
Tiroide - docvadis - sito creato dai medici per i pazienti è interessato da un carcinoma della...
29
TIROIDE SEZIONE 2
Transcript of Tiroide - docvadis - sito creato dai medici per i pazienti è interessato da un carcinoma della...

Tiroide
Sezione 2

Tavola 2.1 Apparato endocrino
36 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
AnAtomiA dellA ghiAndolA tiroideA e delle ghiAndole pArAtiroidi
Situata tra la laringe e la trachea medialmente e la guaina carotica e i muscoli sternomastoidei lateralmente, la ghiandola tiroidea pesa circa 15-25 g. I lobi tiroidei hanno una lunghezza di 3-4 cm e una larghezza di 1,5-2 cm; l’istmo è lungo 1,2-2 cm e largo 2 cm e attraversa la trachea tra gli anelli I e II. Nella Tavola 2.1 (disegno in alto), la cute, il tessuto adiposo sottocutaneo e il muscolo platisma sono stati asportati, lasciando esposta, sulla metà destra del collo, la fascia cervicale anteriore o superficiale, che avvolge le vene giugulari esterna e anteriore, e i nervi cervicali trasversi. Il tessuto adiposo sottocutaneo e il muscolo platisma sono riccamente vasco-larizzati: ciò consente di ottenere ampie esposizioni chirurgiche senza sacrificare la cute, sollevando lembi di cute, di tessuto adiposo sottocutaneo e di platisma. Le vene e i nervi così esposti vengono inizialmente lasciati in situ per essere rimossi successivamente insieme ai muscoli sottostanti.
Sul lato sinistro del collo (Tavola 2.1, disegno in alto) sono stati asportati la fascia cervicale superficiale, la vena giugulare esterna, i nervi trasversi e il muscolo sternocleidomastoideo, mostrando il muscolo omoioideo, l’ansa del nervo ipoglosso, l’importante inser-zione limitativa del muscolo pretracheale interno più corto, il mu-scolo sternotiroideo e l’intero decorso del muscolo tiroioideo lungo. La stessa fascia è stata incisa lungo la linea mediana, mettendo in evidenza i margini mediali del muscolo sternoioideo. Questi muscoli, che normalmente si congiungono sulla linea mediana, sono stati parzialmente dislocati per mostrare la cartilagine tiroidea e cricoidea, l’istmo della tiroide e, al di sotto, la parte superiore della trachea.
Le vene giugulari anteriori insieme alla vena giugulare esterna drenano il sangue della faringe e della parte superiore del collo. Inoltre esse ricevono vasi collaterali per tutta la loro lunghezza: in primo luogo dal platisma, disposto superficialmente rispetto alle vene giugulari anteriori; poi dai muscoli pretracheali (sternoioideo, sternotiroideo e omoioideo), che si trovano in profondità rispetto a esse; e infine a livello della laringe, in particolare nell’incisura, vicino alla linea mediana, da numerosi piccoli vasi che provengono dalla parte superiore della laringe. Nell’esporre la ghiandola tiroidea, le paratiroidi e la trachea, è importante preservare il maggior numero possibile di questi vasi spostandoli, piuttosto che sezionandoli, per evitare un edema residuo della parte superiore del collo e della laringe. Qualora tumori della tiroide o delle strutture del collo esercitino una pressione su una delle vene giugulari interne, le vene giugulari anteriori possono andare incontro a dilatazione.
I nervi cervicali trasversi sensoriali se recisi sono in grado di rigenerarsi. Ciò non avviene se vengono recisi i due rami inferiori del nervo faciale: la sezione del ramo mandibolare marginale è seguita dall’abbassamento del labbro inferiore del lato omolaterale la lesione. L’ansa del nervo ipoglosso, che si trova lungo la faccia antero-mediale della guaina carotica, deve essere preservata, per-ché la lesione di questo nervo causa problemi di deglutizione nel postoperatorio. Nell’esporre il nervo è utile ricordare che, di fronte a esso, scende un piccolo ramo dell’arteria tiroidea superiore che invia diramazioni sia al margine posteriore del muscolo, sia al nervo stesso.
I vasi linfatici della fascia superficiale, anteriori ai muscoli preti-roidei, non sono prominenti. I linfonodi sono rari; il primo linfonodo che si incontra si trova subito di fronte all’istmo della tiroide sulla linea mediana tra i muscoli pretracheali, in profondità rispetto alla fascia anteriore e in superficie rispetto alla seconda fascia cervicale, o fascia cervicale media, detta anche falsa capsula della tiroide. Questo linfonodo drena la faringe o la laringe, ma non la ghiandola tiroidea e i tessuti profondi sottostanti. Pertanto esso risulta in-grossato nei pazienti affetti da faringite e laringite acute, ma non in quelli che soffrono di tiroidite o tracheite.
L’esposizione della ghiandola tiroidea, delle ghiandole paratiroidi e del timo si ottiene facendo arretrare i muscoli pretracheali o pretiroidei. L’esposizione massima si ottiene tagliando i muscoli trasversalmente e spostando le estremità verso l’alto e verso il basso. Una buona veduta del polo superiore della tiroide spesso comporta la sezione trasversale del muscolo più interno.
La posizione dell’esofago, visibile leggermente a destra della linea mediana (Tavola 2.1, disegno in basso), è adiacente al lobo destro della tiroide, di solito più grande del controlaterale.
La Tavola 2.2 (disegno in alto) ritrae gli organi del collo e il mediastino antero-superiore dopo la rimozione dei muscoli ante-
riori del collo e delle ossa della parte superiore del torace. Quando la ghiandola tiroidea, le ghiandole paratiroidi e il timo sono esposti, le loro superfici anteriore, laterale e posteriore sono avvolte da una fascia non ben definita di tessuto areolare lasso (anche chiamata falsa capsula della tiroide), che consente alle ghiandole, alla laringe e alla trachea di sollevarsi e abbassarsi durante la deglutizione. Infatti, quando al paziente viene chiesto di deglutire, è possibile palpare quasi tutta la superficie anteriore di entrambi i lobi tiroidei.
La ghiandola tiroidea normale è quasi sempre asimmetrica, con il lobo destro che può essere anche il doppio del sinistro. Il polo
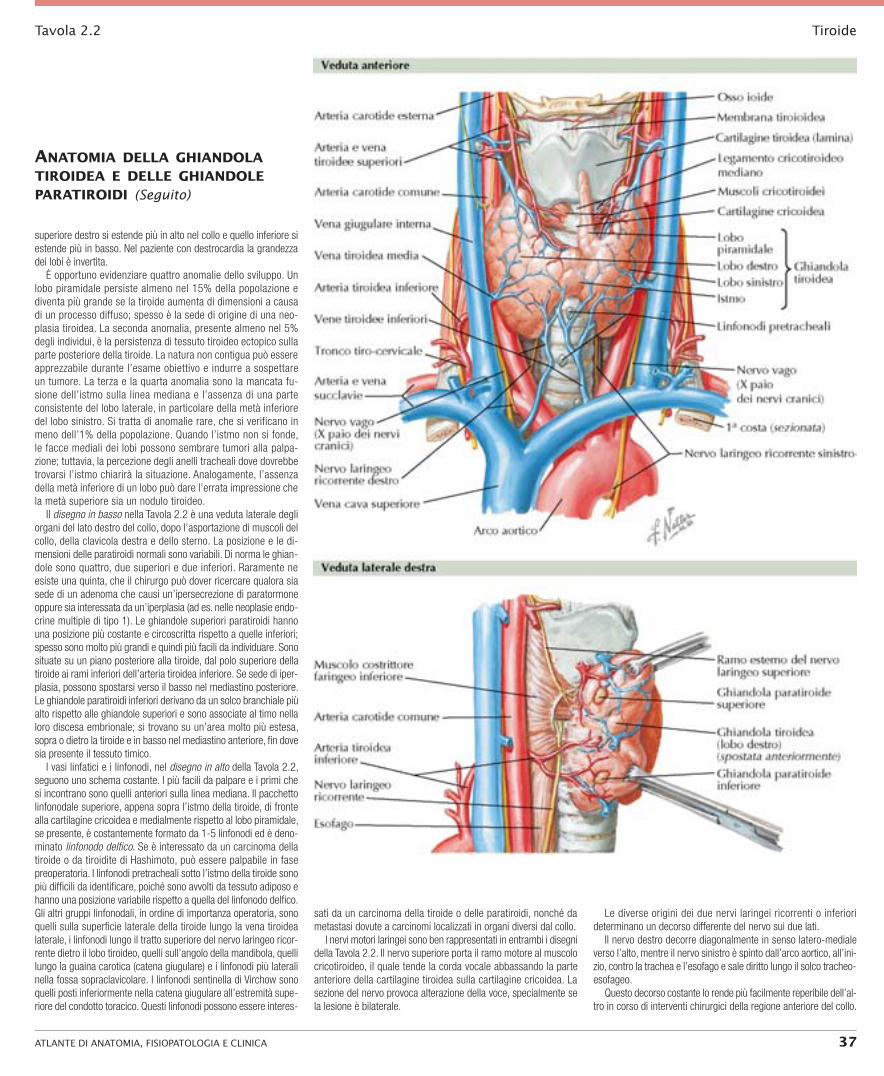
Tavola 2.2 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 37
superiore destro si estende più in alto nel collo e quello inferiore si estende più in basso. Nel paziente con destrocardia la grandezza dei lobi è invertita.
È opportuno evidenziare quattro anomalie dello sviluppo. Un lobo piramidale persiste almeno nel 15% della popolazione e diventa più grande se la tiroide aumenta di dimensioni a causa di un processo diffuso; spesso è la sede di origine di una neo-plasia tiroidea. La seconda anomalia, presente almeno nel 5% degli individui, è la persistenza di tessuto tiroideo ectopico sulla parte posteriore della tiroide. La natura non contigua può essere apprezzabile durante l’esame obiettivo e indurre a sospettare un tumore. La terza e la quarta anomalia sono la mancata fu-sione dell’istmo sulla linea mediana e l’assenza di una parte consistente del lobo laterale, in particolare della metà inferiore del lobo sinistro. Si tratta di anomalie rare, che si verificano in meno dell’1% della popolazione. Quando l’istmo non si fonde, le facce mediali dei lobi possono sembrare tumori alla palpa-zione; tuttavia, la percezione degli anelli tracheali dove dovrebbe trovarsi l’istmo chiarirà la situazione. Analogamente, l’assenza della metà inferiore di un lobo può dare l’errata impressione che la metà superiore sia un nodulo tiroideo.
Il disegno in basso nella Tavola 2.2 è una veduta laterale degli organi del lato destro del collo, dopo l’asportazione di muscoli del collo, della clavicola destra e dello sterno. La posizione e le di-mensioni delle paratiroidi normali sono variabili. Di norma le ghian-dole sono quattro, due superiori e due inferiori. Raramente ne esiste una quinta, che il chirurgo può dover ricercare qualora sia sede di un adenoma che causi un’ipersecrezione di paratormone oppure sia interessata da un’iperplasia (ad es. nelle neoplasie endo-crine multiple di tipo 1). Le ghiandole superiori paratiroidi hanno una posizione più costante e circoscritta rispetto a quelle inferiori; spesso sono molto più grandi e quindi più facili da individuare. Sono situate su un piano posteriore alla tiroide, dal polo superiore della tiroide ai rami inferiori dell’arteria tiroidea inferiore. Se sede di iper-plasia, possono spostarsi verso il basso nel mediastino posteriore. Le ghiandole paratiroidi inferiori derivano da un solco branchiale più alto rispetto alle ghiandole superiori e sono associate al timo nella loro discesa embrionale; si trovano su un’area molto più estesa, sopra o dietro la tiroide e in basso nel mediastino anteriore, fin dove sia presente il tessuto timico.
I vasi linfatici e i linfonodi, nel disegno in alto della Tavola 2.2, seguono uno schema costante. I più facili da palpare e i primi che si incontrano sono quelli anteriori sulla linea mediana. Il pacchetto linfonodale superiore, appena sopra l’istmo della tiroide, di fronte alla cartilagine cricoidea e medialmente rispetto al lobo piramidale, se presente, è costantemente formato da 1-5 linfonodi ed è deno-minato linfonodo delfico. Se è interessato da un carcinoma della tiroide o da tiroidite di Hashimoto, può essere palpabile in fase preoperatoria. I linfonodi pretracheali sotto l’istmo della tiroide sono più difficili da identificare, poiché sono avvolti da tessuto adiposo e hanno una posizione variabile rispetto a quella del linfonodo delfico. Gli altri gruppi linfonodali, in ordine di importanza operatoria, sono quelli sulla superficie laterale della tiroide lungo la vena tiroidea laterale, i linfonodi lungo il tratto superiore del nervo laringeo ricor-rente dietro il lobo tiroideo, quelli sull’angolo della mandibola, quelli lungo la guaina carotica (catena giugulare) e i linfonodi più laterali nella fossa sopraclavicolare. I linfonodi sentinella di Virchow sono quelli posti inferiormente nella catena giugulare all’estremità supe-riore del condotto toracico. Questi linfonodi possono essere interes-
sati da un carcinoma della tiroide o delle paratiroidi, nonché da metastasi dovute a carcinomi localizzati in organi diversi dal collo.
I nervi motori laringei sono ben rappresentati in entrambi i disegni della Tavola 2.2. Il nervo superiore porta il ramo motore al muscolo cricotiroideo, il quale tende la corda vocale abbassando la parte anteriore della cartilagine tiroidea sulla cartilagine cricoidea. La sezione del nervo provoca alterazione della voce, specialmente se la lesione è bilaterale.
Le diverse origini dei due nervi laringei ricorrenti o inferiori determinano un decorso differente del nervo sui due lati.
Il nervo destro decorre diagonalmente in senso latero-mediale verso l’alto, mentre il nervo sinistro è spinto dall’arco aortico, all’ini-zio, contro la trachea e l’esofago e sale diritto lungo il solco tracheo-esofageo.
Questo decorso costante lo rende più facilmente reperibile dell’al-tro in corso di interventi chirurgici della regione anteriore del collo.
AnAtomiA dellA ghiAndolA tiroideA e delle ghiAndole pArAtiroidi (Seguito)
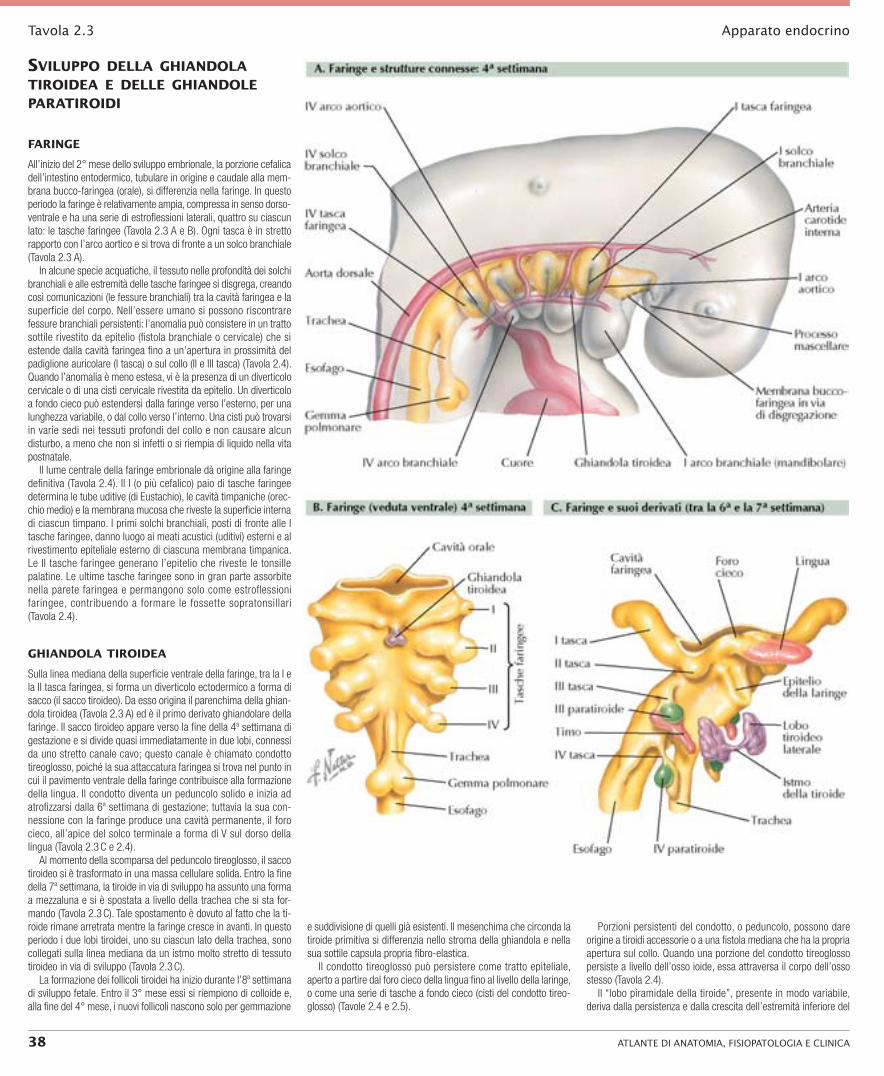
Tavola 2.3 Apparato endocrino
38 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
Sviluppo dellA ghiAndolA tiroideA e delle ghiAndole pArAtiroidi
Faringe
All’inizio del 2° mese dello sviluppo embrionale, la porzione cefalica dell’intestino entodermico, tubulare in origine e caudale alla mem-brana bucco-faringea (orale), si differenzia nella faringe. In questo periodo la faringe è relativamente ampia, compressa in senso dorso-ventrale e ha una serie di estroflessioni laterali, quattro su ciascun lato: le tasche faringee (Tavola 2.3 A e B). Ogni tasca è in stretto rapporto con l’arco aortico e si trova di fronte a un solco branchiale (Tavola 2.3 A).
In alcune specie acquatiche, il tessuto nelle profondità dei solchi branchiali e alle estremità delle tasche faringee si disgrega, creando così comunicazioni (le fessure branchiali) tra la cavità faringea e la superficie del corpo. Nell’essere umano si possono riscontrare fessure branchiali persistenti: l’anomalia può consistere in un tratto sottile rivestito da epitelio (fistola branchiale o cervicale) che si estende dalla cavità faringea fino a un’apertura in prossimità del padiglione auricolare (I tasca) o sul collo (II e III tasca) (Tavola 2.4). Quando l’anomalia è meno estesa, vi è la presenza di un diverticolo cervicale o di una cisti cervicale rivestita da epitelio. Un diverticolo a fondo cieco può estendersi dalla faringe verso l’esterno, per una lunghezza variabile, o dal collo verso l’interno. Una cisti può trovarsi in varie sedi nei tessuti profondi del collo e non causare alcun disturbo, a meno che non si infetti o si riempia di liquido nella vita postnatale.
Il lume centrale della faringe embrionale dà origine alla faringe definitiva (Tavola 2.4). Il I (o più cefalico) paio di tasche faringee determina le tube uditive (di Eustachio), le cavità timpaniche (orec-chio medio) e la membrana mucosa che riveste la superficie interna di ciascun timpano. I primi solchi branchiali, posti di fronte alle I tasche faringee, danno luogo ai meati acustici (uditivi) esterni e al rivestimento epiteliale esterno di ciascuna membrana timpanica. Le II tasche faringee generano l’epitelio che riveste le tonsille palatine. Le ultime tasche faringee sono in gran parte assorbite nella parete faringea e permangono solo come estroflessioni faringee, contribuendo a formare le fossette sopratonsillari (Tavola 2.4).
ghiandola tiroidea
Sulla linea mediana della superficie ventrale della faringe, tra la I e la II tasca faringea, si forma un diverticolo ectodermico a forma di sacco (il sacco tiroideo). Da esso origina il parenchima della ghian-dola tiroidea (Tavola 2.3 A) ed è il primo derivato ghiandolare della faringe. Il sacco tiroideo appare verso la fine della 4a settimana di gestazione e si divide quasi immediatamente in due lobi, connessi da uno stretto canale cavo; questo canale è chiamato condotto tireoglosso, poiché la sua attaccatura faringea si trova nel punto in cui il pavimento ventrale della faringe contribuisce alla formazione della lingua. Il condotto diventa un peduncolo solido e inizia ad atrofizzarsi dalla 6a settimana di gestazione; tuttavia la sua con-nessione con la faringe produce una cavità permanente, il foro cieco, all’apice del solco terminale a forma di V sul dorso della lingua (Tavola 2.3 C e 2.4).
Al momento della scomparsa del peduncolo tireoglosso, il sacco tiroideo si è trasformato in una massa cellulare solida. Entro la fine della 7a settimana, la tiroide in via di sviluppo ha assunto una forma a mezzaluna e si è spostata a livello della trachea che si sta for-mando (Tavola 2.3 C). Tale spostamento è dovuto al fatto che la ti-roide rimane arretrata mentre la faringe cresce in avanti. In questo periodo i due lobi tiroidei, uno su ciascun lato della trachea, sono collegati sulla linea mediana da un istmo molto stretto di tessuto tiroideo in via di sviluppo (Tavola 2.3 C).
La formazione dei follicoli tiroidei ha inizio durante l’8a settimana di sviluppo fetale. Entro il 3° mese essi si riempiono di colloide e, alla fine del 4° mese, i nuovi follicoli nascono solo per gemmazione
e suddivisione di quelli già esistenti. Il mesenchima che circonda la tiroide primitiva si differenzia nello stroma della ghiandola e nella sua sottile capsula propria fibro-elastica.
Il condotto tireoglosso può persistere come tratto epiteliale, aperto a partire dal foro cieco della lingua fino al livello della laringe, o come una serie di tasche a fondo cieco (cisti del condotto tireo-glosso) (Tavole 2.4 e 2.5).
Porzioni persistenti del condotto, o peduncolo, possono dare origine a tiroidi accessorie o a una fistola mediana che ha la propria apertura sul collo. Quando una porzione del condotto tireoglosso persiste a livello dell’osso ioide, essa attraversa il corpo dell’osso stesso (Tavola 2.4).
Il “lobo piramidale della tiroide”, presente in modo variabile, deriva dalla persistenza e dalla crescita dell’estremità inferiore del

