
CORSO INTEGRATO DI GENETICA - medgen.univr.itmedgen.univr.it/didattica/genetica10/lez09_10_espress...
18
1 CORSO INTEGRATO DI GENETICA a.a.2010-2011 Prof. Pier Franco Pignatti 13.10.2010 Lezioni N. 9 e 10 Espressione di caratteri monogenici Malattie legate alla X (Neri-Genuardi cap. 5 e 6) Codominanza, Modificatori, Oligogeni, Epigenetica, Inattivazione Connor e Ferguson-Smith, Principi di Genetica Medica, Esculapio 1986 DOMINANZA e RECESSIVITA’
Transcript of CORSO INTEGRATO DI GENETICA - medgen.univr.itmedgen.univr.it/didattica/genetica10/lez09_10_espress...

1
CORSO INTEGRATO DI GENETICA
a.a.2010-2011Prof. Pier Franco Pignatti
13.10.2010Lezioni N. 9 e 10
Espressione di caratteri monogeniciMalattie legate alla X
(Neri-Genuardi cap. 5 e 6)Codominanza, Modificatori, Oligogeni,
Epigenetica, Inattivazione
Connor e Ferguson-Smith, Principi di Genetica Medica, Esculapio 1986
DOMINANZA e RECESSIVITA’

2
ESPRESSIONE DI UN CARATTERE MONOGENICO
� Dominanza� Recessività1. Dominanza intermedia o incompleta
o semidominanza2. Codominanza3. Livello di analisi del fenotipo4. Geni modificatori del fenotipo5. Ereditarietà oligogenica6. Epigenetica
DOMINANZA INCOMPLETA in Ipercolesterolemia Familiare (FH)
HH
Hh
hh
Curtoni et al, Manuale di genetica, UTET 1991
1

3
Semidominanza in acondroplasia
Neri-Genuardi Gen Um Med II ed Elsevier-Masson 2010, Fig.5-10
La forma omozigote èpiù grave: gli omozigoti muoiono prima della nascita
CODOMINANANZA in gruppo sanguigno ABO
Bodmer e Cavalli, Genetica Evoluzione Uomo, 2°vol, Mondadori 1997
N-acetilgalattosammina
Galattosio
2
Gruppo AB

4
Connor e Ferguson-Smith, Principi di Genetica Medica, Esculapio 1986
Caratteri codominanti
e altri polimorfismi del DNA
Espressione dei caratteri monogenici
ABORivela entrambe le proprietà degli omozigoti
Codominante
FHIntermedio Effetto intermedio
FCNon affettoRecessiva
PKDAffettoDominante
Es.FENOTIPO di un ETEROZIGOTE
EREDITARIETA’

5
FALCEMIA
Read Donnai Genetica clinica, Zanichelli 2007
+ infarti cerebrali e sindrome toracica acuta
3
LIVELLO ANALISI
Clinico
Cellularebassa pO2
Molecolare
EREDITARIETA’
Recessiva
(DominanteIntermedia)
(Codominante)
FENOTIPO dell’ETEROZIGOTE (falcemia)
Stern, Principi di Genetica Umana, Zanichelli 1977

6
IL CONCETTO DI DOMINANZAE’ un concetto operativo e non riflette alcuna proprietà intrinseca del gene: si potrà dire che uno stesso gene controlla un carattere recessivo o dominante a seconda del livello di analisi del fenotipo.Perciò è corretto parlare di dominanza e di recessività riferiti ai caratteri e non ai geni.(NOTA: ciononostante è invalsa l’abitudine di definire dominanti e recessivi i geni)
LA DETERMINAZIONE DI UN FENOTIPO MENDELIANO
Malattia genetica
Gene/i causale/i
Gene/i modificatore/iAmbiente
Il gene modificatore modifica le caratteristiche cliniche della malattia nei diversi individui che hanno le stesse mutazioni del gene causale
4

7
Geni modificatori in falcemia
Meschia e Pankratz 2005
Polimorfismi in 12 geni predicono ictus in falcemici con una accuratezza del 98%
Geni modificatori in Fibrosi Cistica
• Studio di 5 geni candidati in 24 pazienti e 843 controlli (pazienti FC senza malattia epatica grave con ipertensione portale)
• L’allele Z (associato a rischio aumentato di cirrosi epatica) del gene SERPINA1 è un fattore di rischio per malattia epatica in FC. I pazienti che portano l’allele Z sono a rischio aumentato di sviluppare una grave epatopatia con ipertensione portale (OR=5)
Bartlett JR, 2009

