Tecnologie Ricombinanti - webalice.it · grosse molecole di DNA ricombinante. La cellula si dice...
48
Tecnologie Ricombinanti Dr. Giulio Piluso Dipartimento di Biochimica, Biofisica e Patologia Generale Tel. 081-5665685 Email [email protected] http://www.webalice.it/giulio.piluso
Transcript of Tecnologie Ricombinanti - webalice.it · grosse molecole di DNA ricombinante. La cellula si dice...

Tecnologie
Ricombinanti
Dr. Giulio Piluso
Dipartimento di Biochimica, Biofisica e Patologia Generale
Tel. 081-5665685
Email [email protected]
http://www.webalice.it/giulio.piluso

Libri di testo consigliati
T. Strachan, A.P. Read
Genetica Umana Molecolare (3ª Edizione)
UTET
E. Boncinelli, A. Simeone
Ingegneria genetica (2ª Edizione)
IDELSON-GNOCCHI

www.webalice.it/giulio.piluso

Di cosa parleremo?
Le tecnologie ricombinanti sono l’insieme delle metodiche, sviluppatesi all’incirca negli ultimi 20-25 anni, che consentono l’analisi, lo studio e la manipolazione di specifiche sequenze all’interno di una popolazione complessa di molecole di DNA.
Esse trovano applicazione in moltissimi campi della biologia molecolare: Identificazione e caratterizzazione di nuovi geni
Diagnostica molecolare
Analisi di espressione
Studi funzionali
Produzione di modelli animali
Terapia genica
Applicazioni industriali


Clonaggio del DNA (1)
Un frammento di DNA d’interesse rappresenta solo
una piccolissima parte di un genoma complesso.
Il gene della -globina corrisponde al 0.00005%
dell’intero genoma umano
Il gene della distrofina corrisponde al 0.08%
dell’intero genoma umano.
La tecnologia del clonaggio del DNA consente
l’amplificazione selettiva dei frammenti di DNA
desiderati, producendo un forte aumento
programmato del numero di copie della sequenza di
DNA selezionato.

Clonaggio del DNA (2)
Le metodiche normalmente utilizzate si basano sulla
replicazione ciclica del DNA, catalizzata da DNA
polimerasi specifiche.
Si distinguono:
Sistemi di clonaggio che utilizzano cellule (in vivo),
in cui molecole di DNA ricombinante sono trasferite
in cellule adatte, di cui si sfruttano i sistemi
biosintetici per amplificare selettivamente il
frammento di DNA d’interesse.
Sistemi di clonaggio senza cellule (in vitro), in cui la
tecnica di elezione è sicuramente la Polymerase
Chain Reaction (PCR).

