
Frontiere · La presenza dello pseudogene complica l’analisi molecolare, ... sposta a stress...
10
Transcript of Frontiere · La presenza dello pseudogene complica l’analisi molecolare, ... sposta a stress...


77
Gennaio-Marzo 2016 • Vol. 46 • N. 181 • Pp. 77-85 Prospettive in Pediatria
Frontiere
Sindrome adrenogenitale: più comune di quanto si pensi
Paola Carrera1, 2 Chiara Di Resta3
Maurizio Ferrari1-3
1 Laboratorio Biologia Molecolare Clinica, IRCCS Ospedale San Raffaele, Milano; 2 Unità di
Genomica per la Diagnosi delle Patologie Umane, IRCCS
Ospedale San Raffaele, Milano; 3 Università Vita-Salute San
Raffaele, Milano
La sindrome adrenogenitale (SAG) comprende un gruppo di patologie ereditarie che cau-sano una serie di deficit enzimatici della steroidogenesi del surrene. Tra questi, il deficit di 21-idrossilasi è il più frequente (circa 90% dei casi). Le caratteristiche più distintive della patologia sono: i) la virilizzazione dei genitali esterni nelle femmine con la forma severa classica; ii) l’incapacità di mantenere l’equilibrio salino a causa dell’inadeguata sintesi di aldosterone nei pazienti con la forma classica con perdita di sali; condizione potenzial-mente fatale in assenza di terapia. La malattia è ereditata come carattere recessivo; è causata da varianti del gene CYP21A2 che codifica per l’enzima 21-idrossilasi. Le varianti patogenetiche più frequentemente osservate sono la conseguenza di eventi di crossing over ineguale durante la meiosi o di eventi di conversione genica tra il gene CYP21A2 e lo pseudogene CYP21A1P, duplicato in tandem e altamente omologo al gene funzionale. La presenza dello pseudogene complica l’analisi molecolare, richiedendo speciali accor-gimenti per la selezione del gene. Grazie a numerosi studi, si è visto che per le varianti patogenetiche, il grado di inattivazio-ne enzimatica correla con la severità della malattia.Durante gli ultimi venti anni molti progressi sono stati fatti nella conoscenza e nella cura del deficit di 21-idrossilasi, nella possibilità di contenere la virilizzazione nelle femmine affette e nell’identificazione precoce dei neonati affetti. La gestione di questa malattia rappresenta, a nostro parere, uno degli esempi più evoluti di interazione multidisciplinare in medicina.
Riassunto
SummaryCongenital adrenal hyperplasia (CAH) refers to a group of inherited disorders causing enzyme deficiencies that impair steroid synthesis in the adrenal cortex. More than 90% of cases of CAH are caused by 21-hydroxylase deficiency. Hallmarks of the disease are: i) virilisation of external genitalia in females in the severe, classic form; ii) inability to main-tain sodium balance because of insufficient aldosterone synthesis in patients with the severe, salt wasting form, which is a potentially fatal condition if not treated. The disease is recessively inherited and is caused by variants in the CYP21A2 gene encoding the 21-hydroxylase enzyme. The most frequently observed pathogenic variants are due to unequal crossing over during meiosis or to gene conversions between CYP21A2 and the highly homologous in tandem duplicated pseudogene CYP21A1P. The presence of the CYP21A1P pseudogene complicates molecular analysis, requiring special attention in gene selection. Based on several studies, the degree to which pathogenic variants compromise enzymatic activity correlates to clinical severity of the disease. Over the last two decades, many improvements have been made in knowledge and treatment of 21-hy-droxylase deficiency, in minimising virilisation in affected females and in early identifica-tion of affected newborns. In our opinion, management of the disease is one of the most evolved examples of multidisciplinary interaction in medicine.

