punto di vista tecnico, si ricorda chel’NIPPV per definizione è un sistema di ventilazio-ne con “perdite” che richiede, per un correttofunzionamento, il controllo del tono mandibolarenel sonno per prevenire perdite aeree eccessivedurante la fase di insufflazione del ventilatore. Atal fine, si usano spesso le mentoniere che per-mettono di evitare le perdite eccessive e consen-tono una corretta ventilazione.L’NIPPV rappresenta un supporto alla ventilazio-ne spontanea ed è pertanto necessaria l’intera-zione tra paziente e ventilatore. Un’interazioneperfetta non è realizzabile e possono sopraggiun-gere effetti collaterali che, se non gestiti corretta-mente, possono determinare eventi respiratori(asincronie, perdite, apnee ostruttive, miste e cen-trali). Tali eventi derivanti dall’interazione paziente-ventilatore sono stati recentemente descritti e leloro conseguenze cliniche analizzate [16]. Emergeche nei pazienti pediatrici affetti da MNM, l’even-to respiratorio più frequente in NIPPV sono leasincronie senza una ripercussione clinica [14], adifferenza invece del secondo evento più fre-quente che è rappresentato le perdite aeree lequali, invece, si associano a desaturazioni in quan-to invalidano una corretta ventilazione.In definitiva, l’NIPPV è una tecnica che richiedemolta compliance da parte del paziente e dellafamiglia che però è spesso semplice da ottenerein quanto comporta un miglioramento soggettivoe oggettivo immediato. L’NIPPV efficace correggel’ipoventilazione notturna, l’ipercapnia diurna eriduce i sintomi associati.Nei bambini con un’insufficienza muscolare seve-ra, l’insufficienza respiratoria con ipercapnia puòesordire durante il sonno, dunque l’NIPPV not-turna non è sufficiente. Quando l’NIV è necessa-ria per più di sedici/ventuno ore, una tracheosto-mia con ventilazione invasiva è la scelta migliore.Nei pazienti con MNM si possono utilizzaremodalità di ventilazione sia pressumetrica sia

Chiarini Testa, et al. 10
volumetrica; non esiste al momento nessuna indi-cazione di una modalità specifica per ogni tipo dipatologia. Le modalità utilizzate più di frequente sono:• quelle in cui l’atto inspiratorio è innescato dalpaziente tramite un sensore (trigger) e poi l’attorespiratorio è supportato dal ventilatore per sop-perire all’insufficienza ventilatoria senza però con-trollarne la durata inspiratoria (spontaneous/timed,assist/control oppure pressure-control ventilation);• oppure quelle in cui anche la durata dell’attoinspiratorio è gestita dal paziente (pressure-supportventilation).La disponibilità di più modalità di ventilazione per-mette di trovare quella migliore per ogni pazienteottenendo la migliore sincronia possibile trapaziente e ventilatore, riducendo al minimo il lavo-ro muscolare respiratorio del paziente.L’NIPPV dovrebbe erogare una differenza di pres-sione tra i due livelli inspiratori ed espiratori (pres-sione inspiratoria, Inspiratory Positive AirwayPressure, IPAP e pressione di fine espirazione,Expiratory Positive Airway Pressure, EPAP) di almeno10 cm H2O, alcuni studi suggeriscono la possibilitàdi arrivare fino a 25 cm H2O [13] per fornire un’a-deguata ventilazione con massimo reclutamentoalveolare con prevenzione delle atelettasie emigliorata compliance polmonare.
La ventilazione meccanica invasiva
La ventilazione domiciliare può essere fornita oltreche con l’NIPPV anche via tracheostomia. Anche seoggi si tenta di evitare e/o procrastinare il più possi-bile il ricorso a questa metodica, in passato era l’in-terfaccia più utilizzata per la ventilazione anche per ipazienti pediatrici. Con l’avvento dell’NIV il ricorso aquesto tipo di ventilazione si è ridotto.Per ricorrere all’NIV è necessaria la presenza diuna tosse efficace spontanea o assistita e il pazien-te deve poter chiudere la glottide che dipendedall’integrità della funzione bulbare. Se è presentela compromissione di questi come nelle formebulbari della SLA, l’NIV spesso non si dimostra effi-cace e bisogna prendere in considerazione la tra-cheotomia precoce.
Conclusioni
Non esiste, al momento, una terapia in grado didebellare l’MNM. Ma è possibile rallentarne l’evo-luzione, solo con un trattamento multidisciplinaredi supporto, diretto all’assistenza respiratoria,nutrizionale, ortopedico ed alla disostruzione dellevie aeree, mediante la fisioterapia respiratoria e laventilazione. Queste misure hanno permesso dimigliorare sensibilmente la qualità di vita deipazienti limitando le conseguenze della malattia eprolungandone la sopravvivenza.

11Manifestazioni respiratorie nelle patologie neuromuscolari
1. Mercuri E, Bertini E, Iannaccone S. Childhood spinalmuscular atrophy: controversies and challenges. LancetNeurol 2012; 11 (5): 443-452.
2. Mesfin A, Sponseller PD, Leet AI. Spinal MuscularAtrophy: Manifestations and Management. J Am AcadOrthop Surg 2012; 20: 393-401.
3. D’Amico A, Mercuri E, Tiziano FD, et al. Spinal mus-cular atrophy. Orphanet J Rare Dis 2011, 6: 71.
4. Hamilton G, Gillingwater TH. Spinal muscular atrophy:going beyond the motor neuron.Trends Mol Med 2013; 19(1): 40-50.
5.Hull J, Aniapravan R, Chan E, et al. British Thoracic Societyguideline for respiratory management of children with neuromuscular weakness.Thorax 2012; 67 (Suppl. 1): i1-i40.
6. Eagle M, Bourke M, Bullock R, et al. ManagingDuchenne muscular dystrophy – The additive effect ofspinal surgery and home nocturnal ventilation in improv-ing survival. Neuromuscul Disord 2007; 17 (6): 470-475.
7. Perrin C, Unterborn JN, Ambrosio CD, et al.Pulmonary complications of chronic neuromuscular dis-eases and their management. Muscle Nerve 2004; 29: 5.
8.Wang CH, Finkel RS, Bertini ES, et al. Participants ofthe International Conference on SMA Standard of Care. JChild Neurol 2007; 22 (8): 1027-1049.
9. Gregoretti C, Ottonello G, Chiarini Testa MB, et al.Survival of Patients With Spinal Muscular Atrophy Type 1.Pediatrics 2013; 131 (5): e1509-e1514.
10. Dohna-Schwake C, Podlewski P, Voit T, et al. Non-invasive ventilation reduces respiratory tract infections inchildren with neuromuscular disorders. Pediatr Pulmonol2008; 43: 67-71.
11. Katz S, Selvadurai H, Keilty K, et al. Outcome of non-invasive positive pressure ventilation in paediatric neuro-muscular disease. Arch Dis Child 2004; 89: 121-124.
12. Chatwin M, Bush A, Simonds AK. Outcome of goal-directed non-invasive ventilation and mechanical insuffla-tion/exsufflation in spinal muscular atrophy type I. ArchDis Child 2011; 96: 426-432.
13. Ottonello G, Mastella C, Franceschi A, et al. SpinalMuscular Atrophy Type 1: Avoidance of Hospitalization byRespiratory Muscle Support. Am J Phys Med Rehabil2011; 90 (11): 895-900.
14. Caldarelli V, Borel JC, Khirani S, et al. Polygraphic res-piratory events during sleep with noninvasive ventilation inchildren: description, prevalence, and clinical consequences.Intensive Care Med 2013; 39 (4): 739-746.
15. Petrone A, Pavone M, Chiarini Testa MB, et al.Noninvasive ventilation in children with spinal muscularatrophy types 1 and 2. Am J Phys Med Rehabil 2007; 86:216-221.
16. Ramirez A, Delord V, Khirani S, et al. Interfaces forlong-term noninvasive positive pressure ventilation in chil-dren. Intensive Care Med 2012; 38: 655-662.
BIBLIOGRAFIA
Bibliografia

Epidemiologia e eziologia
La bronchiolite obliterante (BO) o bronchiolitecostrittiva è un disordine cronico secondario a undanno severo a carico delle basse vie aeree.Una varietà di fattori possono indurre lesioni epi-teliali e causare lo sviluppo della BO, come l’inala-zione di fumi/gas tossici (soprattutto NO2) e dis-ordini sistemici quali le malattie del tessuto con-nettivo. Inoltre, può insorgere come grave compli-canza a seguito di trapianto polmonare e di allo-trapianto di midollo osseo considerati, rispettiva-mente, come una forma di rigetto cronico e direazione del trapianto verso l’ospite. Altre condi-zioni associate alla BO sono la sindrome diSteven-Johnson e le bronchiectasie [1-5]. Leforme idiopatiche colpiscono in particolare il sessofemminile in età adulta e sono molto rare, ma pro-babilmente sottostimate perché diagnosticatecome broncopneumopatia cronica ostruttiva [6].Nel bambino, la BO si riscontra più comunemente
dopo un episodio di polmonite e/o bronchioliteacuta ed è considerata una sequela a lungo termi-ne di un’infezione delle basse vie aeree, solitamen-te virale, contratta entro i primi anni di vita, oppu-re dopo trapianto di cellule staminali emopoieti-che (Hematopoietic Stem Cell Transplantation,HSCT) come espressione polmonare della reazio-ne del trapianto verso l’ospite (Graft-Versus-HostDisease, GVHD). I dati relativi a trapianti d’organosu casistiche in età pediatrica indicano un’inciden-za variabile compresa tra il 2 e l’8% [7-9].Nel bambino immunocompetente, la BO post-infettiva (PBO) è tra le pneumopatia diffuse piùfrequenti ed è la forma più comune di BO [3].Sebbene la prevalenza non sia nota, il Registro perle Malattie Polmonari Orfane della SocietàToracica Britannica (British Thoracic Society) stimaun’incidenza di 1,3 casi per 100.000 bambini al disotto dei sedici anni di età [10].
Salvatore Cazzato, Luca Bertelli, Emanuela di Palmo, Andrea Pession
Unità Operativa di Pediatria, Azienda Ospedaliero-Universitaria di Bologna, Policlinico “Sant’Orsola-Malpighi”, Bologna
12
La bronchiolite obliteranteBronchiolitis obliterans
Parole chiave: bronchiolite obliterante, tosse cronica, bronchiectasie, test di funzionalità respiratoria, tomografia computerizzata
Keywords: bronchiolitis obliterans, chronic cough, bronchiectasis, respiratory function tests, computed tomography
Riassunto. La bronchiolite obliterante (BO) o costrittiva è un disordine cronico secondario a un danno severo a carico dellebasse vie aeree. Nel bambino, la BO si riscontra più comunemente dopo una polmonite e/o bronchiolite acuta, oppure dopotrapianto di cellule staminali emopoietiche come espressione polmonare della reazione del trapianto verso l’ospite. Il patoge-no universalmente riconosciuto come la causa principale nell’indurre la BO post-infettiva è l’Adenovirus. L’esordio della malat-tia non differisce da una bronchiolite acuta. La progressione verso un’insufficienza respiratoria con esito fatale è relativamenterara, più spesso evolve verso una forma respiratoria cronica di moderata severità caratterizzata dalla persistenza di tachipnea,wheezing, tosse e rantoli crepitanti all’auscultazione. L’ossigeno-dipendenza, quando presente, si risolve solitamente entro iprimi anni di vita e i sintomi respiratori migliorano con l’accrescimento. Tuttavia, la ridotta tolleranza all’esercizio fisico e le riacu-tizzazioni infettive tendono a persistere nella maggior parte dei casi che con il declino progressivo della funzione respiratoriarendono incerta la prognosi a lungo termine della malattia.
Accettato per la pubblicazione il 30 luglio 2013.
Corrispondenza: Salvatore Cazzato, Unità Operativa di Pediatria, Azienda Ospedaliero-Universitaria di Bologna,Policlinico “Sant’Orsola-Malpighi”, via Massarenti 11, 40138 Bologna
e-mail: [email protected]
Pneumologia Pediatrica 2013; 51: 12-19

La bronchiolite obliterante 13
Il patogeno universalmente riconosciuto come lacausa principale nell’indurre la BO è l’Adenovirused in particolare i sierotipi 3, 7, e 21 [2-3, 11].In uno studio caso-controllo [12] su un’ampia casi-stica costituita da 109 bambini è stata documentatauna precedente infezione da Adenovirus in oltre il70% dei casi. La severità dell’evento acuto è un ulte-riore fattore che contribuisce allo sviluppo dellamalattia. Infatti, il numero di bambini con bronchioli-te acuta sottoposti a ventilazione meccanica erasignificativamente superiore nel gruppo che evolveverso una successiva BO postinfettiva rispetto a chiguarisce completamente dopo l’iniziale infezioneacuta (rispettivamente 34 versus 3%, p<0,001) [12].Altri microrganismi implicati dal punto di vistaetiologico includono influenza virus, parainfluenzavirus, virus del morbillo e Mycoplasma pneumo-niae. Benché, il virus respiratorio sinciziale (VRS)sia spesso associato a bronchiolite acuta e nonmanchino descrizioni di casi che hanno sviluppatosuccessivamente la malattia, in realtà si trattava dico-infezione VRS-Adenovirus. Pertanto a tutt’ogginon vi sono evidenze per un ruolo causale del VRSnel determinare una PBO [2, 13].La PBO è considerata una condizione rara, anchese la prevalenza della malattia è stata recentemen-te stimata maggiore di quanto precedentementeriportato, in particolare nei paesi del Sud Americacome Argentina, Brasile, Cile e Uruguay [1].Inoltre, è descritta anche in Europa, Stati Uniti,India, Corea del Sud, Taiwan, Malesia, NuovaZelanda e Australia [2-5].È verosimile l’azione combinata di diversi fattori dirischio nello sviluppo della PBO; tra di essi una pre-disposizione genetica, come documentato dall’as-sociazione tra l’aplotipo HLA DR8-DQB1*0302isolato in elevata frequenza tra le popolazioni delSud America [14]. Inoltre, l’aumento della concen-trazione sierica di interleuchine (IL-6 e IL-8) e fat-tore di necrosi tumorale a (Tumor necrosis factor,TNF-a), e la riduzione del numero dei linfociti T neibambini affetti da polmoniti severe da Adenovirussono tutti elementi che suggeriscono un impor-tante ruolo della risposta immunologica nello svi-luppo della malattia [15].
Patogenesi
La BO è caratterizzata da un’obliterazione parzia-le o completa da parte di tessuto fibro-cicatrizialedel lume dei bronchioli terminali e respiratori.
L’insulto iniziale determina un danno all’epiteliodelle piccole vie aeree che induce, da parte dellasottomucosa, una proliferazione di miofibroblasti ela conseguente deposizione di tessuto di granula-zione endobronchiale con successiva evoluzioneverso lo stadio finale di fibrosi concentrica e obli-terazione delle piccole vie aeree [2, 16].Il meccanismo patogenetico non è ancora chiaroma sembra che neutrofili e linfociti abbiano un ruolochiave nella genesi della BO come documentatodalla loro presenza nel liquido di lavaggio broncoal-veolare (BronchoAlveolar Lavage, BAL) [17]. In parti-colare sono aumentati i linfociti attivati (CD3+HLA-DR+) ed il sottotipo linfocitario CD8+ [18].A tal proposito sono interessanti le osservazioniriguardanti la BO da trapianto di polmone chehanno suggerito un ruolo centrale della rispostaimmunitaria T-mediata nei confronti del collagenedi tipo V (ColV) [19].È verosimile che l’esposizione del ColV, dovuto aldanno epiteliale da chemioterapia e/o da irradiazio-ne con il suo successivo riconoscimento da partedelle cellule T possa svolgere un ruolo patogeneticonella genesi della BO dopo il trapianto. L’attivazionedelle cellule T è avviata dalle cellule presentanti l’an-tigene (APC) e la differenziazione delle cellule T in Thelper 1 (Th1), Th2, Th17 e/o cellule T regolatorie(Treg) è determinata dal profilo delle citochine pre-senti nel microambiente [20]. È stato suggerito chele cellule CD4+ si differenzino in cellule Th17 o inTreg, a seconda della presenza (Th17) o assenza(Treg) rispettivamente di IL-6 [21].Mentre le cellule Th17 sono importanti nello svi-luppo dell’infiammazione e dell’autoimmunità, leTreg, al contrario, possiedono un’attività inibitorianei confronti del rigetto dell’organo trapiantato[22]. A supporto di queste considerazioni vi è ladimostrazione di un livello più basso diCD4+CD25+Treg circolanti nei pazienti con BO[23] e di un aumento di IL-17 con riduzione deilivelli ematici periferici di Treg in modelli animalicon BO [22]. L’IL-17 induce la secrezione di IL-8[24-25] che a sua volta determina il reclutamentodi neutrofili nelle vie aeree [26-27].Il ruolo dei neutrofili nella genesi della BO è con-siderato centrale. Sono associati sia alla manife-stazione clinica della BO [28] che allo sviluppodella malattia quando presenti precocemente nelfluido del BAL [29]. In aggiunta, una loro diminu-zione si associa ad un miglioramento dei flussiespiratori [27].
Cazzato, et al. 14
Clinica e diagnosi
L’esordio della BO post-infettiva non differiscenella presentazione dei segni e sintomi da quelladi una bronchiolite acuta da VRS o altri virus. Aseguito dell’evento acuto, il distress respiratorio, isegni di ostruzione delle vie aeree e l’ipossiemiapossono persistere per settimane o addiritturaper mesi [11-12, 30]. Fattori di rischio associati adevoluzione della malattia verso una BO sono rap-presentati principalmente dall’ospedalizzazioneprolungata, dall’infezione acuta da Adenovirus, dallapresenza di aeree di consolidazione multifocalialla radiografia del torace, dall’ipossiemia, ipercap-nia e trattamento in terapia intensiva mediante laventilazione meccanica [11]. Comunque, la pro-gressione della malattia verso un’insufficienzarespiratoria con esito fatale è relativamente rara,più spesso evolve verso una forma respiratoria dimoderata severità caratterizzata dalla persistenzadi tachipnea, wheezing, tosse e rantoli crepitantiall’auscultazione [4-5] (Tabella 1). L’ipossiemia,quando presente, può persistere anche per anni.In una recente casistica su bambini con patologiacronica, la PBO è risultata essere, dopo la fibrosi
cistica (FC) e la broncodisplasia, la causa più fre-quente per trattamento domiciliare a lungo termi-ne con ossigeno, con una mediana di durata in ossi-geno-dipendenza di 2,3 anni [31]. L’evoluzioneverso l’ipertensione polmonare e il cor polmonaresecondari ad ipossiemia cronica è riportata nelleforme particolarmente severe di BO [4-5].Raramente sono riportati clubbing e ritardo o arre-sto dell’accrescimento staturo-ponderale [32].Quest’ultimo, quando presente, è dipendente dallaseverità del distress respiratorio e dallo stato nutri-zionale. In generale il recupero dell’accrescimentoavviene entro pochi anni dall’evento acuto [33].Nelle prime fasi della malattia la radiografia deltorace spesso dimostra la presenza d’ispessimen-to peribronchiale bilaterale, l’accentuazione deldisegno interstiziale, l’intrappolamento aereo edaeree focali di consolidazione. Le zone di atelec-tasia possono nel tempo andare incontro aregressione [18].Lo studio della funzione polmonare in lattanti chehanno contratto nei primi mesi di vita una graveinfezione delle basse vie aeree da Adenovirus,dimostra, già a distanza di pochi mesi (range di 3-14 mesi) un’ostruzione bronchiale severa ed irre-versibile [34]. Questo sta ad indicare che l’evolu-zione verso l’obliterazione cicatriziale dei bron-chioli è un processo estremamente rapido chelascia poco spazio per intraprendere, allo statoattuale, un eventuale trattamento efficace permodificare il decorso della malattia.La diagnosi di BO si basa su un’accurata valutazio-ne dei dati anamnestici e clinico-funzionali, in asso-ciazione alle alterazioni evidenziabili all’esameradiologico, dopo aver escluso malattie quali fibro-si cistica, discinesia ciliare, broncodisplasia e altrepatologie croniche ostruttive [35].La tomografia computerizzata ad alta risoluzione(High Resolution Computed Tomography, HR-CT)riveste un ruolo importante nella diagnosi dellamalattia (Figura 1). Il quadro classico è caratteriz-zato da zone di oligoemia a mosaico ed ispessi-menti peribronchiali associati o meno ad atelecta-sia e bronchiectasie [18, 32, 36] (cfr. Tabella 1).Quest’ultime, presenti dal 15-20% sino all’80% deicasi a seconda delle casistiche studiate, sono soli-tamente a distribuzione segmentaria. In una recen-te casistica di 79 bambini coreani affetti da bron-chiectasie, la bronchiolite obliterante è stata osser-vata nel 33% dei casi risultando come la principa-le condizione sottostante identificata [37].
Tabella 1 Rilievi clinici e radiologici alla diagnosi di bronchioli-te obliterante post-infettiva. HRCT, tomografia computerizzataad alta risoluzione. Modificata da [18].
Sintomi %
Wheezing 82%
Tosse 64%
Infezioni respiratorie ricorrenti 55%
Ridotta tolleranza allo sforzo 45%
Ipossiemia 18%
Segni %
Rantoli 100%
Wheezing 64%
Tachipnea 9%
Clubbing 9%
Scarso accrescimento 9%
HRCT %
Perfusione a mosaico 100%
Air trapping 100%
Ispessimento bronchiale 91%
Bronchiectasie 36%
Atelectasie 27%
La bronchiolite obliterante 15
Benché la specificità diagnostica dell’HR-CT siaelevata non possiede una buona sensibilità e, inol-tre, non è del tutto dirimente nella diagnosi diffe-renziale tra asma severa persistente e BO. Tuttavia,rispetto ai soggetti con asma severa, la perfusionea mosaico è significativamente associata alla BOdiagnosticata con biopsia polmonare [36]. La sin-drome di Swyer-James o di McLeod è una varian-te della PBO caratterizzata da iperdiafania preva-lentemente unilaterale del polmone, che imponel’esclusione di lesioni o corpi estranei ostruenti ibronchi principali.È stato elaborato uno score predittivo per la dia-gnosi di PBO basato su criteri selettivi che com-prende una storia clinica tipica (0/4), una prece-dente infezione delle basse vie aeree daAdenovirus (0/3) e la presenza di un pattern diperfusione a mosaico alla HR-CT (0/4). Uno scoremaggiore di 7 è predittivo per PBO con una spe-cificità del 100% e una sensibilità del 67%. Tuttavia,uno score inferiore a 7 non esclude del tutto ladiagnosi, poiché lo studio includeva solo le formepiù severe di PBO [38].L’indicazione principale all’esecuzione di una fibro-broncoscopia è la ricerca sul liquido del BAL dieventuali patogeni responsabili della fase acutadella malattia. A distanza dall’evento acuto le inda-gini microbiologiche sono spesso negative, men-tre la citologia su liquido del BAL è caratterizzatada un aumento della cellularità totale, marcata-mente di neutrofili, da un lieve aumento di linfoci-ti [17-18] e da un significativo incremento dellaconcentrazione di IL-8, un potente fattore che-miotattico dei neutrofili [39].Nel bambino più grande, durante la fase cronicadella malattia, la sintomatologia è caratterizzata daepisodi ricorrenti di tosse e wheezing e da unapersistente ridotta tolleranza all’esercizio fisicodovuta alla compromissione della funzione respi-ratoria (Tabella 2). È tipico un quadro disventilato-rio ostruttivo con marcata riduzione dei flussi espi-ratori prevalentemente a carico delle piccole vieaeree [5, 18, 30] e un aumento del volume residuoe del suo rapporto con la capacità polmonaretotale [5, 18]. La capacità di diffusione alveolo-capillare del CO (DLCO) è generalmente ridotta,mentre la DLCO corretta per il volume alveolare(DLCO/VA) è di solito nella norma [18].Nonostante, alcuni soggetti dimostrino un certogrado di risposta al broncodilatatore, la marcataostruzione tipica della malattia continua a persistere
[5, 18]. È stata descritta la presenza d’iperreattivitàbronchiale nella PBO, ma con caratteristiche diffe-renti rispetto agli asmatici. Infatti, molti soggettiaffetti sono iperresponsivi alla broncostimolazionecon metacolina, ma non con l’adenosina 5-mono-fosfato (AMP), a differenza di quanto accade aisoggetti asmatici che rispondono ad entrambe lesostanze [40].La BO post-infettiva, in maniera simile alle bron-chiectasie e alla fibrosi cistica, si comporta comeuna malattia cronica progressiva. Infatti, nel corsodegli anni si osserva un progressivo peggioramen-to della funzione respiratoria con un declinomedio annuo dell’1% del FEV1 (volume espirato-rio forzato al primo secondo), FEF25-75% (flussoespiratorio forzato tra il 25 e il 75%) e delFEV1/FVC (volume espiratorio forzato al primosecondo / capacità vitale forzata) [18]. È ipotizzabi-le che il processo infiammatorio riscontrato adistanza di anni dall’evento infettivo acuto sia lacausa di un persistente rimodellamento delle vieaeree e del conseguente deterioramento dellafunzione respiratoria.Nell’iter diagnostico la biopsia polmonare è rara-mente necessaria ed è riservata ai casi più compli-cati. Tale procedura è scarsamente sensibile acausa della distribuzione non omogenea delle tipi-che alterazioni morfologiche caratterizzate dabronchioli con fibrosi obliterante. Per di più lelesioni non sembrano correlare con la severitàdella malattia [1]. Pertanto, nell’ambito di un
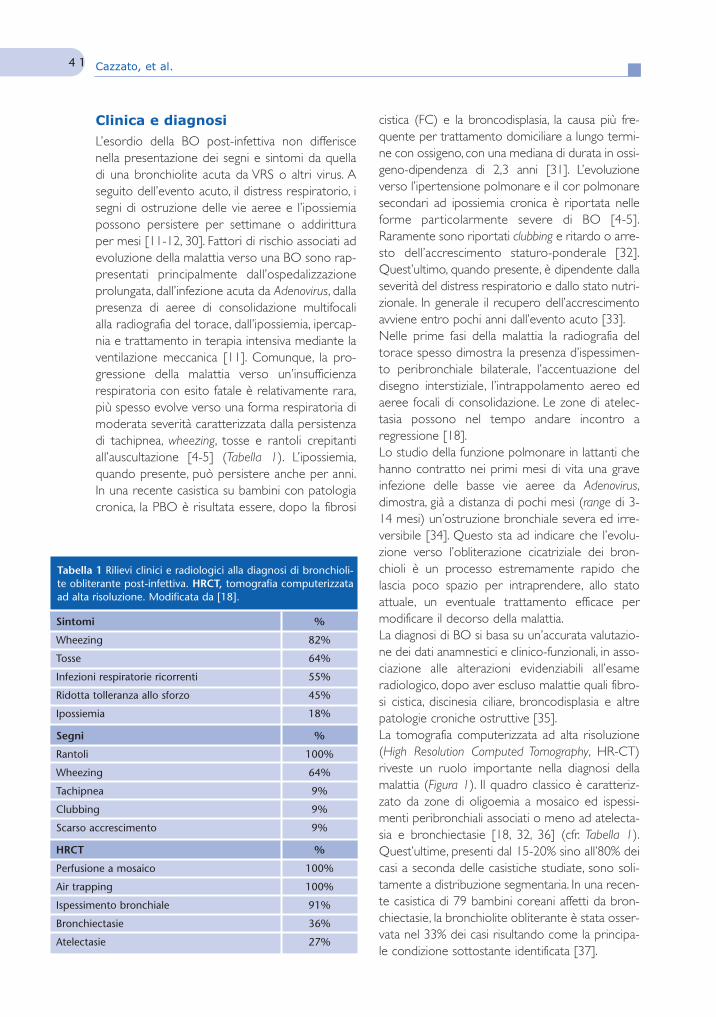
Figura 1 Tomografia computerizzata ad alta risoluzione del tora-ce. Bambina di sei anni affetta da bronchiolite obliterante post-infettiva successiva a bronchiolite acuta severa contratta nei primitre mesi di vita. Sono evidenti le tipiche alterazione della bron-chiolite obliterante caratterizzate da zone patologiche di ridottaattenuazione che si alternano a zone di aumentata densità (per-fusione a mosaico), ispessimento bronchiale e air trapping.

