ALLINEAMENTO DI SEQUENZE -...
49
1 ALLINEAMENTO DI SEQUENZE
Transcript of ALLINEAMENTO DI SEQUENZE -...

1
ALLINEAMENTO DI SEQUENZE

2
DATABASE DI SEQUENZE
RICERCA
SIMILARITA’Ricerca dei record che hanno le sequenze più “simili” ad una sequenza fornita come query
TESTUALERicerca dei record i cui campi soddisfano determinati criteri (hanno certi valori)
Abbiamo già visto nelle lezioni precedenti Oggi iniziamo studiare questo metodo
di ricerca

3
Acidi nucleici e proteine sono costituiti da sequenze lineari rispettivamente di nucleotidi e di aminoacidi; entrambi possono essere rappresentati da singole lettere.
È quindi possibile rappresentare acidi nucleici e proteine come stringhe di lettere e perciò, usando programmi informatici, trattarli come qualsiasi stringa di caratteri.
La stringa di caratteri è soltanto una rappresentazione semplificata del corrispondente acido nucleico o proteina.
CONSIDERAZIONI GENERALI
In questa parte del corso cercheremo di dare risposte alle domande:
Cosa significa il termine ‘similarità’ tra due sequenze ?
A cosa servono le ricerce di sequenze ‘simili’ ?

4
A) Le due sequenze hanno ambedue 11 A, 9 T, 13 C, 7 G; (sono simili ?)ACTTCACCCTAGCTTTCTCGACTGCAGAGAAACTAGCCGACAGCACGTGGCTTACTCACTACCAGTTCTCACAGAATGCA
B) Queste due seq. hanno lo stesso contenuto in basi e se sovrapposte presentano 22 basi allineate (più del 50%) è casuale o possono essere considerate simili?ACTTCACCCTAGCTTTCTCGACTGCAGAGAAACTAGCCGA| | | | | | | || | | | | || | || | | ||ATTCCCCACGATCGTTTCCTAGTCCGGAAACACAACCGGA
C) Anche queste due sequenze presentano 22 basi allineate sovrapposte, come le consideriamo?ACTTCACCCTAGCTTTCTCGACTGCAGAGAAACTAGCCGA | || |||||||||||||||||| | TAGTAGCCGACCATTTCTCGACTGCAGAGAATTACTTCAC
D) Due sequenze sovrapposteACTTTCACCCTAGCTTTCTCGACTGCAGAGAAACTAGCCGA | ||||| | || || |CAAAGCGCGAAAACTTTCACCCTAGCTTTCTCGACTGCAGAG
le due stesse sequenze traslate e sovrapposte TTCACCCTAGCTTTCTCGACTGCAGAGAAACTAGCCGA |||||||||||||||||||||||||||CAAAGCGCGAAAACTTTCACCCTAGCTTTCTCGACTGCAGAG
Possiamo ricavare alcune considerazioni sulla similarità tra queste coppie di sequenze ?

5
Prima di affrontare l’allineamento di sequenze, cerchiamo di capire perché è importante ricercare sequenze simili.
Cos’è che determina le variazioni di sequenze ?
Cosa sono le sequenze omologhe, paraloghe e ortologhe ?
Dagli esempi riportati si intuisce che per determinare quanto due sequenze siano simili, è necessario trovare un modo per allineare le due sequenze e che, per ogni allineamento significativo trovato, fornisca dei dati per la stima della similarità.
E’ evidente che l’allineamento non può avvenire con la semplice sovrapposizione delle due sequenze.
In questo corso, prima vedremo un semplice metodo di allineamento di sequenze (dot-matrix) e poi altri metodi più complessi (blast, blat)

6
Mutazioni puntiformi - gap (Delezioni o inserzioni)
Mutazioni puntiformi (sNPS)
Mutazione senso CysSer
Mutazione silente
Met Glu Pro Cys Leu Arg Glu eccATG GAG CCT TGT TTG CGT CAG ecc
ATG GAA CCT TCT TTG CGT CAG eccMet Glu Pro Ser Leu Arg Glu ecc
ATG GAG CCT TGA TTG CGT CAG eccMet Glu Pro Ter
Mutazione ‘non senso’
Evoluzione GenicaPrincipali motori responsabili dell’evoluzione genica
-Duplicazione dei geni (nelle cellule germinali, intere regioni genomiche (anche contenenti geni) possono essere duplicate. Inizialmente, l’organismo che si svilupperà, può avere due geni che producono la stessa proteina (a volte questo è conveniente spesso è letale).
-Mutazione del codice genetico (mutazioni puntiformi, inversioni, delezioni o inserzioni fanno variare il codice genetico dei singoli geni)