78
P. Carrera et al.
La sindrome adrenogenitaleLa sindrome adrenogenitale (SAG) è una malattia a trasmissione autosomica recessiva, dovuta all’alte-razione della sintesi di alcuni enzimi coinvolti nella steroidogenesi, nella corteccia del surrene. Sono noti alcuni deficit enzimatici a carico di questa via meta-bolica, tutti a carattere autosomico recessivo. La for-ma più frequente è il deficit di 21-idrossilasi (OMIM-201910), che causa oltre il 90% dei casi di SAG. In questa rassegna ci concentreremo quindi sulla SAG causata dal deficit di 21-idrossilasi. Il deficit di 21-idrossilasi si presenta in varie forme cliniche. Il fenotipo clinico e biochimico dipende dal grado di compromissione dell’attività enzimatica, a sua volta correlato alla combinazione genotipica del-le varianti geniche, dall’instaurarsi di meccanismi di compenso extra-surrenalici non ancora chiaramente definiti e da differenze individuali in risposta alle alte-razioni metaboliche. I diversi quadri si riassumono in due forme cliniche: la forma classica e la forma non-classica.La forma classica, è la più severa. Studi epidemiolo-gici hanno indicato che l’incidenza nel mondo della forma classica del deficit di 21-idrossilasi è variabile in diverse popolazioni, come illustrato nella tabella I (White e Speiser, 2000; Balsamo et al., 1996). I pa-zienti affetti mostrano sintomi severi di iper-androge-nismo. In epoca pre-natale e durante le fasi del diffe-renziamento sessuale, l’esposizione a potenti andro-geni (testosterone, alpha4-androstenedione), provoca la virilizzazione dei genitali esterni nelle femmine, con casi di ambiguità genitale alla nascita. La forma classica è suddivisa nelle forme virilizzante sempli-ce (simple-virilizing – SV) e con perdita di sali (salt-wasting – SW). Nella forma con perdita di sali (SW), oltre al deficit di cortisolo e all’aumento di androgeni, si osserva un’inadeguata produzione di aldosterone (Fig. 1), che causa insufficiente riassorbimento di sodio a livello
del tubulo renale distale. Nei casi con virilizzazione semplice (SV) invece, il livello dell’aldosterone è ade-guato. Nei neonati con la forma SW si osserva perdita di peso, ritardo della crescita, vomito, disidratazione, pressione bassa, bassi livelli di sodio e acidosi meta-bolica dovuta ad alti livelli di potassio, situazione che può portare a crisi surrenalica acuta tra la prima-se-conda e la quarta settimana di vita. Queste crisi surre-naliche possono essere fatali se non trattate adegua-tamente (White e Speiser, 2000). I pazienti con SW rappresentano circa il 75% dei casi con forma classica, mentre il rimanente 25% dei pa-zienti presenta la forma SV (New, 1998). La forma non-classica, (non-classic – NC), è una tra le più frequenti patologie a trasmissione autosomica recessiva. L’incidenza della forma non-classica è di circa 1:1000, con una frequenza di 1:27 tra gli ebrei Ashkenazi (Speiser et al., 1985). Gli individui con la forma NC del deficit di 21-idrossilasi hanno solo un deficit enzimatico parziale. La forma non-classica si può presentare a qualsiasi età, con una vasta rosa di sintomi dell’iperandrogenismo, tranne l’ambiguità genitale. Perciò le femmine con la forma NC non pre-sentano virilizzazione alla nascita. Nei maschi, poi-ché la distinzione clinica tra forma SV e forma NC non è sempre facile, sono utili alla diagnosi l’analisi genetica e il test dopo stimolazione con ACTH (ormo-ne adrenocorticotropo) (New, 1998; White e Speiser, 2000). Una parte dei soggetti con la forma NC è completa-mente asintomatica (criptica).In Tabella II riportiamo le caratteristiche cliniche di-stintive per le forme classica e non-classica.La relativa alta frequenza dello stato di portatore in una malattia, che se non trattata può avere esiti an-che molto gravi, ha portato alcuni studiosi a propor-re l’ipotesi del vantaggio dell’eterozigote, secondo la quale l’elevata e veloce risposta al cortisolo, osser-vata nei portatori, potrebbe garantire una rapida ri-
Tabella I. Frequenza del deficit di 21-idrossilasi, forma classica in alcune popolazioni (dati ottenuti da screening neo-natali).
Regione Incidenza Riferimenti bibliografici
Alaska, Esquimesi 1:280 Pang, 1982
Isola La Reunion 1:2100 Pang, 1988
Svezia 1:9800 Thil’en, 1998
Stati Uniti (Wisconsin) 1:11000 Allen, 1997
Francia 1:13000 Cartigny, 1999
Giappone 1:18000 Tajima, 1997
Stati Uniti (Texas) 1:16000 Therrel, 1998
Scozia 1:17000 Pang, 1988
Italia 1:18000 Balsamo A, 1996
Nuova Zelanda 1:23000 Cutfield, 1995
79
Sindrome adrenogenitale: più comune di quanto si pensi
sposta a stress infiammatori, infettivi o ambientali in genere, offrendo un vantaggio selettivo nel prevenire risposte immunitarie eccessive o inappropriate, come avviene nelle malattie autoimmunitarie.
