
Principali Database biologici -...
48
1 Acidi nucleici: - Sequenze DNA genomico - Sequenze di trascritti (mRNA) - Sequenze EST (corte sequenze di trascritti) Proteine: - Sequenze delle proteine ottenute in modo diretto (degradazione di Edman) - Sequenze proteiche ricavate dalle sequenze nucleotidiche (traduzione) - Studi di espressione proteica (gel bidimensionale e spettrometria di massa) - Cristallografia e determinazione delle strutture secondarie e terziarie Principali Database biologici La maggior quantità di dati biologici presenti nei database è rappresentata da sequenze di acidi nucleici Noi ci occuperemo soprattutto delle sequenze nucleotidiche ed in parte delle sequenze proteiche
Transcript of Principali Database biologici -...
1
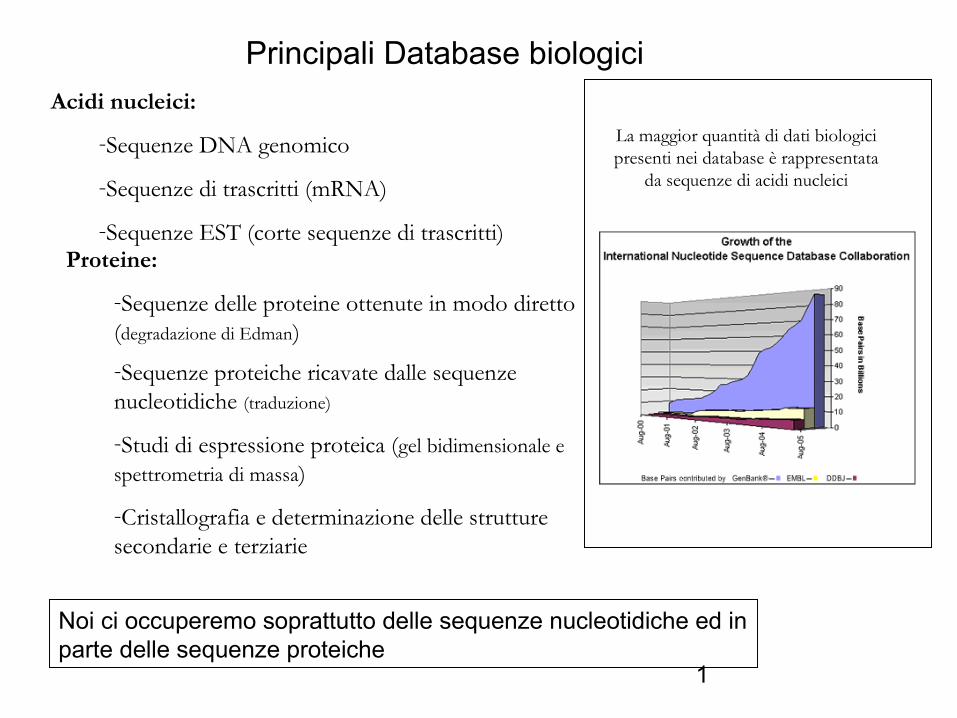
Acidi nucleici:
-Sequenze DNA genomico
-Sequenze di trascritti (mRNA)
-Sequenze EST (corte sequenze di trascritti)Proteine:
-Sequenze delle proteine ottenute in modo diretto (degradazione di Edman)-Sequenze proteiche ricavate dalle sequenze nucleotidiche (traduzione)
-Studi di espressione proteica (gel bidimensionale e spettrometria di massa)
-Cristallografia e determinazione delle strutture secondarie e terziarie
Principali Database biologici
La maggior quantità di dati biologici presenti nei database è rappresentata
da sequenze di acidi nucleici
Noi ci occuperemo soprattutto delle sequenze nucleotidiche ed in parte delle sequenze proteiche

2
Alla fine degli anni 70’ Maxam-Gilbert e Sanger hanno ideato due differenti tecniche per il sequenziamento del DNA basato sulla sintesi del DNA in vitro in presenza di opportuni terminatori marcati.Le sequenze che si ricavano hanno la direzione 5’ 3’ (domanda: lo stampo per la sintesi di queste sequenze che direzione avrà ?)
Oggi sono disponibili delle nuovissime tecniche con le quali è possibile ottenere più di mezzo milione di sequenze in un singolo esperimento (queste nuove tecniche produrranno una nuova rivoluzione nella ricerca genomica)
Sequenze di acidi nucleici
ATTENZIONE: Con le attuali tecniche di sequenziamento si ottengono solo corte sequenze (inferiori a 1000 bp): all’aumentare della lunghezza si perde in risoluzione ed in qualità. Le basi non risolte vengono indicate con ‘n’

3
Esempio: Le sequenze lunghe hanno una scarsa qualità al 3’>CF5530xx.0GgagcccggacgtccaagagatgtcttctgggagccactgggcaattgccagggctccaggaagggctctggctcaggtTgcagacagctgagaaaagatggccctgtcagccaccctctctcagtctgaaacatccaacatccccagaaggcttagc-----------------ecc. ecc. ---------------------- TgaagtagaggggccttcaaactactttatactagtgatagtttgagttaggtaagcatnttaaagctgnntggtgatAaagaaggcagcttangattctgtggttgggaaacaagtgtagtccgcttccccttttttangaaagccctgttaaaatangctnatttgnnaacat
Cromatogrammi della sequenza
Se si vogliono conoscere lunghe sequenze di DNA, è necessario sequenziare frammenti del DNA e poi assemblare le corte sequenze in modo che si sovrappongano tra loro
Sequenze parziali
Sequenza assemblata

4
Come si ottengono le sequenze di DNA Il DNA viene frammentato e poi amplificato con tecniche di biologia molecolare (es. inserimento dei frammenti all’interno di cloni batterici che replicandosi riproducono anche il DNA esogeno). I differenti frammenti vengono poi sequenziati.
Solo con l’assemblaggio delle sequenze ottenute da questi frammenti si ottengono le lunghe sequenze di DNA presenti nei database.
(Ricordate che, se nei DB trovate record contenenti lunghi sequenze (maggiori di un migliaio di basi) , queste sicuramente sono il frutto di un assemblaggio di corte sequenze.)
Come si ottengono le sequenze di mRNAL’mRNA (meno stabile del DNA) deve essere preventivamente trasformato in cDNA (da una molecola di mRNA si ottiene prima una copia complementare di DNA (per questo si chiama cDNA) a singolo filamento che poi viene resa a doppia elica. Si procede poi come per il DNA
Nota: l’insieme dei batteri contenenti gli inserti di DNA esogeno viene detto ‘libreria di DNA’ (o libreria di cDNA)

