Tumori piccolo Intestino
61
SMALL INTESTINE TUMOURS
Transcript of Tumori piccolo Intestino

SMALL INTESTINE TUMOURS

Avvertenza per gli studenti del corso di Anatomia Patologica del corso Integrato Malattie dell’apparato Gastroenterico e
Infettive.
Questi appunti sono solo una traccia di ciò che ho svolto a lezione. Non possono e non vogliono sostituire la trattazione degli stessi argomenti sui libri di testo e non esimono dallo
studio degli altri argomenti del programma.Infine, devono essere utilizzati solo per uso personale del
singolo studente.
Buon studio
Achille PICH

Small intestine tumoursWHO Histological
classification
Epithelial tumours BenignMalignant
Non-epithelial tumours BenignMalignant
Malignant lymphomas
Secondary tumoursPolyps

Adenoma
WHO histological classification
Epithelial tumours
• Tubular• Villous• Tubulovillous
Intraepithelial neoplasia (dysplasia) associated with chronic inflammatory diseases - Low-grade glandular intraepithelial neoplasia - High-grade glandular intraepithelial neoplasia

Carcinoma• Adenocarcinoma• Mucinous adenocarcinoma• Signet-ring cell carcinoma• Small cell carcinoma• Squamous cell carcinoma• Adenosquamous carcinoma• Medullary carcinoma• Undifferentiated carcinoma
WHO histological classificationEpithelial tumours

Epithelial tumours
Carcinoid (well differentiated endocrine neoplasm)
• Gastrin cell tumour, functioning (gastrinoma) or non-functioning
• Somatostatin cell tumour
• EC-cell, serotonin-producing neoplasm
• L-cell, glucagon-like peptide and PP/PYY producing tumour
WHO histological classification

Epithelial tumours
Mixed carcinoid-adenocarcinoma
Gangliocytic paraganglioma
Others
WHO histological classification

• Lipoma• Leiomyoma• Gastrointestinal stromal tumour• Leiomyosarcoma• Angiosarcoma• Kaposi sarcoma• Others
Non-epithelial tumours
WHO histological classification

Malignant Lymphomas• Immunoproliferative small intestine disease (includes -heavy chain disease)• Western type B-cell lymphoma of MALT• Mantle cell lymphoma
• Burkitt lymphoma
• T-cell lymphoma• Others
- enteropathy associated- unspecific
• Diffuse large B-cell lymphoma
• Burkitt-like / atypical Burkitt-lymphoma
WHO histological classification

Secondary tumours
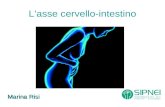
PolypsHyperplastic (metaplastic)
Peutz-JeghersJuvenile
WHO histological classification

Adenoma
Small intestine tumoursWHO Histological
classificationEpithelial tumoursBenign
TubularVillous
Tubulovillous

The frequency is minuscule.The duodenum and jejunum are involved more often than the ileum.
They can be single or multiple, pedunculated or sessile, and they have the microscopic appearance of an adenomatous polyp (tubular adenoma), villoglandular polyp (tubulovillous adenoma), or villous adenoma.
Malignant transformation can occur in them; as for their colorectal counterparts, the incidence of this complication is greater if the lesions are villous, large, or multiple.
Adenomas

Adenoma
Small intestine tumoursWHO Histological
classificationEpithelial tumoursBenign
TubularVillous
Tubulovillous

