LA FLESSIBILITA' NELLA VALUTAZIONE DELLE INTERAZIONI...
118
UNIVERSITA' DEGLI STUDI DI PARMA FACOLTA' DI FARMACIA CORSO DI LAUREA SPECIALISTICA IN CHIMICA E TECNOLOGIA FARMACEUTICHE LA FLESSIBILITA' NELLA VALUTAZIONE DELLE INTERAZIONI TRA MOLECOLE BIOLOGICHE: IL RECETTORE DEGLI ESTROGENI Relatore: Chiar.mo Prof. ANDREA MOZZARELLI Correlatori: Dott. PIETRO COZZINI Dott. ALESSIO AMADASI Laureanda: ELENA ESPOSITO ANNO ACCADEMICO 2004-2005
-
Upload
nguyendung -
Category
Documents
-
view
215 -
download
0
Transcript of LA FLESSIBILITA' NELLA VALUTAZIONE DELLE INTERAZIONI...

UNIVERSITA' DEGLI STUDI DI PARMA
FACOLTA' DI FARMACIA
CORSO DI LAUREA SPECIALISTICA
IN CHIMICA E TECNOLOGIA FARMACEUTICHE
LA FLESSIBILITA' NELLA VALUTAZIONE DELLE
INTERAZIONI TRA MOLECOLE BIOLOGICHE:
IL RECETTORE DEGLI ESTROGENI
Relatore:
Chiar.mo Prof. ANDREA MOZZARELLI
Correlatori:
Dott. PIETRO COZZINI
Dott. ALESSIO AMADASI
Laureanda:
ELENA ESPOSITO
ANNO ACCADEMICO 2004-2005


Alle mie nonne


Indice
INTRODUZIONE.................................................................................................. 1
1.1 Gli estrogeni.................................................................................................. 1
1.2 Meccanismo d’azione degli estrogeni.................................................. 2
1.2.1 Le isoforme recettoriali α e β...................................................... 3
1.2.2 Il legame dell’estradiolo a ERα.................................................. 4
1.3 Principali effetti benefici e avversi della stimolazione estrogenica............................................................................................................
5
1.4 L’elica 12......................................................................................................... 7
1.5 I SERM............................................................................................................ 7
1.5.1 Il legame del raloxifene a ERα................................................... 9
1.5.2 Meccanismo d’azione dei SERM................................................ 10
1.6 I fitoestrogeni............................................................................................... 11
1.7 Endocrine disruptors.................................................................................. 13
1.8 Virtual screening.......................................................................................... 14
1.9 Flessibilità e docking................................................................................. 17
1.10 La flessibilità delle proteine.................................................................. 18
1.11 Incorporazione della flessibilità proteica nel docking................ 20
HINT......................................................................................................................... 23
2.1 Il software HINT......................................................................................... 23
2.1.1 Relazione tra LogP e ∆G°........................................................... 27
2.1.2 Applicazioni del software HINT................................................ 28
SCOPI........................................................................................................................ 29
MATERIALI E METODI..................................................................................... 31
4.1 Tripos............................................................................................................ 31
4.2 Analisi di composti cristallografici proteina-ligando-acqua: protocollo di lavoro........................................................................................... 32
4.3 Docking di complessi cristallografici: protocollo di lavoro........ 36
4.4 Docking di additivi alimentari: protocollo di lavoro..................... 37
4.5 GRID................................................................................................................ 38

Indice
RISULTATI E DISCUSSIONI............................................................................. 41
5.1 Flessibilità del recettore degli estrogeni............................................ 42
5.2 Analisi preliminari....................................................................................... 46
5.2.1 Scelta della struttura cristallografica del recettore............... 46
5.2.2 Ruolo della molecola d’acqua conservata................................. 47
5.3 Docking di SERM e agonisti................................................................... 49
5.4 Analisi del farmacoforo............................................................................. 56
5.5 Docking di additivi alimentari............................................................... 59
5.5.1 Delfinidina....................................................................................... 62
5.5.2 Capsaicina........................................................................................ 64
5.5.3 Bisdemetossicurcumina................................................................ 67
5.5.4 Propil p-idrossibenzoato.............................................................. 69
5.5.5 Acido nordiidroguaiaretico.......................................................... 71
5.5.6 Eugenolo.......................................................................................... 73
5.5.7 o-Fenilfenolo................................................................................... 74
5.5.8 Chinina............................................................................................. 75
5.5.9 Etilvanillina..................................................................................... 78
5.5.10 Acido colico................................................................................... 79
5.5.11 Valutazione dei risultati ottenuti dall’analisi degli additivi........................................................................................................
80
CONCLUSIONI....................................................................................................... 85
BIBLIOGRAFIA..................................................................................................... 87

Introduzione


Introduzione
1
1.1 GLI ESTROGENI
Gli estrogeni appartengono alla famiglia degli ormoni sessuali e presentano la tipica
struttura steroidea a 18 atomi di carbonio. Il 17 beta-estradiolo è il capostipite degli
ormoni estrogenici e viene sintetizzato principalmente a livello ovarico in risposta a
stimoli ipofisari. L’estradiolo è prodotto anche dalla placenta durante la gravidanza
e in piccola percentuale dai testicoli e dalle ghiandole surrenaliche. L’azione
dell’estradiolo è sostenuta da altri due estrogeni: l’estrone e l’estriolo. Tali ormoni
sono caratterizzati da una bassa affinità recettoriale e sono sintetizzati,
principalmente, a livello epatico in seguito a conversione dell’estradiolo stesso
(Figura 1.1.1).
Le proprietà idrofobiche del nucleo steroideo permettono agli estrogeni di
oltrepassare le membrane e svolgere la loro azione fisiologica a livello intracellulare.
Per le stesse ragioni, questi ormoni si trovano in circolo fortemente coniugati con
una globulina plasmatica chiamata SHBG (sex-hormone binding globulin). Ne deriva
che solo una piccola aliquota è libera e disponibile per la diffusione all’interno delle
cellule.
La funzione primaria degli estrogeni è di promuovere lo sviluppo e il mantenimento
dei caratteri sessuali secondari femminili, ma, in realtà, gli estrogeni sono coinvolti
in un ampio spettro di attività fisiologiche che riguardano la regolazione
omeostatica di molteplici eventi biochimici e cellulari (Vaya et al. 2004).
Figura 1.1.1: Metabolismo dell’estradiolo (B. G. Katzung, Farmacologia generale e clinica, Piccin, Padova 2001).
Introduzione
2
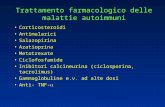
1.2 MECCANISMO D’AZIONE DEGLI ESTROGENI
Gli estrogeni mediano la loro azione fisiologica attraverso la stimolazione di una
classe recettoriale costituita dai recettori degli estrogeni (ERs), i quali
appartengono alla superfamiglia dei recettori nucleari e presentano domini
strutturali comuni a quelli dei recettori per testosterone, progesterone, corticoidi,
ormoni tiroidei vitamine A e D. I recettori ERs sono in grado di attraversare la
membrana nucleare e raggiungere il citoplasma, ma, per lo più, risiedono nel nucleo
complessati con heat shock protein, che mantengono quiescente il recettore fino
all’arrivo di un appropriato agonista. Il legame con l’estradiolo genera una serie di
variazioni conformazionali tali per cui le proteine stabilizzatrici vengono
allontanate e il recettore dimerizza, esponendo la propria superficie all’aggancio dei
coattivatori. Il complesso che si genera presenta un elevata affinità per specifiche
sequenze palindromiche di DNA, chiamate elementi di risposta agli estrogeni
(ERE), localizzate a monte di promotori di diversi geni di cui regolano la
trascrizione (Figura 1.2.1).
Questo meccanismo d’azione è alla base degli effetti promossi dagli estrogeni.
Figura 1.2.1: Meccanismo trasduzionale mediato dal recettore degli estrogeni (Osborne et al. 2000)

Introduzione
3
Alcune delle azioni mediate dall’estradiolo e dai suoi analoghi sembrano essere
legate anche ad effetti indiretti, dovuti alla liberazione, per azione delle proteine
neosintetizzate, di mediatori lipidici, citochine e fattori di crescita.
1.2.1 LE ISOFORME RECETTORIALI α E β
Nel 1986 fu clonato per la prima volta il recettore degli estrogeni, identificato con la
sigla ERα, che per anni rimase l’unico recettore conosciuto. Nel 1996 Gustafsson e
collaboratori riuscirono ad isolare e clonare un secondo sottotipo recettoriale a cui
fu dato il nome ERβ. Tale scoperta sollevò numerose domande riguardo al diverso
impatto dei due recettori nel regolare le molteplici attività mediate dagli estrogeni,
alcune delle quali rimangono ancora irrisolte.
Studi successivi hanno dimostrato che il sottotipo α svolge un ruolo predominante
nello sviluppo dei caratteri sessuali secondari e nella regolazione della densità ossea,
mentre la stimolazione del recettore β sembra essere necessaria nello sviluppo
cerebrale e cognitivo e nei processi di plasticità neuronale, che rendono il recettore
un potenziale target farmacologico nel trattamento del morbo di Alzheimer (Liquin
Zhao et al 2004). Infine, ERβ è espresso in molti altri tessuti, quali il sistema
cardiovascolare, immunitario, gastrointestinale, urogenitale, renale. Non è escluso
che rappresenti la popolazione recettoriale maggiormente presente nell’organismo
(Gustafsson et al 1999).
I recettori degli estrogeni sono formati da sei domini strutturali (A-F),
diversamente coinvolti nel meccanismo di trasduzione del segnale (Figura 1.2.2).
Nella porzione amminoterminale della proteina si trova una sequenza altamente
variabile (dominio A/B), direttamente implicata nella trasduzione del segnale, in
quanto interagisce con i coattivatori. La regione C, localizzata centralmente e
fortemente conservata, rappresenta il dominio di legame al DNA e contiene due
motivi leganti zinco. La regione D contribuisce alla flessibilità e alla dimerizzazione
del recettore. Il dominio (E) di legame degli agonisti endogeni (LBD) è conservato e
coopera nell’aggancio dei coattivatori e dei corepressori trascrizionali. Infine, nella
porzione carbossiterminale si trova il dominio F, che partecipa all’attivazione
trascrizionale. I recettori α e β presentano analogie strutturali a livello dei domini
deputati al legame del DNA (95% di omologia) e del ligando (58% di omologia), che

Introduzione
4
rappresentano le regioni conservate, mentre A/B, D e F non sono necessariamente
simili (Jordan et al 2003). All’interno di LBD (ligand binding domain) i due residui
amminoacidici Leu 384 e Met 421 di ERα corrispondono, rispettivamente, a Met
336 e Ile 373 nel recettore β. Questa sottile diversità potrebbe essere la chiave per la
costruzione di ligandi in grado di discriminare tra le due isoforme recettoriali (Liqin
Zhao et al. 2004), anche se estremamente difficile..
1.2.2 IL LEGAME DELL’ESTRADIOLO A ERα
La struttura cristallografica del complesso ERα-estradiolo fu determinata, per la
prima volta, da Brzozowski e collaboratori nel1997. Il sito di legame dell’estradiolo
è collocato in una cavità profonda del recettore ed è costituito, in larga parte, da
amminoacidi idrofobici. La complementarietà tra il carattere apolare degli agonisti
endogeni e l’idrofobicità del sito contribuisce, unitamente alla formazione di specifici
legami idrogeno, al riconoscimento tra proteine e ligando. L’estradiolo presenta,
nella propria struttura, due gruppi ossidrilici, le cui interazioni sono di
fondamentale importanza nell’attivazione del recettore. In particolare, l’ossidrile
fenolico instaura legami idrogeno con lo ione carbossilato del Glu 353, con il
gruppo guanidinico dell’Arg 394 e con una molecola d’acqua conservata, mentre il
17-β ossidrile forma un unico legame ad idrogeno con l’anello imidazolico dell’His
524. La restante parte della molecola non contrae legami diretti con il recettore, ma
Figura 1.2.2: Domini strutturali del recettore degli estrogeni (COT Report, Phytoestrogens and Health, Food Standards Agency, 2003)

Introduzione
5
prende parte a numerose interazioni idrofobiche (Figura 1.2.3). L’estradiolo lega
Erα con una Ki pari a 0.13 nM (George et al. 1997).
1.3 PRINCIPALI EFFETTI BENEFICI E AVVERSI DELLA
STIMOLAZIONE ESTROGENICA
Tessuto osseo
E’ stato dimostrato che l’aumentata incidenza di osteoporosi e fratture ossee nel
periodo postmenopausale è strettamente correlata alla diminuzione della produzione
di estrogeni (Anthony et al. 2002). Una delle principali terapie nella cura
dell’osteoporosi consiste, infatti, nella somministrazione di ormoni estrogenici
oppure di modulatori selettivi del recettore degli estrogeni che riducono il
riassorbimento del tessuto osseo e ne promuovono la rimineralizzazione.
Sistema cardiovascolare
Gli estrogeni esercitano importanti effetti benefici sul sistema cardiocircolatorio
dovuti, in gran parte, alla riduzione della formazione di placche aterosclerotiche. Le
Figura 1.2.3: Interazione dell’estradiolo con il sito attivo del recettore (Brzozowski et al. 1997).

Introduzione
6
arterie sono parzialmente protette dalla capacità degli estrogeni di limitare il
rilascio di lipoproteine a bassa densità (LDL) e aumentare quello delle lipoproteine
ad alta densità (HDL). Inoltre, tali ormoni, promuovono la vasodilatazione e
riducono i fenomeni di ischemia miocardica (Anthony et al. 2002).
Effetti proliferativi
Probabilmente gli estrogeni non sono i diretti responsabili dei cambiamenti cellulari
che generano una neoplasia, ma favoriscono, comunque, la proliferazione
incontrollata di una popolazione cellulare già mutata (Jordan 1998). L’esposizione
agli estrogeni diventa un fattore di rischio nello sviluppo di tumori e limita la
terapia a base di tali ormoni durante la menopausa. I tessuti maggiormente colpiti
sono l’utero e il seno, che rappresentano le principali sedi dell’azione fisiologica.
Diversamente, gli estrogeni sembrano mostrare effetti favorevoli sul tratto
gastrointestinale, svolgendo un ruolo di protezione dallo sviluppo del cancro al
colon (Enmark et al. 1999).
Sistema immunitario
Molte malattie autoimmuni sono più comuni nella donna che nell’uomo e
frequentemente originano in seguito a cambiamenti drastici dei livelli ormonali,
come durante la pubertà, la gravidanza e la menopausa. Questo fa ipotizzare che gli
estrogeni esercitino azioni rilevanti anche nel sistema immunitario (Gustafsson et
al. 1999).
Centri cerebrali
Gli estrogeni possono esercitare diversi effetti neuroprotettivi sul sistema nervoso
centrale, che comprendono inibizione della formazione delle placche di β-amiloide,
stimolazione colinergica, riduzione dello stress ossidativo e protezione dalle
patologie vascolari. Studi clinici hanno dimostrato che le donne in cura con ormoni
estrogenici presentano una minor incidenza nello sviluppo del morbo di Alzheimer,
stimolando le ricerche in tale direzione (Ohmichi et al. 2005).

Introduzione
7
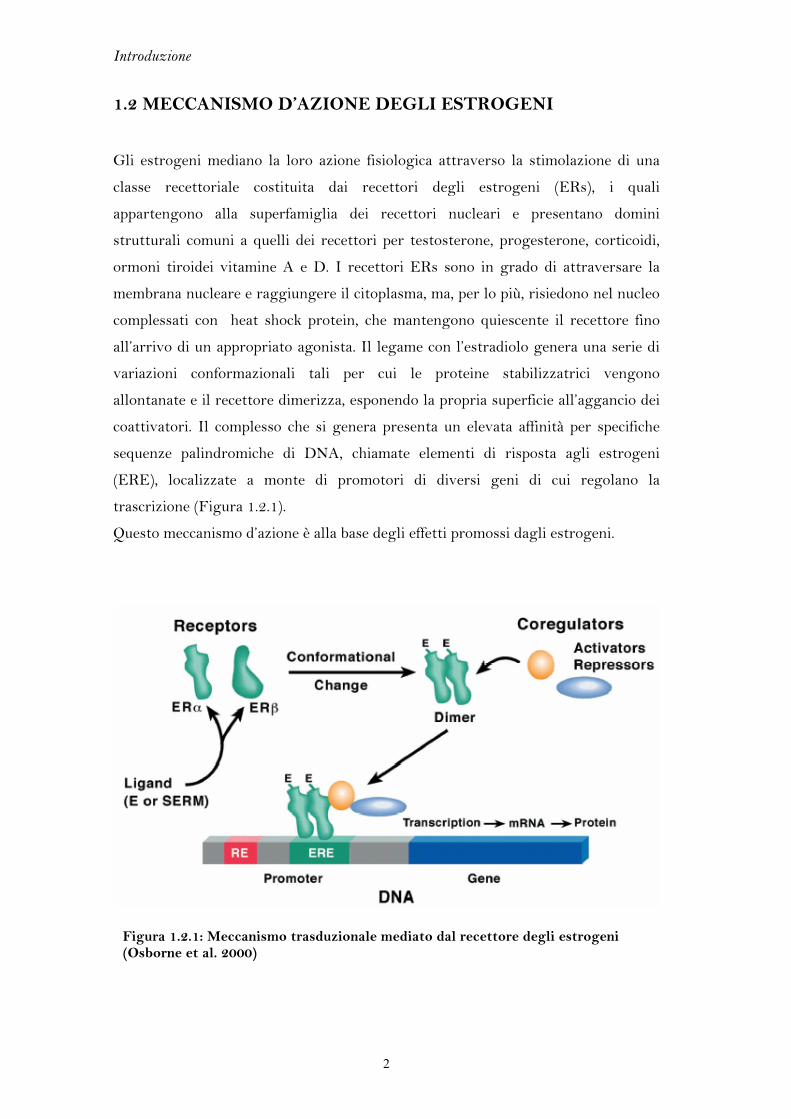
1.4 L’ELICA 12
Erα presenta due regioni chiamate activation functions (AFs) che presiedono
all’attivazione trascrizionale. AF-1, situata nel dominio A/B, è regolata dallo stato
di fosforilazione di alcune serine e non dal legame del recettore con il ligando. Al
contrario la conformazione di AF-2, collocata nel dominio carbossiterminale,
dipende dalla presenza e dal tipo di ligando. AF-2 è meglio conosciuta come elica
12.
Attualmente esistono diverse strutture del recettore Erα complessato con diversi
ligandi, sia di natura agonista che antagonista. L’elica 12 rappresenta una delle
porzioni maggiormente flessibili del recettore e assume orientazioni spaziali
differenti in base alla conformazione recettoriale selezionata dal ligando (Figura
1.4.1).
Quando il recettore è libero da ogni ligando, assume una struttura tridimensionale
affine ai corepressori che ne spengono l’attività. In presenza di un agonista l’elica 12
si dispone in posizione antiparallela rispetto all’elica 11, chiudendo la tasca di
legame e permettendo l’esposizione di una fessura idrofobica adatta all’aggancio dei
coattivatori (Figura 1.4.1A). Gli antagonisti e gli agonisti parziali sono
generalmente caratterizzati da catene laterali lunghe che non possono essere
contenute interamente nei confini del sito di legame e protendono al di fuori di esso.
Questo modello di antagonismo impone un riposizionamento dell’elica 12, che ruota
di 130° rispetto alla conformazione assunta nel complesso dell’agonista, inserendosi
tra le eliche 3, 4 e 5 (Figura 1.4.1B). Questa variazione conformazionale, che
impedisce il reclutamento dei coattivatori, è alla base dell’attività dei SERM
(modulatori selettivi del recettore per gli estrogeni).
1.5 I SERM
La terapia sostitutiva a base di estrogeni (estrogen replacement therapy HRT) è stata
utilizzata per molti anni, nelle donne in menopausa, per alleviare i sintomi associati
alla diminuzione dei livelli di estrogeni circolanti, quali vampate di calore e
decalcificazione delle ossa.
Introduzione
8
A
B
Sfortunatamente, però, gli effetti collaterali di tale terapia sono legati ad un
aumento dell’incidenza del cancro all’utero e al seno, che ne hanno limitato
fortemente l’utilizzo.
La scoperta dell’esistenza di una relazione tra la stimolazione del recettore degli
estrogeni e lo sviluppo di neoplasie, spostò l’attenzione del mondo scientifico verso
la ricerca di antagonisti per il trattamento del tumore mammario. Gli studi che
seguirono portarono alla nascita di una classe di farmaci, nota come SERM, il cui
Figura 1.4.1: Orientazione dell’elica 12 in presenza di un agonista (A) e in presenza di un agonista parziale (B). Il recettore α è rappresentato a sinistra con una proiezione “a cilindri” (elica 12 in rosso) e a destra mediante superficie di Connolly (elica 12 in fucsia).

Introduzione
9
nome riflette la capacità delle molecole di agire da agonisti in alcuni tessuti e da
antagonisti in altri.
I capostipiti di questa famiglia di molecole non steroidee sono due (Figura 1.5.1): il
primo è tamoxifene, che mostra effetti agonisti sulle ossa e sul sistema
cardiocircolatorio, mentre agisce da antagonista nelle ghiandole mammarie. Nel
1986 il tamoxifene era ampiamente prescritto come coadiuvante nella terapia del
cancro al seno, quando studi di tossicità dimostrarono che la molecola aumentava la
probabilità di tumore all’endometrio. Il secondo farmaco, scoperto successivamente,
è il raloxifene, che pur mantenendo tutti i vantaggi del tamoxifene svolge un’azione
antagonista anche a livello uterino, quindi non favorisce l’insorgenza di neoplasie.
Per tale motivo il raloxifene è diventato il prototipo nella ricerca di nuovi SERM.
1.5.1 IL LEGAME DEL RALOXIFENE A ERα
Nella struttura del complesso tra raloxifene e ERα è possibile distinguere gruppi
funzionali importanti per l’aggancio al recettore: la prima è rappresentata dai due
gruppi ossidrilici che, analogamente all’estradiolo, instaurano legami idrogeno con
Glu 353 e Arg 394 da un lato e His 524 dall’altro. I due ossidrili sono separati da un
corpo centrale rigido, che mima la struttura steroidea degli agonisti endogeni,
contraendo interazioni idrofobiche con il sito. Il raloxifene, infine, a differenza
dell’estradiolo, presenta una catena laterale ingombrante che impedisce, come
Figura 1.5.1: Strutture dei due SERM tamoxifene e raloxifene.

Introduzione
10
sottolineato in precedenza, il normale posizionamento dell’elica 12 e la formazione
della conformazione attiva. La catena laterale termina con un anello piperidinico
che, a pH fisiologico, si presenta, per lo più, nella forma protonata e forma un
legame idrogeno con la funzione carbossilica dell’ Asp 351, contribuendo ad
aumentare l’affinità del raloxifene per il recettore (Figura 1.5.2).
1.5.2 MECCANISMO D’AZIONE DEI SERM
Non è ancora del tutto chiaro come i SERM possano agire da antagonisti degli
estrogeni in alcune cellule e da agonisti in altre, ma sono state proposte ipotesi
diverse. Probabilmente questi farmaci interagiscono con il recettore in modo
identico in tutti i tessuti, ma la risposta cambia in base a qualche differenza
dipendente dalla cellula stimolata.
Una prima ipotesi riguarda la natura dei coregolatori: alcuni tessuti potrebbero non
essere in grado di produrre coattivatori capaci di legarsi al complesso SERM-
recettore, mentre altre cellule potrebbero presentare un set di coattivatori differenti
Figura 1.5.2: A sinistra sovrapposizione del sito di legame del raloxifene (blu) con quello dell’estradiolo (verde). A destra interazione del raloxifene con gli amminoacidi chiave della cavità di legame (Brzozowski et al. 1997).