7
Notare che, in caso di inserzioni o delezioni di lunghezza non multipla di 3, la sequenza della proteina a valle del gap è completamente differente dall’originale
I gap introducono delle discontinuità e creano una maggior difficoltà negli allineamenti; gli algoritmi di allineamento devono analizzare in modo esaustivo tutte le possibili inserzione dei gap.
Gap: delezioni o inserzioni (sfasano il frame di traduzione se non sono multipli di 3)
Met Glu Pro Cys Leu Arg GluATG GAG CCT TGT TTG CGT CAG
ATG GAG CCT TGG TTT GCG TCA GMet Glu Pro Trp Phe Ala Ser ecc.
ATG GAG CCT TGA TTT GCG TCA GMet Glu Pro Ter
Inserzione di ‘G’
Inserzione di ‘A’
Si crea uno stop prematuro
Met Glu Pro Cys Leu Arg GluATG GAG CCT TGT TTG CGT CAG
ATG GAG CCT TGT TTC GTC AGMet Glu Pro Cys Phe Val ecc.
Delezione della ‘G’
Cambiano tutti gli aminoacidi a valle

8
Organismo XGene A-Gene B-Gene C-ecc
Organismo XGene A-Gene B-Gene C-ecc
Organismo YGene A2-Gene B-Gene C-ecc
Speciazione(gene A2 da A)
Organismo YGene A2-Gene B-Gene C-ecc
Organismo ZGene A3-Gene B-Gene C-ecc
Speciazione (gene A3 da A2)
Evoluzione genica e omologia
In biologia il termine omologia ha il significato particolare di indicare che due strutture, ad esempio due organi, hanno un'origine evolutiva comune
L'omologia presuppone quindi l'esistenza di un organismo ancestrale comune da cui le strutture omologhe si sono evolute
La speciazione (origine di una nuova specie da una già esistente) implica il cambiamento di alcune funzioni geniche le quali derivato dal ‘cambiamento’ dei rispettivi geni.
Due geni omologhi devono avere un gene ancestrale da cui sono entrambi originati
Nella figura, i geni A, A2, A3 sono omologhi

9
Nel caso di due geni omologhi derivati da duplicazione genica si parla di geni paraloghi, viceversa, quando due geni omologhi sono derivati da speciazione (ossia duplicazione di specie) allora si parla di geni ortologhi. Deve comunque essere chiaro che sia i paraloghi che gli ortologhi sono omologhi.
Geni A e B sono ortologhi
C e D sono paraloghi
La duplicazione genica è l'evento più frequente nell'evoluzione di nuovi geni o funzioni
Geni paraloghiGeni ortologhi
Gene A Gene A
Gene C Gene D
Gene A
Gene A’
Gene B Gene A’
Duplicazione genica
Duplicazione (1% dei geni / milione anni)
Divergenza per mutazioni (0.1% / milione di anni)

10
L’analogia indica che due strutture hanno una funzione simile, ma che si sono evolute indipendentemente: ad esempio l'ala di un uccello e quella di una farfalla. In questo caso la similarità è determinata da una convergenza adattativa anzichè dalla stessa origine evolutiva.
Similarità, Omologia e Analogia (spesso si fanno confusione tra questi termini)
L’omologia è un aspetto qualitativo che riguarda più propriamente la “funzione” delle sequenze ed indica un’origine filogenetica comune Ad esempio l'ala di un uccello e la pinna anteriore di una foca sono omologhi perchè si suppone che si siano entrambi evoluti dall'arto anteriore di un rettile ancestrale.
La similarità è un aspetto quantitativo che indica (fissato un criterio comparativo, % identità, % mutazioni conservative...) un livello di somiglianza tra le sequenze.
Il termine omologia si applica anche a sequenze di acidi nucleici e proteine.
La similarità tra due sequenze è generalmente dovuta ad una origine evolutiva comune per cui molto spesso i termini "similarità" e "omologia" si confondono.
Quando non si è certi di un'origine evolutiva comune si dovrebbe parlare di similarità.
Un errore ancora più grave è di parlare di ‘percentuale di omologia’, che non ha nessun senso.

