
EMATOLOGIA 1 - Aileonlus · staminali ottenuto da un donatore sano capace di ricostituire il...
75
1 EMATOLOGIA direttori della collana Franco Mandelli, Giuseppe Avvisati IL TRAPIANTO DI CELLULE STAMINALI EMOPOIETICHE ALLOGENICHE William Arcese, Anna Paola Iori Dipartimento di Biotecnologie Cellulari ed Ematologia Università “La Sapienza” - Roma 8
Transcript of EMATOLOGIA 1 - Aileonlus · staminali ottenuto da un donatore sano capace di ricostituire il...

1EMATOLOGIAdirettori della collanaFranco Mandelli, Giuseppe Avvisati
IL TRAPIANTO DI CELLULE STAMINALIEMOPOIETICHE ALLOGENICHE
Wil l iam Arcese, Anna Paola Ior i
Dipart imento di Biotecnologie Cel lu lar i ed EmatologiaUniversità “La Sapienza” - Roma
8

ACCADEMIA NAZIONALE DI MEDICINA
REDAZIONE
P.zza della Vittoria, 15/1 - 16121 Genova
Tel. 010/5458611 - Fax 010/541761
E-mail: [email protected]
http: //www.accmed.net
DIREZIONE
Luigi Frati - Stefania Ledda
COORDINAMENTO EDITORIALE
Gabriella Allavena
PROGETTO GRAFICO
Giorgio Prestinenzi
IMPAGINAZIONE
Maria Grazia Granata, Giuliana Vaglio
PROMOZIONE
Luisa Baggiani
SERVIZIO STAMPA
EFFE - Via Cesiolo, 10 - 37126 Verona
© 1998 Forum Service Editore s.c.a r.l.
P.zza della Vittoria, 15/1 - 16121 Genova
Distributore unico per l’Italia:
Del Porto S.p.A. - Via Meucci, 17 - 43015 Noceto (PR)
Tel. 0521/620544 - Fax 0521/627977
Tutti i diritti sono riservati. Nessuna parte del libro può
essere riprodotta o diffusa senza il permesso scritto dell'editore
EMATOLOGIADIRETTORI DELLA COLLANAFranco Mandelli, Giuseppe AvvisatiDipartimento di Biotecnologie Cellulari ed EmatologiaUniversità “La Sapienza”, Roma

GENERALITÀ 1
TRAPIANTO DI MIDOLLO OSSEO ALLOGENICO DA DONATORE FAMILIARE 2
TRAPIANTO DI MIDOLLO OSSEO ALLOGENICO DA DONATORE NON CORRELATO 3
TRAPIANTO DI CELLULE STAMINALI ALLOGENICHE DA SANGUE PERIFERICO 4
TRAPIANTO DI CELLULE STAMINALI EMOPOIETICHEDA SANGUE DI CORDONE OMBELICALE 5
LE COMPLICANZE POST-TRAPIANTO 6
LA GRAFT VERSUS HOST DISEASE 7
LA TERAPIA DI SUPPORTO 8
LA RECIDIVA LEUCEMICA POST-TRAPIANTO 9
BIBLIOGRAFIA GENERALE 10
LE DIAPOSITIVE
INDICE

ADA adenosindeaminasi AR anemia refrattaria AREB AR con eccesso di blastiAREB-T AREB in trasformazione BMDW Bone Marrow Donor WorldwideBUS busulfanoCB crisi blasticaCMV CytomegalovirusCSA ciclosporinaCSE cellula staminale emopoieticaCTX ciclofosfamideCVC catetere venoso centraleDFS sopravvivenza libera da malattiaEFS sopravvivenza libera da eventiEPN emoglobinuria parossistica notturna FA fase accelerataFC fase cronicaG-CSF granulocyte colony stimulating factorGITMO Gruppo Italiano per il Trapianto di Midollo OsseoGM-CSF granulocyte-macrophage colony stimulating factorGVHD graft versus host diseaseGVL graft versus leukemia HLA human leukocyte antigenIFN interferone LAK lymphokine activated killerLDH lattico-deidrogenasiLFS sopravvivenza libera da leucemiaLH linfoma di HodgkinLLA leucemia linfoide acuta LLC leucemia linfoide cronicaLMA leucemia mieloide acutaLMC leucemia mieloide cronicaLMMC leucemia mielomonocitica cronicaLNH linfoma non HodgkinMDS sindrome mielodisplasticaMHC complesso maggiore di istocompatibilitàMM mieloma multiploMTX metotrexatoMUD donatore di midollo non correlatoNK cellule natural killerNR non rispondentiPBSC cellule staminali allogeniche da sangue perifericoPCR polymerase chain reactionPI polmonite interstizialePMN polimorfonucleati PV policitemia veraRC remissione completaRP remissione parzialeSAL siero anti-linfocitarioSCO sangue di cordone ombelicaleTAI toracol abdominal irradiationTBC tubercolosiTBI total body irradiationTMO trapianto di midollo osseoTNI total nodal irradiationTRM mortalità correlata al trapianto VNTR variable number tandem repeatsVOD malattia veno-occlusiva
ABBREVIAZIONI

1
1
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
GENERALITÀ
1.1 L’OBIETTIVO DEL TRAPIANTO ALLOGENICODI CELLULE STAMINALI
Al la base di numerose patologie ematologiche vi è un’alterazioneacquisita (es. aplasia midollare, neoplasie ematologiche) o congenita(emoglobinopatie) del compartimento delle cellule staminali. L’obiettivodel trapianto allogenico di cellule staminali è quello di sostituire ilcompartimento alterato del paziente con un patrimonio di cellulestaminali ottenuto da un donatore sano capace di ricostituire ilsistema emopoietico e immunitario del ricevente. Questo obiettivosi identifica pertanto con la guarigione, e i l suo raggiungimentodipende dalla realizzazione di tre fattori principali:
1. la scomparsa totale del compartimento di cellule staminali totipo-tenti del paziente per mezzo di una chemio-radioterapia pre-tra-pianto (detta di “condizionamento”) il più possibile eradicante per“creare spazio”
2. i l superamento, ai fini dell’attecchimento, della barriera immunologi-ca rappresentata dalle cellule immunocompetenti del paziente chesono responsabili del rigetto
3. i l superamento della barriera immunologica, rappresentata dalle cel-lule immunocompetenti attive del donatore presenti nella sospensio-ne di cellule staminali infuse, responsabili della malattia del trapian-to contro l’ospite (graft versus host disease, GVHD).
Quindi, nel trapianto di cellule staminali allogeniche, a differenza diqualunque altro tipo di trapianto, la barriera immunologica da supe-rare è doppia: del ricevente verso donatore (rigetto) e del donatoreverso ricevente (GVHD).
1.2 CENNI STORICI
Il primo tentativo di impiego di midollo osseo nel trattamento di unapatologia ematologica risale al 1891 quando Brown-Sequard sommini-strò midollo rosso per via orale a un paziente affetto da leucemia acuta(1). Altri tentativi successivi, sporadicamente segnalati, si devono con-siderare più che pionieristici, fino ad arrivare al 1939 quando fu ese-

guita la prima infusione endovenosa di midollo osseo: in quell’anno suAnnals of Internal Medicine fu pubblicato il caso di un paziente affettoda aplasia midollare trattato con infusione endovenosa di midollo otte-nuto da fratello (2).Tuttavia l’inizio della moderna trapiantologia risale alla metà del ‘900con gli studi del 1951 di Jacobson e coll. (3); questi dimostrarono chei topi potevano guarire da un’irradiazione mortale se le aree emopoieti-che del loro femore venivano schermate, avendo osservato in prece-denza che l’aplasia midollare nei topi irradiati poteva essere reversibileschermando la milza. In seguito si osservò che topi in cui erano statesomministrate dosi potenzialmente letali di radiazioni risultavano pro-tetti da un’infusione di midollo e nel 1952 Lorenz e coll. (4) dimostraro-no che la guarigione era dovuta alle cellule contenute nel midollo tra-piantato.Subito dopo la seconda guerra mondiale, gli effetti ematologici osser-vati in seguito all’irradiazione nei sopravvissuti alle bombe atomiche diHiroshima e Nagasaki stimolarono la ricerca sulla potenziale capacitàdel midollo osseo di conferire una radioprotezione.I più gravi problemi che i clinici si trovarono ad affrontare furono quelliimmunologici del rigetto e della reazione del trapianto contro l’ospite,descritta per la prima volta nell’uomo da Mathè e coll. (5).Una parte consistente del lavoro sullo sviluppo del trapianto di midolloosseo (TMO) è stata inoltre svolta da Donall Thomas, premio Nobel perla Medicina nel 1990, che ha usato il cane come modello sperimentaleper sviluppare schemi efficaci di irradiazione “total body” e ha introdot-to il metotrexato (MTX) come mezzo per prevenire la GVHD (6).
Questi progressi tecnici e la caratterizzazione del sistema di istocompa-tibilità (HLA) hanno definitivamente aperto la strada a una nuova eratrapiantologica, portando alla realizzazione del primo trapianto sullabase delle nuove conoscenze in un paziente affetto da sindrome diWiskott-Aldrich, esperienza pubblicata nel 1968 da Bach e coll. (7),
1.3 IL CONCETTO DI CHIMERA
La sostituzione del compartimento staminale del paziente con le celluledel donatore determina la convivenza nello stesso individuo delpatrimonio genetico di due soggetti differenti; il ricevente in questocaso diventa genotipicamente una chimera (termine mutuato dal lamitologia classica per definire una creatura con parti anatomiche deri-vate da individui differenti).Inoltre, la cellula staminale non è presente solo a livello midollare o nelsangue periferico di un individuo, ma è rappresentata in numerosi altri
2E M A T O L O G I A

1
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
3
tessuti: da essa derivano, infatti, i macrofagi degli alveoli polmonari, lecellule del Kupffer del fegato, gli osteoclasti, le cellule del Langheransdella cute, le cellule microgliari del cervello e, come dimostrato deltutto recentemente, anche le cellule muscolari striate (8).Questa caratteristica rende ragione dell’impiego del trapianto allogeni-co di cellule staminali emopoietiche (CSE) anche in pazienti affetti daerror i congenit i del metabol ismo (tesaurismosi l isosomiale). Infatt iquando si esegue un trapianto allogenico in un ricevente sottoposto aregime ablativo, le CSE trapiantate danno origine a tutte le discenden-ze emopoietiche, inclusa la l inea dei monociti come precursori deimacrofagi tissutali che si trovano a livello polmonare, cutaneo o epati-co. Sulla base di questa acquisizione è stato proposto che un trapian-to allogenico di CSE possa servire da fonte permanente dell’enzimamancante e correggere il difetto metabolico o per sostituzione dellecellule fagocito-macrofagiche del fegato portatrici del deficit enzimati-co con cellule normali, o trasferendo l’enzima per contatto diretto cel-lula-cellula dalle cellule derivate dal midollo osseo enzimaticamentenormale in quelle patologiche, o mediante rilascio dell’enzima nel pla-sma con successiva captazione da parte delle cellule carenti.
Di fatto quindi il trapianto allogenico di CSE può essere conside-rato un trapianto sistemico.L’analisi del chimerismo post-trapianto può essere condotta attra-verso l’uso di anal is i citogenetiche classiche (sesso dif ferente tradonatore e ricevente, polimorfismi di bandeggio all’analisi cariotipi-ca, eventuale presenza nei pazienti con patologie clonali di anomaliecitogenetiche caratteristiche), oppure mediante determinazione deigruppi eritrocitari o tipizzazione HLA per trapianti tra soggetti nonidentici. Più recentemente vengono impiegate metodiche di biologiamolecolare, attraverso tecniche di reazione a catena della polimerasi(polymerase chain reaction, PCR), che amplificano regioni del geno-ma umano altamente polimorfiche (quali ad esempio variable numbertandem repeats, VNTR). Attraverso queste metodiche, applicabili siasu cellule midollari che su sangue periferico, è possibile stabil ire i lchimerismo post-trapianto e soprattutto seguirne l’andamento neltempo. In base al la persistenza o meno di cel lule del r icevente al ivel lo midollare o periferico si distinguono tre possibili differentistati chimerici:
• chimerismo completo (assenza di residuo cellulare emopoieticodel paziente)
• chimerismo misto (concomitante presenza di cellule del donatoree del ricevente)
• assenza di chimerismo (ricostituzione emopoietica autologa).

1.4 IL SISTEMA HLA
I geni che esercitano un effetto primario sulle reazioni umorali e cellu-lari determinanti la compatibilità tessutale sono raggruppati in uncomplesso cromosomico che prende il nome di complesso mag-giore di istocompatibil ità (MHC) (nell’uomo viene siglato HLA:human leukocyte antigen) localizzato sul braccio corto del cromo-soma 6. Si conoscono oggi diverse famiglie di geni HLA che si rag-gruppano in regioni distinte del complesso. Si distinguono: la regioneABC, dove trovano sistemazione i geni di classe I; la regione DR, dovesono localizzati i geni di classe II; e infine la regione di classe III checomprende i geni che codificano per alcune frazioni del complemento.Il complesso HLA, salvo l’evenienza del crossing over che interviene incirca il 3% dei casi, si trasmette come un blocco unico di informa-zione genetica secondo la I legge di Mendell. La combinazione deigeni sullo stesso cromosoma si chiama “aplotipo”; i l genotipoconsta dei due aplotipi parentali (paterno e materno) e viene stabili-to esclusivamente con indagine familiare.I prodotti dei geni HLA caratterizzati inizialmente con metodiche sie-rologiche e cellulari, vengono più recentemente identificati con tecni-che di biologia molecolare. Questi prodotti, conosciuti comunementecome “antigeni HLA”, presentano le caratteristiche di un polimor-fismo molto elevato , i l più esteso noto nel l ’uomo. Attualmente i lnumero dei possibili geni e quindi degli antigeni nei vari loci HLA sono:25 nel locus A, 51 nel locus B, 12 nel locus C, 18 nel locus DR, 10 nellocus DQ e infine 6 nel locus DP. Ciò comporta un altissimo numero dicombinazioni aplotipiche e un numero illimitato di genotipi. Il polimor-fismo è tuttavia ristretto in ambito familiare dove esiste una pro-babilità di identità HLA in circa il 30% dei fratelli.Gli antigeni di classe I sono glicoproteine di membrana, altamente poli-morfe, cost ituite da una catena pesante saldamente inserita nel lamembrana cellulare e da una catena leggera rappresentata dalla b2-microglobulina, un polipeptide non polimorfo, e non glicosilato, codifi-cato da un gene situato sul cromosoma 15. Sono presenti su tutte lecellule nucleate e sulle piastrine, ma la loro espressione varia nei diver-si tessuti e nelle differenti categorie di cellule. La massima espressionesi ha sui linfociti dove rappresentano l’1% circa di tutte le proteine dimembrana. Dal punto di vista funzionale gli antigeni di classe I sonoantigeni di trapianto che rappresentano il bersaglio per i linfociticitotossici T nelle reazioni di rigetto.Gli antigeni di classe II hanno distribuzione ristretta, sono infatti pre-senti solo su certe linee cellulari: linfociti B, macrofagi, cellule dell’epi-telio timico, alcuni progenitori delle cellule mieloidi, una certa quota dilinfociti T attivati, cellule del Langherans. Sono composti anch’essi da
4E M A T O L O G I A

1
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
due catene polipeptidiche glicosilate: una catena più lunga e una piùcorta legate tra loro in modo non covalente. Sono state individuate trefamiglie di antigeni di classe II: antigeni HLA-DR, DQ e DP. Gli antige-ni di classe II sono essenzialmente coinvolti nelle cooperazioni frale varie popolazioni di cellule immunocompetenti per la regolazio-ne della risposta immune. I linfociti T, infatti, possono riconoscere unantigene estraneo soltanto se esso forma un complesso con un anti-gene HLA sulla cellula presentante l’antigene (tipicamente un macrofa-go). La funzione di indurre il riconoscimento di un antigene daparte dei linfociti T è propria degli antigeni di classe II per quantoriguarda la popolazione T4 mentre sarebbe svolta dalle molecoledi classe I per la sottopopolazione T8.
1.5 INDICAZIONI
Nella Tabella 1 sono schematicamente rappresentate le patologieneoplastiche e non neoplastiche per le quali è stato impiegato il tra-pianto allogenico di CSE.
1.6 LE FONTI DI CELLULE STAMINALI ALLOGENICHE
Il trapianto di cellule staminali allogeniche rappresenta ormai una pro-cedura terapeutica consolidata nel trattamento di numerose emopatiesistemiche sia neoplastiche che non neoplastiche in pazienti di età<60 anni.Tuttavia la possibilità di reperire un donatore HLA compatibile nel-l’ambito familiare è di circa il 30%, e tale probabilità può essereestesa a un ulteriore 10% dei casi se si includono anche donatorifamiliari incompatibili per un solo locus.A un’ampia proporzione di pazienti, eleggibili per un trapianto di cellu-le staminali allogeniche, calcolata nell’ordine del 70% circa dei casi,rimarrebbe pertanto preclusa la possibilità di usufruire di tale procedu-ra terapeutica.La disponibilità di donatori volontari di cellule staminali da midol-lo osseo reperibili nell’ambito dei Registri Internazionali dei donatori dimidollo (Bone Marrow Donor Worldwide) ha permesso di risponderealla richiesta di un ulteriore 40% circa dei pazienti. Tuttavia, iltempo per la r icerca di un donatore volontar io nei registr i èmediamente di circa 4-6 mesi, spesso troppo lungo per le esigen-ze cliniche del paziente. Inoltre le frequenze HLA rappresentate nei
5

6E M A T O L O G I A
Patologie neoplasticheLeucemia mieloide acutaLeucemia linfoide acutaLeucemia mieloide cronicaLeucemia mielomonocitica cronicaLeucemia mieloide cronica giovanileSindrome mielodisplasticaMielofibrosiOsteomielosclerosiPolicitemia veraLinfoma non HodgkinLinfoma di HodgkinLeucemia a cellule capelluteMieloma multiplo Leucemia linfoide cronica
Patologie non neoplasticheAnemia aplastica graveEmoglobinuria parossistica notturnaAnemia di FanconiAnemia di Blakfan-DiamondTalassemieAnemia falciformeAltre emoglobinopatieImmunodeficienza grave combinataCarenza di adenosindeaminasiDiscinesia reticolareAtassia teleangiectasiaSindrome di Wiskott AldrichMalattia di De GeorgeMalattia granulomatosa cronicaSindrome di Chediak-HigashiSindrome linfoproliferativa legata al cromosoma XDeficienza di adesione dei leucocitiOsteopetrosi
Disordini metabolici geneticiMucopolisaccaridosiSindrome di HurlerSindrome di ScheieSindrome di HunterSindrome di San FilippoSindrome di MorquioSindrome di Maroteaux-LamiDeficienza di beta-glucuronidasiMalattia di GaucherLeucodistrofia metacromaticaMalattia di KrabbeMalattia di Nieman-PickTesaurismosi lisosomialiIstiocitosi XEmofagocitosi linfoistiocitosi familiareEmofagocitosi
Tabella 1 Patologie nelle quali è stato impiegato il trapianto allogenico di cellule staminali emopoietiche
1
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
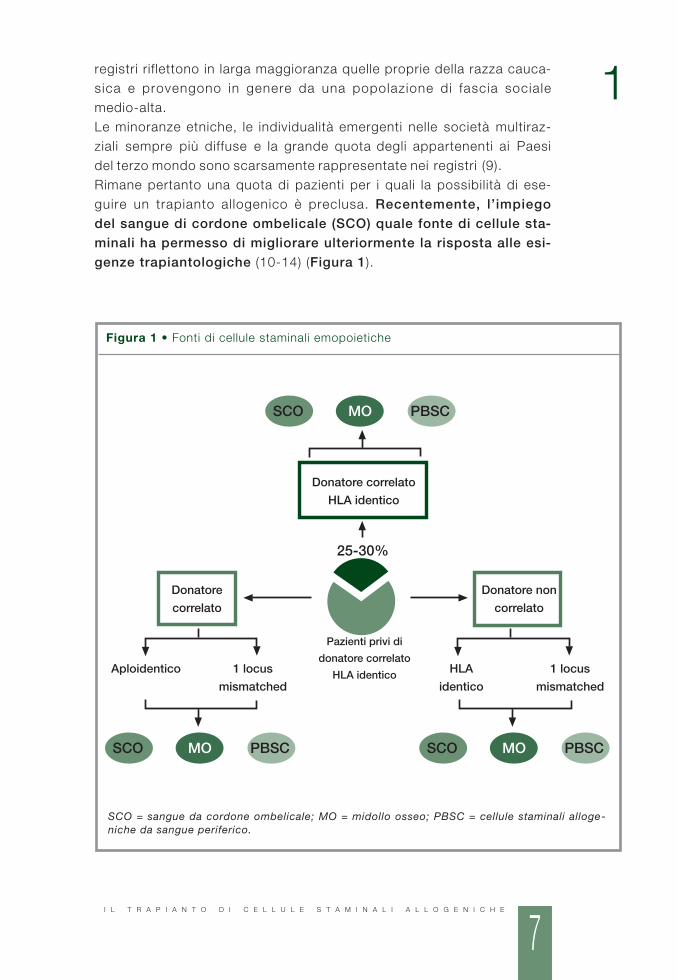
registri riflettono in larga maggioranza quelle proprie della razza cauca-sica e provengono in genere da una popolazione di fascia socialemedio-alta.Le minoranze etniche, le individualità emergenti nelle società multiraz-ziali sempre più diffuse e la grande quota degli appartenenti ai Paesidel terzo mondo sono scarsamente rappresentate nei registri (9).Rimane pertanto una quota di pazienti per i quali la possibilità di ese-guire un trapianto allogenico è preclusa. Recentemente, l’impiegodel sangue di cordone ombelicale (SCO) quale fonte di cellule sta-minali ha permesso di migliorare ulteriormente la risposta alle esi-genze trapiantologiche (10-14) (Figura 1).
7
Donatore correlato
HLA identico
25-30%
Pazienti privi di
donatore correlato
HLA identico
Donatore
correlato
Donatore non
correlato
Aploidentico 1 locus
mismatched
SCO MO PBSC
SCO MO PBSC SCO MO PBSC
1 locus
mismatched
HLA
identico
Figura 1 • Fonti di cellule staminali emopoietiche
SCO = sangue da cordone ombelicale; MO = midollo osseo; PBSC = cellule staminali alloge-niche da sangue periferico.

