
una metodica per l’estrazione di DNA da materiale patologico ... · istologiche presentano...
36
Microdissezione laser: una metodica per l’estrazione di DNA da materiale patologico scarsamente rappresentativo Vincenzo Tranchina Formazione Tecnico in Analisi Biomediche Scuola superiore medico‐tecnica, Locarno Gennaio – Giugno 2011 Laboratorio di Diagnostica Molecolare Istituto Cantonale di Patologia, Locarno Responsabile: Dott. Milo Frattini
Transcript of una metodica per l’estrazione di DNA da materiale patologico ... · istologiche presentano...

Microdissezione laser:
una metodica per l’estrazione di
DNA da materiale patologico
scarsamente rappresentativo
Vincenzo Tranchina
Formazione Tecnico in Analisi Biomediche
Scuola superiore medico‐tecnica, Locarno
Gennaio – Giugno 2011
Laboratorio di Diagnostica Molecolare
Istituto Cantonale di Patologia, Locarno
Responsabile: Dott. Milo Frattini

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 1
1 INDICE
1 Indice ........................................................................................................................................................ 1
2 Abstract .................................................................................................................................................... 1
3 Introduzione ............................................................................................................................................. 3
3.1 Materiale patologico scarsamente rappresentativo ........................................................................ 3
3.2 Il microdissettore laser ..................................................................................................................... 4
3.3 Giustificazione della scelta ............................................................................................................... 6
3.4 Obiettivo ........................................................................................................................................... 6
3.5 Strategia di realizzazione .................................................................................................................. 7
4 Materiali e metodi .................................................................................................................................... 8
4.1 Materiale patologico utilizzato ......................................................................................................... 8
4.2 Macrodissezione manuale ................................................................................................................ 9
4.3 Microdissezione laser ..................................................................................................................... 11
4.4 Quantificazione ............................................................................................................................... 15
4.5 Amplificazione e sequenziamento .................................................................................................. 15
5 Risultati................................................................................................................................................... 17
5.1 Risultati da macrodissezione manuale ........................................................................................... 17
5.2 Confronto macrodissezione manuale‐microdissezione ................................................................. 19
5.3 Ottimizzazione dell’analisi dei microestratti .................................................................................. 19
5.4 Risultati da microdissezione laser .................................................................................................. 20
6 Discussione ............................................................................................................................................. 22
6.1 Tecnica di microdissezione laser .................................................................................................... 22
6.2 Campioni da macrodissezione manuale ......................................................................................... 23
6.3 Campioni da microdissezione laser ................................................................................................ 23
7 Conclusioni ............................................................................................................................................. 24
8 Ringraziamenti ....................................................................................................................................... 25
9 Bibliografia ............................................................................................................................................. 26
10 Allegati ................................................................................................................................................... 27
2 ABSTRACT

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 2
L’estrazione di cellule tumorali da sezioni di tessuto piccole o eterogenee (materiale scarsamente rappresentativo) rappresenta una procedura difficile che viene eseguita sezionando il taglio sul vetrino con un bisturi (macrodissezione manuale), richiedendo notevole precisione. Nel 2007, l’Istituto Cantonale di Patologia di Locarno (ICP) ha acquistato un microdissettore laser per aggirare tali difficoltà. Si è reso perciò necessario stabilire se lo strumento possa essere utile ai tecnici in analisi biomediche, biologi e patologi per quanto riguarda l’estrazione di cellule da materiale patologico scarsamente rappresentativo. Le domande a cui abbiamo voluto dare risposta sono state due: 1. La microdissezione laser è un sistema valido per l’estrazione di DNA genomico dalle cellule? 2. È necessario modificare i protocolli di estrazione di DNA e amplificazione in adattamento alla tecnica? A questo fine, sono stati analizzati tre campioni di adenocarcinoma colorettale (un caso per la macrodissezione manuale e due casi per la microdissezione laser) estratti da varie superfici. Sono stati amplificati e sequenziati i geni KRAS ed EGFR dopo estrazione di cellule con entrambe le tecniche (macrodissezione manuale e microdissezione laser). Dopo macrodissezione, è stato possibile amplificare e sequenziare da una superficie di 20 mm2. Dopo microdissezione sono stati amplificati e sequenziati campioni con superficie di 0,1 mm2, nella maggior parte dei casi con successo. Abbiamo valutato che una superficie di 0,1 mm2 è sufficiente per analizzare i campioni, a patto che il materiale sia di buona qualità (non vecchio, né fissato per periodi troppo lunghi). Inoltre, durante l’estrazione del DNA, un’incubazione di solo 2 ore con ATL Buffer e Proteinasi K (invece di 16 ore) si è rivelata sufficiente.
Tumor cells extraction from a heterogeneous or small section of tissue (non‐representative material) is a difficult procedure which is carried out by cutting with a surgical blade (manual macrodissection) and one which requires a high level of skill. In 2007, the Pathology Institute of Locarno (ICP) bought a Laser Microdissector to bypass these difficulties. Therefore it was necessary to determine if this instrument could help biomedical technicians, biologists and pathologists in extraction of cells from non‐representative sections. The questions we want to answer were: 1. Is laser microdissection a valid procedure to extract genomic DNA from cells? 2. Shall we modify current laboratory’s procedures of DNA extraction and amplification when the laser microdissection technique is used? For this purpose, we investigated three different samples of colorectal adenocarcinoma (one for the manual macrodissection and two for the laser microdissection technique), extracted from different surfaces. We tried to amplify and to sequence the KRAS and EGFR genes after both cells‐extraction techniques (manual and laser dissection). After macrodissection, it was possible to amplify and sequence from a surface of 20 mm2. After microdissection we amplified from a surface of 0,1 mm2, in the majority of cases with success. We found that a surface of 0,1 mm2 is enough to investigate samples, but only if the material is of good quality (not old and not fixed for too much time). Furthermore, we demostrated that during DNA extraction, an incubation treatment with ATL Buffer and Proteinase K of just 2 hours (instead of 16 hours) is enough.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 3
3 INTRODUZIONE
3.1 MATERIALE PATOLOGICO SCARSAMENTE RAPPRESENTATIVO
Per comprendere questo lavoro è necessario partire dal presupposto che non sempre le sezioni istologiche presentano un’abbondanza di cellule tumorali, il principale bersaglio delle analisi molecolari, sia in ricerca che in diagnostica.
Le fasi che portano al compimento di queste analisi richiedono tecniche particolari e grande cura accuratezza del lavoro e, non secondariamente, sono molto numerose:
dissezione del tessuto
lisi cellulare
isolamento del DNA
quantificazione del DNA
amplificazione del DNA
elettroforesi del DNA
eventuali altre analisi (RFLP, sequencing, …)
interpretazione dei risultati
Si denota dalla lista che la fase di dissezione del tessuto è a monte di tutta la catena che occorre seguire per giungere all’interpretazione dei risultati della ricerca di una mutazione o di un polimorfismo e, in quanto tale, influenza tutte le fasi successive. Le metodiche successive alla dissezione del tessuto sono basate sull’utilizzo di kit commerciali o su protocolli ampiamente validati e riproducibili, e non presentano quindi particolari problemi. A livello della dissezione, invece, si possono presentare vari ostacoli, principalmente rappresentati dalla scarsa qualità e dalla ridotta quantità di materiale.
Se sul primo fattore non possiamo intervenire – poiché il materiale subisce il trattamento fissativo con formalina tamponata 4% e la processazione in altre sedi – siamo invece in grado di porre rimedio al secondo.
Qualora il materiale patologico della sezione istologica non sia adeguato per le analisi molecolari (es. scarso tessuto nelle biopsie o poche cellule tumorali in un contesto di ampie zone di tessuto normale, quindi un tessuto molto eterogeneo nella sua struttura) si pone la necessità di estrarre con precisione esclusivamente le cellule di interesse.
In questo lavoro utilizzerò il termine di materiale patologico scarsamente rappresentativo per indicare proprio quel materiale istologico dove le cellule di interesse, intatte e di buona qualità, sono poche o isolate.
Rispetto alla tecnica tradizionale – macrodissezione manuale – le cellule possono essere selezionate in maniera più mirata con un microdissettore laser (vedi capitolo “Materiali e Metodi”). A tal fine, tuttavia, devono essere definite delle procedure per poter effettuare una selezione mirata delle cellule con l’apparecchio.
È inoltre possibile che l’estratto di DNA genomico ottenuto dopo microdissezione laser necessiti di condizioni per l’amplificazione in PCR diverse da quelle ordinarie. Si potrebbe quindi verificare la possibilità di dover adattare i protocolli delle fasi successive alla dissezione per ottenere i risultati sperati.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 4
3.2 IL MICRODISSETTORE LASER
3.2.1 Introduzione alla microdissezione laser
Il microdissettore laser (Figura 1) è uno strumento di precisione utilizzato in istopatologia per effettuare l’estrazione di cellule di interesse da una qualunque sezione istologica montata su vetrino. Esso vuole conciliare un dominio più tecnico e grossolano – quello istologico – con uno più complesso e per molti versi fragile, come quello molecolare.
Lo strumento è costituito un microscopio associato a un dispositivo laser controllato da un software. La selezione delle cellule da estrarre viene effettuata proprio con un ausilio informatico, mostrando al laser dove e come tagliare.
Figura 1. Microdissettore laser Zeiss PALM Microbeam. Questo modello è stato acquistato nel 2007 dall’Istituto Cantonale di Patologia (ICP).
È proprio questa la principale funzione del laser: tagliare il tessuto. Il fascio laser UV A [3] – focalizzato sul vetrino e delle dimensioni di 0,5 µm [4] – passa sulla sezione istologica e interagisce con le cellule che colpisce, “bruciandole”. Il processo fisico che si determina è la fotoframmentazione, che trasforma il materiale biologico in atomi che vengono eliminati a forte velocità [5]. Viene utilizzata una radiazione del tipo UV A in quanto tali raggi, contrariamente a quelli di tipo UV C, non interagiscono ottimamente con le componenti cellulari (fra cui il DNA) permettendone una migliore conservazione (Figura 2). La fotoframmentazione determina quindi la creazione di un anello di vuoto fra le cellule selezionate ed il resto della sezione istologica, così da isolare la parte di tessuto di interesse [4].
Figura 2. Impatto dei raggi UV A in rapporto a quello dei raggi UV C sul materiale cellulare.
Nella tecnologia LPC (Laser Pressure Catapulting) adottata dalla Zeiss, il tessuto tagliato viene colpito sotto il piano della sezione (quindi nello spessore del vetrino) dal fascio laser, per la durata di appena 1 ns [6]. Questo tempo è sufficiente a catapultare la sezione isolata nel dispositivo di raccolta
SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 5
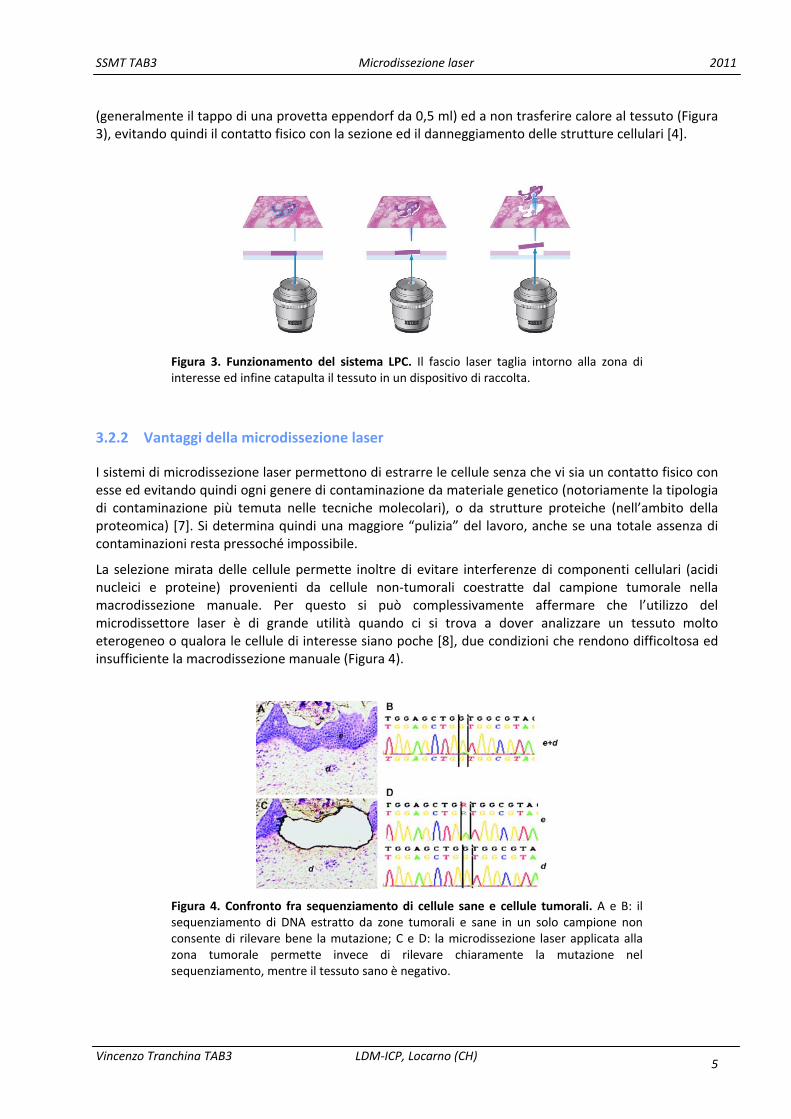
(generalmente il tappo di una provetta eppendorf da 0,5 ml) ed a non trasferire calore al tessuto (Figura 3), evitando quindi il contatto fisico con la sezione ed il danneggiamento delle strutture cellulari [4].