Clonaggio del DNA
utilizzando cellule (1) Le tecniche di clonaggio che utilizzano cellule prevedono
quattro passaggi principali:
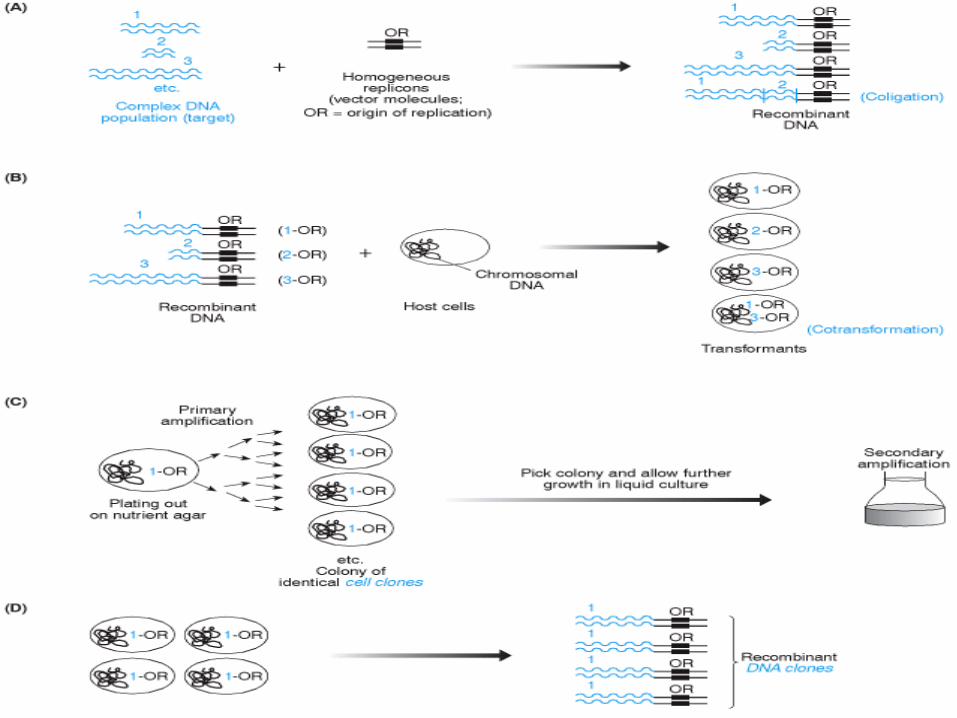
1. Costruzione di molecole di DNA ricombinante,
mediante saldatura dei frammenti di DNA
d’interesse ad un replicone (una qualsiasi
sequenza di DNA capace di replicarsi
autonomamente)
2. Trasformazione, con cui le molecole di DNA
ricombinante vengono trasferite in cellule ospiti
(batteri o lieviti) in cui i repliconi siano in grado di
compiere la replicazione del DNA
indipendentemente dal genoma della cellula ospite.

Clonaggio del DNA
utilizzando cellule (2) 3. Propagazione selettiva dei cloni cellulari.
• Le cellule trasformate vengono piastrate
distribuendole su di una superficie di agar per
favorire la crescita di colonie isolate
• Le colonie isolate possono essere recuperate
per essere cresciute in mezzo di coltura
liquido
4. Isolamento dei cloni cellulari ed estrazione del
DNA ricombinate.

Repliconi extracromosomici
come molecole vettrici I frammenti di DNA estraneo per potersi replicare
all’interno di una cellula devono contenere un’origine di
replicazione capace di funzionare in quel tipo cellulare.
I vettori di clonaggio sono appunto molecole di DNA
opportunamente ingegnerizzate così da assolvere a
questa funzione in cellule specifiche.
Le cellule batteriche sono le più utilizzate per la loro
velocità di replicazione. I vettori più comuni sono:
I plasmidi: piccole molecole di DNA circolare a
doppio filamento.
I batteriofagi: virus che infettano cellule batteriche.

Endonucleasi di Restrizione
L’avvento della tecnologia del DNA ricombinante è strettamente legato alla scoperta delle nucleasi di restrizione di tipo II, enzimi che tagliano il DNA in tutti i punti che contengono specifiche sequenze di riconoscimento.
Questi enzimi proteggono il batterio dall’infezione da parte dei virus (batteriofagi) il cui DNA, non specificamente metilato, è tagliato da queste nucleasi di restrizione.
Metilasi del DNA, con specificità di sequenza, metilano il DNA batterico.
Endonucleasi di restrizione, con specificità di sequenza, agiscono sul DNA del virus, non metilato, ma non sul DNA genomico della cellula batterica.

Caratteristiche degli enzimi
di restrizione
La sequenza riconosciuta dalla maggior parte degli E.R. è palindromica (uguale su entrambi i filamenti quando letta in direzione 5’→3’).
In genere i punti di taglio non coincidono con l’asse di simmetria della palindrome, generando estremità coesive (sticky ends), sporgenti al 5’ o 3’ (5’ or 3’ overhang)
I punti di taglio possono anche cadere sull’asse di simmetria della palindrome, generando estremità tronche (blunt ends).
E.R. diversi che riconoscono la stessa sequenza bersaglio sono detti isoschizomeri.