Tavola 2.4 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 39
peduncolo. Un legamento o una striscia di muscolo, solitamente localizzato a sinistra della linea mediana, può collegare il lobo pira-midale alla cartilagine tiroidea o all’osso ioide. Il lobo piramidale gradualmente va incontro ad atrofia, pertanto si riscontra più spesso nei bambini che negli adulti.
La ghiandola tiroidea può presentare altre anomalie. Ad esempio, l’istmo può essere voluminoso, rudimentale o assente. I lobi laterali possono avere dimensioni differenti, o possono essere entrambi assenti, con la sola presenza della porzione dell’istmo. La forma della ghiandola può essere più simile alla lettera “H” che a un “ferro di cavallo”. Raramente essa può trovarsi alla base della lingua (tiroide linguale) o in profondità vicino allo sterno. L’assenza com-pleta della ghiandola o un suo mancato funzionamento raramente si notano prima che siano trascorse alcune settimane dalla nascita, poiché i feti ricevono, attraverso la placenta, quantità sufficienti di ormone tiroideo materno che consentono uno sviluppo normale. Se dopo la nascita non viene instaurata una terapia ormonale sostitu-tiva, la conseguenza è l’ipotiroidismo congenito.
ghiandole paratiroidi e timo
Durante la 5a e la 6a settimana di sviluppo, l’epitelio ectodermico delle porzioni dorsali delle estremità distali della III e della IV tasca faringea si differenzia negli abbozzi delle ghiandole paratiroidi. Contemporaneamente le porzioni ventrali delle estremità distali delle III tasche differenziano negli abbozzi del timo (Tavola 2.3 C). Le porzioni ventrali delle estremità distali delle IV tasche possono dare luogo ad abbozzi timici, che andranno in atrofia senza concorrere alla formazione del timo definitivo.
Normalmente si formano due paia di ghiandole paratiroidi; alla fine della 6a settimana di gestazione, gli abbozzi delle paratiroidi e del timo perdono il loro collegamento con le tasche faringee. In questo periodo il lume della III e della IV tasca si chiude. Il tessuto paratiroideo derivante dalla III tasca e gli abbozzi timici migrano, durante la 7a settimana, in direzione infero-mediale. Nell’8a setti-mana le estremità inferiori degli abbozzi del timo aumentano di volume e si uniscono superficialmente lungo la linea mediana. Questa estremità inferiore bilobata continua ad abbassarsi e si posiziona nel mediastino superiore, posteriormente al manubrio. Nel corso di questa discesa, le estremità superiori degli abbozzi del timo formano estensioni simili a code, che di solito scompaiono; talvolta esse persistono come frammenti inglobati nella ghiandola tiroidea o come nidi o cordoni timici isolati.
Il tessuto paratiroideo proveniente dalla III tasca migra insieme agli abbozzi del timo e generalmente si ferma a livello caudale della ghiandola tiroidea per formare le ghiandole paratiroidi inferiori dell’adulto. Queste ghiandole, ciascuna con una capsula propria, sono situate all’interno della guaina tiroidea derivata dalla fascia cervicale, attaccate alla parte dorsale della capsula propria di cia-scun lobo tiroideo. A volte il tessuto paratiroideo scende, insieme agli abbozzi timici, a un livello inferiore e si localizza nel torace, vicino al timo.
Le paratiroidi derivanti dalla IV tasca faringea non cambiano posizione in modo rilevante, quindi le paratiroidi provenienti dalla III tasca le oltrepassano nella loro migrazione verso il basso. Quindi, le paratiroidi che originano dalla IV tasca diventano nell’adulto le
ghiandole paratiroidi superiori, situate nella guaina peritiroidea e connesse alla parte dorsale della capsula propria di ciascun lobo tiroideo a livello del margine inferiore della cartilagine cricoidea. Comuni sono le variazioni di numero, dimensione e posizione delle paratiroidi. Le paratiroidi regolari o accessorie possono trovarsi anche a distanza dalla tiroide. Le paratiroidi producono il parator-mone, che regola l’omeostasi del calcio e del fosforo.
Il timo è un organo di dimensioni notevoli nei bambini: all’età di circa 2 anni raggiunge le sue massime dimensioni continuando a crescere fino alla pubertà. Dopo questo periodo subisce una gra-duale involuzione in cui il tessuto timico è sostituito da tessuto adiposo. Quindi nell’adulto il timo ha più o meno le stesse dimensioni e la stessa forma che ha nei primi anni di vita, ma è costituito principalmente da tessuto adiposo.
Sviluppo dellA ghiAndolA tiroideA e delle ghiAndole pArAtiroidi (Seguito)
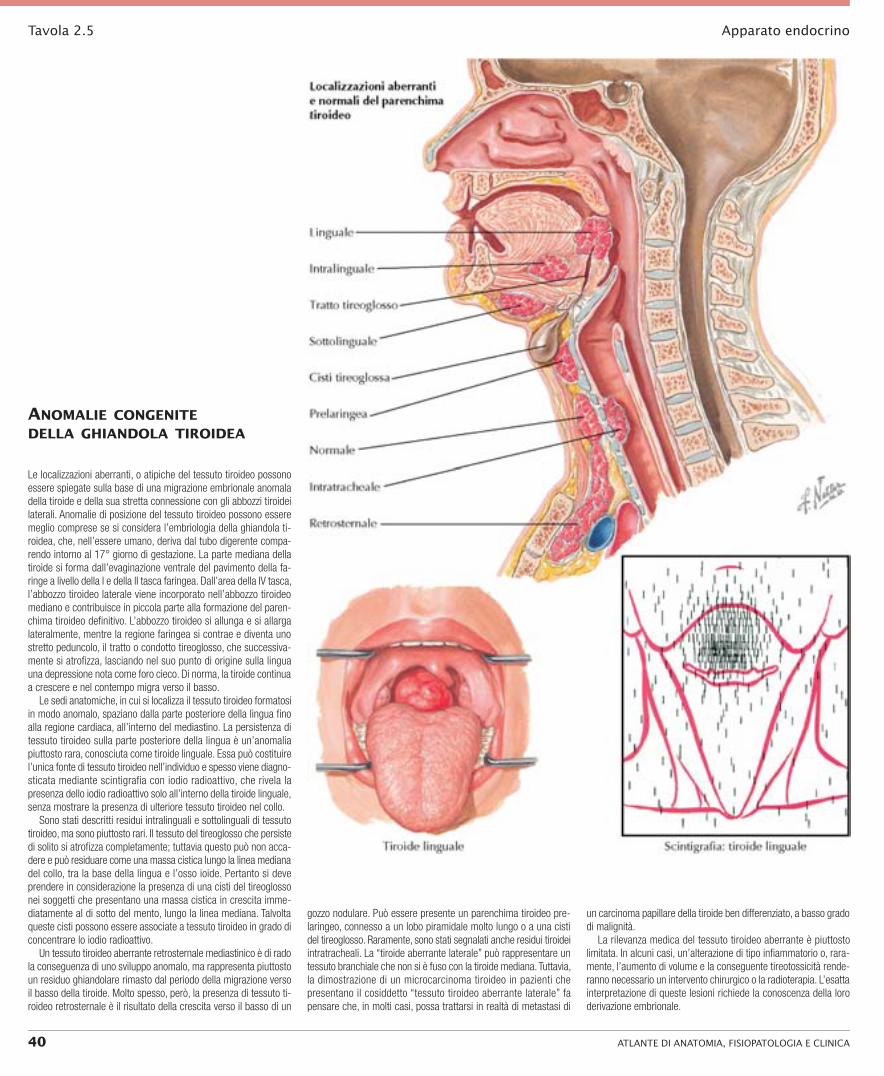
Tavola 2.5 Apparato endocrino
40 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
AnomAlie congenite dellA ghiAndolA tiroideA
Le localizzazioni aberranti, o atipiche del tessuto tiroideo possono essere spiegate sulla base di una migrazione embrionale anomala della tiroide e della sua stretta connessione con gli abbozzi tiroidei laterali. Anomalie di posizione del tessuto tiroideo possono essere meglio comprese se si considera l’embriologia della ghiandola ti-roidea, che, nell’essere umano, deriva dal tubo digerente compa-rendo intorno al 17° giorno di gestazione. La parte mediana della tiroide si forma dall’evaginazione ventrale del pavimento della fa-ringe a livello della I e della II tasca faringea. Dall’area della IV tasca, l’abbozzo tiroideo laterale viene incorporato nell’abbozzo tiroideo mediano e contribuisce in piccola parte alla formazione del paren-chima tiroideo definitivo. L’abbozzo tiroideo si allunga e si allarga lateralmente, mentre la regione faringea si contrae e diventa uno stretto peduncolo, il tratto o condotto tireoglosso, che successiva-mente si atrofizza, lasciando nel suo punto di origine sulla lingua una depressione nota come foro cieco. Di norma, la tiroide continua a crescere e nel contempo migra verso il basso.
Le sedi anatomiche, in cui si localizza il tessuto tiroideo formatosi in modo anomalo, spaziano dalla parte posteriore della lingua fino alla regione cardiaca, all’interno del mediastino. La persistenza di tessuto tiroideo sulla parte posteriore della lingua è un’anomalia piuttosto rara, conosciuta come tiroide linguale. Essa può costituire l’unica fonte di tessuto tiroideo nell’individuo e spesso viene diagno-sticata mediante scintigrafia con iodio radioattivo, che rivela la presenza dello iodio radioattivo solo all’interno della tiroide linguale, senza mostrare la presenza di ulteriore tessuto tiroideo nel collo.
Sono stati descritti residui intralinguali e sottolinguali di tessuto tiroideo, ma sono piuttosto rari. Il tessuto del tireoglosso che persiste di solito si atrofizza completamente; tuttavia questo può non acca-dere e può residuare come una massa cistica lungo la linea mediana del collo, tra la base della lingua e l’osso ioide. Pertanto si deve prendere in considerazione la presenza di una cisti del tireoglosso nei soggetti che presentano una massa cistica in crescita imme-diatamente al di sotto del mento, lungo la linea mediana. Talvolta queste cisti possono essere associate a tessuto tiroideo in grado di concentrare lo iodio radioattivo.
Un tessuto tiroideo aberrante retrosternale mediastinico è di rado la conseguenza di uno sviluppo anomalo, ma rappresenta piuttosto un residuo ghiandolare rimasto dal periodo della migrazione verso il basso della tiroide. Molto spesso, però, la presenza di tessuto ti-roideo retrosternale è il risultato della crescita verso il basso di un
gozzo nodulare. Può essere presente un parenchima tiroideo pre-laringeo, connesso a un lobo piramidale molto lungo o a una cisti del tireoglosso. Raramente, sono stati segnalati anche residui tiroidei intratracheali. La “tiroide aberrante laterale” può rappresentare un tessuto branchiale che non si è fuso con la tiroide mediana. Tuttavia, la dimostrazione di un microcarcinoma tiroideo in pazienti che presentano il cosiddetto “tessuto tiroideo aberrante laterale” fa pensare che, in molti casi, possa trattarsi in realtà di metastasi di
un carcinoma papillare della tiroide ben differenziato, a basso grado di malignità.
La rilevanza medica del tessuto tiroideo aberrante è piuttosto limitata. In alcuni casi, un’alterazione di tipo infiammatorio o, rara-mente, l’aumento di volume e la conseguente tireotossicità rende-ranno necessario un intervento chirurgico o la radioterapia. L’esatta interpretazione di queste lesioni richiede la conoscenza della loro derivazione embrionale.

Tavola 2.6 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 41
effetti dell’ormone tireotropo SullA ghiAndolA tiroideA
L’unità ipotalamo-ipofisi svolge un ruolo indispensabile nella rego-lazione della funzione tiroidea. Una disfunzione ipotalamica o un’insufficienza adenoipofisaria causano la diminuzione del volume tiroideo e della produzione e secrezione degli ormoni tiroidei. L’or-mone ipofisario avente come bersaglio la ghiandola tiroidea è l’ormone tireotropo (TSH), una glicoproteina secreta dalle cellule tireotrope ipofisarie. Il TSH è il regolatore principale della struttura e della funzione della ghiandola tiroidea. Esso è composto da una subunità a e da una subunità b: la subunità a è costituita da 92 aminoacidi ed è identica alla subunità a dell’ormone luteiniz-zante, dell’ormone follicolo-stimolante e della gonadotropina corio-nica umana. La specificità degli ormoni glicoproteici è data dalla subunità b. La subunità b, sintetizzata all’interno delle cellule tireo-trope, è una proteina costituita da 112 aminoacidi. L’ormone ipota-lamico di rilascio dell’ormone tireotropo (TRH) è un tripeptide mo-dificato (piroglutamil-istidil-prolinamide) che incrementa la tra-scrizione di entrambe le subunità; gli ormoni tiroidei (tiroxina [T4] e tri-iodotironina [T3]), invece, ne sopprimono la trascrizione. Nell’in-dividuo sano, la concentrazione sierica di TSH è compresa tra 0,3 e 5,0 mIU/L. I livelli di TSH aumentano nell’ipotiroidismo primario e nell’ipertiroidismo secondario (ad es. tumore ipofisario TSH-secernen-te), mentre diminuiscono nell’ipertiroidismo primario. Le concentrazioni ematiche di TSH variano sia in modo pulsatile, sia secondo un ritmo circadiano: un incremento notturno precede l’inizio del sonno.
Entrambi gli ormoni T4 e T3 esercitano un feedback sulla secre-zione di TRH e di TSH. Esiste una relazione lineare inversa tra la concentrazione di T4 libera (FT4) nel siero e la concentrazione del TSH; quindi la concentrazione sierica di TSH è un indicatore molto sensibile della funzione tiroidea in pazienti con normale funzione ipotalamo-ipofisaria.
Il recettore del TSH è espresso sulle cellule tiroidee. Il recettore del TSH è un membro della famiglia dei recettori accoppiati a proteine G: la proteina G genera un segnale mediante la fosfolipasi C e i canali intracellulari del calcio che regolano l’efflusso di ioduro, la produzione di H2O2 e la iodinazione della tireoglobulina. Il segnale della protein-chinasi A, mediata dall’adenosina monofosfato ciclico (AMPc), regola la captazione di iodio e la trascrizione di tireoglobulina, della tireope-rossidasi e degli mRNA del cotrasportatore sodio-ioduro, determinan-do la produzione di ormone tiroideo. Oltre al TSH, il recettore del TSH lega anche l’anticorpo tireostimolante (presente in quantità elevata nella malattia di Graves) e gli anticorpi bloccanti la tiroide (presenti in quantità elevata nella tiroidite di Hashimoto). A concentrazioni elevate, gli ormoni glicoproteici strettamente correlati – ormone luteinizzante e gonadotropina corionica – si legano anch’essi al recettore del TSH e ne attivano il segnale, provocando talvolta l’ipertiroidismo fisiologico nella fase iniziale della gravidanza.
L’asse ipotalamo-ipofisi-tiroide è intatto quando il volume della ghiandola tiroidea è normale, le cellule follicolari della tiroide hanno una forma cubica, le concentrazioni di TSH e degli ormoni tiroidei rientrano nell’intervallo di riferimento, e la captazione dello iodio
radioattivo è normale. Nel quadro di una disfunzione ipofisaria o ipotalamica, l’ipotiroidismo secondario si manifesta con la riduzione del volume della tiroide (che può non essere palpabile all’esame obiettivo), con cellule follicolari tiroidee dalla forma piatta, bassa concentrazione di TSH (o impropriamente bassa visti i bassi livelli di ormoni tiroidei), concentrazioni di FT4 e di FT3 ( T3 totale) inferiori all’intervallo di riferimento e una bassa captazione di iodio radioattivo.
In un paziente affetto da tumore ipofisario TSH-secernente, tuttavia, la ghiandola tiroidea è ingrossata e di solito è rilevabile all’esame obiettivo come un gozzo compatto, le cellule follicolari tiroidee hanno una forma cilindrica e la colloide è ridotta, la concentrazione di TSH è all’interno o appena al di sopra dell’intervallo di riferimento, mentre le concentrazioni di FT4 e di FT3 sono superiori all’intervallo di rife-rimento con elevata captazione di iodio radioattivo.
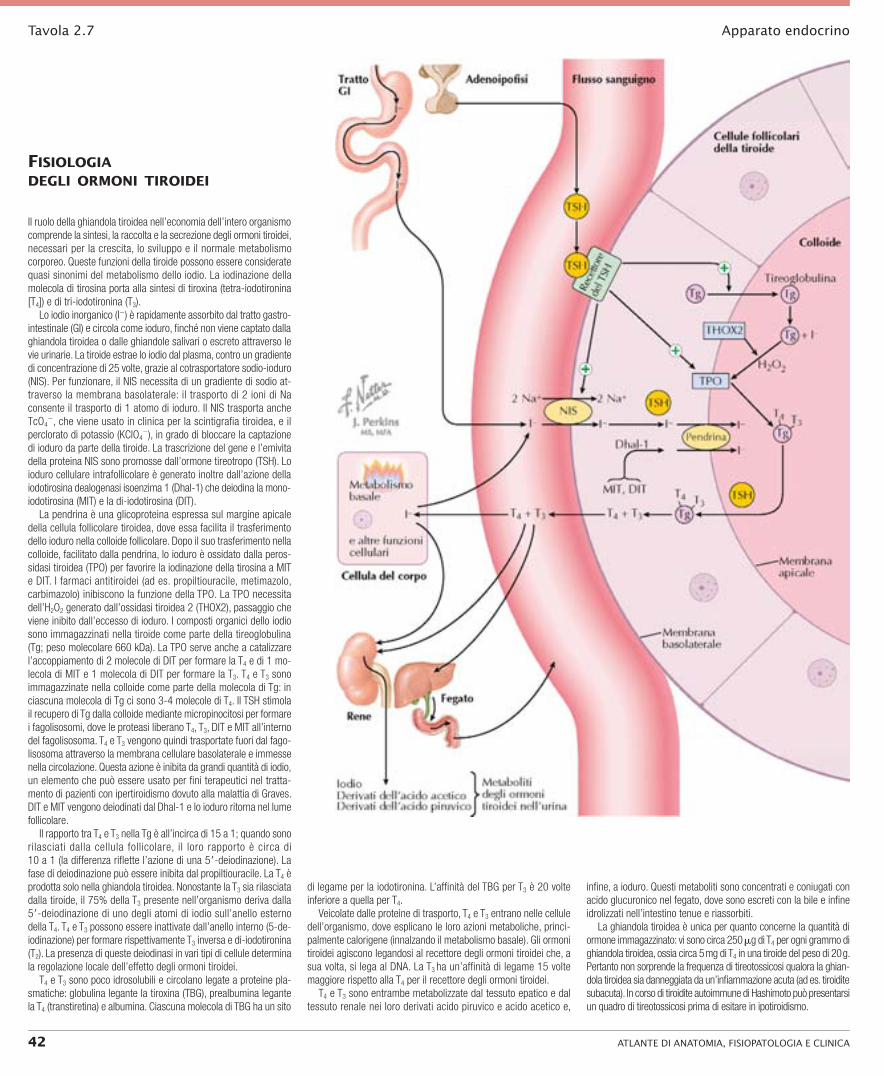
Tavola 2.7 Apparato endocrino
42 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
fiSiologiA degli ormoni tiroidei
Il ruolo della ghiandola tiroidea nell’economia dell’intero organismo comprende la sintesi, la raccolta e la secrezione degli ormoni tiroidei, necessari per la crescita, lo sviluppo e il normale metabolismo corporeo. Queste funzioni della tiroide possono essere considerate quasi sinonimi del metabolismo dello iodio. La iodinazione della molecola di tirosina porta alla sintesi di tiroxina (tetra-iodotironina [T4]) e di tri-iodotironina (T3).
Lo iodio inorganico (I−) è rapidamente assorbito dal tratto gastro-intestinale (GI) e circola come ioduro, finché non viene captato dalla ghiandola tiroidea o dalle ghiandole salivari o escreto attraverso le vie urinarie. La tiroide estrae lo iodio dal plasma, contro un gradiente di concentrazione di 25 volte, grazie al cotrasportatore sodio-ioduro (NIS). Per funzionare, il NIS necessita di un gradiente di sodio at-traverso la membrana basolaterale: il trasporto di 2 ioni di Na consente il trasporto di 1 atomo di ioduro. Il NIS trasporta anche TcO4
−, che viene usato in clinica per la scintigrafia tiroidea, e il perclorato di potassio (KClO4
−), in grado di bloccare la captazione di ioduro da parte della tiroide. La trascrizione del gene e l’emivita della proteina NIS sono promosse dall’ormone tireotropo (TSH). Lo ioduro cellulare intrafollicolare è generato inoltre dall’azione della iodotirosina dealogenasi isoenzima 1 (Dhal-1) che deiodina la mono-iodotirosina (MIT) e la di-iodotirosina (DIT).
La pendrina è una glicoproteina espressa sul margine apicale della cellula follicolare tiroidea, dove essa facilita il trasferimento dello ioduro nella colloide follicolare. Dopo il suo trasferimento nella colloide, facilitato dalla pendrina, lo ioduro è ossidato dalla peros-sidasi tiroidea (TPO) per favorire la iodinazione della tirosina a MIT e DIT. I farmaci antitiroidei (ad es. propiltiouracile, metimazolo, carbimazolo) inibiscono la funzione della TPO. La TPO necessita dell’H2O2 generato dall’ossidasi tiroidea 2 (THOX2), passaggio che viene inibito dall’eccesso di ioduro. I composti organici dello iodio sono immagazzinati nella tiroide come parte della tireoglobulina (Tg; peso molecolare 660 kDa). La TPO serve anche a catalizzare l’accoppiamento di 2 molecole di DIT per formare la T4 e di 1 mo-lecola di MIT e 1 molecola di DIT per formare la T3. T4 e T3 sono immagazzinate nella colloide come parte della molecola di Tg: in ciascuna molecola di Tg ci sono 3-4 molecole di T4. Il TSH stimola il recupero di Tg dalla colloide mediante micropinocitosi per formare i fagolisosomi, dove le proteasi liberano T4, T3, DIT e MIT all’interno del fagolisosoma. T4 e T3 vengono quindi trasportate fuori dal fago-lisosoma attraverso la membrana cellulare basolaterale e immesse nella circolazione. Questa azione è inibita da grandi quantità di iodio, un elemento che può essere usato per fini terapeutici nel tratta-mento di pazienti con ipertiroidismo dovuto alla malattia di Graves. DIT e MIT vengono deiodinati dal Dhal-1 e lo ioduro ritorna nel lume follicolare.
Il rapporto tra T4 e T3 nella Tg è all’incirca di 15 a 1; quando sono rilasciati dalla cellula follicolare, il loro rapporto è circa di 10 a 1 (la differenza riflette l’azione di una 59-deiodinazione). La fase di deiodinazione può essere inibita dal propiltiouracile. La T4 è prodotta solo nella ghiandola tiroidea. Nonostante la T3 sia rilasciata dalla tiroide, il 75% della T3 presente nell’organismo deriva dalla 59-deiodinazione di uno degli atomi di iodio sull’anello esterno della T4. T4 e T3 possono essere inattivate dall’anello interno (5-de-iodinazione) per formare rispettivamente T3 inversa e di-iodotironina (T2). La presenza di queste deiodinasi in vari tipi di cellule determina la regolazione locale dell’effetto degli ormoni tiroidei.
T4 e T3 sono poco idrosolubili e circolano legate a proteine pla-smatiche: globulina legante la tiroxina (TBG), prealbumina legante la T4 (transtiretina) e albumina. Ciascuna molecola di TBG ha un sito
di legame per la iodotironina. L’affinità del TBG per T3 è 20 volte inferiore a quella per T4.
Veicolate dalle proteine di trasporto, T4 e T3 entrano nelle cellule dell’organismo, dove esplicano le loro azioni metaboliche, princi-palmente calorigene (innalzando il metabolismo basale). Gli ormoni tiroidei agiscono legandosi al recettore degli ormoni tiroidei che, a sua volta, si lega al DNA. La T3 ha un’affinità di legame 15 volte maggiore rispetto alla T4 per il recettore degli ormoni tiroidei.
T4 e T3 sono entrambe metabolizzate dal tessuto epatico e dal tessuto renale nei loro derivati acido piruvico e acido acetico e,
infine, a ioduro. Questi metaboliti sono concentrati e coniugati con acido glucuronico nel fegato, dove sono escreti con la bile e infine idrolizzati nell’intestino tenue e riassorbiti.
La ghiandola tiroidea è unica per quanto concerne la quantità di ormone immagazzinato: vi sono circa 250 mg di T4 per ogni grammo di ghiandola tiroidea, ossia circa 5 mg di T4 in una tiroide del peso di 20 g. Pertanto non sorprende la frequenza di tireotossicosi qualora la ghian-dola tiroidea sia danneggiata da un’infiammazione acuta (ad es. tiroidite subacuta). In corso di tiroidite autoimmune di Hashimoto può presentarsi un quadro di tireotossicosi prima di esitare in ipotiroidismo.