Figura 3. Funzionamento del sistema LPC. Il fascio laser taglia intorno alla zona di interesse ed infine catapulta il tessuto in un dispositivo di raccolta.
3.2.2 Vantaggi della microdissezione laser
I sistemi di microdissezione laser permettono di estrarre le cellule senza che vi sia un contatto fisico con esse ed evitando quindi ogni genere di contaminazione da materiale genetico (notoriamente la tipologia di contaminazione più temuta nelle tecniche molecolari), o da strutture proteiche (nell’ambito della proteomica) [7]. Si determina quindi una maggiore “pulizia” del lavoro, anche se una totale assenza di contaminazioni resta pressoché impossibile.
La selezione mirata delle cellule permette inoltre di evitare interferenze di componenti cellulari (acidi nucleici e proteine) provenienti da cellule non‐tumorali coestratte dal campione tumorale nella macrodissezione manuale. Per questo si può complessivamente affermare che l’utilizzo del microdissettore laser è di grande utilità quando ci si trova a dover analizzare un tessuto molto eterogeneo o qualora le cellule di interesse siano poche [8], due condizioni che rendono difficoltosa ed insufficiente la macrodissezione manuale (Figura 4).
Figura 4. Confronto fra sequenziamento di cellule sane e cellule tumorali. A e B: il sequenziamento di DNA estratto da zone tumorali e sane in un solo campione non consente di rilevare bene la mutazione; C e D: la microdissezione laser applicata alla zona tumorale permette invece di rilevare chiaramente la mutazione nel sequenziamento, mentre il tessuto sano è negativo.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 6
Un terzo aspetto sicuramente molto vantaggioso è l’estrazione di cellule in tempo reale su schermo. Se nella macrodissezione manuale il TAB seleziona con un bisturi, in modo sommario e “ad occhio”, piccole aree di tessuto – confrontando la sezione istologica colorata in HE (ematossilina ed eosina) con quella in bianco (non colorata) – al microdissettore laser le cellule che si vogliono estrarre sono visibili su schermo e si ha maggiore consapevolezza di ciò che si sta selezionando [4], oltre che, ovviamente, maggiore precisione.
La colorazione in HE può essere d’aiuto nel creare il sufficiente contrasto, necessario per identificare con sicurezza le cellule. È stato dimostrato [4] che la colorazione in HE non influisce (se non minimamente) sulla resa e sulla qualità di DNA, mentre quest’ultima è influenzata dalle colorazioni di immunoistochimica.
Durante le prime prove pratiche sul microdissettore, la Dott.ssa Kerstin Hagen‐Mann (Zeiss Germany) ha consigliato l’eventuale colorazione con Violetto di Cresile, che andrebbe ad inficiare in minor misura la resa di DNA e la sua qualità.
Per questo lavoro si è deciso di adoperare sezioni in bianco, in quanto il contrasto generato è già sufficiente a distinguere con sicurezza le aree tumorali da quelle sane. Inoltre i casi utilizzati per questo lavoro presentavano aree tumorali vaste e ben distinte da quelle sane.
3.3 GIUSTIFICAZIONE DELLA SCELTA
All’Istituto Cantonale di Patologia di Locarno (ICP) i Tecnici in Analisi Biomediche (TAB) e i biologi del Laboratorio di Diagnostica Molecolare (LDM), diretti dal Dott. Milo Frattini, devono spesso analizzare sezioni istologiche in cui il tessuto ha una superficie molto ridotta, che oltretutto è solo in parte (talvolta in minima parte) tumorale. Si tratta ad esempio di biopsie midollari, rettali, gastriche, prostatiche o frustoli bioptici del fegato, del polmone e del seno.
Pertanto, l’estrazione delle cellule tumorali con l’attuale tecnica di dissezione, il successivo ottenimento di DNA e la sua amplificazione possono risultare difficoltosi.
Questi sono i motivi sottesi la scelta di questo lavoro. L’auspicio del personale dell’ICP‐LDM è che la tecnica di microdissezione laser possa ovviare alle difficoltà tecniche finora riscontrabili nell’estrazione da tessuto scarsamente rappresentativo.
Dal punto di vista personale, questo lavoro mi ha caricato di una grande motivazione, poiché ho la possibilità di confrontarmi con una tecnica di dissezione particolare e poco nota che potrà portare grandi benefici all’ICP‐LDM.
In questo lavoro sono stato assistito dal Dott. Milo Frattini e dal gruppo di Tecnici in Analisi Biomediche e biologi del ICP‐LDM. Inoltre ho seguito lezioni di formazione sull’uso del microdissettore laser da parte di Stefan Wahl e della Dott.ssa Kerstin Hagen‐Mann, tecnici specializzati della ditta produttrice e fornitrice del microdissettore laser (Zeiss, Germany).
3.4 OBIETTIVO
L’obiettivo di questo lavoro è di definire le condizioni per le quali si possa ottenere un estratto di DNA quantitativamente e qualitativamente soddisfacente – tale da permettere le analisi molecolari – anche da campione poco rappresentativo, lavorando sull’utilizzo del microdissettore laser e sui protocolli di estrazione di DNA genomico e di PCR.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 7
3.5 STRATEGIA DI REALIZZAZIONE
Il mio lavoro ha seguito concettualmente e cronologicamente tre fasi principali, che per questioni organizzative e temporali sono state intercalate o sovrapposte:
1. Macrodissezione manuale
Estrazione di DNA genomico con la tecnica tradizionale da campioni quantitativamente rappresentativi, passando gradualmente a campioni sempre più ridotti, questo per esercitare la manualità nella tecnica di macrodissezione, per verificare quanto DNA genomico è possibile ottenere da porzioni di sezione di tessuto sempre più ridotte e per capire personalmente dove si riscontra il problema per cui questo lavoro si rende necessario.
2. Microdissezione laser
Definire le modalità di utilizzo del microdissettore laser, al fine di ottenere una buona selezione delle cellule (evitando di danneggiarle con il laser, ma selezionandole comunque in modo mirato) in modo da avere una sufficiente quantità di DNA da cellule tumorali.
3. Ottimizzazione dell’estrazione e dell’amplificazione
Rivedere le condizioni di estrazione e PCR per le quali, anche da poco materiale, si possano determinare senza difficoltà i genotipi indagati.
Come controllo interno dell’effettiva amplificabilità del DNA estratto, viene amplificato l’esone 2 del gene KRAS perché molto raramente subisce duplicazioni e delezioni [1]. KRAS è un oncogene della famiglia dei geni Ras che codifica per una proteina di 189 aminoacidi con attività GTPasica. Essa è coinvolta nella trasduzione dei segnali di mitosi e induce quindi una proliferazione cellulare. Il cambiamento di un solo aminoacido in posizioni precise (codoni 12 e 13) rende la proteina costitutivamente attiva, e in grado di concorrere significativamente alla cancerogenesi. La ricerca di mutazioni nell’esone 2 di KRAS è richiesta per individuare, nei pazienti affetti da carcinoma colorettale o polmonare, i soggetti che potranno trarre giovamento dalla somministrazione di nuove terapie bersaglio‐specifiche, in particolare dalle terapie anti‐EGFR: in presenza di una mutazione nel gene KRAS, il paziente è resistente a tali trattamenti, e deve essere indirizzato, piu’ utilmente, verso altri schemi chemioterapici.
Similarmente, si è deciso di amplificare anche il gene EGFR (esoni 18, 19, 20 e 21). La presenza di mutazioni in tali esoni conferisce sensibilità (esoni 18, 19 e 21) o resistenza (esone 20) a terapie farmacologiche specificamente indirizzate contro tale recettore. Anche l’analisi mutazionale del gene EGFR è quindi richiesta dai medici oncologi prima della somministrazione della terapia per i pazienti affetti da tumori polmonari e colorettali. I quattro esoni fanno parte del dominio tirosinchinasico del gene EGFR. In breve, la proteina codificata dal gene EGFR è una proteina trans‐membrana della famiglia delle chinasi. Si tratta di un recettore di fattori di crescita che accoppiandosi al ligando induce la proliferazione cellulare. Se ne conoscono moltissime isoforme, responsabili di varie patologie di origine neoplastica [2].

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 8
4 MATERIALI E METODI
4.1 MATERIALE PATOLOGICO UTILIZZATO
Prima fase – Macrodissezione manuale
Il materiale che è stato utilizzato per la prima fase del lavoro (macrodissezione manuale) proviene da un paziente di sesso maschile, di 70 anni, operato per un adenocarcinoma moderatamente differenziato del terzo medio del colon destro. Il materiale è del 2004 ed è identificato come “#1”
Il blocchetto di materiale incluso in paraffina ritenuto idoneo per le analisi che ho eseguito è il 00F (#1‐00F). Si tratta del margine di una massa neoplastica stenosante di dimensioni 6x6 cm, infiltrante il tessuto adiposo pericolico.
Ho preparato 20 sezioni dello spessore di 3 µm su vetrini Thermo Scientific Superfrost Plus (Thermo Fischer Scientific, Braunschweig, Germany) effettuando poi la macrodissezione manuale, come da protocollo in umido ICP‐LDM.
Il blocchetto 00F, esaurito, è stato scartato per la seconda fase del lavoro.
Seconda fase – Microdissezione laser
Dello stesso paziente sopraccitato sono state preparate a più riprese delle sezioni dello spessore di 3 µm, dal blocchetto 00E (#1‐00E), scelto perché facente parte della stessa massa tumorale del caso appena descritto, con condizioni istologiche sovrapponibili al blocco 00F.
Ecco la sequenza temporale della preparazione di sezioni per la microdissezione laser.
05.02.2011: 3 sezioni su vetrino Thermo Scientific Superfrost e 2 sezioni su vetrino Thermo Scientific Superfrost Plus (Thermo Fischer Scientific), per prove di taglio sul microdissettore con confronto fra le due tipologie di vetrini della Thermo Fischer Scientific, nell’ipotesi di riuscire a lavorare anche con vetrini sprovvisti di membrana in PEN.
07.03.2011: 2 sezioni su vetrini Zeiss con membrana di PEN, come da setup definito per obiettivo 40x (vedi sottocapitolo “Metodi”, per spiegazioni sui setup del microdissettore laser).
04.04.2011: 2 sezioni su vetrini Zeiss con membrana di PEN, come da setup definito per obiettivo 10x (vedi sottocapitolo “Metodi”, per spiegazioni sui setup del microdissettore laser).
Nel corso del lavoro sono state eseguite amplificazioni anche su altri materiali, al fine di accertare la riproducibilità dei metodi utilizzati.