5
Perché si sequenzia anche l’mRNA (non è sufficiente conoscere solo le sequenze di DNA) ?
Risposte:
- Per conoscere le sequenze codificanti (nota: negli eucarioti superiori, solo il 3% del genoma è codificante), quindi individuare le sequenze geniche e distingure gli esoni dagli introni.
- Per conoscere le sequenze fiancheggianti dei geni e quindi le regolazioni della trascrizione.
- Per conoscere la sequenza proteica (traducendo la sequenza nucleotidica) e studiare quindi la relativa proteina.
- Per conoscere varianti (splicing alternativi) dello stesso gene e quindi probabili funzioni differenti
- Sequenziando mRNA in tessuti differenti o momenti differenti si può conoscere l’espressione genica: determinare quando (sviluppo o momento particolare) e dove (quale tessuto) un particolare gene viene espresso
L’insieme degli mRNA (RNA messaggeri o trascritti) espressi in un organismo viene definito trascrittoma

6
Importanze delle Sequenze EST (Expressed Sequence Tag) Per individuare un trascritto non serve conoscere tutta la sua sequenza, ma è sufficiente identificarne una parte.
Da questo presupposto sono stati sviluppati progetti di sequenziamento di corte sequenze di cDNA chiamate EST (Expressed Sequence Tag) che hanno permesso di tracciare numerosi profili trascrizionali (espressione genica di un particolare tessuto o in un particolare momento o in presenza di una particolare malattia genetica).
Attualmente nei database esistono più di 30 milioni di sequenze di EST di cui circa 8 milioni relative a Homo sapiens (human) e più di 4 milioni relative a topo
ATG AAAAAAAAATAA
5’UTR Seq. codificante 3’UTR polyA
EST 5’ EST 3’

7
I database primari
Cosa sono i database primari?Sono i contenitori di tutte le sequenze prodotte nel mondo e rese disponibili alla comunità scientifica.Memorizzano essenzialmente le sequenze e poche altra informazioni generiche correlate (laboratorio dove è avvenuto il sequenziamento, data, specie, descrizione …)
EMBL datalibrary Europa GenBank USA DDBJ Giappone
I tre database si aggiornano quotidianamente scambiandosi i dati ricevuti durante la giornata, in modo che sia sufficiente interrogare solo uno dei tre.

8
EBI European Bioinformatics Institute (Hinxton – Cambridge, UK) http://www.ebi.ac.uk/embl/

9
NCBIhttp://www.ncbi.nih.gov/Genbank/index.html

10
DDBJ
http://www.ddbj.nig.ac.jp/

11
Un secondo grande aggregato di banche dati è quello per le sequenze proteiche, le quali possono essere ottenute in seguito a:- determinazione diretta della sequenza proteica- traduzione di sequenze nucleotidiche per le quali sia stata individuata o predetta la funzione di gene codificante la proteina
UniProtKB banca dati di riferimento (protein knowledgebase) sviluppata a Ginevra. Si divide in due sezioni:
SWISS-PROT Contiene informazioni accuratamente annotate, spesso a mano.
TrEMBL (TRanslated EMBL) risultato della traduzione automatica in aminoacidi di tutte le sequenze annotate nella banca dati EMBL come codificanti proteine; supplemento a SWISS-PROT.
PIR (Protein Information Resource, Georgetown University); soprattutto indirizzato a definire gli standard di annotazione, con ridondanza minima (produces a non-redundant annotated protein sequence database). http://pir.georgetown.edu/ NCBI-Protein The Protein database is a collection of sequences from several sources, including translations from annotated coding regions in GenBank, RefSeq and TPA, as well as records from SwissProt, PIR, PRF, and PDB.
Banche Dati proteiche

12
UniProtKB (EBI) http://expasy.org/sprot/
UniProt Knowledgebase Release 2011_05 consists of:
- UniProtKB/Swiss-Prot : 528.048 entries
- UniProtKB/TrEMBL: 15.062.837 entries

13
Banche Dati derivate
Le banche dati primarie contengono tutte le sequenze conosciute, di tutti gli organismi, genomiche di mRNA ecc., per rendere organica la ricerca sono state costruite delle banche dati derivate che raggruppano solo dati relativi a specifici argomenti.
Esempi: - Database sequenze genomiche: GDB (uomo) , MGI (topo), SGD (lievito)- Database di geni e trascritti: UniGene, LocusLink, dbEST, ecc.- Inoltre database dei fattori di trascrizione, dbSNP (di polimorfismi) e molti altri.
Esistono poi dei database integrati che raggruppano i dati provenienti da differenti database fornendo informazioni particolareggiate di argomenti specifici
Allegato alla prima esercitazione troverete un elenco (non completo) di questi database

14
Esistono dei sistemi integrati che permettono di interrogare, attraverso il web, in modo semplice ed intuitivo le banche dati biologiche. I tre sistemi principali sono: ENTREZ → associato a GENBANK SRS → associato a EMBL DBGET → associato a DDBJI sistemi integrati forniscono una interfaccia WEB omogenea a tutti i database gestiti dal sistema.
FORM DI QUERY
PAGINA DI RISPOSTA
SISTEMA INTEGRATO
DB1
DBn
…
12
3
45
COMPUTER „SERVER“ REMOTOPC UTENTE RETE
Sistemi di interrogazione alle banche dati (sistemi di "retrieval“)

15
SRS è un sistema aperto, può essere installato su calcolatori differenti (server) e può integrare banche dati strutturate su altri server SRS o altre banche dati previa strutturazione o indicizzazione nel sistema SRS.http://srs.ebi.ac.uk/
Come SRS, anche ENTREZ è un sistema disponibile via web per la ricerca e l’estrazione dei dati da banche dati di sequenze nucleotidiche, proteiche, dalla banca dati bibliografica MEDLINE, dalla banca dati delle malattie mendeliane OMIM, e da ogni banca dati sviluppata dall’NCBI.
E’ un sistema chiuso e non è possibile ottenere il software che gestisce il sistema.http://www.ncbi.nlm.nih.gov/Entrez/index.html