1.7 I TIPI DI TRAPIANTO
Il trapianto di cellule staminali allogeniche viene definito dalle seguenticaratteristiche:• fonte di cellule staminali (midollo osseo, sangue periferico, sangue
da cordone ombelicale)• familiarità con il donatore o disponibilità di un donatore non familia-
re (o non correlato) • grado di compatibilità che dipende dal numero di antigeni A, B e
DR uguali tra donatore e ricevente.
Sulla base di queste caratteristiche, e utilizzando come fonte di cellulestaminali il midollo osseo, il sangue periferico o il sangue da cordoneombelicale, si distinguono i seguenti tipi di trapianto allogenico:• trapianto singenico (da gemello monocoriale)• trapianto allogenico da donatore familiare HLA compatibile• trapianto da donatore familiare HLA non compatibile• trapianto da donatore non familiare HLA compatibile• trapianto da donatore non familiare HLA non compatibile.
1.8 LE IMPLICAZIONI IMMUNOLOGICHE
Dopo l’infusione di cellule staminali allogeniche possono manifestarsitre effetti immunomediati: il rigetto, la GVHD e la graft versus leukemia(GVL).Il fenomeno del rigetto si instaura allorché le cellule midollari deldonatore, riconosciute come non proprie (“non self”), vengonoattaccate e distrutte dalle cellule immunocompetenti del riceven-te. La profonda immunosoppressione indotta dalle alte dosi di radio-chemioterapia pre-trapianto riduce, tuttavia, l’incidenza del rigetto nelTMO allogenico non T depleto da donatore HLA compatibile all’1-2%dei casi. Il rigetto rappresenta un problema maggiore nei trapiantida donatore non familiare o nei trapianti non compatibili. La reattività delle cellule immunocompetenti allogeniche contro itessuti dell’ospite determina l’effetto GVHD.Inoltre è ben noto l’effetto immunomediato di reazione del tra-pianto verso la leucemia (GVL) che interviene in associazione alregime di condizionamento pre-trapianto, nel prevenire la recidivaleucemica.Numerose sono le evidenze, sia sull’uomo che su modelli animali, chericonoscono una stretta correlazione tra il fenomeno della GVHD e l’ef-fetto GVL (15-17). La probabilità di recidiva leucemica risulta significa-tivamente ridotta nei pazienti con GVHD acuta e cronica rispetto ai
8E M A T O L O G I A

1
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
pazienti senza GVHD. In letteratura sono riportati singoli casi di recidi-va leucemica post-trapianto nei quali la comparsa della GVHD, insortaspontaneamente o dopo la sospensione della terapia immunosoppres-siva, risultava associata alla remissione della malattia leucemica (18,19). Inoltre, la comparazione tra trapianto singenico e allogenicoha fornito molti dati indiretti sul rapporto GVL-GVHD: l’incidenzadella recidiva leucemica risulta più alta nei pazienti sottoposti atrapianto singenico, che non sono a rischio di GVHD, rispetto aipazienti che ricevono trapianto allogenico (20). Esiste comunqueun effetto GVL legato esclusivamente alla natura allogenica del tra-pianto: è stato infatti osservato che, rispetto ai riceventi trapianto sin-genico, l’incidenza della recidiva leucemica risulta ridotta nei pazientitrapiantati con midollo allogenico anche in assenza di GVHD (21).A differenza di quanto riportato in modelli animali nei quali è possibileottenere cellule di origine del donatore con esclusiva attività antileuce-mica e prive di reattività verso i tessuti normali dell’ospite, nell’uomonon sono ancora perfettamente note le popolazioni cellulari coinvoltenel meccanismo fisiopatologico della GVL e della GVHD. Van Locheme coll. nel 1992 (22), esaminando le popolazioni linfocitarie di pazienticon GVHD post-trapianto, hanno distinto tre cloni funzionalmente dif-ferenti di linfociti T citotossici:1. cloni del donatore diretti sia contro antigeni minori del sistema HLA
dell’ospite sia contro le cellule leucemiche2. cloni che riconoscono solo i linfociti del sangue periferico dell’ospi-
te ma non le cellule leucemiche3. cloni d i ret t i esc lus ivamente contro le ce l lu le neoplast iche del
paziente.
Questi risultati deporrebbero per la presenza di cellule effettrici siadistinte che comuni nell’esprimere le due attività GVL e GVHD. L’azione citotossica antileucemica sembrerebbe espressa da clonidi linfociti T sia CD4+ che CD8+, ad attività ristretta per gli antigenidi I e di II classe dell’MHC. Gli antigeni target potrebbero essere anti-geni minori del sistema di istocompatibilità presenti sulle cellule leuce-miche ma anche neo-peptidi prodotti dalle traslocazioni cromosomicheo proteine glicosilate o fosforilate in maniera anomala (22-24).È probabile che siano responsabili dell’attività GVL anche altre popo-lazioni cellulari quali natural killer (NK), cellule LAK, e che siano inoltrecoinvolte alcune citochine o con meccanismo diretto antileucemico omediante reclutamento di cellule accessorie o potenziando la citotos-sicità cellulare (25-27).L’identificazione fenotipica e funzionale delle cellule responsabili del-l’effetto antileucemico potrebbe consentire manipolazioni tali da inten-sificare la GVL rispetto alla GVHD.Il tentativo di rimuovere i linfociti T dal midollo del donatore (tra-
9

pianto T depleto) ha nettamente ridotto l’incidenza della GVHD,favorendo però un netto aumento della recidiva leucemica (28-30).Tuttavia, come riportato da Champlin e coll. (31), la deplezione seletti-va di cellule CD8+ dal midollo infuso associata alla somministrazionedella ciclosporina (CSA), sembra determinare una significativa riduzio-ne del la GVHD acuta senza incrementare la recidiva leucemica inpazienti trapiantati con leucemia mieloide cronica (LMC). Tuttavia ulte-riori studi sono necessari per il miglioramento dell’applicazione clinicadi tali procedure.
1.9 IL CONDIZIONAMENTO PRE-TRAPIANTO
Il regime di condizionamento pre-trapianto, detto anche regime ablati-vo, ha l’obiettivo di eradicare la malattia di base e immunosoppri-mere il paziente. Tale scopo si ottiene mediante l’associazione di far-maci chemioterapici o impiegando regimi di condizionamento cheassocino la chemioterapia a trattamenti radianti.
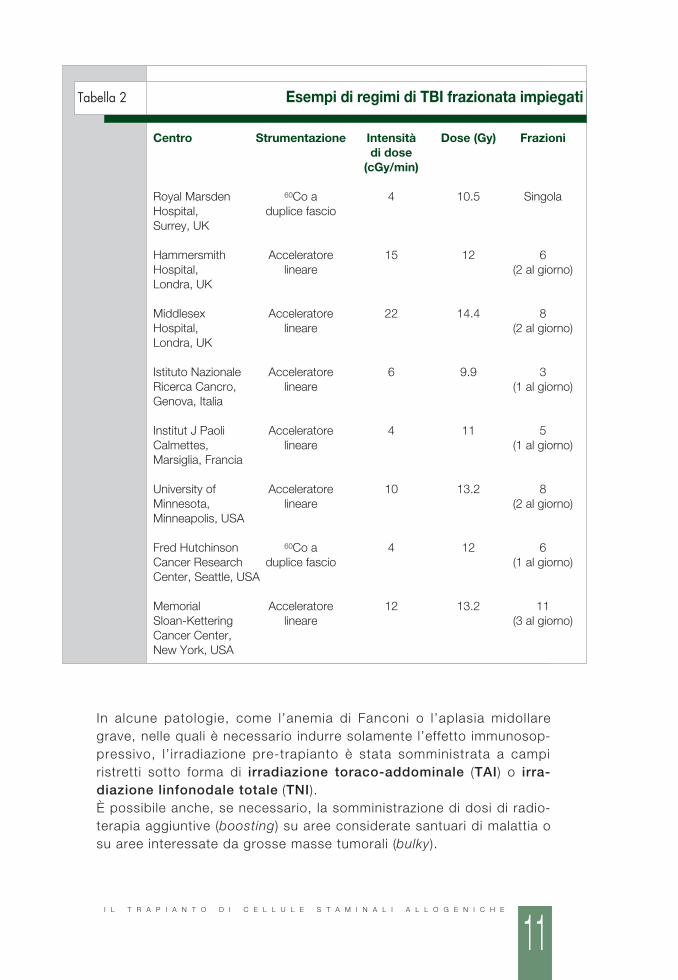
■ IRRADIAZIONE CORPOREA TOTALEL’impiego dell’irradiazione corporea totale (total body irradiation, TBI)nel regime di condizionamento pre-trapianto ha un effetto immunosop-pressivo e antitumorale; in particolare la TBI agisce anche sulle cellulein fase G0 del ciclo cellulare e sulle cellule del sistema nervoso centralee dei testicoli (considerati santuari di malattia).La TBI può essere in dose singola, quando la quantità di radiazioniviene somministrata in un’unica volta, oppure frazionata, quando ladose totale di radiazioni viene somministrata suddivisa in più giorni.La dose totale e la sua intensità sono variabili. In generale i valori sonopiù a l t i ne l la TBI f raz ionata r ispet to a que l la in f raz ione s ingo la(Tabella 2). Quando la TBI si uti l izza per emopatie non maligne, ladose totale è solitamente più bassa, non essendo necessaria l’elimina-zione di cellule tumorali, ma solo l’effetto immunosoppressivo.
Molti centri impiegano la schermatura del polmone, in modo da ridurrela dose totale erogata su questi organi e quindi i danni da radiazione.Durante le prime ore della TBI gli effetti collaterali più facilmente osser-vabili sono la nausea e il vomito, nelle 24-48 ore successive si posso-no osservare eritema cutaneo, dolore mascellare (parotide), mucosite;più tardiva è l’insorgenza dell’alopecia, della sindrome da sonnolenzao della malattia veno-occlusiva (VOD). I possibili effetti tardivi della TBIsono la steril ità, l’ipotiroidismo, la cataratta, l’insorgenza di seconditumori e la polmonite interstiziale (PI).
10E M A T O L O G I A
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
In alcune patologie, come l’anemia di Fanconi o l’aplasia midollaregrave, nelle quali è necessario indurre solamente l’effetto immunosop-pressivo, l’ irradiazione pre-trapianto è stata somministrata a campiristretti sotto forma di irradiazione toraco-addominale (TAI) o irra-diazione linfonodale totale (TNI).È possibile anche, se necessario, la somministrazione di dosi di radio-terapia aggiuntive (boosting) su aree considerate santuari di malattia osu aree interessate da grosse masse tumorali (bulky).
11
Centro Strumentazione Intensità Dose (Gy) Frazionidi dose
(cGy/min)
Royal Marsden 60Co a 4 10.5 SingolaHospital, duplice fascioSurrey, UK
Hammersmith Acceleratore 15 12 6 Hospital, lineare (2 al giorno)Londra, UK
Middlesex Acceleratore 22 14.4 8Hospital, lineare (2 al giorno)Londra, UK
Istituto Nazionale Acceleratore 6 9.9 3Ricerca Cancro, lineare (1 al giorno)Genova, Italia
Institut J Paoli Acceleratore 4 11 5Calmettes, lineare (1 al giorno)Marsiglia, Francia
University of Acceleratore 10 13.2 8Minnesota, lineare (2 al giorno)Minneapolis, USA
Fred Hutchinson 60Co a 4 12 6Cancer Research duplice fascio (1 al giorno)Center, Seattle, USA
Memorial Acceleratore 12 13.2 11Sloan-Kettering lineare (3 al giorno)Cancer Center, New York, USA
Tabella 2 Esempi di regimi di TBI frazionata impiegati

■ FARMACI CHEMIOTERAPICIAnche per la chemioterapia pre-trapianto, come per la TBI, gli obiettivipr incipal i sono creazione di spazio, eradicazione del la malatt ia eimmunosoppressione.Ovviamente l’ablazione pre-trapianto è fondamentale per le patologiecon midollo iperplastico, come nelle forme leucemiche o nelle talasse-mie, mentre è di minore importanza nei casi di midollo ipoplasticocome nel caso dell’aplasia midollare, dove è fondamentale invece l’ef-fetto immmunosoppressivo. I farmaci solitamente impiegati per l’abla-zione midollare sono il busulfano (BUS), l’etoposide, la citosina-arabi-noside e i l melphalan, mentre la ciclofosfamide (CTX), ampiamenteusata in molti regimi di condizionamento, ha prevalentemente un effettoimmunosoppressivo, ma non è in genere suff iciente a determinareun’ablazione midollare dell’ospite tranne che in presenza di uno statomidollare ipoplasico.In relazione alla patologia di base, per ottenere l’effetto sperato dal regi-me di condizionamento (ablazione + immunosoppressione), è fonda-mentale, rispetto all’impiego di una monochemioterapia, l’associazionestrategica di più farmaci in modo da ridurre la probabilità di una resi-stenza nei confronti di qualcuna delle sostanze impiegate. Inoltre l’im-piego di associazioni chemioterapiche può ridurre la morbilità globalelegata alla tossicità dei farmaci rispetto all’equivalente morbilità che siavrebbe utilizzando un unico agente per ottenere lo stesso effetto tera-peutico. Infatti, i chemioterapici impiegati nei regimi di condizionamentovengono somministrati a dosi sovramassimali e gli effetti tossici a essiassociati possono essere particolarmente gravi (PI, cardiotossicità, epa-totossicità, VOD, cistite emorragica, crisi convulsive, ecc.).In relazione al tipo di trapianto è fondamentale, in alcuni casi, privilegia-re o eventualmente incrementare, mediante l’impiego di farmaci aggiun-tivi, l’effetto immunosoppressivo per ridurre il rischio del rigetto o delnon attecchimento, come nel caso dei trapianti da SCO o da MUD com-patibile o da donatore nei quali alla terapia citoriduttiva vengono aggiun-ti il siero anti-linfocitario (SAL) o la fludarabina.
1.10 ATTECCHIMENTO
La ripresa emopoietica dopo trapianto di cellule staminali alloge-niche dipende da vari fattori quali: la malattia di base, il regime dicondizionamento pre-trapianto, la profilassi della GVHD, la com-parsa di eventuali infezioni virali (Cytomegalovirus, CMV), il nume-ro di cellule infuse. Anche se la cellularità midollare incrementa rapidamente dopo circa 2-4 settimane dal trapianto e morfologicamente sono presenti tutte le
12E M A T O L O G I A

1
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
componenti emopoietiche, occorrono 6-12 mesi prima che la cellula-rità ritorni normale (32). L’attecchimento viene definito dal valore dei polimorfonucleati(PMN) delle piastrine e dei reticolociti a livello del sangue periferi-co. Convenzionalmente l’attecchimento per la serie granulocitaria èdefinito dal numero dei PMN, >500/mm3 per almeno tre giorni conse-cutivi, mentre per le piastrine da una conta superiore a 50 000/mm3 eper la serie rossa da un numero di reticolociti superiore a 25 000/mm3
sempre su tre controlli consecutivi in tre giorni successivi.La perdita dell’attecchimento è definita dalla riduzione dei PMN al disotto dei 200/mm3 e dalla cellularità midollare <5% dopo il raggiungi-mento di un normale attecchimento granulocitario.Non ci sono linee guida ben definite per l’impiego dei fattori dicrescita dopo trapianto.
13

I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
TRAPIANTO DI MIDOLLOOSSEO ALLOGENICO DADONATORE FAMILIARE
2.1 INTRODUZIONE
■ CRITERI DI SCELTA DEL DONATOREIn Tabella 3 sono elencati i criteri di scelta solitamente impiegati per ildonatore di midollo osseo allogenico.
■ SCREENING PRE-TRAPIANTO DEL DONATORE DI MIDOLLO OSSEO
Poiché per eseguire il prelievo di midollo osseo il donatore viene sot-toposto ad anestesia generale o epidurale le indagini pre-espiantonecessarie per il donatore sono per lo più quelle richieste per l’aneste-sia generale oltre a uno screening infettivologico (Tabella 4).
15
2
Compatibilità HLA • Gemello omozigote• HLA compatibile genotipicamente identico• HLA compatibile apparentato fenotipicamente identico• HLA compatibile non apparentato fenotipicamente identico• HLA incompatibile per un locus
Fenotipo eritrocitario AB0 Rh • compatibile• incompatibilità minore• incompatibilità maggiore
Età giovaneCompatibilità di sessoGrado di disponibilità alla donazione
Tabella 3 Donatore di midollo osseo: criteri di selezione

La morbilità del donatore derivante dall’intervento di prelievo è moltobassa e si limita generalmente a un temporaneo fastidio a livello dellecreste iliache posteriori dove solitamente viene eseguito il prelievo dimidollo; più rari sono i casi di difficoltà alla deambulazione che posso-no durare da qualche giorno fino ad alcuni mesi.Inoltre, considerando che la quantità di midollo prelevato è dell’ordinedi circa 15 ml/kg, è solitamente necessaria una trasfusione di sangue;per garantire la massima sicurezza al donatore si esegue una trasfu-sione autologa di sangue salassato alcuni giorni prima.
■ SCREENING PRE-TRAPIANTO PER IL PAZIENTEFondamentale, per sottoporre un paziente a trapianto di midollo, è lavalutazione del suo stato clinico. Particolare importanza assume loscreening infettivologico ; infatt i le complicanze infett ive del TMOsono direttamente legate all’esistenza di infezioni inapparenti nel dona-tore e nel ricevente prima del trapianto. Inoltre, alcune infezioni preesi-stenti al TMO sia nel donatore sia nel ricevente (epatite B, C, infezioneda CMV, HSV e da Aspergillo, TBC), possono rappresentare un rischiodurante il decorso post-trapianto anche se il significato e l’util ità dialcune indagini sono tuttora oggetto di dibattito.
16E M A T O L O G I A
• Anamnesi• Esame obiettivo• Emocromo• Esami ematochimici (azotemia, glicemia, creatinina, colinesterasi, elettro-
liti sierici, bilirubina totale e frazionata, transaminasi, gGT, fosfatasi alcali-na, proteine totali con elettroforesi, dosaggio immunoglobuline, sidere-mia, ferritina)
• Screening coagulativo (PT, PTT, fibrinogeno, ATIII)• VES, TAS• Virologia (HSV, HVZ, EBV)• Toxotest• Sierodiagnosi (tifo, paratifo A e B , brucellosi)• Sierologia epatite (HBV, HCV, HIV)• Sierologia per CMV• Prove immunoematologiche• Esame urine• Radiografia del torace • Visita cardiologica con ECG• Valutazione anestesiologica
Tabella 4 Indagini di screening pre-trapianto del donatore di midollo osseo

2
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
17
In Tabella 5 sono elencate le indagini di screening sia generale siainfettivologico previste per il paziente.Fondamentale nella preparazione del paziente al trapianto è l’in-serimento del catetere venoso centrale (CVC), indispensabile perla somministrazione della chemioterapia, terapia di supporto,nutrizione parenterale e per eseguire i numerosi prelievi di sanguenecessari per il monitoraggio giornaliero del paziente.
■ TECNICA DEL PRELIEVO DI MIDOLLO E MANIPOLAZIONI MIDOLLARI
Il midollo osseo viene prelevato in anestesia generale o spinale, dallacresta iliaca anteriore, da quella posteriore e dallo sterno. Solitamenteperò è sufficiente eseguire le aspirazioni solamente dalla cresta poste-riore o anteriore che offrono una quantità di cellule midollari nucleatesufficienti per l’intero prelievo. Il prelievo avviene mediante aspirazioni
• Anamnesi• Esame obiettivo• Emocromo• Esami ematochimici (azotemia, glicemia, creatinina, colinesterasi, elettro-
liti sierici, bilirubina totale e frazionata, transaminasi, gGT, fosfatasi alcali-na, proteine totali con elettroforesi, dosaggio immunoglobuline, sidere-mia, ferritina)
• Screening coagulativo (PT, PTT, fibrinogeno, ATIII)• VES, TAS• Virologia (HSV, HVZ, EBV)• Toxotest• Sierodiagnosi (tifo, paratifo A e B, brucellosi)• Sierologia epatite (HBV, HCV, HIV )• Sierologia per CMV• Prove immunoematologiche• Esame urine• Radiografia del torace • Visita cardiologica con ECG ed ecocardiografia• Emogasanalisi e prove di funzionalità respiratoria• TAC total body con mezzo di contrasto• Ecografia epatosplenica• Radiografia ortopanoramica
Se donna: visita ginecologica con PAP test
Tabella 5 Indagini di screening pre-trapianto del ricevente midollo osseo

multiple in più sedi: si possono eseguire diverse penetrazioni della cor-teccia ossea, attraverso un unico foro di ago nella cute, cercando diridurre al minimo traumi o formazioni di lesioni cicatriziali. In genere,minore è il volume aspirato per ogni penetrazione, più ricca è laconta delle cellule formanti colonie.La dose di midollo per prel ievo è solitamente espressa comenumero di cellule nucleate per Kg di peso corporeo del ricevente.Per un trapianto allogenico è consigliabile, ai fini dell’attecchi-mento un numero di cellule nucleate >3x108/Kg del ricevente.In caso di trapianto da donatore non familiare o incompatibile, o nelcaso sia prevista una manipolazione in laboratorio del midollo preleva-to (T deplezione, rimozione dei globuli rossi, deplasmazione, ecc.), sideve prelevare un numero maggiore di cel lule. Per i donatori conbasso peso corporeo rispetto al ricevente, può essere necessario ese-guire due prelievi di midollo, di cui il primo viene criopreservato.Il midollo, una volta prelevato, viene raccolto in sacche trasfusionali ereinfuso direttamente al paziente mediante il CVC. Solitamente si som-ministrano clorfenamina e idrocortisone prima dell’infusione di midolloper prevenire reazioni allergiche.
In caso di incompatibilità AB0 tra donatore e ricevente (presenzanel plasma del paziente di isoagglutinine contro i globul i rossi deldonatore) è indicata la rimozione degli eritrociti dal midollo prele-vato (meno frequentemente si usa rimuovere le isoagglutinine dal pla-sma del ricevente) per evitare reazioni trasfusionali emolitiche durantel’infusione del midollo. La deplezione eritrocitaria del midollo prelevatosi può ottenere mediante tecniche di eritrosedimentazione per gravitào centrifugazione differenziale mediante separatori cellulari a flussocontinuo o discontinuo (COBE 2991, Haemonetics 30, COBE SPEC-TRA, FENWALL CS 3000, ecc.). Dopo la deplezione eritrocitaria si puòottenere un prodotto finale contaminato solo per il 2% da eritrociti eun massimo recupero di cellule mononucleate. L’incompatibilità AB0tra donatore e ricevente non influisce sull’attecchimento, poiché le cel-lule staminali sono prive degli antigeni AB0 e pertanto non possonovenire distrutte da anticorpi anti-A o anti-B del ricevente, né sulla inci-denza della GVHD o sulla sopravvivenza; tuttavia si possono osservareun ritardo nell’attecchimento della serie rossa con un prolungamentodel fabbisogno trasfusionale e reazioni emolitiche dovute alla continuaproduzione post-trapianto di anticorpi anti-A o anti-B contro gli eritro-citi del donatore da parte dei linfociti residui del ricevente.
La deplezione midollare di cellule T attualmente viene eseguita soloin casi di trapianti ad alto r ischio di GVHD; si ott iene sol i tamentemediante aggiunta di anticorpi monoclonali f issanti i l complementodiretti contro gli antigeni delle cellule T o mediante altre tecniche far-macologiche o fisiche (Tabella 6).
18E M A T O L O G I A