Cazzato, et al. 16
appropriato contesto clinico, la tomografia com-puterizzata del polmone fornisce evidenze suffi-cienti per una corretta diagnosi senza ricorrere alprelievo bioptico [32].Quando eseguito, all’esame istologico si osservain associazione alle tipiche alterazioni fibrotiche,un infiltrato infiammatorio di entità variabile cheinteressa sia la parete bronchiolare sia il tessutoperibronchiale, e un accumulo di macrofagi schiu-mosi nel lume bronchiale e nel parenchimaalveolare circostante [1-2]. L’infiltrato cellulare ècaratterizzato da linfociti T in cui prevale il sotto-tipo CD8+ [39].L’entità chiamata bronchiolite obliterante-polmo-nite organizzativa (Bronchiolitis Obliterans OrganizingPneumonia, BOOP), attualmente classificata tra lemalattie interstiziali, è un’entità distinta caratteriz-zata dall’obliterazione degli spazi aerei centrolobu-lari mediante un tessuto di granulazione prolife-rante che si estende anche alle piccole vie aereeadiacenti con la preservazione dell’architetturaparenchimale [41].Si tratta di un processo di riparazione aspecificopresente in diversi contesti clinici in parte sovrap-ponibili a quelli responsabili di una BO di tipocostrittivo (infezioni virali, malattie autoimmuni etrapianto d’organo). Nel bambino è estremamen-te rara. Sebbene siano descritti casi con formaidiopatica/criptogenetica (Cryptogenic Organisingpneumonia. COP), è quasi sempre una complican-za tardiva del trapianto di midollo osseo (OP
secondaria) [2-3]. Oltre che per le alterazioni ana-tomopatologiche si distingue dal tipo costrittivo diBO sia per le caratteristiche cliniche (quadrosimil-influenzale, tosse secca, dispnea, ipossiemia efebbre), funzionali (pattern prevalentementerestrittivo) e radiologiche (opacità alveolari easpetti infiltrativi tipo “ground glass”), sia per labuona risposta alla terapia con corticosteroidi.
Terapia e prognosi
Il trattamento della BO è principalmente di sup-porto ed include ossigenoterapia, adeguato sup-porto nutrizionale al fine di compensare leaumentate richieste caloriche dovute al lavororespiratorio, fisioterapia respiratoria e vaccinazio-ne antinfluenzale [42-43].Alcuni pazienti richiedono un periodo di ossige-noterapia a domicilio dopo l’evento infettivo acuto[11]. La fisioterapia respiratoria per la mobilizza-zione delle secrezioni è indicata soprattutto neicasi in cui è documentata la presenza di bron-chiectasie. Il ricorso a terapie per il controllo delreflusso gastro-esofageo si rende spesso necessa-rio per evitare un ulteriore insulto da acido, consi-derato che si tratta di una condizione non di radoassociata alla PBO [43].A tutt’oggi non esistono trial clinici controllati [44]e l’approccio farmacologico tende a differire tra idiversi centri. Gli steroidi sistemici sono stati utiliz-zati nelle fasi iniziali della malattia con l’obiettivo di
Tabella 2 Funzionalità respiratoria in bambini con bronchiolite obliterante post infettiva valutata in diverse casisti-che e popolazioni. BD, broncodilatatore; FEF25-75%, flusso espiratorio forzato tra il 25 e il 75%; FEV1, volume espi-ratorio forzato al primo secondo; FVC, capacità vitale forzata; n.d., non disponibile. Modificata da [49].
Autore (nazione/i, anno) [ref] n FVC (%) FEV1 (%) FEF25-75% (%) Risposta al BD, n (%)
Hardy KA, et al. (USA, 1988) [50] 7 69 41 44 n.d.
Chang AB, et al. (Australia, 1998) [51] 9 79 58 n.d. 0 (0)
Zhang L, et al. (Brasile, 2000) [30] 8 67 64 47 3 (38)
Kim CK, et al. (Corea e USA, 2001) [5] - - - - 3 (21)
Corea 5 62 35 18 -
USA 9 51 31 14 -
Mocelin HT, et al. (Brasile, 2004) [42] 19 75 57 n.d. 5 (26)
Linares M, et al. (Cile, 2004) [52] 17 59 74 33 2 (12)
Cazzato S, et al. (Italia, 2008) [18] 10 74 64 40 3 (30)
Mattiello R, et al. (Brasile e Cile, 2010) [53] - - - - n.d.
Brasile 41 62 43 20 -
Cile 36 73 50 23 -

La bronchiolite obliterante 17
attenuare il rimodellamento fibrotico nelle formesevere ossigeno-dipendenti e in corso di riacutiz-zazione infettiva. Allo scopo di attenuare gli effetticollaterali degli steroidi, sono stati proposti schemiterapeutici comprendenti la somministrazioneintravenosa di boli di metilprednisolone (30mg/kg/die sino a dose massima di 1 gr/die) per tregiorni consecutivi, ripetuti mensilmente per tre-seimesi sulla base della risposta al trattamento [45].Il ruolo delle citochine proinfiammatorie nellapatogenesi della malattia ha comportato l’uso dianticorpi monoclonali anti-TNF-a (infliximab)come trattamento di seconda linea, ottenendo lacompleta risoluzione dei sintomi in un bambinocon BO post-trapianto di midollo osseo refratta-ria agli steroidi [46]. Tuttavia, l’utilizzo di tale tera-pia non è raccomandabile, tenuti in debito conto iseri effetti collaterali del farmaco e il decorsomeno aggressivo della BO post-infettiva.La terapia antibiotica è da utilizzare in corso di riacu-tizzazioni infettive ed, in presenza di wheezing, inassociazione al broncodilatatore inalatorio nel casodi una risposta su base clinica e/o funzionale [4].
Alcuni studi supportano l’utilizzo di azitromicina,ritenuto un modulatore della risposta immunologi-ca e infiammatoria per le sue proprietà di inibire laneutrofilia delle vie aeree e la produzione di IL-8messaggera di RNA (mRNA) [47], con l’obiettivo diattenuare il declino della funzione respiratoria [48].Nei casi in cui si osserva un miglioramento dell’o-struzione delle vie aeree dopo l’uso di broncodilata-tore vi può essere indicazione a un trattamento cro-nico in associazione con uno steroide inalatorio. Iltrapianto polmonare è indicato nelle forme ossige-no-dipendenti, con notevole limitazione dell’attivitàfisica e riduzione severa dei flussi espiratori [5, 43].La mortalità è riportata nel 10% dei casi, neirestanti l’ossigeno-dipendenza, quando presente, sirisolve solitamente entro i primi anni di vita e i sin-tomi respiratori migliorano con l’accrescimento[30]. Tuttavia, in particolare la ridotta tolleranzaall’esercizio fisico e le riacutizzazioni infettive acarico delle basse vie aeree tendono a persisterenella maggior parte dei casi che con il declino pro-gressivo della funzione respiratoria rendono incer-ta la prognosi a lungo termine della malattia [18].

Cazzato, et al. 18
1. Mauad T, Dolhnikoff M. Histology of childhood bron-chiolitis obliterans. Pediatr Pulmonol 2002; 33: 466-474.
2. Kurland G, Michelson P. Bronchiolitis obliterans in chil-dren. Pediatr Pulmonol 2005; 39: 193-208.
3. Cazzato S, Bernardi F. Bronchiolar disorders in child-hood. Curr Pediatr Rev 2005; 1: 103-114.
4. Chiu CY, Wong KS, Huang YC, et al. Bronchiolitis oblite-rans in children: clinical presentation, therapy and long-termfollow-up. J Paediatr Child Health 2008; 44: 129-133.
5. Kim CK, Kim SW, Kim JS, et al. Bronchiolitis obliteransin the 1990s in Korea and the United States. Chest2001; 120: 1101-1106.
6. Ryu JH, Myers JL, Swensen SJ. Bronchiolar disorders.Am J Respir Crit Care Med 2003; 168: 1277-1292.
7. Griese M, Rampf U, Hofmann D, et al. Pulmonarycomplications after bone marrow transplantation in chil-dren: twenty-four years of experience in a single pediatriccenter. Pediatr Pulmonol 2000; 30: 393-401.
8. Wieringa J, van Kralingen KW, Sont JK, et al.Pulmonary function impairment in children followinghematopoietic stem cell transplantation. Pediatr BloodCancer 2005; 45: 318-323.
9. Duncan CN, Buonanno MR, Barry EV, et al.Bronchiolitis obliterans following pediatric allogeneichematopoietic stem-cell transplantation. Bone MarrowTransplantat 2008; 41: 971-975.
10. Laverty A, Jaffé A, Cunningham S. Establishment ofa web-based registry for rare (orphan) pediatric lung dis-eases in the United Kingdom: the BPOLD registry. PediatrPulmonol 2008; 43: 451-456.
11. Murtagh P, Giubergia V, Viale D, et al. Lower respira-tory infections by adenovirus in children. Clinical featuresand risk factors for bronchiolitis obliterans and mortality.Pediatr Pulmonol 2009; 44: 450-456.
12. Colom AJ, Teper AM, Vollmer WM, et al. Risk factorsfor the development of bronchiolitis obliterans in childrenwith bronchiolitis. Thorax 2006; 61: 503-506.
13. Massie R., Armstrong D. Bronchiectasis and bron-chiolitis obliterans post respiratory syncytial virus infection:Think again. J Paediatr Child Health 1999; 35: 497-498.
14. Teper AM, Marcos CY, Theiler G, et al. Associationbetween HLA and the incidence of bronchiolitis obliterans(BO) in Argentina. Am J Respir Crit Care Med 2004;169: A382.
15. Mistchenko AS, Koch ERR, Kajon AE, et al.Lymphocyte subsets and cytokines in adenoviral infectionin children. Acta Paediatr 1998; 87: 933-939.
16. Costa CL, Spilborghs GM, Martins MA, et al. Nitricacid-induced bronchiolitis in rats mimics childhoodBronchiolitis obliterans. Respiration 2005; 72: 642-649.
17. Bernardi F, Cazzato S, Poletti V, et al. Swyer-Jamessyndrome: bronchoalveolar lavage findings in two patients.Eur Respir J 1995; 8: 654-657.
18. Cazzato S, Poletti V, Bernardi F, et al. Airway inflam-mation and lung function decline in childhood post-infec-tious bronchiolitis obliterans. Pediatr Pulmonol 2008; 43:381-390.
19. Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predi-sposes to obliterative bronchiolitis in human lung trans-plants. J Clin Invest 2007; 117: 3498-3506.
20. Afzali B, Lombardi G, Lechler RI, et al. The role of Thelper 17 (Th17) and regulatory T cells (Treg) in humanorgan transplantation and autoimmune disease. Clin ExpImmunol 2007; 148: 32-46.
21. Bettelli E, Carrier Y, Gao W, et al. Reciprocal deve-lopmental pathways for the generation of pathogeniceffector TH17 and regulatory T cells. Nature 2006; 441:235-238.
22. Nakagiri T, Inoue M, Morii E, et al. Local IL-17 pro-duction and a decrease in peripheral blood regulatory Tcells in an animal model of bronchiolitis obliterans.Transplantation 2010; 89: 1312-1319.
23. Meloni F, Vitulo P, Bianco AM, et al. RegulatoryCD4+CD25+ T cells in the peripheral blood of lung trans-plant recipients: correlation with transplant outcome.Transplantation 2004; 77: 762-766.
24. Laan M, Linden. IL-17 as a potential target for modu-lating airway neutrophilia. Curr Pharm Des 2002; 8:1855-1861.
25. Vanaudenaerde BM, Wuyts WA, Dupont LJ, et al.Interleukin-17 stimulates release of interleukin-8 byhuman airway smooth muscle cells in vitro: a potential rolefor interleukin-17 and airway smooth muscle cells in bron-chiolitis obliterans syndrome. J Heart Lung Transplant2003; 22: 1280-1283.
26. Zheng L, Whitford HM, Orsida B, et al. The dyna-mics and associations of airway neutrophilia post lungtransplantation. Am J Transplant 2006; 6: 599-608.
27.Verleden GM, Vanaudenaerde BM, Dupont LJ, et al.Azithromycin reduces airway neutrophilia and interleukin-8 in patients with bronchiolitis obliterans syndrome. Am JRespir Crit Care Med 2006; 174: 566-570.
28. Verleden GM, Vos R, De Vleeschauwer SI, et al.Obliterative bronchiolitis following lung transplantation:from old to new concepts?Transpl Int 2009; 22: 771-779.
29. Reynaud-Gaubert M, Thomas P, Badier M, et al.Early detection of airway involvement in obliterative bron-chiolitis after lung transplantation: functional and bron-choalveolar lavage cell findings. Am J Respir Crit CareMed 2000; 161: 1924-1929.
30. Zhang L, Irion K, Kozakewich H, et al. Clinical cour-se of postinfectious bronchiolitis obliterans. PediatrPulmonol 2000; 29: 341-350.
31. Munhoz AS, Adde FV, Nakaie CM, et al. Long-termhome oxygen therapy in children and adolescents: analysis
BIBLIOGRAFIA
Bibliografia

La bronchiolite obliterante 19
of clinical use and costs of a home care program. J Pediatr(Rio J) 2011; 87: 13-18.
32. Chan PW, Muridan R, Debruyne JA. Bronchiolitisobliterans in children: clinical profile and diagnosis.Respirology 2000; 5: 369-375.
33. del Pino M, Bauer G, González Pena H, et al. Growthin post-viral chronic lung disease. Eur J Pediatr 2006; 165:845-849.
34. Teper AM, Kofman CD, Maffey AF, et al. Lung func-tion in infants with chronic pulmonary disease after seve-re adenoviral illness. J Pediatr 1999; 134: 730-733.
35. Jones MH, Pitrez PM, Stein RT, et al. Post-InfectiousBronchiolitis Obliterans. Pediatr Pulmonol 2004; 26S: 64-65.
36. Jensen SP, Lynch DA, Brown KK, et al. High resolu-tion CT features of severe asthma and bronchiolitis oblite-rans. Clin Radiol 2002; 57: 1078-1085.
37. Kim HY, Kwon JW, Seo J, et al. Bronchiectasis in chil-dren: 10-year experience at a single institution. AllergyAsthma Immunol Res 2011; 3: 39-45.
38. Colom AJ, Teper AM. Clinical prediction rule to dia-gnose post-infectious bronchiolitis obliterans in children.Pediatr Pulmonol 2009; 44: 1065-1069.
39. Koh YY, Jung da E, Koh JY, et al. Bronchoalveolar cel-lularity and interleukin-8 levels in measles bronchiolitisobliterans. Chest 2007; 131: 1454-1460.
40. Yoo Y, Yu J, Kim DK et al. Methacholine and adeno-sine 5-monophosphate challenges in children withpostinfectious bronchiolitis obliterans. Eur Respir J 2006;27: 36-41.
41. Cazzato S, Zompatori M, Baruzzi G, et al.Bronchiolitis obliterans-organizing pneumonia: an Italianexperience. Respir Med 2000; 94: 702-708.
42. Mocelin HT, Fischer GB, Iriar KL, et al. A clinical fol-low-up on Bronchiolitis Obliterans in children. Rev ChilPediatr 2004; 75: S12-S17.
43. Fischer GB, Sarria EE, Mattiello R, et al. Post infec-tious bronchiolitis obliterans in children. Paediatr RespirRev 2010; 11: 233-239.
44. Lenney W, Boner AL, Bont L, et al. Medicines usedin respiratory diseases only seen in children. Eur Respir J2009; 34: 531-551.
45. Santamaria F, Montella S, Cazzato S. Bronchiolitis obli-terans. In: Eber E, Midulla F (eds). “ERS Handbook:Paediatric Respiratory Medicine”. Sheffield (GranBretagna): European Respiratory Society 2013: 570-576.
46. Fullmer JJ, Fan LL, Dishop MK, et al. Successful treat-ment of bronchiolitis obliterans in a bone marrow trans-plant patient with tumor necrosis factor-alpha blockade.Pediatrics 2005; 116: 767-770.
47.Verleden GM, Vanaudenaerde BM, Dupont LJ, et al.Azithromycin reduces airway neutrophilia and interleukin-8 in patients with bronchiolitis obliterans syndrome. Am JRespir Crit Care Med 2006; 174: 566-570.
48. Gottlieb J, Szangolies J, Koehnlein T, et al. Long-termazithromycin for bronchiolitis obliterans syndrome afterlung transplantation. Transplantation 2008; 85: 36-41.
49. Champs NS, Lasmar LM, Camargos PA, et al. Post-infectious bronchiolitis obliterans in children. J Pediatr (RioJ) 2011; 87: 187-198.
50. Hardy KA, Schidlow DV, Zaeri N. Obliterative bron-chiolitis in children. Chest 1988; 93: 460-466.
51. Chang AB, Masel JP, Masters B. Post-infectious bron-chiolitis obliterans: clinical, radiological and pulmonaryfunction sequelae. Pediatr Radiol 1998; 28: 23-29.
52. Linares M, Meyer R, Soto G. Assessment of broncho-dilator response in post- adenovirus infection patients. RevChil Pediatr 2004; 75; S37-S44.
53. Mattiello R, Sarria EE, Stein R, et al. Functional capacityassessment in children and adolescents with post-infectiousbronchiolitis obliterans. J Pediatr (Rio J) 2008; 84: 337-343.
BIBLIOGRAFIA

Epidemiologia e fattori di rischio
Con il termine di “aspergillosi” sono indicate variecondizioni cliniche correlate ad alcune specie delgenere Aspergillus. Gli aspergilli sono funghi ubiqui-tari a lenta crescita, assai diffusi sia in ambienti out-door (terreno, fieno, paglia, foraggi, cereali) cheindoor (ospedali, condizionatori) [1-2]; essi costitui-scono le comuni muffe, di abituale osservazionenegli ambienti umidi e scarsamente soleggiati.Sebbene siano state identificate oltre 185 diversespecie, solo una ventina sono correlati allo stato dimalattia nell’uomo [3-4]. In particolare, l’Aspergillusfumigatus e l’Aspergillus niger sono le specie più fre-quentemente responsabili di patologie umane [2].Gli aspergilli colonizzano l’organismo prevalente-mente per via inalatoria; tuttavia in alcuni casi, peresempio in bambini sottoposti a cateterismovenoso, utilizzano anche la via gastrointestinale ocutanea [4-5]. In tali casi il patogeno più spesso
implicato è l’Aspergillus flavus [6]. Quando i conididel fungo vengono inalati, i macrofagi alveolari rico-noscono componenti della parete del fungomediante recettori quali toll-like, lectina legante ilmannosio e dectin-1. Inoltre rilasciano citochineche inducono il reclutamento dei neutrofili checostituiscono la principale linea di difesa contro l’a-spergillo [1, 7-9]. Nei soggetti immunocompetentiil sistema immunitario è in grado di controllare l’oc-casionale colonizzazione, ma nei soggetti immuno-compromessi si può sviluppare un’infezione invasi-va che interessa primariamente il polmone, ma conpossibilità di disseminazione anche al sistema ner-voso centrale fino al 30% dei casi [2, 5]. È pertantolo stato immunitario del paziente, insieme allecaratteristiche del parenchima polmonare, a condi-zionare lo sviluppo dello stato di malattia a livellodi quest’organo [1] (Figura 1 e Tabella 1).
Vito Terlizzi, Federica Improta, Valeria Raia
Dipartimento di Scienze Mediche Traslazionali, Università degli Studi “Federico II”, Napoli
20
La gestione dell’aspergillosi polmonarein età pediatrica
Management of pulmonary aspergillosisin children
Parole chiave: aspergillosi, terapia, gestione
Keywords: aspergillosis, terapy, management
Riassunto. L’aspergillo è un fungo ubiquitario responsabile nella specie umana di diverse condizioni cliniche, dipendenti dallostato immunitario dell’ospite e dalla struttura del parenchima polmonare. L’incremento di bambini con forme di immunodefi-cienza primaria o acquisita ha determinato un aumento della prevalenza di aspergillosi polmonare invasiva. Allo stato attualela scarsità di studi randomizzati e controllati in età pediatrica ne rendono difficile la diagnosi e il trattamento. La maggior partedei dati disponibili è elaborata da studi effettuati in età adulta. Risulta, pertanto, fondamentale la profilassi ambientale e medi-ca in caso di pazienti con fattori di rischio. In tali casi, il sospetto clinico e radiologico è sufficiente per iniziare immediatamen-te una terapia empirica con agenti antifungini. Mancano dati certi sulla terapia ottimale, l’esatta durata del trattamento e gliindici di risposta. Studi di confronto tra le diverse opzioni terapeutiche sono auspicabili in età pediatrica.
Accettato per la pubblicazione il 2 agosto 2013.
Corrispondenza: Valeria Raia, Università degli Studi “Federico II”, Dipartimento di Scienze Mediche TraslazionaliVia Pansini, 5, 80131 Napoli
e-mail: [email protected]
Pneumologia Pediatrica 2013; 51: 20-28

La gestione dell’aspergillosi polmonare in età pediatrica 21
Figura 1 Lo spettro dell’aspergillosi polmonare. Dati tratti da [1]. ° Tubercolosi, sarcoidosi, bronchiectasie, cisti bron-chiali, spondilite anchilosante, infezioni polmonari [1]. * Malnutrizione, diabete, malattia polmonare cronica ostrut-tiva, terapia corticosteroidea a basso dosaggio [1].
Nessuna sequela Aspergilloma Aspergillosinecrotizzante cronica
Aspergillosipolmonare invasiva
Aspergillosibroncopolmonare
allergica
Ospiteimmunocompetente
Presenza di cavitàpolmonari°
Malattia polmonarecronica o lieve
immunodepressione*
Ospiteimmunodepresso
AsmaFibrosi cistica
Atopia
Inalazione di spore di Aspergillus
Tabella 1 Definizioni delle principali forme di aspergillosi polmonare in età pediatrica.
Aspergillosi necrotizzante cronicaSi tratta di una condizione rara, descritta solo in alcuni casi clinici. Spesso è difficilmente distinguibile dall’asper-gilloma o può verificarsi un quadro di overlap. La diagnosi si basa sulla combinazione del quadro clinico (simileall’aspergillosi polmonare invasiva), radiologico (aree di addensamento o lesioni cavitarie) e sierologico (positivi-tà per immunoglobulina (Ig) G ed E, prick test ed espettorato positivo per aspergillo). La diagnosi di certezzarichiede la biopsia polmonare (tipiche aree di necrosi e presenza di ife). La terapia è a base di voriconazolo / amfo-tericina endovena o per os. La percentuale di mortalità è inferiore al 10%.
Aspergillosi allergica broncopolmonareÈ una condizione dovuta ad un’ipersensitività al fungo. Il sospetto clinico è d’obbligo in caso di infiltrato polmo-nare acuto in pazienti a rischio (asmatici o affetti da fibrosi cistica), associato a quadro clinico di “wheezing”, per-dita di peso, positività di IgE totali (>1.000 IU/ml) e specifiche per aspergillo. L’isolamento del fungo nell’espet-torato non è indispensabile per la diagnosi. Spesso i prick test per aspergillo risultano positivi e si associa ipereo-sinofilia. La broncoscopia non è necessaria. La terapia, rappresentata dal prednisone per os per due settimane, siassocia a miglioramento clinico, risoluzione del quadro radiografico e riduzione dei livelli di IgE ed Ig specifiche.In caso di mancata risposta è indicato l’utilizzo di itraconazolo o voriconazolo. Terapie alternative prevedonol’amfotericina B, l’omalizumab, o il metilprednisolone endovena.
AspergillomaÈ la condizione patologica più comune provocata da specie del genere Aspergillus. Consiste nella formazione diun accumulo di ife, cellule infiammatorie, fibrina e muco in una pre-esistente cavità. Il fungo raramente invade ilcircostante parenchima polmonare o i vasi sanguigni. Molto spesso il quadro è asintomatico, ma possono verifi-carsi anche casi di emottisi massiva, specialmente in pazienti immunocompromessi. Gli esami radiologici con-sentono la visualizzazione della lesione. Al contrario l’isolamento del fungo nell’espettorato si ottiene in circa il50% dei casi, mentre l’utilità del lavaggio broncoalveolare è variabile. La lesione rimane stabile nella maggiorparte dei casi, ma talvolta può diminuire di dimensioni o anche risolversi spontaneamente nel 10% dei casi. Iltrattamento medico (instillazione di agenti antifungini o itraconazolo per os) è indicato solo in casi sintomatici.In presenza di emottisi severa e ricorrente è necessaria l’embolizzazione dell’arteria bronchiale, in attesa dell’in-tervento di rimozione chirurgica (cfr. Figura 2).

Terlizzi, et al. 22
L’aspergillosi polmonare invasiva (API) rappresen-ta una problematica emergente, la cui incidenza siè molto incrementata negli ultimi anni, infatti costi-tuisce la principale causa di morbidità e mortalitànei pazienti immunocompromessi [1-2, 4-5]. Datiepidemiologici aggiornati al 2000 riportano un’in-cidenza del 3,7% in bambini con leucemia mieloi-de acuta (LMA) e del 4,5% in caso di trapianto dicellule staminali [10] negli USA. Nel 2012, invece,l’incidenza era superiore al 10% in bambini conimmunodeficienza primaria o acquisita, fino al 5%
in casi di leucemia linfatica acuta (LLA) e del 2-5%nei soggetti sottoposti a trapianto di polmone [5,11]. La neutropenia, soprattutto per valori di neu-trofili inferiori a 500 cellule/mm3 per più di diecigiorni [1-2, 5, 11], rappresenta il principale fattoredi rischio per lo sviluppo di API (Tabella 2).Inoltre, pazienti sottoposti a terapia prolungata(maggiore di tre settimane) con alte dosi di glu-cocorticoidi per patologie immunologiche o che-mioterapia per malattie tumorali [1] risultano par-ticolarmente vulnerabili all’infezione da Aspergillus.Un esteso studio multicentrico ha evidenziatocome il 60-75% dei casi di API si sviluppi in bam-bini oncologici [10-12] con tassi di mortalità cheraggiungono l’85%.Il rischio è maggiore in caso di trapianto allogeni-co rispetto al trapianto autologo e nei casi dimalattia del trapianto contro l’ospite (Graft VersusHost Disease, GVHD) severa. A tal proposito sononoti due picchi di maggiore incidenza: uno nelprimo mese successivo al trapianto ed un altro nelcorso del trattamento immunosoppressivo dellaGVHD [4]. Negli stessi pazienti, fattori che incre-mentano ulteriormente il rischio sono la nutrizio-ne parenterale, l’associazione di più antibiotici e laprolungata ospedalizzazione [4].L’API è una possibile complicanza anche in pazien-ti con infezione da virus dell’immunodeficienzaumana (Human Immunodeficiency Virus, HIV),soprattutto se la conta dei linfociti CD4 è inferiore
Tabella 2 Fattori di rischio per l’aspergillosi polmonare invasiva.
• Neutropenia prolungata (<500 cellule/mm3 per più di dieci giorni)
• Trapianti (rischio maggiore per trapianto polmonare e di cellule staminali emopoietiche)
• Terapia corticosteroidea prolungata (più di tre settimane) ad alto dosaggio
• Terapia immunosoppressiva
• Tumori ematologici (rischio maggiore per leucemia)
• Chemioterapia
• AIDS avanzata
• Malattia granulomatosa cronica
• Basso peso alla nascita
• Immunodeficienze primarie ed acquisite
• Deficit dei fagociti
• Sindromi con insufficienza midollare
• Malattie acute
• Traumi
• Malattie respiratorie croniche
D
Figura 2 Radiografia al torace evidenziante un aspergilloma pol-monare in paziente con fibrosi cistica.