16
Esistono molti tipi di siti e/o database biologici, in questo corso ci soffermeremo solo su alcuni
Qui sono riportati i link di Entrez (NCBI) , all’EBI esistono siti corrispondenti

17
Qui invece sono riportati i link presenti all’ABI con EB-eye la semplice interfaccia grafica
All’EBI è comunque possibile accedere direttamente al sistema SRS per formulare query complesse
18
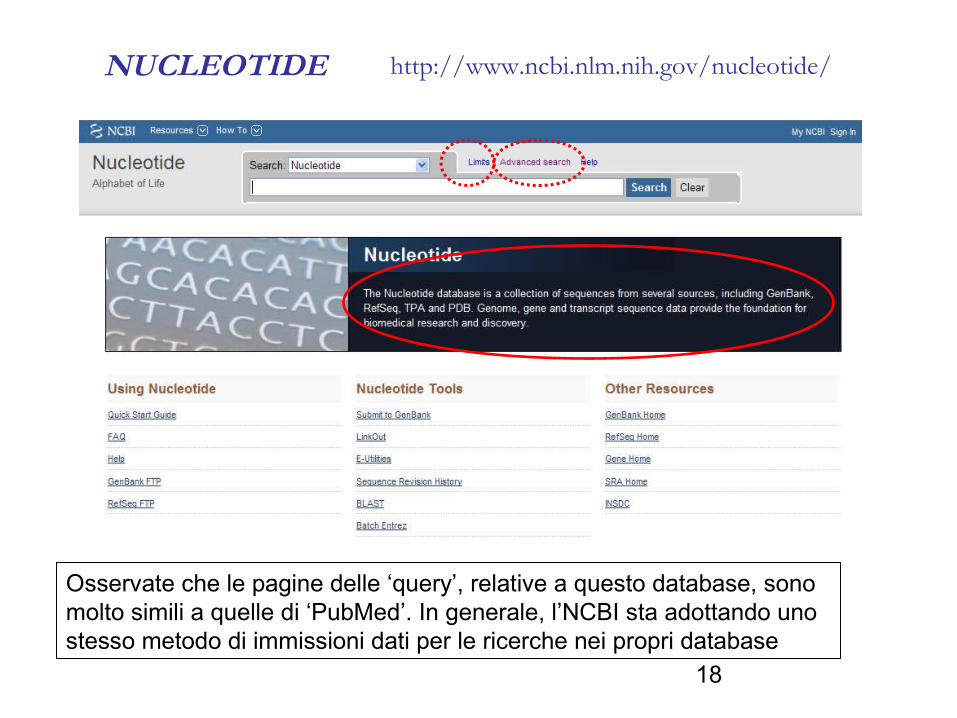
NUCLEOTIDE http://www.ncbi.nlm.nih.gov/nucleotide/
Osservate che le pagine delle ‘query’, relative a questo database, sono molto simili a quelle di ‘PubMed’. In generale, l’NCBI sta adottando uno stesso metodo di immissioni dati per le ricerche nei propri database
19
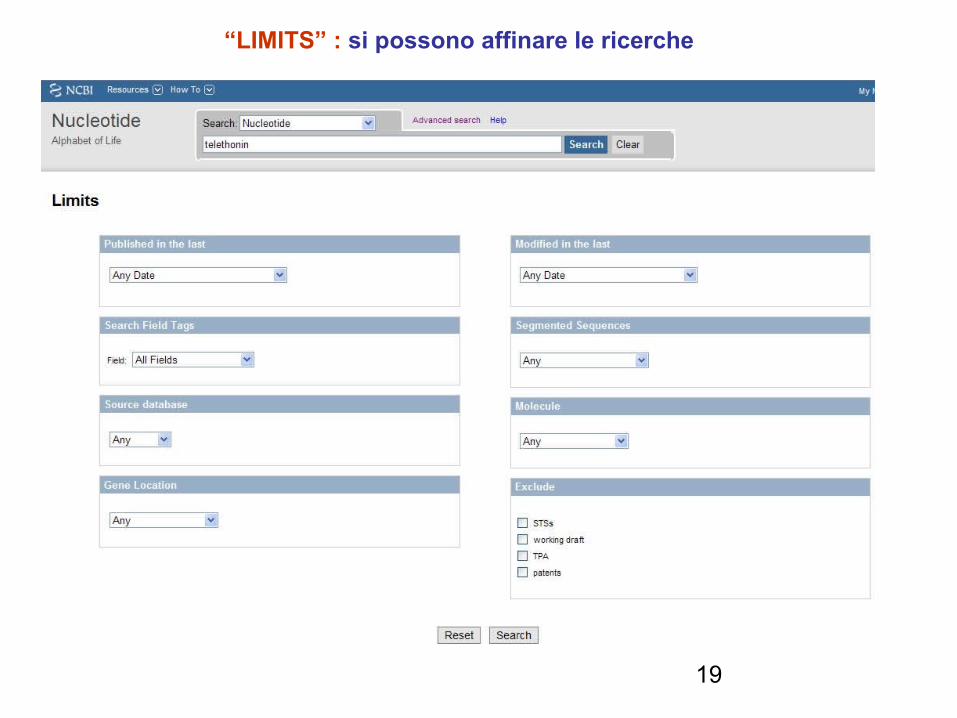
“LIMITS” : si possono affinare le ricerche
20
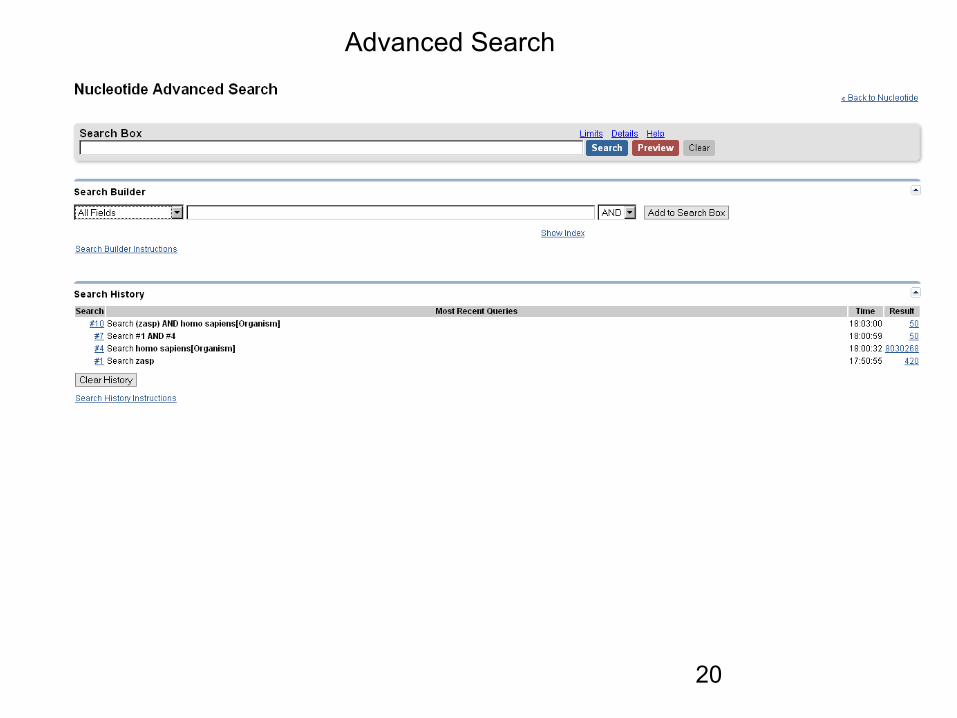
Advanced Search