I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
2.2 INDICAZIONI
■ LEUCEMIA MIELOIDE CRONICABenché sia noto che il TMO allogenico rappresenta un approccioterapeutico curativo nella LMC, tuttavia, a tutt’oggi rimangonoinsoluti alcuni problemi: selezione dei pazienti eleggibili, sceltadel donatore ottimale, tempi della procedura trapiantologica,migl iore tecnologia trapiantologica applicabile, monitoraggiodella malattia post-trapianto.Relativamente al paziente eleggibile, dato che la mortalità da trapiantoincrementa con l’età, solitamente molti Centri trapianto limitano l’età a50 anni.Per la scelta del donatore, essa è in primo luogo legata al livello dicompatibil ità HLA e all’età del donatore stesso, infatti l’ impiego didonatori più giovani correla con una riduzione della TRM. Per quantoriguarda il momento più adeguato della storia della malattia per proce-dere a un trapianto sappiamo che la probabilità di recidiva e la TRMsono più elevate se la procedura è impiegata in fase avanzata dimalattia. Inoltre, per i pazienti in fase cronica (FC) si è osservato unaminore TRM e una migliore sopravvivenza l ibera da leucemia (LFS)quando il TMO viene effettuato entro un anno dalla diagnosi. Questodato comunque è riferibi le a pazienti precedentemente trattati conBUS o idrossiurea, e non è certo se la stessa correlazione possaessere fatta nei pazienti pretrattati con interferone (IFN).Non sono state rilevate sostanziali differenze nei regimi di condiziona-mento r isultando l’associazione TBI+CTX equivalente in termini dirisultati alla combinazione BUS + CTX.
19
Metodi fisici Metodi farmacologici
Microsfere magnetiche Incubazione con complemento rivestite con polistirene e anticorpi monoclonali
Colonna di perle di anticorpi Incubazione con ricino - monoclonali biotina - avidina - anticorpi monoclonalipoliacrilamide
Sedimentazione dopo formazione Incubazione con farmaci di rosette con eritrociti di pecora citotossici
Sedimentazione dopo trattamentocon agglutinina di soia
Tabella 6 Tecniche per la T deplezione del midollo osseo

L’associazione della CSA + MTX è il trattamento maggiormente impie-gato nella profilassi della GVHD. La T deplezione, che viene applicataormai in una minoranza di Centri, come sappiamo riduce il rischio diGVHD, ma incrementa la recidiva leucemica (28-30).Globalmente, dalle analisi più recenti risulta che il trapianto di cellulestaminali da fratello HLA identico nella LMC in FC determina, a secon-da delle casistiche, una probabilità di sopravvivenza, LFS e recidiva a5 anni rispettivamente del 50-70%, 30-60%, 15-30%. Variabile statisti-camente significativa in termini di migliore LFS risulta essere l’età <30anni (33, 34). In considerazione dell’incremento della TRM con l’età edei risultati favorevoli ottenuti con l’impiego dell’IFN, nella LMC in FCpuò essere indicato intervenire con la procedura trapiantologica neipazienti di età <30 anni, mentre nei pazienti di età >30 anni può esse-re indicato in prima istanza l’impiego dell’IFN e successivamente deltrapianto in caso di assente o ridotta risposta citogenetica (Ph >75%)dopo un anno di terapia.
■ LEUCEMIA MIELOIDE ACUTANei pazienti con leucemia mieloide acuta (LMA) si può ottenere la gua-rigione con il trapianto allogenico che tuttavia, se applicato negli stadipiù avanzati o nelle leucemie refrattarie, cura solo il 10% della popola-zione. Sopravvivenza a lungo termine e una probabilità di guarigionedel 20-40% si osservano invece in pazienti trattati in II o >II RC fino adarrivare a valori del 40-70% per i pazienti trapiantati in I RC. Come linee guida si può dire che il trapianto allogenico in pazientisotto i 50 anni, con donatore compatibile, e con malattia in ³ IIremissione o in recidiva precoce ha indicazione elettiva conside-rando che circa il 30% di questi pazienti diventano lunghi soprav-viventi mentre la chemioterapia non offre chance per il controllodella malattia; il trapianto ha inoltre indicazione assoluta ancheper quei pazienti resistenti alla prima linea di trattamento.Più complessa è la scelta terapeutica per la LMA in I RC. Infatti,sebbene gli studi comparativi dimostrino una riduzione, con il trapian-to, della probabilità di recidiva, in alcuni di essi non si osserva un realemigl ioramento del la DFS a causa del la più elevata TRM anche se,comunque è presente un trend a favore del TMO; in soggetti adulti<50 anni è comunque una procedura adeguata soprattutto con i lmiglioramento delle strategie volte a ridurre la TRM (35-37). Sarebbeperò utile realizzare degli studi che possano permettere di individuarequali caratteristiche biologiche e quali fenotipi molecolari indicano lanecessità di un trapianto in I RC; certamente una precedente fasemielodisplastica e alterazioni citogenetiche sfavorevoli rappresen-tano delle indicazioni ben precise al trapianto in I RC.Un discorso a parte merita la LMA FAB M3 per la quale gli ottimi
20E M A T O L O G I A

2
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
risultati ottenuti con l’acido retinoico associato alla chemioterapianon rendono ragione di un approccio trapiantologico in I RC senon nel caso di una remissione solo ematologica e non molecolaredella malattia (38).
■ LEUCEMIA LINFATICA ACUTAI risultati della chemioterapia convenzionale in età pediatrica sonocosì soddisfacenti che il trapianto in I RC in questa categoria dipazienti trova spazio solo in sottogruppi particolari quali pazientiche presentino alla diagnosi fattori riconosciuti ad alto rischio direcidiva (leucemia linfoide acuta (LLA) Ph+, t(4;11)) (39).Sono invece candidati all’allotrapianto bambini con recidiva pre-coce di malattia o recidivati dopo programmi terapeutici partico-larmente intensivi, o refrattari al la prima l inea di trattamento. In questi casi infatti le possibil ità terapeutiche con la chemioterapiasono praticamente assenti. Oggetto di discussione può essere l’impie-go del trapianto nei casi di recidiva tardiva (off therapy) o dopo pro-grammi terapeutici definiti a basso rischio.Il trapianto di midollo allogenico nell’adulto, quando effettuato in I RCha dato DFS del 40-70% a lungo temine (40, 41); tuttavia il ruolo deltrapianto in questa fase è ancora discutibile (42, 43). Il trapianto èsenz’altro una procedura di elezione nei pazienti in I RC di malat-tia ad alto rischio di recidiva, con caratteristiche citogenetichesfavorevoli, o che abbiano ottenuto la I RC dopo terapie di “salva-taggio”, o nei pazienti in fase più avanzata di malattia.
■ APLASIA MIDOLLARENell’aplasia midollare il TMO allogenico determina una DFS superio-re al 50% (44). I risultati migliori si osservano nei pazienti giovani chehanno avuto una malattia di breve durata e un numero limitato di tra-sfusioni. I l regime di condizionamento di scelta comprende la solaCTX. I pazienti con più di 30 anni presentano maggiori complicanzelegate al trapianto e i risultati tra allotrapianto e terapia immunosop-pressiva sono simil i (45). In part icolare è consigliabile la terapiaimmunosoppressiva in pazienti più anziani (>40 anni) o in pazienticon aplasia moderata.
■ EMOGLOBINOPATIEL’efficacia del trapianto allogenico nei pazienti con talassemia èormai chiaramente dimostrata da più di 10 anni di studi effettuatisoprattutto in Italia (46-48).La più grossa casistica finora pubblicata si riferisce a 802 pazienti trat-
21

tati presso il Centro di Pesaro (48). In questa casistica fattori progno-stici sfavorevoli per la sopravvivenza e la DFS sono risultati essere l’e-patomegalia, la fibrosi portale, le complicanze dovute alla terapia fer-rochelante. In part icolare, suddividendo i pazient i in tre gruppi dirischio (I, II o III) in base al numero dei fattori di rischio presenti, laDFS varia dal 90%, all’82%, al 57%, mentre per ciascun gruppo lasopravvivenza è rispettivamente del 95%, 84% e 79%. In particolarenei pazienti appartenenti al gruppo a rischio più elevato correlano conuna migliore sopravvivenza l’età inferiore a 17 anni e la minore inten-sità del regime di condizionamento. Sembra che i pazienti appartenen-ti alla I e II classe di rischio, ovvero le classi con minore compromis-sione di organo, il regime di condizionamento più adeguato sia rappre-sentato da BUS (16 mg/kg) + CTX (200 mg/Kg) ± SAL o deplezionedei linfociti T dal midollo. Per i pazienti a più alto rischio, in particolarese di età >17 anni, si può ridurre la dose della CTX (120-160 mg/Kg).I l successo nei soggetti giovani ha portato a estendere i l trapiantoanche a soggetti adulti. In 106 pazienti adulti, per lo più appartenentialla II e III classe di rischio, sottoposti a trapianto presso il Centro diPesaro, la sopravvivenza globale e l’EFS sono state del 68 e 65%rispettivamente. Tuttavia, l’alta mortalità da trapianto osservata devesuggerire una grossa cautela nella selezione dei pazienti adulti contalassemia da sottoporre a TMO allogenico.Un’altra forma di emoglobinopatia per la quale l’approccio tra-piantologico risulta curativo è l’anemia drepanocitica. Tuttavia, irecenti progressi nel trattamento medico hanno permesso di migliorarela qualità di vita dei pazienti e le complicanze della malattia, riducendocosì l’impiego del trapianto, già comunque limitato a forme partico-larmente a rischio. In Tabella 7 sono elencati i criteri di inclusioneselezionati dal British Paediatric Haematology Forum per il trapianto dimidollo nell’anemia drepanocitica.
■ ERRORI CONGENITI DEL METABOLISMONonostante il numero di trapianti eseguiti in pazienti con tesaurismosil isosomiale, è ancora poco chiaro per molte patologie, i l realebeneficio di tale procedura. Infatti i danni d’organo non sono reversi-bili e dopo trapianto non migliorano né le deformità scheletriche, né idanni neurologici a causa della barriera emato-encefalica che non per-mette alle cellule neuronali di essere raggiunte dall’enzima in circolo.Quello che emerge dagli studi è che l’attività enzimatica determinatanei linfociti e nel tessuto epatico dopo trapianto riflette un attecchi-mento stabile e una sostituzione dei macrofagi tissutali. Infatti la ridu-zione del materiale accumulato nel fegato e nella milza è dovuto allasostituzione dei macrofagi dell’ospite ricchi di prodotto accumulatocon le cellule enzimaticamente competenti del donatore che possono
22E M A T O L O G I A

I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
promuovere anche una clearance del materiale accumulato dalle cellu-le vicine, compresi gli epatociti.Tuttavia un reale miglioramento clinico è stato osservato solo in pochisoggetti sopravvissuti a lungo termine; inoltre, considerata l’estremavariabilità del decorso di tali patologie anche all’interno della stessafamiglia, occorre valutare con cautela i possibili effetti benefici deltrapianto nei singoli pazienti.
■ ANEMIA DI FANCONIAnche se l’anemia di Fanconi può rispondere in modo transitorio allaterapia medica, il trapianto rimane l’unico approccio terapeuticorealmente curativo in casi di pancitopenia. È oggetto di discussione ilmomento ottimale per l’esecuzione del trapianto che comunque vaeseguito tempestivamente in caso di aplasia grave con trasfusionedipendenza o complicanze infettive (49). È da notare come l’anemia di Fanconi si associ a un’alta incidenzadi complicanze trapiantologiche, in particolare mucosite (fino a scol-lamenti di grandi frammenti di mucosa), tossicità cutanea, insorgenzadi secondi tumori dovuti all’estrema sensibilità dei pazienti ad agentialchilanti e alla radioterapia. Anche la GVHD e la cistite emorragica
23
Criteri di inclusione1. Età <16 anni con donatore familiare HLA identico e consenso informato2. Presenza di una o più delle seguenti complicazioni correlate alla malattia:
• compromissione del SNC• compromissione polmonare acuta ricorrente o malattia polmonare
cronica falciforme stadio I/II• dolori ricorrenti gravi con debilitazione (>3 ricoveri annui in 3-4 anni)
3. Problemi relativi al futuro terapeutico del paziente
Criteri di esclusione1. Donatore affetto da emoglobinopatia grave2. Uno o più delle seguenti caratteristiche:
• Karnofsky <70%• fibrosi portale (moderata o severa)• compromissione renale (FGR < 30%)• compromissione intellettiva grave• malattia polmonare falciforme di grado III o IV• cardiomiopatia• infezione da HIV
Tabella 7 Selezione dei pazienti affetti da anemia falciforme per iltrapianto di midollo osseo allogenico: criteri
del British Paediatric Haematologic Forum

sono solitamente gravi. Tuttavia, la probabilità di sopravvivenza è risul-tata essere intorno al 75% nei due maggiori studi fino ad ora pubblicati(49, 50).
■ IMMUNODEFICIENZA PRIMARIAI primi trapianti di midollo allogenico sono stati effettuati in bambinicon immunodeficienza congenita (7, 51). La procedura trapiantologicapermette la sostituzione della cellula staminale alterata con una cellulanormalmente funzionante capace di ricostituire il sistema emopoieticoe immunitario dell’ospite. Quindi la disponibilità di un donatore HLAidentico per un bambino con immunodeficienza rende il trapiantoil trattamento di elezione. In assenza di un donatore compatibilesono stat i esegui t i con successo t rap iant i da donator i HLAmismatched o da donatori non correlati (52, 53) previa deplezionemidollare delle cellule T.
■ EMOGLOBINURIA PAROSSISTICA NOTTURNA L’emoglobinur ia parossist ica notturna (EPN) è una patologia raracaratterizzata da un disordine acquisito della cellula staminale conanemia emolit ica cronica, neutropenia, trombocitopenia ed episoditrombotici. L’unico approccio realmente curativo per questa patologia è il TMOallogenico, tuttavia, tale procedura è gravata da un’alta mortalità emorbilità (54). Considerando la storia naturale della malattia e lapossibilità anche di remissioni spontanee, la scelta terapeuticadel trapianto è da valutare attentamente. Poiché la trombocitopenia, la presenza di trombosi alla diagnosi o unaprecedente diagnosi di aplasia midollare sono fattori prognosticamentesfavorevoli in termini di sopravvivenza, in questi casi potrebbe esseregiustificato l’approccio trapiantologico. Considerato l’esiguo numero dipazienti trapiantati non ci sono delle reali linee guida se non un gravestato di aplasia midollare con dipendenza trasfusionale.
■ SINDROMI MIELOPROLIFERATIVELe sindromi mieloproliferative costituiscono un gruppo di patologiecaratterizzate da una lenta ma progressiva espansione clonale di cellu-le emopoietiche, che possono andare incontro a una evoluzione blasti-ca. Comprendono la LMC (già esaminata in dettaglio), la LMC giovani-le, la policitemia vera (PV), la trombocitemia essenziale e la mielofibrosiidiopatica.Nella PV il ruolo del trapianto è più ipotetico che reale consideratoche la sopravvivenza con la chemioterapia convenzionale è di circa 10
24E M A T O L O G I A

2
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
anni e che l ’età al la diagnosi è sol i tamente super iore ai 60 anni.Essendo tuttavia una malattia della cellula staminale il trapianto trovaovviamente indicazione solo in pazienti giovani non rispondenti altrattamento.Anche per la t rombocitemia essenzia le i l t rapianto è più unapproccio virtuale che reale riferibile solo a pazienti giovani congrave rischio trombotico o episodi emorragici ricorrenti.Per la mielof ibrosi id iopat ica invece i l ruolo del t rapianto èsenz’altro fondamentale anche se l’esperienza per questa formapatologica è molto limitata. L’ostacolo principale all’approccio tera-peutico è sempre l’età del paziente e il grado di fibrosi che non deveessere tuttavia vista come una controindicazione al trapianto, anchese nei casi di grave fibrosi midollare può rappresentare senz’altro unrischio aggiuntivo per l’attecchimento. In un’esperienza riportata daRajantie e coll. (55) il mancato attecchimento è stato osservato nel 6%dei pazienti con fibrosi media o moderata e nel 33% dei pazienti congrave fibrosi midollare.Nella LMC giovanile l’indicazione al trapianto è assoluta conside-rata l’estrema aggressività della patologia e l’assenza di altriapprocci curativi.
■ SINDROMI MIELODISPLASTICHELe sindromi mielodisplastiche (MDS) sono caratterizzate da un disordi-ne clonale dell’emopoiesi con emopoiesi inefficace e citopenia periferi-ca. Sebbene la storia naturale della malattia dipenda dal tipo di mielo-displasia: anemia refrattaria (AR), AR con eccesso di blasti (AREB),AREB in trasformazione (AREB-T), leucemia mielomonocitica cronica(LMMC), i trattamenti convenzionali non sono curativi e la mediana disopravvivenza globalmente è di 15 mesi. Attualmente, il TMO allogeni-co sembra essere potenz ia lmente curat ivo. In part ico lare, in 59pazienti trapiantati dal gruppo di Seattle (56), la sopravvivenza liberada eventi (EFS) a 3 anni è risultata del 45%.Recentemente sono stati pubblicati i dati relativi a 93 pazienti, 64 tra-piantat i da frate l l i HLA ident ic i , i r imanent i da donatore fami l iaremismatched o da MUD (57). Tutti presentavano prima del trapiantoneutropenia o piastrinopenia o una quota blastica superiore al 5% nelmidollo o nel sangue periferico. La probabilità di sopravvivenza liberada malattia (DFS) a 4 anni, la recidiva e la mortalità trapiantologicasono state, rispettivamente, del 41%, 28% e 48%. Fattori prognosticifavorevoli per la DFS sono risultati l’età del paziente (DFS 48% a 4anni per età <40 anni vs 17% per età >40 anni), e una minore duratadel tempo intercorso dalla diagnosi della malattia al trapianto. La reci-diva è stata osservata solamente nel gruppo dei pazienti con eccessodi blasti (51% a 4 anni). La DFS per i pazienti di età inferiore a 40 anni
25

e senza eccesso di blasti al trapianto è risultata del 62% a 4 anni.La procedura trapiantologica sembra pertanto indicata in pazienticon età inferiore a 40 anni se impiegata precocemente prima dellaprogressione blastica o delle citopenie gravi. Certamente è difficileintervenire con una procedura aggressiva come il trapianto in pazienticon AR, senza pancitopenia o anomalie citogenetiche complesse osenza fabbisogno trasfusionale; in questo gruppo potrebbe essereindicata solo un’attenta sorveglianza per intervenire in caso di segni dievoluzione. Per i pazienti di età superiore ai 40 anni o con eccessodi blasti l’impiego della procedura trapiantologica è discutibile. Inquest’ultimo caso può essere indicata una polichemioterapia pre-tra-pianto per sottoporre il paziente alla procedura trapiantologica in RCdi malattia (DFS a 2 anni del 60%) (58).
■ LEUCEMIA LINFATICA CRONICALa leucemia linfatica cronica (LLC) è una patologia che interessa perlo più pazienti anziani, quindi l’approccio trapiantologico con cellulestaminali allogeniche da donatore familiare identico è riservato solo auna ridotta quota di pazienti idonei per età. Fino ad ora il TMO alloge-nico è stato effettuato in piccoli gruppi di pazienti con LLC a prognosisfavorevole (59-61).I risultati preliminari non permettono attualmente di fornire linee guidaper TMO allogenico nella LLC; tuttavia possiamo considerare indica-to il trapianto allogenico in pazienti giovani con malattia a pro-gnosi sfavorevole.
■ MIELOMA MULTIPLOIl trapianto di midollo allogenico da donatore familiare identico è statoutilizzato per la prima volta in questa patologia nel 1982 dal gruppo diSeatt le (62). Si trattava di un trapianto singenico, cui hanno fattoseguito altri casi aneddotici. Dati relativi a un’ampia casistica relativa all’impiego del trapianto dimidollo allogenico in pazienti affetti da mieloma multiplo (MM) sonostati pubblicati nel 1991 da Gahrton e coll. (63). La probabilità attua-riale di sopravvivenza è stata globalmente del 40% con un follow-upmassimo di 78 mesi. È stato osservato un trend statisticamente nonsignif icat ivo a favore dei pazienti trapiantati in stadio I r ispetto aipazienti in stadio II o III di malattia e nei pazienti trapiantati in remis-sione completa (RC) rispetto ai pazienti in remissione parziale (RP) onon rispondenti (NR) o in progressione di malattia. È risultato inoltresignificativo ai fini della sopravvivenza l’ottenimento di uno stato di RCdopo l’attecchimento. La mortalità correlata al trapianto (TRM) è stata
26E M A T O L O G I A