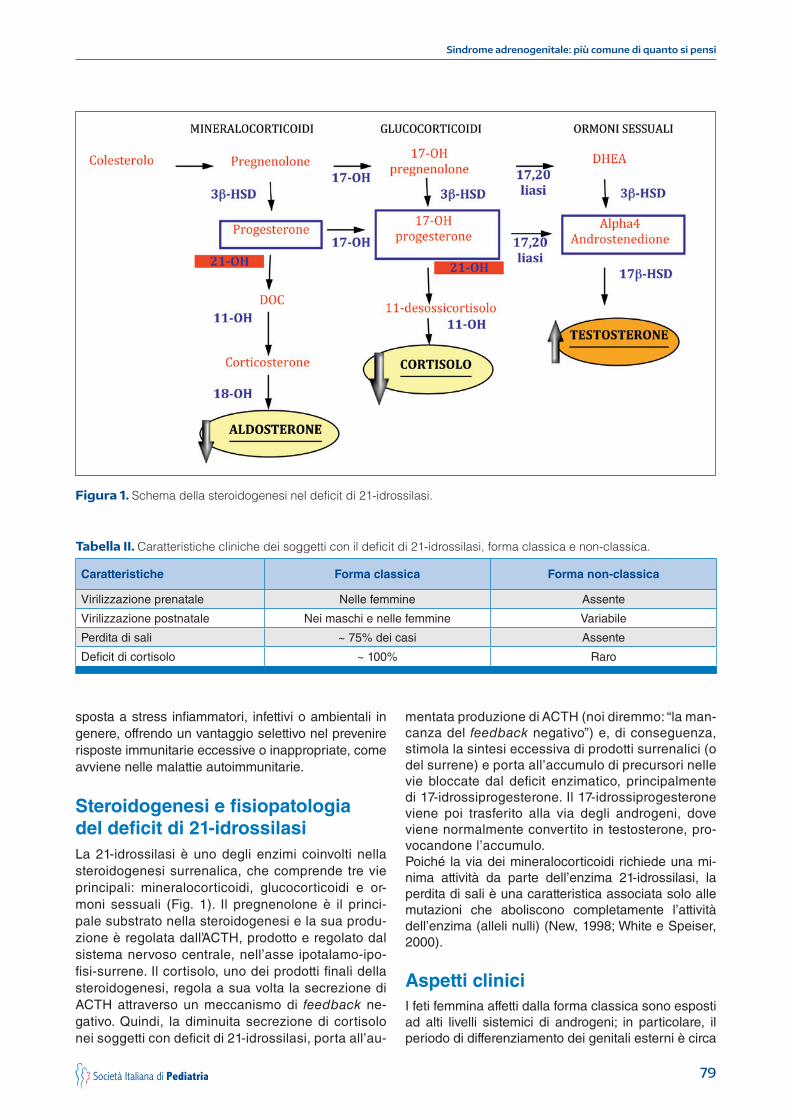
Steroidogenesi e fisiopatologia del deficit di 21-idrossilasiLa 21-idrossilasi è uno degli enzimi coinvolti nella steroidogenesi surrenalica, che comprende tre vie principali: mineralocorticoidi, glucocorticoidi e or-moni sessuali (Fig. 1). Il pregnenolone è il princi-pale substrato nella steroidogenesi e la sua produ-zione è regolata dall’ACTH, prodotto e regolato dal sistema nervoso centrale, nell’asse ipotalamo-ipo-fisi-surrene. Il cortisolo, uno dei prodotti finali della steroidogenesi, regola a sua volta la secrezione di ACTH attraverso un meccanismo di feedback ne-gativo. Quindi, la diminuita secrezione di cortisolo nei soggetti con deficit di 21-idrossilasi, porta all’au-
mentata produzione di ACTH (noi diremmo: “la man-canza del feedback negativo”) e, di conseguenza, stimola la sintesi eccessiva di prodotti surrenalici (o del surrene) e porta all’accumulo di precursori nelle vie bloccate dal deficit enzimatico, principalmente di 17-idrossiprogesterone. Il 17-idrossiprogesterone viene poi trasferito alla via degli androgeni, dove viene normalmente convertito in testosterone, pro-vocandone l’accumulo. Poiché la via dei mineralocorticoidi richiede una mi-nima attività da parte dell’enzima 21-idrossilasi, la perdita di sali è una caratteristica associata solo alle mutazioni che aboliscono completamente l’attività dell’enzima (alleli nulli) (New, 1998; White e Speiser, 2000).
Aspetti cliniciI feti femmina affetti dalla forma classica sono esposti ad alti livelli sistemici di androgeni; in particolare, il periodo di differenziamento dei genitali esterni è circa
Figura 1. Schema della steroidogenesi nel deficit di 21-idrossilasi.
Tabella II. Caratteristiche cliniche dei soggetti con il deficit di 21-idrossilasi, forma classica e non-classica.
