
Anemie Piastrinopenie Leucemie e Linfomi I tumori solidi · Classificazione morfologica delle...
35
- Anemie - Piastrinopenie - Leucemie e Linfomi - I tumori solidi
Transcript of Anemie Piastrinopenie Leucemie e Linfomi I tumori solidi · Classificazione morfologica delle...

- Anemie
- Piastrinopenie
- Leucemie e Linfomi
- I tumori solidi

Anemie
Barone C, Piraino B, Meduri S, Moschella E, Chirico V, Randazzo A, Di Bella C, La Rosa M,
Munafò C, Rigoli L
L’anemia è la patologia del sangue più comune e sintomo di numerose patologie ematologiche e
non, più o meno gravi.
Definizione
Ridotta massa ematica e concentrazione di Hb= < 2DS con conseguente ridotto trasporto di O2 ai
tessuti.
Principali cause di anemia
Definire il tipo di anemia è essenziale da un punto di vista diagnostico e terapeutico, anche se può
essere complesso. “Pallore” ed “ittero” costituiscono i segni che colpiscono a prima vista. Ognuno
di essi può essere espressione di una emopatia. Indubbiamente talune anemie sono proprie di
determinati periodi dell’età infantile come la malattia emolitica del neonato, l’ anemia dovuta ad
un’ allergia al latte vaccino, o l’anemia sideropenica da insufficiente apporto alimentare di ferro.
Per un corretto approccio diagnostico sono importanti: anamnesi, esame obiettivo, analisi di
laboratorio.

Classificazione morfologica delle anemie
Anemie microcitiche
(MCV < 80 fL)
Anemie normocitiche
(MCV da 80 a 94 fl)
Anemie macrocitiche
(MCV >94 fL)
•
Anemia sideroblastica
congenita
Anemia da carenza di ferro
Anemia delle malattie
croniche
Sindromi talassemiche
Anemia saturnina
•
Anemia emolitiche congenite
(difetti di membrana, difetti
enzimatici eritrocitari, hb instabili)
Anemia molitiche acquisite (da
anticorpi, a.emolitica
microangiopatica)
Emorragia
Sequestro splenico
•
Senza midollo megaloblastico
(anemia aplastica, Blackfan-
diamond, Ipotiroidismo,
Malattie epatiche, A.
diseritropoietiche)
Con midollo megaloblastico
(Deficit B12, Deficit ac.
Folico)
Esami di laboratorio necessari in presenza di bambino anemico
Esami di I° livello
Emocromo con reticolociti, sideremia, transferrina, ferritina, elettroforesi Hb
Esami di II° livello
G6PD, Aptoglobina, Test di Coombs, AGA, tTG, Ig, ricerca dell’HP, vitamina B12, acido folico,
piombemia, calcio, fosforo, fosfatasi alc, funzionalità renale
Esami di III° livello
eritropoietina, ricerca ac antivirus, biopsia midollare ed ossea

Anemia Sideropenica
Cause di carenza di ferro più frequenti in età pediatrica
Rapida crescita
Insufficiente assorbimento: ridotta introduzione; malassorbimento (celiachia)
Perdite aumentate: microemorragie croniche, intolleranza proteine latte vaccino, celiachia,
uso prolungato aspirina
Cause perinatali: emorragie feto-materno e gemello-gemello
Altre: diverticolo di Meckel , duplicazione intestinale
Terapia:
1) solfato ferroso per bocca
2) ferro carbonato per bocca
3) ferro destrano EV
Durata della terapia:
1) almeno 6 mesi per bocca
2) almeno 1 mese se EV
Controllo dell’efficacia:
1) controllare crisi reticolocitaria (1 settimana)
2) controllare incremento Hb (1 mese)
3) controllare ferritinemia (6 mesi)

Anemie Emolitiche
Le anemie emolitiche (A.E.) sono condizioni, eterogenee sotto il profilo eziopatogenetico,
caratterizzate da una riduzione della vita media dei globuli rossi (G.R.) circolanti rispetto ai normali
100-120 giorni. La diminuita sopravvivenza delle emazie (iperemolisi), comunque determinata,
comporta l’attivazione di meccanismi di compenso a livello midollare, con incremento dell’attività
eritropoietica fino a 7-8 volte i valori basali. Quando l’aumentata produzione di G.R. ne bilancia
l’accelerata distruzione, i livelli di emoglobina restano entro i limiti della norma (stato emolitico
compensato). Se, invece, l’intensità del processo emolitico è tale da superare le capacità di
compenso del midollo, eventualmente ridotte per la concomitante presenza di fattori che ne limitano
la risposta, si determina uno stato anemico (anemia emolitica).
Classificazione
In base ai meccanismi eziopatogenetici, le A.E. vengono classificate in forme ereditarie, causate da
difetti intrinseci dei G.R. (che possono interessare la membrana, il corredo enzimatico, le catene
globiniche) determinati geneticamente, e forme acquisite. Queste ultime, ad eccezione
dell’emoglobinuria parossistica notturna dovuta ad una mutazione somatica della cellula staminale
con produzione di un clone eritrocitario anomalo, sono causate da noxae esterne che esercitano la
loro azione lesiva su emazia metabolicamente e strutturalmente normali (Tabella I).
In numerose altre condizioni anemiche (associate a neoplasie diffuse, a leucemie, a linfomi, a
epatopatie, a insufficienza renale, ad artrite reumatoide ecc.), generalmente a patogenesi
multifattoriale, può essere presente uno stato iperemolitico, di solito evidenziabile esclusivamente
mediante studi di sopravvivenza eritrocitaria e poco rilevante ai fini del determinismo dell’anemia il
cui meccanismo patogenetico principale risiede nella ridotta produzione midollare di G.R. Tali
situazioni vengono definite di anemia con componente emolitica.

ANEMIE EMOLITICHE EREDITARIE
ANEMIE EMOLITICHE ACQUISITE
ANEMIE EMOLITICHE DA DIFETTO DELLA
MEMBRANA
• sferocitosi ereditaria
• ellissocitosi e piropoichilocitosi ereditarie
• altre forme ereditarie (stomatocitosi,
acantocitosi)
ANEMIE EMOLITICHE DA DIFETTO
ENZIMATICO
• della via glicolitica di Embden-Meyerhof
• dello shunt degli esoso-monofosfati
ANEMIE EMOLITICHE DA DIFETTO DELLE
CATENE GLOBINICHE
• varianti emoglobiniche stabili
• varianti emoglobiniche instabili
ANEMIE EMOLITICHE AUTOIMMUNI
• da anticorpi caldi
• da anticorpi freddi
• da agglutinine fredde
• da emolisine bifasiche
• “mixed”
• con test dell’antiglobulina diretto negativo
ANEMIE EMOLITICHE IMMUNOMEDIATE DA
FARMACI
• con meccanismo dell’immunocomplesso
• con meccanismo dell’adsorbimento del farmaco
• con meccanismo di induzione dell’autoimmunità
ANEMIE EMOLITICHE DA ALLOANTICORPI
• malattia emolitica del neonato
• reazioni trasfusionali emolitiche
EMOGLOBINURIA PAROSSISTICA NOTTURNA
ANEMIE EMOLITICHE MECCANICHE
• microangiopatiche
• da traumatismo cardiaco
• emoglobinuria da marcia
ANEMIE EMOLITICHE DA AGENTI CHIMICI
ANEMIE EMOLITICHE DA AGENTI INFETTIVI

Anemie Emolitiche da Difetto della Membrana Eritrocitaria
Le A.E. ereditarie da difetto della struttura e della funzione della membrana eritrocitaria
comprendono varie forme tradizionalmente raggruppate in base ai peculiari aspetti morfologici dei
G.R. patologici. La membrana eritrocitaria è costituita da un doppio strato di fosfolipidi, con
intercalate molecole di colesterolo non esterificato, e da varie proteine. Alcune di tali proteine (α- e
β-spettrina, actina, ankirina, proteine 4.1, 4.2, 4.9, p55, adducina ecc.), definite “periferiche” o
“estrinseche”, costituiscono il citoscheletro, applicato in forma di reticolo esagonale contro la
superficie interna della membrana, mentre altre (banda 3, glicoforine A, B e C, stomatina ecc.),
denominate “integrali” o “intrinseche”, sono situate nel suo contesto.
Sferocitosi Ereditaria
Con il termine di Sferocitosi Ereditaria (HS) si indica un gruppo eterogeneo di A.E. ereditarie,
dovute a varie mutazioni genetiche trasmesse come carattere autosomico dominante o recessivo, che
causano alterazioni qualitative o quantitative, di differente tipo ed entità, delle diverse proteine di
membrana del G.R., periferiche o integrali (anchirina e/o β-spettrina, banda 3, α-spettrina, banda
4.2). La riduzione della superficie della membrana, l’aumento della sua permeabilità al Na (con
formazione di stomatociti e di sferociti) e la diminuita deformabilità delle emazie che ne
conseguono provocano un’emolisi extravascolare per rimozione macrofagica che si realizza quasi
esclusivamente a livello splenico, a causa dell’ostacolo meccanico rappresentato per gli sferociti
poco deformabili dal tipo di microcircolo di tale organo, dell’ambiente metabolicamente
sfavorevole (per una minore concentrazione di glucosio e di ATP, per un pH più basso, per una
maggiore presenza di radicali liberi), e del contatto più intimo tra emazie e macrofagi
che qui si verifica. Il quadro clinico varia in relazione al tipo e alla gravità del difetto biochimico e
al conseguente grado di alterazione della membrana, potendosi osservare oltre alle forme tipiche,
con modesta anemia, ittero e splenomegalia, forme asintomatiche o lievi, con stato emolitico
compensato, forme gravi, con anemia intensa, e forme complicate da crisi aplastiche, da crisi
emolitiche, da litiasi biliare, da ulcere malleolari ecc. La diagnosi si basa, oltre che sui dati clinico-
anamnestici e sul rilievo degli indici di emolisi extravascolare e di compenso midollare, sulla
evidenziazione di sferociti e di stomatociti nello striscio periferico e sull’aumento delle fragilità
osmotiche eritrocitarie a fresco e dopo incubazione. Il test di lisi al glicerolo acidificato ed il pink
test sono stati proposti in alternativa a quello per le resistenze globulari, mentre il test
dell’autoemolisi viene raramente effettuato per la scarsa specificità. Una precisa caratterizzazione
della forma e dell’anomalia biochimica che la sostiene, con eventuale documentazione del difetto

