
Termodinamica e teoria cinetica dei gas -...
53
1 Termodinamica e teoria cinetica dei gas “L’insieme di relazioni tra le proprieta’ macroscopiche che servono a specificare lo stato interno di un sistema quando interviene il calore”. proprieta’ macroscopiche: quelle direttamente percepibili dai nostri sensi (a contrasto di quelle microscopiche ). anche dette: variabili termodinamiche es.: P,V,T sistema/ ambiente / universo Si distingue tra il “sistema” (termodinamico) e “l’ambiente” che lo circonda il sistema ambiente L’insieme di “sistema” + “ambiente” viene detto “l’universo” La meccanica statistica: Le variabili termodinamiche dipendono dai valori medi delle proprieta’ microscopiche del sistema. La meccanica statistica stabilisce le relazioni tra le proprieta’ microscopiche e le variabili termodinamiche. La teoria cinetica dei gas e’ una parte (semplice) della meccanica statistica.
-
Upload
nguyenngoc -
Category
Documents
-
view
226 -
download
0
Transcript of Termodinamica e teoria cinetica dei gas -...

1
Termodinamica e teoria cinetica dei gas
“L’insieme di relazioni tra le proprieta’ macroscopiche che servono a specificare lo stato interno di un sistema quando interviene il calore”.
proprieta’ macroscopiche: quelle direttamente percepibili dai nostri sensi (a contrasto di quelle microscopiche ).
anche dette: variabili termodinamiche es.: P,V,T
sistema/ ambiente / universo Si distingue tra il “sistema” (termodinamico) e “l’ambiente” che lo circonda
il sistema
ambiente L’insieme di “sistema” + “ambiente” viene detto “l’universo”
La meccanica statistica: Le variabili termodinamiche dipendono dai valori medi delle proprieta’ microscopiche del sistema. La meccanica statistica stabilisce le relazioni tra le proprieta’ microscopiche e le variabili termodinamiche. La teoria cinetica dei gas e’ una parte (semplice) della meccanica statistica.

2
Nozioni e terminologie Un sistema aperto: ammette scambi di energia e di materia Un sistema chiuso: esclude scambi di materia ma ammette scambi di energia Un sistema isolato: esclude entrambi
Un sistema si trova in: equilibrio termodinamico: se lo stato del sistema non cambia spontaneamente Le variabili termodinamiche *descrivono lo stato (di equilibrio) del sistema. di stato di un sistema *Non sono definite se il sistema non e’ in equilibrio (es. PVT per gas ideale) *sono il minimo numero di variabili termodinamiche
necessarie per descrivere lo stato del sistema. *dipendono unicamente dallo stato del sistema ( e non dal
cammino seguito per raggiungerlo) Si distingue tra: varabili estensive(es. M,V) proprieta’ addittive del intero sistema variabili intensive(es. P,T) possono variare da punto in punto e non sono addittive Un equazione di stato di un sistema mette il relazione le varabili di stato del sistema, riducendo Cosi il numero di variabili indipendenti
3
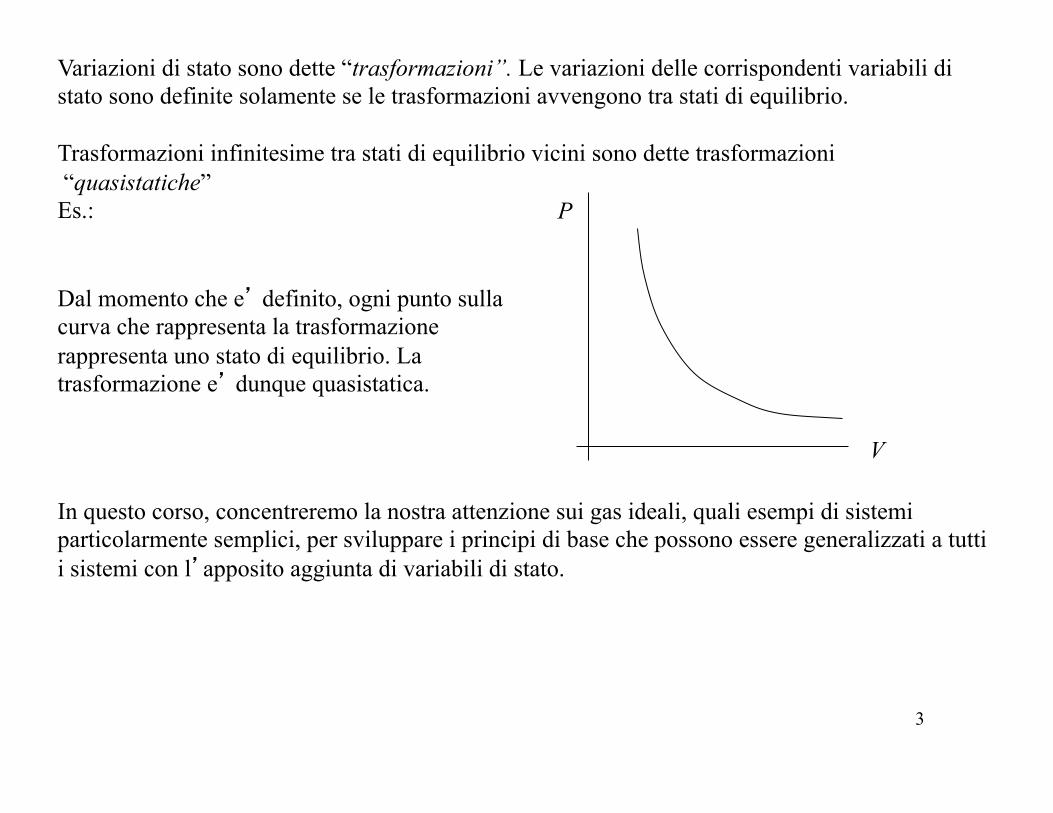
Variazioni di stato sono dette “trasformazioni”. Le variazioni delle corrispondenti variabili di stato sono definite solamente se le trasformazioni avvengono tra stati di equilibrio. Trasformazioni infinitesime tra stati di equilibrio vicini sono dette trasformazioni “quasistatiche” Es.:
In questo corso, concentreremo la nostra attenzione sui gas ideali, quali esempi di sistemi particolarmente semplici, per sviluppare i principi di base che possono essere generalizzati a tutti i sistemi con l’apposito aggiunta di variabili di stato.
P
V
Dal momento che e’ definito, ogni punto sulla curva che rappresenta la trasformazione rappresenta uno stato di equilibrio. La trasformazione e’ dunque quasistatica.
4
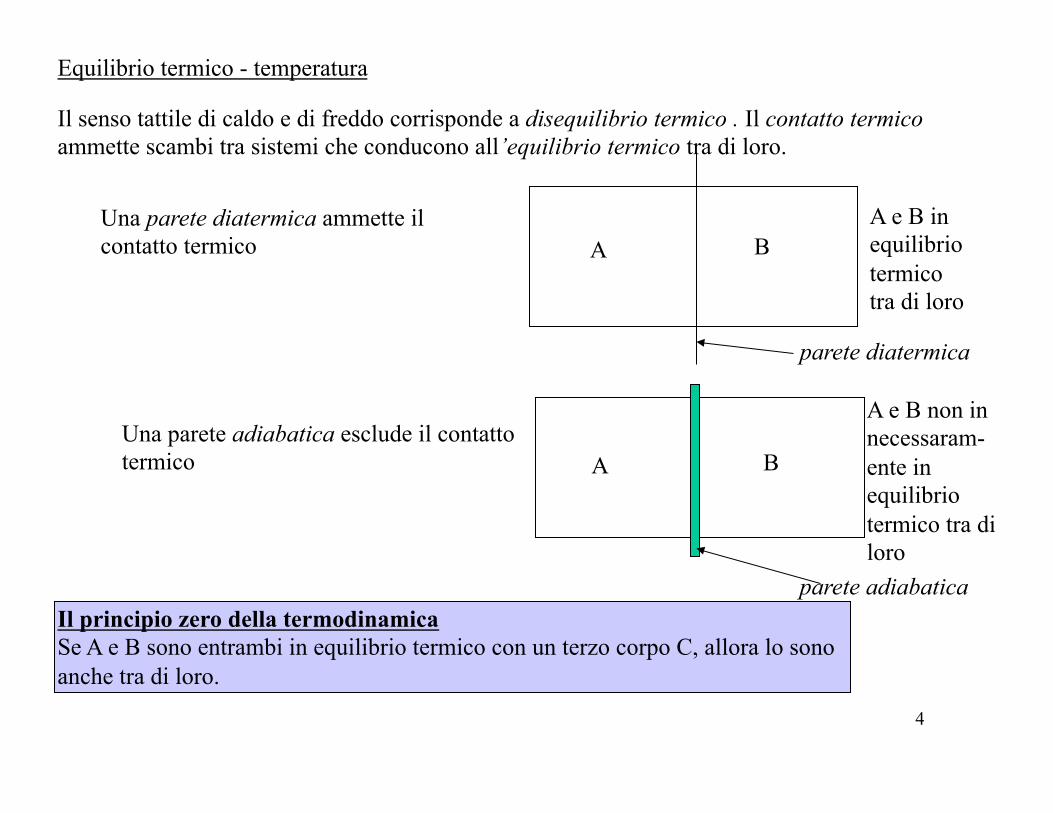
Equilibrio termico - temperatura
Il senso tattile di caldo e di freddo corrisponde a disequilibrio termico . Il contatto termico ammette scambi tra sistemi che conducono all’equilibrio termico tra di loro.
parete diatermica
A B Una parete diatermica ammette il contatto termico
A e B in equilibrio termico tra di loro
parete adiabatica
A B
A e B non in necessaram- ente in equilibrio termico tra di loro
Una parete adiabatica esclude il contatto termico
Il principio zero della termodinamica Se A e B sono entrambi in equilibrio termico con un terzo corpo C, allora lo sono anche tra di loro.