2
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
del 38%; tra le cause principali di decesso sono state osservate PI,recidive, GVHD, infezioni ed emorragie.Da questi dati è evidente come il trapianto allogenico per mieloma ègravato da un’alta mortalità trapiantologica, che varia dal 40 al50% ma si riduce per trapianti effettuati in fase meno avanzata dimalattia. Questo in parte è legato all’età media dei pazienti trapiantatiper mieloma che è superiore a quella dei pazienti trapiantati per leuce-mia, ma probabilmente è anche dovuto all’alta incidenza di malattiaattiva al momento del trapianto e alle alterazioni renali cliniche e sub-cliniche presenti nel MM, nonché alla predisposizione alle infezioni.Non ci sono linee guida ben precise sull’impiego del trapianto nelMM anche se ci potrebbe essere indicazione nei pazienti più gio-vani già precedentemente trattati, con buona risposta alla chemio-terapia. Tuttavia, considerando l’alta mortalità trapiantologica puòanche essere ragionevole aspettare una seconda linea di tratta-mento. Inoltre, potrebbe essere indicato il trapianto in pazienti dietà <50 anni resistenti alla chemioterapia di prima linea con dona-tore compatibile; questi pazient i , infatt i , non hanno possibi l i tà disopravvivenza a lungo termine.
■ LINFOMA DI HODGKINLa buona risposta alla chemioterapia non rende necessario perquesta patologia l’approccio trapiantologico se non in casi parti-colari (64). Il trapianto allogenico è stato finora impiegato in un ridottonumero di pazienti in recidiva di malattia resistente o sensibile al lachemioterapia, criterio quest’ultimo che correla con una minore proba-bilità di recidiva post-trapianto (65-68).
■ LINFOMA NON HODGKINGli studi relativi al TMO allogenico in pazienti affetti da linfoma nonHodgkin (LNH) non si riferiscono a casistiche numerose di pazienti.Tuttavia, da questi studi si evince un’attività di GVL che non si traducein un incremento dell’EFS per i pazienti allotrapiantati, rispetto a quellitrattati con autotrapianto, a causa della maggiore mortalità da allotra-pianto. È comunque consigliabile il trapianto allogenico in pazienticon LNH aggressivo, l infoma di Burkitt o l infoma linfoblasticosoprattutto in soggetti giovani con basso rischio di sviluppareGVHD, nei quali si può sfruttare al meglio l’effetto di GVL.Per i linfomi a basso grado (indolenti) non si possono fare delleconsiderazioni relative all’impiego dell’allotrapianto; sicuramentel’età del paziente, la storia naturale della malattia, la TRM non larendono una procedura di elezione.
27

■ TRAPIANTO APLOIDENTICOIl trapianto di cellule staminali da donatore aploidentico è una proce-dura terapeutica gravata da un elevato rischio di GVHD e di man-cato attecchimento. Recentemente, per aumentare la probabilità disuccesso di questa procedura, sono stati impiegati regimi di condizio-namento più intensivi, deplezione delle cellule T dall’inoculo, incremen-to del numero delle cellule staminali infuse mediante la combinazionecellule midollari + cellule staminali da sangue periferico previa stimola-zione con fattore di crescita.Di fondamentale importanza in questo campo è l’esperienza del grup-po di Perugia che in pazienti in fase avanzata di malattia ha associatola T deplezione a un regime di condizionamento costituito da TBI +CTX (100-120 mg/Kg) + th iotepa (10 mg/Kg) e SAL (20 mg/Kg) . Le cellule staminali infuse erano ottenute sia dal midollo osseo che dalsangue perifer ico del donatore dopo stimolazione con granulocytecolony stimulating factor (G-CSF): sia i l midollo che le leucoaferesivenivano previamente sottoposte a rimozione dei linfociti T (69). L’esperienza di questo gruppo ha evidenziato una probabilità di attec-chimento del 75%, con una riduzione della GVHD acuta. Tuttavia conquesto regime di preparazione pre-trapianto r imane ancora alta laTRM. Una migliore selezione dei pazienti, la modificazione del regimedi condizionamento e i l migl ioramento delle procedure di profi lassidella GVHD sono le direttive verso cui si muove il gruppo di Perugia e irisultati preliminari sembrano promettenti.
28E M A T O L O G I A

3
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
TRAPIANTO DI MIDOLLOOSSEO ALLOGENICODA DONATORE NONCORRELATO
3.1 BANCHE DEI DONATORI VOLONTARI DI MIDOLLO OSSEO
Come già detto il TMO allogenico è una procedura ormai consolidatanel trattamento di numerose emopatie, tuttavia solo il 30% dei pazien-ti, eleggibili per età o malattia di base possono usufruire di un donato-re HLA compatibile nell’ambito della fratria. Tale probabilità si accre-sce del 10% se si considera la possibilità di individuare un familiarecon fenotipo HLA diverso solo per un locus.Di fatto, a circa il 60-70% dei pazienti eleggibili rimarrebbe preclusa lapossibilità di usufruire delle procedure trapiantologiche.Le conoscenze sempre più approfondite del sistema HLA e l’estensio-ne degli studi HLA alla genetica delle popolazioni hanno permesso distabilire la possibilità di esistenza, per ogni singolo individuo, nell’am-bito della popolazione mondiale, di uno o più soggetti fenotipicamenteHLA compatibili.Tale acquisizione ha costituito i l presupposto per la creazione deiRegistr i Internazional i e Nazional i d i donator i volontar i d i midol loosseo.Il Bone Marrow Donor Worldwide (BMDW) costituito a Leiden sotto lapresidenza del Prof. J. van Rood, raccoglie le tipizzazioni HLA prove-nienti da tutti i Registri Nazionali, a ognuno dei quali vengono ritra-smesse con aggiornamenti periodici.Un limite fondamentale di questi registri è che la frequenza HLA riflettein larga maggioranza quella propria della razza caucasica provenienteda una fascia sociale medio alta. Pertanto le minoranze etniche, leindividualità emergenti nelle società multirazziali sempre più diffuse e lagrande quota degli appartenenti ai Paesi a economia non avanzatasono scarsamente o affatto rappresentate nei registri.Il criterio etnico comunque non è la sola variabile che influenza la pro-babil i tà di trovare un donatore compatibi le nel l’ambito dei Registr iInternazionali.
29
Ulteriori fattori sono rappresentati da: 1. dimensione del pool dei donatori 2. f requenza aplot ipica HLA del paziente nel l ’ambito del pool dei
donatori 3. diagnosi e condizione clinica del paziente.
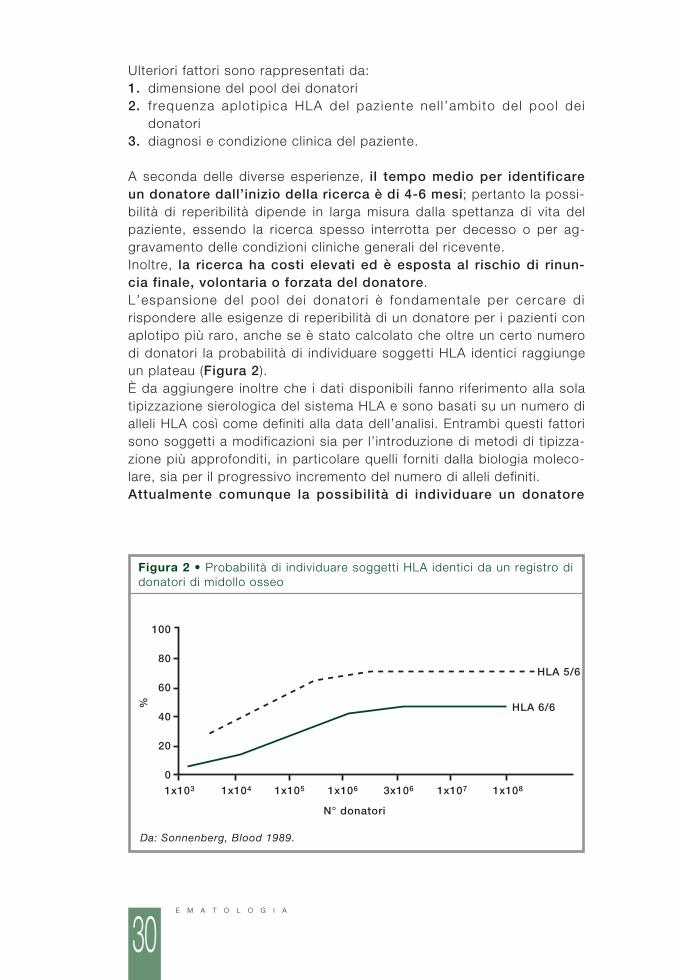
A seconda delle diverse esperienze, il tempo medio per identificareun donatore dall’inizio della ricerca è di 4-6 mesi; pertanto la possi-bilità di reperibilità dipende in larga misura dalla spettanza di vita delpaziente, essendo la ricerca spesso interrotta per decesso o per ag-gravamento delle condizioni cliniche generali del ricevente.Inoltre, la ricerca ha costi elevati ed è esposta al rischio di rinun-cia finale, volontaria o forzata del donatore.L’espansione del pool dei donatori è fondamentale per cercare dirispondere alle esigenze di reperibilità di un donatore per i pazienti conaplotipo più raro, anche se è stato calcolato che oltre un certo numerodi donatori la probabilità di individuare soggetti HLA identici raggiungeun plateau (Figura 2).È da aggiungere inoltre che i dati disponibili fanno riferimento alla solatipizzazione sierologica del sistema HLA e sono basati su un numero dialleli HLA così come definiti alla data dell’analisi. Entrambi questi fattorisono soggetti a modificazioni sia per l’introduzione di metodi di tipizza-zione più approfonditi, in particolare quelli forniti dalla biologia moleco-lare, sia per il progressivo incremento del numero di alleli definiti. Attualmente comunque la possibilità di individuare un donatore
30E M A T O L O G I A
Figura 2 • Probabilità di individuare soggetti HLA identici da un registro didonatori di midollo osseo
Da: Sonnenberg, Blood 1989.
100
80
60
40
20
0
N° donatori
HLA 5/6
HLA 6/6
1x103 1x104 1x105 1x106 3x106 1x107 1x108
%

3
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
compatibile, contando i registr i oltre 4 000 000 di donatori, è del40%. Fino a oggi si calcola che siano stati effettuati più di 5 000 tra-pianti da MUD per malattie ematologiche grazie a donatori reperit iattraverso il network mondiale dei registri.
3.2 RISULTATI
Si calcola che globalmente la probabilità di ottenere l’attecchimen-to in un trapianto MUD sia dell’ordine dell’80-98% a secondadelle diverse casistiche con una mediana di 22 giorni per PMN>500/mm3 (70, 71). Studi recenti hanno evidenziato che un più altonumero di cellule infuse si correla con una riduzione della graft failuree con un acco rc i amen to de i t emp i d i a t t ecch imen to ( 72 , 73 ) .L’impiego dei fattori di crescita non migliora l’andamento clinico deipazienti e non riduce le percentuali di mancato attecchimento, masembra agire solamente sulla velocità di risalita dei PMN. Fattori cheinfluenzano l’attecchimento sono inoltre la deplezione T cellulare delmidollo (20% di insuccessi), il l ivello di compatibilità HLA, l’intensitàdel regime di condizionamento e l’immunosoppressione post-trapianto(74-77).La GVHD acuta rappresenta la maggior causa di insuccesso post-trapianto MUD. La sua incidenza e gravità incrementano in base algrado di incompatibilità HLA (78). I due principali approcci per ridurre la GVHD acuta sono la T deplezio-ne de l m ido l lo e la te rap ia immunosoppress iva post - t rap ian to .Tuttavia, la T deplezione riduce la GVHD senza migliorare la sopravvi-venza, perché incrementa i l rischio di rigetto e la recidiva. Sono incorso studi volti a individuare rimozioni selettive o parziali delle celluleT. Sebbene la somministrazione del MTX con la CSA per la profilassidella GVHD nei trapianti da donatore famil iare abbia mostrato unamaggiore efficacia rispetto alle singole sostanze, tale combinazionenei trapianti MUD previene la GVHD acuta in meno del 25% dei casi ela sua efficacia è ancora più bassa se valutata nei trapianti incompati-bili per un locus. Globalmente l’incidenza della GVHD acuta è del 79% nei pazientiche hanno ricevuto MTX+CSA con donatore HLA identico non cor-relato rispetto al 35% del donatore consanguineo (79). Sembra tut-tavia che in individui con meno di 36 anni, con donatore compatibilenon correlato, l’incidenza della GVHD sia più bassa rispetto a donatorifamiliari incompatibili per un locus (71). Inoltre l’incidenza della GVHDaumenta nel caso che il donatore sia donna con gravidanze preceden-ti rispetto alle nullipare o a donatori maschi. L’incidenza della GVHD cronica risulta significativamente più bassa nei
31

pazienti che hanno ricevuto midollo T depleto (70). Tra i pazienti tra-piantati con midollo non manipolato e che hanno ricevuto profi-lassi della GVHD con MTX e CSA, la GVHD cronica è di gradoesteso nel 77% dei casi, subclinica nell’8%, e solo il 15% deipazienti non presenta GVHD cronica. Globalmente i l 25-30% deipazienti muore per complicanze da GVHD cronica estesa durante laterapia immunosoppressiva (78).
In un’analisi multivariata volta a definire i fattori di rischio associati conuna migliore sopravvivenza in 267 pazienti affetti da emopatia malignasottopost i a TMO da donatore non fami l iare correlavano con unaminore sopravvivenza i seguenti fattori: diagnosi diversa da LMC, sta-dio avanzato di malattia, età >20 anni, sierologia pre-trapianto positivaper CMV, irregolare somministrazione della terapia impiegata per laprofilassi della GVHD (78).In una recente analisi condotta su 333 pazienti affetti da LMC, trapian-tati da donatore non compatibile dal Maggio 1985 al Dicembre 1994dal gruppo di Seattle, la probabilità di sopravvivenza a 3 anni è statadel 59%, 39%, 32% e 7% rispettivamente per i pazienti trapiantati in IFC, FA, II FC e crisi blastica (CB) (80).Sempre il gruppo di Seattle (73) ha condotto un’analisi su 174 pazientiaffetti da leucemia acuta linfoide e mieloide ad alto rischio: la DFS a 3anni è stata del 37% per i pazienti trapiantati in II RC, rispetto al 13%per i pazienti trapiantati in fase più avanzata di malattia. La sopravvi-venza globale r isulta r idotta nei pazienti resistenti o in recidiva dimalattia al trapianto; con blasti nel sangue periferico o più del 30% diblasti nel midollo. Inoltre, dall’analisi multivariata è risultato che unnumero di cellule infuse >3.65x108 cellule/Kg del ricevente correla conuna migliore sopravvivenza.Recentemente è stata pubblicata una casistica relativa a 57 pazientiaffetti da MDS trapiantati a Seattle dall’Ottobre 1987 al Luglio 1995(76); per questo gruppo di pazienti la probabilità di DFS a 2 anni èstata del 38%.
3.3 INDICAZIONI
Le indicazioni a un trapianto da MUD dipendono dalla patologia dibase, dall’età e ovviamente dall’urgenza clinica del paziente.
Secondo il GITMO (Gruppo Italiano per il Trapianto di Midollo Osseo)esse sono:
a. indicazioni di tipo sperimentale, accettabili sulla base dei dati e deiprotocoll i pubblicati, che siano già state sottoposte e approvatedalla Commissione GITMO (ad esempio la talassemia major)
32E M A T O L O G I A

3
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
b. patologie per le quali è già codificato il trapianto da donatore nonconsanguineo:• aplasia midollare grave dopo 6 mesi di terapia inefficace con SAL
o CSA in pazienti al di sotto dei 20 anni• anemia di Fanconi in pazienti al di sotto dei 20 anni• LLA in II RC in pazienti al di sotto dei 40 anni• LMA in II RC in pazienti al di sotto dei 40 anni• LMC in I FC in pazienti con età inferiore a 45 anni non rispondenti
all’IFN• MDS ad alto rischio in pazienti al di sotto dei 45 anni.
Ogni indicazione in deroga a questi criteri deve essere preventivamen-te sottoposta alla Commissione GITMO-MUD prima di procedere a untrapianto allogenico da donatore non compatibile.
33

4
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
TRAPIANTO DI CELLULESTAMINALI ALLOGENICHEDA SANGUE PERIFERICO
Negli ultimi due anni si è assistito a un progressivo incremento nell’im-piego delle cellule staminali allogeniche ottenute da sangue perifericoper il trapianto di pazienti affetti da patologie ematologiche.
I presupposti per la diffusione di tale procedura sono stati i seguenti: 1. comparsa di una ricostituzione emopoietica completa e permanen-
te con chimerismo completo del donatore in esperienze preclinichesu animali
2. possibilità di aumentare considerevolmente il numero di cellule sta-minali circolanti per mezzo di citochine
3. buona tol lerabi l i tà da parte del donatore del G-CSF impiegatoquale citochina per la mobilizzazione delle cellule staminali
4. vantaggi per il donatore al quale vengono evitati i rischi e i disagilegat i a l l ’anestesia, al l ’ospedal izzazione, al l ’autotrasfusione, aldolore nella sede del prelievo e, in alcuni casi, la diff icoltà nelladeambulazione che può durare da qualche giorno fino, sia puremolto raramente, a qualche mese
5. una più rapida risalita dei neutrofili e delle piastrine 6. l ’assenza di un aumentato r ischio di GVHD acuta nonostante i l
numero di linfociti T nel sangue periferico sia da 7 a 10 volte mag-giore rispetto al midollo osseo
7. i l numero maggiore dei linfociti T e delle cellule NK presenti nell’i-noculo, che potrebbe incrementare l’effetto GVL, anche se non visono ancora dati sufficienti al riguardo.
4.1 PRELIEVO
Attualmente la citochina più largamente impiegata per ottenere lamobilizzazione di cellule staminali nel donatore di cellule staminali allo-geniche da sangue periferico (PBSC) è il G-CSF. In uno studio condot-to da Korbling e coll. (81), in 41 donatori sottoposti a trattamento conG-CSF alla dose di 12 mg/Kg per 3 giorni, la concentrazione di globuli
35

rossi, PMN e linfociti è incrementata, rispetto al valore di base, di 6.4,8.0 e 2.2 volte rispettivamente. Inoltre, cellule CD34+ e sottotipi piùimmaturi quali le cellule CD34+ Thy-1dim e CD34+ Thy-1dim CD38– incre-mentano di 16.3, 24.2 e 23.2 volte rispettivamente evidenziando unamobilizzazione selettiva da parte del G-CSF dei progenitori emopoietici ein particolare dei sottotipi più primitivi di cellule staminali.Il granulocyte-macrophage colony stimulating factor (GM-CSF) sembraessere meno efficace in termini di mobilizzazione delle cellule staminalianche se i dati sono ancora scarsi. La combinazione delle due citochi-ne non sembra migliorare il livello di mobilizzazione rispetto all’impiegodel G-CSF da solo e limitate sono le esperienze con le altre citochine. Dati recentemente pubblicati sulla tollerabilità del G-CSF hanno messoin evidenza la comparsa di dolori ossei, cefalea, astenia e nausea. Talieffetti collaterali sono risultati dose dipendente e possono risolversientro pochi giorni dalla sospensione del farmaco; in ogni caso sonoben controllati dall’impiego di analgesici. Gravi effetti collaterali tali dadeterminare la sospensione del farmaco sono rari. In alcuni casi il G-CSF può indurre incremento dei valori della fosfatasi alcalina (anche didue-tre volte), del la latt ico-deidrogenasi (LDH) o più raramente undecremento del potassio e del magnesio sierico.Solitamente la citochina viene impiegata alla dose di almeno 10 mg/Kgdel donatore e somministrata per via sottocutanea per i 4-5 giorni con-secutivi che precedono l’inizio delle leucoaferesi. Infatti, sembra che ilgiorno migliore per la raccolta delle cellule staminali dopo una sommini-strazione giornaliera di 10 mg/Kg di G-CSF sia il 4° o il 5°; continuandoa somministrare ulteriormente il fattore di crescita, si osserva una pro-gressiva riduzione nella mobilizzazione dei progenitori CD34+. Natu-ralmente, è fondamentale poter disporre di un buon accesso venosoper poter procedere alla leucoaferesi ed è da evitare, a eccezione dicasi particolari, un accesso venoso centrale.Le PBSC sono raccolte mediante sedute singole o multiple di leucoa-feresi effettuate mediante separatori cellulari a flusso continuo. Il volu-me di sangue totale processato per ogni seduta è solitamente due-trevolte il volume ematico del donatore; il numero di cellule mononucleate(MNC) raccolto varia da 3 a 5x108/Kg. L’obiettivo, per ottenere unadeguato attecchimento, è raggiungere un totale di cellule CD34+ >3-4x106/Kg del ricevente. Questo obiettivo si raggiunge con una sola leu-coaferesi nell’80% dei donatori e con due leucoaferesi nel rimanente20%. La concentrazione di cel lule CD34+ nel sangue periferico deldonatore può essere predittiva della quantità di cellule staminali pre-sent i nel l ’aferesi . Al l ’anal is i mult ivar iata, tra i fattor i che possonoinfluenzare la resa nella produzione di cellule staminali da donatoresano (82), sembra che l’età del donatore >55 anni correl i con unaridotta mobilizzazione di CD34+.A volte nel donatore si può assistere a una riduzione del numero delle
36E M A T O L O G I A

4
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
37
piastrine con normalizzazione dei valori durante la successiva settima-na; tuttavia, questo inconveniente si può evitare reinfondendo, alla finedell’aferesi, plasma autologo con piastrine. Alterazioni nei l ivel l i dimagnesio o potassio vengono corrette con una adeguata integrazionedi elettroliti.
4.2 RISULTATI
In una recente analisi condotta da Przepiorka e coll. (83), sono stateanalizzate tre coorti di pazienti (TMO + MTX-CSA, TMO + CSA-PDN,trapianto da PBSC + CSA-PDN) al f ine di valutare la mortalità e lamorbilità correlata al trapianto da PBSC rispetto al TMO. La tossicitàlegata al regime di condizionamento (in particolare la mucosite) è statameno grave nei pazient i trapiantat i con PBSC. Anche la degenzaospedaliera in questo gruppo di pazienti è stata più breve di circa 4giorni rispetto al trapianto da midollo. La sopravvivenza calcolata a seimesi è stata più alta nel gruppo PBSC. Tali dati sono stati confermatida Azvedo e coll. (84) e da Russell e coll. (85) che hanno ri levatoanche un ridotto numero di giorni di terapia antibiotica e antifungina eun ridotto numero di trasfusioni di piastrine nei pazienti trapiantati conPBSC.Nonostante i l numero di l infocit i T e cel lule NK sia superiore nel lePBSC rispetto al midollo osseo, numerosi studi non hanno osservatoun aumento nel l ’ incidenza del la GVHD acuta r ispetto al trapiantomidol lare. Sono ancora prel iminari i dati relat ivi al l ’ incidenza del laGVHD cronica, tuttavia in uno studio condotto da Anderlini e coll. (86)sembra che l’incidenza della GVHD cronica sia maggiore nel trapiantoda PBSC rispetto al gruppo dei pazienti sottoposti a TMO. Tuttavia,tale incremento non si traduce in un incremento di mortalità grazie auna più bassa incidenza di recidiva; se ciò si può correlare a un effettoGVL potenziato è ancora da definire.In uno studio condotto da Bacigalupo e coll. (87), relativamente allaricostituzione ematologica dopo trapianto da PBSC, la ripresa dellecellule CD3+ è paragonabile a quella del TMO, d’altro canto, la ripresadelle cellule CD4+ e CD8+ sembra più veloce nel trapianto da PBSC;questo può tradursi in una ricostituzione immunologica più rapida conconseguente riduzione della mortalità e morbilità dovuta alle infezioni.