Il confronto dei risultati delle sezioni ricavate dal blocchetto 00E con quelle degli altri casi analizzati in parallelo, ha fatto emergere un problema nella qualità del DNA del blocchetto (probabilmente dell’intero caso) scelto. Si è quindi deciso di scartare il caso #1‐00E proseguendo le analisi esclusivamente su altri due casi di cancro colorettale utilizzati a scopi di ricerca dal LDM (non di proprietà dell’ICP): 09‐I‐9711‐A6 (anno 2009) e 185‐B1 (anno 2008).

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 9
4.2 MACRODISSEZIONE MANUALE
In questa prima fase si vuole raccogliere il materiale patologico direttamente dal vetrino sparaffinato, grattandolo con la lama sterile di un bisturi e trasportandolo nella provetta per l’estrazione. Si è scelto di utilizzare la metodica di macrodissezione manuale in umido, poiché la raccolta del materiale dal vetrino è facilitata ed il rischio di perderlo per le sue proprietà volatili è ridotto. Inoltre è la metodica più simile a quella richiesta per la microdissezione laser e ci permette di introdurre un minor numero di variabili nel flusso di lavoro. Il protocollo originale di estrazione del DNA genomico, prevede la sparaffinatura dopo macrodissezione manuale, quindi già direttamente nella provetta.
4.2.1 Materiale e strumenti per la macrodissezione
I seguenti materiali e strumenti sono stati utilizzati per la macrodissezione da sezioni in bianco:
Tissue QIAamp DNA MiniKit 250 (Qiagen, USA), contenente
o buffers ATL, AL, AW1, AW2, AE
o colonne per l’isolamento del DNA genomico
o tubi sterili da 2 ml
etanolo 99,8% (Fluka Analytical, Switzerland)
xilolo 98,5% (Carlo Erba Reagents, Italy)
acqua distillata Water purification System Milli‐Q Biocel (Billerica, USA)
vetrini Thermo Scientific Superfrost Plus (Thermo Fischer Scientific, Germany)
bisturi Braun Sterile Surgical Blades (Braun, Germany)
tubi Sarstedt Eppendorf 2,0 e 1,5 ml (Sarstedt AG, Germany)
microtomo Microm HM 440E Microtome (GMI Inc., USA)
centrifuga Eppendorf Centrifuge 5415 R (Vaudaux‐Eppendor, Switzerland)
stufa Heraeus Instruments TL 2436 (Heraeus, Switzerland)
vortex Heidolph REAX 2000 (Heidolph Instruments, Germany)
termoblocco Bio Labo CH‐100 Thermoblock (Châtel‐St.Denis, Switzerland )
stufa Binder FD‐53 (Binder GmbH, Germany )
ruota Stuart Rotator SB3 (Instrumenten‐Gesellschaft AG, Switzerland)
4.2.2 Protocollo per la macrodissezione
Seguire il protocollo a pagina seguente, ponendo attenzione al cambio regolare di guanti fra un campione e l’altro, fra un passaggio e l’altro ed ogni qualvolta si esca dalla cappa o si tocchi qualcosa di potenzialmente contaminato da DNA. Eseguire inoltre un ciclo di sterilizzazione con UV per 15 minuti della cappa per l’estrazione prima di utilizzarla.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 10
La fase di sparaffinatura può essere effettuata direttamente sul vetrino prima dello “scraping” (grattare con il bisturi sul vetrino per rimuovere il materiale), come da seguente protocollo:
vetrini in stufa per 45 min a 60°C
passaggio in xilolo per 5 min (2 volte)
passaggio in etanolo 100% per 5 min
passaggio in etanolo 96% per 5 min
passaggio in etanolo 80% per 5 min
passaggio in acqua distillata per 5 min, asciugare i vetrini
passare alla macrodissezione (lama bisturi nuova o ben pulita con etanolo)
Sovrapporre la sezione in bianco sparaffinata a quella HE e tratteggiare sul retro della sezione in bianco l’area da estrarre. Grattare con un bisturi (lama nuova e sterile, o pulita con etanolo) e depositare il materiale sul fondo di una provetta Eppendorf precedentemente identificata.
Aliquotare 180 µl di tampone di lisi ATL e 20 µl di proteinasi K sul campione, vortexare e sigillare il tappo della provetta con parafilm. Incubare over‐night in stufa a 57°C su Stuart Rotator SB3.
Impostare il termoblocco a 70°C per 10’.
Centrifugare brevemente il campione ed aggiungere 200 µl di buffer AL. Sigillare il tappo della provetta con parafilm e vortexare brevemente. Alloggiare il campione nel termoblocco e far partire il programma preimpostato. Dopodiché centrifugare brevemente e, sotto cappa, aliquotare 200 µl di etanolo. Infine vortexare e centrifugare brevemente.
Inizia ora la fase di isolamento del DNA su colonna. Preparare tre tubi Qiagen da 2 ml ed una colonna (kit QIAamp DNA MiniKit Qiagen Tissue), una provetta Eppendorf senza tappo da 2 ml ed una provetta Eppendorf con tappo da 1,5 ml, per raccogliere l’eluato finale.
Pipettare tutto l’estratto sulla colonna situata nel primo tubo Qiagen da 2 ml
centrifugare a 8’000 rpm per 1 min
spostare la colonna sul 2° tubo da 2 ml ed aggiungere 500 µl di wash buffer AW1
centrifugare a 8’000 rpm per 1 min
spostare la colonna sul 3° tubo da 2 ml ed aggiungere 500 µl di wash buffer AW2
centrifugare a 13’000 rpm per 3 min
spostare la colonna sulla provetta Eppendorf senza tappo da 2 ml
centrifugare a 13’000 rpm per 1 min
spostare la colonna sulla provetta Eppendorf con tappo da 1,5 ml
aliquotare 50 µl di buffer AE
incubare 5 min a temperatura ambiente (TA) e centrifugare a 8’000 rpm per 1 min
ricaricare la colonna con l’eluato centrifugato (questo passaggio permette di aumentare la resa finale di DNA).
incubare 5 min a TA e centrifugare a 8’000 rpm per 1 min
eliminare la colonna e conservare la provetta Eppendorf con l’eluato

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 11
4.2.3 Strategia di realizzazione del lavoro – Parte macrodissezione
Partendo da cinque sezioni, ho gradualmente ridotto la massa di tessuto da cui ho estratto il DNA, passando a due sezioni, poi una ed in seguito estraendo frazioni di sezione fino a 1/16. In questo modo ho potuto rendermi conto di come possa variare la resa di DNA in rapporto alla superficie dissezionata.
Inoltre ho capito che risulta difficile scendere ad aree di tessuto ancora più piccole con la macrodissezione manuale, infatti non si potrebbe più essere sicuri di aver realmente isolato del materiale. Infatti il volume di materiale isolabile da 1/16 di sezione può essere stimato a 0,03 mm3, grossomodo il 3% di 1 mm3, ed è facile comprendere quanto possa essere difficile accertarsi della sua presenza.
4.3 MICRODISSEZIONE LASER
La microdissezione laser sostituisce alla lama del bisturi utilizzata dall’operatore, il sistema automatizzato di dissezionamento a laser. Le nozioni acquisite nei primi due giorni di pratica e messa a punto del microdissettore laser, effettuate il 14 e 15 febbraio 2011, sono qui descritte.
4.3.1 Introduzione al software e calibrazione del microscopio
Il Software permette di effettuare numerosi setup personalizzati al fine di migliorare la selezione delle cellule in funzione dell’ingrandimento, del tipo e dello spessore del tessuto e di qualsiasi condizione possa influire sul risultato. Per ogni variabile citata bisogna infatti impostare i livelli di messa a fuoco e di energia del raggio UV tramite un’apposita mascherina che viene salvata.
Il microdissettore laser si è mostrato molto preciso ma non certo di semplice utilizzo, in quanto complesso nella sua calibrazione e gestione giornaliera. Per la calibrazione ed il successivo uso, bisogna distinguere i due meccanismi di funzionamento del laser:
Cut (taglia): presupposto necessario è che la sezione sia a fuoco a schermo e negli oculari. L’ideale è intervenire ad ingrandimenti maggiori (400x) in modo da eseguire una regolazione fine, ma è necessario calibrare il laser per tutti gli ingrandimenti. Un setup ottimale include:
o Focus: il fascio laser deve essere condotto nella sezione e non sotto o sopra di essa (intervenire nell’apposita mascherina o cercare il livello giusto durante la fase di taglio con i tasti PgUp e PgDn).
o Energia: il fascio laser deve essere sufficientemente aggressivo da causare la fotoframmentazione del tessuto colpito ed al contempo delicato per evitare di bruciare e danneggiare il tessuto selezionato (intervenire con i tasti Home e End).
Pressure Catapulting (catapulta): in questo caso il laser non deve colpire il tessuto (ne causerebbe la fotoframmentazione) ma deve solamente fornire un impulso per far catapultare la sezione tagliata nel dispositivo di raccolta. Si devono modificare:
o Focus: il fascio laser deve essere condotto al di sotto della sezione (quindi nel vetrino). Se il fascio incide troppo lontano dalla sezione il tessuto non sarà catapultato; se troppo vicino trasferirà una quantità di energia eccessiva alle cellule, danneggiandole.
o Energia: un fascio laser troppo poco aggressivo non consentirà alla sezione di essere catapultata nel dispositivo di raccolta; uno troppo forte trasferirà energia alle cellule, danneggiandole.
Si ricerca quindi il corretto (o migliore) compromesso fra messa a fuoco ed energia trasmessa, nei due sistemi di funzionamento del laser.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 12
Ad un ingrandimento di 400 x, il delay ideale per il focus (Δ Focus) è di +2/+3 unità fra Focuscut e Focuscatapult, mentre quello ideale per l’energia (Δ Energy) è di ‐20 unità fra Energycut ed Energycatapult.
Una volta calibrati i livelli di focus ed energy si effettua un taglio di prova, per verificare che il laser sia calibrato a dovere. Si eseguono le seguenti prove:
1. Ricerca del piano di cut: si traccia una linea nel tessuto con l’utility di disegno libero e si imposta la velocità del laser al minimo possibile. Si fa partire il laser e si verifica che il taglio sia visibile e netto, eventualmente si ottimizzano focus ed energy.
2. Ricerca del piano di catapult: si ripete l’operazione precedente ma con l’utility di disegno a cerchio e la funzione cut&catapult attiva. Se il laser non riesce a catapultare la sezione si ottimizzano i livelli di focus e energy. Si verifica che il laser abbia colpito almeno 10 µm al di sotto del tessuto.
Settimanalmente deve essere controllata la calibrazione delle lenti (degli obiettivi) e del laser, per ciascun ingrandimento (50x, 100x, 200x, 400x). In pratica si esegue la manutenzione verificando che il laser tagli esattamente dove il software raffigura la linea di taglio e che questo avvenga nel medesimo punto a tutti gli ingrandimenti voluti.
La procedura di calibrazione delle lenti e del laser non è descritta in quanto lunga, complessa e presupposto minimo per il corretto funzionamento dello strumento.
Effettuati questi test, il microdissettore è pronto a lavorare nella maniera migliore sulla sezione di nostro interesse. Nelle prossime pagine è descritto l’utilizzo giornaliero del microdissettore.
4.3.2 Scelta del materiale per la microdissezione laser
Per la microdissezione laser possono essere utilizzati appositi vetrini della Zeiss, provvisti di una membrana in polietilene naftalato (PEN) che consente di catapultare ampie zone di tessuto in un colpo solo. Inoltre la ditta fornisce tubi in plastica sterili da 0,5 ml contenenti una matrice di silicone nella parte interna del tappo. Quest’ultima è inerte (non influisce in alcun modo sulle analisi seguenti) ed ha lo scopo di catturare le cellule catapultate e di permettere la visualizzazione dell’estratto direttamente nel tappo del tubo di raccolta.
Il microscopio permette infatti la focalizzazione della luce sul tappo del tubo di raccolta, in questo modo ci si può accertare che la zona catapultata sia effettivamente giunta nel tappo.
