
INVECCHIAMENTO PRECOCE NELLA …dad2011.pinp.it/.../article/53/invecchiamento_precoce.pdf2 suo...
133
UNIVERSITA’ DEGLI STUDI DI PADOVA FACOLTA’ DI SCIENZE DELLA FORMAZIONE CORSO DI LAUREA IN Scienze dell’Educazione Indirizzo educatore professionale socio-sanitario TESI DI LAUREA INVECCHIAMENTO PRECOCE NELLA SINDROME DI DOWN: RUOLO DELL’EDUCATORE RELATORE: PROF. PAOLO TESSARI LAUREANDA: GAIO ILENIA MATRICOLA N° 440018/ED ANNO ACCADEMICO: 2006-2007
Transcript of INVECCHIAMENTO PRECOCE NELLA …dad2011.pinp.it/.../article/53/invecchiamento_precoce.pdf2 suo...

UNIVERSITA’ DEGLI STUDI DI PADOVA
FACOLTA’ DI SCIENZE DELLA FORMAZIONE
CORSO DI LAUREA IN
Scienze dell’Educazione
Indirizzo educatore professionale socio-sanitario
TESI DI LAUREA
INVECCHIAMENTO PRECOCE NELLASINDROME DI DOWN: RUOLO
DELL’EDUCATORE
RELATORE: PROF. PAOLO TESSARI
LAUREANDA: GAIO ILENIA
MATRICOLA N° 440018/ED
ANNO ACCADEMICO: 2006-2007


Alla mia nonna,che per prima mi ha incoraggiato
ad iniziare quest’avventura.
GRAZIE NONNA


INDICE
INTRODUZIONE....................... ................................ ................................ ................................ .....1
CAPITOLO 1 ....................... ................................ ................................ ................................ ............3
STORIA CLINICA DELLA SINDROME DI DOWN....................... ................................ ..........3
EZIOPATOGENESI....................... ................................ ................................ .............................6
DIAGNOSI....................... ................................ ................................ ................................ ...........8
TERAPIA ....................... ................................ ................................ ................................ ...........10
CAPITOLO 2 ....................... ................................ ................................ ................................ ..........13
DETERIORAMENTO COGNITIVO....................... ................................ ................................ .13
STADI DEL DETERIORAMENTO COGNITIVO ....................... ................................ ...........14
CAPITOLO 3 ....................... ................................ ................................ ................................ ..........19
QUALITÀ E ASPETTATIVA DI VTA NELLA SINDROME DI DOWN....................... ...... 19
ASPETTATIVA DI VITA....................... ................................ ................................ ..................21
CAPITOLO 4 ....................... ................................ ................................ ................................ ......... 27
DIAGNOSI PRECOCE ....................... ................................ ................................ ......................27
CAPITOLO 5 ....................... ................................ ................................ ................................ ..........35
RICERCAZIONE PER VALUTARE IL DECLINO COGNITIVO NEI SOGGETTI CON
SINDROME DI DOWN....................... ................................ ................................ .....................35
PROGETTO DEL CENTRO INTEGRATO: LA MERIDIANA ....................... ....................... 39
CAPITOLO 6 ....................... ................................ ................................ ................................ ..........49
RUOLO DELL’EDUCATORE ALL’INTERNO DL PROGETTO “LA MERIDIANA”........49
ALLEGATI ....................... ................................ ................................ ................................ .............53
ALLEGATO 1: GRIGLIA OSSERVAZIONE CENTRO ....................... ................................ .53
ALLEGATO 2: GRIGLIA OSSERVAZIONE COMUNITÁ....................... .............................54
ALLEGATO 3: GRIGLIE RIASSUNTIVE....................... ................................ ....................... 55
ALLEGATO 4: TEST....................... ................................ ................................ .........................59
BIBLIOGRAFIA ....................... ................................ ................................ ................................ ..121

VI

1
INTRODUZIONE
L’innegabile miglioramento delle condizioni e della qualità della vita delle
persone diversamente abili ha portato ad un graduale innalzamento
dell’aspettativa di vita, a titolo esemplificativo la mortalità delle persone con
sindrome di Down, che negli ultimi vent’anni, grazie alle migliorate cure generali
e al più attento trattamento dei bambini con cardiologia congenita, è sensibilmente
diminuita nei confronti di quanto avveniva 30-40 anni fa (dal 50-60% di mortalità
entro i primi dieci anni di vita siamo passati a poco meno del 20%). Tale
miglioramento permette di stimare una vita media (cioè del 50% dei nati) di 65
anni1 con un’aspettativa di vita sempre più lunga.
I bisogni e le esigenze della popolazione sono soggette a cambiamento, almeno in
funzione dell’avanzamento anagrafico; la persona infatti si trova al centro di un
processo dinamico che si attua durante tutto l’arco della vita e i risultati di questo
sviluppo dipendono fortemente anche dalle modificazioni ambientali che si
verificano con il passare del tempo che va dal micro al macro livello e che passa
dall’individuo fino ad arrivare al sistema sociale.
Uno dei dati più rilevanti della variabile tempo del processo di sviluppo personale
è legato al fatto che ad un avanzamento dell’età corrisponda un maggiore tasso di
disabilita e questo è un fattore generale nella popolazione dei paesi sviluppati.
E’ diventata oramai una priorità assoluta progettare un percorso di vita per queste
persone quando diventano anziane per evitare che la grande conquista di civiltà
raggiunta finora con l’innalzamento della vita media si traduca in una cocente
sconfitta legata all’incapacità di gestire con nuove risposte le sfide emergenti.
Bisogna innanzitutto evidenziare come stia emergendo in modo notevole nelle
letteratura scientifica l’attenzione per il problema dell’innestarsi del problema
dell’invecchiamento su persone affette da deficit intellettivo.
I casi più eclatanti sono legati all’elevata incidenza di persone affette da sindrome
di Down con una degenerazione neurologica del sistema nervoso centrale che ha il
1Contardi A., Un giorno dopo l’altro. Bambini e adulti con la sindrome di Down, Edizioni Guaraldi, Rimini,
1996

2
suo esordio già a partire dai 35 anni2, poiché la sindrome di Down rappresenta un
modello di invecchiamento celebrale precoce, mentre nelle altre forme
patologiche questo decadimento avviene negli anni successivi.
Questo significa che si manifestano dei repenti e drastici cali prestazioni a livello
di tutte le prassie e delle autonomie personali, il degrado pare imputabile alla
maggiore incidenza del morbo di Alzheimer o forme similari3 nelle persone affette
da celebropatie4e all’insorgenza di diverse forme di demenza senile ben prima
della normale età genetica.
2Cfr. Frigerio E., Gallucci M., Folin M., Similarities in the decline of visual-spatial and attentional abilities
between Downs syndrome and Alzheimer’s disease, Abstract dal Congresso Internazionale di San Marino, 9-11 maggio 2002
3 Di Luca P., Pastorino L. , Cattabeni F., Zanardi R., Scarone S., Racagni G., Smeraldi E. and Perenz J.,Abnormal Pattern of Platelet APP Isoforms in Alzheimer Disease and Down Sindome, Archives ofNeurology, 1996, ol.53, pag. 1162-1166.4 Biguera s., Pati T., Vigolo S.,Forlin M., Analysis of the impact of some Alzheimer’s disease risk factors inDown’s sindrome. Abstract dal Congresso Intenazionale di San Marino 9-11 maggio 2002.

3
CAPITOLO 1
STORIA CLINICA DELLA SINDROME DI DOWN
Sembra incredibile che una condizione relativamente comune come quella della
Sindrome di Down, con caratteristiche cliniche così tipiche, possa essere stata
identificata come entità specifica poco più di un secolo fa.
La Sindrome di Down, viene descritta per la prima volta nel 1838 quando
Esquirol individua particolari caratteristiche somatiche tipiche di questi soggetti:
fessura palpebrale obliqua, radice del naso depressa, testa piccola e bassa statura.
Si potrebbe credere, quindi, che questa sindrome sia relativamente giovane, ma
alcuni manufatti, addirittura antecedenti l’era Cristiana, sembrano spostarne la
comparsa molto più indietro nel tempo. Figure votive in pietra e in argilla risalenti
a oltre 3000 anni fa, attribuite alla cultura degli “Olmec” abitanti del Messico e
dell’America Centrale, presentano caratteristiche somatiche tipiche della sindrome
di Down. Inoltre durante gli scavi archeologici presso Saxon è stato ritrovato un
cranio di un bambino Down vissuto nel VII secolo.
Tra i primi studiosi a ricercare il significato e la causa della variazione specifica
che si manifestava nelle razze fu J.F. Blumenbach nel 1776 nel testo De generis
umani variegate nativa liber5. Blumenbach nella sua tesi aveva classificato,
attraverso l’applicazione di esami morfologici, una varietà di razze tra le quali la
caucasica, la mongolica, l’etiopica, l’americana e la malese. Tale classificazione è
stata considerata il punto di partenza della moderna antropologia. Fu Longdon
Down, medico inglese, nel 1866 a sostenere la necessità di strumenti teorici
efficaci affinché, di fronte al soggetto in difficoltà, lo specialista potesse rilasciare
non “una cauta diagnosi e una prognosi altrettanto cauta”6 ma un autorevole
giudizio per poter avviare una terapia adeguata. Ripartendo dalla classificazione
dei deboli di mente, Blemenbach pone di nuovo l’accento sulla tipologia
mongola. Down dà alla classificazione un fine pratico per poter fare più precise
5 Trad. it., Cura mentale, igene ed educazione degli idioti e di altri fanciulli ritardati, Armando,Roma 1970, pp.5406 John Langdon Down, “Observations on an ethnic classication of idiots” in “Clinical Lectures andRepoits by the Medical Surcical Staff of the London Hospital”, London, J. Churchill & sons, 1866vol. III, p. 260

4
distinzione nella vasta gamma di condizioni patologiche aventi origini e gradi di
gravità diverse. In “Observation on an ethnic classification of idiots” del 1866
Down scrive: “della grande famiglia mongola è che io desidero in questo scritto,
richiamare una speciale attenzione.[…] La capigliatura non è di colore nero, come
nel mongolo reale, ma di colore bruno, liscia e rada. Le guance sono rotonde ed
estese ai lati. Gli occhi sono posti obliquamente, e sono distanziati più del
normale l’uno dall’altro. La fessura delle palpebre è molto stretta. La fronte è
corrugata trasversalmente dal costante movimento dei muscoli che alzano le
palpebre derivano dal muscolo occipito-frontale durante l’apertura degli occhi. Le
labbra sono grandi e ispessite da fenditure trasversali. La lingua è lunga,
ingrossata e assai ruvida. Il naso è piccolo. La pelle possiede un sottile color giallo
sporco, è poco elastica, dando l’impressione di essere troppo abbondante rispetto
al corpo.[…] Il tipo mongoliano di idiozia è presente nel più del dieci per cento
dei casi che mi sono stati presentati. Sono sempre idioti congeniti, e non
dipendono mai da incidenti dopo la vita uterina. Sono per la maggior parte esempi
di degenerazione derivanti dalla tubercolosi dei genitori. Vi sono casi migliorati
moltissimo attraverso un appropriato apprendimento. Richiedono cibo con un alto
contenuto di azoto e con una considerevole quantità di sostanze oleaginose. […]
Sono usualmente abili nel parlare; il linguaggio è stretto e indistinto, ma possono
essere guidati egregiamente ad uno schema ben diretto di ginnastica linguale.[…]
Dall’allenamento sistematico può essere ottenuto un considerevole effetto di
progresso.”7
Down conia i termini di “Mongolian type” o “Mongols” e nasce la definizione di
mongolismo; tali termini vengono assunti anche nel campo scientifico e adottati
per più di un secolo8.
Dobbiamo aspettare fino agli anni trenta, con Bleyer e Waardenburg, perché
comincino a svilupparsi i presupposti che permettano di formulare l’ipotesi che il
mongolismo possa derivare da una aberrazione cromosomica. Per tutta la prima
metà del ‘900, l’analisi dei cromosomi e il loro conteggio risultano spesso
7 Ivi, pag. 260-2618 Anna Nesi, Dal mongolismo alla trisomia 21, in “Pedagogia clinica”, luglio-dicembre 2000, n°2pag. 15

5
imprecisi date le molte difficoltà tecniche nel fissaggio dei preparati che causano
l’aggregazione dei cromosomi.
Nel gennaio del 1959, grazie all’affinamento di nuove tecniche citologiche,
Turpin e Lejeune accertarono che l’anomalia del mongolismo è dovuta alla
presenza di un cromosoma in più. Nel rendiconto depositato all’accademia delle
Scienze di Parigi gli studiosi riportano “Presso nove bambini mongoloidi lo
studio della mitosi dei fibroblasti di recente coltura ci ha permesso di constatare
regolarmente la presenza di 47 cromosomi. […] L’analisi del margine
cromosomico delle cellule “perfette” rivela presso i maschi mongoloidi la
presenza di 6 piccoli telocentrici e presso le femmine mongoloidi di 5 piccoli
telocentrici. Le cellule “perfette” di individui non-mongoloidi non presentano mai
queste caratteristiche, ci sembra legittimo concludere che esiste presso i
mongoloidi un piccolo telocentrico soprannumerario tenuto conto della cifra
anomala di 47”9.
Infine nel 1961, nel periodico, “Lancet” viene pubblicata una lettera, sottoscritta
da 19 studiosi, con la quale si chiedeva l’abolizione del termine “mongolismo” e
l’adozione del termine “Sindrome di Down” o “trisomia 21”, motivando tale
cambiamento col fatto che agli studiosi cinesi e giapponesi viene imposto l’uso di
un termine per loro piuttosto imbarazzante.
La sua causa è a tutt’oggi sconosciuta mentre si sa che la malformazione
congenita è dovuta ad un’anomalia cromosomica: 3 cromosomi 21 anziché 2,
derivante da una non-disgiunzione meiotica o, più raramente, da una mitosi post-
zigotica. La modalità più comune per la nascita di un bambino down e' una
trisomia 21 libera, o trisomia da non disgiunzione, e il cromosoma in più fluttua
libero. La trisomia da traslocazione si verifica invece quando il cromosoma 21 “in
più” è legato a un altro cromosoma. Una forma ancora più rara di Sindrome Down
è rappresentata dalla trisomia a mosaico, in cui nello stesso individuo sono
presenti cellule di due tipi diversi e cioè alcune con 46 e altre con 47 cromosomi.
Le caratteristiche fisiche più comuni sono: la forma degli occhi, che sono piegati
verso l’alto; alcune piccole pieghe di pelle all’interno dell’occhio; un naso piccolo
5 J.Lejeune, M. Gautier, M. R. Turpin, « Etude des chromosomes somatiques de neuf enfantsmongoliens », in : « Comptes endus hebdomadaires des séances de Académie des Sciences »,Paris, t. 248, n. 11 p.1721-1722.

6
e qualche volta un po' “schiacciato”; le orecchie minute, che possono essere
leggermente a sventola e situate in basso; la bocca piccola.
EZIOPATOGENESI10
La sindrome di Down è una condizione genetica caratterizzata dalla presenza di
un cromosoma in più nelle cellule di chi ne è portatore: invece di 46 cromosomi
nel nucleo di ogni cellula ne sono presenti 47, vi è cioè un cromosoma 21 in più;
da qui anche il termine trisomia 21. Genetico non vuol dire ereditario, infatti nel
98 % dei casi la sindrome di Down non è ereditaria. La conseguenza di questa
alterazione cromosomica è un handicap caratterizzato da un variabile grado.
Esistono tre tipi di anomalie cromosomiche nella sindrome di Down, il loro
effetto finale è comunque identico: nelle cellule dei vari organi i geni del
cromosoma 21 sono in triplice dose.Ci sono tre tipi di sindrome Down:
La trisomia 21 libera, la più comune, si verifica infatti nel 95 % dei casi. Il
cromosoma in più è presente nello sperma o nell’uovo, oppure nella prima
divisione cellulare e, come conseguenza, ogni cellula che si forma
successivamente avrà tre cromosomi alla coppia 21.
La trisomia 21 da traslocazione, è invece molto rara e si verifica dal 2 al 3 % dei
casi. In questo tipo di trisomia, una parte del cromosoma 21 si spezza durante la
divisione cellulare, Le possibilità di prevenzione della degenerazione cerebrale
pertanto sono basate sugli antiossidanti (vitamina E, selegilina ecc.) ed in futuro,
forse, l’immunizzazione con beta-proteina che si è osservato negli animali essere
capace di bloccare l’accumulo di amiloide trasloca e si attacca a un altro
cromosoma, di solito al cromosoma 14 oppure all’altro cromosoma 21. In ognuno
dei casi è presente del materiale genetico in più. Circa due terzi delle traslocazioni
avvengono spontaneamente durante la fertilizzazione, mentre un terzo è ereditato
da uno dei due genitori. In questo caso abbiamo l’unica forma di sindrome Down
10 Rosa Ferri Amedeo spagnolo, La sindrome di Down in Il pensiero scientifico, Roma 1989Richard Newton, Conoscere e capire la Sindrome di Down in Tea Milano 1998Elena Zupi, La sindrome di Down: note epidemiologiche e cliniche, Il pensiero scientifico, Roma1986Jean Rondal,, Juan Perera, Lynn Nadel, La sindrome di Down: conoscenze attuali e prospettive,Erip Pordenone 2003

7
che è legata a un aspetto genetico del padre o della madre. Il genitore che ne è il
portatore è normale, ma due dei suoi cromosomi sono uniti, in modo che il
numero totale dei suoi cromosomi è di 45 invece che 46. È importante individuare
attraverso il cariotipo del bambino la presenza o meno della trisomia da
traslocazione, perché se uno dei genitori è il portatore, la possibilità che nascano
in quella famiglia altri bambini con sindrome Down è maggiore. È quindi
consigliabile che anche i genitori, gli altri figli e i consanguinei del genitore
portatore si facciano fare la mappa cromosomica.
Il mosaicismo, che è la forma meno comune: si verifica intorno al 2 % dei casi. In
questa forma di trisomia 21 la divisione cellulare avviene in modo difettoso dopo
la fertilizzazione, durante la seconda o la terza divisione, o durante quelle
successive. Come conseguenza, non tutte le cellule dell’embrione che si sta
formando conterranno il cromosoma in più. Il bambino potrà avere meno aspetti
legati alla sindrome, sia da un punto di vista fisico che mentale. Ma questo varierà
da bambino a bambino, e a seconda della sua percentuale di cellule trisomiche.
La sindrome Down esiste in ogni paese, in ogni razza e in ogni classe sociale. Non
è stato trovato dagli studiosi alcun legame tra questo tipo di anomalia
cromosomica e fattori ambientali. Né il tipo di dieta, né malattie particolari, né il
clima di diverse zone geografiche sono collegabili a una maggiore o minore
incidenza della sindrome.
Non conosciamo affatto quali siano le cause che determinano le anomalie
cromosomiche, tuttavia sappiamo che:
a) le anomalie cromosomiche, soprattutto le trisomie, sono un evento abbastanza
frequente che interessa circa il 9 % di tutti i concepimenti (alla nascita però solo lo
0,6 % dei nati presenta un'anomalia cromosomica a causa dell'elevatissima quota
di embrioni che va incontro ad un aborto spontaneo);
b) l'incidenza delle anomalie cromosomiche in generale, e quelle della trisomia 21
in particolare, è assolutamente costante nelle diverse popolazioni, nel tempo e
nello spazio;
c) tutte le possibili ipotesi eziologiche fino ad oggi formulabili (agenti chimici,
radiazioni ionizzanti, infezioni virali, alterazioni metaboliche o endocrine
materne) non sono state mai avvalorate dalle molte ricerche condotte.