genetico specifico, si può ottenere con valutazioni strutturali e funzionali delle proteine di
membrana (mediante tecniche elettroforetiche e di immunoblotting, eventualmente dopo digestione
triptica limitata della spettrina, tramite studi di trasporto ionico ovvero di rigidità e di fragilità della
membrana ecc.) e con appropriate indagini genetiche e di biologia molecolare (studio del DNA
genomico, del DNA “complementare” ecc.)
TERAPIA
SPLENECTOMIA: migliora nettamente la sopravvivenza eritrocitaria. Praticata dopo i 20
anni se anemia moderata (aumenta rischio di sepsi da meningococco, pneumococco,
hemophilus, soprattutto in età infantile). Splenectomia nell’ infanzia solo se anemia grave,
previa vaccinazione contro i suddetti batteri.
Anemie Emolitiche da Difetti Enzimatici
Al fine di conservare integre le proprie caratteristiche morfologiche e funzionali i G.R. devono
produrre una quantità adeguata di energia da utilizzare in vari processi metabolici ATP-dipendenti
(tra i quali la pompa Na/K che, estrudendo Na, impedisce l’iperidratazione cellulare) e devono
mantenere allo stato ridotto il Fe emoglobinico, la globina e le proteine di membrana. Tali esigenze
vengono soddisfatte metabolizzando il glucosio attraverso due vie principali:
- la glicolisi aerobia di Embden-Meyerhof, con la quale da ogni molecola di glucosio vengono
ricavate due molecole di ATP, utilizzate per soddisfare il fabbisogno energetico cellulare, e due
molecole di NADH, coenzima della metaemoglobina reduttasi principale che mantiene il Fe
emoglobinico allo stato bivalente;
- lo shunt degli esoso-monofosfati, attraverso il quale viene prodotto NADPH, coenzima sia della
metaemoglobina reduttasi accessoria sia, principalmente, della glutatione reduttasi nel processo di
rigenerazione del glutatione ridotto (GSH), il quale a sua volta funge da substrato per la glutatione
perossidasi che neutralizza le noxae ossidative, proteggendo dall’ossidazione la globina e le
proteine strutturali del G.R.
Deficit di glucosio-6-fosfato-deidrogenasi (G6PD)
Le A.E. da deficit di glucosio-6-fosfato-deidrogenasi (G-6-PD) costituiscono un gruppo eterogeneo
di forme dovute a svariate mutazioni del gene codificante per l’enzima posto sul cromosoma X, per
cui vengono trasmesse secondo la modalità ginecoforo-diaginica, con femmine solitamente
portatrici e maschi emizigoti affetti. Le mutazioni note sono oltre 350. Quadro di anemia emolitica
normocitica ad insorgenza acuta, l’emolisi è determinata da noxae ossidanti (farmaci o sostanze

chimiche, alimenti tipo fave, H2O2 rilasciata dai fagociti in corso di infezioni ecc.). Il difetto
enzimatico determina diminuita produzione di NADPH e, conseguentemente, scarsa disponibilità di
glutatione ridotto, per cui le noxae ossidative, non venendo neutralizzate, causano ossidazione dei
gruppi tiolici dell’Hb, delle proteine strutturali del G.R. e del glutatione che si complessano a
costituire i disolfuri misti. Si formano in tal modo i corpi di Heinz, inclusioni cellulari dislocate in
posizione epilemmatica, che comportano intrappolamento delle emazie nel filtro splenico e
successiva rimozione dal circolo.
La sintomatologia, come anche l’età a cui si manifesta l’emolisi, varia notevolmente in relazione
alla gravità del difetto enzimatico, potendosi osservare quadri che vanno una rapida insorgenza di
sintomi di anemia, dolori lombari, subittero, urine ipercromiche per alcuni giorni a febbre,
emoglobinuria e insufficienza renale acuta oppure a una condizione di emolisi cronica con crisi
parossistiche scatenate a distanza di ore o giorni dall’esposizione ad agenti ossidanti, a situazioni
ematologiche apparentemente normali con episodi emolitici intercorrenti.
La diagnosi si basa sui dati clinico-anamnestici, sul rilievo dei consueti indici di emolisi e di
compenso midollare, sulla dimostrazione dei corpi di Heinz (evidenziabili in vivo dopo lo stress
ossidativo e prima della crisi emolitica o indotti in vitro mediante incubazione delle emazie con
acetilfenilidrazina), sulla documentazione della sensibilità dei G.R. alle noxae ossidative (con il test
di riduzione della metaHb mediante blu di metilene, con il test di stabilità del glutatione ecc.), ma
soprattutto con lo studio della G6PD eritrocitaria, da eseguire a distanza dalla crisi emolitica. A tal
fine, oltre ai test di screening, possono essere utili le indagini citochimiche che evidenziano la
doppia popolazione di emazie, normale e carente, presente nelle donne eterozigoti in seguito
all’inattivazione random del cromosoma X. Diagnostico è il dosaggio dell’enzima, mentre indagini
più approfondite possono essere necessaire per l’identificazione delle sue varianti (elettroforesi, Km
per G-6-PD e NADP, stabilità termica, pH ottimale, risposta agli inibitori ecc.). La caratterizzazione
biochimica può essere affiancata o sostituita dall’analisi del DNA mediante tecniche di biologia
molecolare.
PROFILASSI: evitare esposizione a farmaci ossidanti e fave.
TERAPIA CRISI EMOLITICA: idratazione, diuretici, trasfusioni se anemia molto grave

Farmaci che possono causare anemia emolitica in
soggetti con deficit di G6PD
Farmaci che possono essere somministrati a
soggetti con deficit di G6PD e senza NSHA
Antimalarici
Pirimetamina con sulfadossina (Fansidar)
Pirimetamina con dapsone (Maloprim)
Primachina
Clorochina
Sulfonamidi
Sulfametossazolo, Altri sulfonamidi
Sulfoni
Dapsone, Tiazolosulfone
Altri composti antibatterici
Nitrofurantoina, Acido nalidixico
Antielmintici
Beta-naftolo
Miscellanea
Vitamina K, Naftalene (palline antitarma)
Blu di metilene Doxorubicina, rasburicas
Acido ascorbico
Aspirina
Colchicina
Isoniazide
Menadiolo
Fenitoina
Probenecid
Procainamide
Pirimetamina
Chinidina
Chinino
Trimetoprima
Deficit di piruvato-chinasi (PK)
Il deficit di piruvato-chinasi (PK) costituisce il difetto enzimatico meno raro della via glicolitica di
Embden- Meyerhof e viene trasmesso come carattere autosomico recessivo. Il malfunzionamento di
tale via metabolica comporta una ridotta produzione di ATP, uno squilibrio elettrolitico con perdita
del potassio cellulare, anemia di gravità variabile, normocromica e normocitica. Alle oltre 100
mutazioni descritte fa, riscontro una sintomatologia molto varia che si manifesta solo negli
omozigoti. Sono presenti i caratteristici indici laboratoristici di emolisi extravascolare e di compenso
midollare, mentre per la diagnosi vengono utilizzati vari test di screening, restando però conclusivo
il dosaggio dell’attività dell’enzima.
TERAPIA: splenectomia nei casi gravi