5
Lo stato di equilibrio termico puo’ dunque essere una proprieta’, o funzione di stato, commune a tutti i corpi. La funzione di stato che specifica lo stato di equilibrio termico del sistema e’ detta la temperatura. Il corpo C potrebbe essere il nostro termomentro. Per misurare la temperatura, si fa riferimento alla relazione tra la temperatura (o stato di equilibrio termico) ed un altra proprieta’ macroscopica (variabile o funzione di stato) del termometro – una facilmente misurabile, es; la lunghezza di una colonna di mercurio, o la pressione di un gas a volume costante.
6
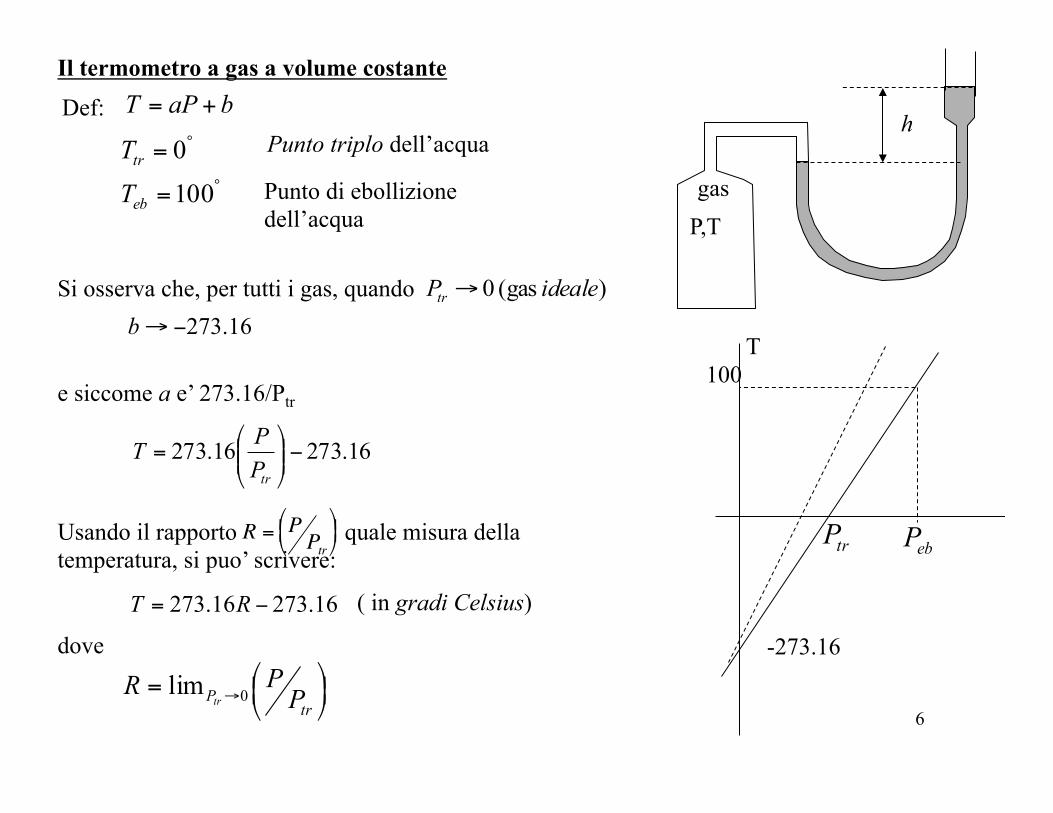
Il termometro a gas a volume costante
h Def: baPT +=
°
°
=
=
100
0
eb
tr
TT Punto triplo dell’acqua
Punto di ebollizione dell’acqua
gas
Si osserva che, per tutti i gas, quando ) (gas 0 idealePtr →16.273−→b
P,T
100
ebPtrP
-273.16
e siccome a e’ 273.16/Ptr
16.27316.273 −⎟⎟⎠
⎞⎜⎜⎝
⎛=
trPPT
Usando il rapporto quale misura della temperatura, si puo’ scrivere:
16.27316.273 −= RT
dove
⎟⎠⎞
⎜⎝⎛= →
trP P
PRtr 0lim
( in gradi Celsius) !
R = PPtr
" # $ %
& '
T

7
Questa e’ la scala di temperatura Celsius. Aggiungendo 273.16 si ottiene la scala assoluta, o la scala Kelvin.
K)(in 16.273 °= RT
La temperatura -273.16 oC = 0 oK e’ detta lo “zero assoluto” N.B.
( ) 16.273)( −°=° KTCT
La scala di temperatura Farenheit
)(59)(32)(
21232
CTFFT
FTFT
e
tr
°+°=°
°=
°=
Altri termomentri sfruttano altre correlazioni tra lo stato di equilibrio termico del Termometro e un altra proprieta’ macroscopica che dipende solo da lo stato di equilibrio termico., es. la lunghezza l di una colonna di fluido (es. Mercurio, alcool etilico, toluolo etc.)
balT +=
La “International practical Temperature Scale” (IPTS) (ultimo aggiornameto nel 1968). E’ costituita da un numero di punti fissi e di regole ( cioe’ termometri ) per interpolare tra di loro. Gamma di applicazione: 13.81 < T <13357.6
Punto triplo di 2H Punto di fusione di Au

8
Avendo definito un altra proprieta’ macroscopica (temperatura), siamo in grado di esprimere alcune relazioni termodinamiche (tra variabili di stato)
Equazione di stato dei gas perfetti (o ideali) Per un gas perfetto ( un gas a bassa pressione s’avvicina a questo stato), si osserva che:
tetancos=TPV (per una determinata massa di gas)
Dove: costante = nR n e’ il numero di moli del gas. Una mole di gas contiene
(il Numero di Avogadro ) di molecole. 230 10022045.6 ×=N
K. J/mol 314.8 °=R ( la “costante universale dei gas perfetti”)
= 1.99 cal/mol oK
Def: un gas perfetto e’ un gas che rispetta:
nRTPV = Equazione di stato dei gas perfetti
9
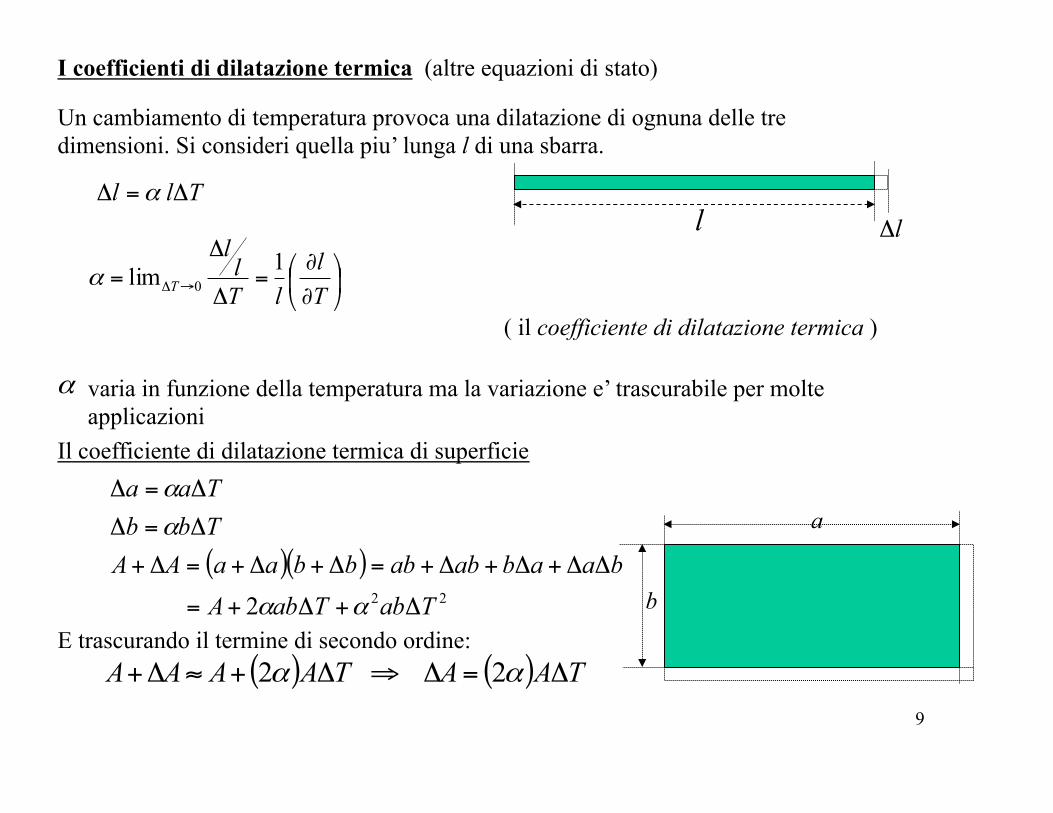
I coefficienti di dilatazione termica (altre equazioni di stato)
Un cambiamento di temperatura provoca una dilatazione di ognuna delle tre dimensioni. Si consideri quella piu’ lunga l di una sbarra.
lΔlTll Δ=Δ α
⎟⎠
⎞⎜⎝
⎛∂
∂=
Δ
Δ= →Δ T
llT
ll
T1lim 0α
α varia in funzione della temperatura ma la variazione e’ trascurabile per molte applicazioni
Il coefficiente di dilatazione termica di superficie
( il coefficiente di dilatazione termica )
a
b ( )( )
222 TabTabAbaabababbbaaAA
TbbTaa
Δ+Δ+=
ΔΔ+Δ+Δ+=Δ+Δ+=Δ+
Δ=Δ
Δ=Δ
αα
α
α
E trascurando il termine di secondo ordine: ( ) ( ) TAATAAAA Δ=Δ⇒Δ+≈Δ+ αα 2 2

10
Il coefficiente di dilatazione termica di volume
a
b
c
( )
TVVTabcabc
ccbbaaVV
Δ+≈
+Δ+=
Δ+Δ+Δ+=Δ+
α
α
3 )0(3
))((
Cioe’
TVV Δ=Δ α3
Il coefficiente di dilatazione termica di volume : β
αβ 311lim 0 =∂
∂=
Δ
Δ= →Δ T
VVT
VVT
Il coefficiente di dilatazione termica di superficie e’ dunque dato da:
α21lim 0 =∂
∂→Δ T
AAT