8
In definitiva si ritiene che l'insorgenza delle anomalie cromosomiche sia un
fenomeno "naturale", in qualche modo legato alla fisiologia della riproduzione
umana, e anche molto frequente.
DIAGNOSI
La sindrome può essere identificata attraverso lo studio della mappa cromosomica
di una persona. L’analisi citogenetica dunque è l’unico esame che permette di
definire la diagnosi. Essa viene eseguita in epoca postatale su cultura di linfociti
del sangue periferico, o in epoca prenatale, nel I o nel II trimestre di gravidanza,
su culture di cellule derivate dai villi coriali o dal liquido amniotico.
La diagnosi clinica può essere posta sulla base dei tratti dismorfici, non tutti
contemporaneamente presenti, ma che nel loro insieme caratterizzano il fenotipo
Down. È importante però sottolineare che i singoli segni morfologici non sono
assolutamente patognomonici: ciascuno di essi può essere presente anche in
soggetti normali; pertanto la trisomia 21 può essere definita con certezza solo
dall’indagine citogenetica.
La consulenza genetica si basa sulla valutazione del rischio procreativo della
singola coppia. Per la sindrome di Down non è attuabile una prevenzione primaria
ma, attraverso la diagnosi prenatale, è possibile diagnosticare il cariotipo fetale
nelle gravitanze ad alto rischio.
L’età materna avanzata costituisce il principale fattore predisponente la non
disgiunzione del cromosoma 21. La possibilità di avere un bambino con sindrome
Down infatti aumenta con l’aumentare dell’età della madre e cresce in modo
evidente dopo i 35 anni.
Numerose indagini epidemiologiche, eseguite su nati vivi, nati morti, aborti
spontanei e i feti nel I e II trimestri di gravidanza confermano in modo univoco
che l’incidenza della sindrome è strettamente dipendente dall’età materna
avanzata, come riportato nella seguente tabella:
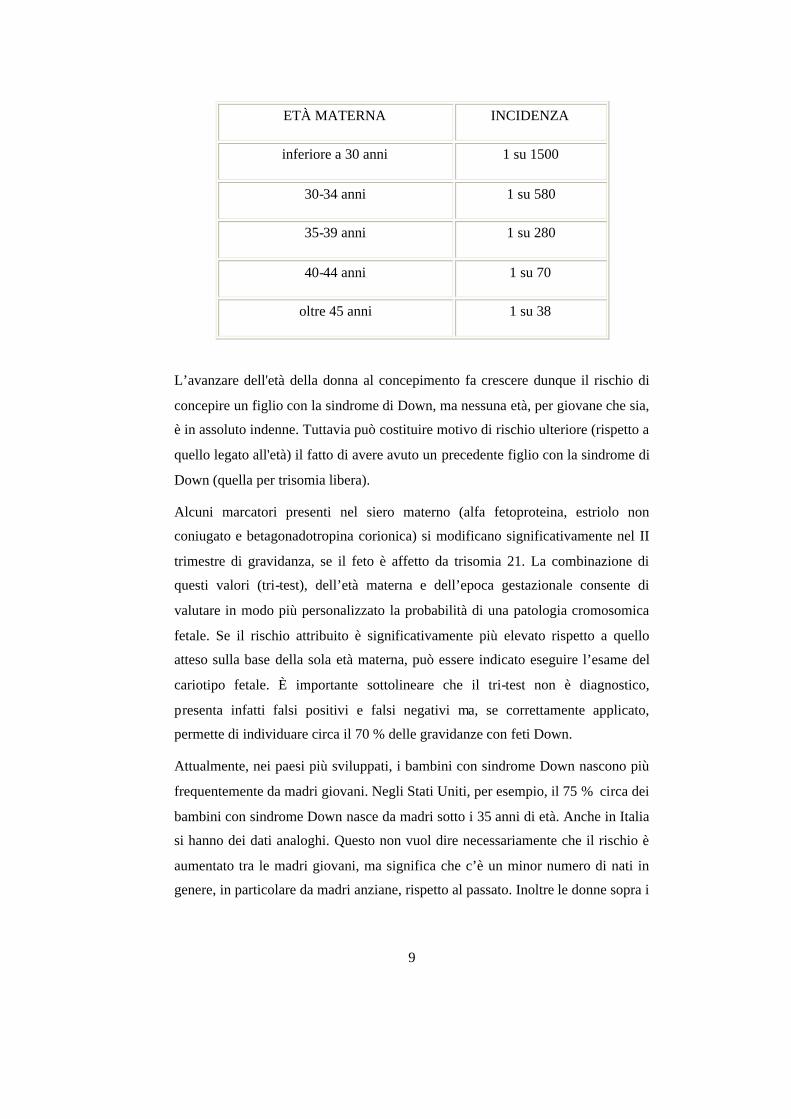
9
ETÀ MATERNA INCIDENZA
inferiore a 30 anni 1 su 1500
30-34 anni 1 su 580
35-39 anni 1 su 280
40-44 anni 1 su 70
oltre 45 anni 1 su 38
L’avanzare dell'età della donna al concepimento fa crescere dunque il rischio di
concepire un figlio con la sindrome di Down, ma nessuna età, per giovane che sia,
è in assoluto indenne. Tuttavia può costituire motivo di rischio ulteriore (rispetto a
quello legato all'età) il fatto di avere avuto un precedente figlio con la sindrome di
Down (quella per trisomia libera).
Alcuni marcatori presenti nel siero materno (alfa fetoproteina, estriolo non
coniugato e betagonadotropina corionica) si modificano significativamente nel II
trimestre di gravidanza, se il feto è affetto da trisomia 21. La combinazione di
questi valori (tri-test), dell’età materna e dell’epoca gestazionale consente di
valutare in modo più personalizzato la probabilità di una patologia cromosomica
fetale. Se il rischio attribuito è significativamente più elevato rispetto a quello
atteso sulla base della sola età materna, può essere indicato eseguire l’esame del
cariotipo fetale. È importante sottolineare che il tri-test non è diagnostico,
presenta infatti falsi positivi e falsi negativi ma, se correttamente applicato,
permette di individuare circa il 70 % delle gravidanze con feti Down.
Attualmente, nei paesi più sviluppati, i bambini con sindrome Down nascono più
frequentemente da madri giovani. Negli Stati Uniti, per esempio, il 75 % circa dei
bambini con sindrome Down nasce da madri sotto i 35 anni di età. Anche in Italia
si hanno dei dati analoghi. Questo non vuol dire necessariamente che il rischio è
aumentato tra le madri giovani, ma significa che c’è un minor numero di nati in
genere, in particolare da madri anziane, rispetto al passato. Inoltre le donne sopra i

10
35 anni di età si sottopongono sempre più frequentemente ad esami prenatali e,
nel caso che nel feto si evidenzi un’anomalia cromosomica, possono decidere di
interrompere la gravidanza.
In Italia un bambino su 1000 nasce con la sindrome Down, ossia ne nascono
approssimativamente due al giorno. Non sappiamo esattamente quante persone
con sindrome di Down esistano attualmente in Italia, ma si presume che siano
49.000 circa.
TERAPIA
Non esiste alcun trattamento farmacologico. L’unico terapia che permette di
ottenere uno sviluppo armonico e buon inserimento scolastico, sociale e
lavorativo è quella riabilitativa. Questo trattamento deve essere iniziato fin dai
primi mesi di vita. I primi tre anni infatti sono molto significativi per quanto
concerne la successiva organizzazione delle abilità cognitive e di socializzazione
delle persone con sindrome di Down.
Lo sviluppo del bambino Down avviene con un certo ritardo, ma secondo le stesse
tappe dei bambini normali.
I bambini Down crescendo possono raggiungere, sia pure con tempi più lunghi,
conquiste simili a quelle dei bambini normali: cammineranno, inizieranno a
parlare, a correre a giocare. Rimane invece comune a tutti un variabile grado di
ritardo mentale che si manifesta anche nella difficoltà di linguaggio frequente tra
le persone Down.
Dal punto di vista riabilitativo non si tratta per loro di compensare o recuperare
una particolare funzione, quanto di organizzare un intervento educativo globale
che favorisca la crescita e lo sviluppo del bambino in una interazione dinamica tra
le sue potenzialità e l'ambiente circostante. È importante inoltre ricordare che ogni
bambino è diverso dall'altro e necessita quindi di interventi che rispettino la
propria individualità e i propri tempi.
La sindrome di Down è una delle principali cause di ritardo mentale associato a
delle caratteristiche facciali e fisiche distintive e da anomalie congenite al cuore e
al tratto gastrointestinale; comporta un maggior rischio di leucemia, deficit del

11
sistema immunitario e del morbo di Alzheimer e il sistema immunitario presenta
delle deficienze.

12

13
CAPITOLO 2
DETERIORAMENTO COGNITIVO
L’aspettativa di vita di un soggetto con Sindrome di Down è minore di quella del
resto della popolazione, anche se negli ultimi anni vi è stato un aumento dell’età
media. Questa nuova spettanza di vita fa emergere nuove problematiche, fino ad
ora mai prese in considerazione, collegate alla vecchiaia, come disfunzioni
tiroidee, autoimmunità e lesioni neuropatologiche tipiche del morbo di Alzheimer.
Infatti, nei soggetti con Sindrome di Down, a partire dai 40 anni di età, si
riscontrano problemi neuropsichiatrici, che diventano dominanti con l’età,
comprese le crisi epilettiche. Negli adulti si osserva un costante declino
dell’intelligenza. Con l’avanzare dell’età, ma sempre prima di quanto avviene
nelle persone normali, si può verificare una riduzione della capacità di elaborare il
pensiero, in particolare nel ragionamento astratto e nelle performance logica.
Caratteristica dell’invecchiamento nella Sindrome di Down è anche la demenza,
che presenta delle sorprendenti analogie con il morbo di Alzheimer e si manifesta
in diversi soggetti dopo i cinquant’anni. Dal punto di vista clinico, le persone
colpite presentano un deterioramento delle risposte mentali ed emotive, apatia o
eccitamento, irritabilità, un temperamento collerico, la perdita del vocabolario
precedentemente acquisito e una diminuzione delle abitudini personali e di
pulizia. La progressione è spesso molto rapida. Le crisi epilettiche possono essere
un sintomo precoce del morbo di Alzheimer.
Il deterioramento cognitivo ha inizio con l’insorgere di particolari disturbi, che
non sono da ritenere fatti occasionali, ma che si pongono stabilmente in contrasto
con il comportamento precedente.
Le funzioni cognitive ci permettono di mantenere nel tempo le informazioni sia
quelle a breve termine (memoria a breve termine) che quelle a lungo termine
(memoria a lungo termine). Esse inoltre ci permettono la padronanza del mondo
interno, dell’ambiente e delle reciproche interazioni. Infine le funzioni cognitive
hanno un notevole impatto su aspetti del funzionamento psichico quali l’affettività
e la personalità. Il deterioramento cognitivo della malattia porta al deterioramento

14
di tutte le funzioni, partendo dalle più complesse fino alle più semplici. Le prime
alterazioni riguardano in genere la memoria per gli eventi recenti, spesso
accompagnate da da disturbi del comportamento e dell’orientamento spazio-
temporale. La progressione è lenta e graduale e può prolungarsi per molti anni.
Possono essere presenti disturbi cognitivi, quali afasia (disturbo del linguaggio),
amnesia (disturbo della memoria), agnosi (disturbo della
percezione/riconoscimento), aprassia (disturbo di attività motorie specifiche) ed
infine disturbi della funzione esecutiva, che limitano così la capacità della persona
ad impegnarsi nelle attività necessarie per una funzionalià autonoma e per una
buona qualità della vita. Nelle fasi più avanzate i sintomi si fanno più evidenti: il
paziente può manifestare un aumento generalizzato del tono muscolare
accompagnato da un impaccio motorio e da una marcia a piccoli passi; i disturbi
afasici e della memoria peggiorano e si possono verificare crisi epilettiche. Il
processo degenerativo e irreversibile e alla fine il paziente muore a causa di
malattie intercorrenti in uno stato di totale mancanza di autosufficienza.
STADI DEL DETERIORAMENTO COGNITIVO
Sono stati identificati sette stadi attraverso i quali avviene il deterioramento
cognitivo nei soggetti affetti dal morbo di Alzheimer. Questo percorso di
decadenza si presenta anche nei soggetti con la sindrome di Down:
STADIO 1 e STADIO 2: in questa fase iniziano i primi difetti di memoria: il
paziente non ricorda dove vengono messi gli oggetti familiari o i nomi, comuni o
propri, in precedenza noti. Non si manifestano ancora lacune sul lavoro o nelle
situazioni sociali.
STADIO 3: durante questo periodo insorgono le prime lacune ben evidenti; ad
esempio il soggetto si perde trovandosi in luoghi non familiari, ha sempre
maggiori difficoltà nel trovare nomi e parole, smarrisce facilmente oggetti di
valore e, nel contesto lavorativo, ha sempre un minor rendimento.
STADIO 4: in questa fase è ormai evidente il disturbo di memoria legato ad una
crescente mancanza di attenzione. La capacità del paziente nella gestione del
denaro disunisce notevolmente, così come si riducono le capacità nello svolgere

15
compiti complessi associati alla rinuncia di situazioni impegnative. A questi
sintomi si associa spesso un‘ansia lieve e moderata e cambiamenti della
personalità, quali apatia, appiattimento emozionale, irascibilità.
STADIO 5: in questo periodo il soggetto può, occasionalmente, non ricordare il
nome del famigliare che si prende cura di lui, il proprio indirizzo o numero di
telefono, essere ignaro degli avvenimenti recenti e delle esperienze della propria
vita, dal momento che ha perso la memoria a breve termine. Quasi sempre il
paziente manifesta disorientamento spazio-temporale e può avere difficoltà a
contare all’indietro. Viene mantenuto, invece, il ricordo di fatti importanti
riguardanti se stessi a gli altri. Il malato inoltre, presenta spesso qualche difficoltà
a (s)-vestirsi, ma completa autosufficienza nella cura della persona e nel mangiare.
STADIO 6: in questa fase il paziente necessita di aiuto in alcune attività della vita
quotidiana come la minzione o l’evacuazione e negli spostamenti fuori casa. Nel
soggetto inoltre, si riscontrano frequenti disturbi del ritmo circadiano (sindrome
del tramonto, inversione sonno-veglia). Sempre in questo stadio possono essere
presenti cambiamenti della personalità: sintomi psicotici (ad esempio considera il
coniuge un impostore o può parlare con persone immaginarie), sintomi ossessivi
(ad esempio attività motoria afinalistica ripetitiva), sintomi di ansia e di agitazione
con occasionali episodi di aggressività fisica inusuale, abulia cognitiva (ad
esempio perdita di forza di volontà perché incapace di tenere a mente le iniziative
pensate sufficientemente a lungo per completare l’azione).
STADIO 7: quest’ultimo stadio è caratterizzato dalla perdita delle competenze
linguistiche; nella fase iniziale di questo stadio il paziente si esprime ancora con
qualche parola o con semplici frasi, ma l’eloquio è molto limitato;
successivamente scompare completamente la capacità di linguaggio articolato.
L’incontinenza doppia diventa permanente e il paziente richiede assistenza anche
nell’igiene e nell’alimentazione. Le capacità psicomotorie di base (come la
semplice deambulazione) sono perse con il progredire della malattia. In questa
fase il cervello sembra “sconnesso” dal proprio corpo.
Le cause sottostanti al deterioramento cognitivo e al successivo insorgere del
morbo di Alzheimer sono ancora sconosciute. Tuttavia a questa patologia sono
state associate delle particolari modificazioni celebrali. Tra queste troviamo la