Introduzione
11
o leggermente modificati in grado di agganciare recettori in conformazione anomala
e dare il via ad un complesso di trascrizione funzionante.
Un’altra teoria si basa sull’ipotesi che i complessi formati dai SERM siano incapaci
di legare gli elementi di risposta agli estrogeni nei geni. Tuttavia, in alcuni tessuti,
alcuni geni potrebbero possedere siti di legame alternativi, che permettono al
processo di trascrizione di continuare come se fosse legato l’estrogeno.
Infine, non si può sottovalutare l’ipotesi che le isoforme α e β del recettore
rispondano in maniera differente al legame dei SERM e, che a seconda della loro
distribuzione tissutale, vari la risposta generata.
1.6 I FITOESTROGENI
Composti naturali presenti nelle piante, definiti fitoestrogeni, hanno mostrato di
possedere attività estrogeno mimetica. Nel 2003 la Food Standards Agency ha
pubblicato un rapporto dal titolo “Phytoestrogens and health” in cui i fitoestrogeni
sono definiti come: “qualsiasi sostanza o metabolita di origine vegetale che induce
risposte biologiche nei vertebrati e che può mimare o modulare l’azione degli
estrogeni endogeni legandosi, di norma, al recettore degli estrogeni stessi”.
I fitoestrogeni sono principalmente suddivisi in quattro classi chimiche differenti,
più o meno potenti a seconda delle somiglianze strutturali con l’estradiolo (Figura
1.6.1). Il gruppo più consistente e più studiato è rappresentato dai flavonoidi
(ulteriormente classificati in flavanoni, flavonoli, isoflavoni), a cui appartengono
composti come la genesteina, la daidzeina, la rutina, la miricetina, l’apigenina.
Elevate concentrazioni di isoflavoni sono stati riscontrati in alimenti come la soia, di
largo consumo nei paesi orientali. Meno popolate e meno indagate sono le altre
classi di fitoestrogeni: lignani (ad es. secoisolariciresinolo), coumestani (come il
coumestrolo) e calconi (tra cui la floretina). La classificazione dei fitoestrogeni può
essere integrata con una quinta classe costituita dai micoestrogeni, il cui capostipite
è lo zearalenone. Tali composti sono meno rilevanti dal punto di vista nutrizionale
poichè, essendo i prodotti di una contaminazione fungina, non sono presenti
costitutivamente negli alimenti.
I fitoestrogeni svolgono le loro azioni attraverso svariati meccanismi biologici.
Principalmente interagiscono con il recettore, modulando l’espressione dei geni

Introduzione
12
responsivi degli estrogeni. A tal proposito, Kuiper e collaboratori hanno messo in
evidenza come agiscano principalmente sul recettore β, pur non mancando una loro
azione sul recettore α.
HO
OH
O OHO
O
OHO
OH
O
O
HO
HO
I fitoestrogeni, inoltre, sono coinvolti nell’inibizione di enzimi per la sintesi e il
metabolismo degli estrogeni, nella modulazione della biosintesi degli ormoni
tiroidei, in reazioni antiossidanti e nell’inibizione di proteine chinasi.
L’osservazione che nel Sud-Est asiatico le donne presentassero una minor incidenza
di tumori ormone-dipendenti, di patologie cardiovascolari e di sintomi
classicamente legati alla menopausa incrementò l’ipotesi che un ruolo di protezione
fosse giocato dal tipo di regime alimentare e che una dieta ricca di fitoestrogeni
potesse rappresentare una possibile opzione nella prevenzione dell’osteoporosi e del
cancro.
La valutazione dell’impatto dei fitoestrogeni è complicata da alcune evidenze:
innanzitutto, molte delle relazioni sui loro effetti benefici si basano su osservazioni
epidemiologiche e non su misure dirette dell’effetto osservato. E’ necessario
sottolineare che molti dei dati raccolti da questi studi riguardano popolazioni
orientali, quindi l’estrapolazione dei risultati è inficiata da differenze genetiche,
O
O
Isoflavoni Estradiolo Coumestani
Calconi Lignani Micoestrogeni
Figura 1.6.1: Strutture chimiche dell’estradiolo e delle principali classi di fitoestrogeni

Introduzione
13
metaboliche, negli stili di vita, nella dieta. Inoltre, spesso gli studi in vivo sono
effettuati utilizzando elevate concentrazioni di fitoestrogeni e questo rende le
condizioni sperimentali non aderenti ai reali livelli di esposizione. Infine, non si può
escludere la possibilità che, in un sistema complesso come un alimento, ci siano altri
componenti attivi che contribuiscano agli effetti osservati (COT report, Foods
Standards Agency, 2003).
1.7 ENDOCRINE DISRUPTORS
Il recettore degli estrogeni è una proteina versatile, che, grazie all’elevata flessibilità
del sito attivo, è in grado di legare molti composti di natura steroidea e non. Il
termine xenoestrogeni è utilizzato per indicare tutte le sostanze chimiche esogene,
sintetiche o naturali, che mostrano, in seguito al legame con ER, attività estrogena
e/o antiestrogenica. Gli xenoestrogeni sono in grado di alterare la normale
funzionalità del sistema endocrino e riproduttivo, imitando o inibendo l’azione degli
ormoni endogeni, modulandone la produzione o alterando la popolazione
recettoriale. Proprio grazie all’abilità di questi composti di interferire con il sistema
endocrino, gli xenoestrogeni sono stati definiti “endocrine disruptors”. Infatti, dato
che ER è direttamente coinvolto nella proliferazione e differenziazione cellulare,
qualsiasi perturbazione del segnale a questo livello può contribuire allo sviluppo di
anomalie, quali infertilità e cancro.
Gli xenoestrogeni comprendono numerose classi chimiche differenti, caratterizzate
da notevoli diversità strutturali: inquinanti ambientali, ad esempio pesticidi
organoclorurati che, grazie alla loro lipofilia, sono altamente persistenti e si
accumulano nei tessuti; composti provenienti dall’industria alimentare, come
additivi e contaminanti, ad esempio plastificanti, quali ftalati e alchilfenoli, che
possono accidentalmente contaminare gli alimenti; difenilalcani, tra cui il bisfenolo
A, utilizzato nella produzione di resine.
Le evidenze sperimentali sugli effetti avversi promossi dagli xenoestrogeni sono in
continuo aumento, tanto che la Commissione Europea li ha identificati come
possibili fattori di rischio prioritari e la crescente attenzione al problema ha portato,
nel 1996, il Congresso USA ad incaricare l’autorità regolatoria EPA (Enviromental
Protection Agency) a sviluppare strategie di screening in grado di stabilire i rischi

Introduzione
14
associati all’esposizione di xenoestrogeni. Nello sviluppo del programma di
screening la commissione istituita da EPA, EDSTAC (Endocrine Disruptor Screening
and Testing Advisory Committee), ha suddiviso il progetto in tre fasi sequenziali, la
prima delle quali dovrebbe definire le priorità sperimentali. Più di 10000 composti
chimici in uso, infatti, richiederebbero studi sulla loro capacità di deregolare le
funzioni endocrine e il numero è destinato ad aumentare (Seong Jae Yu et al. 2002).
Le classiche metodologie di misura dell’attività estrogenica si basano su procedure
lunghe ed elaborate che non sono adatte allo screening di un vasto numero di
composti. E’ necessario, perciò, sviluppare metodi rapidi e a basso costo capaci di
selezionare tra gli innumerevoli composti in uso quelli potenzialmente pericolosi. La
selezione permette di diminuire notevolmente il numero di sostanze su cui effettuare
studi approfonditi di tossicità, focalizzando gli sforzi e le risorse solo ove necessario.
Tra i metodi proposti per definire le priorità operative ci sono sia approcci
sperimentali, come l’high throughput assay, sia metodologie computazionali, come
QSAR e docking, applicabili al virtual screening di potenziali xenoestrogeni (Blair
et al. 2000).
1.8 VIRTUAL SCREENING
I metodi sperimentali che consentono l’analisi di intere librerie di composti in breve
tempo sono estremamente costosi. Un approccio complementare agli studi
sperimentali è il virtual screening, un metodo computazionale in grado di selezionare,
all’interno di un database elettronico, i composti che necessitano di saggi di attività
e/o di tossicità, con lo scopo di creare nuovi farmaci o di identificare sostanze
nocive. Esistono diverse tecniche per condurre analisi computazionali, classificabili,
approssimativamente, in due categorie: ligand-based virtual screening e protein-
based virtual screening. I primi utilizzano le informazioni provenienti dallo studio
di composti che legano il target biologico per identificare altre molecole con simili
proprietà. Quando, invece, è nota la struttura tridimensionale della proteina, è
possibile eseguire il docking di ogni ligando nel sito di legame del target, predire la
conformazione con cui il composto interagisce con la proteina e stimare la bontà del
legame. Questo processo prende il nome di structure based virtual screening ed è

Introduzione
15
caratterizzato da tre fasi: preparazione dei database, docking dei diversi ligandi nel
sito della proteina e analisi postdocking (Figura 1.8.1).
La prima operazione in un virtual screening è avere a disposizione una banca dati in
cui siano raccolti i composti da analizzare.
Nell’ambito di uno studio farmaceutico il database iniziale è parzialmente ridotto
dall’applicazione di diversi filtri fisici e chimici, consentendo di eliminare a priori
composti che, in ogni modo, a causa di proprietà indesiderate, non supererebbero i
test clinici e di ridurre i tempi di calcolo. I più comuni filtri di screening si basano
sulla valutazione del peso molecolare, del bilancio idrofilo/lipofilo e del numero di
gruppi donatori/accettori di legami idrogeno, come per la legge di Lipinski. Altri
Figura 1.8.1: Fasi del processo di virtual screening (Lyne, 2002)

Introduzione
16
importanti filtri, invece, rimuovono i composti caratterizzati da gruppi funzionali
tossici o da scarsa stabilità chimica.
“In silico” screening di tipo tossicologico presentano finalità molto diverse e, di
conseguenza, una scrematura iniziale di diverso tipo, ma si tratta di un campo
ancora piuttosto inesplorato in letteratura.
Il processo di docking consiste nel riprodurre, virtualmente, l’associazione tra una
proteina e un ligando, selezionando, tra tutti quelli possibili, il complesso
energeticamente favorito. Riprodurre lo spazio conformazionale accessibile a una
macromolecola è complicato e quindi necessità di approssimazioni. In commercio
sono disponibili numerosi programmi di docking che differiscono per il tipo di
algoritmo usato, per la valutazione della flessibilità del ligando e della proteina e per
la funzione di scoring impiegata (Lyne, 2002).
Esistono numerose tecniche per valutare l’energia di legame associata a piccole
molecole nella formazione del complesso proteina ligando. In un virtual screening le
funzioni di scoring sono utilizzate, durante il processo di docking, per selezionare la
miglior conformazione della molecola all’interno del sito e, al termine del processo,
per stimare l’affinità di legame dei complessi formati dai vari ligandi del database.
Le funzioni di scoring possono essere classificate in tre categorie (Lyne, 2002):
force field-based scoring functions basate su equazioni ricavate dai principi della
meccanica classica, che cercano di rappresentare nel modo più corretto
possibile le molecole e le loro proprietà chimico-fisiche. Alcuni dei force field
più utilizzati sono Amber, Tripos e Charm;
empirical-based scoring functions in cui lo score di formazione del complesso è
ottenuto sommando i contributi relativi ai diversi tipi di interazioni non
covalenti (legame idrogeno, interazioni di van der Waals, interazioni
idrofobiche ecc…). I coefficienti assegnati ad ogni termine della somma sono
ottimizzati grazie a regressioni multilineari costruite su un set di differenti
complessi proteina-ligando ad affinità nota;
knowledge-based scoring functions costruite mediante metodi statistici. Questa
analisi non fa riferimento a misure sperimentali di affinità di legame, ma si
basa sul concetto che la frequenza di comparsa di uno specifico contatto,
nelle strutture ottenute con diffrazione dei raggi X o spettroscopia NMR,
può essere considerata indicativa del suo contributo al binding.
Spesso le scoring functions utilizzate dal programmi di docking sono inadeguate a
predire realmente l’affinità di legame proteina-ligando. Per superare questo ostacolo

Introduzione
17
una possibile strategia si basa sul concetto di consensus scoring. Tale approccio si basa
sull’utilizzo di molteplici funzioni di scoring per selezionare i ligandi più affini.
Esistono poi diverse analisi post docking per ridurre al minimo il numero di falsi
positivi che si generano tra le centinaia di risultati.
Infine, è spesso necessaria un’ispezione visuale per accertare che i ligandi siano
posizionati nel sito di legame e non in periferia, assumano una conformazione
realistica e interagiscano con i resuidi chiave del sito attivo.
Ad oggi le principali difficoltà negli studi di virtual screening si riscontrano nella
capacità di stimare correttamente l’energia di legame del complesso e, soprattutto,
nello sviluppo di algoritmi di docking che prendano in considerazione la flessibilità
proteica.
1.9 FLESSIBILITA’ E DOCKING
Le procedure di docking possono essere classificate in 3 categorie (Höltje, Sippl,
Rognan, Folkers, Molecular Modeling, 2003):
rigid body docking: sia il ligando che la proteina sono trattati come entità
rigide;
semi-flexible docking: viene presa in considerazione solo la flessibilità del
ligando;
fully flexible docking: la proteina e il ligando sono entrambi considerati
molecole flessibili.
La conformazione con cui una molecola si lega alla proteina target può essere
differente dalla conformazione che essa assume allo stato libero in soluzione. Infatti,
in soluzione, il ligando esiste in un numero di conformazioni molto maggiori
rispetto a quelle capaci di legare la macromolecola. E’ quindi di fondamentale
importanza considerarne la flessibilità conformazionale durante il processo di
docking.
L’approccio più semplice consiste nel creare, per ogni composto presente nel
database, un set di possibili conformazioni e poi nel trattare ogni conformero
generato come entità rigida nel posizionarlo all’interno del sito di legame. I più
comuni software che lavorano utilizzando insiemi conformazionali sono DOCK,

Introduzione
18
Fred e Slide. Alternativamente è possibile considerare una sola conformazione e
trattare il ligando come molecola flessibile durante il processo di docking. Una
strategia di questo tipo è il metodo di costruzione incrementale, che divide il
composto in frammenti e lo ricostruisce, per aggiunte successive, all’interno del sito
attivo. Questo metodo è utilizzato, ad esempio, dal programma FlexX e dalle
versioni più recenti di Slide e DOCK. Un’altra via per esplorare lo spazio
conformazionale all’interno del processo di docking è servirsi di algoritmi genetici,
come fanno i programmi GOLD e AutoDock, dove i conformeri derivano da
operazioni genetiche successive, quali mutazioni o crossing over.
La valutazione della flessibilità del ligando, risulta, tutto sommato, semplice rispetto
alle difficoltà di incorporare nei programmi di docking una funzione che tenga conto
della flessibilità della proteina.
1.10 LA FLESSIBILITA’ DELLE PROTEINE
Al fine di stimare quantitativamente la flessibilità delle proteine Betts and
Sternberg sovrapposero le strutture tridimensionali di uguali proteine, non
complessate, risolte da gruppi di ricerca differenti e ne calcolarono i valori di RMS
(root mean square). Mentre gli atomi di carbonio (Cα) del backbone mostravano
spostamenti compresi tra 0.1 e 0.5 Å, le catene laterali, soprattutto i resuidi di
superficie, erano caratterizzate da valori di RMS molto maggiori (Carlson, 2002).
Il sito di legame può essere caratterizzato da regioni ad alta flessibilità e da regioni
ad alta stabilità. I residui catalitici sono normalmente collocati in aree relativamente
stabili della struttura, così da mantenere la specificità del sistema. Le regioni rigide
di una macromolecola biologica sono frequentemente posizionate nelle porzioni
centrali della proteina, dove il compatto impacchettamento ne riduce la libertà di
movimento.
L’interazione tra una proteina e il suo substrato è un fenomeno ampiamente studiato
che ha prodotto numerose teorie sulle modalità di tale riconoscimento. Fischer
spiegò le relazioni strutturali tra ligando e proteina con il modello “chiave-
serratura”, in cui una molecola è adatta al legame se la sua struttura è
complementare al sito attivo biologico. Tale teoria fu superata nel 1958 da
Koshland, il quale propose un modello di “adattamento indotto” in cui il sito attivo

Introduzione
19
si mostra complementare al ligando in seguito al legame con esso. Attualmente la
tesi più accreditata descrive le proteine come molecole estremamente flessibili che
esistono, in soluzione, in diversi stati conformazionali separati l’uno dall’altro da
barriere energetiche basse (Figura 1.10.1). Entro certi limiti, una proteina è in
grado di riarrangiare la propria struttura a seconda del tipo di ligando presente.
Tale riorganizzazione può essere minima, come piccoli movimenti torsionali delle
catene laterali, o consistente, se coinvolge spostamenti di interi domini della
proteina.
Figura 1.10.1: Una proteina può esistere in differenti conformazioni, ad ognuna delle quali è associato uno stato energetico (Carlson, 2002)

Introduzione
20
1.11 INCORPORAZIONE DELLA FLESSIBILITA’ PROTEICA
NEL DOCKING
La maggior parte dei software che eseguono analisi di docking è concepita
considerando la proteina fissa nella sua conformazione cristallografica. Questa, pur
essendo una notevole approssimazione, è generalmente necessaria per non
aumentare eccessivamente la complessità e i tempi di calcolo. Per tentare di
restringere il numero di stati conformazionali da prendere in considerazione una
strategia è limitare le regioni che possono cambiare conformazione. Esistono,
infatti, alcuni programmi che modificano esclusivamente la posizione delle catene
laterali della proteina in risposta al tipo di ligando presente nel sito attivo. Per far
questo, un primo approccio può essere quello di utilizzare una libreria di rotameri
creata a partire da un insieme di stati conformazionali discreti delle catene laterali
degli amminoacidi, che derivano direttamente da studi di cristallografia ai raggi X e
di spettroscopia NMR. Sfruttando le informazioni contenute nella libreria,
l’algoritmo cerca di trovare la combinazione a minor energia tra gli amminoacidi e il
ligando, mantenendo rigidi gli atomi del backbone (Leach, 1994). Alternativamente,
programmi come SLIDE, consentono piccoli spostamenti delle catene laterali, per
minimizzare le eventuali collisioni che si generano nel posizionare un ligando in un
sito rigido. A differenza dell’approccio precedente, nel modificare l’orientamento
delle catene laterali sono prese in considerazione anche conformazioni che non
appartengono a librerie rotameriche.
Una seconda strada per incorporare la flessibilità nelle analisi di docking è di
utilizzare un programma, come FlexE, che permetta l’introduzione nell’analisi di
più strutture della stessa proteina in conformazioni diverse. L’insieme discreto delle
strutture proteiche viene sovrapposto dal software e la conformazione migliore per
l’interazione con il ligando selezionata. Le strutture tridimensionali del target
provengono da studi cristallografici e spettroscopici, ma, nel caso in cui non siano
direttamente disponibili le informazioni sperimentali, il set conformazionale può
essere generato da studi di dinamica molecolare.
Infine, un ultimo approccio parziale al complesso problema flessibilità, è il “soft
docking”. Tutti i programmi di docking contengono, all’interno del loro algoritmo,
una funzione che tiene conto degli effetti repulsivi e sterici per evitare che il ligando
si disponga in modo non realistico. Assumendo, però, che la proteina, grazie a

Introduzione
21
piccoli spostamenti, si adatta all’ingresso del ligando, nel soft docking sono tollerati
alcuni piccoli urti sterici grazie all’incremento della permissività del potenziale di
Lennard-Jones. Il principale svantaggio di questa tecnica è di essere applicabile solo
per piccoli cambiamenti conformazionali.


Hint


HINT
23
Per valutare la bontà dell’interazione di una proteina con un ligando sono
disponibili numerosi programmi di meccanica molecolare, che si basano su approcci
e strategie diverse. La maggior parte di questi è in grado di quantificare
efficacemente il contributo entalpico dovuto alle forze di interazione atomo-atomo
(legami idrogeno, forze di van der Waals, forze elettrostatiche, ecc.), ma valuta solo
parzialmente il contributo entropico dato, ad esempio, dal processo di
desolvatazione.
Nel 1991 Abraham e Kellogg hanno sviluppato il programma HINT (Hydropathic
INTeractions), con lo scopo di creare uno strumento che tenesse conto del
contributo entropico associato alla formazione di un complesso per descrivere in
modo realistico le interazioni biologiche.
2.1 IL SOFTWARE HINT
HINT è un force field cosiddetto “naturale” ed “intuitivo”, basato su misure
sperimentali di LogPo/w. Il coefficiente di ripartizione di un soluto tra le fasi 1-
ottanolo e acqua (Po/w) è una grandezza termodinamica direttamente correlata
all’energia libera di trasferimento di una molecola da un solvente all’altro e come
tale racchiude in sé informazioni sia di tipo entalpico che di tipo entropico. Le forze
che intervengono in un processo di ripartizione sono le stesse che regolano le
interazioni tra molecole biologiche. Queste ultime sono normalmente costituite da
regioni idrofobiche e da regioni idrofiliche, rispettivamente comparabili ad una fase
apolare ed una acquosa, per cui la formazione di un complesso tra una proteina ed
un ligando può essere realisticamente comparata alla ripartizione del ligando stesso
tra le fasi ottanolo ed acqua.
HINT valuta esclusivamente le interazioni non covalenti, quali legami ad idrogeno,
interazioni acido-base, attrazioni di Coulomb e interazioni idrofobiche, che sono
collettivamente definite “idropatiche” (Kellogg et al. 2000).
Ogni interazione atomo-atomo è quantificata dal force field di HINT attraverso
l’equazione:
bij = ai Si aj Sj Tij Rij + rij

HINT
24
dove:
bij è lo score attribuito all’interazione tra l’atomo i e l’atomo j. Quando b
assume valori positivi l’interazione tra i due atomi è favorevole, al
contrario valori negativi di b indicano che i e j interagiscono
sfavorevolmente;
a è la costante idrofobica atomica;
S rappresenta la superficie accessibile al solvente (SASA);
Tij è una funzione logica, che determina il segno dei valori parziali di
score;
Rij e rij sono funzioni della distanza tra l’atomo i e j.
Il punteggio finale assegnato da HINT al sistema è dato dalla somma delle
interazioni di tutte le coppie di atomi:
∑i ∑j bij = ∑i ∑j (ai Si aj Sj Tij Rij + rij)
L’ HINT score totale può essere scomposto nei singoli contributi relativi ai diversi
tipi di interazione:
HSTOT = HSHH + HSAB + HSHB + HSAA + HSBB + HSHP
Gli HINT score relativi all’effetto idrofobico (HSHH), all’interazione acido-base
(HSAB) e ai legami idrogeno (HSHB), concorrono ad aumentare il punteggio finale del
sistema, mentre i contributi dei contatti acido-acido, base-base e idrofobico-polare,
rispettivamente HSAA, HSBB, HSHP, diminuiscono lo score totale, in quanto
caratterizzati da valori negativi (Tabella 2.1.1).
La costante idrofobica atomica
Leo (Hansch et al. 1979) e Rekker (Rekker et al. 1977) furono i primi a sviluppare
un set di costanti frammentarie, sulla base di misure sperimentali effettuate negli
anni precedenti. Ogni atomo e gruppo chimico è caratterizzato da una determinata
costante e il LogP dell’intera molecola è ottenuto sommando questi contributi
parziali. Questo approccio, quindi, sfrutta il concetto di additività del LogP.
Negli anni successivi Abraham e Leo integrarono il dataset calcolando le costanti
idrofobiche frammentarie relative a molecole di natura peptidica.

