Corso di “Farmacologia” - fvcalabria.unicz.it · – FANS – warfarin – ceftiofur –...
155
Corso di “Farmacologia” Legame Farmacoproteico, metabolismo dei farmaci ed eliminazione
-
Upload
trinhkhanh -
Category
Documents
-
view
214 -
download
0
Transcript of Corso di “Farmacologia” - fvcalabria.unicz.it · – FANS – warfarin – ceftiofur –...

Corso di “Farmacologia”
Legame Farmacoproteico, metabolismo dei farmaci ed eliminazione
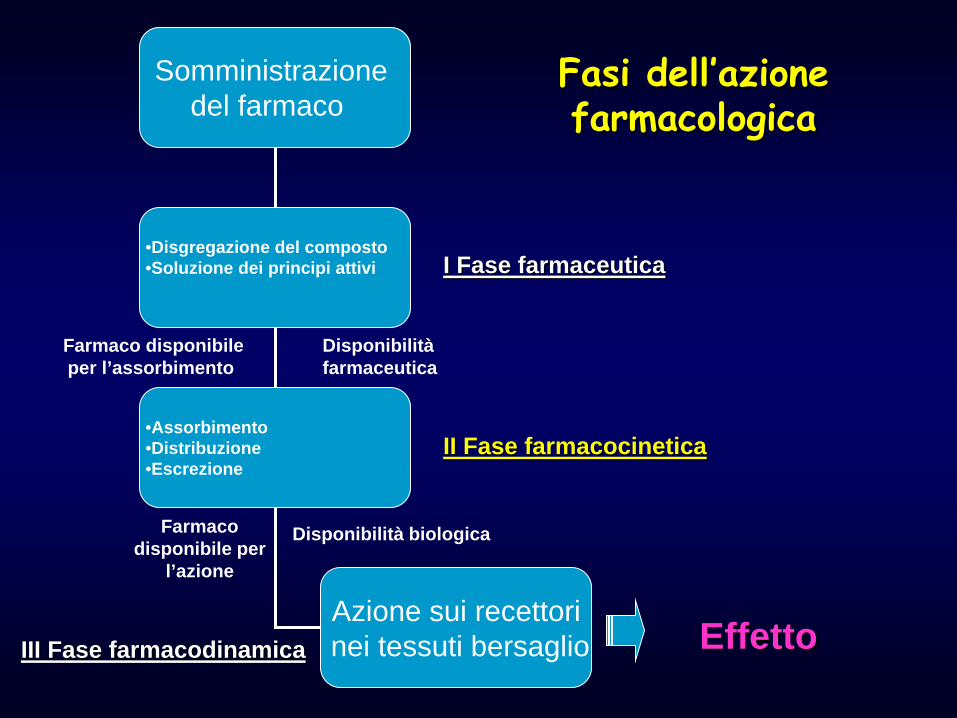
Fasi dell’azione farmacologica
Somministrazione del farmaco
•Disgregazione del composto •Soluzione dei principi attivi
•Assorbimento •Distribuzione •Escrezione
Azione sui recettori nei tessuti bersaglio
I Fase farmaceutica
II Fase farmacocinetica
III Fase farmacodinamica Effetto
Farmaco disponibile per l’assorbimento
Disponibilità farmaceutica
Farmaco disponibile per
l’azione
Disponibilità biologica

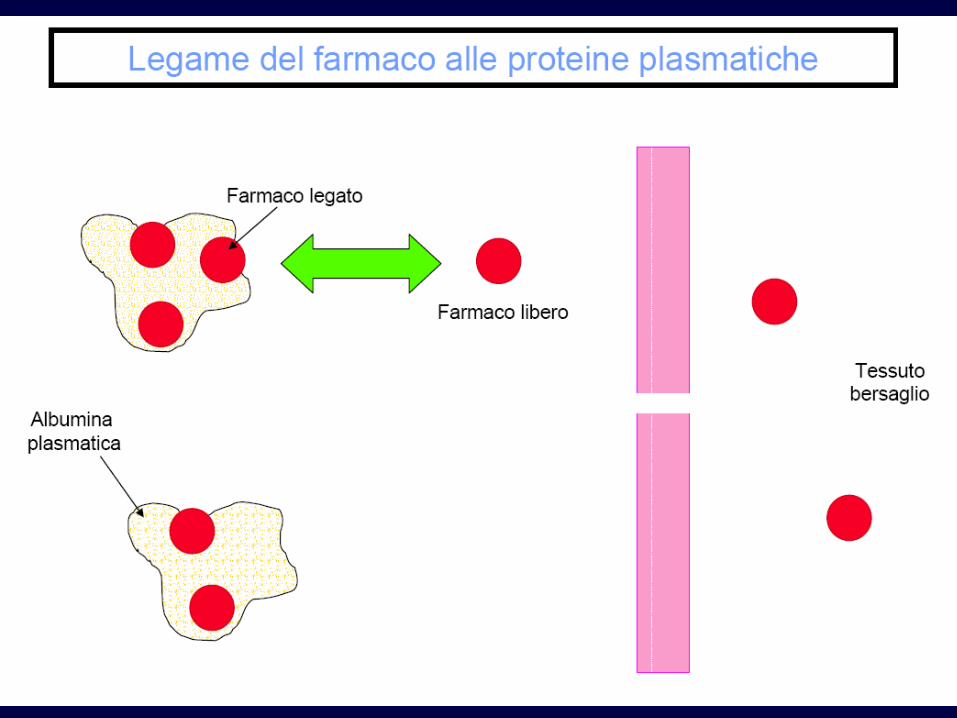
Legame alle proteine
• Soprattutto alle albumine
• Il farmaco legato non attraversa le membrane
• Equilibrio continuo tra parte libera e legata

Legame farmaco-proteine
50% legato 90% legato
Farmaco libero (5)
Farmaco legato (5)
Farmaco totale (10)
Farmaco libero (1)
Farmaco legato (9)
Farmaco totale (10)
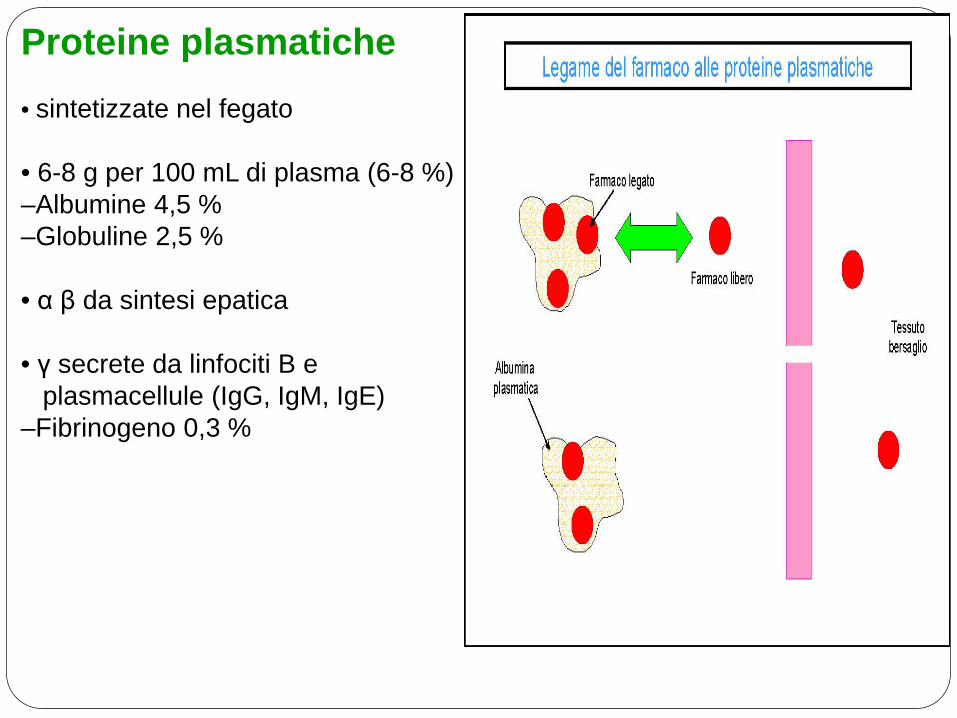
Proteine plasmatiche
• sintetizzate nel fegato • 6-8 g per 100 mL di plasma (6-8 %) –Albumine 4,5 % –Globuline 2,5 % • α β da sintesi epatica • γ secrete da linfociti B e plasmacellule (IgG, IgM, IgE) –Fibrinogeno 0,3 %

Farmaci molto legati... • Legati alle albumine o alle glicoproteine alfa:
– FANS – warfarin – ceftiofur – doxiciclina – furosemide – chinidina – diazepam – propranololo

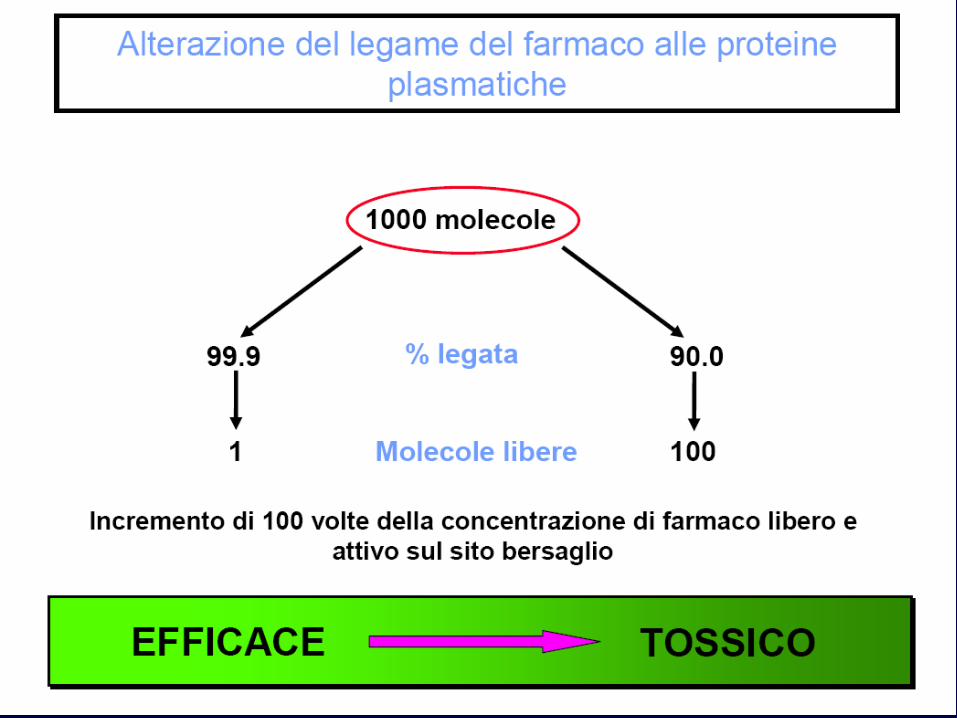
Fattori che modificano il legame farmaco-proteico
• Ogni modificazione del tasso di proteine plasmatiche: – Insufficienza epatica – Insufficienza renale – Enteropatie – Parassitosi – Ustioni
• Se aumenta la quota libera: – Aumento dell’effetto – Aumento della velocità di eliminazione

I farmaci possono legarsi anche con costituenti cellulari tissutali quali proteine, fosfolipidi, nucleoproteine.
Si possono così avere dei siti di deposito a livello di alcuni tessuti nei cui confronti un farmaco ha un particolare TROPISMO
Esempi di tropismo: Tetracicline (antibatterici) verso il tessuto osseo Tiopentale (anestetico) verso il tessuto adiposo Clorochina (antimalarico) verso il fegato Amiodarone (antiaritmico) verso la tiroide
Siti di deposito cellulare

Giocheranno un ruolo importante nel processo di competizione di legame con le proteine i seguenti fattori:
•concentrazione di farmaco • affinità di legame (il farmaco a maggiore affinità spiazza il farmaco a minore affinità)
Così sulfamidici, fenilbutazone, salicilati spiazzano bilirubina, warfarin, tolbutamide di conseguenza aumenta
l’ effetto farmacologico e quindi anche la tossicità l’eliminazione renale e quindi si ha una diminuzione dell’effetto farmacologico.
Fattori che influenzano il legame con le proteine plasmatiche
Ipoalbuminemia, Uremia, Età

Altri siti di accumulo sono costituiti: 2) Dal grasso
ove i farmaci liposolubili possono depositarsi in quantità non trascurabili. Il tiopentale, farmaco lipososolubile si lega preferenzialmente al tessuto adiposo.
3) Dai globuli rossi l’ emoglobina ha un eccesso di cariche cationiche e lega preferenzialmente anioni (es. clortalidone).
4) Dalle ossa le tetracicline formano dei complessi con gli ioni calcio delle ossa e così diminuiscono le loro concentrazioni libere.
5) Dai polmoni Ad esempio la concentrazione di antidepressivi triciclici nei polmoni supera quella del plasma.

Altri siti di accumulo sono costituiti:
5) Dal fegato ove i farmaci liposolubili come la chinacrina possono depositarsi in quantità notevoli. I farmaci antimalarici (p. es., la clorochina) producono concentrazioni nei Gl. Bianchi e nelle cellule epatiche migliaia di volte superiori a quelle plasmatiche. Il farmaco accumulato è in equilibrio con il farmaco presente nel plasma e si sposta nel compartimento intravascolare man mano che procede la sua eliminazione dall'organismo. Esistono determinati antibiotici che possono causare la formazione di calcoli biliari: uno di questi è il ceftriaxone, utilizzato nella cura di numerose infezioni, i farmaci estroprogestinici, etc. Allo stesso modo, i tiazidi, farmaci diuretici, possono provocare malattie della colecisti in pazienti già sofferenti di calcoli biliari. L’octreotide, un farmaco delle nuove generazioni di “statine”, impedisce alla colecisti di svuotarsi dopo un pasto ricco di grassi, lasciando una grande quantità di bile che dà origine a calcoli.

Volume di distribuzione (Vd) Attraverso il “volume di distribuzione” si può interpretare la capacità di ciascun farmaco di distribuirsi di più o di meno nell’organismo Il Vd riflette la quantità di farmaco che rimane nel sangue dopo il suo asssorbimento
– Più farmaco nel sangue minore il volume di distribuzione
– Meno farmaco nel sangue maggiore il volume di distribuzione

Formula per calcolare il volume di distribuzione
Vd = D
C
Vd = volume of distribuzione D = dose (assumendo 100% assorbimento) C = concentrazione farmaco nel sangue
La conoscenza del volume di distribuzione di un farmaco consente di calcolare la sua concentrazione plasmatica in base alla dose somministrata

Supponiamo che il farmaco XXX dia una concentrazione plasmatica di 0,1 mg/ml dopo somministrazione di 1 g per via e.v.
In genere il Vd si esprime come litri/kg peso corporeo. Se la persona dell’esempio pesa 50 kg il volume di distribuzione sarà: Vd = 10L/50kg = 0,2 L/Kg
Volume di distribuzione
Vd = 1000 mg
0,1 mg/ml = 10000 ml = 10 litri
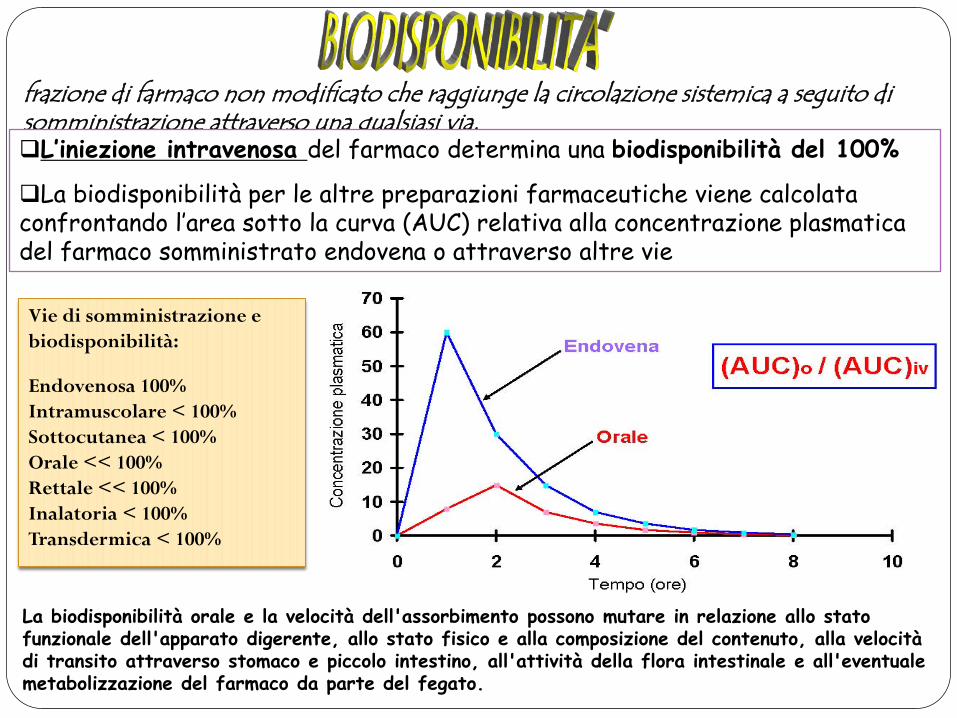
frazione di farmaco non modificato che raggiunge la circolazione sistemica a seguito di somministrazione attraverso una qualsiasi via. L’iniezione intravenosa del farmaco determina una biodisponibilità del 100%
La biodisponibilità per le altre preparazioni farmaceutiche viene calcolata confrontando l’area sotto la curva (AUC) relativa alla concentrazione plasmatica del farmaco somministrato endovena o attraverso altre vie
Vie di somministrazione e biodisponibilità:
Endovenosa 100% Intramuscolare < 100% Sottocutanea < 100% Orale << 100% Rettale << 100% Inalatoria < 100% Transdermica < 100%
La biodisponibilità orale e la velocità dell'assorbimento possono mutare in relazione allo stato funzionale dell'apparato digerente, allo stato fisico e alla composizione del contenuto, alla velocità di transito attraverso stomaco e piccolo intestino, all'attività della flora intestinale e all'eventuale metabolizzazione del farmaco da parte del fegato.

Parametri Farmacocinetici Sono parametri che permettono di prevedere
e studiare il comportamento cinetico dei
farmaci; in particolare, essi consentono di
descrivere quantitativamente i processi della
farmacocinetica (ADME).