La gestione dell’aspergillosi polmonare in età pediatrica 23
a cento cellule per mm3 o se è presente neutro-penia [1, 5, 11]. Per quanto riguarda le immunode-ficienze primarie, l’API può rappresentare la mani-festazione di presentazione o una frequente com-plicanza nelle prime due decadi di vita in bambinicon malattia granulomatosa cronica, condizionealla cui base vi è una disfunzione qualitativa a cari-co dei neutrofili. In tali pazienti l’aspergillo puòdeterminare, oltre al danno polmonare, ascessicerebrali o osteomielite [11, 13].Forme invasive sono state anche descritte in casodi sindrome da Iper-IgE, sindrome di Di George oimmunodeficienza comune variabile [11]. L’API,comunque, non è da considerarsi una condizioneesclusiva del bambino immunocompromesso.Sono a rischio anche pazienti con broncopneu-mopatia cronica ostruttiva o enfisema, bambiniricoverati presso unità di terapia intensiva o affet-ti da patologie croniche, anche epatiche [9, 11]. Il primo gruppo, in particolare, presenta molteplicifattori di rischio, quali una difettosa clearancemuco-ciliare, alterazioni strutturali nell’architetturadel polmone o un deficit delle proteine del surfac-tante, che sono indipendenti dall’uso cronico difarmaci o dall’ospedalizzazione prolungata [9, 11].
Diagnosi
La diagnosi di API rappresenta ancora oggi unasfida per il clinico. Nonostante l’introduzione dinuove tecniche diagnostiche, l’alto tasso di morta-lità di tale condizione è dovuto, in larga parte, al
ritardo diagnostico. Soprattutto nel bambinoimmunocompromesso, la diagnosi precoce e un’im-mediata terapia risultano fondamentali per la pro-gnosi [11, 14-15]. Una delle prime regole alle qualiattenersi è quella di elaborare un alto indice disospetto in pazienti con molteplici fattori di rischio.In caso di febbre poco responsiva alla terapia anti-biotica, tosse con espettorato e dispnea in bambiniad alto rischio, è necessario un approfondimentodiagnostico [1-2, 4, 9]. Dolore toracico con interes-samento pleurico (dovuto a piccoli infarti polmo-nari) ed emottisi, talvolta anche severa, possonoassociarsi al quadro clinico [1-2, 4, 9] di API.Quando l’infezione si dissemina al sistema nervosocentrale possono manifestarsi convulsioni o altera-zioni radiologiche compatibili con infarti cerebrali,emorragie intracraniche o ascessi epidurali [16].Non esistono specifici esami biochimici e/o stru-mentali che consentano una diagnosi di certezza.Di conseguenza, i test diagnostici dovrebberoessere effettuati in sequenza partendo da quellimeno invasivi. Il risultato di ogni esame andrà con-siderato in base allo stato clinico e immunitario delpaziente. I criteri diagnostici attualmente validatiper l’adulto [17] e utilizzati anche in recenti studipediatrici [18-19] distinguono tra API “provata”,“probabile” e “possibile” (Tabella 3).L’isolamento dell’aspergillo da campioni di espet-torato o aspirato faringeo profondo è di scarsorilievo e non si associa a conseguenze cliniche nelsoggetto immunocompetente [1]. In uno studioeffettuato su 66 adulti ospedalizzati, soltanto il
Tabella 3 Criteri diagnostici per l’aspergillosi polmonare invasiva. * Saggio antigenico positivo: riscontro di galat-tomannano nel plasma, siero, lavaggio broncoalveolare o liquor; oppure ß-D-glucano nel siero. ** Criteri clinicicompatibili con l’infezione: caratteristici infiltrati alla tomografia computerizzata (lesioni dense, ben circoscritte cono senza halo sign, air crescent sign o cavità), tracheobronchite diagnosticata mediante broncoscopia oppure infil-trati non caratteristici ma associati a specifici sintomi o segni polmonari (per es. dolore pleurico, emottisi).
Diagnosi Criteri
Certa • Valutazione istologica o citologica del tessuto polmonare con riscontro di ife all’ago aspiratoo danno tissutale alla biopsia; oppure
• Esame colturale positivo per Aspergillus su campione polmonare prelevato con procedura sterile; e
• Anomalie cliniche o radiologiche compatibili con l’infezione.
Probabile • Fattori dell’ospite (Tabella 2); e
• Presenza del fungo (microscopia positiva per Aspergillus o coltura dell’espettorato o lavaggiobroncoalveolare o saggio antigenico positivo*); e
• Criteri clinici compatibili con l’infezione**.
Possibile • Fattori dell’ospite (Tabella 2); e
• Criteri clinici compatibili con l’infezione**.

Terlizzi, et al. 24
4,5% dei soggetti con espettorato positivo peraspergillo sviluppava malattia invasiva [20]. Per talemotivo tale reperto non richiede terapia antifun-gina; tuttavia, l’esecuzione di esami diagnostici diapprofondimento sarebbe raccomandabile. In casodi pazienti immunocompromessi, invece, la positi-vità per aspergillo ha un valore predittivo positivofino al 90%, ma la negatività dell’espettorato nonesclude lo sviluppo di una forma invasiva [1].Per quanto riguarda altri parametri biochimici,quali IgE totali, IgE specifiche per aspergillo o contadegli eosinofili periferici, questi risultano di scarsosupporto per la diagnosi di API.Gli esami radiologici risultano fondamentali per ladiagnosi di API. Nonostante ciò, recenti studidimostrano come gli attuali criteri radiologici uti-lizzati in età adulta (cfr. Tabella 3) non siano appli-cabili nel bambino [12, 18]. La radiografia del tora-ce è talvolta nella norma; in caso di presenza dimultipli noduli aspecifici (fino al 35% dei casi) [5,12] si impone una diagnosi differenziale con pol-moniti virali (da Citomegalovirus o Adenovirus) obatteriche (Nocardia). Allo stesso modo la tomo-grafia assiale computerizzata (TAC) al torace adalta risoluzione, che rappresenta l’esame diImaging più utile, soprattutto per la diagnosi pre-coce, è in grado solo in una minoranza di casi diidentificare nel bambino lesioni caratteristiche, manon patognomoniche di API, quali halo sign (areadi opacità a vetro smerigliato che circonda unnodulo), air crescent sign (area di radiotrasparenzacrescente in una regione di opacità nodulare) olesioni cavitarie [12, 18, 21]. Quindi, i risultati sonospesso insoddisfacenti e sono necessari ulterioriesami per la conferma diagnostica.L’esame meno invasivo per l’identificazione d’in-fezione da aspergillo è rappresentato dal dosag-gio ematico dell’antigene galattomannano (GM).
Si tratta di un componente della parete del fungoche viene rilasciato precocemente in circolo inconseguenza della crescita dell’aspergillo [11, 22].Le Linee Guida 2009 dell’Infectious DiseasesSociety of America (IDSA) raccomandano di pra-ticare il dosaggio ematico del GM e la TAC toraceper la diagnosi precoce in soggetti ad alto rischio[23]. Il GM può essere dosato anche in altri fluidibiologici, quali il liquido di lavaggio broncoalveolare(Broncho Alveolar Lavage, BAL) o il fluido cerebro-spinale, nei rari casi di coinvolgimento neurologico,risultando più attendibile rispetto al valore emati-co [18-19]. La sensibilità e specificità del dosaggioematico dipendono, infatti, da diverse componenti,quali patologia di base, terapie in atto o cut-off uti-lizzato dal laboratorio, non esistendo al momentoun valore univoco di normalità (Tabella 4) [2].Uno studio recente ha dimostrato come il dosag-gio ematico del GM sia più attendibile in bambinioncologici (sensibilità: 91,3%; specificità: 81,7%; falsipositivi: 18%) [18]. I risultati falsamente positivisono da ricondursi all’uso di farmaci quali amoxi-cillina-acido clavulanico o piperacillina-tazobactame infezione da altri funghi quali istoplasmosi o bla-stomicosi [1-2, 11, 22]. Invece, risultati falsamantenegativi possono verificarsi in pazienti non neutro-penici, con infezioni da specie a lenta crescita qualiAspergillus terreus, in terapia antifungina o conmalattia granulomatosa cronica [1-2, 11, 22]. Èimportante sottolineare che il GM può essere tro-vato nel cibo o addirittura in alcune formule ali-mentari per lattanti. Un altro antigene di recenteutilizzo è il b-D-glucano, che però può risultarepositivo anche in caso di infezioni da Candida oforme di pneumocisti. I dati disponibili in età pedia-trica sono tuttora scarsi.Il dosaggio combinato dei due componenti diparete può dare risultati più attendibili [22, 24].
Tabella 4 Test diagnostici sierologici per la diagnosi di aspergillosi. GM, (antigene) galattomannano; PCR, reazio-ne a catena della polimerasi (Polymerase Chain Reaction); BAL, lavaggio broncoalveolare.
Test GM adulti GM bambini GM bambini PCR adulti PCR bambini (1,3)-b-glucano(%) (%) BAL (%) (%) (%) (%)
Sensibilità 71-100 79-87 82,4 67-100 63-100 63-93
Specificità 85-100 48-90 87,5 64-100 37-92 71-96
Valore predittivo positivo 54-100 15-54 82,4 46-64 17-84 52-79
Valore predittivo negativo 97-99 83 87,5 90-100 87-95 87-98
Tasso di falsi positivi 3-10 10-44 <10 45 8 10

La gestione dell’aspergillosi polmonare in età pediatrica 25
La reazione a catena della polimerasi (PolymeraseChain Reaction, PCR) su sangue o BAL potrebbeessere aggiunta agli esami per la diagnosi di asper-gillosi, sebbene non sia inclusa nei criteri diagno-stici [24]. Ciò è dovuto in parte alle diverse tecni-che utilizzate nei vari laboratori e in parte allabassa specificità che determina un alto tasso difalsi positivi (cfr. Tabella 3). Per quanto attiene l’e-secuzione di esami colturali su sangue, l’uso è limi-tato dall’alta percentuale di risultati falsamentenegativi, anche in pazienti con aspergillosi dissemi-nata [23]; solo nei casi in cui sia presente unacomponente allergica può essere utile il monito-raggio dei valori di IgE totali e specifiche peraspergillo.Metodiche più invasive ma anche più attendibilisono il BAL (su cui effettuare l’esame colturale o laricerca di GM) e la biopsia polmonare che resta almomento il gold standard per la diagnosi [1, 23].Tuttavia, quest’ultima risulta spesso difficilmentepraticabile in caso di compromesse condizioni cli-niche del paziente e per la frequente trombocito-penia associata alle forme avanzate di API. Nellamaggior parte dei casi, la biopsia polmonare vieneeffettuata per via transbronchiale una TAC-guidata,contemporaneamente alla raccolta del BAL [1, 11],con possibili complicanze quali pneumotorace,effusione pleurica e prolungata intubazione [11].Dal momento che il campione raccolto è spessoquantitativamente insufficiente e la sensibilità degliesami colturali è scarsa. Sono state introdotte tec-niche di PCR per l’identificazione su campioneistologico del DNA di aspergillo [11, 22, 24].L’analisi istologica effettuata su diciassette bambiniimmunocompromessi, in cui esami ematici, radio-logici e sul BAL non erano stati conclusivi, confer-mava la diagnosi di API in nove casi [11] mediantebiopsia polmonare. In un altro studio, la biopsiapolmonare consentiva la diagnosi nell’80% dei casi[11]. Tuttavia, la biopsia polmonare non va effet-tuata in tutti i casi ma soltanto quando proceduremeno invasive non sono state di utilità diagnostica.In definitiva, la presenza di ife nel tessuto polmona-re o una coltura positiva per aspergillo sullo stessosito o su fluidi biologici normalmente sterili consen-te la diagnosi di API in pazienti con quadro clinico oradiologico suggestivo. La diagnosi è invece sola-mente “probabile” in pazienti con molteplici fattoridi rischio ed evidenza clinica e microbiologica diinfezione. In bambini a rischio con un quadro clini-co e radiologico sospetto, la positività dell’antigene
GM su siero o BAL e la crescita del fungo nel BALsono sufficienti per diagnosi di API; in tale caso nonè richiesta l’esecuzione della biopsia polmonare.
Trattamento
La mortalità dell’API in pazienti pediatrici non trat-tati è vicina al 100% e rimane molto elevata anchein caso di terapia farmacologica aggressiva. Per talemotivo è univoca la raccomandazione di iniziare ilprima possibile una terapia antifungina su baseempirica in bambini ad alto rischio di sviluppareforme invasive di aspergillosi o comunque in corsodi valutazione diagnostica [1-2, 5, 11]. La maggiorparte degli Autori consiglia il trattamento con anti-fungini in tutti i casi di febbre di natura non chiara,persistente o ricorrente nonostante una terapiaantibiotica ad ampio spettro per oltre novantaseiore [11]. Invece, studi effettuati in età adulta riser-vano la terapia antimicotica solo ai pazienti in cui ilquadro clinico, l’imaging e il dosaggio dell’antigeneGM suggeriscono una possibile infezione fungina, alfine di limitare i costi e gli importanti effetti collate-rali dei singoli farmaci attualmente disponibili [11].In età pediatrica le maggiori difficoltà nella sceltaterapeutica sono correlate alla mancanza di studirandomizzati e controllati su ampie casistiche e didati univoci riguardo la classe di farmaco da utiliz-zare come prima scelta, gli esiti per valutare larisposta clinica e la durata della terapia. In Tabella 5sono riportati i dosaggi e gli effetti collaterali deiprincipali farmaci utilizzati nella pratica clinicapediatrica. La maggior parte dei dati tuttora dis-ponibili sono riferiti a studi effettuati in età adulta,e risultano di difficile applicazione nella pratica cli-nica in età pediatrica, considerato che il bambinopresenta caratteristiche peculiari riguardo ai fatto-ri predisponenti, al sito di infezione e soprattuttoalla farmacocinetica, agli eventi avversi e al dosag-gio dei farmaci [1-2, 11].A conferma di ciò, già nel 2007 una revisione dellaLetteratura evidenziava come solo in una minoran-za di casi la terapia antifungina utilizzata in etàpediatrica era in linea con i dati tratti dalla stessaLetteratura [2]. Per tale motivo risulta fondamenta-le effettuare una corretta profilassi sia ambientaleche medica in pazienti ad alto rischio di sviluppareAPI [4, 7, 9]. La durata della terapia antifungina nonè stata stabilita e spesso va individualizzata, soprat-tutto in base allo stato immunitario del bambino. Inuna popolazione pediatrica esaminata nel 2007 ladurata media della terapia era di 93 giorni (range

Terlizzi, et al. 26
1-880), ma sono stati descritti anche casi di pazien-ti gravemente immunocompromessi in cui la tera-pia è stata continuata indefinitamente [25].Le problematiche più rilevanti in età pediatricarestano la sequenza di farmaci da utilizzare e i para-metri clinici a cui attenersi per valutare l’efficaciadella terapia. Allo stato attuale, le opzioni per il trat-tamento di prima scelta in bambini di età uguale omaggiore ai due anni sono il voriconazolo e, piùrecentemente, le nuove formulazioni di amfoterici-na B liposomiale [2, 5, 11]. Quest’ultima è preferitaalle forme classiche di amfotericina per la riduzio-ne d’importanti effetti collaterali, e resta l’unico far-maco approvato in prima istanza per bambini dietà inferiore ai due anni [2, 9]. In caso di reazioneallergica in risposta all’infezione da aspergillo è indi-cata la terapia con steroidi a dosaggi e durata varia-bili. In età adulta le Linee Guida IDSA raccomanda-no l’utilizzo del voriconazolo, preferendo la sommi-nistrazione endovenosa [23]. Un ampio studio multicentrico randomizzato dimo-strava infatti che il gruppo in terapia con voricona-zolo presentava una più alta percentuale di risposta,maggiore sopravvivenza alla dodicesima settimana
di trattamento e minori effetti collaterali rispetto algruppo che assumeva l’amfotericina B [26].In età pediatrica invece non esiste una chiara evi-denza per preferire uno dei due farmaci, mancandostudi controllati e randomizzati di confronto.Nonostante ciò il British National Formulary forChildren consiglia l’utilizzo della amfotericina B lipo-somiale solo in caso d’intolleranza al voriconazolo[2]. Inoltre, un ampio studio effettuato su 62.842bambini in terapia con farmaci antifungini e affettida patologie tumorali, ematologiche, immunologi-che o cardiache, mostra come, nel tempo, si siaincrementato l’uso di voriconazolo rispetto all’am-fotericina B [27]. Non esistono comunque dati certisull’esatto dosaggio del voriconazolo in bambini dietà compresa tra due e dodici anni [11]. Inoltre, talefarmaco è stato approvato dall’European MedicineAgency, ma non ancora dalla U.S. Food and DrugAdministration [11]. È univoca, invece, la necessitàdi monitorare i livelli plasmatici del voriconazolo[1-2, 11] in corso di terapia.Per quanto riguarda le forme di API non responsiveai farmaci suddetti, mancano studi pediatrici su gran-di casistiche e risulta difficile stabilire il corretto
Tabella 5 Principali farmaci antifungini di utilizzo in Pediatria. BLC, Amfotericina B complesso lipidico; n.d., non disponibi-le; e.v., endovena; L-AMB, Amfotericina B liposomiale; ABCD, Amfotericina B dispersione colloidale.
Farmaco Dosaggio per fasce d’età Eventi avversi
Principio attivo 13-18 anni 2-12 anni 1-24 mesi Neonati
Voriconazolo e.v. (mg/kg/die) 8 (in 2 dosi) 14 (in 2 dosi) n.d. n.d.
Voriconazolo per os (mg/die) 400 (in 2 dosi) 400 (in 2 dosi) n.d. n.d.
Amfotericina convenzionale 1-1,5 1-1,5 1-1,5 1-1,5 • Nefrotossicitàe.v. (mg/kg/die) • Disturbi elettrolitici
• Ipersensibilità
Amfotericina formulazione 5 5 5 5 • Lieve nefrotossicitàlipidica (mg/kg/die) 3-5 3-5 3-5 3-5 • Disturbi elettrolitici
3-4 3-4 3-4 n.d.
Caspofungina (mg/m2/die) 50 (max 70); 50 (max 70); 50 (max 70); 25 • Febbregiorno 1 su 70 giorno 1 su 70 • Epatotossicità
giorno 1 su 70 • Rash cutaneo• Tachicardia• Cefalea
Posaconazolo per os (mg/die) 800 n.d. n.d. n.d. • Epatotossicità(in 2 o 4 dosi) • Nausea e vomito
• Cefalea
Itraconazolo per os (mg/kg/die) 5 (in 2 dosi) 5 (in 2 dosi) n.d. n.d. • Dolore addominale• Nausea• Rash cutaneo
• Disturbi visivi• Epatotossicità• Ipersensibilità• Rash cutaneo

La gestione dell’aspergillosi polmonare in età pediatrica 27
dosaggio sulla base di studi effettuati in età adulta. Lacaspofungina è stata approvata per il trattamentodell’API in pazienti pediatrici refrattari o intolleran-ti alla terapia di prima scelta [4, 23] ed è stata uti-lizzata anche in bambini di età inferiore ai due anni[4, 23]. Non esiste invece, al momento, il dosaggiopediatrico per la formulazione endovenosa dell’i-traconazolo e dati limitati, seppur promettenti,riguardano solo il posaconazolo [25, 28]. Ciò cherisulta evidente è che, in caso di forme refrattariedi API, è consigliabile la sostituzione con farmaci diclasse diversa, mentre la terapia combinata (caspo-fungina in associazione con voriconazolo o amfo-tericina B lisosomiale) è indicata solo come salva-vita, seppure i dati in proposito non sono univoci[2, 11].Per quanto riguarda gli outcome per valutare l’effica-cia della terapia antifungina, sono stati effettuati trestudi clinici randomizzati in età pediatrica [29-31].Nonostante i risultati attesi siano diversi, tutti con-cordano nel considerare il miglioramento clinico elo sfebbramento per almeno tre giorni consecutivicome i migliori indici di risposta alla terapia. Dati dis-cordanti riguardano invece l’utilità dell’andamentodelle lesioni radiologiche; infatti, secondo alcuniAutori, esso è indipendente dalla terapia in atto [4].Dosaggi seriati dell’antigene GM possono essereutili per il monitoraggio terapeutico in adulti e bam-bini con valori positivi alla diagnosi, mentre la dura-ta della terapia antifungina non può essere deter-minata solo dalla scomparsa dell’antigene in circolo[1, 19]. Un ulteriore supporto può derivare dalla
graduale riduzione degli indici di flogosi. Prerequisitiper sospendere la terapia includono la risoluzioneclinica, il controllo della risposta microbiologica e lareversione dello stato immunosoppressivo [1].Recentemente è stata anche valutata l’utilità di unaterapia immunostimolante con fattori di crescitaper granulociti / macrofagi o interferone gamma,combinata con la terapia antifungina [1, 11]. Non ci sono, però, allo stato attuale studi rando-mizzati che ne dimostrino l’efficacia. Infine, la rese-zione chirurgica è limitata ai casi d’invasione del-l’osso o di ascessi epidurali [32]. Può, inoltre, esse-re presa in considerazione nei casi resistenti allaterapia medica, in particolare quando la malattiacoinvolge strutture vascolari maggiori con altorischio di sanguinamenti massivi [32].Per quanto riguarda la profilassi medica il farma-co per il quale sono disponibili più evidenze inetà adulta è il posaconazolo, utilizzabile solo inpazienti di età uguale o maggiore ai tredici anni,neutropenici, affetti da forme de novo o ricorren-ti di LMA, forme ricorrenti di LLA, che hannoricevuto trapianto di cellule staminali o affetti daGVHD.Per bambini di età compresa tra due e dodici anniil voriconazolo o l’itraconazolo per via orale rap-presentano i farmaci di scelta. In pazienti di etàinferiore ai due anni o incapaci di assumere il far-maco per os, si può utilizzare l’amfotericina B liso-somiale, oppure il voriconazolo per via endove-nosa se sono invece di età uguale o superiore aidue anni [5, 11].

Terlizzi, et al. 28
1. Kousha M, Tadi R, Soubani AO. Pulmonary aspergillo-sis: a clinical review. Eur Respir Rev 2011; 20: 156-174.
2. Thomas L, Baggen L, Chisholm J, et al. Diagnosis andtreatment of aspergillosis in children. Expert Rev AntiInfect Ther 2009; 7: 461-472.
3. Lass-Florl C. The changing face of epidemiology ofinvasive fungal disease in Europe. Mycoses 2009; 52:197-205.
4. Steinbach WJ. Invasive aspergillosis in pediatricpatients. Curr Med Res Opin 2010; 26: 1779-1787.
5. Tragiannidis A, Roilides E, Walsh TJ, et al. Invasiveaspergillosis in children with acquired immunodeficiencies.Clin Infect Dis 2012; 54: 258-267.
6. Steinbach WJ, Walsh TJ. Mycoses in pediatric patients.Infect Dis Clin North Am 2006; 20: 663-678.
7. Segal BH. Aspergillosis. N Engl J Med 2009; 360:1870-1884.
8. Gersuk GM, Underhill DM, Zhu L, et al. Dectin-1 andTLRs permit macrophages to distinguish between differentAspergillus fumigatus cellular states. J Immunol 2006;176: 3717-3724.
9. Sherif R, Segal BH. Pulmonary aspergillosis: clinicalpresentation, diagnostic tests, management and complica-tions. Curr Opin Pulm Med 2010; 16: 242-250.
10. Zaoutis TE, Heydon K, Chu JH, et al. Epidemiology,outcomes, and costs of invasive aspergillosis in immuno-compromised children in the United States, 2000.Pediatrics 2006; 117: e711-e716.
11. Groll AH, Schrey D, Tragiannidis A, et al. Invasiveaspergillosis in children and adolescents. Curr Pharm Des2013; 19: 3545-3568.
12. Burgos A, Zaoutis TE, Dvorak CC, et al. Pediatricinvasive aspergillosis: a multicenter retrospective analysisof 139 contemporary cases. Pediatrics 2008; 121:e1286-e1294.
13. Van den Berg JM, van Koppen E, Ahlin A, et al.Chronic granulomatous disease: the European experience.PLoS One 2009; 4: e5234.
14. Lass-Flörl C, Roilides E, Löffler J, et al. Minireview:host defence in invasive aspergillosis. Mycoses 2013; 56:403-413.
15. Roilides E. Early diagnosis of invasive aspergillosisin infants and children. Med Mycol 2006; 44 (Supp 1):199-205.
16. Dotis J, Iosifidis E, Roilides E. Central nervous systemaspergillosis in children: a systematic review of reportedcases. Int J Infect Dis 2007; 11: 381-393.
17. De Pauw B, Walsh TJ, Donnelly JP, et al. Revised defini-tions of invasive fungal disease from the EuropeanOrganization for Research and Treatment of Cancer/InvasiveFungal Infections Cooperative Group and the NationalInstitute of Allergy and Infectious Diseases Mycoses StudyGroup (EORTC/MSG) Consensus Group. Clin Infect Dis2008; 46: 1813-1821.
18. Choi SH, Kang ES, Eo H, et al. Aspergillus galactoman-nan antigen assay and invasive aspergillosis in pediatric can-cer patients and hematopoietic stem cell transplant recipi-ents. Pediatr Blood Cancer 2013; 60: 316-322.
19. De Mol M, de Jongste JC, van Westreenen M, et al.Diagnosis of invasive pulmonary aspergillosis in childrenwith bronchoalveolar lavage galactomannan. PediatrPulmonol 2013; 48: 789-796.
20. Soubani AO, Khanchandani G, Ahmed HP. Clinicalsignificance of lower respiratory tract Aspergillus culture inelderly hospitalized patients. Eur J Clin Microbiol InfectDis 2004; 23: 491-494.
21. Thomas KE, Owens CM, Veys PA, et al. The radio-logical spectrum of invasive aspergillosis in children: A 10-year review. Pediatr Radiol 2003; 33: 453-460.
22. Roilides E, Pana ZD. Application of diagnostic mark-ers to invasive aspergillosis in children. Ann N Y Acad Sci2012; 1272: 1-8.
23.Walsh TJ, Anaissie EJ, Denning DW, et al. Treatmentof aspergillosis: clinical practice guidelines of the InfectiousDiseases Society of America. Clin Infect Dis 2008; 46:327-360.
24. Badiee P, Alborzi A, Karimi M, et al. Diagnostic poten-tial of nested PCR, galactomannan EIA, and beta-D-glucanfor invasive aspergillosis in pediatric patients. J Infect DevCtries 2012; 6: 352-357.
25. Krishna G, Sansone-Parsons A, Martinho M, et al.Posaconazole plasma concentrations in juvenile patientswith invasive fungal infection. Antimicrob AgentsChemother 2007; 51: 812-818.
26. Herbrecht R, Denning DW, Patterson TF, et al.Voriconazole versus amphotericin B for primary therapy ofinvasive aspergillosis. N Engl J Med 2002; 347: 408-415.
27. Prasad PA, Coffin SE, Leckerman KH, et al. Pediatricantifungal utilization: new drugs, new trends. Pediatr InfectDis J 2008; 27: 1083-1088.
28. Lehrnbecher T, Attarbaschi A, Duerken M, et al.Posaconazole salvage treatment in paediatric patients: a mul-ticentre survey. Eur J Clin Microbiol Infect Dis 2010; 29:1043-1045.
29. Prentice HG, Hann IM, Herbrecht R, et al. A random-ized comparison of liposomal versus conventional ampho-tericin B for the treatment of pyrexia of unknown origin inneutropenic patients. Br J Haematol 1997; 98: 711-718.
30. Maertens JA, Madero L, Reilly AF, et al. A random-ized, double-blind, multicenter study of caspofungin versusliposomal amphotericin B for empiric antifungal therapy inpediatric patients with persistent fever and neutropenia.Pediatr Infect Dis J 2010; 29: 415-420.
31. Sandler ES, Mustafa MM, Tkaczewski I, et al. Use ofamphotericin B colloidal dispersion in children. J PediatrHematol Oncol 2000; 22: 242-246.
32. Limper AH, Knox KS, Sarosi GA, et al. An officialAmerican Thoracic Society statement: Treatment of fungalinfections in adult pulmonary and critical care patients.Am J Respir Crit Care Med 2011; 183: 96-128.
BIBLIOGRAFIA
Bibliografia