È comunque possibile utilizzare il microdissettore laser con normali vetrini e tubi sterili, con le seguenti limitazioni:
Vetrini Thermo Scientific Superfrost o Superfrost Plus (Thermo Fischer Scientific, Braunschweig, Germany): l’assenza della membrana non consente di catapultare ampie zone di tessuto. Si deve procedere shot by shot, cioè cellula dopo cellula (o a piccoli clusters). Le funzioni del software consentono comunque di eseguire questa procedura in maniera automatica una volta selezionata l’area di tessuto da colpire.
Tubi sterili 0,5 ml: il tappo del tubo deve contenere un fluido per agevolare la cattura delle cellule. È stata effettuata l’applicazione all’interno del tappo del primo reattivo utilizzato durante l’estrazione di DNA, ossia il Buffer ATL (Tissue QIAamp DNA MiniKit 250, Qiagen, Chatsworth, USA), in un volume di 20 µl. Ovviamente l’aliquota di Buffer ATL restante, per giungere al volume complessivo di 180 µl, verrà aggiunta all’inizio della procedura di estrazione di DNA genomico.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 13
4.3.3 Gestione del software nell’uso giornaliero
Per la microdissezione laser sono stati utilizzati vetrini provvisti di membrana di PEN (Figura 5). Abbiamo riscontrato che la procedura di sparaffinamento e reidratazione in umido del tessuto (vedi paragrafi “Macrodissezione”) provoca un più facile distacco della sezione dal vetrino, rispetto a quanto accadeva con i vetrini Thermo Scientific Superforst Plus (Braunschweig, Germany). Questo è stato dovuto a una minore forza d’interazione della sezione con il vetrino, proprio a causa della presenza della membrana, di natura idrofobica.
La Dott.ssa Hagen‐Mann ha consigliato di sottoporre i vetrini ad un trattamento con raggi UV. Questa procedura ha trovato riscontro anche all’interno di un protocollo open source (Allegato 8), che specifica la durata del trattamento: 30 minuti. La procedura è stata testata il 07.03.2011 ed ha permesso di migliorare l’aderenza del tessuto al vetrino, che di fatto non si è più staccato dalla membrana PEN.
Figura 5. Polimero PEN. Struttura chimica di un unità del polimero PEN che costituisce la membrana che ricopre i vetrini utilizzati per la microdissezione laser.
Per contro, si è deciso di utilizzare tubi da 0,5 ml senza la matrice di silicone nel tappo, utilizzando il Buffer ATL (aliquota da 20 µl) come fluido di raccolta.
Dopo numerose prove ed insuccessi, descritti nel capitolo “Risultati” (“5.3 Ottimizzazione dell’analisi dei microestratti”), ha avuto inizio la cattura di cellule sulle sezioni dei casi scelti per questo lavoro, al fine di acquisire manualità con lo strumento. Descrivo brevemente come questo è avvenuto:
Accendere il computer, il Power Supply (unità di trasmissione dati fra computer e microscopio) e infine il microscopio.
Aprire il software attraverso l’icona PALM@RoboV4 presente sul desktop.
Cliccare sull’icona Load Position ed inserire i vetrini sul supporto.
Inserire un tubo di raccolta nel dispositivo di cattura e riporlo sul RoboMover (ala rimovibile del dispositivo di raccolta).
Selezionare 3 slide holder e confermare su Return to working area.
Cliccare sull’icona Capture Device e selezionare il tappo del tubo di raccolta in modo che questo si sposti sotto il percorso della luce (quindi al di sopra della sezione, per raccogliere le cellule).
Cliccare sull’icona Navigator e selezionare il vetrino su cui lavorare.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 14
Ricercare l’area del tessuto desiderata e l’ingrandimento a cui si desidera effettuare la microdissezione laser.
Caricare il setup personalizzato in funzione del caso su cui si lavora.
Con uno strumento di disegno tracciare la/e linea/e di taglio del laser sul tessuto (prestare attenzione alla corretta messa a fuoco in questa fase).
Selezionare la funzione di taglio desiderata (Cut, LPC, Cut+autoLPC, ecc.).
Cliccare sull’icona Element List per selezionare quali aree tagliare di quelle disegnate.
Cliccare sull’icona Cut per tagliare il tessuto e/o catapultarlo.
Infine cliccare sull’icona Cap viewer per visualizzare il tessuto raccolto nel tappo della Eppendorf (meglio con gli obiettivi 5x e 10x).
L’immagine seguente (Figura 6) mostra la schermata principale del software associato al microdissettore.
Figura 6. Schermata principale del software PALM@RoboV4. Legenda: 1 = elements list; 2 = Cap viewer; 3 = Capture device; 4 = Load Position; 5 = Navigator; 6 = Objective; 7 = Start Laser; 8 = Cut function; 9 = Draw tools; 10 = Laser energy levels; 11 = Laser focus levels; 12 = Cut lines viewer.
Prestare la massima attenzione nel rimuovere il tubo di raccolta dal RoboMover, poiché qualsiasi movimento brusco può far sì che il tessuto catapultato si stacchi dal fluido di raccolta nel tappo, andando perso o contaminando il piano di lavoro.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 15
4.4 QUANTIFICAZIONE
Dopo aver estratto il campione, con una o con l’altra tecnica, e prima di poter effettuare l’amplificazione, occorre determinare la quantità e la qualità del DNA genomico ottenuto. Tale analisi viene eseguita con lo spettrofotometro Thermo Scientific NanoDrop ND‐1000 Spectrophotometer, fornito dalla ditta Witec (Luzern, Switzerland).
Dopo gli opportuni lavaggi (3 lavaggi delle mini‐cavità con acqua sterile e carta monouso) e la calibrazione con il Blank (buffer AE usato per l’eluizione nell’ultima fase dell’estrazione), si utilizzano 2 µl di eluato che vengono pipettati sulla mini‐cavità dell’apparecchio. L’apparecchio fornisce lo spettro di assorbimento da 220 nm a 350 nm e ricava la concentrazione di DNA in ng/µl dell’eluato in funzione dell’assorbanza rilevata a 230 nm. Inoltre vengono calcolati i rapporti di assorbanza A260/230 e A260/280 indicativi per eventuali contaminazioni rispettivamente da alcoli (ad esempio da etanolo, utilizzato nell’estrazione) e da materiale proteico. I rapporti di assorbanza devono essere indicativamente intorno al 2, ma questo valore – come pure quello della concentrazione della sospensione di DNA eluito – non è decisionale e l’amplificabilità del DNA genomico ottenuto è valutata solo dopo aver analizzato il gene di controllo [9].
Ho quantificato in questa maniera tutti i campioni estratti sia con la macrodissezione manuale che con la microdissezione laser (vedi capitolo “Risultati”).
4.5 AMPLIFICAZIONE E SEQUENZIAMENTO
Dopo la quantificazione, tutti i campioni in esame hanno seguito lo stesso flusso di analisi di amplificazione e sequenziamento. Questa scelta è stata fatta per poter valutare l’impatto della tecnica di microdissezione laser sia sulla PCR che sul sequenziamento.
4.5.1 Amplificazione
Tutti i campioni estratti, quantificati e opportunamente diluiti a 25 ng/µl (o non diluiti se la concentrazione era inferiore a 25 ng/µl), sono stati amplificati per l’esone 2 del gene KRAS per verificare che il DNA non fosse qualitativamente scarso e quindi non amplificabile. Successivamente sono stati amplificati anche gli esoni 18, 19, 20 e 21 del gene EGFR, con una nested PCR. La nested PCR (una metodica che prevede due PCR in serie al posto di una soltanto) comporta un alto rischio di contaminazione in quanto, aumentando i cicli rispetto ad una PCR normale, più facilmente si osservano amplificati anche nei bianchi di reazione. Per ovviare a tale inconveniente, il LDM sta mettendo a punto un’analisi basata su un normale protocollo di amplificazione, utilizzando primers più interni, che forniscono un prodotto di amplificazione più piccolo (200 bp circa contro 400 bp della nested), senza peraltro perdere informazioni rispetto all’intersa sequenza nucleotidici esonica.
Il materiale utilizzato e la procedura di amplificazione nel dettaglio sono consultabili nell’Allegato 1.
4.5.2 Migrazione su gel d’agarosio 1,8% e colorazione
Sia gli amplificati di KRAS che di EGFR sono stati fatti migrare in gel d’agarosio 1,8%, preparato con tampone TBE 1x. Infine il gel è stato colorato in una soluzione di Gel Red, preparata con tampone TBE 0,5x e fotografato con scanner a UV rilevatore di fluorescenza Fusion Fx5.
Il materiale utilizzato e la procedura di migrazione e colorazione del gel sono consultabili negli Allegati 2 e 3.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 16
4.5.3 Purificazione amplificato PCR
È stato utilizzato il kit HiYield Gel/PCR DNA Fragments Extraction YDF 300 (RBC Bioscience, Taipei, Taiwan). La purificazione avviene su colonna. L’intento è quello di rimuovere dall’amplificato di PCR tutto ciò che non sia DNA sintetizzato, ossia i primers ed i deossinucleotidi in eccesso.
Il materiale utilizzato e la procedura di purificazione sono consultabili nell’Allegato 4
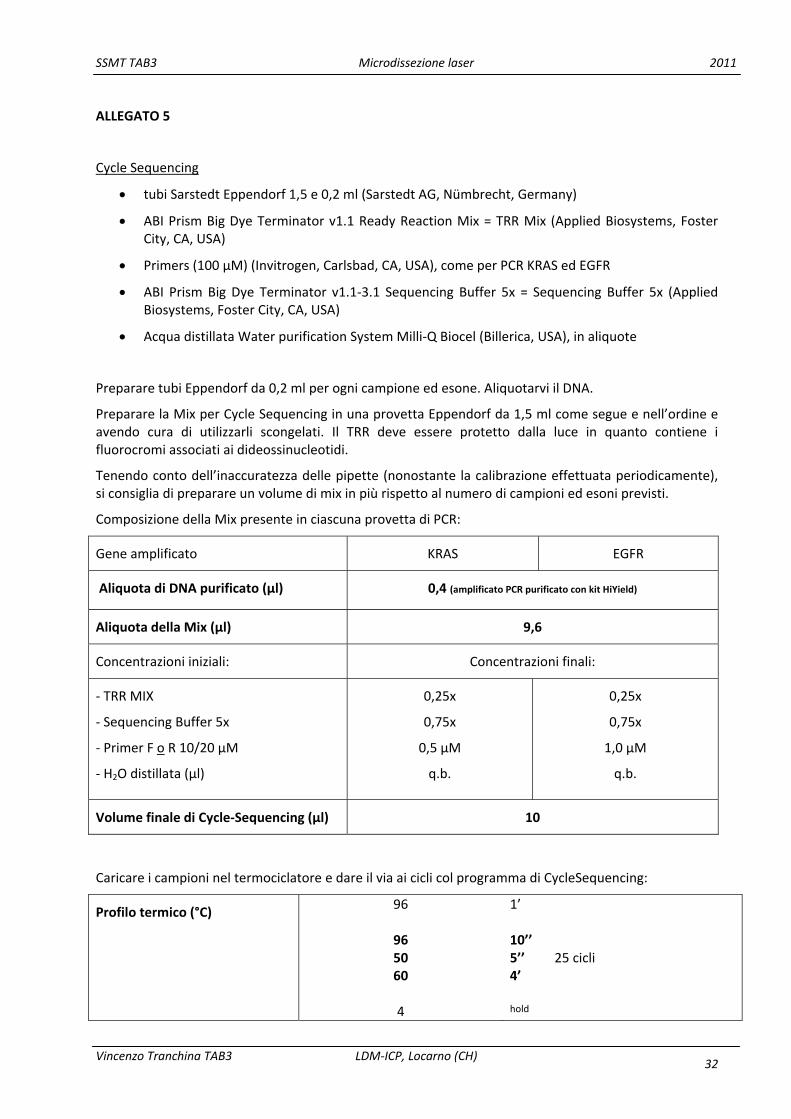
4.5.4 Cycle Sequencing
È una PCR asimmetrica effettuata con un solo primer ed una mix contenente dideossinucleotidi marcati con un fluorocromo (oltre ai deossinucleotidi e la polimerasi). L’intento è quello di amplificare il filamento di DNA introducendo in maniera aleatoria un dideossinucleotide fluorocromato (che interrompe la polimerizzazione, e pertanto detto anche terminatore), in modo da creare frammenti di diversa lunghezza. Solitamente si sceglie di amplificare entrambi gli strands quando si sa che il primer utilizzato lega il DNA in una zona prossima a hot‐spots di mutazione. Infatti i primi deossinucleotidi (e dideossinucleotidi) polimerizzati dopo il primer generano tracciati anomali nel sequenziamento e per questo si richiede la controprova di un eventuale sospetto di mutazione sull’altro strand, a cui corrisponde la zona terminale della polimerizzazione dell’esone.