Enzyme Source Sequence cut Average expected fragment
size (kb) in human DNAa
AluI Arthrobacter luteus AGCT 0.3
HaeIII Hemophilus aegyptus GGCC 0.6
TaqI Thermus aquaticus TCGA 1.4
MnlI Moraxella nonliquefaciens CCTC/GAGG 0.4
HindIII Hemophilus influenzae Rd AAGCTT 3.1
EcoRI Escherichia coli R factor GAATTC 3.1
BamHI Bacillus amyloliquefaciens H GGATCC 7.0
PstI Providencia stuartii CTGCAG 7.0
MstI Microcoleus species CCTNAGGc 7.0
SmaI Serratia marcescens CCCGGG 78
BssHII Bacillus stearothermophilus GCGCGC 390b
NotI Norcadia otitidis-caviarum GCGGCCGC 9766b
a Assuming 40% G + C, and a CpG frequency 20% of that expected. b Observed average sizes are often lower than these estimates. c N = A, C, G or T.
Note: Names are normally derived from the first letter of the genus and the first two letters of the species name, e.g. PstI is the first restriction nuclease to have been
isolated from Providenciastuartii. MnlI is an example of an enzyme whose recognition sequence is not palindromic. So-called rare-cutters often have recognition
sequences containing one or more CpG dinucleotides and cut vertebrate DNA comparatively infrequently.
Alcuni esempi di Enzimi di
Restrizione


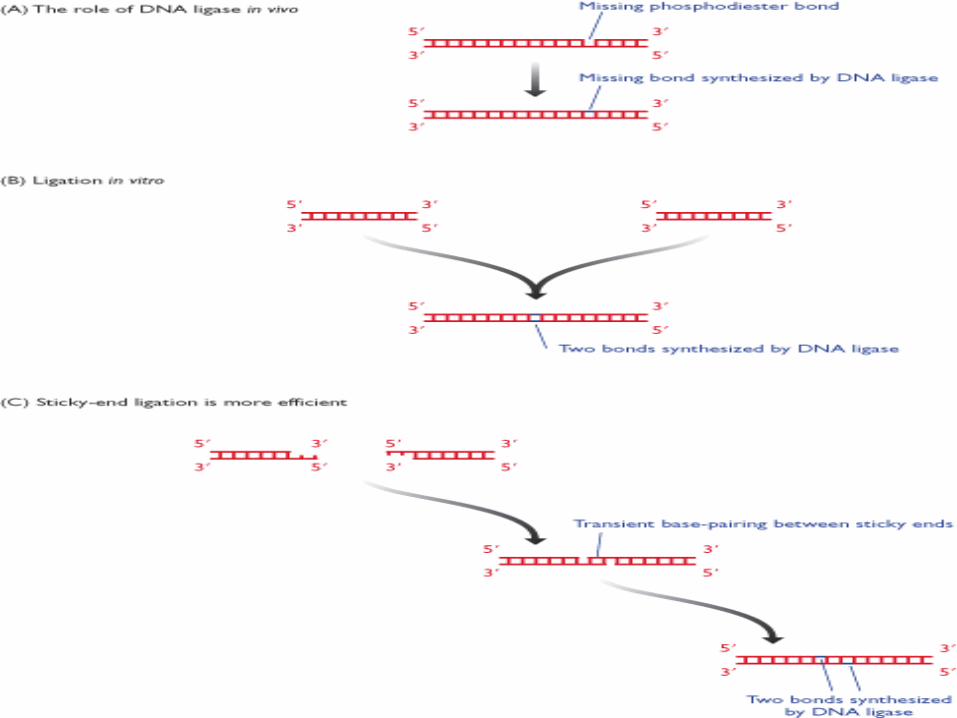
DNA Ligasi
I frammenti di DNA ottenuti per digestione con un E.R. possono essere riuniti insieme per azione di una DNA Ligasi che catalizza la formazione di ponti fosfodiesterici tra i nucleotidi di due frammenti di DNA.
La reazione richiede ATP come fonte di energia.
La più comune DNA Ligasi utilizzata è la T4 DNA Ligasi.


