Dott.ssa Maria Antonietta Lozzi Dipartimento di sanità ... · frammenti di DNA attraverso una...
71
“DAGLI ESORDI DELLA MEDICINA MOLECOLARE ALL’EVOLUZIONE DELL’AUTOMAZIONE” Dott.ssa Maria Antonietta Lozzi Dipartimento di sanità Pubblica e Malattie Infettive Sapienza Università di Roma
Transcript of Dott.ssa Maria Antonietta Lozzi Dipartimento di sanità ... · frammenti di DNA attraverso una...

“DAGLI ESORDI DELLA MEDICINA MOLECOLARE ALL’EVOLUZIONE DELL’AUTOMAZIONE”
Dott.ssa Maria Antonietta Lozzi Dipartimento di sanità Pubblica e Malattie Infettive
Sapienza Università di Roma

• La scoperta che il DNA è alla base di tutte le funzioni della cellula ha aperto la strada allo sviluppo di una disciplina denominata biologia molecolare • Lo sviluppo delle metodologie e delle tecniche per isolare, analizzare, manipolare e utilizzare gli acidi nucleici ha determinato lo sviluppo di interessanti settori come le biotecnologie, le mappature dei genomi, la medicina molecolare e la terapia genica. • Molte tecniche utilizzate in biologia molecolare imitano le funzioni naturali degli acidi nucleici

Alla fine degli anni ‘70 e all’inizio degli anni ’80 le tecnologie di clonaggio, sequenza di acidi nucleici e di identificazione di sequenze specifiche del DNA o di RNA erano appannaggio esclusivo di alcuni laboratori attrezzati e impegnati esclusivamente in progetti di ricerca.
Le tecniche, all’epoca, non erano standardizzate, presentavano grande variabilità a seconda dei laboratori con ovviamente risultati difformi. Inoltre l’esecuzione richiedeva apparecchiature sofisticate e competenze non sempre disponibili nei laboratori di diagnostica

Le tecnologie allora disponibili erano le tecniche di ibridazione degli acidi nucleici attraverso le quali si poteva determinare la presenza e la quantità di acido nucleico, utilizzando una sequenza specifica (sonda) corrispondente al DNA bersaglio nel campione biologico analizzato La prima di queste tecniche, il Southern blot fu sviluppata da Edwin Southern nel 1975. La tecnica prevede, prima dell’ibridazione, la digestione del DNA bersaglio mediante il taglio con enzimi di restrizione, separazione dei frammenti di DNA attraverso una corsa elettroforetica su gel di agarosio, trasferimento su membrana di nylon o nitrocellulosa che costituisce il supporto per l’ibridazione, utilizzo quindi di una sonda marcata. Il metodo è diventato molto importante dopo lo sviluppo del clonaggio molecolare, che ha reso possibile l’uso di una miriade di sonde specifiche per i geni.
Il Northern blot variante del Southern inventata nel 1977 da alcuni scienziati dell’Università di Stanford. Il bersaglio è l’RNA, che viene separato tramite elettroforesi su gel di agarosio in condizioni denaturanti

1955 Severo Ochoa scopre RNA polimerasi Arthur Kornberg scopre DNA polimerasi
1952 Rosalind Franklin usa la diffrazione di raggi X per studiare la struttura del DNA e suggerisce che la struttura portante è formata da zuccheri e fosfati ed è situata all'esterno della molecola
1953 Dopo aver esaminato i risultati, non pubblicati, di Rosalind Franklin, James Watson e Francis Crick propongono la struttura a doppia elica per il DNA
1960 Arthur Kornberg sintetizza DNA in vitro, dimostrando che un enzima DNA polimerasi produce nuovi segmenti di DNA utilizzando precursori, un fonte di energia e un "template" di DNA
Nascita della biologia molecolare
……nel corso degli anni

1975 •Edwin Southern inventa la tecnica del Southern blot
1970 scoperta degli enzimi di restrizione → diventa possibile tagliare il DNA in siti specifici e riunire molecole di DNA di diversa origine elettroforesi su gel → diventa possibile separare DNA,RNA e proteine sfruttando un campo elettrico
1977 •James Alwine, David Kemp e George Stark dell’Università di Stanford inventano la tecnica del Northern blot •Maxam e Gilbert introducono il sequenziamento con metodo chimico Il DNA è tagliato chimicamente in corrispondenza di basi specifiche: i prodotti di taglio sono analizzati su gel elettroforesi •Sanger: introduce il sequenziamento con metodo a terminazione di catena Il Dna da sequenziare è replicato a partire da un primer innesco con una polimerasi; il filamento neosintetizzato è radiomarcato e la reazione è interrotta per incorporazione di dideossi-nucleotidi. Il metodo di Sanger è rimasto alla base dei protocolli di sequenziamento automatico