16
globale atrofia ( una riduzione della massa celebrale: come vediamo nella fig.1
nell’immagine più a destra la massa celebrale più ritirata rispetto alla fig. di
sinistra), la perdita di neuroni, la perdita di sinapsi, grovigli neurofibrillari,
l’angiopatia amiloide e la formazione di placche β-amiloide.
Figura 1. Rappresentazione dell’atrofia cerebrale
Tra le numerose modificazioni celebrali riscontrabili nel morbo di Alzheimer vi
sono le placche senili, formazioni extracellulari nella cui parte centrale è
depositata la proteina amiloide e nella periferia vi sono detriti assonali. Il
cromosoma che codifica questa proteina è il cromosoma 21 e i grovigli
neurofibrillari sono attualmente impiegati come caratteristico elemento
diagnostico. Nei soggetti con Sindrome di Down, sin dall’età di 10 anni si può
riscontrare nel Sistema Nervoso Centrale la formazione di aggregati di questa
proteina; inoltre tutti i pazienti oltre i 35 anni sviluppano un quadro istopatologico
tipico della Demenza di Alzheimer. I depositi della proteina β-amiloide
costituiscono la principale modificazione patologica osservata nel cervello degli
adulti con sindrome di Down una volta raggiunti i trent’anni. Da un punto di vista
quantitativo, gli addensamenti della proteina β-amiloide nel cervello sono

17
chiaramente superiori ai livelli tipicamente riscontrati nel morbo di Alzheimer
senza sindrome di Down, e alcuni studiosi hanno osservato che gli addensamenti
della proteina β-amiloide sono significativamente associati alla demenza in questi
pazienti11.
11 Cummings B., Pike C., Shanakle R., Cotman C. (1996). Β-amyloid depositino and othermeasure of neuropathology predict cognitive status in Alzheimer disease. Neurobiology of Aging17, 921-933

18

19
CAPITOLO 3
QUALITÀ E ASPETTATIVA DI VTA NELLA SINDROME DI DOWN
Nei paesi sviluppati, il concetto di qualità della vita come parametro importante
nella programmazione e nella valutazione dei servizi per persone disabili è stato
utilizzato e studiato per la prima volta solo dieci anni fa12. Nei paesi in via di
sviluppo, è praticamente impossibile parlare di qualità della vita quando troppo
frequentemente le preoccupazioni si focalizzano sulla sopravvivenza quotidiana,
sulla lotta per il riconoscimento dei diritti umani e sull’ottenimento delle
fondamentali cure sanitarie, dell’educazione, della sicurezza personale, del lavoro
e dei mezzi di sostentamento quotidiani.13
Il termine “qualità della vita” è stato progressivamente introdotto nel mondo delle
persone con sindrome di Down a causa di tre fattori principali:
1. applicazione del principio di normalizzazione e la sua conseguenza, che è
l’inserimento;
2. una più lunga aspettativa di vita per le persone con la sindrome di Down;
3. i progressi nella conoscenza scientifica sull’identità del cromosoma extra
21 e sulle conseguenze specifiche che derivano dalla sua presenza.
Parlare di qualità della vita non avrebbe avuto senso cinquant’anni fa, quando
l’aspettativa di vita per le persone con la sindrome di Down non era superiore ai
ventitrè anni, e ci si concentrava prevalentemente sull’affrontare la quotidianità .
Al giorno d’oggi, nei paesi sviluppati, la maggioranza delle persone con sindrome
di Down è costituita da giovani o da adulti che hanno avuto cura della propria
salute, hanno beneficiato di precoci programmi di attenzione, hanno frequentato
scuole inclusive e hanno affrontato la vita adulta con gli stessi desideri e le stesse
aspirazioni di qualsiasi giovane persona della loro età14. Questo è il motivo per cui
12 Goode D.,Quality of life:Perspective and Issues,American, Association on Mental Retardation,Washington 1990, pp.41-58.13 Fabian E. Using quality of life indicator in rehabilitation program evaluation, RehabilitationCounseling Bulletin 34 (4),1991,pp.334-35614 Steele J. , Epidemiology: incidence, prevalence and size of the Down sindrome population, inB.Stratford e P.Gunn (Eds.), New approaches to Down syndrome, London: Lussel 1996

20
oggi ha senso parlare della qualità della vita alla luce di un obiettivo
fondamentale: il raggiungimento della massima autonomia personale.
Nella nostra società siamo oggi di fronte alla sfida della vita da adulto che il
soggetto con sindrome di Down si trova ad affrontare. Non appena sono stati
soddisfatti i bisogni sanitari di base è necessario per la società sviluppare dei
servizi educativi e sociali rivolti, gradualmente, alla vita adulta dei soggetti.
Perciò è necessario integrare i servizi già esistenti con nuove proposte educative
volte al miglioramento della qualità della vita.
E’ importante essere consapevoli del fatto che efficaci programmi per la qualità
della vita delle persone con la sindrome di Down devono avere una prospettiva
che contempli l’intera vita dell’individuo e specialmente quella dell’adulto.
Questa cosa non si può improvvisare perché non si può chiedere ad un adulto di
fare ciò che questi non ha avuto la possibilità di fare da bambino o da ragazzo;
quello che si acquisisce durante le prime fai della vita determina, fino ad un certo
grado, quello che succede in seguito. Ad esempio se un bambino è stato
superprotetto durante i primi anni di vita e non ha beneficiato di una vasta gamma
di stimolazioni, quel bambino raggiungerà, da adulto, più bassi livelli di
adattamento e di autonomia15.
Una cosa è chiara: con migliori opportunità per esprimere i loro desideri e le loro
preferenze, e con l’abilità di prendere delle decisioni in un ambiente normalizzato,
le persone con sindrome di Down sono più preparate per la vita da adulti.
L’opportunità di scegliere è un criterio fondamentale nei modelli della qualità
della vita.16
La realtà è che oggi, nei paesi industrializzati, i validi programmi relativi alla
qualità della vita per le persone con la sindrome di Down stanno creando una
situazione in cui essi godono di una buona salute e di una vita più lunga. La realtà
è che queste persone hanno un riconoscimento sociale, vengono educate in una
scuola, vengono formate per il un lavoro, vivono integrate nella comunità, sono in
15 Perera J. , Social and laboru integration for people with Down’s syndrome, in J.A.Rondal,J.Perera, L.Nadel e A. Comblain (Eds.), Down’s syndrome: Psychological, psychobiological andsocio – educational perspectives, London 1997, pp. 219-233.16 Brown R.I., Quality of life for people with disabilities, Models, Research and Practice,Chltenham: Stantley Thones 1997
21
grado di autotutelarsi e hanno i propri progetti per il futuro. Nei paesi in via di
sviluppo, dove molto di questo deve essere ancora fatto, è importante che essi
abbiano un chiaro modello di sviluppo, degli obbiettivi da raggiungere e un
sostegno incondizionato dalle società più avanzate.
ASPETTATIVA DI VITA
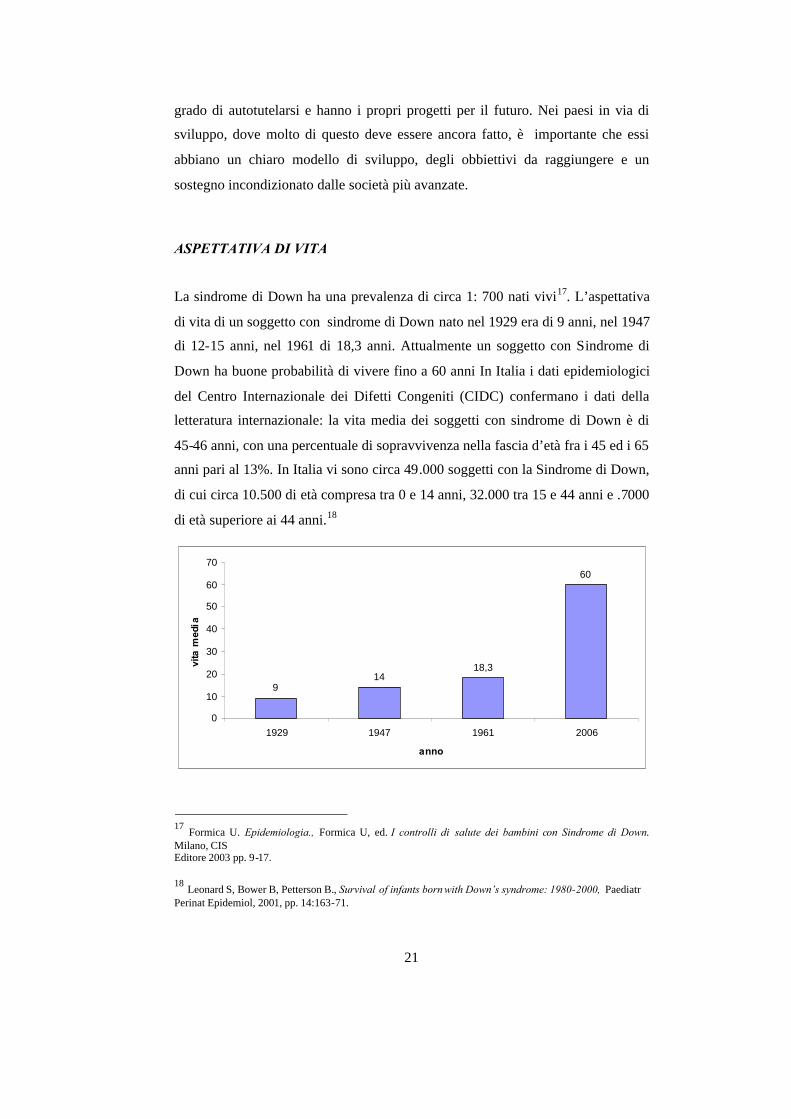
La sindrome di Down ha una prevalenza di circa 1: 700 nati vivi17. L’aspettativa
di vita di un soggetto con sindrome di Down nato nel 1929 era di 9 anni, nel 1947
di 12-15 anni, nel 1961 di 18,3 anni. Attualmente un soggetto con Sindrome di
Down ha buone probabilità di vivere fino a 60 anni In Italia i dati epidemiologici
del Centro Internazionale dei Difetti Congeniti (CIDC) confermano i dati della
letteratura internazionale: la vita media dei soggetti con sindrome di Down è di
45-46 anni, con una percentuale di sopravvivenza nella fascia d’età fra i 45 ed i 65
anni pari al 13%. In Italia vi sono circa 49.000 soggetti con la Sindrome di Down,
di cui circa 10.500 di età compresa tra 0 e 14 anni, 32.000 tra 15 e 44 anni e .7000
di età superiore ai 44 anni.18
914
18,3
60
0
10
20
30
40
50
60
70
1929 1947 1961 2006
anno
vita
med
ia
17Formica U. Epidemiologia., Formica U, ed. I controlli di salute dei bambini con Sindrome di Down.
Milano, CISEditore 2003 pp. 9-17.
18Leonard S, Bower B, Petterson B., Survival of infants born with Down’s syndrome: 1980-2000, Paediatr
Perinat Epidemiol, 2001, pp. 14:163-71.

22
Sulla base di quanto pubblicato dall’ISTAT (2000) emerge che in Italia le persone
disabili sono 2 milioni e 615 mila ( il 5% della popolazione complessiva
escludendo i bambini di età inferiore ai 6 anni che non sono compresi in tali
indagini), e il 73,2% pari a un milione e 900 mila persone sono concentrate tra gli
anziani.
In linea con la popolazione generale, la popolazione affetta da disabilita, la
popolazione affetta da disabilita intellettiva in generale e da sindrome di Down in
particolare, sta rapidamente invecchiando, con maggior rischio di sviluppare una
demenza simile alla malattia di Alzheimer.
Il significativo aumento della sopravvivenza è stato attribuito alla riduzione della
mortalità infantile, ottenuta tramite interventi di cardiochirurgia per la correzione
di difetti cardiaci congeniti (presenti in circa il 40% dei soggetti), miglioramento
delle cure mediche e provvedimenti di carattere sociale. Si ritiene che non vi sia
una differenza nell’aspettativa di vita fra i due sessi.
Programmi di “life-planning” hanno favorito la tendenza a ridurre
l’istituzionalizzazione dei soggetti con sindrome di Down. Ciò è importante
poiché gli anni di sopravvivenza dei soggetti istituzionalizzati sono inferiori del
10-15% rispetto a quelli dei non istituzionalizzati.
Anche programmi di stimolazione intellettiva e motoria precoce incrementano la
sopravvivenza nei soggetti con sindrome di Down.
Oggi si richiedono ulteriori acquisizioni che possano fornire indicazioni in merito
all’aspettativa di vita dei soggetti con sindrome di Down al fine di programmare
le dimensioni della famiglia, di organizzare le abitudini di vita all’interno dei
nuclei familiari dei soggetti con sindrome di Down e di favorire supporti socio-
sanitari.

23
27,10
42,10
18,87
11,08
0,00
5,00
10,00
15,00
20,00
25,00
30,00
35,00
40,00
45,00
donne < 34 anni uomini < 34 anni donne > 35 anni uomini > 35 anni
età
freq
uenz
a(%
)
E’ stata condotta una ricerca analizzando i dati relativi a una popolazione di
soggetti con sindrome di Down di diversa età appartenenti all’Associazione
ViviDown di Milano19.
Sono stati presi in considerazione 173 soggetti, 80 donne e 93 uomini: l’età media
delle donne era di 34,3 anni (range da 22 a 60anni), quella degli uomini di 32,1
anni (range da 18 a 65 anni).
Oltre all’età dei soggetti, è stato valutato il grado di scolarità, suddividendo i
soggetti in quattro gruppi (0 anni, scuola elementare: 5 anni, scuola media: 8 anni,
scuola superiore: 11 anni). Sono state inoltre considerate l’attività abituale svolta e
la partecipazione a Centri Socio-Educativi .
Il 6,3% dei soggetti ha frequentato la scuola elementare, il 54,9% ha frequentato
la scuola media ed il 37,5% ha frequentato la scuola superiore. Solo l’1,1% dei
soggetti non ha frequentato alcuna scuola. Il 12,7% dei soggetti con sindrome di
Down svolge una attività prevalentemente di tipo manuale. Il 40,5% dei soggetti
con sindrome di Down frequenta un centro socio-educativo.
Contrariamente fra i soggetti di età ≤ 35 anni prevalgono le donne rispetto agli
uomini.
Il numero dei soggetti con sindrome di Down con età ≥ 34 anni che hanno
frequentato una scuola superiore è significativamente più alto rispetto a quello
19 www.vividown.org.

24
degli individui con età ≤ 35 anni. Ciò indica che i soggetti più giovani frequentano
più a lungo i corsi di istruzione rispetto al passato. Tra i soggetti con sindrome di
Down di età ≥34 anni non vi è alcun individuo senza alcun grado di istruzione. La
percentuale di coloro che indipendentemente dall’età non frequentano centri di
riabilitazione (59,5%) è comunque maggiore rispetto a quella degli individui che
li frequentano (40,5%).
I soggetti con sindrome di Down ≥ 34 anni frequentano di più i centri che
promuovono l’integrazione sociale rispetto ai soggetto ≤35 anni.
Emerge tra gli individui più giovani la tendenza ad “uscire di casa” ed a
frequentare ambiti sociali idonei dei quali c’è, per altro, più disponibilità rispetto
al passato.
Sono stati individuati diversi fattori sociali e ambientali che si correlano con la
durata e la qualità della vita, in particolare con la compromissione cognitiva. Essi
comprendono lo stato sociale, la disponibilità di servizi sul territorio, l’interazione
con l’ambiente e lo stimolo intellettivo.
La durata della vita dipende per il 30 % circa dai geni e per il 70 % circa
dall’ambiente. Da questa ricerca emerge che i soggetti con sindrome di Down più
giovani frequentano più a lungo la scuola e hanno maggiore possibilità di
accedere ai centri socio-educativi 20. Si ritiene pertanto che, pur in presenza di una
componente genetica qual è quella connessa con la trisomia 21, la disponibilità di
un ambiente idoneo possa influenzare favorevolmente non solo la durata della vita
ma anche la performance intellettiva dei soggetti con sindrome di Down.
20Roizen NJ, Patterson D., Down’s Syndrome. Lancet 2003, pp.361

25

26

27
CAPITOLO 4
DIAGNOSI PRECOCE
La popolazione affetta da disabilità intellettive (Intellectual disabilities) in generale, e
da sindrome di Down in particolare, sta rapidamente invecchiando con un maggiore
rischio di sviluppare una demenza simile alla malattia di Alzheimer (Dementia in
Alzheimer’s Disease).
L’età media di esordio delle manifestazioni di una Dementia in Alzheimer’s Disease in
soggetti affetti da sindrome di Down si situa tra i 51 –54 anni di età21, e dalla letteratura
disponibile risulta che all’esame autoptico dei casi finora esaminati, quasi la totalità
delle persone con sindrome di Down di età superiore a 40 anni, mostra le tipiche
alterazioni neuropatologiche della Dementia in Alzheimer’s Disease22 anche se non
sempre è evidente la demenza in vita23.
Alcuni studi riportano che dal 50 al 75 % delle persone con sindrome di Down
sviluppano Dementia in Alzheimer’s Disease nella mezz’età 24, ma rimane da chiarire se
la neuropatologia alzheimeriana o le sequenze degli eventi coinvolte nello sviluppo
della Dementia in Alzheimer’s Disease e della Alzheimer Disease nella popolazione
generale siano le stesse25.
21Lay F. Clinicopathologic features of Alzheimer disease in Down syndrome. In L. Nadel & C.Epstein C. (Eds)
Down syndrome and Alzheimer disease. New York: Wiley-Liss, 1992, pp. 15-34.
22 Wisniewski HM, Silverman W. Alzheimer’s disease neuropathology and dementia in Down’s Syndrome. In JA.Ronald, J. Perera, L. Nadel, & A. Comblain (Eds.) Down syndrome. Psychological, psychobiological, and socio-educational perspectives. London: Whurr, 1996, pp. 43-50.
23Brugge KI, Nichols SL, Salmon DP et al. Cognitive impairment in adults with Down’s syndrome: similarities to
early cognitive changes in Alzheimer’s disease. Neurology 1994;44:232-238.
24Devenny DA, Silverman WP, Hill AL et al. Normal ageing in adults with Down’s syndrome: a longitudinal study.
J Intellect Disabil Res 1996;40:208–221.
25 Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome. Arch Neurol 1989;46:849–853.Aylward EH, Burt DB, Thorpe LU, Lai F, Dalton AJ. Diagnosis of dementia in individuals with intellectualdisability. J Intell Disab Res 1997;41:152-164.