21
Nome ufficiale del gene
22
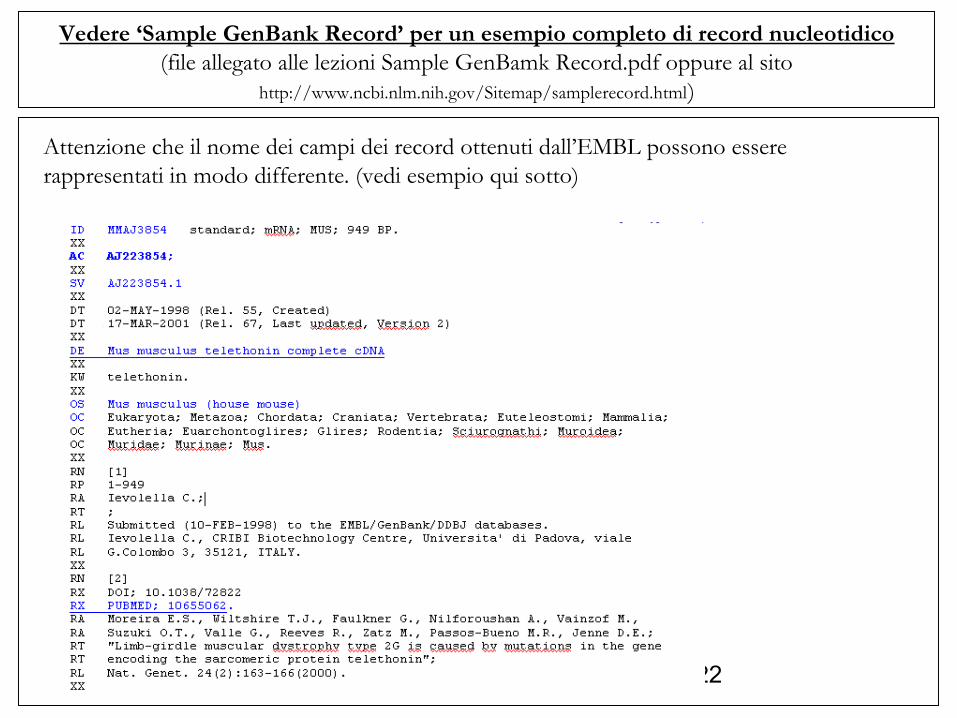
Vedere ‘Sample GenBank Record’ per un esempio completo di record nucleotidico(file allegato alle lezioni Sample GenBamk Record.pdf oppure al sito
http://www.ncbi.nlm.nih.gov/Sitemap/samplerecord.html)
Attenzione che il nome dei campi dei record ottenuti dall’EMBL possono essere rappresentati in modo differente. (vedi esempio qui sotto)

23
In questo corso ci limiteremo ad approfondire solo alcuni contenuti di particolari campi (fra parentesi gli ‘headers’ dei campi utilizzati dall’EMBL)
ACCESSION (AC): codice identificativo del record.SOURCE (OS): abbreviazione del nome dell’organismo (specificato poi meglio qui sotto). -ORGANISM (OC): The formal scientific name for the source organism (genus and species, where appropriate) and its lineage, based on the phylogenetic classification scheme used in the NCBI Taxonomy Database.REFERENCE (RN): riferimenti bibliografici (nei relativi sottocampi).
FEATURES (FT): Regioni o siti della sequenza considerati interessanti. Descritti in più ‘sottocampi’. I più importanti: - source: in un record, può essere riportata una lunga sequenza. E’ possibile scrivere delle annotazioni a parti specifiche della sequenza facendo riferimento alla localizzazione seguita da una o più righe che iniziano con ‘/’ - gene: dati del relativo gene (se esiste ed è conosciuto): inizio e fine della sequenza, poi negli altri ‘sottocampi’, nome del gene ed eventuali link (db_xref). - 5’UTR: la sequenza 5’UTR (inizio e fine). - CDS: la sequenza codificante (inizio e fine) e poi negli altri sottocampi link al DB (protein_ID) (ad altri DB (db_xref), da ricordare link ad OMIM: /db_xref=‘MIM xx’ , traduzione (se conosciuta), - 3’UTR: la sequenza 3’UTR (inizio e fine).
ORIGIN (SQ) : la sequenza scritta come stringa di caratteri.