Clonaggio di un frammento di
DNA esogeno in un vettore
plasmidico.


Come favorire la formazione
di molecole ricombinati utili
E’ utile linearizzare il plasmide (vettore) con E.R. le cui
estremità coesive siano compatibili con quelle del
frammento di DNA da clonare.
E’ utile defosforilare il vettore. Il trattamento con una
fosfatasi (Fosfatasi alcalina) rimuovendo i gruppi
fosfato alle estremità del vettore linearizzato, impedisce
al plasmide di richiudersi su se stesso nella reazione
ligazione.
Il frammento di DNA da clonare ed il vettore sono
utilizzati nella reazione di ligazione in rapporto
equimolare o in leggero eccesso dell’inserto.

Come trasferire il DNA
ricombinante nella cellula
ospite La membrana cellulare è permeabile in modo selettivo e,
normalmente, non lascia transitare grosse molecole come
frammenti di DNA.
Opportuni trattamenti possono rendere la membrana
plasmatica permeabile consentendo l’ingresso anche di
grosse molecole di DNA ricombinante. La cellula si dice
allora competente.
Trattamenti con Sali ad elevata forza ionica (CaCl)
Brevi shock elettrici (elettroporazione)
L’introduzione di DNA ricombinante in una cellula resa
competente (es. cellula batterica) è detta trasformazione.

Come selezionare le cellule che
contengono le molecole di DNA
ricombinante Un problema rilevante è selezionare le cellule che, dopo la trasformazione, contengono la molecola di DNA ricombinante e che, poste in coltura, consentiranno di ottenere da esse una notevole quantità del frammento di DNA d’interesse.
Questo avviene comunemente in due modi:
Geni per la resistenza ad antibiotici (AmpR, KanR, TetR)
Le cellule batteriche contenenti il plasmide sono selezionate per la loro capacità di crescere su terreni contenenti uno specifico antibiotico.
Complementazione del gene per la -galattosidasi.
Consente alle cellule contenenti il vettore di produrre la -galattosidasi e di convertire una sostanza incolore, X-
gal (5-bromo-4-chloro-3-indolyl- -D-galactopyranoside) in un composto che colora di blu le colonie.

Come è fatto un vettore
plasmidico

La selezione dei cloni
contenenti l’inserto di DNA


Altri tipi di vettori: il
batteriofago
Il principale limite dei vettori plasmidici è che
non si possono clonare frammenti superiori a
5-10 Kb.
Vettori virali basati sul batteriofago
consentono in parte di superare questo limite
arrivando a 20 Kb.
In questi vettori parte del genoma virale, non
essenziale per la replicazione del
batteriofago, viene sostituito dall’inserto di
DNA che deve essere clonato.

Modalità di replicazione del
batteriofago in E. Coli

Il genoma del batteriofago

Clonaggio nel batteriofago

Limiti dei diversi vettori di
clonaggio oggi disponibili
Cloning vector Size of insert
Standard high copy number plasmid vectors 0-10 kb
Bacteriophage insertion vectors 0-10 kb
Bacteriophage replacement vectors 9-23 kb
Cosmid vectors 30-44 kb
Bacteriophage P1 70-100 kb
PAC (P1 artificial chromosome) vectors 130-150 kb
BAC (bacterial artificial chromosome) vectors up to 300 kb
YAC (yeast artificial chromosome) vectors 0.2-2.0 Mb

Librerie di DNA
I moderni metodi di clonaggio consentono di creare vaste collezioni di cloni di DNA (DNA libraries), rappresentative di una complessa popolazione di DNA. Principalmente si distinguono:
Librerie di DNA genomico, rappresentative dell’intero genoma di una determinata specie.
Librerie di cDNA, rappresentative dei geni espressi da un tessuto o in un particolare momento funzionale.

