
Corso di Scienza e Tecnologia dei Materiali · Università di Roma – Tor Vergata Corso di Scienza...
86
Università di Roma – Tor Vergata Corso di Scienza e Tecnologia dei Materiali per Ing. Energetica (II emisemestre: Corrosione e Protezione dei Materiali) Prof. G. Montesperelli
-
Upload
nguyenkhanh -
Category
Documents
-
view
222 -
download
1
Transcript of Corso di Scienza e Tecnologia dei Materiali · Università di Roma – Tor Vergata Corso di Scienza...

Università di Roma – Tor Vergata
Corso di Scienza e Tecnologia dei Materiali
per Ing. Energetica (II emisemestre: Corrosione e Protezione dei Materiali)
Prof. G. Montesperelli
2
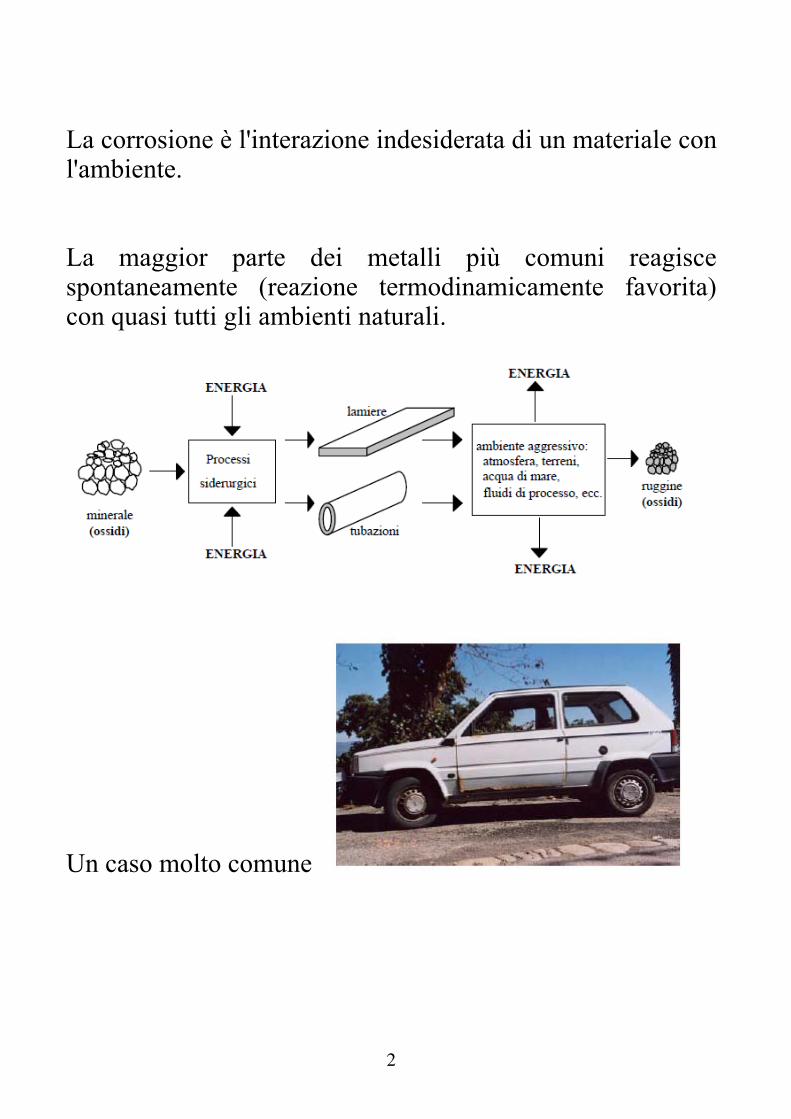
La corrosione è l'interazione indesiderata di un materiale con l'ambiente. La maggior parte dei metalli più comuni reagisce spontaneamente (reazione termodinamicamente favorita) con quasi tutti gli ambienti naturali.
Un caso molto comune

3
Tubazione in rame
Calcestruzzo armato
Paletta per turbina
4
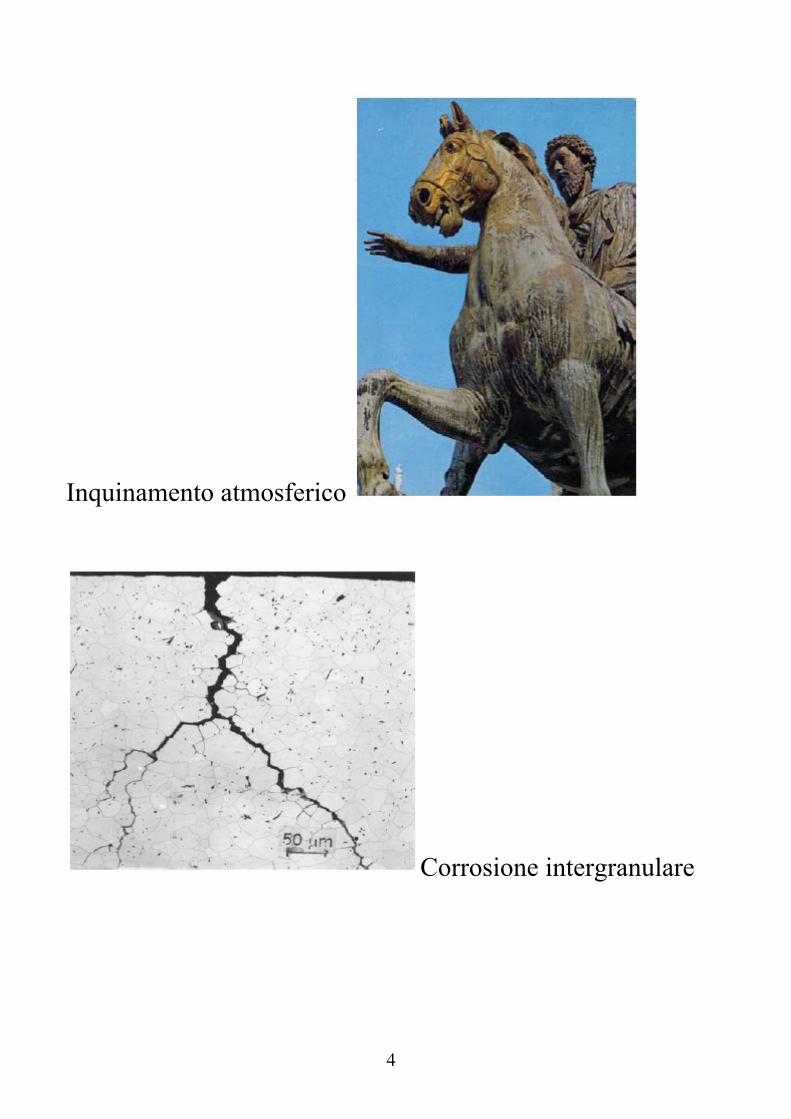
Inquinamento atmosferico
Corrosione intergranulare
5
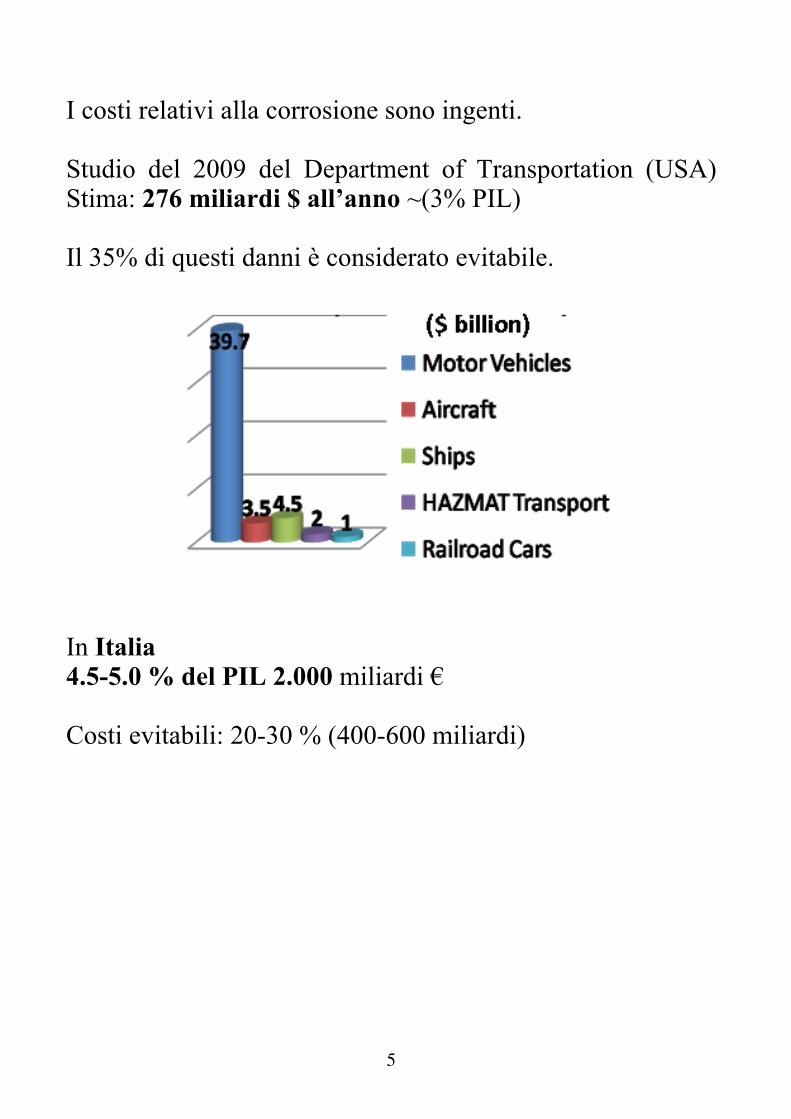
I costi relativi alla corrosione sono ingenti. Studio del 2009 del Department of Transportation (USA) Stima: 276 miliardi $ all’anno ~(3% PIL) Il 35% di questi danni è considerato evitabile.
In Italia 4.5-5.0 % del PIL 2.000 miliardi € Costi evitabili: 20-30 % (400-600 miliardi)

6
I danni si dividono in diretti ed indiretti. Diretti: sostituzione delle parti danneggiate, verniciature protettive, rivestimenti metallici (zincature), protezione elettrica, scelta di materiali più costosi. Indiretti: interruzione della produzione, perdite di prodotti, diminuzione di rendimenti degli impianti, inquinamento, sovraprogettazione, danni al personale. Le stime U.S. per il 2012 sui costi diretti: $468 miliardi (3.1% del PIL)

7
La corrosione può essere ad umido ed a secco. La corrosione ad umido (e a bassa temperatura) ha un meccanismo di tipo elettrochimico ed è regolata da leggi di tipo termodinamico e di tipo cinetico. Può essere quindi schematizzata da una reazione di ossido- riduzione (ovvero può essere divisa in due semireazioni):
Reaz. Anodica (Ossidazione):
Fe Fe2 2e
Zn Zn 2 2e
M Mn ne
Reaz. Catodica (Riduzione):
3
2
2 2
2 2
2
32 7 2
2H 2e H
O 4H 4e 2H O (pH acido)
O 2H O 4e 2OH
(pH neutro o alcalino)
NO 4H 3e NO 2H O
Cr O 14H 6e 2Cr 7H O
Nella maggior parte dei casi esisteranno delle zone dove avverrà di preferenza la reazione anodica e delle zone dove avverrà di preferenza la reazione catodica.
8
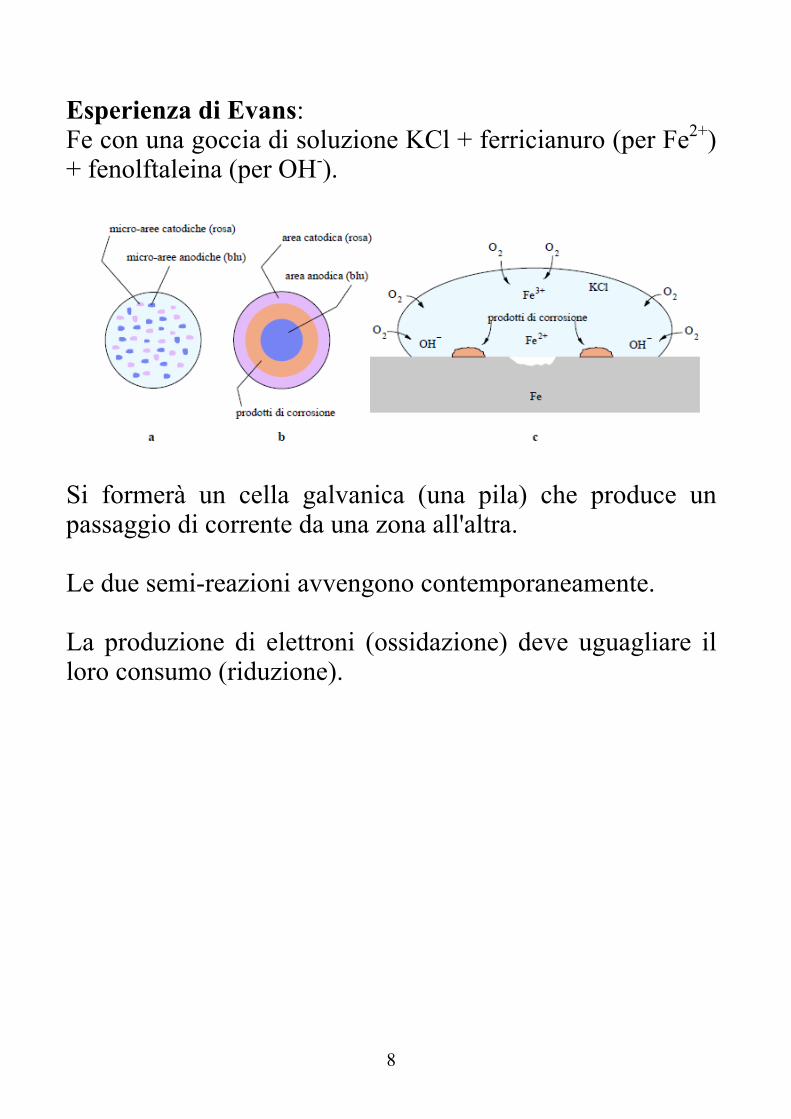
Esperienza di Evans: Fe con una goccia di soluzione KCl + ferricianuro (per Fe2+) + fenolftaleina (per OH-).
Si formerà un cella galvanica (una pila) che produce un passaggio di corrente da una zona all'altra. Le due semi-reazioni avvengono contemporaneamente. La produzione di elettroni (ossidazione) deve uguagliare il loro consumo (riduzione).