Anemia Megaloblastica
Nell'anemia megaloblastica il difetto principale risiede nell'alterata sintesi del DNA, quasi sempre
imputabile a una carenza di vitamina B12 e/o acido folico. L'anemia perniciosa o malattia di
Addison-Biermer rappresenta il 75% circa dei casi di ipovitaminosi B12: consiste nell'associazione
fra anemia megaloblastica e gastrite cronica atrofica autoimmune con achilia gastrica. La
megaloblastosi midollare è identica nel deficit di folati e in quello della vitamina B12 (cobalamina).
Il fabbisogno quotidiano di acido folico è stimato intorno ai 50 mg per un adulto, nei bambini e
negli adolescenti si raccomanda un'assunzione giornaliera variabile da 50 a 200 mg/24 h a seconda
dell'età. L'assorbimento della vitamina B12 è un processo complesso: legata alle proteine della
dieta la vitamina B12 è liberata nello stomaco in presenza di HCl e pepsina, legata al fattore
proteico R che ne impedisce l'assorbimento gastrico. Nell'ambiente alcalino del duodeno la
vitamina B12 è liberata dal fattore R dalle proteasi pancreatiche, legata quindi al fattore intrinseco
(FI), proteina prodotta dalle cellule parietali della mucosa gastrica. La carenza di vitamina B12 si
manifesta clinicamente dopo anni di bilancio negativo della vitamina. L'anemia da carenza dietetica
è rara nei paesi occidentali, ma possibile nei vegetariani di stretta osservanza, a meno che non
facciano uso di supplementi vitaminici. La gastrectomia totale costituisce un'indicazione per il
trattamento sostitutivo con vitamina B12 per tutta la vita. Nell'anemia perniciosa l'insufficiente
secrezione gastrica è dovuta alla distruzione della mucosa da parte di autoanticorpi anti-cellule
parietali gastriche, presenti in circa il 65-70% dei pazienti: gli autoanticorpi, diretti contro una
ATPasi di membrana deputata allo scambio tra ioni H+, secreti nel lume gastrico, e ioni K+,
innescano un processo flogistico cronico con conseguente distruzione delle ghiandole gastriche.
Nel 90% dei pazienti affetti da anemia perniciosa sono presenti autoanticorpi anti-FI; questi
anticorpi sono tuttavia presenti anche in alcuni soggetti sani e possono comparire in altre patologie
autoimmunitarie. L'anemia perniciosa è frequentemente associata ad altre patologia autoimmuni
come il morbo di Graves (30% dei casi), la tiroidite di Hashimoto (10% dei casi); la vitiligine, il
morbo di Addison, l'ipoparatiroidismo. L'anemia perniciosa è anche associata ad un aumento
dell'incidenza di carcinoma e carcinoidi gastrici. Le manifestazioni cliniche del deficit di vitamina
B12 sono suddivise in ematologiche (piastrinopenia e granulocitopenia), ed extraematologiche
(psichiatriche parestesie, disturbi dell'equilibrio, diminuzione o perdita della sensibilità,
cardiomegalia, glossite, iperpigmentazione, ingrigimento precoce dei capelli, di rado infertilità e
sterilità). La diagnosi di anemia megaloblastica poggia sulla dimostrazione di una diminuzione
dell'ematocrito e dell'emoglobina.. Il volume eritrocitario medio può raggiungere valori molto
elevati, anche superiori a 125 fL; l'MCH è solitamente aumentato mentre l'MCHC è diminuito;
spesso sono anche ridotti di numero le piastrine e i leucociti.

Drepanocitosi
La drepanocitosi è caratterizzata dalla combinazione di una grave anemia emolitica cronica
ereditaria con crisi vaso-occlusive, a carattere recessivo. L’anemia falciforme è caratterizzata dalla
presenza di un’ emoglobina anomala: HbS dovuta ad una mutazione puntiforme al codone 6 del
gene b globinico che determina la sostituzione dell’Ac glutammico in valina (GAG GTG Glu
Val). L’Hb S è scarsamente solubile e polimerizza quando deossigenata. I polimeri assumono
una forma allungata e determinano distorsione a falce (donde il nome di falcemia) e rigidità del
globulo rosso, che a sua volta è determinante dell’emolisi e delle crisi vasocclusive . Portatori del
tratto falciforme (eterozigoti) hanno una certa resistenza al spesso fatali malaria causata da
Plasmodium falciparum. Il gene S si riscontra con maggiore frequenza nei paesi dove
l’immigrazione di persone di origine africana o mediterranea è stata più intensa (Americhe, Canada,
Australia e più recentemente nel Nord Europa, dove gli immigrati costituiscono il 3% della
popolazione residente). In Italia sono presenti più di 2 milioni di immigrati, che nei prossimi anni
sicuramente raddoppieranno; molti di loro sono originari da regioni ad alta prevalenza di
emoglobinopatie, pertanto la distribuzione e la presenza della malattia drepanocitica è destinata ad
aumentare ed infatti trovare soggetti con malattia drepanocitica tra gli immigrati è un’evenienza
sempre più frequente, anche in regioni dove la malattia era quasi sconosciuta.
Il soggetto con anemia drepanocitica può manifestare disturbi estremamente eterogenei, variando da
forme asintomatiche a forme gravi (talassodrepanocitosi, drepanocitosi omozigote). Le
manifestazioni cliniche che caratterizzano la malattia sono l’anemia, il dolore causato dalle crisi
vaso-occlusive e le infezioni. Nel lattante la malattia si manifesta nel 30% dei casi, con alterazioni a
carico delle mani e dei piedi (sindrome mani-piede) che appaiono caldi, tumefatti e dolenti.
L'anemia generalmente si manifesta dopo il 4° mese di vita, cioè quando l'emoglobina fetale
(emoglobina normalmente nel feto) lascia il posto all'Hb S. I livelli di emoglobina oscillano tra i 7
ed i 10 g%, ma non occorre praticare trasfusioni perché l'Hb S cede l'ossigeno ai tessuti meglio
dell'emoglobina normale.

Altre condizioni in cui le trasfusioni sono necessarie: anemia marcata (Hb <6 g%); scompenso
cardiocircolatorio; gravi processi infettivi (meningiti, sepsi); episodi dolorosi che durino da più di 7
giorni; priapismo; ulcere agli arti inferiori; complicazioni a carico del sistema nervoso; perdita di
sangue con le urine ed anemizzazione grave; interventi chirurgici che richiedono anestesia generale;
esami radiologici con mezzo di contrasto; gravidanza; disfunzione cronica d’organo (cuore,
polmone, rene, fegato). Il dolore rappresenta "l’incubo" del soggetto drepanocitico. Le crisi
dolorose possono essere scatenate da: infezioni; febbre elevata; perdita di grandi quantità di liquidi
(vomito incoercibile, diarrea, sudorazione profusa); chetosi; soggiorno in alta montagna; freddo;
intensa fatica; traumi; uso di farmaci diuretici ed anestetici; condizioni in cui vi è diminuzione della
disponibilità di ossigeno.
Terapia
Analgesia (dal Paracetamolo per il dolore di intensità lieve fino ad arrivare alla Morfina per il
dolore con intensità severa); Idratazione; Profilassi antibiotica (le infezioni - broncopolmoniti,
osteomieliti, meningiti, sepsi etc, devono essere trattate tempestivamente, pertanto sono importanti
le vaccinazioni, sia quelle obbligatorie che quelle facoltative); Trasfusioni (sporadiche, regolari,
Exsanguinotrasfusioni); Sostegno psicologico.
Se le crisi colpiscono ripetutamente lo stesso organo con il passare del tempo si provoca un
deterioramento dell’organo colpito: osso/articolazioni (sindrome mani-piedi nel lattante; lo
scheletro può andare incontro ad osteomielite e necrosi asettiche); addome ( addome acuto,
infarti splenici,epatici, calcoli della colecisti); polmone (Chest syndrome caratterizzata da dolori
toracici, spesso violenti, tosse, e febbre); rene (isostenuri-necrosi papillare); SNC (accidenti
cerebrovascolari: nei bambini prevalgono gli infarti cerebrali, mentre negli adolescenti e negli adulti
le emorragie cerebrali); cute (ulcere malleolari); retina (emorragie –retinopatia proliferativi); corpi
cavernosi (priapismo).
L’unica possibilità concreta di guarigione è data dal trapianto di midollo osseo donato da un fratello
compatibile. Altra possibilità terapeutica è rappresentata dalla regolare eritroexchange o terapia
trasfusionale che, da un lato riduce i livelli di Hb S e quindi il rischio di crisi vaso-occlusive,
dall’altro lato si ha il rischio di contrarre malattie infettive, siderosi o alloimmunizzazione.
Un’alternativa terapaeutica farmacologica è l’ idrossiurea, un chemioterapico che ha la capacità di
incrementare l’emoglobina fetale riducendo notevolemente le crisi vasoccluisive. I pazienti in
trattamento con idrossiurea devono essere sottoposti a periodici controlli ematologici, poiché può
causare di una transitoria aplasia midollare (leucopenia, piastrinopenia e anemia), che si risolve con
la sospensione del trattamento.