11
Sequenziamento geni Database di sequenze geniche (o proteiche) con funzione conosciuta
Ricerca di similarità
Assegnazione e annotazione della probabile funzione del gene
Determinazione della funzione di nuove proteine mediante allineamento
Il sequenziamento sistematico di interi organismi e di interi trascrittomi, hanno permesso di identificare migliaia di geni, molti dei quali codificano per proteine con funzione ignota. L’analisi di similarità (e quindi l’allineamento) con proteine già note può fornire la comprensione della loro funzione.
PERCHE’ CERCARE SEQUENZE SIMILI ?
Alcuni esempi:- Individuare i geni in genomi ‘neo’ sequenziati- Scoprire nuovi geni- Assegnare particolari funzioni geniche - ricostruire la filogenesi di organismi- ecc. ecc.

12
L'analisi comparata classica confronta l’anatomia degli organismi per determinare la loro evoluzione biologica.Esempio: confrontando l'anatomia dei vertebrati, si può dedurre che i mammiferi sono nati da un gruppo di rettili nati a loro volta da un gruppo di anfibi nati a loro volta da un gruppo di pesci.Applicando metodi comparativi, in molti casi, si può riconoscere anche l'origine degli organi; esempio i polmoni che si sono originati dalle vesciche natatorie di alcuni pesci, ecc. ecc.
Analisi comparata a livello molecolare Confrontando sequenze proteiche in organismi diversi è possibile ricostruire passo dopo passo l'evoluzione delle proteine nel corso della filogenesi e capire come esse abbiano affinato le loro funzioni, oppure, come siano nate funzioni nuove.
Analisi comparata ed evoluzione biologica e filogenesi

13
Il problema dell'allineamento di sequenze è molto complesso e costituisce un campo di ricerca della bioinformatica in continua evoluzione.
I principali algoritmi (metodi computazionali) per allineare sequenze saranno affrontati in Bioinformatica 2, che è un corso fondamentale del corso di laurea in Biologia Molecolare. In questo corso ci limiteremo a spiegare in modo semplice i principali metodi di allineamento.
Allineamento mediante DOT MATRIX
Il metodo della dot matrix consiste nel creare una matrice in cui vengono confrontati tutti i possibili appaiamenti di ogni carattere delle due sequenze da allineare.
In termini pratici, una sequenza viene scritta orizzontalmente sul lato superiore della matrice, da sinistra a destra, ponendo ogni carattere in corrispondenza di ogni colonna. Similmente, la seconda sequenza viene scritta verticalmente sul lato sinistro della matrice, dall'alto in basso ponendo ogni carattere in corrispondenza di ogni riga. Nella figura la direzione delle sequenze è indicata dalle frecce.
Si riempie la matrice, annerendo le caselle che hanno nella corrispondente riga e colonna la stessa lettera. Si faranno N(righe) x M (colonne) operazioni di comparazione.

14
Se analizzassimo due sequenze identiche (cioè usiamo la stessa sequenza sia come sequenza orizzontale che come sequenza verticale) allora otterremmo una diagonale continua che parte dall'angolo in alto a sinistra per arrivare a quello in basso a destra
Analisi della matrice
Ovviamente oltre alla diagonale troveremmo molti altri puntini: per le sequenze proteiche ci dovremmo aspettare una casella positiva ogni 20, una ogni 4 invece in caso di sequenze nucleotidiche. Questo crea un notevole rumore di fondo.
Un modo per ridurre il rumore è di considerare la media dei valori presenti sulla diagonale all’interno di una determinata finestra (window) invece che considerare le singole caselle della matrice
Esempio: variando l’ampiezza della windows

15
Cerchiamo ora di spiegare la piccola diagonale che si può distinguere in prossimità dell'angolo in alto a destra: una diagonale posta in quella posizione indica che la parte finale della sequenza orizzontale è simile alla parte iniziale della sequenza verticale.Inoltre, considerando che la stessa parte della sequenza verticale è simile anche alla parte iniziale della sequenza orizzontale, possiamo dedurre che la prima parte e l'ultima parte della sequenza orizzontale sono simili, probabilmente generate da una duplicazione parziale del gene.In generale, quando si osservano due diagonali parallele si deve pensare ad una porzione della sequenza che è ripetuta.