28
Inoltre alcuni recenti studi hanno confermato che individui con n alto QI, un alto livello
di educazione, di stato occupazionale o partecipazione in attività piacevoli hanno un
rischi ridotto di sviluppare Alzheimer’s Disease, condizioni che si rovesciano in
presenza di persone con disabilita intellettiva.
La diagnosi di una Demenitia in Alzheimer’s Disease è difficile26, non solo per la
carenza di strumenti diagnostici affidabili, ma anche per la presenza di condizioni
progressive invecchiamento-dipendenti (ad esempio, cifosi, malattia osteo-articolare), di
comorbilità somatica (ad esempio, malattie endocrine) e psichiatrica (ad esempio,
depressione) che possono singolarmente od in associazione provocare un quadro clinico
simile a quello di una Dementia in Alzheimer’s Disease irreversibile.27
Quello che emerge con forza sembra indicare che una diagnosi ritardata di una
Dementia in Alzheimer’s Disease ha delle conseguenze estremamente negative sia per il
malato che per tutti coloro che gli stanno attorno, con un immediato effetto sul
peggioramento della qualità di vita e ulteriore aggravamento degli oneri di cura,
assistenza e sorveglianza da parte della famiglia o dei servizi. Tutto questo perché la
gestione quotidiana dei soggetti affetti da Dementia in Alzheimer’s Disease è quasi
sempre complicata dalla insorgenza di sintomi non cognitivi dirompenti28, definiti
sintomi comportamentali e psicologici della demenza, che aggravano lo stress non solo
dei malati, ma anche dei caregiver formali e non, che spesso si vedono costretti a
ricorrere a dei servizi residenziali per il loro caro, con un inevitabile aumento della
sofferenza psicologica e dei costi economici per la collettività.
26Deb S, Braganza J. Comparison of rating scales for the diagnosis of dementia in adults with Down’s syndrome.
J Intellect Disabil Res 1999;43:400–407.
27Janicki MP, Heller T, Seltzer G, Hogg J. Practice guidelines for the clinical assessment and care management of
Alzheimer and other dementias among adults with mental retardation. Washington: American Association on MentalRetardation, 1995.
28 Millichap D, Oliver C, McQuillan S, Kalsy S, Lloyd V, Hall S. Descriptive functional analysis of behavioralexcesses shown by adults with Down syndrome and dementia. Int J Geriatr Psychiatry 2003;18:844-854.Jozsvai E. Behavioural and Psychological Symptoms of Dementia in Individuals With Down Syndrome, J IntellectDisabil Res 2005;12:31-40.

29
Essendo la Dementia in Alzheimer’s Disease una malattia attualmente inguaribile, una
corretta gestione di questi malati dovrebbe mirare a diminuire lo stress e ad aumentare il
benessere raggiungendo così una loro funzionalità ottimale come essere sociale e nel
rispetto della loro personalità, fatta non solo di funzioni cognitive, ma anche di
sentimenti, azione, appartenenza, attaccamento alle persone e di identità. Tale
particolare attenzione metodologica implica a livello organizzativo una massima
flessibilità nei tempi e modi di attuazione degli interventi: riattivativi nella Intellectual
disabilities o sindrome di Down in età avanzata, migliorativi nelle fasi iniziali della
Dementia in Alzheimer’s Disease, protesici ed orientati al benessere nelle fasi avanzate
della malattia. In questo contesto l’elasticità organizzativa viene considerata uno degli
elementi terapeutici essenziali nella relazione tra malato e caregiver.
Come di solito accade in medicina, anche nel caso della Dementia in Alzheimer’s
Disease, è evidente che per ottenere al paziente i maggiori benefici consentiti è di
essenziale importanza che la diagnosi venga posta dallo specialista il più precocemente
possibile, quando ovviamente ciò risulti un'operazione realizzabile29. Gli adulti con
sindrome di Down sono ritenuti una categoria a rischio per lo sviluppo della demenza di
tipo Alzheimer ma, dato il loro deficit intellettivo congenito, è difficile individuare i
sintomi più precoci e caratteristici di un declino correlato all’età e di un quadro di
demenza.
La demenza si definisce come una patologia caratterizzata da un deterioramento globale
delle funzioni superiori, tale da interferire con le normali attività quotidiane. Sebbene il
declino della memoria sia a breve che a lungo termine sia un tipico segno della
demenza, la diagnosi definitiva richiede la presenza di deficit in altre funzioni cognitive
oltre a quelle legate alla memoria (per esempio, un deficit della capacità di pensiero
astratto, di giudizio, di linguaggio, di coordinazione, pianificazione e organizzazione).
29 De Vreese LP, Belloi L. L’esperienza del Nucleo Specialistico per le Demenze: da un approccio distile di vita (LifeStyle Approach) a una qualità di vita migliore del malato di demenza. ProTerza Età 2002;8:4-11

30
La demenza è facilmente diagnosticabile negli stadi più avanzati, ma da numerosi studi
risulta che gli operatori sanitari spesso trascurano i segni iniziali della malattia30.
La visita medica di routine e l’anamnesi del paziente non sono abbastanza sensibili per
formulare una diagnosi di demenza, specie se i caregiver non sono presenti a
convalidare ciò che il soggetto riferisce durante l’esame. Molti medici includono
nell’esame fisico e nella raccolta dei dati solo un esame superficiale dello stato mentale
del paziente.
I più comuni standard diagnostici per la demenza comprendono una valutazione
dettagliata dello stato mentale del paziente, associata a un’attenta visita che possa
escludere altre cause di declino cognitivo.
I segni precoci di demenza sono spesso trascurati nelle visite di routine e sono
disponibili vari test per indagare lo stato mentale e permettere al medico una
valutazione più accurata delle funzioni cognitive dei soggetti con sindrome di
Down. Basandosi esclusivamente sui risultati dei test, la diagnosi può risultare in molti
casi scorretta. Per una valutazione qualitativa di eventuali deficit cognitivi, è buona
norma far precedere la somministrazione di test da un breve colloquio, il quale se
condotto in modo adeguato già può fornire indicazioni in merito alle aree cognitive
disturbate31. Il colloquio non deve essere eccessivamente prolungato, per non stancare il
paziente e alterare quindi le successive prove del test. Le aree da indagare con il
colloquio possono essere quelle che riguardano la quotidianità e la sfera personale, ad
esempio "Con chi vive, come si chiamano le sue sorelle, che lavoro fanno, quanti anni
hanno, dove vive, da quanto tempo vive in questo paese,…". Può essere altresì utile
riproporre le medesime domande in due momenti diversi dello stesso colloquio, al fine
di valutare se il paziente fornisce risposte sempre uguali o diverse. Eventuali risposte
date dal paziente dovranno poi essere verificate con un familiare di riferimento.
Il colloquio, così come la somministrazione di eventuali test, deve essere condotto in un
momento della giornata in cui il paziente non risulta particolarmente affaticato, ad
30 Gedey A. Manual for the Dementia Scale for Down Syndrome: Gedey Research and Counseling,Vancouver,Canada, 199531
Evenhuis HM, Kengen MMF, Eurlings HAL, Dementia Questionnaire for Mentally Retarded Persons (DMR). JIntellect Disabil Res 2004; 40:369–373

31
esempio durante le prime ore della giornata o dopo il riposo pomeridiano ed è
indispensabile instaurare una buona alleanza di lavoro. Questo vuol dire spiegare alla
persona con cui lavoriamo chi siamo e che cosa dobbiamo fare, utilizzando una
terminologia adeguata alla persona che abbiamo di fronte e soprattutto senza
squalificare la sue risposte e prestazioni. Creare una buon "contatto" con il paziente è la
condizione essenziale per poter lavorare con lui. Può capitare che un paziente si rifiuti
di sottoporsi alle prove di un test; insistere può essere controproducente per la nostra
relazione con quella persona, se possibile vale la pena riproporre il lavoro in un'altra
occasione, rispettando i tempi di chi abbiamo di fronte.
Da non trascurare inoltre sono gli esami ematobiochimici che comprendono un esame
emocromocitometrico completo, prove che valutano la funzione epatica e renale, test
dietologici per la sifilide, il dosaggio degli ormoni tiroidei, i livelli di vitamina B12 e di
acido folico, degli elettroliti, l'esame del liquor cefalorachidiano,
l'elettroencefalogramma, ed altri esami, tutti spesso necessari per identificare forme di
demenza secondaria a patologie concomitanti reversibili32. La demenza di Alzheimer
deve infatti essere differenziata dalle varie forme di demenza secondaria.
Il percorso diagnostico per arrivare alla diagnosi di Dementia in Alzheimer’s Disease,
nell'evenienza di uno stadio iniziale, deve quindi prendere sempre l'avvio da un'attenta
raccolta e valutazione dei dati anamnestici, da un preciso esame dello stato fisico e
mentale (esame obiettivo generale e neurologico), avendo cura di vagliare il livello
funzionale sino ad arrivare all'effettuazione di indagini ematologiche, biochimiche e
strumentali che si propongano, in modo particolare, di stabilire anzitutto se i disturbi
rilevati siano correlabili ad un declino cognitivo dovuto all'età (invecchiamento
cerebrale non patologico), a condizioni cliniche quali depressione o delirium, o ad una
vera demenza in fase di esordio, evenienza in cui si prospetta sempre anche la necessità
di una diagnosi differenziale.
32 Brugge KI, Nichols SL, Salmon DP et al. Cognitive impairment in adults with Down’s syndrome:similarities to early cognitive changes in Alzheimer’s disease. Neurology 1994;44:232-238.

32
Si comprende quindi l'importanza di una definizione dei confini nosografici tra queste
due realtà rappresentate dall'invecchiamento cerebrale "fisiologico" e da un processo
dementigeno in fase iniziale, al fine anche di una utile distinzione tra semplici fattori di
rischio e la fase iniziale di un vero processo patologico, con tutte le conseguenze che
una diagnosi precoce, in questa seconda evenienza, potrebbe avere sul trattamento
preventivo e sulla terapia.
Si sottolinea l'importanza di una somministrazione periodica del test, effettuata da
personale qualificato, per monitorare l'eventuale evoluzione del decadimento cognitivo
o il mantenimento della cognitività stessa33.
Anche l'osservazione quotidiana del soggetto e la raccolta di informazioni relative alla
sua quotidianità riferite da chi si prende cura di quel paziente forniscono preziose
indicazioni per poter effettuare una corretta diagnosi34.
Presso il centro “La Meridiana” (vedi cap.6) lo screening e la diagnosi sono tra gli
obbiettivi primari. Per poter valutare l’evoluzione clinica dei soggetti si utilizza una
sequenza di test i quali sono innanzitutto stati convalidati poiché in lingua italiana non
erano ancora disponibili. Qui di seguito riporto i test con una breve didascalia sull’area
di valutazione e in allegato 4 i test integrali:
Il DMR ha la funzione di diagnosticare eventuali declini mnestici o di altre
funzioni cognitive attraverso un’ intervista ai caregiving formali o informali.
Il TSI come il DMR riconoscere i declini delle funzioni cognitive attraverso
quesiti sulla performance motoria, sulla comprensione e produzione orale, sulle
conoscenze generali , sulla memoria immediata e quella ritardata, sulle
performance motoria. A differenza del DMR questo test va somministrato al
soggetto a valutare.
33 Gélinas I, Gauthier L, McIntyre M, Gauthier S. Development of a functional measure for persons withAlzheimer's disease: the disability assessment for dementia. Am J Occupat Ther Janicki MP, Dalton AJ.Alzheimer disease in a select population of older adults with mental retardation. Irish J Psychol1993;14:38-4734
Brugge KI, Nichols SL, Salmon DP et al. Cognitive impairment in adults with Down’s syndrome: similarities toearly cognitive changes in Alzheimer’s disease. Neurology 1994;44:232-238.

33
L’AFAS-IT sempre attraverso un intervista ai caregiving ha funzione di
valutare le abilità funzionali come per esempio la deambulazione,la capacità di
lavarsi, di mangiare da solo, la consapevolezza dell’ambiente nel quale è
inserito.
Il TINETTI serve per valutare le abilità funzionali ed in particolare la scala
verifica la capacità di equilibri e di andatura (ad esempio l’equilibrio da seduto,
di girarsi, lunghezza del passo, traiettoria, camminata).
Il CMAI concorre insieme all’AFAS-IT e al TINETTI alla verifica della
gestione quotidiana; in particolare viene stabilito il comportamento aggressivo
verbale e non e il comportamento agitato.
Il BEAM-D si colloca accanto al CMAI per la valutazione del comportamento.
Il test si divide in una prima parte vengono analizzati i comportamenti sotto
esame (aggressività, comportamento distruttivo, interferenza in altre attività,
comportamento asociale, richiesta di attenzione, girovagare, comportamento
sessuale inadeguato, comportamento di accumulo)e i comportamenti
desunti(depressione, deliri, allucinazioni, ansia, stabilità affettiva, insonnia,
aumento o diminuzione dell’appettito).
Il RUOCCO invece fa una valutazione clinica generica (anamnesi, terapie,
ricoveri ospedalieri, disturbi particolari in qualche apparato ecc.)compreso
l’esame fisico generale e neurologico.
L’ AA-DD valuta l’adeguatezza dell’ambiente nel quale è inserito ad esempio
se il soggetto è agitato, vagabonda durante il giorno o la notte, piange, si
appropria di cose altrui, non è cooperativo, aggredisce fisicamente le altre
persone, trova difficoltà nel ricordare parole o simboli, disinteresse nelle attività
abituali.
Il CIRS è un indice di comorbilità sullo stato di salute fisica generale .Tra i
fattori che vengono presi in considerazione c’è l’ipertensione, patologie
dell’apparato GL superiore e di quello inferiore, patologie ai vari apparati 35.
35 www.validazione.eu/dad

34
Nella fase di screening e di diagnosi di demenza dei soggetti sperimentali, nel progetto
di ricerca “La Meridiana”, sono stati coinvolti circa 60 persone, di entrambi i sessi, di
età compresa fra i > 50 e 70 anni. I soggetti inclusi nello studio verranno seguiti per un
periodo minimo di 30 mesi, con follow-up successivi ogni 6 mesi, nel corso dei quali
verranno sottoposti ad una serie di scale e prove oggettive volti ad evidenziare
l’eventuale evoluzione clinica del processo dementigeno.
La prima taratura di valutazione dei dati raccolti sulle scale di valutazione ed eventuale
implementazione delle stesse o costruzione di nuovi di nuovi strumenti di misura (ad es.
un nuovo test di screening per la valutazione oggettiva dello stato cognitivo globale)
avverrà nell’agosto del 2007 ad un anno dall’inizio della raccolta dei dati.
L’importanza della valutazione e della taratura degli strumenti è data dal fatto che i dati
raccolti ci consentono anche di essere consapevoli dei limiti e delle potenzialità della
propria attività a più livelli, evitando così il rischio di autoreferenzialità della propria
che si può attuare nel dialogo tra operatori dei servizi ed inoltre può essere uno
strumento essenziale per ricollocare la persona come protagonista dello stesso processo
di valutazione. Il processo di valutazione è molto importante anche per gli operatori in
quanto è un feedback sul proprio lavoro, facilita la presa di decisioni o la modifica di
comportamenti, favorisce un lavoro d’equipe anche tra le diverse professionalità,
protegge dallo stress e dalla routine. MA tale processo risulta importante anche per
l’ospite perché promuove un atteggiamento partecipativo ed aumenta il potere di
intervento/controllo.