Librerie di DNA genomico
Il materiale di partenza è il DNA genomico estratto dai nuclei di cellule facilmente accessibili (es. cellule del sangue).
Il DNA è frammentato in genere per digestione con un E.R.
Per limitare la frammentazione del DNA si ricorre spesso a digestioni parziali (riducendo la quantità di enzima o i tempi di digestione).
Questo tipo di trattamento rende la frammentazione casuale e i frammenti possono sovrapporsi rendendo possibile il riordino dei cloni.
Il numero di cloni indipendenti è definito in termini di genoma-equivalenti (GE).
Un GE è il num. dei cloni indipendenti pari al rapporto tra dimensione del genoma e dimensione media dei cloni.
Per una libreria genomica umana con cloni di circa 40 Kb GE=3000Mb/40Kb= 75.000 cloni
Esistono anche librerie sub-genomiche, ottenute da specifici cromosomi od anche porzioni di essi.
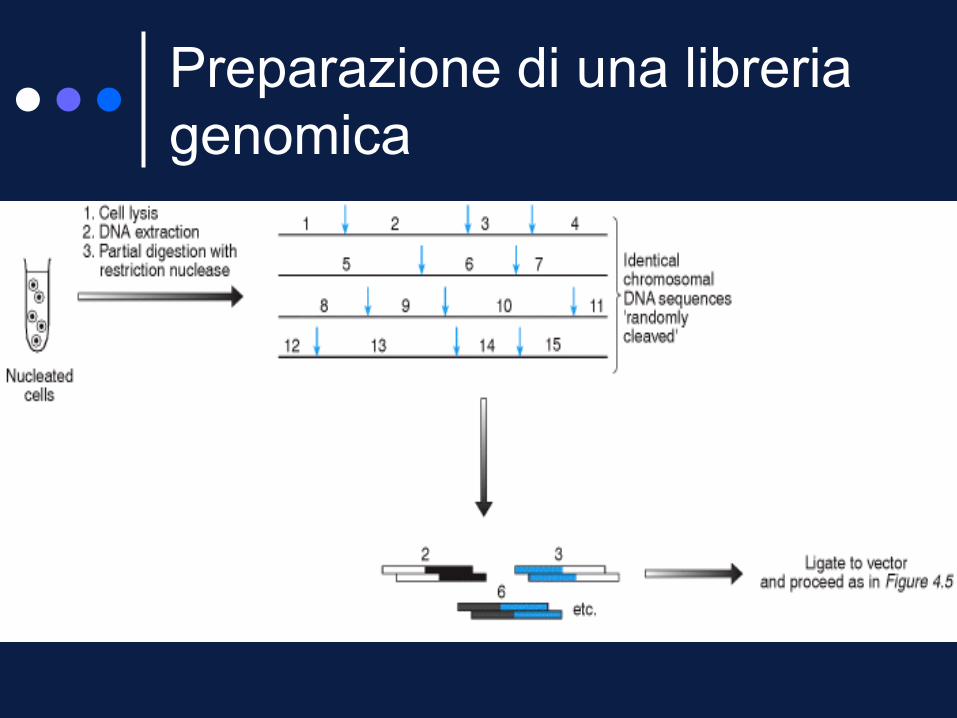
Preparazione di una libreria
genomica

Librerie di cDNA
Il materiale di partenza è l’RNA totale estratto da uno specifico tessuto o stadio di sviluppo di cui la libreria deve essere rappresentativa.
E’ possibile selezionare gli mRNA poli (A) mediante cromatografia su colonne che legano oligo(dT)
L’RNA è retrotrascritto (RNA→DNA) utilizzando specifiche DNA-polimerasi RNA dipendendenti. Si ottiene così il cDNA (una molecola di DNA complementare ad un RNA).
Per facilitare il clonaggio del cDNA vengono aggiunti degli specifici linkers, contenenti siti di restrizione che favoriscono il clonaggio.