Adenoma
Small intestine tumoursWHO Histological
classificationEpithelial tumoursBenign
TubularVillous
Tubulovillous
Small intestine tumoursWHO Histological
classification
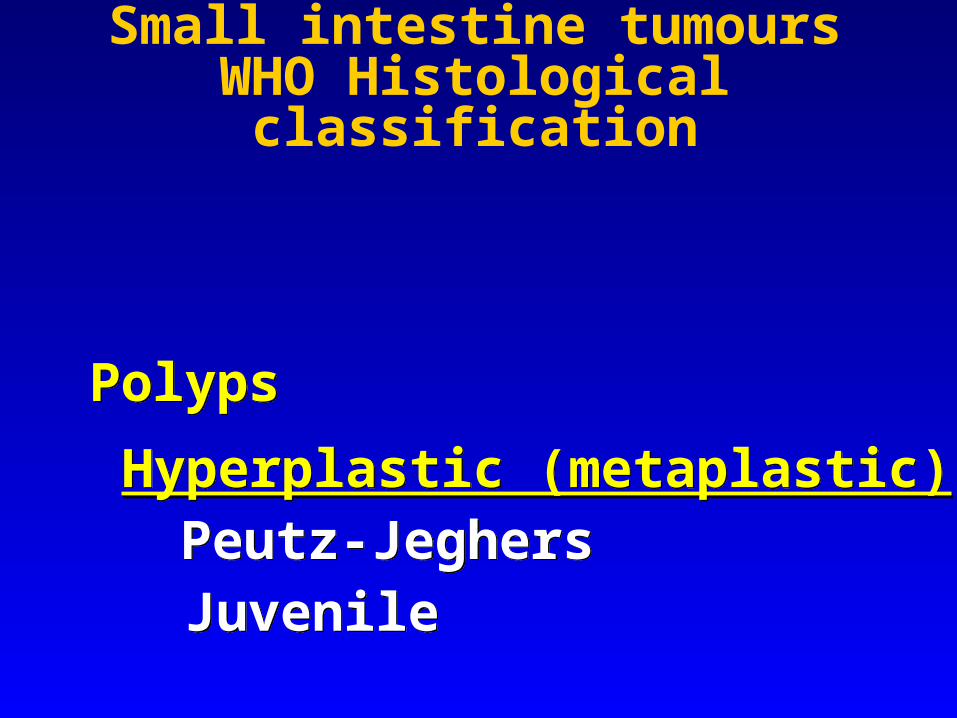
PolypsHyperplastic (metaplastic)
Peutz-JeghersJuvenile

Small intestine tumoursWHO Histological
classification
PolypsHyperplastic (metaplastic)
Peutz-JeghersJuvenile

Hamartomatous polyps of the jejunoileum are usually seen as a component of the Peutz-Jeghers syndrome, a familial disorder transmitted as a mendelian autosomal dominant trait with variable degrees of penetrance.
Similar polyps can also be present in the stomach, duodenum, and large bowel, and the patients have a typical pigmentation of the lips, oral mucosa, digits, palms, and soles.
Peutz-Jeghers

Several cases of gastrointestinal adenocarcinoma have been reported in patients with Peutz-Jeghers syndrome.
Patients with Peutz-Jeghers syndrome may also develop the distinctive ovarian neoplasm known as sexcord tumor with annular tubules so-called adenoma malignum of uterine cervix, ovarian mucinous tumors, breast carcinoma (often bilateral), and other types of malignancy.

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Malignant
Carcinoma– Adenocarcinoma– Mucinous adenocarcinoma– Signet-ring cell carcinoma– Small cell carcinoma– Squamous cell carcinoma– Adenosquamous carcinoma– Medullary carcinoma– Undifferentiated carcinoma

Adenocarcinoma of the small bowel is forty to sixty times less common than its counterpart in the large bowel. Most patients are elderly, without sex predilection. It is more common in the upper portions (40% to 50% of the cases occur in the duodenum, mostly in the region of the ampulla).
Adenocarcinomas of the small bowel can occur in the Lynch syndrome II, or as a complication of Peutz-Jeghers syndrome and Crohn's disease, in bowel duplication, at ileostomy sites, in surgically bypassed duodenum, and in association with Recklinghausen's disease.
ADENOCARCINOMA

Gross anatomy
Duodenal carcinoma tends to have a papillary configuration. Adenocarcinomas located more distally usually have a napkin-ring appearance and produce partial intestinal obstruction with marked dilatation of the proximal bowel; however, about 20% have a predominantly polypoid or fungating appearance, perhaps caused in some instances by their origins in pre-existing adenomatous polyps or villous adenomas. Occasionally these carcinomas present as multiple tumors or in association with primary malignant neoplasms in other sites.