Tavola 2.8 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 43
mAlAttiA di grAveS
L’eponimo “malattia di Graves” descrive una sindrome autoimmune della tiroide caratterizzata da ipertiroidismo, gozzo, oftalmopatia e, in alcuni casi, dermopatia infiltrativa (mixedema pretibiale o loca-lizzato). Malattia di Graves e ipertiroidismo non sono sinonimi, poiché alcuni pazienti affetti da malattia di Graves presentano l’oftalmopatia ma non l’ipertiroidismo; inoltre l’ipertiroidismo riconosce diverse altre cause oltre alla malattia di Graves. In questa malattia, l’iperti-roidismo è causato da autoanticorpi contro il recettore dell’ormone tireotropo (TSH) che lo attivano e stimolano la sintesi e la secrezione di ormoni tiroidei (tiroxina [T4] e tri-iodotironina [T3]), nonché la crescita della ghiandola tiroidea.
La malattia di Graves si manifesta più comunemente nella donna che nell’uomo (8:1) e con maggiore frequenza durante l’età fertile, benché possa insorgere sia durante l’infanzia, sia in età molto avanzata. Sebbene i segni principali di questa patologia siano l’in-grossamento della tiroide e gli occhi sporgenti, uniti a sintomi car-diovascolari, di fatto essa interessa l’intero organismo; si tratta pertanto di una malattia sistemica. La tiroide è diffusamente in-grandita (gozzo) ed è, in ogni suo punto, da due a diverse volte più grande della sua normale dimensione. Si può osservare una certa asimmetria, essendo il lobo destro più grande del sinistro; anche il lobo piramidale è di solito ingrossato. Di rado accade che, in un paziente con malattia di Graves, non vi sia un ingrossamento palpa-bile della ghiandola tiroidea. La ghiandola presenta un aumento della vascolarizzazione, evidenziato da un soffio udibile con uno steto-scopio e talvolta da un fremito percepibile alla palpazione, apprez-zabili sui poli superiori della ghiandola. Dal punto di vista istologico, si riscontra un’iperplasia follicolare con una marcata riduzione della colloide nei follicoli e la presenza di alte cellule acinose cilindriche che possono mostrare introflessioni papillari nei follicoli stessi. In una fase avanzata della malattia, può verificarsi un’infiltrazione lin-focitaria multifocale (principalmente cellule T) in tutta la ghiandola tiroidea e, in alcuni casi, è possibile osservare persino follicoli linfatici (principalmente cellule B) all’interno del parenchima tiroideo.
La tiroide iperplastica funziona a un ritmo marcatamente acce-lerato, documentato dall’aumento della captazione e del ricambio di iodio radioattivo e da un incremento dei livelli di T4 e T3, che aumentano la velocità del consumo dell’ossigeno o il metabolismo basale. Inoltre provocano la riduzione delle concentrazioni sieriche di colesterolo totale e di colesterolo legato alle lipoproteine ad alta densità. I livelli elevati di T4 e T3 danno origine a una serie di manife-stazioni fisiche e psicologiche. I pazienti affetti da questa patologia presentano abitualmente nervosismo, agitazione, irrequietezza, insonnia, cambiamenti di personalità e instabilità emotiva. I reperti comportamentali includono difficoltà di concentrazione, confusione e scarsa memoria immediata.
All’esame obiettivo, i pazienti con malattia di Graves presentano un tremore lieve, che può non essere evidente ma che è obiettivabile ponendo un foglio di carta sulle dita estese. L’aumento dei livelli di T4 e T3 e del consumo di ossigeno, con concomitante vasodilatazione generalizzata, determina l’aumento della gittata cardiaca, che si manifesta con cardiopalmo e tachicardia sinusale. L’ulteriore sti-molazione dell’attività cardiaca può indurre fibrillazione atriale e insufficienza cardiaca.
La cute dei pazienti affetti da questa malattia è calda e vellutata (a causa dell’assottigliamento dello strato cheratinico); inoltre può essere arrossata ed è spesso associata a marcata sudorazione per l’aumento della calorigenesi. In alcuni casi si osserva vitiligine, un’altra manifestazione autoimmune. L’onicolisi (nota come “unghia di Plummer”) – caratterizzata dall’ammorbidimento delle unghie e dal loro distacco dal letto ungueale – si verifica in un numero esiguo di pazienti affetti da malattia di Graves. La dermopatia infiltrativa (mixedema pretibiale) è un’alterazione cutanea che talvolta interessa gli arti inferiori o gli avambracci dei pazienti con oftalmopatia pro-gressiva grave. Essa è associata a un forte ispessimento cutaneo senza fovea presentandosi come un edema gommoso, senza fovea, dei tessuti cutanei e sottocutanei, con colorazione violacea della cute nel terzo inferiore della gamba. Di solito è predominante nella
metà esterna della gamba. I noduli (di 1 cm di diametro) sopra la tibia, che si estendono fino all’altezza del ginocchio, possono essere associati al classico mixedema pretibiale localizzato. Questa lesione può colpire anche gli avambracci e sono stati segnalati casi di coinvolgimento dei piedi e persino delle dita dei piedi. Una caratte-ristica di questi siti colpiti da mixedema è l’assenza di peli, ma la presenza occasionale di follicoli piliferi, che producono peluria in questi siti, non esclude la diagnosi. Il mixedema localizzato si verifica quasi sempre in pazienti affetti da oftalmopatia grave e progressiva. La malattia di Graves è inoltre associata a ippocratismo delle dita delle mani e dei piedi (acropachia tiroidea) (Tavola 2.9).
L’iperattività del sistema simpatico determina fissità dello sguardo e la retrazione delle palpebre nella maggior parte dei pazienti con ipertiroidismo. La retrazione delle palpebre viene dimostrata chie-dendo al paziente di seguire con gli occhi il dito dell’esaminatore mentre traccia un arco verticale: di solito è possibile vedere la sclera sopra l’iride quando il paziente guarda verso il basso. Caratteristica peculiare della malattia di Graves è l’oftalmopatia (Tavola 2.10).
L’aumento del tasso metabolico e della calorigenesi in questi pazienti determina una perdita di peso nonostante un aumento dell’appetito, nonché il deperimento di alcuni muscoli, associato a debolezza muscolare. L’ipertiroidismo ha vari effetti sul metabolismo
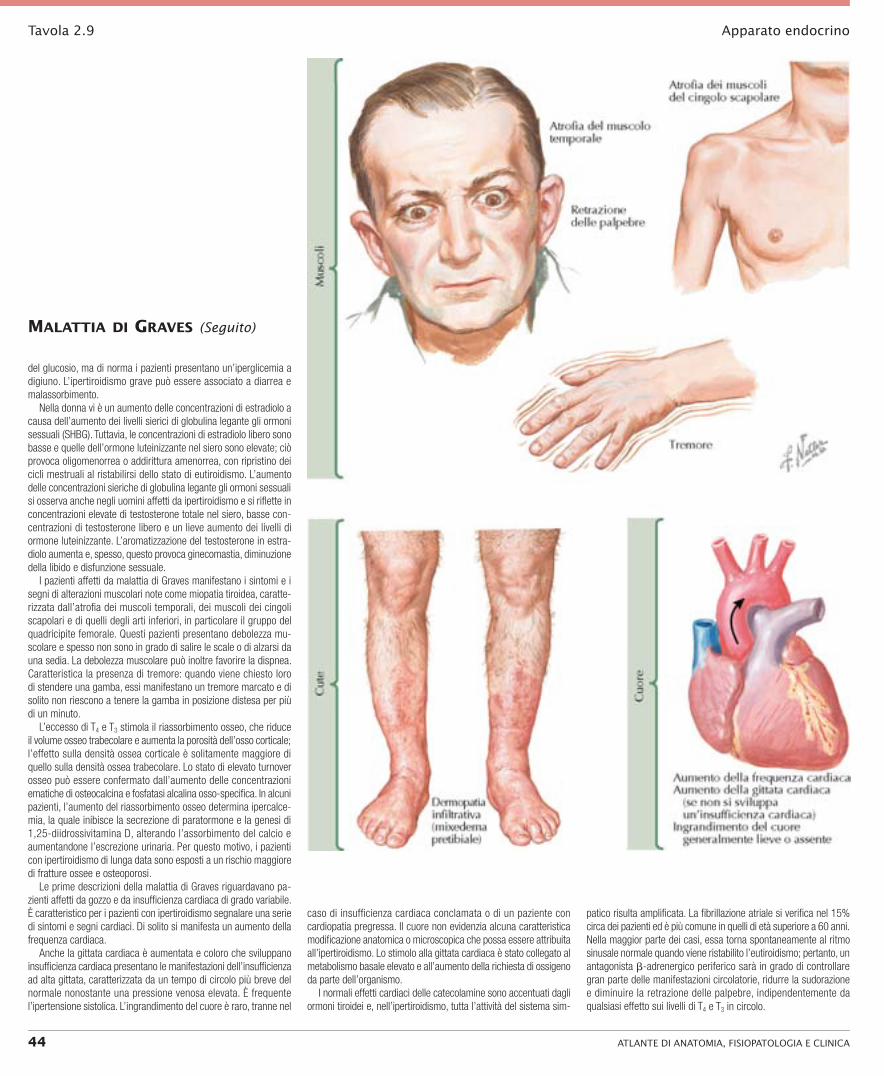
Tavola 2.9 Apparato endocrino
44 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
del glucosio, ma di norma i pazienti presentano un’iperglicemia a digiuno. L’ipertiroidismo grave può essere associato a diarrea e malassorbimento.
Nella donna vi è un aumento delle concentrazioni di estradiolo a causa dell’aumento dei livelli sierici di globulina legante gli ormoni sessuali (SHBG). Tuttavia, le concentrazioni di estradiolo libero sono basse e quelle dell’ormone luteinizzante nel siero sono elevate; ciò provoca oligomenorrea o addirittura amenorrea, con ripristino dei cicli mestruali al ristabilirsi dello stato di eutiroidismo. L’aumento delle concentrazioni sieriche di globulina legante gli ormoni sessuali si osserva anche negli uomini affetti da ipertiroidismo e si riflette in concentrazioni elevate di testosterone totale nel siero, basse con-centrazioni di testosterone libero e un lieve aumento dei livelli di ormone luteinizzante. L’aromatizzazione del testosterone in estra-diolo aumenta e, spesso, questo provoca ginecomastia, diminuzione della libido e disfunzione sessuale.
I pazienti affetti da malattia di Graves manifestano i sintomi e i segni di alterazioni muscolari note come miopatia tiroidea, caratte-rizzata dall’atrofia dei muscoli temporali, dei muscoli dei cingoli scapolari e di quelli degli arti inferiori, in particolare il gruppo del quadricipite femorale. Questi pazienti presentano debolezza mu-scolare e spesso non sono in grado di salire le scale o di alzarsi da una sedia. La debolezza muscolare può inoltre favorire la dispnea. Caratteristica la presenza di tremore: quando viene chiesto loro di stendere una gamba, essi manifestano un tremore marcato e di solito non riescono a tenere la gamba in posizione distesa per più di un minuto.
L’eccesso di T4 e T3 stimola il riassorbimento osseo, che riduce il volume osseo trabecolare e aumenta la porosità dell’osso corticale; l’effetto sulla densità ossea corticale è solitamente maggiore di quello sulla densità ossea trabecolare. Lo stato di elevato turnover osseo può essere confermato dall’aumento delle concentrazioni ematiche di osteocalcina e fosfatasi alcalina osso-specifica. In alcuni pazienti, l’aumento del riassorbimento osseo determina ipercalce-mia, la quale inibisce la secrezione di paratormone e la genesi di 1,25-diidrossivitamina D, alterando l’assorbimento del calcio e aumentandone l’escrezione urinaria. Per questo motivo, i pazienti con ipertiroidismo di lunga data sono esposti a un rischio maggiore di fratture ossee e osteoporosi.
Le prime descrizioni della malattia di Graves riguardavano pa-zienti affetti da gozzo e da insufficienza cardiaca di grado variabile. È caratteristico per i pazienti con ipertiroidismo segnalare una serie di sintomi e segni cardiaci. Di solito si manifesta un aumento della frequenza cardiaca.
Anche la gittata cardiaca è aumentata e coloro che sviluppano insufficienza cardiaca presentano le manifestazioni dell’insufficienza ad alta gittata, caratterizzata da un tempo di circolo più breve del normale nonostante una pressione venosa elevata. È frequente l’ipertensione sistolica. L’ingrandimento del cuore è raro, tranne nel
caso di insufficienza cardiaca conclamata o di un paziente con cardiopatia pregressa. Il cuore non evidenzia alcuna caratteristica modificazione anatomica o microscopica che possa essere attribuita all’ipertiroidismo. Lo stimolo alla gittata cardiaca è stato collegato al metabolismo basale elevato e all’aumento della richiesta di ossigeno da parte dell’organismo.
I normali effetti cardiaci delle catecolamine sono accentuati dagli ormoni tiroidei e, nell’ipertiroidismo, tutta l’attività del sistema sim-
patico risulta amplificata. La fibrillazione atriale si verifica nel 15% circa dei pazienti ed è più comune in quelli di età superiore a 60 anni. Nella maggior parte dei casi, essa torna spontaneamente al ritmo sinusale normale quando viene ristabilito l’eutiroidismo; pertanto, un antagonista b-adrenergico periferico sarà in grado di controllare gran parte delle manifestazioni circolatorie, ridurre la sudorazione e diminuire la retrazione delle palpebre, indipendentemente da qualsiasi effetto sui livelli di T4 e T3 in circolo.
mAlAttiA di grAveS (Seguito)
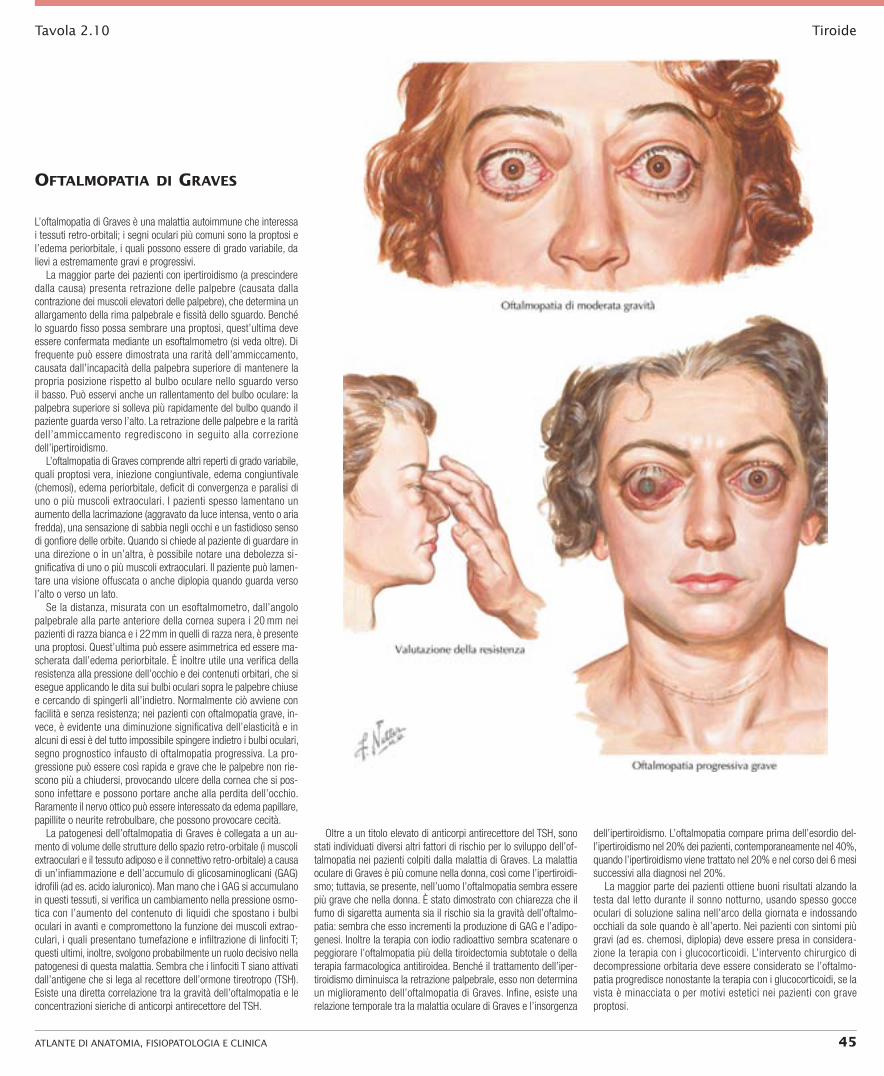
Tavola 2.10 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 45
oftAlmopAtiA di grAveS
L’oftalmopatia di Graves è una malattia autoimmune che interessa i tessuti retro-orbitali; i segni oculari più comuni sono la proptosi e l’edema periorbitale, i quali possono essere di grado variabile, da lievi a estremamente gravi e progressivi.
La maggior parte dei pazienti con ipertiroidismo (a prescindere dalla causa) presenta retrazione delle palpebre (causata dalla contrazione dei muscoli elevatori delle palpebre), che determina un allargamento della rima palpebrale e fissità dello sguardo. Benché lo sguardo fisso possa sembrare una proptosi, quest’ultima deve essere confermata mediante un esoftalmometro (si veda oltre). Di frequente può essere dimostrata una rarità dell’ammiccamento, causata dall’incapacità della palpebra superiore di mantenere la propria posizione rispetto al bulbo oculare nello sguardo verso il basso. Può esservi anche un rallentamento del bulbo oculare: la palpebra superiore si solleva più rapidamente del bulbo quando il paziente guarda verso l’alto. La retrazione delle palpebre e la rarità dell’ammiccamento regrediscono in seguito alla correzione dell’ipertiroidismo.
L’oftalmopatia di Graves comprende altri reperti di grado variabile, quali proptosi vera, iniezione congiuntivale, edema congiuntivale (chemosi), edema periorbitale, deficit di convergenza e paralisi di uno o più muscoli extraoculari. I pazienti spesso lamentano un aumento della lacrimazione (aggravato da luce intensa, vento o aria fredda), una sensazione di sabbia negli occhi e un fastidioso senso di gonfiore delle orbite. Quando si chiede al paziente di guardare in una direzione o in un’altra, è possibile notare una debolezza si-gnificativa di uno o più muscoli extraoculari. Il paziente può lamen-tare una visione offuscata o anche diplopia quando guarda verso l’alto o verso un lato.
Se la distanza, misurata con un esoftalmometro, dall’angolo palpebrale alla parte anteriore della cornea supera i 20 mm nei pazienti di razza bianca e i 22 mm in quelli di razza nera, è presente una proptosi. Quest’ultima può essere asimmetrica ed essere ma-scherata dall’edema periorbitale. È inoltre utile una verifica della resistenza alla pressione dell’occhio e dei contenuti orbitari, che si esegue applicando le dita sui bulbi oculari sopra le palpebre chiuse e cercando di spingerli all’indietro. Normalmente ciò avviene con facilità e senza resistenza; nei pazienti con oftalmopatia grave, in-vece, è evidente una diminuzione significativa dell’elasticità e in alcuni di essi è del tutto impossibile spingere indietro i bulbi oculari, segno prognostico infausto di oftalmopatia progressiva. La pro-gressione può essere così rapida e grave che le palpebre non rie-scono più a chiudersi, provocando ulcere della cornea che si pos-sono infettare e possono portare anche alla perdita dell’occhio. Raramente il nervo ottico può essere interessato da edema papillare, papillite o neurite retrobulbare, che possono provocare cecità.
La patogenesi dell’oftalmopatia di Graves è collegata a un au-mento di volume delle strutture dello spazio retro-orbitale (i muscoli extraoculari e il tessuto adiposo e il connettivo retro-orbitale) a causa di un’infiammazione e dell’accumulo di glicosaminoglicani (GAG) idrofili (ad es. acido ialuronico). Man mano che i GAG si accumulano in questi tessuti, si verifica un cambiamento nella pressione osmo-tica con l’aumento del contenuto di liquidi che spostano i bulbi oculari in avanti e compromettono la funzione dei muscoli extrao-culari, i quali presentano tumefazione e infiltrazione di linfociti T; questi ultimi, inoltre, svolgono probabilmente un ruolo decisivo nella patogenesi di questa malattia. Sembra che i linfociti T siano attivati dall’antigene che si lega al recettore dell’ormone tireotropo (TSH). Esiste una diretta correlazione tra la gravità dell’oftalmopatia e le concentrazioni sieriche di anticorpi antirecettore del TSH.
Oltre a un titolo elevato di anticorpi antirecettore del TSH, sono stati individuati diversi altri fattori di rischio per lo sviluppo dell’of-talmopatia nei pazienti colpiti dalla malattia di Graves. La malattia oculare di Graves è più comune nella donna, così come l’ipertiroidi-smo; tuttavia, se presente, nell’uomo l’oftalmopatia sembra essere più grave che nella donna. È stato dimostrato con chiarezza che il fumo di sigaretta aumenta sia il rischio sia la gravità dell’oftalmo-patia: sembra che esso incrementi la produzione di GAG e l’adipo-genesi. Inoltre la terapia con iodio radioattivo sembra scatenare o peggiorare l’oftalmopatia più della tiroidectomia subtotale o della terapia farmacologica antitiroidea. Benché il trattamento dell’iper-tiroidismo diminuisca la retrazione palpebrale, esso non determina un miglioramento dell’oftalmopatia di Graves. Infine, esiste una relazione temporale tra la malattia oculare di Graves e l’insorgenza
dell’ipertiroidismo. L’oftalmopatia compare prima dell’esordio del-l’ipertiroidismo nel 20% dei pazienti, contemporaneamente nel 40%, quando l’ipertiroidismo viene trattato nel 20% e nel corso dei 6 mesi successivi alla diagnosi nel 20%.
La maggior parte dei pazienti ottiene buoni risultati alzando la testa dal letto durante il sonno notturno, usando spesso gocce oculari di soluzione salina nell’arco della giornata e indossando occhiali da sole quando è all’aperto. Nei pazienti con sintomi più gravi (ad es. chemosi, diplopia) deve essere presa in considera-zione la terapia con i glucocorticoidi. L’intervento chirurgico di decompressione orbitaria deve essere considerato se l’oftalmo-patia progredisce nonostante la terapia con i glucocorticoidi, se la vista è minacciata o per motivi estetici nei pazienti con grave proptosi.