16
La diagonale principale della figura mostra un'evidente interruzione nella parte centrale per poi continuare su una diagonale diversa, un po' più bassa.Questi salti di diagonale sono dovuti alla presenza di "gap", ossia di "buchi" in una delle due sequenze, cioè a segmenti di sequenza che sono presenti in una sequenza, ma non nell'altra. Nel caso del nostro esempio abbiamo una delezione nella sequenza orizzontale o, se si preferisce, una inserzione nella sequenza verticale.
In generale possiamo dire che:
- Gli allineamenti di una certa lunghezza appaiono come segmenti diagonali
-Le sequenze ripetute appaiono come segmenti diagonali paralleli
-I gap appaiono come salti in diagonale.
-Segmenti diagonali ortogonali rappresentano delle inversioni

17
Durante le esercitazioni utilizzerete il programma ‘DOTLET’ sviluppato da Marco Pagni e Thomas Junier, che essenzialmente utilizza l'approccio della Dot Matrix in un modo elegante e di facile uso.Il programma DotLet è accessibile al sito http://www.isrec.isb-sib.ch/java/dotlet/Dotlet.html

18
Con l'approccio della dot matrix viene semplicemente mostrata una matrice che deve essere interpretata visivamente (soggettivamente).
Abbiamo bisogno di un sistema che sia anche in grado di estrarre i dati dalla matrice e trovare automaticamente gli allineamenti migliori, cioè identificare il "percorso" migliore all'interno della matrice.
In pratica è necessario disporre di programmi che applichino appositi ALGORITMI DI ALLINEAMENTO (cioè metodi di come allineare le sequenze) ed applicare un CRITERIO DI SIMILARITA’ per stimare quanto ‘simili’ sono le sequenze allineate.
Quantificazione della SIMILARITA’ tra due sequenzeQuantificazione della SIMILARITA’ tra due sequenze
Un ALGORITMO E’ caratterizzato dall’insieme di tutte le operazioni (finite) applicate per risolvere il problema (determinazione di tutti i possibili allineamenti) e dalla Complessità, cioè da come varia il numero delle operazioni da effettuare al variare della dimensione del problema.
Il tempo di esecuzione di un programma che implementa l’algoritmo è proporzionale alla complessità dell’algoritmo.

19
si fa scorrere una sequenza sull’altra contando, per ogni spostamento, le identità tra le due sequenze
30 confronti (6x5)1+2+3+4....
4 identità:
Notare che lo scorrimento semplice non permette di individuare questo particolare allineamento:
AAKKQWAA-KQWLa complessità aumenta, se si considerano anche i possibili gap (inserzioni e delezioni).(l’algoritmo di scorrimento non è un buon algoritmo per allineare due sequenze)
Esempio: nella ricerca di un particolare record, in un database ordinato, col metodo dicotomico, l’algoritmo è dividere in due l’insieme e scegliere il sotto insieme in cui è presente il record voluto, reiterando questa operazione fino alla fine. La max. complessità in questo caso è data da (log2(n.record) +1)
Banale esempio di un algoritmo che determina la similarità tra due sequenze, scorrendo una sequenza sull’altra (algoritmo di scorrimento)

20
Funzione di score:
L’allineamento delle sequenze dovrebbe fornire un punteggio (score), col quale è possibile valutare (secondo i criteri impostati) il grado di similarità.
Ad esempio, con l’algoritmo di scorrimento, utilizzando una banale funzione di score che assegni ‘1’ se le lettere tra le due sequenze sono uguali o ‘0’ se diverse; per ogni allineamento, si ricava uno score dato dalla somma ottenuta da tutti gli appaiamenti. Il ‘miglior’ allineamento potrebbe essere quello che ha ottenuto il maggior punteggio.
Questa funzione di score però non rappresenta un criterio molto valido: non ha senso penalizzare tutti i mismatch allo stesso modo (in genere nelle proteine, la sostituzione tra aminoacidi carichi non comporta grandi cambiamenti strutturali, mentre la sostituzione di aminoacidi aromatici con altri carichi comporta spesso pesanti cambiamenti conformazionali.
Per questo sono state introdotte le matrici di sostituzione (vedi prossima slide)
Inoltre possono essere adottate particolari penalità (score) per l’apertura o l’estensione di un gap che si sommano allo ‘score’ totale
2) Criterio di Similarità
Adozione di criteri differenti funzioni di ‘score differenti score finali differenti
Come si può affermare che una sequenza è simile ad un’altra? Come possiamo ‘misurare’ la similarità ?