35
CAPITOLO 5
RICERCAZIONE PER VALUTARE IL DECLINO COGNITIVO NEI
SOGGETTI CON SINDROME DI DOWN
Come già descritto nei capitoli precedenti, l'aspettativa di vita delle persone con
sindrome Down è aumentata moltissimo. Attualmente l'aspettativa di vita è di
oltre 50 anni, e una persona su dieci raggiunge i 70 anni36. Nella misura in cui
diminuiscono le nascite di persone con sindrome Down e cresce l'aspettativa di
vita, il declino cognitivo è destinato ad essere sempre più frequente tra le persone
Down anziane.
Queste nuove esigenze fanno emergere problematiche fino ad ora mai riscontrate
e al contempo rende molto difficile mantenere queste persone nelle attuali
strutture. La ricercazione ha inizio nel Marzo del 2003 con l’osservazione
strutturata di sei Down, di età comprese tra i 52 e 56 anni, per sei mesi e i risultati
ottenuti sono stati la base del progetto, di una struttura integrata “La meridiana”,
iniziato nel Marzo del 2006 a Trento.
Nel primo gruppo di osservazione sono state coinvolte sei persone con sindrome
di Down di età superiore ai 52 anni. A causa proprio della loro età molti hanno
vissuto in istituti, per cui queste osservazioni non possono essere applicate
direttamente alle nuove coorti di giovani adulti attuali. Bisogna infatti sottolineare
che è estremamente importante mettere in relazione le percezioni delle persone nei
confronti del contesto in cui hanno vissuto.
Sebbene la presenza di un cromosoma in più dia origine ad uno sbilanciamento
genetico, che comporta una serie di svantaggi biochimici, come per esempio
l'accumulo di sostanza amiloide, proteina associata alla malattia di Alzheimer37,
non dobbiamo dimenticare che esiste un’interazione tra natura e ambiente. Esiste
36 Baird, P.A. & Sadovnik, P.D. (1987). Life expectancy in Down syndrome. Journal ofPaediatrics, 110, 849-54.
37 Wishart, J.G. (1996). Avoidant learning styles and cognitive development in young children. InB. Stratford & P. Gunn (Eds.) New Approaches to Down Syndrome. London: Cassell. pp 173-205.

36
tuttavia la possibilità di diminuire l'influenza genetica creando delle difese
ambientali.
Nello studio da noi effettuato, sono state utilizzate due griglie di osservazione
(vedi allegato 1 e 2): una per i centri socio-educativi e l’altra per le strutture
residenziali che ospitano i ragazzi una volta conclusa la giornata al centro diurno;
tali schede venivano somministrate quotidianamente dagli operatori che
interagivano con i soggetti coinvolti nell’osservazione. Questo strumento è stato
utilizzato in quattro diversi centri ANFFAS trentini (Primiero, Borgo Valsugana,
Trento, Tione) dove complessivamente sono state raccolte circa 350 griglie
durante un arco di tempo di sei mesi .
Le griglie presentano una trentina di quesiti atti a verificare la presenza delle
principali aprassie, delle afasie , delle agnosie e di disturbi della personalità (il “si
pianta” indica proprio la principale problematica verificata in queste persone; a
volte anche le persone più miti manifestavano atteggiamenti aggressivi e
immotivati verso un compagno o un operatore) e venivano somministrate
quotidianamente sia presso i centri diurni che nelle comunità alloggio.
Ovviamente il linguaggio usato nel questionario è importante. Si è cercato quindi
di utilizzare un linguaggio semplice e diretto per facilitare coloro che redigono la
griglia di non avere difficoltà. A tal proposito le domande sono state collocate in
ordine temporale (dall’arrivo al centro al ritorno in comunità alloggio, dalla
sveglia mattutina al coricarsi serale).
Il questionario doveva fornire le risposte alle numerose domande che, come
operatori, ci ponevamo per poter migliorare la vita ai soggetti Down anziani. Già
da qualche mese avevamo notato cambiamenti comportamentali, che spesso
sfociavano in atteggiamenti aggressivi apparentemente immotivati, difficoltà nel
controllo degli sfinteri, a volte riscontravamo difficoltà a spostarsi nei luoghi
famigliari, nell’uso delle posate, nel compiere le prassie di tutti i giorni. In seguito
ad una serie di ricerche su manuali o siti che si interessano alla sindrome di Down
non eravamo arrivati a trovare la risposta che cercavamo: cosa determina questo
cambiamento? Sarà reversibile o definitivo?
Partendo da questi presupposti, in collaborazione col Medico di Medicina
Generale Ulrico Mantesso di Trento e il pedagogista Tiziano Gomiero di Trento
per la formulazione di una griglia di osservazione, ci siamo posti una serie di

37
obbiettivi: primo fra tutti la ricerca di una correlazione tra gli atteggiamenti
oppositivi e l’ambiente nel quale il soggetto è inserito, valutazione della modalità
e dei tempi di perdita delle prassie principali, valutazione del setting adeguato nel
quale sono inseriti i soggetti per poter garantire uno stato di “pace e benessere” ai
soggetti presi in esame.
L’osservazione è iniziata a marzo 2003 e si è conclusa nell’agosto 2003 per un
totale di sei mesi. In seguito ho riportato (allegato 3), a titolo esemplificativo, il
riassunto del mese di aprile ,del centro e della comunità alloggi, P. e R. in due
stadi differenti; il primo in una fase ancora iniziale con sporadici comportamenti
problema, il secondo in uno stadio più avanzato .
I soggetti coinvolti denunciavano tutti un decadimento cognitivo ma in stadi
differenti; infatti se per alcuni era già perso l’uso del linguaggio in altri vi era
ancora una buona comunicazione; si notava inoltre una perdita della memoria a
breve termine mentre quella a lungo termine resisteva.
Non vi erano dati sul quoziente di intelligenza prima dell’inizio del processo
degenerativo e quindi era difficoltoso valutare a il livello raggiunto dal soggetto.
Non vi era una modalità valida per valutare il decadimento cognitivo soprattutto
nella fase iniziale, quando vi sono episodi sporadici - come lacune
apparentemente accidentali - spesso non letti nella maniera adeguata, e che una
somministrazione giornaliera della griglia non permetteva di cogliere. Una
somministrazione mensile forse sarebbe stata sufficiente.
Si è invece rivelata adeguata e corretta la somministrazione giornaliera per
valutare il decadimento neurofisiologico e comportamentale di quei soggetti che si
trovano negli stadi più avanzi del decadimento.
Nei soggetti più avanzati si è potuta osservare l’importanza del controllo del
setting per limitare lo stress ambientale riducendo, di conseguenza, i blocchi
oppositivi. Si può notare, nell’allegato 3, come cambi notevolmente il
comportamento del soggetto R. tra il centro diurno e la comunità alloggio. Questa
differenza è stata giustificata dal setting differente dei due ambienti; nel centro
diurno confluiscono molte persone aumentano così gli stimoli uditivi, i tempi
sono più frenetici e c’è meno flessibilità rispetto alle esigenze di taluni ed infine
l’ambiente è ricco di colori e stimoli visivi che creavano nel soggetto ancora più
motivo di turbamento, nella comunità alloggio, invece, vi era un setting più

38
protetto con maggiore flessibilità e minori stimoli visivi ed uditivi e di
conseguenza R. manifestava meno atteggiamenti oppositivi. E’importante creare
un ambiente protetto per dare un senso di sicurezza al paziente, come ad esempio
fissare dei corrimano, diminuire i rumori forti che possono provenire dall’esterno
o dell’interno dell’edificio, disporre un arredamento per facilitare gli spostamenti
del malato, mettere sotto chiave qualunque cosa possa rappresentare un pericolo,
assicurarsi che stanze e corridoi siano ben illuminati. Diverse possono essere le
cause che scatenano atteggiamenti oppositivi, tuttavia la causa più comune è la
paura; si tratta quindi di una naturale reazione difensiva contro la falsa percezione
di un pericolo o di una minaccia.
Si è potuto riscontrare una riduzione della latenza e dell’ampiezza di alcune onde
di risposta ai potenziali evocati. Non si presenta più la velocità sinaptica normale,
ma le risposte rispetto uno stimolo possono avvenire anche dopo parecchi minuti;
dunque il ripetere più volte un’informazione o una richiesta invece di aiutare il
soggetto lo pone in difficoltà, perché aumenta le informazioni da codificare e non
la velocità di risposta.
Dalle griglie emergono anche i suggerimenti su come interagire e comunicare in
maniera efficacie con questi soggetti. Ad esempio si è visto come poche
informazioni siano più facili da recepire che un lungo discorso: non dirò più “ciao,
come va? È ora di alzarci o di mangiare” ma semplicemente “alzati” o “mangia”.
Con la prima frase chiedo tre cose: di rispondere al mio saluto, di dirmi come va e
di alzarsi o mangiare, con la seconda frase chiedo solo una cosa di alzarsi o di
mangiare.
Si è vista l’importanza di una visione olistica della Dementia in Alzheimer’s
Disease. L’approccio multidisciplinare, vale a dire il considerare che il
comportamento normale o patologico sia il frutto di una relazione “mente-corpo”;
vanno quindi considerati sia l’aspetto psicologico sia quello medico che quello
sociale. E solo attraverso questo sguardo d’insieme si può arrivare alla “pace e
benessere del soggetto”, ovvero la tranquillità e la buona salute del paziente
Down.
Da questa prima ricerca si è preso atto dell’emergenza sempre maggiore di offrire
assistenza e strutture adeguate alle nuove esigenze di questi soggetti. Si è iniziato
a progettare un centro integrato per Anziani con Ritardo Mentale (centro diurno +

39
centro residenziale) in collaborazione con il centro diurno Alzheimer di Trento e
l’ANFFAS Trentino.
PROGETTO DEL CENTRO INTEGRATO: LA MERIDIANA
Oggi le risposte che vengono offerte alle persone non autosufficienti sono di tipo
assistenziale-custodistico ( RSA, Reparti di Lungo Degnza e assistenti private
famigliari), mentre la poca prevenzione della disabilità è legata quasi
esclusivamente agli screening e agli interventi di tipo medico-sanitario; la ricerca
è prevalentemente orientata in termini biomedici e si deve far ricorso ancora a
molti interventi rivolti alla contenzione fisica, chimica e ambientale.
Il sopraggiungere delle polipatologie dell’età anziana che si innestano su un
quadro già fortemente compromesso, rende molto difficile mantenere queste
persone sia nei centri diurni o nelle comunità alloggio per il prevalere di necessità
medico-sanitarie specifiche che si aggiungono al bisogno di assistenza e lo
modificano. Si è reso necessario un percorso alternativo che preveda la possibilità
di intervento costante di personale sanitario accanto ad educatori e a operatori
assistenziali appositamente formati, di spazi appositamente strutturati e di attività
mirate con un taglio progettuale che unisca le competenze delle attività relative ai
disabili con quelle legate alla neurologia, geriatrie e alla gerontologia.
Tutto questo allo scopo di mantenere il più a lungo possibile le competenze e le
autonomie, combattere il deterioramento neurofisiologico e favorire il benessere
di queste persone, in modo da prolungare il periodo di autosufficienza delle
persone in oggetto ed evitando di ricorrere prematuramente ad una assistenza solo
di tipo “custodialistico”.
Nel mese di novembre 2005 ha preso il via l’attività presso ANFFAS trentino
onlus del centro pilota “La Meridiana” di Trento dedicato a soggetti Down e con
disabilità intellettiva di età adulta avanzata. In questo centro gli interventi sono
orientati alla funzionalità globale e specifica (abilità della vita quotidiana)
seguono i principi di due approcci di tipo globale, ossia la “gestione centrata sulla
persona” e la “Gentle Care”.
Il primo approccio richiede agli operatori l’acquisiione della personalità, della
biografia e della psicologia sociale di ciascun utente prima di impostare qualsiasi

40
intervento orientato al mantenimento dell’intelligenza sociale e delle abilitò
funzionali residue.
Il secondo individua come obbiettivo non tanto il rendimento quanto il benessere
sia dell’utente che di coloro che gi stanno attorno, compresi gli operatori, e come
metodo l’approccio protesico che costruisce dall’esterno tutte le funzioni
celebrali che il malato ha perso o sta per perdere. La protesi comprende sia lo
spazio fisico ( sicurezza, confort, semplicità, ect.) sia le persone i cui
atteggiamenti sono caratterizzati da flessibilità, pazienza, e uso del linguaggio del
corpo, che le attività di gruppo ed individuali che valorizzano i bisogni primari,
necessari ed essenziali di ciascun ospite.
Affianco a questi vi sono altri obbiettivi più specifici che l’ANFFAS Trentino
onlus ha nella Meridiana :
1. validare gli strumenti di valutazione per la diagnosi differenziale tra
demenza potenzialmente reversibile - la cosiddetta pseudo-demenza -
dovuta a un peggioramento di tutte quelle condizioni psico-fisiche
inficiando la funzionalità cerebrale già di per sé subottimale per la
disabilità intellettiva congenita, ed una demenza irreversibile e progressiva
in soggetti di età adulta avanzata con disabilità intellettiva o sindrome di
Down. Di conseguenza, è necessario ipotizzare i criteri per l’erogazione
dei servizi e dei processi assistenziali più adeguati possibili, con un
monitoraggio che copra i quattro domini principali della demenza secondo
le recenti linee-guida del trattamento della demenza Alzheimer nella
popolazione generale38: funzioni cognitive e abilità funzionali, emotività e
comportamento, famiglia e qualità della vita.
2. Verificare l’impatto di diverse modalità d’intervento prevalentemente di
tipo non farmacologico in un ambiente protesico secondo i principi del
Gentlecare39, per un’assistenza alle persone con o senza Dementia in
Alzheimer’s Disease orientata al mantenimento delle capacità cognitive,
sociali, funzionali residue ed al miglioramento della sfera affettiva e del
38Caltagirone C, Bianchetti A, Di Luca M et al., Guidelines for the treatment of Alzheimer’s disease from
the Italian Association of Psychogeriatrics. Drugs Aging 2005;22 (suppl1): S1-S26.
39Jones M. GentleCare. Carocci Editore, Roma, 2005.

41
comportamento per un benessere psicologico globale, accettabile con il
grado di disabilità intellettiva.
3. Individuare attraverso un monitoraggio longitudinale, le variabili bio-
psico-sociali in grado di predire lo sviluppo di una Dementia in
Alzheimer’s Disease in soggetti di età adulta avanzata con disabilità
intellettiva o sindrome di Down .
Per poter avere una raccolta di dati più affidabile possibile si testerà l’utilizzo
della cartella sanitaria assistenziale ATLANTEÓ (Studio Vega vedi allegato 4)
che permette, oltre ad una facile raccolta dati, controlli periodici visto il continuo
mutamento del quadro di gravità e della rilevanza dei vari gruppi sintomatologici
nelle diverse fasi della Dementia in Alzheimer’s Disease. Le informazioni raccolte
dalla cartella ATLANTEÓ hanno lo scopo di programmare le attività sanitarie e la
gestione dei problemi sanitari intercorrenti. Infatti, vi è una difficoltà specifica nel
riconoscimento e nel trattamento delle patologie organiche concomitanti o
intercorrenti in questa tipologia di malati: ciò è necessario per impedire che la
demenza divenga una condizione che copre tutto, tale per cui ogni sintomo viene
attribuito alla psicopatologia comportamentale premorbosa legata alla disabilità
intellettiva o alla Dementia in Alzheimer’s Disease in atto, senza più attenzione
alle situazioni trattabili che spesso l’accompagnano40.
A questi obbiettivi generali si sono uniti ad altri sei obbiettivi più specifici:
1. Valutazione multidimensionale:
Ho più volte ribadito il concetto dell’importanza della visione olistica del
problema e non solamente a segmenti. Di seguito sono riportate in modo
sintetico gli strumenti preferenziali utilizzati in ambito diagnostico (tabella 1)
e quelli relativi agli interventi riattivativi, migliorativi, protesici e orientati alle
emozioni (tabella 2). Queste schede di valutazione sono state redatte sia a
scopo di prevenzione nei soggetti con disabilità intellettiva non ancora affetti
da demenza che di contenimento sintomatico dei disturbi cognitivi e non
cognitivi in coloro con una diagnosi clinica di disabilità intellettiva.
40 De Vreese LP, Cester A. Da “Demenza complicata da co-morbilità” a “Comorbilità complicata dademenza”. Riv Medico Pratico, Marzo 2003, 44-48.
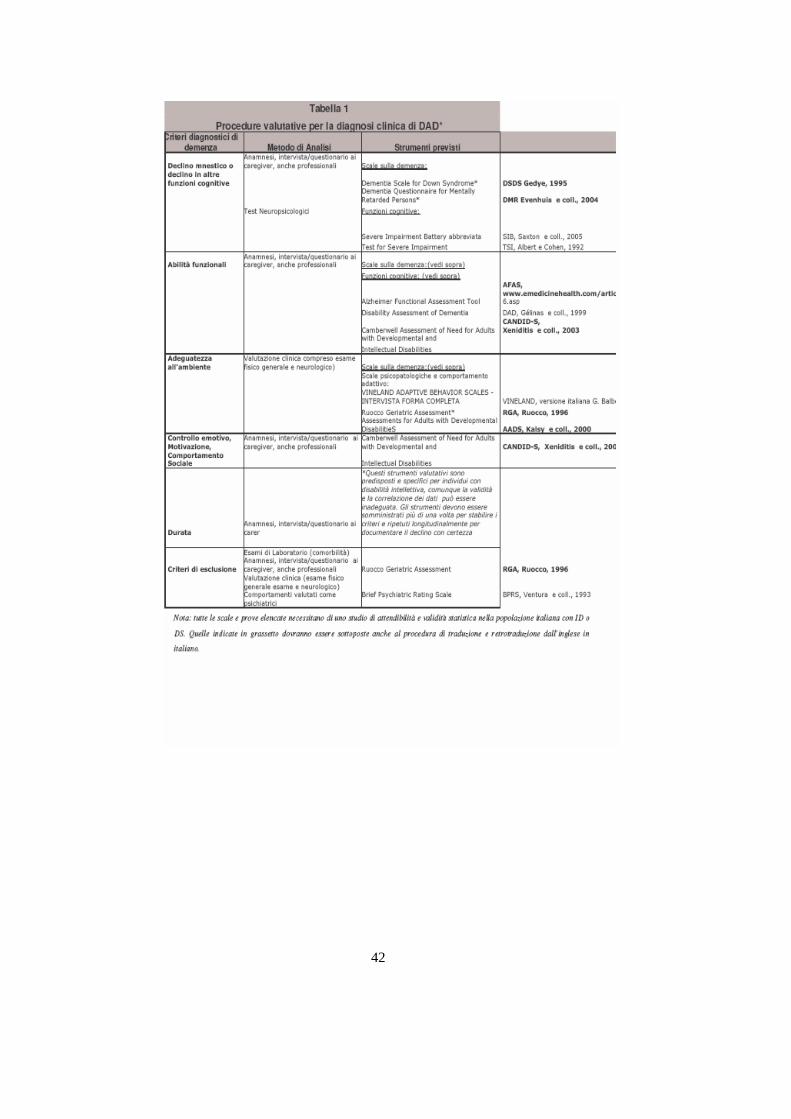
42