24
LOCUS MMAJ3854 949 bp mRNA linear ROD 19-MAR-2001DEFINITION Mus musculus telethonin complete cDNA.ACCESSION AJ223854VERSION AJ223854.1 GI:3115294KEYWORDS telethonin.SOURCE Mus musculus (house mouse) ORGANISM Mus musculus Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Glires; Rodentia; Sciurognathi; Muroidea; Muridae; Murinae; Mus.REFERENCE 1 AUTHORS Valle,G., Faulkner,G., De Antoni,A., Pacchioni,B., Pallavicini,A., Pandolfo,D., Tiso,N., Toppo,S., Trevisan,S. and Lanfranchi,G. TITLE Telethonin, a novel sarcomeric protein of heart and skeletal muscle JOURNAL FEBS Lett. 415 (2), 163-168 (1997) PUBMED 9350988REFERENCE 2 AUTHORS Moreira,E.S., Wiltshire,T.J., Faulkner,G., Nilforoushan,A., Vainzof,M., Suzuki,O.T., Valle,G., Reeves,R., Zatz,M., Passos-Bueno,M.R. and Jenne,D.E. TITLE Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin JOURNAL Nat. Genet. 24 (2), 163-166 (2000) PUBMED 10655062REFERENCE 3 (bases 1 to 949) AUTHORS Ievolella,C. TITLE Direct Submission JOURNAL Submitted (10-FEB-1998) Ievolella C., CRIBI Biotechnology Centre, Universita' di Padova, viale G.Colombo 3, 35121, ITALY

25
Struttura delle FEATURES o FT (Feature Table) (Regioni o siti della sequenza considerati interessanti):
Possono essere riportate più regioni particolari. Ognuna è caratterizzata dalla definizione (es. source, gene, 5’UTR ecc.) seguita dalla localizzazione (location) punto di inizio e di fine della regione, seguite da una o più righe che iniziano con ‘/’ e che riportano note caratteristiche di tale regione (Qualifiers).
Traduzione
Cross-Ref.

26
La regione della sequenza identificata come gene (inizio- fine) (in questo caso corrisponde alla sequenza completa)
CDS (coding sequence): la sequenza codificante inizia in 15 e finisce in 518
GENE ONTOLOGYhttp://www.geneontology.org/
Sequenza Proteina
Link al DB delle proteine
Se nella sequenza esistono regioni geniche, allora vengono riportati anche dati relativi al ‘gene’, alle regioni codificanti (CDS) e alla sequenza proteica
Link ad OMIM (database di malattie genetiche

27
Altri dettagli delle ‘Features’Source: in un record può essere riportata una lunga sequenza. E’ possibile scrivere delle annotazioni a parti specifiche della sequenza facendo riferimento alla localizzazione seguita da una o più righe che iniziano con ‘/’ con riportate particolari annotazioni specifiche.
5’ UTR: Qui è riporta (se si conosce) la localizzazione della sequenza NON codificante posta a monte dell’mRNA. In questo caso 1-36
3’ UTR: Qui è riporta (se si conosce) la localizzazione della sequenza NON codificante posta a valle dell’mRNA
CDS (coding sequence): la sequenza codificante inizia in 37e finisce in 540

28
IL FORMATO “FASTA” Spesso i programmi che effettuano analisi bioinformatiche sulle sequenze richiedono
che esse vengano date come input in questo formato particolare: FASTA è un formato per la descrizione di una sequenza “grezza”. Consiste
essenzialmente in una parte iniziale di intestazione, di solito limitata a una linea di testo, e da una o più linee che riportano una sequenza di DNA o di amminoacidi usando l’alfabeto standard. Ecco un esempio:
L’intestazione (la prima riga del file precedente) `e riconoscibile perchè ha inizio con il simbolo ‘>’.
Il testo che segue tale simbolo nella stessa riga può essere strutturato liberamente: di solito, la prima cosa che si trova scritta `e un accession number, ossia l’identificatore della sequenza che ne permette il reperimento
>37463.f1 g83244 telethonin ecc.ACGTGACTGCTACGTACGGGCGTTACGACTGCTACGACGCATGCTATGTCGTAGCAGCCGTGTACACGTGTTTATTCGTAGGGCTTCTA
‘>’ Simbolo d’inizio della riga di intestazione
Sequenza
Riga di intestazione
Interruzione di riga

29
SEQUENZAPer recuperare la sequenza nucleotidica in formato FASTA

30
Database ‘NON RIDONDANTI’ : RefSeq, UniGene, Gene
Come già detto: nei database primari sono inserite tutte le sequenze conosciute ottenute sperimentalmente e/o ricostruite.
La stessa regione genomica o lo stesso trascritto possono essere stati sequenziati più volte. Quindi ci aspettiamo, in molti casi, che la stessa sequenza sia presente più volte.
Per evitare problemi di ridondanza sono stati creati dei database ‘semplificati’ senza ripetizioni di informazioni. In particolare:
In RefSeq sono rappresentate, in modo non ridondante, tutte le sequenze genomiche, sequenze di mRNA e di proteine.
In UniGene Sono rappresentate in modo non ridondante , le sequenze ottenute dal sequenziamento dei trascritti (mRNA)
Gene: è un sottoinsieme di RefSeq con rappresentate solo le sequenze geniche.

31
UniGene: An Organized View of the Transcriptome.Each UniGene entry is a set of transcript sequences that appear to come from the same transcription locus (gene or expressed pseudogene), together with information on protein similarities, gene expression, cDNA clone reagents, and genomic location.
The Reference Sequence (RefSeq) collection aims to provide a comprehensive, integrated, non-redundant set of sequences, including genomic DNA, transcript (RNA), and protein products. RefSeq is a baseline for medical, functional, and diversity studies; they provide a stable reference for genome annotation, gene identification and characterization, mutation and polymorphism analysis, expression studies, and comparative analyses RefSeq are derived from GenBank records but differ in that each RefSeq is a synthesis of information, not an archived unit of primary research data. Similar to a review article in the literature, a RefSeq represents the consolidation of information by a particular group at a particular time.