Vie enterali
Vie parenterali
Via Biodisponibilità % Caratteristiche
Via Biodisponibilità % Caratteristiche

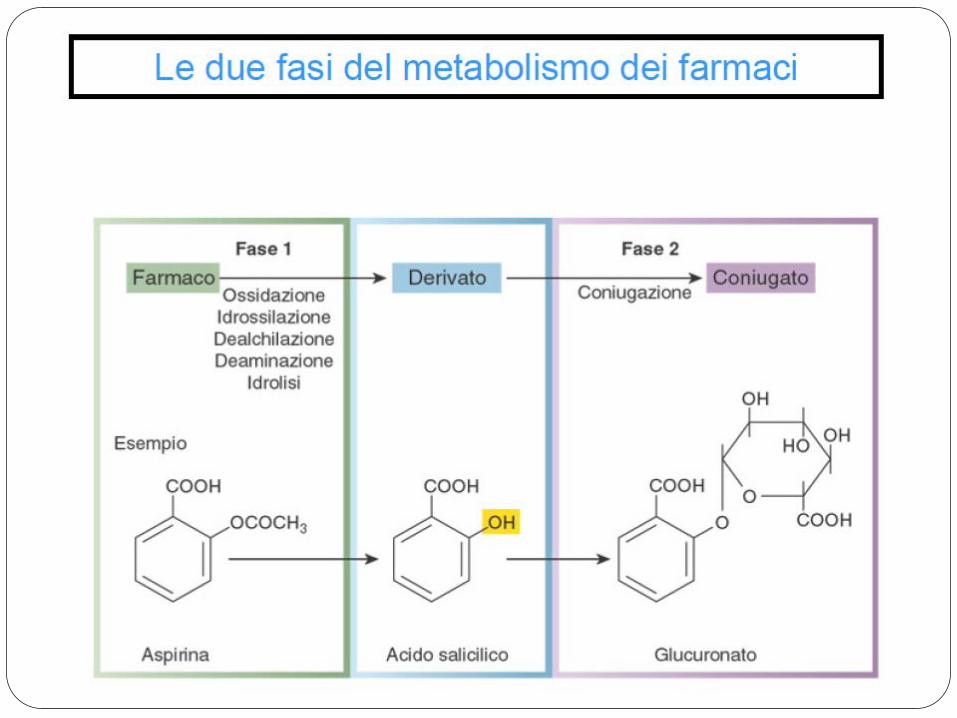
Con il termine metabolismo si intendono le modificazioni chimiche che un farmaco subisce nell’organismo.
Sede principale dei processi metabolici è il FEGATO per l’azione degli enzimi microsomiali delle cellule epatiche.
Altre sedi di metabolizzazione di minore importanza sono il rene, il polmone, l’intestino (anche per azione della flora batterica).
METABOLISMO O BIOTRASFORMAZIONE DEI FARMACI

Le reazioni chimiche con le quali si attua il metabolismo dei farmaci sono: OSSIDAZIONE RIDUZIONE FASE I (Citocromo P450) IDROLISI
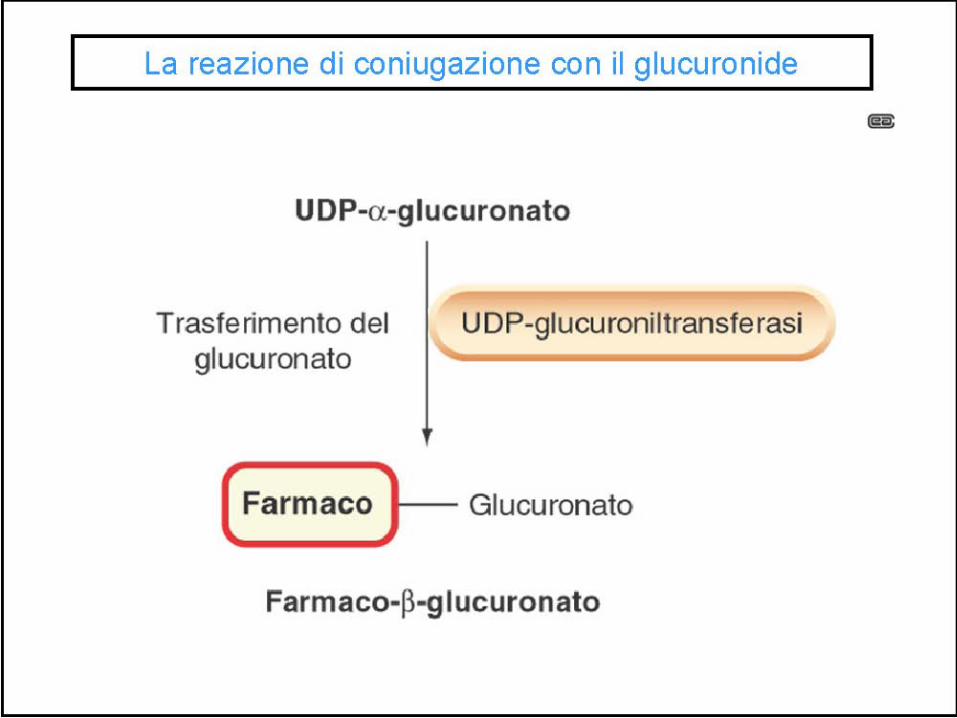
CONIUGAZIONE FASE II
Pazienti con patologie epatiche possono avere dei
problemi di metabolizzazione di farmaci.
METABOLISMO O BIOTRASFORMAZIONE DEI FARMACI
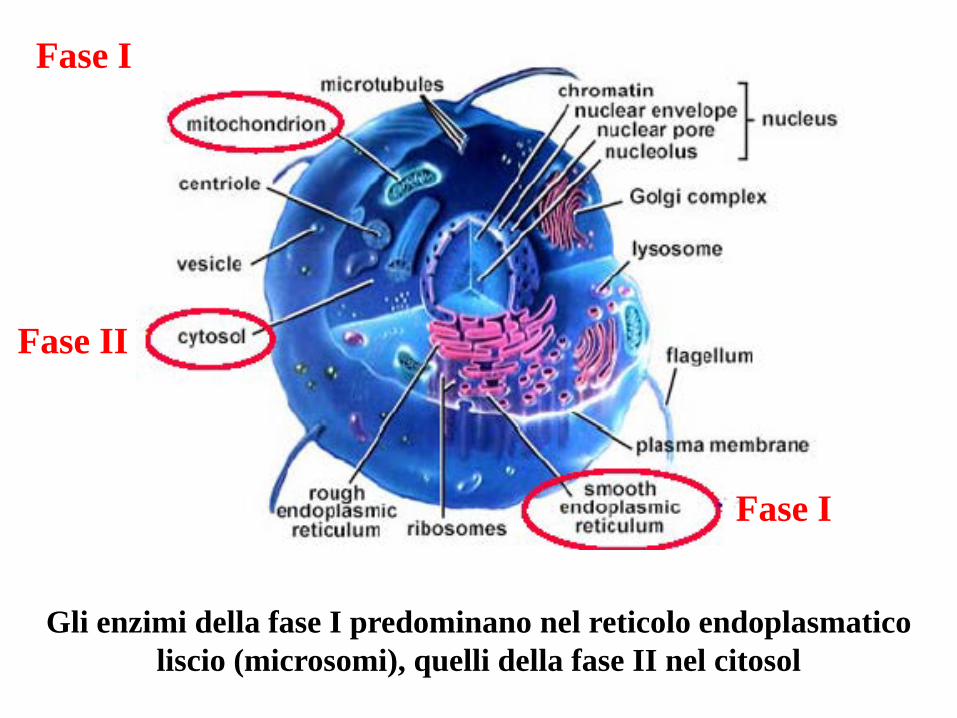
Fase I
Fase I
Fase II
Gli enzimi della fase I predominano nel reticolo endoplasmatico liscio (microsomi), quelli della fase II nel citosol
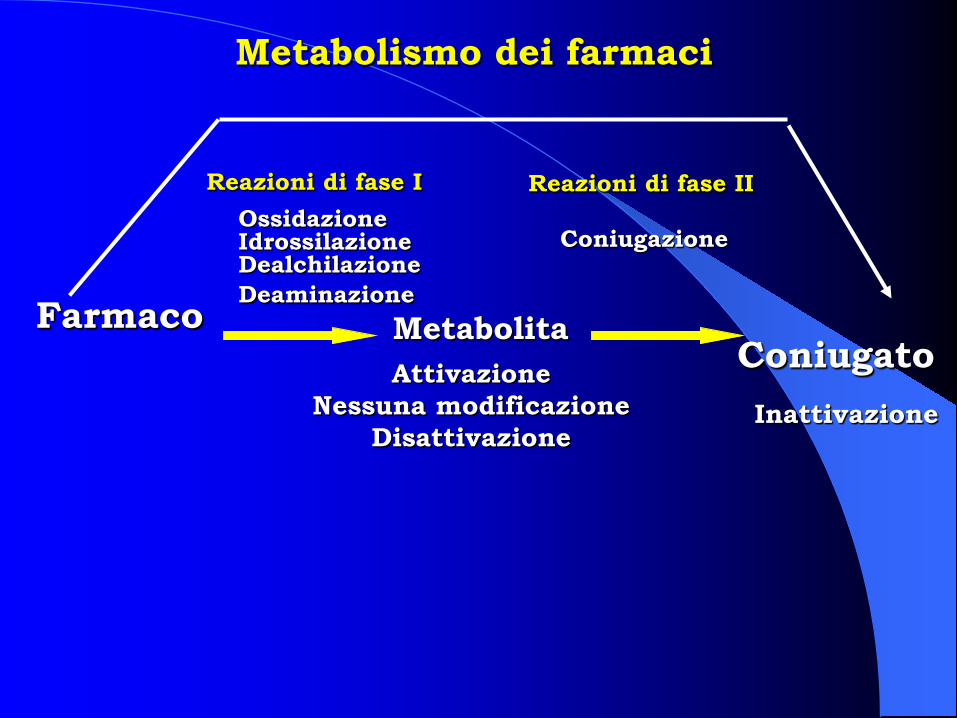
Metabolismo dei farmaci
Farmaco Coniugato
Metabolita
Reazioni di fase I Reazioni di fase II Ossidazione Idrossilazione Dealchilazione Deaminazione
Coniugazione
Attivazione Nessuna modificazione
Disattivazione Inattivazione

Le caratteristiche lipofile, che promuovono il passaggio dei farmaci attraverso le membrane biologiche e il conseguente accesso ai siti d’azione, ostacolano la loro eliminazione dall’organismo.
La biotrasformazione dei farmaci ha un’importanza fondamentale per la cessazione della loro attività biologica e per l’eliminazione dall’organismo.
Generalmente le reazioni di biotrasformazione danno origine a composti più polari, metaboliti inattivi che vengono più facilmente escreti dall’organismo.
METABOLISMO O BIOTRASFORMAZIONE DEI FARMACI
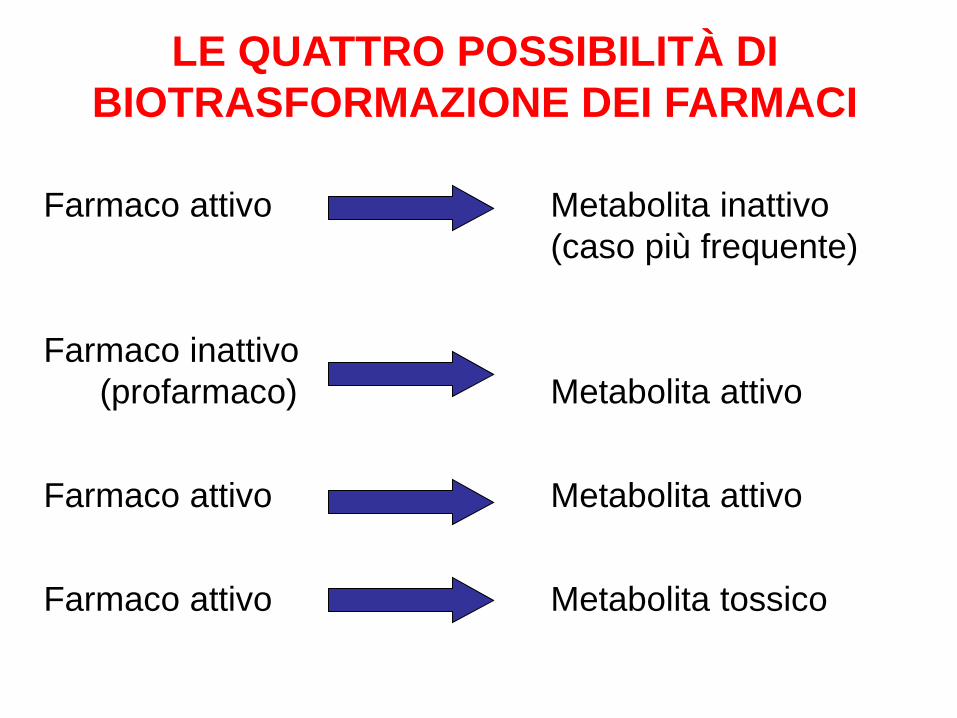
Farmaco attivo Metabolita inattivo (caso più frequente)
Farmaco inattivo (profarmaco) Metabolita attivo
Farmaco attivo Metabolita attivo
Farmaco attivo Metabolita tossico
LE QUATTRO POSSIBILITÀ DI BIOTRASFORMAZIONE DEI FARMACI

LEVODOPA Utilizzata nel morbo di Parkinson è il precursore inerte della dopamina. La conversione metabolica (decarbossilazione) avviene nel SNC, principalmente entro i terminali presinaptici dei neuroni dopaminergici nello striato. Nella pratica clinica la levodopa viene somministrata assieme alla carbidopa o alla benserazide, inibitori periferici della decarbossilasi, per impedire che venga inattivata prima di raggiungere il SNC.
CODEINA Analgesico oppiaceo (contenuto nell’oppio) che esplica la sua azione antidolorifica dopo trasformazione nell’organismo in morfina.
ENALAPRIL-QUINAPRIL-FOSINOPRIL-RAMIPRIL ACE-inibitori che diventano attivi quando convertiti, dalle esterasi epatiche, rispettivamente a enalaprilato, quinaprilato, fosinoprilato, ramiprilato.
ALCUNI ESEMPI DI PROFARMACI

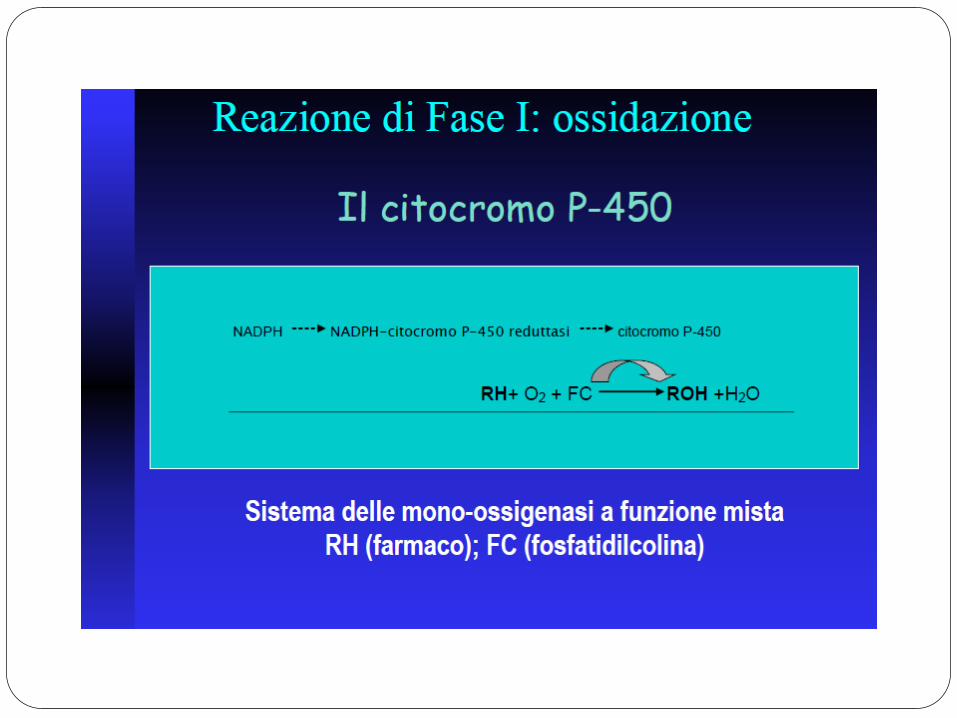
E’ costituito da proteine di membrana, contenenti un gruppo eme, localizzate nel reticolo endoplasmatico liscio, prevalentemente a livello epatico. Producono una caratteristica banda di assorbimento spettrofotometrico a 450 nM.
La famiglia del gene P450 (CYP) si è differenziata (in miliardi di anni) garantendo il metabolismo di un numero sempre crescente di composti chimici ambientali, tossine alimentari, farmaci.
La superfamiglia di enzimi che ne è derivata catalizza una varietà enorme di reazioni (ossidazione, riduzione) nei confronti di diversi substrati, differenti dal punto di vista chimico.
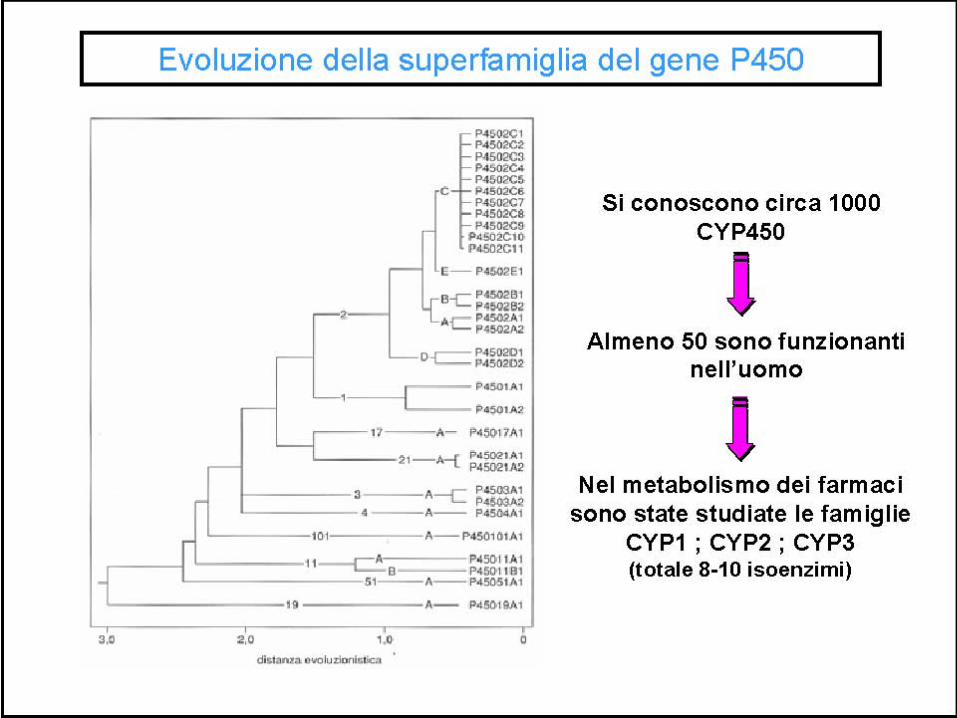
A seconda della somiglianza nella catena di aminoacidi gli isoenzimi sono raggruppati in famiglie e subfamiglie. Attualmente nell’uomo conosciamo 18 famiglie di CYP450, con 42 sottofamiglie e 57 geni codificanti.
SISTEMA CITOCROMO P450 MONOOSSIGENASI

Nomenclatura dei citocromi P450, esempio: CYP2D6
CYP = citocromo P450 2 = famiglia D = sub-famiglia 6 = specifico isoenzima (specifico gene) La nomenclatura è basata sui geni e non ha implicazioni funzionali
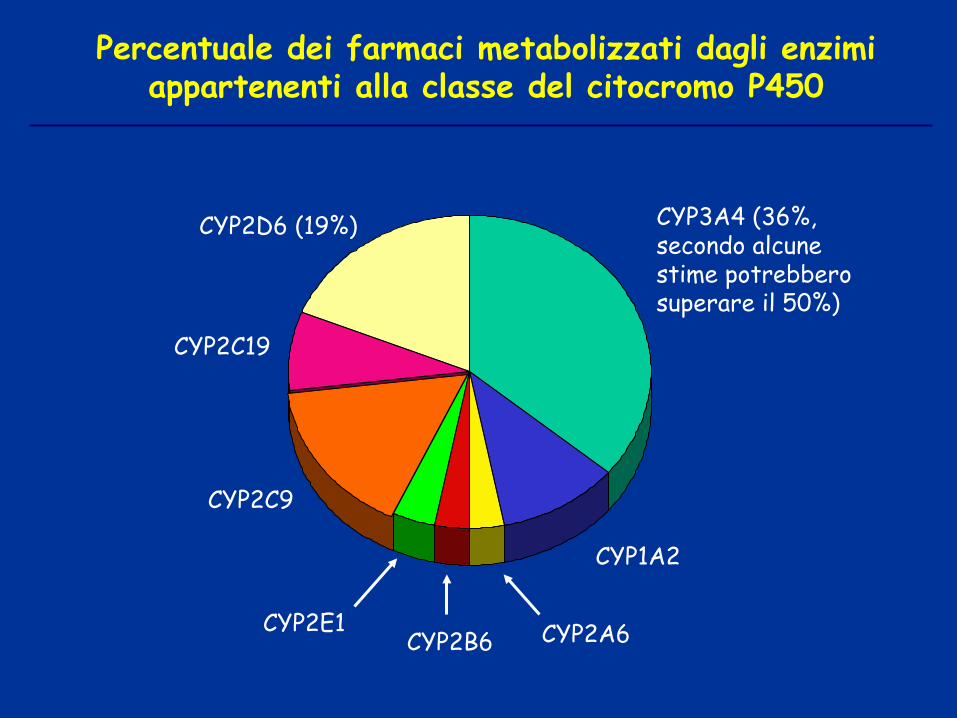
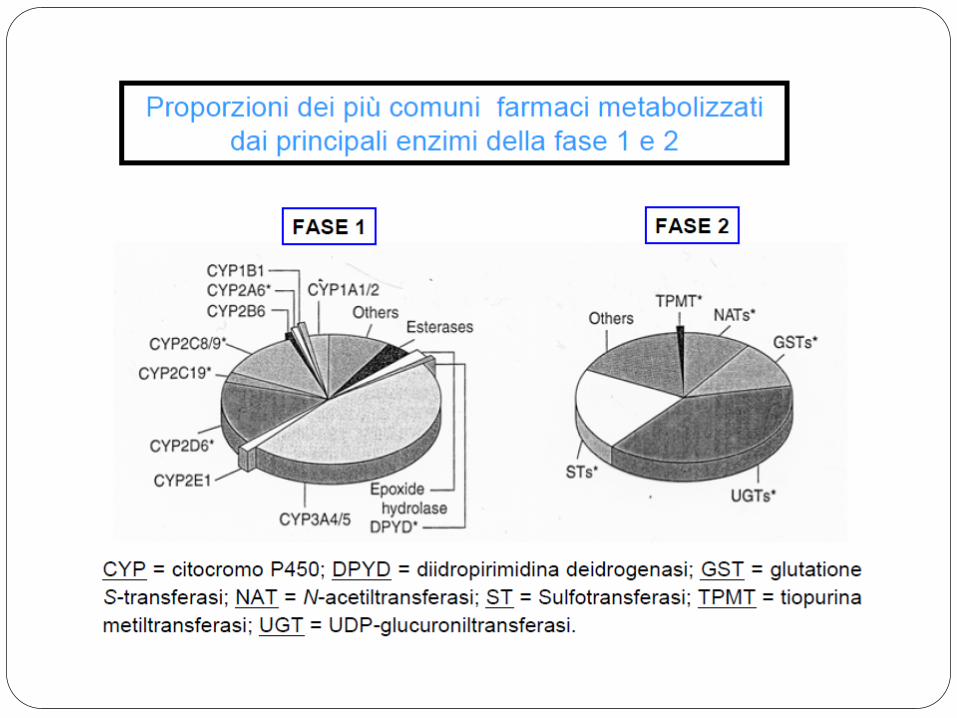
CYP3A4 (36%, secondo alcune stime potrebbero superare il 50%)
CYP2E1 CYP2B6 CYP2A6
CYP1A2
CYP2D6 (19%)
CYP2C9
CYP2C19
Percentuale dei farmaci metabolizzati dagli enzimi appartenenti alla classe del citocromo P450