21
Le matrici di sostituzioneNel caso dell’allineamento di aminoacidi è opportuno applicare dei criteri di similarità che non si limitano a verificare l’identità assoluta ma tengano conto del fatto che gli aminoacidi possono essere più o meno simili tra loro. Aminoacidi molto simili possono essere indifferentemente sostituiti in una proteina senza alcuna variazione apprezzabile nella struttura della proteina.Per esempio acido aspartico (D) e acido glutammico (E) sono molto simili e molto spesso nel corso dell’evoluzione prendono il posto l’uno dell’altro nelle proteine. Al contrario acido aspartico (D) e triptofano (W) sono molto diversi e non sono assolutamente interscambiabili. E’ quindi ragionevole valutare differentemente la sostituzione (in generale il confronto) di D con E e di D con W.Ciò viene descritto in matrici quadrate di 20*20 caselle in cui si attribuisce un punteggio ad ogni possibile coppia di aminoacidi. Quanto più alto è il punteggio tanto più interscambiabili sono gli aminoacidi. Punteggi negativi penalizzano invece aminoacidi molto differentiEsitono due tipi di matrici di sostituzione: Matrici PAM e matrici BLOSUM (in questo corso non approfondiamo il loro significato, è sufficiente ricordare la loro funzione in generale)
Matrici PAM: si basano sulla frequenza con cui ciascun aminoacido può subire un evento di sostituzione calcolato mediante uno studio di filogenesi molecolare(esistono matrici PAM1, PAM10, ecc che si differenziano per i ‘passo evolutivi’ di cui noi non ci occuperemo e che approfondirete nei corsi successivi)
Matrici BLOSUM: sono invece basate su una banca dati (BLOCKS) di allineamenti multipli di segmenti proteici senza GAP.(anche in questo caso esistono differenti matrici BLOSUM adatte per allineamenti tra sequenze con differenti distanze filogenetica)

22
Esempio: matrice di sostituzione BLOSUM 50
Alanina Ala A
Arginina Arg R
Asparagina Asn N
Ac. aspartico Asp D
Asn + Asp Asx B
Cisteina Cys C
Glutamina Gln Q
Ac.glutammico Glu E
Gln + Glu Glx Z
Glicina Gly G
lstidina His H
lsoleucina Ile I
Leucina Leu L
Lisina Lys K
Metionina Met M
Fenilalanina Phe F
Prolina Pro P
Serina Ser S
Treonina Thr T
Triptofano Trp W
Tirosina Tyr Y
Valina Val V

23
Consideriamo i seguenti due differenti allineamenti delle stesse sequenze
Nel primo caso si hanno 14 identità (evidenziate dalle linee verticali che uniscono aminoacidi uguali) e tre sostituzioni conservative (es. A-I, S-T) distribuite su tutta la lunghezza della sequenza. Nel secondo caso si hanno 13 identità ed una sostituzione conservativa su una regione di 14 aminoacidi.Quale dei due allineamenti è da considerarsi migliore?
ALLINEAMENTI GLOBALI E LOCALIALLINEAMENTI GLOBALI E LOCALI

24
La ricerca di similarità di sequenze in banche dati è diventata un'operazione molto importante in diversi settori della biologia.I programmi che effettuano questa operazione si basano sul confronto sistematico di una sequenza di partenza (generalmente chiamata query) con ognuna delle sequenze del database.
Ricerca di similarità di sequenza in banche dati
Database di sequenze
Sequenza query Confronti
Abbiamo bisogno di un sistema in grado di trovare automaticamente gli allineamenti migliori utilizzando un opportuno algoritmo e valutando la similarità tra le coppie di sequenze.
Esempio con l’algoritmo di Smith & Waterman: per ogni coppia di sequenze, costruzione della matrice di allineamento ed identificazione del "percorso" migliore.
Generalmente, le operazioni eseguite da un programma di allineamento sistematico, per ogni sequenza del database, sono: * effettua l'allineamento con la sequenza query; * attribuisce un punteggio; * se il punteggio è alto lo memorizza; * ripete fino alla fine delle sequenze; * mostra la lista dei migliori allineamenti trovati.