11
Si puo’ definire il calore cosi’: “Il calore e’ cio’ che viene trasferito da un sistema all’ambiente ( o da un sistema all’altro ) solo a causa della differenza di temperatura (cioe’ di equilibrio termico) tra il sistema e l’ambiente ( o l’altro sistema ).” oppure: “Il solo scambio di calore tra un sistema e l’ambiente porta ad un cambiamento di temperatura.” Si definiscono quindi unita’ di calore nel modo seguente: 1 caloria (cal) e’ la quantita’ di calore necessaria per in alzare la temperatura di un grammo di acqua da 14.5 aq 15.5 oC 1 British thermal unit (Btu) e’ la quantita’ di calore necessaria per aumentare la Temperatura di una libbra d’acqua da 63 oF a 64 oF. 1 caloria alimentare = 1000 cal = 1 kcal = 3.968 Btu
Calore – Capacita’ Termica – trasferimento di calore

12
La Capacita Termica C di un corpo e’ definita:
dTdQ
TQC T =⎟⎠
⎞⎜⎝
⎛Δ
Δ= →Δ 0lim
dove =ΔQ Quantita’ di calore assorbita dal corpo
=ΔT Corrispondente aumento di temperatura
Il calore specifico c di una materia viene definito:
dTdQ
mmCc 1/ ==
Si definisce anche una capacita’ termica molare C’ ( anche detto calore specifico molare ):
mole una di massa la e' moli di numero il e'
//'
Mndove
McnmcnCC ===
13
VV
PP
dTdQC
dTdQC
⎟⎠
⎞⎜⎝
⎛=
⎟⎠
⎞⎜⎝
⎛=
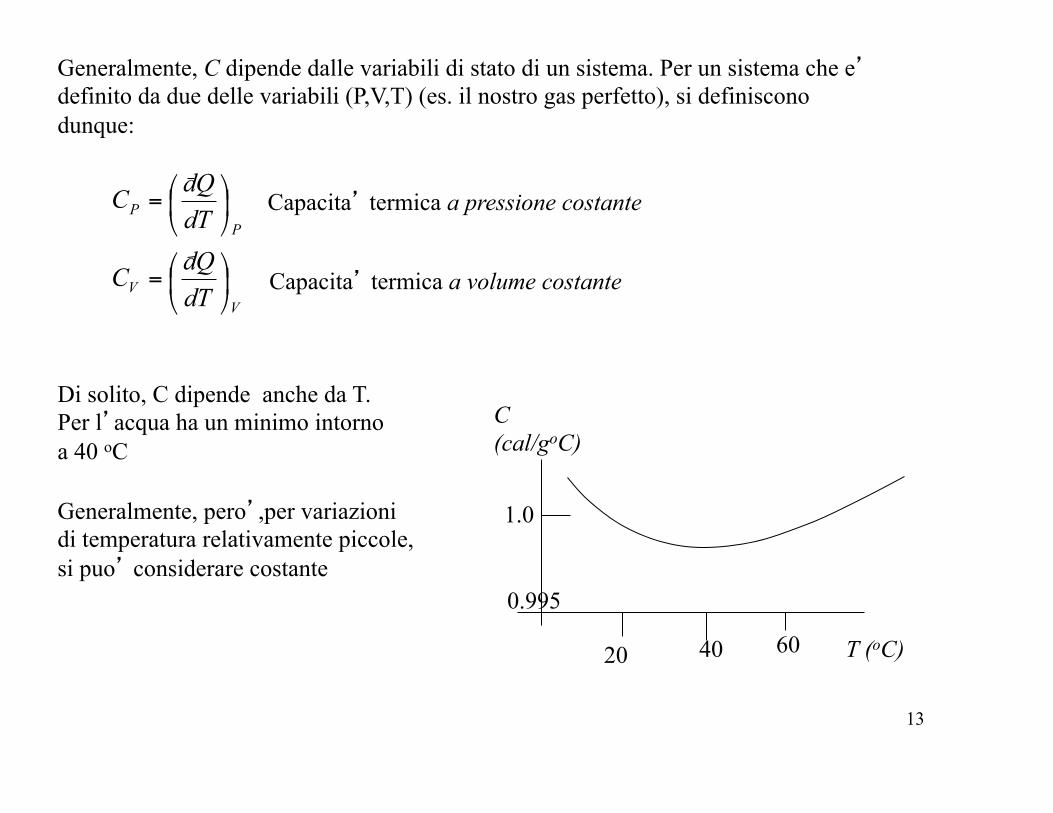
Generalmente, C dipende dalle variabili di stato di un sistema. Per un sistema che e’ definito da due delle variabili (P,V,T) (es. il nostro gas perfetto), si definiscono dunque:
Capacita’ termica a pressione costante
Capacita’ termica a volume costante
Di solito, C dipende anche da T. Per l’acqua ha un minimo intorno a 40 oC
C (cal/goC)
T (oC)
1.0
0.995
20 40 60
Generalmente, pero’,per variazioni di temperatura relativamente piccole, si puo’ considerare costante
14
( ) ( ) ( )aFRaFaaFxxx TTCTTcmTTcm −+−=−
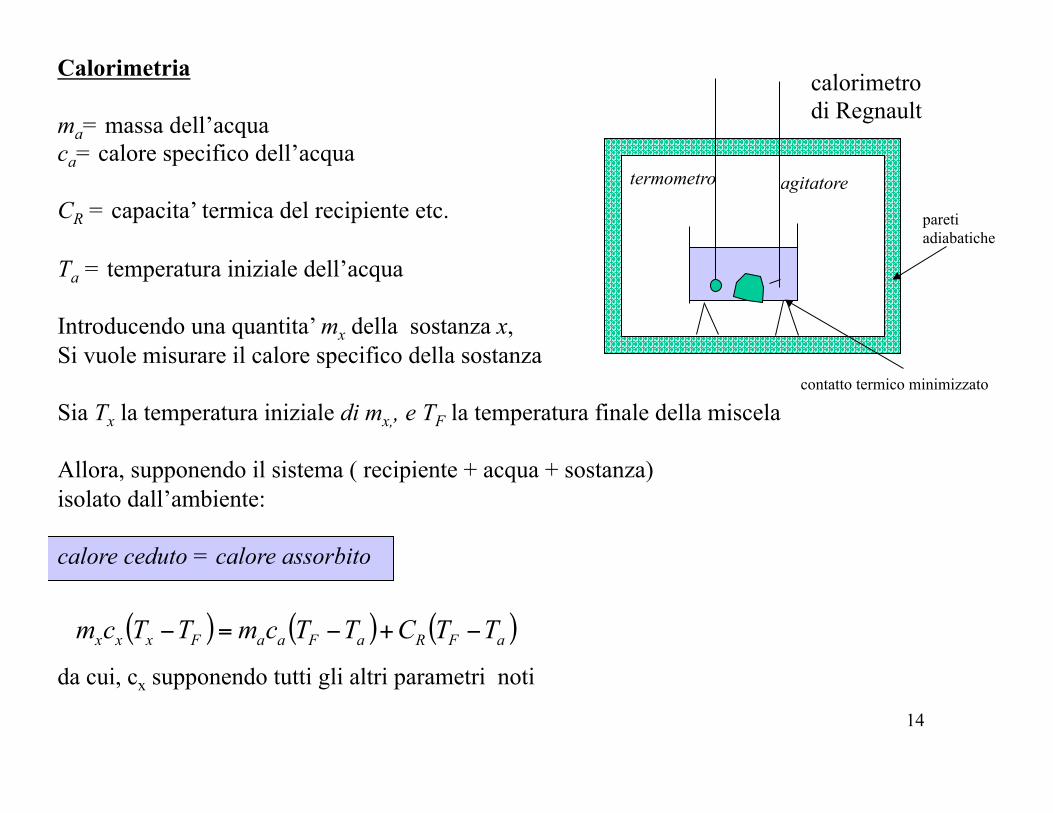
calorimetro di Regnault
termometro agitatore
pareti adiabatiche
contatto termico minimizzato
Calorimetria ma= massa dell’acqua ca= calore specifico dell’acqua CR = capacita’ termica del recipiente etc. Ta = temperatura iniziale dell’acqua Introducendo una quantita’ mx della sostanza x, Si vuole misurare il calore specifico della sostanza Sia Tx la temperatura iniziale di mx,, e TF la temperatura finale della miscela Allora, supponendo il sistema ( recipiente + acqua + sostanza) isolato dall’ambiente: calore ceduto = calore assorbito
da cui, cx supponendo tutti gli altri parametri noti
15
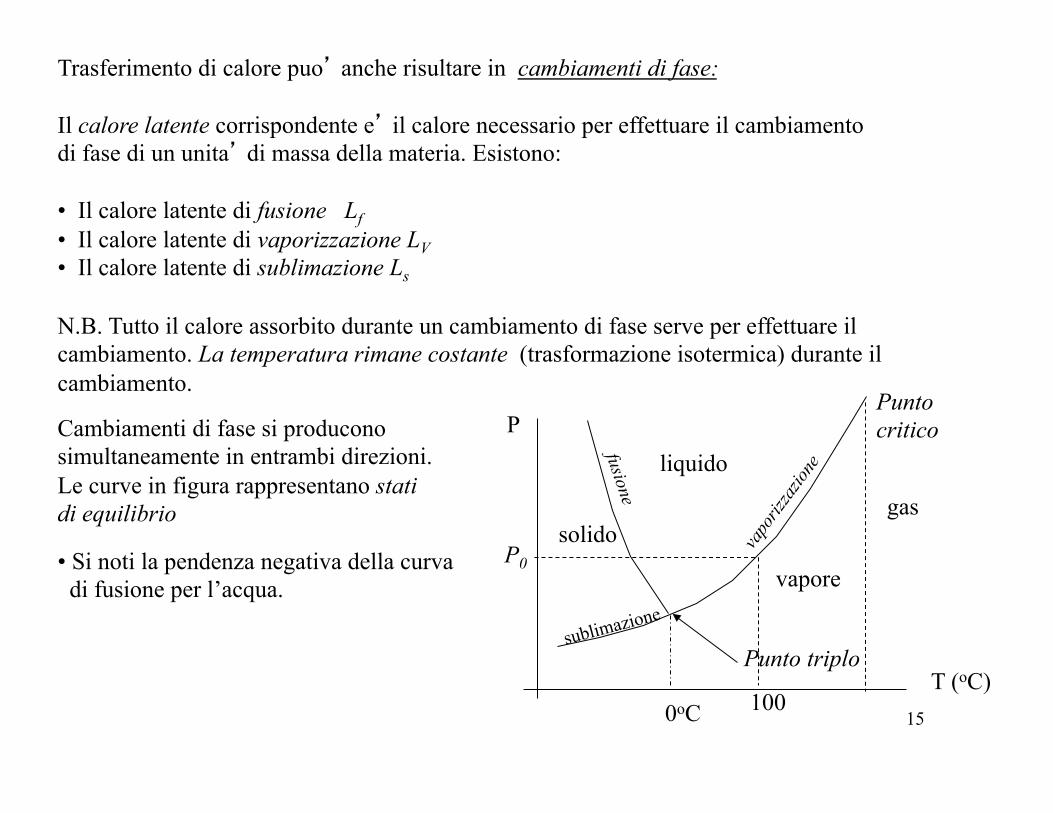
Trasferimento di calore puo’ anche risultare in cambiamenti di fase: Il calore latente corrispondente e’ il calore necessario per effettuare il cambiamento di fase di un unita’ di massa della materia. Esistono:
• Il calore latente di fusione Lf • Il calore latente di vaporizzazione LV • Il calore latente di sublimazione Ls
N.B. Tutto il calore assorbito durante un cambiamento di fase serve per effettuare il cambiamento. La temperatura rimane costante (trasformazione isotermica) durante il cambiamento.
solido
liquido
vapore
Cambiamenti di fase si producono simultaneamente in entrambi direzioni. Le curve in figura rappresentano stati di equilibrio
sublimazione
fusione
Punto triplo
gas
Punto critico P
T (oC)
• Si noti la pendenza negativa della curva di fusione per l’acqua.
0oC 100
P0