Quando preparazioni di enzimi epatici per il metabolismo dei farmaci lega il monossido di carbonio (gruppo eme ridotto più monossido di carbonio) il complesso assorbe la luce nello spettro visibile e diventa di colore BLUVIOLETTO. Picco a 0,45 pm = 450 nm = P450
Quando il ferro presente nel gruppo eme dell’emoglobina è ridotto e lega il monossido di carbonio (o l’ossigeno) il complesso assorbe la luce nello spettro del visibile e diventa ROSSO (700-750 nm).



succinilcolina agente miorilassante
la presenza di sostituenti che sottraggono elettroni indebolisce il legame amidico rendendo la molecola più suscettibile all’idrolisi enzimatica
durata d’azione anestetico
carbossilesterasi sierica nota come pseudocolinesterasi
circa il 2% tra i caucasici è geneticamente carente dell’enzima
prolungato blocco neuromuscolare apnea
CARBOSSILESTERASI Reazione di Fase 2
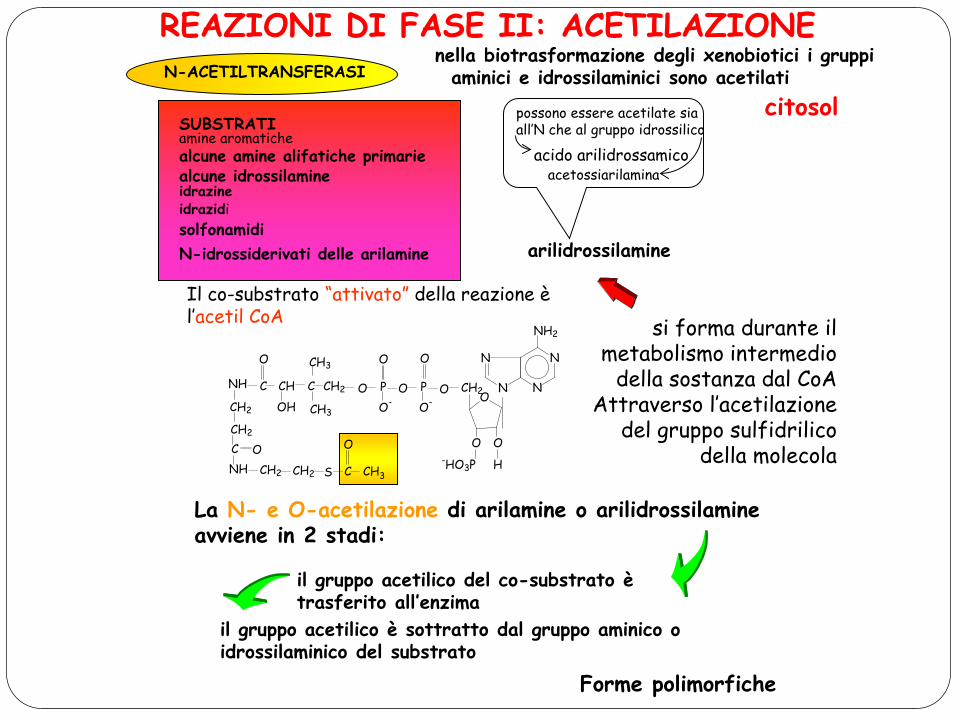
REAZIONI DI FASE II: ACETILAZIONE N-ACETILTRANSFERASI
nella biotrasformazione degli xenobiotici i gruppi aminici e idrossilaminici sono acetilati
citosol SUBSTRATI amine aromatiche alcune amine alifatiche primarie alcune idrossilamine idrazine idrazidi solfonamidi N-idrossiderivati delle arilamine arilidrossilamine
possono essere acetilate sia all’N che al gruppo idrossilico
acido arilidrossamico acetossiarilamina
Il co-substrato “attivato” della reazione è l’acetil CoA si forma durante il
metabolismo intermedio della sostanza dal CoA
Attraverso l’acetilazione del gruppo sulfidrilico
della molecola
N
NN
N
NH2
O
O
HHO3P
O-
CH2
O
OPO-
O OO-P
O
CH2C
CH3
CH3
CHOH
C
O
NH
CH2
CH2
C ONH CH2 CH2 S
O
C CH3
La N- e O-acetilazione di arilamine o arilidrossilamine avviene in 2 stadi:
il gruppo acetilico del co-substrato è trasferito all’enzima
il gruppo acetilico è sottratto dal gruppo aminico o idrossilaminico del substrato
Forme polimorfiche

FATTORI CHE INFLUENZANO LA CAPACITA’ METABOLICA DEL FEGATO
•Fattori legati all’età
• Fattori patologici





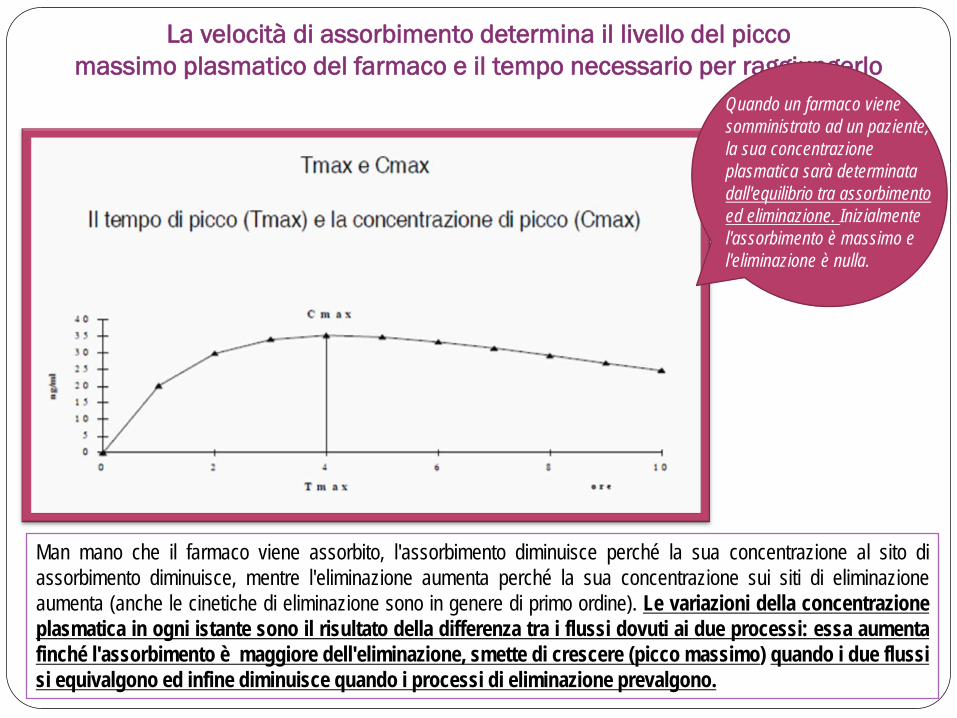
Man mano che il farmaco viene assorbito, l'assorbimento diminuisce perché la sua concentrazione al sito di assorbimento diminuisce, mentre l'eliminazione aumenta perché la sua concentrazione sui siti di eliminazione aumenta (anche le cinetiche di eliminazione sono in genere di primo ordine). Le variazioni della concentrazione plasmatica in ogni istante sono il risultato della differenza tra i flussi dovuti ai due processi: essa aumenta finché l'assorbimento è maggiore dell'eliminazione, smette di crescere (picco massimo) quando i due flussi si equivalgono ed infine diminuisce quando i processi di eliminazione prevalgono.
La velocità di assorbimento determina il livello del picco massimo plasmatico del farmaco e il tempo necessario per raggiungerlo
Quando un farmaco viene somministrato ad un paziente, la sua concentrazione plasmatica sarà determinata dall'equilibrio tra assorbimento ed eliminazione. Inizialmente l'assorbimento è massimo e l'eliminazione è nulla.

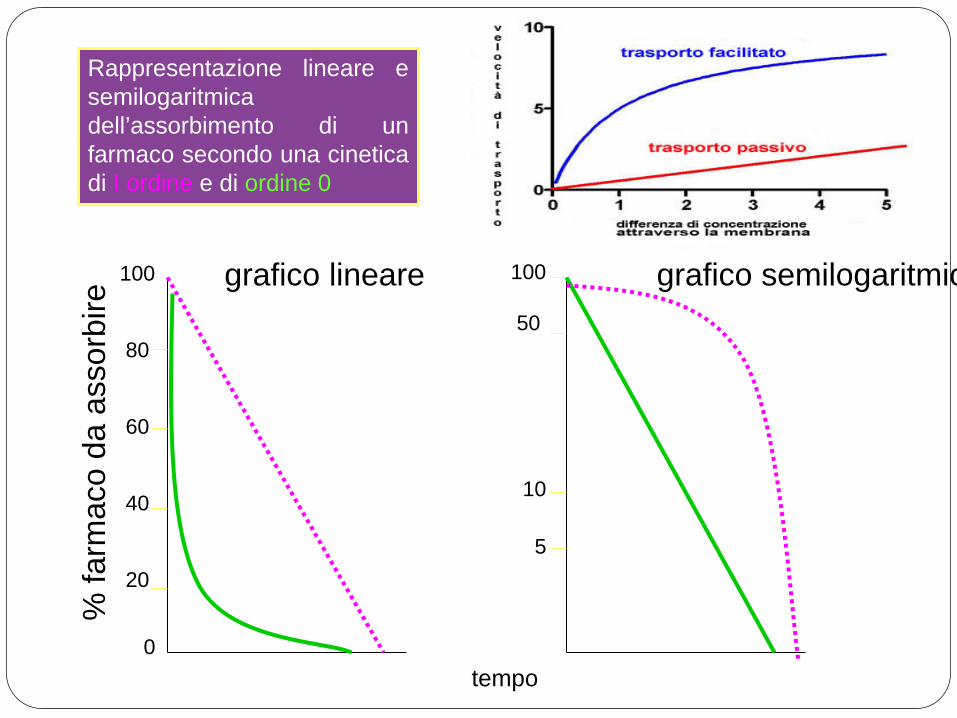
CINETICA DI I ORDINE: LA QUANTITA’ DI FARMACO ASSORBITA NELL’UNITA’ DI TEMPO E’ UNA % COSTANTE DI QUELLA CHE RIMANE DA ASSORBIRE
La maggior parte dei farmaci diffonde per diffusione passiva, seguendo una cinetica di I ordine.
CINETICA DI ORDINE 0: LA QUANTITA’ DI FARMACO ASSORBITA NELL’UNITA’ DI TEMPO E’ COSTANTE
Per alcuni farmaci l’assorbimento avviene attraverso meccanismi di trasporto attivo saturabili (la velocità di assorbimento dipende dal numero di trasportatori), seguendo una cinetica di ordine 0.
100 80 60 40 20
0
% fa
rmac
o da
ass
orbi
re grafico lineare grafico semilogaritmic 100
50 10 5
tempo
Rappresentazione lineare e semilogaritmica dell’assorbimento di un farmaco secondo una cinetica di I ordine e di ordine 0

Studio della velocità di un processo
dove: k: costante di
velocità di ordine n
n: ordine del processo cinetico
nABA Ckv ⋅=→
In farmacocinetica: processi di ordine primo (n = 1) = dose-indipendenti processi di ordine zero (n = 0) = dose-dipendenti processi misti
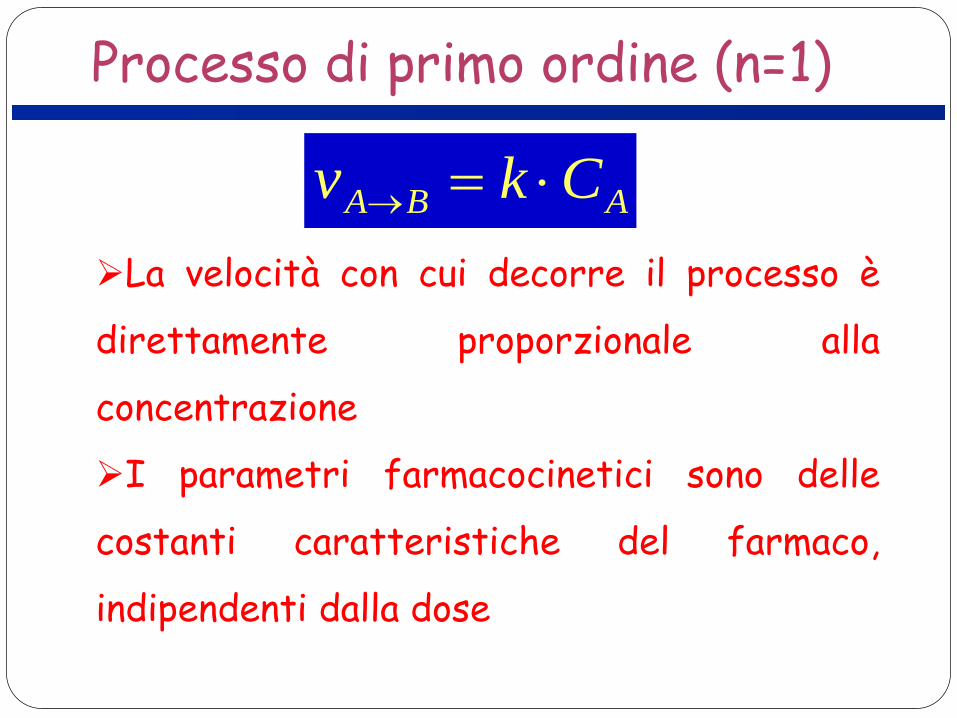
Processo di primo ordine (n=1)
ABA Ckv ⋅=→
La velocità con cui decorre il processo è
direttamente proporzionale alla
concentrazione
I parametri farmacocinetici sono delle
costanti caratteristiche del farmaco,
indipendenti dalla dose

Processo di ordine zero (n=0)
kv BA =→
La velocità con cui decorre il processo è
costante ed indipendente dalla
concentrazione (cinetica di saturazione)
I parametri farmacocinetici non sono
costanti caratteristiche del farmaco, ma
sono dipendenti dalla dose

La concentrazione al picco di una dose di farmaco somministrata in un singolo bolo per via endovenosa è sempre più alta di quella ottenibile somministrando la stessa dose per una qualunque via extravascolare. La velocità di assorbimento di un farmaco dipende da variabili legate al farmaco stesso e/o alle caratteristiche funzionali dell'area assorbente.
Il picco di concentrazione plasmatica di un farmaco dipende dalla velocità di assorbimento: più lento è l’assorbimento, più basso è il picco plasmatico

Metaboliti attivi dotati di spettro farmacologico uguale a quello del composto di origine


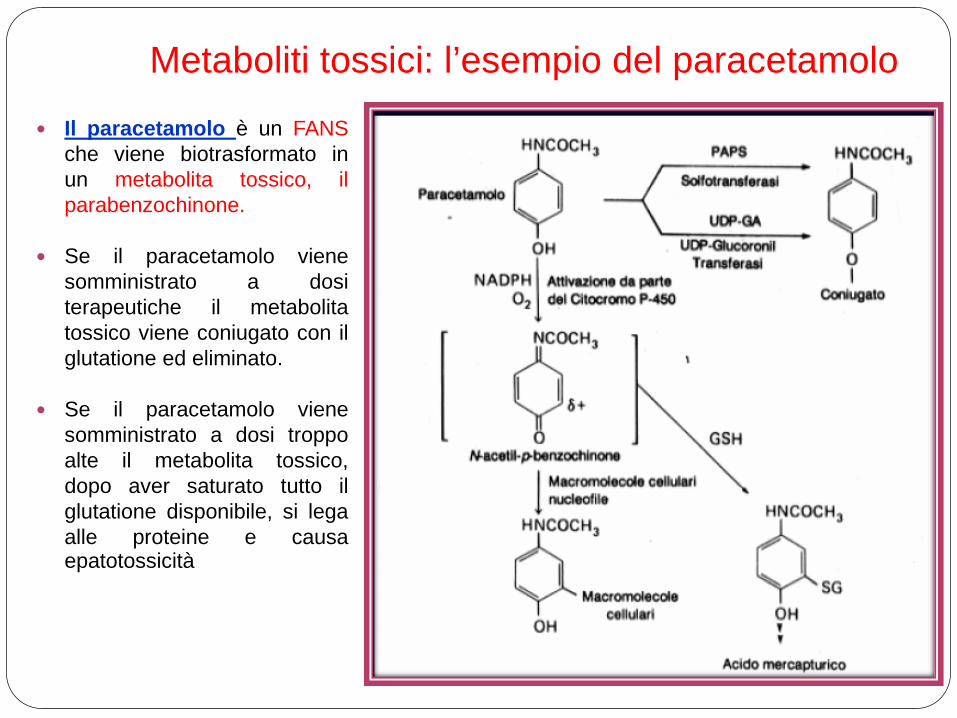
Il paracetamolo è un FANS che viene biotrasformato in un metabolita tossico, il parabenzochinone.
Se il paracetamolo viene somministrato a dosi terapeutiche il metabolita tossico viene coniugato con il glutatione ed eliminato.
Se il paracetamolo viene somministrato a dosi troppo alte il metabolita tossico, dopo aver saturato tutto il glutatione disponibile, si lega alle proteine e causa epatotossicità
Metaboliti tossici: l’esempio del paracetamolo
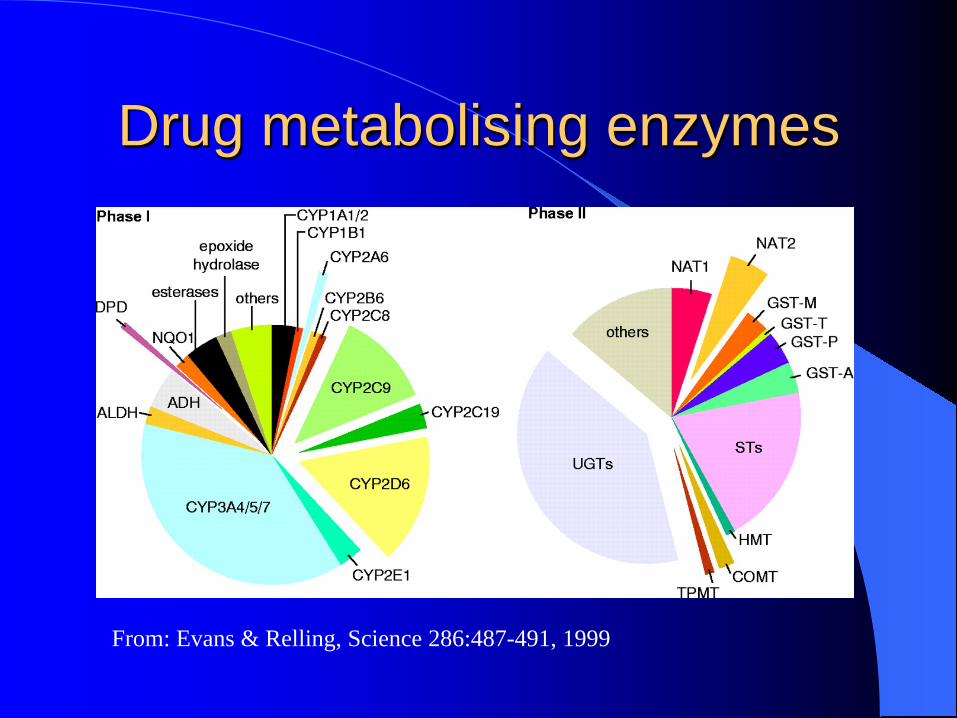
Drug metabolising enzymes
From: Evans & Relling, Science 286:487-491, 1999

Controllo genetico degli enzimi metabolizzanti i farmaci
controllo poligenico o multifattoriale
controllo monogenico - rari fenotipi - polimorfismo genetico

Cytochrome P-450 as a crossroad
1A2 2C 2D6 3A
Induction Inhibition
Endobiotics
Therapeutic failure Toxicity

CONDIZIONI CHE RENDONO UN POLIMORFISMO GENETICO CLINICAMENTE RILEVANTE
l’enzima polimorfo deve svolgere un ruolo quantitativamente importante nell’eliminazione del farmaco e/o dei suoi metaboliti
la risultante variabilità farmacocinetica è importante per l’efficacia e la sicurezza del farmaco
non è possibile modificare lo schema di dosaggio sulla base della risposta clinica o sulla base di esami di laboratorio e/o strumentali

Metabolizzatori lenti (PM)
diminuita inattivazione esagerata risposta accumulo del farmaco tossicità ridotta formazione di inefficacia terapeutica
metaboliti attivi Metabolizzatori rapidi (EM)
somministrazione di substrati, interazioni farmacocinetiche inibitori o induttori dell’enzima Metabolizzatori ultrarapidi (UM) aumentata inattivazione inefficacia terapeutica aumentata formazione di tossicità metaboliti attivi
Potenziali conseguenze farmacocinetiche e cliniche del polimorfismo genetico