9
Per stimare l'intensità dell'attacco corrosivo si utilizzano due grandezze chiamate velocità di perdita di massa e velocità di penetrazione o velocità di corrosione (utilizzabili solo nel caso di corrosione generalizzata)
velocità di perdita di massa: v m pAt
(mdd)
velocità di corrosione: vc vm
pAt
(mm/a, mpy)
Δp = perdita in peso A = superficie esposta t = tempo ρ = densità

10
Legge di Faraday La quantità di materia che si deposita o che passa in soluzione in una reazione di ossido-riduzione è proporzionale alla quantità di carica che passa nella cella. p n q
n MzF
q it; I iA
q IAt
p MIAt
zF
Δp = variazione di peso (g) n = numero di equivalenti q = carica (Coulomb) M =peso molecolare z = numero di elettroni scambiati F = Faraday = carica di una mole di elettroni = 96500 C I =densità di corrente (A/cm
2)
A = area elettrodo (cm2)
t = tempo (sec)

11
m
mc
3
3c 2
2 2
pV velocità di perdita di massa
AtV p M I
V velocità di corrosioneA t z F
Mmassa g
z
massa gg A cm cm mm
V 10volume cmcm g C sec sec
Ampere CI
cm seccmF Coulomb
10 365 24
6mm mm60 60 315 10
a a

12
Termodinamica dei processi corrosivi Per una qualsiasi reazione chimica:
A B C D si definisce l’energia libera G, una funzione di stato che, con la sua diminuzione, misura il lavoro motore disponibile per lo sviluppo della reazione. La condizione ΔG < 0 è quindi la condizione necessaria per lo svolgimento di una reazione. ΔG = 0 la reazione è in equilibrio ΔG > 0 la reazione non avverrà
(avverrà la reazione opposta)

13
Il ΔG per la reazione soprascritta si può esprimere come:
0 C D
A B
a aG G RTln
a a
Dove ΔG
0 è la variazione di energia libera della reazione
fatta avvenire in condizioni standard e cioè: con attività unitaria per le specie in fase liquida e solida, e fugacità 1 atm per le specie gassose R = 8,314 J/mole·K T = temperatura assoluta in K a
i sono le attività delle specie i elevate al rispettivo
coefficiente stechiometrico.

14
La quantità di energia chimica che può essere trasformata in elettricità tramite una semireazione redox generica
nM M ne
è data da:
GnEF G E
nF
Dove E è la tensione di equilibrio della semireazione M/Mn+ (detta anche tensione di Galvani). Non è possibile misurare E !! Si aggira il problema accoppiando l’elettrodo di cui si vuole misurare la tensione con un opportuno elettrodo di riferimento → Elettrodo a idrogeno. L’elettrodo a idrogeno è costituito da una lamina di Pt immersa in una soluzione ad attività (concentrazione) unitaria di H+ nella quale gorgoglia H2 a fugacità (pressione) unitaria.
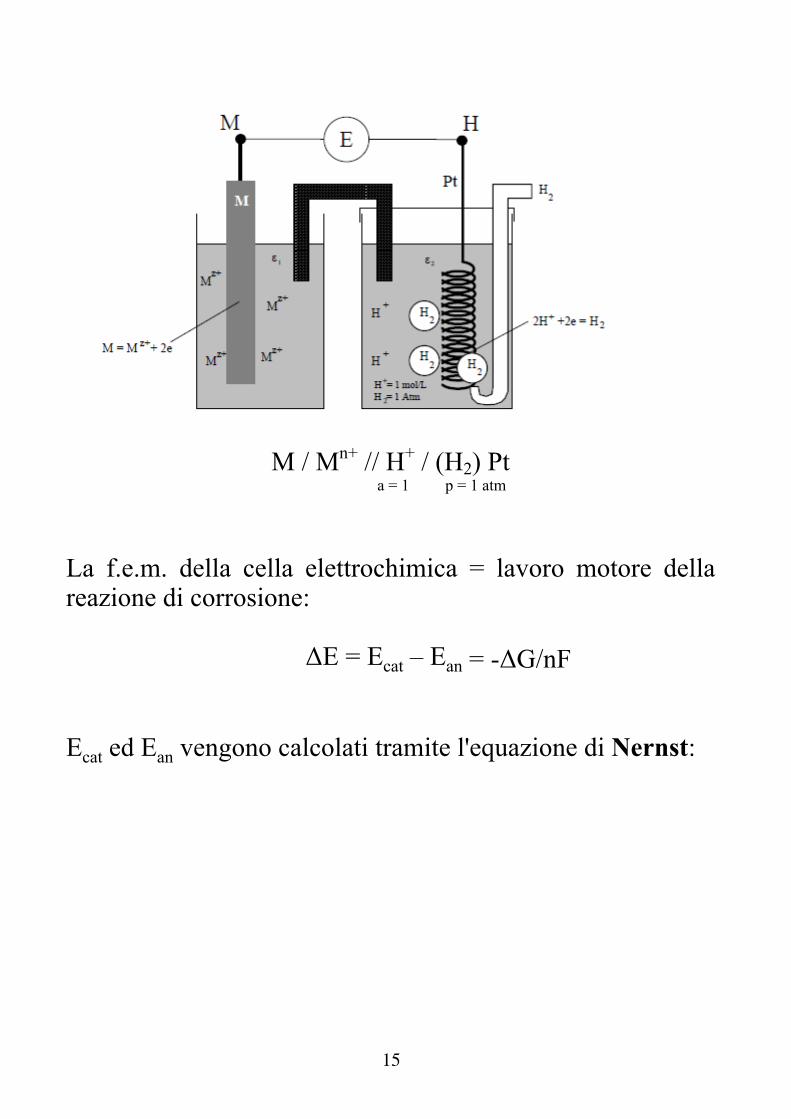
15
M / Mn+ // H+ / (H2) Pt a = 1 p = 1 atm
La f.e.m. della cella elettrochimica = lavoro motore della reazione di corrosione:
ΔE = Ecat – Ean = -ΔG/nF Ecat ed Ean vengono calcolati tramite l'equazione di Nernst:

16
0 0ox ox
rid rid
0
RT a 0.059 aE E ln E log
nF a n a
Ox0.059E E log
n Rid
aox = attività della specie ossidata (p.es. M
n+, H
+, O2)
arid = attività della specie ridotta (p. es. M, H2, H2O) α, β = coefficienti stechiometrici R = 8.31 JK-1 T = 298.14 K (25ºC) 2.303 = costante per il passaggio ln→log Nel caso in esame:
nM M ne
n
0M M
M0.059E E log
n M
per definizione M 0

17
22H H 2e
2 2
2
0
H / H H / HH2
H0.059E E log
2 P
E
H /H2
0 e EM/ Mn0 sono i potenziali redox in condizioni
standard ovvero ad attività unitaria dei reagenti H2P 1 H 1 .
Per convenzione 2
0
H / HE 0 →
2H / HE 0
Il segno di ΔE determina a sua volta quello di ΔG e quindi la spontaneità della reazione.
Catodo polo + Anodo polo - ΔE > 0 ΔG < 0 reazione spontanea corrosione ΔE < 0 ΔG > 0 reazione impossibile immunità

18
Considerato come unitarie le attività (o le pressioni parziali) delle specie allo stato di ossidazione zero, abbiamo che la tensione della cella è:
n n
n n
0H M H M/ M M
0
M/ M M
0.059E E E 0.059loga E loga
n0.059
0.059pH E logan
La disponibilità di lavoro motore dipende dal pH. Per basse concentrazioni, l’attività si può sostituire con la concentrazione.
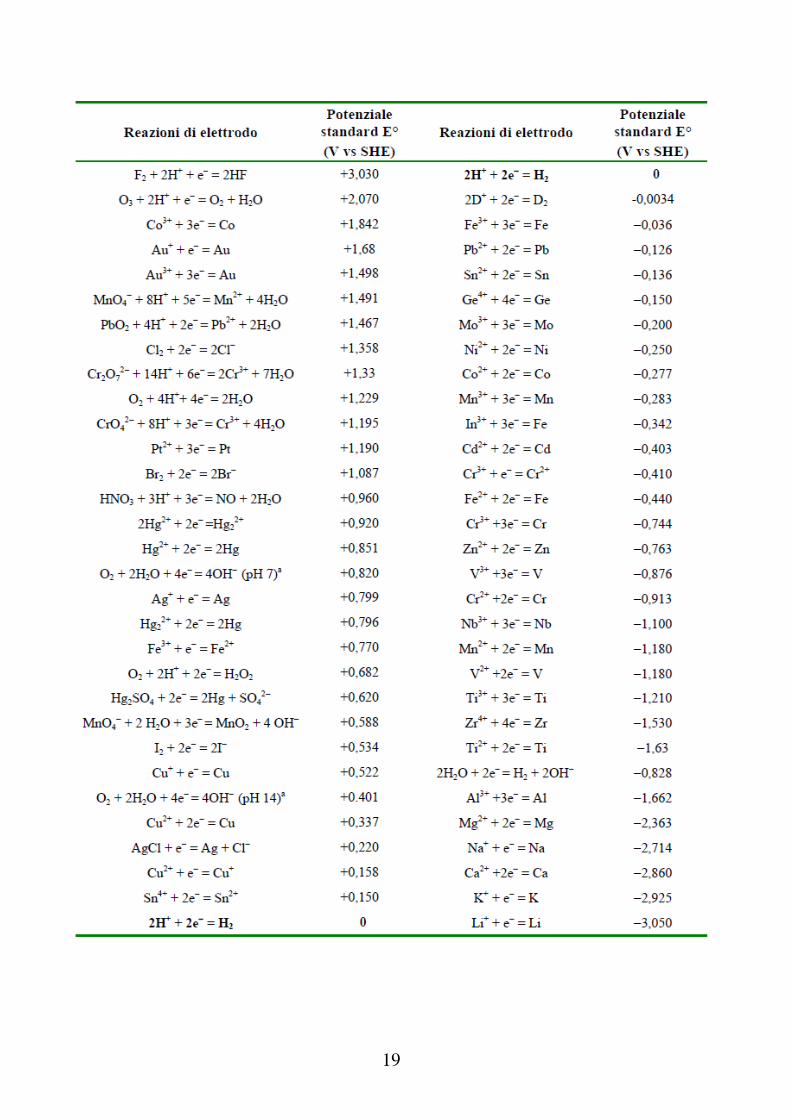
19
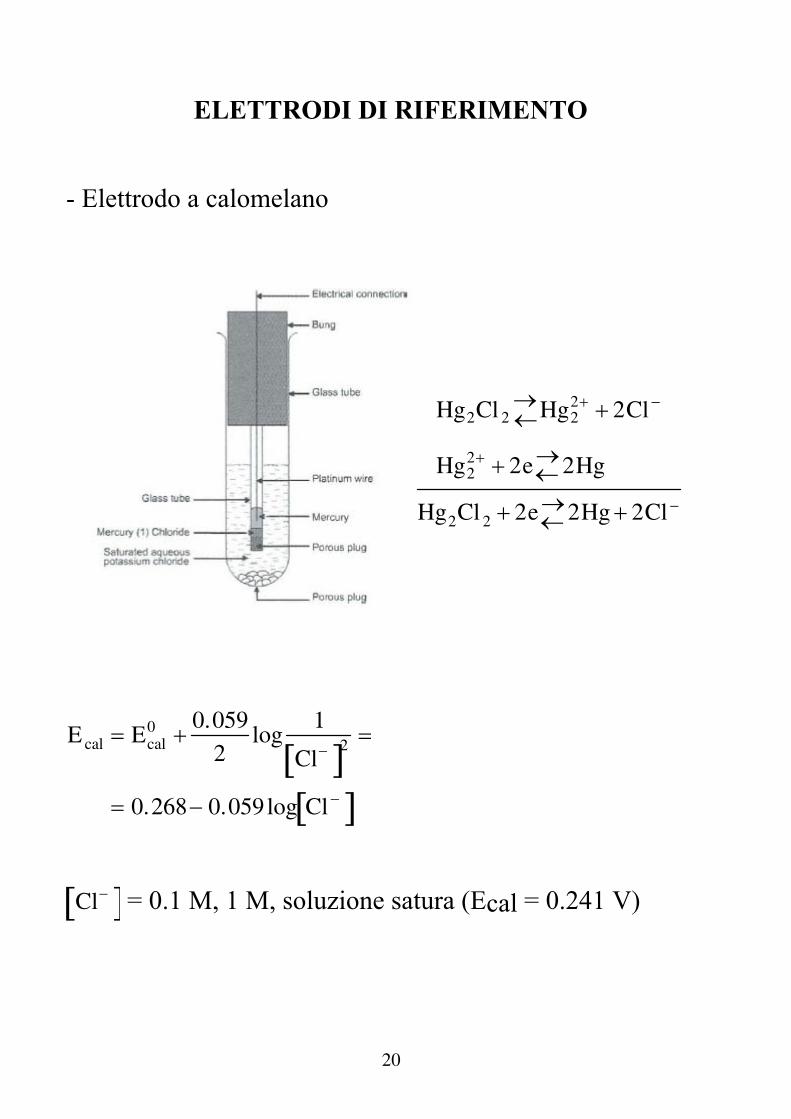
20
ELETTRODI DI RIFERIMENTO - Elettrodo a calomelano
Hg2Cl 2Hg2
2 2Cl
Hg22 2e2Hg
Hg2Cl 2 2e2Hg 2Cl
E cal Ecal0 0.059
2log
1
Cl 2
0.268 0.059log Cl
Cl = 0.1 M, 1 M, soluzione satura (Ecal = 0.241 V)
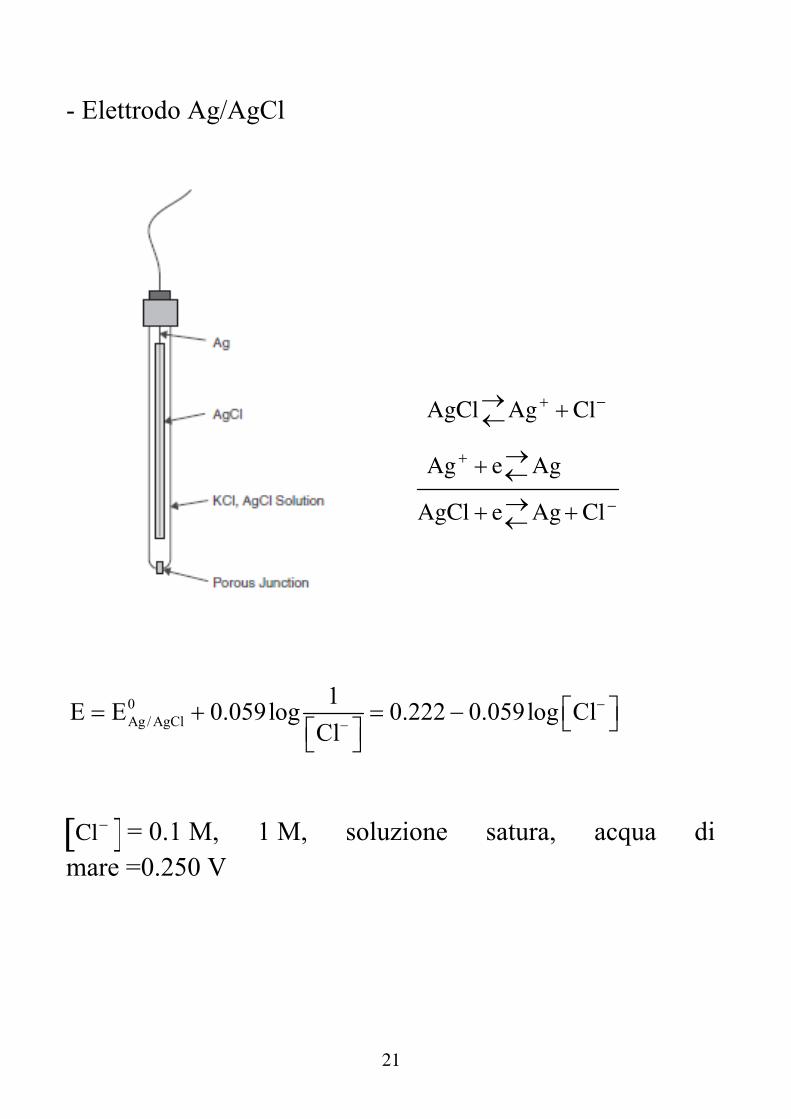
21
- Elettrodo Ag/AgCl
AgClAg Cl
Ag eAg
AgCl eAgCl
0Ag / AgCl
1E E 0.059log 0.222 0.059log Cl
Cl
Cl = 0.1 M, 1 M, soluzione satura, acqua di
mare =0.250 V
22
- Elettrodo a Cu/CuSO4
Cu2 2eCu
E ECu/Cu20
0.0592
log Cu2 0.340 0.0295log Cu2
E = 0.318 V - Elettrodo di Zn/acqua di mare Ec = -0.800 V