25
UCSC genome browser• Questo browser genomico è stato sviluppato per fronteggiare il problema
dell’enorme aumento di dati genomici derivanti in particolare dai progetti di sequenziamento dei vertebrati.
• Ovviamente è poco utile avere lunghe stringhe di basi corrispondenti ai singoli cromosomi dei vari organismi.
• Per questo motivo l’UCSC genome browser fornisce una rapida visualizzazione grafica di ogni regione di genoma di qualsivoglia lunghezza assieme ad una grande quantità di informazioni come:
• geni noti, geni predetti, ESTs (expressed sequence TAGs), mRNA, geni omologhi di altri organismi, ecc..

26

27
Organismo di cui si vuole visualizzare la regione genomica
Durante il sequenziamento sitematico di un genoma, spesso vengono rilasciate versioni successive specialmente nella fase conclusiva del progetto; esse possono essere più o meno definitive. Qui si fa riferimento a varie versioni (release) del genoma umano.
Gruppo di organismi di interesse
Genome Browse

28
Pulsanti di spostamento sul genoma
Pulsanti per ingrandire o rimpicciolire l’area di interesse
Posizione attuale sul genoma Permette di “saltare” sulla
posizione digitata sulla finestra di sinistra
‘Cromosoma’, rappresentazione schematica e posizione attuale

29
ESTs
Grado di conservazione della sequenza nei differenti organismi
Varie sequenze allineate
Sequenze ‘ufficiali’ del gene presenti nei differenti database
esone 3’UTR
Fine sequenza codificante
Notare le regioni maggiormente conservate.(domanda: le regioni codificanti sono ben conservate?)

30
Le sequenze dei trascritti sono caratterizzate dagli esoni. Questi sono rappresentati con line spesse.
Ricordando che la sequenza codificante di un gene può risiedere nel filamento ‘+’ (forward) o nel filamento ‘-’ (reverse).
Il verso della sequenza codificante è mostrato tramite i segni ‘ >>> ‘ (forward) oppure ‘<<<‘ (reverse)
Durante l’esercitazione osservate, negli esempi che vi verranno dati, le regioni conservate. Noterete che le regioni codificanti sono generalmente molto conservate. Esistono, però, anche regioni intergeniche altamente conservate (quale potrebbe essere il motivo?).
esoniEcc.
introniEcc.
>>>>>Il verso della sequenza è forward (filamento +)

31
Moltissimi sono i campi a disposizione, essi possono essere visualizzati (dense-full o pack) o nascosti (hide) utilizzando le opzioni presenti nella parte inferiore
della pagina
Ci sono varie possibilità di visualizzazione di ogni informazione sul genome browser, si possono fare varie prove ad esercitazione

32
Genome Browser: Descrizione del gene scelto e link ai differenti tool e database

33
Genome Browser: Link allla sequenza genomica e ai dati di microarray

34
Blast è il programma più usato per fare ricerche di similarità in banche dati, è molto efficiente ed è in grado di portare a termine una ricerca di similarità con molte sequenze in pochi secondi
BLAST
In questo corso non entreremo in merito alla strategia computazionale applicata da questo programma (e da altri), ma ci limiteremo ad utilizzarlo e a capire il significato dei risultati.
Il programma Blast può essere installato e fatto funzionare in un PC locale (nel proprio PC) oppure è accessibile dalla rete ad esempio al sito http://www.ncbi.nlm.nih.gov/BLAST/ dove può essere direttamente utilizzato per effettuare ricerche di similarità in banche dati.
E' inoltre disponibile un dettagliato "Blast tutorial" dove possono essere trovate informazioni dettagliate sull'uso del programma e dell'algoritmo.
Considerando che molti database di sequenze, come ad esempio GenBank, contengono milioni di sequenze diverse è necessario disporre di programmi informatici in grado di portare avanti queste ricerche in modo automatico e veloce.