16
Il segno negativo indica che il verso del flusso e’ contrario a quello della variazione di T
Trasferimento di calore: Tre meccanismi (a) Conduzione : se, in una sostanza, la temperatura varia con spostamento, si osserva un flusso di calore. Supponendo che la materia sia uniforme e che T non vari nel piano (y,z) ( e dunque solo in direzione x ). Supponendo, inoltre, che la situazione sia stazionaria, allora la quantita di calore che passa attraverso area di superficie nel piano (y,z) per unita’ di tempo e:
T TT Δ+
AΔ ⇐xΔ
Q A
xTk
tQ
Δ⎟⎠
⎞⎜⎝
⎛Δ
Δ−≈
Δ
Δ
Dove: • k e’ la conduttivita’ termica • direzione del flusso ⊥Δ superficie di areal' e'A
Nel limite infinitesimo, si scrive
AΔ
AdxdTk
dtdQ
Δ−=
x Superfici isoterme
Legge di Fourier di conduttivita’ termica
Dove corrisponde al gradiente di temperatura (tasso di variazione di temp nella direzione in cui e’ massimo).
dxdT
Unita’ di k (“coefficiente di conduttivita’ o di conducibilita’ termica” Dalla legge di Fourier, [k] = [(cal/sec)/(oK/m)].[1/m2]=[cal/ oK.m.s] k puo’ dipendere, generalmente, dal tempo, le coordinate e anche dalla temperatura*.
x⊥

17
Nel caso generale, T(t) varia in funzione del tempo, e la soluzione dell’equazione di Fourier diventa un problema di trasporto. Qui’, ci siamo limitati al considerare: le situazioni stazionarie che si stabiliscono di solito dopo un intervallo finito di tempo.
xΔ
A 1T 2T
d
tAdxdTkQ Δ−≈Δ
dove, siccome lo stato e’ stazionario, lo stesso vale per ogni spessore ( Δx ) di area uguale, cioe’:
k dTdx
=dQdt
= costan te
Supponendo, inoltre, che k sia costante (materia omogenea), allora:
( ) tdTTkAQ Δ
−−≈Δ 21
Una tale situazione si puo’ approssimare collocando uno strato di materia ( cioe’ una lastra) omogenea, di spessore d, a contatto termico con due sorgenti di calore a temperature costanti (T1 e T2 ), detti termostati, e isolandone i lati per ridurre al minimo le perdite di calore laterali. Dopo un intervallo di tempo si stabilira’ una situazione stazionaria (indipendente da t ) e il calore trasmesso in tempo Δt, attraverso un spessore Δx verra’ dato da:
dQ_

18
Definendo la resistenza termica R
Def: kdR =
si puo’ scrivere la legge di Fourier per lo stato stazionario :
RTTA
dtdQ )( 21 −−
=
Si considerino ora, due lastre infinite, a contatto termico.
1QΔ 2QΔ
1k 2k
1d 2d( )
( ) td
TTAkQ
tdTTAkQ
x
x
Δ−
=Δ
Δ−
=Δ
2
222
1
111
In situazione stazionaria:
21 QQ Δ=Δcioe’,
( ) ( )
2
22
1
11
2
2
1
1
2
22
1
11
dTk
dTk
dk
dkT
dTTk
dTTk
x
xx
+=⎟⎟⎠
⎞⎜⎜⎝
⎛+
−=
−
21
1221
RRRTRTTx +
+=
e sostituendo nell’eq. di Fourier
( ) ( )RTTA
RRTTA
RRRTRTT
RA
dtdQ
21
21
21
21
12211
1
−=
+
−=
⎟⎟⎠
⎞⎜⎜⎝
⎛
+
+−=
La resistenza termica e’ additiva
2
2
1
1
21
11RT
RT
RRTx +=⎟⎟
⎠
⎞⎜⎜⎝
⎛+
A

19
(b) Convezione
Il trasferimento di calore corrispondente al trasferimento di materia es. convezione in un fluido contenuto tra due termostati
T1
T2
Fluido caldo
Fluido freddo
Conseguenze naturali importanti: - variabilita’ climatica - il mare si gela in superficie.
Aquile - alianti -
(c) Irraggiamento: trasferimento di calore nel vuoto per irraggiamento elettromagnetico Il potere emissivo di una superficie e’ dato dalla “legge di Stefan-Boltzmann”:
41 TdtdQ
Ae ασ=
A Dove Qe e’ il calore emesso, α e’ l’emissivita‘ della superficie, A e’ l’area e σ e’ la costante di Stefan-Boltzmann:
( ) sKmcal
402
8
187.4
106703.5 −×=σ

20
mg
termometro Pareti adiabatiche
Equivalente meccanico del calore: (James Prescott Joule 1818 - 1894) Aumentando la temperatura dell’acqua adiabaticamente, si dimostra che
1 cal = 4.187 Joule dove la costante 4.187 e’ detta “l’equivalente meccanico del calore” Joule dimostro’ inoltre che il rapporto lavoro/calore non dipende dal metodo (adiabatico) utilizzato per generare il lavoro. Le implicazioni sono:
• il calore e’ una forma di energia • l’energia interna di un sistema e’ correlata alla temperatura
Equivalenza calore - energia meccanica - Primo Principio della Termodinamica

21
Il primo principio della termodinamica
afferma che: Per ogni sistema termodinamico, esiste una funzione di stato U detta “energia interna”, tale che:
LQUUU if −=Δ=−
Dove: • Q e’ il calore assorbito dal sistema • L e’ il lavoro eseguito dal sistema
Si noti che mentre Q ed L dipendono dal metodo impiegato (la trasformazione) per portare il sistema dallo stato iniziale allo stato finale mentre U, in quanto funzione di stato, non dipende dalla trasformazione.
P
V
i f
Il primo principio e’ la legge di conservazione dell’energia per sistemi T.D. La forma differenziale e’:
dLdQdU −=
non differenziali esatti
22
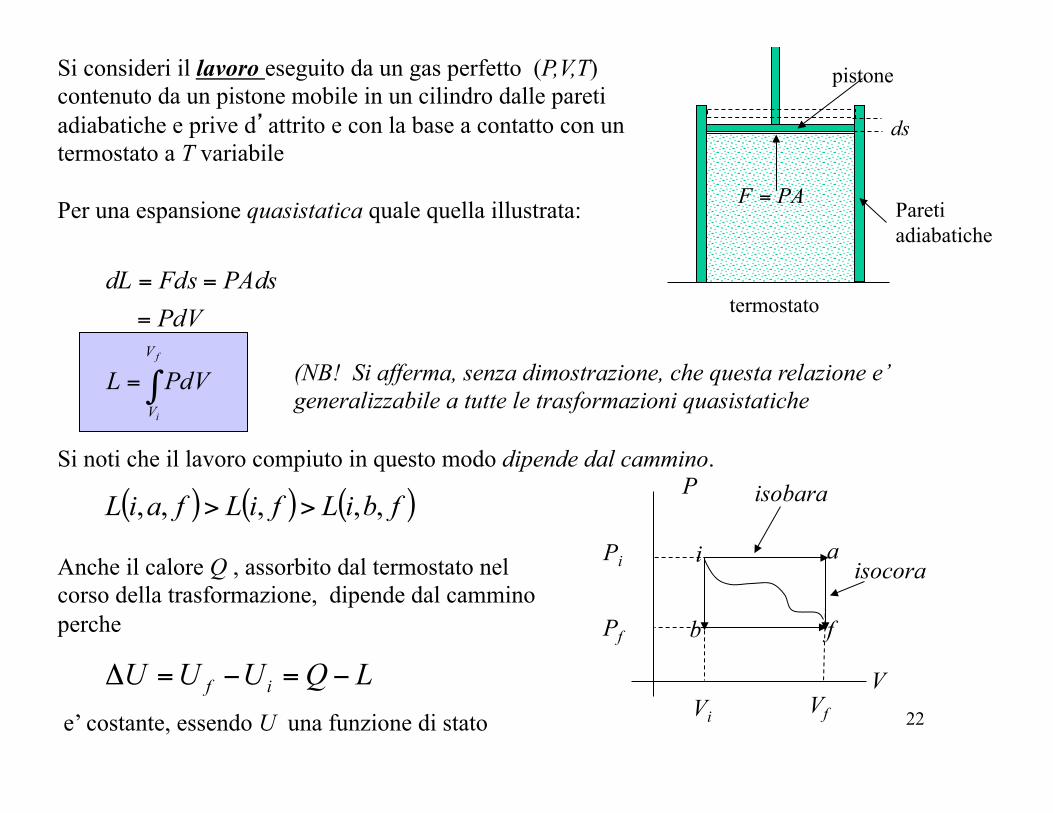
Si consideri il lavoro eseguito da un gas perfetto (P,V,T) contenuto da un pistone mobile in un cilindro dalle pareti adiabatiche e prive d’attrito e con la base a contatto con un termostato a T variabile Per una espansione quasistatica quale quella illustrata:
ds
PAF = Pareti adiabatiche
termostato
pistone
∫=
=
==
f
i
V
V
PdVL
PdVPAdsFdsdL
Si noti che il lavoro compiuto in questo modo dipende dal cammino.
i a
f b
P
V
Pi
Pf
Vi Vf
isobara
isocora
( ) ( ) ( )fbiLfiLfaiL ,,,,, >>
Anche il calore Q , assorbito dal termostato nel corso della trasformazione, dipende dal cammino perche
LQUUU if −=−=Δ
e’ costante, essendo U una funzione di stato
(NB! Si afferma, senza dimostrazione, che questa relazione e’ generalizzabile a tutte le trasformazioni quasistatiche