La tecnologia ideata da Kary Mullis e sviluppata da un gruppo di scienziati della Cetus Corporation, è stata pubblicata per la prima volta nel 1985 e largamente utilizzata poi come uno degli strumenti più potenti della biologia molecolare.
2003 Annuncio completamento del genoma (2 anni prima del previsto!) Cinquantesimo anniversario della scoperta della doppia elica da parte di Watson e Crick
1983 Kary Mullis inventa la reazione a catena della polimerasi(PCR)
1985 Primo impiego dell’impronta genetica (fingerprinting) in una investigazione criminale
1990 Lancio ufficiale del Progetto internazionale genoma umano (tempo previsto 15 anni) 2000 Pronta una bozza del genoma

“Sometimes a good idea comes to you when you are
not looking for it”
Kary B. Mullis

La PCR (Polymerase Chain Reaction) nasce da un’idea di Kary B. Mullis in una notte di Aprile del 1983, mentre è alla guida della
sua auto “Cominciando da una singola molecola di DNA, mediante una reazione a catena, si possono generare 100 bilioni di molecole simili in un solo pomeriggio. La reazione è facile : non richiede altro che una provetta, pochi semplici reagenti e una sorgente di calore”.
Per questa scoperta rivoluzionaria, il Dr. Mullis ha ricevuto, nel
1993, il premio Nobel per la Chimica.
Dalla sua introduzione, nel 1985, la tecnologia PCR ha modificato il modo in cui l’analisi del DNA viene condotta nei laboratori sia di ricerca che di diagnostica.

L’introduzione quindi di sistemi basati sulla rivelazione diretta di sequenze genomiche specifiche in un campione biologico nasce dall’esigenza di avere a disposizione,nel laboratorio diagnostico metodologie con un’elevata soglia di sensibilità e specificità. Sorprendentemente la PCR è in grado di soddisfare le più diverse necessità analitiche.
APPLICAZIONI DELLA PCR
• Diagnostica clinica ricerca di uno specifico virus o batterio • Oncologia diagnosi e prevenzione dei tumori • Agricoltura identificazione OGM • Ricerche sul genoma • Studio delle malattie ereditarie • Medicina legale • Archeologia

all’interno della cellula, i due filamenti di una doppia elica si svolgono e si separano per permettere la sintesi di nuove catene. Ogni filamento originario agisce da stampo per la formazione di un nuovo filamento complementare. In questo modo, la replicazione del DNA è semiconservativa ed ogni molecola figlia riceve un filamento dalla molecola madre
La reazione a catena della polimerasi (PCR) si basa sul principio naturale della replicazione del DNA:

PER UNA REAZIONE DI PCR SONO NECESSARI:
Una piccola quantità di DNA stampo ( Target )
Due oligonucleotidi sintetici specifici ( Primers )
Un enzima ( Taq Polimerasi )
Una idonea concentrazione di Sali (MgCl2)
Una miscela di nucleotidi
Soluzione tampone
La PCR si può definire come : una reazione di amplificazione in vitro di un segmento specifico di
DNA,sequenza “Target”, per mezzo di una DNA polimerasi.

Questo processo ha luogo nel thermal cycler, uno strumento che controlla ed alterna in modo automatico le temperature, per periodi di tempo programmati e per il numero di cicli di PCR adeguati (20-40)
Come il processo di replicazione naturale, il processo PCR crea copie multiple della sequenza scelta:
Un ciclo PCR è composto dalle seguenti fasi:
1. Denaturazione 2. Ibridazione 3. Estensione

PCR FASE PER FASE
PCR Fase 1: Denaturazione termica (94°C)
PCR Fase 2: Ibridazione del primer al bersaglio (annealing 55-65°C) 5’
5’
5’
3’
3’
5’
3’
3’
PCR Fase 3: Estensione (72°C)
5’
5’
3’
3’
5’
5’
3’
3’

Formazione di DUE nuove catene e
ripetizione del ciclo
No. cicli No. Ampliconi copie del target
1 2
2 4
3 8
4 16
5 32
6 64
20 1,048,576
30 1,073,741,824

Cinetica della PCR: 3 fasi
Esponenziale Dopo ogni ciclo si verifica un raddoppiamento del prodotto accumulato. La reazione è molto specifica e precisa
Lineare Continua la crescita esponenziale della reazione espressa come una fase logaritmica lineare.
Plateau (punto finale) La reazione si arresta e non da più luogo a prodotti di reazione. La rilevazione degli ampliconi in questa fase è definita rilevazione al punto finale.
Tecnica end-point

VANTAGGI DELLA PCR
1) Velocità In poche ore si possono ottenere milioni di copie della sequenza di
DNA target 2) Sensibilità Capacità di amplificare copie singole della sequenza bersaglio 3) Robustezza Amplificazione possibile anche utilizzando DNA di bassa qualità 4) Specificità La selezione di primers idonei aumenta la specificità della reazione

SVANTAGGI-PROBLEMATICHE
L’estrema sensibilità della PCR, in grado di generare una quantità enorme di copie di DNA, a partire da una sequenza bersaglio, rappresenta il maggior problema per la sua applicazione, soprattutto a fini diagnostici
Una fonte di contaminazione crociata può avvenire quando si prepara un campione positivo che contamina un campione negativo
Un terzo tipo di contaminazione, che dà falsi positivi, si può verificare durante la rilevazione
Contaminazione
Il saggio è pericolosamente sensibile alla presenza nell’ambiente, sugli strumenti o sull’operatore stesso, di molecole di DNA
proveniente da amplificazioni precedenti. CARRY-OVER!!!