35
Due parole su come lavora BlastLe sequenze, all’interno dei database, sono viste come lunghe parole.I database sono preventivamente scanditi alla ricerca di corte parole (normalmente lunghe 3 aa per le proteine e 11 basi per i nucleotidi)Tutte le parole trovate sono memorizzate in modo ordinato in particolari file (‘database di parole’), inoltre, ad ogni parola sono associati tutti i relativi riferimenti (sequenza e posizione).
Come avviene la ricerca di similarità:La sequenza query viene vagliata in tutte le possibili corte parole trovando in maniera veloce le eventuali corrispondenze presenti nel database di parole. Trovati gli ‘hit’ (sequenze e posizioni dove esistono le corte parole) il programma cerca di allungare l’allineamento, verso sinistra e verso destra, ‘al meglio’, in base ai parametri impostati,
parola posizione (e sequenza) dove esiste la parola
AAA data, data, data
AAC data, data, data, data
Scansione e
memorizzazione dati
DatabaseCon molte sequenze
In questo caso non si utilizza lo scorrimento tra due sequenze, ma si utilizza indici e puntatori a corte parole che rende meno complesso l’algoritmo e quindi più veloce la ricerca

36
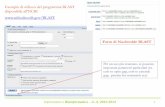
Pagina iniziale di Blast
Sono disponibili ricerche ‘particolari’ e/o database ‘particolari’
Differenti metodi e programmi di Blast

37
Sono state sviluppate diverse versioni del programma BLAST che consentono di ricercare sequenze sia a livello di acidi nucleici che di proteine:
blastp→cerca similarità in una banca dati di sequenze di aminoacidi a partire da una sequenza query di aminoacidi;
blastn→cerca similarità in una banca dati di sequenze nucleotidiche a partire da una sequenza query di nucleotidi;
blastx→cerca similarità in una banca dati di sequenze di aminoacidi a partire da una sequenza query di nucleotidi, dopo aver tradotto automaticamente la query in aminoacidi utilizzando tutti i possibili frame di lettura;
tblastn→cerca similarità in una banca dati di sequenze di nucleotidi a partire da una sequenza query di aminoacidi, traducendo automaticamente ogni sequenza della banca dati utilizzando tutti i possibili frame di lettura;
tblastx→cerca similarità in una banca dati di sequenze di nucleotidi a partire da una sequenza query nucleotidica, traducendo sia la query che le sequenze subject in aminoacidi, in tutte le possibili fasi di lettura.
In breve:

38
Alcuni criteri per la scelta del tipo di blast da adottare
Se si ha a disposizione una nuova sequenza nucleotidica e si vuole cercare eventuali sequenze di geni omologhi ‘conosciuti’ (sono disponibili le sequenze proteiche) si usa blastx (traduzione della seq. nucleotidica e ricerca di similarità in un database di proteine)
Nota: le sequenze nucleotidiche omologhe potrebbero essere ‘molto’ differenti per il codice genetico degenere
Per ricercare se una sequenza esiste già in un database si usa generalmente blastn o blastp rispettivamente per sequenze nucleotidiche e proteiche
Se si ricercano omologie con geni ancora sconosciuti (potrebbero esistere le sequenze nucleotidiche, ma non le proteiche) si usa tblastx: traduzione della sequenza query nucleotidica in proteina, traduzione di tutte le sequenze nucleotidiche del database in proteine e poi ricerca di similarità.
Esempio: un eventuale allineamento ‘significativo’ ottenuto con tblastx di una nuova sequenza nucleotidica contro un esteso database nucleotidico, potrebbe far scoprire nuovi domini o geni (attenzione però che questo programma è lento .. Perché ?...).
Ecc. ecc. ecc.

39
Nelle nostre esperienze in laboratorio utilizzeremo il programma Blast disponibile all’NCBI
(scegliendo le opzioni descritte in Basic Blast)http://www.ncbi.nlm.nih.gov/blast/Blast.cgi
Form relativa a nucleotide blast
Per un uso più avanzato, possono essere impostati parametri particolari (es. cost to open gap, cost to extende gap, penalty for mismatch ecc)
Program Selection: tre differenti modi (solo per le sequenze nucleotidiche).
1) Blastn classico
2) Megablast utilizza lunghe parole per cercare gli hits (adatto per ricercare lunghe sequenze molto simili) molto veloce
3) Discontiguous megablast: utilizza lunghe parole per gli hits, ma cerca anche parole non perfettamente uguali (veloce e permette ricerche di ‘scarse’ similarità