Caratteristiche Forma classica Forma non-classica
Virilizzazione prenatale Nelle femmine Assente
Virilizzazione postnatale Nei maschi e nelle femmine Variabile
Perdita di sali ~ 75% dei casi Assente
Deficit di cortisolo ~ 100% Raro

80
P. Carrera et al.
dalla 9 alla 15 settimana di gestazione. Ciò determina uno sviluppo dei genitali esterni con fenotipo maschi-le a vari livelli di gravità, dalla semplice clitoridomega-lia alla scrotalizzazione delle strutture labiali e a seni urogenitali, che possono portare a errori nell’identifi-cazione del sesso. Il grado di virilizzazione genitale è classificato in cinque stadi, secondo la scala di Prader (Prader e Gurtner, 1955). I genitali femminili interni (utero, tube di Falloppio e ovaie) sono normali. L’alta frequenza di queste forme porta a riconoscere il defi-cit di 21-idrossilasi come la causa singola più comune di pseudo-ermafroditismo femminile. I neonati maschi non mostrano anormalità alla nascita; si evidenzia una virilizzazione precoce attorno al 18°-24° mese. Nei pazienti con la forma classica trattati adeguata-mente, la pubertà si presenta come atteso (Ghali et al., 1977), o con un po’ di anticipo rispetto alla popo-lazione generale, sia nelle femmine che nei maschi (Trinh et al., 2006). Se non trattato rapidamente, in entrambi i sessi, l’eccesso di androgeni può portare a pubarca precoce e a un anticipato sviluppo nell’accre-scimento. I soggetti affetti mostreranno una precoce comparsa dei peli del pube e delle ascelle, saranno ad alta statura durante l’infanzia ma rimarranno bassi, come risultato della prematura chiusura dei centri di crescita epifisari delle ossa, circa 1,1-1,5 deviazioni standard sotto la media dell’altezza dei genitori (Bru-nelli et al., 2003). Nelle femmine, i segni dell’iperan-drogenismo possono includere anche alopecia, acne, irsutismo, infertilità, amenorrea o disfunzione del ciclo mestruale. Inoltre è stata descritta l’associazione tra SAG e sindrome dell’ovaio policistico (Barnes et al., 1994).
Identità di genereI maschi, al contrario delle femmine, non mostrano alterazioni del comportamento per quanto riguarda l’identità di genere, l’orientamento sessuale e il gio-co nell’infanzia (Hines et al., 2004). Nelle femmine, è stata identificata una correlazione tra orientamento sessuale, esposizione agli androgeni in epoca prena-tale e comportamento mascolino nell’infanzia (Meyer-Bahlburg et al., 2008).
FertilitàLe femmine affette da sindrome adrenogenitale pos-sono avere problemi di fertilità per varie ragioni, in-clusa non-ovulazione, sindrome dell’ovaio policistico, irregolarità mestruali, alti livelli di progesterone non soppressi dalla terapia. La fertilità è ridotta special-mente nelle femmine con la forma classica (Mulaikal et al., 1987). Studi recenti hanno dimostrato che l’a-deguatezza della terapia con i glucocorticoidi è una variabile importante, capace di migliorare i livelli di fertilità individuale (Premawardhana, et al., 1997). Anche nei maschi affetti dalla forma classica, studi a
lungo termine hanno indicato che, se trattati adegua-tamente, essi mostrano uno sviluppo puberale nor-male, così come normale spermatogenesi e fertilità (Trinh et al., 2006). Al contrario, se la patologia non è ben controllata con la terapia, si possono osservare un ridotto numero di spermatozoi e testicoli di ridot-te dimensioni o con noduli iperplasici (Cabrera et al., 2001).
TerapiaL’obiettivo della terapia nella sindrome adrenogenitale è di correggere il deficit di cortisolo e di abolire l’iper-produzione di ACTH. Un trattamento adeguato con corticosteroidi riduce la stimolazione della sintesi de-gli androgeni, prevenendo un’ulteriore virilizzazione e normalizzando le fasi dello sviluppo. Lo scopo della terapia è di somministrare la minor dose richiesta per un controllo ottimale. Nei bambini la terapia deve es-sere monitorata spesso nelle prime fasi, più raramen-te in seguito. Negli adulti, è consigliato il monitoraggio a lungo termine per problemi di sovrappeso, densità ossea, fertilità e rischio cardiovascolare. Nei periodi di malattia o particolare stress o in occasione di un inter-vento chirurgico la dose di mantenimento può essere aumentata.
Trattamento chirurgicoNel passato, nei neonati con genitali ambigui era rac-comandata la correzione chirurgica precoce. Più re-centemente, questa pratica è diventata controversa a causa della mancanza di dati sugli effetti funzionali a lungo termine. Pertanto per accompagnare le scelte dei genitori dei pazienti nel caso di assegnazione del sesso e correzione chirurgica, è consigliato un ap-proccio multidisciplinare, che coinvolga l’endocrinolo-go pediatra, l’urologo, il genetista, e lo psicologo nella valutazione del singolo caso (Schnitzer e Donahoe, 2001).