HINT
25
IDROFOBICO POLARE (acido di Lewis)
POLARE (base di Lewis)
IDROFOBICO interazione idrofobica
idrofobico-polare (desolvatazione)
idrofobico-polare (desolvatazione)
POLARE (acido di Lewis)
idrofobico-polare (desolvatazione)
repulsione coulombiana
interazione acido-base (legame
idrogeno)
POLARE (base di Lewis)
idrofobico-polare (desolvatazione)
interazione acido-base (legame
idrogeno)
repulsione coulombiana
Tabella 2.1.1: Matrice delle interazioni atomo-atomo quantificate da HINT (Kellogg et al. 2000). In turchese sono indicate le interazioni positive, in nero quelle negative.
Le costanti idrofobiche atomiche sono parametri adimensionali, ricavate da HINT
grazie a progressive riduzioni delle costanti frammentarie di Leo e adattate per
introduzione di una serie di fattori di correzione che tengono conto dell’intorno
degli atomi e delle loro connessioni.
Un atomo i è idrofobico, quindi solubile nella fase organica (1-ottanolo), quando ai
assume valore positivo, mentre è idrofilico, perciò solubile nella fase polare (acqua),
se la sua costante atomica è minore di zero. Questa regola generale è sempre valida,
tranne che per gli atomi di idrogeno polari, che, per convenzione, assumono valori
di ai sempre positivi.
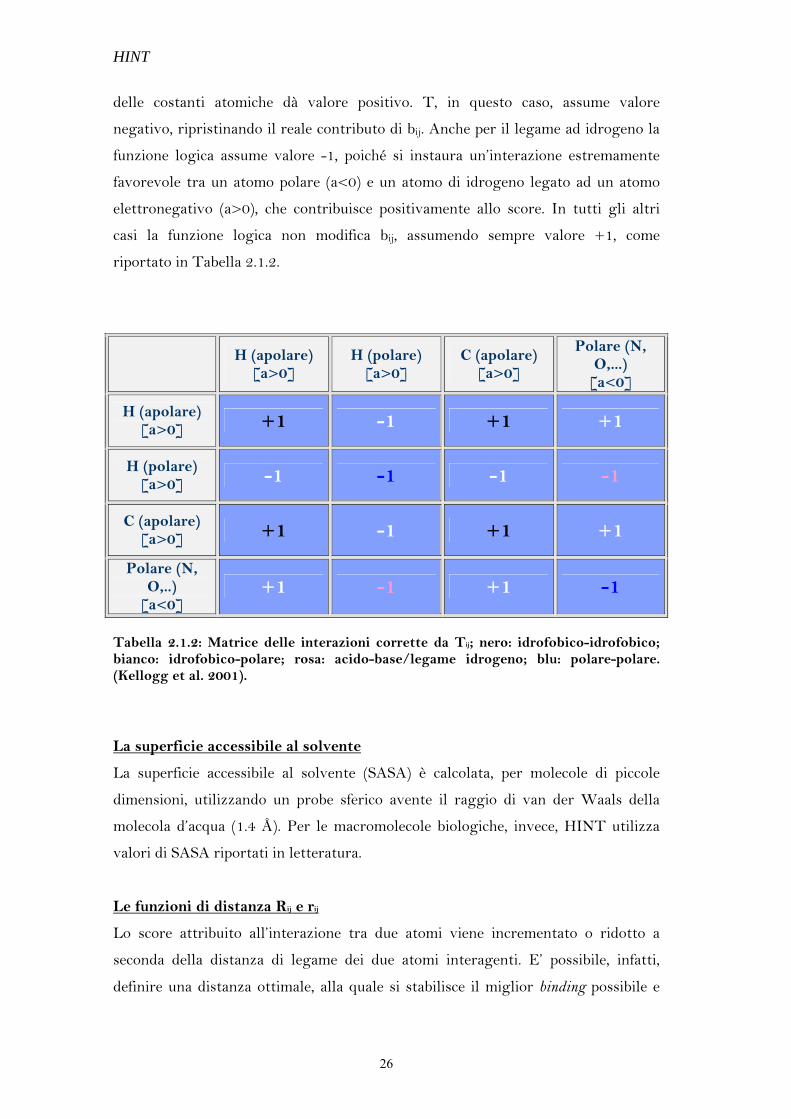
La funzione logica Tij
HINT stima favorevolmente l’interazione fra due atomi i e j quando questi sono
solubili nella stessa fase, cioè quando le costanti ai e aj presentano lo stesso segno
(ai>0 e aj>0 oppure ai<0 e aj<0). Se le costanti idrofobiche atomiche dei due atomi
hanno segno opposto ( ai>0 e aj<0 oppure ai<0 e aj>0) l’associazione risulterà
sfavorita.
Esistono, tuttavia, delle eccezioni che hanno portato Abraham e Kellogg ad
introdurre un fattore di correzione, che modificasse, all’occorrenza, il segno dello
score. Si tratta della funzione logica T che assume valore +1 o -1 a seconda del tipo
di interazione.
Atomi polari della stessa natura, acidi o basi di Lewis, danno origine ad interazioni
repulsive quando si generano dei contatti acido-acido o base-base, ma il prodotto
HINT
26
delle costanti atomiche dà valore positivo. T, in questo caso, assume valore
negativo, ripristinando il reale contributo di bij. Anche per il legame ad idrogeno la
funzione logica assume valore -1, poiché si instaura un’interazione estremamente
favorevole tra un atomo polare (a<0) e un atomo di idrogeno legato ad un atomo
elettronegativo (a>0), che contribuisce positivamente allo score. In tutti gli altri
casi la funzione logica non modifica bij, assumendo sempre valore +1, come
riportato in Tabella 2.1.2.
H (apolare) [a>0]
H (polare) [a>0]
C (apolare) [a>0]
Polare (N, O,...)
[a<0]
H (apolare) [a>0] +1 -1 +1 +1
H (polare) [a>0] -1 -1 -1 -1
C (apolare) [a>0] +1 -1 +1 +1
Polare (N, O,..)
[a<0] +1 -1 +1 -1
Tabella 2.1.2: Matrice delle interazioni corrette da Tij; nero: idrofobico-idrofobico; bianco: idrofobico-polare; rosa: acido-base/legame idrogeno; blu: polare-polare. (Kellogg et al. 2001).
La superficie accessibile al solvente
La superficie accessibile al solvente (SASA) è calcolata, per molecole di piccole
dimensioni, utilizzando un probe sferico avente il raggio di van der Waals della
molecola d’acqua (1.4 Å). Per le macromolecole biologiche, invece, HINT utilizza
valori di SASA riportati in letteratura.
Le funzioni di distanza Rij e rij
Lo score attribuito all’interazione tra due atomi viene incrementato o ridotto a
seconda della distanza di legame dei due atomi interagenti. E’ possibile, infatti,
definire una distanza ottimale, alla quale si stabilisce il miglior binding possibile e

HINT
27
oltre la quale diminuisce l’affinità fra gli atomi coinvolti, a causa di un aumento delle
forze repulsive o di una riduzione dell’effetto attrattivo.
Nell’ equazione di HINT sono presenti due parametri che tengono conto delle
distanze di legame: Rij e rij.
Il primo termine descrive una funzione esponenziale tale per cui le energie di
interazione si affievoliscono all’aumentare della distanza r tra due atomi:
Rij = e-r
rij, invece, è un’implementazione del potenziale di Lennard-Jones ed apporta un
contributo negativo allo score perché identifica le interazioni repulsive che si
generano quando la distanza r è troppo piccola. L’equazione che descrive rij è la
seguente:
rij = A εij [ ( rvdw / r)12 – 2 (rvdw / r) 6]
dove A è un fattore di correzione che correla il potenziale di Lennard-Jones alla
funzione esponenziale e-r , εij rappresenta l’energia associata all’effetto di dispersione
tra i due atomi i e j e rvdw è la somma dei loro raggi di van der Waals (Abraham et
al. 1993).
2.1.1 RELAZIONE TRA LogP e ∆G°
Il coefficiente di ripartizione del soluto Po/w è la costante di equilibrio del processo
di trasferimento di un soluto dalla fase 1-ottanolo alla fase acquosa ed è correlato al
∆G° secondo l’equazione:
LogPo/w = -∆G° / 2.303 RT
dove R è la costante dei gas e T è la temperatura assoluta. Entrambe assumono
valori costanti, in condizioni normali (1 atm, 25 °C), per cui si può riscrivere:
LogPo/w = k ∆G°

HINT
28
alla temperatura di 298 K la costante k è pari a -0.733 Kcal/mol.
Come detto in precedenza il LogP è calcolato da HINT sommando le costanti
atomiche idrofobiche assegnate ai vari atomi e direttamente correlato all’energia
libera di legame tramite l’equazione:
∑ ai = k ∆G°
Da questa relazione è possibile affermare, quindi, che l’HINT score è funzione
dell’energia libera di legame e per questo contiene informazioni sia di natura
entalpica che entropica.
2.1.2 APPLICAZIONI DEL PROGRAMMA HINT
HINT è un software versatile, che può essere applicato nell’ambito dello structure
base drug design per scopi diversi:
calcolo delle costanti idrofobiche atomiche degli atomi appartenenti a ligandi
e proteine;
costruzione di mappe idrofobiche di molecole e macromolecole biologiche;
determinazione della natura chimica del sito farmacoforico di un recettore a
partire dalla struttura del ligando e viceversa;
predizione dell’affinità di legame e calcolo dello score relativo ad un
complesso biologico;
costruzione grafica della mappa d’interazioni idrofobiche e polari di un
complesso;
determinazione della struttura secondaria e terziaria di una proteina;
localizzazione dei residui suscettibili di mutazione allo scopo di individuare
interazioni idrofobiche e polari, positive e negative;
valutazione del ruolo delle molecole d’acqua presenti nelle strutture
proteiche.

Scopi


Scopi della ricerca
29
Negli ultimi decenni l’attenzione del mondo scientifico nei confronti del recettore
degli estrogeni è aumentata esponenzialmente grazie alle evidenze, sempre più
stringenti, di una relazione tra gli ormoni estrogenici e l’insorgenza di neoplasie.
Attualmente gli studi sono condotti principalmente e parallelamente lungo due
filoni: da un lato si cerca di utilizzare il recettore degli estrogeni come target
farmacologico per lo sviluppo di farmaci antitumorali e post-menopausali, dall’altro
l’elevata versatilità recettoriale impone l’identificazione di sostanze che, pur
legandosi debolmente, possono mediare modificazioni endocrine a lungo termine
(xenoestrogeni).
Questo lavoro di tesi si inserisce all’interno di un progetto di ricerca più ampio,
volto alla caratterizzazione sempre più approfondita del sistema recettoriale
estrogenico e allo sviluppo di una procedura di “in silico” screening per
l’identificazione di sostanze che mimano l’azione degli estrogeni o agiscono da
antagonisti. In particolare, il lavoro qui presentato ha avuto come obiettivo l’analisi
della flessibilità di ERα tramite lo studio delle interazioni del sito di legame con
due principale tipologie di composti: agenti ad attività nota (agonisti, SERM) e
sostanze che potenzialmente possono venire a contatto con il recettore degli
estrogeni (additivi di origine alimentare). Nell’affrontare il problema della
versatilità del recettore, particolare attenzione è stata posta nell’individuazione di
un metodo che permettesse l’inserimento della flessibilità negli studi di docking su
grande scala. Il lavoro è stato così articolato in tre parti:
1. verifica della capacità dei softwares utilizzati negli studi computazionali di
riprodurre i dati sperimentali, tramite la costruzione di una retta di taratura
che correla il valore di HINT score con il ∆G° sperimentale. La retta di
regressione lineare costituisce uno strumento fondamentale nella predizione
della forza di legame di composti potenzialmente interagenti con il recettore;
2. messa a punto di un protocollo di lavoro con cui condurre le analisi di virtual
screening. In questa fase si è posta particolare attenzione alla risoluzione dei
problemi e alla definizione delle variabili che caratterizzano gli studi di un
grande numero di composti, prima fra tutte la valutazione della flessibilità
negli studi di docking. Per ottenere rapidamente risultati omogenei e
riproducibili è necessario, infatti, trovare un compromesso tra semplicità e
capacità predittiva del metodo;
3. studi predittivi di affinità per un set di additivi alimentari naturali e di sintesi
utilizzati normalmente nella produzione di alimenti.


Materiali e Metodi


Materiali e metodi
31
Le analisi computazionali eseguite in questo lavoro di tesi sono state effetuate col
supporto del software di modellistica molecolare SYBYL della Tripos Associates
(Tripos, Inc., StLouis, MO; www.tripos.com). Sybyl versione 7.0 è stato installato
sulla workstation Silicon Graphics Irix modello OCTANE, il cui sistema operativo
è IRIX 64 versione 6.5. HINT, nella versione 3.09 β-test, è stato utilizzato come
modulo aggiuntivo di Sybyl. Questo software è gentilmente fornito dal Prof. Glen
E. Kellogg dell’Institute for Structural Biology and Drug Discovery della Virginia
Commonwealth University, Richmond, VA, USA e sviluppato congiuntamente
nell’ambito del progetto di collaborazione con l’Università di Parma.
4.1 TRIPOS
Il software Sybyl contiene diversi force field di meccanica molecolare. Tra i tanti è
stato scelto Tripos, particolarmente adatto all’analisi di composti differenti sia dal
punto di vista chimico che da quello strutturale (Clark et al., 1989). In una
procedura computazionale eseguita con un programma di meccanica molecolare, ad
ogni atomo viene assegnato un atom type descritto nel force field. Nella versione 6.5
di Tripos sono presenti 53 atom types differenti, caratterizzati dal punto di vista della
valenza, della geometria, del raggio di van der Waals e della carica formale degli
atomi. Esistono, inoltre, otto tipi diversi di bond types, cinque dei quali rappresentano
i legami classici come il legame singolo, doppio, triplo, amidico e aromatico, mentre
gli altri tre (dummy, unknown e not connected) sono legami atipici o non riconosciuti.
L’avere a disposizione un numero di atom type limitato costituisce
un’approssimazione rispetto alla realtà, che è molto più variegata, per cui non è
sempre possibile caratterizzare tutti gli atomi nel modo più adeguato.

Materiali e metodi
32
4.2 ANALISI DI COMPLESSI CRISTALLOGRAFICI
PROTEINA-LIGANDO-ACQUA: PROTOCOLLO DI LAVORO
1. Ricerca della struttura cristallografica
Le strutture tridimensionali dei complessi proteina-ligando analizzate in questo
lavoro di tesi derivano da studi di diffrazione ai raggi X. I file provengono dalla
Brookhaven Protein Databank (PDB), che raccoglie strutture tridimensionali di
macromolecole biologiche; ad oggi sono depositate presso questa banca dati quasi
35000 strutture ottenute non solo tramite raggi X, ma anche attraverso tecniche di
NMR e di diffrazione a neutroni. Ogni struttura è contraddistinta da un codice. Una
volta identificato il codice corrispondente al complesso desiderato i file PDB sono
importati in Sybyl per essere studiati e analizzati.
2. Controllo degli atom name e atom type dei residui terminali della proteina
Il valore di HINT score relativo all’interazione proteina-ligando è influenzato da
tutti i residui amminoacidici compresi nell’intorno di 10 Å dal sito catalitico.
Nell’ipotesi in cui anche i residui terminali amminico e carbossilico siano inclusi in
questo intorno occorre controllare che lo stato di ionizzazione sia tale da avere
un’estremità amminica carica positivamente (ione ammonio) e un’estremità
carbossilica carica negativamente (ione carbossilato). Questa operazione permette di
utilizzare un modello più aderente alla realtà fisiologica.
3. Correzione degli atom type e dei bond type del ligando
I file PDB non contengono informazioni relative alla natura e all’ordine dei legami
fra i diversi atomi, per cui è probabile che il force field assegni atom type e bond type
errati e che la molecola risultante non corrisponda esattamente alla struttura
riportata in letteratura.
4. Addizione degli atomi di idrogeno
Le strutture cristallografiche risolte ai raggi X contengono informazioni sulla
posizione degli atomi “pesanti” (come ossigeno, carbonio, zolfo, azoto, alogeni, ecc..),
ma non danno indicazioni sulla localizzazione degli atomi di idrogeno, la cui densità
elettronica è troppo bassa per essere rilevata con precisione. Gli atomi di idrogeno
vengono quindi aggiunti mediante un opzione del programma.

Materiali e metodi
33
5. Minimizzazione degli idrogeni
Sybyl addiziona gli idrogeni sugli atomi “pesanti” senza tenere conto delle
interazioni repulsive e attrattive o degli eventuali ingombri sterici tra gli atomi. Il
processo di minimizzazione permette di abbassare l’energia associata al complesso
proteina-ligando, in modo da generare un modello dove le interazioni degli idrogeni
con gli altri atomi sia minima. La posizione degli idrogeni è ottimizzata scegliendo
il comando Minimize dal menù Compute di Sybyl.
Il processo di minimizzazione dipende da molteplici parametri. Le condizioni
standard utilizzate in questo lavoro di tesi sono:
Method → Powell, Simplex
Simplex opera l’ottimizzazione iniziale, mentre Powell calcola le derivate
prima e seconda, così da stabilire l’effettiva posizione dei minimi.
Gradient → 0.5
E’ un parametro limite e indica il valore che deve assumere la derivata prima.
Charge → Gasteiger-Huckel
E’ il metodo con cui vengono assegnate le cariche atomiche.
Iteration → 1500
Indica il numero massimo di cicli di minimizzazione che possono essere
effettuati.
Il processo di minimizzazione coinvolge solo gli atomi di idrogeno, tutti i rimanenti
atomi devono rimanere fissi nelle loro posizioni determinate cristallograficamente.
Tramite l’opzione Aggregate, tutti gli atomi “pesanti” vengono inclusi in un insieme
che resta escluso dal processo.
6. Rotazione degli idrogeni ossidrilici del ligando
Durante il processo di minimizzazione gli atomi di idrogeno possono rimanere
intrappolati in un minimo energetico relativo e l’orientazione assunta dagli idrogeni
del ligando può non essere ottimale per l’interazione con il sito catalitico della
proteina. La mancanza, ad esempio, di un legame idrogeno tra una molecola e una
proteina, dovuta ad un’errata orientazione degli atomi, può portare ad riduzione del
valore di HINT score proteina-ligando e quindi ad una sottostima dell’affinità di
legame. E’ necessario, a volte, ruotare manualmente i legami tra gli atomi così da
ottenere posizioni più realistiche. Nei complessi presi in esame in questo lavoro di

Materiali e metodi
34
tesi è stato a volte necessario ruotare i gruppi ossidrilici del ligando per favorire le
interazioni con i residui chiave della proteina.
7. Divisione in aree
Un file PDB è composto da più oggetti: la macromolecola biologica, il ligando, le
molecole d’acqua ed eventuali metalli e cofattori. Sybyl ragiona secondo il concetto
di aree differenti in cui è possibile isolare ognuna di queste componenti. Le diverse
strutture possono essere visualizzate una alla volta oppure contemporaneamente,
pur rimanendo confinate ognuna nella propria area di appartenenza.
La divisione in aree è necessaria per il calcolo del Log P. HINT, infatti, partiziona il
contenuto di ogni area ed è quindi indispensabile che ognuna di esse sia occupata da
un solo tipo di struttura. Come procedura operativa, collochiamo la proteina
nell’area M1, il ligando nell’area M2 e le molecole d’acqua nell’area M3. Ad ogni
area è attribuito un nome specifico in modo che HINT la possa riconoscere
selettivamente.
8. Calcolo del Log P della proteina
Prima di procedere alla stima dell’HINT score occorre calcolare il coefficiente di
ripartizione ottanolo/acqua di proteina, ligando e molecole d’acqua. Dai valori di
Log P HINT determina le costanti idrofobiche atomiche necessarie per il calcolo
dello score.
Per le macromolecole, come le proteine si utilizza l’opzione Dictionary. Tale opzione,
pur essendo un’approssimazione permette di calcolare velocemente e con buona
precisione il Log P di molecole complesse, utilizzando un dizionario che associa ad
ogni atomo un valore di Log P prestabilito.
La misura del Log P è influenzata anche dalle condizioni del solvente. Esistono
quattro differenti possibilità di trattamento del solvente: acid, basic, neutral e inferred.
E’ stata utilizzata l’opzione neutral, che dovrebbe riprodurre al meglio le condizioni
fisiologiche. Ne deriva che i residui aspartato e glutammato saranno carichi
negativamente, mentre lisina e arginina positivamente. Il Log P può essere
calcolato in funzione di tutti gli idrogeni presenti (opzione All), solo di quelli legati
ad atomi pesanti polari (opzione Essential) o facenti parte degli atomi pesanti stessi
(opzione United). Gli idrogeni dei complessi analizzati sono stati trattati utilizzando
l’opzione Essential.

Materiali e metodi
35
L’opzione +20-NH-SASA è un fattore di correzione che viene selezionato per
evitare che HINT sottostimi la capacità del gruppo amidico, impegnato nel legame
peptidico, di formare legami ad idrogeno.
9. Calcolo del Log P del ligando
La partizione del ligando è analoga a quella della proteina. Non viene utilizzata
l’opzione Dictionary, ma l’opzione Calculate essendo la molecola di dimensioni molto
minori. HINT analizza accuratamente gli atom type e i bond type del ligando e
calcola il LogP sulla base delle connessioni fra i vari atomi della molecola, dando un
risultato meno approssimayo rispetto a quello ottenuto nella partizione della
proteina.
10. Ottimizzazione delle molecole d’acqua
All’interno di una struttura cristallografica alcune molecole d’acqua possono
rivestire un’importanza maggiore di altre. Queste molecole sono generalmente
localizzate all’interfaccia del complesso e possono, talvolta, svolgere un ruolo
essenziale per l’interazione proteina-ligando. Si utilizza HINT per individuare ed
ottimizzare automaticamente le molecole d’acqua posizionate all’interno di un certo
range di distanza dalla proteina e dal ligando. E’ possibile anche in questo caso
impostare alcuni restrizioni:
Contact distance → 4 Å
Permette di ottimizzare solo le acque che si trovano ad una distanza
massima di 4 Å sia dal ligando che dalla proteina.
Traslation Limit → 0.1 Å
Impone che lo spostamento dell’atomo di ossigeno dell’acqua non sia
superiore a 0.1 Å, così da non alterare significativamente i dati
cristallografici.
11. Calcolo dell’HINT score
Viene calcolato l’HINT score relativo all’interazione proteina-ligando o ligando-
acqua. Nel calcolo dell’HINT score è possibile scegliere quali forze considerare e
quali escludere; si possono valutare, ad esempio, i contributi idrofobici e trascurare
quelli idrofilici o viceversa. Nell’analisi dei complessi cristallografici, analizzati in
questo lavoro di tesi, tutte le forze coinvolte (legami a idrogeno, interazioni

Materiali e metodi
36
acido/base, interazioni idrofobiche, interazioni acido/acido, interazioni base/base,
interazioni idrofobico/polare) rientrano nello score finale assegnato da HINT.
Il valore complessivo dell’HINT score ed i contributi relativi ad ogni tipo di
interazione sono riportati, in modo analitico, nel tab file corrispondente, nel quale
sono individuabili anche le costanti atomiche, le superfici accessibili al solvente, le
interazioni instaurate da ogni atomo, la distanza tra gli atomi e il loro raggio di van
der Waals.
L’HINT score finale è dato dalla somma dell’HINT score relativo all’interazione
proteina-ligando e di quello associato all’interazione ligando-acqua. Il valore
complessivo è messo in relazione con l’energia di legame determinata
sperimentalmente.
4.3 DOCKING DI COMPLESSI CRISTALLOGRAFICI:
PROTOCOLLO DI LAVORO
Simulazioni di docking sono state effettuate su complessi cristallografici proteina-
ligando al fine di verificare l’effettiva validità dei software sul sistema oggetto di
studio. I ligandi sono stati estratti dalla proteina e successivamente rinseriti tramite
procedure di docking. La sovrapposizione tra il complesso generato dal programma
e quello cristallografico fornisce una stima diretta della precisione con cui il
software posiziona la molecola all’interno del sito di legame.
1. Docking
Per effettuare le simulazioni di docking è stato utilizzato il programma GOLD
versione 2.2 (CCDC, Cambridge, UK). Tale software è stato installato su un dual
pentium processor, il cui sistema operativo è Linux Red Hat Enterprise 3.0.
I parametri che controllano l’algoritmo genetico di GOLD, sono stati mantenuti
come stabiliti di default dal programma stesso. Il numero totale delle operazioni
effettuate dall’algoritmo è 100.000, mentre il numero delle “isole” e degli “individui”
è fissato rispettivamente a 5 e 100. La “niche size” è fissata a due, mentre il
crossover, la mutazione e la migrazione rispettivamente a 95, 95 e 10. Il box del
docking viene costruito a partire dalle coordinate di un punto localizzato al centro

Materiali e metodi
37
del sito di legame, impostando come raggio una distanza di 20 Å. GOLD genera di
default 10 conformeri per ogni ligando utilizzato. Questo parametro è stato
incrementato da 10 a 50, così da consentire una migliore esplorazione dello spazio
conformazionale.
2. Analisi idropatica
I 50 conformeri generati da GOLD sono stati analizzati con HINT, portando ad
individuare il conformero con il miglior punteggio.
4.4 DOCKING DI ADDITIVI ALIMENTARI: PROTOCOLLO DI
LAVORO
1. Ricerca degli additivi alimentari
Gli additivi alimentari presi in esame sono classificati nel database sviluppato da
JECFA (Joint FAO/WHO Expert Committee on Food Additives), consultabile on
line al sito http://apps3.fao.org/jecfa/additive_specs/foodad-q.jsp. Tale
commissione è nata da una collaborazione tra FAO (Food and Agriculture
Organization of the United Nations) e WHO (World Health Organization)
nell’ambito del programma internazionale sulla salute alimentare. Ad oggi JECFA
ha analizzato e classificato più di 1500 additivi. Il database è suddiviso in categorie
funzionali, cioè in base all’utilizzo degli additivi stessi. Ogni sostanza è
accompagnata da una scheda descrittiva che contiene informazioni generali
sull’additivo e sulle sue proprietà chimico-fisiche, inoltre è riportata la struttura
bidimensionale del composto.
E’ possibile consultare on-line anche la banca dati creata da FDA (Food and Drug
Administration), che contiene più di 3000 sostanze direttamente addizionate ai cibi.
Il database EAFUS (“Everything” Added to Food in the United States) non è stato
utilizzato a fini di questo lavoro, in quanto non sono riportate le strutture chimiche
bidimensionali dei composti classificati.

Materiali e metodi
38
2. Preparazione del ligando
Le strutture tridimensionali dei ligandi sono state costruite con Sybyl. Gli idrogeni
vengono aggiunti e minimizzati, come descritto in precedenza per le proteine, con la
sola differenza che tutti gli atomi, non solo quelli di idrogeno, sono sottoposti ad un
processo di minimizzazione.
3. Procedura di docking
Il docking dei ligandi è stato effettuato analogamente a quanto descritto
precedentemente per i complessi cristallografici.
4. Analisi dei conformeri generati
La scoring function di GOLD seleziona tra gli innumerevoli conformeri generati i
50 caratterizzati da più alto valore energetico. Successivamente, l’interazione tra la
proteina target ed i 50 conformeri generati è stata valutata utilizzando il force field
di HINT. Alla fine sarà possibile valutare e confrontare con particolare attenzione i
due conformeri scelti dalla funzione di scoring di GOLD e da HINT.
GRID
Grid, versione 22a è stato installato sulla workstation Sgi modello FUEL.
Grid è un software di modellistica molecolare progettato da Goodford nei primi
anni ottanta (Goodford et al. 1985). Questo software si basa sull’utilizzo di una
sonda (probe), che ispeziona virtualmente il sito attivo di una proteina individuando
le posizioni energeticamente e stericamente compatibili con il probe stesso. I probe
utilizzabili sono molteplici e possono essere costituiti da atomi, piccole molecole o
da porzioni di esse. In particolare, in questo lavoro di tesi, sono stati impiegati un
probe idrofobico (Dry), un probe donatore (N1) e un probe accettore di legami
idrogeno (O), al fine di delineare con maggior precisione le caratteristiche chimiche
del sito di legame della proteina.
Grid opera attraverso la generazione di una griglia energetica che delimita lo spazio
all’interno del quale il probe si può muovere. In ogni punto della griglia viene
calcolata l’energia potenziale Ex,y,z, la cui entità dipende da tre diversi contributi
secondo l’equazione:

Materiali e metodi
39
Ex,y,z = ∑ Elj + ∑ Eel + ∑ Ehb
Elj rappresenta il potenziale di Lennard-Jones, Eel aggiunge il contributo delle
ineterazioni elettrostatiche e Ehb quello dei legami idrogeno. I punti in cui l’energia
è positiva o negativa vengono visualizzati graficamente da Contour Create in Sybyl,
attraverso l’opzione Map Contours del sottomenù Hint.