Introduzione e classificazione
Le deformità della gabbia toracica sono malforma-zioni frequenti, con un’incidenza stimata di uno sutrecento, e con una spiccata prevalenza nel sessomaschile [1]. L’età dello sviluppo (preadolescenza,adolescenza) è quella in cui tali malformazioni sirendono più manifeste, ma alcune di esse sonopresenti alla nascita, a volte in forma lieve.Nonostante la loro frequenza, le deformità toraci-che spesso non vengono adeguatamente prese inconsiderazione né conosciute da parte della clas-se medica e raramente sono incluse nei program-mi universitari e post-universitari.Come conseguenza di ciò, può essere frequenteuna diagnosi ritardata e a volte una presa in cariconon corretta del problema. Ciò può essere in partedovuto alla convinzione che le deformità toracicherappresentino un problema puramente estetico enon funzionale. Effettivamente, la ragione più comu-ne per la quale i pazienti con deformità toracica si
rivolgono allo specialista è il disagio psicologicodovuto alla malformazione. Una parte di questipazienti, tuttavia, presenta disturbi più o menoaccentuati di tipo respiratorio o cardiologico. Inqualche caso, come nella sindrome di Jeune, si trat-ta di quadri molto severi e a volte fatali. In altri casi,come nella maggior parte dei pazienti con PectusExcavatum (PE), i sintomi sono più sfumati.Negli ultimi quindici anni abbiamo assistito ad unaumento vertiginoso dell’interesse sulle deformitàtoraciche, testimonato dall’incremento esponen-ziale delle pubblicazioni scientifiche su questoargomento, in parte dovuto alle nuove tecnichechirurgiche che sono state proposte. In questolavoro vengono presentati i diversi tipi di deformi-tà toracica che sono stati osservati negli ultimiventi anni presso il nostro Istituto, che rappresentaun Centro di riferimento nazionale, con particola-re riferimento ai pazienti con disturbi respiratori.
29Pneumologia Pediatrica 2013; 51: 29-38
Michele Torre, Vincenzo Jasonni
Chirurgia Pediatrica, Istituto “G. Gaslini”, Genova
Le deformità toracicheChest wall deformities
Parole chiave: pectus excavatum, pectus carinatum, sindrome di Poland, sindrome di Jeune, toracoplastica, deformità toracica,malformazione toracica, parete toracica
Keywords: pectus excavatum, pectus carinatum, Poland syndrome, Jeune syndrome, thoracoplasty, thoracic deformity, thoracic malformation, chest wall
Riassunto. Le deformità toraciche sono uno spettro molto ampio di frequenti anomalie congenite o acquisite della gabbiatoracica. In qualche caso le deformità toraciche possono causare sintomi respiratori gravi, più spesso alterazioni più lievi oppu-re solo un disagio di tipo cosmetico. Le cause delle deformità toraciche possono essere in parte legate a fattori genetici o fami-liari, ma per lo più sono sconosciute. L’interesse per questo tipo di malformazioni, in particolare per il pectus excavatum, si èmolto accresciuto negli ultimi anni anche in seguito all’introduzione di nuove tecniche chirurgiche che hanno rivoluzionato iltrattamento. In Centri di riferimento per queste deformità giungono molti pazienti con le più diverse forme, di qui nasce l’im-portanza di una precisa classificazione e definizione. Il trattamento chirurgico è indicato per molte di queste forme, e può darerisultati estremamente soddisfacenti, sia sul piano cosmetico che su quello funzionale. In questo lavoro vengono esposte, sullabase dell’esperienza di un Centro di riferimento nazionale, le più comuni deformità toraciche, presentate con particolare rife-rimento alle implicazioni respiratorie ed al possibile trattamento.
Accettato per la pubblicazione il 6 agosto 2013.
Corrispondenza: Michele Torre, Via Pirandello 12/1, 16145 Genovae-mail: [email protected]
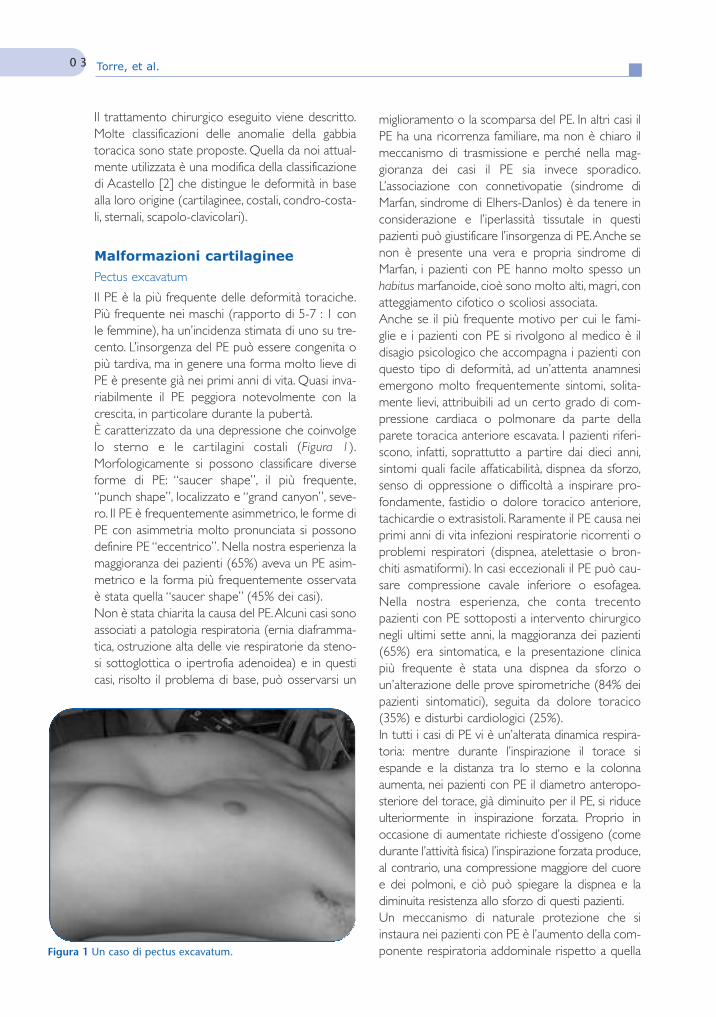
Il trattamento chirurgico eseguito viene descritto.Molte classificazioni delle anomalie della gabbiatoracica sono state proposte. Quella da noi attual-mente utilizzata è una modifica della classificazionedi Acastello [2] che distingue le deformità in basealla loro origine (cartilaginee, costali, condro-costa-li, sternali, scapolo-clavicolari).
Malformazioni cartilaginee
Pectus excavatum
Il PE è la più frequente delle deformità toraciche.Più frequente nei maschi (rapporto di 5-7 : 1 conle femmine), ha un’incidenza stimata di uno su tre-cento. L’insorgenza del PE può essere congenita opiù tardiva, ma in genere una forma molto lieve diPE è presente già nei primi anni di vita. Quasi inva-riabilmente il PE peggiora notevolmente con lacrescita, in particolare durante la pubertà.È caratterizzato da una depressione che coinvolgelo sterno e le cartilagini costali (Figura 1).Morfologicamente si possono classificare diverseforme di PE: “saucer shape”, il più frequente,“punch shape”, localizzato e “grand canyon”, seve-ro. Il PE è frequentemente asimmetrico, le forme diPE con asimmetria molto pronunciata si possonodefinire PE “eccentrico”. Nella nostra esperienza lamaggioranza dei pazienti (65%) aveva un PE asim-metrico e la forma più frequentemente osservataè stata quella “saucer shape” (45% dei casi).Non è stata chiarita la causa del PE. Alcuni casi sonoassociati a patologia respiratoria (ernia diaframma-tica, ostruzione alta delle vie respiratorie da steno-si sottoglottica o ipertrofia adenoidea) e in questicasi, risolto il problema di base, può osservarsi un
miglioramento o la scomparsa del PE. In altri casi ilPE ha una ricorrenza familiare, ma non è chiaro ilmeccanismo di trasmissione e perché nella mag-gioranza dei casi il PE sia invece sporadico.L’associazione con connetivopatie (sindrome diMarfan, sindrome di Elhers-Danlos) è da tenere inconsiderazione e l’iperlassità tissutale in questipazienti può giustificare l’insorgenza di PE. Anche senon è presente una vera e propria sindrome diMarfan, i pazienti con PE hanno molto spesso unhabitus marfanoide, cioè sono molto alti, magri, conatteggiamento cifotico o scoliosi associata.Anche se il più frequente motivo per cui le fami-glie e i pazienti con PE si rivolgono al medico è ildisagio psicologico che accompagna i pazienti conquesto tipo di deformità, ad un’attenta anamnesiemergono molto frequentemente sintomi, solita-mente lievi, attribuibili ad un certo grado di com-pressione cardiaca o polmonare da parte dellaparete toracica anteriore escavata. I pazienti riferi-scono, infatti, soprattutto a partire dai dieci anni,sintomi quali facile affaticabilità, dispnea da sforzo,senso di oppressione o difficoltà a inspirare pro-fondamente, fastidio o dolore toracico anteriore,tachicardie o extrasistoli. Raramente il PE causa neiprimi anni di vita infezioni respiratorie ricorrenti oproblemi respiratori (dispnea, atelettasie o bron-chiti asmatiformi). In casi eccezionali il PE può cau-sare compressione cavale inferiore o esofagea.Nella nostra esperienza, che conta trecentopazienti con PE sottoposti a intervento chirurgiconegli ultimi sette anni, la maggioranza dei pazienti(65%) era sintomatica, e la presentazione clinicapiù frequente è stata una dispnea da sforzo oun’alterazione delle prove spirometriche (84% deipazienti sintomatici), seguita da dolore toracico(35%) e disturbi cardiologici (25%). In tutti i casi di PE vi è un’alterata dinamica respira-toria: mentre durante l’inspirazione il torace siespande e la distanza tra lo sterno e la colonnaaumenta, nei pazienti con PE il diametro anteropo-steriore del torace, già diminuito per il PE, si riduceulteriormente in inspirazione forzata. Proprio inoccasione di aumentate richieste d’ossigeno (comedurante l’attività fisica) l’inspirazione forzata produce,al contrario, una compressione maggiore del cuoree dei polmoni, e ciò può spiegare la dispnea e ladiminuita resistenza allo sforzo di questi pazienti.Un meccanismo di naturale protezione che siinstaura nei pazienti con PE è l’aumento della com-ponente respiratoria addominale rispetto a quella
Torre, et al. 30
Figura 1 Un caso di pectus excavatum.

Le deformità toraciche 31
toracica, che riduce l’effetto negativo di cui sopra.Ciò è stato elegantemente dimostrato da studicondotti con la pletismografia optoelettronica [3],che dimostrano un aumento delle escursionirespiratorie addominali e una riduzione di quelletoraciche nei pazienti con PE prima dell’interven-to chirurgico. Questa situazione viene modificatadall’intervento chirurgico, che fa tornare i pazientiad una dinamica respiratoria più fisiologica.Nelle femmine con PE, l’asimmetria può causare almomento della pubertà un’asimmetria mammaria,dovuta principalmente alla rotazione dello sternoe al conseguente infossamento di una mammella(più spesso la destra) rispetto alla controlaterale.Qualche ragazza presenta, associata, un’ipoplasiavera e propria di una o entrambe le ghiandolemammarie.La valutazione strumentale e radiologica deipazienti con PE si basa sulla spirometria basale osotto sforzo, l’ecocardiografia o la tomografia com-puterizzata (TC). Questi esami possono metterein evidenza un quadro restrittivo (solitamentelieve), ostruttivo o misto. Nella casistica più nume-rosa al mondo [4] i valore di FVC (Forced VitalCapacity, capacità vitale forzata) e FEV1 (ForcedExpiratory Volume in the 1st second, volume espira-torio forzato nel primo secondo) medi erano piùbassi nei pazienti con PE (88% e 83% rispettiva-mente). L’ecocardiografia può evidenziare com-pressione cardiaca, prolasso della valvola mitrale.Le prove cardiorespiratorie sotto sforzo possonorisultare alterate, in particolare mostrare un indicecardiaco inferiore alla norma [5].La TC è l’esame radiologico più importante neipazienti con PE, perché oltre alla morfologia dellosterno e delle cartilagini (che può essere rico-struita in 3D) è in grado di documentare la com-pressione dello sterno sul cuore o il dislocamen-to del cuore verso sinistra. L’indice di Haller [6]che corrisponde al rapporto tra diametro toraci-co trasverso e anteroposteriore (Figura 2), è statoproposto per misurare in maniera oggettiva laprofondità della deformità. Si è determinato chenella popolazione normale l’indice di Haller sianormalmente inferiore a 2,5 e che un indice diHaller superiore a 3,2 costituisca un’indicazionealla correzione chirurgica. Nella nostra casistical’indice di Haller è stato dcompreso tra 2,7 e 39,con una media di 5,2.Il trattamento chirurgico del PE mira a riposizio-nare in posizione corretta lo sterno e le cartilagini
costali depresse, eliminando la compressione car-diaca o sugli altri visceri e aumentando il diametrotoracico antero-posteriore (Figura 3). L’interventoha anche uno scopo cosmetico, che si basa sulripristino di un normale aspetto della parete tora-cica anteriore e nelle femmine di un miglioramen-to dell’eventuale asimmetria mammaria.Diverse tecniche chirurgiche sono state propostenel corso degli anni. Schematicamente le più diffu-se sono di due tipi: a cielo aperto (con l’interven-to di Ravitch e sue successive modifiche) e mini-invasivo (con l’intervento di Nuss).Il principio dell’intervento di Ravitch [7] e delle suemodifiche è quello di asportare le cartilagini ano-male in modo da mobilizzare lo sterno anterior-mente. Lo sterno viene fissato nella nuova posi-zione con dispositivi metallici o protesici non rias-sorbibili, che in seguito potranno essere rimossi
Figura 2 Il rapporto tra diametro trasverso e diametro anteriore deltorace alla tomografia computerizzata determina l’indice di Haller.
Figura 3 Un caso di pectus excavatum al termine dell’intervento(stesso caso della figura 1).