Diagnosi ormonaleLa diagnosi biochimica del deficit di 21-idrossilasi può essere confermata dalla valutazione ormonale. Misu-rando la concentrazione di 17-idrossiprogesterone (il precursore dell’enzima) su un prelievo di sangue, si può fare diagnosi di forma classica. Il test di elezio-ne per la diagnosi ormonale nella forma non-classica è il test da stimolo con l’ormone adrenocorticotropo (ACTH-test), nel quale i livelli di 17-idrossiprogestero-ne e di alpha4-androstenedione vengono misurati al tempo zero e dopo 60’ dalla stimolazione endoveno-sa con ACTH. Per definire la gravità clinica, i valori ottenuti sono confrontati con quelli di riferimento, ri-portati in letteratura (nomogramma di New) (New et al., 1983).

81
Sindrome adrenogenitale: più comune di quanto si pensi
GeneticaIl gene codificante per la 21-idrossilasi è un citocromo P450 (citocromo P450, famiglia 21, subfamiglia A, po-lipeptide 21, CYP21A2), localizzato sul braccio corto del cromosoma 6, all’interno della regione dei geni di classe III del complesso maggiore di istocompatibilità (MHC) (Fig. 2) (Dupont et al., 1977). CYP21A2 e il suo omologo, lo pseudogene CYP21A1P, sono alter-nati ai geni C4B e C4A, che codificano due isoforme del quarto componente del sistema del complemento. CYP21A2 e il suo pseudogene contengono 10 esoni ciascuno e sono omologhi al 98% negli esoni, e al 96% negli introni (White et al., 1986).Nel gene CYP21A2 sono state descritte più di 200 va-rianti patogenetiche, tra queste, sostituzioni nucleoti-diche puntiformi, piccole delezioni e inserzioni e riar-rangiamenti complessi (Human Cytocrome P450 Alle-le Nomenclature Committee Institute of Environmen-tal Medicine, Karolinska Institutet, Stockholm, Swe-den – www.cypalleles.ki.se/cyp21.htm) (The Human Gene Mutation Database (HGMD), Cardiff – www.hgmd.cf.ac.uk/ac/all.php) (New et al., 2013). Le va-rianti patogenetiche più comuni, identificate in circa il 95% dei pazienti, sono il risultato di due tipi di riar-rangiamento tra il gene CYP21A2 e lo pseudogene CYP21A1P: i) crossing over diseguale, in seguito a un anomalo appaiamento durante la meiosi, che ha come conseguenza la generazione di estese delezio-ni; ii) eventi di conversione genica che provocano il trasferimento nel gene CYP21A2 di varianti patoge-netiche puntiformi o di piccola-media scala presenti
nello pseudogene CYP21A1P (Fig. 3A). Le varianti patogenetiche derivate dallo pseudogene sono le più frequenti e mostrano una distribuzione variabile nelle varie etnie (Wilson et al., 2007; Carrera et al., 1996; Bobba et al., 1999; White e Speiser, 2000; New et al., 2013). Il rimanente 5% delle varianti patogenetiche sono varianti puntiformi, rare o private, più spesso lo-calizzate nella regione 3’ del gene. In figura 4 sono riportate le varianti patogenetiche co-muni raggruppate in base all’attività enzimatica resi-dua, misurata attraverso studi di espressione in vitro (White e Speiser, 2000). Poiché il deficit di 21-idros-silasi è una malattia autosomica recessiva, è neces-saria la combinazione di due varianti patogenetiche per dare il fenotipo affetto e, generalmente, è la va-riante meno severa che determina il fenotipo (Fig. 4). Possiamo quindi affermare che la forma clinica del deficit di 21-idrossilasi è dipendente dalla combina-zione delle varianti alleliche nel genotipo del soggetto in esame. Anche se la predizione del fenotipo è ri-sultata inizialmente non del tutto accurata (Speiser et al., 1992), studi recenti hanno permesso di stabilire con più precisione la correlazione genotipo-fenotipo e struttura proteica-fenotipo nel deficit di 21-idrossilasi (Haider et al., 2013; New et al., 2013).
Diagnosi molecolareL’identificazione delle varianti patogenetiche è pos-sibile attraverso il test molecolare. Esso prevede la combinazione di due protocolli: I) per l’identificazione dei grossi riarrangiamenti, come la delezione genica
Figura 2. Viene mostrata la localizzazione del gene CYP21A2 all’interno dei geni HLA del complesso maggiore di isto-compatibilità sul cromosoma 6.