Bibliografia
1. Autoimmune Hemolytic Anemia (AIHA) By J.L. Jenkins. The Regional Cancer Center. 2001
2. Worldwide prevalence of anaemia 1993–2005.World Health Organization (2008). Retrieved
2009-03-25.
3. Iron Deficiency Anaemia: Assessment, Prevention, and Control: A guide for programme
managers" Retrieved 2010-08-24
4. Sickle cell disease in children and adolescents: diagnosis, guidelines for comprehensive care,
and care paths and protocols for management of acute and chronic complications**revised at
the annual meeting of the sickle cell disease care Consortium, Sedona, AZ, November 10-12,
2001)

Piastrinopenie
Gallizzi R, Vicchio P, Deak A, Barone C, Manti S, Chirico V, Piraino B, Salpietro DC
Le piastrine sono piccole cellule anucleate (1-3 µm), derivanti dai megacariociti maturi e deputate
all’emostasi primaria. Il range ematico di normalità delle piastrine varia dai 150.000 ai 400.000 µ/L
e la loro emivita in circolo è di circa 9 giorni. Il mantenimento di tale range è il risultato di un
costante equilibrio tra trombopoiesi e senescenza/consumo. Il principale regolatore di tale equilibrio
è la trombopoietina (TPO), un fattore di crescita emopoietico sintetizzato dal fegato in maniera
costitutiva, ciò significa che, nel momento in cui i livelli ematici di piastrine e megacariociti maturi
si riducono, il TPO viene rimosso dal plasma in minore quantità, stimolando in questo modo la loro
produzione.
Si definisce piastrinopenia un numero di piastrine <150.000 µ/L. Innanzitutto, specie in assenza di
sintomatologia clinica suggestiva, andrebbe esclusa la pseudopiastrinopenia, fenomeno causato
dall’aggregazione piastrinica in presenza di EDTA (acido etilenediaminotetracetico), responsabile
di ridotti livelli piastrinici nel 15-20% dei casi [1]. Sebbene la maggior parte dei nuovi contatori di
cellule sono programmati per rilevare aggregati piastrinici, la conta manuale su striscio di sangue
periferico rimane il test diagnostico più accurato.
In relazione all’età di insorgenza, le cause di piastrinopenia possono essere varie. In epoca
neonatale la soppressione midollare dovuta a infezioni, la trombocitopenia alloimmune neonatale
(NAIT) e il passaggio di anticorpi da madre affetta da porpora trombocitopenica autoimmune (PTI)
risultano essere le cause più frequenti di trombocitopenia. Nelle epoche successive le infezioni
virali e la PTI rappresentano le situazioni di più comune riscontro. Le cause di trombocitopenia
sono riassunte nella tabella 1, suddivise in cause immuni e non immuni (aumentato consumo,
ridotta produzione, alterata distribuzione).
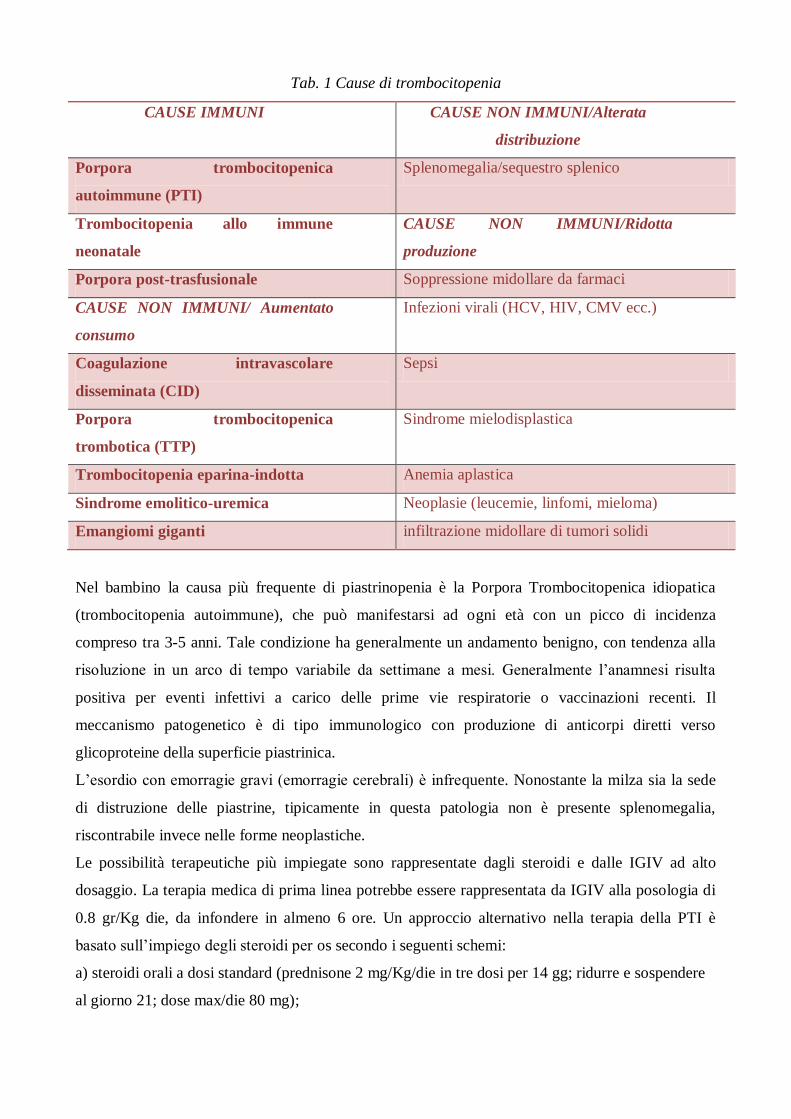
Tab. 1 Cause di trombocitopenia
CAUSE IMMUNI CAUSE NON IMMUNI/Alterata
distribuzione
Porpora trombocitopenica
autoimmune (PTI)
Splenomegalia/sequestro splenico
Trombocitopenia allo immune
neonatale
CAUSE NON IMMUNI/Ridotta
produzione
Porpora post-trasfusionale Soppressione midollare da farmaci
CAUSE NON IMMUNI/ Aumentato
consumo
Infezioni virali (HCV, HIV, CMV ecc.)
Coagulazione intravascolare
disseminata (CID)
Sepsi
Porpora trombocitopenica
trombotica (TTP)
Sindrome mielodisplastica
Trombocitopenia eparina-indotta Anemia aplastica
Sindrome emolitico-uremica Neoplasie (leucemie, linfomi, mieloma)
Emangiomi giganti infiltrazione midollare di tumori solidi
Nel bambino la causa più frequente di piastrinopenia è la Porpora Trombocitopenica idiopatica
(trombocitopenia autoimmune), che può manifestarsi ad ogni età con un picco di incidenza
compreso tra 3-5 anni. Tale condizione ha generalmente un andamento benigno, con tendenza alla
risoluzione in un arco di tempo variabile da settimane a mesi. Generalmente l’anamnesi risulta
positiva per eventi infettivi a carico delle prime vie respiratorie o vaccinazioni recenti. Il
meccanismo patogenetico è di tipo immunologico con produzione di anticorpi diretti verso
glicoproteine della superficie piastrinica.
L’esordio con emorragie gravi (emorragie cerebrali) è infrequente. Nonostante la milza sia la sede
di distruzione delle piastrine, tipicamente in questa patologia non è presente splenomegalia,
riscontrabile invece nelle forme neoplastiche.
Le possibilità terapeutiche più impiegate sono rappresentate dagli steroidi e dalle IGIV ad alto
dosaggio. La terapia medica di prima linea potrebbe essere rappresentata da IGIV alla posologia di
0.8 gr/Kg die, da infondere in almeno 6 ore. Un approccio alternativo nella terapia della PTI è
basato sull’impiego degli steroidi per os secondo i seguenti schemi:
a) steroidi orali a dosi standard (prednisone 2 mg/Kg/die in tre dosi per 14 gg; ridurre e sospendere
al giorno 21; dose max/die 80 mg);
b) steroidi orali ad alte dosi (prednisone 4 mg/Kg/die in tre dosi per 7 gg; scalare del 50% nella
settimana successiva; ridurre gradatamente in seguito fino al gg 21; dose max 180 mg/die nella
prima settimana). In casi selezionati in bambini con grave piastrinopenia dopo fallimento delle
terapie farmacologiche di prima linea , sono previsti protocolli terapeutici alternativi:
Rituximab 375 mg/m2/dose, settimanalmente
AZA 2-3 mg/Kg/die, CYS 5 mg/Kg in 2 somministrazioni
Splenectomia, utile vaccinazione anti Meningococco, H. Influenzae, Pneumococco
Esistono forme congenite di trombocitopenia classificate in base all’età di insorgenza, severità del
quadro clinico, modalità di trasmissione, associazione ad altre anomalie di sviluppo, alterazione
genetica, dimensione delle piastrine. La Tabella 2 riassume le principali trombocitopenie congenite
suddivise per modalità di trasmissione [2].
Tab.2 Trombocitopenie congenite/modalità di trasmissione
Autosomiche dominanti Autosomiche recessive X-linked
Anomalia May-Heggling Trombocitopenia
amegacariocitica congenita
(CAMT)
Sindrome di Wiskott-Aldrich
Sindrome di Fetchner Trombocitopenia e assenza del
radio
Trombocitopenia X-linked
Sindrome di Epstein Sindrome di Bernard-Soulier Mutazioni GATA1
Sindrome di Sebastian
Trombocitopenia
mediterranea
Sindrome di Di George
Disordini piastrinici
familiari/leucemia mieloide
acuta
Cromosoma 10/THC2
Sindrome di Jacobsen
Sindrome di Gray
Trombocitopenia e sinostosi
radiale