Risultati e Discussioni


Risultati e discussioni
41
Per verificare la validità dell’associazione tra il programma di docking GOLD e la
funzione di scoring HINT, utilizzati in questo lavoro di tesi per studiare il sistema
degli estrogeni, ligandi cristallografici, appartenenti a classi strutturali differenti,
sono stati estratti e poi reinseriti all’interno della proteina. La sovrapposizione tra il
complesso prescelto da HINT e quello cristallografico fornisce una stima della
precisione con cui il ligando è posizionato all’interno del sito di legame dalla
procedura di docking. Il parametro che permette di quantificare l’abilità della
tecnica utilizzata nel riprodurre il dato cristallografico è l’r.m.s. (root mean square).
Nella Tabella 5.1 sono riportate le misure di r.m.s. e i codici PDB dei complessi
cristallografici analizzati. Valori di r.m.s. inferiori a 2.0 Å sono considerati indicativi
di una buona capacità predittiva (Kontoyianni et al. 2004 e Brooijmans et al. 2003).
Ligando Codice PDB r.m.s. (Å)
Estradiolo 1ERE 0.20
Genisteina 1X7E 0.20
Dietilstilbestrolo 3ERD 0.26
4-OH tamoxifene 3ERT 1.43
Raloxifene 1ERR 1.46
1SJ0 0.66
1XP1 0.28
1XP6 0.68
1XP9 0.32
Ligandi benzotiopiranici
1XPC 1.47
1YIN 0.54 Ligandi benzopiranici
1YIM 0.95
Tabella 5.1: Valori di r.m.s dei complessi cristallografici analizzati

Risultati e discussioni
42
5.1 FLESSIBILITA’ DEL RECETTORE DEGLI ESTROGENI
L’elevata flessibilità del recettore degli estrogeni è l’elemento chiave che ha
permesso di discriminare l’azione agonista da quella antagonista, portando allo
sviluppo di una classe di farmaci di grande interesse terapeutico come i SERM. Per
lo stesso motivo, però, il recettore si presta al legame di molteplici composti di
natura chimica anche molto varia, i quali possono mediare una perturbazione degli
equilibri endocrini.
Gli studi di docking, come esposto precedentemente, sono in parte limitati
dall’assenza di tecniche che trattino in modo esaustivo la flessibilità delle proteine.
In un precedente lavoro di tesi si è cercato di tenere conto della versatilità del
recettore tramite un approccio differente da quelli trattati nel capitolo introduttivo.
Per superare il problema della flessibilità, i processi di docking sono stati compiuti
utilizzando quattro conformazioni caratteristiche del recettore: 1ERE, 3ERD,
1ERR e 3ERT. Le prime due sono in complesso con agonisti, rispettivamente
estradiolo e dietilstilbestrolo, mentre le altre sono legate a SERM, rispettivamente
raloxifene e tamoxifene. A queste due classi principali, agonisti e SERM, sono
associate le due conformazioni limite del recettore, caratterizzate da un evidente
differenza nell’orientazione dell’elica 12 (Figura 1.4.1). All’interno di ognuna delle
due categorie esistono molteplici popolazioni conformazionali, selezionate dai
ligandi stessi, che differiscono tra loro per modesti spostamenti degli amminoacidi e
non per variazioni di interi domini. Ad esempio, la conformazione selezionata dal
raloxifene si diversifica da quella imposta dal tamoxifene nell’orientazione dell’His
524, uno dei residui chiave del sito di legame (Figura 5.1.1). All’interno di questo
lavoro di tesi sono stati effettuati studi di docking incrociato per indagare l’effettiva
necessità di utilizzare quattro conformazioni differenti del recettore. Tramite il
programma GOLD il raloxifene è stato introdotto nella struttura 3ERT e il
tamoxifene nel recettore 1ERR; analogamente è avvenuto lo scambio tra estradiolo
e dietilstilbestrolo. Gli score ottenuti per i nuovi complessi si discostano in modo
poco significativo da quelli relativi ai complessi cristallografici. Questo ha fatto
ipotizzare che l’utilizzo di quattro conformazioni recettoriali sia superfluo nella
prospettiva del virtual screening. In questo studio, quindi, la flessibilità è stata
inserita nelle analisi di docking prendendo in considerazione solo le due
conformazioni limite. In particolare, gli agonisti sono stati inseriti nella
conformazione recettoriale corrispondente, viceversa, composti simil raloxifene

Risultati e discussioni
43
sono stati introdotti nella struttura di riferimento per i SERM. Per le sostanze il cui
profilo di attività è ancora sconosciuto sono stati compiuti, parallelamente su
entrambe le conformazioni, studi predittivi di affinità.
Utilizzando solo le due conformazioni limite, ovviamente, le informazioni relative ai
piccoli spostamenti vengono perse. Questa approssimazione si rende necessaria
nella prospettiva di messa a punto di un protocollo di screening virtuale, per
limitare la complessità del sistema. E’ accettabile considerando che lo scopo
dell’indagine è la predizione di legame su larga scala di molecole sconosciute dalle
svariate caratteristiche chimiche, per le quali è desiderabile, ma non necessario,
apprezzare variazione di energia al di sotto di una Kcal/mole. Studi di dinamica
(Sivanesan et al. 2005) e analisi computazionali di docking (Anstead et al. 1997)
hanno dimostrato, inoltre, che il legame idrogeno tra un ossidrile e l’His apporta
all’energia libera di legame un contributo di 0.6 Kcal/mol, molto inferiore rispetto
Figura 5.1.1: Sovrapposizione dei quattro amminoacidi essenziali che costituiscono il sito di legame di ERα per i complessi 1ERR (verde), 3ERT (rosso) e 1XPC (giallo). Gli amminoacidi si orientano in maniera simile in tutti e tre i siti e le variazioni più marcate si osservano a carico dell’istidina.

Risultati e discussioni
44
all’apporto fornito dal legame idrogeno tra l’anello fenolico e il Glu353 (1.9
Kcal/mol).
Quando le differenze tra le conformazioni sono esigue la risoluzione della struttura
cristallografica assume maggiore peso nella capacità di discriminare la disposizione
degli amminoacidi e non sempre è possibile disporre di cristalli ad elevata
risoluzione. Utilizzare un numero limitato di strutture ad alta risoluzione, quindi,
permette anche di ridurre al minimo gli errori che dipendono esclusivamente dalla
qualità della tecnica cristallografica, che varia da laboratorio a laboratorio.
Per descrivere l’attendibilità di una struttura cristallografica, oltre alla risoluzione,
si possono considerare i valori di B factor associati ad ogni atomo della proteina. Il
fattore B misura quantitativamente la dispersione della nuvola elettronica intorno
alla posizione media definita cristallograficamente. Tanto maggiore è tale valore
tanto più la reale collocazione dell’atomo oscillerà intorno al punto medio. Valori
elevati di B factor, inoltre, possono dipendere dalla flessibilità della proteina. Infatti,
se la mobilità delle catene laterali è considerevole, sarà più difficile localizzarne con
esattezza la posizione.
Sono stati analizzati i valori di B factor degli amminoacidi chiave del sito di legame
(Asp 351, Glu 353, Arg 394, His 524) del recettore ERα in complesso con diversi
SERM. E’ interessante notare come in tutti i casi considerati la posizione dell’His
524 è quella maggiormente indeterminata, in quanto presenta sempre valori
superiori al B factor medio della proteina. Queste osservazioni suggeriscono che la
catena laterale dell’istidina è particolarmente flessibile e per questo è ostacolata la
sua precisa collocazione. Questa ipotesi è supportata anche da studi di dinamica
molecolare presenti in letteratura. (Sivanesan et al. 2005). A titolo di esempio sono
riportati i dati di B factor relativi al recettore complessato con il raloxifene, che, tra
tutte le strutture cristallografiche analizzate, mostra per gli atomi dell’His 524 i
valori più elevati (Tabella 5.1.1).
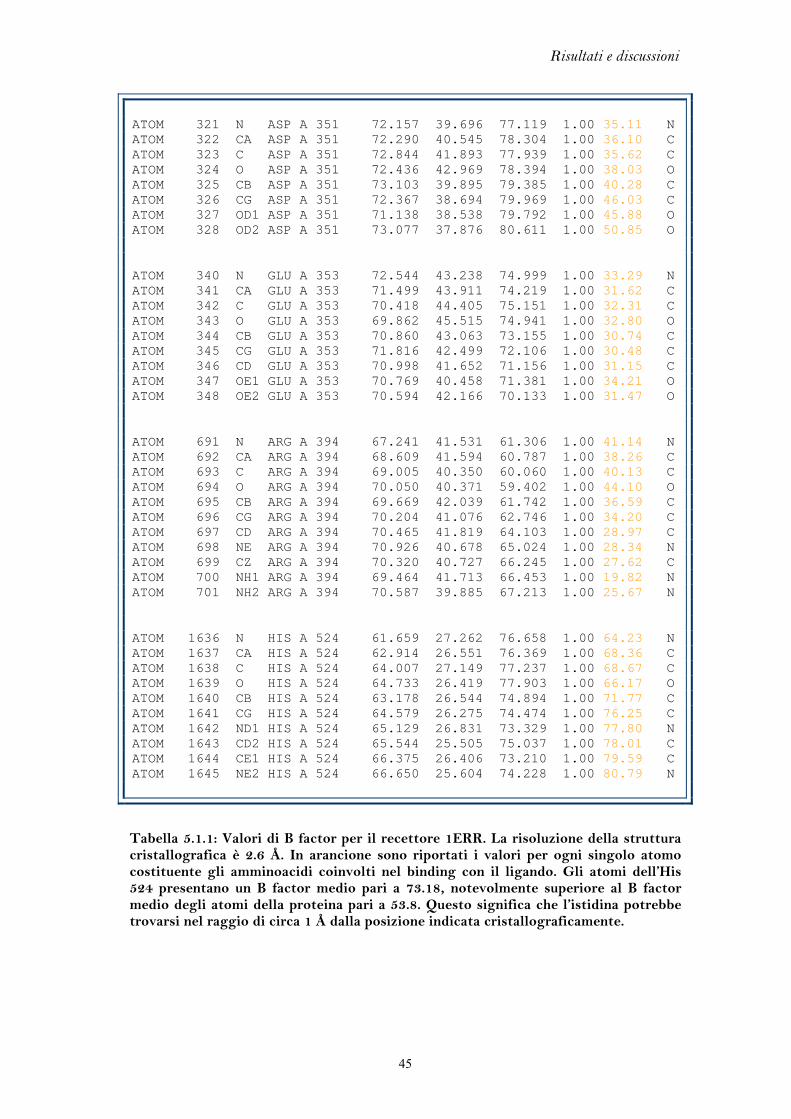
Risultati e discussioni
45
ATOM 321 N ASP A 351 72.157 39.696 77.119 1.00 35.11 N ATOM 322 CA ASP A 351 72.290 40.545 78.304 1.00 36.10 C ATOM 323 C ASP A 351 72.844 41.893 77.939 1.00 35.62 C ATOM 324 O ASP A 351 72.436 42.969 78.394 1.00 38.03 O ATOM 325 CB ASP A 351 73.103 39.895 79.385 1.00 40.28 C ATOM 326 CG ASP A 351 72.367 38.694 79.969 1.00 46.03 C ATOM 327 OD1 ASP A 351 71.138 38.538 79.792 1.00 45.88 O ATOM 328 OD2 ASP A 351 73.077 37.876 80.611 1.00 50.85 O ATOM 340 N GLU A 353 72.544 43.238 74.999 1.00 33.29 N ATOM 341 CA GLU A 353 71.499 43.911 74.219 1.00 31.62 C ATOM 342 C GLU A 353 70.418 44.405 75.151 1.00 32.31 C ATOM 343 O GLU A 353 69.862 45.515 74.941 1.00 32.80 O ATOM 344 CB GLU A 353 70.860 43.063 73.155 1.00 30.74 C ATOM 345 CG GLU A 353 71.816 42.499 72.106 1.00 30.48 C ATOM 346 CD GLU A 353 70.998 41.652 71.156 1.00 31.15 C ATOM 347 OE1 GLU A 353 70.769 40.458 71.381 1.00 34.21 O ATOM 348 OE2 GLU A 353 70.594 42.166 70.133 1.00 31.47 O ATOM 691 N ARG A 394 67.241 41.531 61.306 1.00 41.14 N ATOM 692 CA ARG A 394 68.609 41.594 60.787 1.00 38.26 C ATOM 693 C ARG A 394 69.005 40.350 60.060 1.00 40.13 C ATOM 694 O ARG A 394 70.050 40.371 59.402 1.00 44.10 O ATOM 695 CB ARG A 394 69.669 42.039 61.742 1.00 36.59 C ATOM 696 CG ARG A 394 70.204 41.076 62.746 1.00 34.20 C ATOM 697 CD ARG A 394 70.465 41.819 64.103 1.00 28.97 C ATOM 698 NE ARG A 394 70.926 40.678 65.024 1.00 28.34 N ATOM 699 CZ ARG A 394 70.320 40.727 66.245 1.00 27.62 C ATOM 700 NH1 ARG A 394 69.464 41.713 66.453 1.00 19.82 N ATOM 701 NH2 ARG A 394 70.587 39.885 67.213 1.00 25.67 N ATOM 1636 N HIS A 524 61.659 27.262 76.658 1.00 64.23 N ATOM 1637 CA HIS A 524 62.914 26.551 76.369 1.00 68.36 C ATOM 1638 C HIS A 524 64.007 27.149 77.237 1.00 68.67 C ATOM 1639 O HIS A 524 64.733 26.419 77.903 1.00 66.17 O ATOM 1640 CB HIS A 524 63.178 26.544 74.894 1.00 71.77 C ATOM 1641 CG HIS A 524 64.579 26.275 74.474 1.00 76.25 C ATOM 1642 ND1 HIS A 524 65.129 26.831 73.329 1.00 77.80 N ATOM 1643 CD2 HIS A 524 65.544 25.505 75.037 1.00 78.01 C ATOM 1644 CE1 HIS A 524 66.375 26.406 73.210 1.00 79.59 C ATOM 1645 NE2 HIS A 524 66.650 25.604 74.228 1.00 80.79 N
Tabella 5.1.1: Valori di B factor per il recettore 1ERR. La risoluzione della struttura cristallografica è 2.6 Å. In arancione sono riportati i valori per ogni singolo atomo costituente gli amminoacidi coinvolti nel binding con il ligando. Gli atomi dell’His 524 presentano un B factor medio pari a 73.18, notevolmente superiore al B factor medio degli atomi della proteina pari a 53.8. Questo significa che l’istidina potrebbe trovarsi nel raggio di circa 1 Å dalla posizione indicata cristallograficamente.

Risultati e discussioni
46
5.2 ANALISI PRELIMINARI
Prima di procedere con le analisi computazionali è stato necessario definire le
condizioni operative con cui condurre le indagini per rendere i risultati attendibili,
omogenei e riproducibili. In particolare, la caratterizzazione di due variabili è stato
oggetto di un attento studio:
scelta delle strutture cristallografiche con cui condurre le procedure di
docking;
determinazione del contributo apportato dalla molecola d’acqua conservata
nel binding con il ligando.
5.2.1 SCELTA DELLA STRUTTURA CRISTALLOGRAFICA DEL
RECETTORE
Esistono numerose strutture cristallografiche del recettore degli estrogeni in
complesso con svariati ligandi. Per condurre simulazioni di docking verosimili e
confrontabili occorre selezionarne due (una per gli agonisti e una per i SERM) a cui
riferirsi per ogni analisi. In entrambi i casi la scelta è ricaduta sul complesso
cristallografico a più alta risoluzione. I SERM sono stati introdotti nella struttura
1XPC (Blizzard et al 2005), caratterizzata da un’ottima risoluzione (R = 1.6 Å) e dai
migliori valori di B factor. Il sito di legame è stato sovrapposto con il sito dei
complessi ERα-raloxifene (1ERR) e ERα-tamoxifene (3ERT), al fine di
determinarne le differenze (Figura 5.1.1). L’unica variazione interessante riguarda la
posizione dell’His 524, sostenendo ulteriormente l’ipotesi che, trovandosi in una
zona flessibile, i suoi piccoli spostamenti siano condizionati dal tipo di ligando, ma
trascurabili in una prospettiva di screening su larga scala. Per quanto riguarda gli
agonisti, è stata preferita la conformazione recettoriale selezionata dal
dietilstilbestrolo (3ERD, Shiau et al. 1998), risolta a 2.0 Å, piuttosto che il
complesso dell’estradiolo (1ERE), caratterizzato da bassa risoluzione (R = 3.1 Å). In
questo caso la sovrapposizione dei siti di legame mostra variazioni ancora più
piccole. Essendo le due conformazioni confrontabili è giustificata la scelta della
struttura a miglior risoluzione.

Risultati e discussioni
47
5.2.2 RUOLO DELLA MOLECOLA D’ACQUA CONSERVATA
I composti che accedono al sito di legame interagiscono, oltre che con i quattro
amminoacidi chiave, con una molecola d’acqua situata tra il Glu 353 e l’Arg 394
(Brzozowski et al. 1997), che non è spiazzata dall’ingresso del ligando e come tale è
conservata in tutti i complessi cristallografici.
Studi precedenti, condotti in questo laboratorio, hanno dimostrato che nel sistema
recettoriale degli estrogeni il contributo all’energia di legame apportato da questa
specifica molecola d’acqua è poco rilevante, come si osserva nei due grafici sotto
riportati. Le rette con e senza il contributo della molecola d’acqua, ottenute
analizzando 6 ligandi cristallografici e 27 ligandi ad affinità nota, sono
caratterizzate dai seguenti valori: R = 0.53, R2 = 0.28 ed errore standard = 1.9
Kcal/mol, per il Grafico 5.2.1; R = 0.56, R2 = 0.31 ed errore standard=1.85
Kcal/mol per il Grafico 5.2.2.
Generando la superficie di Connolly, è stato, inoltre, osservato che la molecola
d’acqua conservata è sepolta in una cavità del recettore, dalla quale contrae
interazioni con la proteina e, più difficilmente, con il ligando, poichè non riesce ad
esporsi completamente all’interno del sito di legame (Figura 5.2.1).
Figura 5.2.1: Superficie di Connolly della cavità di legame del complesso 1XPC. La molecola d’acqua conservata è esclusa dal sito di legame.
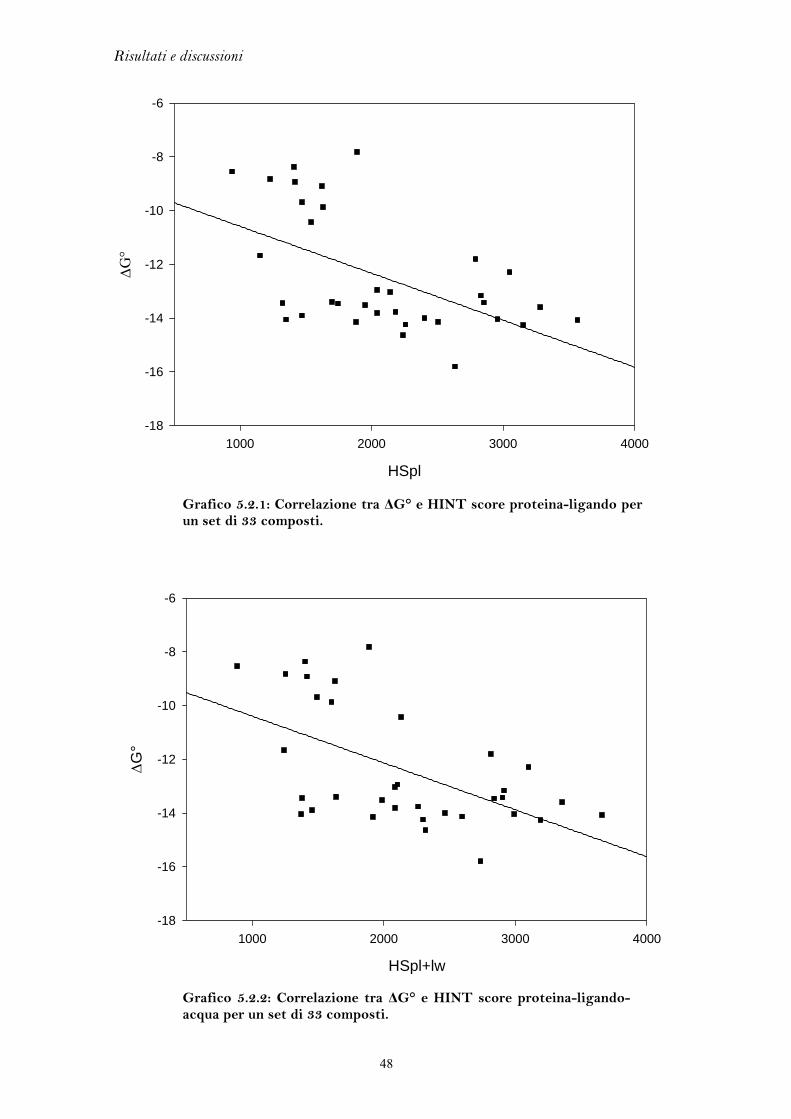
Risultati e discussioni
48
HSpl
1000 2000 3000 4000
∆G°
-18
-16
-14
-12
-10
-8
-6
Grafico 5.2.1: Correlazione tra ∆G° e HINT score proteina-ligando per un set di 33 composti.
Grafico 5.2.2: Correlazione tra ∆G° e HINT score proteina-ligando-acqua per un set di 33 composti.
HSpl+lw
1000 2000 3000 4000
∆ G°
-18
-16
-14
-12
-10
-8
-6