Torre, et al. 32
una volta che le cartilagini neoformate si sianonuovamente unite con lo sterno. Nell’intervento diNuss [8], al contrario, non si esegue alcuna rimo-zione di tessuto, bensì si inserisce al di dietro dellosterno una barra concava che viene ruotata di 180gradi ed è in grado di spingere anteriormente losterno. La barra viene poi fissata alla parete toraci-ca e dopo alcuni anni rimossa.La differenza tra i due interventi è notevole: l’inter-vento a cielo aperto si esegue con un’ampia cica-trice anteriore, è piuttosto invasivo e cruento acausa dell’estesa dissezione della parete anterioredel torace, delle resezioni multiple di cartilagini, edell’osteotomia sternale; necessita di posizionamen-to di tubi di drenaggio ed ha un recupero posto-peratorio gravato da dolore e alcuni giorni di decu-bito a letto. Offre però il vantaggio di poter model-lare la parete toracica secondo quanto necessario,per cui può trattare bene casi asimmetrici, misti diPE e pectus carinatum (PC), e pazienti adulti.L’intervento di Nuss è minimamente invasivo inquanto si realizza attraverso piccole cicatrici late-rali, evitando la dissezione muscolare, la resezionedelle cartilagini e l’incisione sternale. La ripresapostoperatoria è più rapida; non consente però unottimale trattamento delle forme molto asimme-triche e dei casi adulti. Inoltre sono stati segnalatioccasionali casi di perforazione cardiaca durante ladissezione del mediastino.Quale che sia il trattamento chirurgico, pur se idati non sono univoci in tutti i lavori, la maggio-ranza degli studi recenti dimostra un beneficoeffetto della toracoplastica sulla volumetria pol-monare e nei test da sforzo cardiopolmonare, inparticolare a lungo termine, una volta rimossa labarra [5, 9-12].
Pectus carinatum
Il PC è la seconda, per frequenza, delle deformitàtoraciche, sebbene in alcune aree geografiche siafrequente quanto il PE.I disturbi respiratori in questo caso non sono quasimai presenti, sebbene il PC possa teoricamentealterare la dinamica respiratoria. In qualche caso ilPC è però una conseguenza di altre deformitàtoraciche o pneumopatie, per esempio quando ipolmoni, compressi lateralmente da una rigiditàdella gabbia toracica di altra origine (sindrome diJeune, deformità postchirurgiche, etc.), spinganoanteriormente lo sterno causandone un rimodel-lamento graduale verso una forma di PC.
Il PC classico solo in pochi casi si manifesta neiprimi anni di vita. È più comune l’insorgenza di PCdopo i dieci anni di età, in particolare durante ilpicco di crescita peripuberale. Come per il PE, lacausa del PC è sconosciuta, ma gli stessi fattori(familiari, connetivopatia, iperlassità ligamentosa,etc.) possono essere chiamati in causa. Ciò è sug-gerito dall’esistenza di famiglie con casi di PE e altridi PC, e da studi che dimostrano alterazioni cartil-ginee sia in pazienti con PE che in pazienti con PC.Il PC ha una gamma di presentazioni clinichemolto varia (Figura 4). Si distinguono da un puntodi vista della morfologia del PC le seguenti forme:• PC condrogladiolare (o tipo 1). È la forma piùcomune, caratterizzata dalla protrusione delle car-tilagini inferiori e del processo xifoideo;• PC condromanubriale (o tipo 2). È una formamolto rara, a volte confusa con la seguente, moltopiù comune;• PC con sindrome di Currarino-Silverman [13]. Piùche un’anomalia cartilaginea la componente ano-mala prevalente è a carico dello sterno, che è piùcorto e morfologicamente anomalo, simile ad una“S”, con protrusione del manubrio e depressionedella metà inferiore dello sterno. In questa formaoccorre escludere anomalie cardiache congenite;• PC unilaterale. Si presenta come una protrusio-ne di tutte o quasi le articolazioni condrosternalidi un lato.Il trattamento del PC può essere chirurgico o orto-tico. Il trattamento con bustini compressivi è effica-ce se iniziato quando il torace sia ancora compri-mibile. In questi pazienti, l’applicazione di un bustino
Figura 4 Un caso di pectus carinatum.
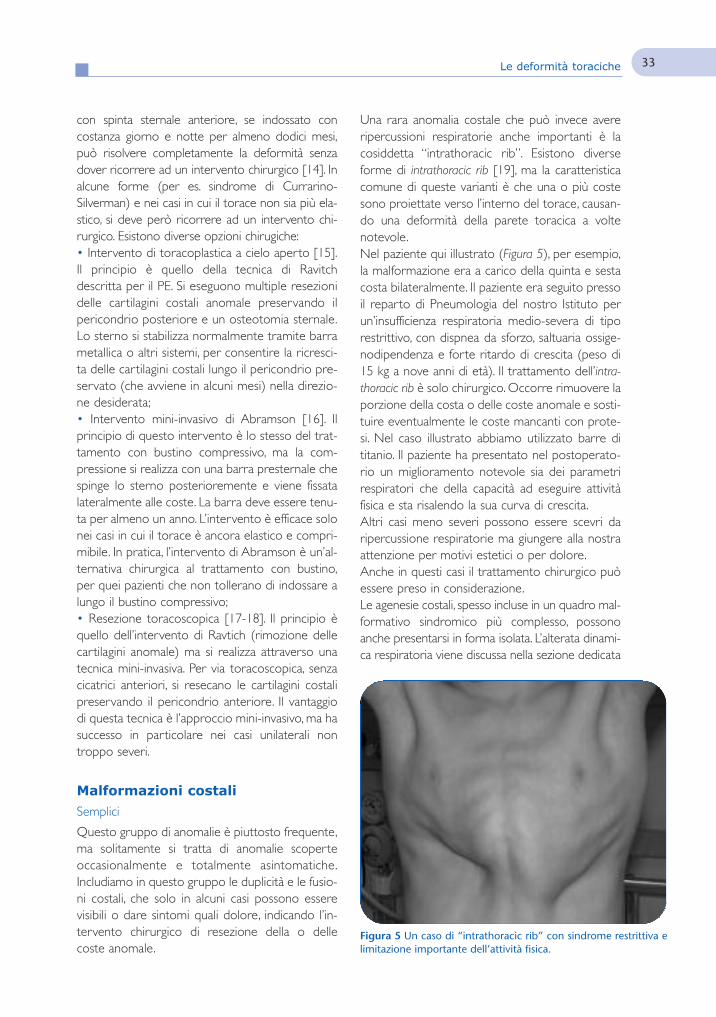
Le deformità toraciche 33
con spinta sternale anteriore, se indossato concostanza giorno e notte per almeno dodici mesi,può risolvere completamente la deformità senzadover ricorrere ad un intervento chirurgico [14]. Inalcune forme (per es. sindrome di Currarino-Silverman) e nei casi in cui il torace non sia più ela-stico, si deve però ricorrere ad un intervento chi-rurgico. Esistono diverse opzioni chirugiche:• Intervento di toracoplastica a cielo aperto [15].Il principio è quello della tecnica di Ravitchdescritta per il PE. Si eseguono multiple resezionidelle cartilagini costali anomale preservando ilpericondrio posteriore e un osteotomia sternale.Lo sterno si stabilizza normalmente tramite barrametallica o altri sistemi, per consentire la ricresci-ta delle cartilagini costali lungo il pericondrio pre-servato (che avviene in alcuni mesi) nella direzio-ne desiderata;• Intervento mini-invasivo di Abramson [16]. Ilprincipio di questo intervento è lo stesso del trat-tamento con bustino compressivo, ma la com-pressione si realizza con una barra presternale chespinge lo sterno posterioremente e viene fissatalateralmente alle coste. La barra deve essere tenu-ta per almeno un anno. L’intervento è efficace solonei casi in cui il torace è ancora elastico e compri-mibile. In pratica, l’intervento di Abramson è un’al-ternativa chirurgica al trattamento con bustino,per quei pazienti che non tollerano di indossare alungo il bustino compressivo;• Resezione toracoscopica [17-18]. Il principio èquello dell’intervento di Ravtich (rimozione dellecartilagini anomale) ma si realizza attraverso unatecnica mini-invasiva. Per via toracoscopica, senzacicatrici anteriori, si resecano le cartilagini costalipreservando il pericondrio anteriore. Il vantaggiodi questa tecnica è l’approccio mini-invasivo, ma hasuccesso in particolare nei casi unilaterali nontroppo severi.
Malformazioni costali
Semplici
Questo gruppo di anomalie è piuttosto frequente,ma solitamente si tratta di anomalie scoperteoccasionalmente e totalmente asintomatiche.Includiamo in questo gruppo le duplicità e le fusio-ni costali, che solo in alcuni casi possono esserevisibili o dare sintomi quali dolore, indicando l’in-tervento chirurgico di resezione della o dellecoste anomale.
Una rara anomalia costale che può invece avereripercussioni respiratorie anche importanti è lacosiddetta “intrathoracic rib”. Esistono diverseforme di intrathoracic rib [19], ma la caratteristicacomune di queste varianti è che una o più costesono proiettate verso l’interno del torace, causan-do una deformità della parete toracica a voltenotevole.Nel paziente qui illustrato (Figura 5), per esempio,la malformazione era a carico della quinta e sestacosta bilateralmente. Il paziente era seguito pressoil reparto di Pneumologia del nostro Istituto perun’insufficienza respiratoria medio-severa di tiporestrittivo, con dispnea da sforzo, saltuaria ossige-nodipendenza e forte ritardo di crescita (peso di15 kg a nove anni di età). Il trattamento dell’intra-thoracic rib è solo chirurgico. Occorre rimuovere laporzione della costa o delle coste anomale e sosti-tuire eventualmente le coste mancanti con prote-si. Nel caso illustrato abbiamo utilizzato barre dititanio. Il paziente ha presentato nel postoperato-rio un miglioramento notevole sia dei parametrirespiratori che della capacità ad eseguire attivitàfisica e sta risalendo la sua curva di crescita.Altri casi meno severi possono essere scevri daripercussione respiratorie ma giungere alla nostraattenzione per motivi estetici o per dolore.Anche in questi casi il trattamento chirurgico puòessere preso in considerazione.Le agenesie costali, spesso incluse in un quadro mal-formativo sindromico più complesso, possonoanche presentarsi in forma isolata. L’alterata dinami-ca respiratoria viene discussa nella sezione dedicata
Figura 5 Un caso di “intrathoracic rib” con sindrome restrittiva elimitazione importante dell’attività fisica.
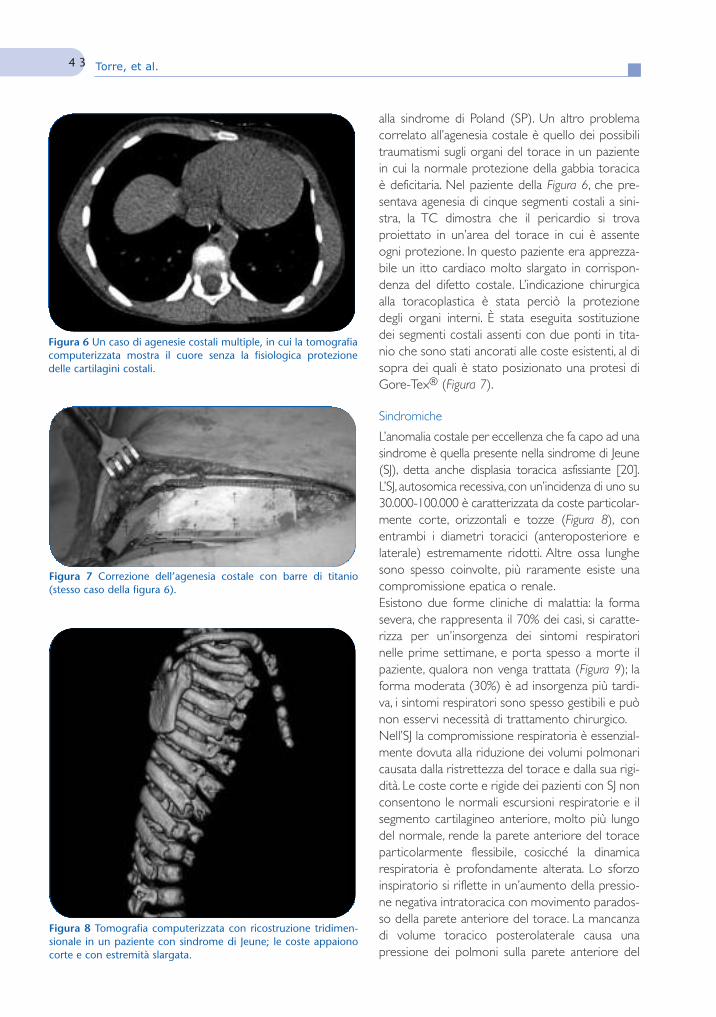
Torre, et al. 34
alla sindrome di Poland (SP). Un altro problemacorrelato all’agenesia costale è quello dei possibilitraumatismi sugli organi del torace in un pazientein cui la normale protezione della gabbia toracicaè deficitaria. Nel paziente della Figura 6, che pre-sentava agenesia di cinque segmenti costali a sini-stra, la TC dimostra che il pericardio si trovaproiettato in un’area del torace in cui è assenteogni protezione. In questo paziente era apprezza-bile un itto cardiaco molto slargato in corrispon-denza del difetto costale. L’indicazione chirurgicaalla toracoplastica è stata perciò la protezionedegli organi interni. È stata eseguita sostituzionedei segmenti costali assenti con due ponti in tita-nio che sono stati ancorati alle coste esistenti, al disopra dei quali è stato posizionato una protesi diGore-Tex® (Figura 7).
Sindromiche
L’anomalia costale per eccellenza che fa capo ad unasindrome è quella presente nella sindrome di Jeune(SJ), detta anche displasia toracica asfissiante [20].L’SJ, autosomica recessiva, con un’incidenza di uno su30.000-100.000 è caratterizzata da coste particolar-mente corte, orizzontali e tozze (Figura 8), conentrambi i diametri toracici (anteroposteriore elaterale) estremamente ridotti. Altre ossa lunghesono spesso coinvolte, più raramente esiste unacompromissione epatica o renale.Esistono due forme cliniche di malattia: la formasevera, che rappresenta il 70% dei casi, si caratte-rizza per un’insorgenza dei sintomi respiratorinelle prime settimane, e porta spesso a morte ilpaziente, qualora non venga trattata (Figura 9); laforma moderata (30%) è ad insorgenza più tardi-va, i sintomi respiratori sono spesso gestibili e puònon esservi necessità di trattamento chirurgico.Nell’SJ la compromissione respiratoria è essenzial-mente dovuta alla riduzione dei volumi polmonaricausata dalla ristrettezza del torace e dalla sua rigi-dità. Le coste corte e rigide dei pazienti con SJ nonconsentono le normali escursioni respiratorie e ilsegmento cartilagineo anteriore, molto più lungodel normale, rende la parete anteriore del toraceparticolarmente flessibile, cosicché la dinamicarespiratoria è profondamente alterata. Lo sforzoinspiratorio si riflette in un’aumento della pressio-ne negativa intratoracica con movimento parados-so della parete anteriore del torace. La mancanzadi volume toracico posterolaterale causa unapressione dei polmoni sulla parete anteriore del
Figura 6 Un caso di agenesie costali multiple, in cui la tomografiacomputerizzata mostra il cuore senza la fisiologica protezionedelle cartilagini costali.
Figura 7 Correzione dell’agenesia costale con barre di titanio(stesso caso della figura 6).
Figura 8 Tomografia computerizzata con ricostruzione tridimen-sionale in un paziente con sindrome di Jeune; le coste appaionocorte e con estremità slargata.
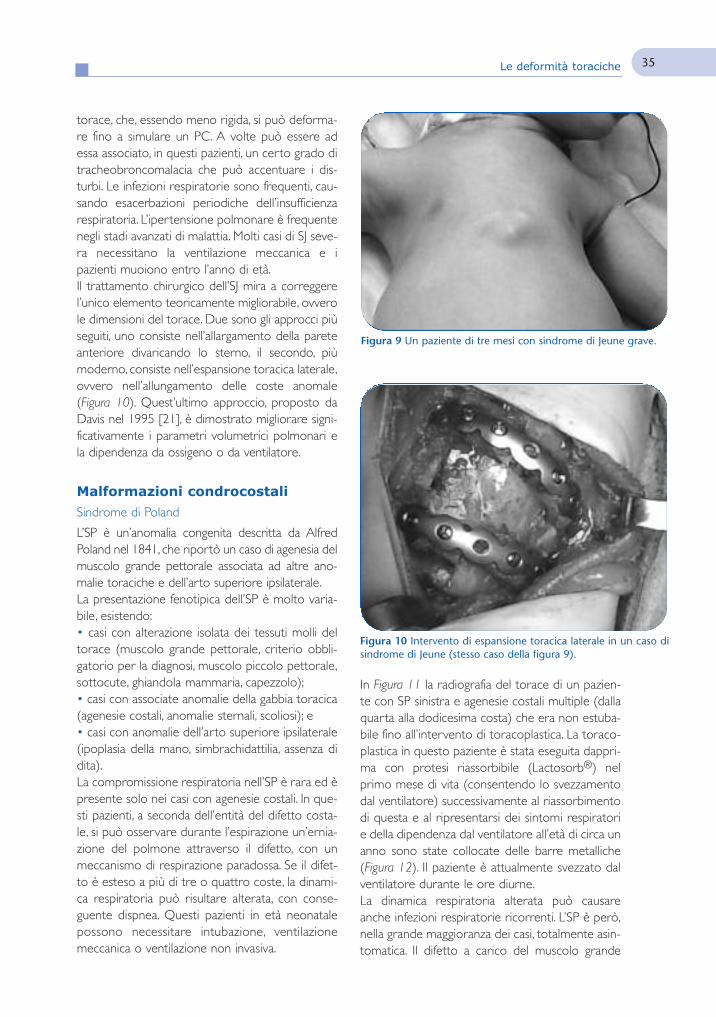
Le deformità toraciche 35
torace, che, essendo meno rigida, si può deforma-re fino a simulare un PC. A volte può essere adessa associato, in questi pazienti, un certo grado ditracheobroncomalacia che può accentuare i dis-turbi. Le infezioni respiratorie sono frequenti, cau-sando esacerbazioni periodiche dell’insufficienzarespiratoria. L’ipertensione polmonare è frequentenegli stadi avanzati di malattia. Molti casi di SJ seve-ra necessitano la ventilazione meccanica e ipazienti muoiono entro l’anno di età.Il trattamento chirurgico dell’SJ mira a correggerel’unico elemento teoricamente migliorabile, ovverole dimensioni del torace. Due sono gli approcci piùseguiti, uno consiste nell’allargamento della pareteanteriore divaricando lo sterno, il secondo, piùmoderno, consiste nell’espansione toracica laterale,ovvero nell’allungamento delle coste anomale(Figura 10). Quest’ultimo approccio, proposto daDavis nel 1995 [21], è dimostrato migliorare signi-ficativamente i parametri volumetrici polmonari ela dipendenza da ossigeno o da ventilatore.
Malformazioni condrocostali
Sindrome di Poland
L’SP è un’anomalia congenita descritta da AlfredPoland nel 1841, che riportò un caso di agenesia delmuscolo grande pettorale associata ad altre ano-malie toraciche e dell’arto superiore ipsilaterale.La presentazione fenotipica dell’SP è molto varia-bile, esistendo:• casi con alterazione isolata dei tessuti molli deltorace (muscolo grande pettorale, criterio obbli-gatorio per la diagnosi, muscolo piccolo pettorale,sottocute, ghiandola mammaria, capezzolo);• casi con associate anomalie della gabbia toracica(agenesie costali, anomalie sternali, scoliosi); e• casi con anomalie dell’arto superiore ipsilaterale(ipoplasia della mano, simbrachidattilia, assenza didita).La compromissione respiratoria nell’SP è rara ed èpresente solo nei casi con agenesie costali. In que-sti pazienti, a seconda dell’entità del difetto costa-le, si può osservare durante l’espirazione un’ernia-zione del polmone attraverso il difetto, con unmeccanismo di respirazione paradossa. Se il difet-to è esteso a più di tre o quattro coste, la dinami-ca respiratoria può risultare alterata, con conse-guente dispnea. Questi pazienti in età neonatalepossono necessitare intubazione, ventilazionemeccanica o ventilazione non invasiva.
In Figura 11 la radiografia del torace di un pazien-te con SP sinistra e agenesie costali multiple (dallaquarta alla dodicesima costa) che era non estuba-bile fino all’intervento di toracoplastica. La toraco-plastica in questo paziente è stata eseguita dappri-ma con protesi riassorbibile (Lactosorb®) nelprimo mese di vita (consentendo lo svezzamentodal ventilatore) successivamente al riassorbimentodi questa e al ripresentarsi dei sintomi respiratorie della dipendenza dal ventilatore all’età di circa unanno sono state collocate delle barre metalliche(Figura 12). Il paziente è attualmente svezzato dalventilatore durante le ore diurne.La dinamica respiratoria alterata può causareanche infezioni respiratorie ricorrenti. L’SP è però,nella grande maggioranza dei casi, totalmente asin-tomatica. Il difetto a carico del muscolo grande
Figura 9 Un paziente di tre mesi con sindrome di Jeune grave.
Figura 10 Intervento di espansione toracica laterale in un caso disindrome di Jeune (stesso caso della figura 9).
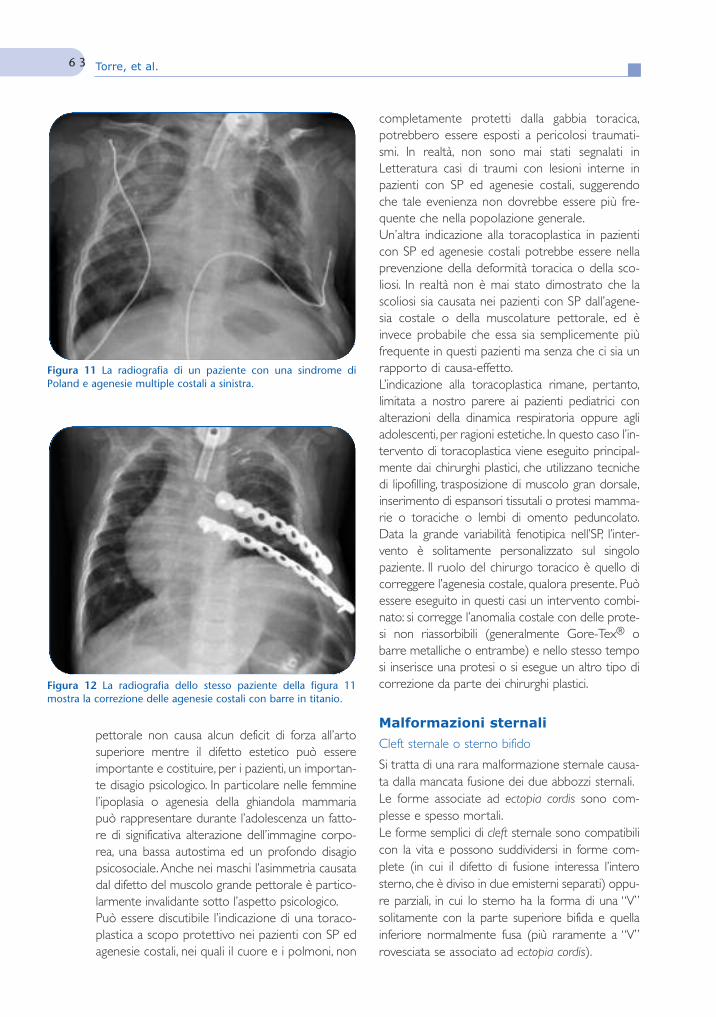
Torre, et al. 36
pettorale non causa alcun deficit di forza all’artosuperiore mentre il difetto estetico può essereimportante e costituire, per i pazienti, un importan-te disagio psicologico. In particolare nelle femminel’ipoplasia o agenesia della ghiandola mammariapuò rappresentare durante l’adolescenza un fatto-re di significativa alterazione dell’immagine corpo-rea, una bassa autostima ed un profondo disagiopsicosociale. Anche nei maschi l’asimmetria causatadal difetto del muscolo grande pettorale è partico-larmente invalidante sotto l’aspetto psicologico.Può essere discutibile l’indicazione di una toraco-plastica a scopo protettivo nei pazienti con SP edagenesie costali, nei quali il cuore e i polmoni, non
completamente protetti dalla gabbia toracica,potrebbero essere esposti a pericolosi traumati-smi. In realtà, non sono mai stati segnalati inLetteratura casi di traumi con lesioni interne inpazienti con SP ed agenesie costali, suggerendoche tale evenienza non dovrebbe essere più fre-quente che nella popolazione generale.Un’altra indicazione alla toracoplastica in pazienticon SP ed agenesie costali potrebbe essere nellaprevenzione della deformità toracica o della sco-liosi. In realtà non è mai stato dimostrato che lascoliosi sia causata nei pazienti con SP dall’agene-sia costale o della muscolature pettorale, ed èinvece probabile che essa sia semplicemente piùfrequente in questi pazienti ma senza che ci sia unrapporto di causa-effetto.L’indicazione alla toracoplastica rimane, pertanto,limitata a nostro parere ai pazienti pediatrici conalterazioni della dinamica respiratoria oppure agliadolescenti, per ragioni estetiche. In questo caso l’in-tervento di toracoplastica viene eseguito principal-mente dai chirurghi plastici, che utilizzano tecnichedi lipofilling, trasposizione di muscolo gran dorsale,inserimento di espansori tissutali o protesi mamma-rie o toraciche o lembi di omento peduncolato.Data la grande variabilità fenotipica nell’SP, l’inter-vento è solitamente personalizzato sul singolopaziente. Il ruolo del chirurgo toracico è quello dicorreggere l’agenesia costale, qualora presente. Puòessere eseguito in questi casi un intervento combi-nato: si corregge l’anomalia costale con delle prote-si non riassorbibili (generalmente Gore-Tex® obarre metalliche o entrambe) e nello stesso temposi inserisce una protesi o si esegue un altro tipo dicorrezione da parte dei chirurghi plastici.
Malformazioni sternali
Cleft sternale o sterno bifido
Si tratta di una rara malformazione sternale causa-ta dalla mancata fusione dei due abbozzi sternali.Le forme associate ad ectopia cordis sono com-plesse e spesso mortali.Le forme semplici di cleft sternale sono compatibilicon la vita e possono suddividersi in forme com-plete (in cui il difetto di fusione interessa l’interosterno, che è diviso in due emisterni separati) oppu-re parziali, in cui lo sterno ha la forma di una “V”solitamente con la parte superiore bifida e quellainferiore normalmente fusa (più raramente a “V”rovesciata se associato ad ectopia cordis).
Figura 11 La radiografia di un paziente con una sindrome diPoland e agenesie multiple costali a sinistra.
Figura 12 La radiografia dello stesso paziente della figura 11mostra la correzione delle agenesie costali con barre in titanio.
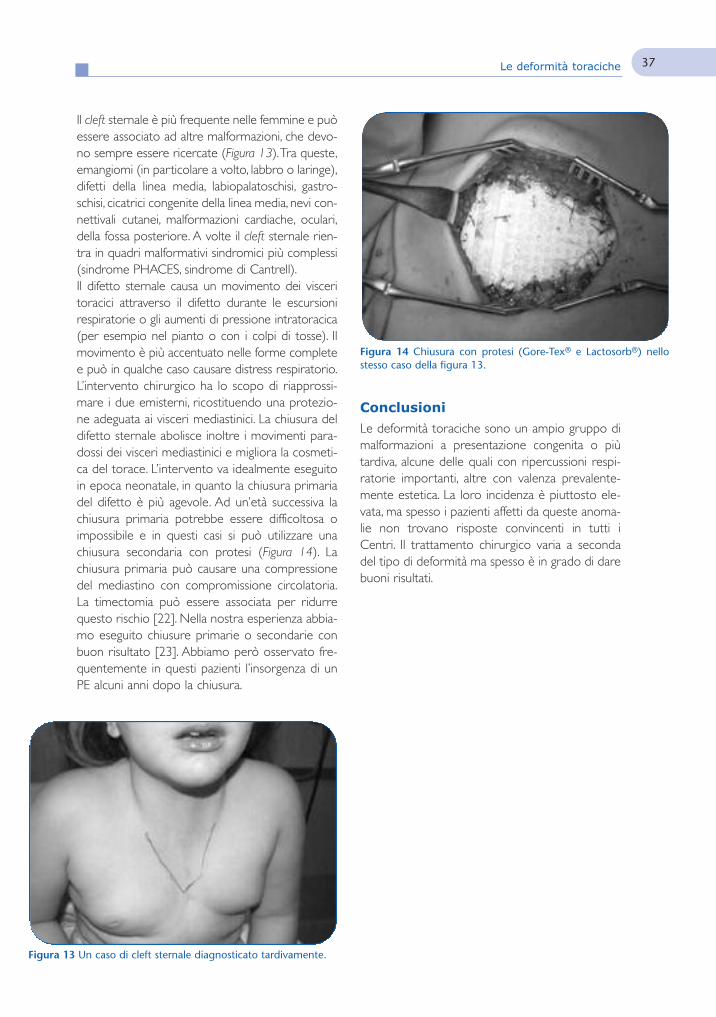
Le deformità toraciche 37
Il cleft sternale è più frequente nelle femmine e puòessere associato ad altre malformazioni, che devo-no sempre essere ricercate (Figura 13). Tra queste,emangiomi (in particolare a volto, labbro o laringe),difetti della linea media, labiopalatoschisi, gastro-schisi, cicatrici congenite della linea media, nevi con-nettivali cutanei, malformazioni cardiache, oculari,della fossa posteriore. A volte il cleft sternale rien-tra in quadri malformativi sindromici più complessi(sindrome PHACES, sindrome di Cantrell).Il difetto sternale causa un movimento dei visceritoracici attraverso il difetto durante le escursionirespiratorie o gli aumenti di pressione intratoracica(per esempio nel pianto o con i colpi di tosse). Ilmovimento è più accentuato nelle forme completee può in qualche caso causare distress respiratorio.L’intervento chirurgico ha lo scopo di riapprossi-mare i due emisterni, ricostituendo una protezio-ne adeguata ai visceri mediastinici. La chiusura deldifetto sternale abolisce inoltre i movimenti para-dossi dei visceri mediastinici e migliora la cosmeti-ca del torace. L’intervento va idealmente eseguitoin epoca neonatale, in quanto la chiusura primariadel difetto è più agevole. Ad un’età successiva lachiusura primaria potrebbe essere difficoltosa oimpossibile e in questi casi si può utilizzare unachiusura secondaria con protesi (Figura 14). Lachiusura primaria può causare una compressionedel mediastino con compromissione circolatoria.La timectomia può essere associata per ridurrequesto rischio [22]. Nella nostra esperienza abbia-mo eseguito chiusure primarie o secondarie conbuon risultato [23]. Abbiamo però osservato fre-quentemente in questi pazienti l’insorgenza di unPE alcuni anni dopo la chiusura.
Conclusioni
Le deformità toraciche sono un ampio gruppo dimalformazioni a presentazione congenita o piùtardiva, alcune delle quali con ripercussioni respi-ratorie importanti, altre con valenza prevalente-mente estetica. La loro incidenza è piuttosto ele-vata, ma spesso i pazienti affetti da queste anoma-lie non trovano risposte convincenti in tutti iCentri. Il trattamento chirurgico varia a secondadel tipo di deformità ma spesso è in grado di darebuoni risultati.
Figura 13 Un caso di cleft sternale diagnosticato tardivamente.
Figura 14 Chiusura con protesi (Gore-Tex® e Lactosorb®) nellostesso caso della figura 13.

Torre, et al. 38
1. Fokin AA, Steuerwald NM, Ahrens WA, et al.Anatomical, histologic and genetic characteristics of con-genital chest wall deformities. Semin Thorac CardiovascSurg 2009; 21: 44-57.
2. Acastello E. Patologias de la pared toracica en pedia-tria. Buenos Aires (Argentina): Editorial El Ateneo 2006.
3. Redlinger R, Wootton A, Kelly R, et al. Optoelectronicplethysmography demonstrates abrogation of regionalchest wall motion dysfunction in patients with pectusexcavatum after Nuss repair. J Pediatr Surg 2012; 47:160-164.
4. Kelly RE, Goretsky MJ, Obermeyer R, et al. Twenty-one years of experience with minimally invasive repair ofpectus excavatum by the Nuss procedure in 1215patients. Ann Surg 2010; 252: 1072-1081.
5. Tang M, Moller Nielsen HH, Lesbo M, et al. Improvedcardiopulmonary exercise function after modified Nussoperation for pectus excavatum. Eur J Cardiothorac Surg2012; 41: 1063-1067.
6. Haller JA, Kramer SS, Lietman SA. Use of CT scans inselection of patients for pectus excavatum surgery: a pre-liminary report. J Pediatr Surg 1987; 22: 904-906.
7. Ravitch MM. The operative treatment of PectusExcavatum. Ann Surg 1949; 129: 429-444.
8. Nuss D, Kelly RE, Croitoru DP, et al. A 10 year reviewof a minimally invasive technique for the correction of pec-tus excavatum. J Pediatr Surg 1998; 33: 545-552.
9. Lawson M, Mellins R, Tabangin M, et al. Impact ofpectus excavatum on pulmonary function before andafter repair with the Nuss procedure. J Pediatr Surg2005; 40: 174-180.
10. Johnson J, Hartman T, Pianosi P, et al.Cardiorespiratory function after operation for PectusExcavatum. J Pediatr 2008; 153: 359-364.
11. Sigalet D, Montgomery M, Harder J, et al. Long termcardiopulmonary effects of closed repair of pectus exca-vatum. Pediatr Surg Int 2007; 23: 493-497.
12. Neviere R, Montaigne D, Benhamed L, et al.Cardiopulmonary response following surgical repair of pec-tus excavatum in adult patients. Eur J Cardiothorac Surg2011; 40: e77-e82.
13. Currarino G, Silverman FN. Premature obliteration ofthe sterna sutures and pigeon-breast deformity. Radiology1958; 70: 532-540.
14. Martinez-Ferro M, Fraire C, Bernard S. Dynamiccompression system for thecorrection of pectus carinatum.Semin Pediatr Surg 2008; 17: 194-200.
15. Robicsek F. Surgical treatment of pectus carinatum.Chest Surg Clin N Am 2000; 10: 357-376.
16. Abramson H. A minimally invasive technique to repairpectus carinatum. Preliminary report. Arch Bronconeumol2005; 41: 349-351.
17. Kim S & Idowu O. Minimally invasive thoracoscopicrepair of unilateral pectus carinatum. J Pediatr Surg 2009;44: 471-474.
18. Varela P, Torre M. Thoracoscopic cartilage resection withpartial perichondrium preservation in unilateral pectus cari-natum: preliminary results. J Pediatr Surg 2011; 46: 263-266.
19. Kamano H, Ishihama T, Ishihama H, et al. Bifidintrathoracic rib: a case report and classification ofintrathoracic ribs. Intern Med 2006; 45: 627-630.
20. Jeune M, Carron R, Beraud C, et al.Polychondrodystrophie avec blocage thoracique d’évolutionfatale. Pediatrie 1954; 9: 390-392.
21. Davis JT, Ruberg RL, Leppink DM, et al. Lateral tho-racic expansion for Jeune’s asphyxiating dystrophy: a newapproach. Ann Thorac Surg 1995; 60: 694-696.
22. Torre M, Rapuzzi G, Guida E, et al. Thymectomy toachieve primary closure of total sternal cleft. J PediatrSurg 2008; 43: e17-e20.
23. Torre M, Rapuzzi G, Carlucci M, et al. Phenotypicspectrum and management of sternal cleft: literaturereview and presentation of a new series. Eur JCardiothorac Surg 2012; 41: 4-9.
BIBLIOGRAFIA
Bibliografia

Introduzione
L’empiema pleurico è una raccolta di liquido puru-lento nello spazio pleurico. Esso rappresenta unainfrequente complicanza della polmonite battericadell’età pediatrica. Uno studio recente riporta,peraltro, un incremento di tale complicanza: neipazienti tra due e quattro anni l’incidenza negliUSA sarebbe passata da 3,5 su 100.000 nel perio-do 1996-1998, a 10,3 su 100.000 nel periodo2005-2007 [1].Il coinvolgimento pleurico in corso di polmoni-te, viene classicamente suddiviso in tre stadiingravescenti [2]:
• Fase essudativa. Presenza di modesto versamen-to pleurico limpido con basso numero di globulibianchi (versamento parapneumonico semplice).Può risolversi con opportuna terapia o evolverein tre-quattro giorni nel successivo stadio;• Fase fibrinopurulenta. La quantità di liquidoaumenta, con una più alta concentrazione di glo-buli bianchi, il liquido è macroscopicamente opa-lescente e più denso fino al pus franco.Compaiono sottili tralci fibrinosi che evolvononella formazione di concamerazioni (versamentoparapneumonico complicato);
39Pneumologia Pediatrica 2013; 51: 39-45
Aurelio Porreca
Struttura Complessa di Chirurgia Pediatrica e d’Urgenza, Azienda Ospedaliera di Rilievo Nazionale“Santobono-Pausilipon”, Napoli
La chirurgia toracoscopicavideo-assistita nella gestione
dell’empiema pleuricoVideo Assisted Thoracic Surgery
in the managementof parapneumonic pleural empyema
Parole chiave: toracoscopia, empiema, versamento pleurico
Keywords: thoracoscopy, empyema, pleural effusion
Riassunto. L’empiema pleurico è una rara complicanza della polmonite batterica. Il trattamento di questa condizione negli ulti-mi anni è stato oggetto di numerosi studi. Data per certa l’inadeguatezza della sola terapia medica e la necessità di procederead un trattamento più aggressivo, capace di rimuovere il materiale fibrino-purulento accumulato nello spazio pleurico, sonoemerse due opzioni ugualmente efficaci. Una si basa sulla detersione chimica del cavo mediante l’uso di fibrinolitici introdottiattraverso il drenaggio pleurico, e l’altra su una detersione meccanica effettuata sotto controllo visivo in corso di toracoscopia(Video Assisted Thoracic Surgery,VATS). Numerosi sono i sostenitori delle due opzioni, ma alcuni recenti studi prospettici rando-mizzati hanno meglio chiarito le relative indicazione delle due metodiche. La fibrinolisi, in considerazione della maggiore sempli-cità di impiego e del poter essere praticata con la sola sedazione necessaria all’introduzione del drenaggio pleurico, sembra possaconsiderarsi come la metodica da mettere in atto in prima istanza. La VATS, che rappresenta una vera procedura chirurgica anchese mini-invasiva, troverebbe le sue indicazioni nei casi d’insuccesso della fibrinolisi. Non mancano peraltro i sostenitori dell’utiliz-zo precoce della VATS. Ulteriori studi sono auspicabili per chiarire ancor meglio le rispettive indicazioni.
Accettato per la pubblicazione il 3 agosto 2013.
Corrispondenza: Aurelio Porreca, via G. Gigante 204, 80128 Napoli;e-mail: [email protected]
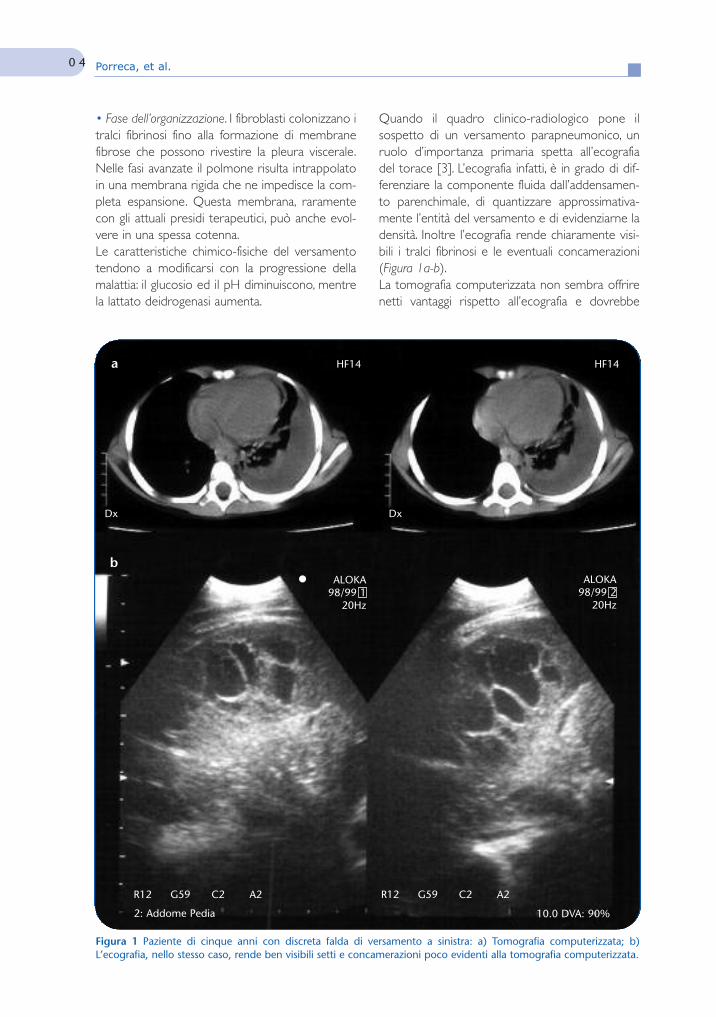
Porreca, et al. 40
• Fase dell’organizzazione. I fibroblasti colonizzano itralci fibrinosi fino alla formazione di membranefibrose che possono rivestire la pleura viscerale.Nelle fasi avanzate il polmone risulta intrappolatoin una membrana rigida che ne impedisce la com-pleta espansione. Questa membrana, raramentecon gli attuali presidi terapeutici, può anche evol-vere in una spessa cotenna.Le caratteristiche chimico-fisiche del versamentotendono a modificarsi con la progressione dellamalattia: il glucosio ed il pH diminuiscono, mentrela lattato deidrogenasi aumenta.
Quando il quadro clinico-radiologico pone ilsospetto di un versamento parapneumonico, unruolo d’importanza primaria spetta all’ecografiadel torace [3]. L’ecografia infatti, è in grado di dif-ferenziare la componente fluida dall’addensamen-to parenchimale, di quantizzare approssimativa-mente l’entità del versamento e di evidenziarne ladensità. Inoltre l’ecografia rende chiaramente visi-bili i tralci fibrinosi e le eventuali concamerazioni(Figura 1a-b).La tomografia computerizzata non sembra offrirenetti vantaggi rispetto all’ecografia e dovrebbe
Figura 1 Paziente di cinque anni con discreta falda di versamento a sinistra: a) Tomografia computerizzata; b)L’ecografia, nello stesso caso, rende ben visibili setti e concamerazioni poco evidenti alla tomografia computerizzata.
ALOKA98/99 2
20Hz
ALOKA98/99 1
20Hz
R12
2: Addome Pedia 10.0 DVA: 90%
G59 C2 A2 R12 G59 C2 A2
HF14 HF14
b
a
DxDx