23
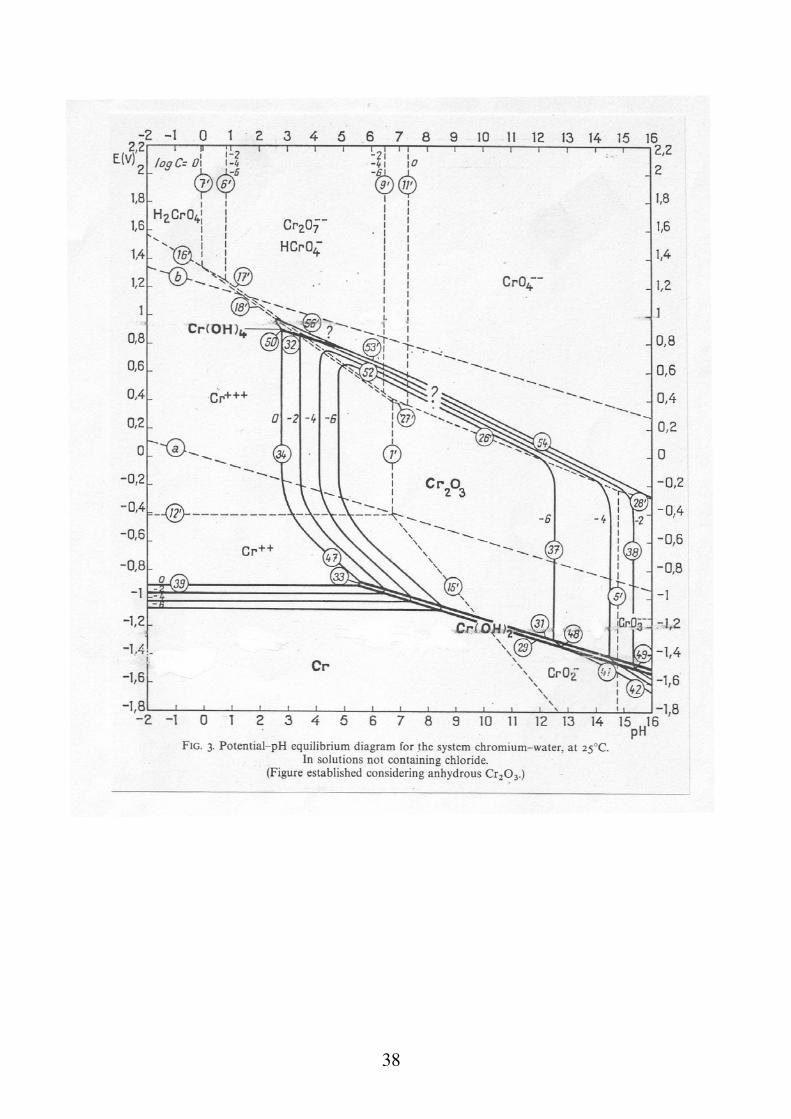
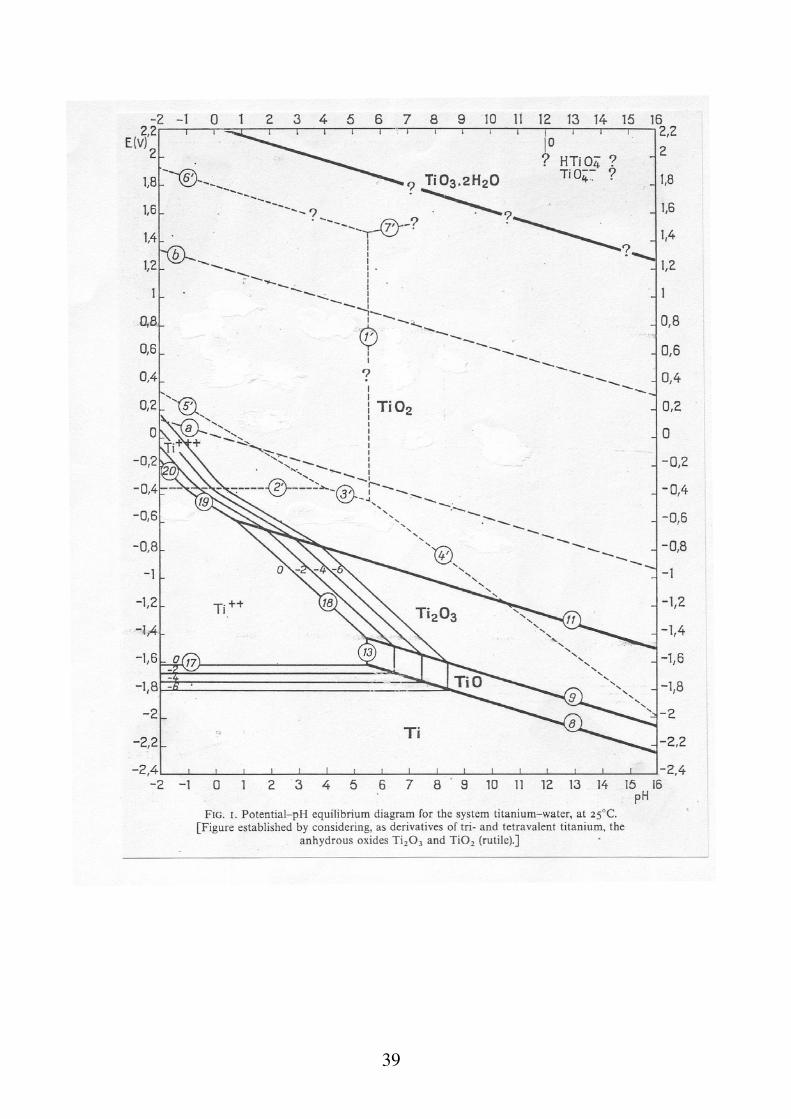
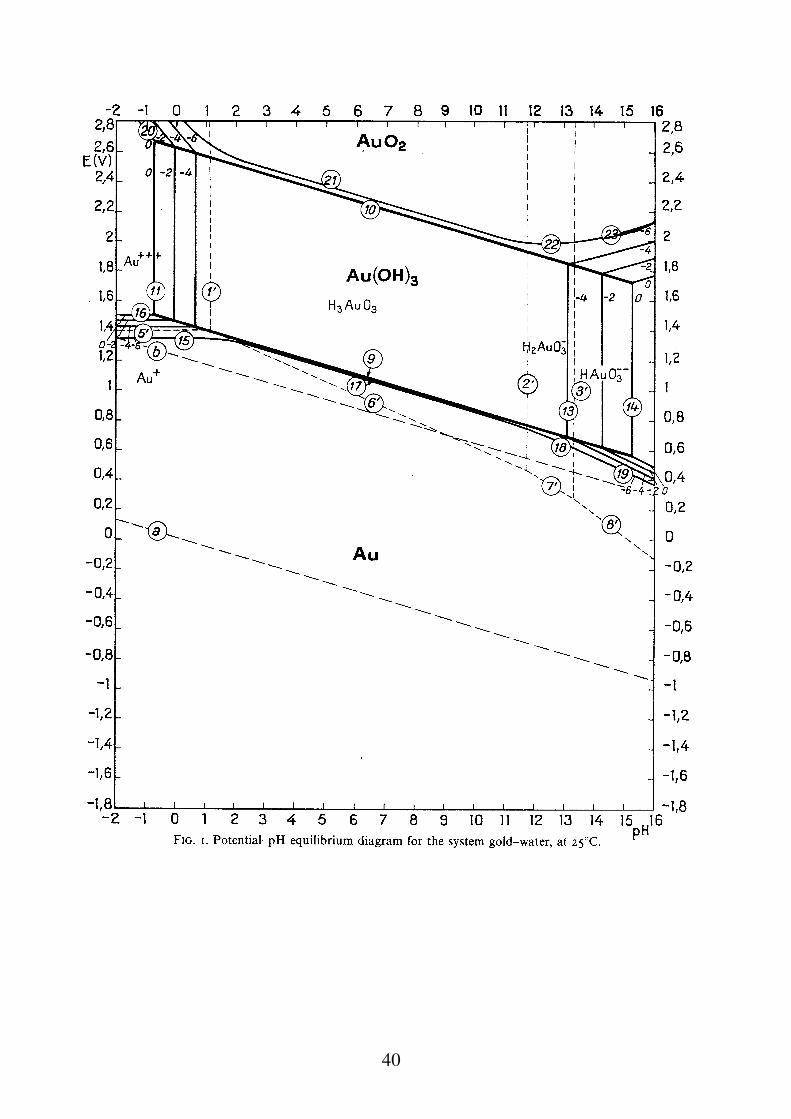
Diagrammi tensione - pH Sono diagrammi che riportano la tensione elettrodica in funzione del pH dell'ambiente, tenendo conto di tutte le reazioni possibili per un dato metallo. Permettono di stimare per ogni metallo, la presenza o meno di un lavoro motore disponibile per la reazione di corrosione in relazione ad una data reazione catodica che, ad ogni valore di pH. La costruzione teorica dei diagrammi, si effettua tenendo conto di tutti gli equilibri relativi alle reazioni che possono avvenire in un dato ambiente acquoso. In maniera molto approssimata, le reazioni si possono dividere in tre casi: a) reazioni in cui partecipano H+ o OH- ma non elettroni b) reazioni in cui partecipano elettroni ma non H+ o OH- c) reazioni in cui partecipano H+ o OH- ed elettroni