Tavola 2.11 Apparato endocrino
46 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
AnAtomiA pAtologicA dellA tiroide nellA mAlAttiA di grAveS
Nei pazienti affetti da malattia di Graves, le alterazioni anatomiche più marcate interessano la ghiandola tiroidea, sebbene si verifichino anche modificazioni caratteristiche in altri organi. La tiroide, che nell’adulto sano pesa 15-20 g, in questi pazienti è di solito due-quattro volte le sue normali dimensioni. In casi estremi può arrivare a essere 10 volte la sua grandezza normale. Raramente, questi pazienti non presentano un ingrandimento significativo della ghian-dola tiroidea. L’ingrossamento e la congestione diffusa della tiroide avvengono in modo più o meno simmetrico. Queste caratteristiche possono essere evidenziate molto bene mediante una scintigrafia tiroidea, previa somministrazione di una dose test di iodio radioattivo. Come mostra la figura, la tiroide di questi pazienti concentra lo iodio radioattivo in maniera diffusa e uniformemente. Nonostante la ge-neralizzazione del processo e l’apparente simmetria della tiroide, alcuni chirurghi hanno richiamato l’attenzione sul fatto che un lobo possa essere più grande, anche se di poco, rispetto all’altro. Carat-teristicamente, il lobo piramidale, che si estende sopra l’istmo sull’uno o sull’altro lato della trachea, è abbastanza ingrandito da essere facilmente palpabile. La ghiandola tiroidea ingrossata è compatta, liscia e gommosa alla palpazione; di solito è molto va-scolarizzata, come evidenziato da un soffio udibile (che può essere percepito solitamente sopra i poli superiori dell’uno o dell’altro lobo) e, in alcuni casi, da un fremito palpabile sopra i lobi. La ghiandola tiroidea non trattata in questa patologia, essendo vascolarizzata e fragile, può essere origine di un grave sanguinamento durante l’intervento chirurgico.
L’esame istologico della tiroide non trattata rivela un’immagine microscopica molto caratteristica di iperplasia diffusa. Di solito il follicolo è completamente privo di colloide; quella che eventualmente rimane è di colore pallido e mostra margini frastagliati e vacuoliz-zazione. Le cellule tiroidee sono ipertrofiche e iperplastiche: le cellule acinose, che normalmente sono cubiche basse, diventano cubiche alte o cilindriche alte e, se misurate, possono essere alte più del doppio di quelle presenti nella ghiandola tiroidea normale. In alcuni casi l’iperplasia delle cellule acinose è così massiccia da dare luogo a introflessioni papillari intra-acinose.
Insieme all’iperplasia marcata, vi è un aumento notevole dell’avi-dità per lo iodio radioattivo: mentre la captazione di iodio normale è del 3-16% dopo 6 ore e dell’8-25% dopo 24 ore, nei pazienti con malattia di Graves essa supera quasi sempre il 50% e può anche arrivare all’80 o 90%.
In un piccolo numero di pazienti con malattia di Graves di lunga data, l’iperplasia è accompagnata da infiltrazione linfocitaria (in gran parte linfociti T) significativa o estesa del parenchima tiroideo, tal-volta con presenza di grandi follicoli linfatici. Il grado di infiltrazione linfocitaria può essere ridotto dalla terapia farmacologica antitiroi-dea. La dimensione delle cellule epiteliali follicolari è correlata al-l’intensità dell’infiltrazione linfocitaria locale, che implica la stimo-lazione delle cellule tiroidee locali da parte degli anticorpi antirecet-tore dell’ormone tireotropo (TSH).
Le altre modificazioni anatomiche e funzionali sono quelle che riguardano gli occhi, la cute, i muscoli scheletrici, il sistema nervoso, il cuore, il fegato, il timo e i tessuti linfatici. Gli occhi presentano di frequente una proptosi, con muscoli extraoculari ingranditi ed edematosi, con aumento del liquido e del tessuto adiposo nello spazio retro-orbitale (Tavola 2.10). Questi muscoli, così come i muscoli scheletrici, mostrano edema, infiltrazione di cellule tondeg-gianti, ialinizzazione, frammentazione e distruzione. L’ipertiroidismo
può interessare il sistema nervoso centrale e periferico, modulando effetti diretti o indiretti della tireotossicosi. Gli altri effetti sul sistema nervoso sono legati alla natura autoimmune della malattia di Graves (ad es. miastenia grave). Il cuore può essere alquanto ingrossato, ma non presenta alcuna alterazione patologica caratteristica o tipica. Tipicamente, il timo e i tessuti linfatici sono ingranditi, evidenziando un’ipertrofia semplice. In corso di ipertiroidismo possono comparire, inoltre, alterazioni psichiche come ansia e agitazione.