PRECAUZIONI
Il rischio di contaminazione è ridotto al minimo attraverso accorte procedure di laboratorio
•Ambienti separati: preparazione campione, preparazione reagenti,
rivelazione prodotto
•Vetreria sterilizzata, materiale usa e getta
•Pulizia superfici lavoro e apparecchiature (Ipoclorito ed etanolo 70%)
•Utilizzo reagenti pre-aliquotati
•Uso di pipette distinte nelle varie aree di lavoro
•Uso di puntali con filtro
•Uso di controlli sia positivi che negativi per verifica dei reagenti ed
esecuzione corretta della procedura
• Controlli di estrazione e di amplificazione

La tecnica di PCR è stata la prima tecnica di amplificazione di DNA riportata in letteratura e quella più ampliamente sviluppata e applicata attualmente.
Dopo l’introduzione della PCR si sono sviluppati altri metodi alternativi o complementari
ANNI ‘90
METODI DI AMPLIFICAZIONE DEL “TARGET” METODI DI AMPLIFICAZIONE DEL SEGNALE

E’ una variante della tecnica di PCR end-point. Prevede due distinte e successive reazioni di amplificazione: 1) Nella prima amplificazione si utilizzano primers più esterni al frammento di DNA da amplificare 2) Nella seconda amplificazione si utilizzano primers che amplificano un frammento interno a quello amplificato nella I reazione.
PCR NESTED
…ALTRE TECNICHE SVILUPPATE SULLA SCIA DELLA PCR
Miglioramento della sensibilità. Se il prodotto di amplificazione fosse aspecifico la seconda PCR non andrebbe a buon fine. Questa tecnica, però, trova sempre minore applicazione in ragione degli alti rischi di contaminazione.
Amplificazione del target

Non è termoresistente ( è attiva 37-42°C). Recentemente sono state isolate e clonate delle RT mutanti che resistono a temperatura fino a 60°C per aumentare specificità.
Il prodotto della reazione è un prodotto ibrido RNA/DNA. Dopo allontanamento dell’RNA, il cDNA può essere utilizzato
come templato per l’amplificazione enzimatica (PCR)
Tecnica di amplificazione genica sviluppata per amplificare l’RNA target.
REVERSE TRANSCRIPTASE PCR ( RT-PCR)
L’RNA target viene inizialmente convertito in cDNA ad opera di un enzima, la Trascrittasi Inversa. E’ una DNA polimerasi RNA dipendente, inizialmente identificata nei retrovirus.
Amplificazione del target

MULTIPLEX PCR
Reazione di amplificazione in cui una o più coppie di “primers” specifici per differenti “target”, vengono introdotte nello stesso tubo di reazione, permettendo l’amplificazione di molteplici sequenze.
LIGASE CHAIN REACTION (LCR)
Tecnica di amplificazione genica simile alla PCR ma che utilizza due coppie di primers e due enzimi:DNA polimerasi, DNA ligasi. I primers sono complementari ai due filamenti della sequenza target. Dopo l’ibridazione dei primers, la ligasi unisce i due primers adiacenti. Tecnica che offre una elevata specificità.
Amplificazione del target

TMA (Transcription-Mediated Amplification) Utilizza due “primers” e due enzimi:
• T7RNA polimerasi che sintetizza RNA dal DNA • Trascrittasi inversa che converte l’RNA in cDNA e ha attività RNAasica Amplificazione isotermica a 41,5°C estremamente veloce in grado di produrre in 15-30 minuti una quantità di amplificato che, con la PCR convenzionale, non può essere ottenuta prima di 3-4 ore.
NASBA (Nucleic Acid Sequence-Based Amplification) Utilizza due “primers” e tre enzimi:
• T7RNA polimerasi che sintetizza RNA dal DNA • Trascrittasi inversa che converte l’RNA in cDNA e ha attività Rnasica • RNAasi H Amplificazione isotermica a 41,5°C
Il principale vantaggio di questi due sistemi è che tutte le reazioni avvengono in un’unica provetta. Solo alla fine del processo l’operatore ha modo di rivelare gli amplificati formatisi. La rivelazione finale in chemiluminescenza dei due metodi è un concetto innovativo, mentre i passaggi che portano alla formazione degli ampliconi di RNA appartengono alle metodiche molecolari classiche.
Minimizzata contaminazione da
carry over
Amplificazione del target
Tecniche che possono essere completamente automatizzate

RIVELAZIONE DEGLI ACIDI NUCLEICI
L’ultima fase di un saggio di PCR riguarda la rivelazione del prodotto di PCR
Le prime amplificazioni di PCR erano effettuate con il frammento di Klenow (enzima) di DNA polimerasi di E.coli e non avevano un alto livello di specificità. Per analizzare il DNA amplificato era necessaria una ibridazione con una sonda specifica marcata con radioisotopi (sonda calda)
Con l’utilizzo della Taq polimerasi (1988) la sensibilità della PCR ha raggiunto un livello elevato tanto da essere sufficiente una semplice elettroforesi su gel di agarosio per visualizzare il DNA amplificato.