Risultati e discussioni
49
A partire da queste considerazioni tutte le molecole d’acqua, comprese quelle
conservate, sono state escluse dal recettore nelle analisi di docking. Tale
approssimazione permette di ridurre i tempi delle simulazioni computazionali e di
eliminare un’ulteriore variabile che, se non significativa, può essere fonte di errori.
5.3 DOCKING DI SERM E AGONISTI
Il Grafico 5.2.1 mostra la correlazione tra HINT score proteina-ligando e ∆G°
sperimentale ottenuta per un set di composti a cui appartengono sia agonisti che
SERM. Gli studi sono stati compiuti utilizzando più conformazioni del recettore e
per ogni composto è stata scelta, per confronto con i ligandi cristallografici, la
struttura più idonea. Questo approccio oltre che rallentare i tempi delle analisi,
rende, come spiegato in precedenza, i risultati poco confrontabili, in quanto si affida
a qualità risolutive anche molto diverse.
Per rendere omogenee le analisi, il docking dei 33 composti è stato ripetuto
utilizzando la struttura 3ERD per gli agonisti e 1XPC per gli agonisti parziali. La
nuova correlazione è mostrata nel Grafico 5.3.1. La retta non si discosta di molto da
quella del Grafico 5.2.1, presentando valori di correlazione leggermente migliori: R
= 0.58, R2 = 0.34, errore standard = 1.85 Kcal/mol.
Per affrontare uno studio di virtual screening occorre avere a disposizione una retta
di taratura solida che validi il metodo di analisi nei confronti del sistema oggetto di
studio. Per queste ragioni una parte di questo lavoro di tesi è stata dedicata allo
studio della correlazione tra HINT score e ∆G° sperimentale. Ai 33 composti già
analizzati ne sono stati aggiunti altri 19 appartenenti a classi chimiche e funzionali
differenti, di cui 8 cristallografici e 11 ad affinità nota. E’ stata prestata particolare
attenzione nella selezione dei ligandi, in modo da costituire un set di composti
eterogeneo e rappresentativo delle varibilità chimiche attualmente più indagate.
Nell’analisi sono stati inseriti: ligandi cristallografici (SERM), agonisti steroidei,
agonisti non steroidei, fitoestrogeni e inquinanti. Le strutture chimiche delle
sostanze utilizzate sono riportate nella Tabella 5.3.1.
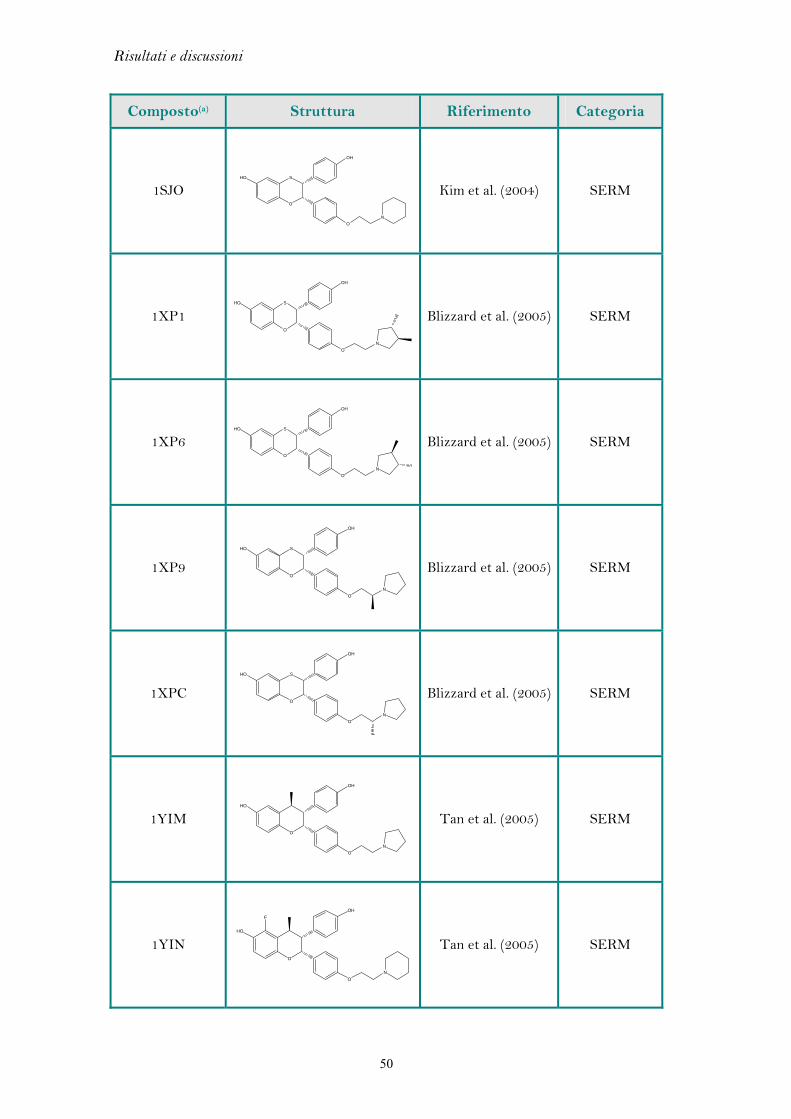
Risultati e discussioni
50
Composto(a) Struttura Riferimento Categoria
1SJO O
S
ON
OH
HO
Kim et al. (2004) SERM
1XP1 O
S
O
OH
HO
N
Blizzard et al. (2005) SERM
1XP6 O
S
O
OH
HO
N
Blizzard et al. (2005) SERM
1XP9 O
S
O
OH
HO
N
Blizzard et al. (2005) SERM
1XPC O
S
O
OH
HO
N
Blizzard et al. (2005) SERM
1YIM O
O
OH
HO
N
Tan et al. (2005) SERM
1YIN O
O
OH
HO
N
F
Tan et al. (2005) SERM
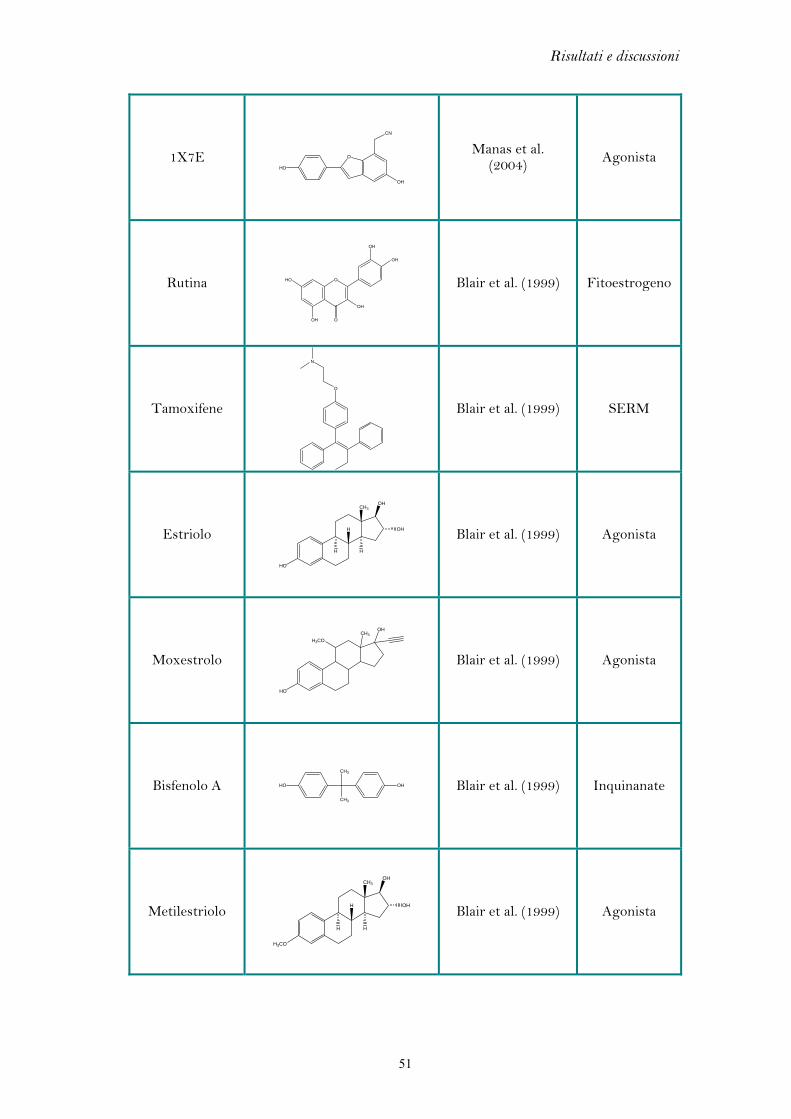
Risultati e discussioni
51
1X7E O
CN
OH
HO
Manas et al. (2004) Agonista
Rutina O
OOH
HO
OH
OH
OH
Blair et al. (1999) Fitoestrogeno
Tamoxifene
O
N
Blair et al. (1999) SERM
Estriolo H H
OH
OHCH3
H
HO
Blair et al. (1999) Agonista
Moxestrolo
HO
OH
H3COCH3
Blair et al. (1999) Agonista
Bisfenolo A HO OH
CH3
CH3 Blair et al. (1999) Inquinanate
Metilestriolo H H
OH
OHCH3
H
H3CO
Blair et al. (1999) Agonista
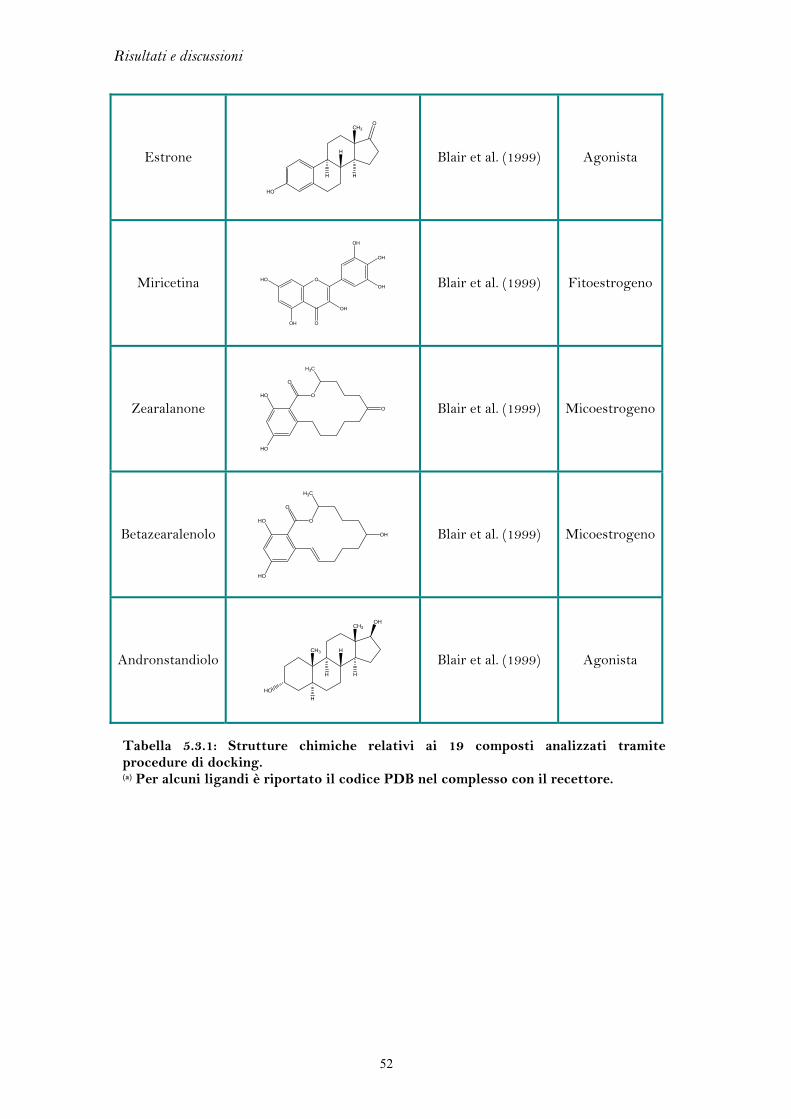
Risultati e discussioni
52
Estrone H H
CH3
H
HO
O
Blair et al. (1999) Agonista
Miricetina O
OOH
HO
OH
OH
OH
OH
Blair et al. (1999) Fitoestrogeno
Zearalanone OHO
HO
O
H3C
O
Blair et al. (1999) Micoestrogeno
Betazearalenolo OHO
HO
O
H3C
OH
Blair et al. (1999) Micoestrogeno
Andronstandiolo H H
OHCH3
HCH3
HHO
Blair et al. (1999) Agonista
Tabella 5.3.1: Strutture chimiche relativi ai 19 composti analizzati tramite procedure di docking. (a) Per alcuni ligandi è riportato il codice PDB nel complesso con il recettore.
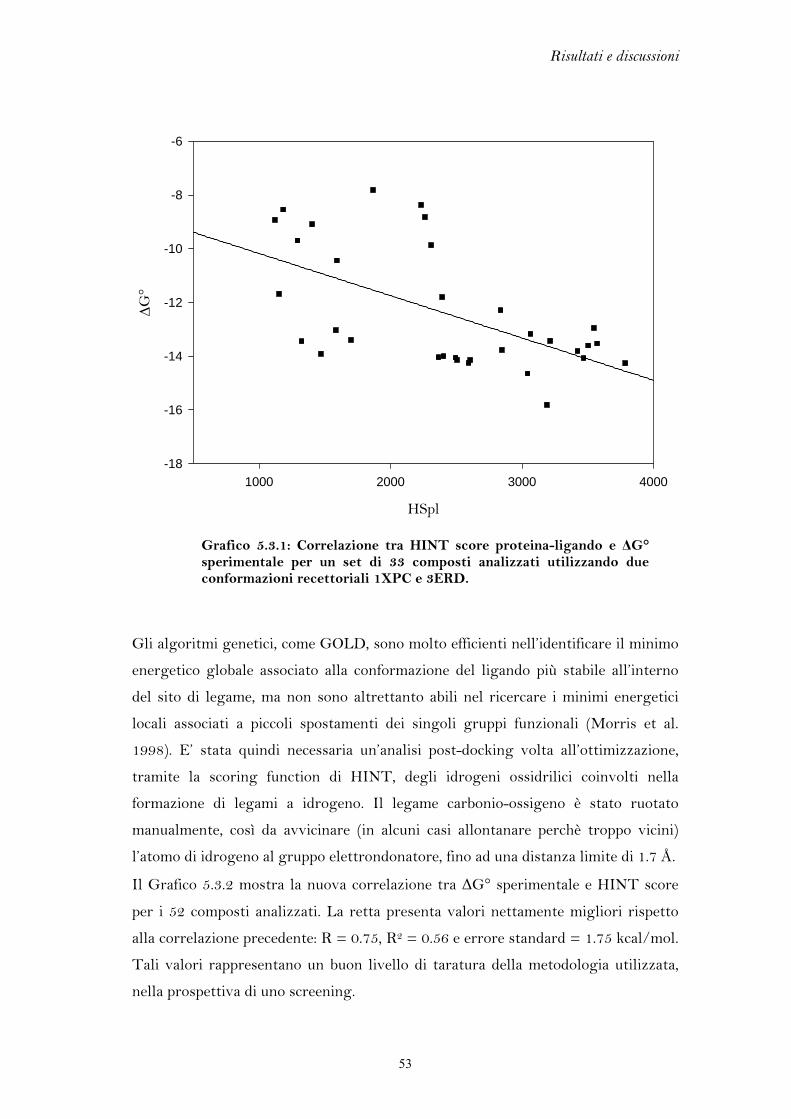
Risultati e discussioni
53
HSpl
1000 2000 3000 4000
∆G°
-18
-16
-14
-12
-10
-8
-6
Gli algoritmi genetici, come GOLD, sono molto efficienti nell’identificare il minimo
energetico globale associato alla conformazione del ligando più stabile all’interno
del sito di legame, ma non sono altrettanto abili nel ricercare i minimi energetici
locali associati a piccoli spostamenti dei singoli gruppi funzionali (Morris et al.
1998). E’ stata quindi necessaria un’analisi post-docking volta all’ottimizzazione,
tramite la scoring function di HINT, degli idrogeni ossidrilici coinvolti nella
formazione di legami a idrogeno. Il legame carbonio-ossigeno è stato ruotato
manualmente, così da avvicinare (in alcuni casi allontanare perchè troppo vicini)
l’atomo di idrogeno al gruppo elettrondonatore, fino ad una distanza limite di 1.7 Å.
Il Grafico 5.3.2 mostra la nuova correlazione tra ∆G° sperimentale e HINT score
per i 52 composti analizzati. La retta presenta valori nettamente migliori rispetto
alla correlazione precedente: R = 0.75, R2 = 0.56 e errore standard = 1.75 kcal/mol.
Tali valori rappresentano un buon livello di taratura della metodologia utilizzata,
nella prospettiva di uno screening.
Grafico 5.3.1: Correlazione tra HINT score proteina-ligando e ∆G° sperimentale per un set di 33 composti analizzati utilizzando due conformazioni recettoriali 1XPC e 3ERD.
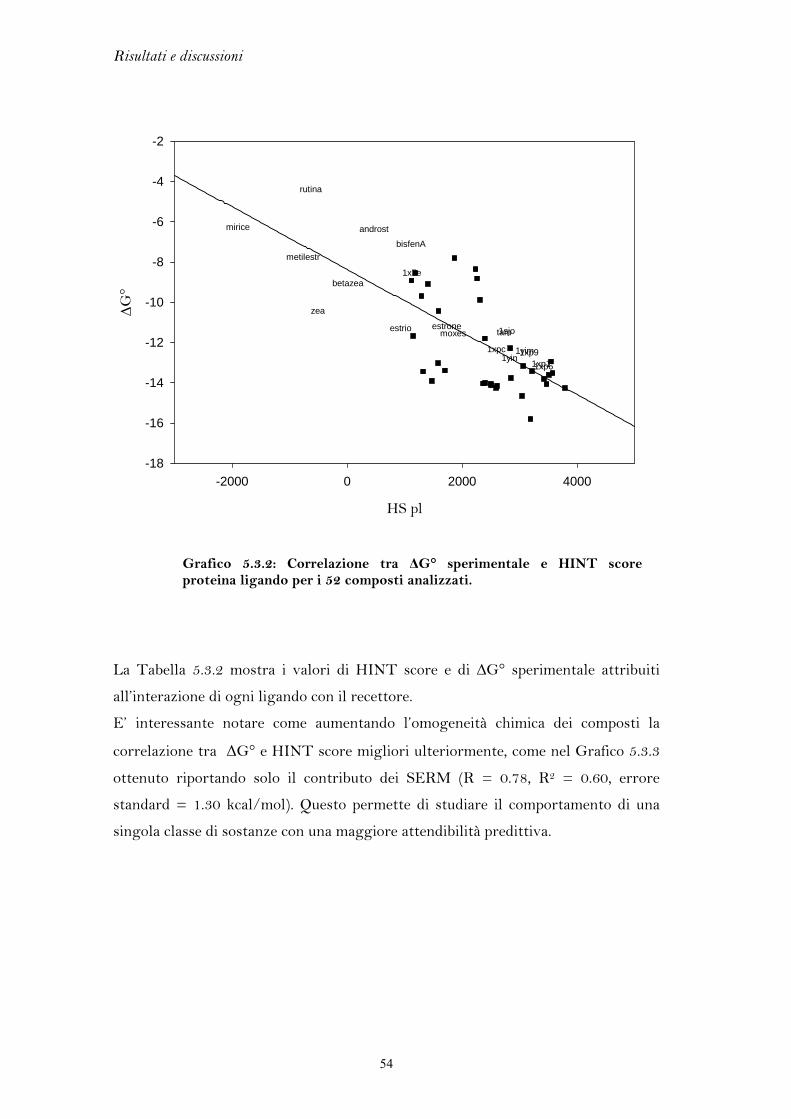
Risultati e discussioni
54
HS pl
-2000 0 2000 4000
∆G
°
-18
-16
-14
-12
-10
-8
-6
-4
-2
1sjo
1xp11xp61xp91xpc 1yim
1yin
1x7e
rutina
tamestrio moxes
bisfenAmetilestr
estrone
mirice
zea
betazea
androst
La Tabella 5.3.2 mostra i valori di HINT score e di ∆G° sperimentale attribuiti
all’interazione di ogni ligando con il recettore.
E’ interessante notare come aumentando l’omogeneità chimica dei composti la
correlazione tra ∆G° e HINT score migliori ulteriormente, come nel Grafico 5.3.3
ottenuto riportando solo il contributo dei SERM (R = 0.78, R2 = 0.60, errore
standard = 1.30 kcal/mol). Questo permette di studiare il comportamento di una
singola classe di sostanze con una maggiore attendibilità predittiva.
Grafico 5.3.2: Correlazione tra ∆G° sperimentale e HINT score proteina ligando per i 52 composti analizzati.
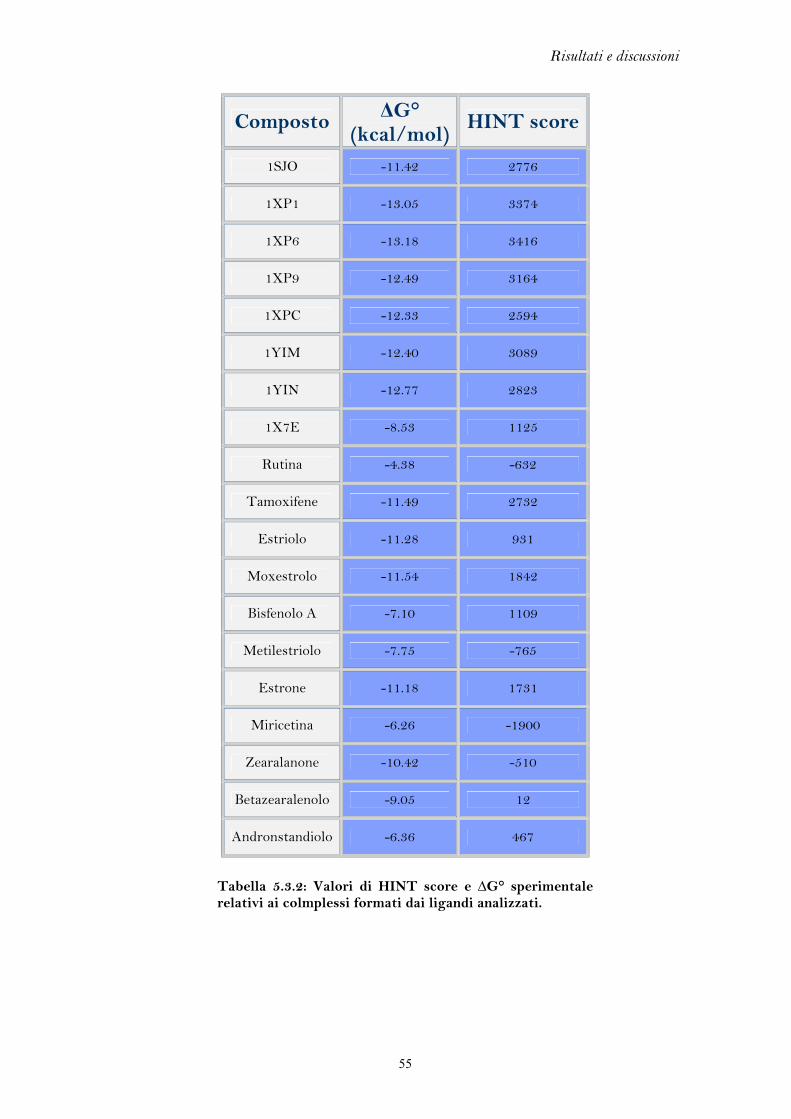
Risultati e discussioni
55
Composto ∆G° (kcal/mol)
HINT score
1SJO -11.42 2776
1XP1 -13.05 3374
1XP6 -13.18 3416
1XP9 -12.49 3164
1XPC -12.33 2594
1YIM -12.40 3089
1YIN -12.77 2823
1X7E -8.53 1125
Rutina -4.38 -632
Tamoxifene -11.49 2732
Estriolo -11.28 931
Moxestrolo -11.54 1842
Bisfenolo A -7.10 1109
Metilestriolo -7.75 -765
Estrone -11.18 1731
Miricetina -6.26 -1900
Zearalanone -10.42 -510
Betazearalenolo -9.05 12
Andronstandiolo -6.36 467
Tabella 5.3.2: Valori di HINT score e ∆G° sperimentale relativi ai colmplessi formati dai ligandi analizzati.
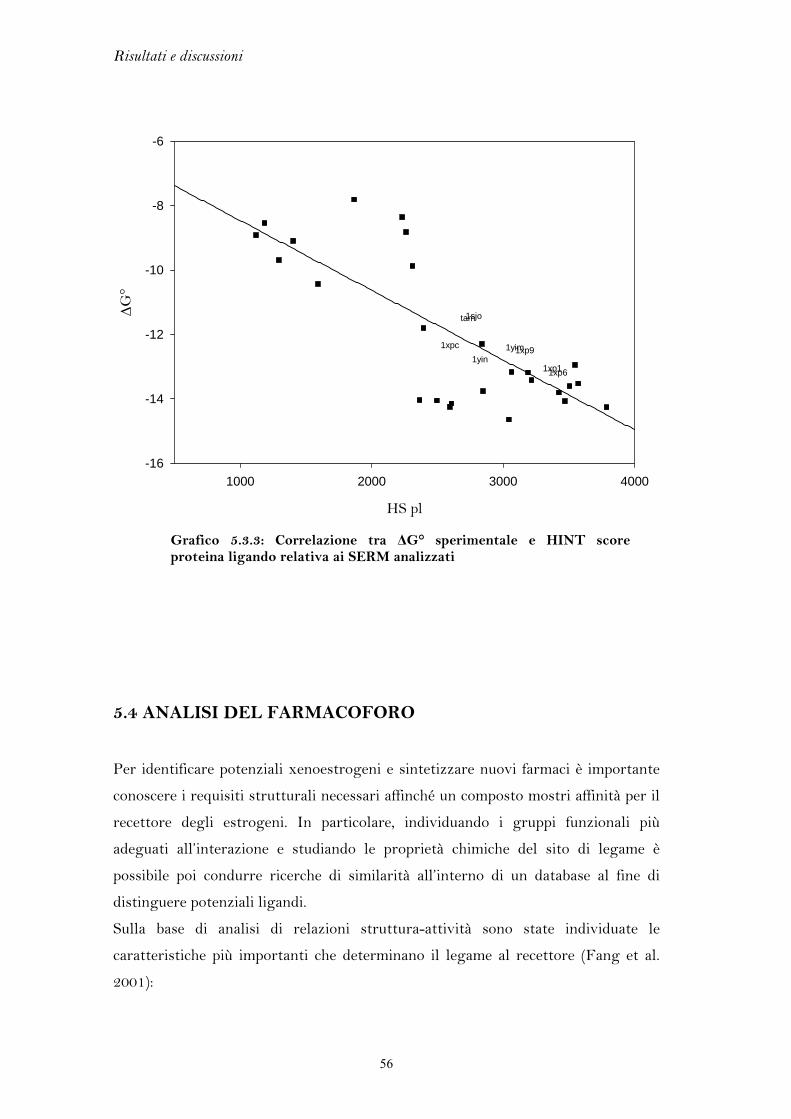
Risultati e discussioni
56
HS pl
1000 2000 3000 4000
∆G
°
-16
-14
-12
-10
-8
-6
1sjo
1xp11xp6
1xp91xpc 1yim1yin
tam
5.4 ANALISI DEL FARMACOFORO
Per identificare potenziali xenoestrogeni e sintetizzare nuovi farmaci è importante
conoscere i requisiti strutturali necessari affinché un composto mostri affinità per il
recettore degli estrogeni. In particolare, individuando i gruppi funzionali più
adeguati all’interazione e studiando le proprietà chimiche del sito di legame è
possibile poi condurre ricerche di similarità all’interno di un database al fine di
distinguere potenziali ligandi.
Sulla base di analisi di relazioni struttura-attività sono state individuate le
caratteristiche più importanti che determinano il legame al recettore (Fang et al.
2001):
Grafico 5.3.3: Correlazione tra ∆G° sperimentale e HINT score proteina ligando relativa ai SERM analizzati

Risultati e discussioni
57
1. presenza di un gruppo ossidrilico in grado di mimare la funzione 3-idrossi
dell’estradiolo mediante la formazione di un legame idrogeno con il Glu353
e l’Arg 394. Qualsiasi fattore capace di modificare il contributo di tale
legame, come ingombri sterici, effetti induttivi di risonanza, o sostituzione
del gruppo OH con un donatore di idrogeno debole quale NH, riduce la forza
del binding;
2. formazione di un legame ad idrogeno tra l’His 524 e un secondo gruppo
ossidrilico posto ad una distanza ottimale di 11 Å dal primo, come
nell’estradiolo. Per i ligandi con una differente distanza tra i due atomi di
ossigeno, il gruppo imidazolico, grazie alla flessibilità della catena, può
compensare tale distanza;
3. strutture caratterizzate da un certo grado di idrofobicità si prestano meglio
all’interazione con il sito essenzialmente apolare;
4. presenza di un anello, di norma aromatico, che contribuisce alla rigidità della
molecola. Ad oggi non sono conosciuti ligandi attivi sul recettore degli
estrogeni che non presentino almeno una struttura ad anello;
5. per quanto riguarda agonisti parziali e antagonisti, presenza di una funzione
amminica protonata in grado di mettere a disposizione l’idrogeno per
formare in legame idrogeno con Asp 351.
Lo studio del sito di legame, effettuato in questo lavoro di tesi con il software
GRID, mostra risultati perfettamente in accordo con le valutazioni precedenti. Per
l’analisi farmacoforica sono stati utilizzati quattro probe al fine di identificare
all’interno del sito i gruppi accettori/donatori di legami idrogeno e disponibili ai
contatti idrofobici. Il programma, inoltre, non si limita ad individuare strutture
funzionali complementari al probe, ma da una stima quantitativa dell’energia
associata all’interazione della sonda con il sito.
Probe donatore di legami idrogeno
La mappa della cavità recettoriale mostra che le zone favorevoli ad accettare un
legame idrogeno sono localizzate in corrispondenza del Glu 353, dell’His 524 e
dell’Asp 351 e, infatti, la maggior parte dei SERM è caratterizzata dalla presenza di
due gruppi fenolici che interagiscono con il glutammato e l’istidina e da una
funzione amminica eterociclica protonata che lega l’aspartato. In corrispondenza del
Glu 353, GRID evidenzia la porzione energeticamente più favorita, in accordo con
gli studi di SAR decritti.