24
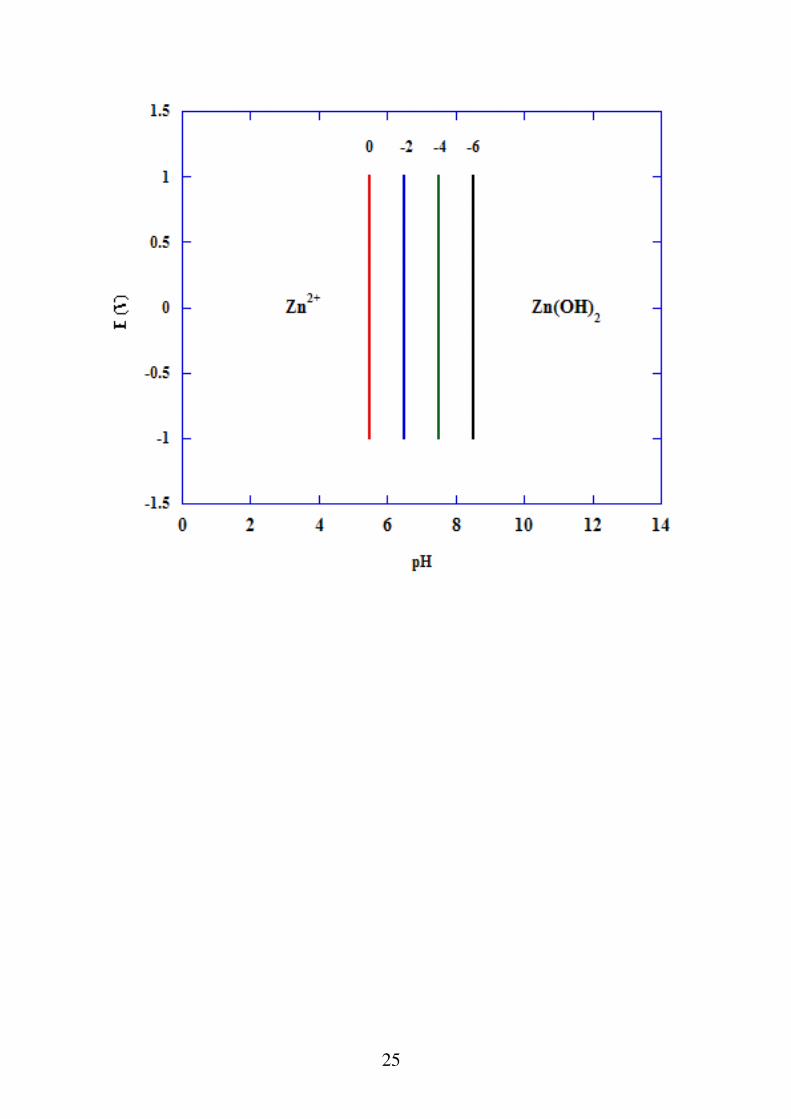
a) reazioni in cui partecipano H+ o OH- ma non elettroni
Zn 2 2OH Zn OH 2
In questo caso l'andamento della reazione è indipendente dalla tensione. L'equilibrio non è governato dall'equazione di Nernst, ma da una normale costante di equilibrio, che in questo caso è il prodotto di solubilità dell'idrossido di Zn.
K ps Zn 2 OH 2
Zn OH 2 10 17
logK ps log Zn 2 2log OH 17
pH 12
11 log Zn 2 Sul diagramma E-pH è un fascio di rette perpendicolare
all'asse dei pH, dipendente dalla concentrazione di Zn2+
.
25

26
b) reazioni in cui partecipano elettroni ma non H+ o OH-
Cd Cd 2 2e
L'equilibrio è governato dall'equazione di Nernst:
E Cd ECd0
0.0592
log Cd2 0.402 0.0295log Cd2 Sul diagramma E-pH è un fascio di rette parallele all'asse
dei pH, dipendente dalla concentrazione di Cd2+
. L'equilibrio è indipendente dal pH.
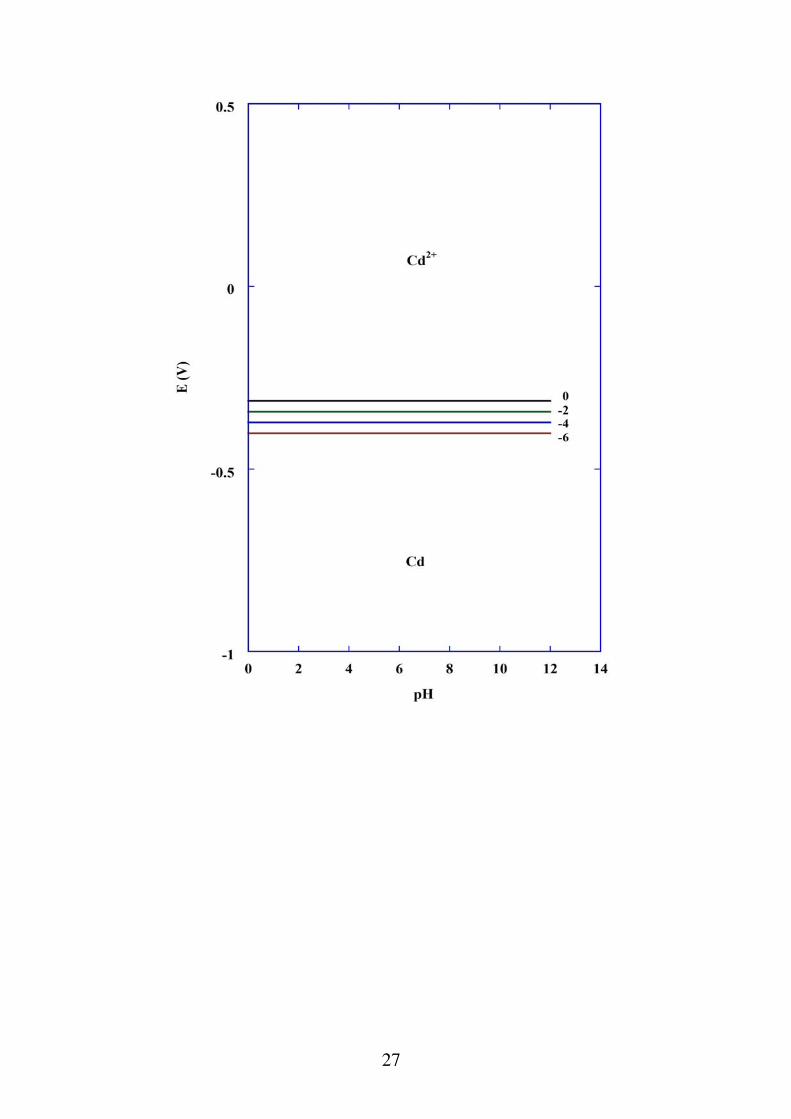
27

28
c) reazioni in cui partecipano H
+ o OH
- ed elettroni
Fe 2 3H2O Fe OH 3 3H e
E E 0 0.059logH 3Fe2 E 0 0.059log Fe2 0.177pH
Rappresenta un fascio di rette con pendenza negativa (-0.177). Il parametro che distingue le rette è la concentrazione degli ioni Fe2+ (normalmente si sceglie pari a 10-6 M che rappresenta il limite analitico).
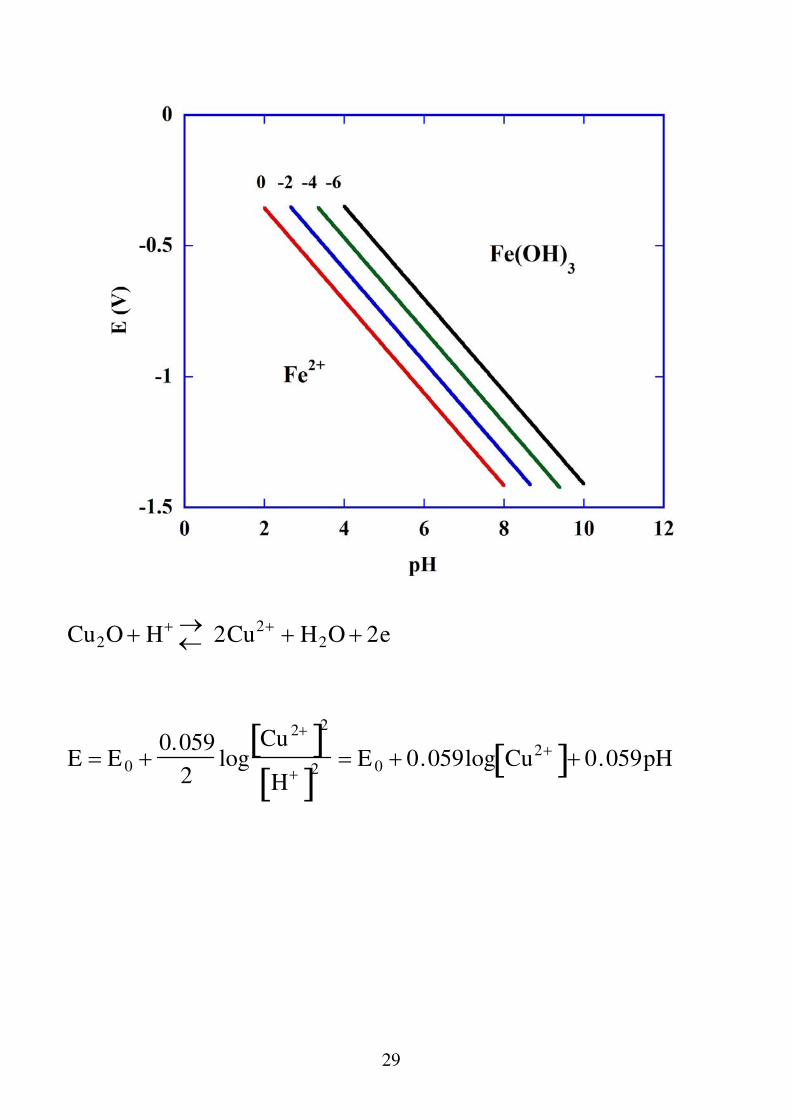
29
Cu2O H
2Cu2 H2O 2e
E E 0 0.059
2log
Cu 2 2
H 2 E 0 0.059log Cu2 0.059pH

30
Tutti i diagrammi tensione-corrente vengono completati dalle rette corrispondenti alle due reazioni catodiche:
2H 2e H2
E 0.059
2log
H 2P H2
0.059pH 0.295logP H2
È l'equazione di un fascio di rette parallele con pendenza -0.059 aventi come parametro discriminante la pressione parziale di H2 (normalmente si riporta in grafico PH2 = 1). Analogamente per la reazione catodica di riduzione dell'ossigeno:
O2 4H 4e 2H2O
E 1.23 0.059
4logP O2
H 4 1.23 0.059pH 0.015logP O2
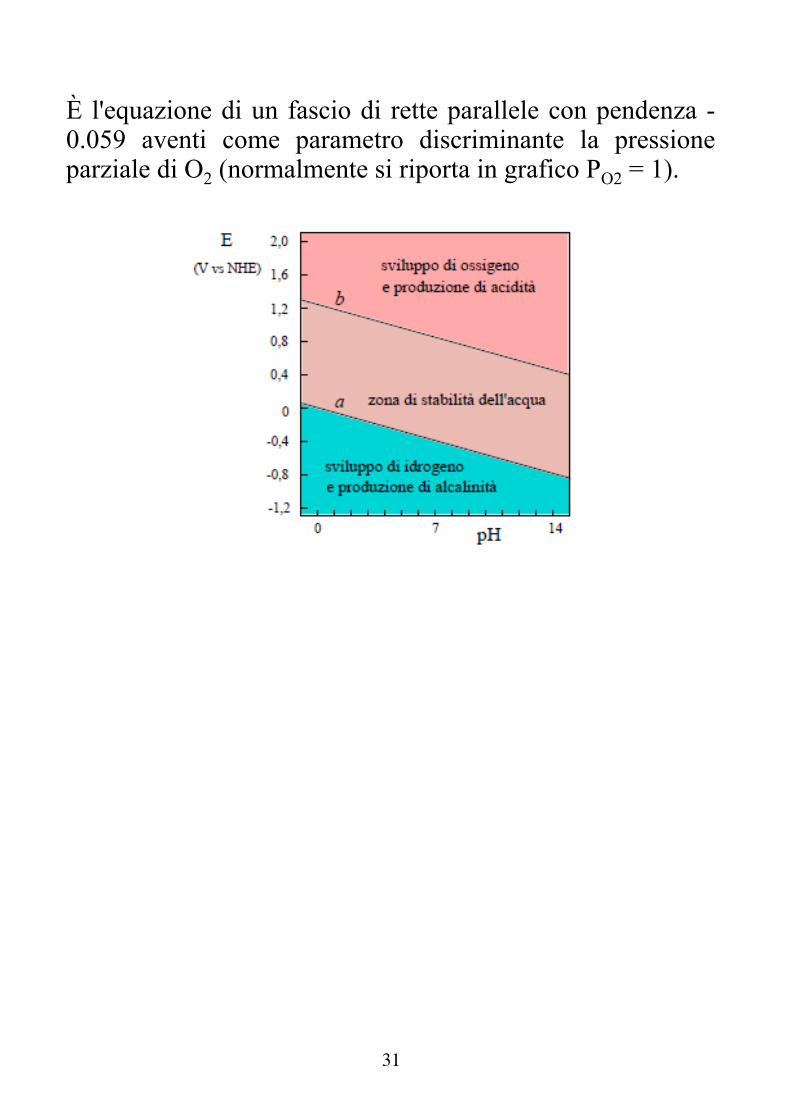
31
È l'equazione di un fascio di rette parallele con pendenza -0.059 aventi come parametro discriminante la pressione parziale di O2 (normalmente si riporta in grafico PO2 = 1).