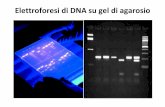
1) Gel di agarosio al 2% + EtBr 2) Elettroforesi: 100~150 volt. 3) Analizzare su un transilluminatore
UV la banda del prodotto di amplificazione
Le bande di DNA si visualizzano alla luce ultravioletta
Il prodotto dell’amplificazione viene introdotto in un pozzeto ricavato in un gel, attraverso il quale viene applicato un campo elettrico
L’elettroforesi su gel separa i frammenti di DNA in base alle loro dimensioni: i frammenti più piccoli migrano più velocemente
ELETTROFORESI SU GEL 1

IBRIDAZIONE PER LA RIVELAZIONE DEL MATERIALE AMPLIFICATO
Fine anni ‘80 vengono sviluppati metodi di ibridazione che utilizzano specifiche sonde o primers di amplificazione , marcati con enzimi (fosfatasi alcalina), molecole fluorescenti, chemiluminescenti (estere di acridinio) o con biotina.
Nel corso degli anni si sono messe a punto tecniche rapide, semplici e adeguate alla diagnostica di routine. Si sono resi disponibili sul mercato Kit che permettevano la rivelazione del DNA amplificato in breve tempo, con l’impiego di traccianti non radioattivi e con procedure molto semplici e automatizzabili.
2

Le sonde possono essere adese al pozzetto di una micropiastra o a biglie magnetiche e sono specifiche per il prodotto di
amplificazione.
CATTURA DEL PRODOTTO DI AMPLIFICAZIONE
Nella fase di PCR vengono utilizzati dei primers biotinilati. Il DNA biotinilato si lega alla sonda adesa al pozzetto e, dopo alcuni lavaggi per allontanare i prodotti non specifici, si aggiunge substrato. La reazione colorimetrica viene letta allo spettrofotometro.
RIVELAZIONE MEDIANTE TEST HPA (Hybridization Protection Assay)
Gli ampliconi di RNA, generati con il TMA, sono ibridati con sonde di DNA a singola elica marcate con estere di acridinio. Le sonde non ibridate sono idrolizzate dall’azione di un reagente di idrolisi, mentre l’estere di acridinio, dopo l’ibridazione, risulta protetto. I tubi di reazione vengono introdotti in un luminometro che, dopo aggiunta di un acido e una base, rileva il segnale chemiluminescente e lo converte in RLU (unità luminose relative).
3
4

Con lo sviluppo dei test commerciali, le procedure si sono semplificate, migliorando il livello di accuratezza e riproducibilità
Numerosi kit disponibili contengono tutti i reagenti per l’intera metodica:
Preparazione del campione
Mix per PCR
Rivelazione del prodotto amplificato
Nascono i primi sistemi completamente automatici per l’esecuzione del test PCR, accessibili a tutti i laboratori di
routine.
Fine anni ‘90

Si tratta di metodiche basate sull’utilizzo di: Primers biotinilati Sonde di ibridazione Rivelazione colorimetrica Lettura al fotometro (densità ottica)
Viene definito un valore di cut-off che rende semplice il risultato analogalmente ai comuni test immunoenzimatici.
Ogni fase del test presuppone, da parte della ditta produttrice, una approfondita messa a punto per garantire la massima efficienza e la qualità dei reagenti utilizzati.
Determinazione qualitativa

BRANCHED DNA
Si tratta del legame di una grande numero di sonde specificamente e direttamene alla sequenza di acido nucleico da identificare, creando una struttura a “branched DNA(bDNA), cioè DNA ramificato.
Amplificazione del segnale
Tecnica usata in alternativa alla PCR e alla RT-PCR per rilevare piccole quantità di sequenze specifiche di DNA o RNA
L’aggiunta di sonde marcate con fosfatasi alcalina e di un substrato chemiluminescente genera un’emissione di luce. La reazione è letta da un luminometro e il segnale è direttamente proporzionale alla quantità di acido nucleico.
Metodica di ibridazione di ultima generazione effettuata su uno
strumento che permette di automatizzare i passaggi operativi di ibridazione e
rivelazione
Determinazione quantitativa

Si evitano problemi di contaminazione, ma la sensibilità è inferiore alle metodiche di amplificazione del target

Questo metodo è basato sul legame tra biotina e streptavidina e consta di tre passaggi: 1) Il DNA amplificato marcato con
biotina viene ibridato con la sonda 2) l’ibrido biotinilato viene incubato
con la streptavidina legata all’enzima 3) il substrato viene messo a contatto
con il complesso DNA-biotina-streptavidina-enzima e sviluppa una reazione colorimetrica
Il metodo chiamato IBRIDAZIONE INVERSA (REVERSE DOT-BLOT) utilizza sonde legate a una membrana di nitrocellulosa che “catturano” le eventuali sequenze di DNA specifiche.
FINE ANNI ’90 il metodo è automatizzato