La chirurgia toracoscopica video-assistita nella gestione dell’empiema pleurico 41
essere riservata ai casi più complessi (Figura 2a-b),per quantificare la compromissione parenchimale,per evidenziare raccolte ascessuali intraparenchima-li, o se vi è il sospetto di patologia tumorale [4-5].Un versamento pleurico iniziale e di modesta enti-tà può essere efficacemente trattato con la solaterapia medica, come riportato in un ampio studioretrospettivo [6]. Peraltro lo stesso studio usaindifferentemente il termine di “versamento pleu-rico” ed “empiema”. Verosimilmente è il versa-mento allo stadio essudativo quello che più pro-babilmente può essere risolto con un’adeguataterapia medica.I criteri da tenere presente per individuare lanecessità di affiancare alla terapia medica un trat-tamento più aggressivo sono sostanzialmente [7]:• il quadro clinico;• l’entità del versamento;• la presenza di concamerazioni all’Imaging.In tutti i casi in cui il quadro clinico resta compro-messo malgrado un’ottimale terapia medica, quan-do il versamento è di notevole entità (maggiore al50% di opacamento dell’emitorace alla radiografiain ortostatismo) o quando l’ecografia dimostrachiaramente la presenza di concamerazioni indivi-duate da setti multipli, è necessario ricorrere ad untrattamento più invasivo.
La chirurgia toracica video-assistita
La toracoscopia nel trattamento dell’empiemapleurico pediatrico è stata utilizzata con successoper la prima volta da Rodgers e collaboratori [8].In nove bambini, dopo il fallimento della terapiamedica e del drenaggio pleurico, la detersione intoracoscopia ha consentito di evitare il ricorsoalla toracotomia. L’Autore concludeva che un pre-coce uso della detersione toracoscopica potevaevitare la necessità di ricorrere ad un interventochirurgico più aggressivo. Non di rado in passatoera infatti necessario ricorrere alla decorticazionetoracotomica: asportazione a torace aperto delmateriale fibrinopurulento accumulato nel cavopleurico ed escissione della membrana fibrosache, adesa alla pleura viscerale, impedisce l’espan-sione polmonare.Con lo sviluppo della video-endoscopia ad altadefinizione, applicata in prima istanza alla laparo-scopia e quindi alla toracoscopia, la chirurgia tora-cica video-assistita (Video Assisted Thoracic Surgery,VATS) ha conosciuto una notevole diffusioneanche in età pediatrica [9].
Sono comparsi pertanto in Letteratura numerosicontributi che riportavano eccellenti risultati conl’uso precoce della VATS nell’empiema pleurico[10-13]. Per lo più in tali studi, l’uso precoce dellaVATS (entro tre-sette giorni, dalla mancata rispo-sta alla terapia medica) si dimostrava capace,rispetto all’utilizzo del solo drenaggio pleurico, dirisolvere la patologia in tempi più brevi e di evita-re inoltre una decorticazione toracotomica neicasi più avanzati.Dal punto di vista tecnico la VATS consiste in unvero e proprio intervento chirurgico, ancorchémini-invasivo. Viene infatti effettuata in anestesiagenerale in sala operatoria. Posizionato il pazientein decubito laterale ed aspirato parzialmente ilversamento prelevandone un campione per lavalutazione microbiologica, chimico-fisica e citolo-gica, si introducono tre cannule nel cavo pleurico(Figura 3).Lo spazio di lavoro viene ottenuto con l’insuffla-zione a flusso e la pressione controllata di anidri-de carbonica. Attraverso una cannula si inserisce
Dx
Figura 2 Paziente di sei anni: a) Radiografia in ortostatismo eviden-ziante un versamento ed ampio livello idroaereo; b) La tomografiacomputerizzata rende più chiara la situazione del piopneumotoraceiperteso con sbandamento del mediastino.
Dx
b
a
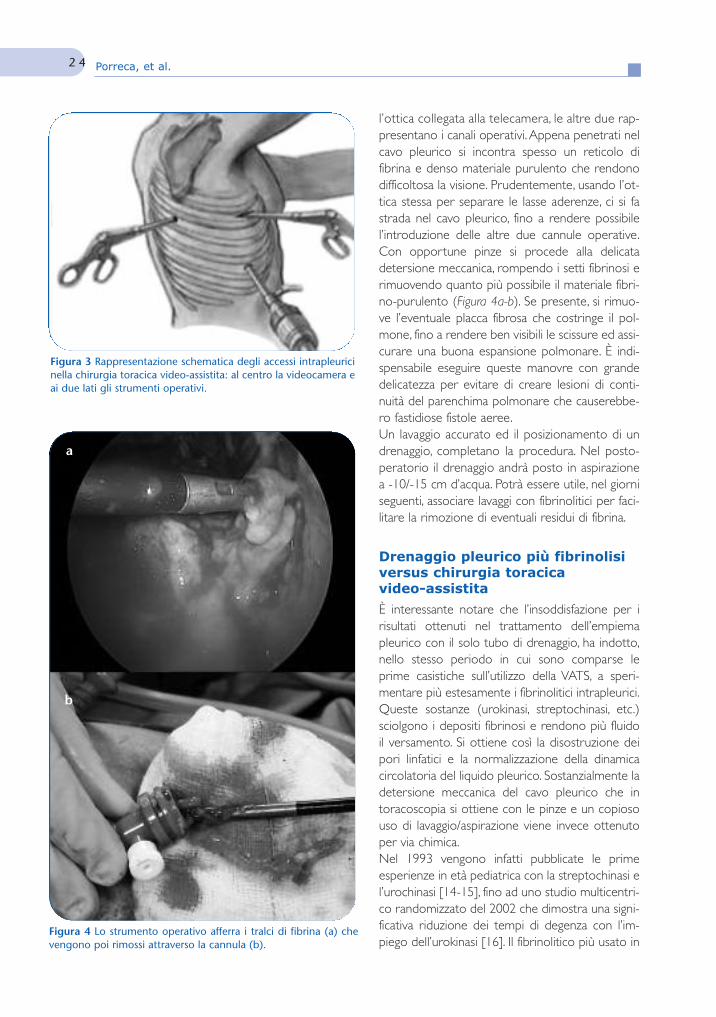
Porreca, et al. 42
l’ottica collegata alla telecamera, le altre due rap-presentano i canali operativi. Appena penetrati nelcavo pleurico si incontra spesso un reticolo difibrina e denso materiale purulento che rendonodifficoltosa la visione. Prudentemente, usando l’ot-tica stessa per separare le lasse aderenze, ci si fastrada nel cavo pleurico, fino a rendere possibilel’introduzione delle altre due cannule operative.Con opportune pinze si procede alla delicatadetersione meccanica, rompendo i setti fibrinosi erimuovendo quanto più possibile il materiale fibri-no-purulento (Figura 4a-b). Se presente, si rimuo-ve l’eventuale placca fibrosa che costringe il pol-mone, fino a rendere ben visibili le scissure ed assi-curare una buona espansione polmonare. È indi-spensabile eseguire queste manovre con grandedelicatezza per evitare di creare lesioni di conti-nuità del parenchima polmonare che causerebbe-ro fastidiose fistole aeree.Un lavaggio accurato ed il posizionamento di undrenaggio, completano la procedura. Nel posto-peratorio il drenaggio andrà posto in aspirazionea -10/-15 cm d’acqua. Potrà essere utile, nel giorniseguenti, associare lavaggi con fibrinolitici per faci-litare la rimozione di eventuali residui di fibrina.
Drenaggio pleurico più fibrinolisiversus chirurgia toracicavideo-assistita
È interessante notare che l’insoddisfazione per irisultati ottenuti nel trattamento dell’empiemapleurico con il solo tubo di drenaggio, ha indotto,nello stesso periodo in cui sono comparse leprime casistiche sull’utilizzo della VATS, a speri-mentare più estesamente i fibrinolitici intrapleurici.Queste sostanze (urokinasi, streptochinasi, etc.)sciolgono i depositi fibrinosi e rendono più fluidoil versamento. Si ottiene così la disostruzione deipori linfatici e la normalizzazione della dinamicacircolatoria del liquido pleurico. Sostanzialmente ladetersione meccanica del cavo pleurico che intoracoscopia si ottiene con le pinze e un copiosouso di lavaggio/aspirazione viene invece ottenutoper via chimica.Nel 1993 vengono infatti pubblicate le primeesperienze in età pediatrica con la streptochinasi el’urochinasi [14-15], fino ad uno studio multicentri-co randomizzato del 2002 che dimostra una signi-ficativa riduzione dei tempi di degenza con l’im-piego dell’urokinasi [16]. Il fibrinolitico più usato in
Figura 4 Lo strumento operativo afferra i tralci di fibrina (a) chevengono poi rimossi attraverso la cannula (b).
Figura 3 Rappresentazione schematica degli accessi intrapleuricinella chirurgia toracica video-assistita: al centro la videocamera eai due lati gli strumenti operativi.
b
a

La chirurgia toracoscopica video-assistita nella gestione dell’empiema pleurico 43
Figura 5 Algoritmo terapeutico dell’empiema pleurico.
Italia è l’urokinasi, che è facilmente reperibile ecomporta scarsi rischi di reazioni allergiche.Un contributo molto importante riguardo allascelta tra le due opzioni terapeutiche (VATSoppure fibrinolisi), è venuto da due importantistudi prospettici randomizzati pubblicati indipen-dentemente in Inghilterra ed USA [17-18].Entrambi gli studi hanno dimostrato una sostan-ziale pari efficacia delle due metodiche con alcunisvantaggi a carico della VATS:• costi più elevati;• necessità di una vera e propria narcosi a frontedi una semplice sedazione;• necessità di una équipe chirurgica specificamen-te competente.Alla luce di questi lavori, un possibile algoritmo peril trattamento dell’empiema in età pediatrica èmostrato nella Figura 5.
Da tale algoritmo risulta evidente che il ruolo dellaVATS sarebbe limitato alle situazioni in cui la terapiafibrinolitica non riesce ad aver ragione del coinvol-gimento pleurico nell’infezione, situazione riporta-ta con un’incidenza del 16,6% [17-18].La persistenza della compromissione clinica inassenza di persistente malattia pleurica, richiede-rebbe solo la continuazione della terapia medica.Va peraltro sottolineato che questa posizione nonè universalmente condivisa.Uno studio retrospettivo in cui l’esito della terapiaveniva comparata con lo stadio ecografico del ver-samento [20], ha mostrato un successo della terapiacon urokinasi nel 100% dei pazienti con versamen-to al secondo stadio, ma del 60% in quelli con ver-samento al terzo stadio, mentre la VATS aveva otte-nuto il 100% di successo in entrambi i casi. Gli Autoripertanto concludono che la VATS dovrebbe essere
Riduzione della quantità di liquido drenata senza miglioramento clinico
Versamento resistente a terapia medica opresenza di setti o concamerazioni
Ecografia o tomografia computerizzata
Drenaggio pleurico (possibilmente con tubo 12 Fr)+
Urokinasi: 4000 UI/kg/die in due ml/kg di soluzione fisiologica [19]
Empiema
Chirurgia toracicavideo-assistita
Continuare la terapiamedica
Versamento persistente(setti/concamerazioni)
Versamento assenteo molto ridotto
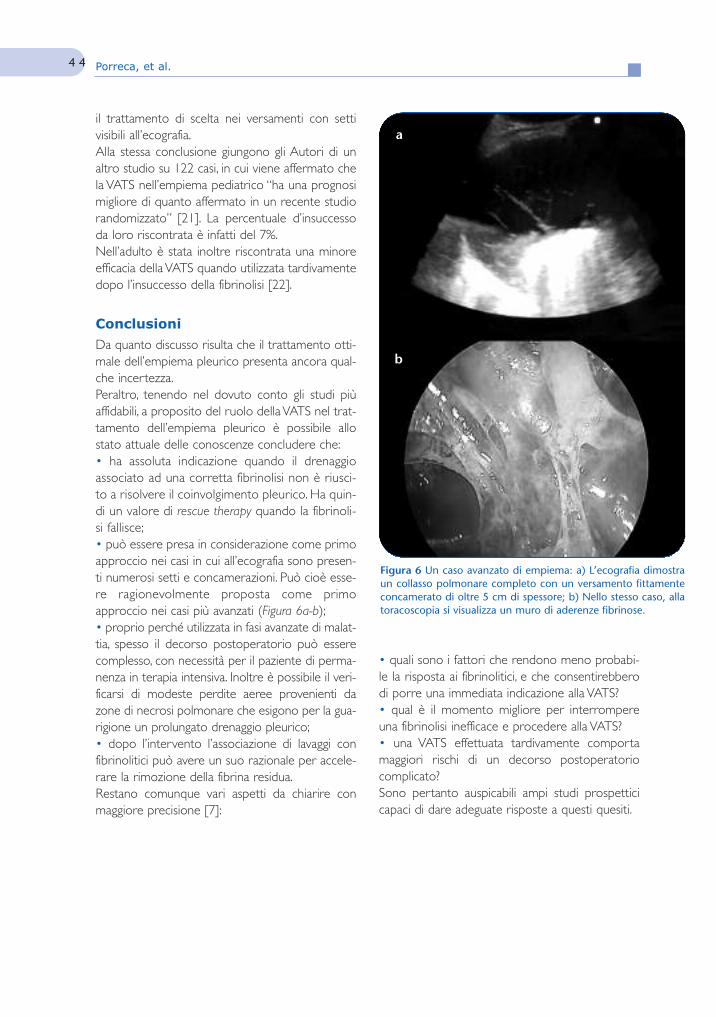
Porreca, et al. 44
il trattamento di scelta nei versamenti con settivisibili all’ecografia.Alla stessa conclusione giungono gli Autori di unaltro studio su 122 casi, in cui viene affermato chela VATS nell’empiema pediatrico “ha una prognosimigliore di quanto affermato in un recente studiorandomizzato” [21]. La percentuale d’insuccessoda loro riscontrata è infatti del 7%.Nell’adulto è stata inoltre riscontrata una minoreefficacia della VATS quando utilizzata tardivamentedopo l’insuccesso della fibrinolisi [22].
Conclusioni
Da quanto discusso risulta che il trattamento otti-male dell’empiema pleurico presenta ancora qual-che incertezza.Peraltro, tenendo nel dovuto conto gli studi piùaffidabili, a proposito del ruolo della VATS nel trat-tamento dell’empiema pleurico è possibile allostato attuale delle conoscenze concludere che:• ha assoluta indicazione quando il drenaggioassociato ad una corretta fibrinolisi non è riusci-to a risolvere il coinvolgimento pleurico. Ha quin-di un valore di rescue therapy quando la fibrinoli-si fallisce;• può essere presa in considerazione come primoapproccio nei casi in cui all’ecografia sono presen-ti numerosi setti e concamerazioni. Può cioè esse-re ragionevolmente proposta come primoapproccio nei casi più avanzati (Figura 6a-b);• proprio perché utilizzata in fasi avanzate di malat-tia, spesso il decorso postoperatorio può esserecomplesso, con necessità per il paziente di perma-nenza in terapia intensiva. Inoltre è possibile il veri-ficarsi di modeste perdite aeree provenienti dazone di necrosi polmonare che esigono per la gua-rigione un prolungato drenaggio pleurico;• dopo l’intervento l’associazione di lavaggi confibrinolitici può avere un suo razionale per accele-rare la rimozione della fibrina residua.Restano comunque vari aspetti da chiarire conmaggiore precisione [7]:
• quali sono i fattori che rendono meno probabi-le la risposta ai fibrinolitici, e che consentirebberodi porre una immediata indicazione alla VATS?• qual è il momento migliore per interrompereuna fibrinolisi inefficace e procedere alla VATS?• una VATS effettuata tardivamente comportamaggiori rischi di un decorso postoperatoriocomplicato?Sono pertanto auspicabili ampi studi prospetticicapaci di dare adeguate risposte a questi quesiti.
Figura 6 Un caso avanzato di empiema: a) L’ecografia dimostraun collasso polmonare completo con un versamento fittamenteconcamerato di oltre 5 cm di spessore; b) Nello stesso caso, allatoracoscopia si visualizza un muro di aderenze fibrinose.
b
a

La chirurgia toracoscopica video-assistita nella gestione dell’empiema pleurico 45
1. Grijalva CG, Nuorti JP, Zhu Y, et al. Increasing Incidenceof empyema complicating childhood community acquiredpneumonian the United States. CID 2010; 50: 805-813.
2. Balfour-Lynn IM, Abrahamson E, Cohen G, et al. BTSguidelines for the management of pleural infection in chil-dren. Thorax 2005; 60 (Suppl 1): i1-i21.
3. Davies CH, Gleeson FV, Davies RJO. BTS guidelineson the management of pleural infection. Thorax 2003; 58(Suppl II): ii18-ii28.
4. Bradley JS, Byington CL, Shah SS, et al. ExecutiveSummary: The management of community acquired pneu-monia in infants and children older than 3 months of age:Clinical practice guidelines by the Pediatric InfectiousDisease Society and the Infectious Disease Society ofAmerica. Clin Infect Dis 2011; 53: 617-630.
5. Jaffé A, Balfour-Lynn IM. Management of empyema inchildren. Pediatr Pulmonol 2005; 40: 148-156.
6. Carter E, Waldhausen J, Zhang W, et al. Managementof children with empyema: pleural drainage is not alwaysnecessary. Pediatr Pulmonol 2010; 45: 475-480.
7. Islam S, Calkins CM, Goldin AB, et al. The diagnosisand management of empyema in children: a comprehen-sive review from the APSA Outcomes and Clinical TrialsCommittee. J Pediatr Surg 2012; 47 (11): 2101-2110.
8. JA Kern, BM Rodgers. Thoracoscopy in the manage-ment of empyema in children. 1993; 28 (9): 1128-1132.
9. Rothenberg SS. Thoracoscopy in infants and children.Semin Pediatr Surg 1998; 7 (4): 194-201.
10. Davidoff AM, Hebra A, Kerr J, et al. Thoracoscopicmanagement of empyema in children. J LaparoendoscSurg 1996; 6: S51-S54.
11. Grewal H, Jackso RJ, Wagner CW, et al. Early Video-assisted Thoracic Surgery in the Management of empye-ma. Pediatrics 1999; 103: e63.
12. Cohen G, Hjortdal V, Ricci M, MD, et al. Primarythoracoscopic treatment of empyema in children. J ThoracCardiovasc Surg 2003; 125 (1): 79-83.
13. Wurnig PN, Wittmer V, Pridun NS, et al. Video-assisted thoracic surgery for pleural empyema. AnnThorac Surg 2006; 81: 309-313.
14. Rosen H, Nadkarni V, Theroux M, et al. Intrapleuralstreptokinase as adjunctive treatment for persistentempyema in pediatric patients. Chest 1993; 103:1190-1193.
15. Handman HP, Reuman PD. The use of urokinase forloculated thoracic empyema in children: a case reportand review of the literature. Pediatr Infect Dis J 1993;12: 958-959.
16. Thomson AH, Hull J, Kumar MR, et al. Randomisedtrial of intrapleural urokinase in the treatment of childhoodempyema. Thorax 2002; 57: 343-347.
17. Sonnappa S, Cohen G, Owens CM, et al.Comparison of urokinase and video-assisted thoracoscop-ic surgery for treatment of childhood empyema. Am JRespir Crit Care Med 2006; 174: 221-227.
18. St Peter SD, Tsao K, Spilde TL, et al. Thoracoscopicdecortication vs tube thoracostomy with fibrinolysis forempyema in children: a prospective, randomized trial. JPediatr Surg 2009; 44: 106-111.
19. Barbato A, Panizzolo C, Monciotti C, et al. Use ofurokinase in childhood pleural empyema. PediatrPulmonol 2003; 35: 50-55.
20. Chiu CY, Wong KS, Huang YC, et al. Echo-guidedmanagement of complicated parapneumonic effusion inchildren. Pediatr Pulmonol 2006; 41: 1226-1232.
21. Bishay M, Short M, Shah K, et al. Efficacy of video-assisted thoracoscopic surgery in managing childhoodempyema: a large single-centre study. J Pediatr Surg 2009;44 (2): 337-342.
22. Petrkis IE, Kogerakis NE, Drositis IE, et al. Video-assi-sted thoracoscopic surgery for thoracic empyema: prima-rily, or after fibrinolytic therapy failure? Am J Surg 2004;187 (4): 471-474.
BIBLIOGRAFIA
Bibliografia

Introduzione
Il primo trapianto polmonare risale al 1987 in unragazzo di 16 anni con diagnosi di fibrosi polmo-nare familiare presso l’Università di Toronto, inCanada. Da allora, il trapianto polmonare è statoaccettato come possibile opzione terapeutica perpazienti pediatrici con patologie polmonari in sta-dio avanzato di malattia. Nello specifico, si è osser-vato un aumento esponenziale del numero di tra-pianti polmonari pediatrici negli anni Novanta,rimasto poi costante negli anni successivi. A diffe-renza che negli adulti, per i quali il numero dei tra-pianti è però diminuito nei primi anni 2000 rima-nendo poi costante. Attualmente, più di 3.500 tra-pianti polmonari vengono effettuati ogni annonella popolazione adulta, e poco più di cento inquella pediatrica [1-2]. Fin dall’inizio sono presentidifferenze fra le due coorti nella diagnosi iniziale dimalattia che porta al trapianto polmonare: negli
adulti più frequentemente si osservavano casi dimalattie ostruttive polmonari croniche, piuttostoche fibrosi polmonare idiopatica o fibrosi cistica(FC) che rimane la principale, se non l’unica, indi-cazione in età pediatrica.Le patologie che più frequentemente richiedono iltrapianto variano fortemente con l’età comemostrato dalla Tabella 1. In età neonatale la mag-gior indicazione a trapianto è costituita dallemalattie interstiziali polmonari, che includono dis-ordini delle proteine B e C del surfactante e il tra-sportatore ABCA3. Secondariamente, le cardiopa-tie congenite e l’ipertensione polmonare idiopati-ca (IPAH). L’IPAH seguìta dalla fibrosi polmonare edalla bronchiolite obliterante costituiscono le mag-giori indicazioni in bambini di età compresa trauno e cinque anni, mentre l’FC è la maggiore tra isei e i diciotto anni [1] (cfr. Tabella 1).
Francesco Parisi, Sara Alfieri, Maria Assunta Castelluzzo
Unità di Funzione Trapianti e Gestione delle Cardiopatie Congenite Complesse, Ospedale Pediatrico“Bambino Gesù”, Roma
46
Trapianto polmonare in età pediatricaPediatric lung transplantation
Parole chiave: trapianto polmonare, età pediatrica
Keywords: lung transplantation, pediatric
Riassunto. Il trapianto polmonare è stato accettato come possibile opzione terapeutica per pazienti pediatrici con patologiepolmonari in stadio avanzato di malattia. Le patologie che più frequentemente richiedono il trapianto variano fortemente conl’età. In età neonatale la maggior indicazione a trapianto è costituita dalle malattie interstiziali polmonari. L’ipertensione pol-monare idiopatica seguita dalla fibrosi polmonare e la bronchiolite obliterante costituiscono le maggiori indicazioni in bambinidi età compresa tra uno e cinque anni, mentre la fibrosi cistica è la maggiore tra i sei e i diciotto anni. La scarsità di donazionicontinua a limitare il numero di trapianti di polmone anche in età pediatrica comportando così lunghi tempi di attesa ed ele-vata mortalità per i bambini in lista di trapianto. L’outcome del trapianto di polmone in età pediatrica è notevolmente miglio-rato rispetto agli anni Ottanta quando iniziarono ad essere effettuati i primi trapianti di polmone. Attualmente la sopravviven-za a cinque anni è del 50% risultando così sovrapponibile a quella per gli adulti. Tuttavia la sopravvivenza del trapianto di pol-mone rimane comunque inferiore a quella degli altri organi solidi. La prima causa di insuccesso del trapianto a lungo termineè costituita dalla Bronchiolite Obliterante, che va interpretata come la principale espressione clinica di rigetto cronico. La tera-pia definitiva dell’insufficienza respiratoria acuta dopo il primo trapianto è il ritrapianto.
Accettato per la pubblicazione il 8 agosto 2013.
Corrispondenza: Francesco Parisi, Unità di Funzione Trapianti e Gestione delle Cardiopatie Congenite Complesse,Ospedale Pediatrico “Bambino Gesù”, Piazza Sant’Onofrio 4, 00165 Roma;
e-mail: [email protected]
Pneumologia Pediatrica 2013; 51: 46-51