5
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
TRAPIANTO DI CELLULESTAMINALI EMOPOIETICHEDA SANGUE DI CORDONEOMBELICALE
Risale al 1974 la prima dimostrazione della presenza nel SCO di CSE(88) il cui potenziale uso a fini trapiantologici è stato successivamenteprecisato da numerosi studi, in particolare dal gruppo della IndianaUniversity (89) e confermato definitivamente nel 1989 dal primo tra-pianto di SCO eseguito con successo in un paziente effetto da anemiadi Fanconi (10).
5.1 CARATTERISTICHE BIOLOGICHE DELLA CELLULA STAMINALE EMOPOIETICA DA SANGUE DI CORDONE OMBELICALE
Secondo un ordine ontogenetico, la CSE origina primariamente nelsacco vitellino, per migrare successivamente nel fegato fetale e quindinel midollo osseo, che ne costituisce dopo la nascita la fonte principa-le. La generale immaturità tessutale alla nascita e la particolarità ana-tomo-funzionale del circolo materno-fetale contribuiscono a conferirecaratteristiche specifiche alla componente cellulare del sangue placen-tare sia sotto il profilo emopoietico che immunologico.
5.2 CARATTERISTICHE EMOPOIETICHE
Sulla base di numerosi dati sperimentali, la CSE del SCO risulta fenoti-picamente diversa, funzionalmente più immatura e dotata di poten-ziale proliferativo maggiore rispetto a quella del midollo osseo o delsangue periferico.L’entrata in ciclo delle cellule cordonali CD34+ per stimolazione con lostem cell factor avviene più rapidamente rispetto alle cellule midollari,
39

mentre la crescita cellulare in vitro sembra essere indipendente dal-l’aggiunta dei fattori di crescita prodotti per via autocrina o paracrina.Queste specificità ematopoietiche conferiscono alla CSE del cordoneombelicale proprietà peculiari che la rendono particolarmente indicataper la manipolazione in vitro sia ai fini della terapia genica (già pratica-ta in bambini affetti da deficienza di adenosindeaminasi) sia ai fini del-l’espansione del pool delle cellule staminali per un loro impiego nel tra-pianto dei pazienti adulti (9).
5.3 CARATTERISTICHE IMMUNOLOGICHE
Rispetto al sangue periferico, il SCO contiene linfociti in numero asso-luto più elevato, rappresentati, nell’ambito della sottopopolazione T,da cellule più immature e fenotipicamente distinte, a elevata attivitàsoppressoria, scarsamente alloreattive, capaci comunque di esprimereintensa citotossicità di t ipo NK e LAK dopo stimolazione con IL-2.Inoltre, sono state r iconosciute funzionalmente immature le cel luledendritiche del SCO, accessorie della risposta T cellulare. Questi datidi laboratorio supportano sul piano biologico l’osservazione clinica diun aumentato rischio per l’attecchimento e di una ridotta incidenza egravità della GVHD nei trapianti di SCO. Tuttavia, solo un numero piùelevato di pazienti trapiantati e un più lungo follow-up potranno rispon-dere al quesito se a una diminuzione del la GVHD corr isponderà omeno una riduzione dell’effetto GVL da parte delle cellule cordonali. Glistudi in vitro sembrerebbero tuttavia evidenziare il mantenimento del-l’attività citotossica anti-leucemica (9).
5.4 TECNICA DEL PRELIEVO
Il sangue contenuto nel cordone ombelicale e nella placenta può esse-re facilmente prelevato dopo l’espletamento del parto sia spontaneoche cesareo. Per il recupero di maggiori quantità di sangue, sono statiadottati vari metodi di raccolta: sistemi aperti o chiusi, impiegati primao dopo l’espulsione della placenta con o senza l’ausilio di una soluzio-ne di lavaggio anticoagulante. Attualmente si ritiene che un sistemachiuso sia da preferire poiché si associa a una minore incidenza dicontaminazione. Fondamentale ai fini del prelievo è la rapidità del clam-paggio del funicolo dopo la nascita del neonato, viene quindi incannu-lata la vena ombelicale e i l sangue viene fatto defluire attraverso i lsistema chiuso. Il momento migliore per il prelievo, nel parto sponta-neo, sembra essere prima del secondamento in quanto le contrazioni
40E M A T O L O G I A

5
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
uterine a placenta in situ permettono un più efficace svuotamento equindi il recupero di un maggior volume di sangue (Figura 3).
In Tabella 8 sono elencati i criteri di esclusione per la raccolta.
Successivamente vengono ef fettuat i studi infett ivologic i sul s ieromaterno (HBsAg, anti-HCV, anti-HIV 1-2, anti-CMV, TPHA) e un test
41
Figura 3 • Prelievo di CSE da sangue di cordone ombelicale
• Età gestazionale < 35 settimane• Rottura delle membrane > 12 ore• Patologie della gravidanza e/o distociche• Sofferenza fetale e/o malformazioni fetali• Malattie familiari genetiche• Malattie trasmissibili per via ematogena• Assenza di consenso informato
Tabella 8 Criteri di esclusione per la raccolta

di ster i l i tà su l SCO. Su ogni un i tà SCO v iene esegui to lo studiodell’HLA, del gruppo sanguigno e della composizione cellulare.L’unità di SCO viene quindi congelata e resa disponibile solo dopo chea 6 mesi di distanza sia stata confermata la negatività del test per l’HIVin un nuovo campione di siero materno.
5.5 RISULTATI CLINICI
■ TRAPIANTO DI SCO DA DONATORE CORRELATOIn un’analisi condotta da Wagner e coll. nel 1995 (11) relativa all’espe-rienza generale su 44 bambini riceventi trapianto SCO, per malattianeoplastica o non neoplastica, da fratello HLA identico o incompatibileper uno o più loci, la probabilità di sopravvivenza globale a 1.6 anni èrisultata essere del 72%. L’attecchimento è stato ottenuto nell’86% deicasi con un tempo mediano di recupero in PMN (>500/mm3) e piastri-ne (>50 000/mm3) rispettivamente di 22 e 49 giorni, mentre l’incidenzadella GVHD acuta di grado > I è stata pari al 3% dei casi. Il numerolimitato dei pazienti, l’eterogeneità delle patologie di base e dei regimidi condizionamento pre-trapianto conferiscono all’analisi i limiti propridi uno studio retrospettivo e multicentrico.
Tuttavia alcuni risultati appaiono indicativi:1. i l mancato attecchimento è stato osservato esclusivamente nei
pazienti con malattia non neoplastica o riceventi SCO HLA incom-patibile per 2-3 loci
2. nessuna correlazione è stata osservata tra capacità e rapidità diattecchimento, impiego terapeutico dei fattori di crescita emopoieti-ci e dose cellulare sia in termini di cellule nucleate infuse che diCFU-GM
3. l ’ incidenza del la GVHD acuta e cronica, pur considerando l’etàpediatrica della casistica, appare particolarmente limitata.
Recentemente Gluckman e coll. (14) hanno presentato i dati dell’espe-rienza europea relativa a 143 pazienti trapiantati con SCO dal 1988 al1996 in 45 Centri Trapianto. Nei 78 pazienti sottoposti a trapianto diSCO da donatore correlato, l’età mediana era di 5 anni (0.2-20) il pesodi 19 Kg (5-50); 46 pazienti erano affetti da malattie neoplastiche (44ematologiche, 2 neuroblastomi), 17 da sindromi da insufficienza midol-lare, 8 da emoglobinopatie, 7 da errori congeniti del metabolismo. Laprobabilità di sopravvivenza è stata del 63% a un anno. Fattori pro-gnostici favorevoli per la sopravvivenza sono risultati l’età più giovanedel paziente, il più basso peso corporeo, il grado di compatibilità HLAe la sieronegatività per CMV. L’incidenza della GVHD di grado ³ II è
42E M A T O L O G I A

5
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
stata del 9% nei 60 pazienti riceventi SCO HLA identico e del 50% in18 pazienti sottoposti a trapianto di SCO HLA non compatibile.In questa casistica una correlazione è stata trovata tra attecchimento,età, peso corporeo e numero di cellule nucleate infuse/Kg del ricevente.
■ TRAPIANTO SCO DA DONATORE NON CORRELATOSe i risultati clinici del trapianto SCO da donatore correlato sono daconsiderarsi ancora preliminari, ancora più lo sono i dati relativi ai tra-pianti da donatore non correlato.Wagner e coll. (13) hanno riportato i dati relativi a 18 pazienti con etàmediana di 2.7 anni (0.1-21.3) e peso di 15 Kg (3-78) riceventi SCOproveniente da donatore non correlato. Dei 18 pazienti, 13 erano affettida una patologia oncoematologica e 5 da malattia non neoplastica. I gradi di compatibilità HLA erano di 6/6 loci in 7 pazienti, 3-5/6 locinei rimanenti 11. Tutti i pazienti esaminati hanno presentato attecchi-mento per neutrofili con un tempo mediano di 24 giorni (16-53), mentremo l t o p i ù t a rd i vo è s t a t o l ’ a t t ecch imen to pe r l e p i a s t r i n e(>50 000/mm3) osservato dopo una mediana di 67 giorni (55-120). Laprobabilità di sviluppare GVHD di grado III-IV è stata dell’11%. Conuna mediana di follow-up di 6 mesi la probabilità di sopravvivenza èstata del 65%. In questa casistica non è stata trovata nessuna correla-zione tra il numero di cellule nucleate infuse/Kg del ricevente e attec-chimento.Un’a l t ra impor tan te cas is t i ca re la t i va a l l ’ esper ienza de l l a DukeUniversity di Kurtzenberg e coll. (12) comprende 25 pazienti con un’etàmediana di 7 anni (0.8-23.5); peso corporeo mediano 19.4 Kg (7.5-79),trapiantati per patologie neoplastiche e non neoplastiche. Impiegandometodiche di tipizzazione molecolare ad alta risoluzione, in 9 pazienti viera un’incompatibilità per un solo locus con l’unità cordonale, mentrein 15 casi l’incompatibilità era relativa a 2-3 loci HLA. Il mancato attecchimento o rigetto è stato osservato in circa il 10% deicasi, mentre la GVHD acuta di grado I-II è stata osservata nel 50% deipazienti. Un solo paziente ha presentato GVHD acuta di grado >II.Considerata la fase avanzata di malattia, solo 7 pazienti risultavanosopravviventi tra 1 e 24 mesi post-trapianto.Nell’esperienza europea riportata da Glukman e coll. (14), 65 pazientihanno r icevuto trapianto di SCO da donatore non correlato. L’etàmediana era di 9 anni (0.3-45), peso mediano 30 Kg (4-90). Una patolo-gia oncoematologica era presente in 49 pazienti, 9 avevano un’insuffi-cienza midollare e 7 erano affetti da errori congeniti del metabolismo. L’attecchimento in termini di PMN >500/mm3 è stato globalmentedell’87% e del 94% per i pazienti che hanno ricevuto un numero di cel-lule nucleate >3.7x107/Kg. La GVHD acuta di grado >II è stata osserva-ta in 21 dei 65 pazienti. Globalmente la sopravvivenza a un anno è statadel 29%. Fattori prognostici favorevoli correlati alla sopravvivenza sono
43

risultati la sierologia negativa per CMV del ricevente, fattore quest’ulti-mo correlato anche con una ridotta incidenza della GVHD acuta, e lafase favorevole di malattia al momento del trapianto.
5.6 BANCHE DI SANGUE DI CORDONE OMBELICALE
Sebbene preliminari, i risultati clinici delle esperienze precedentementeriportate sembrano promettenti e indicano alcuni sostanziali vantaggiofferti dall’impiego del SCO nel trapianto allogenico da donatore noncorrelato. Per tale motivo in numerosi Centri sia americani che europeisono state costituite o stanno per essere attivate banche di SCO com-plementari e per alcuni versi alternative ai Registri Internazionali dimidollo.Infatti, come abbiamo già precedentemente detto, nonostante la gran-de espansione del numero dei donatori volontari di midollo osseo, soloil 40% dei pazienti privi di donatore HLA identico ha la possibilità direperire un donatore HLA compatibile nell’ambito dei registri. Inoltre iltempo mediano di 4-6 mesi per l’ identif icazione del donatore, puòessere eccessivo rispetto alle esigenze cliniche del paziente. Bisognaanche tener presente che i donatori volontari possono essere portatoridi infezioni latenti o croniche, in particolare di natura virale e che laricerca ha costi elevati ed è esposta al rischio di rinuncia finale alladonazione che è stata calcolata nell’ordine dell’1% al mese. Infine, lefrequenze HLA dei registri riflettono in larga maggioranza quelle pro-prie della razza caucasica provenienti da una popolazione di fasciasociale medio-alta; le minoranze etniche sono scarsamente rappresen-tate nell’ambito dei registri. Il SCO costituisce una fonte illimitata di rifornimento, la raccolta ètecnicamente semplice e non comporta alcun rischio né per lamadre né per il bambino. Una Banca di SCO è programmabile sullabase delle frequenze HLA della popolazione e le unità, una volta rag-giunta la copertura della domanda corrente, possono essere selezio-nate concentrandone la raccolta su gruppi HLA più rari. La completatipizzazione HLA, l’immediata disponibilità alla richiesta, l’assenza delrischio di rinuncia finale alla donazione e la facile trasportabilità si tra-ducono in un drastico accorciamento degli intervalli di tempo tra l’ini-zio della ricerca e il trapianto. Sul piano infettivo il SCO è generalmen-te esente da contaminazioni batteriche o virali trasmissibili.Tuttavia vanno considerati anche i limiti delle banche di SCO: neces-sità di coordinamento operativo tra competenze diverse (ostetrico-gine-cologiche, pediatriche, ematologico-trasfusionali); necessità di reperireampi spazi per lo stoccaggio dei campioni; reperibilità della madre e delbambino per il controllo infettivologico e clinico a distanza di almeno 6mesi dalla donazione del cordone; rischio di mantenimento indefinito di
44E M A T O L O G I A

5
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
unità non utilizzabili. La creazione di banche parallele di DNA, cellule,siero e plasma per ogni singola unità criopreservata costituisce unaparte integrante del progetto di creazione di una banca di SCO.Attualmente banche di SCO sono presenti negli Stati Uniti, in partico-lare presso il New York Blood Center e in Paesi europei quali Ger-mania, Belgio, Gran Bretagna e Francia. In Italia è particolarmente atti-va la banca di Milano ma anche altre banche sono state istituite aTorino, Firenze e Roma. Un lavoro di coordinamento è in atto al fine distandardizzare le procedure di raccolta, manipolazione e criopreserva-zione, incrementando così il livello di qualità delle singole banche.
5.7 INDICAZIONI
Si possono considerare eleggibili per un trapianto di cellule cor-donali da donatore non correlato i pazienti di età ²45 anni privi didonatore familiare compatibile per almeno 5/6 loci HLA o di dona-tore di midollo HLA identico nell’ambito dei Registri Internazionali.
Le patologie per le quali il trapianto SCO è una possibile indicazioneterapeutica sono le seguenti:• leucemia linfoblastica acuta in II o III RC (in seconda remissione si
possono considerare eleggibili pazienti recidivati precocemente)• leucemia mieloide acuta in I RC ad alto rischio di recidiva• leucemia mieloide acuta in II RC• leucemia acuta promielocitica in II RC o I RC ematologica con per-
sistenza di malattia molecolare• mielodiplasia ad alto rischio• leucemia mieloide cronica in FC, senza risposta citogenetica alla
terapia con IFN, dopo almeno 6 mesi di ricerca nei Registri Inter-nazionali di donatori di midollo o LMC in FA
• anemia di Fanconi• anemia aplastica acquisita non rispondente alla terapia immunosop-
pressiva dopo almeno 2 cicli di terapia e un precario compenso clinico• errori congenit i del sistema immunitar io che rendono urgente e
indifferibile l’esecuzione del trapianto.
Sono fondamental i per un trapianto con SCO la presenza deiseguenti parametri nell’unità di sangue placentare:
1. compatibilità di almeno 4/6 loci HLA dopo tipizzazione DRB1 adalta risoluzione;
2. numero di cellule contenute nell’unità cordonale prima dellacriopreservazione >10x106/Kg di peso corporeo del ricevente.
45

Inoltre, tutti i pazienti devono avere un’unità criopreservata di midolloosseo o sangue periferico autologo contenente un numero di celluleCD34+ ³ 2x106/kg. In generale, per quel che riguarda l’alternativa tral’uso di cellule staminali da SCO o da midollo osseo da donatore noncorrelato, è opportuno sottolineare che a favore del primo vi è l’osser-vazione che i dati attualmente disponibili (seppure su casistiche limita-te) indicano che esiste una r iduzione del r ischio di svi luppare unaGVHD acuta di grado elevato, mentre a favore del TMO esiste ormaiuna casistica consolidata dal punto di vista numerico sui risultati otte-nuti. La decisione di usare ai fini trapiantologici l’una o l’altrafonte di cellule staminali dovrebbe essere adottata da ogni singo-lo Centro tenendo conto del grado di compatibilità tra donatore oricevente, del numero di cellule dell’unità placentare disponibile,del rischio di complicanze immunomediate (GVHD) dei due diffe-renti tipi di trapianto e del tempo che mediamente intercorre dal-l’inizio della ricerca all’esecuzione del trapianto in funzione dellapatologia. È infatti chiaro che per pazienti affetti da leucemia acuta oda malattie in equilibrio ematologico precario il tempo a disposizioneper poter eseguire un trapianto in condizioni favorevoli è relativamentebreve.
46E M A T O L O G I A

6
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
LE COMPLICANZEPOST-TRAPIANTO
6.1 COMPLICANZE PRECOCI
Si definiscono complicanze precoci quelle che intervengono nei primi100 giorni post-trapianto; vengono definite tardive quelle che si mani-festano successivamente.
■ COMPLICANZE INFETTIVELa neutropenia e il danno alla barriera mucosa indotto dalla chemio-radioterapia di condizionamento rappresentano fattori di rischio chepredispongono il paziente alle infezioni. La durata della neutropenia è variabile e dipende dal tipo di trapianto,dal numero di cellule infuse, dalla profilassi della GVHD, dall’uso di cito-chine; tuttavia mediamente è dell’ordine di 2-3 settimane. Il danno allemucose dipende solitamente dal tipo di regime di condizionamento: far-maci quali il BUS, l’etoposide, il melphalan, la citarabina e la TBI si asso-ciano a un danno maggiore. Questo danno è presente non solo a caricodel cavo orale, ma anche a livello del tratto gastrointestinale e l’impiegodel MTX per la profilassi della GVHD peggiora il danno alle mucose.Alla citopenia e al danno alle mucose vanno aggiunti quali fattori dirischio per le complicanze infettive l’ impiego del CVC, la nutrizioneparenterale e anche le alterazioni dell’ integrità della cute dovute airipetuti prelievi del sangue, agli aspirati midollari e alle biopsie ossee ecutanee. Inoltre, alla comparsa delle complicanze infettive contribui-scono anche il periodo di profonda immunosoppressione cui il pazien-te va incontro. La durata e la gravità di questo periodo dipendono daltipo di trapianto, dal grado di incompatibilità donatore-ricevente, dallaT deplezione, dal tipo e dalla durata della profilassi per la GVHD, dallapresenza di infezione da CMV e di GVHD. Naturalmente, con il tempoc’è un recupero dell’immunità cellulare e umorale che verosimilmente èpiù rapido dopo un trapianto da donatore familiare compatibile che inaltre condizioni trapiantologiche. Tuttavia, in presenza di GVHD croni-ca lo stato immunodepressivo può persistere per mesi o anche peranni; solitamente in condizioni ottimali il tempo di recupero immunolo-gico è di circa un anno.In funzione della sequenza di eventi legati a tali fattori di rischio sidistinguono diversi periodi di comparsa di complicanze infettive nelpaziente trapiantato.
47