23
Si consideri ora il calore assorbito dal sistema. Per una trasformazione quasistatica:
( ) ( )dTTCndTTcmdTTCQf
i
f
i
f
i
T
Tx
T
Tx
T
Txx ∫∫∫ ʹ′=== )(
massa calore specifico
capacita’ termica Capacita’ termica
molare dove
PP
VV
VPx
dTdQ
nC
dTdQ
nC
CCC
⎟⎠
⎞⎜⎝
⎛=ʹ′⎟⎠
⎞⎜⎝
⎛=ʹ′
ʹ′ʹ′=ʹ′
1 e 1 o
Per cambiamenti di fase (trasformazioni isoterme):
λmQ =
massa
Calore latente

24
Applicazioni del Primo Principio a delle trasformazioni semplici .
Dal momento che le variabili di stato sono definite solo per stati di equilibrio, siamo sempre limitati a considerare trasformazioni quasistatiche. Si conderi anzitutto una: (a)Trasformazione Isobara di un gas ideale
P
V
i f
Vi Vf
TPnRV
PnR
TV
nRTPV
Δ⎟⎠
⎞⎜⎝
⎛=Δ
=
=
(costante)
( ) VPVVPdVPPdVLf
i
f
i
V
Vif
V
V
Δ=−=== ∫∫Dall’equazione di stato dei gas perfetti:
e sostituendo nell’espressione per il lavoro L:
( )if TTnRL −=

25
Il calore assorbito dal sistema nella trasformazione isobara e’ dato da:
dTCnQf
i
T
Tp∫ ʹ′=
e supponendo costante per un gas ideale, pCʹ′
( )ifP TTCnQ −ʹ′=
Applicando il primo principio,si ottiene:
( ) ( )( ) TRCnU
TTnRTTCnLQU
P
ififp
Δ−ʹ′=Δ
−−−ʹ′=−=Δ
da questa relazione, si puo concludere che (per gas ideali):
RTU
nC
PP +⎟
⎠
⎞⎜⎝
⎛∂
∂=ʹ′1
Vale dire che la capacita’ termica molare (a pressione costante) corrisponde alla variazione di energia interna per unita’ di variazione di temperatura + R. Vedremo riproporsi questa relazione quando intraprenderemo un analisi microscopica della capacita’ termica.

26
(b) Trasformazione isocora di un gas ideale
i
f
Pi
Pf
V
( )ifV
V
V
TTCnQ
PdVLf
i
−ʹ′=
== ∫ 0
Si noti che:
VV T
Un
C ⎟⎠
⎞⎜⎝
⎛∂
∂=ʹ′1
Anche questa relazione si vedra’ riproporsi successivamente
TCnLQU VΔʹ′=−=Δ
Segue dal primo principio che:
27
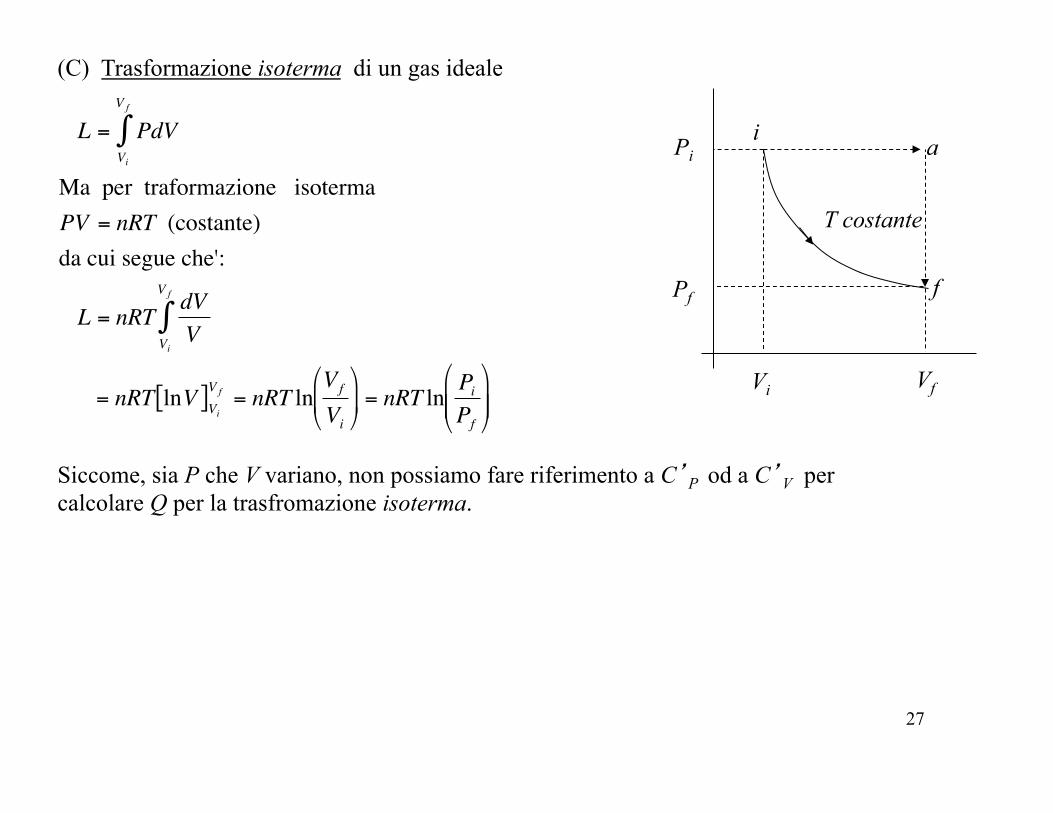
(C) Trasformazione isoterma di un gas ideale
i
f
T costante
Pi
Pf
Vi Vf
Siccome, sia P che V variano, non possiamo fare riferimento a C’P od a C’V per calcolare Q per la trasfromazione isoterma.
a
!
L = PdVVi
V f
"
Ma per traformazione isotermaPV = nRT (costante)da cui segue che':
L = nRT dVVVi
V f
"
= nRT lnV[ ]ViV f = nRT ln
Vf
Vi
#
$ %
&
' ( = nRT ln
PiPf
#
$ % %
&
' ( (

28
Teoria cinetica dei gas Limitiamoci a considerare gas ideali o perfetti. Le caratteristiche microscopiche dei gas ideali sono le seguenti: (1) sono composti di molecole identiche (2) il numero di queste molecole e’ grande (3) le molecole sono in moto casuale (4) obbediscono alle leggi di Newton (5) il loro volume e’ trascurabile rispetto a quello che le contiene (6) le sole interazioni tra di loro e con le pareti sono urti elastici
Si consideri un recipiente cubico: y
x
Nel corso di un urto elastico, con una delle pareti (prive di attrito), il cambiamento della q.d.m. di una molecola e:
( ) imvimvmvqqq xxxifˆ2ˆ −=−−=−=Δ
Il tempo tra urti successivi (trascurando eventuali urti tra le molecole) e’:
xvlt 2=Δ
La variazione della q.d.m per unita’ di tempo che avviene per effetto di urti con una parete e’:
( ) ⎟⎠
⎞⎜⎝
⎛−=⎟⎠
⎞⎜⎝
⎛−=ΔΔ lmv
lvmvtq xx
x
2
22/
l
l

29
Tenendo conto di tutte le molecole, la variazione totale della q.d.m. (distribuita uniformemente nel tempo) e’:
! m / l( ) vx,i2" = !F
Questa e' la forza media esercitata dalla parete sulle molecole.-F e' dunque la forza esercitata dalle molecole sulla parete e
PA1= F A = m / l3( ) vx,i
2" =nNmV
v2x,i"
nN
#
$%%
&
'((=
nMV
vx,i2"
nN
#
$%%
&
'((
dove M e’ la massa di una mole del gas e N e’ il numero di Avogadro e n e’ il numero di moli di gas. Ma (dove ρ e’ la densita’ del gas) e, ρ=VnM
vx,i2!
nN= vx
2 il valore medio di vx2
vale dire che: 2
1 xA vP ρ=
Ora, per ogni molecola, 2222zyx vvvv ++=
e siccome il movimento delle molecole e’ casuale, 2222
31 vvvv zyx === Segue che
!
P = 13( )"v 2 (1)

30
Si noti che, lo stesso risultato si ottiene ammettendo gli urti (elastici) tra molecole, e per qualsiasi forma di recipiente.
La velocita quadratica media
ρPvv mq32
.. ==
Esempi: per H2 a 0oC vq.m.=1838 m/s per O2 a 0oC vq.m.= 461 m/s per aria a 0oC vq.m.= 485 m/s per aria a 0oC la velocita’ del suono e’ 331 m/s
Moltiplicando (1) per V, si ottiene:
!
PV = 13( )"Vv 2 = 1
3( )nMv 2Dove: M e’ la massa molare e n e’ il numero di moli
Si puo riscrivere l’equazione nella forma seguente:
!
PV = 23( ) 12 nMv
2"
# $
%
& '
E confrontandola con l’equazione di stato dei gas perfetti: nRTPV =
Si conclude che:
!
32"
# $ %
& ' RT =
12Mv 2 = N 1
2mv 2
"
# $
%
& ' = Ktr
l’energia cinetica di traslazione di una mole

31
Inoltre,
!
32( ) RN( )T = 1
2( )mv 2 L’energia cinetica media di traslazione per ogni molecola
oppure,
!
12( )mv 2 = 3
2( )kTdove
NRk = la costante di Boltzmann
!
k =8.314 J mol.°K( )
6.022 "1023 molecolemol( )
=1.381"10#23 Jmolecola.°K( )
L’equazione di stato dei gas perfetti si puo’ dunque scrivere:
nNkTPV =