Qualunque sia la causa, i segni e i sintomi di sospetto di una trombocitopenia comprendono
manifestazioni emorragiche muco-cutanee (porpora, gengivorragia, epistassi). La trombocitopenia
grave, definita come una conta piastrinica inferiore a 50x109/L, comporta un incrementato rischio di
sanguinamento durante procedure invasive. Una conta piastrinica inferiore a 10x109/L può indurre
delle gravi emorragie e causare di conseguenza la morte del paziente (emorragia intracranica).
Il riconoscimento dell’eziologia alla base della trombocitopenia permette l’impostazione di un
regime terapeutico appropriato che risulta essere parzialmente diverso per le diverse forme di
piastrinopenia. La trasfusione di concentrati piastrinici è indicata per la profilassi e la terapia delle
emorragie nei pazienti affetti da piastrinopenie iporigenerative e in quelli affetti da CID in presenza
di emorragie (in quest’ultimo caso è necessaria la contemporanea correzione di altri deficit
coagulativi). In ogni caso la soglia trasfusionale è di 10x109 piastrine/L. È possibile la trasfusione
anche per conte comprese fra 10x 109 e 20x10
9/L. Va comunque tenuto presente che emorragie
spontanee maggiori, sostenute da una piastrinopenia isolata, sono infrequenti per conte superiori a
20x109/L.
La trasfusione di concentrati piastrinici non è indicata, salvo casi particolari, nei soggetti affetti da
porpora trombocitopenica trombotica, sindrome emolitico-uremica, trombocitopenia autoimmune
(salvo in caso di sanguinamenti maggiori), porpora post-trasfusionale e nei deficit funzionali
piastrinici. Nella trombocitopenia alloimmune neonatale la trasfusione piastrinica è efficace solo se
il donatore è negativo per gli alloantigeni piastrinici in causa.
Importante ricordare che la trasfusione di concentrati piastrinici comporta una frequente, più o
meno rapida, comparsa di alloimmunizzazione, con conseguente refrattarietà trasfusionale, per la
difficoltà di reperire donatori compatibili.
Bibliografia
1. Silvestri F, Virgolini L, Savignano C, Zaja F, Velisig M, Baccarani M. Incidence and
diagnosis of EDTA-dependent pseudothrombocytopenia in a consecutive outpatient
population referred for isolated thrombocytopenia. Vox Sang. 1995;68: 35-39.
2. Jonathan G. Drachman. Inherited thrombocytopenia: when a low platelet count does not
mean ITP. Blood, 15 january 2004 _ volume 103, number 2.

Leucemie e linfomi
Rigoli L, Loddo I, Procopio V, La Rosa M, Amorini G, Di Bella, Salpietro A, Munafò C, Piraino B
Le malattie linfoproliferative rappresentano un gruppo eterogeneo di neoplasie che originano dai
sistemi reticoloendoteliale e linfatico.
Le forme più importanti sono i linfomi di Hodgkin (LH) e i linfomi non Hodgkin (LNH).
Le cause più frequenti di linfadenopatia sono le seguenti:
1. Infezioni (EBV, Toxoplasmosi, Cytomegalovirus, Febbre da graffio di gatto, Faringite,
Tubercolosi, Scarlattina, HBV, HIV, Rosolia, Parotite, Varicella Zoster Virus ).
2. Tumori linfatici (linfoma di Hodgkin, linfoma non-Hodgkin, LLC, LLA,LMA).
3. Tumori metastatici (melanoma, carcinoma mammario, carcinoma polmonare, carcinoma
gastrico, carcinoma prostatico,carcinoma renale, tumori del collo).
4. Malattie del collagene e vasculiti (RA,LES).
Nell’inquadramento diagnostico, è molto importante l’anamnesi (graffio da gatto, ingestione di
carne cruda, puntura di zecca, esposizione alla TBC, tossicodipendenze, trasfusioni, Leishmania,
febbre tifoide, viaggi in Africa, Asia, Australia). Inoltre, la diagnosi si avvale di esami
ematochimici (emocromo, sierologia per HIV, EBV, Toxoplasma, CMV, HBV, HCV), Rx torace,
ecografia addome e biopsia linfonodale.
Linfoma di Hodgkin
Nel 1832, il patologo inglese Thomas Hodgkin descrisse per la prima volta la malattia che porta il
suo nome: il morbo di Hodgkin. Si tratta di un processo neoplastico di verosimile origine linfoide o
monocito-macrofagica contrassegnato da un tipico marker citologico: la cellula di Reed-Sternberg.
Ha una frequenza di circa 5-6 nuovi casi/anno/1.000.000 e colpisce spesso soggetti di età inferiore
ai 15 anni. L’incidenza dell’HD presenta un andamento bimodale, in relazione all’età. Un picco si
ha intorno ai 20 anni e il secondo dopo i 50 anni (Fig 1). Il rapporto maschio/femmina è pari a
2,3:1.
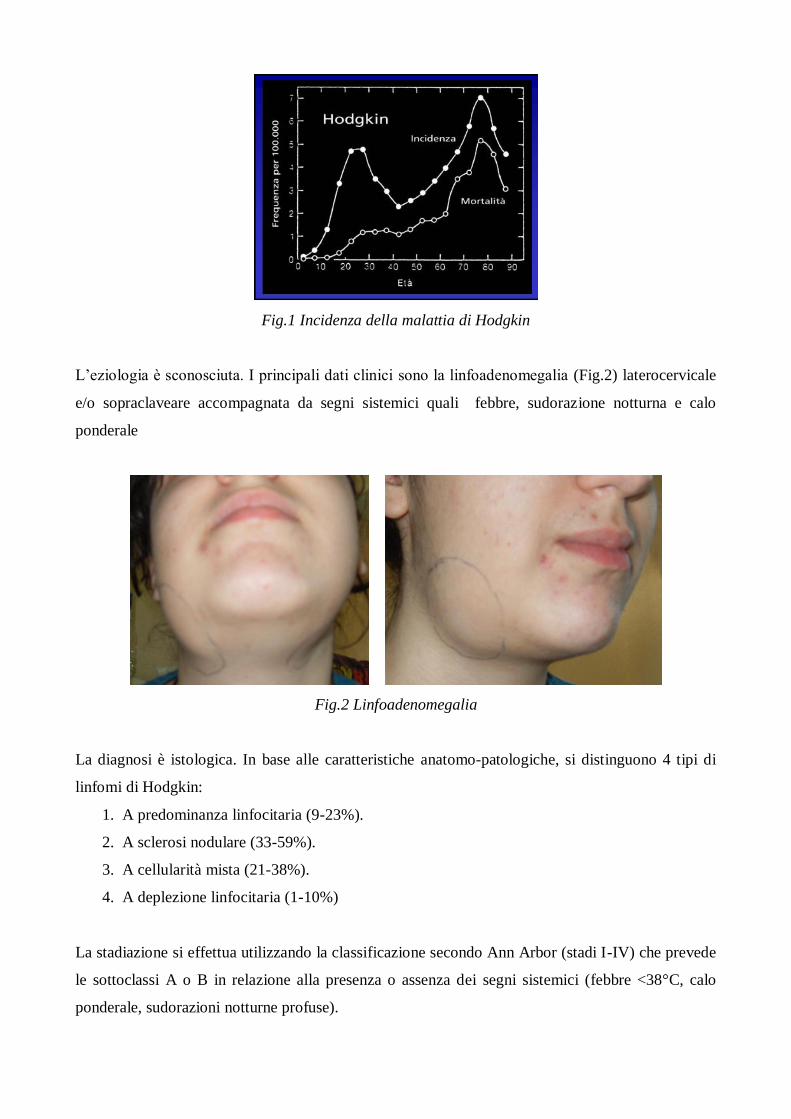
Fig.1 Incidenza della malattia di Hodgkin
L’eziologia è sconosciuta. I principali dati clinici sono la linfoadenomegalia (Fig.2) laterocervicale
e/o sopraclaveare accompagnata da segni sistemici quali febbre, sudorazione notturna e calo
ponderale
Fig.2 Linfoadenomegalia
La diagnosi è istologica. In base alle caratteristiche anatomo-patologiche, si distinguono 4 tipi di
linfomi di Hodgkin:
1. A predominanza linfocitaria (9-23%).
2. A sclerosi nodulare (33-59%).
3. A cellularità mista (21-38%).
4. A deplezione linfocitaria (1-10%)
La stadiazione si effettua utilizzando la classificazione secondo Ann Arbor (stadi I-IV) che prevede
le sottoclassi A o B in relazione alla presenza o assenza dei segni sistemici (febbre <38°C, calo
ponderale, sudorazioni notturne profuse).