Entro le prime tre settimane dal trapianto sono frequenti le infe-zioni batteriche e fungine (neutropenia + lesioni mucose).Le infezioni da virus erpetico si sviluppano solitamente entro ilprimo mese (riattivazione del virus latente).Entro i primi tre mesi si osservano il maggior numero di infezionida CMV.Le infezioni da Aspergil lo, toxoplasma e P. carini i si osservano neiprimi 6 mesi da trapianto o anche successivamente se insorge GVHDcronica e persiste il trattamento immunosoppressivo.Di più raro riscontro sono le infezioni da adenovirus, rotavirus e daEBV. Il terzo periodo di rischio infettivo fa seguito al terzo mese daltrapianto in corrispondenza del la GVHD cronica. In tale periodo siosservano soprattutto infezioni respiratorie: Haemophilus influenzae,Streptococcus pneumoniae, germi capsulati, queste ultime soprattuttoin assenza di profilassi con penicillina. Il paziente trapiantato, in questafase tardiva, può andare incontro a infezioni batteriche, virali o fungineinsorte ex novo o per riattivazione di infezioni pregresse.Frequenti le infezione causate dal virus della Varicella zoster a partiredal sesto mese (30% dei pazienti) (90-92). In Figura 4 sono riportate schematicamente le fasi infettive post-tra-pianto correlate ai fattori di rischio e ai periodi di rischio.
48E M A T O L O G I A
Polmonite
Virus
Funghi
Batteri
Fattoridi rischio
Batterica Interstiziale non batterica
VHS CMV ADENO VVZ
Candida
Gram +Gram –
Capsulati
neutropenia GVHD cronicaGVHD acuta + Rx
0 50 100 12mesigiorni post TMO
Aspergillus
Periododi rischio
Primo Secondo Terzo
Figura 4 • Fasi di rischio infettivo dopo trapianto di midollo osseo allogenico

6
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
■ COMPLICANZE GASTROINTESTINALINegli ultimi 20 anni l’incidenza delle complicanze gastrointestinali pre-coci post-trapianto è rimasta per lo più invariata, anche se è cambiatal’origine. Infatti, negli anni ‘70 le cause principali di queste complican-ze erano la GVHD epatica e intestinale e le infezioni erpetiche; succes-sivamente, con il miglioramento dei regimi di profilassi anti-GVHD edegli agenti antivirali le cause principali sono diventate quelle legate alregime di condizionamento. Recentemente, con l’incremento dei tra-pianti HLA incompatibili sta riemergendo, tra le complicanze gastroin-testinali precoci, la GVHD intestinale. Di fatto, ancora negli anni ‘90 lecomplicanze epatiche e intestinali costituiscono una causa considere-vole di morbilità post-trapianto. Gli effetti tossici legati al regime di condizionamento durante il periodoprecoce post-trapianto sono rappresentati da nausea, vomito e ano-ressia. I meccanismi principali che li determinano sono l’effetto dellachemioterapia sui centri del vomito, probabilmente gli elevati livelli dicitochine e la presenza di mucosite.Sempre entro i primi 100 giorni, sembra che la GVHD acuta intervenganel determinare perdita dell’appetito, nausea e vomito. In uno studiocondotto da Weinstorf e coll. nel 1990 (93), il 60% dei pazienti connausea e vomito erano positivi per GVHD a livello dello stomaco e delduodeno agli esami bioptici.Inoltre, possono contribuire all’insorgenza di queste complicanze preco-ci l’impiego degli antibiotici, della CSA, della nutrizione parenterale (lipidie alti livelli di glucosio o aminoacidi) e le infezioni virali.I l regime di condizionamento, la GVHD, gli agenti infettivi, i farmaciimpiegati nella profilassi della GVHD, possono favorire l’insorgenza dialtre due complicanze precoci: la mucosite e la diarrea.Il regime di condizionamento pre-trapianto è inoltre responsabile dellaVOD, una grave complicanza a carico del fegato, dovuta a un dannoche interessa la zona 3 dell’acino epatico e che si manifesta, a secon-da delle diverse casistiche, dall’1 al 54% dei pazienti. Questa grandedisparità nella percentuale di incidenza è dovuta sia alla tossicità deidiversi regimi di condizionamento, sia al la selezione dei pazienti esoprattutto ai criteri diagnostici impiegati per definire una VOD (94).Clinicamente è una sindrome caratterizzata da iperbilirubinemia, epa-tomegal ia associata a sintomatologia dolorosa e r i tenzione idr ica:secondo i criteri utilizzati dal gruppo di Seattle, la diagnosi è definitadalla presenza di almeno due dei tre criteri elencati. Per il Centro diBaltimora una diagnosi di VOD richiede la presenza di iperbilirubinemia(>2.0 mg/dl) e di due dei seguenti segni, epatomegalia con dolore,ascite o incremento del peso corporeo >5%. La patogenesi della VOD è dovuta a un’obliterazione fibrotica dellevenule epatiche terminali e delle vene sublobulari, dilatazione e fibrosi
49

dei sinusoidi centrolobulari e necrosi degli epatociti della zona 3.La sindrome si può manifestare precocemente, anche prima dell’infu-sione stessa del midollo; tuttavia, più frequentemente i segni di labora-torio si manifestano tra il 6° e 7° giorno post-trapianto con picco neisuccessivi 10 giorni nei pazienti che vanno incontro a guarigione, ivalori della bilirubina tendono a normalizzarsi dopo ulteriori 10 giorni. La mortalità per VOD in base alle diverse casistiche varia dal 3 al 67%e la mortalità entro i primi 100 giorni dipende dalla gravità della sindro-me: 9% nei pazienti con VOD di grado lieve, 23% per le forme modera-te, 98% nei pazienti con VOD di grado grave.
■ COMPLICANZE POLMONARILa PI da CMV è una delle più temibili complicanze polmonari che siosservano precocemente nel periodo post-trapianto. Si caratterizzaper un quadro di compromissione interstiziale evidente a livello radiolo-gico con concomitante dimostrazione della presenza del virus nel liqui-do del broncolavaggio. Si manifesta, in genere, tra i 70 e i 100 giornipost-trapianto e si caratterizza per la presenza di febbre, tosse, tachi-pnea e occasionalmente dolore toracico. Prima dell’introduzione deitrattamenti preventivi, la probabil ità di insorgenza era, in base al lediverse casist iche, del 15-30% con una morta l i tà del l ’80% circa.Attualmente sia l’ insorgenza che la mortalità della PI da CMV sononotevolmente ridotte. In caso di comparsa di una PI da CMV, il trattamento di elezione èl’ impiego del gancyclovir in associazione al le immunoglobuline; neicasi di tossicità midollare o di resistenza al gancyclovir è indicato l’im-piego del foscarnet, meno mielotossico, anche se responsabile di alte-razioni renali dose dipendenti, comunque reversibili. Sebbene il CMV sia il principale responsabile della PI dopo trapianto,la PI può essere determinata anche da altri agenti infettivi o dacause sconosciute: in questo caso la polmonite viene definita idio-patica.
I principali fattori di rischio per la polmonite idiopatica sono:• il regime di condizionamento• l’età del paziente• l’impiego della TBI• l’uso del MTX nella profilassi della GVHD• la GVHD.
Nella polmonite idiopatica, i test di funzionalità polmonare evidenzianoriduzione dei volumi polmonari e ipossiemia; estremamente elevata è lamortalità soprattutto per i pazienti che richiedono intubazione mecca-nica (90%).
50E M A T O L O G I A

6
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
■ CISTITE EMORRAGICALa cist ite emorragica rappresenta una grave complicanza precocepost-trapianto. Si manifesta a una mediana di circa 20 giorni e ha unaincidenza di circa il 15-25%.I fattori di rischio che correlano con l’insorgenza della cistite emorragi-ca sono la presenza a livello urinario di papovavirus (BK virus) e ade-novirus e l’impiego della CTX nel regime di condizionamento pre-tra-pianto (95, 96).Comunemente, la profilassi per la cistite emorragica si avvale, duranteil regime di condizionamento pre-trapianto, di regimi di iperdiuresi o dilavaggio vescicale continuo associato o meno al MESNA.
6.2 COMPLICANZE TARDIVE
Si definiscono tardive quelle complicanze che intervengono dopo100 giorni dal trapianto. Alcune di esse sono direttamente correlateal trapianto (effetti da GVHD cronica o immunodeficienza), altre sonodovute all’intensità del regime di condizionamento, molte hanno unapatogenesi multifattoriale.
■ COMPLICANZE OCULARIGli occhi possono essere sede di complicanze tardive post-trapiantoper effetto della GVHD cronica, della terapia steroidea impiegata per iltrattamento, per infezioni o per sequele dovute al regime di condizio-namento, in particolare la TBI, più raramente i chemioterapici. È statariferita un’incidenza di cataratta post-TBI pari, in alcune casistiche, al75% a 5-6 anni post-trapianto dopo irradiazione singola; tale incidenzasi riduce nel caso della TBI frazionata al 50% per dosaggi superiori a1200 rad, fino a percentuali del 30-35% per dosi di 1200 rad o inferio-ri. L’incidenza post-chemioterapia è dell’ordine del 20%. La catarattapuò cominciare a insorgere già dopo un anno dal trapianto.Un’altra complicanza oculare tardiva è la “sindrome degli occhi sec-chi” dovuta a una minore produzione di lacrime in seguito a radiazionio a “sindrome SICCA” (tipo Sjögren) da GVHD cronica.Il danno determinato dalla GVHD cronica può inoltre causare sinechie,ectropion e anche perforazioni corneali, inoltre sono stati segnalatiostruzioni del dotto nasolacrimale.
■ COMPLICANZE OSSEEFrequentemente, dopo trapianto, è possibile osservare osteoporosi.Questa complicanza può essere dovuta alla menopausa precoce nelledonne, alla GVHD cronica o all’uso prolungato dei corticosteroidi.
51

Un’altra conseguenza della terapia corticosteroidea è la necrosi aset-tica della testa del femore che si manifesta nel 10% circa dei pazien-ti; può insorgere non solo dopo trattamenti prolungati, ma anche dopocicli di terapia di breve durata ad alte dosi, come nel trattamento dellaGVHD acuta.
■ ACCRESCIMENTOL’accrescimento è un fenomeno determinato durante l’infanzia preva-lentemente dallo stato nutrizionale, quindi dall’ormone della crescita(GH) e durante la pubertà dall’azione combinata dell’ormone della cre-scita e degli ormoni sessuali.Le radiazioni possono indurre un ritardo della crescita. Anche lo svi-luppo della dentizione e dello scheletro facciale risultano alterati inbambini sottoposti a radioterapia prima dei 6 anni. L’irradiazionedel SNC si associa infatti a una riduzione del GH in correlazione all’etàdel paziente, alla dose di radiazione e al tipo di frazionamento. Questodeficit si può osservare in particolare quando alla TBI è associata unaradioterapia craniale prima del trapianto e può non svilupparsi nel casodella sola TBI. Sembra che il ritardo della crescita possa essere note-volmente contenuto con l’ impiego della TBI frazionata rispetto al ladose unica.L’alterazione dell’accrescimento nei bambini con GVHD cronica puòessere in parte attribuita all’ impiego dei corticosteroidi o all’effettocatabolico della GVHD cronica. La somministrazione dell’ormone dellacrescita può migliorare la velocità dell’accrescimento e la sua secre-zione può venire stimolata mediante somministrazione dell’ormone dirilascio del GH a indicare che dopo radioterapia l’ipotalamo può subireun danno superiore all’ipofisi stessa (39).
■ EFFETTI SULLA TIROIDELa chemioterapia convenzionale non determina solitamente danni allatiroide, mentre la radioterapia pre-trapianto può determinare a cari-co della ghiandola problemi di ipotiroidismo. Dopo TBI il danno fun-zionale, anche se ben compensato, può intervenire dal 28 al 56% deicasi e successivamente convertirsi in ipotiroidismo clinico nel 9-13%dei pazienti sopravvissuti a lungo termine, anche se con minor fre-quenza dopo TBI frazionata. Il danno tiroideo non sembra correlarecon l’età del paziente al trapianto, con la GVHD acuta o cronica, o conil sesso. Nell’asse ipotalamo - ghiandola pituitaria - ghiandola tiroideaquest’u l t ima appare maggiormente danneggiata dal la radiaz ione. È stato inoltre descritto il trasferimento dal donatore al ricevente ditiroidite autoimmunitaria (39).
52E M A T O L O G I A

6
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
53
■ COMPLICANZE TARDIVE DEL TRATTO GASTROINTESTINALELa GVHD cronica è la causa principale di complicanze epatiche post-trapianto; essa è caratterizzata da un quadro colestatico con incre-mento della fosfatasi alcal ina, del le transaminasi e del la bi l irubina. I l paziente può essere asintomatico o presentare prurito, astenia operdita di peso. Sono stati descritti casi di progressione in cirrosi. Ladiagnosi risulta facile quando ai segni di colestasi si associa anche unimpegno cutaneo o mucoso o di altri tessuti tipicamente compromessinel la GVHD cronica. La diagnosi diventa più complessa in caso dilocalizzazione isolata al fegato; in questi casi è dirimente, per iniziareuna terapia immmunosoppressiva, la biopsia epatica.Un trial condotto da Fried e coll. nel 1992 (97) ha mostrato un miglio-ramento dei parametri di colestasi con l’impiego dell’acido ursodesos-sicolico.Tra le complicanze intestinali tardive vanno segnalate anche diarrea eperdita di peso per sindrome da malassorbimento.
■ COMPLICANZE POLMONARI A LUNGO TERMINEA carico del polmone sono stati segnalati sia deficit restrittivi cheostruttivi, quali sequenze a distanza del trapianto. In uno studio con-dotto da Springmeyer e coll. (98) il 20% dei pazienti mostrava un defi-cit restrittivo dopo un anno dal trapianto indipendentemente dal regi-me di condizionamento o dalla GVHD cronica, con miglioramento dopoil 3° o 4° anno. Poco conosciuti sono i meccanismi che determinano ildeficit ostruttivo che si osserva dal 10 al 15% dei pazienti con GVHDcronica, che sono in generale i più esposti allo sviluppo di deficit pol-monari gravi. Questi possono favorire l’insorgenza di infezioni con ulte-riore peggioramento della funzionalità polmonare fino a quadri di PI obronchiolite obliterante.
■ FERTILITÀ Dopo trapianto, a causa della radio-chemioterapia sovramassimale delregime di condiz ionamento, la pubertà spontanea è r itardata oassente nelle ragazze e solo una quota di esse giunge al menarcaspontaneamente. Molte r ichiedono terapia ormonale sostitut iva abase di ormoni sessuali. I ragazzi frequentemente recuperano la fun-zione delle cellule del Leydig e producono testosterone a meno chenon abbiano ricevuto dosi supplementari di radioterapia sui testicoli esolitamente non necessitano di terapia ormonale sostitutiva.Negli adulti l’infertilità è quasi la norma. Dopo TBI nelle donne lagravidanza è un’evenienza rarissima, in letteratura sono stati riferit isolo casi sporadici di maternità post-trapianto. Tutte le donne sottopo-ste a irradiazione vanno incontro a insufficienza ovarica primitiva, in

particolare l’incidenza aumenta con l’età della paziente ed è richiestaterapia ormonale sostitutiva.Dopo TBI più del 90% degli uomini va incontro ad azospermia per-manente. In alcuni casi è stata osservata ripresa della spermogenesi adistanza di anni dal trapianto. Sicuramente più frequente è la ripresadella fertilità (64%) dopo un condizionamento con l’impiego dellasola CTX.
■ SECONDO TUMOREIn una recente analisi relativa a 19 229 pazienti condotta da Rochelle ecoll. (99) i pazienti sottoposti a TMO allogenico hanno un rischiopiù elevato, rispetto alla popolazione generale di sviluppare tumo-ri solidi. In particolare il rischio è 8.3 volte superiore per quelli chesopravvivono oltre 10 anni dal trapianto. La probabilità di sviluppare unsecondo tumore è del 2.2% a 10 anni e 6.7% a 15 anni. I tumori piùfrequentemente osservat i sono i l melanoma mal igno, tumori del lacavità buccale, del SNC, della tiroide, del tessuto osseo e tessutoconnettivo. In particolare il rischio di sviluppare un secondo tumoresembra più alto per i pazienti trapiantati in giovane età rispetto aglialtr i (p<0.001). Il fattore di rischio che all’analisi multivariata siassocia a una maggiore incidenza di secondo tumore risulta esse-re la TBI. La GVHD cronica e il sesso maschile sembrano correlarecon un aumentato rischio di tumori squamocellulari della cavità bucca-le e della cute. Questa aumentata incidenza di secondi tumori rende ragione, per i lpaziente trapiantato, di una stretta sorveglianza, anche a distanza daltrapianto, per il monitoraggio di tali eventuali complicazioni.
54E M A T O L O G I A

7
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
55
LA GRAFT VERSUSHOST DISEASE
La malattia del trapianto contro l’ospite viene comunemente chia-mata GVHD. Già nel 1956 Barnes intuì l’esistenza di questo effetto deltrapianto. Nei suoi studi ormai storici, topi leucemici ricevevano unadose letale di irradiazione totale più midollo singenico o allogenicoHLA compatibile. Mentre i riceventi di midollo singenico morivano tuttiper recidiva di malattia, parte dei topi allotrapiantati guariva dalla leu-cemia, anche se quasi tutti morivano per la “malattia del trapianto con-tro l’ospite” (GVHD) (100).
La GVHD rappresenta a tutt’oggi una delle più frequenti complicanzedel trapianto di cellule staminali allogeniche, particolarmente nei casidi trapianto incompatibile.Dalle diverse casistiche, per il trapianto di midollo HLA identico, l’inci-denza della GVHD è del 30-50%; tale incidenza aumenta al 50-80% deicasi nei trapianti da MUD o familiari HLA parzialmente compatibili.La sopravvivenza a lungo termine nei pazienti con GVHD di grado >IIrisulta inferiore al 30%.Ovviamente, non solo il tipo di trapianto (il trapianto di SCO correlainfatti con una più bassa probabilità di incidenza della GVHD) e il gradodi compatibilità donatore-ricevente influenzano l’incidenza della GVHD,ma anche il regime di profilassi impiegato e numerosi altri fattori.
L’esatta identificazione della popolazione cellulare responsabile dellaGVHD come precedentemente già riferito resta poco chiara ed esisto-no prove che sia i linfociti T CD4+ che CD8+ possano giocare un ruoloin questo fenomeno insieme a cellule NK. Possono inoltre contribuirealla GVHD le citochine, compresi gli IFN, il tumor necrosis factor e ilGM-CSF. In particolare, nell’immediato post-trapianto gli alti livelli dicitochine e molecole di adesione possono rendere maggiormente reat-tivi i linfociti T infusi verso gli antigeni HLA del ricevente e in tal modocontribuire al danno tessutale della GVHD.
7.1 GVHD ACUTA E CRONICA
Sono distinguibili due differenti “sindromi” distinte di GVHD, denominateGVHD acuta e GVHD cronica. La GVHD acuta compare entro i primi

100 giorni mentre la forma cronica compare successivamente.La GVHD acuta si manifesta mediamente intorno al 15° giorno dopol’infusione del midollo allogenico. Le sue caratteristiche cliniche ruota-no intorno al la tr iade rash cutaneo, diarrea, disfunzione epatica(ittero colestatico). Non necessariamente tutti e tre gli organi vengo-no interessati, ma in relazione al livello di compromissione d’organo ealle manifestazioni cliniche, alla GVHD acuta viene dato un grado com-plessivo di gravità che va dal I al IV (Tabella 9).La GVHD cronica ha un tempo di comparsa successivo ai primi100 giorni dal TMO allogenico e può far seguito a una GVHD acutao insorgere de novo.Può interessare le stesse sedi della GVHD acuta, ma solitamente lasua estensione è più sistemica con impegno di quasi tutti gli organi e
56E M A T O L O G I A
Stadio Cute Fegato Intestino
+ Rash maculopapulare Bilirubina Diarrea in <25% della 2-3 mg/dl 500-1000
superficie corporea ml/die
++ Rash maculopapulare sul Bilirubina Diarrea 25-50% della 3-6 mg/dl 1000-1500
superficie corporea ml/die
+++ Eritrodermia Bilirubina Diarrea generalizzata 6-15 mg/dl >1500
ml/die
++++ Desquamazione Bilirubina Dolore e formazioni >15 mg/dl o ileo
bollose
Grado della GVHD acuta
Grado Cute Fegato Intestino Compromissione funzionale
0 0 0 0 0I +/++ 0 0 0II +/+++ + + +III ++/+++ ++/+++ ++/+++ ++IV ++/++++ ++/++++ ++/++++ +++
Tabella 9 Criteri di stadiazione della GVHD acuta

7
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
57
apparat i (cute, occhi, mucosa orale o esofagea, fegato, polmone,apparato neuromuscolare, intestino; può assumere le caratteristichedella sclerodermia, della cirrosi biliare o della bronchiolite obliterante).In base alla compromissione d’organo la GVHD cronica può esserelimitata o estesa (Tabella 10).È da sottolineare che entrambe le forme di GVHD aumentano la ten-denza alle complicanze infettive, sia per la loro natura immunosop-pressiva che per l’effetto immunosoppressore delle terapie impiegateper il suo trattamento. In questa fase diventa pertanto particolarmenteimportante il monitoraggio delle complicanze infettive e la loro even-tuale profilassi.
7.2 PROFILASSI
I farmaci impiegati nella profilassi della GVHD sono farmaci cito-tossici o immunosoppressori quali il MTX, la CSA e i corticosteroidi;sono inoltre state impiegate tecniche di manipolazioni del midollo infu-so quali la rimozione dei linfociti T.Il MTX agisce come farmaco citotossico che interferisce sulla sintesinucleotidica della timidina e purina. Inizialmente impiegato in modelli di trapianto canino, successivamenteè stato utilizzato sull’uomo. In particolare il gruppo di Seattle nel 1977(101) ha utilizzato il MTX a basse dosi (15 mg/m2 giorno +1 post-TMO,quindi 10 mg/m2 nei giorni +3, +6, +11 e poi ogni settimana fino al100 giorno) riducendo l’incidenza di GVHD a circa il 50%.Le principali complicanze legate all’impiego del MTX sono la mucositee l’ipoplasia midollare.La CSA è un peptide cicl ico idrofobico di origine micotica. Agiscecome immunosoppressore riducendo la produzione di interleuchina 2
LimitataCompromissione localizzata della cute e/o compromissione epatica; nessun’altra compromissione di organo
EstesaCompromissione generalizzata o localizzata della cute e/o compromissione multipla di organi
Tabella 10 Grado GVHD cronica (IBMTR)