3'5'

82
P. Carrera et al.
e le conversioni geniche estese sono utilizzati proto-colli basati su Multiple Ligation Probe Assay (MLPA) oppure Southern blotting; in figura 3B un esempio di uno studio familiare per l’identificazione dei grossi riarrangiamenti; II) per l’identificazione delle varia-zioni puntiformi e di piccole indels (riarrangiamenti che sono costituiti da inserzioni o delezioni nucleo-tidiche), la metodica di elezione è il sequenziamento diretto con il metodo Sanger. Affinché il risultato del sequenziamento sia corretto, è cruciale selezionare in modo specifico la sequenza del gene CYP21A2, ad esempio mediante amplificazione con Polymerase Chain Reaction (White e Speiser, 2000).Per quanto riguarda il test genetico per il deficit di 21-idrossilasi, l’accesso al test è sempre preceduto da consulenza genetica. Generalmente, essendo l’esor-dio neonatale o pediatrico, la consulenza coinvolge i genitori. Quando viene identificato un soggetto affetto, in base alla diagnosi clinica o in seguito a screening neonatale, si esegue il test genetico per conferma-re la diagnosi e vedere quali varianti sono presenti. Il test è richiesto anche nei familiari e nei partner dei soggetti affetti o portatori, allo scopo di sapere se si è portatori sani e quindi stabilire se esiste un rischio riproduttivo.
Diagnosi e terapia prenataleNelle famiglie con un precedente figlio affetto da for-ma classica, e nella quale sono state identificate le due varianti patogenetiche, si può offrire la diagno-si prenatale e la terapia con desametasone (Fig. 5). La terapia prenatale prevede una somministrazione orale di desametasone alle madri a rischio per ri-durre la virilizzazione nelle femmine affette; l’indica-zione è di iniziare il trattamento prima della settima ed entro l’ottava settimana di gestazione, (Forest et al., 1989). Dopo il risultato della diagnosi prenatale, il trattamento è sospeso in caso di feto maschio o di femmina sana, mentre continuerà per tutta la gra-vidanza in caso di feto femmina affetta. Sono stati riportati risultati contrastanti sull’effetto della terapia prenatale sulle madri e sul feto (New et al., 2003; Pang et al., 1992). Gli effetti collaterali riportati in-cludono un eccessivo aumento di peso, un aumento delle strie gravidiche, una risposta iperglicemica al carico orale di glucosio.Più recentemente, alcuni studi sugli effetti a lungo ter-mine nei bambini trattati con desametasone non han-no evidenziato in generale criticità tranne un effetto a lungo termine a livello cognitivo (New et al., 2003;
Figura 3. A. Assetti genomici osservati in alleli normali e in alleli con riarrangiamenti patogenetici. B. Esempio di uno studio familiare per l’analisi dei grandi riarrangiamenti mediante Southern blotting. I genitori sono portatori (eterozigoti) di una delezione estesa di 30 kb che coinvolge I geni CYP21A2 e C4B; il soggetto affetto (SAG) ha ereditato da entrambi i genitori la delezione estesa; il fratello è sano-non portatore, avendo ereditato da entrambi i genitori l’allele non-mutato.

83
Sindrome adrenogenitale: più comune di quanto si pensi
Figura 4. Le varianti patogenetiche più comuni del gene CYP21A2 sono raggruppate in base all’attività enzimatica re-sidua. In rosso, le varianti severe (nulle); in arancione, le varianti intermedie; in giallo, le varianti meno gravi. La variante c.293-13A-C > G è un caso particolare in quanto si associa più spesso alla forma SW ma anche alla forma SV.
Figura 5. Studio familiare: famiglia con due figli affetti da forma classica SW (II.1, II.2), inviata al test genetico per con-ferma diagnostica. Identificate le mutazioni: padre asintomatico con genotipo criptico (combinazione di una variante severa, CV, con una variante lieve, p.Val281Leu). Madre eterozigote per la mutazione severa CV. Figli affetti entrambi omozigoti per la variante severa CV (conversione genica estesa). Questo caso evidenzia l’importanza dello studio familia-re; se avessimo fatto il test genetico solo ai due soggetti affetti non avremmo definito correttamente il rischio riproduttivo della famiglia.Diagnosi prenatale (II.3): in una gravidanza successiva, la madre ha iniziato la terapia per la prevenzione della virilizza-zione. La DPN: risultato genotipo non-classico (combinazione della variante paterna lieve, p.Val281Leu, con la variante materna severa, CV); fenotipo atteso: forma non-classica, quindi sospensione terapia desametasone.