Preparazione di una libreria
di cDNA


Saggi d’ibridazione con gli
acidi nucleici
L’ibridazione degli acidi nucleici è una tecnica largamente utilizzata in biologia molecolare per il riconoscimento di specifiche sequenze in una miscela complessa di frammenti di DNA o RNA, sfruttando la specifica complementarietà delle basi.
Le sonde utilizzate sono molecole di DNA o RNA che devono essere opportunamente marcate.
Le sonde più comunemente utilizzate possono essere a singolo/doppio filamento e di DNA o RNA.


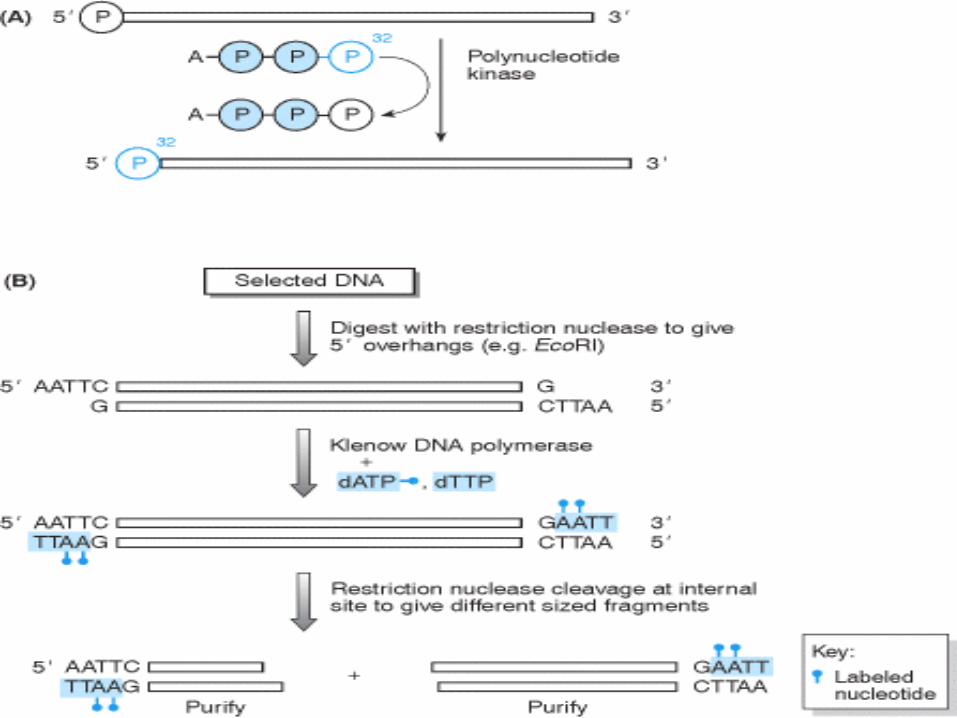
Modalità di marcatura delle
sonde
Per la marcatura delle sonde si utilizzano prevalentemente tecniche di polimerizzazione in vitro, durante le quali le molecole di DNA/RNA neosintetizzato incorporano nucleotidi opportunamente marcati (normalmente mediante radioisotopi).
Per la marcatura di sonde a DNA a doppio filamento le tecniche prevalentemente utilizzate sono:
Nick- translation
Random priming
Per la marcatura di sonde a RNA si utilizzano tecniche di trascrizione in vitro.
Per la marcatura di sonde oligonucleotidiche si utilizzano tecniche di marcatura terminale.