Microscopy
These tumors are usually moderately or well-differentiated adenocarcinomas. Mucin production and CEA reactivity are the rule; there may also be immunoreactivity for lysozyme, suggesting focal differentiation toward Paneth cells. In addition, it is common to find scattered endocrine cells, particularly in ileal tumors. These cells are positive for chromogranin and serotonin and may show immunoreactivity for somatostatin, YY peptide, neurotensin, glucagon, and glycentin. Ultrastructurally, there is a prominent development of microvilli.

Prognosis
At the time of diagnosis, the majority of the tumors have extended deeply into the wall and may have already metastasized to regional lymph nodes. The prognosis mainly depends on the presence or absence of regional lymph node involvement. In one series, 88% of the patients with positive nodes died of tumors, contrasted with 45% of those with negative nodes.

TNM Clinical Classification
TX Primary tumour cannot be assessedT0
Lamina propria, muscularis mucosae or submucosaT1a Lamina propria or muscularis mucosaeT1b Submucosa
T2 Muscularis propria
T3 Tumour invades into subserosa or into non-peritonealized perimuscular tissue for 2 cm or less
T4Tumour perforates visceral peritoneum or directly invades other organs or structures (loops of small intestine, mesentery, retroperitoneum for more than 2 cm) or pancreas
T - Primary Tumour
No evidence of primary tumour
T1Tis Carcinoma in situ

TNM Clinical Classification
N0 No regional lymph node metastasisN1 Metastasis in 1-3 regional lymph nodes
M0 No distant metastasisM1 Distant metastasis
N - Regional Lymph Nodes
NX Regional lymph nodes cannot be assessed
M - Distant MetastasisMX Distant metastasis cannot be assessed
N2 Metastasis in 4 or more regional lymph nodes
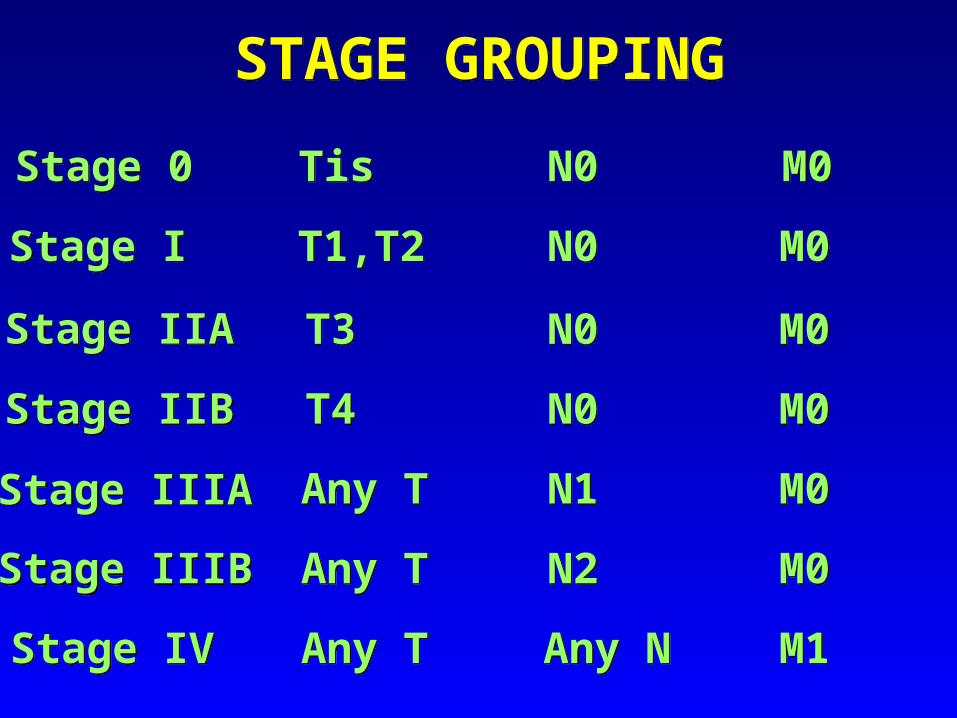
STAGE GROUPING
Stage I
Stage IIB
Stage IIIB
Stage IV
T1,T2
T3
T4
Any T
Any T
Any T
N0
N0
N0
N1
N2
Any N
M0
M0
M0
M0
M0
M1
Stage 0 Tis N0 M0
Stage IIA
Stage IIIA