Tavola 2.12 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 47
mAnifeStAzioni cliniche dell’AdenomA toSSico e del gozzo multinodulAre
L’ipertiroidismo associato agli adenomi tossici e ai gozzi multino-dulari tossici è causato da adenoma/i iperfunzionante/i, che rap-presenta la causa più comune di ipertiroidismo dopo la malattia di Graves. L’adenoma iperfunzionante è dovuto a un’iperplasia no-dulare delle cellule follicolari della tiroide che non dipende dalla regolazione dell’ormone tireotropo (TSH). Il quadro clinico di questo tipo di ipertiroidismo presenta importanti differenze rispetto a quello che si osserva nei pazienti affetti da malattia di Graves. I pazienti con gozzo adenomatoso e ipertiroidismo hanno di solito un’età superiore a 40 anni; spesso dalla loro anamnesi risulta una tireopatia multinodulare o uninodulare di lunga data. Di regola i pazienti presentano sintomi cardiovascolari e spesso sono stati indirizzati dal cardiologo prima che dall’endocrinologo. Questi pa-zienti descrivono un’importante dispnea e tachicardia, con frequen-te fibrillazione atriale. Se hanno un’insufficienza cardiaca, essi manifestano tutti i segni e i sintomi di questa malattia tranne per il fatto che, solitamente, non presentano un aumento del tempo di circolo, come avviene nella malattia di Graves. Una caratteristica di questi pazienti è che non sono affetti da oftalmopatia; raramente si può osservare una retrazione palpebrale minima o anche una rarità trascurabile dell’ammiccamento. Non vi sono segni di acropa-chia tiroidea, né di mixedema pretibiale. La debolezza muscolare, caratteristica della malattia di Graves, è presente in misura minore nei pazienti con questo tipo di ipertiroidismo. Il metabolismo basale non è così marcatamente elevato come nella malattia di Graves e i soggetti non sono particolarmente nervosi o eccitabili. Tipica-mente non si ha una notevole perdita di peso o deperimento mu-scolare, come invece avviene nella malattia di Graves. Poiché un’alta percentuale delle pazienti colpite si trova nel periodo po-stmenopausale, non sono presenti le alterazioni del ciclo mestruale spesso riscontrate nella malattia di Graves.
La patogenesi di un adenoma tossico e di un gozzo multinodulare tossico è frequentemente associata a mutazioni somatiche attivanti del gene che codifica per il recettore del TSH. Il gozzo multinodulare tossico tende a essere più comune nelle aree geografiche dove l’assunzione di iodio è relativamente bassa, ma l’incidenza di adenomi solitari tossici della tiroide non sembra essere influenzata dall’assunzione di iodio.
I pazienti con questa malattia hanno un moderato innalzamento dei livelli sierici di tiroxina (FT4) e di tri-iodotironina (FT3). Si verifica un leggero abbassamento delle concentrazioni sieriche di cole-sterolo totale e di colesterolo legato alle lipoproteine ad alta densità (HDL).
Le indagini con iodio radioattivo sono estremamente utili per studiare questi pazienti, specialmente se il sito di concentrazione dello iodio radioattivo è localizzato. Benché la captazione di iodio radioattivo possa non essere così elevata come quella osservata nella malattia di Graves classica, in questa patologia lo iodio radio-attivo è di solito concentrato primariamente nell’adenoma iperfun-zionante, senza che ve ne sia praticamente alcuna traccia nel resto della ghiandola tiroidea. Tuttavia, nei pazienti con gozzo multinodu-lare tossico, è tipico riscontrare una o più aree focali che presentano una maggiore captazione di radioiodio; in alcuni di questi pazienti sono inoltre evidenti noduli non funzionanti (o “freddi”).
Il trattamento efficace dell’ipertiroidismo ha come obiettivo sia la regressione dei sintomi, sia la diminuzione della produzione ec-cessiva di ormone tiroideo. Gli antagonisti b-adrenergici controllano molti dei sintomi di tipo ipermetabolico dell’ipertiroidismo. Le opzioni terapeutiche per normalizzare l’eccesso di T4 e T3 comprendono la somministrazione di tionamide, di radioiodio o l’intervento chirurgico.
Le tionamidi (metimazolo e propiltiouracile) sono usate frequen-temente come trattamento iniziale di elezione nei pazienti anziani con malattia cardiovascolare pre-esistente. Tuttavia, a differenza dell’ipertiroidismo di Graves, che può andare incontro a una re-
missione a lungo termine dopo la sospensione della tionamide, l’ipertiroidismo associato a noduli tossici e a gozzo multinodulare tossico si ripresenta quando la terapia con tionamide viene so-spesa. Lo scopo di tale terapia è quello di stabilire uno stato eutiroideo prima della terapia definitiva (ad es. radioiodio o inter-vento chirurgico). I pazienti giovani e sani di solito non necessitano del trattamento con tionamide prima della terapia definitiva. Una cura definitiva si ottiene con il radioiodio; esso causa un esteso
danno tissutale e distrugge l’adenoma o i focolai autonomi entro 2-4 mesi dopo il trattamento, ma poiché il radioiodio è assorbito principalmente dai noduli iperfunzionanti e il tessuto tiroideo normale tra di essi è quiescente, la maggior parte dei pazienti è eutiroidea dopo la terapia con radioiodio. Il tasso di guarigione di questa terapia diminuisce nel caso di gozzi multinodulari tossici molto grandi; per questo sottogruppo di pazienti, il trattamento di elezione è l’intervento chirurgico.
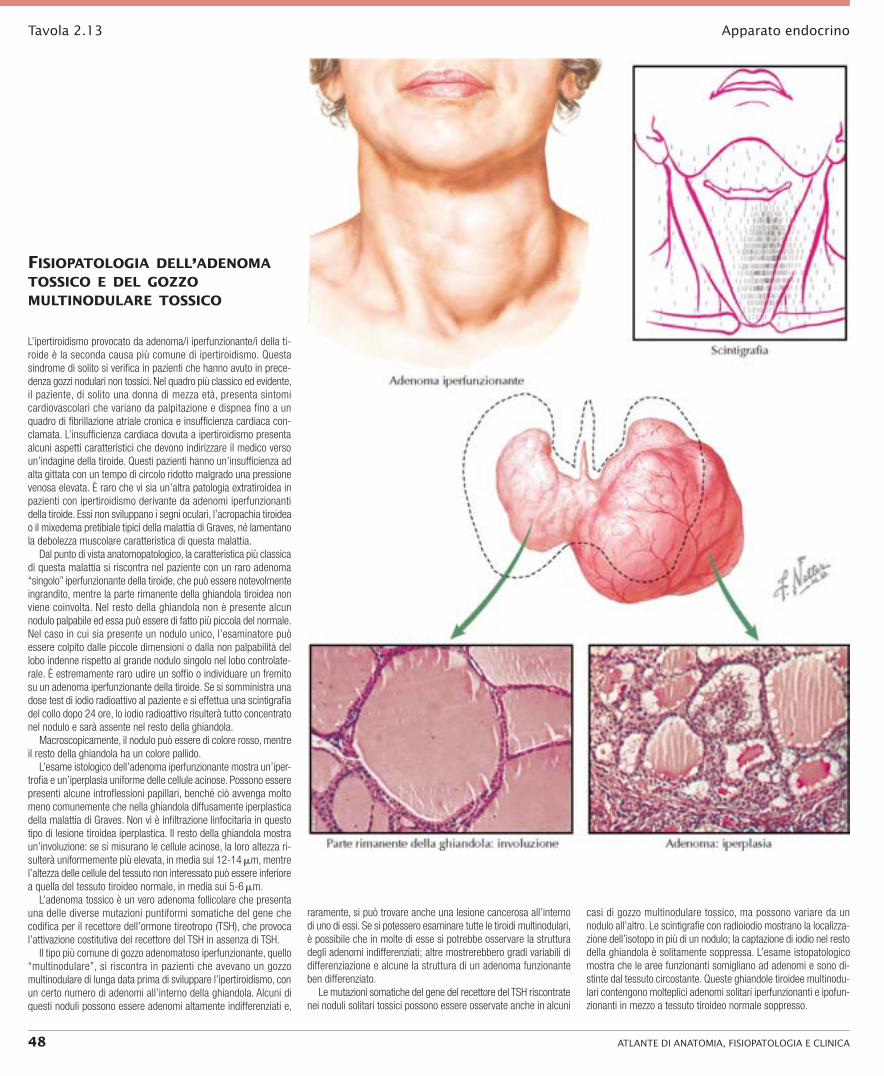
Tavola 2.13 Apparato endocrino
48 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
fiSiopAtologiA dell’AdenomA toSSico e del gozzo multinodulAre toSSico
L’ipertiroidismo provocato da adenoma/i iperfunzionante/i della ti-roide è la seconda causa più comune di ipertiroidismo. Questa sindrome di solito si verifica in pazienti che hanno avuto in prece-denza gozzi nodulari non tossici. Nel quadro più classico ed evidente, il paziente, di solito una donna di mezza età, presenta sintomi cardiovascolari che variano da palpitazione e dispnea fino a un quadro di fibrillazione atriale cronica e insufficienza cardiaca con-clamata. L’insufficienza cardiaca dovuta a ipertiroidismo presenta alcuni aspetti caratteristici che devono indirizzare il medico verso un’indagine della tiroide. Questi pazienti hanno un’insufficienza ad alta gittata con un tempo di circolo ridotto malgrado una pressione venosa elevata. È raro che vi sia un’altra patologia extratiroidea in pazienti con ipertiroidismo derivante da adenomi iperfunzionanti della tiroide. Essi non sviluppano i segni oculari, l’acropachia tiroidea o il mixedema pretibiale tipici della malattia di Graves, né lamentano la debolezza muscolare caratteristica di questa malattia.
Dal punto di vista anatomopatologico, la caratteristica più classica di questa malattia si riscontra nel paziente con un raro adenoma “singolo” iperfunzionante della tiroide, che può essere notevolmente ingrandito, mentre la parte rimanente della ghiandola tiroidea non viene coinvolta. Nel resto della ghiandola non è presente alcun nodulo palpabile ed essa può essere di fatto più piccola del normale. Nel caso in cui sia presente un nodulo unico, l’esaminatore può essere colpito dalle piccole dimensioni o dalla non palpabilità del lobo indenne rispetto al grande nodulo singolo nel lobo controlate-rale. È estremamente raro udire un soffio o individuare un fremito su un adenoma iperfunzionante della tiroide. Se si somministra una dose test di iodio radioattivo al paziente e si effettua una scintigrafia del collo dopo 24 ore, lo iodio radioattivo risulterà tutto concentrato nel nodulo e sarà assente nel resto della ghiandola.
Macroscopicamente, il nodulo può essere di colore rosso, mentre il resto della ghiandola ha un colore pallido.
L’esame istologico dell’adenoma iperfunzionante mostra un’iper-trofia e un’iperplasia uniforme delle cellule acinose. Possono essere presenti alcune introflessioni papillari, benché ciò avvenga molto meno comunemente che nella ghiandola diffusamente iperplastica della malattia di Graves. Non vi è infiltrazione linfocitaria in questo tipo di lesione tiroidea iperplastica. Il resto della ghiandola mostra un’involuzione: se si misurano le cellule acinose, la loro altezza ri-sulterà uniformemente più elevata, in media sui 12-14 mm, mentre l’altezza delle cellule del tessuto non interessato può essere inferiore a quella del tessuto tiroideo normale, in media sui 5-6 mm.
L’adenoma tossico è un vero adenoma follicolare che presenta una delle diverse mutazioni puntiformi somatiche del gene che codifica per il recettore dell’ormone tireotropo (TSH), che provoca l’attivazione costitutiva del recettore del TSH in assenza di TSH.
Il tipo più comune di gozzo adenomatoso iperfunzionante, quello “multinodulare”, si riscontra in pazienti che avevano un gozzo multinodulare di lunga data prima di sviluppare l’ipertiroidismo, con un certo numero di adenomi all’interno della ghiandola. Alcuni di questi noduli possono essere adenomi altamente indifferenziati e,
raramente, si può trovare anche una lesione cancerosa all’interno di uno di essi. Se si potessero esaminare tutte le tiroidi multinodulari, è possibile che in molte di esse si potrebbe osservare la struttura degli adenomi indifferenziati; altre mostrerebbero gradi variabili di differenziazione e alcune la struttura di un adenoma funzionante ben differenziato.
Le mutazioni somatiche del gene del recettore del TSH riscontrate nei noduli solitari tossici possono essere osservate anche in alcuni
casi di gozzo multinodulare tossico, ma possono variare da un nodulo all’altro. Le scintigrafie con radioiodio mostrano la localizza-zione dell’isotopo in più di un nodulo; la captazione di iodio nel resto della ghiandola è solitamente soppressa. L’esame istopatologico mostra che le aree funzionanti somigliano ad adenomi e sono di-stinte dal tessuto circostante. Queste ghiandole tiroidee multinodu-lari contengono molteplici adenomi solitari iperfunzionanti e ipofun-zionanti in mezzo a tessuto tiroideo normale soppresso.
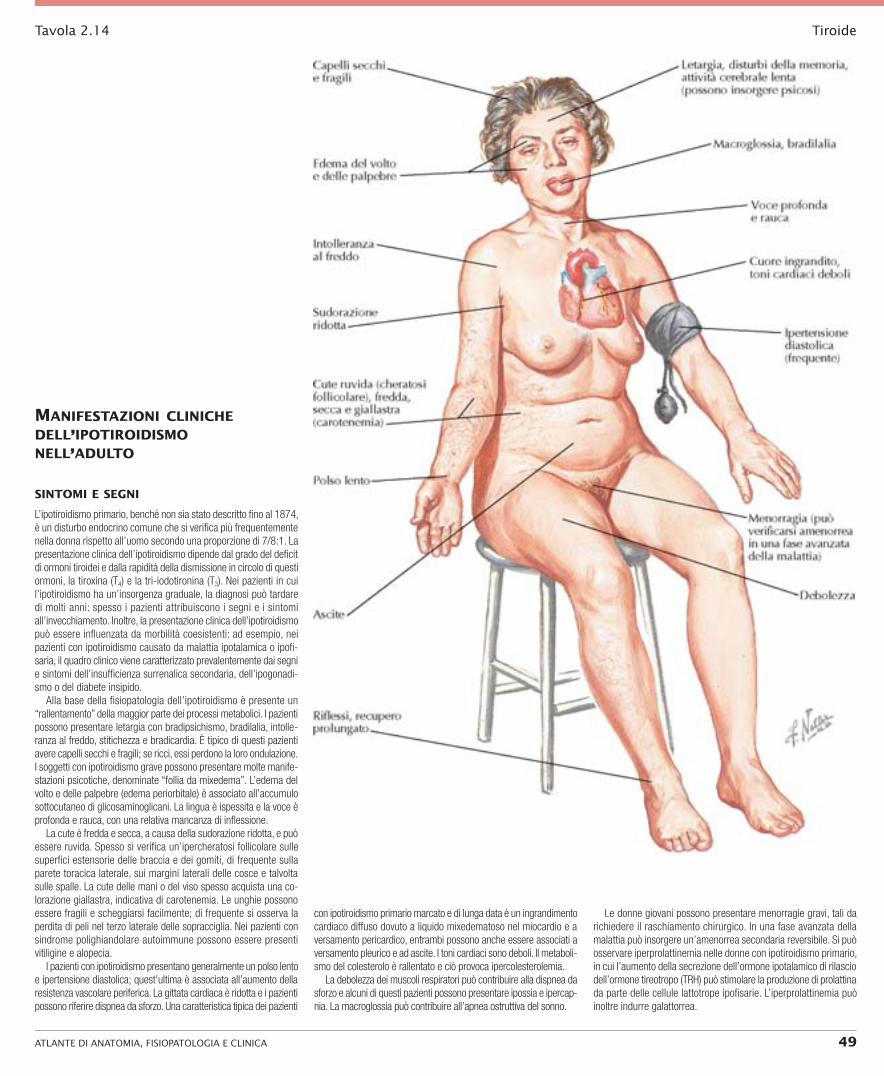
Tavola 2.14 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 49
mAnifeStAzioni cliniche dell’ipotiroidiSmo nell’Adulto
Sintomi e Segni
L’ipotiroidismo primario, benché non sia stato descritto fino al 1874, è un disturbo endocrino comune che si verifica più frequentemente nella donna rispetto all’uomo secondo una proporzione di 7/8:1. La presentazione clinica dell’ipotiroidismo dipende dal grado del deficit di ormoni tiroidei e dalla rapidità della dismissione in circolo di questi ormoni, la tiroxina (T4) e la tri-iodotironina (T3). Nei pazienti in cui l’ipotiroidismo ha un’insorgenza graduale, la diagnosi può tardare di molti anni: spesso i pazienti attribuiscono i segni e i sintomi all’invecchiamento. Inoltre, la presentazione clinica dell’ipotiroidismo può essere influenzata da morbilità coesistenti: ad esempio, nei pazienti con ipotiroidismo causato da malattia ipotalamica o ipofi-saria, il quadro clinico viene caratterizzato prevalentemente dai segni e sintomi dell’insufficienza surrenalica secondaria, dell’ipogonadi-smo o del diabete insipido.
Alla base della fisiopatologia dell’ipotiroidismo è presente un “rallentamento” della maggior parte dei processi metabolici. I pazienti possono presentare letargia con bradipsichismo, bradilalia, intolle-ranza al freddo, stitichezza e bradicardia. È tipico di questi pazienti avere capelli secchi e fragili; se ricci, essi perdono la loro ondulazione. I soggetti con ipotiroidismo grave possono presentare molte manife-stazioni psicotiche, denominate “follia da mixedema”. L’edema del volto e delle palpebre (edema periorbitale) è associato all’accumulo sottocutaneo di glicosaminoglicani. La lingua è ispessita e la voce è profonda e rauca, con una relativa mancanza di inflessione.
La cute è fredda e secca, a causa della sudorazione ridotta, e può essere ruvida. Spesso si verifica un’ipercheratosi follicolare sulle superfici estensorie delle braccia e dei gomiti, di frequente sulla parete toracica laterale, sui margini laterali delle cosce e talvolta sulle spalle. La cute delle mani o del viso spesso acquista una co-lorazione giallastra, indicativa di carotenemia. Le unghie possono essere fragili e scheggiarsi facilmente; di frequente si osserva la perdita di peli nel terzo laterale delle sopracciglia. Nei pazienti con sindrome polighiandolare autoimmune possono essere presenti vitiligine e alopecia.
I pazienti con ipotiroidismo presentano generalmente un polso lento e ipertensione diastolica; quest’ultima è associata all’aumento della resistenza vascolare periferica. La gittata cardiaca è ridotta e i pazienti possono riferire dispnea da sforzo. Una caratteristica tipica dei pazienti
con ipotiroidismo primario marcato e di lunga data è un ingrandimento cardiaco diffuso dovuto a liquido mixedematoso nel miocardio e a versamento pericardico, entrambi possono anche essere associati a versamento pleurico e ad ascite. I toni cardiaci sono deboli. Il metaboli-smo del colesterolo è rallentato e ciò provoca ipercolesterolemia.
La debolezza dei muscoli respiratori può contribuire alla dispnea da sforzo e alcuni di questi pazienti possono presentare ipossia e ipercap-nia. La macroglossia può contribuire all’apnea ostruttiva del sonno.
Le donne giovani possono presentare menorragie gravi, tali da richiedere il raschiamento chirurgico. In una fase avanzata della malattia può insorgere un’amenorrea secondaria reversibile. Si può osservare iperprolattinemia nelle donne con ipotiroidismo primario, in cui l’aumento della secrezione dell’ormone ipotalamico di rilascio dell’ormone tireotropo (TRH) può stimolare la produzione di prolattina da parte delle cellule lattotrope ipofisarie. L’iperprolattinemia può inoltre indurre galattorrea.

Tavola 2.15 Apparato endocrino
50 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
I reperti neurologici includono una riduzione del riflesso achilleo e debolezza generalizzata. La sindrome del tunnel carpale è piuttosto comune in questi pazienti. Il coma da mixedema, una rara com-plicanza, deve essere tenuto in considerazione nei pazienti che manifestano iponatriemia, ipercapnia e ipotermia; nei pazienti con ipotiroidismo grave, esso può essere provocato dalla sommini-strazione di oppiacei, da un’infezione o da un trauma.
L’anemia ipocromica, se presente, può essere di qualsiasi tipo, microcitica o normocitica. Talvolta si riscontra un’anemia macrocitica normocromica. Se il paziente ha una sindrome polighiandolare, può essere presente un’anemia perniciosa. La menorragia osservata nelle donne ipotiroidee in premenopausa può causare un’anemia da carenza di ferro (anemia sideropenica). La riduzione della clea-rance dell’acqua libera può determinare iponatriemia.
L’ipotiroidismo primario (dovuto a una patologia della ghiandola tiroidea) deve essere distinto dall’ipotiroidismo centrale (dovuto a una patologia dell’ipofisi o dell’ipotalamo). Alcuni segni e sintomi possono fornire degli indizi circa la causa dell’ipotiroidismo. L’anam-nesi delle pazienti con ipotiroidismo centrale spesso include un’emorragia post partum seguita da assenza di lattazione e mancata ricomparsa del ciclo mestruale dopo il recupero dal puerperio. Di solito il quadro di mixedema non si sviluppa fino a qualche tempo dopo il primo segno di insufficienza ipofisaria (ad es. amenorrea senza vampate di calore). Questi soggetti normalmente lamentano marcata astenia, sonnolenza, intolleranza al freddo, disturbi della memoria e bradipsichismo. All’esame obiettivo essi differiscono dai pazienti con ipotiroidismo primario se hanno un deficit di altri ormoni ipofisari; pertanto, i pazienti con ipotiroidismo centrale possono avere anche capelli più sottili e soffici, perdita dei peli pubici e ascellari, cuore di dimensioni ridotte (al contrario dei pazienti con mixedema primario che presentano un ingrossamento del cuore), un certo grado di ipotensione e una cute meno secca e non squamosa.
Benché l’anamnesi e l’esame obiettivo forniscano al medico indizi in merito al tipo di ipotiroidismo (primario o centrale), gli esami diagnostici di laboratorio sono rappresentati dalla misurazione delle concentrazioni sieriche di TSH e FT4. Nell’ipotiroidismo primario, la concentrazione di TSH nel siero è più elevata rispetto ai valori di riferimento e il livello ematico di FT4 è abitualmente al di sotto del limite inferiore dell’intervallo di riferimento. Nell’ipotiroidismo cen-trale causato da disfunzione ipotalamica o ipofisaria, la concen-
trazione di TSH nel siero è troppo bassa per il basso livello di FT4.La captazione di iodio radioattivo è bassa in entrambi i tipi di ipotiroidismo.
eziologia
L’ipotiroidismo primario, caratterizzato da secrezione insufficiente degli ormoni tiroidei T4 e T3, è la causa più comune di ipotiroidismo. Esso può derivare dalla distruzione o asportazione della ghiandola
tiroidea o da atrofia della ghiandola con conseguente fibrosi. L’ipo-tiroidismo primario può, inoltre, anche svilupparsi in gozzi in cui la sintesi degli ormoni tiroidei è impedita in seguito all’assunzione di sostanze esogene che inibiscono l’organificazione dello iodio oppure, quando presente, a un deficit degli enzimi necessari per l’ormono-sintesi. Può anche essere la conseguenza di una tiroidite cronica autoimmune, come la tiroidite di Hashimoto. L’ipotiroidismo centrale è causato da un processo che inibisce la liberazione dell’ormone di rilascio del TSH dall’ipotalamo o del TSH dall’ipofisi.
mAnifeStAzioni cliniche dell’ipotiroidiSmo nell’Adulto (Seguito)