Conseguenze cliniche di eventuali polimorfismi in sistemi enzimatici che metabolizzano i farmaci anti-ipertensivi
Istit. di Farmacologia- Università di CZ
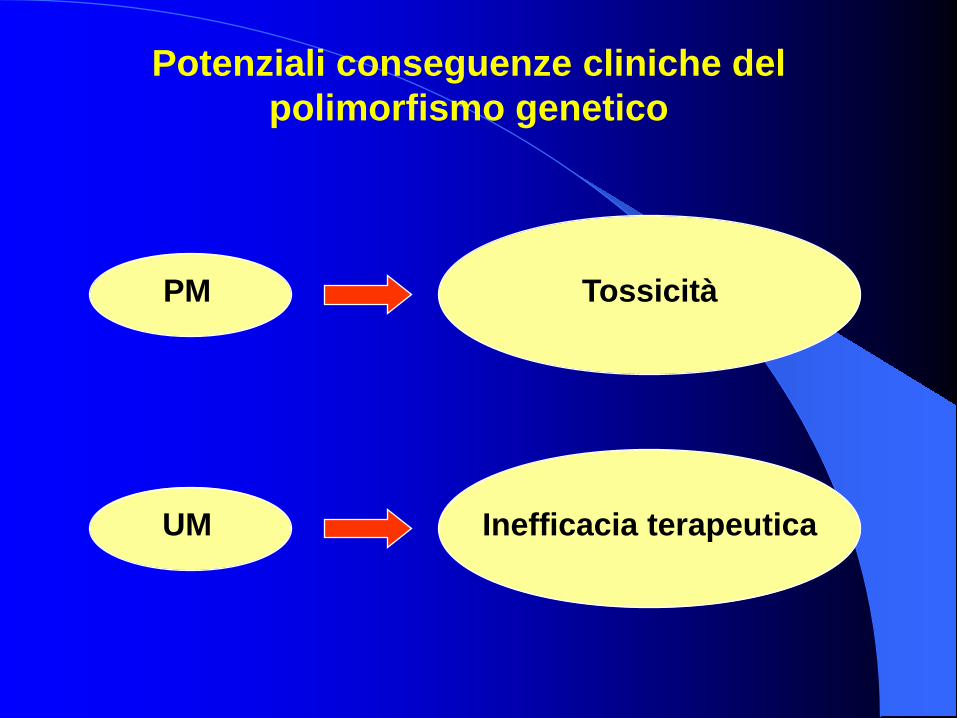
PM Tossicità
UM Inefficacia terapeutica
Potenziali conseguenze cliniche del polimorfismo genetico
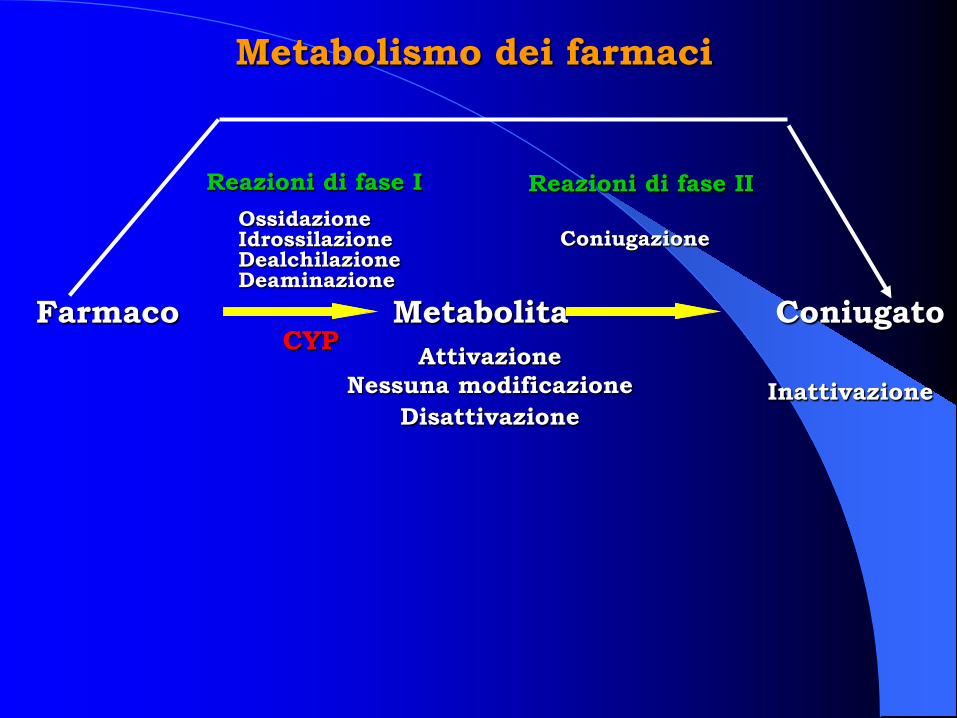
Metabolismo dei farmaci
Farmaco Coniugato Metabolita
Reazioni di fase I Reazioni di fase II Ossidazione Idrossilazione Dealchilazione Deaminazione
Coniugazione
CYP Attivazione Nessuna modificazione
Disattivazione Inattivazione

Cytochrome P450 system
57 human CYP-genes, 33 pseudogenes CYP1A2
18 families
(>40% amino acid homology)
43 subfamilies (>55% amino acid homology)
Specific isoform

Enzimi del citocromo P450 (CYP) Espressi prevalentemente a livello
epatico, ma presenti anche in altri tessuti
Catalizzano le reazioni metaboliche di fase I degli xenobiotici e di molti composti endogeni
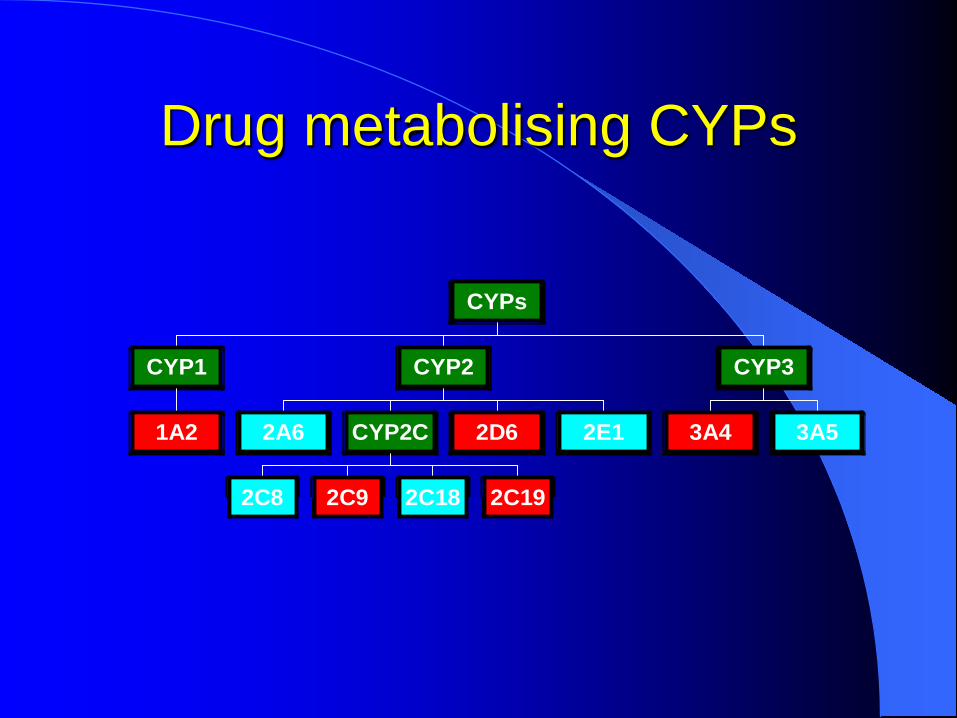
Drug metabolising CYPs
1A2
CYP1
2A6
2C8 2C9 2C18 2C19
CYP2C 2D6 2E1
CYP2
3A4 3A5
CYP3
CYPs
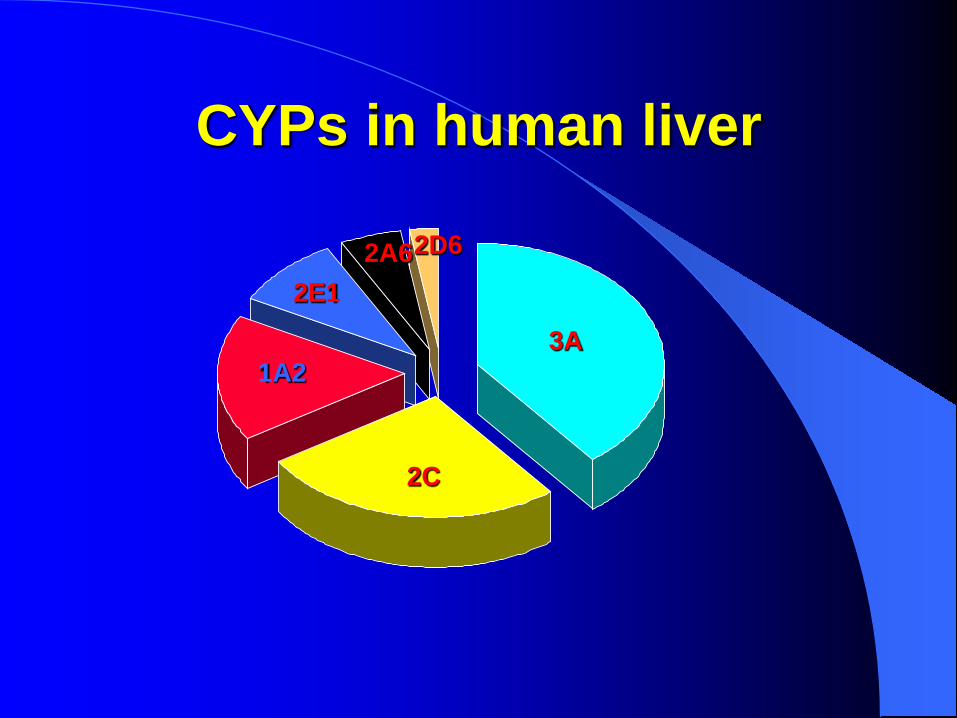
CYPs in human liver
3A
2C
1A2
2E1 2A6 2D6

Proportion of drugs metabolised by the different CYPs
CYP3A4 36%
CYP2E1 CYP2B6 CYP2A6 CYP1A2
CYP2D6 19%
CYP2C9
CYP2C19

CYP2D6
• Enzima • Locus genico • % PMs • Eredità • Substrati • Fenotipizzazione
• Genotipizzazione
• CYP2D6 • cromosoma 22 • 3 - 10 % • autosomica recessiva • ~ 20% dei farmaci • debrisochina, sparteina, destrometorfano • tecniche di biologia
molecolare

SSuubbssttrraattii IInniibbiittoorrii IInndduuttttoorrii
AAnnttiiddeepprreessssiivvii AAmmiittrriippttiilliinnaa CClloommiipprraammiinnaa DDeessiipprraammiinnaa FFlluuooxxeettiinnaa PPaarrooxxeettiinnaa AAnnttiiaarriittmmiiccii EEnnccaaiinniiddee FFlleeccaaiinniiddee SSppaarrtteeiinnaa
OOppppiiaacceeii CCooddeeiinnaa DDeessttrroommeettoorrffaannoo TTrraammaaddoolloo
NNeeuurroolleettttiiccii AAllooppeerriiddoolloo PPeerrffeennaazziinnaa RRiissppeerriiddoonnee TTiioorriiddaazziinnaa
ββ --bbllooccccaannttii PPrrooppaannoolloolloo MMeettoopprroolloolloo TTiimmoolloolloo PPiinnddoolloolloo
CChhiinniiddiinnaa TTiioorriiddaazziinnaa FFlluuooxxeettiinnaa PPaarrooxxeettiinnaa AAmmiiooddaarroonnee CCiimmeettiiddiinnaa IInnddiinnaavviirr RRiittoonnaavviirr MMiibbeeffrraaddiill
NNeessssuunn iinndduuttttoorree nnoottoo
Substrati ed inibitori del CYP2D6

MMuuttaatteedd aalllleelleess MMuuttaattiioonn CCoonnsseeqquueenncceess FFrreeqquueenncciieess
CCYYPP22DD66**33 FFrraammee--sshhiifftt IInnaaccttiivvee eennzzyymmee 11--22%%
CCYYPP22DD66**44 DDeeffeeccttiivvee sspplliicciinngg IInnaaccttiivvee eennzzyymmee 1122--2211 %%
CCYYPP22DD66**55 GGeennee ddeelleettiioonn NNoo eennzzyymmee 22--77%%
CCYYPP22DD66**66 FFrraammee--sshhiifftt ggeenneerraattiinngg ssttoopp ccooddoonn
IInnaaccttiivvee eennzzyymmee 11--22%%
CCYYPP22DD66**22xxNN GGeennee dduupplliiccaattiioonn IInnccrreeaasseedd eennzzyymmee aaccttiivviittyy
22--1100%%
CCYYPP22DD66**1100 PPrroo3344SSeerr,, SSeerr448866TThhrr UUnnssttaabbllee eennzzyymmee 11--22%% CCaauuccaassiiaannss 5511%% AAssiiaannss
CCYYPP22DD66**1177 TThhrr110077IIllee,, AArrgg229966CCyyss,, SSeerr448866TThhrr
RReedduucceedd aaffffiinniittyy ffoorr ssuubbssttrraatteess
00%% CCaauuccaassiiaannss 3344%% AAffrriiccaannss
Polimorfismo genetico del CYP2D6

CYP2D6 Alleles and genotypes frequencies in Italians
FFrreeqquueenncciieess CCoonnffiiddeennccee iinntteerrvvaall CCYYPP22DD66**11 00..775500 00..771188 –– 00..779922 CCYYPP22DD66**33 00..000077 00..000011 –– 00..001133 CCYYPP22DD66**44 00..115533 00..112277 –– 00..117799 CCYYPP22DD66**55 00..003344 00..002211 –– 00..004477 CCYYPP22DD66**66 00..001144 00..000055 –– 00..002233
CCYYPP22DD66**22xxnn
00..004422 00..002277 –– 00..005577
GGeennoottyyppeess **11//**11 5533..33%% HHoommoozzyyggoouuss EEMM **11//**33 **11//**44 **11//**55
3355..00%%
HHeetteerroozzyyggoouuss EEMM
**11//**66 **33//**44;; **44//**44;; **44//**55 33..44%% PPMM **22xxnn//**11 88..33%% UUMM
Scordo et al. Pharmacol Res (2004)

Interethnic differences in frequency of CYP2D6 PM and UM genotypes (%)
Population PM UM Swedish 7 1-2 Italian, Spanish 5-10 7-10 Oriental 1-2 ? African (Ethiopian) 2-10 -29

CYP2C19
• Enzima • Locus genico • % PMs • Eredità • Substrati • Fenotipizzazion
e • Genotipizzazion
e
• CYP2C19 • cromosoma 10 • 2 - 5 % • autosomica recessiva • > 10 composti • mefenitoina, omeprazolo • tecniche di biologia
molecolare

SSuubbssttrraatteess IInnhhiibbiittoorrss IInndduucceerrss
AAnnttiiddeepprreessssaannttss:: aammiittrriippttyylliinnee,, cclloommiipprraammiinnee,, iimmiipprraammiinnee,, cciittaalloopprraamm,, mmoocclloobbeemmiiddee,, fflluuooxxeettiinnee,, sseerrttrraalliinnee MMiisscceellllaanneeoouuss:: pphheennyyttooiinn,, ddiiaazzeeppaamm,, oommeepprraazzoollee,, pprroopprraannoollooll,, pprroogguuaanniill,, SS--mmeepphheennyyttooiinn,, RR--wwaarrffaarriinn
FFlluuooxxeettiinnee FFlluuvvooxxaammiinnee OOmmeepprraazzoollee
PPhheennoobbaarrbbiittaall PPhheennyyttooiinn
Substrati, iduttori ed inibitori del CYP2C19

CYP2C19 polymorphism
MMuuttaatteedd aalllleelleess MMuuttaattiioonn CCoonnsseeqquueenncceess FFrreeqquueenncciieess CCYYPP22CC1199**22 AAbbeerrrraanntt sspplliiccee ssiittee IInnaaccttiivvee eennzzyymmee 1111--1155%% ((CCaauuccaassiiaannss))
2255--3300%% ((OOrriieennttaallss))
CCYYPP22CC1199**33 PPrreemmaattuurree ssttoopp ccooddoonn IInnaaccttiivvee eennzzyymmee 00--11%% ((CCaauuccaassiiaannss)) 55--1100%% ((OOrriieennttaallss))
5-10% (orientals )
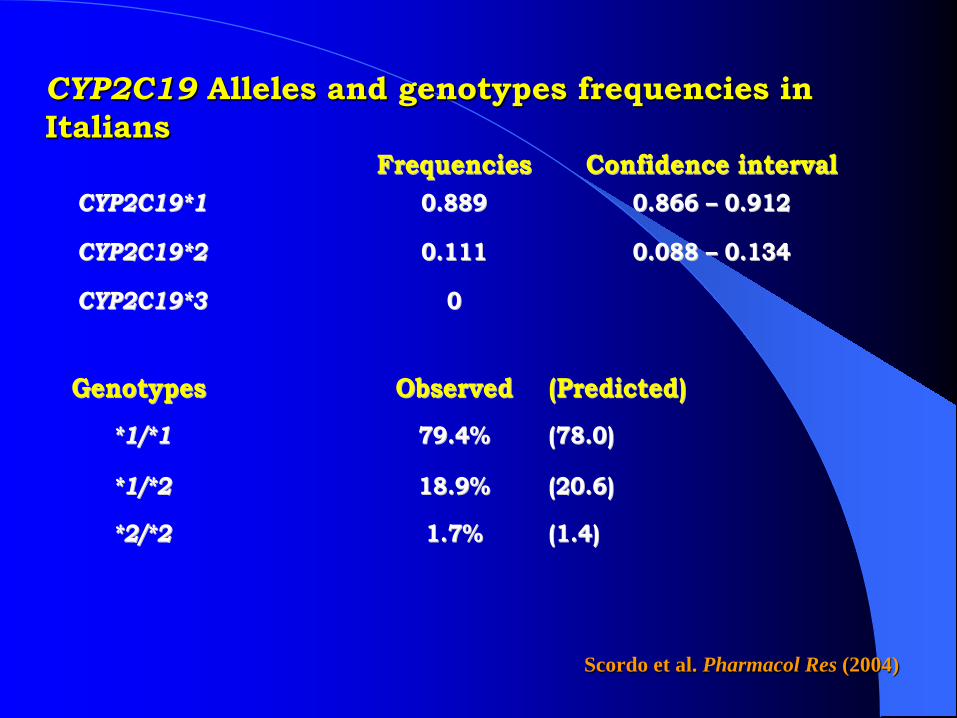
CYP2C19 Alleles and genotypes frequencies in Italians
FFrreeqquueenncciieess CCoonnffiiddeennccee iinntteerrvvaall CCYYPP22CC1199**11 00..888899 00..886666 –– 00..991122
CCYYPP22CC1199**22 00..111111 00..008888 –– 00..113344
CCYYPP22CC1199**33 00
GGeennoottyyppeess OObbsseerrvveedd ((PPrreeddiicctteedd))
**11//**11 7799..44%% ((7788..00))
**11//**22 1188..99%% ((2200..66))
**22//**22 11..77%% ((11..44))
Scordo et al. Pharmacol Res (2004)

CYP2C9
Enzima locus genico % PMs Eredità Substrati Fenotipizzazione Genotipizzazione
CYP2C9 cromosoma 10 2 - 6 % autosomica recessiva > 20 composti S-warfarina, losartan,
diclofenac tecniche di biologia
molecolare

SSuubbssttrraatteess IInnhhiibbiittoorrss IInndduucceerrss
NNSSAAIIDDssCCeelleeccooxxiibbDDiiccllooffeennaaccIIbbuupprrooffeennNNaapprrooxxeennPPiirrooxxiiccaamm
MMiisscceellllaanneeaaPPhheennyyttooiinnSS--wwaarrffaarriinnLLoossaarrttaannFFlluuooxxeettiinneeTToollbbuuttaammiiddeeTToorraasseemmiiddee
SSuullpphhaapphheennaazzoolleeAAmmiiooddaarroonneeFFlluuccoonnaazzoollee
PPhheenniillbbuuttaazzoonneeKKeettooccoonnaazzoolleeMMeettrroonniiddaazzoollee
RRiittoonnaavviirr
RRiiffaammppiinnPPhheennoobbaarrbbiittaall
PPhheennyyttooiinn
Substrati, induttori ed inibitori del CYP2C9