NECESSITA’ DELLA STANDARDIZZAZIONE
L’utilizzo di kit comuni migliora la qualità dei risultati rispetto all’impiego di
protocolli diversi (metodiche “in house”).
Indagini multicentriche periodiche con l’utilizzo di pannelli di campioni
“standard” sono necessarie per monitorare la performance del laboratorio
(riproducibilità e sensibilità)
Studi collaborativi a livello internazionale hanno sottolineato la necessità di standardizzazione della PCR per poterla considerare
completamente integrata nel laboratorio clinico.
Nonostante la semplicità delle metodologie e la facilità di lettura del risultato, l’attendibilità della PCR in ambito diagnostico è vincolata dal
suo inserimento nella routine diagnostica e dall’esecuzione di personale competente ed esperto.
Sviluppo dei kit commerciali
Standardizzazione dei protocolli
Apparecchiature automatizzate
Rapidità di esecuzione
Diagnostica clinica più precisa
EVOLUZIONE DELLA PCR

I marcatori fluorescenti fungono da indicatori diretti del numero di ampliconi sintetizzati durante l’amplificazione PCR.
Nasce la Real Time PCR
Prima pubblicazione sul metodo della Real-Time PCR da parte dei ricercatori della Cetus (Holland et. al. 1991).
La PCR è stata migliorata notevolmente con l’introduzione di coloranti fluorescenti che possono essere usati per marcare sonde o primer e permettere la rilevazione dei prodotti di PCR durante o
dopo l’amplificazione.

PCR “REAL TIME”
1. Permette di visualizzare l’amplificato man mano che si forma, sin dalle prime fasi di PCR
2. La concentrazione dell’amplicone è misurata in tempo reale
nel tubo di reazione mentre l’amplificazione è ancora in corso
3. Permette quindi una valutazione quantitativa del DNA presente(alta sensibilità analitica: fino a poche copie)
4. La PCR Real Time consente di abbandonare tutte le manipolazioni successive all’amplificazione, potenziali fonti di inquinamento del campione (carry-over)

PCR “REAL TIME” : principio
Il principio alla base della maggior parte delle Real-Time PCR è il trasferimento di energia fluorescente per risonanza FRET
(Fluorescence Resonance Energy Transfer)
CHE COS’È LA FRET ?
E’ il trasferimento di energia tra due molecole adiacenti: una funge da donatore e l’altra da accettore di energia. L’accettore può essere anch’esso una molecola fluorescente o una molecola non fluorescente in grado di spegnere il segnale del donatore ( in questo caso si chiama Quencer). Le molecole fluorescenti sono attaccate a sonde di DNA specifiche per la sequenza target L’efficienza del trasferimento di energia tra i due marcatori dipende dalla distanza che li separa e dal grado di sovrapposizione tra lo spettro di emissione del donatore e quello di assorbimento dell’accettore/quencer

Nella Real-Time PCRsi utilizza un solo sistema che funge sia da termociclatore, sia da rivelatore del segnale di amplificazione.
I risultati quantitativi sono disponibili immediatamente dopo l’amplificazione senza ulteriori amplificazioni o analisi. Non essendo necessarie altre fasi post PCR, la possibilità di contaminazioni incrociate da parte del prodotto di PCR è ridotta al minimo.
E’ la vera rivoluzione nel campo della diagnostica molecolare
COMPONENTI DELLA REAZIONE
DNA target DNA polimerasi Due primers dNTPs Sonda fluorescente/ Colorante fluorescente

La Real-Time PCR si può realizzare mediante l’impiego: COLORANTI FLUORESCENTI (es. SYBR Green) che si intercalano nella doppia elica del DNA
SYBR green
Il SYBR Green è un colorante fluorescente che si lega al solco minore del dsDNA. La fluorescenza emessa è letta a 530 nm. La rilevazione del prodotto di PCR utilizzando il SYBR Green è ottenuta misurando l’aumento della fluorescenza provocato dal legame del colorante al dsDNA.
Il SYBR Green non discrimina tra diverse specie di DNA a doppia catena presenti in una soluzione di reazione. Di conseguenza ogni artefatto del primer ed altri prodotti di reazione non specifici, contribuiranno alla misurazione dell’intensità del segnale fluorescente.
REAZIONE ASPECIFICA
Ciò produrrà una sovrastima della concentrazione della sequenza PCR bersaglio per cui è necessario ottimizzare la metodica utilizzando primers ben definiti.