Marcatura di una sonda a
RNA (ribosonda)
La preparazione di una sonda a RNA si ottiene per trascrizione in vitro a partire da un frammento di DNA clonato in un vettore plasmidico.
Il vettore deve contenere, in genere a monte del MCS la sequenza di un promotore fagico. Quelle più utilizzate sono:
SP6
T3
T7
L’RNA polimerasi dello specifico batteriofago (es. SP6 RNA polimerasi) è utilizzata in presenza di nucleotidi di cui almeno uno opportunamente marcato.
E’ possibile ottenere sonde senso e antisenso

Schema per sonde a RNA

Isotopi radioattivi utilizzati
Nella marcatura di sonde a DNA/RNA si possono
utilizzare diversi isotopi radioattivi.
L’intensità del segnale autoradiografico dipende da:
Intensità della radiazione emessa dal radioisotopo.
Dal tempo di esposizione (ore, giorni, settimane).
R adioisotope H alf-life D ecay type E nergy of em ission
3H 12.4 years
- 0 .019 M eV
32P 14.3 days
- 1 .710 M eV
33P 25.5 days
- 0 .248 M eV
35S 87.4 days
- 0 .167 M eV

Marcatura non isotopica
Esistono tecniche di marcatura delle sonde che non prevedono l’utilizzo di radioisotopi.
Questi sistemi di marcatura possono essere:
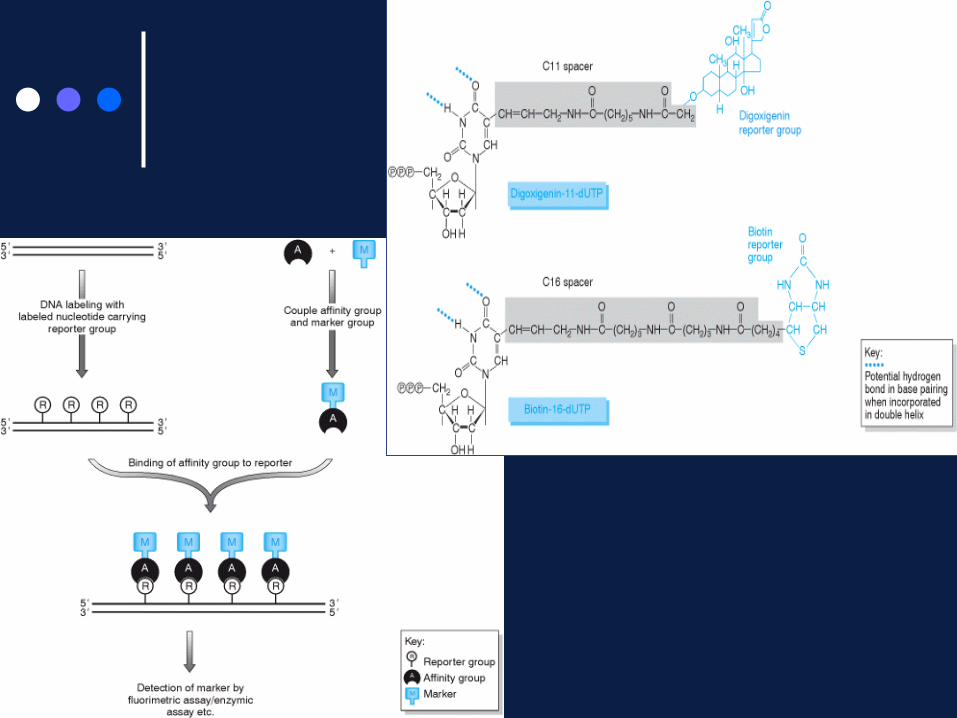
Diretti – In cui sono incorporati nucleotidi opportunamente modificati (es. contenenti fluorofori).
Indiretti – In cui nucleotidi modificati incorporano indicatori che hanno elevata affinità con molecole complessate a marcatori evidenziabili con uno specifico saggio.
• Sistema Biotina-streptavidina, in cui la sonda biotinilata lega ad elevata affinità la streptavidina marcata con un saggio colorimetrico
• Sistema della digossigenina, che sfrutta un anticorpo specifico per questa molecola