(IL-2) da parte dei linfociti T helper e interferendo con il suo recettorecellulare, bloccando in tal modo l’amplificazione della risposta alloim-munitaria.L’impiego di tale farmaco può associarsi a tossicità renale, special-mente quando è previsto il concomitante impiego di altri farmaci nefro-tossici (antibiotici, antifungini, ecc.) o in presenza di altre complicanzequali sepsi o VOD. È pertanto fondamentale monitorizzare i l valoredella creatinina sierica per avere una misura della tossicità renale delfarmaco o eventualmente i livelli sierici di CSA.La CSA può inoltre determinare irsutismo, lesioni retiniche, ipertensio-ne, disturbi neurologici, disfunzione epatica, ipomagnesiemia, emolisimicroangiopatica e mialgia. Tuttavia, rispetto al MTX, non determinamucosite né ipoplasia midollare e non interferisce perciò con l’attec-chimento.Uno schema posologico comune che previene la comparsa di GVHDprevede l’impiego della CSA dal giorno -1 (alla dose di 3 mg/kg per viaendovenosa) seguito, a risoluzione dei problemi di mucosite del pazien-te, da una somministrazione per via orale alla dose di 12.5 mg/Kg, conriduzione graduale settimanale del 5% della dose a partire dal giorno+50 fino a sospensione in assenza di segni di GVHD al sesto mese.Sono stati proposti numerosi altri schemi di profilassi con la CSA condosi variabili da 1 mg/kg a 5 mg/kg allo scopo di individuare uno sche-ma adeguato per ridurre l’ incidenza della GVHD senza aumentare ilrischio di recidiva leucemica.Studi randomizzati che hanno paragonato l’uso della CSA e quello delMTX in pazienti leucemici sottoposti ad allotrapianto hanno dato risul-tati equivalenti.Successivamente, in uno studio condotto su cani aploidentici è statoimpiegato con successo un regime di profilassi della GVHD che preve-deva l’impiego del MTX per 11 giorni e della CSA per 180 giorni. Studicontrollati sull’uomo hanno confrontato questo regime di associazioneverso il MTX alle dosi standard in pazienti affetti da aplasia midollare, everso CSA da sola in pazienti affetti da leucemia ottenendo una signifi-cativa riduzione nell’ incidenza della GVHD e un miglioramento dellasopravvivenza in quei pazienti trattati con l’associazione MTX + CSA.Attualmente questo schema di prof i lassi è largamente impiegato,soprattutto nei trapianti a maggior rischio di GVHD, in particolare daMUD o nei trapianti non compatibil i. L’associazione del MTX con laCSA è invece discutibile nei trapianti di SCO da donatore non correlatoin quanto l’impiego del MTX potrebbe prolungare i tempi di attecchi-mento con danno a carico dei progenitori emopoietici e con possibileriduzione della capacità di ripopolamento midollare. Tuttavia, pur inpresenza di un ridotto rischio di GVHD acuta e cronica nei trapianti daSCO non correlati, la sola CSA non sembra sufficiente a evitare l’insor-genza di complicanze immunomediate. Per questo motivo, in questi
58E M A T O L O G I A

7
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
59
pazienti è opportuno associare alla CSA, al dosaggio di 3 mg/Kg evdie, una terapia corticosteroidea. Un effetto preventivo sullo sviluppo della GVHD è svolto anche daltrattamento con SAL che è previsto nei regimi di condizionamento deltrapianto con SCO (15 mg/Kg/die per 4 giorni) allo scopo di ridurre ilrischio del rigetto. Nei trapianti da SCO da donatore correlato HLA identico in pazientiaffetti da emopatia maligna, il regime di profilassi della GVHD, visto ilbasso rischio della GVHD stessa, può essere sicuramente meno inten-sivo, con l’impiego di CSA alla dose di 1 mg/Kg ev die seguito da undosaggio per os di 6 mg/Kg al giorno dal momento in cui le condizionicliniche del paziente lo consentano. Nei pazienti sottoposti a trapiantocon SCO da donatore correlato HLA identico affetti da patologie nonneoplastiche, nelle quali l’effetto GVL non è richiesto, i regimi di profi-lassi della GVHD potrebbero prevedere l’impiego della CSA alla dosedi 3 mg/Kg ev die seguita dalla dose di 10 mg/Kg per os.Diversi studi sulla rimozione ex vivo dei linfociti T dall’inoculo di cellulestaminali (mediante l’impiego di anticorpi monoclonali o di metodi fisicio della combinazione dei due), hanno dimostrato una netta riduzionedell’incidenza della GVHD acuta di gran lunga superiore a qualunquealtro regime di profilassi. Tuttavia i brillanti risultati ottenuti sulla ridu-zione della GVHD sono gravati dall’incremento del rischio di recidivaleucemica e del rigetto (28-30).Interessanti sono gli studi di Champlin e coll. (31) che hanno adottato,in alternativa a una deplezione pan-T, una deplezione selett iva deilinfociti CD8+ dal midollo infuso in associazione alla somministrazionedi CSA, riducendo l’ incidenza della GVHD acuta al 22% rispetto al58% dei pazienti che avevano ricevuto midollo non T depleto e al 5%del gruppo storico dei pazienti che avevano ricevuto una deplezionepan-T. In questo studio non è stata osservata alcuna recidiva neipazienti e la DFS a 3 anni è stata del 68% rispetto al 45% per i pazien-ti riceventi midollo non manipolato e al 35% per i pazienti riceventimidol lo T depleto. Questi studi, come già precedentemente detto,necessitano di ulteriori conferme. Degli altri farmaci attualmente in studio per la profilassi della GVHDquali i l SAL, la tal idomide, i l succini lacetone, i l tracol imus (FK506)sembra essere il più promettente.
7.3 TERAPIA DELLA GVHD ACUTA
La terapia di prima linea nel trattamento della GVHD è rappresenta-ta dai corticosteroidi. Sebbene siano state osservate delle rispostecon dosi variabili da 1 a 60 mg/Kg/die di metilprednisolone, le mega

dosi sono state associate a un’alta incidenza di infezioni mortali. Difatto, il dosaggio di metil-prednisolone attualmente impiegato in moltiCentri di trapianto è 2 mg/Kg/die. I pazienti che non rispondono allaprima linea di terapia sono eleggibili per trattamenti più aggressivi anchese in prima battuta l’atteggiamento è quello di incrementare la dose disteroidi.I farmaci di seconda linea impiegati agiscono con meccanismi diversi ei più usati sono: la globulina anti-linfocitaria; gli anticorpi monoclonalianti-cellule T (OKT3); gli anticorpi anti-recettore dell’IL-2; l’FK506 (far-maco immunosoppressore che blocca l’attivazione delle cellule T); lesostanze antiossidanti (che determinano una modulazione negativa dialcune risposte immunitarie) quali l’N-acetil cisteina; la deossispergua-lina (farmaco capace di inibire la funzione dei macrofagi, i l infociti Tcitotossici e la maturazione dei linfociti B).Studi su casistiche più numerose di pazienti con maggiore follow-upsono tuttavia necessari per valutare la reale efficacia di questi farmacie di altri ancora in corso di sperimentazione.
60E M A T O L O G I A

8
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
61
LA TERAPIA DI SUPPORTO
8.1 PROFILASSI INFETTIVA
Il trapianto di cellule staminali allogeniche comporta sempre un’elevataimmunosoppressione che è responsabile di complesse problematicheinfett ivologiche per le quali è fondamentale un’adeguata profi lassi.Infatti, le infezioni batteriche, fungine, virali e protozoarie sono ancoragravate dal 30-50% di letalità. Inoltre, le complicanze infettive sono piùfrequenti e gravi nel caso in cui il trapianto non sia HLA compatibile, ilmidollo sia stato T depleto e sia presente GVHD acuta o cronica.Pertanto, la profilassi antimicrobica rappresenta un importante presidioper ridurre il tasso di morbilità e di letalità da infezioni nel paziente sot-toposto a trapianto al logenico. Tuttavia, malgrado tale prat ica siaampiamente impiegata in tutti i Centri trapiantologici, in considerazionedella non provata efficacia e della tossicità di alcuni farmaci utilizzati,sono tuttora non risolte molte problematiche.
8.2 PROFILASSI ANTIBATTERICA
Per la profilassi antibatterica sono stati impiegati vari antibiotici oralinon assorbibili quali vancomicina, gentamicina, neomicina, colestina,polimixina B in varie combinazioni. Nonostante i numerosi studi clinicicontrollati, la scarsa compliance dei pazienti, gli alti costi e soprattuttol’emergenza di resistenze hanno reso necessario l’impiego di altri anti-biotici. Attualmente durante il periodo della neutropenia post-trapianto è con-sigliabile l’impiego dei chinolonici, preferibilmente i nuovi fluorchino-lonici. In particolare la ciprofloxacina sembra preferibile rispetto allanorfloxacina (i due tipi di chinolonici più largamente impiegati) perchéraggiunge alti l ivell i plasmatici, risulta più attiva contro alcuni germigram positivi e verso lo Pseudomonas spp.Di contro la ciprofloxacina non è attiva contro molti stafilococchi meti-cillino-resistenti, che rappresentano la maggior parte degli stafilococ-chi isolati.Da considerare il problema della possibile insorgenza di resistenze aichinolonici. Tuttavia in uno studio prospettico effettuato su una coorte

di 500 pazienti presso il Johns Hopkins Oncology Center, non è statamessa in evidenza l’insorgenza di resistenza in pazienti trattati in viaprofilattica con la norfloxacina. Inoltre i chinolonici hanno una scarsaatt iv i tà contro gl i anaerobi, quindi la f lora intest inale anaerobica,durante il loro impiego non viene alterata; questo potrebbe essere unfattore protettivo nei confronti di altri germi patogeni più virulenti.Le dosi consigliate nella maggior parte degli studi per la cipro-floxacina sono 500 mg x 2/die anche se vanno verif icati eventualidosaggi minori. La somministrazione di immunoglobuline endovena ha dato risultaticontrastanti e in ogni caso è gravata da un elevato rapporto costobeneficio.
8.3 PROFILASSI ANTITUBERCOLARE
La reale incidenza della tubercolosi nei pazienti sottoposti a trapiantoallogenico non è nota. È quindi difficile dare delle linee guida in merito,così come, del resto, per altre categorie di pazienti immunocompro-messi. È indicata la profi lassi nei pazienti con anamnesi positiva o concutireazione positiva alla tubercolina. Il farmaco da utilizzare è l’iso-niazide al dosaggio di 300 mg/die che va continuato per almenotre mesi dopo la sospensione della terapia immunosoppressiva.
8.4 PROFILASSI DELLA POLMONITE DA PNEUMOCISTIS CARINII
La profilassi contro lo Pneumocistis carinii viene attuata con il co-tri-mossazolo (trimetropim-sulfametossazolo): 160-800 mg per os pertre volte alla settimana dall’attecchimento e per la durata del trat-tamento immunosoppressivo.Per i soggetti allergici al co-trimossazolo viene usata la pentamidinaaerosol (300 mg una volta al mese) o il dapsone per os (50 mg x2volte a settimana).
8.5 PROFILASSI ANTIFUNGINA
Varie sostanze somministrate per via orale sono state impiegate nellaprofilassi antifungina delle infezioni da Candida (nistatina, clotrimazolo,miconazolo, amfotericina B) senza dare tuttavia risultati incoraggianti.
62E M A T O L O G I A

8
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
L’impiego dell’amfotericina B per via endovenosa, somministrata abassi dosaggi, non si è mostrata chiaramente efficace mentre è daconsiderare la possibile tossicità del farmaco. Alcuni trial controllati sull’impiego del ketoconazolo hanno dato buonirisultati sulla riduzione delle mucositi da Candida, ma nessuno studioha mostrato l’efficacia nel ridurre le infezioni sistemiche. Il miconazolosomministrato per via endovenosa ha dato buoni risultati nel ridurre leinfezioni sistemiche in una sola esperienza, ma non è risultato attivocontro l’Aspergillo e inoltre ha creato numerosi problemi di tossicità.Attualmente i triazolici sembrano avere una buona attività antifungina euna ridotta tossicità. Il primo farmaco di questo gruppo impiegato negliStati Uniti è stato il fluconazolo, disponibile sia per via orale che endo-venosa; ha un’escrezione prevalentemente renale, possiede una buonacapacità di penetrazione nel f luido cerebrospinale, ha un rapido edelevato assorbimento per via orale anche in pazienti con mucosite, einoltre può essere somministrato una sola volta durante la giornata. Ilfarmaco sembra interferire con altre sostanze che vengono metaboliz-zate a livello del sistema enzimatico citocromo P450, come la CSA.Tuttavia, le dosi di farmaco solitamente impiegate (100-400 mg/die)non sembrano determinare interferenze clinicamente significative. Duestudi controllati (102, 103) ne hanno evidenziato l’efficacia nel ridurrele infezioni disseminate e le mucositi da Candida e nel migliorare lasopravvivenza rispetto al gruppo placebo. Tuttavia il fluconazolo non èefficace contro l’Aspergil lo e contro alcune specie di Candida qualiCandida krusei e Candida glabrata. Pochi sono i dati sull’impiego inprofilassi dell’itraconazolo.Relat ivamente al la prof i lassi contro le infezioni da Aspergi l lo sononecessari studi clinici per definire l’efficacia dei triazolici, in particolaredell’itraconazolo che, in uno studio non randomizzato, sembrerebberidurre l’ incidenza delle infezioni da Aspergil lo rispetto al gruppo dipazienti trattato con ketoconazolo. Tuttavia l’itraconazolo è un farma-co il cui assorbimento per via orale dipende dall’acidità gastrica e lacui efficacia è strettamente legata ai livelli plasmatici, quindi è fonda-mentale valutarne la reale biodisponibi l i tà dopo assunzione orale.Importante sarà la valutazione dell’impiego della formulazione endove-nosa di prossima produzione.
8.6 PROFILASSI ANTIVIRALE
Le infezioni da virus erpetico si osservano per lo più nel primo mese post-trapianto per la riattivazione del virus latente nei soggetti sieropositivi.È consigliabile usare nella profilasi antivirale (HSV, VZV) acyclovir alladose di 250 mg/m2 ev ogni 8 ore durante i periodi in cui la mucosi-
63

te non permette l’assunzione per os del farmaco iniziandone l’as-sunzione già durante il condizionamento. Quindi la profilassi antivi-rale può essere continuata per os alla dose di 400 mg x 5/die perun anno, a meno di terapia immunosoppressiva ancora in atto.
8.7 PROFILASSI DELLE INFEZIONI DA CYTOMEGALOVIRUS
L’infezione da CMV viene definita dalla presenza del virus o degli anti-geni virali nel sangue o nei tessuti, o dall’incremento del titolo anticor-pale di 4 volte o più. La malattia da CMV viene definita dalla presenzadi manifestazioni cliniche legate al virus. I principali fattori di rischioper la malattia da CMV sono l’età del paziente, la sieropositività pre-trapianto, il grado di compatibilità HLA, la presenza della GVHD.L’associazione CMV-GVHD è determinata probabilmente dall’induzionedell’attivazione di linfociti citotossici da parte del virus e a sua volta laGVHD si associa a un aumentato rischio di infezione da CMV.I pazienti sieronegativi con donatore sieronegativo per CMV nonnecessitano di nessuna profilassi, ma devono essere trasfusi conemoderivati sieronegativi. L’impiego di emoderivati filtrati per rimuo-vere i globuli bianchi dai prodotti ematici sembrerebbe, in assenza didonatori CMV negativi, ridurre l’incidenza dell’infezione virale. Tuttaviasono necessari ulteriori studi per valutare la reale validità di questaprocedura e sono necessari studi randomizzati per confrontare l’impie-go di donatori sieronegativi verso prodotti filtrati.L’uso di acyclovir ad alte dosi si è dimostrato solo modestamente effi-cace nella prevenzione delle infezioni da CMV. La somministrazione di gancyclovir a tutti i pazienti sieropositiviper CMV è molto costosa e ingiustificata a causa dell’elevata tossi-cità midollare. In uno studio condotto da Goodrich e coll. (104), i lgancyclovir somministrato in via profilattica alla dose di 5 mg/Kg x 2/diefino al 100° giorno, ha ridotto l’incidenza della PI da CMV, della malattiada CMV e dell’escrezione virale. Tuttavia non c’è stata nessuna differen-za in termini di mortalità tra il gruppo che ha ricevuto gancyclovir rispet-to al gruppo placebo sia durante il trattamento che a 180 giorni.Il trattamento si è mostrato infatti mielotossico e ha incrementato le com-pl icanze infett ive legate al la neutropenia. Uno studio condotto daWinston e coll. (105) che hanno impiegato il gancyclovir a dosi più basse,ha ridotto il rischio di infezioni da CMV, ma non la malattia da CMV.Sembra essere più giustificato trattare con gancyclovir (5 mg/Kg x2/die) i pazienti che presentano viremia o antigenemia positiva peril virus o presenza di CMV nel broncolavaggio: è necessario a talescopo un controllo virologico settimanale.
64E M A T O L O G I A

8
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
L’impiego del foscarnet in profilassi è ancora da definire. Il foscar-net è indicato nelle forme resistenti al gancyclovir e nei pazientiche presentano indagini virologiche positive nella fase di attecchi-mento midollare.Nei trapianti da donatore incompatibile familiare o non correlato, gra-vati da un maggior rischio di infezione da CMV, l’impiego del gancyclo-vir in profilassi è oggetto di studio.
8.8 PROFILASSI DELLE INFEZIONI BATTERICHE TARDIVE
La morbilità e la mortalità da GVHD cronica sono spesso dovute alleinfezioni. Si rende pertanto necessaria una profilassi antibiotica som-ministrata anche nelle fasi più tardive del trapianto. Sia in presenzache in assenza di GVHD cronica deve essere presa in considera-zione la terapia antibiotica con penicillina per ridurre l’incidenzadelle infezioni da Streptococcus pneumoniae e da batteri capsula-ti. Tali infezioni sono legate all’asplenia funzionale nel pazientetrapiantato e all’incapacità di produrre anticorpi opsonizzanti.La disponibil ità di vaccini contro pneumococco, Haemophilusinfluenzae e meningococco è particolarmente utile, purtroppo inquesta categoria di pazienti è poco efficace la risposta alle vacci-nazioni, soprattutto in presenza di GVHD cronica. Per i pazient i con r iduzione del le immunoglobul ine l ’ impiego del leimmunoglobuline endovena potrebbe essere utile, ma anche in questocaso è discutibile il rapporto costo beneficio.
8.9 ALIMENTAZIONE PARENTERALE
L’elevata tossicità del regime di condizionamento con gli effetti collate-rali a esso legati (anoressia, nausea, vomito, mucositi, diarrea), lo svi-luppo di infezioni e la GVHD non consentono un adeguato apporto ali-mentare nel paziente trapiantato e i l mantenimento di un adeguatostato nutrizionale. Si rende pertanto necessario in corso di trapiantol’impiego di un supporto nutrizionale parenterale. Inoltre, dati speri-mentali hanno dimostrato gli effetti negativi della malnutrizione protei-co-calorica sull’attecchimento di cellule emopoietiche infuse in animalida esperimento precedentemente sottoposti a irradiazione. Il ruolo di un’adeguata alimentazione parenterale sulla sopravvivenza diun paziente sottoposto a trapianto è mostrato in Figura 5 (106).
65
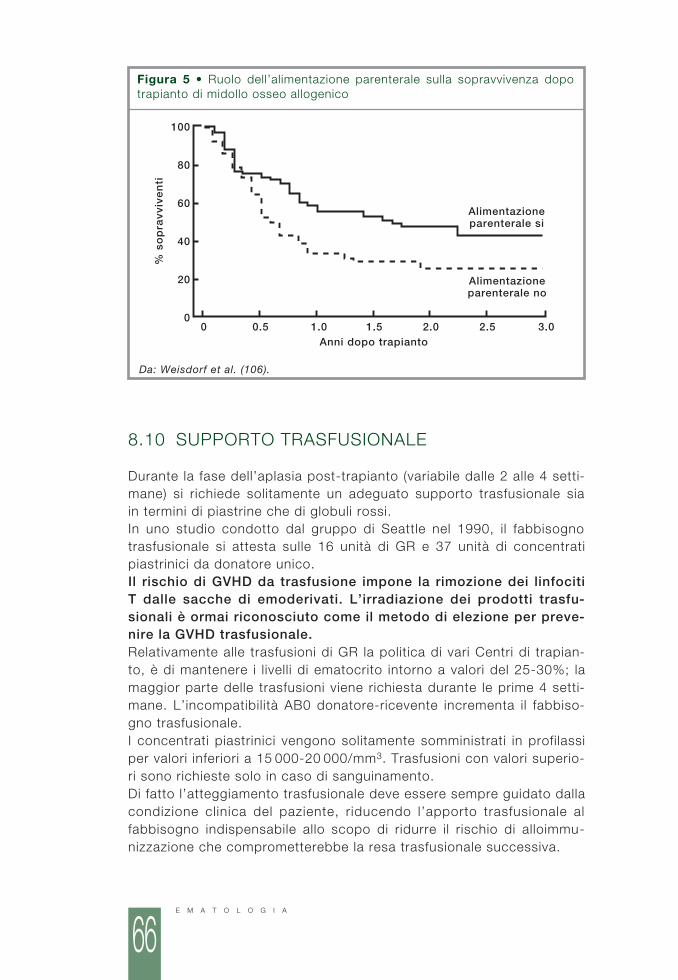
8.10 SUPPORTO TRASFUSIONALE
Durante la fase dell’aplasia post-trapianto (variabile dalle 2 alle 4 setti-mane) si richiede solitamente un adeguato supporto trasfusionale siain termini di piastrine che di globuli rossi. In uno studio condotto dal gruppo di Seattle nel 1990, il fabbisognotrasfusionale si attesta sulle 16 unità di GR e 37 unità di concentratipiastrinici da donatore unico.Il rischio di GVHD da trasfusione impone la rimozione dei linfocitiT dalle sacche di emoderivati. L’irradiazione dei prodotti trasfu-sionali è ormai riconosciuto come il metodo di elezione per preve-nire la GVHD trasfusionale. Relativamente alle trasfusioni di GR la politica di vari Centri di trapian-to, è di mantenere i livelli di ematocrito intorno a valori del 25-30%; lamaggior parte delle trasfusioni viene richiesta durante le prime 4 setti-mane. L’incompatibilità AB0 donatore-ricevente incrementa il fabbiso-gno trasfusionale.I concentrati piastrinici vengono solitamente somministrati in profilassiper valori inferiori a 15 000-20 000/mm3. Trasfusioni con valori superio-ri sono richieste solo in caso di sanguinamento.Di fatto l’atteggiamento trasfusionale deve essere sempre guidato dallacondizione clinica del paziente, riducendo l’apporto trasfusionale alfabbisogno indispensabile allo scopo di ridurre il rischio di alloimmu-nizzazione che comprometterebbe la resa trasfusionale successiva.
66E M A T O L O G I A
Figura 5 • Ruolo dell’alimentazione parenterale sulla sopravvivenza dopotrapianto di midollo osseo allogenico
Da: Weisdorf et al. (106).
100
80
60
40
20
00 0.5 1.0 1.5 2.0 2.5 3.0
Anni dopo trapianto
Alimentazione parenterale si
% s
op
ravv
ive
nti
Alimentazione parenterale no