Tavola 2.16 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 51
La causa più comune di ipotiroidismo primario è la tiroidite di Hashimoto, mentre la seconda causa più comune è iatrogena. La maggior parte dei pazienti sottoposti a tiroidectomia nel trattamento del gozzo non tossico o della malattia di Graves sviluppa ipotiroidi-smo primario. La terapia più comune della malattia di Graves è lo iodio radioattivo, che ha come obiettivo la completa distruzione della ghiandola tiroidea e quindi l’ipotiroidismo primario.
La tiroidite di Hashimoto è la causa più comune di ipotiroidismo primario. In seguito a tiroidite acuta o subacuta, può insorgere un ipotiroidismo primario transitorio (Tavola 2.22). Un’elevata concen-trazione sierica di anticorpi antiperossidasi tiroidea è compatibile con la tiroidite di Hashimoto.
L’ipotiroidismo centrale è il risultato di una serie di processi che interessano l’adenoipofisi e provocano un deficit della secrezione di TSH. Il deficit di TSH può presentarsi in modo isolato (ad es. nel caso dell’ipofisite linfocitaria) o, più comunemente, nel quadro dell’insufficienza adenoipofisaria completa (Tavola 1.16). Que-st’ultima può avere diverse origini: infiammazione, infarto (ad es. apoplessia post partum), neoplasie primarie, malattia metastatica, malattie infiltrative (ad es. sarcoidosi, istiocitosi a cellule di Langer-hans, emocromatosi), intervento chirurgico, trauma cranico o radioterapia (Tavole 1.12-18). La risonanza magnetica dell’ipofisi è l’esame indicato in questi pazienti per distinguere tra queste molte-plici cause.
trattamento
Il trattamento dell’ipotiroidismo, sia esso a eziologia primaria o secondaria, consiste nella somministrazione giornaliera di levotiro-xina per via orale. Nei pazienti con ipotiroidismo primario viene
misurata la concentrazione di TSH nel siero per ottenere indicazioni sull’aggiustamento del dosaggio della levotiroxina; l’obiettivo è portare il livello di TSH a metà dell’intervallo di riferimento. Nei pazienti con ipotiroidismo centrale la misurazione del TSH è super-flua: il dosaggio di levotiroxina viene aggiustato per ottenere una concentrazione di T4 libera che sia a metà dell’intervallo di riferi-
mento. Tuttavia, prima di iniziare la terapia con levotiroxina in pazienti con ipotiroidismo centrale, è essenziale un esame dell’asse ipota-lamo-ipofisi-ghiandola surrenale. La levotiroxina, che può accelerare il metabolismo del cortisolo, se somministrata a un paziente affetto da insufficienza surrenalica concomitante non in terapia sostitutiva, può provocare una crisi surrenalica.
mAnifeStAzioni cliniche dell’ipotiroidiSmo nell’Adulto (Seguito)
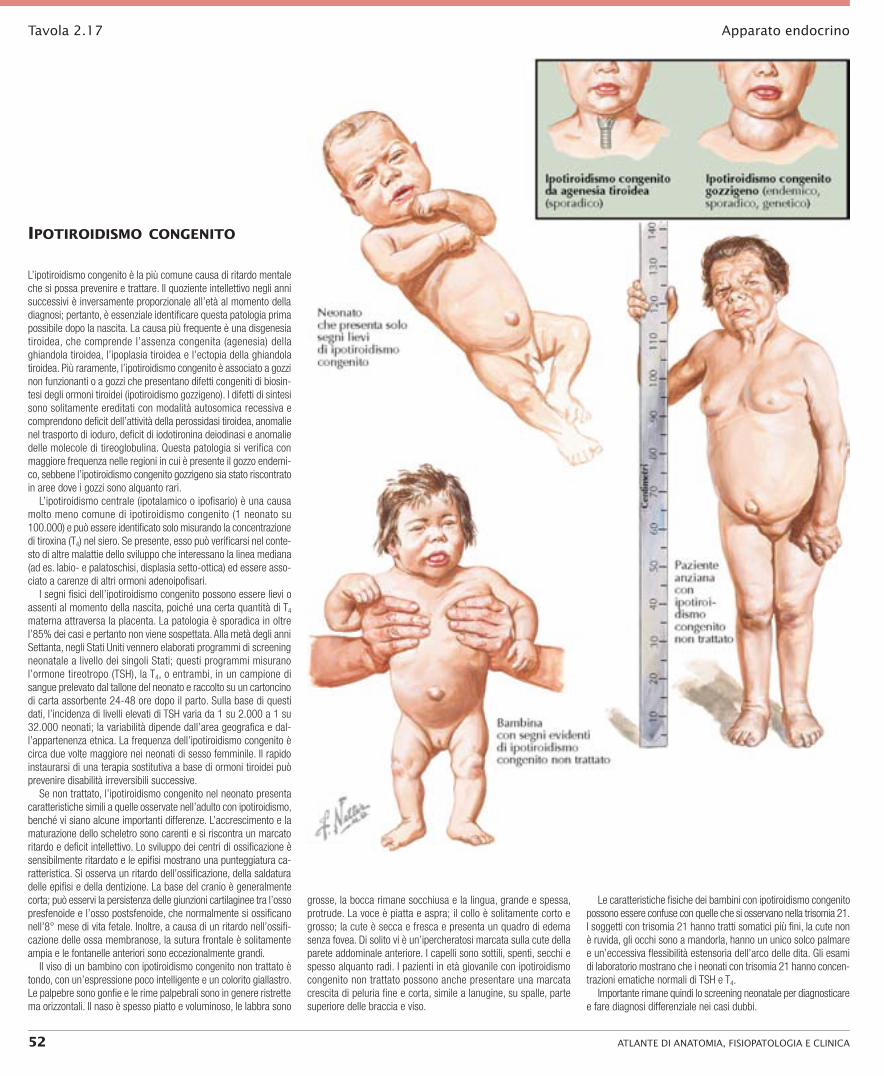
Tavola 2.17 Apparato endocrino
52 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
ipotiroidiSmo congenito
L’ipotiroidismo congenito è la più comune causa di ritardo mentale che si possa prevenire e trattare. Il quoziente intellettivo negli anni successivi è inversamente proporzionale all’età al momento della diagnosi; pertanto, è essenziale identificare questa patologia prima possibile dopo la nascita. La causa più frequente è una disgenesia tiroidea, che comprende l’assenza congenita (agenesia) della ghiandola tiroidea, l’ipoplasia tiroidea e l’ectopia della ghiandola tiroidea. Più raramente, l’ipotiroidismo congenito è associato a gozzi non funzionanti o a gozzi che presentano difetti congeniti di biosin-tesi degli ormoni tiroidei (ipotiroidismo gozzigeno). I difetti di sintesi sono solitamente ereditati con modalità autosomica recessiva e comprendono deficit dell’attività della perossidasi tiroidea, anomalie nel trasporto di ioduro, deficit di iodotironina deiodinasi e anomalie delle molecole di tireoglobulina. Questa patologia si verifica con maggiore frequenza nelle regioni in cui è presente il gozzo endemi-co, sebbene l’ipotiroidismo congenito gozzigeno sia stato riscontrato in aree dove i gozzi sono alquanto rari.
L’ipotiroidismo centrale (ipotalamico o ipofisario) è una causa molto meno comune di ipotiroidismo congenito (1 neonato su 100.000) e può essere identificato solo misurando la concentrazione di tiroxina (T4) nel siero. Se presente, esso può verificarsi nel conte-sto di altre malattie dello sviluppo che interessano la linea mediana (ad es. labio- e palatoschisi, displasia setto-ottica) ed essere asso-ciato a carenze di altri ormoni adenoipofisari.
I segni fisici dell’ipotiroidismo congenito possono essere lievi o assenti al momento della nascita, poiché una certa quantità di T4 materna attraversa la placenta. La patologia è sporadica in oltre l’85% dei casi e pertanto non viene sospettata. Alla metà degli anni Settanta, negli Stati Uniti vennero elaborati programmi di screening neonatale a livello dei singoli Stati; questi programmi misurano l’ormone tireotropo (TSH), la T4, o entrambi, in un campione di sangue prelevato dal tallone del neonato e raccolto su un cartoncino di carta assorbente 24-48 ore dopo il parto. Sulla base di questi dati, l’incidenza di livelli elevati di TSH varia da 1 su 2.000 a 1 su 32.000 neonati; la variabilità dipende dall’area geografica e dal-l’appartenenza etnica. La frequenza dell’ipotiroidismo congenito è circa due volte maggiore nei neonati di sesso femminile. Il rapido instaurarsi di una terapia sostitutiva a base di ormoni tiroidei può prevenire disabilità irreversibili successive.
Se non trattato, l’ipotiroidismo congenito nel neonato presenta caratteristiche simili a quelle osservate nell’adulto con ipotiroidismo, benché vi siano alcune importanti differenze. L’accrescimento e la maturazione dello scheletro sono carenti e si riscontra un marcato ritardo e deficit intellettivo. Lo sviluppo dei centri di ossificazione è sensibilmente ritardato e le epifisi mostrano una punteggiatura ca-ratteristica. Si osserva un ritardo dell’ossificazione, della saldatura delle epifisi e della dentizione. La base del cranio è generalmente corta; può esservi la persistenza delle giunzioni cartilaginee tra l’osso presfenoide e l’osso postsfenoide, che normalmente si ossificano nell’8° mese di vita fetale. Inoltre, a causa di un ritardo nell’ossifi-cazione delle ossa membranose, la sutura frontale è solitamente ampia e le fontanelle anteriori sono eccezionalmente grandi.
Il viso di un bambino con ipotiroidismo congenito non trattato è tondo, con un’espressione poco intelligente e un colorito giallastro. Le palpebre sono gonfie e le rime palpebrali sono in genere ristrette ma orizzontali. Il naso è spesso piatto e voluminoso, le labbra sono
grosse, la bocca rimane socchiusa e la lingua, grande e spessa, protrude. La voce è piatta e aspra; il collo è solitamente corto e grosso; la cute è secca e fresca e presenta un quadro di edema senza fovea. Di solito vi è un’ipercheratosi marcata sulla cute della parete addominale anteriore. I capelli sono sottili, spenti, secchi e spesso alquanto radi. I pazienti in età giovanile con ipotiroidismo congenito non trattato possono anche presentare una marcata crescita di peluria fine e corta, simile a lanugine, su spalle, parte superiore delle braccia e viso.
Le caratteristiche fisiche dei bambini con ipotiroidismo congenito possono essere confuse con quelle che si osservano nella trisomia 21. I soggetti con trisomia 21 hanno tratti somatici più fini, la cute non è ruvida, gli occhi sono a mandorla, hanno un unico solco palmare e un’eccessiva flessibilità estensoria dell’arco delle dita. Gli esami di laboratorio mostrano che i neonati con trisomia 21 hanno concen-trazioni ematiche normali di TSH e T4.
Importante rimane quindi lo screening neonatale per diagnosticare e fare diagnosi differenziale nei casi dubbi.
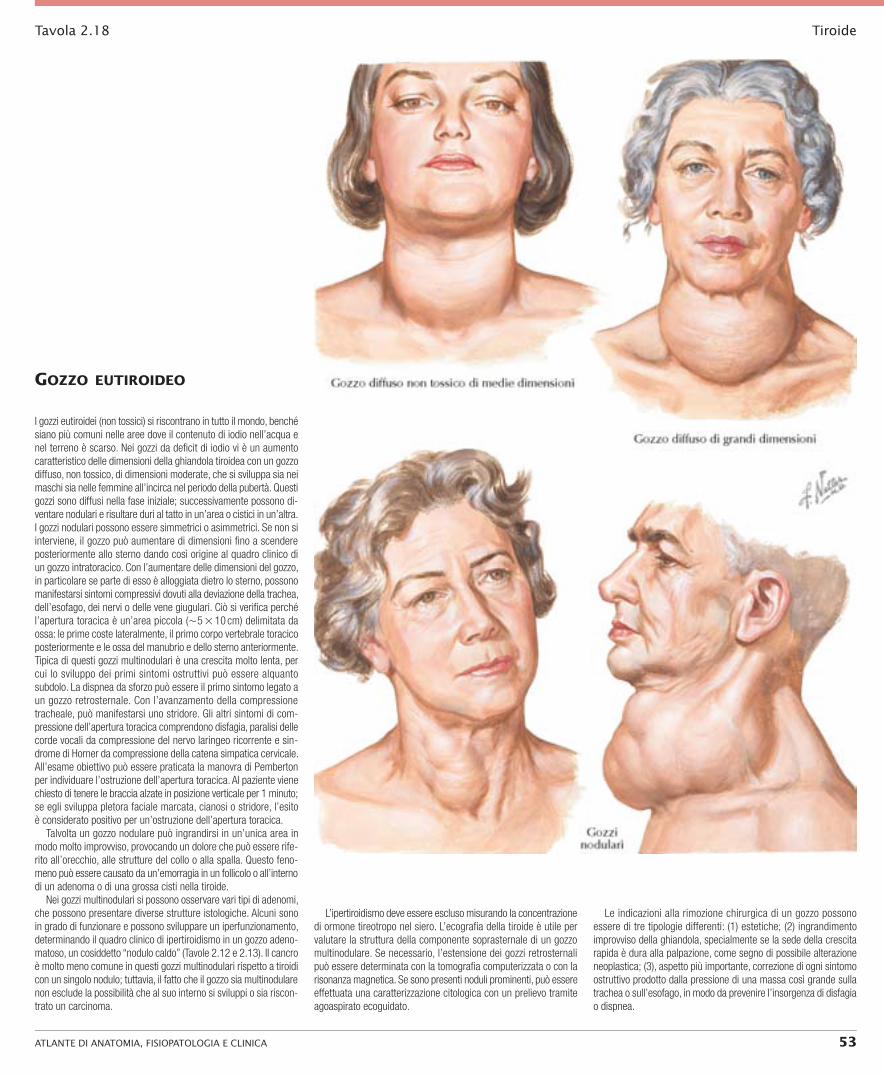
Tavola 2.18 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 53
gozzo eutiroideo
I gozzi eutiroidei (non tossici) si riscontrano in tutto il mondo, benché siano più comuni nelle aree dove il contenuto di iodio nell’acqua e nel terreno è scarso. Nei gozzi da deficit di iodio vi è un aumento caratteristico delle dimensioni della ghiandola tiroidea con un gozzo diffuso, non tossico, di dimensioni moderate, che si sviluppa sia nei maschi sia nelle femmine all’incirca nel periodo della pubertà. Questi gozzi sono diffusi nella fase iniziale; successivamente possono di-ventare nodulari e risultare duri al tatto in un’area o cistici in un’altra. I gozzi nodulari possono essere simmetrici o asimmetrici. Se non si interviene, il gozzo può aumentare di dimensioni fino a scendere posteriormente allo sterno dando così origine al quadro clinico di un gozzo intratoracico. Con l’aumentare delle dimensioni del gozzo, in particolare se parte di esso è alloggiata dietro lo sterno, possono manifestarsi sintomi compressivi dovuti alla deviazione della trachea, dell’esofago, dei nervi o delle vene giugulari. Ciò si verifica perché l’apertura toracica è un’area piccola (∼5 × 10 cm) delimitata da ossa: le prime coste lateralmente, il primo corpo vertebrale toracico posteriormente e le ossa del manubrio e dello sterno anteriormente. Tipica di questi gozzi multinodulari è una crescita molto lenta, per cui lo sviluppo dei primi sintomi ostruttivi può essere alquanto subdolo. La dispnea da sforzo può essere il primo sintomo legato a un gozzo retrosternale. Con l’avanzamento della compressione tracheale, può manifestarsi uno stridore. Gli altri sintomi di com-pressione dell’apertura toracica comprendono disfagia, paralisi delle corde vocali da compressione del nervo laringeo ricorrente e sin-drome di Horner da compressione della catena simpatica cervicale. All’esame obiettivo può essere praticata la manovra di Pemberton per individuare l’ostruzione dell’apertura toracica. Al paziente viene chiesto di tenere le braccia alzate in posizione verticale per 1 minuto; se egli sviluppa pletora faciale marcata, cianosi o stridore, l’esito è considerato positivo per un’ostruzione dell’apertura toracica.
Talvolta un gozzo nodulare può ingrandirsi in un’unica area in modo molto improvviso, provocando un dolore che può essere rife-rito all’orecchio, alle strutture del collo o alla spalla. Questo feno-meno può essere causato da un’emorragia in un follicolo o all’interno di un adenoma o di una grossa cisti nella tiroide.
Nei gozzi multinodulari si possono osservare vari tipi di adenomi, che possono presentare diverse strutture istologiche. Alcuni sono in grado di funzionare e possono sviluppare un iperfunzionamento, determinando il quadro clinico di ipertiroidismo in un gozzo adeno-matoso, un cosiddetto “nodulo caldo” (Tavole 2.12 e 2.13). Il cancro è molto meno comune in questi gozzi multinodulari rispetto a tiroidi con un singolo nodulo; tuttavia, il fatto che il gozzo sia multinodulare non esclude la possibilità che al suo interno si sviluppi o sia riscon-trato un carcinoma.
L’ipertiroidismo deve essere escluso misurando la concentrazione di ormone tireotropo nel siero. L’ecografia della tiroide è utile per valutare la struttura della componente soprasternale di un gozzo multinodulare. Se necessario, l’estensione dei gozzi retrosternali può essere determinata con la tomografia computerizzata o con la risonanza magnetica. Se sono presenti noduli prominenti, può essere effettuata una caratterizzazione citologica con un prelievo tramite agoaspirato ecoguidato.
Le indicazioni alla rimozione chirurgica di un gozzo possono essere di tre tipologie differenti: (1) estetiche; (2) ingrandimento improvviso della ghiandola, specialmente se la sede della crescita rapida è dura alla palpazione, come segno di possibile alterazione neoplastica; (3), aspetto più importante, correzione di ogni sintomo ostruttivo prodotto dalla pressione di una massa così grande sulla trachea o sull’esofago, in modo da prevenire l’insorgenza di disfagia o dispnea.

Tavola 2.19 Apparato endocrino
54 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
AnAtomiA pAtologicA mAcroScopicA del gozzo
Il termine “gozzo” si riferisce a un ingrossamento della ghiandola tiroidea. In generale, la prevalenza del gozzo dipende dall’assunzione di iodio con l’alimentazione: pertanto i gozzi possono essere ende-mici nelle aree geografiche che presentano scarsità di iodio. Nella fase precoce dello sviluppo di un gozzo non tossico, la ghiandola è abitualmente ingrandita in modo diffuso e uniforme, con un aumento delle dimensioni del lobo piramidale. Questo quadro è conosciuto come gozzo non tossico diffuso, o colloide. I gozzi non tossici sono più comuni nella donna con una proporzione di 8:1 e spesso diven-tano evidenti nell’adolescenza o durante una gravidanza. Queste ghiandole tiroidee possono essere due-tre volte più grandi rispetto alla norma o anche di più. Il paziente può accorgersi della condizione perché altri commentano il gonfiore del collo, perché i colletti delle camicie sono troppo stretti o perché ha difficoltà a deglutire. I gozzi grandi possono comprimere la trachea e determinare uno stridore. Può verificarsi una congestione venosa dovuta al restringimento dell’apertura toracica. I gozzi semplici e multinodulari sono in gran parte associati a uno stato eutiroideo.
All’esame obiettivo, la ghiandola appare compatta ma non dura. Con il progredire del processo e l’avanzare dell’età del paziente la tiroide può diventare asimmetrica e multinodulare, condizione evi-dente all’esame macroscopico della ghiandola. Si manifestano cambiamenti significativi nelle dimensioni e nella struttura dei noduli. Nel caso di gozzi nodulari di lunghissima durata, è probabile osser-vare emorragie in vari punti della ghiandola, formazione di cisti, fi-brosi e anche calcificazione. Nelle radiografie del torace, i gozzi asimmetrici causano tipicamente lo spostamento laterale della trachea; inoltre qualsiasi estensione retrosternale di un gozzo di questo tipo può, se calcificata, simulare inizialmente calcificazioni intrapolmonari.
La sezione istologica di un gozzo colloide mostra un’uniforme colorazione ambrata dall’aspetto traslucido. Il peso dei gozzi colloidi può variare da 40 a 1.000 g o più. La ghiandola tiroidea ha una forma distorta e nodulare; alcuni noduli sono parzialmente o com-pletamente separati dalla ghiandola. L’anatomia patologica macro-scopica della sezione mostra tipicamente aree di nodularità, fibrosi, emorragia e calcificazione. Alcuni noduli possono presentare alte-razioni cistiche, altri possono avere una capsula di tessuto connettivo fibroso ispessito e avere l’aspetto complessivo di una neoplasia follicolare.
L’esame citologico mediante agoaspirato dei noduli colloidi evi-denzia di norma la presenza di colloide e popolazioni cellulari miste, con una quantità relativamente esigua di cellule nel materiale aspirato. I tipi di cellule abitualmente riscontrati all’esame citologico sono cellule follicolari con nuclei uniformi, cellule infiammatorie e cellule di Hürthle. Focolai ipercellulati all’interno di un gozzo multi-nodulare possono simulare una neoplasia follicolare.
L’esame microscopico del gozzo nodulare colloide può rivelare ogni possibile tipo di adenoma benigno, compreso un pattern tra-becolare altamente indifferenziato o lo stadio più precoce di diffe-renziazione di struttura tubulare, la struttura dei microfollicoli, o il quadro di un adenoma iperplastico. I follicoli, solitamente rivestiti da epitelio appiattito con modificazioni involutive, possono avere dimensioni variabili e raggiungere i 2 mm di diametro. Grandi follicoli dilatati possono fondersi e creare aree cistiche.
Raramente si possono osservare, all’interno di questi noduli, vari tipi di formazioni cancerose, come i carcinomi tiroidei differenziati (papillari e follicolari). Tuttavia le alterazioni cancerose in queste tiroidi sono molto meno comuni di quelle nei pazienti che presentano un nodulo singolo della tiroide. I sintomi di ostruzione dell’apertura toracica rappresentano l’indicazione più importante per un intervento terapeutico, ma la rara evenienza di una piccola neoplasia maligna deve sempre essere tenuta in considerazione.

Tavola 2.20 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 55
eziologiA del gozzo non toSSico
Lo sviluppo di un gozzo non tossico – ingrandimento della tiroide (diffuso o nodulare) – non è associato a ipertiroidismo o ipotiroidismo conclamati, o essere secondario a neoplasia o infiammazione ma può essere attribuito solitamente a fattori genetici o ambientali che determinano un deficit della secrezione di ormoni tiroidei, a cui l’ipofisi risponde aumentando la produzione dell’ormone tireotropo (TSH). Ad esempio, il deficit di iodio può far sì che la ghiandola tiroidea produca una quantità minore di tiroxina (T4) e tri-iodotironina (T3), provocando un aumento della secrezione di TSH che a sua volta stimola la crescita della ghiandola tiroidea. Il deficit di iodio è di solito caratteristico di aree geografiche dove questo elemento scarseggia nel terreno e nell’acqua, in particolare nelle regioni montuose e in passato ricoperte da ghiacciai. Circa un miliardo di persone vive in regioni del mondo carenti di iodio ed è esposto al rischio di gozzo endemico. Lo ioduro plasmatico è in parte reintegrato mediante lo ioduro liberato attraverso la deiodinazione delle iodotironine nei tessuti periferici. In definitiva, però, è la dieta la fonte più importante di iodio. La tiroide richiede 75 mg di iodio al giorno; nell’America Settentrionale l’assunzione quotidiana di iodio con l’alimentazione varia da 150 a 300 mg. L’impiego di sale da tavola iodato ha ridotto note-volmente, in quest’area, l’incidenza del gozzo da carenza di iodio. Pertanto, un’interferenza o un danno alla sintesi degli ormoni tiroidei è la causa più comune di gozzo negli Stati Uniti.
Viene riconosciuto un numero crescente di entità cliniche in cui deficit congeniti di un passaggio necessario per il metabolismo intratiroideo dello iodio, o per la sintesi degli ormoni tiroidei, spie-gano l’ipotiroidismo congenito e lo sviluppo dei gozzi non tossici. Ad esempio, sono state ora identificate alcune famiglie affette da gozzo con ipotiroidismo congenito nelle quali sono evidenti varie mutazioni del gene del cotrasportatore sodio-ioduro (NIS), nonché un difetto di trasporto dello ioduro.
Qualsiasi mutazione dei geni responsabili della sintesi di tireo-globulina (Tg), perossidasi tiroidea (TPO) e del recettore del TSH può causare il gozzo non tossico. Ciononostante, nella maggior parte dei pazienti con questa patologia non si riscontrano mutazioni geniche predisponenti.
La sindrome di Pendred è l’associazione della sintesi alterata di ormone tiroideo e sordità neurosensoriale. La pendrina, una proteina essenziale per il trasporto dello ioduro nel lume della cellula follico-lare tiroidea, è necessaria anche per il trasporto di ioni e liquidi nell’apparato cocleare.
Difetti congeniti dell’organificazione dell’ormone tiroideo causano il gozzo; ad esempio, l’assenza congenita di TPO, o la produzione insufficiente di perossido di idrogeno da parte dell’ossidasi tiroidea 2 (THOX2), è gozzigena.
I pazienti con sensibilità ridotta agli ormoni tiroidei possono avere mutazioni del gene che codifica per il recettore di questi ormoni
(resistenza agli ormoni tiroidei, che generalmente è causata da mutazioni del dominio di legame a T3 del gene del recettore b tiroi-deo), o difetti dei trasportatori transmembrana di ormoni tiroidei o delle deiodinasi responsabili dell’attivazione intracellulare di T4 a T3. I pazienti con resistenza agli ormoni tiroidei presentano normalmente un gozzo non tossico, sono eutiroidei dal punto di vista clinico e hanno concentrazioni sieriche di T4 e T3 che superano l’intervallo di riferimento.
Dopo un periodo prolungato di iperplasia tiroidea, si formano, all’interno della tiroide iperplastica, noduli non completamente capsulati di vario tipo. Infine, dopo un periodo di iperplasia marcata si verifica l’esaurimento o l’involuzione. L’epitelio si appiattisce, i follicoli si riempiono di colloide viscosa e, alla fine, possono formarsi cisti emorragiche, alcune delle quali diventano fibrose o calcificate. Raramente si può formare un carcinoma all’interno di una ghiandola iperplastica.