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Malignant
Carcinoma– Adenocarcinoma– Mucinous adenocarcinoma– Signet-ring cell carcinoma– Small cell carcinoma– Squamous cell carcinoma– Adenosquamous carcinoma– Medullary carcinoma– Undifferentiated carcinoma

Small cell carcinoma is a rare type of small bowel malignancy composed of small, round, or oval cells of scanty cytoplasm and hyperchromatic nucleus.
The appearance is very similar to that of pulmonary small cell carcinoma. As in the latter, dense core granules of neurosecretory type can be identified ultrastructurally and neuroendocrine markers immunohistochemically. These tumors can present in a pure form or mixed with ordinary adenocarcinoma.
They are deeply invasive, highly prone to metastasize, and associated with a very poor prognosis.
SMALL CELL CARCINOMA

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Malignant
Carcinoma– Adenocarcinoma– Mucinous adenocarcinoma– Signet-ring cell carcinoma– Small cell carcinoma– Squamous cell carcinoma– Adenosquamous carcinoma– Medullary carcinoma– Undifferentiated carcinoma

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Malignant
Carcinoma– Adenocarcinoma– Mucinous adenocarcinoma– Signet-ring cell carcinoma– Small cell carcinoma– Squamous cell carcinoma– Adenosquamous carcinoma– Medullary carcinoma– Undifferentiated carcinoma

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Malignant
Carcinoma– Adenocarcinoma– Mucinous adenocarcinoma– Signet-ring cell carcinoma– Small cell carcinoma– Squamous cell carcinoma– Adenosquamous carcinoma– Medullary carcinoma– Undifferentiated carcinoma

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Carcinoid (well differentiated endocrine neoplasm)
– Gastrin cell tumour, functioning (gastrinoma) or non-functioning
– Somatostatin cell tumour
– EC-cell, serotonin-producing neoplasm
– L-cell, glucagon-like peptide and PP/PYY producing tumour

CARCINOID Carcinoid tumor is the generic term applied to low-grade malignant neoplasms originating from the diffuse endocrine system outside of the pancreas and the thyroid C-cell.
They represent a group of neoplasms rather than a single pathologic entity, as the digestive tract in general and the small bowel in particular contain a large number of endocrine types, any of which can be represented in these neoplasms, singly or in combination.
Carcinoid tumor comprises about one third of all neoplasms of the small bowel.

The majority occur in adults. Most are located in the ileum (including Meckel's diverticulum), followed in frequency by the jejunum and distal duodenum. They are multiple in 15% to 35% of the cases and are sometimes associated with malignant gastrointestinal tumors of other microscopic types or with tumors of endocrine differentiation in other locations.
Grossly, they appear as nodules protruding through the ileal mucosa.

Microscopically, the pattern is that of solid masses of monotonous cells with small, round nuclei; a moderate amount of finely granular cytoplasm; and fine nucleoli. The mucosa is often intact over the tumor. Infiltration of the submucosa is the rule, and extension into the muscularis externa is common.

Microscopic types Based on their patterns of growth, carcinoid tumors may be subdivided into several types:
A = insular (the most common form)
B = trabecular
C = glandular
D = undifferentiated
E = mixed
The various types correlate with behavioral differences. Carcinoid tumors with a predominantly glandular pattern are referred to as adenocarcinoids. On occasion, features of endocrine and exocrine secretion can be seen in the same cell (so-called amphicrine cell).