Risultati e discussioni
58
Probe accettore di legami idrogeno
L’anello imidazolico dell’His 524 e, soprattutto, la funzione imidazolica dell’Arg 394
costituiscono i due gruppi donatori di legami idrogeno principali. Infatti, l’His 524
ha un carattere duplice, grazie alla tautomeria dell’anello imidazolico che può
comportarsi da H-donatore o H-accettore a seconda del ligando presente nel sito. Il
contributo energetico che GRID associa all’istidina è meno rilevante dei contributi
apportati da Arg 394 e Glu353, in accordo con altri studi computazionali effettuati
(Sivanesan et al.2005). Il software, inoltre, mostra una zona energeticamente
favorita, alla formazione di legami idrogeno, delimitata dagli amminoacidi Met 343 ,
Cys 530 e Thr 347, che non era stata ancora evidenziata da studi di SAR.
Probe idrofobico
Il sito del recettore degli estrogeni si presenta come una cavità essenzialmente
apolare, che predilige ligandi caratterizzati da un alto grado di idrofobicità. La
conformazione recettoriale selezionata dai SERM è caratterizzata dalla presenza di
un canale nel quale essi alloggiano la catena laterale. All’interno di tale canale si
sono osservate due piccole cavità energeticamente favorevoli a contatti idrofobici: la
prima è delinetata dagli amminoacidi Trp 383, Leu 354, Leu 356 ed interagisce,
molto probabilmente, con l’anello eterociclico delle catene laterali dei SERM; la
seconda, invece, è caratterizzata da un profilo energetico minore ed è localizzata in
corrispondenza dei residui Tyr 526 e Cys 530.
Ossidrile fenolico
Un’ulteriore analisi è stata effettuata utilizzando come probe un gruppo ossidrilico
di natura fenolica, cha mostra carattere ambivalente di accettore e donatore di
legame idrogeno. La valutazione energetica realizzata da GRID stabilisce che il
probe fenolico instaura interazioni migliori rispetto a tutte le altre sonde utilizzate,
rimarcando l’importanza che tale gruppo riveste nell’instaurare il legame (Figura
5.4.1).
E’ necessario notare l’esistenza di composti che, pur non possedendo un ossidrile
fenolico nella struttura, mostrano una buona affinità recettoriale. L’ospemifene, ad
esempio, pur non possedendo l’idrossile fenolico è attualmente in fase clinica per il
trattamento dell’osteoporosi e dell’atrofia urogenitale (Henke et al. 2005).
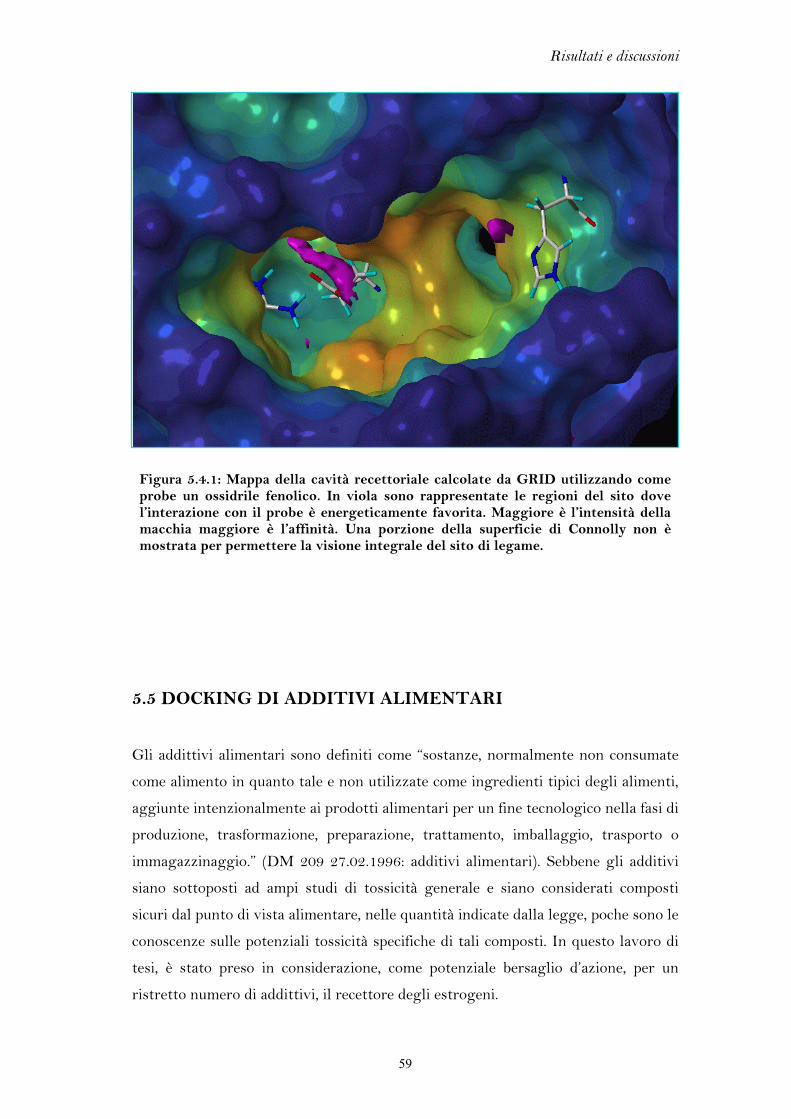
Risultati e discussioni
59
5.5 DOCKING DI ADDITIVI ALIMENTARI
Gli addittivi alimentari sono definiti come “sostanze, normalmente non consumate
come alimento in quanto tale e non utilizzate come ingredienti tipici degli alimenti,
aggiunte intenzionalmente ai prodotti alimentari per un fine tecnologico nella fasi di
produzione, trasformazione, preparazione, trattamento, imballaggio, trasporto o
immagazzinaggio.” (DM 209 27.02.1996: additivi alimentari). Sebbene gli additivi
siano sottoposti ad ampi studi di tossicità generale e siano considerati composti
sicuri dal punto di vista alimentare, nelle quantità indicate dalla legge, poche sono le
conoscenze sulle potenziali tossicità specifiche di tali composti. In questo lavoro di
tesi, è stato preso in considerazione, come potenziale bersaglio d’azione, per un
ristretto numero di addittivi, il recettore degli estrogeni.
Figura 5.4.1: Mappa della cavità recettoriale calcolate da GRID utilizzando come probe un ossidrile fenolico. In viola sono rappresentate le regioni del sito dove l’interazione con il probe è energeticamente favorita. Maggiore è l’intensità della macchia maggiore è l’affinità. Una porzione della superficie di Connolly non è mostrata per permettere la visione integrale del sito di legame.

Risultati e discussioni
60
Il database sviluppato da FAO/WHO è organizzato in due cataloghi: da una parte
sono classificati gli aromatizzanti e nell’altra rientrano tutte le restanti classi
funzionali, compresi anche alcuni aromatizzanti stessi. In questa tesi sono stati
valutati additivi provenienti dal secondo gruppo, rimandando l’analisi del catalogo
completo degli aromatizzanti a studi successivi. Il database considerato contiene
842 entries, dalle quali sono state selezionate, sulla base delle dimensioni e del
modello farmacoforico sviluppato, circa 60 sostanze potenzialmente affini al
recettore. Tuttavia il filtro applicato è stato permissivo per evitare di escludere
potenziali ligandi dall’analisi. In particolare nessun composto inorganico è stato
preso in considerazione e tra le molecole organiche sono stati scartati a priori gli
zuccheri, gli acidi grassi e i polimeri. Tra i composti che hanno superato la fase di
filtro ne sono stati analizzati 10, le cui strutture sono riportate nella Tabella 5.5.1 I
restanti additivi saranno valutati in studi seguenti.
Ogni potenziale ligando è stato inserito in entrambe le conformazioni scelte del
recettore, 3ERD e 1XPC, tranne nei casi in cui, data la caratteristiche chimiche e
geometriche del composto, non è realistico pensare che esso sia in grado di
selezionare la struttura recettoriale aperta tipica del complesso tra ERα e SERM.
Infatti, ad oggi, non sono conosciuti modulatori selettivi del recettore degli
estrogeni che non posseggano una catena laterale ingombrante capace di legare
l’Asp 351. E’, quindi, ipotizzabile che, a meno di evidenti ragioni steriche, che
impongano al recettore di assumere la conformazione aperta, i potenziali ligandi
selezionino la struttura chiusa tipica degli agonisti. In questa conformazione, infatti,
un buon ligando è in grado, entrando nel sito, di escludere completamente dalla
cavità idrofobica le molecole di solvente, contribuendo ad un notevole guadagno
entropico.
Dopo aver effettuato il docking, a partire dall’espressione analitica della retta di
taratura:
∆G° (kcal/mol) = -0.0016 HS – 8.38
è stato possibile calcolare, conoscendo il valore di HINT score (HS), la variazione di
energia libera, predetta dal programma, associata alla formazione dei diversi
complessi recettore-addittivo.
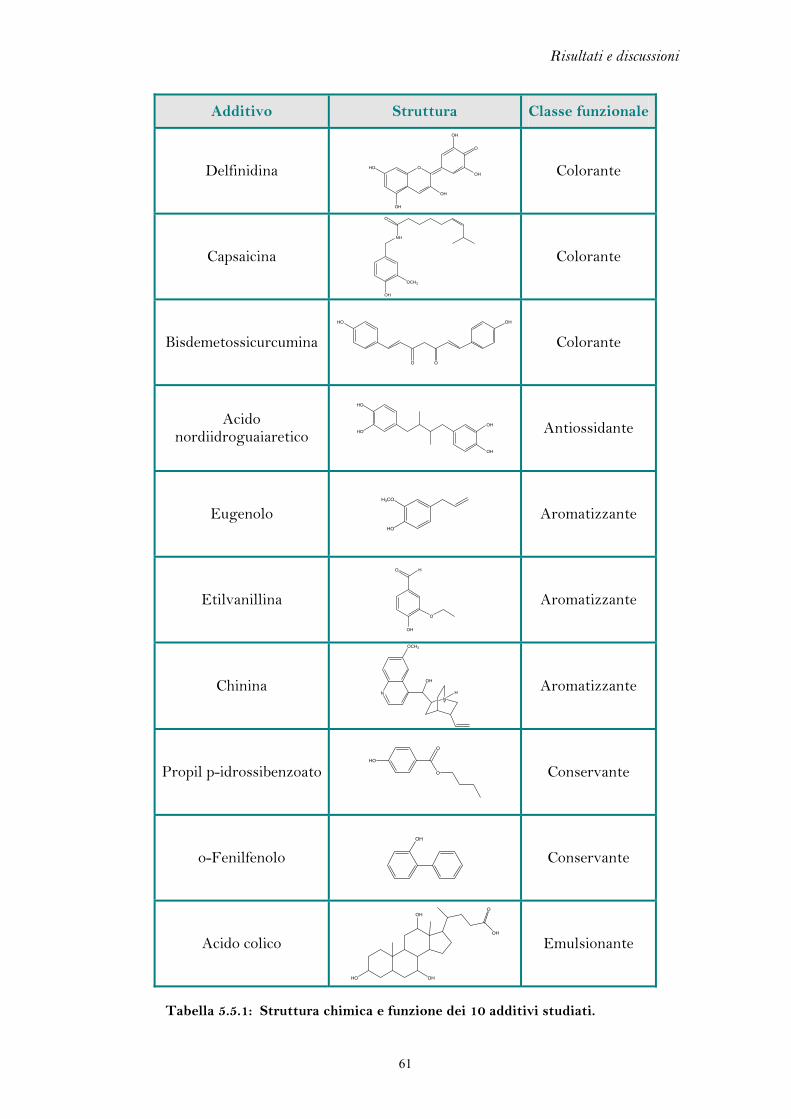
Risultati e discussioni
61
Additivo Struttura Classe funzionale
Delfinidina
OH
HO O
OH
O
OH
OH
Colorante
Capsaicina
OH
OCH3
NH
O
Colorante
Bisdemetossicurcumina HO OH
O O Colorante
Acido nordiidroguaiaretico
HO
HO
OH
OH
Antiossidante
Eugenolo H3CO
HO Aromatizzante
Etilvanillina
OH
O H
O
Aromatizzante
Chinina N
OCH3
NH
OH
Aromatizzante
Propil p-idrossibenzoato HO
O
O
Conservante
o-Fenilfenolo OH
Conservante
Acido colico
OHHO
OH
OH
O
Emulsionante
Tabella 5.5.1: Struttura chimica e funzione dei 10 additivi studiati.

Risultati e discussioni
62
5.5.1 DELFINIDINA
La delfinidina appartiene alla famiglia delle antocianidine, molto diffuse nel mondo
vegetale e particolarmente concentrate nella buccia dell’uva da cui sono estratte. Le
antocianidine sono utilizzate come coloranti naturali nell’industria alimentare,
anche se la loro applicazione è piuttosto limitata dalla scarsa stabilità chimica.
Questa classe di composti fa capo al vasto insieme dei flavonoidi, come la genisteina
e vista proprio la somiglianza con quest’ultima la delfinidina è stata inserita solo
nella conformazione recettoriale dell’agonista.
In soluzione acquosa, le antocianidine sono in equilibrio tra diverse forme, più o
meno predominanti a seconda del pH del mezzo (Figura 5.5.1 ). Ammesso che la
delfinidina arrivi integra al recettore, dovrebbe presentarsi nella struttura
predominante a pH 6-7, per cui ai fini del docking, è stata utilizzata quest’ultima.
OOH
OH
OR
OHOH
OOH
OH
OR
OH
OHOH
OOH
OH
OR
OH
OHOH
OOH
OH
OR
OHO
OO
OH
OR
OHO
+
pH < 1, rosso pH = 4-5, incolore
pH = 6-7, viola
pH = 7-8, blu scuro pH = 7-8, giallo
-
Figura 5.5.1: Equilibri in soluzione acquosa per le antocianidine.

Risultati e discussioni
63
L’Hint score associato alla formazione del complesso è pari a – 341, quindi
applicando l’equazione riportata precedentemente il ∆G° predetto è pari a -7.83
Kcal/mol.
La delfinidina si orienta nel sito di 3ERD, come mostrato in Figura 5.5.2. L’additivo
è caratterizzato dalla presenza di cinque gruppi ossidrilici e un ossigeno carbonilico,
che aumentano significativamente la polarità della molecola. I diversi gruppi
idrossilici formano legami idrogeno con gli amminoacidi importanti del sito Glu
353, Arg 394, His 524 e con due residui non fondamentali per l’attivazione, Met 421
e Leu 387. Tali interazioni sono state ottimizzate manualmente ruotando i legami
carbonio-ossigeno laddove necessario. Nonostante i numerosi contributi positivi
apportati dai legami idrogeno lo score è significativamente basso. Questo è
imputabile all’elevata polarità della molecola che va ad interagire con un sito
essenzialmente idrofobico generando numerose incompatibilità chimiche. Tale
risultato è in accordo con molti studi effettuati sui fitoestrogeni, in cui è stato
provato che l’esistenza di più di quattro gruppi ossidrilici in una molecola riduce le
proprietà di binding della sostanza stessa (Vaya et al. 2004; Fang et al. 2001).
Questa teoria permette anche di spiegare la diversa affinità di legame della
genisteina (∆G° sperimentale= -11.67), rispetto alla delfinidina. Infatti, la genisteina
presenta solo tre funzioni ossidriliche e un ossigeno carbonilico.
Arg 394
Glu 353
Figura 5.5.2: Orientazione assunta dalla delfinidina nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Leu 387
Glu 353
Arg 394 Met 421
His 524

Risultati e discussioni
64
La delfinidina, come tutte le antocianidine, è scarsamente assorbita nel tratto
gastrointestinale, soprattutto in assenza della porzione glicosidica a cui di norma si
trovano legate in natura. Conoscere i preocessi di biotrasformazione potrebbe
risultare molto importante ai fini dell’analisi sia per individuare la struttura che più
probabilmente arriva al recettore, sia per valutare eventuali metaboliti tossici. Per
quanto riguarda le antocianidine le informazioni sul metabolismo sono piuttosto
limitate in letteratura e riguardano soprattutto processi di metilazione a carico dei
gruppi ossidrilici (Wu et al. 2002).
5.5.2 CAPSAICINA
La capsaicina è una sostanza ad azione pungente contenuta, primariamente, nel
peperoncino e appartiene alla famiglia dei capsaicinoidi, di cui è il principale
esponente. La capsaicina è un comune costituente della dieta ed attualmente è
utilizzato come antimicrobico, nell’analgesia topica, come prodotto omeopatico e
come colorante nell’industria alimentare.
Nella conformazione distesa la distanza tra l’ossidrile fenolico e il metile
isopropilico della capsaicina è di circa 18 Å, mentre nell’estradiolo, che occupa
pienamente il sito, i due OH fenolici distano 11 Å (Vaya et al. 2004). La capsaicina,
quindi, è stata inserita in entrambe le conformazioni limite del recettore, 1XPC e
3ERD, anche se la presenza di numerosi legami ruotabili consente, comunque,
l’accesso al sito dell’agonista. In entrambi i recettori la funzione ossidrilica
dell’additivo forma interazioni favorevoli del tipo legame a idrogeno con il Glu 353
e l’Arg 394, come ci si aspettava dall’analisi farmacoforica effettuata. Nel sito di
1XPC la catena alchilica della capsaicina si distende e protende nella cavità occupata
tipicamente dai SERM, instaurando, tramite l’idrogeno del gruppo ammidico, un
legame idrogeno con la Leu 346 e lasciando il resto del sito di legame non occupato
(Figura 5.5.3). All’interno di 3ERD, invece, il ligando non può distendersi per ovvi
motivi sterici, quindi assume una posa più compatta, che a partire da un’ispezione
visuale sembrerebbe realistica, ma non è detto che dal punto di vista energetico tale
conformazione sia verosimile. Tuttavia la cavità recettoriale risulta pienamente
occupata dalla capasaicina e anche in questo caso si instaura un’interazione positiva
tra la funzione ammidica e il gruppo carbossilico della Leu 346 (Figura 5.5.4).
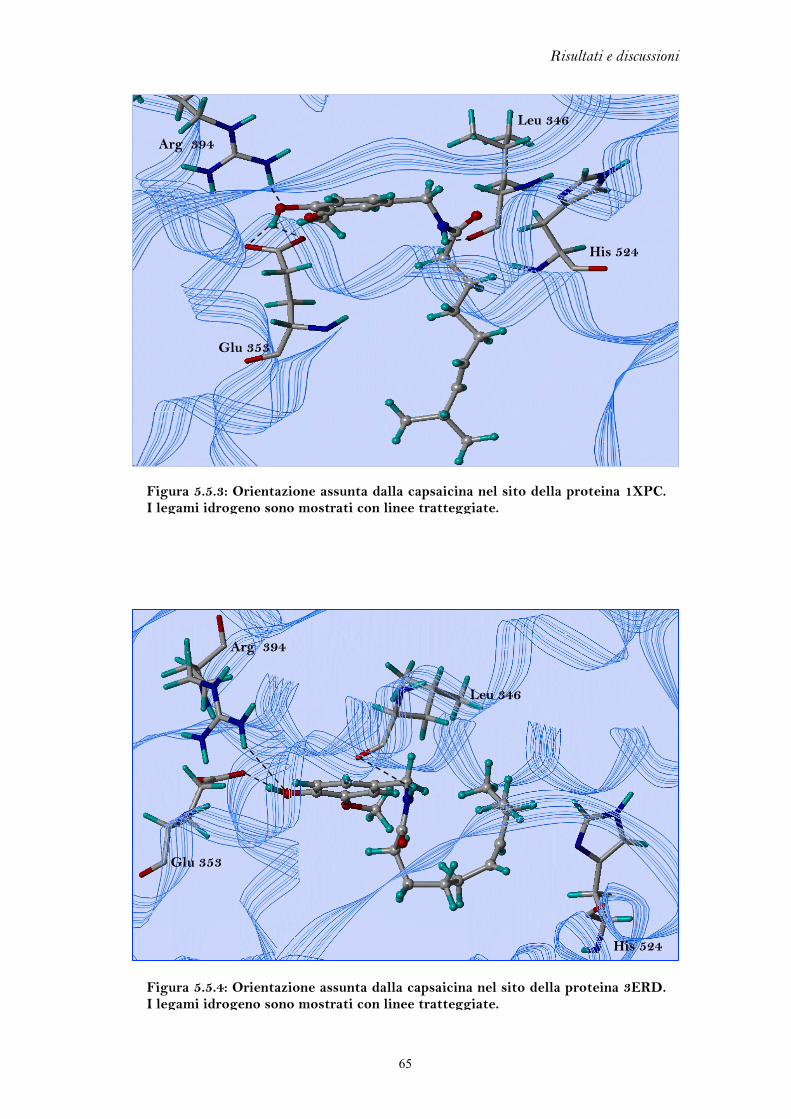
Risultati e discussioni
65
Figura 5.5.3: Orientazione assunta dalla capsaicina nel sito della proteina 1XPC. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524
Leu 346
Figura 5.5.4: Orientazione assunta dalla capsaicina nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Leu 346
Glu 353
His 524

Risultati e discussioni
66
Gli score assegnati da HINT all’interazione proteina-additivo sono 1436 per 3ERD
e 1539 per 1XPC. I contributi più significativi all’innalzamento dello score sono
apportati dalle numerose interazioni idrofobiche instaurate dalla catena alchilica
della capsaicina con il sito. Il valore di ∆G° predetto per la formazione del
complesso tra l’additivo e il recettore degli estrogeni è pari a -10.68 Kcal/mol per
3ERD e -10.84 Kcal/mol per 1XPC.
Il metabolismo microsomiale della capsaicina porta alla formazione di numerosi
composti caratterizzati da un aumento della polarità (Figura 5.5.5). Le principali
reazioni a cui la capsaicina è soggetta sono idrossilazioni, deidrogenazioni e
demetilazioni (Reilly et al. 2003). Studi successivi si occuperanno di indagare la
potenziale affinità dei metaboliti al recettore degli estrogeni.
Figura 5.5.5: Vie metaboliche a cui va incontro la capsaicina in seguito all’assorbimento (Reilly et al. 2003).