CYP2C9 polymorphism
MMuuttaatteedd aalllleelleess MMuuttaattiioonn CCoonnsseeqquueenncceess FFrreeqquueenncciieess CCYYPP22CC99**22 AArrgg114444CCyyss RReedduucceedd aaffffiinniittyy ffoorr
PP445500 rreedduuccttaassee 00..110077 ((SSwweeddiisshh)) 00..110066 ((TTuurrkkiisshh))
00..0088 ((AAmmeerriiccaann--CCaauuccaassiiaann)) 00 ((CChhiinneessee)) 00 ((JJaappaanneessee))
CCYYPP22CC99**33 IIllee335599LLeeuu AAlltteerreedd ssuubbssttrraattee ssppeecciiffiicciittyy
00..007744 ((SSwweeddiisshh)) 00..110000 ((TTuurrkkiisshh))
00..0066 ((AAmmeerriiccaann--CCaauuccaassiiaann)) 00..002266 ((CChhiinneessee)) 00..002211 ((JJaappaanneessee))
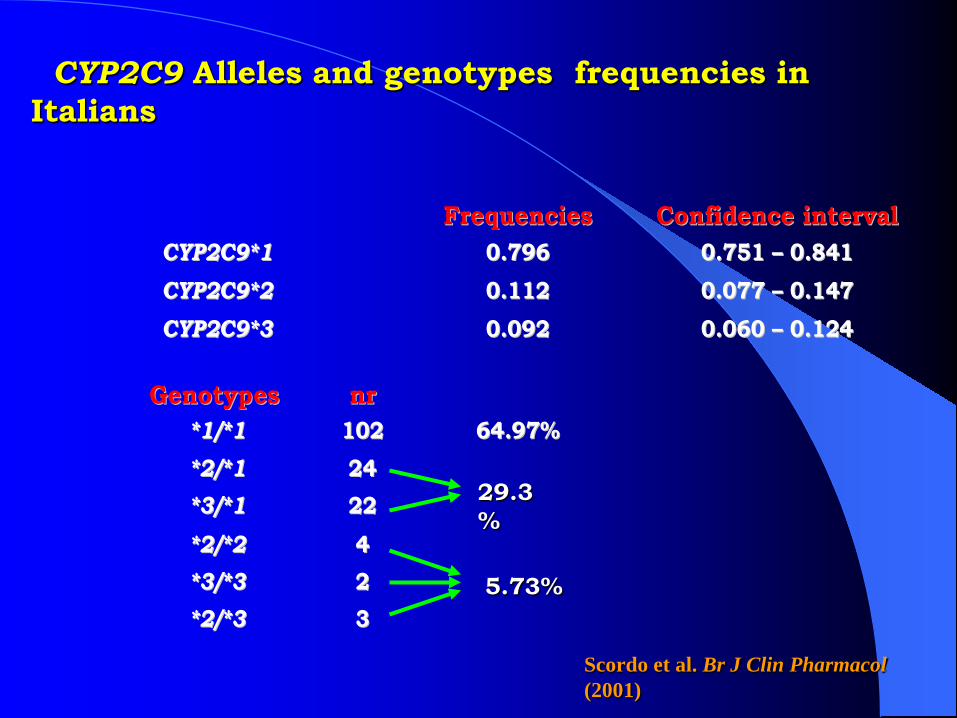
CYP2C9 Alleles and genotypes frequencies in Italians
FFrreeqquueenncciieess CCoonnffiiddeennccee iinntteerrvvaallCCYYPP22CC99**11 00..779966 00..775511 –– 00..884411CCYYPP22CC99**22 00..111122 00..007777 –– 00..114477CCYYPP22CC99**33 00..009922 00..006600 –– 00..112244
GGeennoottyyppeess nnrr**11//**11 110022 6644..9977%%**22//**11 2244**33//**11 2222**22//**22 44**33//**33 22**22//**33 33
29.3%
5.73%
Scordo et al. Br J Clin Pharmacol (2001)

CYP1A2
• Enzima • Locus genico • % PMs • Eredità • Substrati • Fenotipizzazion
e • Genotipizzazion
e
• CYP1A2 • cromosoma 15 • ??? • ??? • ~ 6-8% dei farmaci • caffeina, melatonina • tecniche di biologia
molecolare: -PCR -restriction enzyme analysis

SSuubbssttrraatteess IInnhhiibbiittoorrss IInndduucceerrss
AAnnttiiddeepprreessssaannttss:: aammiittrriippttyylliinnee,, cclloommiipprraammiinnee,, iimmiipprraammiinnee,, fflluuvvooxxaammiinnee,, mmiirrttaazzaappiinnee AAnnttiippssyycchhoottiiccss:: hhaallooppeerriiddooll,, cclloozzaappiinnee,, oollaannzzaappiinnee,, tthhiioorriiddaazziinnee MMiisscceellllaanneeoouuss:: ppaarraacceettaammooll,, pphheennaacceettiinn,, ttaaccrriinnee,, RR--wwaarrffaarriinn,, tthheeoopphhyylllliinnee,, ccaaffffeeiinnee
CCiipprrooffllooxxaacciinn FFlluuvvooxxaammiinnee
SSmmookkiinngg RRiiffaammppiicciinn
BBaarrbbiittuurraatteess PPhheennyyttooiinn
CCaarrbbaammaazzeeppiinnee
Substrati, induttori ed inibitori del CYP1A2

CYP1A2 polymorphism
MMuuttaatteedd aalllleelleess MMuuttaattiioonn CCoonnsseeqquueenncceess FFrreeqquueenncciieess
CCYYPP11AA22**11CC --33886600GG>>AA DDeeccrreeaasseedd aaccttiivviittyy 2200%% ((JJaappaanneessee))
CCYYPP11AA22**11DD --22446677ddeellTT 4422 %% ((OOrriieennttaallss))
CCYYPP11AA22**11FF --116633CC>>AA HHiigghheerr iinndduucciibbiilliittyy 6677..88%% ((CCaauuccaassiiaannss)) 6611..33%% ((OOrriieennttaallss))
CCYYPP11AA22**11KK --773399TT>>GG;; --772299CC>>TT;; --116633CC>>AA
DDeeccrreeaasseedd aaccttiivviittyy 11--33%%
CCYYPP11AA22**77 33553344GG>>AA SSpplliicciinngg ddeeffeecctt
DDeeccrreeaasseedd aaccttiivviittyy

CYP3A4
• Enzima • Locus genico • Substrati • Fenotipizzazion
e
• CYP3A4 • cromosoma 7 • ~ 50% dei farmaci • eritromicina

SSuubbssttrraattii IInniibbiittoorrii IInndduuttttoorrii
AAllpprraazzoollaamm AAmmiiooddaarroonnee CCiicclloossppoorriinnaa
DDiillttiiaazzeemm EErriittrroommiicciinnaa
SSeerrttrraalliinnaa VVeerraappaammiill
TTeerrffeennaaddiinnaa AAsstteemmiizzoolloo
CCeerriivvaassttaattiinnaa CCiissaapprriiddee
EErriittrroommiicciinnaa KKeettooccoonnaazzoolloo
SSuuccccoo ddii ppoommppeellmmoo
BBaarrbbiittuurriiccii CCaarrbbaammaazzeeppiinnaa
FFeenniittooiinnaa
Substrati, inibitori ed induttori del CYP3A4
CYP 1A2 2C9 2D6 3A4
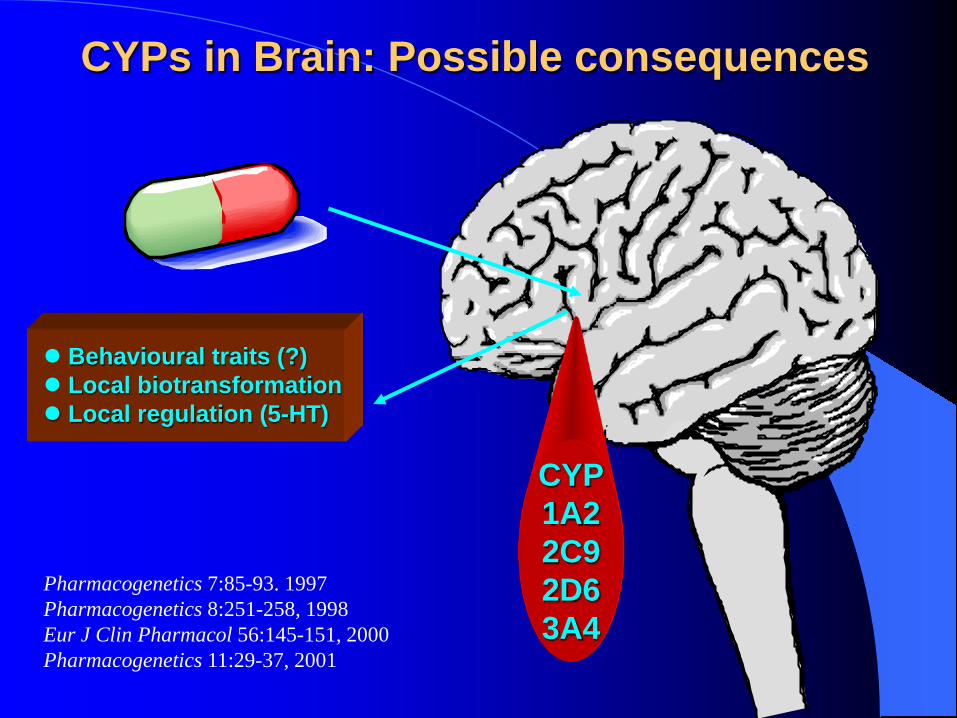
CYPs in Brain: Possible consequences
Behavioural traits (?) Local biotransformation Local regulation (5-HT)
Pharmacogenetics 7:85-93. 1997 Pharmacogenetics 8:251-258, 1998 Eur J Clin Pharmacol 56:145-151, 2000 Pharmacogenetics 11:29-37, 2001

Polimorfismi genetici (variazioni a livello dei geni presenti in >1% popolazione)
Stati fisiologici (età, sesso) Stati patologici Induzione o inibizione da farmaci concomitanti o
fattori ambientali
PRINCIPALI FATTORI RESPONSABILI DELLA VARIABILITÀ NEL METABOLISMO DEI FARMACI

SNPs Si parla di Single Nucleotide Polymorphism (SNPs) quando un sito di un gene presenta diversi nucleotidi (e la proteina diversi
aminoacidi) in individui della stessa specie.
Polimorfismi Genetici
Aplotipo Set di SNPs correlati tra loro che vengono ereditati assieme

Basi molecolari della variabilità umana
(Sachinanandan, Nature 2001)
1.4 milioni di polimorfismi a singolo nucleotide
60.000 in zone codificanti
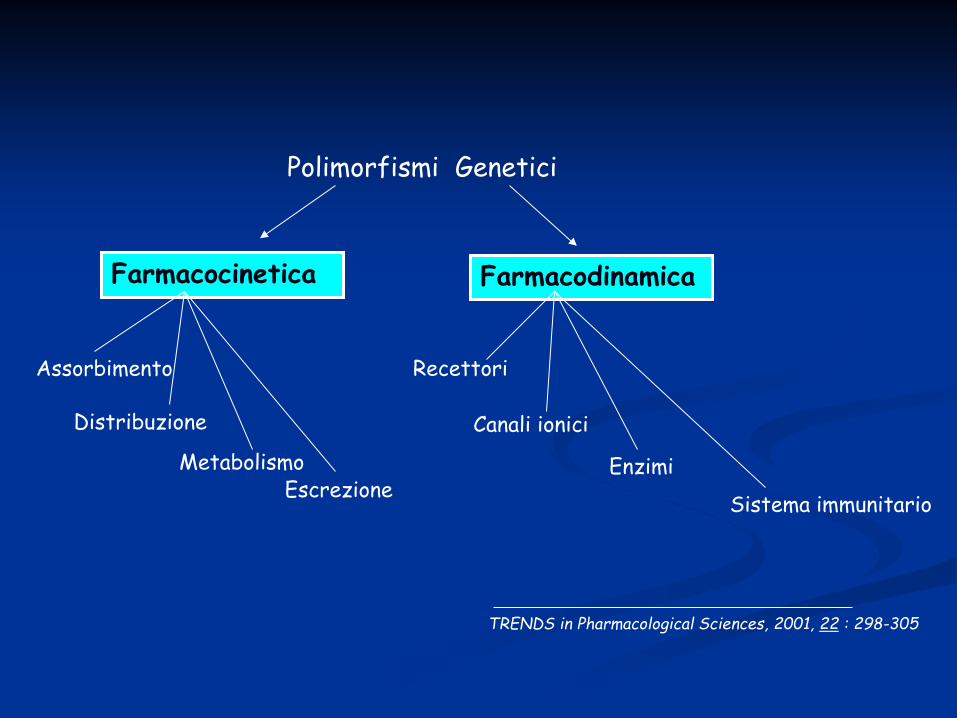
Polimorfismi Genetici
Farmacodinamica Farmacocinetica
Recettori
Canali ionici
Enzimi
Sistema immunitario
Assorbimento
Distribuzione
Metabolismo Escrezione
TRENDS in Pharmacological Sciences, 2001, 22 : 298-305

Evans et al., NEJM 2003; 348: 538-549
Effetto dei polimorfismi: -Farmacocinetica
Metabolismo
-Farmacodinamica
Efficacia/Tossicità

Deficit della pseudocolinesterasi o pseudocolinesterai atipiche Apnea da succinilcolina
Deficit della Glucosio-6-Fosfato Deidrogenasi (G6PD) Anemia emolitica da antimalarici o da altri farmaci ossidanti
Carenza di metaemoglobina reduttasi Metaemoglobinemia da clorochina
Carenza di glutatione Epatotossicità da paracetamolo Acetil-tranferasi (acetilatori lenti) Neuropatie da isoniazide
Esempi di polimorfismi enzimatici non legati al citocromo P450
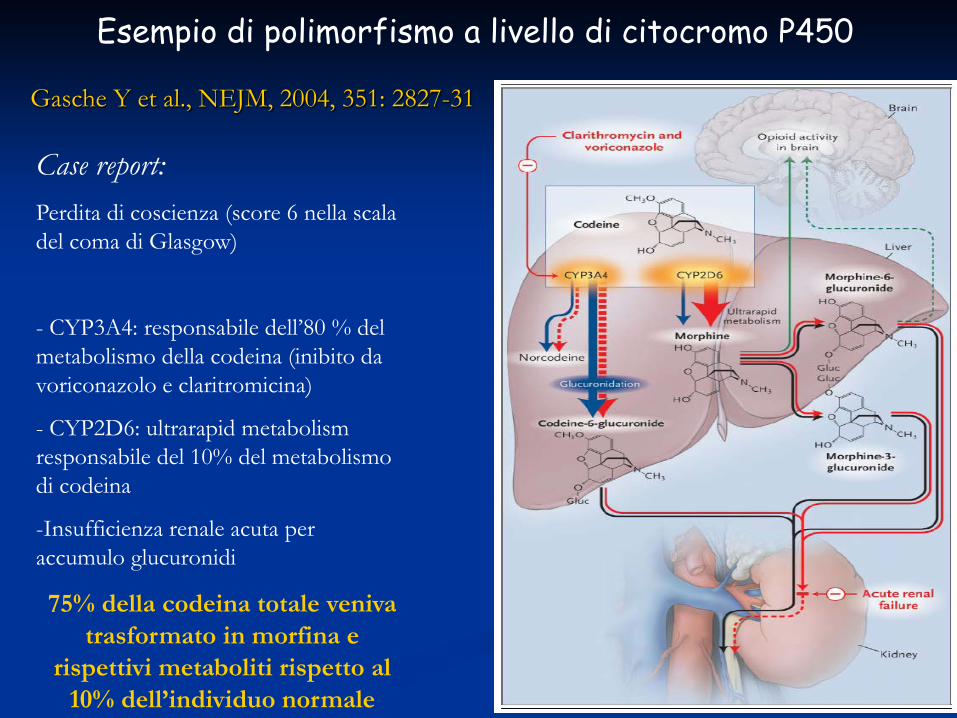
Gasche Y et al., NEJM, 2004, 351: 2827-31
Case report: Perdita di coscienza (score 6 nella scala del coma di Glasgow)
- CYP3A4: responsabile dell’80 % del metabolismo della codeina (inibito da voriconazolo e claritromicina)
- CYP2D6: ultrarapid metabolism responsabile del 10% del metabolismo di codeina
-Insufficienza renale acuta per accumulo glucuronidi
75% della codeina totale veniva trasformato in morfina e
rispettivi metaboliti rispetto al 10% dell’individuo normale
Esempio di polimorfismo a livello di citocromo P450

Pompa di efflusso multifarmaci energia-dipendente Fosfoglicoproteina di surperfice Trovata nell’intestino, rene, fegato, cervello, testicoli,
placenta, surrenali Ambudkar SV et al., Ann Rev Pharmacol Toxicol 1999; 39:361-98
ATP ATP
GLICOPROTEINA-P
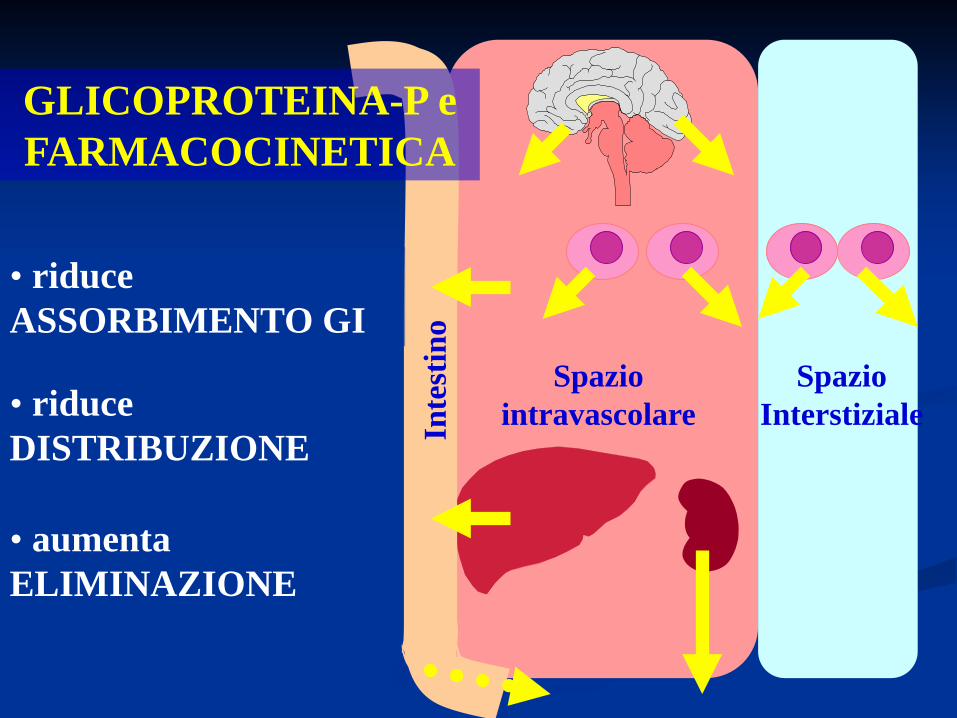
Spazio Interstiziale
Spazio intravascolare
GLICOPROTEINA-P e FARMACOCINETICA
Inte
stin
o
• riduce ASSORBIMENTO GI
• riduce DISTRIBUZIONE
• aumenta ELIMINAZIONE
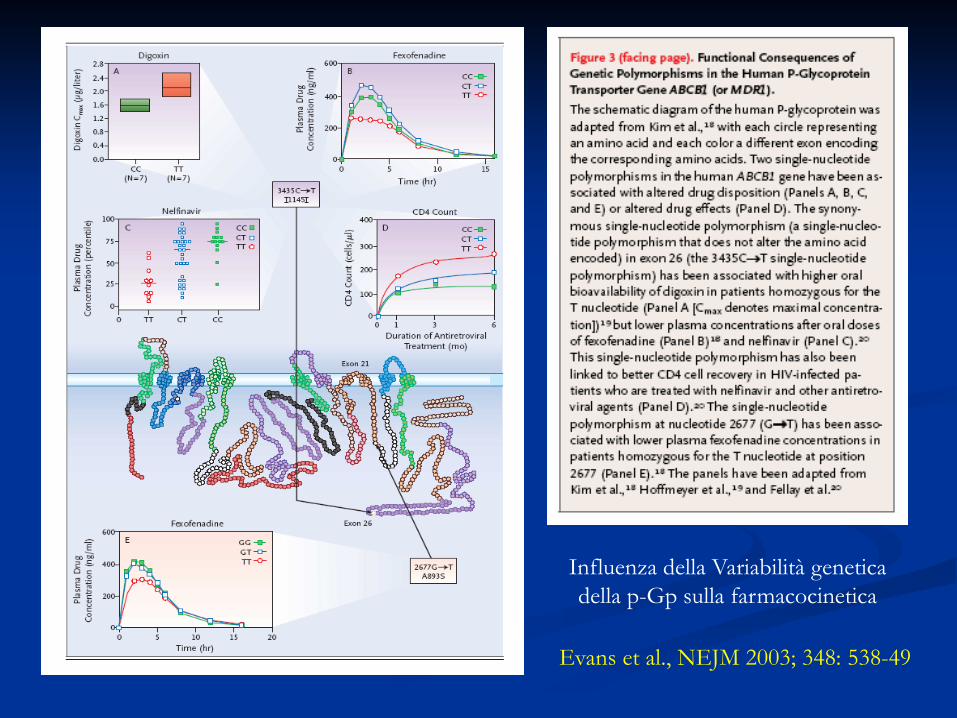
Evans et al., NEJM 2003; 348: 538-49
Influenza della Variabilità genetica della p-Gp sulla farmacocinetica

Recettore (difetto genetico) Rilevanza clinica
Androgeni Risposta alla terapia antiandrogenica
Recettore delle sulfuniluree (SUR) Iperinsulinismo Infantile, diabete (1)
β2-Adrenocettori (mutazioni missense) Alterata risposta ad antiasmatici;
mortalità nell’insufficienza cardiaca
Canali del potassio (mutazioni missense) Sindrome del QT lungo (LQTS)
Canali del sodio (mutazioni missense) Sindrome del QT lungo (LQTS)
Polimorfismi genetici di alcuni recettori e loro rilevanza clinica
Modified from Weber, Mut Res 2001; 479:1