Il materiale utilizzato e la procedura di Cycle Sequencing sono consultabili nell’Allegato 5.
4.5.5 Purificazione amplificato CycleSequencing
È stato utilizzato il kit G50 Dye Terminator Removal (RBC Bioscience, Taipei, Taiwan). La purificazione avviene su colonna contenente una resina, attraverso un procedimento chiamato di gel‐filtrazione. Anche in questo caso si vogliono rimuovere primers e deossinucleotidi (e dideossinucleotidi) non utilizzati nel Cycle Sequencing, ma il principio è diverso: non si effettua l’eluizione, bensì – centrifugando – si provoca la migrazione dell’amplificato attraverso la resina. I frammenti di DNA non sono ritardati dalle maglie della resina e vengono quindi raccolti, mentre i primers rimangono intrappolati nella resina (procedimento di gel‐filtrazione).
Il materiale utilizzato e la procedura di purificazione sono consultabili nell’Allegato 6.
4.5.6 Sequenziamento
È stato utilizzato il sequenziatore automatico Applied Biosystems ABI 3130 (Applied Biosystems, Foster City, CA, USA). L’apparecchio preleva un’aliquota di sospensione di DNA (trattata con 15 µl di formammide) dal pozzetto grazie ad un capillare a cui viene applicato un campo elettrico. I frammenti di DNA si muovono attraverso il capillare più o meno velocemente a seconda delle loro dimensioni, secondo il processo della elettroforesi capillare. Al termine della corsa è posto un laser che eccita i fluorocromi e un detector che raccoglie i segnali di fluorescenza prodotti. Un software riorganizza i dati di lunghezza e fluorescenza dei frammenti, generando la sequenza a schermo.
Il materiale utilizzato e la procedura di sequenziamento sono consultabili nell’Allegato 7.
SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 17
5 RISULTATI
5.1 RISULTATI DA MACRODISSEZIONE MANUALE
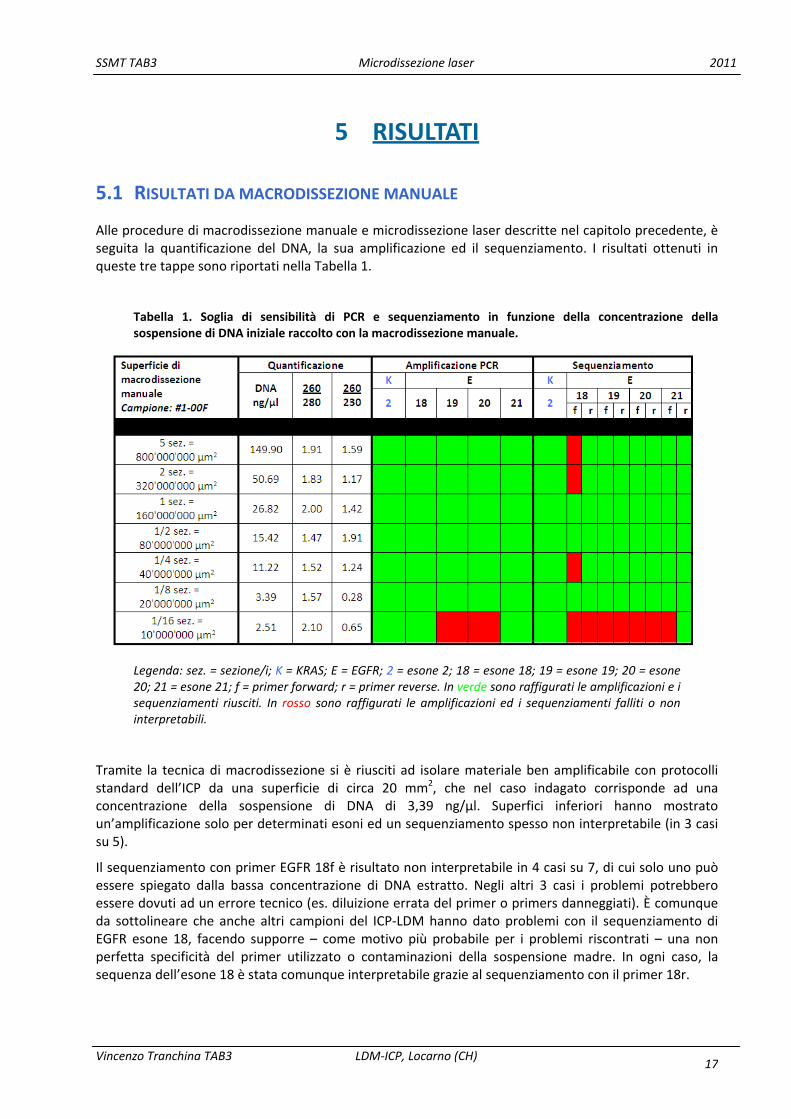
Alle procedure di macrodissezione manuale e microdissezione laser descritte nel capitolo precedente, è seguita la quantificazione del DNA, la sua amplificazione ed il sequenziamento. I risultati ottenuti in queste tre tappe sono riportati nella Tabella 1.
Tabella 1. Soglia di sensibilità di PCR e sequenziamento in funzione della concentrazione della sospensione di DNA iniziale raccolto con la macrodissezione manuale.
Legenda: sez. = sezione/i; K = KRAS; E = EGFR; 2 = esone 2; 18 = esone 18; 19 = esone 19; 20 = esone 20; 21 = esone 21; f = primer forward; r = primer reverse. In verde sono raffigurati le amplificazioni e i sequenziamenti riusciti. In rosso sono raffigurati le amplificazioni ed i sequenziamenti falliti o non interpretabili.
Tramite la tecnica di macrodissezione si è riusciti ad isolare materiale ben amplificabile con protocolli standard dell’ICP da una superficie di circa 20 mm2, che nel caso indagato corrisponde ad una concentrazione della sospensione di DNA di 3,39 ng/µl. Superfici inferiori hanno mostrato un’amplificazione solo per determinati esoni ed un sequenziamento spesso non interpretabile (in 3 casi su 5).
Il sequenziamento con primer EGFR 18f è risultato non interpretabile in 4 casi su 7, di cui solo uno può essere spiegato dalla bassa concentrazione di DNA estratto. Negli altri 3 casi i problemi potrebbero essere dovuti ad un errore tecnico (es. diluizione errata del primer o primers danneggiati). È comunque da sottolineare che anche altri campioni del ICP‐LDM hanno dato problemi con il sequenziamento di EGFR esone 18, facendo supporre – come motivo più probabile per i problemi riscontrati – una non perfetta specificità del primer utilizzato o contaminazioni della sospensione madre. In ogni caso, la sequenza dell’esone 18 è stata comunque interpretabile grazie al sequenziamento con il primer 18r.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 18
Come è possibile osservare dalla Tabella 1, superfici sempre minori di materiale macrodissezionato hanno correlato con rese di DNA progressivamente decrescenti (Figura 7). Nel caso di dissezioni di superfici ridotte, la correlazione è stata meno buona. Ciò è imputabile in primis alla variabilità del tessuto, secondariamente a limitazioni pratiche introdotte dall’operatore e infine a limitazioni tecniche dei materiali utilizzati per l’estrazione che non possono garantire una perfetta riproducibilità del rapporto area/resa.
Figura 7. Correlazione fra resa di DNA e superficie macrodissezionata dal vetrino. I dati sono riportati nella Tabella 1. È evidente a colpo d’occhio la correlazione fra resa di Dna e superficie dissezionata, confermata da un coefficiente di correlazione R = 0.9942.
Il campione #1‐00F in esame ha mostrato una mutazione puntiforme nel gene KRAS. Nello specifico il tessuto tumorale mostra un’adenina (A) al posto della guanina (G), come si nota dal doppio picco in posizione 95 (Figura 8). La sostituzione determina la traduzione del codone in un’aspargina, anziché in una glicina, iperattivando il gene e concorrendo alla proliferazione cellulare.
Figura 8. Mutazione Gly12Asp nel codone 12 dell’esone 2 di KRAS nel campione utilizzato nella macrodissezione manuale (#1‐00F).

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 19
5.2 CONFRONTO MACRODISSEZIONE MANUALE‐MICRODISSEZIONE
In data 07.03.2011 ho eseguito una prima cattura di cellule per l’estrazione di DNA con il microdissettore. Dopo aver osservato con il Dott. Milo Frattini le sezioni colorate in HE, ho potuto visualizzare le aree tumorali anche su sezione in bianco ed orientarmi sulle cellule bersaglio della microdissezione laser.
Ho proceduto a catturare ammassi di cellule quanto più grandi possibile, grossomodo a clusters di 2000‐3000 µm2 l’uno, fino a giungere ad una superficie di tessuto catturato di 100’000 µm2. Se il tessuto è piuttosto omogeneo è possibile usare ingrandimenti inferiori, catturando aree più ampie e riducendo quindi i tempi di microdissezione laser, come accaduto nella microdissezione laser del 04.04.2011 per i due casi ritenuti idonei per il lavoro. Prima di proseguire con estrazioni a quantità di materiale decrescenti, ho deciso di estrarre il DNA dal tessuto già raccolto, per verificare che ci fosse una resa minima accettabile.
Per resa minima accettabile è stata considerato la concentrazione più bassa dalla quale siamo riusciti a estrarre e sequenziare efficacemente il DNA dei macroestratti (vedi in seguito). Abbiamo scoperto con stupore che nonostante la piccola area microdissezionata (0,1 mm2) la resa di DNA è stata comunque piuttosto elevata, in linea con quanto estratto da 40 mm2 con la tecnica di macrodissezione manuale.
5.3 OTTIMIZZAZIONE DELL’ANALISI DEI MICROESTRATTI
In data 09.03.2011, gli estratti ricavati dalla microdissezione laser hanno sorprendentemente dato problemi di amplificazione in KRAS. Non si riusciva infatti a capire come un estratto a concentrazione di DNA di 9,18 ng/µl non riuscisse ad essere amplificato, tanto più che l’estratto di 1/16 di sezione a concentrazione di DNA di 2,51 ng/µl – coamplificato con gli estratti da microdissettore laser – mostrava invece una banda chiara, dopo elettroforesi su gel di agarosio.
Si è deciso di ripetere la microdissezione laser e di estrarre il DNA con solo 2 ore di incubazione a 57°C in moto rotatorio, dopo l’aggiunta di Buffer ATL e Proteinasi K: tale modifica è stata apportata per escludere la possibilità che fosse il trattamento overnight a danneggiare il DNA.
La PCR KRAS è quindi stata eseguita con i microestratti da 20'000 e 100'000 µm2 trattati con ATL e proteinasi K overnight assieme alle rispettive superfici dei microestratti trattati però per solo 2 ore. Si è verificata una chiara amplificazione dei microestratti da 100'000 µm2, mentre quelli da 20'000 µm2 non hanno mostrato bande. Un’incubazione di appena 2 ore non ha influenzato negativamente il procedimento di estrazione di DNA genomico. Pertanto tale modifica al protocollo è stata resa definitiva.
Per verificare la ripetibilità della PCR si è deciso di amplificare nuovamente tutti i microestratti (con e senza trattamento in estrazione ridotto), assieme a due macroestratti a concentrazione standard (25 ng/µl) e limitante (2,51 ng/µl, quindi 1/16 di sezione della macrodissezione manuale) come controlli positivi.