Risultati e discussioni
67
5.5.3 BISDEMETOSSICURCUMINA
La curcumina è il principale pigmento estratto dalla curcuma, comunemente
utilizzata come spezia soprattutto nei paesi asiatici. In studi preclinici condotti su
modelli animali la curcumina ha mostrato un’azione preventiva nei confronti dello
sviluppo di neoplasie intestinali. Comunque, l’attività clinica della curcumina deve
essere ancora confermata (Ireson et al. 2002). Oltre alla curcumina i principali
coloranti estratti dalla curcuma, addizionati agli alimenti sono la
demetossicurcumina e la desmetossicurcumina. Quest’ultima è stata prescelta per
l’analisi di docking, in quanto presenta minore idrofilia e minori ingombri sterici.
Data la dimensione del ligando la simulazione è stata effettuata utilizzando sia il
recettore 3ERD che 1XPC, per essere sicuri di valutare completamente la
potenziale affinità dell’additivo. In entrambe le conformazioni recettoriali,
nonostante la presenza di due ossidrili fenolici, la desmetossicurcumina instaura un
solo legame idrogeno con il Glu353. Il secondo OH non riesce a legarsi all’His 524
perchè la distanza tra le due funzioni fenoliche è maggiore rispetto a quella ideale
Figura 5.5.6: Orientazione assunta dalla bisdemetossicurcumina nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Glu 353
Arg 394
His 524

Risultati e discussioni
68
assunta dall’estradiolo di 11 Å (Vaya et al. 2004). La posa assunta dal ligando nel
complesso 3ERD-desmetossicurcumina selezionato da HINT è rappresentata nella
Figura 5.5.6. Lo score assegnato a tale interazione è di 450 punti e il valore di ∆G°
predetto è -9.10 Kcal/mol. A partire dall’analisi visuale è stato concluso che la
distorsione dei legami è eccessiva e che la struttura della desmetossicurcumina,
generata dal processo di docking, non sembra verosimile.
Nel sito recettoriale di 1XPC l’additivo è meno distorto rispetto alla conformazione
assunta nel recettore 3ERD, perchè la catena alchilica si distende verso l’esterno
promuovendo un’interazione favorevole tra l’Asp 351 e uno dei due ossidrili fenolici
(Figura 5.5.7). Poichè HINT valuta la formazione del complesso con un punteggio
di 2063, la variazione dell’energia libera predetta è di -11.68 Kcal/mol.
La biodisponibilità della curcumina, in seguito ad assunzione per via orale, è
piuttosto limitata, in quanto è scarsamente assorbita nel tratto intestinale ed è
ampiamente metabolizzata dal fegato. I metaboliti principali sono la
tetraidrocurcumina, la esaidrocurcumina e l’esidrocurcuminolo (Figura 5.5.8) e
Figura 5.5.7: Orientazione assunta dalla bisdemetossicurcumina nel sito della proteina 1XPC. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524
Asp 351

Risultati e discussioni
69
sembrano essere fortemente coinvolti nell’azione antineoplastica ipotizzata per la
curcumina. La diidrogenazione dei doppi legami aumenta il numero delle possibili
conformazioni assunte dal ligando perchè genera un aumento della flessibilità della
molecola. Alcuni metaboliti, quindi, saranno analizzati in studi di docking futuri. Il
metabolismo di seconda fase, invece, riguarda soprattutto reazione di
glucuronidazione e solfatazione (Ireson et al. 2002).
5.5.4 PROPIL p-IDROSSIBENZOATO
Il propil p-idrossibenzoato è un composto di origine sintetica, appartenente alla
classe dei parabeni, cioè degli esteri dell’acido 4-idrossibenzoico. Queste sostanze
sono largamente impiegate come conservanti nella fabbricazione di prodotti
alimentari, cosmetici e farmaceutici. In letteratura sono presenti studi sperimentali
che riconoscono nei parabeni ligandi in grado di attivare il recettore degli estrogeni
(Byford et al. 2002; Blair et al. 2000).
Data l’assenza di funzioni ingombranti e le modeste dimensioni del propil p-idrossi
benzoato, l’additivo è stato inserito solo nella conformazione recettoriale 3ERD.
L’orientazione assunta dal ligando nel sito è rappresentata in Figura 5.5.9. La
funzione fenolica, anche in questo caso, come ci si aspetta, forma un legame
idrogeno con Il Glu 353 e l’Arg 394, mentre la catena propilica instaura favorevoli
Figura 5.5.8: Principali metaboliti della curcumina (Ireson et al. 2002).

Risultati e discussioni
70
contatti idrofobici con i residui apolari del sito. L’HINT score assegnato al
complesso Erα-additivo è 1586 e il ∆G° predetto -10.92 Kcal/mol. Il propil p-
idrossibenzoato non occupa interamente la cavità di legame, infatti alcuni studi
valutano l’ipotesi che ai fini dell’attivazione recettoriale siano necessarie due
molecole del ligando in grado di orientare le proprie funzioni ossidriliche
rispettivamente verso il Glu353 e l’His 524. Tali studi dimostrano anche che la
tasca di legame sia in grado di accettare due molecole dell’estere propilico senza
costrizioni steriche (Byford et al. 2002). Siccome una sola molecola di additivo,
probabilmente, non estrude completamente le molecole d’acqua dal sito, è possibile
che HINT, non valutando a pieno tale perdita entropica, sopravvaluti l’interazione
tra recettore e propil benzoato. Tramite il programma GRASP, è stato misurato il
volume dell’additivo, che è pari a 156.45 Å3, mentre il minimo volume conosciuto
per agonisti e SERM è circa 200 Å3. Inoltre, la presenza di analisi sperimentali che
classificano i parabeni come “weak binders” (Blair et al. 2000), rafforza l’ipotesi che
il programma sovrastimi l’affinità della molecola.
Figura 5.5.9: Orientazione assunta del propil benzoato nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524

Risultati e discussioni
71
Come era possibile immaginare il principale metabolita dei parabeni si forma per
idrolisi del gruppo estereo ed è l’acido p-idrossi benzoico, il quale, data la sua
idrofilia, avrà una affinità minore per il recettore rispetto all’estere. Comunque
eventuali studi futuri si occuperanno di stabilire le conseguenze legate alla
biotrasformazione dei parabeni.
5.5.5 ACIDO NORDIIDROGUAIARETICO
L’acido nordiidroguaiaretico (NDGA) è un composto di origine vegetale utilizzato
come conservante nell’industria alimentare per grassi e burro. A tale sostanza sono
state attribuite diverse azioni, come antiossidante, inibitore delle lipoossigenasi,
induttore di nefropatie e, più recentemente, come inibitore dello crescita di cellule
cancerose in vitro (Fujimoto et al. 2004).
Per analizzare il comportamento dell’additivo nei confronti del recettore degli
estrogeni, sono state utilizzate entrambe conformazioni del recettore ERα, 3ERD e
Figura 5.5.10: Orientazione assunta dell’acido nordiidroguaiaretico nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524
Met 343
Thr 347

Risultati e discussioni
72
1XPC. L’acido nordiidroguaiaretico è costituito da una struttura simmetrica in cui
sono presenti quattro ossidrili fenolici. La lunghezza della molecola nella forma
distesa è pari a 14 Å, permettendo al ligando, anche grazie alla presenza di diversi
legami ruotabili, di occupare interamente il sito di legame 3ERD, assumendo una
conformazione meno distorta rispetto, ad esempio, al derivato della curcumina, più
ingombrante e meno flessibile. Due dei gruppi fenolici formano legami ad idrogeno
con gli amminoacidi Glu 353 e Arg 394, mentre gli altri due ossidrili instaurano
legami idrogeno con i residui Thr 347 e Met 343 (Figura 5.5.10). I gruppi metilici
contribuiscono positivamente all’interazione con il recettore poichè generano
contatti idrofobici con gli amminoacidi apolari del sito. Dato lo score assegnato da
HINT pari a 949, la variazione di energia libera predetta per l’interazione tra ERα e
acido nordiidroguaiaretico è -9.90 Kcal/mol. L’orientazione dell’additivo all’interno
del sito di 1XPC è mostrata nella Figura 5.5.11. Analogamente al complesso
precedente, due ossidrili fenolici si legano al glutammato e all’arginina, mentre una
delle restanti funzioni OH riesce a formare un legame ad idrogeno con l’Asp 351. Lo
score assegnato da HINT alla formazione del complesso è più elevato rispetto al
precedente ed è pari a 1795. Il ∆G° predetto è -11.25 Kcal/mol. Alcuni studi recenti,
in effetti, indicano che l’acido diidroguaiaretico possiede attività SERM-like
(Fujimoto et al. 2004).
Figura 5.5.11: Orientazione assunta dell’acido nordiidroguaiaretico nel sito della proteina 1XPC. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524
Asp 351

Risultati e discussioni
73
In letteratura i dati relativi al metabolismo dell’acido nordiidroguaiaretico sono
molto limitati. Studi successivi si occuperanno di indagare approfonditamente tale
aspetto.
5.5.6 EUGENOLO
L’eugenolo è uno dei principali componenti dell’olio di garofano e come tale è molto
utilizzato per esaltare aromi e sapori nell’industria alimentare. Studi farmacologici
hanno dimostrato che questo componente naturale mostra attività
anticonvulsivante e proprietà analgesica locale sfruttata nella pratica dentistica
(Ardjmanda et al. 2006).
Date le caratteristiche strutturali dell’eugenolo le simulazioni di docking sono state
eseguite esclusivamente adottando la conformazione recettoriale dell’agonista
Figura 5.5.12: Orientazione assunta dell’eugenolo nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524

Risultati e discussioni
74
(3ERD). L’eugenolo occupa solo parte della cavità di legame come si osserva nella
Figura 5.5.12. L’ossidrile fenolico, come ci si aspetta, forma legami ad idrogeno con
gli amminoacidi Glu 353 e l’Arg 394. L’His 524 è esclusa dall’interazione con il
ligando, a causa dell’assenza di gruppi funzionali adatti e, soprattutto, a causa delle
piccole dimensioni. Lo score associato alla formazione del complesso tra eugenolo e
recettore degli estrogeni è pari a 1381 e il ∆G° predetto, calcolato a partire dalla
retta di taratura, assume valore -10.59 Kcal/mol. Studi di binding sull’eugenolo
hanno escluso che esso leghi il recettore degli estrogeni (Blair et al. 2000), per cui è
probabile che HINT sopravvaluti tale interazione.
L’eugenolo è rapidamente assorbito e metabolizzato in seguito ad assunzione per via
orale. Nelle urine sono stati individuati nove metaboliti dell’eugenolo, che è escreto
immodificato per meno dello 0.1%. Il metabolismo comprende soprattutto reazioni
di diidrogenzazione, ossidrilazione e ossidazione (Fischer et al. 1990), che saranno
indagate in studi futuri.
5.5.7 o-FENILFENOLO
L’orto-fenilfenolo è utilizzato come fungicida e agente antibatterico nell’industria
agricola e alimentare. Alte esposizioni a tale disinfettante hanno evidenziato lo
sviluppo di carcinomi urinari nei ratti (Murata et al. 1999).
Il fenilfenolo presenta una struttura molto semplice, costituita da due anelli
aromatici e un gruppo ossidrilico. Il miglior conformero selezionato da HINT,
nell’insieme generato da GOLD all’interno del recettore 3ERD, è caratterizzato da
un punteggio di 1707 e il ∆G° predetto è pari a -11.11 Kcal/mol. Dalla Figura
5.5.13 si osserva come, anche in questo caso, l’ossidrile fenolico formi un legame
idrogeno con il Glu 353. Il sostituente fenilico invece è coinvolto in numerose
interazioni idrofobiche-idrofobiche con Leu 384, Thr 347, Ala 350, Leu 540 e
contatti π-π con Trp 383. L’His 524 è troppo lontana per interagire con il
fenilfenolo. La capacità dei composti difenilici di attivare il recettore degli estrogeni
è ben documentata in letteratura e comprende l’analisi di inquinanti, come il
bisfenolo A. In particolare l’o-fenilfenolo è stato classificato come legante debole del
recettore (Blair et al. 2000), supportando l’ipotesi che HINT tenda a sovrastimimare
il legame di piccole molecole costituite da gruppi aromatici.

Risultati e discussioni
75
L’o-fenilfenolo è assorbito in quantità consistente e rapidamente eliminato in
seguito a somministrazione orale nei ratti e dermica nell’uomo. Nelle urine si
ritrovano principalmente coniugati solfati (69%) e glucuronati dell’additivo non
modificato (Timchalk et al. 1998). Inoltre sono stati riportati due metabiliti, il
fenilbenzochinone e il fenilidrochinone, a cui sono attribuite azioni ossidative
carcinogenetiche per formazione di H2O2 (Murata et al. 1999).
5.5.8 CHININA
La chinina è un alcaloide naturale, ricavato dalla corteccia dell’albero di cincona,
utilizzato come farmaco di prima scelta nel trattamento della malaria, specialmente
nei casi gravi. Tale alcaloide costituisce, insieme alla caffeina, un additivo molto
usato nel preparazione di bevande gasate.
La chinina è caratterizzata dalla presenza nella struttura di una anello eterociclico
non aromatico, chiamato chinuclidina, la cui funzione amminica ha pKa pari a circa
Figura 5.5.13: Orientazione assunta dall’orto-fenilfenolo nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524

Risultati e discussioni
76
10. L’azoto è quindi da considerarsi protonato a pH fisiologico. Date le
caratteristiche strutturali della chinina, si sono considerate entrambe le strutture
cristallografiche 3ERD e 1XPC nell’affrontare gli studi di docking. L’orientazione
assunta dalla chinina nel sito di legame della proteina 3ERD è mostrata in Figura
5.5.14. L’ossidrile alcolico non riesce ad instaurare un legame idrogeno con Glu 353
e con His 524, in quanto, i due gruppi ingombranti adiacenti lo limitano
stericamente. Tra recettore e ligando non si sono riscontrati legami significativi,
ma, nonostante questo, lo score attribuito da Hint alla formazione del complesso tra
chinina e Erα è pari a 1130, che equivale a un valore di ∆G° predetto di -10.19
Kcal/mol. Probabilmente ad innalzare lo score sono le numerose interazioni
idrofobiche che stabiliscono sia l’anello chinuclidinico che quello chinolinico.
All’interno della conformazione recettoriale 1XPC il ligando assume una posizione
ribaltata rispetto a quella precedente (Figura 5.5.15), orientando il biciclo in
direzione dell’Asp 351, con il quale, l’ammina protonata non forma un legame a
idrogeno a causa dell’eccessiva distanza. Anche in questo caso, numerosi sono i
Figura 5.5.14: Orientazione assunta dalla chinina nel sito della proteina 3ERD.
Arg 394
Glu 353
His 524

Risultati e discussioni
77
contatti idrofobici tra proteina e chinina. Lo score, pari a 1398, è leggermente, ma
non significativamente, più alto rispetto al precedente e la variazione di energia
libera predetta corrisponde a -10.62 Kcal/mol.
La chinina è un composto ampiamente studiato data la sua applicazione in campo
farmaceutico. Numerosi sono gli effetti tossici legati all’uso dell’alcaloide nella
pratica medica e sono raggruppati sotto il nome di cinconismo, una sindrome
caratterizzata da tinnito, cefalea, nausea, vomito e disturbi visivi. Gli studi della
potenziale attività sul recettore degli estrogeni della chinina e dei suoi metaboliti
sarà continuata in studi successivi. I principali prodotti del metabolismo sono: 3-
idrossichinina, 2’-chinone e 10,11-diidrossidiidrochinone, anche se, per lo più, la
chinina è escreta nella sua forma immodificata (Mirgani et al. 2003).
Figura 5.5.15: Orientazione assunta dalla chinina nel sito della proteina 1XPC.
Arg 394
Glu 353
His 524
Asp 351

Risultati e discussioni
78
5.5.9 ETILVANILLINA
L’Etilvanillina è un prodotto artificiale che presenta caratteristiche aromatiche tre o
quattro volte più intense rispetto alla semplice vanillina e per questo è molto
utilizzata in campo alimentare.
L’additivo è stato inserito solo nella conformazione 3ERD, analogamente a tutti i
ligandi caratterizzati da piccole dimensioni. Nonostante la presenza di un ossidrile
fenolico nella struttura dell’etilvanillina, l’orientazione della molecola non è idonea
alla formazione del legame idrogeno con Glu 353. Questa condizione si verifica in
tutti i complessi generati da GOLD, facendo supporre che la presenza del gruppo
etossilico, in orto alla funzione alcolica, impedisca stericamente l’aggancio. E’
possibile notare, invece, dalla Figura 5.5.16 la formazione di un legame idrogeno tra
l’ossigeno aldeidico dell’etilvanillina e un idrogeno del gruppo guanidinico dell’Arg
394. L’His 524 è esclusa dall’interazione con il ligando, mentre Leu 346 interagisce
tramite contatto idrofobico con il sosituente etilico. L’ HINT score è 977 e il
corrispondente ∆G° di legame è -9.94 Kcal/mol.
Figura 5.5.16: Orientazione assunta dall’etilvanillina nel sito della proteina 3ERD. I legami idrogeno sono mostrati con linee tratteggiate.
Arg 394
Glu 353
His 524

Risultati e discussioni
79
All’interno del nostro organismo, molto probabilmente, una delle principali
modoficazioni a cui va incontro l’etilvanillina è la deetilazione del gruppo
ossidrilico, ricostituendo la vanillina. Studi di farmacocinetica hanno dimostrato che
in 48 ore il 94% di una dose di vanillina viene escreto con le urine sottoforma di:
vanillina non modificata (7%), alcol vanillico (19%), acido vanillico (47%),
vanniloilglicina (10%), catecolo (8%), 4-metilcatecolo (2), guaiacolo (0-5%) e 4-metil
guaiacolo (0-6%) (Strand et al. 1975), che eventuali indagini future si occuperanno
di studiare.
5.5.10 ACIDO COLICO
L’acido colico è un normale componente della bile di tutti i vertebrati e può essere
utilizzato come emulsionante nella fabbricazione di prodotti destinati
all’alimentazione. Gli acidi colico e chenodesossicolico si formano nel fegato in un
rapporto di circa 2:1 e costituiscono l’80% degli acidi biliari. Dopo una completa
coniugazione nell’epatocita, sia con la glicina che con la taurina, gli acidi biliari sono
escreti nell’intestino, dove vanno incontro ad una circolazione enteroepatica da 10 a
12 volte al giorno. Durante ciascun passaggio, una piccola quantità di acidi biliari
raggiunge il colon, dove l’acido colico viene convertito in acido desossicolico,
ampiamente riassorbito.
La struttura molecolare del composto è stata costruita in maniera che il gruppo
carbossilico risulti in forma deprotonata, la condizione più verosimile a pH
fisiologico. L’acido colico presenta una struttura steroidea molto simile
all’estradiolo, ma la molecola è dotata anche di una catena alchilica ingombrante che
non ritroviamo nell’ormone. Per queste ragioni è stato scelto di condurre le analisi
di docking su entrambe le conformazioni recettoriali. Nella confirmazione 3ERD
l’acido colico non ha spazio per collocare la catena laterale, formando numerosissime
interazioni repulsive che fanno crollare lo score assegnato da HINT a valori
inferiori a -4000 punti. Anche nel recettore 1XPC l’acido colico non riesce a trovare
una collocazione idonea ai fini di una buona interazione (Figura 5.5.17). All’interno
del sito di legame il ligando è costretto a posizionare alcuni atomi a distanze
comprese tra 0.9-1.6 Å, inverosimili nella realtà e anche per il programma che
assegna a tale interazione uno score negativo di -1878. Inoltre i sostituenti

Risultati e discussioni
80
ossidrilici non trovano i target giusti per formare dei buoni legami ad idrogeno. Il
∆G° predetto per la formazione del complesso 1XPC-acido colico è -5.38 Kcal/mol.
5.5.11 VALUTAZIONE DEI RISULTATI OTTENUTI
DALL’ANALISI DEGLI ADDITIVI
In Tabella 5.5.2 sono riportati gli score assegnati da HINT e il valore di ∆G°
predetto per ogni interazione additivo-recettore esaminata nel testo. Inoltre, il
Grafico 5.5.1 mostra come, a seconda del valore di HINT score, si dispongono gli
additivi sulla retta di taratura. Per semplicità, laddove i composti siano stati inseriti
in entrambe le conformazioni recettoriali (3ERD e 1XPC), è stato riportato in
grafico solo il complesso con il punteggio più alto, che dovrebbe rappresentare la
condizione energeticamente favorita.
Figura 5.5.17: Orientazione assunta dall’acido colico nel sito della proteina 1XPC.
Arg 394
Glu 353
His 524
Asp 351
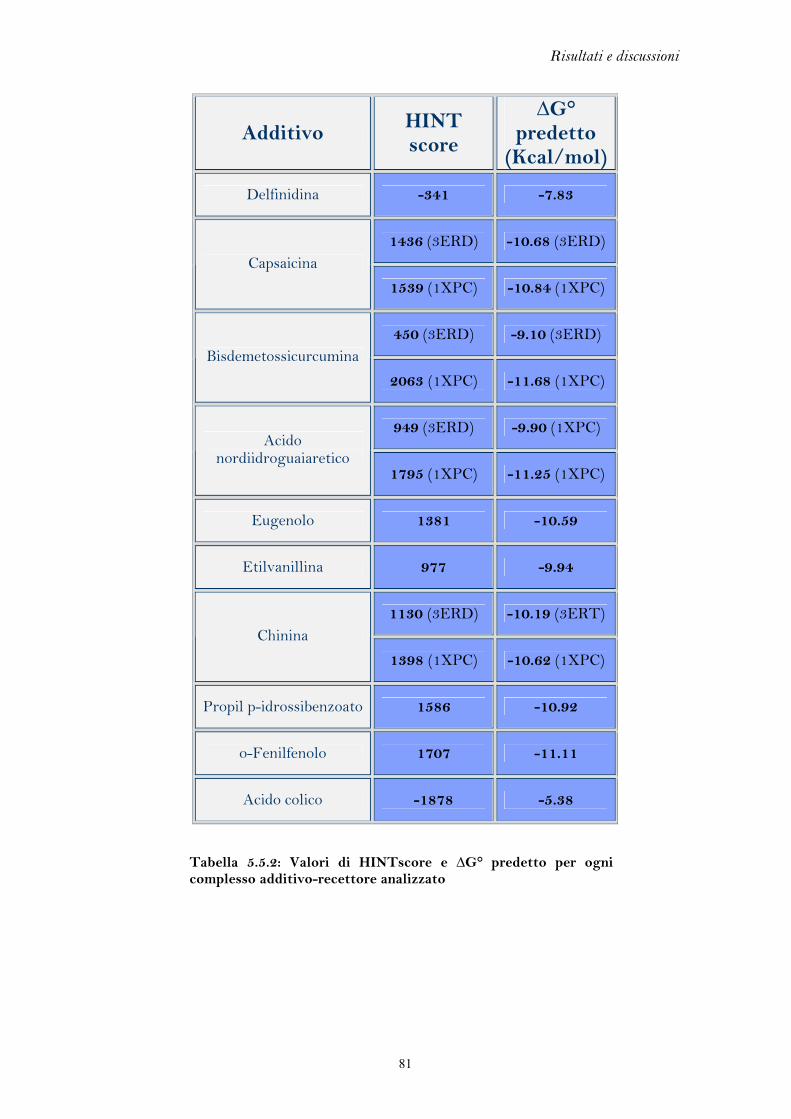
Risultati e discussioni
81
Additivo HINT score
∆G° predetto
(Kcal/mol)
Delfinidina -341 -7.83
1436 (3ERD) -10.68 (3ERD) Capsaicina
1539 (1XPC) -10.84 (1XPC)
450 (3ERD) -9.10 (3ERD) Bisdemetossicurcumina
2063 (1XPC) -11.68 (1XPC)
949 (3ERD) -9.90 (1XPC) Acido
nordiidroguaiaretico 1795 (1XPC) -11.25 (1XPC)
Eugenolo 1381 -10.59
Etilvanillina 977 -9.94
1130 (3ERD) -10.19 (3ERT) Chinina
1398 (1XPC) -10.62 (1XPC)
Propil p-idrossibenzoato 1586 -10.92
o-Fenilfenolo 1707 -11.11
Acido colico -1878 -5.38
Tabella 5.5.2: Valori di HINTscore e ∆G° predetto per ogni complesso additivo-recettore analizzato
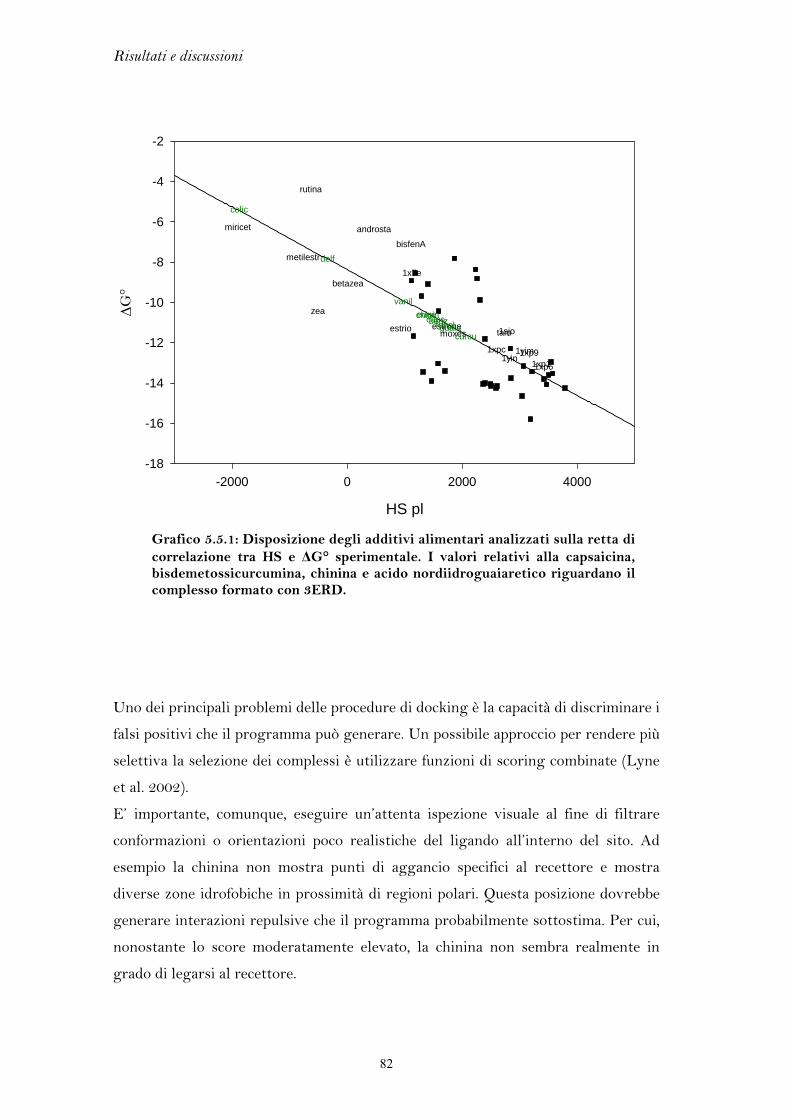
Risultati e discussioni
82
HS pl
-2000 0 2000 4000
∆G°
-18
-16
-14
-12
-10
-8
-6
-4
-2
1sjo
1xp11xp61xp91xpc 1yim
1yin
1x7e
rutina
tamestrio moxes
bisfenAmetilestr
estrone
miricet
zea
betazea
androsta
delf
caps
curcuguaia
eugebenz
chininvanil
fenol
colic
Uno dei principali problemi delle procedure di docking è la capacità di discriminare i
falsi positivi che il programma può generare. Un possibile approccio per rendere più
selettiva la selezione dei complessi è utilizzare funzioni di scoring combinate (Lyne
et al. 2002).
E’ importante, comunque, eseguire un’attenta ispezione visuale al fine di filtrare
conformazioni o orientazioni poco realistiche del ligando all’interno del sito. Ad
esempio la chinina non mostra punti di aggancio specifici al recettore e mostra
diverse zone idrofobiche in prossimità di regioni polari. Questa posizione dovrebbe
generare interazioni repulsive che il programma probabilmente sottostima. Per cui,
nonostante lo score moderatamente elevato, la chinina non sembra realmente in
grado di legarsi al recettore.
Grafico 5.5.1: Disposizione degli additivi alimentari analizzati sulla retta di correlazione tra HS e ∆G° sperimentale. I valori relativi alla capsaicina, bisdemetossicurcumina, chinina e acido nordiidroguaiaretico riguardano il complesso formato con 3ERD.