Evans et al., NEJM 2003; 348: 538-49
Variabilità genetica e risposta agli agonisti
β2

Sindrome del QT Lungo (LQTS)
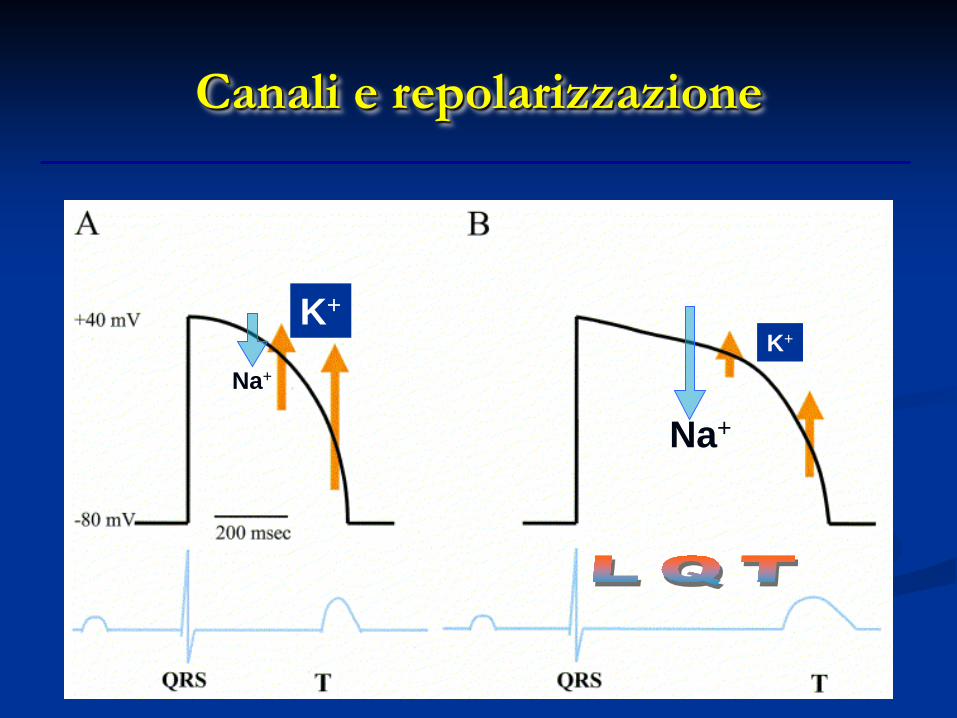
Canali e repolarizzazione
Na+
Na+
K+ K+
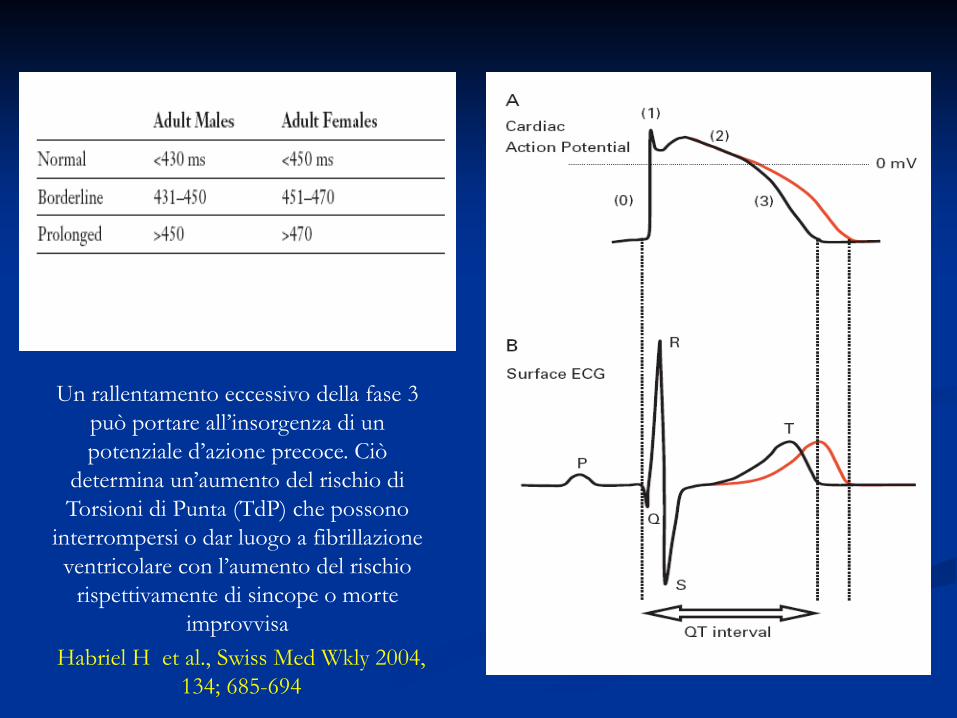
Habriel H et al., Swiss Med Wkly 2004, 134; 685-694
Un rallentamento eccessivo della fase 3 può portare all’insorgenza di un potenziale d’azione precoce. Ciò
determina un’aumento del rischio di Torsioni di Punta (TdP) che possono
interrompersi o dar luogo a fibrillazione ventricolare con l’aumento del rischio
rispettivamente di sincope o morte improvvisa

IPERTERMIA MALIGNA
• Caratterizzata clinicamente da febbre, contratture muscolari, tachicardia, aumento della CO2 espiratoria, lattacidosi e ipertermia
• La mutazione R614C della proteina codificata da ryr1 è responsabile dell’ipertermia maligna umana (Gillard et al., 1991)
• Raro disturbo del muscolo scheletrico, potenzialmente fatale, che può essere scatenato dall’uso di anestetici volatili e miorilassanti depolarizzanti (~ sindrome maligna da neurolettici)
• Il gene ryr1 sul cromosoma 19q13.1 (locus MSH-1) è associato a più del 50% di tutte le forme familiari di ipertermia maligna (McCarthy et al., 2000)
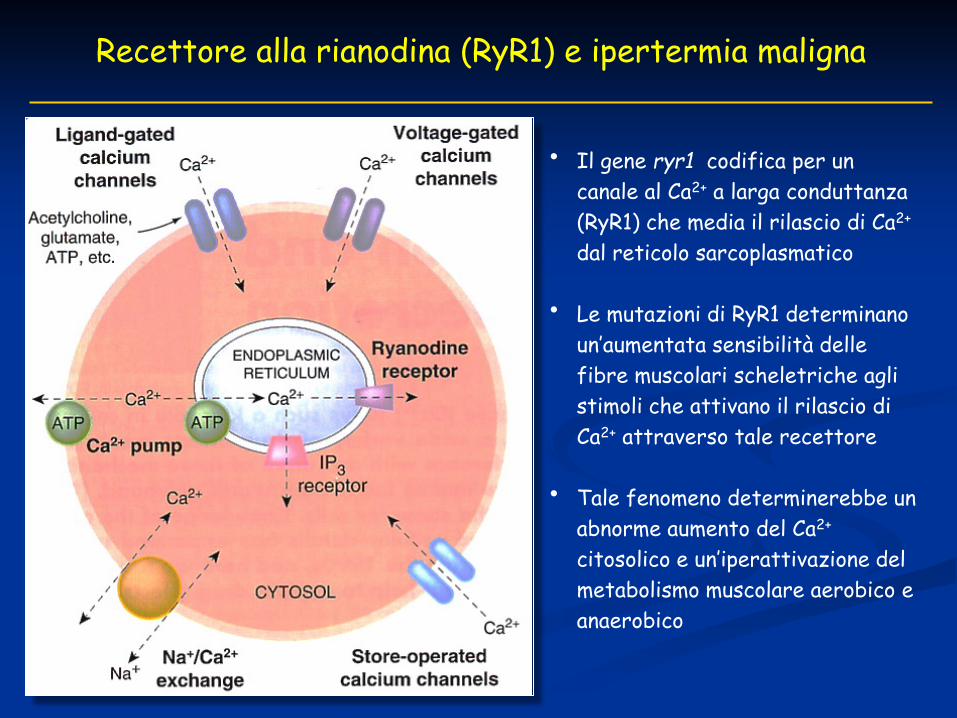
Recettore alla rianodina (RyR1) e ipertermia maligna
• Il gene ryr1 codifica per un canale al Ca2+ a larga conduttanza (RyR1) che media il rilascio di Ca2+ dal reticolo sarcoplasmatico
• Le mutazioni di RyR1 determinano un’aumentata sensibilità delle fibre muscolari scheletriche agli stimoli che attivano il rilascio di Ca2+ attraverso tale recettore
• Tale fenomeno determinerebbe un abnorme aumento del Ca2+ citosolico e un’iperattivazione del metabolismo muscolare aerobico e anaerobico

Le interazioni tra farmaci possono verificarsi a diversi livelli influenzando la farmacocinetica o la farmacodinamica dei farmaci stessi. Le interazioni conosciute sono moltissime tuttavia quelle di rilevanza clinica maggiore (da ricordare) sono relativamente poche.
Le interazioni più frequenti sono quelle a livello del metabolismo dei farmaci, dovute a meccanismi di induzione o inibizione enzimatica
Alcune volte le interazioni tra farmaci possono essere sfruttate per avere un maggiore effetto terapeutico o per contrastare fenomeni di intossicazione. Nella maggioranza dei casi, tuttavia, le interazioni sono alla base della comparsa di reazioni avverse.
I pazienti vanno educati a non aggiungere farmaci (ad esempio per autoprescrizione) alla terapia prescritta dal medico, in modo da evitare interazioni tra farmaci.
INTERAZIONI TRA FARMACI
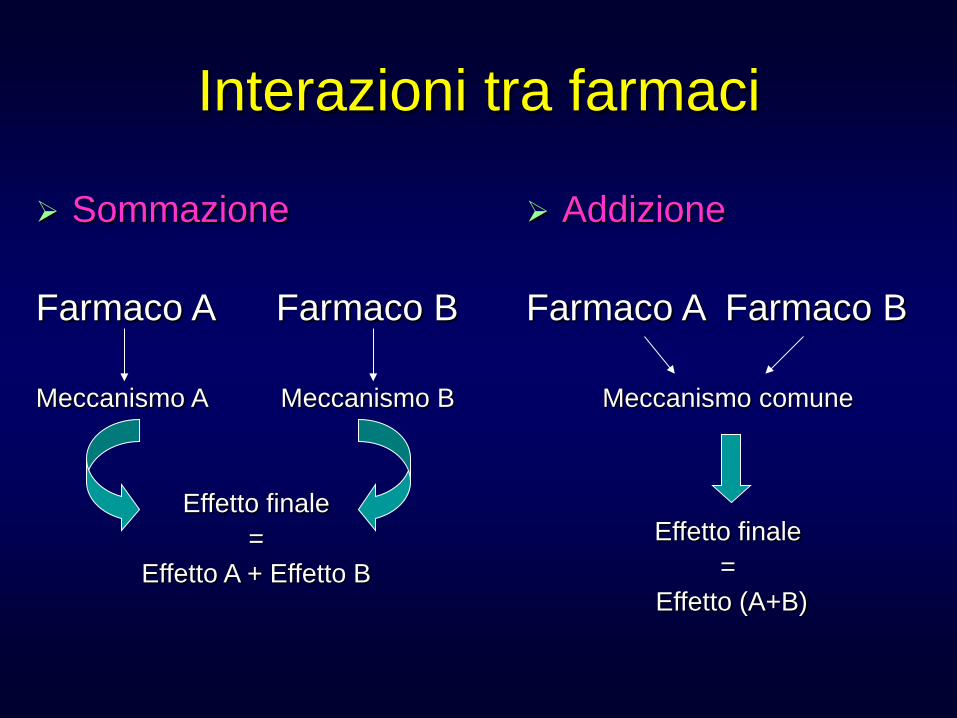
Interazioni tra farmaci
Sommazione Farmaco A Farmaco B Meccanismo A Meccanismo B
Effetto finale =
Effetto A + Effetto B
Addizione Farmaco A Farmaco B
Meccanismo comune
Effetto finale =
Effetto (A+B)

Interazioni tra farmaci Potenziamento
Farmaco A
Recettore Effetto finale > effetto A
•Assorbimento
•Eliminazione
•Spiazzamento dall’accettore
•Inibizione enzimatica
Farmaco B
Sinergismo
Farmaco A
Farmaco B
Risposta terapeutica
Effetto finale >
effetto A + effetto B
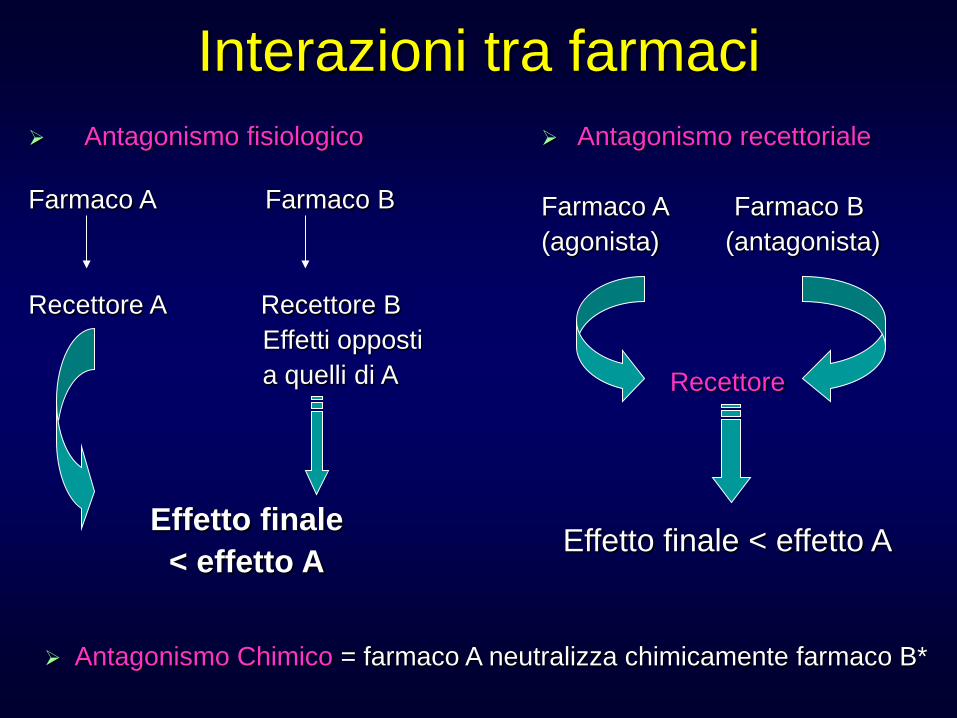
Interazioni tra farmaci Antagonismo fisiologico Farmaco A Farmaco B Recettore A Recettore B Effetti opposti a quelli di A
Effetto finale
< effetto A
Antagonismo recettoriale Farmaco A Farmaco B (agonista) (antagonista)
Recettore
Effetto finale < effetto A
Antagonismo Chimico = farmaco A neutralizza chimicamente farmaco B*

Interazioni tra farmaci
Degradazione
Farmaco A Assorbimento Eliminazione Induzione enzimatica
Recettore Effetto finale < effetto A
Neutralizzazione Farmaco A + Farmaco B
Effetto finale assente
Farmaco B
Inattivazione
Chimico-fisica

I farmaci che sono substrato dello stesso enzima possono inibire reciprocamente il loro metabolismo, ma spesso non ad un livello clinicamente rilevante.
Il meccanismo più comune di inibizione enzimatica è il legame competitivo a un citocromo P450 (CYP), tuttavia alcuni farmaci inibiscono l’attività enzimatica senza essere substrato dell’enzima.
La potenza dell’inibizione può essere più importante del suo meccanismo. Il ketoconazolo e l’itraconazolo, ad esempio, possono inibire quasi completamente il CYP3A4 anche a concentrazioni molto basse. Anche l’eritromicina è un potente inibitore del CYP3A4, ma per un motivo differente; si lega con legame covalente all’enzima e lo inattiva in modo che l’effetto persista anche dopo che il farmaco è stato eliminato.
L’inibizione del metabolismo epatico inizia non appena nel fegato vi siano concentrazioni sufficienti dell’inibitore (in genere dopo poche ore dall’assunzione). L’effetto dell’inibizione sul metabolismo di un altro farmaco perciò è usualmente massimo nelle prime 24 ore.
Tuttavia , nonostante che l’inibizione insorga rapidamente, la comparsa dell’effetto clinico conseguente (generalmente una reazione tossica) può essere più ritardata.
L’inibizione enzimatica generalmente termina più rapidamente rispetto all’induzione enzimatica.
INIBIZIONE ENZIMATICA

Un aumento dell’attività degli enzimi metabolizzanti che determina una riduzione dei livelli serici di un dato farmaco, è generalmente dovuta alla stimolazione della sintesi dell’enzima (da parte degli induttori enzimatici).
Gli induttori enzimatici stimolano il metabolismo di altri farmaci in maniera graduale. Sebbene l’effetto dell’induzione può essere individuato anche entro i primi due giorni di terapia, generalmente occorre una settimana prima che l’effetto massimo compaia.
Il tempo di comparsa del fenomeno dell’induzione dipende comunque anche dall’emività del farmaco inducente. Ad esempio la rifampicina, che ha una emivita relativamente breve, induce gli enzimi più rapidamente del fenobarbitale (induttore con emivita più lunga). Al contrario l’effetto dell’induzione si protrarrà più a lungo se determinata da un induttore con emivita più lunga.
INDUZIONE ENZIMATICA

INIBITORI E INDUTTORI DEL METABOLISMO DI FARMACI
•INIBITORI •Amiodarone
•Antimicotici imidazolici
•Alcuni farmaci anti-HIV
•Cimetidina
•Macrolidi (NO azitr.)
•Isoniazide
•Fluorochinoloni
•Alcuni SSRI
•Inibitori pompa proton.
•Chinidina
•Succo di pompelmo
•INDUTTORI
•Carbamazepina
•Fenobarbital
•Fenitoina
•Rifampicina/rifabutina
•Erba di San Giovanni (iperico)

Succo di pompelmo Il succo di pompelmo, ma non quello d'arancia dolce, aumenta la biodisponibilità di diversi farmaci, in particolare dei Ca-antagonisti. Nel caso della felodipina, che normalmente ha una biodisponibilità del 15% dopo metabolismo di primo passaggio, il succo di pompelmo produce concentrazioni di farmaco circa 3 volte più elevate della norma. Le conseguenze nei pazienti ipertesi borderline sono un'aumentata riduzione della pressione arteriosa ed un incremento della frequenza cardiaca. Le reazioni avverse correlate alla vasodilatazione (es. cefalea) sono di conseguenza più frequenti. Il succo di pompelmo inibisce selettivamente, nel tratto GI, il CYP3A4. L'interazione tra felodipina e succo di pompelmo chiarisce due importanti concetti: l'importanza dell'intestino come sede di farmacometabolismo e che l'interazione dipende dalla via di somministrazione del farmaco. (Il succo di pompelmo non interagisce con farmaci somministrati per via endovenosa).

Succo di pompelmo • La durata dell'inibizione intestinale del CYP3A4 dura fino a 24 ore dopo l'assunzione del succo. Così anche se si ritarda di diverse ore la somministrazione del farmaco l'interazione è ugualmente significativa. • Questa lunga durata d'azione deriva da un'inattivazione intestinale del CYP3A4 di tipo "suicida", intendendo con questo termine che il ripristino dell'attività del CYP3A4 dipende dalla sintesi di nuovo enzima. • La rilevanza dell'interazione è estremamente variabile tra gli individui; in alcuni casi le concentrazioni plasmatiche di felodipina rimangono inalterate e in altri si arriva a livelli 8 volte superiori rispetto ai controlli (felodipina + acqua). Ciò dipende dal contenuto intestinale di CYP3A4: gli individui con i livelli più elevati sono quelli con i maggiori incrementi nelle concentrazioni plasmatiche di felodipina

Farmaci e succo di pompelmo
Classe Ansiolitici Antiaritmici Antidepressivi Antiepilettici Antistaminici Calcioantagonisti
Farmaci Buspirone, diazepam midazolam,triazolam Amiodarone Clomipramina Carbamazepina Terfenadina Amlodipina, felodipina Nifedipina, nimodipina
Possibili eventi avversi ↓ capacità psicomotorie, ↑ della sedazione Aritmie Sonnolenza, depressione resp. Sonnolenza, atassia, nausea Aritmie, prolungamento Q-T Tachicardia, ipotensione


Farmaci “me too”
Il farmaco “me too” è una molecola con caratteristiche molto simili all’originetor (ad es. esomeprazolo che è l’isomero attivo dell’omeprazolo che è un composto racemico). Ranitidina Amlodipina L’atorvastatina è la settima delle statine immesse sul mercato ma è anche la più potente.