La Real-Time PCR si può realizzare mediante l’impiego: SONDE AD IBRIDAZIONE specifiche per il frammento di interesse, marcate con molecole fluorescenti.
Esistono diversi tipi di sonde
• Sonde di idrolisi TaqMan • Sonde di ibridazione FRET • Sonde Molecular Beacons • Sonde Scorpions

SONDA TAQMAN
Presenta all’estremità 5’ un fluoroforo “Reporter” ed all’estremità 3’ una molecola “Quencer”
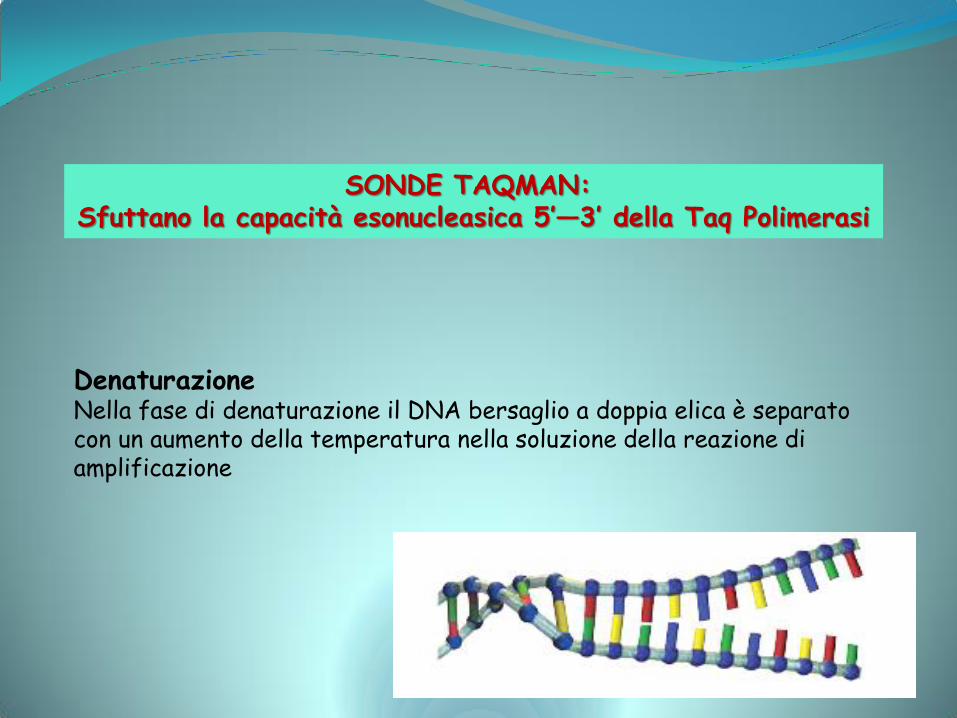
SONDE TAQMAN: Sfuttano la capacità esonucleasica 5’—3’ della Taq Polimerasi
Denaturazione Nella fase di denaturazione il DNA bersaglio a doppia elica è separato con un aumento della temperatura nella soluzione della reazione di amplificazione

Ibridazione Nel corso dell’ibridazione prima le sonde TaqMan e poi i primer ibridizzano in modo specifico con le sequenze complementari del filamento di DNA bersaglio. La sonda TaqMan porta due marcatori fluorescenti: un marcatore FAM (carbossifluoresceina) è il Reporter, l’altro è il Quencer. Quando la sonda è intatta FAM e Q sono molto vicini e il segnale del reporter è smorzato.
SONDE TAQMAN: Sfuttano la capacità esonucleasica 5’—3’ della Taq Polimerasi
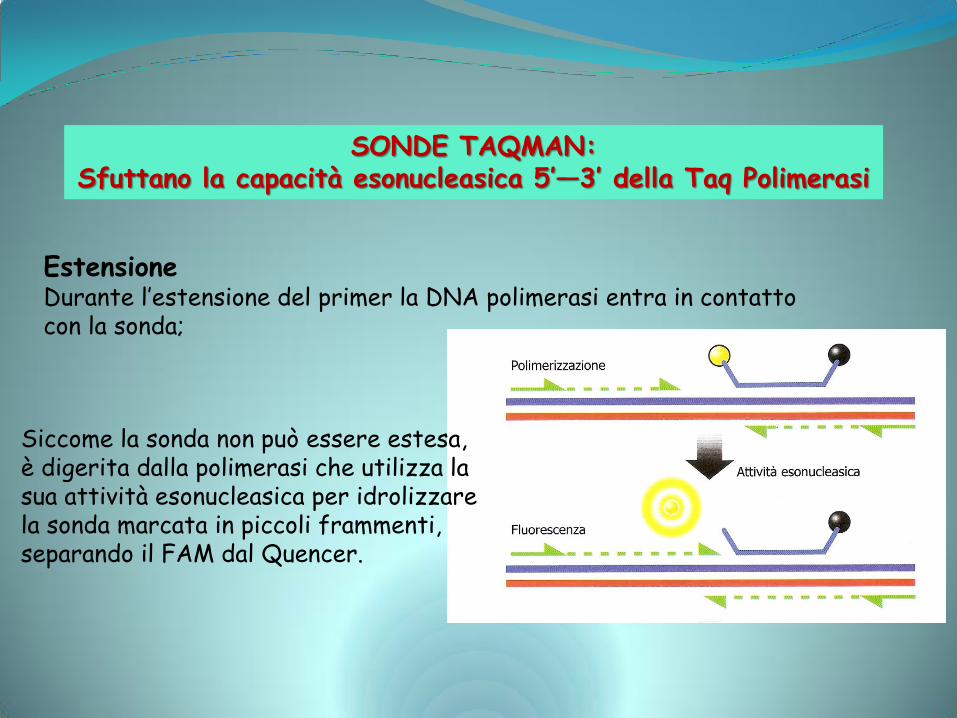
Estensione Durante l’estensione del primer la DNA polimerasi entra in contatto con la sonda;
Siccome la sonda non può essere estesa, è digerita dalla polimerasi che utilizza la sua attività esonucleasica per idrolizzare la sonda marcata in piccoli frammenti, separando il FAM dal Quencer.
SONDE TAQMAN: Sfuttano la capacità esonucleasica 5’—3’ della Taq Polimerasi