At the histochemical level, the tumor is argentaffin (and therefore also argyrophilic). Ultrastructurally, there are numerous dense-core secretory granules. Immunohistochemically, there is both epithelial and neuroendocrine differentiation. The first is manifested by keratin and CEA; the second by a series of "pan-endocrine" markers, such as neuron-specific enolase, chromogranin, synaptophysin, Leu7, PGP 9.5, and serotonin. A variety of peptide hormones has also been demonstrated in these tumors: substance P (the most common), gastrin, somatostatin, glucagon, glycentin, pancreatic polypeptide, bombesin, gastrin-releasing peptide (GRP), and growth hormonereleasing factor (GRF). Aneuploidy is present in over half of the ileal carcinoid tumors.

SPREAD AND METASTASIS Classic carcinoids are low-grade malignant neoplasms, with a slow growth rate, but also are highly invasive and can metastatize. Metastases occur most commonly in the regional lymph nodes and liver (usually multiple), but also in bone, skin, and thyroid.
TREATMENT AND PROGNOSIS The primary treatment of carcinoid is wide surgical removal of the primary tumor and its lymphatic drainage, even in the presence of extensive liver metastasis. Solitary liver metastases are also amenable to surgical excision. The 5-year OS is between 50% and 65%, with a marked difference between the tumors confined to the wall (85% OS) and those invading the serosa or beyond (5% OS). Large tumor size and CEA expression correlate with a more aggressive course.

ASPETTI CLINICI DELLA SINDROME DA CARCINOIDE
1. Disturbi vasomotori Flushes cutanei e cianosi evidente (nella maggior parte dei pazienti)
2. Ipermotilità intestinale Diarrea, crampi, nausea, vomito (nella maggior parte dei pazienti)
3. Attacchi asmatici broncocostrittivi Tosse, sibili, dispnea (in un terzo dei pazienti)
4. Epatomegalia nodulare, correlata a metastasi epatiche (in alcuni pazienti)

5. Fibrosi sistemica
Interessamento cardiaco Ispessimento e stenosi valvolare tricuspidale e polmonare
Fibrosi endocardiaca, principalmente nel ventricolo destro (nei carcinoidi bronchiali è interessato quello sinistro)
Fibrosi retroperitoneale e pelvica
Placche collagene della pleura e dell’intimadell’aorta

DUODENAL ENDOCRINE TUMOURS Endoscopically, duodenal endocrine tumors appear as smooth, round elevations, usually measuring between 5 and 20 mm in diameter. Microscopically, they rarely have the features of classic carcinoids. Many are composed of either G- or D-cells, as demonstrated by their immunoreactivity for gastrin and somatostatin, respectively. The G-cell tumors (gastrinomas) may be associated with the Zollinger-Ellison syndrome and MEN type I, whereas D-cell tumors (somatostatinomas) are hormonally silent at the clinical level. Exceptionally, duodenal endocrine tumors are of pancreatic B-cell type. Metastases were present in 21% of the duodenal endocrine tumors.

Small intestine tumoursWHO Histological
classificationEpithelial tumours
Mixed carcinoid-adenocarcinoma
Gangliocytic paraganglioma
Others

GANGLIOCYTIC PARAGANGLIOMA
Gangliocytic paraganglioma (nonchromaffin paraganglioma; paraganglioneuroma) occurs almost exclusively in the second portion of the duodenum, especially in the proximity of the ampulla of Vater.
The peculiar location of this tumor, its highly organoid arrangement, and the striking predominance of PP cells suggest that this lesion may represent a hamartomatous process derived from the ventral primordium of the pancreas.
Nearly all cases published so far have followed a benign clinical course, but isolated instances associated with lymph node metastases are on record.

Grossly, most lesions are small, pedunculated, and submucosal, with frequent ulceration of the overlying mucosa and bleeding.
Microscopically, three cell components are present: (1) endocrine cells with a carcinoid-like appearance, containing dense-core granules ultrastructurally and exhibiting immunoreactivity for a variety of markers, particularly pancreatic polypeptide (PP); (2) isolated ganglion cells, immunoreactive for neuron-specific enolase (NSE) and other neural markers; and (3) spindle-shaped Schwann cells and/or sustentacular cells, immunoreactive for S-100 protein.