84
P. Carrera et al.
Meyer-Bahlburg et al., 2004; Hirvikoski et al., 2007). Gli autori concludono che questi dati andranno tutta-via verificati in ulteriori studi.
ConclusioneLa conoscenza del deficit di 21-idrossilasi è progre-
dita molto nel corso del tempo, con un impatto molto positivo sulle procedure diagnostiche e terapeutiche. In particolare, grazie a una gestione multidisciplinare integrata (endocrinologo, chirurgo, genetista, psicolo-go), è ora possibile ottenere un normale sviluppo dei pazienti, così come il mantenimento della loro salute nel corso della vita.
Bibliografia
Allen DB, Hoffmann GI, Fitzpatrick P, et al. Improved precision of newborn screening for congenital adrenal hyper-plasia using weight adjusted criteria for 17-hydroxyprogesterone levels. J Pediatr 1997;130:128-33.
Balsamo A, Cacciari E, Piazzi S, et al. Congenital adrenal hyperplasia: neona-tal mass screening compared with clini-cal diagnosis only in the Emilia-Romagna region of Italy, 1980-1995. Pediatrics 1996;98:362-7.
Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilising disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab 1994;79:1328-33.
Bobba A, Marra E, Giannattasio S, et al. 21-hydroxylase deficiency in Italy: a dis-tinct distribution pattern of CYP21 muta-tions in a sample from southern Italy. J Med Genet 1999;36:648-50.
Brunelli V, Russo G, Bertelloni S, et al. Fi-nal height in congenital adrenal hyperplasia due to 21-idroxylase deficiency: the Italian experience. J Pediatr Encocrinol Metab 2003;16:277-83.
Cabrera M, Vogiatzi M, New M. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin En-docrinol Metab 2001;86:3070-80.
Carrera P, Ferrari M, Beccaro F, et al. Mo-lecular characterization of 21-hydroxylase deficiency in 70 Italian families. Hum Hered 1993;43:190-6.
Cartigny-Maciejewski M, Guilley N, Van-derbecken S, et al. Neonatal screening of con-genital adrenal hyperplasia due to 21-hydro-xylase deficiency. Arch Pediatr 1999;6:151-8.
Cutfield WS, Webster D. Newborn scree-ning for congenital adrenal hyperplasia in New Zealand. J Pediatr 1995;126:118-21.
Dupont B, Oberfield SE, Smithwick EM, et al. Close genetic linkage between HLA and congenital adrenal hyperpla-sia (21-hydroxylase deficiency). Lancet 1977;2:1309-12.
Forest MG, Betuel H, David M. Prenatal treatment in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update 88 of the French multicentric study. Endocr Res 1989;15:277-301.
Ghali I, David M, David L. Linear growth and pubertal development in treated con-genital adrenal hyperplasia due to 21-hy-droxylase deficiency. Clin Endocrinol 1977;6:425-36.
Haider S, Islam B, D’Atri V, et al. Struc-ture-phenotype correlations of human CYP21A2 mutations in congenital adre-nal hyperplasia. Proc Natl Acad Sci USA 2013;110:2605-10.
** Non tutte le nuove varianti del gene CYP21A2 sono state caratterizzate a livello biochimico, con studi funzionali. Per cer-care di supplire a questa carenza, gli autori fanno delle predizioni sulla patogenicità delle varianti, modellando i mutanti sulla struttura cristallizzata dell’omologo bovino del gene. Viene proposta la correlazione tra dominio/funzione interessata dalla variante e forma clinica.
Hines M, Brook C, Conway GS. Androgen and psychosexual development: core gen-der identity, sexual orientation and recalled childhood gender role behaviour in women and men with congenital adrenal hyperpla-sia (CAH). J Sex Res 2004;41:75-81.
Meyer-Bahlburg HF, Dolezal C, Baker SW, et al. Sexual orientation in women with classical or non-classical congenital adre-nal hyperplasia as a function of degree of prenatal androgen excess. Arch Sex Behav 2008;37:85-99.
Mulaikal RM, Migeon CJ, Rock JA. Ferti-lity rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med 1987;316:178-82.
New MI, Lorenzen F, Lerner AJ, et al. Genotyping steroid 21-hydroxylase de-ficiency: hormonal reference data. J Clin Endocrinol Metab 1983;57:320-6.
** Dati di correlazione tra dosaggio or-monale e forma clinica, proponendo i valori di riferimento.
New MI. Diagnosis and management of congenital adrenal hyperplasia. Annu Rev Med 1998;49:311-28.
New MI, Carlson A, Obeid J, et al. Prena-tal diagnosis for congenital adrenal hyper-plasia in 595 pregnancies. Endocrinologist 2003;13:233-9.