32
Miscele di gas ideali
Ognuno di due o piu’ gas ideali che occupano lo stesso recipiente si comporta come se gli altri gas non vi fossero, ovvero come se fossero soli ad occupare il recipiente. Segue che:
!
P = Pii" =
13
#ivi2( )
i"
(dove gli indici i si riferiscono ora ai diversi gas) e che
!
PV = 13( ) niMiv i
2
i" = 2
3( ) 12
niMiv i2
i"
#
$ %
&
' (
= ni" 23)
12
Mivi2#
$ %
&
' ( = ni
i"#
$ %
&
' ( RT
da cui si conclude che
!
12
niMiv i2
i"
#
$ %
&
' (
nii"#
$ %
&
' (
= Nni12
miv i2#
$ %
&
' (
i"
nii"#
$ %
&
' (
= N 12
mv 2#
$ %
&
' ( =
32
RT
Dove e’ ora la media delle energie molecolari dei gas 2
21mv

33
Energia interna dei gas perfetti -- equipartizione di energia
Per un gas monatomico si puo’ trascurare la struttura interna delle molecole. Allora l’energia interna corrisponde all’energia cinetica di traslazione delle molecole:
( )RTKU tr 23== per mole
Va notato che questa dipende unicamente dalla temperatura T Va notato inoltre che:
( ) ( )2222
21
21
23
zyx vvvMMvRT ++==
Abbiamo gia’osservato che, siccome il moto e’ casuale; 222zyx vvv ==
Segue che:
RTMvMvMv zyx 21
21
21
21 222 ===
Vale dire che l’energia interna e’ distribuita, in parti uguali, tra i tre gradi di liberta’ disponibili, un grado di liberta’ essendo un modo indipendente per assorbire energia.

34
Si puo’ generalizzare questa deduzione a qualsiasi numero di gradi di liberta’, ottenendo cosi’, il teorema dell’equipartizione di energia.
“A ciascun grado di liberta’ di un gas in equilibrio e associata un energia di per mole“
RT21
Si puo quindi dire che, per qualsiasi gas ideale, l’energia interna dipende solo dalla temperatura ed e’ proporzionale ad essa.
Si consideri ora un gas biatomico. Schematizzandolo come due molecole separate da un asta sottile e leggera, si conclude che
x
y
z
Ix e’ trascurabile perche le dimensioni delle molecole sono trascurabili. Inoltre Iy = Iz = I e supponendo di poter trascurare le vibrazioni:
!
U = 12Mvx2 + 12Mvy
2 + 12Mvz2 + 12 I" y
2 + 12 I" z2 = 52RT per mole
Ammettendo anche le vibrazioni, si ammettono altri due gradi di liberta, ottenendo: RTU 2
7=
(Non e’ facile giusificare, in termini classici, che vi siano solo 2 gradi di liberta’ vibrazionale)

35
Si consideri ora un gas poliatomico. In questo caso vi sono 3 gradi di liberta’per le rotazioni
Trascurando le vibrazioni, ed applicando il principio di equipartizione di energia,
RTRTU 326 ==
Ed ammettendo anche le vibrazioni:
RTU 29=
( + 3 gradi di rotazione)
(+ 3 gradi di vibrazione)

36
Armati dell’informazione ricavata dall’analisi microscopica dei gas ideali, ritorniamo ora alla termodinamica. Ricordiamo, anzitutto che, per trasformazione isocora
da cui seguiva che:
VCndTdU
ʹ′=
vV
nCTU
=⎟⎠
⎞⎜⎝
⎛∂
∂
Sapendo ora che U dipende solo dalla temperatura si puo’ scrivere:
oppure
dTCndU Vʹ′=
TCnQU VΔʹ′==Δ
N.B. Questo vale per tutte le trasformazione quasistatiche di un gas ideale

37
La relazione di Mayer
V T
T+ΔΤ Si consideri la trasformazione isobara tra il punto a sulla isoterma T ed il punto b sull’isoterma T+ΔT. Abbiamo gia’ visto che, per una trasformazione isobara,
( ) TRCnVPTCnU PP Δ−ʹ′=Δ−Δʹ′=Δ
a b
c
Per la trasformazione isocora (a,c) tra le medesime isoterme, sappiamo invece che: TCnU VΔʹ′=Δ
cioe’ che:
La relazione di Mayer
Pero’, trattandosi di trasformazioni tra le medesime isoterme, la variazione di energia interna e’ sempre quella. Segue che: UΔ
( ) VP CnRCn ʹ′=−ʹ′
RCC VP =ʹ′−ʹ′
Conoscendo U per i gas perfetti, e’ dunque possibile esprimerne la capacita’ termica molare a volume costante in base all’equazione sovrastante. Prima di passare ad un elenco di queste capacita’ termiche a volume costante, vediamo che e’ possibile specificare contemporaneamente anche le capacita’ termiche a pressione costante.
P

38
Capacita’ termiche molari
(a) Per un gas monatomico perfetto
Sapendo che: '
TU che e 2
3vCd
dnRTU ==
segue che:
RCV 23=ʹ′
E tenendo conto di Mayer:
RRCC VP 25=+ʹ′=ʹ′
(b) Per un gas biatomico ideale
nRTU
nRTU
2725
=
= (escludendo vibrazioni)
(ammettendo anche le vibrazioni)
Segue che, escludendo vibrazioni:
RC
RC
P
V
2725
=ʹ′
=ʹ′
e ammettendo vibrazioni
RC
RC
P
V
2927
=ʹ′
=ʹ′
Molto prossimo ai valori osservati in un larga gamma di temperatura intorno alla temperatura ambiente, per i gas inerti (He, Ne, Ar)
Prossimo ai valori per H2 , D2 , O2 , N2 NO, CO…a temp amb.

39
Per H2 si osserva l’andamento illustrato, e tenendo conto del fatto che
2
4
6
8 molKcalR ./ 2 °≈
( )R23( )R25
( )R27Si puo’ interpretare i successivi aumenti di in base all’accessibilita’ dei gradi di liberta di rotazione e di vibrazione
Questo e’ spiegabile solo applicando la meccanica quantistica .
Per un gas poliatomico perfetto
nRTU
nRTU
293
=
= escludendo vibrazioni
ammettendo anche le vibrazioni
Segue che:
RCRC
P
V
43
=ʹ′
=ʹ′escludendo vibrazioni, e
RC
RC
P
V
21129
=ʹ′
=ʹ′ammettendo anche le vibrazioni
KT°
( )KmolcalCV
°⋅
ʹ′
100 1000 10000
dissociazione
'VC

40
dTCndQdTCndQ VP ʹ′=ʹ′= o
Segue che:
PdVdTnCdLdTCndQ VV +=+ʹ′= '
VP CC ʹ′ʹ′ e
dLdQdU −=
e siccome:
Ritorniamo, ora il problema di calcolare il calore assorbito da un sistema nel corso di una trasformazione quasistatica qualsiasi di un gas ideale. Abbiamo appurato, di poter calcolare
e sappiamo, inoltre, che sono costanti per uno specifico gas. Tuttavia, nel caso generale, (P,V non necessariamente costanti) non possiamo scrivere:
D’altra parte, sappiamo di poter sempre scrivere:
(primo principio)
dTCndU Vʹ′=
si puo’ anche dimostrare (esercizio) che:
VdPdTCndQ P −ʹ′=
Abbiamo dunque due modi per calcolare dQ nel caso generale.

41
Equazione di stato per trasformazioni adiabatiche di gas ideali
Abbiamo gia’ visto che, per un gas perfetto:
PdVdTCndQ V +ʹ′=
e siccome la trasformazione e’ adiabatica, ( dQ = 0 ),
0=+ʹ′ PdVdTCn V
Sostituendo dall’equazione dei gas perfetti:
!
n " C V dT +nRTV
#
$ %
&
' ( dV = 0
e sostituendo poi per R dalla relazione di Mayer:
!
" C V dT = # " C P # " C V( )T dVV
Riordinando:
( ) VdV
TdT 1−−= γ con
V
P
CCʹ′
ʹ′=γ
ed integrando
!
" lnT[ ]if
= # "1( ) lnV[ ]if cioe’
1−
⎟⎟⎠
⎞⎜⎜⎝
⎛=
γ
i
f
f
i
VV
TT ovvero =−1γTV costante
e, sostituendo T = PVnR =γPV costante

42 !
dU = dQ " PdVcioe'n # C V dT = "PdV
$U = n # C V dTTi
T f
% = n # C V Tf "Ti( ) = " PdVi
f
% = "L < 0( )
(b) Espansione adiabatica P
V
Ti
Tf
Si noti che, per un espansione quasistatica, la variazione di energia interna e’ negativa.
Riprendendo ora: Alcune trasformazioni communi (di gas ideali)
(a)Espansione isoterma (quasistatica) P
V
i •
•f
Q
!
dU = dQ " PdV = 0dQ = PdV
Q = L = PdVVi
V f
# = nRT lnVf
Vi
$
% &
'
( ) > 0( )
Tutto il calore lavoro