L’esito dei campioni è stato negativo mostrando una scarsa qualità del materiale, i controlli sono invece risultati chiaramente positivi. In seguito sono state fatte delle prove di amplificazione cambiando alcune variabili al normale protocollo di KRAS:
Taq polimerasi AmpliTaq Gold (5U/µl) (Applied Biosystems, Foster City, CA, USA) utilizzata da 2 a 5 unità
45 cicli di amplificazione anziché 40
5 unità di Taq polimerasi e 4 µl di sospensione di DNA anziché 2
SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 20
5 unità di Taq polimerasi, 4 µl di sospensione di DNA e 45 cicli di amplificazione
È stato necessario intervenire modificando una variabile per volta in modo da poter identificare l’elemento risolutore del problema.
I risultati ottenuti dalla PCR – discordanti e di difficile lettura – nonché il sequenziamento delle sospensioni che erano state amplificate hanno portato alla conclusione che l’intera serie di campioni estratti dal blocchetto #1‐00E fosse da scartare (ipotizzando un errore nelle fasi a monte dell’estrazione) e una nuova microdissezione laser dai blocchetti 09‐I‐9711‐A6, 185‐B1 e #1‐00E, è stata effettuata il 04.04.2011 con 2 ore di incubazione con Buffer ATL e proteinasi K.
5.4 RISULTATI DA MICRODISSEZIONE LASER
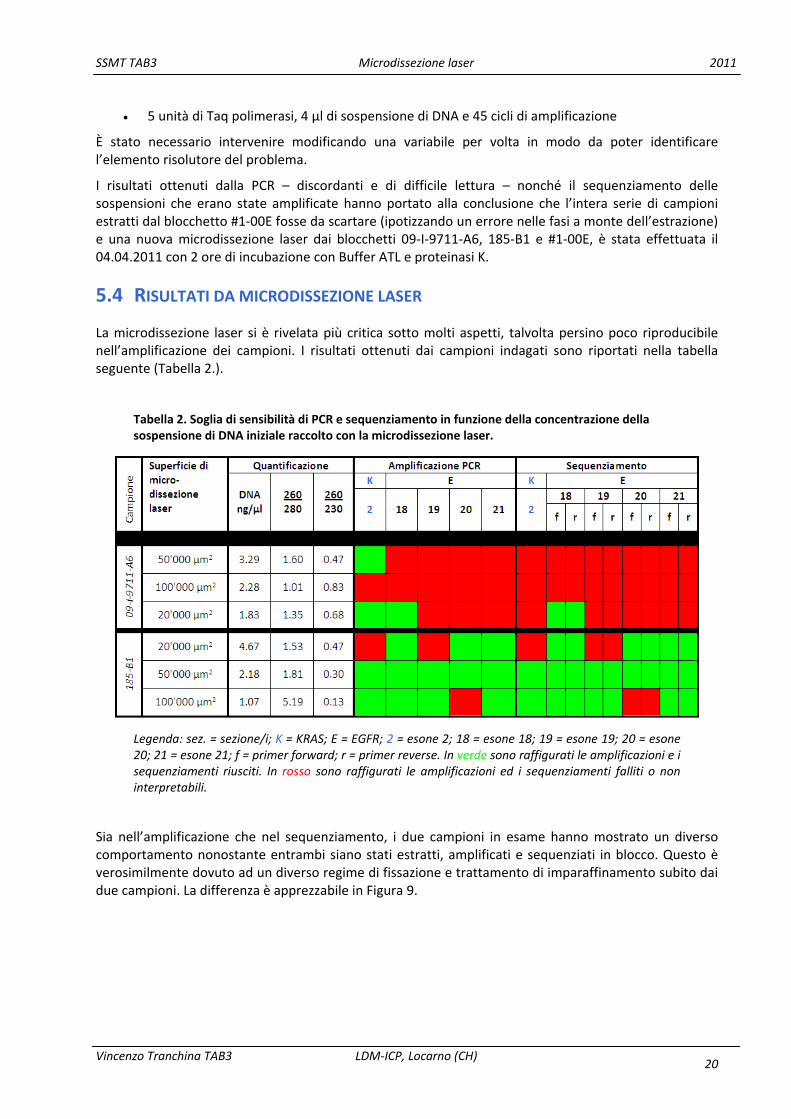
La microdissezione laser si è rivelata più critica sotto molti aspetti, talvolta persino poco riproducibile nell’amplificazione dei campioni. I risultati ottenuti dai campioni indagati sono riportati nella tabella seguente (Tabella 2.).
Tabella 2. Soglia di sensibilità di PCR e sequenziamento in funzione della concentrazione della sospensione di DNA iniziale raccolto con la microdissezione laser.
Legenda: sez. = sezione/i; K = KRAS; E = EGFR; 2 = esone 2; 18 = esone 18; 19 = esone 19; 20 = esone 20; 21 = esone 21; f = primer forward; r = primer reverse. In verde sono raffigurati le amplificazioni e i sequenziamenti riusciti. In rosso sono raffigurati le amplificazioni ed i sequenziamenti falliti o non interpretabili.
Sia nell’amplificazione che nel sequenziamento, i due campioni in esame hanno mostrato un diverso comportamento nonostante entrambi siano stati estratti, amplificati e sequenziati in blocco. Questo è verosimilmente dovuto ad un diverso regime di fissazione e trattamento di imparaffinamento subito dai due campioni. La differenza è apprezzabile in Figura 9.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 21
Figura 9. Amplificazione mediante nested PCR dell'esone 20 di EGFR : i due campioni in esame si comportano diversamente. Legenda: a20‐a100 = aree di microdissezione campione 09‐I‐9711‐A6; b20‐b100 = aree di microdissezione campione 185‐B1; neg = controllo negativo; pA2‐pA25 = controlli positivi a concentrazioni diverse (2,5 e 25 ng/µl) campione 09‐I‐9711‐A6; pB2‐pB25 = controlli positivi a concentrazioni diverse (2,5 e 25 ng/µl) campione 185‐B1; M = marker di taglia.
Il caso 09‐I‐9711‐A6 si è mostrato notevolmente più critico, amplificandosi solo nel 67% dei casi per l’esone 2 del gene KRAS e solo nel 33% dei casi per l’esone 18 di EGFR, indipendentemente dalla concentrazione della sospensione e, anzi, con preferenza per quella più diluita. Verosimilmente sostanze contaminanti sono andate ad inficiare le reazioni di PCR e quando diluite in un determinato volume di eluato hanno perso l’effetto di inibizione della PCR. Tutti gli altri esoni indagati non si sono amplificati. La criticità del campione si è tradotta anche a livello di sequenziamento, dove solo un campione su 15 (6,7%) ha mostrato un sequenziamento interpretabile, contro i 3 campioni su 15 (20%) risultati amplificati.
Il caso 185‐B1 è stato amplificato nel 66,7% dei casi (10/15). Identico è stato il risultato nel sequenziamento.
In questo caso la superficie microdissezionata non ha correlato direttamente con una maggior concentrazione della sospensione di DNA eluita su colonna. È possibile notarlo già dal fatto che non è stato possibile ordinare i campioni sia per superficie microdissezionata che per resa finale di DNA.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 22
6 DISCUSSIONE
6.1 TECNICA DI MICRODISSEZIONE LASER
Considerando che il lavoro si è reso necessario per poter effettuare estrazioni efficaci soprattutto da biopsie del colon e frustoli bioptici polmonari, il microdissettore laser ha mostrato la capacità di estrarre con precisione anche aree molto piccole (20‐30 µm2), con una precisione di circa 1‐2 µm, confermando la validità per cui si è deciso di acquistarlo.
L’estrazione di cellule è risultata molto efficace e le rese di DNA con la nuova tecnica sono state ragguardevoli in rapporto a quanto ottenuto dalla macrodissezione manuale. Il risultato che con la microdissezione laser si sia ottenuto piu’ DNA genomico rispetto alla macrodissezione manuale (a parità di area di tessuto estratta) può essere spiegato dal fatto che non sono stati coestratti tessuti connettivali ma solo zone riccamente cellulari oppure che il trasferimento del materiale con bisturi durante la macrodissezione manuale di sezioni così piccole comporta la perdita del tessuto.
Inoltre si è riusciti a ridurre il rischio di contaminazione del campione, limitare fortemente la dissezione di tessuto connettivo limitandosi alle zone cellulari e migliorare la scelta dell’area da estrarre.
Tuttavia non si è riusciti a stabilire una completa correlazione tra area microdissezionata e concentrazione di DNA eluito al termine dell’estrazione di DNA genomico. Si sospetta che questo sia dovuto ad un rischio intrinseco della procedura di microdissezione laser di perdita di materiale durante la catapulta o nelle fasi successive di manipolazione del tubo di raccolta, oltre che alle variabili introdotte dall’operatore e della non sufficiente sensibilità del sistema di isolamento del DNA utilizzato.
Per mettere in uso il microdissettore è stato necessario trascorrere svariate giornate lavorative ottimizzando i livelli di energia e messa a fuoco in maniera minuziosa. L’uso giornaliero è divenuto semplice solo dopo molti utilizzi dello strumento.
La selezione delle aree da isolare, l’utilizzo dell’ausilio informatico, la calibrazione del microscopio e le procedure di preparazione dei tubi di raccolta e dei vetrini richiedono infatti competenze trasversali ed una buona esperienza. Per questo si può affermare che l’utilizzo del microdissettore laser deve essere di competenza di personale altamente specializzato e ben istruito.
La raccolta di quantitativi molto ridotti di tessuto ha condotto alla riduzione dei tempi d’incubazione con buffer di lisi tissutale (ATL) e proteinasi K da 16 ore ad appena 2 ore. Questo ha permesso di ridurre notevolmente i tempi delle analisi mutazionali. L’impatto della riduzione dei tempi in ambito diagnostico potrebbe rivelarsi di notevole importanza.
L’uso di protocolli di PCR standard si è rivelato critico per molti campioni (in particolare a concentrazioni limitanti) denotando probabili imprecisioni nei profili termici o primers poco adatti ad un’amplificazione specifica e sensibile dei geni indagati in condizioni limite, quali quelle ottenute con l’utilizzo della microdissezione.
Per il futuro, l’ICP‐LDM si è preposto di utilizzare nuovi primers (lo sviluppo di nuovi protocolli è già in corso, ad opera di biologi del gruppo di ricerca del ICP‐LDM) ed effettuare PCR a gradiente per ottimizzare la temperatura di annealing dei primers, così da poterli utilizzare specificatamente per i campioni estratti dopo microdissezione laser. Questo porterà verosimilmente ad un aumento della sensibilità e riproducibilità dell’amplificazione.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 23
6.2 CAMPIONI DA MACRODISSEZIONE MANUALE
Dopo macrodissezione manuale si è riusciti ad amplificare e sequenziare da sospensioni di DNA a concentrazioni ben inferiori a quelle richieste dai protocolli di amplificazione utilizzati in laboratorio (25 ng/µl), dimostrando che anche campioni poco concentrati possono essere amplificati e sequenziati senza alcun problema, a conferma di quanto già sperimentato in laboratorio.
Inoltre, la correlazione fra superficie dissezionata e resa di DNA osservata può fungere d’aiuto ai tecnici in analisi biomediche che devono decidere da quante sezioni estrarre per poter ottenere un quantitativo di DNA soddisfacente.
6.3 CAMPIONI DA MICRODISSEZIONE LASER
Con le attuali procedure di laboratorio, i campioni microdissezionati sono risultati essere molto influenzati da fattori preanalitici (quali la fissazione con formalina, ad esempio), amplificandosi in maniera poco riproducibile e sicuramente denotando falle da colmare in particolare nei protocolli di amplificazione.
L’estrazione di DNA genomico da sezioni di superficie così ridotta è stato comunque un successo.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 24
7 CONCLUSIONI
Il microdissettore laser Zeiss PALM Microbeam (ed. 2007) è stato messo a punto ed è funzionante su sezioni colorettali dello spessore di 3 µm montate su vetrini Zeiss provvisti di membrana di PEN. Vi è ragione di ritenere che tale tecnica possa essere applicata con successo anche ad altri tessuti.