32
Per la costruzione dei diagrammi E-pH bisogna tenere conto di tutti gli equilibri in soluzione e delle costanti (E, K) che li governano: Cd Cd2+ 2e
Cd OH 2 HCdO2
- H
Cd OH 2 Cd2+ 2OH
Cd + 2H 2O Cd OH 2 2e 2H
Cd + 2H 2O HCdO2
- 2e 3H
E
Cd/Cd20 0.403V
E Cd/Cd OH 2
0 0.005V K Cd2 OH 2 1.55 1014 E
Cd/HCdO2
0 0.583V
K HCdO2
H Cd OH 2
2.9 1020
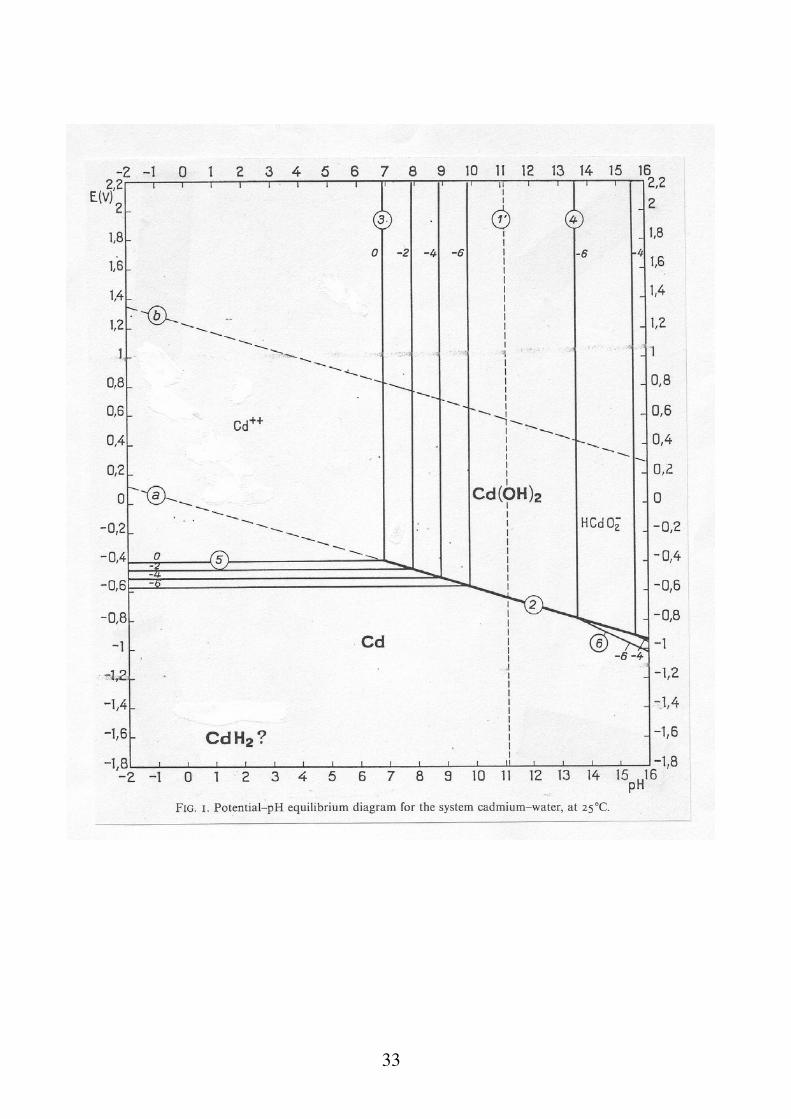
33
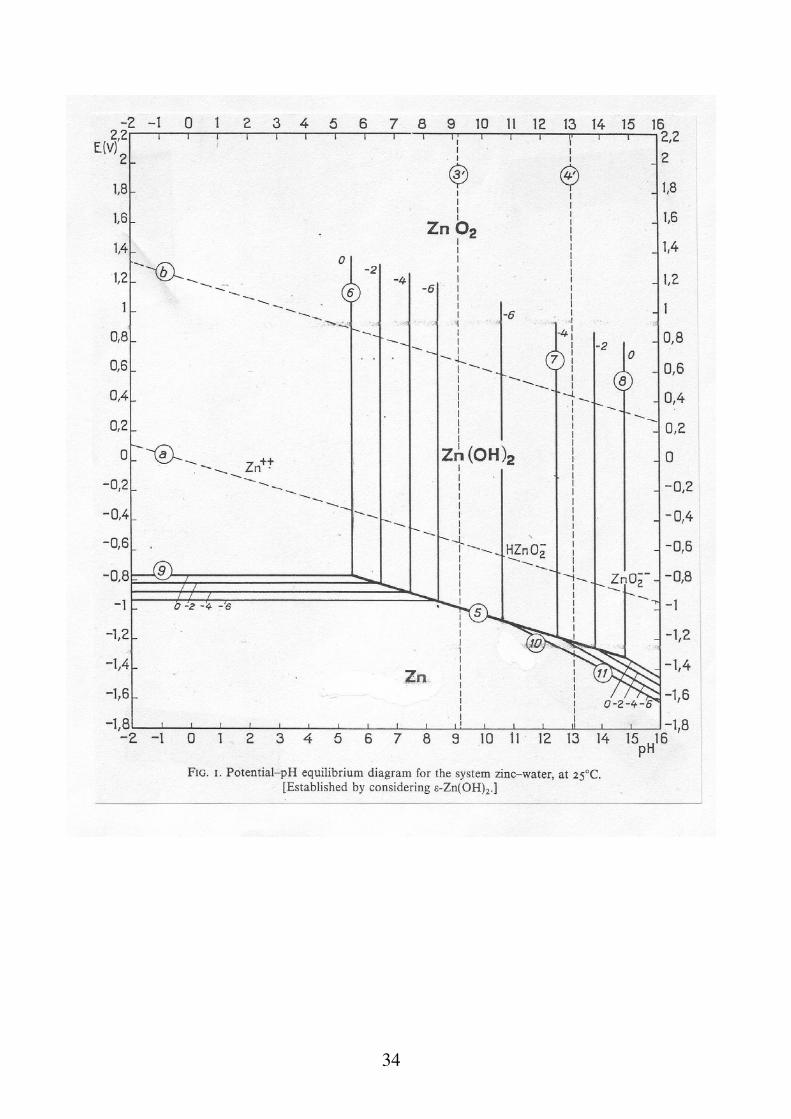
34
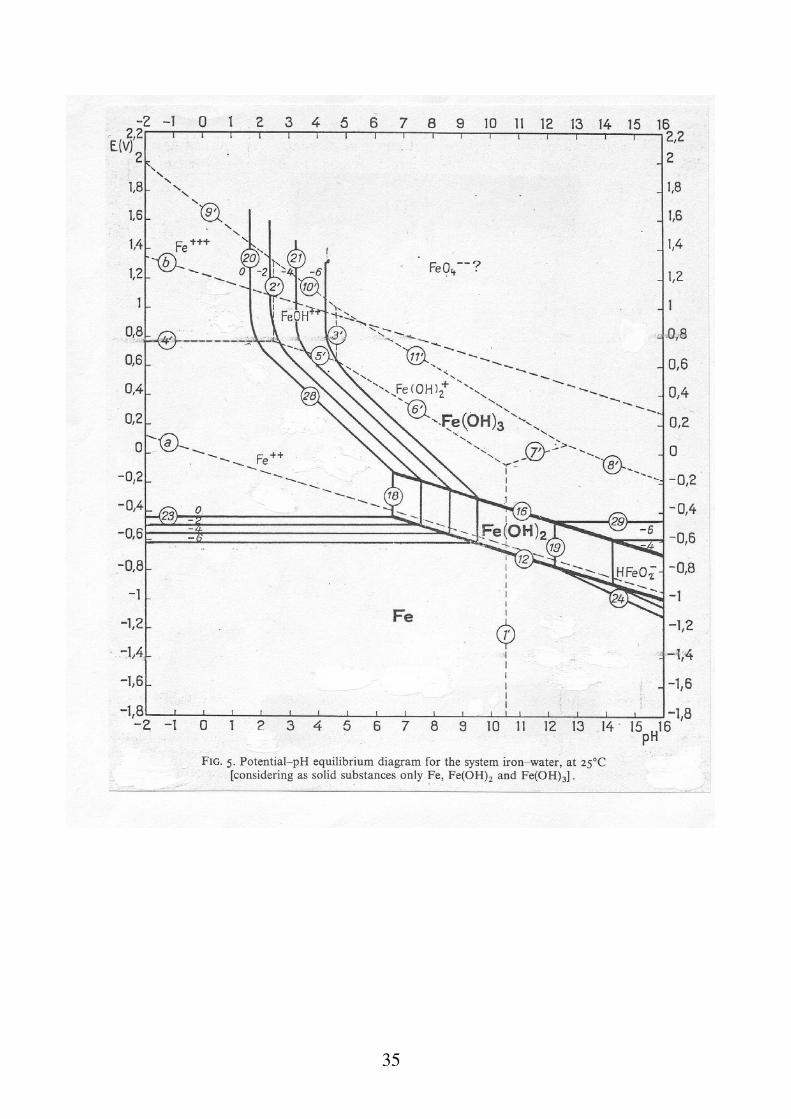
35
36

37
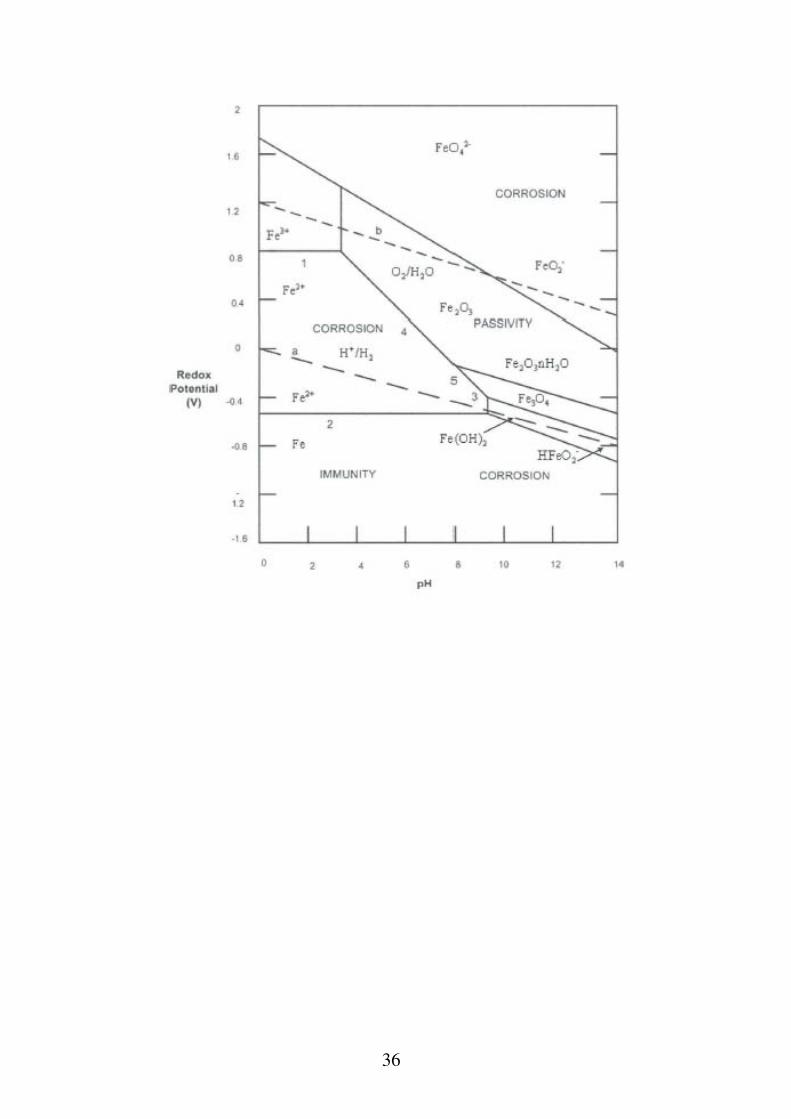
Interpretazione del diagramma E-pH del Ferro In assenza di O2: - pH < 9.5: corrosione, con formazione spontanea di ioni ferrosi; poiché il lavoro motore aumenta con il diminuire del pH è ragionevole supporre che la tendenza alla corrosione segua lo stesso andamento. - 9.5 < pH < 12.5: passivazione, per la formazione di idrossido ferroso.
- pH > 12.5: corrosione, con formazione di HFeO2
-
In presenza di O2: - pH < 4: corrosione, di intensità superiore di quella in assenza di ossigeno. - 4 < pH < 9: corrosione o passivazione: il diagramma non può chiarire questo dubbio. 9 < pH < 12.5: passivazione, per formazione di idrossido ferroso o ferrico. pH > 12.5: corrosione o passivazione
38
39
40