43
Trasformazioni cicliche e macchine termiche Si noti che, per il ciclo,
a
b
P
V
se
CA
LLLQQQ
+=
+=QA
QC
( > 0 )
LS
Le
Mentre, essendo U un funzione di stato, la sua variazione e’ nulla
0=ΔU
Ma, dal primo principio,
LQU −=Δ
Segue che per il ciclo,
LQ =
Vale dire che, nel corso di un ciclo, calore viene trasformato in lavoro. Il processo ciclico costituisce dunque una macchina termica, il cui rendimento meccanico e’ (per definizione):
A
C
A
C
A
CA
A
QQQ
QL
−=
+=+
==
1
1
η
η
44
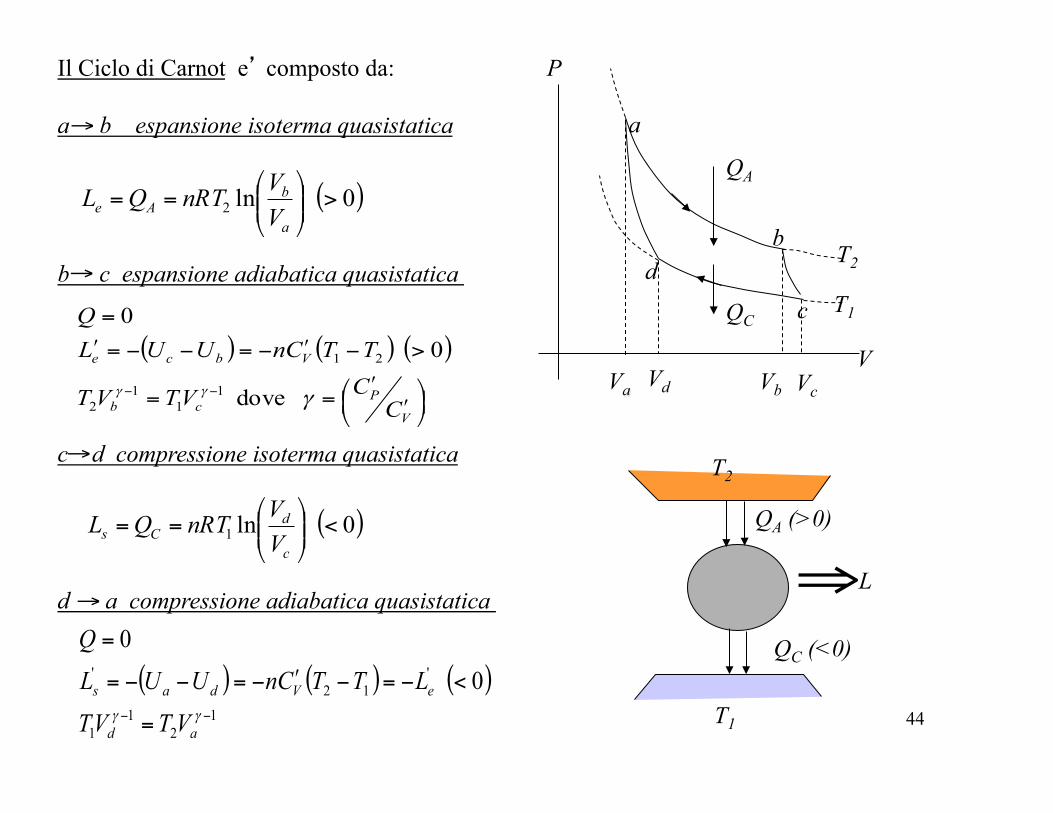
Il Ciclo di Carnot e’ composto da: a b espansione isoterma quasistatica →
( )0 ln2 >⎟⎟⎠
⎞⎜⎜⎝
⎛==
a
bAe V
VnRTQL
a
b d
c
Va Vd Vb Vc
T2
T1
P
V
b c espansione adiabatica quasistatica
( ) ( ) ( )
⎟⎠⎞
⎜⎝⎛
ʹ′ʹ′==
>−ʹ′−=−−=ʹ′
=
−−
V
Pcb
Vbce
CCVTVT
TTCnUULQ
γγγ dove
0 0
11
12
21
c d compressione isoterma quasistatica
( )0 ln1 <⎟⎟⎠
⎞⎜⎜⎝
⎛==
c
dCs V
VnRTQL
→
→
d a compressione adiabatica quasistatica
( ) ( ) ( )1
21
1
'12
' 0 0
−− =
<−=−ʹ′−=−−=
=
γγad
eVdas
VTVTLTTCnUUL
Q→
T2
T1
QA (>0)
QC (<0)
⇒L
QA
QC

45
Per il ciclo,
sseeCA LLLLLQQQ ʹ′++ʹ′+==+=
Il rendimento e’:
A
C
A QQ
QL
+== 1η
e sostituendo: ⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
−=⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
+=
a
b
d
c
a
b
c
d
VVT
VVT
VVnRT
VVnRT
ln
ln1
ln
ln1
2
1
2
1
η
ma
11
12
11
12
−−
−−
=
=γγ
γγ
da
cb
VTVTVTVT
e dividendo queste due equazioni si ottiene:
d
c
a
b
d
c
a
b
VV
VV
VV
VV
=⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛−−
cioe' 11 γγ
Segue che 2
11TT
−=η
La macchina di carnot, pur non essendo realizzabile, e’ utile per stabilire il limite superiore teorico del rendimento. Il teorema di carnot (che vi viene chiesto di prendere per dato) recita :
“Il rendimento di tutte le macchine reversibili operanti tra gli stessi due termostati e’ il medesimo e nessuna macchina operante tra questi due termostati puo’ avere rendimento maggiore”.

46
Il Ciclo Carnot Frigorifero
T2
T1
a
b
d c
QC
QA
P
V
(gas ideale)
Calore assorbito a temperatura bassa
!
QA = Le = nRT1 ln VCVd
" # $ %
& ' > 0( )
Calore ceduto a temperatura alta
( )0 ln2 <⎟⎠⎞
⎜⎝⎛==
b
asC V
VnRTLQ
L’efficienza o coefficiente di prestazione del frigorifero e’ definita:
e = QA
L=
QA
Le + Ls=
nRT1 lnVcVd( )
nRT1 lnVcVd( )! nRT2 ln Vb Va( )
Ed avendo dimostrato che segue che d
c
a
bV
VV
V =
12
1
TTTe−
= e’ il limite superiore dell’efficienza
Abbiamo gia’ dimostrato che il lavoro eseguito nelle trasformazioni adianatiche e’ complessivamente nullo. Segue che se LLL +=

47
Entropia ed il secondo principio della termodinamica - reversibilita’
La variazione di entropia S per trasformazioni quasistatiche e’, per definizione:
∫=Δf
i TdQS
Segue dalla definizione, che l’entropia S e’ una funzione di stato.
Dimostrazione (per un ciclo di Carnot)
1
1
2
2
1212
lnln
00
TV
VnRT
TV
VnRT
TQ
TQ
TQ
TQ
TdQ
TdQ
TdQ
TdQ
TdQ
c
d
a
b
CACA
a
d
d
c
c
b
b
a
⎟⎠⎞
⎜⎝⎛
+⎟⎠⎞
⎜⎝⎛
=
+=+++=
+++= ∫∫∫∫∫
T2
T1
a
b
d c
QA
QC
P
V
Si consideri il ciclo di Carnot per un gas ideale
⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢
⎣
⎡
⎟⎠⎞
⎜⎝⎛
⎟⎠⎞
⎜⎝⎛
=⎥⎦⎤
⎢⎣⎡
⎟⎠⎞
⎜⎝⎛−⎟
⎠⎞
⎜⎝⎛=∫
d
c
a
b
d
c
a
b
VVV
V
nRVV
VVnR
TdQ lnlnln
Ma abbiamo gia’ dimostrato che d
c
a
bV
VV
V =
Segue allora che, per il ciclo di Carnot: 0==∫ ∫ TdQdS

48
Si prenda per dato che questo risultato e’ generalizzabile a tutte le trasformazioni quasistatiche cicliche. Esiste dunque una funzione di stato S detta Entropia
Si ipotizza, inoltre, che per tutte le trasformazioni mai osservate o concepite,
0≥Δ USDove ΔSU e’ la variazione dell’entropia dell’universo (cioe’ del sistema + l’ambiente). Generalizzando, quindi dall’esperienza, si formula il secondo principio della termodinamica:
“Esiste una funzione di stato S, detta Entropia e tale che, per qualsiasi trasformazione tra stati di equilibrio, . 0≥Δ US
Reversibilita Trasformazioni con sono irreversibili.
Questo segue dal secondo principio: supponendo che una trasformazione per la quale possa essere invertita, invertendola si otterrebbe una trasformazione per la quale (perche S e’funzione di stato) . Questo sarebbe in contradizione col secondo principio della TD.
0<Δ US
0>Δ US
0>Δ US
I processi naturali sono sempre irreversibili ( ). Vale dire che procedono in direzione unica .
0>Δ US