I fattori prognostici sfavorevoli sono la presenza di sintomi sistemici alla diagnosi, impegno
mediastinico importante, sesso maschile ed età superiore ai 7 anni.
Per la stadiazione clinica del linfoma di Hodgkin sono utili la valutazione anamnestica ed obiettiva;
esami di laboratorio quali VES, esame emocromocitometrico, LDH, cupremia, fibrinogenemia,
esami della funzionalità epatica e renale, protidogramma, dosaggio Ig, intradermoreazioni,
tipizzazione linfocitaria, Ac anti-HIV; ed infine la diagnostica per immagini (Rx-grafie, TAC,
RMN, Scintigrafie, Linfografia).
Per la stadiazione anatomo-patologica, si fa riferimento alla biopsia osteomidollare, alle biopsie
epatiche e spleniche (laparoscopia) e alle biopsie dei linfonodi addominali.
Qui di seguito è riportata la stadiazione dei linfomi di Hodgkin secondo la classificazione di
Cotswolds (ANN ARBOR modificata) (Fig 3 e 4):
Stadio I: Interessamento di una singola regione linfonodale superficiale (I) o di un singolo organo
extralinfatico (IE).
Stadio II: Interessamento di due o più regioni linfonodali poste tutte o sopra o sotto il diaframma
(II2,3,etc.); eventuale estensione ad un organo extralinfatico contiguo (IIE).
Stadio III: Interessamento di regioni linfonodali poste sia sopra che sotto il diaframma; eventuale
estensione ad un organo extralinfatico contiguo (IIIE).
III- Interessamento di regioni linfonodali dell'ilo epatico, splenico o tripode celiaco.
III2- Interessamento di regioni linfonodali a livello mesenterico, iliaco e para-aortico.
Stadio IV: Interessamento diffuso di uno o più organi extralinfatici, con o senza interessamento
linfonodale.
Fig.3 Fig.4
Nei linfomi di Hodgkin, la presentazione extranodale primitiva è molto rara (< 1% dei casi) eccetto
che nell’AIDS. In tal caso, le localizzazioni preferite sono rappresentate dal polmone, dal S.N.C.,
dalla cute e dal tessuto linfoide del tratto gastrointestinale.
I fattori prognostici sfavorevoli del linfoma di Hodgkin sono rappresentati da:

Stadio: III e IV
Varietà clinica: B (es. presenza di più sintomi)
Istotipo: deplezione linfocitaria (Ki-1 related, CD30)
Età: > 60 anni
Sesso: maschile
VES aumentata
Massa tumorale rilevante: bulky disease
Più sedi linfonodali interessate
Mancata remissione dopo chemioterapia
Ricaduta precoce dopo la remissione (prima di 1 anno)
Trattamento inadeguato (bassa intensità di dose)
Le più frequenti complicanze sono:
Infezioni acute (15% in fase pre-chemioterapica)
Sindrome mediastinica
Versamenti pleurici e pericardici
Compressione del SNC
Insufficienza renale
Insufficienza epatica
Anemia
Problemi connessi con la gravidanza
L’approccio terapeutico del linfoma di Hodgkin varia secondo lo stadio.
Nello stadio limitato (coinvolgimento di non più di 2 sedi contigue) IA e IIA, si effettua la
radioterapia (dopo laparotomia), mentre nello stadio limitato IB e IIB alla radioterapia si associa la
chemioterapia.
Gli stadi IE e IIE (A e B), e lo stadio IIA avanzato, necessitano della chemioterapia e radioterapia
limitata.
Negli stadi III e IV, si effettua l’associazione chemioterapia-radioterapia su bulky.

Linfomi Non-Hodgkin
Sono un gruppo eterogeneo di neoplasie che originano dalla degenerazione maligna di cellule
appartenenti a linee diverse del sistema immunitario. Sono descritti 3-4 nuovi casi/anno/1.000.000
di soggetti di età inferiore ai 15 anni. Il picco d’incidenza è compreso fra 7 e 11 anni, ed il rapporto
maschio/femmina è pari a 3:1. L’eziologia è sconosciuta.
Le localizzazioni più frequenti sono tonsille e cavo orale; stomaco, intestino e retto; cute.
La sintomatologia dipende dalle sedi coinvolte.
Nell’interessamento mediastinico si ha tosse, febbricola, dispnea ingravescente, versamento
pleurico, sindrome della vena cava superiore.
Nell’interessamento addominale prevalgono dolori addominali, vomito, massa addominale con
presenza anche di un quadro di addome acuto.
L’interessamento linfonodale è caratterizzato da linfoadenopatia laterocervicale, più raramente
ascellare o inguinale.
La diagnosi si basa sui reperti istologici e sull’aspirato midollare. L’esame citologico è utile in caso
di versamento pleurico o ascitico.
I linfomi non Hodgkin sono ad alto grado di malignità.
Secondo la classificazione REAL (Tab.1) si distinguono in linfoma maligno (a grandi cellule,
immunoblastico, plasmocitoide, a cellule chiare, polimorfo, con componente a cellule epitelioidi);
linfoma maligno linfoblastico (a cellule convolute, a cellule non convolute); linfoma maligno a
piccole cellule non clivate (linfoma di Burkitt, con aree follicolari).
Si distinguono 3 fenotipi immunologici: T, B, e nonT non B.
La stadiazione si basa su criteri clinico-strumentali; è chirurgica solo in caso di LNH addominale.
Per la stadiazione si utilizza la classificazione secondo Murphy (stadi I-IV).
Tab.1 Classificazione REAL dei linfomi non Hodgkin

Linfomi Non-Hodgkin in età pediatrica
I linfomi non Hodgkin sono rari in età pediatrica, pur rappresentando il terzo tumore in ordine di
frequenza.
La gestione di un bambino con linfoma non Hodgkin è molto complessa e richiede l'intervento
coordinato di ematologi, chirurghi, radioterapisti, neurologi, psicologi ed eventualmente altri
specialisti esperti.
In età pediatrica si ha un’elevata frequenza dei linfomi ad alto grado di malignità.
Vi sono 3 tipi istologici con diversi sottotipi:
1. Linfoma linfoblastico
2. Linfoma di Burkitt o tipo Burkitt
3. Linfoma diffuso a grandi cellule
In caso di interessamento del midollo osseo, si ha difficoltà di classificazione riguardo i primi due
tipi di linfoma. Pertanto, in questi casi, in presenza di una percentuale di cellule neoplastiche
superiore al 25% (o 30% secondo altri Autori), si preferisce inquadrare la malattia come leucemia
linfoblastica acuta.
La terapia è sostanzialmente identica, indipendentemente dal grado di infiltrazione midollare.
La stadiazione dei linfomi non Hodgkin in età pediatrica non differisce da quella dei linfomi degli
adulti, ma è diverso il ruolo della chirurgia.
Infatti, in età pediatrica, se i linfomi si presentano come grosse masse addominali e/o mediastiniche,
è necessaria la laparotomia esplorativa e/o la mediastinoscopia per effettuare prelievi bioptici,
soprattutto in assenza di linfonodi superficiali da sottoporre a biopsia.
Durante l'intervento chirurgico sull'addome si effettua l'asportazione delle masse patologiche, in
quanto secondo molti studi, la prognosi può migliorare.
La terapia dei LNH nei bambini richiede un approccio multidisciplinare.
La chemioterapia è importante praticamente in tutti i casi, poiché la malattia è considerata sempre
disseminata fin dall'inizio, anche in quelli apparentemente localizzati. Sono stati proposti numerosi
protocolli, a volte molto complessi, che richiedono l'utilizzo di 4-10 farmaci diversi.
Nei LNH pediatrici la radioterapia ha un ruolo abbastanza marginale. A volte, si utilizza prima della
chemioterapia per ottenere una rapida riduzione delle masse tumorali mediastiniche.
Studi più recenti suggeriscono l'esclusione della radioterapia dal programma terapeutico, anche
allo scopo di ridurre l'incidenza degli effetti collaterali a lungo termine nei soggetti trattati con
modalità combinata, chemioterapia più radioterapia.

Negli ultimi anni, si è avuto un miglioramento della prognosi dei bambini affetti da LNH. Nel 60-
70% dei casi, si ottiene la remissione a 5 anni; la percentuale può arrivare al 90% in alcuni sottotipi
con stadio limitato (Tab.2).
Il trapianto di midollo allogenico o autologo è una procedura ancora sperimentale, sulla cui
efficacia a lungo termine mancano ancora dati certi.
Tab.2 Fattori prognostici dei LNH

Leucemie infantili
Le leucemie sono le neoplasie più comuni dell’infanzia, rappresentando circa il 41% di tutte quelle
che colpiscono i bambini di età inferiore ai 15 anni.
La Leucemia Linfocitica Acuta (ALL, Acute Lymphoblastic Leukemia) comprende circa il 77% dei
casi di leucemia pediatrica, la Leucemia Mieloide Acuta (AML, Acute Myelogenous Leukemia)
l’11%, la Leucemia Mieloide Cronica (CML, Chronic Myelogenous Leukemia) il 2-3% e la
Leucemia Mieloide Cronica Giovanile (JCML, Juvenile Chronic Myelogenous Leukemia) l’1-2%.
I restanti casi sono provocati da diverse forme acute e croniche che non trovano una collocazione
nelle classiche definizioni.
Le leucemie possono essere definite un gruppo di malattie neoplastiche in cui alterazioni genetiche
a carico della cellula ematopoietica danno origine a un’abnorme proliferazione clonale. Il
progenitore di queste cellule ha un vantaggio sulla crescita rispetto agli elementi cellulari normali, a
causa di un aumento della percentuale di proliferazione e di una riduzione delle apoptosi spontanee,
questo fenomeno provoca un sovvertimento del normale funzionamento del midollo osseo, che con
il passare del tempo porta a un quadro di insufficienza midollare. Gli elementi clinici, le indagini di
laboratorio e le risposte ai trattamenti variano in relazione al tipo di leucemia.
Leucemia Linfocitica Acuta (ALL)
Sono più frequentemente colpiti i maschi delle femmine e si ha un picco di incidenza tra 2-6 anni.
La malattia risulta più frequente nei bambini con particolari anomalie genetiche: sindrome di Down,
atassia-teleangectasia e sindrome di Fanconi.
L’eziologia dell’ALL rimane sconosciuta, anche se diversi fattori genetici ed ambientali (radiazioni
ionizzanti, farmaci, agenti alchilanti ecc) sono stati correlati alla leucemia in età pediatrica.
La classificazione dell’ALL è fatta sulla base della tipizzazione delle cellule maligne del midollo
osseo, in relazione alla morfologia, alle caratteristiche fenotipiche con la valutazione dei marker di
membrana e alle caratteristiche citogenetiche e genetico-molecolari. La morfologia da sola in
genere è sufficiente a stabilire la diagnosi, ma sono indispensabili altre indagini per una corretta
classificazione della malattia, che possono avere notevole influenza sia sulla prognosi sia sulla
scelta della terapia più appropriata. Nella maggior parte dei pazienti con ALL sono state riscontrate
anomalie cromosomiche come ad esempio la traslocazione t(9;22) (cromosoma Philadelphia) che
esprime la proteina di fusione BCR/ABL.
Manifestazioni cliniche
La presentazione iniziale è generalmente aspecifica e di entità relativamente lieve. Sono quasi
sempre presenti anoressia, astenia, irritabilità e una febbricola intermittente. Può presentarsi un