41
I diagrammi E-pH sono basati su considerazioni termodinamiche e quindi non tengono conto della cinetica delle reazioni di corrosioni (no Vc). Sono validi solo in ambiente acquoso contenente ioni provenienti dalla specie in esame. In presenza di particolari
ioni (Cl-, NH3/NH4
+ S
= CO3
=...) le conclusioni non sono più
valide.
Nel caso del ferro in presenza di Cl-, si può avere un attacco corrosivo per "pitting" non prevedibile con i diagrammi E-pH. Nel caso dell'Ag, in presenza di solfuri, la formazione di solfuro di Ag (molto poco solubile), abbassa la retta di inizio corrosione al di sotto della retta dell'idrogeno. Nel caso del Cu, in presenza di ammoniaca, la formazioni di ioni complessi molto stabili cambia completamente la forma del diagramma. Nel caso del Pb, in presenza di CO2, la formazione del carbonato di Pb insolubile, protegge la superficie del metallo dall'attacco corrosivo.
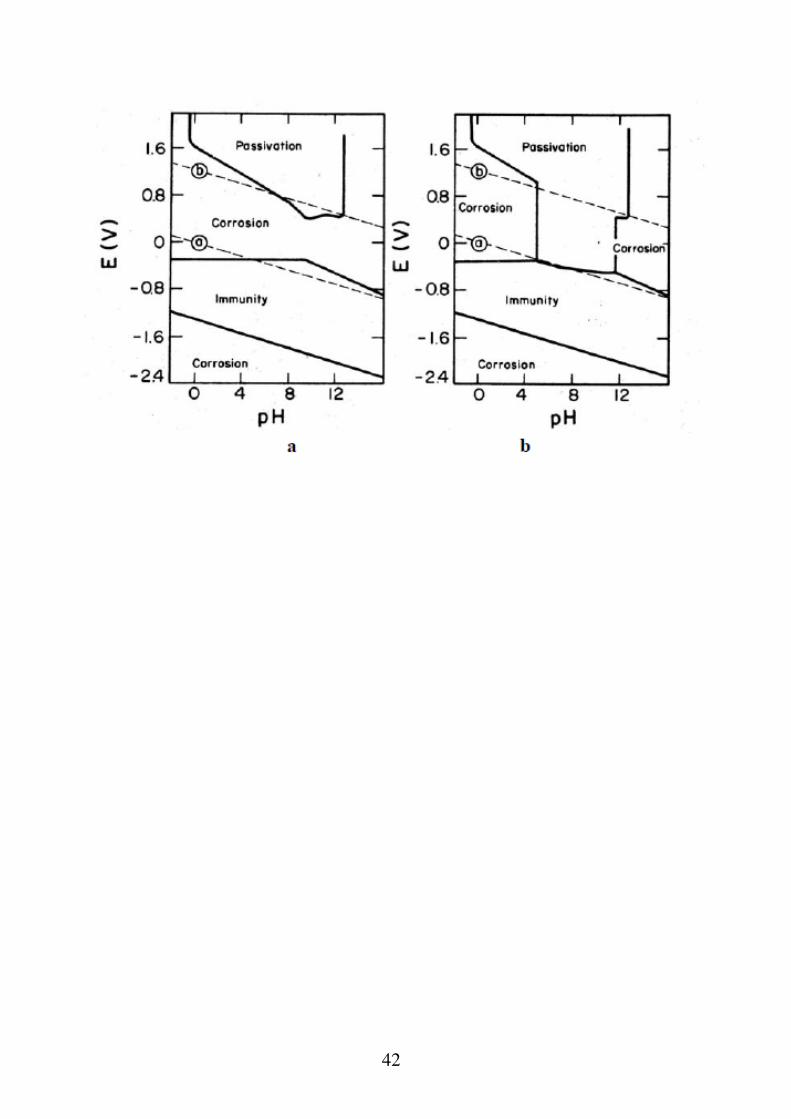
42

43
Cinetica dei processi corrosivi La termodinamica non è in grado di spiegare l'andamento di tutti i fenomeni corrosivi. La presenza di un lavoro motore positivo è una condizione necessaria, ma non sufficiente affinché la reazione di corrosione avvenga. Sono sempre presenti degli "attriti" che dissipano l'energia disponibile chiamati "sovratensioni". L'entità di questi attriti dipende dalla natura dei materiali e delle reazioni elettrodiche, dallo stato superficiale dei materiali e dalla presenza o meno di film superficiali o prodotti di corrosione. La termodinamica tratta di sistemi galvanici in condizioni di equilibrio, ovvero in assenza di passaggio di corrente. Durante il passaggio di corrente in un sistema galvanico, le tensioni possedute dagli elettrodi si spostano dai valori di partenza (di equilibrio) verso valori più positivi (per l'anodo) o verso valori più negativi (per il catodo).

44
Si può pensare di studiare la cinetica dei singoli processi elettrodici costruendo le curve E-I (diagrammi di Evans). In pratica si può operare con un potenziostato al quale sono collegati tre elettrodi: l'elettrodo di lavoro (W) del materiale in esame l'elettrodo di riferimento (R) per il potenziale il controelettrodo (C) per chiudere il circuito e misurare il passaggio di corrente
Il metodo consiste nell'impostare potenziali Ew crescenti in senso anodico ed in senso catodico, a partire dal potenziale di equilibrio e misurare il conseguente passaggio di corrente. La scansione deve essere fatta in modo sufficientemente lento, da permettere al sistema di andare a regime.

45
In un generico processo elettrochimico in cui abbia luogo una sola reazione
MMn ne
si possono distinguere diversi stadi attraverso i quali si deve passare per ottenere il prodotto finale. Ogni stadio sarà caratterizzato da un velocità e la velocità del processo più lento determinerà la velocità globale e quindi il tipo e l'entità delle sovratensioni. Si possono distinguere 3 tipi di sovratensioni: • Sovratensione di trasferimento di carica (o di
attivazione): è dovuta alle barriere energetiche per avere il trasferimento di carica attraverso il doppio strato elettrochimico.
• Sovratensione di diffusione: dovuta alla variazione di
concentrazione in vicinanza dell'elettrodo ed alla velocità con cui la specie che deve reagire si diffonde nella soluzione.
• Sovratensione di cristallizzazione: difficoltà
nell'inserimento (o nell'uscita) nel (dal) reticolo cristallino delle specie adsorbite.

46
Sovratensione di attivazione Nel processo
Van
Vc
M M ne
la reazione di trasferimento di carica nel senso anodico consiste nel passaggio degli ioni metallici dal reticolo cristallino (fase α) alla soluzione (fase β), attraverso il superamento della barriera dell'energia di attivazione Ea. Nel processo opposto di riduzione la specie ossidata reagisce con gli elettroni del metallo, superando la barriera energetica Ec. In condizioni di equilibrio, la velocità (e quindi le correnti anodica e catodica) delle due reazioni è uguale:
Ia k1CrideEa
RT Ic k 2CoxeEc
RT I0 dove k1 e k2 sono delle costanti e Crid e Cox sono le concentrazioni rispettivamente della specie ridotta ed ossidata e I0 è detta densità di corrente di scambio.

47
Nel caso sia presente una sovratensione (η) ovvero nel caso in cui la tensione di un elettrodo si discosti dalle condizioni di equilibrio, la corrente che viene prodotta è data dall’equazione di Butler-Volmer:
aa
a
1 nFnF
RT RTc 0I I I I e e
In questo caso si è immaginato una sovratensione anodica
(che renderà il potenziale dell’elettrodo più positivo e
favorirà la reazione di ossidazione), ovvero che porti allo
sviluppo di una corrente anodica.
In modo analogo per una sovratensione catodica:
c cnF 1 nF
RT RTc a 0I I I I e e
dove β = 1 - α η è la sovratensione di attivazione
E E eq
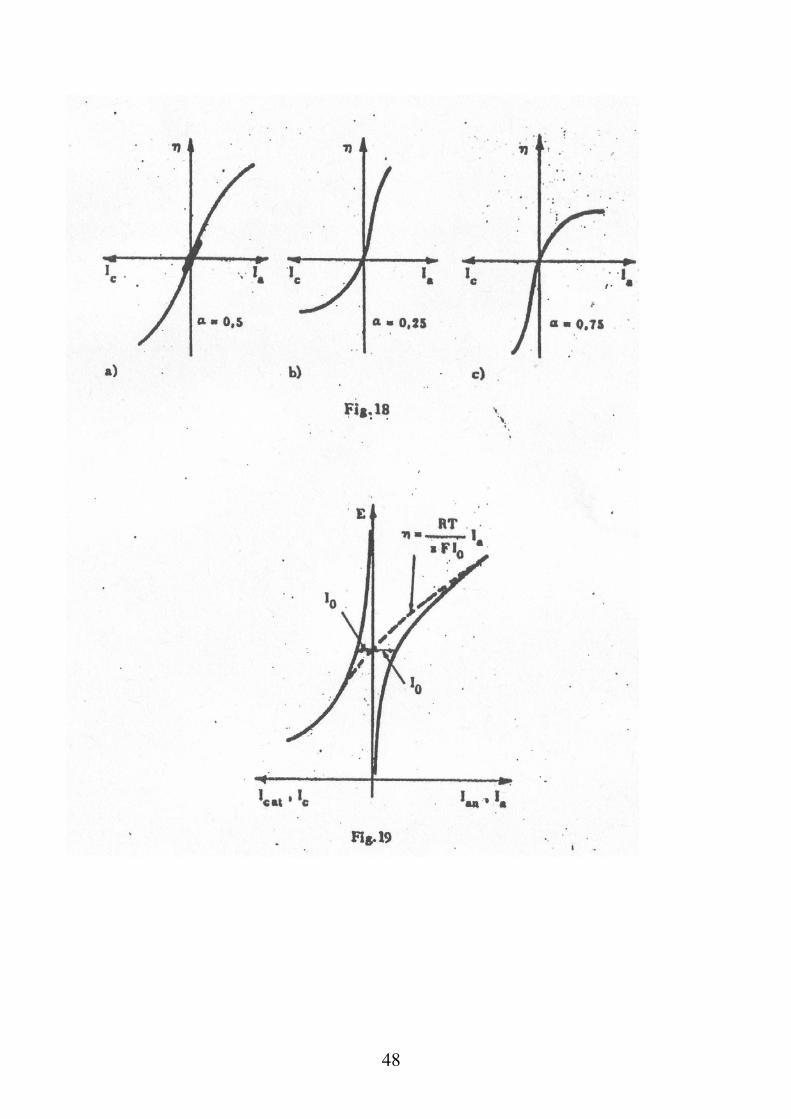
48

49
In genere è considerata in valore assoluto, altrimenti: η > 0 se E > Eeq sovratensione anodica η < 0 se E < Eeq sovratensione catodica Per η > 30 mV in modo che valga la relazione RT
nF
anF
RTc a 0I 0; I I e
da cui:
a 0 a
RT RTln I ln I
nF nF

50
Analogamente
cnF
RTa c 0I 0; I I e
c 0 c
RT RTln I ln I
nF nF
Generalizzando le due equazioni si ottiene l'equazione di Tafel:
A Bln I
blogII0
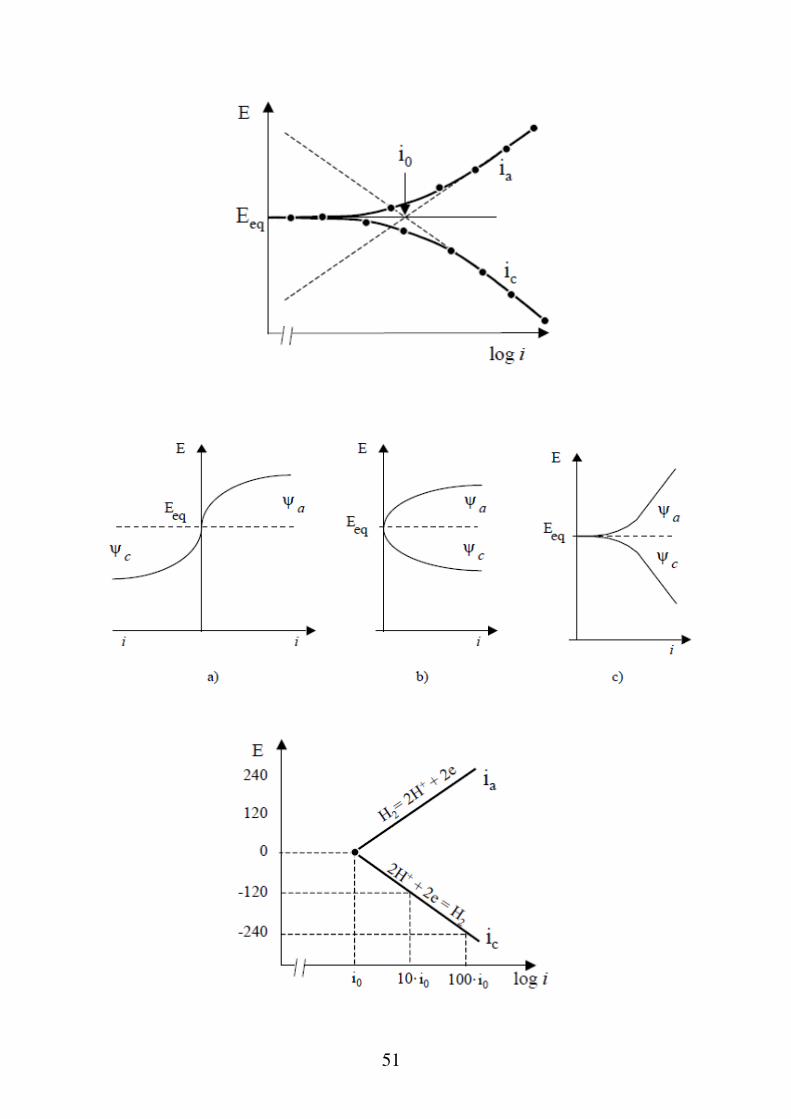
51

52
Sovratensione di diffusione Se durante un processo elettrodico lo stadio lento è la migrazione degli ioni verso l'elettrodo (o viceversa), si instaura la sovratensione di diffusione. Si può supporre che la densità di corrente che corrisponde alla sovratensione di attivazione η sia piccola rispetto alla densità di corrente di scambio (I0) la sovratensione di attivazione sia trascurabile. Se tutte le reazioni del meccanismo di reazione elettrodico sono veloci rispetto alla diffusione delle specie reagenti, la tensione dell'elettrodo, ad ogni istante, potrà essere calcolata dell'equazione di Nernst nella quale, le concentrazioni della specie ossidata e ridotta variano nel tempo. La sovratensione di diffusione è quindi data dalla differenza tra la tensione Eeq, in assenza di passaggio di corrente, ed EI la tensione generica per la quale si ha un passaggio di corrente I.