La separazione delle due molecole, interrompe il trasferimento di energia di risonanza fluorescente eliminando, quindi, lo “spegnimento” del FAM (Reporter) che è libero di emettere un segnale fluorescente
L’attività esonucleasica della Taq si ha solo se la sonda è attaccata al bersaglio. L’aumento di fluorescenza è direttamente proporzionale alla quantità di prodotto di PCR che si è formata.
SONDE TAQMAN: Sfuttano la capacità esonucleasica 5’—3’ della Taq Polimerasi

SONDE MOLECULAR BEACONS:
La vicinanza del Quencer al donatore impedisce a riposo l’emissione di fluorescenza. Durante la fase di annealing, la sonda si lega al DNA bersaglio, c’è un cambio di conformazione per cui il donatore è in grado di emettere fluorescenza. Nella fase di estensione del primer, le sonde si staccano dal DNA bersaglio e non interferiscono con la PCR
Le sonde Molecular Beacons sono formate da una singola catena di DNA ripiegata come una forcina e contengono un fluorocromo e un quencer non fluorescente.

PRIMER SCORPION:
I primer Scorpion sono primer PCR che sono legati all’estremità 5’ a una molecola simile ad una sonda Molecular Becons (stelo-e-ansa) contenente un Fluoroforo e un Quencher. La sonda è separata dal primer per mezzo di un “bloccante PCR”.
Nel corso della PCR, il primer Scorpion è esteso a partire dalla sua estremità3’. Durante la fase di ibridazione del ciclo PCR, la sequenza della sonda nella coda del primer Scorpion si ricurva per ibridare con la sequenza bersaglio del prodotto di PCR, separando il Fluoroforo dal Quencher e permettendo l’emissione di una fluorescenza rilevabile.
5’ 3’

Si hanno due sonde distinte, ciascuna marcata con un fluorocromo FRET: un donatore (Fluoresceina) e un accettore (LC Red)
SONDE FRET:
Quando le due sonde marcate sono libere nella soluzione, la distanza tra loro impedisce il trasferimento di energia tra i coloranti. Le due sonde di acido nucleico marcate sono disegnate per ibridare con determinate sequenze bersaglio in modo che i fluorofori finiscano molto vicini.
Durante la fase di ibridazione le due sonde si legano a regioni adiacenti del DNA bersaglio. Il donatore è eccitato da una fonte luminosa esterna e trasferisce la sua energia di attivazione all’accettore adiacente, che emette un segnale fluorescente.

La fluorescenza emessa in fase di amplificazione è registrata da software dedicati che visualizzano il segnale di fluorescenza come curve di amplificazione. Le curve sono riportate su monitor collegati al termociclatore. L’emissione di fluorescenza è direttamente proporzionale alla quantità di DNA presente. Per elevate concentrazioni di DNA le curve di amplificazione si registrano già dopo i primi cicli di PCR.
PCR “REAL TIME”

SISTEMI AUTOMATIZZATI

Sul monitor del computer è possibile la visualizzazione del profilo termico della reazione di PCR
All’interno dello strumento c’è una parte che funge da thermal cycler e una parte ottica che comprende sia i componenti per l’eccitazione dei fluoroforidonatori, sia un sistema di rivelazione dell’emissione di luce fluorescente.

Sul PC del sistema di ampl/rivel è possibile visualizzare in tempo reale l’andamento dell’amplificazione sull’intera piastra
Oppure c’è la possibilità di visualizzare ogni singolo campione della piastra

Il software legge le unità di fluorescenza in ciascuno dei pozzetti delle piastre e memorizza i dati. Poi utilizza i dati memorizzati per calcolare il ciclo soglia (Ct) per ogni calibratore, controllo e campione.
VISUALIZZAZIONE E VERIFICA DEI RISULTATI

Real-Time PCR : vantaggi
Tempi brevi per ottenere i risultati.
Riduzione del lavoro manuale con diminuzione costo del lavoro.
Uso di sistemi che prevengono il carry-over.
Alto rendimento.
Lunga durata dei probes marcati.