32
Entrez Gene is NCBI's database for gene-specific information. It does not include all known or predicted genes; instead Entrez Gene focuses on the genomes that have been completely sequenced, that have an active research community to contribute gene-specific information, or that are scheduled for intense sequence analysis. The content of Entrez Gene represents the result of curation and automated integration of data from NCBI's Reference Sequence project (RefSeq)
Gene (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene)
Continua
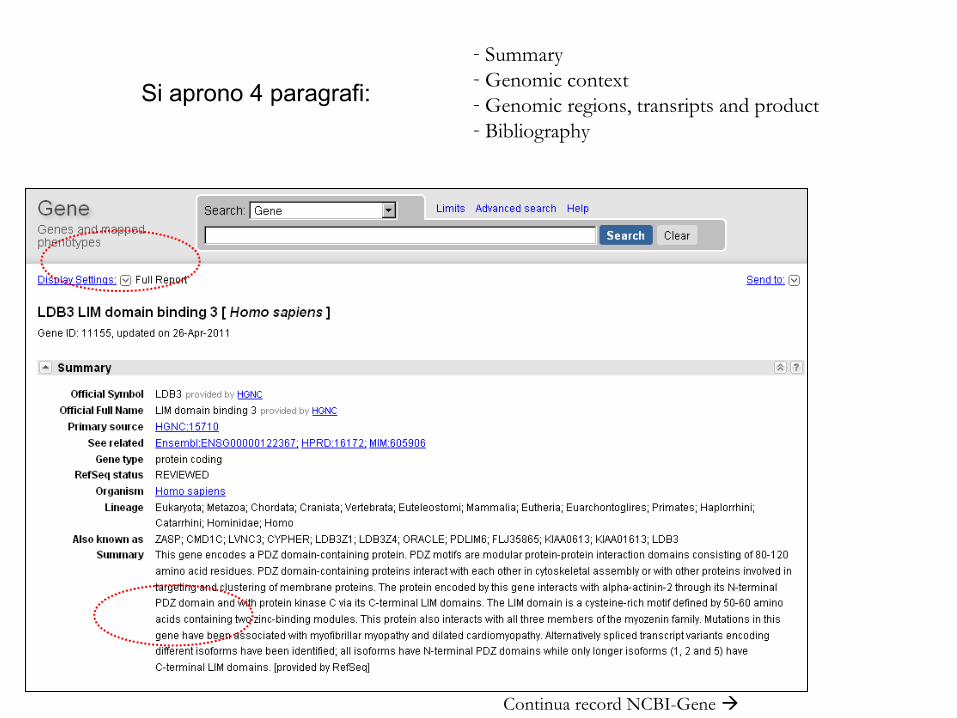
33Continua record NCBI-Gene
- Summary- Genomic context- Genomic regions, transripts and product- Bibliography
Si aprono 4 paragrafi:
34
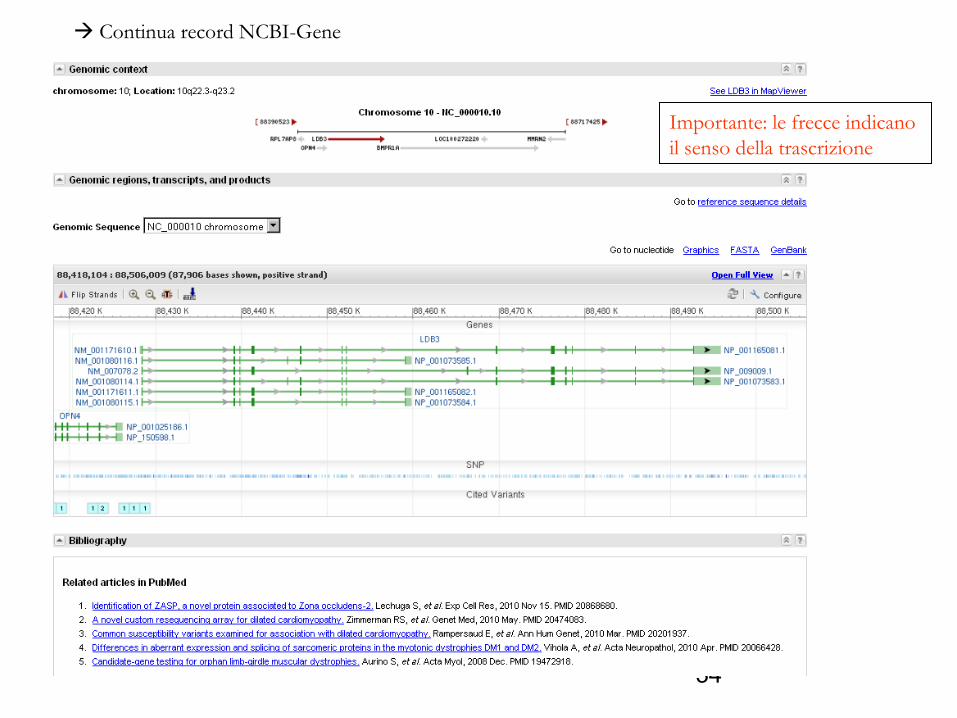
Continua record NCBI-Gene
Importante: le frecce indicano il senso della trascrizione

35
Vengono riportate 6 isoforme (varianti dello stesso gene), dovute a splicing alternativo dello stesso gene
Osservate gli introni e gli esoni, le regioni codificanti e le regioni UTR
Introni: sono rappresentati dalle linee più sottili
Domande: Quanti introni, quanti esoni sono rappresentati ?Il gene è codificato dal filamento ‘+’ (forwars) oppure dal filamento ‘-’ (reverse)?Qual è il senso della trascrizione?Quante isoforme sono visibili ?Le diverse isoforme sono dovute a splicing alternativo o a differenti metodi di traduzione?
Esoni: sono rappresentati dalle linee più spesse
Importante osservare il senso della trascrizione: un gene può essere codificato dal filamento ‘senso’ (detto anche ‘+’ o ‘forward’) o dal filamento ‘antisenso’ (detto anche ‘-’ o reverse)

36
Record simile ai records del DB nucleotide
Ricerca sul DB Protein dell’NCBI
37
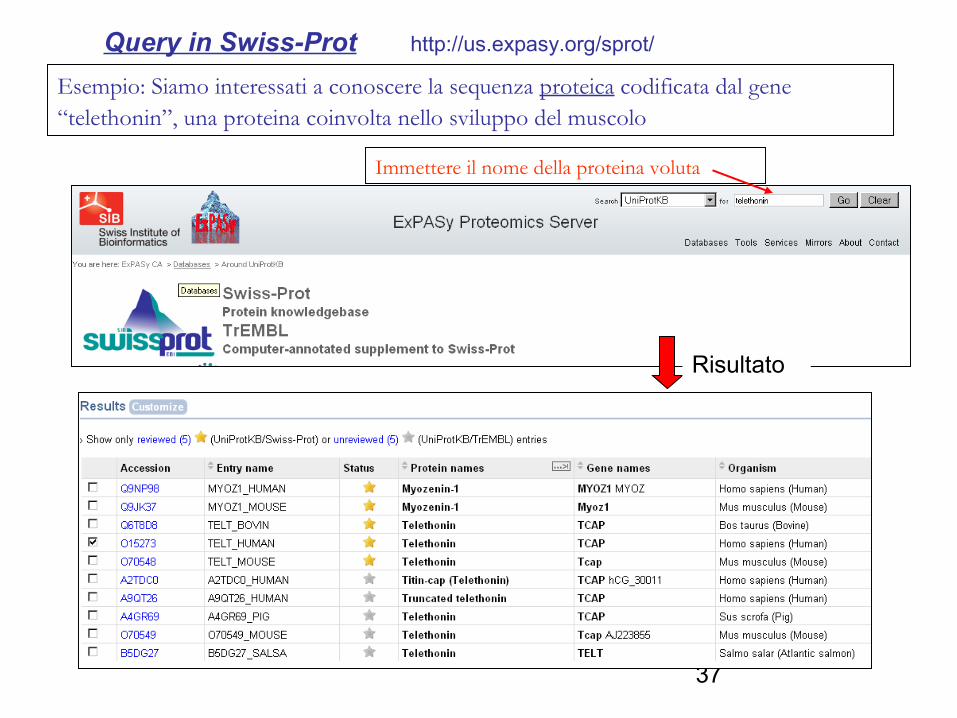
Esempio: Siamo interessati a conoscere la sequenza proteica codificata dal gene “telethonin”, una proteina coinvolta nello sviluppo del muscolo
Query in Swiss-Prot http://us.expasy.org/sprot/
Immettere il nome della proteina voluta
Risultato