53
Poste uguale a Cox0 e Crid
0 le concentrazioni della specie ossidata e ridotta in assenza di passaggio di corrente, e Cox Crid le concentrazioni della specie ossidata e ridotta nel momento in cui si ha passaggio di corrente I, si può scrivere:
0ox rid
I 0rid ox
RT C CE E ln
nF C C
Nel caso della reazione: M
Mn ne , il movimento degli
ioni all'interno della soluzione può avvenire sia per diffusione (in relazione al gradiente di concentrazione) sia per migrazione (l'elettrodo tende a caricarsi negativamente).
Sia δ lo spessore dello strato diffusivo della specie Mn+
nella soluzione. Tale spessore dipende dall'agitazione della soluzione, dalla viscosità del mezzo e dalla temperatura (~ 100 nm in soluzioni stagnanti, 10 nm in soluzioni agitate).

54
Vale la legge di Fick:
dMdt
SD
CMn0 C
M n M =moli di ioni che diffondono S = superficie elettrodo D = coefficiente di diffusione
La densità di corrente in condizioni di in cui non ci sia un apporto sufficientemente elevato della specie Mn+ sulla superficie dell’elettrodo, viene stimata dalla legge di Faraday, imponendo la condizione della diffusione tramite la legge di Fick (e tenendo conto della migrazione)
n n
0
M M
nFDI C C
t '
1

55
Il valore della corrente è massimo quando CMn = 0, ovvero
quando la reazione di trasferimento di carica è molto più veloce del movimento degli ioni in soluzione. Ponendo questa condizione, e trascurando la migrazione si ottiene la corrente limite:
n
0L M
nFDI C
2
Esprimendo il risultato ottenuto in termine di sovratensione:
RTnF
lnCox
Crid
Crid
0
C ox0
RTnF
lnC
Mn
CMn0
ricavando i due valori delle concentrazioni dalle equazioni 1 e 2 e sostituendo, si trova l'espressione della sovratensione di diffusione:
RTnF
lnI L I
IL
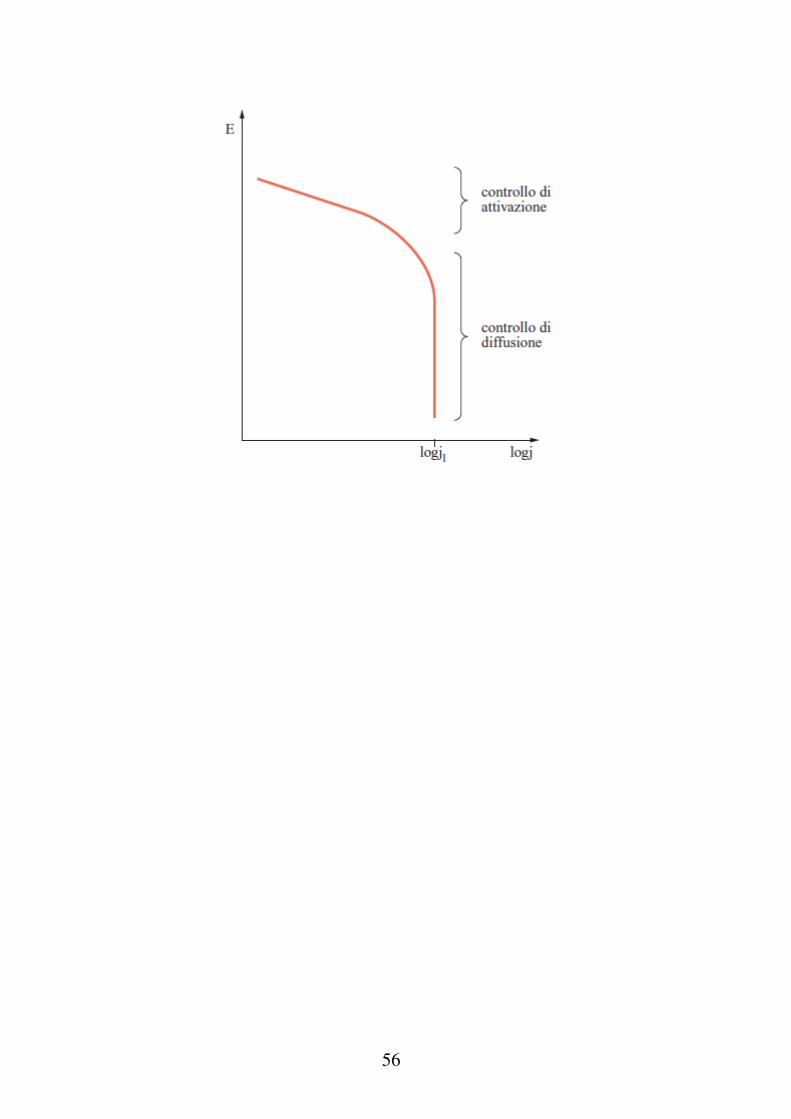
56

57
Caduta ohmica Per consentire il passaggio di una determinata quantità di corrente attraverso un elettrodo può accadere di dover fornire più energia per vincere le cadute ohmiche che si instaurano all'interfaccia elettrodo-elettrolita. Tali cadute ohmiche sono in genere dovute alla scarsa conducibilità della soluzione o alla formazione di film (ossidi, idrossidi, cloruri ecc.) sulla superficie del metallo. Se lo strato superficiale è caratterizzato da una resistività r, da uno spessore L e da una superficie S, lo spessore del film è dato da:
LR
S
In conseguenza al passaggio di una densità di corrente I si ha una polarizzazione ohmica pari a:
ISR I L Ammettendo che L e R non varino con I, la sovratensione ohmica è direttamente proporzionale alla densità di corrente.

58
Passività e caratteristica anodica dei materiali a comportamento attivo-passivo Per molti materiali metallici la resistenza alla corrosione è strettamente dipendente dalle condizioni superficiali. In determinate condizioni si osserva la formazione di film di ossidi (o più genericamente di precipitati) molto stabili, compatti e protettivi. Al, Fe + inox, Cr, Ti, Zr, Ta ... hanno questo tipo di comportamento passività. La passivazione completa di un metallo porta in pratica all'annullamento della velocità di corrosione. Nei casi in cui il film che si forma non è compatto ed uniforme (presenza di difetti e di pori) la reazione continua all'interno dei pori e dei difetti (corrosione localizzata pitting).

59
I parametri che influenzano la formazione dello strato passivo sono molti: - pH ( diagrammi E - pH) - Potere ossidante dell'ambiente (ossigeno, idrogeno ...) - Temperatura In molti casi, per un corretto funzionamento di un'apparecchiatura, è indispensabile effettuare una "messa in opera" del materiale in cui si lo si fa passivare nelle condizioni migliori, prima di poterlo usare normalmente.
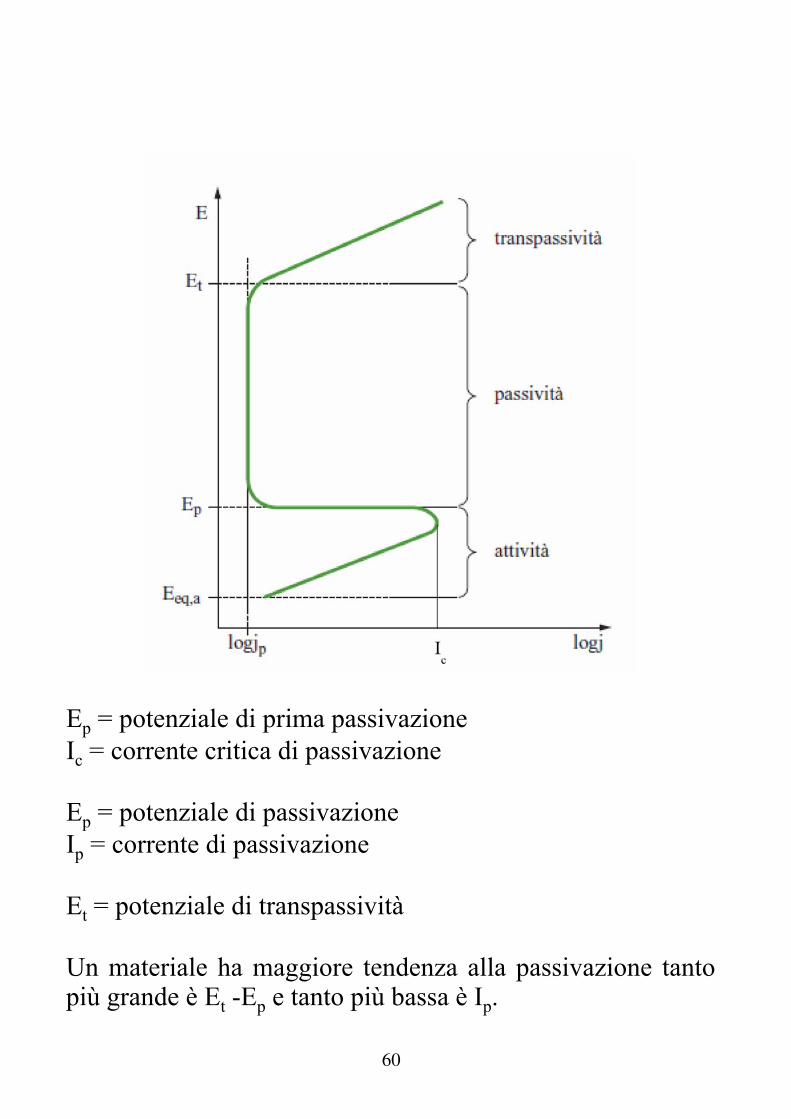
60
Ep = potenziale di prima passivazione Ic = corrente critica di passivazione Ep = potenziale di passivazione Ip = corrente di passivazione Et = potenziale di transpassività Un materiale ha maggiore tendenza alla passivazione tanto più grande è Et -Ep e tanto più bassa è Ip.

61
Le curve I-E possono essere determinate sperimentalmente. È possibile operare in 2 modi: amperostatico e potenziostatico. I metodi sono formalmente equivalenti, ma presentano delle differenze essenziali. - metodo amperostatico: si impongono valori crescenti di corrente, rilevando di volta in volta il potenziale a cui si viene a trovare l'elettrodo. Crea problemi con i metalli a comportamento attivo passivo. - metodo galvanostatico

62
si impongono valori crescenti di potenziale, rilevando di volta in volta la corrente che passa. È di gran lunga il metodo migliore per la determinazione delle curve di polarizzazione (I-V). Si utilizza un potenziostato che garantisce massima precisione (fattore di amplificazione 1 A/mV) e massima velocità nel controllo della tensione dell'elettrodo (~msec).
63