43
2. Interventi orientati alla funzionalità:
Gli interventi orientati alla funzionalità globale e specifica (abilità della vita
quotidiana) seguono i principi di due approcci paradigmatici di tipo globale, ossia
la “Gestione centrata sulla persona” di Tom Kitwood (1997) e la Gentle Care di
Moyra Jones (2005). Il primo approccio richiede dagli operatori l’acquisizione
della personalità, della biografia e della psicologia sociale di ciascun malato prima
di implementare qualsiasi intervento orientato al mantenimento dell’intelligenza
sociale e delle sue abilità funzionali residue. Il secondo individua come obiettivo
non tanto il rendimento quanto il benessere sia del malato che di coloro che gli
stanno attorno, compresi gli operatori, e come metodo l’approccio protesico che
costruisce dall’esterno tutte le funzioni cerebrali che il malato ha perso o sta per

44
perdere. La protesi comprende sia lo spazio fisico (sicurezza, comfort, semplicità,
etc.) sia le persone i cui atteggiamenti sono caratterizzati da flessibilità, pazienza,
e uso del linguaggio del corpo, che le attività di gruppo ed individuali che
valorizzano i bisogni primari, necessari ed essenziali di ciascun malato.
3. Interventi orientati alle funzioni cognitive.
Gli interventi di stimolazione cognitiva mirano al miglioramento o mantenimento
della capacità di elaborare e conservare nel tempo informazioni necessarie per
raggiungere una padronanza del proprio mondo interno, dell’ambiente e delle loro
reciproche interazioni. Sempre tenendo conto dell’obiettivo realistico di garantire
“pace e benessere”, è essenziale una accurata valutazione delle principali funzioni
corticali superiori i cui dati devono essere tradotti in termini sia riabilitativi che
ecologici41. Inoltre, per ridurre il rischio di stress nel malato durante questi
interventi, è fondamentale tenere conto non solo del quoziente d’intelligenza
premorbosa ma anche della stadiazione del processo dementigeno. Questo impone
l’adattamento degli strumenti di valutazione delle funzioni cognitive alla
progressione della malattia42 .
4. Interventi orientati alle emozioni e al controllo comportamentale:
Sempre nel rispetto del principio “pace e benessere”, l’uso degli psicofarmaci per
il controllo dei BPSD (Behavioral and Psychological Symptoms of Dementia)
deve rappresentare l’ultimissima risorsa. Vi sono evidenze recenti ottenute nella
popolazione generale affetta da demenza Alzheimer che alcuni interventi orientati
alle emozioni se applicati con rigore e personalizzati, risultano altrettanto efficaci
con ripercussioni positive anche sulla soddisfazione professionale degli operatori,
in particolare:
41De Vreese LP. Psicoriabilitazione: alternativa sostenibile? Geriatria 2005;27(suppl): S163-166.
42Aylward EH, Burt DB, Thorpe LU, Lai F, Dalton AJ. Diagnosis of dementia in individuals with
intellectual disability. J Intell Disab Res 1997;41:152-164.Prasher VP, Huxley A., Haque MS and the Down syndrome Ageing Study Group. A 24-week, double-blind,placebo-controlled trial of donepezil in patients with Down syndrome and Alzheimer’s diseaee – Pilot study.Int J Geriatr Psychiatry 2002;17:270-278.

45
Musicoterapica recettiva (ascolto passivo e ascolto con ballo) e attiva
(canto o utilizzo di vari strumenti musicali) supervisionata da un
musicoterapeuta43: Queste attività hanno come obiettivo non solo di
evocare ricordi ed esperienza autobiografiche, ma anche di migliorare il
tono dell’umore, l’autostima e il comportamento.
Snoezelen (stimolazione multisensoriale) secondo il metodo proposto da
Van Weert 44: la stimolazione attiva dei sensi con luci, suoni, odori e
gusti, individualizzata tenendo conto dello stile di vita, delle preferenze e
desideri e delle differenze socio-culturali di ciascun malato ed applicata al
bisogno nell’arco delle 24 ore ha come obiettivo di diminuire lo stress e i
conseguenti comportamenti socialmente impropri e di migliorare
l’autostima e il tono dell’umore.
Pet-therapy45: la sua funzione è di indurre il malato a svolgere attività
significative (dalla carezza alla passeggiata al nutrimento al semplice
gioco) ma anche ad evocare ricordi personali senza dimenticare anche la
stimolazione sensoriali e la grande interazione affettiva.
5. Valutazione dello stress nei caregivers formali ed informali:
Per la verifica dell’impatto degli interventi sia di tipo globale che legate alle
tecniche specifiche sopradescritte sullo stress dei caregivers formali e informali si
usano le versioni italiane di:
- Maslach Burnout Inventory (Maslach C.,1992) validata da Sirigatti e Stefanile
(1993).
- Caregiver Burden Inventory (CBI, Novak e Guest, 1989) validata da Zanetti
coll. (1999) in famigliari di malati Alzheimer’s disease (molto) lieve, con verifica
43Raglio A, Manarolo G, Villani D. Musicoterapia e Malattia di Alzheimer. Ed. Cosmopolis, 2001.
44Van Weert JCM, van Dolmen AM, Spreeuwenberg PMM, Ribbe MW, Bensing JM. Behavioral and mood
effects of Snoezelen integrated into 24-hour dementia care. J Am Geriatr Soc 2005;53:24-33.
45Kanamori M, Suzuki M, Yamamoto K. Et al. A day care program and evaluation of animal-assisted
therpay for elderly with senile dementia. Am J Alzheimer Dis Other Demen 2001;16:234-239.

46
della sua validità e della fedeltà in famigliari di soggetti con disabilità intellettiva
o sindrome di Down con Dementia in Alzheimer’s Disease .
6.. Valutazione della Qualità di vita del malato:
Sempre nell’ambito delle verifiche comportamentali si effettuerà una applicazione
in ambito italiano ed estensione in ambito multicentrico della ricerca Excess
Behaviour in Down's syndrome with Alzheimer's disease tramite uno studio
osservazionale da realizzarsi mediante videoriprese e utilizzo del programma
software OBSWIN46.
La diagnosi di una Dementia in Alzheimer’s Disease è difficile47, non solo per la
carenza di strumenti diagnostici affidabili, ma anche per la presenza di condizioni
progressive invecchiamento-dipendenti (ad es., cifosi, malattia osteo-articolare),
di comorbilità somatica (ad es., malattie endocrine) e psichiatrica (ad es.,
depressione) che possono singolarmente od in associazione provocare un quadro
clinico simile a quello di una Dementia in Alzheimer’s Diseas irreversibile48.
Quello che emerge con forza dalle prime osservazioni da noi rilevate sembra
indicare che una diagnosi ritardata di una Dementia in Alzheimer’s Diseas ha
delle conseguenze estremamente negative sia per il malato che per tutti coloro che
gli stanno attorno, con un immediato effetto sul peggioramento della qualità di
vita e ulteriore aggravamento degli oneri di cura, assistenza e sorveglianza da
parte della famiglia o dei servizi.
Tutto questo perché la gestione quotidiana dei soggetti affetti da Dementia in
Alzheimer’s Diseas è quasi sempre complicata dalla insorgenza di sintomi non
46 Millichap D, Oliver C, McQuillan S, Kalsy S, Lloyd V, Hall S. Descriptive functional analysis o behavioralexcesses shown by adults with Down syndrome and dementia. Int J Geriatr Psychiatry 2003;18:844-854.
47Deb S, Braganza J. Comparison of rating scales for the diagnosis of dementia in adults with Down’s
syndrome. J Intellect Disabil Res 1999;43:400–407
48Janicki MP, Heller T, Seltzer G, Hogg J. Practice guidelines for the clinical assessment and care
management of Alzheimer and other dementias among adults with mental retardation. Washington: AmericanAssociation on Mental Retardation, 1995.

47
cognitivi dirompenti49, definiti sintomi comportamentali e psicologici della
demenza, che aggravano lo stress non solo dei malati, ma anche dei caregivers
formali e non, che spesso si vedono costretti a ricorrere a dei servizi residenziali
per il loro caro.
Essendo la Dementia in Alzheimer’s Diseas una malattia attualmente inguaribile,
una corretta gestione di questi malati dovrebbe mirare a diminuire lo stress e ad
aumentare il benessere raggiungendo così una loro funzionalità ottimale come
essere sociale e nel rispetto della loro personalità, fatta non solo di funzioni
cognitive, ma anche di sentimenti, azione, appartenenza, attaccamento alle
persone e di identità. Tale particolare attenzione metodologica implica a livello
organizzativo una massima flessibilità nei tempi e modi di attuazione degli
interventi: riattivativi nella disabilità intellettiva o sindrome di Down in età
avanzata, migliorativi nelle fasi iniziali della Dementia in Alzheimer’s Disease,
protesici ed orientati al benessere nelle fasi severe ed avanzate della malattia. In
questo contesto l’elasticità organizzativa viene considerata uno degli elementi
terapeutici essenziali nella relazione tra malato e caregiver.
.
49Millichap D, Oliver C, McQuillan S, Kalsy S, Lloyd V, Hall S. Descriptive functional analysis of
behavioral excesses shown by adults with Down syndrome and dementia. Int J Geriatr Psychiatry2003;18:844-854.Jozsvai E. Behavioural and Psychological Symptoms of Dementia in Individuals With Down Syndrome, JIntellect Disabil Res 2005;12:31-40.

48

49
CAPITOLO 6
RUOLO DELL’EDUCATORE ALL’INTERNO DL PROGETTO “LAMERIDIANA”
Il ruolo che l’educatore all’interno del progetto “La Meridiana “ è molteplice:
deve infatti monitorare periodicamente attraverso i test, i vari stadi del
deterioramento cognitivo dei soggetti in osservazione
formare e assistere i caregiving siano questi formali che informali
valutare la qualità della vita e lo stato di” pace e benessere” che
protagonisti del progetto hanno raggiunto e provvedere al mantenimento.
Oltre alla somministrazione dei test (vedi cap. diagnosi precoce) l’educatore deve
formare ed informare i caregiver, sia formali che informali, circa il decorso della
malattia, le problematiche correlate, i nuovi bisogni dei soggetti con Dementia in
Alzheimer’s Disease, le strategie per garantite ai soggetti uno stato di “pace e
benessere”.
In seguito ho rappresentato, in forma schematica, le nozioni che l’educatore deve
trasmettere al caregiver rispetto al rapporto con il paziente affetto da Alzheimer’s
Disease:

50
Innanzitutto il caregiver deve essere consapevole dell’importanza del suo ruolo di
“terapia “ e di promotore di setting in ambiente protesico il tutto per garantire una
situazione di “pace e benessere”al paziente.
Per creare un setting di un ambiente protesico bisogna prima di tutto considerare
la persona come un insieme molteplice di fattori (ambientali, biologici
interpersonali e psico spirituali) e alcuni sono direttamente influenzabili dal
caregiver come il fattore interpersonale. La persona, che apparentemente non
comunica più con il mondo esterno, si dimostra sensibile allo stato d’animo del
suo caregiver o all’ambiente nel quale è inserito o alla cura e igiene personale che
gli viene prestata. Troppo spesso il cargiver dimentica di aver di fronte una
persona considerandola invece una persona privo di volontà e sensibilità.
I compiti del caregiver, ed il carico che ad essi si accompagna, si modificano di
pari passo al decorso della malattia, e spaziano dall’aiuto in compiti strumentali,
all’aiuto in compiti personali, al sostegno emotivo e alla gestione delle
problematiche comportamentali con una supervisione continua del soggetto50.
L’educatore deve dare degli utili e semplici conigli su come interagire con i
malati per poter creare un ambiente sicuro e sereno , inoltre deve sostenere i
caregiver a gestire lo stress e a rilassarsi. Spesso i caregiver, sia formali che
informali, non possiedono una specifica preparazione nella gestione del malato e
devono quindi essere supportati e guidati dall’educatore . Diventano cosi
importanti consigli come: “parlargli mettendosi di fronte a lui e alla sua altezza”,
“stabilire un contatto con lo sguardo”,” toccare il suo corpo”,” usare frasi e parole
molto brevi, semplici e concrete”, “mantenere sempre un atteggiamento positivo e
rassicurante”,” rispettare il bisogno di privacy e la dignità del malato”,” garantire
l’assistenza necessaria senza privare il malato dell’indipendenza”,” rispettare le
abitudini del malato”,” aiutarlo a mantenere una certa indipendenza”,” non
preoccuparsi troppo delle buone maniere e della pulizia”, “assicurare un’ adeguata
idratazione” 51.
Spesso il rapporto con il malato può cambiare in maniera veloce senza apparente
motivazione il caregiver deve essere preparato ad accettare i cambiamenti nel
50 Henderson J. H., Holding T., Mountjoy C., I disturbi mentali degli anziani, Aldo Primerano, Roma, 1990.51 Beck J.C. (tr. It. A cura di C. Vergani) Manuale di gerontologia e geriatria, Masson, Milano 1994

51
rapporto con la persona malata e concentrandosi sui lati positivi del
cambiamento.
L’educatore in collaborazione con i caregiver deve cercare di coinvolgere i
soggetti nelle attività quotidiane, anche se il suo aiuto non è realmente necessario
semplificando il più possibile l’attività, enfatizzando il divertimento e non il
risultato. Le attività non devono comunque durare più di 15-20 minuti ed essere
interrotte ai primi segni di stanchezza e possono spaziare dalle passeggiate al ballo
dal dipingere all’ascolto della musica dal giardinaggio al guardare album
fotografici52.
Infine il compito principale dell’educatore nei confronti del caregiver è di
sostenerlo a livello psicologico nel gestire lo stress ,che il contatto con questi
soggetti comporta, attraverso metodi di rilassamento, training autogeno, il
pensiero positivo.
Non va mai dimenticato che il sorriso è un buon modo di alleviare lo stress: una
tensione può a volte sciogliersi se riusciamo a vedere il lato comico della
situazione.
52 Cayton H., Clausen R., Croy A., Georges J., Gove D., Selmes J., Meulenbergs L., manuale per prendersicura del malato di Alzheimer,Alzheimer Europe, Milano 1999

52

53
ALLEGATI
ALLEGATO 1: GRIGLIA OSSERVAZIONE CENTRO
Griglia di osservazione per il centro SI NOScende tranquillamente dal pulminoCollabora nel levarsi la giaccaRichiama l'attenzione di un operatore solo su di séRichiede l'isolamento in una stanza da soloConsuma in maniera corretta la merendaConsuma la merenda
Partecipa alle attività " proposte "Compie attività riconducibili a prassie del suo passatoSi isola rispetto ai compagniPresenta sbalzi d'umoreMostra affetto verso gli operatori e compagniDimostra interesse verso il mondo che lo circondaRimane immobile per lunghi periodo di tempoSi comporta come vedesse o sentisse persone inesistentiPartecipa alle attività ludiche propostegli
PranzoUsa la forchettaUsa il cucchiaioUsa il coltelloResta seduto durate il pastoMangia tutto ciò che gli viene propostoAfferra correttamente il bicchiereRiesce a bere autonomamenteMuta l'interazione con gli altri dopo il pasto
Si " pianta " (manifesta atteggiamenti oppositivi) durante la mattinataQuante volte? 5 5/10 10/15 15/20 più di 20Si " pianta " (manifesta atteggiamenti oppositivi)durante il pomeriggioQuante volte? 5 5/10 10/15 15/20 più di 20OSSERVAZIONI