Con la tecnica di microdissezione laser è stato possibile isolare quantitativi sufficienti di DNA genomico, addirittura al di sopra delle aspettative. La qualità del DNA raccolto è stata variabile e principalmente legata all’impatto della fissazione del tessuto con formalina tamponata 4%. Una casistica più ampia permetterà di poter confermare questi risultati preliminari.
L’estrazione di DNA genomico è stata ottimizzata riducendo il tempo di incubazione con buffer di lisi tissutale e proteinasi K da 16 ore a 2 ore.
I protocolli di PCR non hanno subito modifiche ma saranno oggetto di ulteriori studi per l’ottimizzazione delle temperature di annealing e delle sequenze dei primers. Una prima transitoria soluzione all’amplificabilità difficoltosa dei campioni è stato l’aumento del numero di cicli di PCR, in nessun caso determinando aspecificità del prodotto.
L’ICP‐LDM ha concluso che dopo questo studio il microdissettore laser acquistato nel 2007 può essere una buona soluzione ai problemi di macrodissezione da campioni bioptici. Ulteriori studi, su casistiche più ampie, determineranno l’eventuale impiego dello strumento in diagnostica.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 25
8 RINGRAZIAMENTI
Se questo lavoro ha potuto essere portato a termine lo devo al prezioso aiuto fornitomi dai tecnici in analisi biomediche e dai biologi dell’Istituto Cantonale di Patologia – Laboratorio di Diagnostica Molecolare. Ci tengo ad esprimere tutta la mia gratitudine al Dottor Milo Frattini che ha supervisionato questo lavoro ed un grazie di cuore ad Elena Zanellato che mi ha accompagnato nel mio percorso di formazione e lungo tutto il lavoro, dimostrandosi sempre molto paziente, comprensiva, competente e dimostrandomi completa fiducia, fin dai primi giorni. Nel contempo, desidero ringraziare anche il Prof. Luca Mazzucchelli, direttore dell’ICP, che ha permesso lo svolgimento di questo lavoro di diploma.
Non posso di certo dimenticare il resto del gruppo della ricerca di cui fanno parte Francesca Molinari, Martina Nucifora, Alice Riva, Davide Romanelli, Erica Curcio e Francesca Mancuso, nonché le tecniche in analisi biomediche della diagnosi Morena Ghisletta, Antonella Camponovo, Sara Banfi, Paola Galli e la Dott.ssa Vittoria Martin. Senza di loro, senza i loro consigli e senza la loro solarità questo lavoro non avrebbe avuto le stesse positive conclusioni.
Grazie a Stefan Wahl e alla Dott.ssa Kerstin Hagen‐Mann, tecnici specializzati del reparto di microdissezione laser della Zeiss, che in brevissimo tempo hanno saputo formarmi sull’utilizzo del microdissettore laser.
Un grazie anche al team scolastico di supporto metodologico capitanato dal direttore Dott. Andrea Boffini, affiancato dal Prof. Claudio Naiaretti e dalla tecnica in analisi biomediche Sonja Marci, anche mia accompagnatrice pedagogica sul posto di lavoro. Un grazie pure alle compagne di classe che hanno ascoltato pazientemente per decine di volte gli aggiornamenti del lavoro, esprimendo considerazioni per indirizzarmi al raggiungimento degli obiettivi.
Infine un enorme ringraziamento alla mia compagna Céline Agassis, per il supporto prestatomi nei momenti tesi dello sviluppo del lavoro e per lo spirito critico, volto a incrementare la qualità dello scritto, dimostrato nelle fasi di stesura e correzione di questo studio.
Ancora un grazie di cuore a tutti.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 26
9 BIBLIOGRAFIA
Fonti bibliografiche e informatiche
[1] Camponovo A, Banfi S, Frattini M. 4.1.IO K‐Ras ‐ mutazione ‐ DM, Locarno (CH): ICP LDM; 2008. Procedure operative di laboratorio
[2] EGFR epidermal growth factor receptor. Ultima consultazione 09.03.2011 su: http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=full_report&list_uids=1956
[3] P.A.L.M. Microlaser ‐ Physics behind Laser Microdissection and Pressure Catapulting. Ultima consultazione 08.03.2011 su: http://palm.dasat.ch/dasat/index.php?cid=100148&conid=100160&sid=dasat
[4] Murray GI, Curran S. Laser capture microdissection, Methods in molecular biology, Methods and Protocols, Totowa (New Jersey): Humana Press; 2005; 293: 4,6,12,17‐8
[5] Murray GI, Curran S. Laser capture microdissection, Methods in molecular biology, Methods and Protocols, Totowa (New Jersey): Humana Press; 2005; 293: 152
[6] The PALM Family ‐ A New Dimension in Sample Purity brochure. Ultima consultazione 21.01.2011 su: http://www.zeiss.com/C1256D18002CC306/0/608D7B197ABD1659C1257441004DF73E/$file/60‐3‐0001_palm‐famliy_e.pdf
[7] doc. 2 Lasermikrodissektion ‐ Entwicklung einer Methode zur Gewinnung von homogenem Analyse material. Ultima consultazione 20.01.2011 su: www.photonicnet.de/Aktuelles/partner/2006/05/pn‐leica‐microsystems‐a1.zip/download
[8] Microdissection Technology and Challenger – Slides 5,6,9,14,16‐9,20. Ultima consultazione 20.01.2011 su: http://sigs.nih.gov/biospecimens/Documents/080723_LCM_microdissection.pdf
[9] Ghisletta M, Banfi S, Frattini M. 4.1.IO Estrazione DNA da sezioni in bianco ‐ DM, Locarno (CH): ICP LDM; 2010. Procedure operative di laboratorio
Fonti delle figure
Figura di copertina. http://www.zeiss.com/C1256D18002CC306/0/608D7B197ABD1659C1257441004DF73E/$file/60‐3
0001_palm‐famliy_e.pdf
Figura 1. http://www.zeiss.com/C1256D18002CC306/0/608D7B197ABD1659C1257441004DF73E/$file/60‐3‐0001_palm
famliy_e.pdf
Figura 2. http://palm.dasat.ch/dasat/index.php?cid=100148&conid=100160&sid=dasat
Figura 3. http://www.zeiss.com/C1256D18002CC306/0/608D7B197ABD1659C1257441004DF73E/$file/60‐3‐0001_palm
famliy_e.pdf
Figura 4. http://jmg.bmj.com/content/47/12/859/F2.large.jpg
Figura 5. http://pslc.ws/macrogcss/images/pet04.gif
Figura 6. Print Screen Software PALM@RoboV4
Figura 7. Grafico di correlazione ‐ Software Gnumeric (Proj. Gnome)
Figura 8. Print Screen Software Sequencing Analysis 5.2
Figura 9. Print Screen Software Fusion Fx5

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 27
10 ALLEGATI
ALLEGATO 1
Amplificazione PCR
vaschetta con ghiaccio o blocco refrigerante
kit AmpliTaq gold with GeneAmp (Applied Biosystems, Foster City, CA, USA), contenente:
o PCR Buffer II (10x)
o Soluzione di cloruro‐magnesio (25 mM)
o Enzima Taq polimerasi AmpliTaq Gold (5U/l)
miscela dNTPs (100 mM) (Amersham, Buckinghamshire, United Kingdom)
soluzione dUTP (100 mM) (Amersham, Buckinghamshire, United Kingdom)
Primers (100 µm) (Invitrogen, Carlsbad, CA, USA)
o 17F e 18B (PCR KRAS) diluiti a 10 µM
o 18EF, 18ER, 19EF, 19ER, 20EF, 20ER, 21EF, 21ER (PCR EGFR ext.) diluiti a 20 µM
o 18IF, 18IR, 19IF, 19IR, 20IF, 20IR, 21IF, 21IR (PCR EGFR int.) diluiti a 20 µM
Acqua distillata Water purification System Milli‐Q Biocel (Billerica, USA), in aliquote
Tubi Sarstedt Eppendorf sterili da 1,5 ml e da 0,2 ml (Sarstedt AG, Nümbrecht, Germany)
Preparare tubi Eppendorf da 0,2 ml per ogni campione ed esone. Aliquotarvi il DNA.
Preparare la Mix in una provetta Eppendorf da 1,5 ml come indicato nella pagina seguente e nell’ordine, mantenendo i reagenti in ghiaccio, ma avendo cura di utilizzarli scongelati, vortexati e centrifugati. Prendere la polimerasi Taq gold 5 U/L dal congelatore solo al momento dell’uso e riporla al più presto. Dopo l’aggiunta dell’enzima, terminare ed aliquotare la Mix nel più breve tempo possibile.
Tenendo conto dell’inaccuratezza delle pipette di grosso calibro (nonostante le periodiche calibrazioni), con le quali si aliquota, si consiglia di preparare un volume di mix in più rispetto al numero di campioni (comprensivi dei bianchi di reazione e dei controlli positivi) previsti.
SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 28
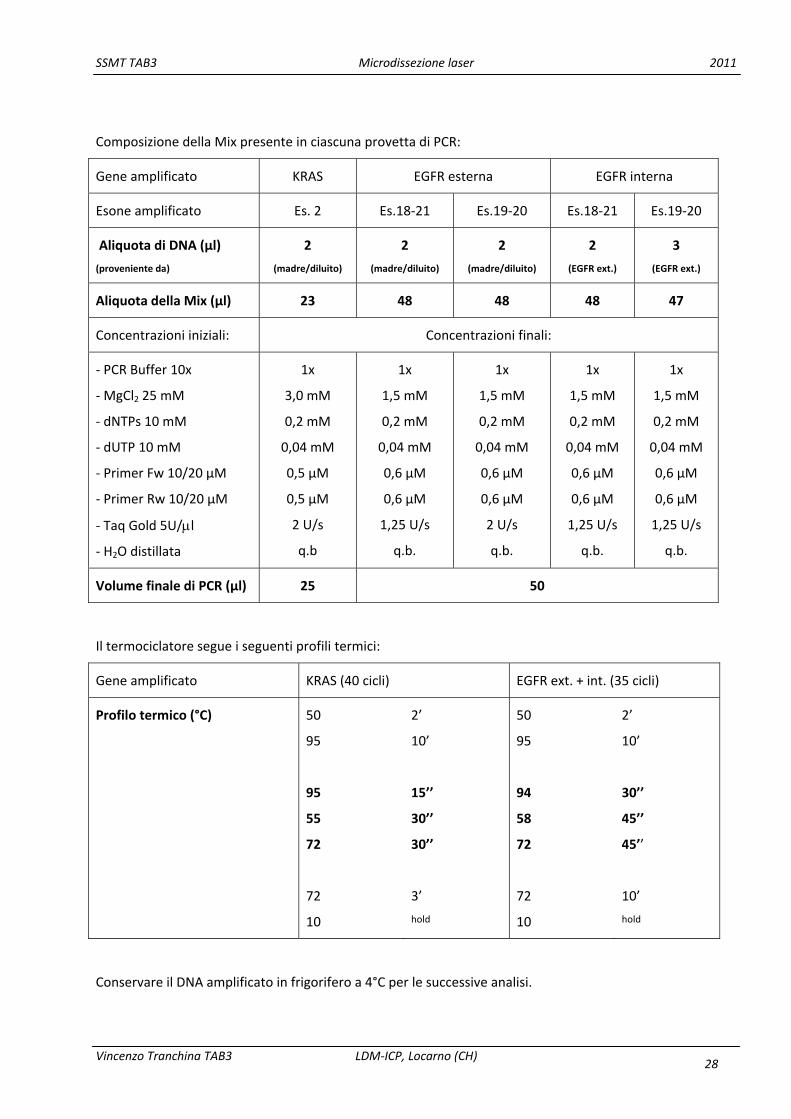
Composizione della Mix presente in ciascuna provetta di PCR:
Gene amplificato KRAS EGFR esterna EGFR interna
Esone amplificato Es. 2 Es.18‐21 Es.19‐20 Es.18‐21 Es.19‐20
Aliquota di DNA (µl)
(proveniente da)
2
(madre/diluito)
2
(madre/diluito)
2
(madre/diluito)
2
(EGFR ext.)