49
Il secondo principio serve dunque a definire la direzione dei processi (trasformazioni) naturali. Es. Espansione libera di un gas perfetto (passa per stati di non-equilibrio) Es. Dissipazione dell’energia meccanica. Es. Conduzione di calore.
Generalmente, nelle trasformazioni irreversibili, si verificano uno o entrambi dei seguenti fenomeni: dissipazione di energia meccanica e/o passaggio attraverso stati di non-equilibrio.
Una trasformazione quasistatica e’ reversibile se e se non intervengono fenomeni dissipativi
!
"SS = 0
Variazione di Entropia di un gas perfetto.
Sappiamo che, nel caso generale:
VdPdTCndQ P −ʹ′=
per cui
dPTV
TdTCn
TdQdS P −ʹ′==
cioe’
PdPnR
TdTCndS P −ʹ′=
50
∫==f
i
TdSQTdSdQ che e
ed integrando
⎟⎟⎠
⎞⎜⎜⎝
⎛−ʹ′=−ʹ′=Δ ∫∫∫
i
ff
iP
f
i
f
iP P
PnR
TdTCn
PdPnR
TdTCnS ln
e, se e’ costante (es. gas ideale) PCʹ′
⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟⎟⎠
⎞⎜⎜⎝
⎛ʹ′=Δ
i
f
i
fP P
PnR
TT
CnS lnln
Partendo da: PdVdTCndQ V +ʹ′= si ottiene
⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟⎟⎠
⎞⎜⎜⎝
⎛ʹ′=Δ
i
f
i
fV V
VnR
TT
CnS lnln
Si noti che
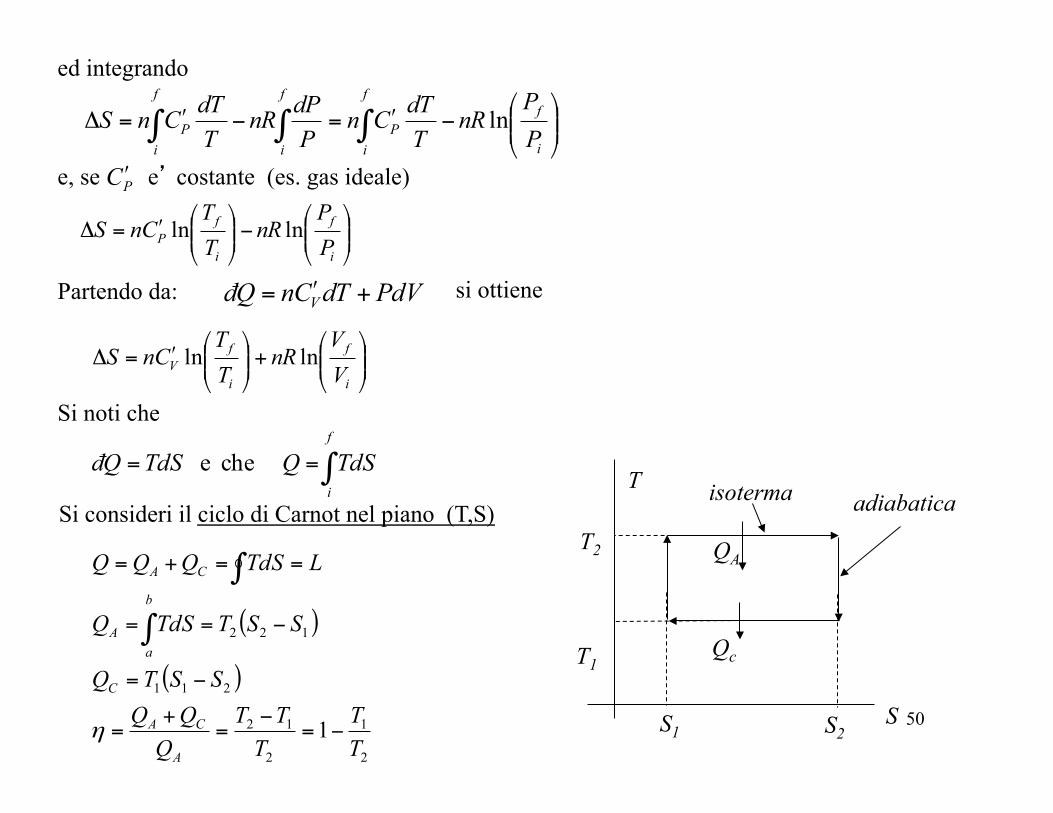
Si consideri il ciclo di Carnot nel piano (T,S) T
S
isoterma adiabatica
( )
( )
2
1
2
12
211
122
1
TT
TTT
QQQSSTQ
SSTTdSQ
LTdSQQQ
A
CA
C
b
aA
CA
−=−
=+
=
−=
−==
==+=
∫
∫
η
T2
T1
QA
Qc
S1 S2
51
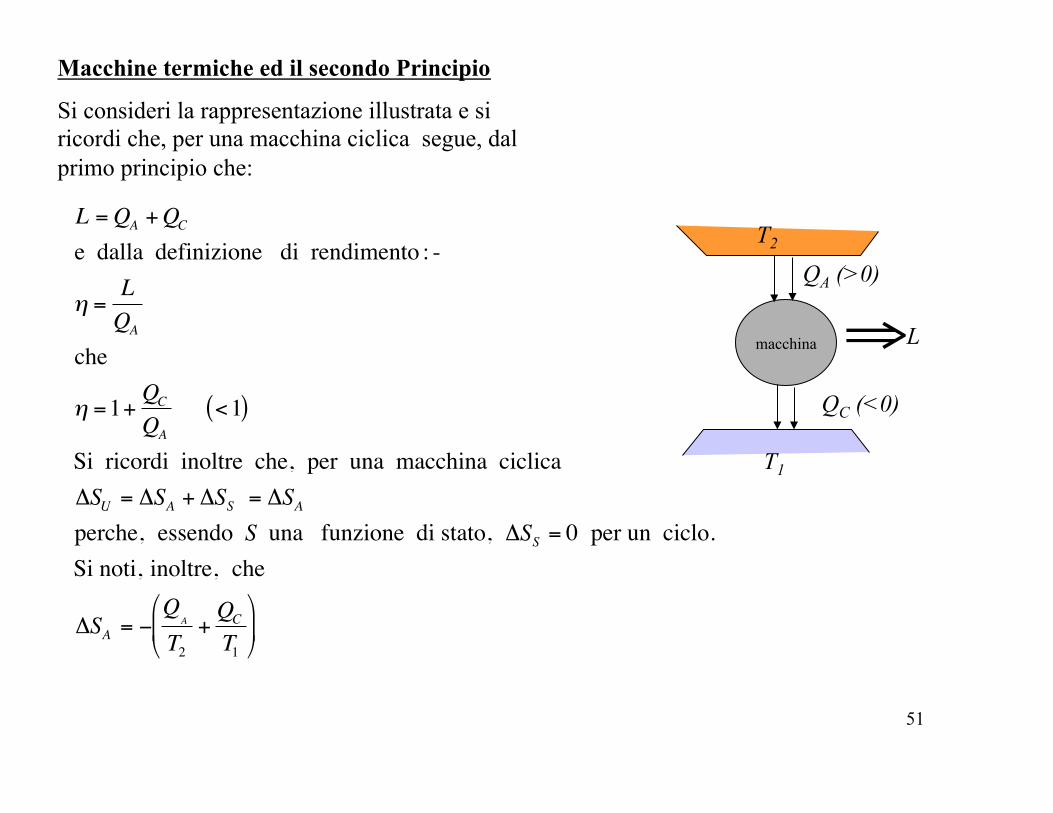
Si consideri la rappresentazione illustrata e si ricordi che, per una macchina ciclica segue, dal primo principio che:
macchina
T2
T1
QA (>0)
QC (<0)
⇒L
Macchine termiche ed il secondo Principio
!
L =QA +QC
e dalla definizione di rendimento : -
" =LQA
che
" =1+QC
QA
<1( )
Si ricordi inoltre che, per una macchina ciclica #SU = #SA + #SS = #SA perche, essendo S una funzione di stato, #SS = 0 per un ciclo.Si noti, inoltre, che
#SA = $Q
A
T2
+QC
T1
%
& '
(
) *

52
⇒QA
L
Si ipotizzi ora una macchina perfetta
1==AQL
η cioe’ 0 1 =⇒=+
CA
CA QQQQ
Segue che: ( ) ⎟⎟⎠
⎞⎜⎜⎝
⎛+−=Δ=Δ
12 TQ
TQScicloS CA
AU
cioe’ che: 0<Δ US
E siccome questa possibilita’ e’ esclusa dal secondo principio, segue che “E’ impossibile realizzare un processo ciclico il cui unico risultato sia quello di assorbire calore da un serbatoio e convertirlo tutto in lavoro”.
Questo e’ “l’Enunciato di Kelvin-Plank” del secondo principio della T.D. Analogamente, con riferimento and una macchina frigorifera (ciclo invertito) si dimostra che: “E’ impossibile realizzare una trasformazione ciclica, il cui unico risultato sia quello di far passare calore da un corpo freddo ad uno piu’ caldo.”
Questo e’ “l’Enunciato di Clausius” del secondo principio della T.D.
T2
T1
53
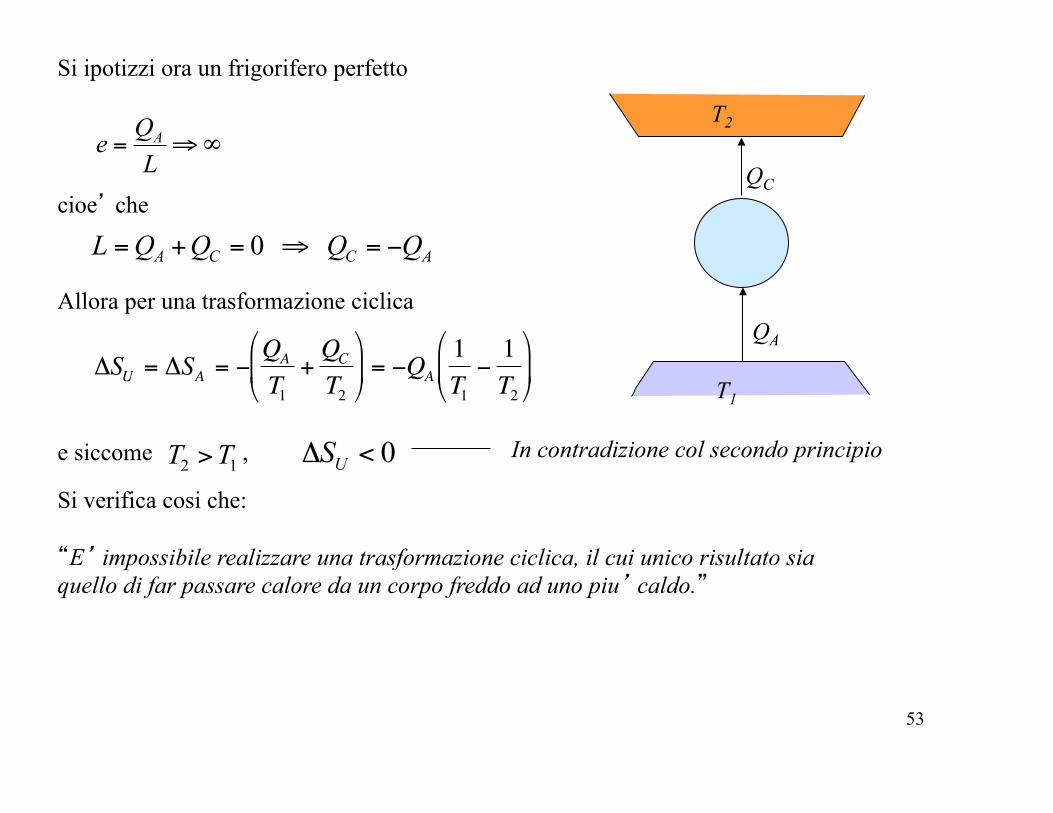
Si ipotizzi ora un frigorifero perfetto
T2
T1
QA
QC ∞⇒=
LQe A
cioe’ che
ACCA QQQQL −=⇒=+= 0
Allora per una trasformazione ciclica
!
"SU = "SA = #QA
T1+QC
T2
$
% &
'
( ) = #QA
1T1#1T2
$
% &
'
( )
e siccome , 12 TT > 0<Δ US In contradizione col secondo principio
Si verifica cosi che: “E’ impossibile realizzare una trasformazione ciclica, il cui unico risultato sia quello di far passare calore da un corpo freddo ad uno piu’ caldo.”


