Tavola 2.21 Apparato endocrino
56 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
tiroidite linfocitAriA cronicA e tiroidite fibroSA
tiroidite linfocitAriA cronicA (di hAShimoto)
Nelle regioni del mondo con abbondanza di iodio, la tiroidite di Hashimoto è la causa più comune di ipotiroidismo primario. Si tratta di una tiroidite autoimmune cronica caratterizzata dalla presenza nella circolazione sanguigna di anticorpi contro gli antigeni tiroidei (perossidasi tiroidea e tireoglobulina) e da un’infiltrazione linfocitaria diffusa della ghiandola tiroidea. Come per altre malattie endocrine autoimmuni, essa è più comune nella donna (8:1 il rapporto donna-uomo) e presenta una predisposizione genetica. Benché la riserva tiroidea consenta di avere livelli normali di ormoni tiroidei per molti anni, si verifica una graduale perdita di funzione della tiroide e infine i pazienti evolvono dall’ipotiroidismo subclinico a quello conclamato. La tiroidite di Hashimoto diventa evidente dal punto di vista clinico nel periodo tra i 20 e i 40 anni di età. All’esame patologico, queste ghiandole tiroidee mostrano una marcata infiltrazione linfocitaria (sia cellule T, sia cellule B), distruzione dei follicoli tiroidei e dei centri germinativi linfatici.
All’esame obiettivo, la maggior parte dei pazienti colpiti eviden-zia un gozzo simmetrico, compatto e asintomatico; i margini sono frastagliati per la presenza di pseudopodi e la superficie è bitorzoluta.
Una volta formulata la diagnosi di tiroidite di Hashimoto, basata su livelli sierici elevati di anticorpi antitireoglobulina e antiperossidasi tiroidea, e di ipotiroidismo primario, basata sulla concentrazione elevata dell’ormone tireotropo (TSH) nel siero, i pazienti con tiroidite di Hashimoto sono sottoposti semplicemente alla terapia sostitutiva con levotiroxina. Di solito non è necessaria una biopsia della tiroide a conferma della diagnosi di questa patologia. L’intervento chirurgico raramente si rende necessario, tranne, ad esempio, nei pazienti con grandi gozzi sintomatici.
tiroidite fibroSA (di riedel)
La tiroidite di Riedel, o tiroidite fibrosa, è una malattia rara che colpi-sce soprattutto i soggetti di sesso maschile. Si tratta di un processo cronico, proliferativo, invasivo e fibrotico che coinvolge la ghiandola tiroidea. Esso può estendersi fino a spostare e/o comprimere la trachea e l’esofago insieme alle fasce e ai muscoli sovrastanti. Benché non si conoscano le cause della tiroidite di Riedel, essa è principalmente una malattia fibrotica e alcuni pazienti possono anche presentare fibrosi retroperitoneale e mediastinica.
All’esame microscopico questa patologia è caratterizzata da fi-brosi diffusa e significativa, con infiltrazione della ghiandola tiroidea da parte di macrofagi ed eosinofili. È caratteristica una consistenza dura e legnosa della ghiandola.
Le porzioni della ghiandola non interessate rivelano una quantità variabile di acini persistenti, che appaiono compressi dal denso stroma fibroso che li circonda.
All’esame obiettivo, la tiroidite di Riedel è caratterizzata da una ghiandola tiroidea ingrandita e dura come un sasso, che aderisce saldamente alle strutture adiacenti ma non alla cute. Spesso la ghiandola è asimmetrica, più ingrandita su un lato che sull’altro.
A livello sintomatologico, i pazienti con tiroidite di Riedel possono lamentare pressione e rigidità al collo, disfagia e raucedine. Come per la tiroidite di Hashimoto, può esservi un aumento degli anticorpi antiperossidasi tiroidea e antitireoglobulina. Tuttavia, di solito questi
pazienti sono clinicamente eutiroidei e la concentrazione di TSH nel siero è nella norma. Quando all’esame obiettivo si sospetta una tiroidite di Riedel, per confermare la diagnosi si esegue la biopsia della tiroide.
La terapia con glucocorticoidi o tamoxifene può arrestare l’avan-zamento del processo fibrotico in atto o contribuire a risolverlo. Può essere necessario l’intervento chirurgico in caso di peggioramento dei sintomi da compressione tracheale.

Tavola 2.22 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 57
tiroidite SubAcutA
La tiroidite subacuta – conosciuta anche come tiroidite granuloma-tosa subacuta, tiroidite non suppurativa acuta o tiroidite di de Quervain – è caratterizzata dall’insorgenza improvvisa di sintomi associati all’ipertiroidismo: febbre, spossatezza, mialgie e un in-grandimento molto dolente della ghiandola tiroidea. Si tratta di una malattia non comune e ha una frequenza cinque volte maggiore nella donna rispetto all’uomo. La ghiandola tiroidea si ingrossa in modo solitamente asimmetrico e la sua dimensione può essere 1,5-2 volte quella normale. Il dolore causato dalla ghiandola può essere riferito alle articolazioni della mandibola o alle orecchie. È evidente una marcata dolenzia della tiroide, nonché dei linfonodi nelle vicinanze, e il paziente può riferire disfagia.
La causa di questa malattia sembra essere legata a infezioni virali; molti pazienti in anamnesi riferiscono episodi recenti di infe-zione alle vie aeree superiori. L’insulto provoca l’infiammazione della tiroide, un danno follicolare e la liberazione della tiroxina (T4) e della tri-iodotironina (T3) immagazzinate, causando ipertiroidismo sinto-matico seguito da una fase ipotiroidea. L’ipertiroidismo prosegue fino a quando non si esauriscono le scorte della ghiandola tiroidea; la durata tipica è 2-8 settimane. Una volta risolta l’infiammazione ghiandolare, i follicoli tiroidei si rigenerano e il normale funziona-mento della tiroide viene infine ripristinato.
All’esame obiettivo, la ghiandola tiroidea è simmetricamente ingrandita e acutamente dolente alla palpazione, al punto che alcuni pazienti rifiutano la palpazione del collo.
Se si esegue la biopsia della tiroide, si osserva una reazione infiammatoria con infiltrazione di linfociti e neutrofili. In varie parti del campione si osserva la necrosi delle cellule follicolari e la di-struzione dei follicoli tiroidei.
Alcuni studi di laboratorio dimostrano l’aumento delle concen-trazioni sieriche di FT4, FT3 e tireoglobulina, insieme a bassi livelli dell’ormone tireotropo (TSH). La velocità di eritrosedimentazione (VES) è di solito superiore a 50 mm/h e può anche essere presente una leucocitosi. Se si esegue una scintigrafia con iodio radioattivo (131I), la ghiandola tiroidea infiammata non concentra quantità si-gnificative di iodio: la captazione nelle 24 ore è solo dell’1-2%. L’insieme dei seguenti fattori è indicativo di una tiroidite subacuta:
bassa captazione di 131I, concentrazioni ematiche normali o elevate di FT4 e FT3, aumento del livello ematico di tireoglobulina, sop-pressione del TSH e aumento della VES. Risultati simili si trovano nella tiroidite silente e post partum ma non sono presenti il dolore al collo e l’aumento della VES.
La terapia deve concentrarsi sul controllo del dolore e sul tratta-mento della sintomatologia ipertiroidea. Normalmente la terapia prevede un ciclo di 2-8 settimane con farmaci antinfiammatori non
steroidei o glucocorticoidi. I sintomi dell’ipertiroidismo (ad es. tre-more, ansia, palpitazioni) possono essere trattati con un antagonista b-adrenergico.
Quando la fase ipertiroidea è risolta, la fase ipotiroidea può essere subclinica, oppure i pazienti possono presentare i sintomi dell’ipo-tiroidismo. Per i pazienti sintomatici può essere prescritta la terapia con levotiroxina per 6-8 settimane. Alla fine la funzione tiroidea torna alla normalità.

Tavola 2.23 Apparato endocrino
58 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
cArcinomA pApillAre dellA tiroide
Il carcinoma papillare della tiroide (PTC) è uno dei tre carcinomi della tiroide di derivazione epiteliale (gli altri due sono il carcinoma follicolare e il carcinoma anaplastico della tiroide). Il PTC è il tumore maligno più comune della ghiandola tiroidea, responsabile all’incirca del 75% dei casi di tumore. L’incidenza del PTC è massima nella quarta e quinta decade di vita ed è 2,5 volte più comune nella donna che nell’uomo. I PTC possono essere molto piccoli oppure possono essere facilmente palpabili. La presentazione più frequente è quella di un nodulo solitario della tiroide; tuttavia, con l’avvento dell’uso generalizzato della tomografia computerizzata (TC) e dell’ecografia, questo tumore può anche presentarsi come nodulo tiroideo scoperto incidentalmente.
Spesso i PTC hanno focolai multipli all’interno della ghiandola tiroidea: è comune trovare due o più lesioni nella ghiandola tiroidea di un paziente che presenta un linfonodo nel collo che all’agoa-spirato si rivela essere una lesione papillare di origine tiroidea. Benché alcuni di questi siti rappresentino metastasi intraghiandolari, almeno la metà ha origini clonali differenti.
Dal punto di vista istologico, il PTC di solito è non capsulato ed è costituito da cordoni papillari, con tessuto connettivo finemente vascolarizzato che è rivestito da uno a molti strati di cellule cubiche e cilindriche. Nel PTC puro, la colloide e i follicoli tiroidei sono as-senti. I nuclei sono alquanto caratteristici per le loro grandi dimen-sioni e la forma ovale con cromatina ipodensa; essi mostrano “pseudoinclusioni” citoplasmatiche (membrana nucleare sovrab-bondante). Nel 50% circa dei PTC si riscontrano resti calcificati e cicatrizzati di papille tumorali (corpi arenacei). Circa il 10% di tutti i PTC appartiene alla variante follicolare, caratterizzata dalla pre-senza di follicoli in aggiunta ai reperti microscopici caratteristici del PTC. La prognosi complessiva è uguale a quella del PTC comune, anche se la variante follicolare ha di solito un diametro inferiore al momento della diagnosi. Tuttavia, la variante a cellule alte del PTC (l’1% del totale) è un tumore più aggressivo: questi carcinomi sono più grandi al momento della diagnosi ed è più probabile che siano invasivi rispetto al tipo comune. Altre varianti meno comuni di PTC, associate a una maggiore aggressività del tumore, sono le varianti a cellule chiare, insulare, a cellule cilindriche, trabecolare, a cellule ossifile e sclerosante diffusa.
Il PTC spesso metastatizza ai linfonodi cervicali e del mediastino superiore. Al momento della diagnosi iniziale, il PTC viene riscontrato oltre i limiti del collo solo nel 2% dei pazienti, di norma nei polmoni e meno comunemente nelle ossa (altre sedi meno comuni di metastasi sono l’encefalo, il fegato, i reni e le ghiandole surrenali). Nel quadro della malattia metastatica ai polmoni, la radiografia o la TC toracica mostra tipicamente noduli miliari che si aprono a ventaglio a partire dall’ilo. Le metastasi scheletriche si verificano di rado e un coinvolgimento osseo si ha di solito nei pazienti più anziani.
Il PTC è uno dei tumori meno aggressivi e maligni che si for-mano nel corpo umano; la maggioranza dei pazienti affetti da PTC non muore a causa di questo tumore. Tuttavia esso può portare alla morte. I tre fattori maggiormente associati a un aumento del rischio di recidiva del PTC e della mortalità a esso correlata sono: paziente di età superiore a 45 anni al momento della diagnosi, tumori più grandi (>7 cm di diametro) e invasione dei tessuti molli (ad es. trachea, esofago). Fattori addizionali che aumentano il ri-schio di recidiva sono l’appartenenza al sesso maschile, PTC multifocale, numero elevato di metastasi linfonodali (>10) ed età inferiore a 7 anni.
Il trattamento di elezione per PTC con un diametro superiore a 1 cm o per PTC con metastasi linfonodali note è la tiroidectomia totale con linfadenectomia del compartimento centrale. Un inter-vento chirurgico più esteso è indicato nei pazienti che presentano
l’invasione di altri tessuti del collo (ad es. trachea, esofago), ma si può prendere in considerazione una chirurgia meno aggressiva (ad es. lobectomia e istmectomia) nei pazienti con PTC solitari di diametro inferiore a 1 cm. La terapia con iodio radioattivo 131 (131I) funge da terapia adiuvante per il PTC, viene somministrata non a tutti e personalizzata. La radioterapia a fasci esterni può essere presa in considerazione nei pazienti che hanno una malattia metastatica non resecabile e refrattaria alla terapia con 131I.
La chemioterapia sistemica può produrre benefici nei pazienti con PTC aggressivo e sintomatico che è refrattario a ogni altra opzione terapeutica. Sono attualmente in fase di studio alcuni farmaci inibitori delle vie molecolari (ad es. inibitori della tirosin-chinasi) per i pazienti con malattia refrattaria. Tutti i pazienti devono essere sottoposti a terapia con levotiroxina dopo l’inter-vento chirurgico per impedire che la secrezione ipofisaria di ormone tireotropo stimoli la crescita del PTC.
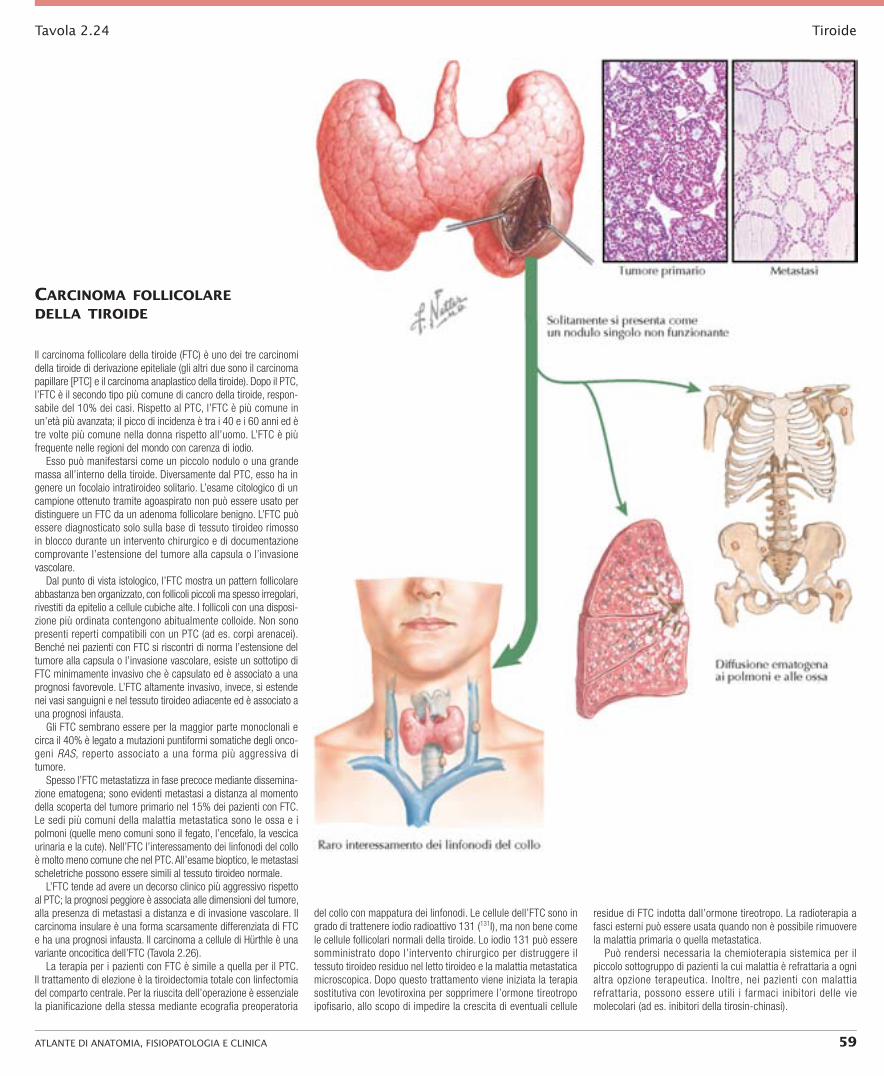
Tavola 2.24 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 59
cArcinomA follicolAre dellA tiroide
Il carcinoma follicolare della tiroide (FTC) è uno dei tre carcinomi della tiroide di derivazione epiteliale (gli altri due sono il carcinoma papillare [PTC] e il carcinoma anaplastico della tiroide). Dopo il PTC, l’FTC è il secondo tipo più comune di cancro della tiroide, respon-sabile del 10% dei casi. Rispetto al PTC, l’FTC è più comune in un’età più avanzata; il picco di incidenza è tra i 40 e i 60 anni ed è tre volte più comune nella donna rispetto all’uomo. L’FTC è più frequente nelle regioni del mondo con carenza di iodio.
Esso può manifestarsi come un piccolo nodulo o una grande massa all’interno della tiroide. Diversamente dal PTC, esso ha in genere un focolaio intratiroideo solitario. L’esame citologico di un campione ottenuto tramite agoaspirato non può essere usato per distinguere un FTC da un adenoma follicolare benigno. L’FTC può essere diagnosticato solo sulla base di tessuto tiroideo rimosso in blocco durante un intervento chirurgico e di documentazione comprovante l’estensione del tumore alla capsula o l’invasione vascolare.
Dal punto di vista istologico, l’FTC mostra un pattern follicolare abbastanza ben organizzato, con follicoli piccoli ma spesso irregolari, rivestiti da epitelio a cellule cubiche alte. I follicoli con una disposi-zione più ordinata contengono abitualmente colloide. Non sono presenti reperti compatibili con un PTC (ad es. corpi arenacei). Benché nei pazienti con FTC si riscontri di norma l’estensione del tumore alla capsula o l’invasione vascolare, esiste un sottotipo di FTC minimamente invasivo che è capsulato ed è associato a una prognosi favorevole. L’FTC altamente invasivo, invece, si estende nei vasi sanguigni e nel tessuto tiroideo adiacente ed è associato a una prognosi infausta.
Gli FTC sembrano essere per la maggior parte monoclonali e circa il 40% è legato a mutazioni puntiformi somatiche degli onco-geni RAS, reperto associato a una forma più aggressiva di tumore.
Spesso l’FTC metastatizza in fase precoce mediante dissemina-zione ematogena; sono evidenti metastasi a distanza al momento della scoperta del tumore primario nel 15% dei pazienti con FTC. Le sedi più comuni della malattia metastatica sono le ossa e i polmoni (quelle meno comuni sono il fegato, l’encefalo, la vescica urinaria e la cute). Nell’FTC l’interessamento dei linfonodi del collo è molto meno comune che nel PTC. All’esame bioptico, le metastasi scheletriche possono essere simili al tessuto tiroideo normale.
L’FTC tende ad avere un decorso clinico più aggressivo rispetto al PTC; la prognosi peggiore è associata alle dimensioni del tumore, alla presenza di metastasi a distanza e di invasione vascolare. Il carcinoma insulare è una forma scarsamente differenziata di FTC e ha una prognosi infausta. Il carcinoma a cellule di Hürthle è una variante oncocitica dell’FTC (Tavola 2.26).
La terapia per i pazienti con FTC è simile a quella per il PTC. Il trattamento di elezione è la tiroidectomia totale con linfectomia del comparto centrale. Per la riuscita dell’operazione è essenziale la pianificazione della stessa mediante ecografia preoperatoria
del collo con mappatura dei linfonodi. Le cellule dell’FTC sono in grado di trattenere iodio radioattivo 131 (131I), ma non bene come le cellule follicolari normali della tiroide. Lo iodio 131 può essere somministrato dopo l’intervento chirurgico per distruggere il tessuto tiroideo residuo nel letto tiroideo e la malattia metastatica microscopica. Dopo questo trattamento viene iniziata la terapia sostitutiva con levotiroxina per sopprimere l’ormone tireotropo ipofisario, allo scopo di impedire la crescita di eventuali cellule
residue di FTC indotta dall’ormone tireotropo. La radioterapia a fasci esterni può essere usata quando non è possibile rimuovere la malattia primaria o quella metastatica.
Può rendersi necessaria la chemioterapia sistemica per il piccolo sottogruppo di pazienti la cui malattia è refrattaria a ogni altra opzione terapeutica. Inoltre, nei pazienti con malattia refrattaria, possono essere utili i farmaci inibitori delle vie molecolari (ad es. inibitori della tirosin-chinasi).