8
EREDITARIETA’ DIGENICA
Sindrome di Bardet-Biedl (BBS): dismorfismi, obesità, polidattilia, distrofia retinica progressiva, deficit cognitivo patologia renale. 8 geni identificati (formazione e funzione cilia). Ereditarietà AR mendeliana o con mutazioni in due geni e tre alleli in 11/259 famiglie (4%)
Beales PL et al, AJHG 2003
5
EREDITA’ DIGENICA TRIALLELICA
Figure 3. Pedigree AR259 carrying three nonsenseBBS mutations. Individual AR259-05 carries twononsense BBS2 mutations but no BBS6 mutationsand is phenotypically normal.

9
EREDITA’ DIGENICA TRIALLELICASingole mutazioni in BBS1, BBS2 o BBS6 possono avere un effetto epistatico in pazienti Bardet-Biedlcon 2 mutazioni in un secondo locus BBS
03 e 04 sono omozigoti per la mutazione R275X del gene BBS2 (a sinistra). 04, più gravemente affetto, ha una mutazione aggiuntiva BBS1 (a destra)
EPIGENETICA
NC Whitelaw & E Whitelaw 2006
Variazioni ereditarie della espressione genica senza modificazione permanente del DNA. Modificazioni trasmissibili del DNA o della struttura della cromatina
6
(imprinting)
10
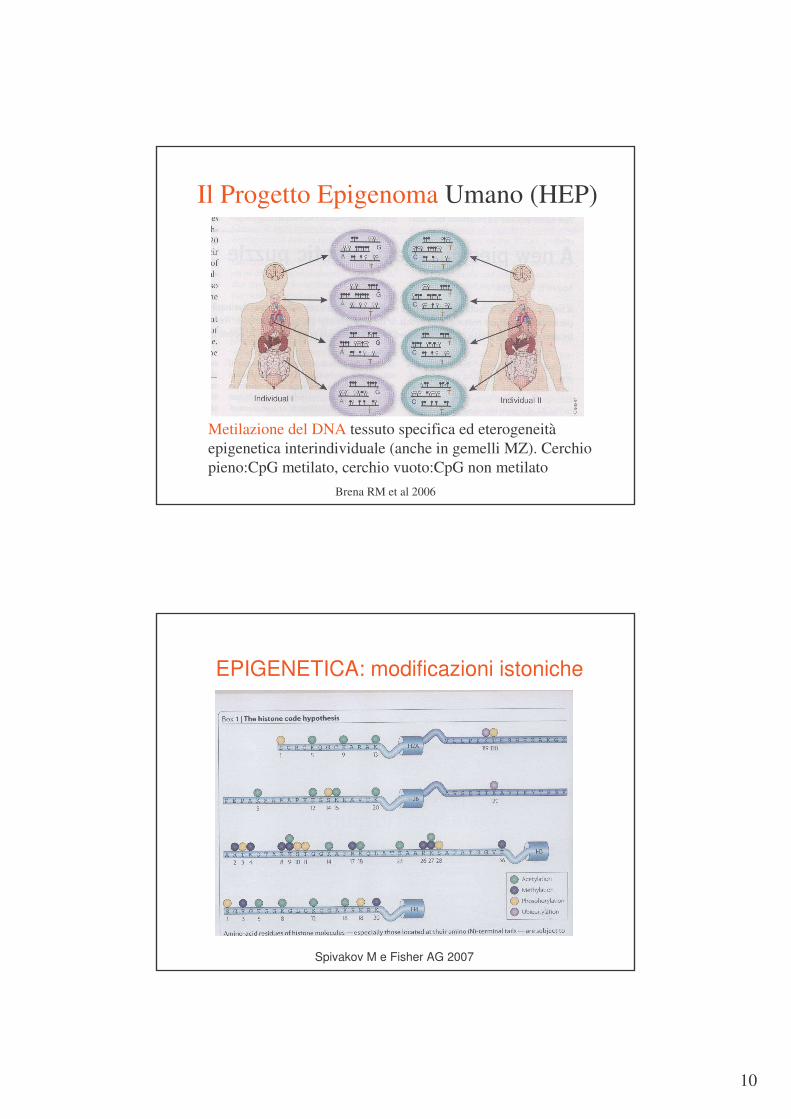
Il Progetto Epigenoma Umano (HEP)
Metilazione del DNA tessuto specifica ed eterogeneitàepigenetica interindividuale (anche in gemelli MZ). Cerchio pieno:CpG metilato, cerchio vuoto:CpG non metilato
Brena RM et al 2006
EPIGENETICA: modificazioni istoniche
Spivakov M e Fisher AG 2007