– Lipoma
Non-epithelial tumours
Small intestine tumoursWHO Histological classification
– Leiomyoma– Gastrointestinal stromal tumour– Leiomyosarcoma– Angiosarcoma– Kaposi sarcoma– Others

– Lipoma
– Gastrointestinal stromal tumour– Leiomyosarcoma– Angiosarcoma– Kaposi sarcoma– Others
Non-epithelial tumours
Small intestine tumoursWHO Histological classification
– Leiomyoma

– Lipoma– Leiomyoma– Gastrointestinal stromal tumour – Angiosarcoma– Kaposi sarcoma– Others
Non-epithelial tumours
Small intestine tumoursWHO Histological classification
– Leiomyosarcoma

Small intestine tumoursWHO Histological classification
Malignant Lymphomas– Immunoproliferative small intestine disease (includes -heavy chain disease)
– Western type B-cell lymphoma of MALT– Mantle cell lymphoma
– Burkitt lymphoma
– T-cell lymphoma– Others
• enteropathy associated• unspecific
– Diffuse large B-cell lymphoma
– Burkitt-like / atypical Burkitt-lymphoma

Immunoproliferative small intestine disease (includes -heavy chain disease)
Immunoproliferative small intestinal disease (IPSID), Mediterranean lymphoma, and Middle Eastern lymphoma is a B-cell lymphoma relatively common among non-European Jews, Arabs in the Middle East, and the black population of South Africa.
Patients often have a short history of diarrhea and malabsorption, but the mucosa is not completely flat in most cases.
The involvement centers in the distal portions of the duodenum and the upper jejunum.

Microscopy
The low-grade form (by far the most common) shows a heavy mature or only slightly immature lymphoplasmacytic infiltration. This is associated with the presence of monoclonal alpha heavy chains of immuno-globulins in their cytoplasm, as well as in the serum and urine, hence the alternate designation ”alpha chain disease.” It has been speculated that this lymphoplasmacytic infiltration is originally reactive in nature and represents a response to a continuous antigenic stimulus of possible infectious nature (some cases respond to tetracycline).
The high-grade form of the disease, which is usually preceded and accompanied by the low-grade form, shows microscopic appearance of highly pleomorphic large cell lymphoma.

The MALT concept Mucosa-associated lymphoid tissue (MALT) is the term proposed by Isaacson et al. for the component of the immune system that has evolved to protect the freely permeable surface of the gastrointestinal tract and other mucosal membranes directly exposed to the external environment.
This comprises lymphoid nodules (which in the ileum form the Peyer's patches), lymphocytes and plasma cells in the lamina propria, and intraepithelial lymphocytes.
The lymphomas arising from it ("MALTomas") have propensity to involve other mucosal sites, which has been explained by the normal homing pattern of MALT lymphocytes.

Western type B-cell lymphoma of MALT
Represent most of B-cell lymphomas of the small bowel. The low-grade form is not as common as it is in the stomach. Its gross, microscopic, and immunohisto-chemical features are similar to those of its gastric counterpart. The key features are the predominance of small lymphoid cells (either centrocyte-like or monocytoid B cells), the formation of lymphoepithelial lesions, and the presence of reactive follicles. The high-grade form of this tumor may be seen in association with the low-grade form or in its absence. This is a large cell lymphoma of large cell type, often associated with plasmacytoid forms.