Trapianto polmonare in età pediatrica 47
Così come per gli adulti, sussistono controindica-zioni assolute e relative (Tabella 2). Le controindi-cazioni chirurgiche includono severa tracheomala-cia, presenza di talcaggio pleurico, marcata scoliositoracica ed insufficienza laringea [4-5]. Molto spes-so, tuttavia, le controindicazioni chirurgiche sonocentro-specifiche e non universalmente riconosciu-te. Anche le controindicazioni mediche risultanoessere molto variabili fra i vari Centri. Ad esempio,prime far tutte: infezioni virali attive, resistenza anti-biotica diffusa (come da Burkholderia cenocepacia,)non aderenza alla terapia, scarsa compliance al fol-low up, scarso controllo della epilessia o del diabe-te mellito nonostante il trattamento specifico.In generale, l’infezione da epatite C è consideratacontroindicazione assoluta, mentre quelle daMycobacterium ascessus sono responsabili di severeinfezioni post-trapianto nel sito chirurgico. Nonsono inoltre da sottovalutare le problematiche chepossono sussistere da parte genitoriale o di chi nefa la funzione: casi di severa instabilità psichiatrica egravi difficoltà socio-economiche devono sempreessere considerate al momento dell’immissione inlista di trapianto (cfr. Tabella 2).
Fibrosi cistica
L’FC rappresenta la maggior indicazione a trapian-to in età pediatrica. Ogni paziente affetto da FC èun ricevente “unico” data la coesistenza di molte-plici patologie multisistemiche e la presenza di infe-zioni cronicizzate delle vie respiratorie. Tali infezio-ni risultano essere frequentemente dovute a germimultiresistenti: in fase post-trapianto, quando ipazienti sono sottoposti a terapia immunosoppres-siva antirigetto, questi germi possono colonizzareanche il polmone trapiantato. Nonostante ciò la
sopravvivenza degli adulti con FC sottoposti a tra-pianto polmonare risulta essere sovrapponibile aquella di altri pazienti con trapianto polmonare condiversa patologia di base [2, 5-7].Una considerazione accurata deve quindi essereffettuata al momento dell’immissione in lista diattesa: mentre infezione da PseudomonasAeruginosa multi resistente, Staphylococcus Aureusmeticillino-resistente ed altri patogeni non sonoconsiderate controindicazioni a trapianto, possonoperciò portare a complesse indicazioni di regime
Tabella 1 Indicazioni al trapianto polmonare in età pediatrica, divisa per gruppi d’età (gennaio 1990-giugno2011). Modificata da [3].
Diagnosi <1 anno 1-5 anni 6-11 anni 12-17 anni
Fibrosi cistica 1,1% 5,0% 53,8% 71,5%
Ipertensione arteriosa polmonare idiopatica 13,2% 22,5% 9,7% 7,2%
Ritrapianto 3,3% 9,1% 5,6% 5,6%
Malattia cardiaca congenita 15,4% 7,5% 1,3% 0,9%
Malattia vascolare polmonare 8,8% 5,8% 1,3% 0,1%
Carenza della proteina B del surfattante 17,6% 2,5% - -
Altre diagnosi 40,6% 47,6% 28,3% 14,7%
Tabella 2 Controindicazioni al trapianto polmonare in etàpediatrica generalmente riconosciute. Modificata da [3].
Chirurgiche
• Severa tracheomalacia
• Pleurodesi
• Marcata scoliosi toracica
• Insufficienza della laringe
Mediche
• Infezione virale attiva
• Infezione polmonare batterica resistente ai pan-antibiotici
• Infezione polmonare con ascesso micobatterico positivoallo “smear”
• Diabete di tipo 2 poco controllato
• Disturbo convulsivo poco controllato
• Neoplasia entro due anni
• Disfunzione d’organo multipla
• Tubercolosi attiva
• Sepsi
Psicosociali
• Non-aderenza refrattaria
• Disturbo mentale incurabile nel paziente o nel suo caregiver

Parisi, et al. 48
antibiotico nella fase post trapianto, in molti centripediatrici, l’infezione da Burkholderia cepacia (geno-movar III) è considerata una controindicazione poi-ché molti studi hanno dimostrato un aumentosignificativo della mortalità in pazienti affetti e suc-cessivamente sottoposti a trapianto [9-11].Un’altra riflessione inerente questa patologia èlegata alla sua natura genetica, con possibile coin-volgimento di più organi. In caso di avanzata insuf-ficienza epatica, la possibilità di trapianto combina-to fegato-polmoni deve essere considerata.Ovviamente prima del trapianto le varie comorbi-dità che possono sussistere (quali il diabete mellito,l’osteoporosi, patologie dei seni mascellari e reflus-so gastroesofageo) devono essere affrontate e pos-sibilmente stabilizzate. Per quanto riguarda, in questipazienti, il timing dell’inserimento in lista questo nonè uniformemente descritto dalle Linee Guida, basa-te sullo studio di Kerem e collaboratori [12].Pazienti con volume espiratorio massimo nelprimo secondo (Forced Expiratory Volume in the 1st
second, FEV1) inferiore al 30%, rapido decremen-to del FEV1, riesacerbazione polmonare cherichieda cure in terapia intensiva, aumento dellafrequenza delle ricadute polmonari, pneumotoracio emottisi ricorrenti, meritano una considerazionecollegiale per un eventuale inserimento in lista diattesa. Molti studi negli ultimi anni hanno tentatodi trovare fattori predittivi che aiutassero i clinicinello stabilire un timing adeguato, ma nessuno diquesti è, attualmente, uniformemente accettato.
Ipertensione polmonare idiopatica
La storia naturale dell’IPAH in tutte le età è dicarattere progressivo e inevitabilmente conduce agrave disabilità e spesso a insufficienza cardiacadestra. Questa è la seconda indicazione a trapian-to polmonare in età pediatrica e la principale inpazienti da uno a cinque anni d’età [1]. Negli ulti-mi anni si è assistito ad un notevole avanzamentoin campo farmacologico del trattamento di questapatologia, soprattutto si è evidenziato un aumentodella sopravvivenza e della qualità di vita in pazien-ti trattati con infusione continua di epoprostenolo;tuttavia, un numero significativo di pazienti conti-nua ad avere la progressione di malattia sino adavere la necessità di un trapianto polmonare. I fat-tori prognostici che possono aiutare il clinico nel-l’ipotesi di trapianto sono la classe NYHA, gene-ralmente di classe III o IV, l’andamento nel tempodel 6 minute walk test ed ovviamente lo studio
emodinamico, generalmente con indice cardiacoinferiore a 2 L/min/m2 e pressione in atrio destromaggiore a 15 mmHg.
Bambini sotto l’anno di età
L’indicazione a trapianto in questa coorte variamoltissimo sebbene le principali indicazioni riman-gono il deficit della proteina B del surfactante, car-diopatie congenite ed IPAH. Uno studio condottopresso il St. Louis Children’s Hospital (Missouri,USA) ha riportato la più estesa coorte di pazien-ti di età inferiore all’anno in cui si è confrontata lasopravvivenza dei pazienti in questione con quelladi bambini di altre età. Tale studio ha evidenziatoche i più piccoli, specialmente se inferiori ad unanno, avevano minor incidenza di rigetto cellulareacuto e, meno frequentemente, sviluppavanobronchiolite obliterante [13].Solo 84 lattanti sono stati sottoposti a trapiantopolmonare negli ultimi venticinque anni [14] e solodue centri negli Stati Uniti hanno eseguito trapian-ti polmonari in questi pazienti prima del 2006.Infine, i bambini di questa età sottoposti a trapian-to polmonare necessitano spesso di assistenzameccanica e, pertanto, come negli adulti, tale pro-cedura incide sfavorevolmente sull’outcome nelperiodo post-trapianto [13].
Management dell’insufficienzarespiratoria e bridge al trapianto
Nei pazienti in lista di attesa e in stadio terminaledi malattia un problema rilevante è rappresentatodalle strategie da utilizzare quale ponte verso iltrapianto. La maggior parte di questi pazienti pre-sentano, infatti, quadri difficilmente trattabili diipossia e ipercapnia. La ventilazione non invasiva apressione positiva (NonInvasive Positive-PressureVentilation, NIPPV) può essere un valido bridge altrapianto, negli adulti affetti fa FC, infatti, è statodimostrato quanto questo tipo di ventilazionecontribuisca a: stabilizzazione di malattia, riduzionedei sintomi e incremento della tolleranza allo sfor-zo. Quando, inoltre, la NIPPV viene affiancata daesercizio quotidiani di fisioterapia, si assiste ad:aumento della saturazione di ossigeno, aumentodel volume tidale, massimo allungamento deimuscoli espiratori e miglioramento della capacitàdi espettorazione [15-17]. Tuttavia, tale presidionon ha aumentato la sopravvivenza e i pazienti siaadulti che pediatrici che, subito prima del trapianto,

Trapianto polmonare in età pediatrica 49
hanno presentato insufficienza respiratoria acutacon l’utilizzo di NIPPV, hanno evidenziato unaumento della morbidità e mortalità post-opera-toria e un allungamento del tempo di degenza interapia intensiva.Talvolta è necessario un tipo di assistenza maggio-re che richiede l’uso di ECMO (Extra CorporealMembrane Oxygenation) in fase pre-trapianto.L’esperienza in questo campo è ancora limitata:tale procedura espone il paziente a comorbiditàquali emorragie, stroke e infezioni nel 63% circa deicasi. È attualmente in corso uno studio multicen-trico volto a confrontare l’outcome di pazienti sot-toposti a ECMO in fase pre-trapianto e pazientiche direttamente hanno effettuato il trapianto.Un ulteriore mezzo di assistenza a trapianto nellapopolazione adulta è il Novalung Pumpless Device(sistema per la rimozione extracorporea dellaCO2); i dati emersi dal suo utilizzo sono moltosoddisfacenti, tuttavia questa tecnica non è stataancora adoperata in campo pediatrico.
Selezione del donatore
La scarsità di donazioni continua a limitare ilnumero di trapianti di polmone anche in etàpediatrica comportando così lunghi tempi di atte-sa ed elevata mortalità per i bambini in lista di tra-pianto.È fondamentale adottare un approccio adeguato,sia clinico che organizzativo, volto all’identificazio-ne dei potenziali donatori e alla corretta gestioneclinica degli stessi prima del trapianto. Negli ultimianni i criteri di selezione per i donatori di polmo-ne sono significativamente cambiati rendendocosì idonei per il trapianto molti polmoni cheprima non venivano considerati tali. Un’adeguataed importante assistenza intensiva dei potenzialidonatori in condizioni cliniche sub-ottimali è fon-damentale per incrementare il numero dei dona-tori e di conseguenza per ridurre la mortalità inlista d’attesa. A tale proposito un ruolo importan-te è rappresentato dall’Ex Vivo Lung Perfusion(EVLP) [18], una tecnica innovativa in grado dipreservare il polmone del donatore riproducen-do la perfusione in vivo mediante l’utilizzo di unliquido di perfusione attraverso un circuito chiuso.Rispetto agli adulti, inoltre, non va trascurato cheil trapianto di polmone in età pediatrica è mag-giormente condizionato dalla compatibilità ditaglia.
Outcome
L’outcome del trapianto di polmone in età pedia-trica è notevolmente migliorato rispetto agli anniOttanta quando iniziarono ad essere effettuati iprimi trapianti di polmone. Attualmente la soprav-vivenza a cinque anni è del 50% risultando cosìsovrapponibile a quella per gli adulti. Tale aumentoè dovuto principalmente alla riduzione della mor-talità precoce [1]. Tuttavia la sopravvivenza del tra-pianto di polmone rimane comunque inferiore aquella degli altri organi solidi.È importante sottolineare che, poiché la principa-le indicazione a trapianto polmonare in età pedia-trica è rappresentata da un’FC in stato avanzato, idati sulla sopravvivenza riguardano una popolazio-ne “ristretta”.I principali fattori che influenzano negativamentel’outcome del trapianto di polmone sono la venti-lazione invasiva prima del trapianto e nel peri-tra-pianto, l’uso cronico di steroidi, l’età del ricevente(maggiore ai dodici anni), il trapianto di singolopolmone e la ridotta esperienza del Centro(meno di cinque trapianti l’anno) [1]. InLetteratura, infine, esistono dati discordanti riguar-danti il ruolo dell’ECMO come bridge al trapiantodi polmone in questi pazienti.La prima causa di insuccesso del trapianto a lungotermine è costituita dalla Bronchiolite Obliterante,che va interpretata come la principale espressioneclinica di rigetto cronico.La terapia definitiva dell’insufficienza respiratoriacronica dopo il primo trapianto è il ritrapianto. Incirca la metà dei casi l’indicazione al ritrapianto èla Bronchiolite Obliterante.
Il reflusso gastroesofageo
La malattia da reflusso gastroesofageo dopo il tra-pianto polmonare è considerata, nell’adulto, unfattore di rischio per lo sviluppo di bronchioliteobliterante.Sebbene in ambito pediatrico i dati in Letteraturasono limitati, è provata la correlazione tra malattiada reflusso gastroesofageo e sindrome della bron-chiolite obliterante.Nessuna terapia è risultata efficace per il tratta-mento dell’insufficienza respiratoria secondaria adaspirazione di materiale gastrico e, nei pazienti arischio, la terapia di scelta è la plastica antireflusso. Ivantaggi di tale terapia sembrano essere rilevantisoprattutto nel periodo post-trapianto ed in alcuni

Parisi, et al. 50
pazienti con bronchiolite obliterante in stadio inizia-le si è assistito ad un recupero della funzione pol-monare dopo l’intervento di plastica antireflusso.
L’esperienza dell’OspedalePediatrico “Bambino Gesù”
Dal 1991 ad oggi all’Ospedale Pediatrico“Bambino Gesù” si sono eseguiti trentotto tra-pianti polmonari (Tabella 3) la cui tipologia è ripor-tata nella Tabella 4. La sopravvivenza è descrittadalla curva di Kaplan Meier riportata in Figura 1.Nella Tabella 5 sono riportate le indicazioni al tra-pianto e l’età media dei pazienti, mentre nellaTabella 6 sono riportati il numero e le cause deidecessi.I risultati sono gravati sia dalla curva di apprendi-mento che dalla scelta istituzionale che tende aduna selezione dei pazienti non particolarmenterigida. Questo aspetto è stato recentementeoggetto di discussione portando ad alcune modi-fiche del protocollo con maggiore rigidità soprat-tutto nella selezione dei pazienti con gravi conta-minazioni infettive.
Tabella 3 Attività della Trapiantologia polmonare dell’OspedalePediatrico “Bambino Gesù” di Roma.
Tipologia n. Sopravvivenza
Totale pazienti 35 -
Totale trapianti 38 -effettuati
Viventi 8 Media dieci anni e sette mesi(minimo un anno e un mese;
massimo diciotto anni e dieci mesi)
Deceduti 27 Media un anno (minimo un mese;massimo cinque anni e sei mesi)
Ritrapianto 3 Solo un vivente
Tabella 5 Indicazioni ed età media al trapianto polmonare effet-tuato presso l’Ospedale Pediatrico “Bambino Gesù” di Roma(età media 14 anni e 9 mesi).
Patologia n.
Fibrosi cistica 15
Sindrome di Eisenmenger 6
Ipertensione polmonare 5
Bronchiolite obliterante 4
Malattia da trapianto contro l’ospite 1
Fibrosi polmonare idiopatica 2
Proteinosi alveolare 1
Cardiomiopatia restrittiva più ipertensione 1polmonare
Ernia diaframmatica più ipertensione 1polmonare
Cardiopatia congenita plurioperata 1
Tabella 4 Tipologia di trapianto polmonare effettuata pressol’Ospedale Pediatrico “Bambino Gesù” di Roma.
Tipologia n.
Doppio polmone 13
Singolo polmone 1
Cuore -polmoni 20
Ritrapianto di doppio polmone 3
Cuore -polmoni - fegato 1
1
0,75
0,5
0,25
00 5 10 15 20
Soprav
vive
nza (%
)
Anni
Figura 1 Curva di sopravvivenza dei trapianti polmonari nella stimaKaplan-Meier (Ospedale Pediatrico “Bambino Gesù” 1991-2012).
Tabella 6 Cause e numero dei decessi dopo trapianto polmonarepresso l’Ospedale Pediatrico “Bambino Gesù” di Roma.
Causa n.
Infezione 9
Sindrome da bronchiolite obliterante 5
Insufficienza renale 1
Complicanze perioperatorie 5
Infezione 4
Linfoma 1
Rigetto acuto anticorpo-mediato 1
Recidiva della malattia di base 1

Trapianto polmonare in età pediatrica 51
1. Benden C, Edwards LB, Kucheryavaya AY, et al. Theregistry of the International society of heart and lungtransplantation: fifteenth pediatric lung and heart trans-plantation report-2012. J Heart Lung Transplant 2012;31: 1087-1095.
2. Christie JD, Edwards LB, Kucheryavaya AY, et al. Theregistry of the International society of heart and lungtransplantation: 29th adult lung transplant report -2012. JHeart Lung Transplant 2012; 31: 1073-1086.
3. Goldfarb S, Benden C, Sweet S, et al. (eds). PediatricLung Transplantation. ISHLT Monograph Series Volume7. Addison (Texas, USA): The International Society forHeart and Lung Transplantation 2013.
4. Su JW, Mason DP, Murty SC, et al. Successful doublelung transplant in 2 patients with severe scoliosis. J HeartLung Transplant 2008; 27: 1262-1264
5. Garcha PS, Santacruz JF, Machuzak MS, et al. Clinicalcourse after successful double lung transplant in a patientwith severe scoliosis. J Heart Lung Transplant 2011; 30:234-235.
6. Khan MS, Heinle JS, Samayoa AX, et al. Is lung trans-plantation survival better in infants? Analysis of over 80infants. J Heart Lung Transplant 2013; 32: 44-49.
7. Hofer M, Benden C, Inci I, et al. True survival benefitof lung transplantation for cystic fibrosis patients: theZurich experience. J Heart Lung Transplant 2009; 28:334-339.
8. Meachery G, De Soyoza A, Nicholson A, et al.Outcomes of lung transplantation for cystic fibrosis in alarge UK cohort.Thorax 2008; 63: 725-731.
9. Egan TM, Detterbeck FC, Mill MR, et al. Long termresults of lung transplantation for cystic fibrosis. Eur JCardiothorac Surg 2002; 22: 602-609.
10. Aris RM, Routh JC, LiPuma JJ, et al. Lung transplan-tation for cystic fibrosis with Burkholderia cepacia com-plex. Survival linked to genomovar type. Am J Respir CritCare Med 2001; 164: 2102-2106.
11. De Soyza A, Meachery G, Hester KL, et al. Lungtransplantation for cystic fibrosis with Burkholderia cepa-cia complex infection: a single centre experience. J HeartLung Transplant 2010; 29: 1395-1404.
12. De Soyza A, McDowell A, Archer L, et al.Burkholderia cepacia complex genomovars and pul-monary transplantation outcomes in patients with cysticfibrosis. Lancet 2001; 358: 1780-1781.
13. Kerem E, Reisman J, Corey M, et al. Prediction ofmortality in patients with cystic fibrosis. Engl J Med 1992;326: 1187-1191.
14. Mason DP, Thuita L, Nowicki ER, et al. Should lungtransplantation be performed for patients on mechanicalsupport? The US experience. J Thorac Surg 2010; 139:765-773.
15. Young AC, Wilson JW, Kotsimbos TC, et al.Randomised placebo controlled trial of non-invasive venti-lation for hypercapnia in cystic fibrosis. Thorax 2008; 63(1): 72-77.
16. Faroux B, Boulé M, Lofaso F, et al. Chest physiother-apy in cystic fibrosis: improved tolerance with nasal pres-sure support ventilation. Pediatrics 1999; 103 (3): E32.
17. Holland AE, Denehy L, Ntoumenopoulos G, et al.Non invasive ventilation assists chest physiotherapy inadults with acute exacerbations of cystic fibrosis. Thorax2003; 58 (10): 880-884.
18. Cypel M, Jeung JC, Liu M, et al. Normothermic ExVivo Lung Perfusion in Clinical Lung Transplantation. N EngJ Med 2011; 364: 1431-1440.
BIBLIOGRAFIA
Bibliografia

Introduzione
Con il termine discinesia ciliare primitiva (DCP) siintende un gruppo di condizioni rare accomunateda alterazioni congenite della struttura e della fun-zionalità del ciglio mobile, caratterizzate dalla suaimmobilità o dismotilità. La DCP è per frequenza laseconda malattia congenita dell’apparato respirato-rio dopo la fibrosi cistica (FC) e la sua incidenza èstimata tra 1 su 10.000 e 1 su 20.000 nati vivi [1-2].
In circa la metà dei casi è presente un’anomala dis-posizione degli organi interni, situs viscerum inver-sus (sindrome di Kartagener, SK) o situs ambiguused è riconosciuta come “malattia rara” (codiceRN0950). La reale incidenza della malattia sembratuttavia essere più alta di quanto riportato.L’insufficiente conoscenza della presentazione clini-ca della malattia, l’aspecificità dei segni e dei sintomi,
Rita Padoan1, Silviana Timpano1, Daniela Zaniboni2, Elide Spinelli3, Maria Margherita De Santi4,Marco Berlucchi5, Alessandro Plebani2
1 Centro di supporto per la Fibrosi Cistica, Clinica Pediatrica, Ospedale dei Bambini, Brescia; 2 ClinicaPediatrica, Università di Brescia; 3 Servizio Malattie Rare, Azienda Ospedaliera “Spedali Civili”, Brescia;4 Unità Operativa Complessa di Anatomia Patologica, Policlinico “Santa Maria alle Scotte”, AziendaOspedaliera Universitaria Senese, Siena; 5 Unità Operativa di Otorinolaringoiatria Pediatrica, Ospedaledei Bambini, Brescia
52
La discinesia ciliare primitiva:esperienza di un percorso diagnostico
in ambulatorioPrimary ciliary dyskinesia:
experience of an ambulatorydiagnostic work-up
Parole chiave: Discinesia ciliare primaria, malattie rare, situs viscerum inversus
Keywords: Primary ciliary dyskinesia, rare diseases, situs viscerum inversus
Riassunto. La discinesia ciliare primitiva (DCP) è dovuta ad alterazioni congenite della struttura del ciglio mobile e alla suaalterata funzione. I sintomi principali sono respiratori cronici delle alte e/o basse vie aeree, associati nel 50% circa dei casi asitus viscerum inversus (sindrome di Kartagener). La DCP sembra essere sottodiagnosticata con diagnosi tardive in età adulta.Nella nostra clinica pediatrica nel 2005 è iniziata l’attività di un ambulatorio per le malattie respiratorie croniche ed è stato ela-borato un work-up diagnostico per la DCP. Allo scopo di valutare le diagnosi di DCP abbiamo analizzato le cartelle cliniche deipazienti che sono stati sottoposti a microscopia elettronica (ME) della mucosa delle vie aeree dal 2005 al 2012. Fra i 51 pazien-ti sottoposti a ME, in nove (18%) è stata identificata una alterazione congenita del ciglio mobile: l’età media dei pazienti alladiagnosi è stata di sei anni, e, in particolare, in quattro (44,4%) soggetti la diagnosi è stata posta ad una età inferiore ai quindi-ci mesi. Il difetto più frequente è stato l’assenza del braccio interno di dineina (5/9). Distress respiratorio neonatale, rinite cro-nica, tosse produttiva cronica, ventricolomegalia e/o Situs Viscerum Inversus sono risultati essere significativamente associati alladiagnosi di DCP. In conclusione, l’apertura di un ambulatorio per i pazienti pediatrici con patologia respiratoria cronica e l’ela-borazione di un work-up diagnostico per la DCP hanno permesso un riconoscimento precoce di questa patologia.
Accettato per la pubblicazione il 3 agosto 2013.
Corrispondenza: Rita Padoan, Centro di Supporto per la Fibrosi Cistica,Clinica Pediatrica, piazzale Spedali Civili 1, 25123 Brescia;
e-mail: [email protected]
Pneumologia Pediatrica 2013; 51: 52-56 R U B R I C A : e a n c o r a . . .

La discinesia ciliare primitiva: esperienza di un percorso diagnostico in ambulatorio 53
e il difficile reperimento delle indagini diagnosticherendono questa patologia sottodiagnosticata inEuropa [3].L’età media alla diagnosi varia nelle diverse casisti-che da tre anni e sette mesi a sei anni e sei mesi,con un ampio intervallo (dalla nascita ai dicianno-ve anni) [4-6]. Il progressivo coinvolgimento pol-monare comporta alterazioni della struttura bron-chiale (bronchiectasie) e conseguentemente undeclino della funzionalità respiratoria [7]. Questapuò essere preservata con una precoce diagnosiseguita dalla presa in carico da parte di CentriSpecialistici [8-9] anche se non esistono al momen-to presente Linee Guida nel trattamento della DCP,per cui i principali indirizzi terapeutici vengonomutuati dall’esperienza consolidata in FC [10].La conferma diagnostica avviene con la documen-tazione delle alterazioni della struttura del cigliomobile alla microscopia elettronica (ME) [11].Presso la nostra Clinica nel 2003 è stato istituitoun Centro di Supporto per la Fibrosi Cistica, a cuiè stato affiancato dal 2005 un Ambulatorio per lemalattie respiratorie croniche dell’età evolutiva. Aquesto ambulatorio vengono inviati, per una defi-nizione diagnostica, pazienti in età pediatrica (finoai diciotto anni) affetti da sintomatologia respira-toria cronica o recidivante.Lo scopo del presente studio è descrivere lanostra esperienza nell’elaborazione e nell’applica-zione di un work-up diagnostico per la DCP otte-nuto attraverso la collaborazione di diverse com-petenze specialistiche del Dipartimento Pediatricodegli Spedali Civili di Brescia, che è presidio per leMalattie Rare della Regione Lombardia.
Metodi
I pazienti inviati all’Ambulatorio per le malattierespiratorie croniche dell’età infantile seguono unpercorso diagnostico volto alla esclusione o con-ferma diagnostica di malattie croniche polmonariquali FC, malformazioni bronco-polmonari, disci-nesia ciliare, inalazione polmonare cronica, osecondari a immunodeficienza.La nostra attività, per quanto riguarda la DCP, è ini-ziata con la rilevazione di quale fosse, all’internodel nostro Istituto, il percorso diagnostico nei casisospetti. Prima del 2005, in presenza di un sospet-to clinico di DCP, i pazienti venivano inviati pressoaltre strutture ospedaliere (sia intra- che extrare-gionali) per l’esecuzione del test di conferma dia-gnostica, quale il brushing della mucosa nasale per
l’analisi alla microscopia elettronica. Data l’afferen-za di soggetti con quadro clinico suggestivo perDCP, sono state formalizzate le collaborazioni congli specialisti in Otorinolaringoiatria pediatrica perimplementare i test di screening e la possibilità dieseguire il brushing della mucosa nasale e/o labiopsia della mucosa tracheale presso la nostrasede. È stata contemporaneamente attivata unacollaborazione con l’Unità Operativa Complessadi Anatomia Patologica del Policlinico “Santa Mariaalle Scotte” - Azienda Ospedaliera UniversitariaSenese, per l’esecuzione dell‘ME a trasmissione.Allo scopo di valutare la nostra esperienza nelladiagnosi della DCP abbiamo analizzato le cartellecliniche di tutti i pazienti che nel periodo 2005-2012 sono stati sottoposti ad un’analisi delle ciliain ME. Sono stati quindi raccolti i dati anagrafici, lapresentazione clinica, e l’esito dello studio ultra-strutturale.È stata condotta un’analisi descrittiva dei dati rac-colti, inoltre sono stati valutati quali sintomi clinicifossero associati significativamente alla diagnosidella DCP usando il test “chi quadro” per il con-fronto fra le frequenze.
Risultati
Nel 2005, all’apertura dell’Ambulatorio, un solopaziente affetto da DCP era seguìto presso gliAmbulatori specialistici della nostra Clinica. Ilpaziente, maschio di quattordici anni, era stato dia-gnosticato nel 1996 (dati clinici non riportati).Dal 2005 il percorso diagnostico prevede che ipazienti, dopo la raccolta di un’accurata anamnesivolta a rilevare la presenza di segni e sintomi sug-gestivi per la DCP (distress respiratorio neonata-le, rinite cronica e infezioni ricorrenti delle alte e/obasse vie respiratorie) e/o l’anomala posizionedegli organi toraco-addominali (Situs ViscerumInversus, SVI o Situs Ambiguus, SA), vengano inizial-mente sottoposti a fibroscopia nasale per valutareil trofismo della mucosa naso-sinusale, per osser-vare le caratteristiche della secrezione nasale (sie-rosa, mucosa, purulenta) e per evidenziare even-tuali anomalie anatomiche naso-sinusali. Per quan-to riguarda i test di screening i pazienti di età supe-riore ai sei anni vengono sottoposti al test alla sac-carina, mentre la misurazione dell’ossido nitriconasale è disponibile presso la nostra strutturaattualmente solo per l’età adulta e lo studio delbattito ciliare viene effettuato presso un’altrastruttura regionale.