54
ALLEGATO 2: GRIGLIA OSSERVAZIONE COMUNITÁ
Griglia di osservazione per la comunità SI NOSi sveglia da soloComincia a fare colazione da soloCambia d'umore dopo colazioneSi veste da soloSi lava mani e viso da soloSi asciuga le mani e il viso dopo averle lavateE' disponibile nelle pratiche quotidiane di igiene intimaScambio verbale " stereotipato "Interagisce con gli altri ragazzi
Cammina autonomamente per un breve percorso
PranzoUsa la forchettaUsa il cucchiaioUsa il coltelloResta seduto durante il pastoMangia tutto ciò che gli viene propostoAfferra correttamente il bicchiereRiesce a bere autonomamenteMuta l'interazione con gli altri dopo il pastoCenaUsa la forchettaUsa il cucchiaioUsa il coltelloResta seduto durante il pastoMangia tutto ciò che gli viene propostoAfferra correttamente il bicchiereRiesce a bere autonomamenteMuta l'interazione con gli altri dopo il pastoRichiama l'attenzione di un operatore solo su di séCollabora durante la docciaCollabora nella igiene dentaleFissa lo sguardo nel vuoto per tempi lunghiAfferra un oggetto con entrambe le mani quando gli è lanciatoLancia oggetti in una direzione definitaHa difficoltà ad addormentarsiEvacua correttamente quando viene portato in bagnoSi bagna durante il giornoSi bagna durante la notteSegnala autonomamente il bisogno di urinareSi " pianta " (manifesta atteggiamenti oppositivi) durante la mattinataQuante volte? 5 5/10 10/15 15/20 più di 20Si " pianta " (manifesta atteggiamenti oppositivi)durante il pomeriggio
Quante volte? 5 5/10 10/15 15/20 più di 20
OSSERVAZIONI

55
ALLEGATO 3: GRIGLIE RIASSUNTIVE

56

57

58

59
ALLEGATO 4: TEST

60

61

62
63
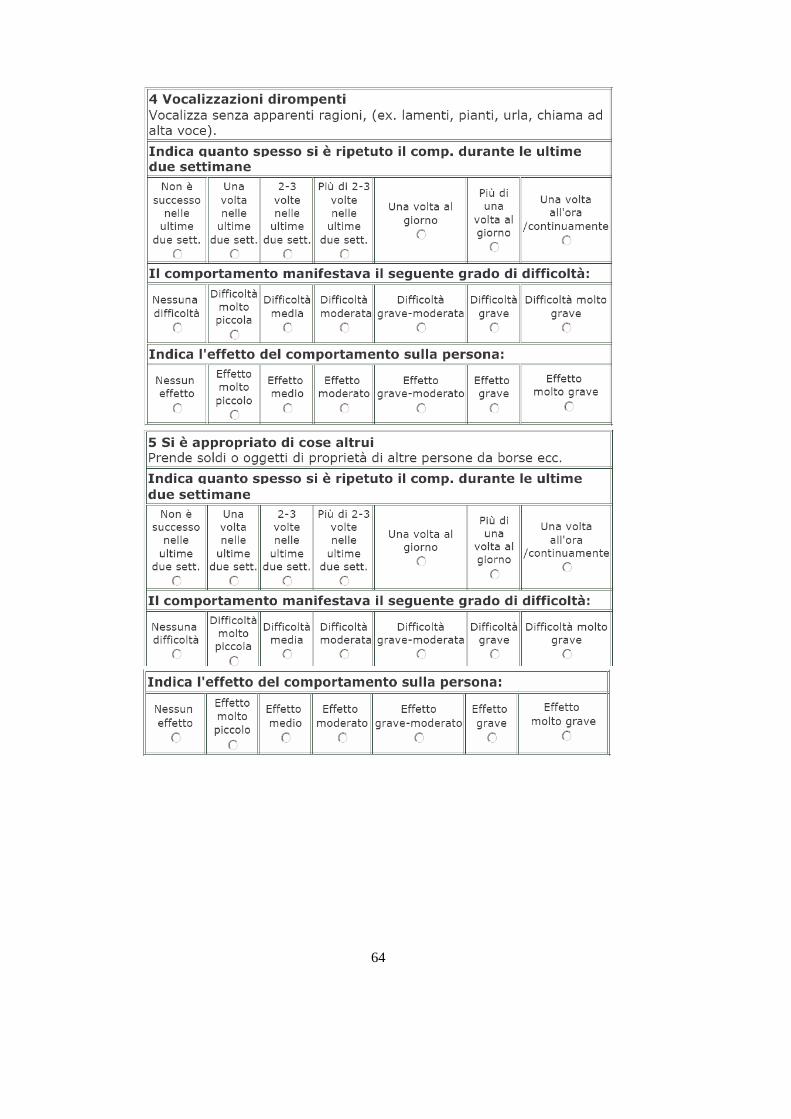
64
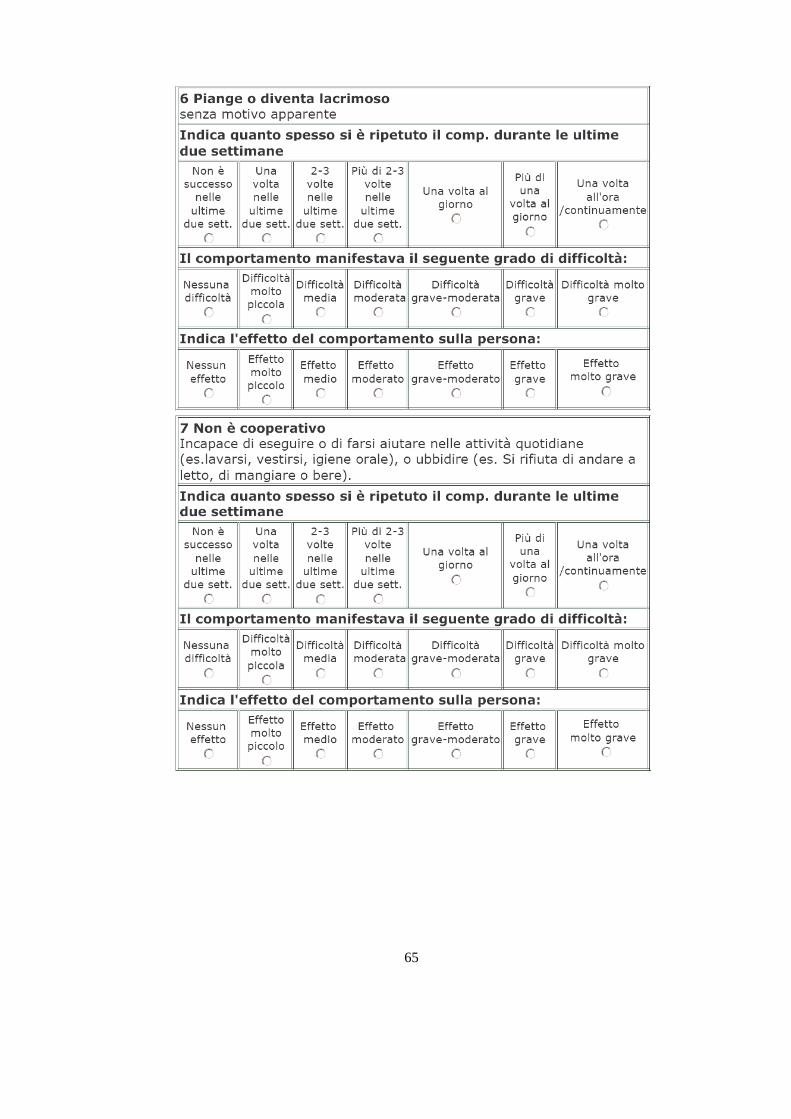
65

66
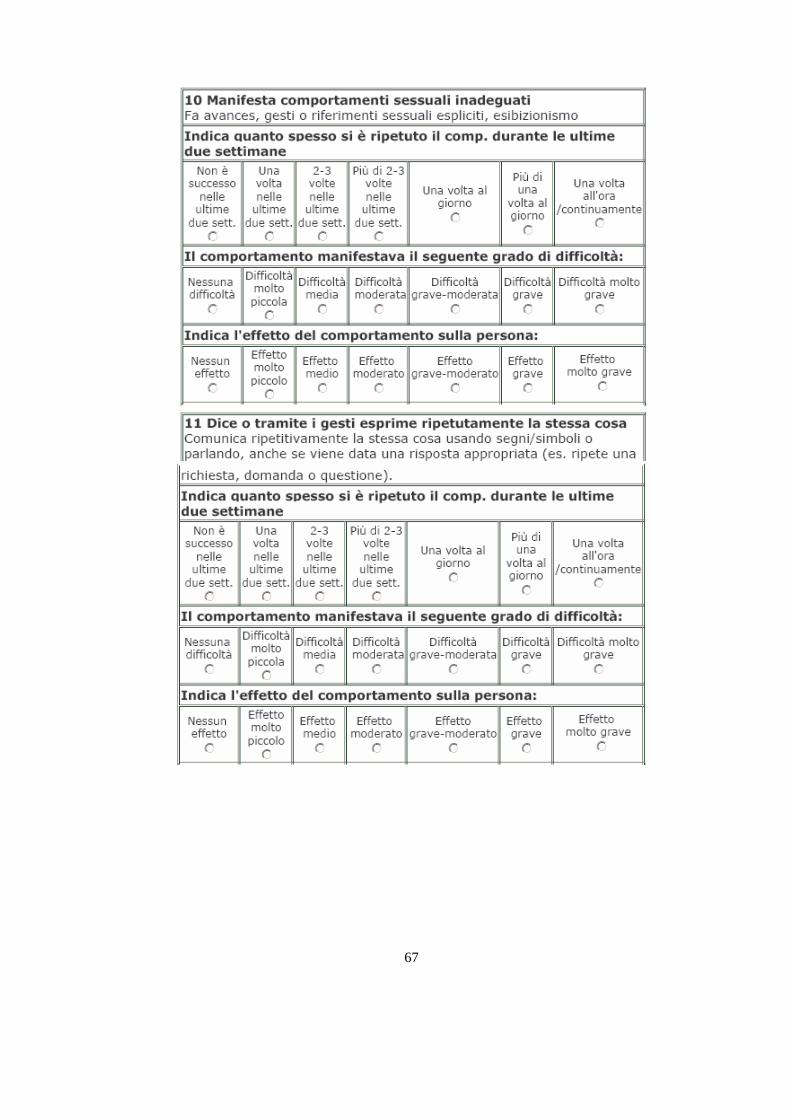
67

68
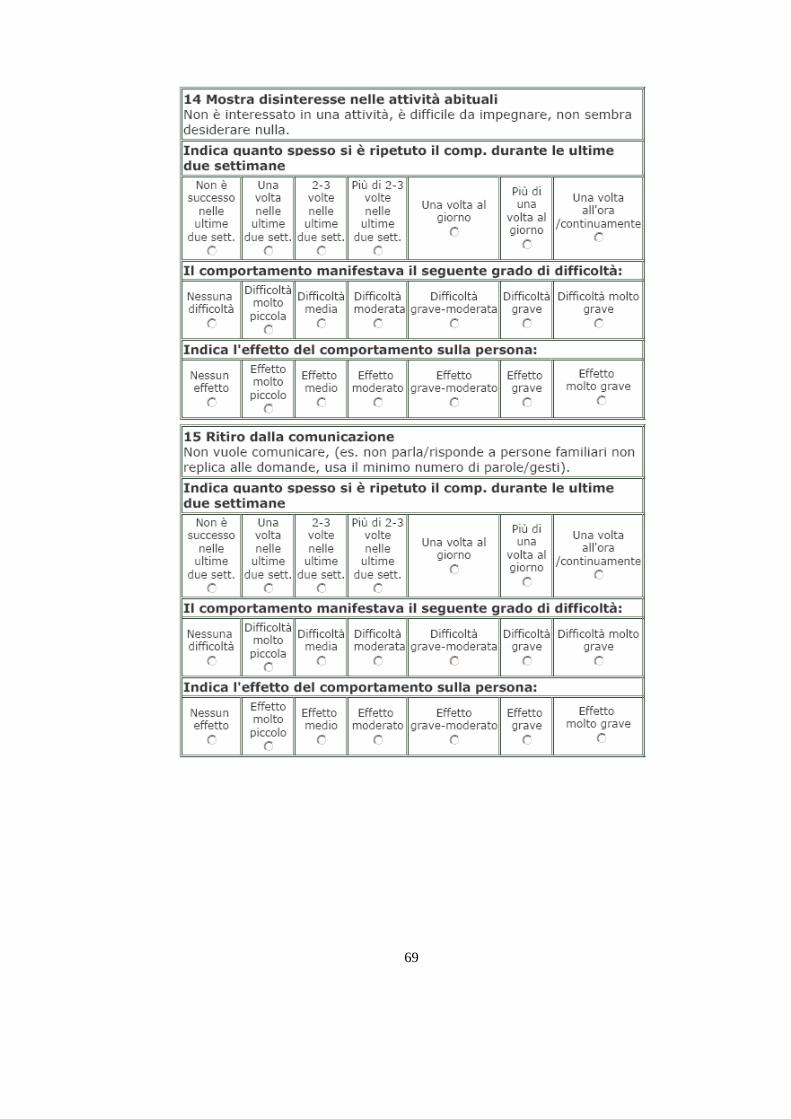
69
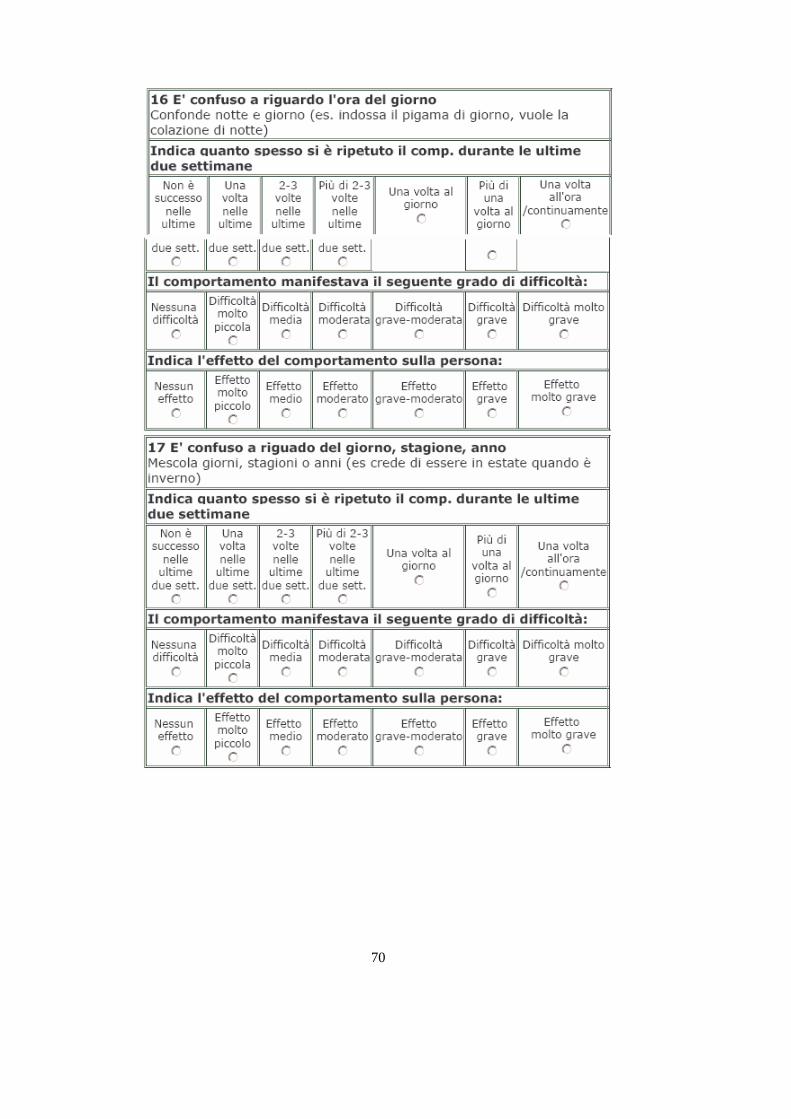
70