38Continua record Swiss-Prot
Si ottiene un foglio html con diversi paragrafi (qui, sono riportati solo alcuni di questi)
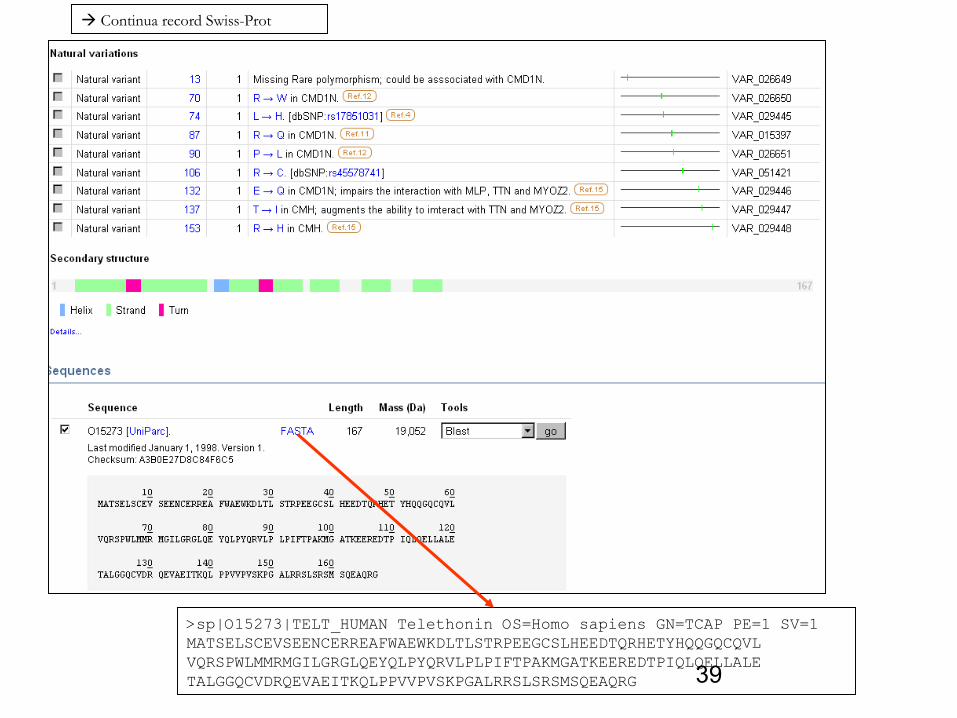
39
Continua record Swiss-Prot
>sp|O15273|TELT_HUMAN Telethonin OS=Homo sapiens GN=TCAP PE=1 SV=1 MATSELSCEVSEENCERREAFWAEWKDLTLSTRPEEGCSLHEEDTQRHETYHQQGQCQVL VQRSPWLMMRMGILGRGLQEYQLPYQRVLPLPIFTPAKMGATKEEREDTPIQLQELLALE TALGGQCVDRQEVAEITKQLPPVVPVSKPGALRRSLSRSMSQEAQRG
40
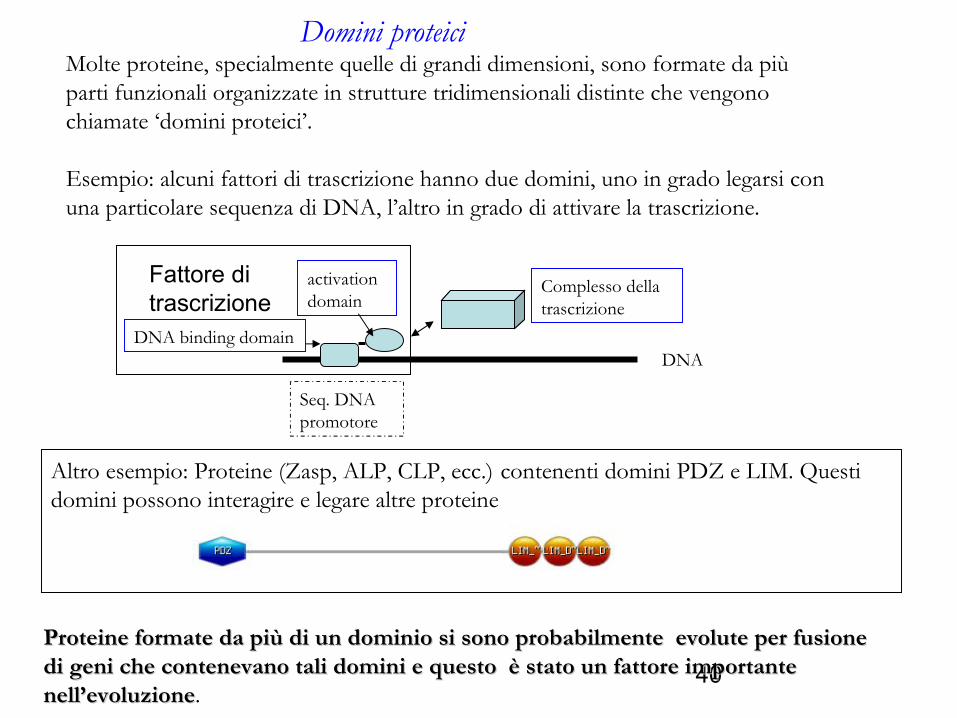
Molte proteine, specialmente quelle di grandi dimensioni, sono formate da più parti funzionali organizzate in strutture tridimensionali distinte che vengono chiamate ‘domini proteici’.
Esempio: alcuni fattori di trascrizione hanno due domini, uno in grado legarsi con una particolare sequenza di DNA, l’altro in grado di attivare la trascrizione.
Domini proteici
Proteine formate da più di un dominio si sono probabilmente evolute per fusione Proteine formate da più di un dominio si sono probabilmente evolute per fusione di geni che contenevano tali domini e questo è stato un fattore importante di geni che contenevano tali domini e questo è stato un fattore importante nell’evoluzionenell’evoluzione.
Altro esempio: Proteine (Zasp, ALP, CLP, ecc.) contenenti domini PDZ e LIM. Questi domini possono interagire e legare altre proteine
Complesso della trascrizione
DNA
Seq. DNA promotore
DNA binding domain
activation domain
Fattore di trascrizione