Padoan, et al. 54
Dal gennaio 2005 al dicembre 2012, sono stati sot-toposti a ME 51 pazienti, dei quali 28 maschi(54,9%), con una età media di cinque anni e diecimesi (± quattro anni e un mese, in un range com-preso tra un mese e diciassette anni) al momentodell’esame diagnostico. Il prelievo del campione dicellule respiratorie è stato eseguito in regimeambulatoriale presso l’Unità Operativa di Otorino-laringoiatria Pediatrica, tramite brushing della muco-sa del turbinato nasale, oppure in regime di day sur-gery, tramite biopsia della mucosa tracheale a livellodella carena tracheale. I frammenti bioptici, dopoessere stati adeguatamente fissati e conservati incondizioni idonee, sono stati inviati per l’indagineultrastrutturale presso la struttura convenzionata.Nella Tabella 1 sono riportate le caratteristiche cli-niche presentate dall’intera popolazione oggettodi studio.Nove pazienti su cinquantuno (18%, di cui ottomaschi) hanno ricevuto la diagnosi di DCP: duenel periodo 2005-2007, tre tra il 2008 e il 2010 e,infine, cinque casi sono stati identificati dal 2011 aldicembre 2012. L’età media dei pazienti alla dia-gnosi è stata di sei anni (range da tre mesi a dicias-sette anni). In particolare, in quattro pazienti(44,4%) l’età alla diagnosi è risultata inferiore aiquindici mesi. Inoltre, in cinque casi era presenteSVI (quattro casi) o SA addominale (un caso).La Tabella 2 mostra i dati clinici dei pazienti affettida DCP e la Figura 1 le diverse alterazioni ultra-strutturali identificate in ognuno di loro.
In ulteriori sei pazienti che presentavano SVI(quattro casi) o SA (due casi) l’ME non ha confer-mato il sospetto diagnostico della DCP per l’as-senza di alterazioni ultrastrutturali.L’analisi statistica ha dimostrato come nella nostracasistica il distress respiratorio neonatale senzaapparente causa, la rinite cronica, la tosse produt-tiva cronica, l’SVI/SA, l’ipoacusia trasmissiva e/o laventricolomegalia/macrocefalia siano significativa-mente associati alla diagnosi di DCP (Tabella 3).
Discussione
In questo studio abbiamo descritto l’esperienzanella diagnosi della DCP in un ambulatorio direcente istituzione dedicato alla patologia polmo-nare cronica dell’età pediatrica. L’inizio dell’attivitàsi è concretizzata nell’elaborazione di un work-updiagnostico, che, coinvolgendo diverse UnitàOperative del nostro Dipartimento Pediatrico, hapermesso in breve tempo di ottenere un incre-mento delle diagnosi di DCP e la presa in caricodi un numero discreto di pazienti. Abbiamo inoltreassistito ad una positiva ricaduta all’interno delDipartimento Pediatrico, osservando un progres-sivo aumento delle richieste di valutazione persospetta DCP/SK, per pazienti con SVI/SA inviati inprecedenza esclusivamente all’ambulatorio diGenetica pediatrica e/o per pazienti con infezionirespiratorie ricorrenti seguiti presso altri ambula-tori specialistici (asma, immunologia, infettivologia,otorinolaringoiatria).Nell’ambito della gestione dei pazienti con malat-tie rare nella nostra Azienda Ospedaliera, ilServizio Malattie Rare attivo nel nostro Presidio hacontribuito all’identificazione degli specialisti(pneumologi e otorinolaringoiatri pediatrici e del-l’età adulta) coinvolti nel work-up diagnostico-tera-peutico dei pazienti con DCP/SK, facilitando lacreazione di un percorso dedicato a tutte le fascedi età. Questa collaborazione consente una miglio-re gestione dei piccoli pazienti e dei loro familiari,che talvolta necessitano di un inquadramento cli-nico, e garantisce la possibilità di un graduale pas-saggio dall’Ambulatorio pediatrico all’Ambulatoriodell’età adulta. La disponibilità di un medico gene-tista attivo presso il Servizio Malattie Rare rende,inoltre, possibile la consulenza genetica per ipazienti e i loro familiari.I test di screening sono stati eseguiti solo in unaminoranza dei pazienti, e questo si è riflesso in unalto numero di richieste di ME, con un numero di
Tabella 1 Caratteristiche cliniche della popolazione oggettodi studio.
Caratteristiche cliniche N. pazienti %
Numero totale dei pazienti 51 -
Infezioni ricorrenti delle alte vie respiratorie 34 67%
Infezioni ricorrenti delle basse vie respiratorie 33 65%
Tosse produttiva cronica 21 41%
Rinite cronica 17 33%
Otite media cronica 15 29%
Distress respiratorio neonatale 11 22%
Situs viscerum inversus / situs ambiguus 11 22%
Sinusite cronica 8 16%
Ventricolomegalia /macrocefalia 7 14%
Ipoacusia trasmissiva 6 12%
Bronchiectasie 5 10%
Poliposi nasale 4 8%

La discinesia ciliare primitiva: esperienza di un percorso diagnostico in ambulatorio 55
diagnosi confermate del 18% (9/51). Sarà comun-que necessario, in futuro, implementare presso lanostra struttura gli esami di screening al momentonon disponibili come la valutazione dell’ossidonitrico nasale, e strutturare un network di Centrispecialistici per offrire al paziente un percorso dia-gnostico all’interno della nostra Regione che pre-veda una più completa valutazione del pazientecon sospetta DCP. L’età media di conferma dia-gnostica nella nostra casistica è in linea con laLetteratura (sei anni), ma nel 44,4% dei nostripazienti la diagnosi è stata effettuata precoce-mente, ad un’età inferiore ai quindici mesi, per-mettendo così una tempestiva presa in caricospecialistica.In prospettiva futura, mettere in atto iniziative diformazione per i pediatri, allo scopo di abbassare
l’età della diagnosi della patologia, per una preco-ce presa in carico da parte di Centri specialistici emigliorare quindi la prognosi dei pazienti con laDCP, deve essere considerata una delle prioritàdella Pneumologia pediatrica italiana. Inoltre, sem-bra necessario il coinvolgimento anche del gineco-logo ostetrico che effettua l’ecografia in corso digravidanza e dei neonatologi, che in presenza diun’anomala disposizione degli organi interni, e/o didistress respiratorio (in neonato, peraltro, sano)avviino il neonato al Centro specialistico per laconferma diagnostica della DCP [12].Anche nella nostra casistica, pur limitata, vieneconfermata la stretta associazione fra alcuni sinto-mi e la diagnosi della DCP: particolarmente inte-ressante è la significatività della presenza di ventri-colomegalia/macrocefalia.
Tabella 2 Caratteristiche cliniche dei pazienti con diagnosi di discinesia ciliare primitiva / sindrome di Kartagener. SVI, SitusViscerum Inversus; SA, Situs Ambiguus; TEM, microscopia elettronica a trasmissione; ODA, Assenza parziale o completa deibracci esterni di dineina; IDA, Assenza parziale o completa dei bracci interni di dineina.
Pz Età alla diagnosi Sesso [ref] SVI/SA Sintomi principali di presentazione Alterazione al TEM
1 6 mesi M SVI Ventricolomegalia, IDA + assonemainfezioni ricorrenti alte vie respiratorie asimmetrico
2 8 mesi M SVI Distress respiratorio neonatale, IDAventricolomegalia, infezioni ricorrenti
delle alte vie respiratorie, tosse produttiva cronica
3 1 anno M (17) – Piomucocele, infezioni ricorrenti delle basse IDAe alte vie respiratorie, tosse produttiva cronica,
otite media cronica
4* 1 anno e 3 mesi M (18) – Distress respiratorio neonatale, Aplasia ciliare ventricolomegalia, infezioni ricorrentidelle alte e basse vie respiratorie,
tosse produttiva cronica
5 6 anni M SA addominale Macrocefalia, infezioni ricorrenti delle alte e IDAbasse vie respiratorie, tosse produttiva cronica,
ipoacusia trasmissiva
6 8 anni e 2 mesi M SVI Distress respiratorio neonatale, infezioni ODAricorrenti delle alte e basse vie respiratorie,tosse produttiva cronica, otite media cronica
7 8 anni e 4 mesi M SVI Otite media cronica, ipoacusia trasmissiva IDA
8* 11 anni e 2 mesi F (18) – Distress respiratorio neonatale, infezioni Aplasia ciliarericorrenti delle alte e basse vie respiratorie,
tosse produttiva cronica,bronchiectasie, macrocefalia
9 17 anni M – Distress respiratorio neonatale, infezioni Cilia corte e/oricorrenti delle alte e basse vie respiratorie, assenti
sinusite cronica, bronchiectasie, ipoacusia trasmissiva
* Fratelli

Padoan, et al. 56
Nella nostra casistica sei soggetti, pur in presenzadi un quadro clinico tipico (SVI/SA) non presenta-no alterazioni strutturali del ciglio mobile all’ME. Lacapacità di diagnosticare correttamente la DCPpuò essere migliorata affiancando all’ME la valuta-zione dell’ossido nitrico nasale e l’identificazionedelle mutazioni nei geni che causano la DCP. La possibilità di questo approccio diagnosticocombinato dimostra che almeno il 30% deipazienti con DCP hanno un’ultrastruttura ciliarenormale [13]. Infatti sono stati segnalati soggetticon DCP, senza alterazioni del ciglio mobile, manei quali sono state riconosciute alterazioni delgene DNAH11 [14]. D’altra parte, è stato stimatoche solo il 25% circa dei soggetti con SVI sonoaffetti da DCP [15]. In questi casi sarà necessariosottoporre i pazienti ad analisi molecolare del
gene DNAH11 per giungere ad una corretta dia-gnosi. Nel nostro Istituto, la recente acquisizione diuno strumento diagnostico di ultima generazionepermetterà anche lo studio molecolare per ipazienti con DCP/SK.
Conclusioni
In conclusione, l’apertura di un Ambulatorio specia-listico per le patologie respiratorie croniche dell’etàpediatrica, e la definizione all’interno della nostrastruttura ospedaliera di un work-up diagnostico perla DCP e la SK hanno avuto come ricaduta unamaggior sensibilità nel sospetto diagnostico daparte degli operatori della nostra Clinica Pediatrica,con un incremento delle conferme diagnostichesoprattutto nell’ultimo biennio, permettendo la
Figura 1 Immagine in microscopia elettronica a trasmissione (1a-d ingrandimento originale x110.00; tutte le imma-gini sono in sezione perfettamente trasversale). a) Assonema ciliare normale, in cui tutte le substrutture sono benrappresentate; b) Assenza del braccio esterno di dineina; c) Assenza del braccio interno di dineina; d) Assenza delbraccio interno di dineina e difetti dei ponti radiali, con conseguente asimmetria dell’assonema; e) Totale assenza dicilia, sostituite da lunghi microvilli (aplasia ciliare), ingrandimento originale x9.800.
Coppia di microtuboliperiferici
Ponti radiali
Coppia centrale
Braccio internodi dineina
Braccio esternodi dineina
a
b c
d e
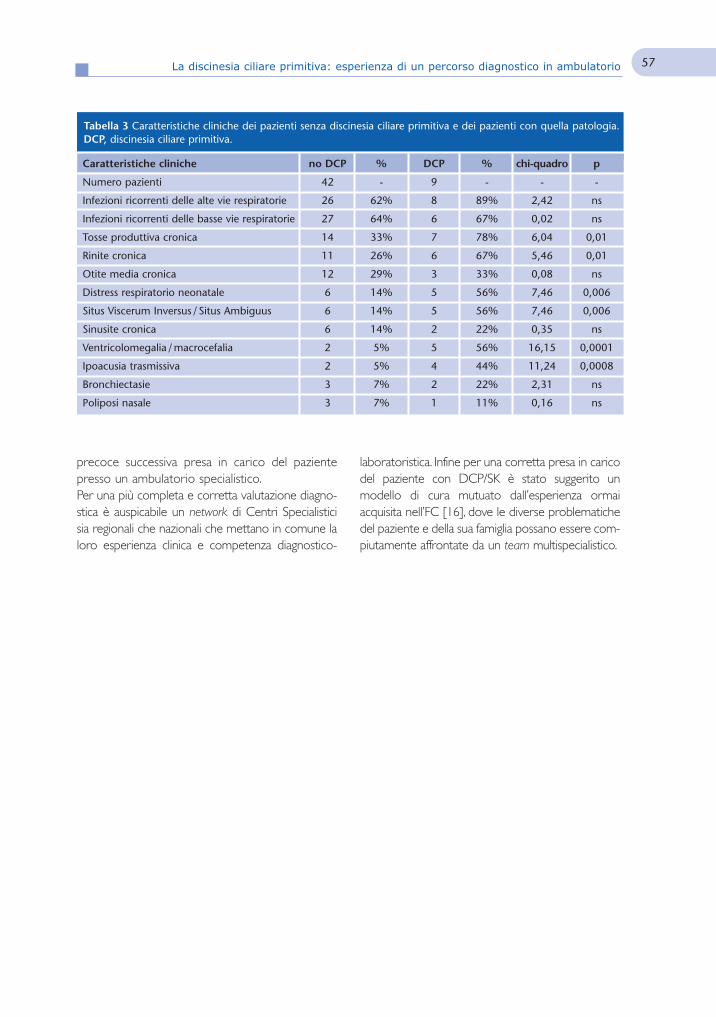
La discinesia ciliare primitiva: esperienza di un percorso diagnostico in ambulatorio 57
Tabella 3 Caratteristiche cliniche dei pazienti senza discinesia ciliare primitiva e dei pazienti con quella patologia.DCP, discinesia ciliare primitiva.
Caratteristiche cliniche no DCP % DCP % chi-quadro p
Numero pazienti 42 - 9 - - -
Infezioni ricorrenti delle alte vie respiratorie 26 62% 8 89% 2,42 ns
Infezioni ricorrenti delle basse vie respiratorie 27 64% 6 67% 0,02 ns
Tosse produttiva cronica 14 33% 7 78% 6,04 0,01
Rinite cronica 11 26% 6 67% 5,46 0,01
Otite media cronica 12 29% 3 33% 0,08 ns
Distress respiratorio neonatale 6 14% 5 56% 7,46 0,006
Situs Viscerum Inversus / Situs Ambiguus 6 14% 5 56% 7,46 0,006
Sinusite cronica 6 14% 2 22% 0,35 ns
Ventricolomegalia /macrocefalia 2 5% 5 56% 16,15 0,0001
Ipoacusia trasmissiva 2 5% 4 44% 11,24 0,0008
Bronchiectasie 3 7% 2 22% 2,31 ns
Poliposi nasale 3 7% 1 11% 0,16 ns
precoce successiva presa in carico del pazientepresso un ambulatorio specialistico.Per una più completa e corretta valutazione diagno-stica è auspicabile un network di Centri Specialisticisia regionali che nazionali che mettano in comune laloro esperienza clinica e competenza diagnostico-
laboratoristica. Infine per una corretta presa in caricodel paziente con DCP/SK è stato suggerito unmodello di cura mutuato dall’esperienza ormaiacquisita nell’FC [16], dove le diverse problematichedel paziente e della sua famiglia possano essere com-piutamente affrontate da un team multispecialistico.

Padoan, et al. 58
1. Bush A, Chodhari R, Collins N, et al. Primary ciliarydyskinesia: current state of the art. Arch Dis Child 2007;92; 1136-1140.
2. Vallet C, Escudier E, Roudot-Thoraval F, et al.Primary ciliary dyskinesia presentation in 60 childrenaccording to ciliary ultrastructure. Eur J Pediatr 2013;172; 1053-1060.
3. Boon M, Jorissen M, Proesmans M, et al. Primaryciliary dyskinesia, an orphan disease. Eur J Pediatr 2012;172; 151-162.
4. Beucher J, Chambellan A, Segalen J, et al. Primaryciliary dyskinesia: a retrospective review of clinical andparaclinical data. Rev Mal Respir 2011; 28; 856-863.
5. Busquets RM, Caballero-Rabasco MA, Velasco M, etal. Primary Ciliary Dyskinesia: Clinical Criteria IndicatingUltrastructural Studies. Arch Broncneumol 2013; 49;99-104.
6. Coren ME, Meeks M, Morrison I, et al. Primary ciliarydyskinesia: age at diagnosis and symptom history. ActaPediatr 2002; 91: 667-669.
7. Kennedy MP, Noone PG, Leigh MW, et al. High-reso-lution CT of patients with primary ciliary dyskinesia. Am JRoentgenol 2007; 188; 1232-1238.
8. Ellerman A, Bisgaard H. Longitudinal study of lungfunction in a cohort of primary ciliary dyskinesia. EurRespir J 1997; 10: 2376-2379.
9. Hellinckx J, Demets M, De Boeck K. Primary ciliarydyskinesia: evolution of pulmonary function. Eur J Pediatr1998; 157; 422-426.
10. Stillwell PC, Wartchow EP, Sagel SD. Primary CiliaryDyskinesia in Children: A Review for Pediatricians, Allergists,and Pediatric Pulmonologists. Pediatr Allergy ImmunolPulmonol 2011; 24; 191-196.
11. Leigh MW, O’Callaghan C, Knowles MR. The chal-lenges of diagnosing primary ciliary dyskinesia. Proc AmThorac Soc 2011; 8; 434-437.
12. Palumbo E, Nasca G, Malorgio C, et al. Diagnosi neo-natale di discinesia ciliare primitiva: descrizione di un caso.Medico e Bambino pagine elettroniche 2008; 11 (2).Disponibile su http://www.medicoebambino.com
13. Knowles MR, Daniels LA, Davis SD, et al. PrimaryCiliary Dyskinesia: recent advances in diagnostics, genetics,and characterization of clinical disease. AJRCCM 2013Jun 24 [Epub ahead of print].
14. Schwabe GC, Hoffmann K, Loges NT, et al. Primaryciliary dyskinesia associated with normal axoneme ultra-structure is caused by DNAH11 mutations. Hum Mutat2008; 29; 289-298.
15. Aylsworth AS. Clinical aspects of defects in the deter-mination of laterality. Am J Med Genet 2001; 101; 345-355.
16.O’Callaghan C, Chilvers M, Hogg C, et al. Diagnosingprimary ciliary dyskinesia. Thorax 2007; 62; 656-657.
17. Berlucchi M, Maroldi R, Aga A, et al. Ethmoid muco-cele: a new feature of primary ciliary dyskinesia. PediatrPulmonol 2010; 45; 197-201.
18. Berlucchi M, de Santi M, Bertoni E, et al. Ciliary apla-sia associated with hydrocephalus: an extremely rare occur-rence. Eur Arch Otorinolaryngol 2012; 269; 2295-2299.
BIBLIOGRAFIA
Bibliografia

59Conferenze e meetingConferences and meetings
OTTOBRE 2013
Corso NIV. Ventilazione Non Invasiva,dalla A alla ZBologna, 1-2 ottobre 2013Segreteria organizzativaStart Promotion - MilanoTel. 02.67071383E-mail: [email protected]
Attualità in Pediatria 2013Matera, 5 ottobre 2013Segreteria organizzativaServizio Eventi Formativi ECM OspedalePediatrico “Bambino Gesù” - RomaTel. 06.68592290-2411E-mail: [email protected]
VIII World Congress of Immunopathology,�Respiratory Allergy & AsthmaDubai (Emirati Arabi Uniti), 12-15 ottobre 2013Segreteria organizzativaWorld Immunopathology OrganizationTel. +7 (495) 735-1414�E-mail: [email protected]
III Corso di Aggiornamento in AllergologiaRoma, 18 ottobre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
Novità in tema di Pneumologiaed Allergologia pediatricaGenova, 18-19 ottobre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
Mixing 2013Tra aggiornamento e formazione.Percorso interattivo per specialistiJesi (AN), 18-19 ottobre 2013Segreteria organizzativaAIM Group International - Firenze Tel. 055.233881Fax 055.2480246E-mail: [email protected]
III Consensus Conference AIMAR in Medicina RespiratoriaCosa definisce l’appropriatezza nellagestione delle cronicità respiratorieRoma, 18-19 ottobre 2013Segreteria organizzativaDynamicom - MilanoTel. 02.89693784-3757Fax 02.201176E-mail: [email protected]
L’urgenza formativa... continuaPadova, 24-26 ottobre 2013Segreteria organizzativa: Dipartimento per la salute della Donna e delBambino (S. Baggio)Tel. 049.8213526Info: www.sdb.unipd.it/urgenzaformativa
VIII Congresso regionale FIMP SiciliaUn grande progetto per diventarebambinoRagusa, 25-27 ottobre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
Conferenze e meetingConferences and meetings

60
114° Congresso nazionale della SocietàItaliana di Medicina InternaRoma, 26 ottobre 2013Segreteria organizzativaAristea Education - RomaTel. 06.845431Fax 06.84543700e-mail: [email protected]
CHEST 2013 Annual CongressChicago (Illinois, USA), 26-31 ottobre 2013Segreteria organizzativaAmerican College of CHEST PhysiciansTel. (800)3432227Fax (800)847498Info: www.chestnet.org
Assistenza al neonato in sala partoe uso della maschera laringeaNapoli, 28 ottobre 2013Segreteria organizzativaMaliga Viaggi ed Organizzazione Eventi - NapoliTel. 081.7282538Fax 081.7663029E-mail: [email protected]
NOVEMBRE 2013
VI Congresso nazionale della Pediatriaospedaliera italianaRoma, 7-9 novembre 2013Segreteria organizzativaPenta Eventi - RomaTel. 06.45491195Fax 06.45435442E-mail: [email protected]
ACCP Capitolo italiano.Corso di Aggiornamentoin Pneumo-CardiologiaFerrara, 8 novembre 2013Segreteria organizzativaVictory Project Congressi - MilanoTel. 02.89053524Fax 02.201395E-mail: [email protected]
XVI Giornata pediatrica irpinaConvegno nazionale di aggiornamentoSerino (AV), 9 novembre 2013Segreteria organizzativaConsorzio ISMESS - SalernoTel. 089.2578642E-mail: [email protected]
Highligths in allergy and respiratorydiseasesGenova, 14-16 novembre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
XVI MasterVASDiagnosi e terapia delle patologierespiratorie. I sintomi Ercolano (NA), 15-17 novembre 2013Segreteria organizzativaBle Consulting - CasertaTel. 0823.301653-361086Fax 0823.363828
XII Giornata mondiale della BPCOVIII Conferenza nazionale della BPCORoma, 20 novembre 2013Segreteria organizzativaEffeti - MilanoTel. 02.3343281Fax 02.38002105E-mail: [email protected]
2° Congresso nazionale AMIETIPEmergenza e terapia intensivae semintensiva del bambinoNapoli, 21-23 novembre 2013Segreteria organizzativaCongress Lab - FirenzeTel. 055.5539739Fax 055.5539741E-mail: [email protected]
Conferenze e meetingConferences and meetings

III focus di aggiornamento in TossicologiapediatricaNapoli, 27 novembre 2013Segreteria organizzativaPenta Eventi - RomaTel. 06.45491195Fax 06.45435442E-mail: [email protected]
DICEMBRE 2013
V° Corso Residenziale“Il pediatra, l’infermiere e il bambino conpatologia grave: La terapia semintensiva”Roma, 4-6 dicembre 2013Segreteria OrganizzativaCenter - Comunicazione & congressi - NapoliInformazioni: [email protected]
Nuove prospettive per la gestionedell'asma e della BPCO. Le nuove Linee Guida GINA e GOLDIII edizionePisa, 6 dicembre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
XXVI Congresso nazionale confronti inPediatria. Il bambino che diventa grande. Cosa deve sapere il pediatra, cosa devesapere il medico dell'adulto Trieste, 6-7 dicembre 2013Segreteria organizzativaQuickline Congressi - TriesteTel. 040.363586Fax 040.7606590E-mail: [email protected]
Corso Classic di Ventilazione ArtificialeRoma, 12-13 dicembre 2013Segreteria organizzativaStart Promotion - MilanoTel. 02.67071383Fax: 02.67072294E-mail: [email protected]
1st International Workshop on Lung Health, COPD: The New ParadigmsMonaco (Principato di Monaco), 12-14 dicembre2013Segreteria organizzativaPubli Créations - Monaco (Principato di Monaco)Tel. +(377)97973555Fax +(377)9797550E-mail: [email protected]
Thesis 2013. Percorsi interattivi e formativi pediatriciNapoli, 13-14 dicembre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
Update in asthma and allergy at Christmas time (and Christmas wishes)Brescia, 14 dicembre 2013Segreteria organizzativaiDea congress - RomaTel. 06.36381573Fax 06.36307682E-mail: [email protected]
Pneumologia in LombardiaMilano, 19-20 dicembre 2013Segreteria organizzativaVictory Project Congressi - MilanoTel. 02.89053524Fax 02.201395E-mail: [email protected]
61Conferenze e meetingConferences and meetings

21-23 November 2013 (Naples, Italy)
Primary ciliary dyskinesia: sharing knowledge and experience across Europe
EDUCATIONAL AIMSThis course aims to provide participants with practical guidance for suspecting, screening anddiagnosing primary ciliary dyskinesia (PCD).
TOPICS• Epidemiology and burden of PCD• Screening tools and diagnostic procedures• PCD genetics• Major clinical presenting features• Diagnostic challenges in PCD (genetics, upper and lower airways disease with particular attention to the differential diagnosis of non-CF bronchiectasis in adults and in children)
• PCD treatment, including chest physiotherapy• Quality of life from physicians, patients and families perspectives• PCD in adult life with particular attention to fertility issues• Future of cilia research in Europe
TARGET AUDIENCE• General paediatricians• Paediatric and adult pulmonologists• Ear, nose and throat specialists• Paediatric and adult allergists• Pathologists
VENUEFederico II University Congress CentreVia Partenope 3680121 Naples, Italy
Per informazioni:http://www.ersnet.org/education/courses/item/4499-primary-ciliary-dyskinesia-sharing-knowledge-and-experience-across-europe.html

Articoli del prossimo numeroForthcoming articles
Novità dal congresso SIMRI 2013
Articoli del prossimo numero 63


