La principale via di eliminazione dei farmaci (e dei loro metaboliti) è il RENE
Altre vie di eliminazione possono essere la VIA RESPIRATORIA, ad esempio per i farmaci gassosi o la VIA BILIARE (escrezione con le feci)
I farmaci che vengono eliminati tramite la bile possono venire in parte riassorbiti a livello intestinale: si viene cosi a creare il cosidetto CIRCOLO ENTERO-EPATICO
Altra via di eliminazione dei farmaci è rappresentata dal LATTE MATERNO. Questo fatto deve essere tenuto in considerazione quando si prescrivono farmaci a madri che allattano, per i possibili rischi di tossicità a cui si può esporre il neonato
Fattori che possono modificare l’eliminazione dei farmaci, con possibile ACCUMULO, sono: presenza di patologie renali (insufficienza renale), l’età del paziente (neonati e anziani), ostacolo al deflusso biliare (per farmaci eliminati per questa via).
ELIMINAZIONE DEI FARMACI
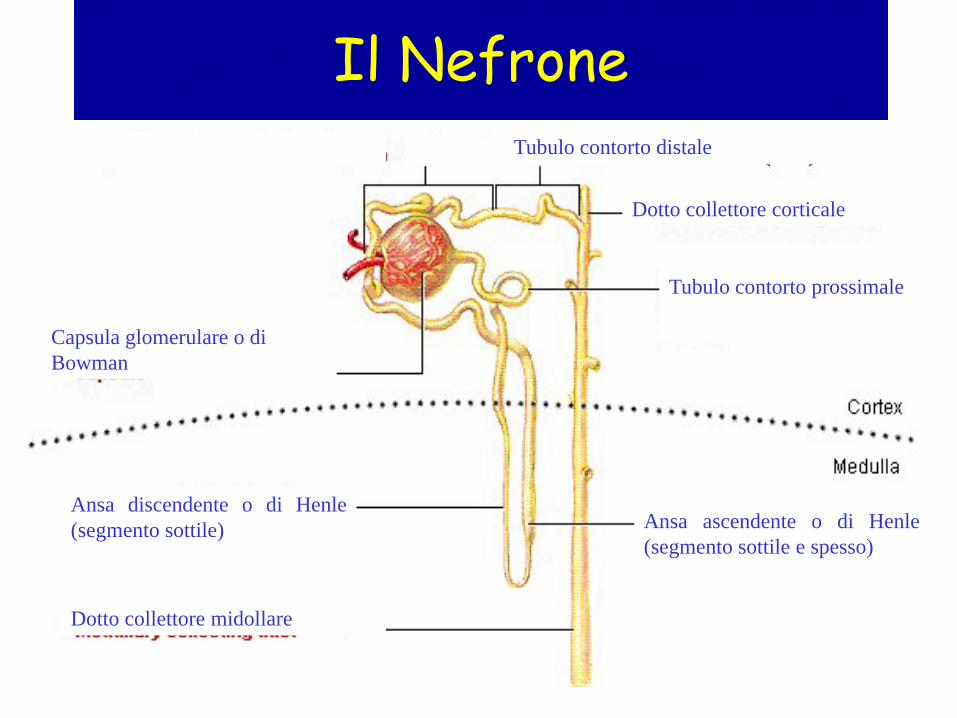
Il Nefrone
Tubulo contorto prossimale
Ansa discendente o di Henle (segmento sottile) Ansa ascendente o di Henle
(segmento sottile e spesso)
Capsula glomerulare o di Bowman
Dotto collettore midollare
Dotto collettore corticale
Tubulo contorto distale
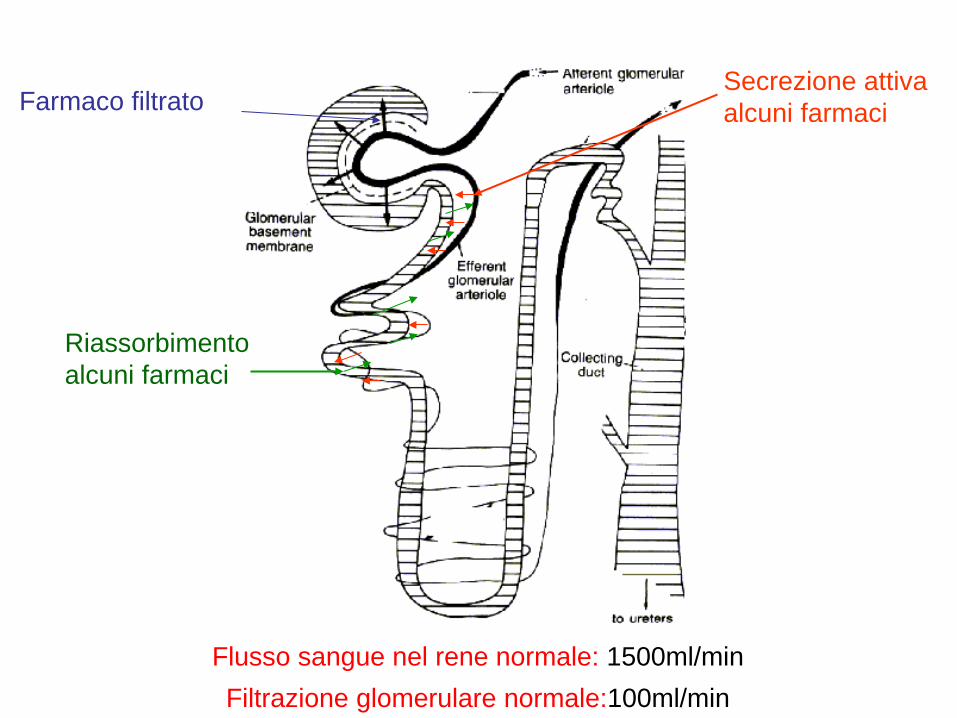
Farmaco filtrato Secrezione attiva alcuni farmaci
Riassorbimento alcuni farmaci
Flusso sangue nel rene normale: 1500ml/min Filtrazione glomerulare normale:100ml/min

ELIMINAZIONE DEI FARMACI PER VIA RENALE
1) I farmaci liposolubili tendono ad essere escreti a concentrazioni simili a quelle presenti nel plasma. La loro concentrazione dipende soprattutto dal volume delle urine 2) I farmaci polari tendono ad essere escreti nelle urine a concentrazioni superiori a quelle presenti nel plasma , quindi la loro escrezione dipende più dal volume del filtrato glomerulare che dal volume delle urine 3) I farmaci coniugati si comportano in maniera simile alle sostanze polari, ma possono essere escreti in misura maggiore perché soggetti a meccanismi di secrezione attiva 4) I farmaci che si ionizzano facilmente, cioè acidi e basi, vengono escreti in maniera pH dipendente


MODIFICANDO IL pH POSSIAMO INFLUENZARE L’ASSORBIMENTO ED L’ELIMINAZIONE DEL FARMACO

CLEARANCE (ml/min) = U x V P
U = Concentrazione del farmaco nell’urina V = Volume urina in 1 min. P = Concentrazione del farmaco nel plasma
Quantità di plasma che in un minuto viene depurata da una sostanza
CLEARANCE
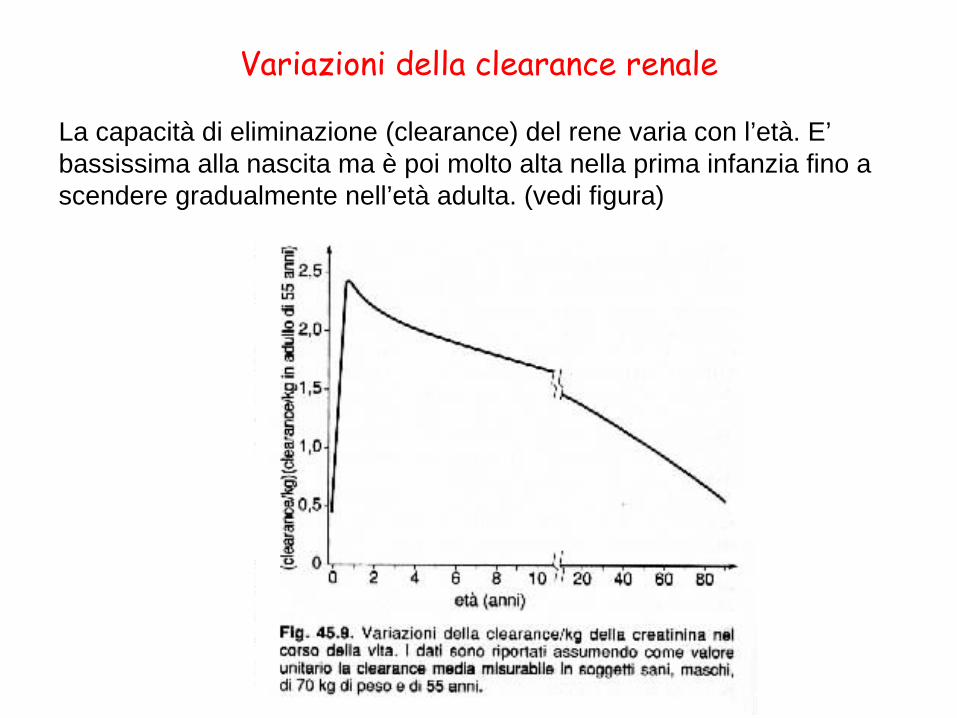
La capacità di eliminazione (clearance) del rene varia con l’età. E’ bassissima alla nascita ma è poi molto alta nella prima infanzia fino a scendere gradualmente nell’età adulta. (vedi figura)
Variazioni della clearance renale

Nella grande maggioranza dei casi, l’eliminazione dei farmaci dal corpo segue una cinetica monoesponenziale con base e. Questo significa che viene eliminata nell’unità di tempo una percentuale fissa del farmaco presente nel corpo Quindi, se un certo farmaco è eliminato al 10% all’ora e la sua concentrazione è di 10 mg/l alle ore 15.00, alle ore 16.00 sarà 9 mg/l; alle 17.00 sarà 8,1 mg/l; alle 18.00 sarà 7,3 mg/l; alle 19.00 sarà 6,6 ecc. ecc Quando una curva monoesponenziale viene disegnata su un grafico semilogaritmico (asse x lineare; asse y logaritmico in cui ogni tacca della stessa lunghezza corrisponde ad un raddoppio del valore) essa diventa una retta Il dato cinetico più informativo di una cinetica monoesponenziale è l’emivita L’emivita di un farmaco è il tempo necessario perché la concentrazione diventi la metà di quella che era al primo punto
ELIMINAZIONE DEI FARMACI
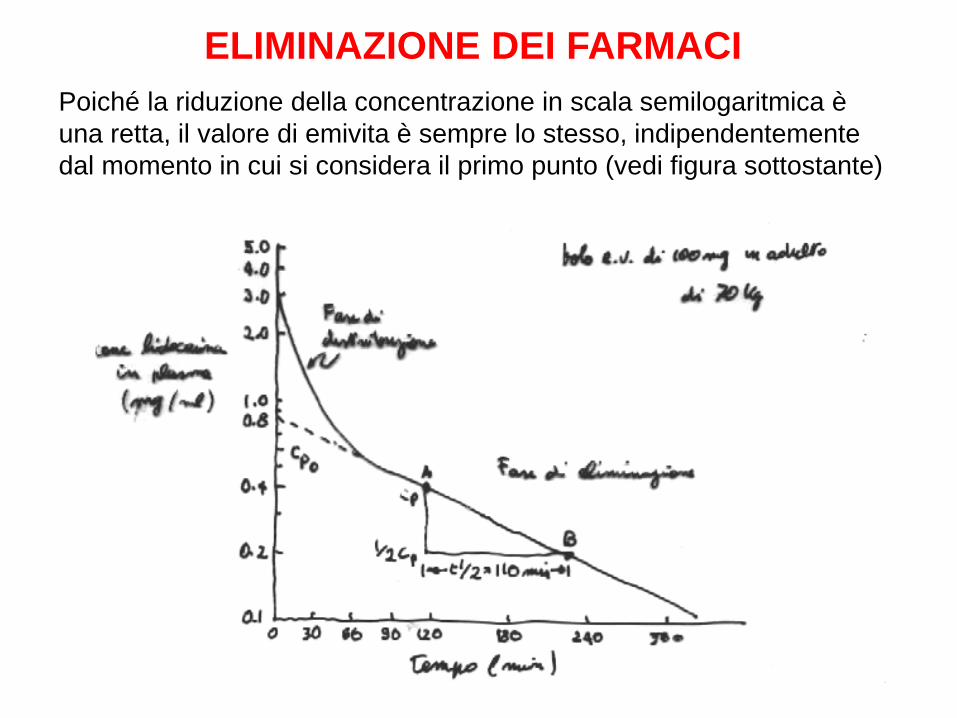
ELIMINAZIONE DEI FARMACI Poiché la riduzione della concentrazione in scala semilogaritmica è una retta, il valore di emivita è sempre lo stesso, indipendentemente dal momento in cui si considera il primo punto (vedi figura sottostante)

Emivita
*** Sono neccessarie 10 emivite per eliminare il 99,9%***
N° di t½ Frazione di farmaco rimanente 0 1 2 3 4 5 6 7 8 9 10
100% 50% 25% 12.5% 6.25% 3.125% 1.56% 0.78% 0.39% 0.195% 0.0975%
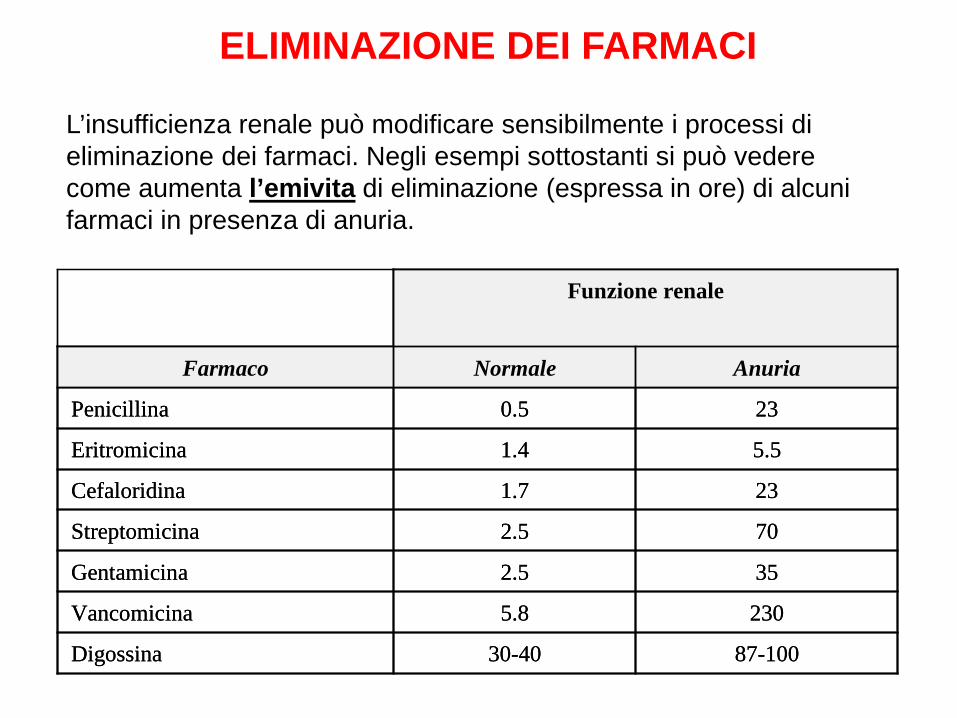
L’insufficienza renale può modificare sensibilmente i processi di eliminazione dei farmaci. Negli esempi sottostanti si può vedere come aumenta l’emivita di eliminazione (espressa in ore) di alcuni farmaci in presenza di anuria.
ELIMINAZIONE DEI FARMACI
87-10030-40Digossina
2305.8Vancomicina
352.5Gentamicina
702.5Streptomicina
231.7Cefaloridina
5.51.4Eritromicina
230.5Penicillina
AnuriaNormaleFarmaco
Funzione renale
87-10030-40Digossina
2305.8Vancomicina
352.5Gentamicina
702.5Streptomicina
231.7Cefaloridina
5.51.4Eritromicina
230.5Penicillina
AnuriaNormaleFarmaco
Funzione renale

Somministrando un farmaco a intervalli di una emivita si ottengono minime oscillazione della concentrazione ematica intorno a quella terapeutica (steady-state)
EMIVITA
Sono necessarie circa 5-7 emivite per raggiungere lo steady-state Sono necessarie almeno 7-10 emivite per ottenere il wash-out del farmaco
L’emivita permette di stimare quanto tempo deve passare perchè buona parte del farmaci sia eliminata. Per un farmaco con emivita di 1 ora, la sua concentrazione diventa il 50% dopo un’ora, il 25% dopo due ore; il 12,5 dopo 3 ore, il 6,25 dopo 4 ore e il 3,12 dopo 5 ore. Se il farmaco ha emivita di 8 ore, la sua concentrazione si sarà ridotto al 3% 40 ore (5 emivite moltiplicato 8 ore)

I farmaci possono avere differenti durate d’azione (si parla di farmaci ad azione breve, talora ultrabreve, intermedia, lunga). La durata d’azione di un farmaco dipende principalmente:
o Dalla velocità di eliminazione o Dai processi di biotrasformazione (metaboliti inattivi o
attivi) La velocità di eliminazione dipende dalla funzionalità degli
organi emuntori, dalle caratteristiche chimico-fisiche del farmaco o dei metaboliti (in particolare l’idrosolubilità), dalla forma farmaceutica, dalla via di introduzione. Normalmente la dose non influenza la velocità di eliminazione tranne che non si somministrino dosi molto elevate, tali da saturare i processi di eliminazione.
DURATA D’AZIONE DEI FARMACI

Rappresentano la quantità di farmaco attivo contenuta nel sangue nel tempo.
Esiste una corrispondenza tra i livelli ematici di un farmaco e la quantità di farmaco che raggiunge la sede d’azione. In altre parole vi è corrispondenza tra i livelli ematici e l’effetto farmacologico. Ad esempio il massimo effetto di un farmaco si avrà nel momento in cui è massima la concentrazione del farmaco nel sangue.
LIVELLI EMATICI (CONCENTRAZIONI EMATICHE) DEI FARMACI