Mantle cell lymphoma MCL is a B-cell lymphoma that tipically involves both spleen and intestine and may present as an isolated mass or multiple polyps (multiple lymphomatous polyposis).
The polyps range in size from 0.5 to 2 cm. The architecture is most frequently diffuse, but a nodular and a less common true mantle-zone pattern are also seen. The lymphoma cells are small to medium sized. They are mature B-cells and express both CD19 and CD20, with the characteristic coexpression of CD5 and CD43. Bcl-1 (cyclin D1) is found in virtually all cases within the nuclei.
MCL is an aggressive lymphoma, which typically presents in advanced stage.
Burkitt lymphomaBurkitt lymphoma occurs in two major forms.
1) Endemic Burkitt is found primarily in Africa and typically presents in the jaw, orbit or paraspinal region, and is strongly associated with Epstein-Barr virus (EBV).In other endemic regions however, it is relatively common for Burkitt lymphoma to present in the small intestine, usuallyinvolving the ileum, with preferential localization to the ileo-caecal region. In parts of the Middle East, primary gastro-intestinal Burkitt lymphoma is a common disease of children.
2) Sporadic or non-endemic Burkitt lymphoma is a rare disease, not associated with EBV infection, that frequently presents as primary intestinal lymphoma. Burkitt lymphomais also seen in the setting of HIV infection when it often involves the gastrointestinal tract.

The histology in all cases is identical and is characterized by a diffuse infiltrate of medium-sized cells with round to oval nuclear outlines, 2-5 small but distinct nucleoli and a small amount of intensely basophilic cytoplasm. Numerous mitotic figures and apoptotic cells are present.
The prominent starry-sky appearance is caused by benign phagocytic histiocytes engulfing the nuclear debris resultingfrom apoptosis.
It is a mature B-cell lymphoma and the neoplastic cells express pan-B-cell antigens CD19, CD20, CD22, and CD79α. In approximately 60-80% of cases, the neoplasticcells co-express CD10, but fail to express CD5 or CD23. Surface immunoglobulin expression is moderately intense and is nearly always IgM with either kappa or lambda light chain restriction.
The growth fraction, as assessed by Ki-67 or the paraffin equivalent MIB-1, is typically in excess of 90% of tumour cells. Burkitt lymphoma cells uniformly fail to express bcl-2.
Burkitt-like lymphomas with ‘dual translocation’ of both bcl-2 and c-myc oncogenes have a markedly shortened overall survival.

Prognostic factors
The main determinants of clinical outcome in small intestinal lymphomas are histological grade, stage, and resectability.
Advanced age at diagnosis, an acute presentation with perforation, and the presence of multifocal tumours havean adverse impact on survival.
The behaviour of diffuse large B-cell lymphoma is not affected by the presence of a low-grade MALT component.
The expression of bcl-2 protein and the presence of TP53 mutations may adversely affect outcome in this group, but a systematic study of small intestinal lymphomas is lacking.

T-cell malignant lymphomas are rare (5% of all GI lymphomas); they represent, in most instances, a compli-cation of long-standing celiac sprue or related malabsorption syndrome. These are called enteropathy-associated T-cell lymphomas. T-cell lymphomas not associated with malabsorption also occur.
The affected bowel is often dilated and oedematous, and shows multiple circumferential ulcers.
The histological appearences are variable, the most frequent type is composed of highly pleomorphic, medium to large cells, followed by a type that shows a morphology most consistent with anaplastic large cell lymphoma.
T- cell lymphoma

Prognosis and predictive factors
The clinical course is very unfavorable due to complications from peritonitis and malnutrition and later from progressive disease typically characterized by intestinal recurrences.
The malabsorption due to underlying coeliac disease is detrimental to these patients, particularly when recovering from surgery or receiving multiagent chemotherapy. Consequently, only one half of the patients is amenable to chemotherapy and only a proportion of these is able tofinish the complete course.
The overall median survival is only 3 months, and 5-year survival ranges from 8-25%. The small group of long-term survivors usually received chemotherapy and none had a previous diagnosis of coeliac disease.

Small intestine tumoursWHO Histological
classification
Secondary tumours

Metastatic tumors can involve the small bowel, often in the form of multiple polypoid tumors. The lesions may result in obstruction or perforation, necessitating palliative resection.
The most common types of primary tumor are malignant melanoma, carcinoma from lung and breast, and choriocarcinoma