Real-Time PCR : svantaggi
Alti costi degli strumenti che utilizzano la real-time PCR.
Costo dei reagenti ( royalties).
Costo della sintesi dei probes

ESTRAZIONE ACIDI NUCLEICI
L’estrazione di acidi nucleici è il primo passo nelle applicazioni di biologia molecolare.
Questa fase del processo è molto importante, in quanto l'efficienza di amplificazione di un tratto di genoma è legata alla purezza del campione che viene utilizzato per l'amplificazione. Proteine e sostanze presenti nel campione interagiscono con l'enzima Taq polimerasi riducendo di molto l'efficienza dell'amplificazione stessa. Pertanto, una buona amplificazione è anche il risultato di una buona estrazione degli acidi nucleici che consente di avere un prodotto di partenza il più puro possibile.

SEPARAZIONE CON SOLVENTI ORGANICI (Metodo di estrazione classico)
Viene sfruttata la diversa solubilità delle molecole presenti in soluzione (acidi nucleici/contaminanti) fra due fasi tra loro non miscibili.
Una delle prime sostanze utilizzate è il fenolo, nel quale gli acidi nucleici non sono solubili. Altre sostanze utilizzate sono il cloroformio o l’etere che permettono di eliminare le tracce di fenolo eventualmente presenti nella fase acquosa.

3 fasi:
1) fase acquosa contenente DNA 2) interfaccia contenente proteine denaturate 3) fase organica contenente i lipidi
Estrazione DNA Trattamento 1:1 con fenolo-cloroformio alcool isoamilico pH 8.0
Lisi del campione SDS (Detergente) EDTA (Ag. chelante) Proteinasi K (Ag. Proteinasico)
DNA

Precipitazione con Etanolo 100% e lavaggio con Etanolo 70%
Evaporazione etanolo (potente inibitore enzimatico), per almeno 15 min a provetta aperta
Risospensione con H2 O distillata ultrapura
Conservazione : DNA 4°C/ -20°C/-80°C

3 fasi:
1) fase acquosa contenente RNA 2) interfaccia contenente proteine denaturate 3) fase organica contenente i lipidi e DNA
Estrazione RNA Trattamento con fenolo-cloroformio pH 5.6
Lisi del campione SDS (Detergente) Proteinasi K (Ag. Proteinasico) Guanidina tiocianato (Ag. Caotropico) β-mercaptoetanolo (Ag. riducente)
RNA

Precipitazione con Isopropanolo e lavaggio con Etanolo 75%
Evaporazione etanolo (potente inibitore enzimatico), per almeno 15 min a provetta aperta
Risospensione con H2 O distillata ultrapura
Conservazione : RNA -80°C

SEPARAZIONE PER BOLLITURA (Metodo di estrazione classico)
Il campione viene riscaldato a 100°C Lasciato raffreddare Centrifugato ad alta velocità
Gli acidi nucleici si trovano liberi nel sovranatante Nel precipitato è presente materiale insolubile
Resa decisamente bassa di estrazione

Negli anni ‘90 a protocolli manuali poco standardizzati cominciarono ad affiancarsi metodi semi-automatizzati, in cui
si riduceva di molto l’intervento da parte dell’operatore.
Era necessario, però,svolgere manualmente alcuni passaggi critici, come la lisi del campione.
Molti dei kit in commercio prevedono l'aggiunta al campione di un tampone di lisi già pronto che contiene GuSCN e detergenti necessari a rompere le cellule e stabilizzare gli acidi nucleici con la distruzione degli enzimi RNA-
asi e DNA-asi.

1 = Lysis Rack
2 = Filter Tube Rack
3 = Waste Container
4 = Elution Rack
5 = Cover Rack
6 = Grippers
Kit per estrazioni manuali

SEPARAZIONE PER AFFINITA’ Il metodo è basato sulla proprietà degli a. nucleici di adsorbirsi alla
silice, a differenza delle proteine e di altri composti organici
Lisi del campione SDS (Detergente) Proteinasi K (Ag. Proteinasico) Guanidina tiocianato (Ag. Caotropico)
56°C per 10 min
DNA/RNA

Adsorbimento DNA al gel di silice contenuto in una colonnina
in presenza di etanolo e sali caotropici il DNA si lega alla silice
Lavaggi con soluzioni contenenti alcool in grado di rimuovere le
proteine denaturate e gli altri componenti cellulari e lasciare il DNA legato alla silice
Eluizione DNA con H2O o soluzioni a bassa
concentrazione salina

Da pochi anni sono disponibili sistemi automatizzati che prevedono l’estrazione degli acidi nucleici con metodiche
standardizzate, procedure rapide e semplificate che riducono sensibilmente i problemi correlati alla manualità
dando così un elevato grado di affidabilità.
AUTOMAZIONE DELL’ESTRAZIONE DEGLI ACIDI NUCLEICI
Per una ulteriore garanzia del processo di estrazione è prevista l’aggiunta di un Controllo Interno, sottoposto agli stessi trattamenti del campione biologico, aumentando la
validità del metodo.

I sistemi automatizzati sfruttano il principio di affinità degli acidi nucleici per la silice.
La silice riveste la superficie di biglie magnetiche.
La lisi del campione, la deproteinizzazione, la separazione e il recupero dell’acido nucleico avviene all’interno della stessa postazione di lavoro