I livelli ematici di un farmaco dipendono da diversi fattori, quali: la via di somministrazione la quantità e velocità dell’assorbimento la velocità di eliminazione la modalità di somministrazione (unica o ripetuta, nel
secondo caso ha rilievo l’intervallo di tempo tra le somministrazioni)
la quantità di farmaco somministrata (DOSE)
LIVELLI EMATICI (CONCENTRAZIONI EMATICHE) DEI FARMACI
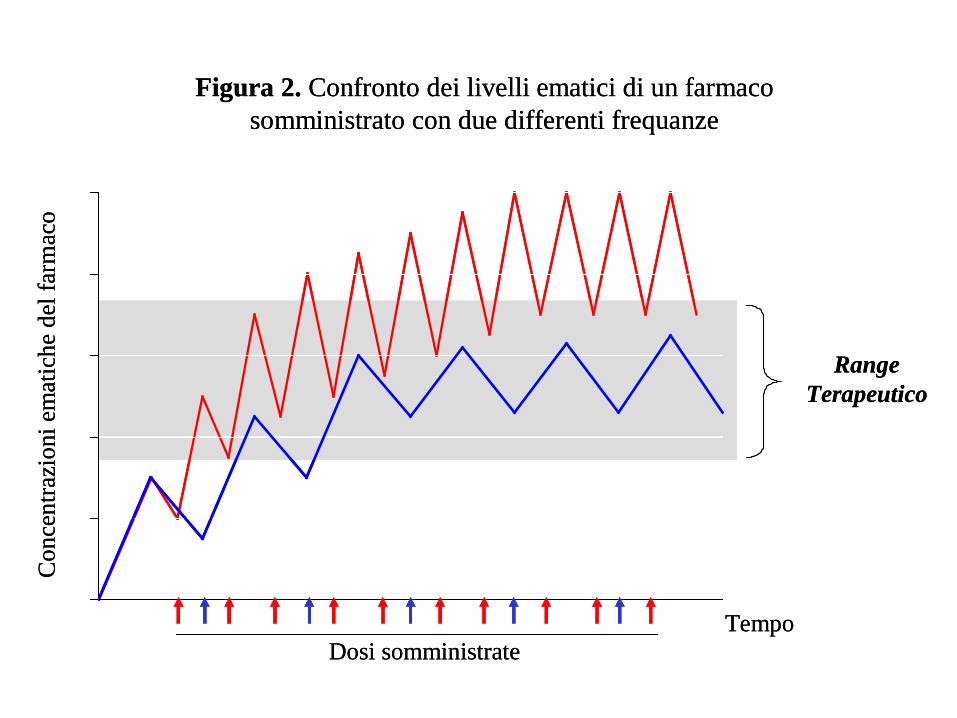
RangeTerapeutico
Con
cent
razi
oni e
mat
iche
del
farm
aco
TempoDosi somministrate
Figura 2. Confronto dei livelli ematici di un farmaco somministrato con due differenti frequanze
RangeTerapeutico
Con
cent
razi
oni e
mat
iche
del
farm
aco
TempoDosi somministrate
Figura 2. Confronto dei livelli ematici di un farmaco somministrato con due differenti frequanze
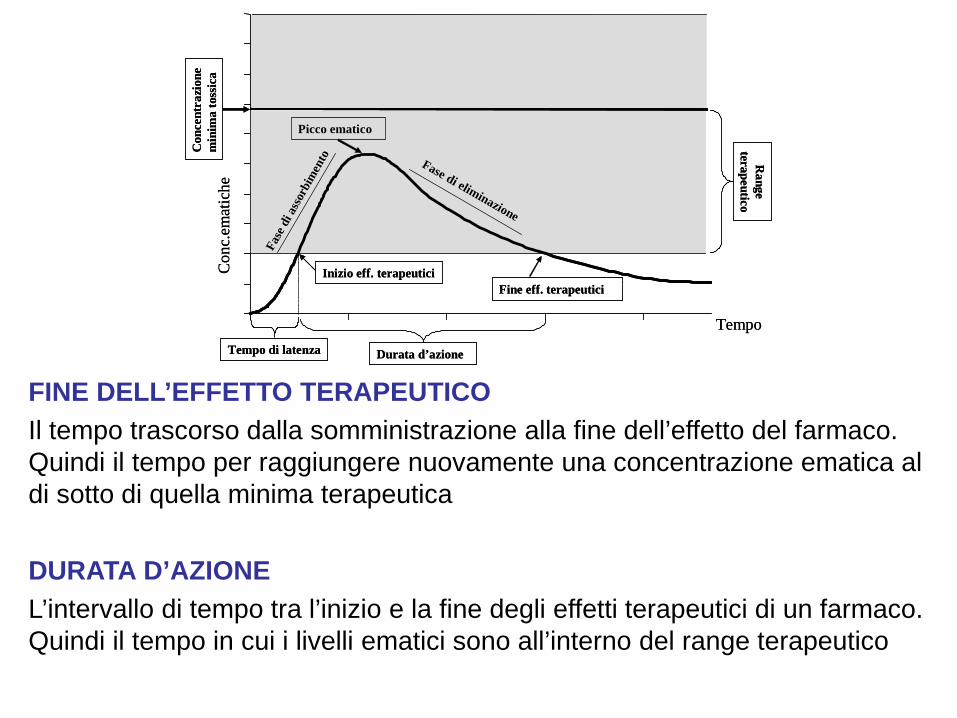
Tempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azioneTempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azione
Tempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azioneTempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azione
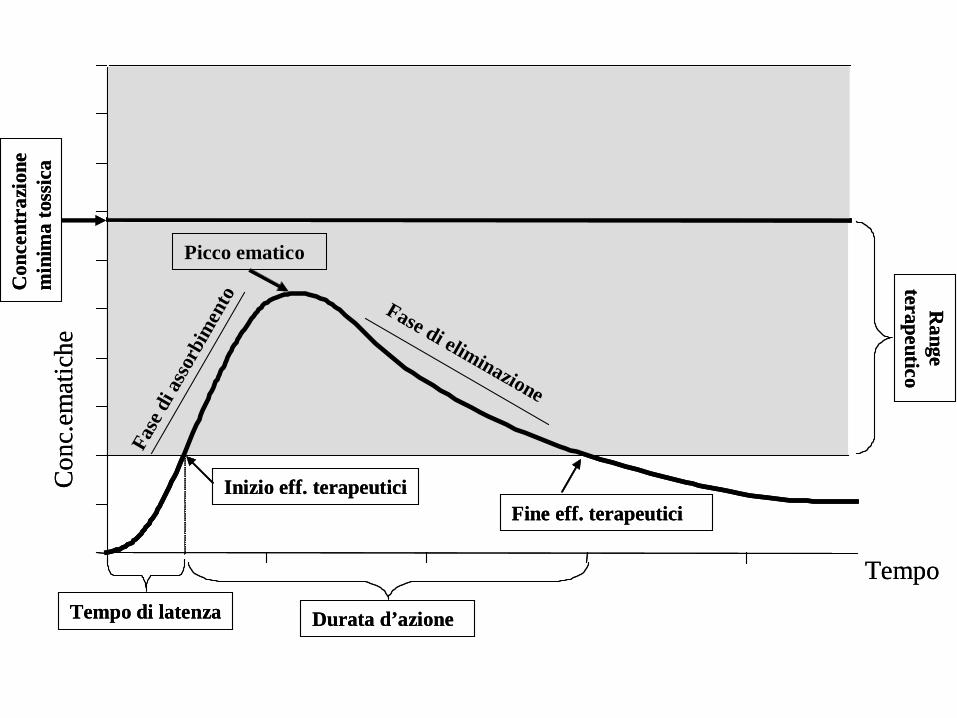
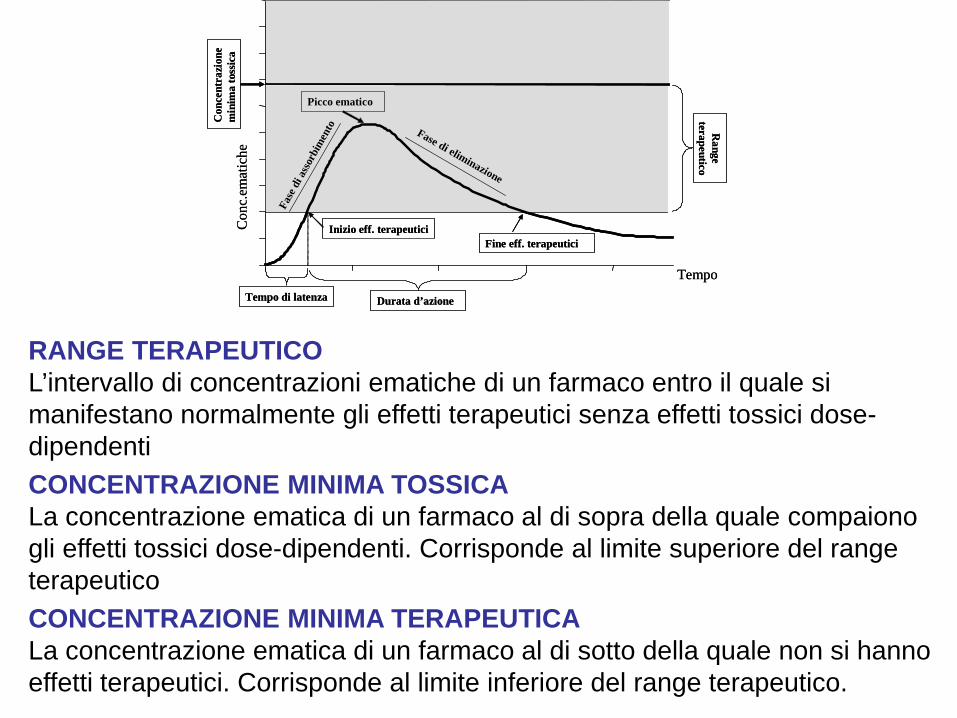
RANGE TERAPEUTICO L’intervallo di concentrazioni ematiche di un farmaco entro il quale si manifestano normalmente gli effetti terapeutici senza effetti tossici dose-dipendenti CONCENTRAZIONE MINIMA TOSSICA La concentrazione ematica di un farmaco al di sopra della quale compaiono gli effetti tossici dose-dipendenti. Corrisponde al limite superiore del range terapeutico CONCENTRAZIONE MINIMA TERAPEUTICA La concentrazione ematica di un farmaco al di sotto della quale non si hanno effetti terapeutici. Corrisponde al limite inferiore del range terapeutico.
Tempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azioneTempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azione
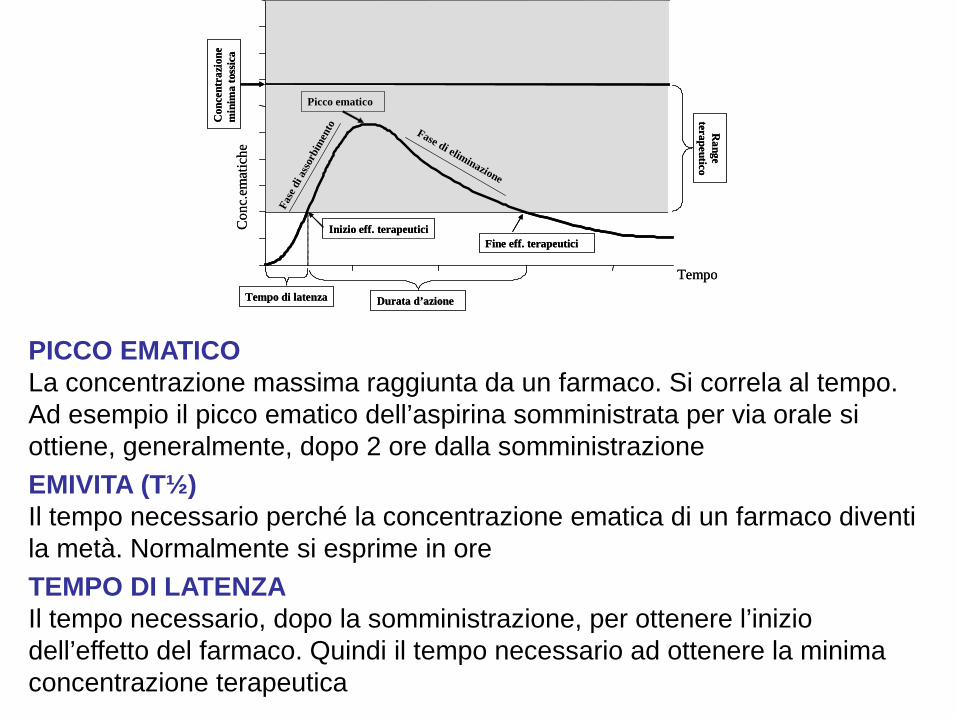
PICCO EMATICO La concentrazione massima raggiunta da un farmaco. Si correla al tempo. Ad esempio il picco ematico dell’aspirina somministrata per via orale si ottiene, generalmente, dopo 2 ore dalla somministrazione EMIVITA (T½) Il tempo necessario perché la concentrazione ematica di un farmaco diventi la metà. Normalmente si esprime in ore TEMPO DI LATENZA Il tempo necessario, dopo la somministrazione, per ottenere l’inizio dell’effetto del farmaco. Quindi il tempo necessario ad ottenere la minima concentrazione terapeutica
Tempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azioneTempo di latenza
Con
c.em
atic
he
Tempo
Con
cent
razi
one
min
ima
toss
ica
Picco ematico
Inizio eff. terapeuticiFine eff. terapeutici
Range
terapeutico
Fase di eliminazione
Fase
di a
ssor
bim
ento
Durata d’azione
FINE DELL’EFFETTO TERAPEUTICO Il tempo trascorso dalla somministrazione alla fine dell’effetto del farmaco. Quindi il tempo per raggiungere nuovamente una concentrazione ematica al di sotto di quella minima terapeutica DURATA D’AZIONE L’intervallo di tempo tra l’inizio e la fine degli effetti terapeutici di un farmaco. Quindi il tempo in cui i livelli ematici sono all’interno del range terapeutico

L’indice terapeutico di un farmaco è rappresentato dal numero derivante dal rapporto tra la dose tossica e la dose terapeutica.
Ad esempio per un farmaco che ha una dose tossica di 10 grammi ed una dose terapeutica di 2 grammi:
Risulta evidente che quanto più l’indice terapeutico di un farmaco è basso (vicino all’unità) tanto più ristretto è il margine di sicurezza nel dosaggio del farmaco.
INDICE TERAPEUTICO
Indice terapeutico: Dose tossica
Dose terapeutica =
10
2 = 5

L’indice terapeutico non rappresenta la valutazione di un farmaco dal punto di vista dell’efficacia e/o della tollerabilità ma ci indica soltanto la vicinanza o meno della dose tossica rispetto a quella terapeutica.
Farmaci con un basso indice terapeutico (ad esempio antiepilettici, teofillina, aminoglicosidi, antitumorali, warfarin) devono essere monitorati. Il monitoraggio si può effettuare direttamente, cioè prelevando dei campioni di sangue e determinando la quantità di farmaco presente, o indirettamente attraverso dei parametri di laboratorio, ad esempio per il warfarin o altri anticoagulanti misurando il tempo di coagulazione del sangue. In base ai risultati ottenuti si aggiusta la dose da somministrare.
INDICE TERAPEUTICO

La valutazione clinica di un farmaco è un processo complesso non esprimibile con un semplice rapporto tra dose tossica e dose terapeutica (indice terapeutico). Si tratta, infatti, di esprimere un giudizio valutando da una parte i benefici che si ottengono e dall’altra i rischi che si corrono utilizzando il farmaco (rapporto beneficio/rischio).
Per stabilire un corretto rapporto beneficio/rischio per un farmaco è necessario conoscere i benefici che si ottengono (quindi conoscere i dati sulla sua efficacia clinica) e i rischi derivanti dal suo uso (quindi conoscere i suoi effetti avversi).
Bisogna tenere presente che il rapporto beneficio/rischio di un farmaco può essere diverso a seconda del paziente e/o della patologia da trattare. Quindi in certe situazioni un farmaco, che ha in generale un rapporto beneficio/rischio favorevole (cioè i benefici superano i rischi), potrebbe avere un rapporto sfavorevole (i rischi superano i benefici).
RAPPORTO RISCHIO/BENEFICIO

DOSAGGIO: DEFINIZIONI
DOSE Quantità di farmaco somministrata per produrre un determinato effetto terapeutico POSOLOGIA Dose, tempi e modalità di somministrazione di un farmaco Esempio di posologia: Rocefin 500 mg due volte al giorno per via i.m. per 7 giorni DOSE MASSIMA La massima quantità di farmaco tollerata, senza cioè che si verifichino effetti tossici DOSE GIORNALIERA La quantità di farmaco somministrata nelle 24 ore

Modalità di somministrazione Peso (per farmaci ad alto rischio con basso indice
terapeutico meglio utilizzare la superficie corporea) Età Patologie concomitanti Gravidanza Contemporanea somministrazione con altri farmaci
che interagiscono
FATTORI DA CONSIDERARE NELLA DETERMINAZIONE DELLA DOSE

Microgrammo (µg o mcg) o gamma (γ): millesima parte del milligrammo Milligrammo (mg): millesima parte del grammo Grammo (g) Esempi:
400 mcg = 0,4 mg; 2 mg = 2000 mcg; 500 mg = 0,5 g; 3 g = 3000 mg Unità internazionali (UI): quantità di farmaco che provoca un
determinato effetto biologico [esempi di farmaci per cui si utilizzano le UI: insulina, eparina, eritropoietina, fattori della coagulazione, penicillina G, interferone, immunoglobuline, calcitonina]
PRINCIPALI UNITA’ DI MISURA DEI FARMACI

Microlitro (µl): millesima parte del millilitro Millilitro (ml): millesima parte del litro Centilitro (cl): centesima parte del litro Decilitro (dl): decima parte del litro Litro (L)
Esempi:400 µl = 0,4 ml; 2 ml = 2000 µl; 500 ml = 0,5 L;
50 cl = 0,5 L; 10 dl = 1 L
MISURE DI CAPACITA’

Soluzione al 5% = 5 grammi di farmaco in 100 ml Soluzione al 2% = 2 grammi di farmaco in 100 ml Soluzione al 9 per mille = 9 grammi in 1000 ml (1 L) questi sono esempi di concentrazioni peso/volume, nel caso
di pomate o unguenti le concentrazioni sono peso/peso ad esempio pomata al 5% = 5 grammi di farmaco in 100 g di pomata
SOLUZIONI DEI FARMACI

Modificazioni della Farmacocinetica indotte dall’attività motoria

CAMBIAMENTI FISIOLOGICI durante attività fisica
• Aumento gittata cardiaca • Ridistribuzione del flusso sanguigno
lontano dall’area splancnica verso i muscoli • Incremento flusso sanguigno polmonare • Riduzione pH di tessuti, sangue, urine. • Riduzione flusso urinario • Riduzione funzione gastro-intestinale • Aumento temperatura corporea, epiteliale e
tasso di sudore

Cambiamenti nel flusso sanguigno indotti
dall’attività fisica
Lenz TL et al. Sports Med 2004; 34: 293-306

• Riduzione svuotamento gastrico intestinale. Brouns et al. Int J Sport Med 1987: Es. strenuo> 70 % VO2max
• Riduzione del tempo di transito intestinale. Cordain et al. J Sports Med 1994: 6 sett.allenamento aerobico, riduzione
del 22,8%.
• Correlazione tra riduzione del flusso sanguigno intestinale e intensità dell’esercizio.
Rowell LB.Physiological Reviews 1974: - 70%VO2max = riduzione flusso del 50% - Intensità max = riduzione flusso del 80%
Cambiamenti a livello gastro-intestinale indotti dall’attività fisica

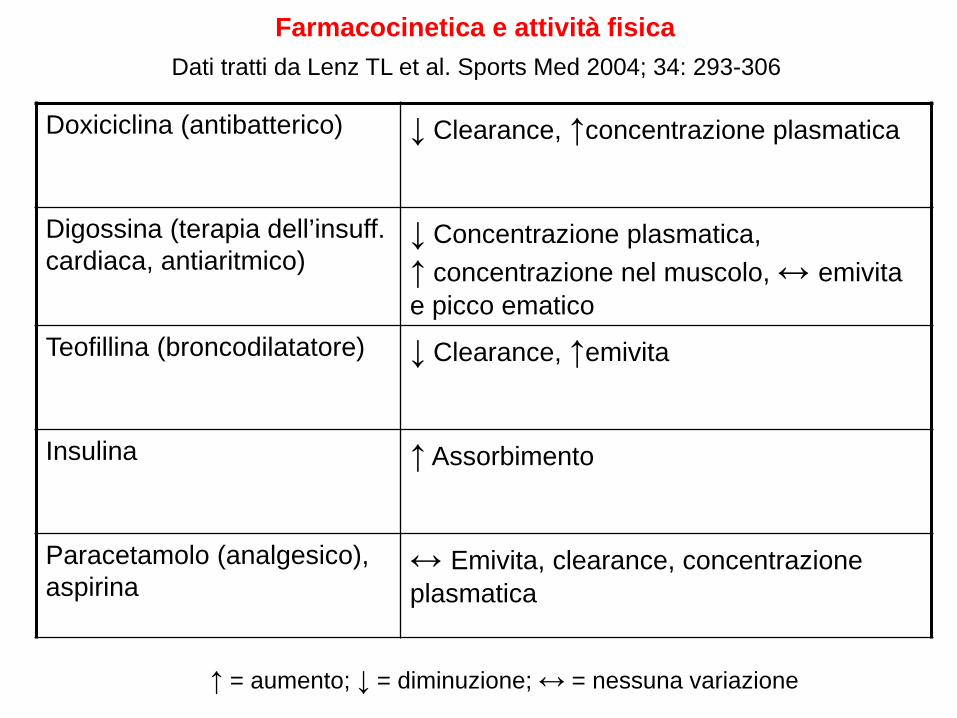
Farmacocinetica e attività fisica Dati tratti da Lenz TL et al. Sports Med 2004; 34: 293-306
Atenololo (beta-bloccante) ↓ Clearance renale
↔ Concentrazione plasmatica
Carvedilolo (alfa- e beta-bloccante)
↔ Concentrazione plasmatica
Propranololo (beta-bloccante), singola sessione di attività fisica
↓ Emivita, ↓ clearance, ↑ concentrazione plasmatica
Propranololo (beta-bloccante), attività fisica continua
↔ Concentrazione plasmatica, emivita, biodisponibilità, legame proteine, volume distribuzione
Verapamil (calcio-antagonista) ↔ Clearance, emivita, volume distribuzione
↑ = aumento; ↓ = diminuzione; ↔ = nessuna variazione
Farmacocinetica e attività fisica Dati tratti da Lenz TL et al. Sports Med 2004; 34: 293-306
Doxiciclina (antibatterico) ↓ Clearance, ↑concentrazione plasmatica
Digossina (terapia dell’insuff. cardiaca, antiaritmico)
↓ Concentrazione plasmatica, ↑ concentrazione nel muscolo, ↔ emivita e picco ematico
Teofillina (broncodilatatore) ↓ Clearance, ↑emivita
Insulina ↑ Assorbimento
Paracetamolo (analgesico), aspirina
↔ Emivita, clearance, concentrazione plasmatica
↑ = aumento; ↓ = diminuzione; ↔ = nessuna variazione

Le evidenze disponibili sugli effetti dell’attività motoria sulla farmacocinetica o sulla farmacodinamica non fanno emergere una problematica rilevante, sia in termini di diminuzione dell’efficacia che di aumento della tossicità dei farmaci, tuttavia alcune considerazioni vanno fatte: •Gli studi che mettono in relazione l’attività motoria con la cinetica dei farmaci sono pochi e su pochi farmaci •La maggioranza degli studi sono stati effettuati su persone sane che assumevano i farmaci appositamente per lo studio, possono esserci differenze rispetto ai malati in terapia con gli stessi farmaci •Non si è verificato se ci sono differenze tra soggetti allenati e soggetti non allenati •Per alcuni farmaci (teofillina, digossina, insulina), sono state osservate delle modificazioni della farmacocinetica che potrebbero essere rilevanti in quanto sono farmaci con un ristretto indice terapeutico.


![DOTT.AMMIRATI [modalità compatibilità] · P-gp inibitori Con cautela Inibitori/induttoriP-gp: Inibitori/induttoridi CYP o P gp •claritromicina •amiodarone •chinidina - : •claritromicina](https://static.fdocumenti.com/doc/165x107/5fac329fcf14a059e9511b09/dottammirati-modalit-compatibilit-p-gp-inibitori-con-cautela-inibitoriinduttorip-gp.jpg)