40
Sequenza query:> PBPRA1929 atgtctgatcacagtgcaatacatacaggacaatcaacaaaagacactgatcttaacaacacggaaaacccagaagcgaaacctgaaataaaaggagggcttgatggctacttctatattacagagcgtggcagtacagtaaaaatagaaatggtagccgggttaactacgtttttagcgatggtttact
La risposta che si ottiene può essere suddivisa in 4 parti:
a) Dati generalib) Allineamento graficoc) Listato delle sequenze con significativo
allineamentod) Dettaglio degli allineamenti ottenuti
a) Dati generali
b) Allineamento grafico

41
c) Listato delle sequenze con allineamento ‘significativo’
Significato delle colonne evidenziate- Max score: punteggio dell’allineamento locale più significativo’ (punteggio allto elevata similarità)- Total score: la somma dei punteggi di tutti gli allineamenti locali trovati tra la sequenza query e la sequenza del database- Query coverage: percentuale della sequenza allineata- E value (Expect): esprime la probabilità che l’allineamento trovato sia casuale. Più basso è maggiore è la probabilità che NON sia casuale. (dipende, oltre che dalla similarità, anche dalla numerosità delle sequenze in database)- Max identit: percentuale di identità dell’allineamento locale più ‘significativo’TTTCTCGACTGCAGAGAAA||||| ||| |||||||| TTTCTAGACTGCAGAGAAAIdentità=82% (16 / 19)
Ricordo che blast è un programma di allineamenti locali, quindi, per ogni confronto tra la sequenza query e una delle sequenza del database, potrebbero essere trovati più allineamenti differenti

42
d) Dettaglio degli allineamenti ottenuti
......continua con i dettagli degli altri allineamenti .....

43
Allineamento in più regioni (due) della sequenza query con la sequenza subjct

44
Risultato ricerca (con la stessa sequenza nucleotidica) tramite BLASTXRicordo che BLASTX cerca similarità in una banca dati di sequenze proteiche a partire da una sequenza query di nucleotidi, dopo aver tradotto automaticamente la query in aminoacidi utilizzando tutti i possibili frame di lettura;

45
E’ possibile allineare due sequenze (nucleotidiche o proteiche) tra loro
ESEMPIO BLAST 2 SEQUENCEESEMPIO BLAST 2 SEQUENCE

46
MultiallineamentoAllineamento contemporaneo di più sequenze
Abbiamo usato Blast per determinare l’allineamento migliore tra due sequenze e/o per ricercare tra le sequenze di un database quelle che possiedono una miglior similarità con una sequenza impostata come query.
Per studiare l'evoluzione delle proteine o dei geni, per eseguire studi di filogenesi è necessario fare una analisi comparata tra più sequenze. Per questo è necessario ottenere il miglior allineamento complessivo di tutte le sequenze relative allo studio.
Uno dei programmi capaci di eseguire multiallineamenti di sequenze (proteiche o nucleotidiche) è ClustalW (disponibile all’EBI)
(E’ possibile anche scaricare il programma ed installarlo nel proprio PC)

47
Qual’è l’algoritmo di ClustalW: allinea tutte le sequenze a coppie (tutte le possibili combinazioni delle due sequenze di input prese 2 alla volta) , poi assembla le coppie di allineamenti ottenendo alla fine un allineamento di tutte le sequenze.
ClustalW fornisce anche il probabile albero filogenetico (osservare anche le stime delle distanze evolutive; ricordo che Danio rerio è un pesce mentre gli altri sono mammiferi)
Es. di multiallineamento ottenuto con le sequenze delle proteine relative al gene tcap (telethonin) di 5 organismi differenti

48
Il confronto tra più sequenze permette anche di individuare eventuali regioni conservate sconosciute (domini, motivi o pattern funzionali)
Abbiamo già visto i domini funzionali (parte di proteina con una propria struttura tridimensionale ed in grado di svolgere una propria funzione).
Motivi funzionali sono invece corte sequenze aminoacidiche, formate da combinazioni di pochi aa, che forniscono alla proteina particolari proprietà. Es. siti di glicosilazione o fosforilazione
SITI CONSERVATISITI CONSERVATII siti funzionalmente più rilevanti mostrano un elevato grado di conservazione o risulteranno invariati in molte delle sequenze considerate
Al contrario i siti corrispondenti a regioni funzionalmente meno importanti mostreranno una maggiore variabilità

49
Dettaglio di un allineamento multiplo ed individuazione di motivi o pattern
In una delle prossime esercitazione simuleremo una ricerca di motivi funzionale sconosciuti