Risultati e discussioni
83
Le analisi eseguite sugli additivi hanno evidenziato come potenziali ligandi
l’eugenolo, l’etilvanillina., il fenilfenolo e il propil p-idrossibenzoato. Effettivamente
studi di binding hanno dimostrato che il fenilfenolo e il benzoato sono relativamente
affini al recettore (Blair et al. 2000), ma, in generale, si nota una tendenza di HINT
a sovrastimare l’interazione di piccole molecole che presentano centri aromatici
come un benzene. Come riportato in precedenza, il volume minimo presentato da
agenti agonisti e SERM conosciuti è pari a circa 200 Å3, mentre i quattro additivi
menzionati, occupando uno spazio minore, non riempiono completamente la cavità
di legame. Questo non significa che i composti non si legano, ma, quantomeno, che
difficilmente lo score sarà paragonabile a quello di SERM o agonisti.
Infine anche l’analisi farmacoforica può essere utile per spiegare la possibilità di un
ligando a formare complessi stabili. Ad esempio la delfinidina è costituita da cinque
gruppi ossidrilici, la cui eccessiva polarità rende ragione dello score basso, mentre la
bisdemetossicurcumina, la capsaicina e l’acido nordiidroguaiaretico, caratterizzati da
strutture paragonabili a ligandi noti, presentano valori di score significativamente
migliori. Questi ultimi tre additivi rappresentano, in effetti, i composti più favoriti
alla formazione di un’interazione stabile con il recettore degli estrogeni e saranno
proposti per indagini sperimentali.


Conclusioni


Conclusioni
85
L’inserimento della flessibilità proteica nelle analisi computazionali è uno dei
problemi predominanti che caratterizzano lo studio “in silico” delle macromolecole
biologiche. In particolare, il virtual screening necessità di strumenti rapidi e poco
complessi in grado, tuttavia, di non trascurare la flessibilità recettoriale. In questo
lavoro di tesi sono state analizzate le modificazioni strutturali a cui va incontro il
recettore degli estrogeni in seguito al legame con composti strutturalmente
differenti. Attraverso l’analisi di numerose strutture cristallografiche e sulla base
delle loro caratteristiche geometriche si sono distinte due conformazioni recettoriali
fondamentali da utilizzare negli studi di docking per poter considerare la flessibilità:
la conformazione selezionata dagli agonisti e la conformazione selezionata dai
SERM. Inoltre, tramite analisi di cross-docking e l’osservazione dei dati di B-factor
sono state individuate le due strutture cristallografiche che sembrano meglio
rappresentare ognuna delle due categorie conformazionali: 1XPC, come esponente
della conformazione selezionata dai SERM e 3ERD come esponente
conformazionale della struttura selezionta dagli agonisti.
L’approccio utilizzato ha consentito di ottenere una buona correlazione tra le
misure di ∆G° sperimentali e i valori di score assegnati da HINT ai complessi
proteina-ligando generati tramite docking.
In questo lavoro di tesi, inoltre, il metodo sviluppato è stato applicato allo screening
di un piccolo gruppo di additivi alimentari. L’utilizzo di due conformazioni
recettoriali molto diverse ha permesso di individuare potenziali ligandi come la
bisdemetossicurcumina, che mostra valori di score significativamente differenti
all’interno delle due conformazioni del recettore. Uno dei problemi più importanti
riscontrati nel tentativo di messa a punto di una procedura di virtual screening è
discriminare i potenziali ligandi dai falsi positivi che il programma potrebbe
generare. L’ispezione visuale è uno degli strumenti indispensabili per determinare
se l’orientazione e le interazioni del ligando sono realistiche. Inoltre, per valutare
più accuratamente l’interazione di piccole molecole con il sito di legame, sono stati
confrontati i volumi degli additivi con quelli di ligandi noti. Saranno, comunque,
necessari studi futuri per identificare le strategie più adeguate ad affrontare le
analisi post-docking. Un possibile approccio è l’utilizzo di funzioni di scoring
combinate.
Infine, in questo studio, è stata rilevata la necessità di conoscere, per ogni ligando
considerato, gli equilibri acido/base, quelli tautomerici e le reazioni metaboliche a
cui esso è soggetto all’interno dell’organismo. Le informazioni di farmacocinetica,

Conclusioni
86
infatti, permettono di valutare, non solo la struttura più probabile con cui un
composto si presenta a livello recettoriale, ma anche di analizzare analisi di
eventuali tossicità legate alla formazione di metaboliti.

Bibliografia


Bibliografia
87
Alberts I. L., Todorov N. P. and Dean P. M. Receptor flexibility in de novo
ligand design and docking. Journal of Medicinal Chemistry, 48, 6585-6596.
2005.
Anstead G. M., Carlson K. E. and Katzenellebogen J. A. The estradiol
pharmacophore: ligand structure-estrogen receptor binding affinity
relationships and model for the receptor binding site. Steroids, 62, 268-303.
1997.
Ardjmand A., Fathollahi Y., Sayyah M., Kamalinejad M. And Omrani A.
Eugenol depresses synaptic transmission but does not prevent the induction
of long-term potentiation in the CA1 region of rat hippocampal slices.
Phytomedicine, 13, 146-151. 2006.
Arun B., Anthony M. and Dunn B. The search for the ideal SERM. Expert
Opinion Pharmacotherapy 3, 681-691. 2002.
Blair R. M., Fang H., Branham W. S., Hass B. S., Dial S. L., Moland C. L.,
Tong W., Shi L., Perkins R. and Sheehan D. M. The estrogen receptor
relative binding affinities of 188 natural and xenochemical: structural
diversity of ligands. Toxicological Sciences, 54, 138-153. 2000.
Blizzard T. A, Dininno F., Morgan J. D. 2nd, Chen H. Y., Wu J. Y., Kim S.,
Chan W., Birzin E. T., Yang Y. T., Pai L. Y., Fitzgerald P. M., Sharma N.,
Li Y., Zhang Z., Hayes E. C., Dasilva C. A., Tang W., Rohrer S. P., Schaeffer
J. M. and Hammond M. L. Estrogen receptor ligands. Part 9:
Dihydrobenzoxathiin SERAMs with alkyl substituted pyrrolidine side
chains and linkers. Bioorganic and Medicinal Chemistry Letters, 15, 107-113.
2005.
Branca F. Dietary phyto-estrogens and bone health. Proceedings of the
Nutrition Society, 62, 877-887. 2003.
Branham W. S., Dial S. L., Moland C. L. Hass B. S., Blair R. M., Fang H.,
Shi L., Tong W., Perkins R. G. and Sheehan D. M. Phytoestrogens and
Mycoestrogens bind to the rat uterine estrogen receptor. The Journal of
Nutrition, 132, 658-664. 2002.
Brooijmans N. and Kunts I. D. Molecular recognition and docking
algorithms. Annual review of biophysics and biomolecular structure, 32, 335-373.
2003.

Bibliografia
88
Brzozowski A. M., Pike A. C. W., Dauter Z., Hubbard R. E., Bonn T.,
Engstrom O., Öhman L., Greene G. L., Gustafsson J. Å. and Carlquist M.
Molecular basis of agonism and antagonism in the oestrogen receptor.
Nature, 389, 753-758. 1997.
Byford J. R., Shaw L. E., Drew M. G. B., Pope G. S., Sauer M. J. and Darbre
P. D. Oestrogenic activity of parabens in MCF7 human breast cancer cells.
Journal of Steroid Biochemistry and Molecular Biology, 80, 49-60. 2002.
Carlson H. A. Protein flexibility and drug design: how to hit a moving
target. Current Opinion in Chemical Biology, 6, 447-452. 2002.
Committee on Toxicity of Chemicals in Food, Consumer Products and the
Enviroment. Phytoestrogens and Health. Food Standards Agency. 2003.
Cozzini P., Fornabaio M., Marabotti A., Abraham D. J., Kellogg G. E. and
Mozzarelli A. Simple, intuitive calculations of free energy binding for
protein-ligand complexes. 1. Models explicit constrained water. Journal of
Medicinal Chemistry, 45, 2469-2483. 2002.
Degen G. H. and Bolt H. M., Endocrine disruptors: update on
xenoestrogens. International Archives of Occupational and Environmental
Health, 73, 433-441. 2000.
Enmark E. and Gustaffson J. Å. Oestrogen recptors – an overview. Journal
of Internal Medicine 246, 133-138. 1999.
Fang H., Tong W., Shi L. M., Blair R., Perkins R., Branham W., Hass B. S.,
Xie Q., Dial S. L., Moland C. L. and Sheehan D. M. Structure-activity
relationship for a large diverse set of natural, synthetic and environmental
estrogens. Chemical Research in Toxicology, 14, 280-294. 2001.
Ferrari A. M., Wei B. Q., Costantino L. and Schoichet B. K. Soft docking and
multiple receptor conformation in virtual screening. Journal of Medicinal
Chemistry, 47, 5076-5084. 2004.
Fischer I. U., von Unruh G. E. And Dengler H. J. The metabolism of
eugenol in man. Xenobiotica, 20, 209-222. 1990.
Fujimoto N., Kohta R., Kitamura S. and Honda H. Estrogenic activity of an
antioxidant, nordihydroguaiaretic acid (NDGA). Life Sciences, 74, 1417-1425.
2004.

Bibliografia
89
Goodford P. J. A computational procedure for determining energetically
favorable binding sites on biologically important macromolecules. Journal of
Medicinal Chemistry, 28, 849-857. 1985.
Henke B. R. and Heyer D. Recent advances in estrogen receptor modulators.
Current Opinion in Drug Discovery and Development, 8, 437-448. 2005.
Höltje H.-D., Sippl W., Rognan D. and Folkers G. Molecular Modeling.
Basic principles and applications. II edition. WILEY-VCH. Weinheim 2003
Ireson CR, Jones DJ, Orr S, Coughtrie MW, Boocock DJ, Williams ML,
Farmer PB, Steward WP, Gescher AJ. Metabolism of the cancer
chemopreventive agent curcumin in human and rat intestine. Cancer
Epidemiology Biomarkers and Prevention, 11, 105-111. 2002.
Jordan V. C. Antiestrogens and selective estrogen receptor modulators as
multifunctional medicines. 1. Receptor interactions. Journal of Medicinal
Chemistry, 46, 883-908. 2003.
Jordan V. G. Gli estrogeni artificiali. Le Scienze, 364 1998.
Katzung B. G. Farmacologia generale e clinica. V edizione. Piccin. Padova
2001.
Kellogg G. E. and Abraham D. J. Hydrophobicity: is LogPo/w more than the
sum of its parts? Journal of Medicinal Chemistry, 35, 651-661. 2000.
Kellogg G. E., Burnett J. C. and Abraham D. J. Very empirical treatment of
solvation and entropy: a force field derived from LogPo/w. Journal of
Computer-Aided Molecular Design, 15, 381-393. 2001.
Kellogg G. E., Fornabaio M., Spyrakis F., Lodola A., Cozzini P., Mozzarelli
A., Abraham D. J. Getting it right: modeling of pH, solvent and “nearly”
everything else in virtual screening of biological targets. Journal of Molecular
Graphics and Modelling 22, 479-486. 2004.
Kim S., Wu J. Y., Birzin E. T., Frisch K., Chan W., Pai L. Y., Yang Y. T.,
Mosley R. T., Fitzgerald P. M., Sharma N., Dahllund J., Thorsell A. G.,
DiNinno F., Rohrer S. P., Schaeffer J. M. and Hammond M. L. Estrogen
receptor ligands. II. Discovery of benzoxathiins as potent, selective estrogen
receptor alpha modulators. Journal of Medicinal Chemistry, 74, 2171-2175.
2004.

Bibliografia
90
Kontoyianni M., McClellan L. and Sokol G. S. Evaluation of docking
performance: comparative data on docking algorithms. Journal of Medicinal
Chemistry, 47, 558-565. 2004.
Lyne P. D. Structure-based virtual screening: an overview. Drug Discovery
Today, 20, 1047-1055. 2002.
Manas E. S., Unwalla R. J., Xu Z. B., Malamas M. S., Miller C. P., Harris
H. A., Hsiao C., Akopian T., Hum W.T., Malakian K., Wolfrom S., Bapat
A., Bhat R. A., Stahl M. L., Somers W. S., Alvarez J. C. Structure-Based
Design of Estrogen Receptor-Beta Selective Ligands. Journal of American
Chemical Society. 126,15106-15119. 2004.
Mirghani R. A., Hellgren U., Bertiisson I., Gusafsson L. L., Erlesson O.
Metabolism and elimination of quinine in healty volunteers. European
Journal of Clinical Pharmacology, 59, 423-427. 2003.
Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R.
K. and Olson A. J. Automated docking using a lamarckian genetic algorithm
and an empirical binding free energy function. Journal of Computational
Chemistry, 19, 1639-1662. 1998.
Mueller S.O. Xenoestrogens: mechanisms of action and detection methods.
Analytical and Bioanalytical Chemistry, 378, 582-587. 2004.
Murata M., Moriya K., Inoue S. and Kawanishi S. Oxidative damage to
cellular and isolated DNA by metabolites of a fungicide ortho-phenylphenol.
Carcinogenesis, 20, 851-857. 1999.
Ohmichi M., Tasaka K.. and Kurachi H., Murata Y. Molecular mechanism of
action of selective estrogen receptor modulator in target tissues. Endocrine
Journal 52, 161-167. 2005.
Olea N., Pazos P. and Exposito J. Inadvertent exposure to xenoestrogens.
European Journal of Cancer Prevention, 7, s17-s23. 1998.
Osborne C. K., Zhao H. and Fuqua S. A. W. Selective estrogen receptor
modulators: structure, function and clinical use. Journal of Clinical Oncology,
18, 3172-3186. 2000.
Reilly C. A., Ehlhardt W. J., Jackson D. A., Kulanthaivel P., Mutlib A. E.,
Espina R. J., Moody D. E., Crouch D. J. and Yost G. S. Metabolism of
capsaicin by cytochrome P450 produces novel dehydrogenated metabolites

Bibliografia
91
and decreases cytotoxicity to lung and liver cells. Chemical Research in
Toxicology, 16, 336-349. 2003.
Sivanesan D., Rajnarayanan R. V., Doherty J. and Pattabiraman N. In-silico
screening using flexible ligand binding pockets: a molecular dynamics-
based approach. Journal of Computer-Aided Molecular Design, 19, 213-228.
2005.
Strand L. P. and Scheline R. R. The metabolism of vanillin and isovanillin in
the rat. Xenobiotica. 5, 49-63. 1975.
Tan Q, Blizzard T. A., Morgan J. D. 2nd, Birzin E. T., Chan W., Yang Y. T.,
Pai L. Y., Hayes E. C, DaSilva C. A, Warrier S., Yudkovitz J, Wilkinson H.
A, Sharma N., Fitzgerald P. M., Li S., Colwell L., Fisher J. E, Adamski S.,
Reszka A. A., Kimmel D., DiNinno F., Rohrer S. P., Freedman L. P.,
Schaeffer J. M. and Hammond M. L. Estrogen receptor ligands. Part 10:
Chromanes: old scaffolds for new SERAMs. Bioorganic and Medicinal
Chemistry Letters, 15, 1675-1681. 2005.
Timchalk C., Selim S., Sangha G. and Bartels M. J. The pharmacokinetics
and metabolism of 14C/13C-labeled ortho-phenylphenolo formation
following dermal application to human volunteers. Human and Experimental
Toxicology, 17, 411-417. 1998.
Vaja J. and Tamir S. The relation between the chemical structure of
flavonoids and their estrogen-like activities. Current Medicinal Chemistry, 11,
1333-1343. 2004.
Wu X., Cao G. and Prior R. L. Absorption and metabolism of anthocyanins
elderly women after consumption of elderberry or blueberry. The Journal of
Nutrition, 132, 1865-1871. 2002.
Yu S. J., Keenan S. M., Tong W. and Welsh W. J. Influence of the structural
diversity of data set on the statistical quality of Three-Dimensional
Quantitative Structure-Activity Relationship (3D-QSAR) models: predicting
the estrogenic activity of xenoestrogens. Chemical Research in Toxicology, 15,
1229-1234. 2002.
Zhao L. and Brinton R. D. Structure-Based Virtual Screening for plant-
based ERβ –selective ligands as potential preventative therapy against age-
related neurodegenerative diseases. Journal of Medicinal Chemistry 48, 3463-
3466. 2004.


Ringraziamenti


Ringraziamenti
Uno degl’incubi più ricorrenti, durante la scrittura della tesi, è stato di non avere il
tempo necessario per salutare e ringraziare tutte le persone a cui voglio bene. Per
fortuna non è andata così.
Non posso non immaginare quanto meno meravigliosa sarebbe stata la mia esistenza
se non avessi avuto accanto la mia famiglia, a cui dedico oggi e sempre tutto quello
che di buono riuscirò a “combinare” nella vita. Ringrazio la mia mamma Maria, i
cui occhi dolci rappresentano per me il significato della parola CASA e ringrazio il
mio papà Alfonso, per essere l’uomo intelligente e premuroso che è. Inoltre
ringrazio anche mia sorella Assunta che, anche se mi fa disperare, non cambierei
mai con nessun’altra.
Un ringraziamento del tutto speciale spetta alle mie nonne, Maria e Assunta, che
stimo molto per la combattività e la tenacia con cui hanno affrontato tutta la loro
vita. Spero, un giorno, che qualcuno dica di me, che ho saputo affrontare la vita
coraggiosamente come hanno saputo fare loro.
Un sentito grazie al Prof. Mozzarelli, per la dote, rara, di saper comunicare la
bellezza di ciò che si conosce da una cattedra universitaria. Grazie anche per avermi
permesso di conoscere il grande mondo della modellistica molecolare, da cui sono
rimasta profondamente affascinata. Mi ritengo privilegiata per aver avuto la
possibilità di muovere una proteina nelle tre dimensioni.
Ringrazio il Dipartimento di Chimica che mi ha ospitato in questi mesi, in
particolare il Prof. Cozzini, non solo per avermi fornito gli strumenti con cui
lavorare, ma per non avermi mai fatto mancare un consiglio e un aiuto mentre mi
avventuravo nel mondo computazionale.
Un grazie infinito alle tre persone che più da vicino hanno seguito il mio lavoro:
Francesca, la “regina della slide”, bella e intelligente; Claudia, o meglio “Santa
Claudia”, a cui è impossibile non volere bene; e, soprattutto, ringrazio quel
“giovanotto” di Alessio, una persona dalla cultura vasta e l’intelligenza fine, che mi
ha “tolto le castagne dal fuoco” più di una volta, che ha sopportato tutte le mie
paranoie e che mi ha fatto morire dal ridere. Grazie davvero per la gentilezza e la
premura con cui mi hai sempre trattata. Una menzione particolare la merita Alice, la
Silicon dei laureandi, a cui sono molto affezionata e guai a chi dice che urla...

Ringraziamenti
Questi anni di università sono stati caratterizzati soprattutto dalla presenza di
quattro amici insostituibili. La Lis, che adoro e che mi ha insegnato soprattutto due
cose: quanto possano essere incredibilmente ricci i capelli e a “vendere cara la pelle
dell’orso”; la Vale, che sa sempre tutto e che tutte le mattine per cinque anni mi ha
accolto con il sorriso sulle labbra; Luca, che ha sempre un sacco di aneddoti
“interessanti” e che ha reso le lezioni un piacere invece che un dovere; Francè, il
cui giro losco e mafioso di appunti “pirata” ha decisamente contribuito a non
allungare ulteriormente i tempi della mia laurea.
E adesso è il momento di ringraziare gli amici di sempre, che, quando si ritrovano
una tesi in mano, si divertono a commentare la copertina, guardare le immagini,
leggere i ringraziamenti e, aprendo una pagina a caso, trovarti un errore di
grammatica. E allora questi ringraziamenti sono tutti per voi!
Grazie a Matteo, il compagno di ogni avventura, la cui presenza, per me, fa la
differenza in ogni cosa. Grazie alla Madi, per l’amicizia duratura e indissolubile che
ci lega e per le tante premure che ha per me.
E poi voglio ringraziare tutti gli amici del sabato sera, ma non solo: Bafo, Alessia,
Stepo, Vascio, Peddy, Alle, Dalla, Vensan (il bello e maledettissimo), Mezza, Gavaz,
Paolo, Prince, Ferra, Erika, Kiappo, Marino, Deno, Silvia e Sara. Una menzione
particolare spetta al Melviu, che devo ringraziare sia perchè sta per rendere la Madi
la donna più felice del mondo, sia per tutto il supporto tecnico e informatico che, con
infinita gentilezza, mi ha sempre dato.
Grazie alla FUCI, Don Marco e Clara per aver arricchito la mia esperienza
universitaria offrendomi uno sguardo sul mondo più ampio di quello tecnico-
scientifico.
Voglio ringraziare i miei nuovi amici di chimica, con cui ho pranzato negl’ultimi sei
mesi: Martina, Botta, Tonni, Losia (che è senza dubbio matta, ma simpaticissima),
Andrea. Grazie per la compagnia e per avermi iniziato allo yogurt Muller.
Grazie a mia cugina Barbara, per essere stata la “pranoterapeuta” dei miei libri.
Vorrei anche ringraziare sentitamente Anna e Rudy Melegari, per avermi ospitato a
pranzo e cena ogni volta (spesso) in cui si presentava l’occasione.
Infine, il ringraziamento a cui tengo in assoluto di più spetta ad Andrea, Mello per
tutti e anche per me. Con lui, che non mi ha MAI fatto sentire seconda a nessuno,
voglio condividere la gioia, la soddisfazione e il merito di questo traguardo. Pur non

Ringraziamenti
essendone cosciente, tu conosci il significato di dedizione e cura più di chiunque
altro io conosca. Grazie per avermi cercata tanto tempo fa e avermi amata da
allora. Sei la mia gioia e il mio tesoro.