dolore osseo, o meno frequentemente articolare, soprattutto agli arti inferiori. Quando la malattia
progredisce, i segni e i sintomi da interessamento midollare diventano più evidenti con la comparsa
del pallore cutaneo, di astenia, ematomi o epistassi, ma anche di febbre, che può risultare secondaria
a un’infezione. All’esame obiettivo, i reperti di pallore, lesioni cutanee tipo porpora e petecchie o
emorragie cutanee, possono essere segni di un’insufficienza midollare. La natura proliferativa della
malattia può manifestarsi con linfadenopatia, splenomegalia o, meno comunemente, epatomegalia.
Diagnosi
L’ipotesi diagnostica viene suggerita dagli elementi riscontrati nel sangue periferico, che sono
indicativi di un’alterazione midollare. L’anemia e la trombocitopenia sono presenti nella maggior
parte dei pazienti. Quando i risultati dell’emocromo suggeriscono la possibilità di una leucemia,
andrebbe subito analizzato il midollo per porre una diagnosi. La diagnosi di ALL è posta quando il
midollo presenta una invasione da parte di una popolazione omogenea di linfociti >25%.
Trattamento
Il fattore prognostico più importante nell’ALL è il trattamento: senza un’adeguata terapia, la
malattia è fatale. Negli ultimi 40 anni, la sopravvivenza dei bambini con l’ALL è aumentata grazie
ai trial clinici, che hanno permesso di migliorare le terapie e l’evoluzione della malattia. La scelta
del trattamento si fonda sulla stima del rischio clinico di ricaduta nel singolo paziente, che varia in
modo significativo tra i diversi sottotipi dell’ALL.
I tre fattori predittivi principali sono:
- L’età del paziente al momento della diagnosi
- Il numero di leucociti all’esordio
- La velocità di risposta al trattamento (cioè quanto rapidamente le cellule leucemiche
vengono eliminate dal midollo o dal sangue periferico)
Rischio medio:
Età compresa tra 1-10 anni
Conta leucocitaria <50000/uL
Rischio elevato:
Età >10 anni
Conta leucocitaria >50000/uL
La prognosi dei pazienti a rischio più elevato può essere migliorata con la somministrazione di una
terapia più intensa, anche se con un maggiore pericolo di tossicità.
I bambini con ALL e quelli con specifiche alterazioni cromosomiche, quali t(9;22) o t(4;11),
presentano un rischio di recidiva ancora maggiore, anche se vengono trattati con terapia intensiva.

Generalmente la terapia iniziale è disegnata per eliminare le cellule leucemiche dal midollo osseo,
nella fase nota come induzione della remissione. Durante questa fase la terapia è solitamente
somministrata per 4 settimane e consiste in Vincristina settimanale, Corticosteroidi quali
Desametasone o Prednisone e dosi ripetute di L- asparginasi o una singola dose di una
preparazione a lunga durata di asparginasi pegilata. Si possono anche utilizzare Citarabina e/o
Methotrexate per via intratecale. Con questo approccio, il 98% dei pazienti va in remissione,
definita per la presenza di <5% di blasti nel midollo e un ritorno dei leucociti e delle piastrine ai
valori nell’intervallo di normalità dopo 4-5 settimane dall’inizio del trattamento. La chemioterapia
intratecale si impiega in genere all’inizio della terapia e poi ancora durante l’induzione.
La seconda fase del trattamento è focalizzata sulla terapia del SNC, nel tentativo di prevenire le
ricadute tardive. La chemioterapia intratecale è somministrata ripetutamente per mezzo di
rachicentesi in concomitanza con la chemioterapia sistemica intensiva. La probabilità di ricaduta
tardiva sul SNC è in questo modo ridotta a <5%. Una piccola percentuale di soggetti con
caratteristiche che predicono un alto rischio di recidiva a livello del SNC può essere sottoposta a
radioterapia cranio spinale, cui sono specialmente candidati coloro che, al momento della diagnosi,
hanno linfoblasti e un aumento dei leucociti nel liquido cerebrospinale o segni di leucemia estesa a
livello del SNC, come la presenza di paralisi dei nervi cranici.
Dopo che è stata indotta la remissione, molti regimi prevedono 14-28 settimane di politerapia con
farmaci e schedule variamente combinate a seconda del gruppo di rischio del paziente.
Per finire, i pazienti assumono quotidianamente la Mercaptopurina e settimanalmente il
Methotrexate, in genere con dosi intermittenti di Vincristina e Corticosteroidi. Questo periodo, noto
come fase di mantenimento della terapia, dura dai 2 ai 3 anni, a seconda del protocollo utilizzato.
Molti pazienti beneficiano della somministrazione di una fase intensiva ritardata del trattamento
(intensificazione ritardata), circa 5-7 mesi dopo l’inizio della terapia e dopo una fase di relativa non
tossicità (durante il mantenimento), per permettere un recupero dall’inizio della terapia intensiva.
Un piccolo numero di soggetti con fattori prognostici particolarmente sfavorevoli, soprattutto quelli
con la traslocazione t(9;22) nota come cromosoma Philadelphia, possono essere sottoposti ad un
trapianto di midollo osseo durante la prima remissione.
Il principale ostacolo alla guarigione è la recidiva della malattia, che si realizza a livello del midollo
osseo nel 15-20% dei pazienti con ALL e si correla a gravi conseguenze, soprattutto se ha luogo
durante o subito dopo il completamento della terapia. Una chemioterapia intensiva con farmaci non
utilizzati in precedenza, seguita da un trapianto allogenico di cellule staminali, può aumentare la
sopravvivenza a lungo termine di alcuni casi con recidiva midollare.

Terapia di supporto
Una particolare attenzione alle terapie mediche di supporto è essenziale per gestire con successo i
programmi di chemioterapia aggressiva. I pazienti con estese masse tumorali sono a rischio di
sviluppare una sindrome da lisi tumorale, nel momento in cui si inizia la terapia. La chemioterapia
spesso provoca una grave mielosoppressione, che può rendere necessario trasfondere emazie e
piastrine e che spesso richiede la somministrazione empirica di potenti antibiotici ad ampio spettro
in caso di sepsi febbrile neutropenica.
L’efficacia della terapia ha trasformato l’ALL da malattia acuta con un elevato indice di mortalità a
malattia cronica. Tuttavia, il trattamento cronico può portare serie conseguenze psicosociali ai
bambini affetti. A causa dell’intensità della terapia si possono verificare inoltre effetti tossici tardivi
e acuti.
Prognosi
Al giorno d’oggi, la maggior parte dei bambini con l’ALL può avere una lunga aspettativa di vita,
con una sopravvivenza a 5 anni dalla diagnosi >80%. Il principale fattore prognostico è la scelta
della terapia strettamente correlata al rischio, mentre gli altri fattori sono il genere di trattamento
scelto in relazione al tipo di ALL, lo stadio della malattia, l’età del paziente e l’entità della risposta
alla prima terapia (favorevole se il paziente risponde in meno di 1 mese).
La Minima Malattia Residua (MRD) può essere valutata con specifiche sonde molecolari per le
traslocazioni e con altri marcatori del DNA contenuto nelle cellule leucemiche. La MRD può essere
quantitativa e fornire una stima di quante cellule leucemiche sono ancora presenti nel midollo.
Sebbene non sia noto quanta MRD possa essere eliminata dai meccanismi di difesa immunitaria del
paziente, la presenza di un suo alto grado al termine dell’induzione è suggestiva per una prognosi
sfavorevole e per un aumentato rischio di recidiva.
Bibliografia
1. Pediatria di Nelson. 18° Edizione

I Tumori Solidi
Briuglia S, Comito D, Salpietro V, Barone C, Colavita L, Deak A, Talenti A, Munafò C, Salpietro DC
I tumori solidi in età pediatrica sono la seconda causa di morte nei pazienti tra i 0 e i 14 anni (con
una prevalenza dell’11%) nei Paesi industrializzati.
La sopravvivenza dipende dal tipo di tumore e dalla precocità della diagnosi.
I tipi di tumore solido più frequenti sono:
- tumori del sistema nervoso centrale
- retinoblastoma
- neuroblastoma
- nefroblastoma o tumore di Wilms
- rari tumori dei tessuti molli
- tumori ossei
- altri più rari
I tumori si originano per un processo di oncogenesi, che riconosce una componente genetica,
talvolta ereditabile, e una componente ambientale (agenti mutageni).
I segni e sintomi di tumori pediatrici sono piuttosto aspecifici e per questo si deve eseguire un
attenta diagnosi differenziale all’insorgere di uno di essi:
- cefalea e vomito mattutino sono molto frequenti in bambini con tumori cerebrali (per la
presenza di ipertensione endocranica), ma possono essere provocati anche da emicrania e
sinusite;
- dolore osseo è presente nel tumore osseo e nelle leucemie, ma anche nelle infezioni, traumi
e durante la crescita;
- una massa addominale può essere causata da tumore di Wilms, linfoma o neuroblastoma ma
anche da stipsi (per la presenza di fecaloma, che è un reperto frequente nei bambini), cisti
renali e altro.