71

72
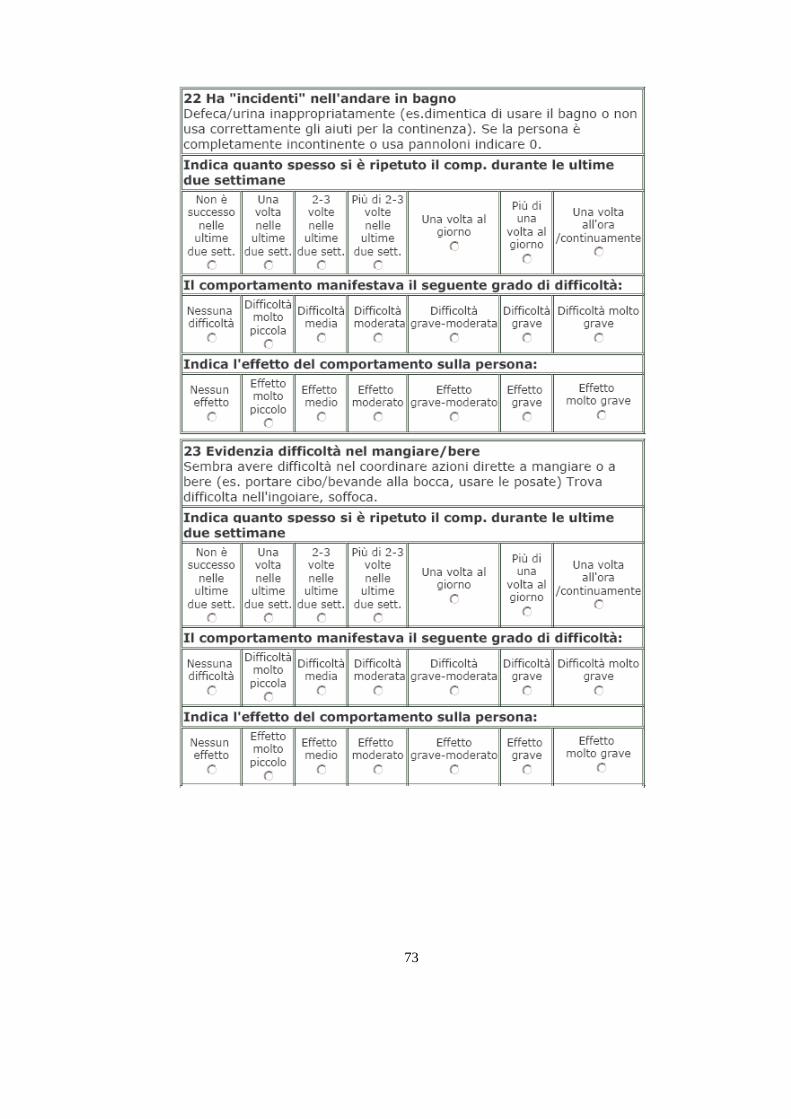
73
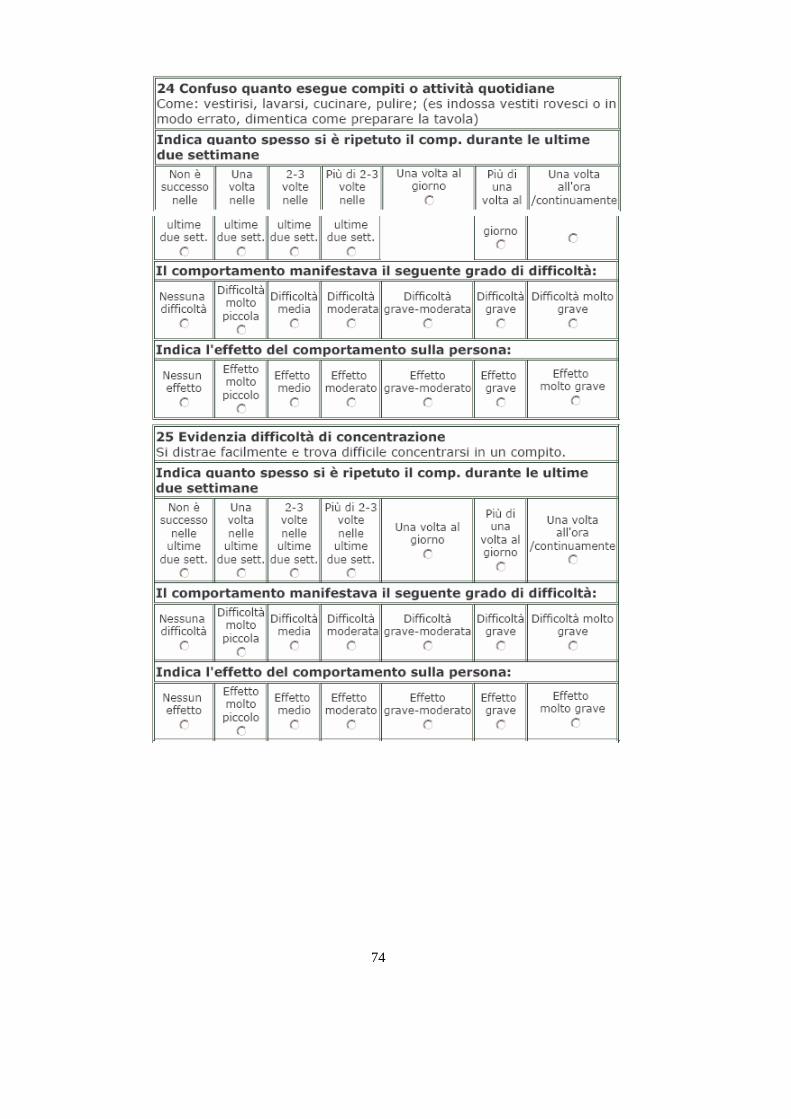
74
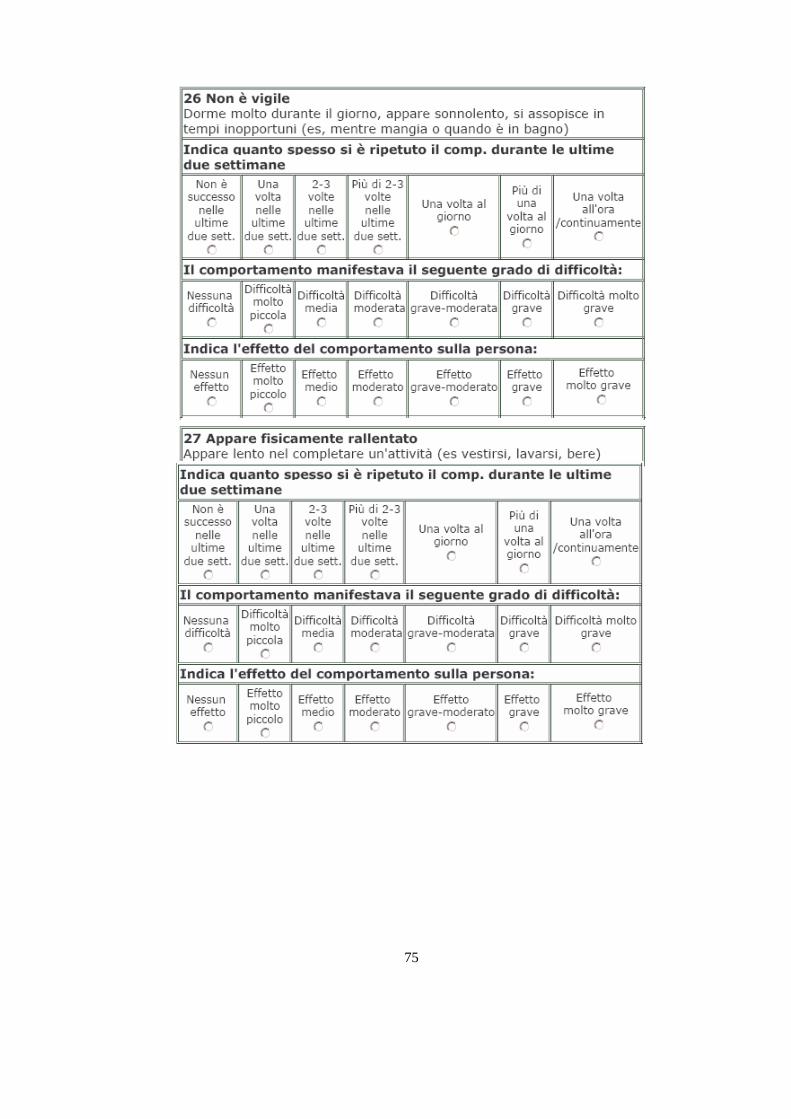
75
76

77

78

79

80

81

82

83

84

85

86

87

88

89

90

91

92

93

94

95

96

97

98

99

100

101

102

103

104

105

106

107

108

109

110

111

112

113

114

115

116

117

118

119

120

121
BIBLIOGRAFIA
Anna Nesi, Dal mongolismo alla trisomia 21, in “Pedagogia clinica”, luglio-dicembre
2000, n°2 pag. 15
Aylward EH, Burt DB, Thorpe LU, Lai F, Dalton AJ. Diagnosis of dementia in
individuals with intellectual disability. J Intell Disab Res 1997;41:152-164.
Baird, P.A. & Sadovnik, P.D. (1987). Life expectancy in Down syndrome. Journal of
Paediatrics, 110, 849-54.
Beck J.C. (tr. It. A cura di C. Vergani) Manuale di gerontologia e geriatria, Masson,
Milano 1994
Biguera s., Pati T., Vigolo S.,Forlin M., Analysis of the impact of some Alzheimer’s
disease risk factors in Down’s sindrome. Abstract dal Congresso Intenazionale di San
Marino 9-11 maggio 2002.
Brown R.I., Quality of life for people with disabilities, Models, Research and Practice,
Chltenham: Stantley Thones 1997
Brugge KI, Nichols SL, Salmon DP et al. Cognitive impairment in adults with Down’s
syndrome: similarities to early cognitive changes in Alzheimer’s disease. Neurology
1994;44:232-238.
Caltagirone C, Bianchetti A, Di Luca M et al., Guidelines for the treatment of
Alzheimer’s disease from the Italian Association of Psychogeriatrics. Drugs Aging
2005;22 (suppl1): S1-S26.
Cayton H., Clausen R., Croy A., Georges J., Gove D., Selmes J., Meulenbergs L.,
manuale per prendersi cura del malato di Alzheimer,Alzheimer Europe, Milano 1999
Contardi A., Un giorno dopo l’altro. Bambini e adulti con la sindrome di Down,
Edizioni Guaraldi, Rimini, 1996
Cfr. Frigerio E., Gallucci M., Folin M., Similarities in the decline of visual-spatial and
attentional abilities between Downs syndrome and Alzheimer’s disease, Abstract dal
Congresso Internazionale di San Marino, 9-11 maggio 2002

122
Cummings B., Pike C., Shanakle R., Cotman C. (1996). Β-amyloid depositino and
other measure of neuropathology predict cognitive status in Alzheimer disease.
Neurobiology of Aging 17, 921-933
De Vreese LP, Belloi L. L’esperienza del Nucleo Specialistico per le Demenze: da un
approccio distile di vita (Life Style Approach) a una qualità di vita migliore del malato
di demenza. ProTerza Età 2002;8:4-11
De Vreese LP, Cester A. Da “Demenza complicata da co-morbilità” a “Comorbilità
complicata da demenza”. Riv Medico Pratico, Marzo 2003, 44-48.
De Vreese LP. Psicoriabilitazione: alternativa sostenibile? Geriatria 2005;27(suppl):
S163-166
Deb S, Braganza J. Comparison of rating scales for the diagnosis of dementia in adults
with Down’s syndrome, J Intellect Disabil Res 1999;43:400–407.
Devenny DA, Silverman WP, Hill AL et al. Normal ageing in adults with Down’s
syndrome: a longitudinal study. J Intellect Disabil Res 1996;40:208–221.
Di Luca P., Pastorino L. , Cattabeni F., Zanardi R., Scarone S., Racagni G., Smeraldi
E. and Perenz J., Abnormal Pattern of Platelet APP Isoforms in Alzheimer Disease
and Down Sindome, Archives of Neurology, 1996, ol.53, pag. 1162-1166.
Elena Zupi, La sindrome di Down: note epidemiologiche e cliniche, Il pensiero
scientifico, Roma 198 Goode D.,Quality of life:Perspective and Issues,American,
Association on Mental Retardation, Washington 1990, pp.41-58.
Evenhuis HM, Kengen MMF, Eurlings HAL, Dementia Questionnaire for Mentally
Retarded Persons (DMR). J Intellect Disabil Res 2004; 40:369–373
Fabian E. Using quality of life indicator in rehabilitation program evaluation,
Rehabilitation Counseling Bulletin 34 (4),1991,pp.334-356
Formica U. Epidemiologia., Formica U, ed. I controlli di salute dei bambini con
Sindrome di Down. Milano, CIS Editore 2003 pp. 9-17.
Gedey A. Manual for the Dementia Scale for Down Syndrome: Gedey Research and
Counseling,Vancouver, Canada, 1995
Gélinas I, Gauthier L, McIntyre M, Gauthier S. Development of a functional measure
for persons with Alzheimer's disease: the disability assessment for dementia. Am J

123
Occupat Ther Janicki MP, Dalton AJ. Alzheimer disease in a select population of
older adults with mental retardation. Irish J Psychol 1993;14:38-47
Henderson J. H., Holding T., Mountjoy C., I disturbi mentali degli anziani, Aldo
Primerano, Roma, 1990
Lay F. Clinicopathologic features of Alzheimer disease in Down syndrome. In L.
Nadel & C.Epstein C. (Eds) Down syndrome and Alzheimer disease. New York:
Wiley-Liss, 1992, pp. 15-34
Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome.
Arch Neurol 1989;46:849–853
Leonard S, Bower B, Petterson B., Survival of infants born with Down’s syndrome:
1980-2000, Paediatr Perinat Epidemiol, 2001, pp. 14:163-71.
Millichap D, Oliver C, McQuillan S, Kalsy S, Lloyd V, Hall S. Descriptive functional
analysis of behavioral excesses shown by adults with Down syndrome and dementia.
Int J Geriatr Psychiatry 2003;18:844-854
Richard Newton, Conoscere e capire la Sindrome di Down in Tea Milano 1998
Trad. it., Cura mentale, igene ed educazione degli idioti e di altri fanciulli ritardati,
Armando, Roma 1970, pp.540
Janicki MP, Heller T, Seltzer G, Hogg J. Practice guidelines for the clinical
assessment and care management of Alzheimer and other dementias among adults
with mental retardation. Washington: American Association on Mental Retardation,
1995.
Jean Rondal,, Juan Perera, Lynn Nadel, La sindrome di Down: conoscenze attuali e
prospettive, Erip Pordenone 2003
John Langdon Down, “Observations on an ethnic classication of idiots” in “Clinical
Lectures and Repoits by the Medical Surcical Staff of the London Hospital”, London,
J. Churchill & sons, 1866 vol. III, p. 260
J.Lejeune, M. Gautier, M. R. Turpin, « Etude des chromosomes somatiques de neuf
enfants mongoliens », in : « Comptes endus hebdomadaires des séances de Académie
des Sciences », Paris, t. 248, n. 11 p.1721-1722.
Jones M. GentleCare. Carocci Editore, Roma, 2005.

124
Jozsvai E. Behavioural and Psychological Symptoms of Dementia in Individuals With
Down Syndrome, J Intellect Disabil Res 2005;12:31-40
Kanamori M, Suzuki M, Yamamoto K. Et al. A day care program and evaluation of
animal-assisted therpay for elderly with senile dementia. Am J Alzheimer Dis Other
Demen 2001;16:234-239
Millichap D, Oliver C, McQuillan S, Kalsy S, Lloyd V, Hall S. Descriptive functional
analysis o behavioral excesses shown by adults with Down syndrome and dementia.
Int J Geriatr Psychiatry 2003;18:844-854.
Perera J. , Social and laboru integration for people with Down’s syndrome, in
J.A.Rondal, J.Perera, L.Nadel e A. Comblain (Eds.), Down’s syndrome:
Psychological, psychobiological and socio – educational perspectives, London 1997,
pp. 219-233.
Prasher VP, Huxley A., Haque MS and the Down syndrome Ageing Study Group. A
24-week, double-blind, placebo-controlled trial of donepezil in patients with Down
syndrome and Alzheimer’s diseaee – Pilot study. Int J Geriatr Psychiatry
2002;17:270-278
Raglio A, Manarolo G, Villani D. Musicoterapia e Malattia di Alzheimer . Ed.
Cosmopolis, 2001
Richard Newton, Conoscere e capire la Sindrome di Down in Tea Milano 1998
Roizen NJ, Patterson D., Down’s Syndrome. Lancet 2003, pp.361
Rosa Ferri Amedeo spagnolo, La sindrome di Down in Il pensiero scientifico, Roma
1989
Steele J. , Epidemiology: incidence, prevalence and size of the Down sindrome
population, in B.Stratford e P.Gunn (Eds.), New approaches to Down syndrome,
London: Lussel 1996
Van Weert JCM, van Dolmen AM, Spreeuwenberg PMM, Ribbe MW, Bensing JM.
Behavioral and mood effects of Snoezelen integrated into 24-hour dementia care. J Am
Geriatr Soc 2005;53:24-33.
Wishart, J.G. (1996). Avoidant learning styles and cognitive development in young
children. In B. Stratford & P. Gunn (Eds.) New Approaches to Down Syndrome.
London: Cassell. pp 173-205

125
Wisniewski HM, Silverman W. Alzheimer’s disease neuropathology and dementia in
Down’s Syndrome. In JA. Ronald, J. Perera, L. Nadel, & A. Comblain (Eds.) Down
syndrome. Psychological, psychobiological, and socio-educational perspectives.
London: Whurr, 1996, pp. 43-50.
SITI INTERNET
www.vividown.org.
www.validazione.eu/dad

126

127
Ringraziamenti
Ringrazio di cuore:
Thomas, mio marito, che mi ha sempre sostenuta e spronata a raggiungere questo
traguardo. Un po’ dottore sei anche tu.
Luca, per essere entrato nella mia vita all’improvviso e averle dato un significato tutto
speciale.
Alla mia mamma un grazie grande come il mondo per l’incoraggiamento che sa
sempre darmi.
Alla Tina ,sorella d’adozione, vera amica, braccio destro e spesso anche sinistro,
consigliatrice fedele, troppo ordinata e perfezionista, brontolona d’eccezione ma se
non ci fosse bisognerebbe inventarla.
Alla Prof.ssa Montani, punto di riferimento, un grazie particolare per la sua
disponibilità, per la passione e l’amore che ci ha trasmesso nel lavoro.
Roberta, amica da una vita, che quando serve spunta sempre e così anche negli anni
dell’università la sua presenza si è rivelata preziosa ed indispensabile.
Angela, Elena, Giovi, Lyuba e Magda insostituibili compagne di studio, pronte sempre
ai full immersion e a darmi una mano, senza di loro non ce l’avrai mai fatta.
A Luca, il mio strano compare, che dal Giappone fa il tifo per me.
Ai colleghi e ragazzi del centro ANFFAS di Primiero che pazientemente mi hanno
supportata e sopportata in questi ultimi mesi e rendono speciale la mia esperienza
lavorativa.


