11
ECCEZIONI A INCROCI EQUIVALENTI
• Ereditarietà legata ai cromosomi sessuali• Ereditarietà mitocondriale (citoplasmatica)• Imprinting genetico (silenziamento genico
genitore specifico)• Disomia uniparentale (trasmissione di
entrambi i cromosomi di una coppia da un solo genitore)
Albero genealogico ereditarietà XR
Counseling aids for geneticists, Greenwood Genetic Center 1955

12
Segregazione di un carattere XR
Counseling aids for geneticists, Greenwood Genetic Center 1955
CRITERI PER EREDITARIETA’ XR
• L’incidenza del carattere è molto maggiore nei maschi che nelle femmine
• Tutte le figlie di un maschio affetto sono portatrici
• I figli maschi delle femmine portatrici hanno il 50% di probabilità di essere affetti
• Non c’è trasmissione del carattere da padre a figlio

13
Famiglia con femmina affetta da malattia XR
Thompson & Thompson Genetics in Medicine, Saunders 1991
Apparente trasmissione da maschio a maschio
Frequenza di alcune malattie XR
Connor e Ferguson-Smith Principi di Geentica Medica, Esculapio 1986

14
EMOFILIA
Read Donnai Genetica clinica, Zanichelli 2007, pg.82
Una famiglia con Emofilia
Luria Gold Singer, Una visione della vita, Zanichelli 1984
portatrice
affetto

15
DISTROFIA MUSCOLARE di DUCHENNE (DMD)
Read Donnai Genetica clinica, Zanichelli 2007, pg.4
N DMD
LA DISTROFINAancora il citoscheletrodelle cellule muscolari alla matrice extracellulare mediante il complesso distrofina-glicoproteine
Read Donnai Genetica clinica, Zanichelli 2007, pg.152

16
NEJM 2003; 349:969
Effetti di mutazioni nel gene della distrofina
Regola della cornice di lettura: in DMD la delezione produce spostamento modulo lettura, in BMD no. Ci sono eccezioni: uno studio indica in BMD 17/56 (30%) delezioni out of frame(Kesari A et al 2008)
DMDBMD
CARDIOMIOPATIA
Possibile terapia per DMD
Dietz H 2010

17
Inattivazione del cr. X
Mary Lyon 1961
Inattivazione: precoce (3-7 gg da fertilizzazione, 16-54 cellule), casuale, permanente, reversibile nei gameti, non comprende regione pseudoautosomica, mantenuta da espressione XIST e metilazione C.Conseguenze: compenso di dose (tranne alcuni geni), espressione variabile in femmine eterozigoti, mosaicismo.
Gelehrter Collins Ginsburg, Genetica Medica, Masson 1999, Fig 8.31
Mappa parziale del cromosoma X umano
DMD: distrofia muscolare DuchenneAR: recettore per gli androgeni HPRT: ipoxantina fosforibosil trasferasiFMR1: ritardo mentale legato all’X, tipo 1 G6PD: glucoso-6-fosfato deidrogenasiSTS: steroido solfatasiXIST: X inactivation specific transcriptXIC: centro di inattivazione dell’X
Inattivati Non inattivati

18
Immunocolorazione della distrofina in DMDFemmina portatrice Femmina non affettaMaschio affetto
Thompson & Thompson, Genetics in Medicine, Saunders 1991x240 x480 x480
INATTIVAZIONE SBILANCIATA DELL’ X
Dallapiccola e Novelli, Genetica Medica Essenziale, Il Minotauro 2006
:X pat inattivo


