New MI, Moolamannil A, Gonzalez B, et al. Genotype-phenotype correlation in 1507 families with congenital adrenal hyperpla-sia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci USA 2013;110:2611-6.
** La più ampia casistica di deficit di 21-idrossilasi, mai riportata (1507 fami-glie), correlazione genotipo-fenotipo dallo storico gruppo di Maria New.
Pang S, Murphey W, Levine LS, et al. A pilot newborn screening for congenital adrenal hyperplasia in Alaska. J Clin Endo-crinol Metab 1982;55:413-20.
Pang S, Wallace MA, Hofman L, et al. Worldwide experience in newborn screen-ing for classical congenital adrenal hyper-plasia due to 21-hydroxylase deficiency. Pediatrics 1988;81:866-74.
Pang S, Clark AT, Freeman LO, et al. Ma-ternal side-effect of prenatal dexametha-sone theraphy for phetal congenital Adre-nal hyperplasia. J Clin Endocrinol Metab 1992;76:249-53.
Pang S, Clark A. Congenital Adrenal hy-perplasia due to 21-hydrpxylase deficien-cy: newborn screening and its relationship to the diagnosis and treatment of the disor-der. Screening 1993;2:105-39.
Prader A, Gurtner HP. The syndrome of pseudohermaphrodism in congenital adrenocortical hyperplasia without over-production of androgens. Helv Paediatr Acta 1955;10:397-412.
Premawardhana L, Hughes I, Read G, et al. Longer term outcome in females with congenital adrenal hyperplasia (CAH): the Cardiff experience. Clin Endocrinol 1997;46:327-32.
Schnitzer J, Donahoe P. Surgical tre-atment of congenital adrenal hyperpla-sia. Endocrinol Metab Clin North Am 2001;30:137-54.
Speiser PW, Dupont B, Rubstein, et al. High frequency of nonclassic steroid 21-hydroxylase deficiency. Am J Hum Ge-net 1985;37:650-67.
Speiser PW, Dupont J, Zhu D, et al. Di-sease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest 1992;90:584-95.
Tajima T, Fujieda K, Nakae J, et al. Mole-cular basis of nonclassical steroid 21-hy-droxylase deficiency detected by neonatal mass screening in Japan. J Clin Endocrinol Metab 1997;82:2350-6.
Therrel BLJ, Berenbaum SA, Manter-Kapanke V, et al. Results of screening 1.9 millio Texas newborns for 21-hydroxylase

85
Sindrome adrenogenitale: più comune di quanto si pensi
deficient congenital adrenal hyperplasia. Pediatrics 1998;101:583-90.
Thil’en A, Nordenstrom A, Hagenfeld I, et al. Benefits of neonatal screening for congenital adrenal hyperplasia (21-hydro-xylase deficiency) in Sweden. Pediatrica 1998;101:E11.
Trinh L, Nimkarn S, Obeid J, et al. Growth and puberal characteristics in patients with congenital adrenal hyper-plasia due to 21-hydroxylase deficiency. ENDO 2006 The Endocrine Society’s 88th
Annual Meeting. Boston, USA, June 24-27, 2006, p. 368.
White PC, New MI, Dupont B. Structure of the human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 1986;83:5111-5.
* Descrizione della struttura e della se-quenza del gene CYP21A2 e del suo pseu-dogene CYP21A1P.
White PC, Speiser PW. Congenital adre-nal hyperplasia due to 21-hydroxylase de-ficiency. Endocrine Rev 2000;21:245-91.
** Anche se non recentissima, rimane una delle più dettagliate e omnicomprensi-ve review sull’argomento.
Wilson RC, Nimkarn S, Dumic M, et al. Ethnic specific distribution of mutations in 716 patients with congenital adrenal hyper-plasia owing to 21-hydroxylase deficiency. Mol Genet Metab 2007;90:414-21.
* Distribuzione delle varianti patogene-tiche del gene CYP21A2 in base all’etnia in 716 pazienti. Evidenziate differenze tra gruppi etnici.
Corrispondenza
Paola CarreraIRCCS Ospedale San Raffaele, via Olgettina 60, 20132 Milano - Tel. +39 02 26434759 - Fax +39 02 26434351 - E-mail: [email protected]
![bruni [Sola lettura] [modalità compatibilità] · ecografica dello sviluppo mammario. ... contributo da stress ... free T3 ) e di attivazione dell’asse ipotalamo-ipofisi surrene](https://static.fdocumenti.com/doc/165x107/5c696d6d09d3f25c6a8d0e72/bruni-sola-lettura-modalita-compatibilita-ecografica-dello-sviluppo-mammario.jpg)

