Tavola 2.25 Apparato endocrino
60 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
cArcinomA midollAre dellA tiroide
Il carcinoma midollare della tiroide (MTC) è una neoplasia delle cellule parafollicolari tiroidee, o “cellule C”. Circa il 3% di tutti i tu-mori maligni della tiroide si rivela essere un MTC. Le cellule C sono localizzate nella porzione superiore di ciascun lobo tiroideo e hanno origine dalla cresta neurale embrionale; pertanto, dal punto di vista clinico e istologico, l’MTC è più un tumore neuroendocrino che una neoplasia tiroidea.
Nell’80% circa dei pazienti, l’MTC è sporadico, ma può essere familiare come parte della sindrome da neoplasie endocrine multi-ple di tipo 2 (MEN 2) o come MTC familiare (FMTC). L’MTC spora-dico si manifesta tipicamente come un nodulo tiroideo solitario tra i 40 e i 60 anni di età, con una lieve preponderanza nella donna. È facilmente diagnosticato mediante agoaspirato di un nodulo tiroi-deo. Al momento della diagnosi, oltre la metà dei pazienti con MTC sporadico presenta malattia metastatica, che di solito interessa i linfonodi regionali.
L’MTC secerne l’ormone calcitonina: i pazienti con MTC possono avere livelli molto elevati di calcitonina, che possono determinare una grave diarrea. A causa della sua origine embrionale neuroendo-crina, inoltre, l’MTC ha la capacità di secernere altri ormoni che possono generare una sintomatologia clinica aggiuntiva. Ad esem-pio, l’MTC può secernere un eccesso di corticotropina e causare la sindrome di Cushing.
All’esame istologico, l’MTC mostra un pattern trabecolare omo-geneo con cellule fittamente ammassate, che presentano notevoli differenze di ipercromatismo e di dimensione dei nuclei. Le cellule di solito sono positive all’immunocolorazione per calcitonina, galec-tina-3 e antigene carcino-embrionario.
L’MTC ereditario è associato a mutazioni del proto-oncogene RET e si presenta come MEN 2A, MEN 2B o FMTC. La penetranza del-l’MTC nei pazienti con MEN 2 è pari al 100%. La frequenza nell’uo-mo è uguale a quella nella donna. I pazienti con MEN 2B hanno una forma più aggressiva di MTC e devono essere sottoposti a tiroidec-tomia profilattica nel primo anno di vita. I pazienti con MTC ereditario che non viene riconosciuto nei primi anni di vita manifestano la malattia in genere tra i 20 e i 30 anni di età. Tuttavia, quando è noto il rischio di MTC familiare, il tumore può essere diagnosticato prima che sia palpabile o clinicamente evidente. Dopo che la mutazione specifica del proto-oncogene RET è stata identificata nel probando, i familiari a rischio possono essere sottoposti a test genetico per la ricerca della mutazione specifica al fine di stabilire il loro rischio di contrarre l’MTC.
Anche i pazienti con MTC apparentemente sporadico devono essere sottoposti al test per la ricerca di mutazioni del proto-oncogene RET, poiché circa il 7% presenta una mutazione. Questo dato può incoraggiare il test genetico dei familiari a rischio per individuare i soggetti con MTC e sottoporli a intervento chirurgico prima che si sviluppino le metastasi.
Su tutti i pazienti con MTC devono essere condotte analisi bio-chimiche per escludere l’iperparatiroidismo primitivo e il feocromo-citoma (componenti di MEN 2). Inoltre in questi pazienti deve essere misurata la concentrazione sierica di calcitonina in fase preopera-toria. Più la concentrazione di calcitonina sierica è elevata, maggiore
è la possibilità che il paziente sia affetto da malattia metastatica e che la tiroidectomia non sarà la cura definitiva.
Il trattamento di elezione è la tiroidectomia totale. La prognosi dipende in parte dall’età al momento della diagnosi (la prognosi peggiora con l’aumentare dell’età del paziente). Nei pazienti con familiarità, la prognosi è determinata dall’età alla quale viene eseguita la tiroidectomia; i tassi di guarigione sono più elevati quando la tiroi-
dectomia è effettuata in età giovanile. La concentrazione sierica di calcitonina deve essere misurata in fase postoperatoria per stabilire se si è ottenuta una guarigione chirurgica. La malattia metastatica può coinvolgere il collo, il mediastino, i polmoni, il fegato, le ossa e i reni. La malattia metastatica persistente, che non può essere rimossa chirurgicamente, può essere trattata con i farmaci inibitori delle vie molecolari (ad es. inibitori della tirosin-chinasi).
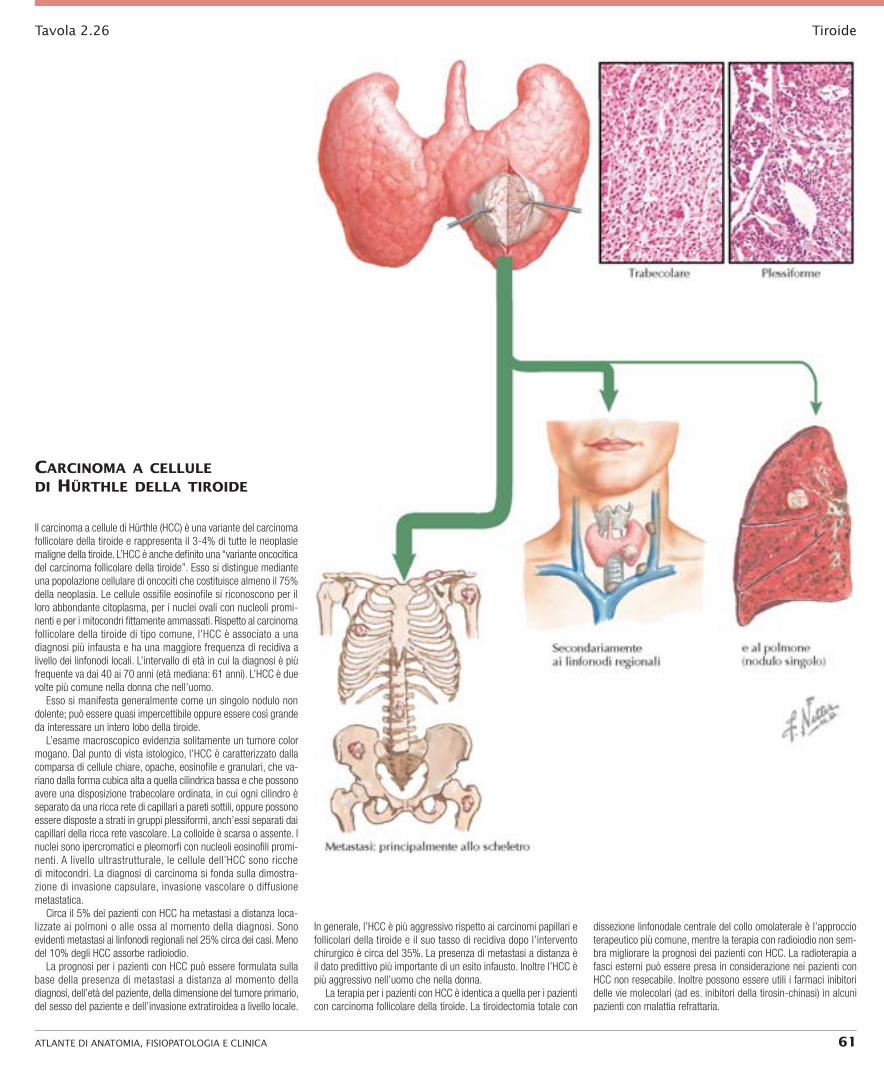
Tavola 2.26 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 61
cArcinomA A cellule di hürthle dellA tiroide
Il carcinoma a cellule di Hürthle (HCC) è una variante del carcinoma follicolare della tiroide e rappresenta il 3-4% di tutte le neoplasie maligne della tiroide. L’HCC è anche definito una “variante oncocitica del carcinoma follicolare della tiroide”. Esso si distingue mediante una popolazione cellulare di oncociti che costituisce almeno il 75% della neoplasia. Le cellule ossifile eosinofile si riconoscono per il loro abbondante citoplasma, per i nuclei ovali con nucleoli promi-nenti e per i mitocondri fittamente ammassati. Rispetto al carcinoma follicolare della tiroide di tipo comune, l’HCC è associato a una diagnosi più infausta e ha una maggiore frequenza di recidiva a livello dei linfonodi locali. L’intervallo di età in cui la diagnosi è più frequente va dai 40 ai 70 anni (età mediana: 61 anni). L’HCC è due volte più comune nella donna che nell’uomo.
Esso si manifesta generalmente come un singolo nodulo non dolente; può essere quasi impercettibile oppure essere così grande da interessare un intero lobo della tiroide.
L’esame macroscopico evidenzia solitamente un tumore color mogano. Dal punto di vista istologico, l’HCC è caratterizzato dalla comparsa di cellule chiare, opache, eosinofile e granulari, che va-riano dalla forma cubica alta a quella cilindrica bassa e che possono avere una disposizione trabecolare ordinata, in cui ogni cilindro è separato da una ricca rete di capillari a pareti sottili, oppure possono essere disposte a strati in gruppi plessiformi, anch’essi separati dai capillari della ricca rete vascolare. La colloide è scarsa o assente. I nuclei sono ipercromatici e pleomorfi con nucleoli eosinofili promi-nenti. A livello ultrastrutturale, le cellule dell’HCC sono ricche di mitocondri. La diagnosi di carcinoma si fonda sulla dimostra-zione di invasione capsulare, invasione vascolare o diffusione metastatica.
Circa il 5% dei pazienti con HCC ha metastasi a distanza loca-lizzate ai polmoni o alle ossa al momento della diagnosi. Sono evidenti metastasi ai linfonodi regionali nel 25% circa dei casi. Meno del 10% degli HCC assorbe radioiodio.
La prognosi per i pazienti con HCC può essere formulata sulla base della presenza di metastasi a distanza al momento della diagnosi, dell’età del paziente, della dimensione del tumore primario, del sesso del paziente e dell’invasione extratiroidea a livello locale.
In generale, l’HCC è più aggressivo rispetto ai carcinomi papillari e follicolari della tiroide e il suo tasso di recidiva dopo l’intervento chirurgico è circa del 35%. La presenza di metastasi a distanza è il dato predittivo più importante di un esito infausto. Inoltre l’HCC è più aggressivo nell’uomo che nella donna.
La terapia per i pazienti con HCC è identica a quella per i pazienti con carcinoma follicolare della tiroide. La tiroidectomia totale con
dissezione linfonodale centrale del collo omolaterale è l’approccio terapeutico più comune, mentre la terapia con radioiodio non sem-bra migliorare la prognosi dei pazienti con HCC. La radioterapia a fasci esterni può essere presa in considerazione nei pazienti con HCC non resecabile. Inoltre possono essere utili i farmaci inibitori delle vie molecolari (ad es. inibitori della tirosin-chinasi) in alcuni pazienti con malattia refrattaria.

Tavola 2.27 Apparato endocrino
62 ATlAnTe di AnATomiA, FisiopATologiA e CliniCA
cArcinomA AnAplAStico dellA tiroide
Il carcinoma anaplastico della tiroide (ATC) è uno dei tre carcinomi della tiroide di derivazione epiteliale (gli altri due sono il carcinoma papillare [PTC] e il carcinoma follicolare [FTC] della tiroide). Mentre il PTC e l’FTC sono considerati carcinomi tiroidei differenziati, l’ATC è un cancro indifferenziato della tiroide. È uno dei carcinomi più maligni e mortali che si possano sviluppare nell’essere umano ed è responsabile all’incirca del 2% di tutti i casi di cancro della tiroide. Solitamente si manifesta dopo i 50 anni (età media: 65 anni) e circa i due terzi degli ATC si verificano nella donna.
L’ATC si sviluppa come un tumore del collo dolente e in rapida crescita, che non mostra mai alcun segno di attività ormonale. Spesso il paziente può fornire la data esatta dell’esordio (di solito una data molto recente) e ne descrive la crescita rapida che causa sintomi da compressione, dispnea, disfagia, raucedine, tosse e anche dolenzia più o meno acuta della massa. Inoltre possono essere presenti sintomi sistemici quali perdita di peso, anoressia, spossatezza e febbre. L’esame del nodulo rivela una massa grande (spesso di diametro superiore a 5 cm), dura, che può essere fissa. Normalmente è dolente; la cute sopra il nodulo può essere calda e anche arrossata. Di frequente si riscontra un’adenopatia cervicale. Possono verificarsi la deviazione della trachea e la paralisi delle corde vocali. Inoltre può evidenziarsi una sindrome della vena cava superiore nei pazienti in cui il tumore occupa gran parte dell’apertura toracica.
Circa il 20% dei pazienti con ATC in anamnesi riferisce un carcinoma tiroideo differenziato (PTC o FTC) e circa il 50% episodi pregressi di gozzo. Pertanto, l’impressione è che l’ATC derivi da neoplasie tiroidee differenziate, probabilmente a causa di una fase di sdifferenziazione (ad es. la perdita di una proteina oncosop-pressiva o una mutazione attivante acquisita).
La diagnosi di ATC può essere confermata mediante agoaspirato o biopsia chirurgica. Dal punto di vista istologico, questo tumore è una neoformazione solida e notevolmente anaplastica, con una preponderanza di cellule fusate, ma con la presenza di molte cellule giganti in tutto il tumore.
Questo cancro, di natura così maligna, difficilmente forma metastasi diffuse. La sua rapida crescita avviene localmente, invadendo le strutture del collo circostanti (ad es. muscoli, linfonodi, laringe, trachea, esofago e grandi vasi del collo) e causando di solito la morte mediante l’invasione diretta della trachea, che determina compressione e asfissia. I polmoni sono la sede più frequente di diffusione a distanza, ma l’ATC può dare metastasi anche alle ossa, alla cute sopra la parete toracica, al fegato, al cuore, ai reni e alle ghiandole surrenali.
La tomografia computerizzata del collo e del torace è utile per pianificare la terapia e monitorare la risposta agli interventi tera-peutici. La durata della sopravvivenza è maggiore nel caso di ATC di diametro inferiore a 6 cm che sono confinati alla ghiandola tiroidea.
L’ATC non è quasi mai curabile chirurgicamente; se la malattia appare localizzata alla ghiandola tiroidea, si deve tentare la resezione completa. Tuttavia l’ATC di solito si ripresenta entro alcuni mesi dalla
rimozione chirurgica anche se la lesione sembrava essere stata completamente estirpata con l’operazione. Dopo l’intervento, può essere presa in considerazione la radioterapia a fasci esterni. Nel caso dell’ATC metastatico non esiste alcuna terapia risolutiva. La chemioterapia con farmaci quali il paclitaxel può dare risultati temporanei. La sopravvivenza a oltre 12 mesi dalla diagnosi è estre-mamente infrequente. Il tasso di mortalità di questa patologia è fondamentalmente del 100%.
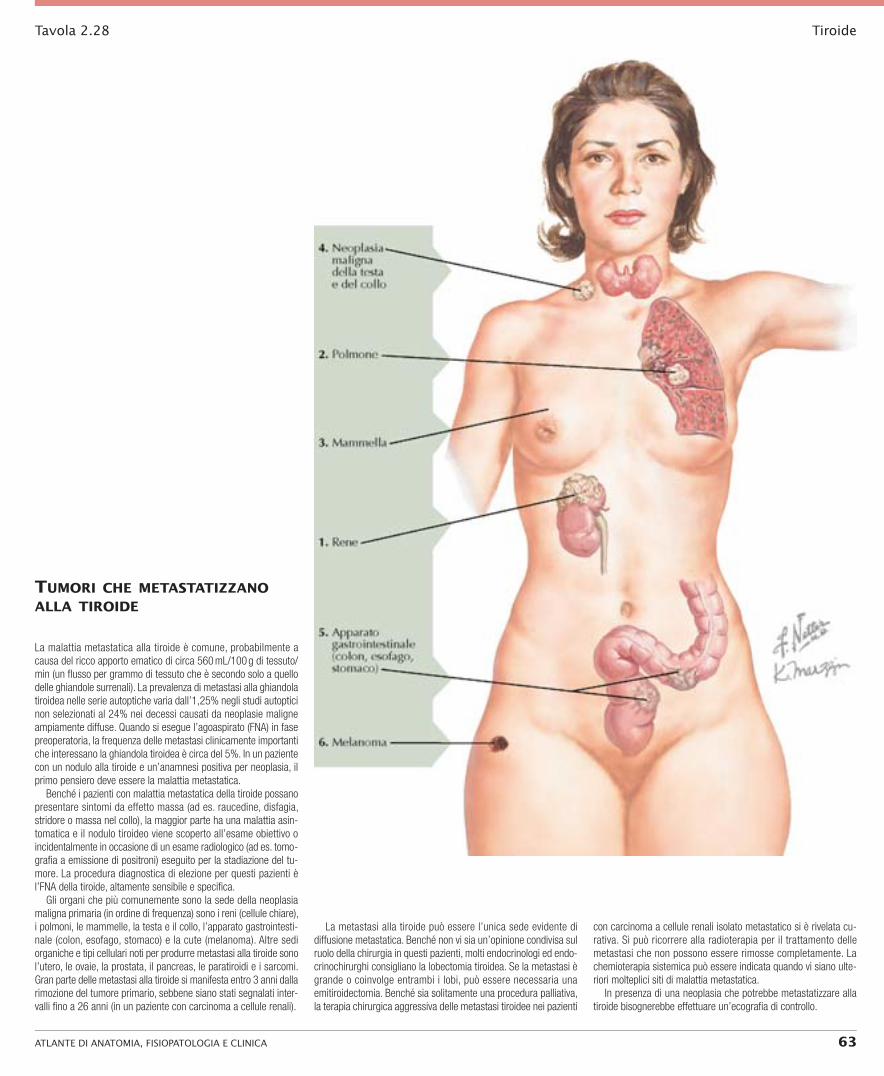
Tavola 2.28 Tiroide
ATlAnTe di AnATomiA, FisiopATologiA e CliniCA 63
tumori che metAStAtizzAno AllA tiroide
La malattia metastatica alla tiroide è comune, probabilmente a causa del ricco apporto ematico di circa 560 mL/100 g di tessuto/min (un flusso per grammo di tessuto che è secondo solo a quello delle ghiandole surrenali). La prevalenza di metastasi alla ghiandola tiroidea nelle serie autoptiche varia dall’1,25% negli studi autoptici non selezionati al 24% nei decessi causati da neoplasie maligne ampiamente diffuse. Quando si esegue l’agoaspirato (FNA) in fase preoperatoria, la frequenza delle metastasi clinicamente importanti che interessano la ghiandola tiroidea è circa del 5%. In un paziente con un nodulo alla tiroide e un’anamnesi positiva per neoplasia, il primo pensiero deve essere la malattia metastatica.
Benché i pazienti con malattia metastatica della tiroide possano presentare sintomi da effetto massa (ad es. raucedine, disfagia, stridore o massa nel collo), la maggior parte ha una malattia asin-tomatica e il nodulo tiroideo viene scoperto all’esame obiettivo o incidentalmente in occasione di un esame radiologico (ad es. tomo-grafia a emissione di positroni) eseguito per la stadiazione del tu-more. La procedura diagnostica di elezione per questi pazienti è l’FNA della tiroide, altamente sensibile e specifica.
Gli organi che più comunemente sono la sede della neoplasia maligna primaria (in ordine di frequenza) sono i reni (cellule chiare), i polmoni, le mammelle, la testa e il collo, l’apparato gastrointesti-nale (colon, esofago, stomaco) e la cute (melanoma). Altre sedi organiche e tipi cellulari noti per produrre metastasi alla tiroide sono l’utero, le ovaie, la prostata, il pancreas, le paratiroidi e i sarcomi. Gran parte delle metastasi alla tiroide si manifesta entro 3 anni dalla rimozione del tumore primario, sebbene siano stati segnalati inter-valli fino a 26 anni (in un paziente con carcinoma a cellule renali).
La metastasi alla tiroide può essere l’unica sede evidente di diffusione metastatica. Benché non vi sia un’opinione condivisa sul ruolo della chirurgia in questi pazienti, molti endocrinologi ed endo-crinochirurghi consigliano la lobectomia tiroidea. Se la metastasi è grande o coinvolge entrambi i lobi, può essere necessaria una emitiroidectomia. Benché sia solitamente una procedura palliativa, la terapia chirurgica aggressiva delle metastasi tiroidee nei pazienti
con carcinoma a cellule renali isolato metastatico si è rivelata cu-rativa. Si può ricorrere alla radioterapia per il trattamento delle metastasi che non possono essere rimosse completamente. La chemioterapia sistemica può essere indicata quando vi siano ulte-riori molteplici siti di malattia metastatica.
In presenza di una neoplasia che potrebbe metastatizzare alla tiroide bisognerebbe effettuare un’ecografia di controllo.



![[Med ITA] (Endocrinologia)TIROIDE](https://static.fdocumenti.com/doc/165x107/56d6c0321a28ab3016995d41/med-ita-endocrinologiatiroide.jpg)










![tumori della tiroide [modalit compatibilit ]) tiroide.pdfCarcinoma della tiroide Microcarcinomi (](https://static.fdocumenti.com/doc/165x107/5b1b4e917f8b9a1e258e9340/tumori-della-tiroide-modalit-compatibilit-tiroidepdfcarcinoma-della-tiroide.jpg)