Tumori del Sistema Nervoso Centrale
Sono un gruppo di neoplasie maligne eterogeneo per sede e tipo istologico. Rappresentano il 21%
di tutte le neoplasie dell’età pediatrica. L’incidenza è di 23-30 nuovi casi/anno/1.000.000 di soggetti
di età < 15 anni, con un picco d’incidenza tra i 5–10 anni. L’eziologia è sconosciuta.
I principali dati clinici sono i segni di ipertensione endocranica con l’insorgenza di cefalea, vomito
a getto, papilledema, strabismo, diplopia; nei pz di età <2 anni si evidenzia: accresciuta irritabilità,
aumento della circonferenza cranica, allargamento e tensione della fontanella, stasi delle vene del
cuoio capelluto, occhi a “sole calante”.
Vi sono inoltre sintomi tipici secondo la sede del tumore:
T. cervelletto e IV ventricolo: astenia, asinergia, nistagmo, ipotonia muscolare, torcicollo o
rigidità nucale
T. del tronco cerebrale: paralisi progressive multiple e bilaterali a carico dei nervi cranici,
atassia
T. emisferi e ventricoli laterali: paresi, episodi comizali
T. sellari, parasellari o soprasellari: ritardo di crescita (o pubertà ritardata e obesità),
progressiva perdita del visus, diabete insipido
La diagnosi è quando possibile istologica attraverso prelievo bioptico mediante stereotassi della
sede interessata o strumentale con esame TAC, RMN, angiografia.

Neuroblastoma
È una neoplasia maligna ad insorgenza dalle cellule della cresta neurale e poichè tali cellule sono
dislocate ubiquitariamente (gangli paravertebrali, surreni, cute,…) la neoplasia può svilupparsi in
qualsiasi organo o apparato.
Ha un’incidenza di 6.5– 10.1 nuovi casi/anno/1.000.000 di soggetti di età < 15 anni, con un picco
d’incidenza tra 0– 2 anni e una lieve prevalenza nel sesso maschile (M/F = 1.2:1). L’eziologia è
sconosciuta.
I sintomi variano secondo la localizzazione del tumore:
Localizzazione addominale: è la più frequente e spesso si manifesta con una massa palpabile nei
quadranti laterali dell’addome o in regione alto costale, abitualmente oltrepassante la linea mediana,
linfoadenomegalia, dolori addominali diffusi, anoressia, vomito,
Localizzazione intratoracica: tosse, dispnea, disfagia, segni compressivi a carico del midollo
spinale.
Segni sistemici: dolore osteo-articolari, astenia, perdita di peso, pallore, febbre ricorrente.
Può dare metastasi in vari organi (fegato, cute, ossa,…).
La diagnosi si avvale di:
- esame obiettivo
- markers tumorali: incremento delle catecolamine urinarie VMA (acido vanil mandelico),
HVA (acido omovanillico), incremento meno specifico di LDH, enolasi, ferritina
- esami strumentali: (ecografia, TAC, RM, scintigrafia ossea) per lo studio morfologico della
lesione e delle eventuali metastasi;
- prelievo istologico della lesione (tumore primario e sue metastasi) per la diagnosi certa.
-
La stadi azione è prevalentemente clinica ed è determinante per la scelta terapeutica e la prognosi.
stadio tipo di trattamento prognosi
I asportazione chirurgica 90-100% sopravvive
II asportazione chirurgica 90% sopravvive; la sopravvivenza non è
peggiorata dalla permanenza di residui tumorali
III chirurgia e chemioterapia contro
le metastasi
30-40% sopravvive
IV chirurgia e radioterapia,
chemioterapia contro le metastasi
10-15% sopravvive
IVs nessun trattamento o solo minimo
supporto
81% sopravvive

Tumore di Wilms
È una neoplasia maligna di tipo embrionario ed è il più frequente tumore in età pediatrica (5-10%).
Origina dal blastema metanefrico primitivo, cioè dall’abbozzo embrionale del rene, quando alcune
cellule perdono la loro capacità di differenziarsi in cellule renali adulte normali e acquisiscono la
capacità di proliferare indefinitamente. L’incidenza è di 5.5 – 7.1 nuovi casi/anno/1.000.000 di
soggetti di età < 15 anni, con un picco d’incidenza tra l’età di 1-5 anni e una prevalenza nel sesso
femminile (M/F = 0.9:2). L’eziologia è sconosciuta ma nel 15-20% dei casi si presenta come una
forma ereditaria, bilaterale o multifocale, spesso associata ad altre anomalie congenite (aniridia,
emi-ipertrofia corporea, anomalie del tratto genito-urinario). E’ determinata nella maggior parte dei
casi da una alterazione del gene WT1 (banda 11p13) situato sul braccio corto del cromosoma 11.
I sintomi e/o segno clinici più frequenti sono:
- il riscontro occasionale di “massa addominale” non oltrepassante la linea mediana,
- macroematuria,
- ipertensione per un’alterazione del sistema renina-angiotensina,
I sintomi aspecifici (stipsi, anoressia, febbre, dolori addominali) spesso mancano, il bambino
presenta buone condizioni cliniche e il sospetto viene posto per il riscontro di asimmetria o massa
palpabile all’addome.
L’iter diagnostico prevede oltre all’anamnesi e all’esame obiettivo, una valutazione con esami
strumentali (ecografia, TC, RMN), mentre non vi sono markers tumorali specifici. La diagnosi
differenziale viene spesso posta con il Neuroblastoma a localizzazione addominale. La diagnosi è
istologica e la stadiazione chirurgica e anatomo-patologica è basata sul grado di estensione macro o
microscopica del tumore (stadio I-V sec. NWTS).

Retinoblastoma
È un tumore raro con un’incidenza di circa 1:20.000 nati, ma è il principale tumore infantile
intraoculare della retina. La diagnosi viene effettuata intorno ai 12 mesi di età, raramente >5 anni.
Circa il 60% dei retinoblastomi sono sporadici (e unilaterale), mentre il 40% è familiare (e
bilaterale). Nel 1971 Knudson scoprì che il retino blastoma è causato da due eventi mutazionali: una
mutazione è ereditata per via germinale, mentre la seconda avviene nelle cellule somatiche. Nella
forma non ereditaria entrambe le mutazioni avvengono nelle cellule somatiche.
Il tumore si definisce endofitico se la massa tumorale si accresce verso l’interno della cavità
oculare, esofitico se si estende verso la porzione esterna della retina.
I sintomi principali sono:
- una massa bianca nel campo pupillare (Leucocoria)
- strabismo,
- glaucoma,
- dilatazione della pupilla
- nistagmo
L’iter diagnostico prevede:
- esame oculare con fundus oculi
- ecografia oculare
- TC e/o RMN
La terapia è efficace in oltre il 90% dei casi senza metastasi e consiste in:
• Chemioterapia (chemioriduzione) seguita da radioterapia, crioterapia o laser
• Radioterapia o enuclaezione
• Nei tumori bilaterali: si procede all’escissione dell’occhio in cui il tumore appare più
sviluppato, mentre nell’altro occhio la malformazione viene curata con fotocoagulazione
laser.

Sarcoma di Edwing
Tumore maligno primitivo dell’osso, caratterizzato da tessuto d’aspetto istologico uniforme
rappresentato da cellule piccole e rotonde con scarso citoplasma. I sintomi principali sono: febbre,
dolore, tumefazione a carico del segmento osseo colpito, e dei tessuti molli circostanti. Esordisce
tipicamente a carico della pelvi, degli arti, delle regioni paraspinali della parete toracica e
metastatizzano alle ossa, al midollo osseo e ai polmoni. La diagnosi è radiologica ed istologica.
Rabdomiosarcoma
Neoplasia maligna ad insorgenza dalle cellule del muscolo scheletrico che rappresenta circa il 50%
dei sarcomi dei tessuti molli del bambino. I sintomi dipendono dalla sede del tumore e sono
determinati solitamente da un effetto massa (compressione). La diagnosi è istologica.
Osteosarcoma
Neoplasia primitiva dell’osso, caratterizzato dalla formazione diretta di osso o di tessuto osteoide da
parte di cellule tumorali. Si presenta come un dolore e tumefazione a livello del segmento osseo
colpito: femore, tibia, omero, mandibola, mascella, fibula. Il polmone è sede elettiva di metastasi.
La diagnosi è radiologica ed istologica.


