3
(EGFR ext.)
Aliquota della Mix (µl) 23 48 48 48 47
Concentrazioni iniziali: Concentrazioni finali:
‐ PCR Buffer 10x
‐ MgCl2 25 mM
‐ dNTPs 10 mM
‐ dUTP 10 mM
‐ Primer Fw 10/20 µM
‐ Primer Rw 10/20 µM
‐ Taq Gold 5U/l
‐ H2O distillata
1x
3,0 mM
0,2 mM
0,04 mM
0,5 µM
0,5 µM
2 U/s
q.b
1x
1,5 mM
0,2 mM
0,04 mM
0,6 µM
0,6 µM
1,25 U/s
q.b.
1x
1,5 mM
0,2 mM
0,04 mM
0,6 µM
0,6 µM
2 U/s
q.b.
1x
1,5 mM
0,2 mM
0,04 mM
0,6 µM
0,6 µM
1,25 U/s
q.b.
1x
1,5 mM
0,2 mM
0,04 mM
0,6 µM
0,6 µM
1,25 U/s
q.b.
Volume finale di PCR (µl) 25 50
Il termociclatore segue i seguenti profili termici:
Gene amplificato KRAS (40 cicli) EGFR ext. + int. (35 cicli)
Profilo termico (°C) 50
95
95
55
72
72
10
2’
10’
15’’
30’’
30’’
3’
hold
50
95
94
58
72
72
10
2’
10’
30’’
45’’
45’’
10’
hold
Conservare il DNA amplificato in frigorifero a 4°C per le successive analisi.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 29
ALLEGATO 2
Preparazione del gel di agarosio 1,8%
vaschetta e pettini in plastica per preparazione gel e pozzetti
nastro adesivo isolante
vetreria
Peqlab peqGOLD Universal Agarose (Biotechnologie GmbH, Erlangen, Germany)
bilancia Mettler Toledo PC2200 Delta Range (Mettler‐Toledo, Greifensee, Switzerland)
tampone Sigma Tris‐Borate‐EDTA buffer 10x Conc. (Sigma‐Aldrich, Steinheim, Germany)
acqua distillata Water purification System Milli‐Q Biocel (Billerica, USA)
Microonde Moulinex Symbio ADM 8.2 (Moulinex, Glattbrugg, Switzerland)
Pesare 1,80 g di agarosio in polvere in una beuta tarata da 250 ml sulla bilancia di precisione.
Versare nella beuta 100 ml di TBE 1x, precedentemente diluito dalla soluzione madre di TBE 10x con acqua deionizzata.
Agitare cautamente alcuni secondi per sospedere l’agarosio nel tampone, in seguito mettere la beuta nel microonde.
Scaldare il gel per 10 minuti a 200W (e fino a quando non è perfettamente limpido), avendo l’accortezza di controllare che non si formino bolle (altrimenti mescolare cautamente e rimettere la beuta nel microonde).
Lasciare raffreddare la soluzione 2 minuti, agitare per omogeneizzare la soluzione e versarla quando è ancora ben liquida in una vaschetta in plastica precedentemente chiusa sui lati con nastro adesivo isolante e provvista di pettine della taglia desiderata.
Far solidificare il gel a temperatura ambiente per almeno un’ora prima di utilizzarlo.
Rimuovere infine il pettine, riporre il gel nella camera elettroforetica e sommergerlo con TBE 1x di circa 5 mm.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 30
ALLEGATO 3
Migrazione e colorazione del gel
Parafilm
Peqlab Marker PeqGOLD 50 bp DNA‐Leiter (Biotecnologie GmbH, Erlangen, Germany)
Soluzione di Blu di Bromofenolo 0,25% in 40% saccarosio (Merck, Withehouse Station, NJ, USA) in acqua distillata
tampone Sigma Tris‐Borate‐EDTA buffer 10x Conc. (Sigma‐Aldrich, Steinheim, Germany)
acqua distillata Water purification System Milli‐Q Biocel (Billerica, USA)
Biotium’s GelRed Nucleic Acid Gel Stain 1 μl/1 ml (Biotium, Hayward, CA, USA)
Camera elettroforetica e generatore di corrente (Bio‐Rad, Philadelphia, PA, USA)
Preparare la camera elettroforetica riempiendola con tampone TBE 1x e depositarvi il gel.
Il gel deve essere sommerso di 5 mm dal TBE 1x. Questo garantisce una migrazione ottimale per i tempi e la tensione elettrica prevista (empirico).
Su una striscia di parafilm aliquotare i reagenti come segue:
Pazienti e controlli (μl) Marker 50 bp (μl)
Blu di bromofenolo 2 2
Sospensione di DNA amplificato 5 1
TBE 1x 0 9
Pipettare 7 μl della goccia di soluzione appena preparata e aliquotarla nei pozzetti del gel, avendo cura di non formare bolle e pipettando l’intero volume previsto.
Chiudere la camera elettroforetica con il coperchio e collegare i connettori secondo la giusta polarità. Far migrare per 35 minuti a una tensione elettrica di 100V.
Colorare infine il gel per 20 min al buio in soluzione di Gel Red a concentrazione di 0,1 μl/ml in TBE 0,5x e risciacquarlo brevemente in TBE 0,5x prima di rilevare le bande.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 31
ALLEGATO 4
Purificazione amplificato PCR
kit HiYield Gel/PCR DNA Fragments Extraction YDF 300 (RBC Bioscience, Taipei, Taiwan), contenente:
o DF Columns con membrana
o DF Buffer
o Wash Buffer
o Eluition Buffer
Eppendorf Centrifuge 5415 R (Vaudaux‐Eppendorf, Basel, Switzerland)
tubi Sarstedt Eppendorf 2,0 e 1,5 ml (Sarstedt AG, Nümbrecht, Germany)
etanolo 99,8% (Fluka Analytical, Buchs, Switzerland)
Al prodotto di PCR da purificare, aggiungere un volume 5 volte superiore di Buffer DF e miscelare (ev. se il prodotto di PCR eccede i 25 μl, eseguire questo passaggio in una provetta Eppendorf da 1,5 ml)
Trasferire l’intero volume (DNA+DF Buffer) su una DF Column alloggiata in una provettaeppendorf da 2 ml
Centrifugare a 13'000 rpm per 30 sec e scartare il flow‐through
Aggiungere 600 μl di Wash Buffer sulla colonna
Centrifugare a 13'000 rpm per 30 sec e scartare il flow‐through
Ricentrifugare a 13'000 rpm per 2 min
Alloggiare la colonna su una provetta eppendorf da 1,5 ml e applicare 30 μl di Eluition Buffer
Incubare 2 min a temperatura ambiente e centrifugare a 13'000 rpm per 2 min
Fasi della purificazione con kit HiYield Fragments Extraction YDF 300 da amplificato PCR (o da gel d'agarosio).
SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 32
ALLEGATO 5
Cycle Sequencing
tubi Sarstedt Eppendorf 1,5 e 0,2 ml (Sarstedt AG, Nümbrecht, Germany)
ABI Prism Big Dye Terminator v1.1 Ready Reaction Mix = TRR Mix (Applied Biosystems, Foster City, CA, USA)
Primers (100 µM) (Invitrogen, Carlsbad, CA, USA), come per PCR KRAS ed EGFR
ABI Prism Big Dye Terminator v1.1‐3.1 Sequencing Buffer 5x = Sequencing Buffer 5x (Applied Biosystems, Foster City, CA, USA)
Acqua distillata Water purification System Milli‐Q Biocel (Billerica, USA), in aliquote
Preparare tubi Eppendorf da 0,2 ml per ogni campione ed esone. Aliquotarvi il DNA.
Preparare la Mix per Cycle Sequencing in una provetta Eppendorf da 1,5 ml come segue e nell’ordine e avendo cura di utilizzarli scongelati. Il TRR deve essere protetto dalla luce in quanto contiene i fluorocromi associati ai dideossinucleotidi.
Tenendo conto dell’inaccuratezza delle pipette (nonostante la calibrazione effettuata periodicamente), si consiglia di preparare un volume di mix in più rispetto al numero di campioni ed esoni previsti.
Composizione della Mix presente in ciascuna provetta di PCR:
Gene amplificato KRAS EGFR
Aliquota di DNA purificato (µl) 0,4 (amplificato PCR purificato con kit HiYield)
Aliquota della Mix (µl) 9,6
Concentrazioni iniziali: Concentrazioni finali:
‐ TRR MIX
‐ Sequencing Buffer 5x
‐ Primer F o R 10/20 µM
‐ H2O distillata (µl)
0,25x
0,75x
0,5 µM
q.b.
0,25x
0,75x
1,0 µM
q.b.
Volume finale di Cycle‐Sequencing (µl) 10
Caricare i campioni nel termociclatore e dare il via ai cicli col programma di CycleSequencing:
Profilo termico (°C) 96
96 50 60 4
1’ 10’’ 5’’ 25 cicli 4’ hold

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 33
ALLEGATO 6
Purificazione amplificato CycleSequencing
kit G50 Dye Terminator Removal (RBC Bioscience, Taipei, Taiwan)
Tubi Sarstedt Eppendorf sterili da 1,5 ml e da 2,0 ml (Sarstedt AG, Nümbrecht, Germany)
Eppendorf Centrifuge 5415 R (Vaudaux‐Eppendorf, Basel, Switzerland)
Centrifuga a vuoto termostatata Speedvac (Vaudaux‐Eppendorf, Basel, Switzerland)
Identificare le colonne per ogni esone e paziente da amplificare
Reidratare la resina dispensando 300 µl di acqua sterile in ogni colonna
Picchiettare con l’unghia la colonna fino a risospendere la resina
Rimuovere il tappo rosso nella parte inferiore della colonna e porla su un tubo da 2,0 ml
Centrifugare a 3’000 rpm per 2 min
Scartare il flow‐thorugh e mettere le colonne su tubi sterili da 1,5 ml
Dispensare l’intero volume dell’amplificato Cycle‐Sequencing sulla colonna
Centrifugare a 3’000 rpm per 3 min
Eliminare le colonne
Porre i tubi sterili da 1,5 ml nello Speedvac a 30°C per 30 min col tappo aperto e fino a completa essiccazione del materiale
Conservare i campioni (pellet invisibile) in congelatore, col tappo chiuso e al buio fino al momento del sequenziamento.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 34
ALLEGATO 7
Sequenziamento
sequenziatore Applied Biosystems ABI 3130 (Applied Biosystems, Foster City, CA, USA)
piastra sequencing Applied Biosystems MicroAmp Optical 96‐Well Reaction Plate (Applied Biosystems, Foster City, CA, USA)
formammide HiDi (Applied Biosystems, Foster City, CA, USA)
Il sequenziamento si articola in 4 fasi:
1) Risospensione dei campioni
Pipettando, risospendere i pellet (essiccati nella purificazione) con 15 µl di formammide
Porre i campioni risospesi nel termoblocco preimpostato a 95°C per 5 min
Porre i campioni in congelatore, al buio e col tappo chiuso fino al momento del caricamento
2) Preparazione della piastra di sequenziamento
Preparare al PC la griglia di sequenziamento
Dispensare l’intero contenuto del tubo (15 µl) nei pozzetti della piastra sequencing
Coprire la piastra sequencing con la piastra di gomma
3) Sequenziamento automatico con Applied Biosystems ABI 3130
Eseguire la calibrazione del sequenziatore sotto il percorso: “GA3130/Spatial Run Scheduler”
Caricare la piastra di sequenziamento sulla guida in plastica, infine inserire la piastra nell’apparecchio
Selezionare il percorso: “GA3130/Run Scheduler”
Dare avvio al sequenziamento
4) Lettura del sequenziamento
Utilizzare il software Sequencing Navigator (Applied Biosystems)
N.B. Per i dettagli sul funzionamento del sequenziatore, consultare le istruzioni o le direttive riassuntive a fianco dell’apparecchio.

SSMT TAB3 Microdissezione laser 2011
Vincenzo Tranchina TAB3 LDM‐ICP, Locarno (CH) 35
ALLEGATO 8


