41
Esempi: Domini LIM associati ad altri domini(Sono riportate solo alcune strutture proteiche contenenti il LIM domain)
PFAM: http://pfam.sanger.ac.uk , PROSITE: http://www.expasy.org/prosite , SMART: http://smart.embl.de/ , InterPro: http://www.ebi.ac.uk/interpro/
sono database contenenti domini funzionali delle proteine

42
Esempio: voglio ricerca i domini presenti nella proteina ZASP
PfamThe Pfam database is a large collection of protein families. Proteins are generally composed of one or more functional regions, commonly termed domains. Different combinations of domains give rise to the diverse range of proteins found in nature. The identification of domains that occur within proteins can therefore provide insights into their function.
Continua

43
Continua da scelta PDZ Domain
Possono essere visualizzati le principali architetture proteiche che possiedono domini PDZ

44
Purtroppo non esiste un modo univoco per indicare un gene (esempio potete trovare scritto ‘subunit 4’ o ‘subunit iv’ (nella prima esercitazione affronterete questo problema)), anche i geni che io ho chiamato telethonin o zasp possono essere scritti in modi differenti (tcap , LDB3) . Questo crea confusione e non facilita la ricerca informatica
The Human Genome Organisation (HUGO) (è una organizzazione scientifica internazionale che promuove e sostiene le collaborazioni internazionali nella genetica umana) ha istituito un comitato allo scopo di dare un unico nome significativo a tutti i geni umani. Con questo intento è stato costruito il database HGNC (HUGO Gene Nomenclature Committee)
45
Oltre che esistere differenti nomi per lo stesso gene/proteina, possono esistere o essere adottati anche differenti modi per descrivere le loro funzioni e le loro localizzazioni.A volte è corretto assegnare differenti funzioni ad una molecola biologica (spesso una proteina svolge più di una funzione), per non generare confusioni è però necessario usare una terminologia univoca per ogni funzione.Per questo motivo è stato fondato il database GeneOntology che fornisce una definizione precisa del ruolo svolto dalle singole proteine tramite un vocabolario (delle ontologie) che consenta di definire in modo corretto e non arbitrario il o i processi biologici cui una proteina partecipa, la/e sue funzioni molecolari e la/e sue localizzazioni cellulare.Troverete spesso link con la sigla ‘GO’ questi rimandano al database della gene ontology

46
Mutazioni (alterazioni della sequenza nucleotidica di un gene) possono riflettersi in alterazioni della funzionalità della proteina da esso codificata. Questo mutazioni possono causare le cosiddette malattie genetiche.
Esempio: una mutazione a carico del gene della β globina fa sì che una particolare base del gene venga sostituita con un’altra, ciò altera il codone e nella proteina ciò si riflette nella sostituzione di un glutamato con una valina e in una ridotta funzionalità della proteina che causa una malattia genetica detta anemia a cellule falciformi (anemia falciforme).
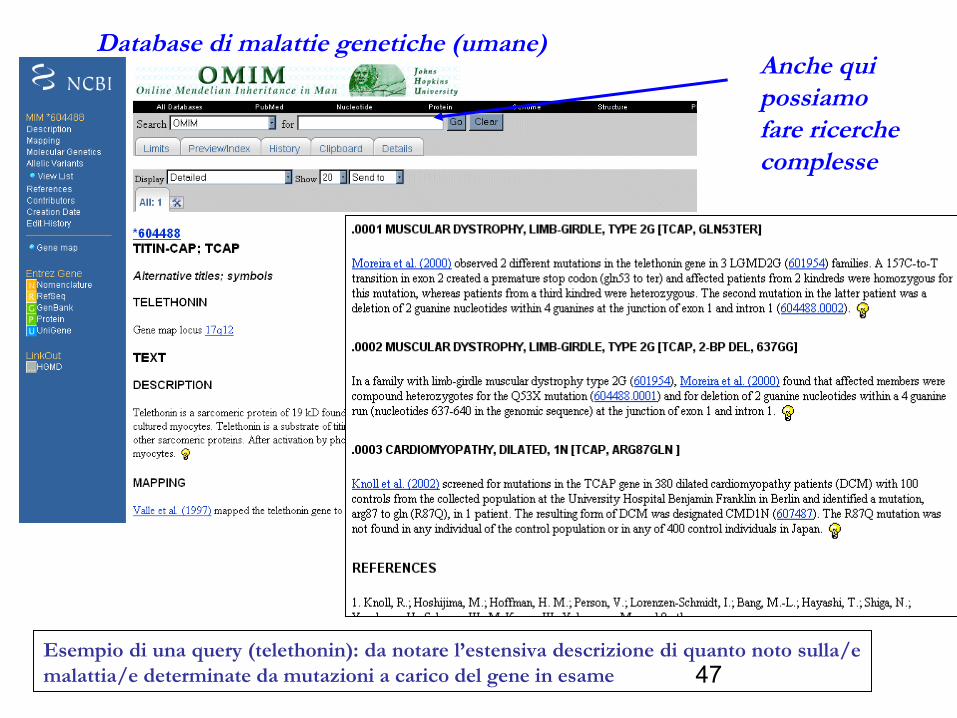
Il database OMIM cataloga le malattie genetiche, fornisce descrizioni particolareggiate delle malattie e delle possibili cause (mutazioni).
47
Database di malattie genetiche (umane)Anche qui possiamo fare ricerche complesse
Esempio di una query (telethonin): da notare l’estensiva descrizione di quanto noto sulla/e malattia/e determinate da mutazioni a carico del gene in esame

48


