9
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
LA RECIDIVA LEUCEMICAPOST-TRAPIANTO
La recidiva leucemica rappresenta tuttora una delle cause più frequentidi insuccesso dopo trapianto di midolloL’aumento della sopravvivenza osservato negli ultimi 10 anni sembraessere dovuto principalmente al miglioramento della terapia di suppor-to, mentre la probabilità di recidiva è rimasta relativamente costante:tra il 10% e il 40% per pazienti trapiantati in fase favorevole di malattia(I RC di leucemia acuta o FC di LMC) e il 50-70% per pazienti con leu-cemia in fase più avanzata (107). La recidiva generalmente originadalle cellule del ricevente, a dimostrazione del fatto che il clone leuce-mico può sopravvivere alle dosi sovramassimali di radio-chemioterapiae sottrarsi all’effetto GVL. Solo in rari casi la malattia, osservata per lopiù tardivamente dopo trapianto, si è r ipresentata nel le cel lule deldonatore. Diverse cause sono state considerate responsabil i di taleevento: l’impiego della TBI, la persistenza dello stimolo leucemogenomicroambientale, il trasferimento di materiale oncogenetico dalle cellu-le leucemiche del paziente e quelle normali del donatore.
Anche i pazienti con GVHD acuta o cronica, pregressa o in atto, pos-sono recidivare a ulteriore indicazione che non sempre la GVHD si tra-duce in un effetto GVL. I tentativi di ridurre la recidiva leucemica sonostati diretti da una parte verso il potenziamento dei regimi di condizio-namento pre-trapianto, dall’altra parte verso la ricerca di amplificazio-ne dell’effetto GVL mediante la riduzione della profilassi per la GVHD.L’aggiunta di nuovi chemioterapici o l’aumento di intensità della radio-terapia, pur dimostratisi maggiormente eradicanti verso il clone leuce-mico, non hanno migliorato i risultati globali in termini di sopravviven-za, essendo correlati a un maggior rischio di mortalità post-trapianto.L’osservazione che anche per i pazienti riceventi midollo non T depletoil rischio di recidiva leucemica si modifica in rapporto al livello e tipo diprofilassi per la GVHD, ha favorito studi clinici basati sulla modulazionedei regimi di profilassi della GVHD senza ottenere tuttavia risultati sod-disfacenti.
Influenzano l’evoluzione della recidiva leucemica post-trapianto il tipodi leucemia, l’intervallo di tempo trascorso dal trapianto alla recidiva, ilperformance status del paziente e il tipo di trattamento.
67

68E M A T O L O G I A
9.1 CHEMIOTERAPIA CONVENZIONALE
Nei pazienti con leucemia acuta in recidiva dopo trapianto la prognosiè senz’altro sfavorevole e, se non viene somministrata alcuna terapia,la mediana di sopravvivenza è di soli 3-4 mesi.Sono segnalati rarissimi casi di RC ottenuta con la sola sospensionedella CSA in pazienti recidivati in corso di trattamento immunosop-pressivo.La probabilità di RC con chemioterapia convenzionale varia dal 30 al40%, con una mediana di sopravvivenza di 8 mesi per la LMA e di 14mesi per la LLA. Nei pazienti che non ottengono RC la mediana disopravvivenza si riduce a 3 mesi.La prognosi per i pazienti recidivati dopo TMO per LMC dipende dalsesso del paziente, dall’intervallo di tempo dal trapianto e soprattuttodalla fase di malattia al momento della recidiva: in analisi univariata laprobabilità di sopravvivenza a 6 anni è del 52% per i pazienti con solarecidiva citogenetica, rispetto al 30% per i pazienti in recidiva ancheematologica; nessun paziente che recidiva dopo trapianto con malattiain fase avanzata ha una sopravv ivenza super iore a 3.5 ann i da lmomento della recidiva (108).Il trattamento con chemioterapia convenzionale, proposto per i pazien-ti con recidiva di LMC in FA o CB non ha dato risultati soddisfacenti: lamediana di sopravvivenza risulta inferiore a 6 mesi. Al contrario, nelcaso in cui la recidiva compaia in corso di trattamento con CSA, l’im-mediata sospensione di quest’ultima può indurre una RC con recuperodell’emopoiesi del donatore. Infine il secondo trapianto, pur permet-tendo una probabil ità di DFS a 4 anni variabile tra i l 20 e i l 30% aseconda delle diverse casistiche, è correlato con un elevato rischio dimortalità precoce e di ulteriore recidiva (109). È consigliabile pertantoche tale procedura sia limitata a pazienti non clinicamente compro-messi nei quali la recidiva sia intervenuta tardivamente dopo il primotrapianto (>6 mesi nei pazienti pediatrici, >12 mesi nei pazienti adulti).
9.2 TRATTAMENTI IMMUNOMODULANTI CON FATTORI DI CRESCITA E CITOCHINE
La patologia per la quale l’impiego di fattori immunomodulanti è megliodefinita è senz’altro la LMC in recidiva post-trapianto distinta in recidi-va molecolare, citogenetica ed ematologica.La tecnica della PCR applicata allo studio della malattia minima resi-dua (MRD) dopo trapianto permette di identificare il trascritto di fusio-ne RNA bcr/abl specifico della malattia fino a una diluizione di 105-106
cellule.

9
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
Sebbene ampiamente praticata, tale tecnica rimane tuttavia una proce-dura particolarmente delicata per i problemi correlati alla sua esecuzio-ne, alla sua specificità e sensibilità.È pertanto difficile un confronto di risultati provenienti da laboratoridiversi e devono essere considerate con cautela le interpretazioni clini-che dei dati che da essi vengono tratte. Inoltre, solo in alcuni studi ipazienti testati presentano un fol low-up suff icientemente lungo pervalutare l’eventuale correlazione con la recidiva citogenetica ed emato-logica, e il numero dei pazienti analizzati spesso non è sufficiente peruna corretta valutazione del test in relazione ad altre variabili. Uno studio del gruppo di Seattle del 1995 (110) ha valutato il significa-to prognostico della PCR in 346 pazienti, analizzati a diversi intervallidi tempo dal TMO.
Le principali conclusioni di questo studio possono essere sintetizzatenei seguenti punti:1. esiste una concordanza di risultato della PCR tra midollo e sangue
periferico nel 91% dei casi;2. la precoce positività della PCR, rilevata entro 3 mesi o oltre 36 mesi
dal trapianto, non è predittiva del rischio di recidiva;3. una singola positività della PCR rilevata tra 6 e 12 mesi e tra 12 e
24 mesi post-trapianto si correla con una probabilità attuariale direcidiva rispettivamente del 42 e 25%, significativamente più eleva-ta del 3 e 1% della probabilità calcolata nei pazienti PCR negativiagli stessi intervalli di tempo;
4. la progressione di malattia interviene con una mediana di 7 mesi neipazienti risultati PCR positivi;
5. in analisi multivariata, la PCR positività rimane il principale fattoreindipendente correlato con il rischio di recidiva; gli altri due fattori aessa associati che mantengono significatività statistica sono il TMOda donatore HLA compatibile e la GVHD acuta di grado 0-I.
Il dibattito ancora aperto sul significato biologico della PCR dopo TMOnon permette di trovare ancora un approccio clinico terapeutico nelcaso di una recidiva molecolare di malattia. Tuttavia, alla luce dei datiprecedentemente esposti, è proponibile che la recidiva molecolare,dopo 6 mesi dal trapianto nei pazienti non riceventi CSA, sia valu-tata in uno studio prospettico randomizzato per la terapia conIFN; ovviamente interventi terapeutici più aggressivi non sono giu-stificati.Part icolarmente complesso è i l quadro del la recidiva citogenetica.Sebbene la ricomparsa di cellule Ph positive dopo trapianto sia spessoseguita da una progressione di malattia verso la fase ematologica, noninfrequente è l’osservazione di metafasi Ph positive transitorie o persi-stenti in assenza di recidiva ematologica.Il significato predittivo di progressione ematologica rappresentato dalla
69

recidiva citogenetica è influenzato da diversi fattori: metafasi Ph positi-ve frequentemente rilevate entro i primi 100 giorni possono rappresen-tare un residuo cellulare maturante destinato a estinguersi; una ridottapercentuale (<10%) di cellule Ph positive, rilevate anche tardivamentedopo trapianto nell’ambito di una normale emopoiesi del donatore,vanno facilmente incontro a una remissione spontanea; un aumentatorischio di progressione ematologica è stato invece segnalato per unaproporzione di cellule Ph positive >25% in associazione con una con-dizione di chimerismo misto presente nel compartimento emopoieticonormale (111). Inoltre, l’evoluzione della recidiva citogenetica è influen-zata dal regime impiegato per la profilassi della GVHD: pazienti rice-venti midollo T depleto anche in presenza di una bassa percentuale dimetafasi Ph positive quasi sempre evolvono in recidiva ematologicafranca.Da un punto di vista terapeutico è ormai confermata l’indicazione tera-peutica alla terapia con IFN. L’analisi retrospettiva eseguita dall’EBMTsu 130 pazienti (108), dimostra che la terapia con IFN ritarda la pro-gressione di malattia e induce pertanto un significativo aumentodella sopravvivenza dei pazienti con LMC in recidiva sia ematolo-gica sia soltanto citogenetica.Tale approccio terapeutico deve tuttavia essere attualmentericonsiderato alla luce delle nuove esperienze provenienti dall’in-fusione dei linfociti del donatore la cui potente attività GVL si tra-duce in elevata efficacia terapeutica come verrà meglio definito nelprossimo paragrafo.Del tutto preliminari sono i risultati relativi all’azione del G-CSF sullarecidiva di leucemia acuta e cronica recentemente riportati dal gruppodell’M.D. Anderson: 3 di 7 pazienti trattati con G-CSF hanno ottenutola RC mantenuta per un follow-up sufficientemente lungo.Infine, anche l’IL-2 sembra un agente potenzialmente attivo nel tratta-mento della recidiva dopo trapianto.
9.3 TRATTAMENTO IMMUNOMODULANTE MEDIANTE INFUSIONE DEI LINFOCITI DEL DONATORE
Nei pazienti con recidiva di malattia dopo TMO il chimerismo e la con-seguente tolleranza immunologica verso le specificità HLA del donato-re costituiscono il presupposto necessario per l’impiego di una immu-noterapia adottiva con i linfociti del donatore il cui obiettivo è quello dievocare un’attività GVL. Nel lo stud io ret rospett ivo, mul t icentr ico de l l ’EBMT (112) su 135pazienti sottoposti a infusione di linfociti del donatore per LMC, LMA,LLA e MDS in recidiva post-TMO, la maggiore efficacia terapeutica in
70E M A T O L O G I A

9
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
termini di percentuale di RC si riscontra per i pazienti con LMC in reci-diva citogenetica (82%) o ematologica in FC (74%). Tali percentuali siriducono al 22% per i pazienti con LMA, al 12% dei pazienti con LMCin FA, mentre nessuno dei pazienti con LLA risulta rispondente. Le complicanze più frequentemente osservate con l’infusione dei linfo-citi del donatore sono state la GVHD e la mielosoppressione osservatenel 50 e 35% rispettivamente dei pazienti.L’ottenimento della RC per effetto GVL indotto dal DLT è significativa-mente associato all’insorgenza della GVHD e/o della mielosoppressione.Allo stato attuale la terapia con DLT è da considerarsi elettiva per ipazienti in recidiva di LMC in FC post-trapianto, mentre per ipazienti con recidiva in fase avanzata è indicato un trattamentocon chemioterapia seguita da infusione di DLT. Fondamentale è lostudio dello stato di chimerismo pre-DLT, infatti, in uno stato di chime-rismo completo o misto l’aplasia midollare risulta transitoria o assente,mentre in assenza di chimerismo è più alto il rischio di sviluppareun’aplasia midollare grave (113). In quest’ultimo caso è indicatopertanto associare all’infusione di DLT anche cellule staminali deldonatore mobilizzate previa somministrazione di G-CSF.Per i pazienti in recidiva citogenetica è proponibile una stratificazione,in base al numero di metafasi Ph+ a ricevere o meno terapia con IFN;se il numero di metafasi Ph+ è elevato (>40%) i pazienti possono esse-re randomizzati a ricevere DLT + IFN.Più aggressivo dovrebbe essere l’atteggiamento terapeutico e il moni-toraggio citogenetico e molecolare per i pazienti che hanno ricevutomidollo T depleto in considerazione dell’elevato rischio di evoluzioneematologica in caso di recidiva citogenetica o molecolare di malattia.
71

10
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
BIBLIOGRAFIA GENERALE
1. Quine WE et al., J Am Med Assoc 26: 1012; 1896.2. Osgood EE et al., Ann Intern Med 13: 357-367; 1939.3. Jacobson LO et al., Science 113: 510; 1951.4. Lorenz E et al., Radiology 58: 863-877; 1952.5. Mathè G et al., Br Med J 2: 1633-1635; 1963.6. Thomas ED et al., N Engl J Med 257: 491; 1958.7. Bach FH et al., Lancet ii: 1364-1366; 1968.8. Ferrari G. et al., Science 279:1528-1530; 1998.9. Arcese W et al., La trasfusione del sangue 41: 443-448; 1996.
10. Gluckman E et al., N Engl J Med 321: 1174-1178; 1989.11. Wagner JE et al., Lancet 346: 214-219; 1995.12. Kurtzenberg J et al., N Engl J Med 335: 157-166; 1996.13. Wagner JE et al., Blood 88: 795-802; 1996.14. Gluckman E et al., N Engl J Med 337: 373-381; 1997.15. Weiden P et al., N Engl J Med 304: 1529; 1981.16. Sullivan K et al., Blood 72: 546-554; 1988.17. Sullivan K et al., Blood 73: 1720-1728; 1989.18. Collins Jr RH et al., Bone Marrow Transplant 10: 391-395; 1992.19. Suzuke R et al., Bone Marrow Transplant 20: 615-617; 1997.20. Gale R et al., Lancet ii: 28; 1984.21. Ringden O et al., Transplant Proc 21: 2989; 1989.22. Van Lochem E et al., Bone Marrow Transplant 10: 181; 1992.23. Neiderwieser D et al., Blood 81: 2200; 1993.24. Chen W et al., Proc Natl Acad Sci USA 89: 1468; 1992.25. Hausch M et al., Blood 75: 2250; 1990.26. Ferrara J et al., N Engl J Med 324: 667; 1991.27. Baron S et al., JAMA 266: 1375; 1991.28. Apperley J et al., Bone Marrow Transplant 1: 53; 1986.29. Maraninchi D et al., Lancet ii: 175; 1987.30. Goldmann JM et al., Ann Intern Med 108: 806; 1988.31. Champlin R et al., Blood 76: 418-423; 1990.32. Fo rman SE e t a l . , Bone Mar row T ransp lan ta t ion , Bos ton :
Blackwell Scientific Publications, 1994.33. Carella AM et al., Haematologica 82: 478-495; 1997.34. Armitage JO, N Engl J Med 330: 827-838; 1994.35. Appelbaum FR et al., Blood 72: 179-184; 1988.36. Schiller GJ et al., J Clin Oncol 10: 41-46; 1992.
73

74E M A T O L O G I A
37. Cassileth PA et al., Blood 79: 1924-1930; 1992.38. Avvisati G et al., Blood 88: 1390-1398; 1996.39. Barret AJ et al., Blood 79: 3067-3070; 1992.40. Doney K et al., Bone Marrow Transplant 2: 355-363; 1987.41. Chao Nj et al., Blood 78: 1923-1927; 1991.42. Horowitz MM et al., Ann Intern Med 115: 13-17; 1991.43. Fiere D et al., J Clin Oncol 11: 1990-1991; 1993.44. Gluckman E et al., Blood 79: 269-275; 1992.45. Bacigalupo A et al., Br J Haematol 70: 177-182; 1988.46. Lucarelli G, N Engl J Med 322: 417-421; 1990.47. Lucarelli G et al., Blood 80: 1603-1607; 1992.48. Galimberti M et al., Bone Marrow Transplant 19 (Suppl. 2): 45-
47; 1997.49. Gluckman E et al., Br J Haematol 45: 557-564; 1980. 50. Flowers MED et al., Bone Marrow Transplant 9: 167-173; 1992.51. Gatti RA et al., Lancet ii: 1366-1369; 1968.52. O’Reilly RJ et al., Immunodefic Rev 1: 273-309; 1989.53. O’Reilly RJ et al., N Engl J Med 297: 1311-1318; 1977.54. Sociè G et al., Lancet 348: 573-577; 1996.55. Rajantie J et al., Blood 67: 1693-1697; 1986.56. Appelbaum FR et al., Ann Intern Med 112: 590; 1990.57. Anderson JE et al., Blood 82: 677-681; 1993.58. De Witte T et al., Br J Haematol 74: 151-157; 1990.59. Michallet M et al., Bone Marrow Transplant 7: 275-279; 1991.60. Rabinowe SN et al., Blood 82: 1366-1376; 1993.61. Khouri IF et al., J Clin Oncol 12: 748-758; 199262. Osserman EF et al., Acta Hematol 68: 215-223; 1982.63. Gahrton G et al., N Engl J Med 325: 1267-1273; 1991.64. Armitage JO et al., Blood 73: 1749-1758; 1989.65. Appelbaum FR et al., J Clin Oncol 5: 1340-1347; 1987.66. Jones RJ et al., Blood 77: 649-653; 1991.67. Lundberg JH et al., J Clin Oncol 9: 1848-1859; 1991.68. Phillips GL et al., J Clin Oncol 7: 1039-1045; 1989.69. Aversa F et al., Blood 84: 3948-3955; 1994.70. Kernan NA et al., N Engl J Med 328: 593-602; 1993.71. Beatty PG et al., Blood 81: 249-253; 1993.72. Balduzzi A et al., Blood 86: 3247-3256; 1995.73. Sierra et al., Blood 89: 4226-4253; 1997.74. Davies SM et al., Bone Marrow Transplant 13: 51-57; 1993.75. Mehta J et al., Bone Marrow Transplant 13: 583-587; 1993.76. Anderson JE et al., Br J Haematol 93: 59-67; 1996.77. Marmont AM et al., Blood 79: 2120-2130; 1991.78. Hansen JA et al., Bone Marrow Transplant 15: 128-139; 1995.79. Beatty PG et al., Transplantation 2: 443-446; 1991.80. Hansen JA et al., Blood 86: 479 (Abs); 1995.

10
I L T R A P I A N T O D I C E L L U L E S T A M I N A L I A L L O G E N I C H E
81. Korbling M et al., Blood 86: 2842-2848; 1995.82. Anderlini P et al., Transfusion 37: 507-512; 1997.83. Przepiorka D et al., Bone Marrow Transplant 19: 455-460; 1997.84. Azvedo WM et al., Bone Marrow Transplant 16: 647-653; 1995.85. Russell JA et al., Bone Marrow Transplant 17: 703-708; 1996.86. Anderlini P et al., Blood 86; 109 (Abs); 1995.87. Bacigalupo A et al., Blood 88: 353-357; 1996.88. Knudtzon S et al., Blood 43: 357-361; 1974.89. Broxmeyer HE et al., Proc Natl Acad Sci USA 86: 3828; 1989.90. Winston DJ et al., Exp Hematol 12: 205-215; 1984.91. Meyers JD, Am J Med 81 (Suppl. 1A): 27-38; 1984.92. Mandell GL, Douglas RG, Bennett JE. Principles and Practice in
Infectious Diseases. Churchill Livingston, Fourth Edition, 1995.93. Weisdorf DJ et al., Blood 76: 624-629; 1990.94. Bearman S, Blood 85: 3005-3020; 1995.95. Sencer SF et al., Transplantation 56: 875-879; 1993.96. Bedi A et al., J Clin Oncol 13: 1103-1109; 1995.97. Fried RH et al., Ann Intern Med 116: 624-629; 1992.98. Springmeyer SC et al., in: Recent advances in bone marrow
transplantation. Gale RP (ed). New York: Alan R Liss, 343-353;1983.
99. Rochelle E et al., N Engl J Med 336: 897-904; 1997.100. Barnes DW et al., Br J Haematol 3: 241; 1967.101. Thomas ED et al., Blood 49: 522-533; 1977.102. Goodman Jl et al., N Engl J Med 326: 845-851; 1992.103. Slavin MA et al. J Infect Dis 171: 1545-1552; 1995.104. Goodrich JM et al., Ann Intern Med 118: 173-178; 1993.105. Winston DJ et al., Ann Intern Med 118: 179-184; 1993.106. Weisdorf SA et al., Transplantation 43: 833-838; 1987.107. Giralt SA et al., Blood 84: 3603; 1994.108. Arcese W et al., Blood 82: 3211; 1993.109 Radich J et al., J Clin Oncol 11: 304; 1993.110. Radich JP et al., Blood 85: 2632; 1995.111. Offitt K et al., Blood 75: 1346; 1990.112. Kolb Hj et al., Blood 86: 2041; 1995.113. Rapanott i MC et a l . , Bone Marrow Transplant 19: 703-707;
1997.
75


