64
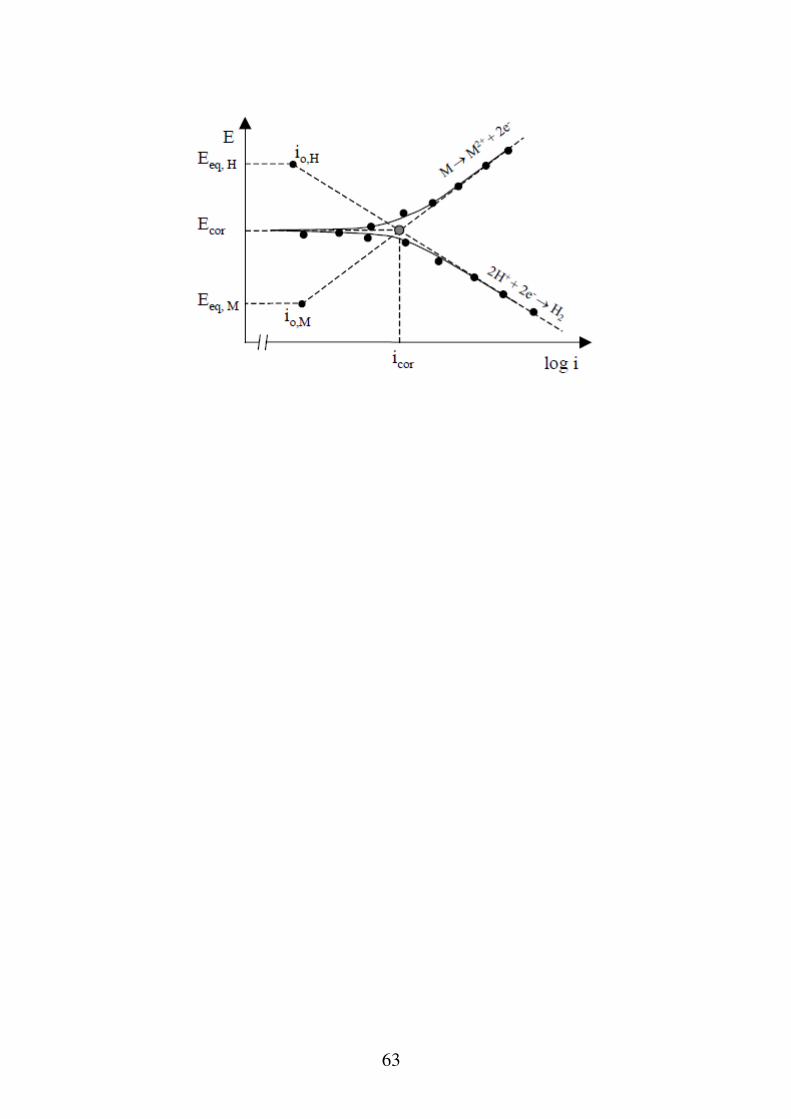
Funzionamento dei sistemi di corrosione Nel caso in cui sullo stesso elettrodo possano svolgersi due (o più) reazioni (come nel caso dei processi corrosivi spontanei, il potenziale elettrodico sarà diverso dai due (o più) potenziali di equilibrio delle singole reazioni. Tale potenziale non è più una tensione di equilibrio (c'è passaggio di corrente) e viene chiamata potenziale misto o di corrosione. L'elettrodo sarà sede, nello stesso tempo di una reazione anodica ed una catodica a cui competono le correnti Ian e Icat uguali in valore assoluto ma di segno contrario. L'ideale chiusura del circuito provoca la dissipazione del lavoro motore che a sua volta determina: - lo sviluppo dei fenomeni di sovratensione provocando la snobilitazione (abbassamento) della tensione catodica e la nobilitazione (innalzamento) della tensione anodica. - le reazioni elettrochimiche provocheranno una modifica delle concentrazioni delle specie nelle vicinanze dell'elettrodo e instaureranno una sovratensione di diffusione.
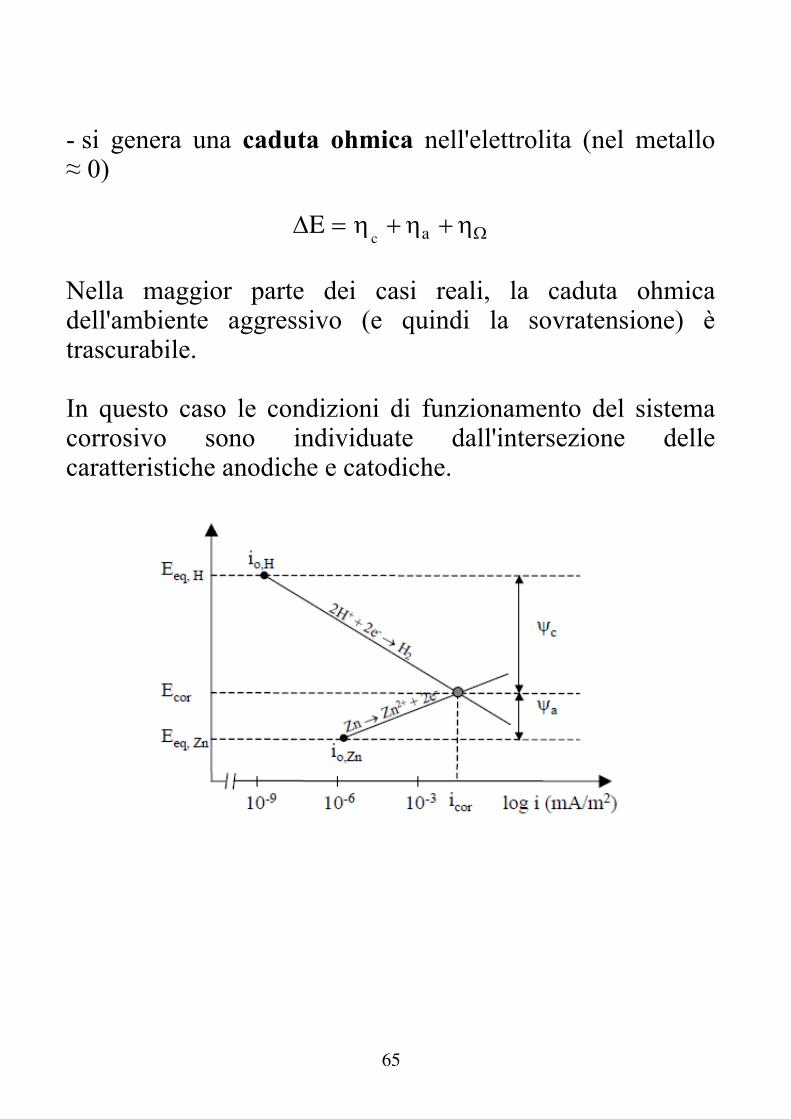
65
- si genera una caduta ohmica nell'elettrolita (nel metallo ≈ 0)
E c a
Nella maggior parte dei casi reali, la caduta ohmica dell'ambiente aggressivo (e quindi la sovratensione) è trascurabile. In questo caso le condizioni di funzionamento del sistema corrosivo sono individuate dall'intersezione delle caratteristiche anodiche e catodiche.

66
Teoria delle “coppie locali”
Ci sono molte evidenze sperimentali che in condizioni di disuniformità nel materiale metallico, le regioni anodiche e catodiche sono separate su scala macroscopica o microscopica e quindi sono distinguibili direttamente o con l'aiuto di un microscopio. In questi casi il meccanismo elettrochimico del fenomeno è direttamente accertabile e trova quindi giustificazione la teoria detta delle “coppie locali”. il meccanismo dei processi di corrosione è
elettrochimico le diverse aree del materiale metallico assumono un
funzionamento anodico o catodico i sistemi di corrosione sono quindi costituiti da “coppie
locali” in corto circuito

67
La teoria dei “potenziali misti”
Esistono però fenomeni corrosivi che si sviluppano anche in totale assenza di eterogeneità sia nell'ambiente che nel materiale metallico (amalgame a contatto con soluzioni omogenee). In questi casi non è possibile immaginare la presenza di coppie locali in senso stretto e quindi ritenerne valida la relativa teoria. Fu allora ipotizzata la possibilità che alla superficie di materiali metallici omogenei a contatto con ambienti pure omogenei si possano svolgere simultaneamente due o anche più processi elettrodici, alcuni dei quali in un senso ed altri in senso opposto e su tale ipotesi basarono la teoria dei fenomeni di corrosione detta “dei potenziali misti”: i fenomeni corrosivi si producono con meccanismo
elettrochimico e procedono in forma di processi parziali anodici e catodici sull'intera superficie metallica
la velocità di ogni processo parziale anodico o catodico
non dipende da quella degli altri processi ma solo dal potenziale
la somma delle velocità dei processi anodici uguaglia la
somma delle velocità dei processi catodici

68
In seguito la teoria fu generalizzata per tenere conto della presenza di disuniformità del materiale metallico e nell'ambiente. In questi casi il comportamento elettrodico delle diverse aree del materiale metallico risulta prevalentemente anodico o catodico mentre la teoria delle coppie locali assume per le diverse aree del materiale metallico, un funzionamento esclusivamente anodico o catodico. La teoria delle coppie locali può quindi essere vista come un caso particolare della teoria più generale dei potenziali misti.
69
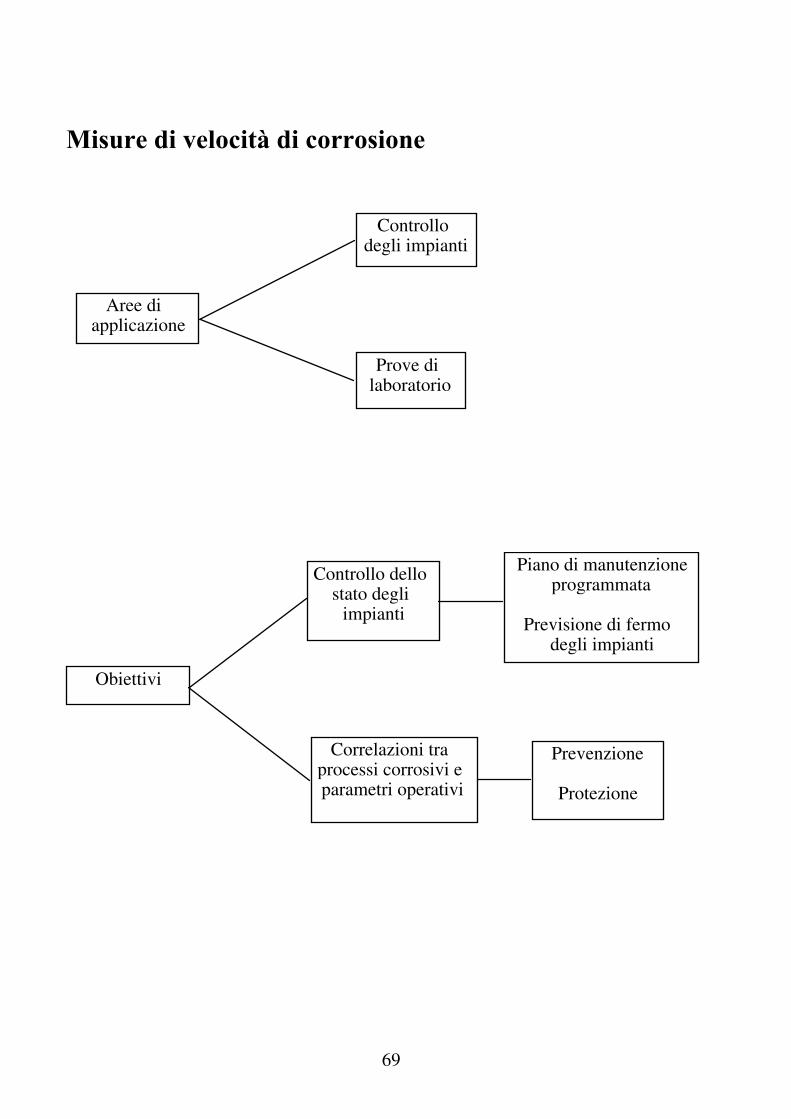
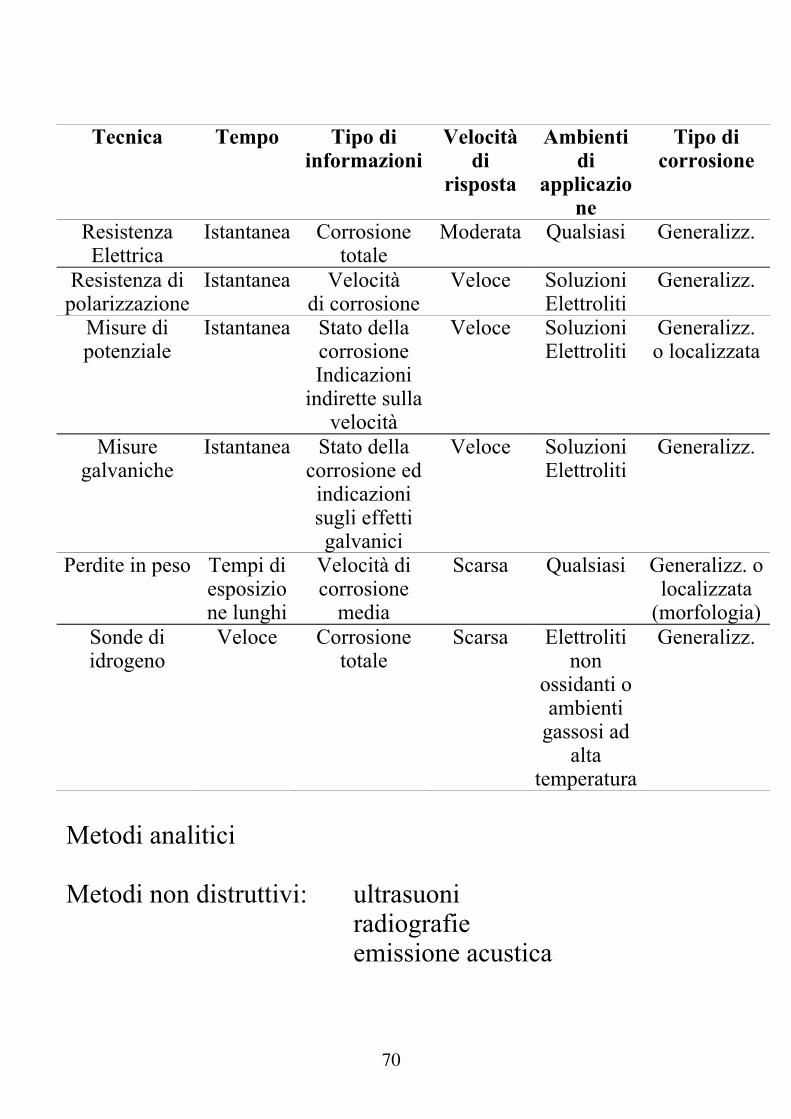
Misure di velocità di corrosione
Prove di laboratorio
Controllo degli impianti
Prevenzione
Protezione
Aree di applicazione
Obiettivi
Controllo dello stato degli
impianti
Piano di manutenzione programmata
Previsione di fermo
degli impianti
Correlazioni tra processi corrosivi e parametri operativi
70
Tecnica Tempo Tipo di
informazioniVelocità
di risposta
Ambienti di
applicazione
Tipo di corrosione
Resistenza Elettrica
Istantanea Corrosione totale
Moderata Qualsiasi Generalizz.
Resistenza di polarizzazione
Istantanea Velocità di corrosione
Veloce Soluzioni Elettroliti
Generalizz.
Misure di potenziale
Istantanea Stato della corrosione Indicazioni
indirette sulla velocità
Veloce Soluzioni Elettroliti
Generalizz. o localizzata
Misure galvaniche
Istantanea Stato della corrosione ed indicazioni sugli effetti galvanici
Veloce Soluzioni Elettroliti
Generalizz.
Perdite in peso Tempi di esposizione lunghi
Velocità di corrosione
media
Scarsa Qualsiasi Generalizz. o localizzata
(morfologia)Sonde di idrogeno
Veloce Corrosione totale
Scarsa Elettroliti non
ossidanti o ambienti
gassosi ad alta
temperatura
Generalizz.
Metodi analitici Metodi non distruttivi: ultrasuoni radiografie emissione acustica

71
Equazione di Stern-Geary (Resistenza di Polarizzazione)
La costruzione delle curve di polarizzazione e l'estrapolazione delle rette di Tafel permette la determinazione della velocità di corrosione con buona precisione. Il metodo non è però pratico, specie quando vi sono più processo anodici e/o catodici concomitanti. Inoltre il grande campo di polarizzazione che si deve impostare può alterare le reazioni di corrosione. Il metodo elettrochimico di determinazione della velocità di corrosione di gran lunga più utilizzato è il metodo di Stern-Geary detto anche della resistenza di polarizzazione. Nel caso in cui le reazioni anodica e catodica siano:
MMn ne
2H 2eH2 ,
in relazione ad una polarizzazione ΔE si avrà il passaggio di una corrente netta pari a: I est I c Ia .
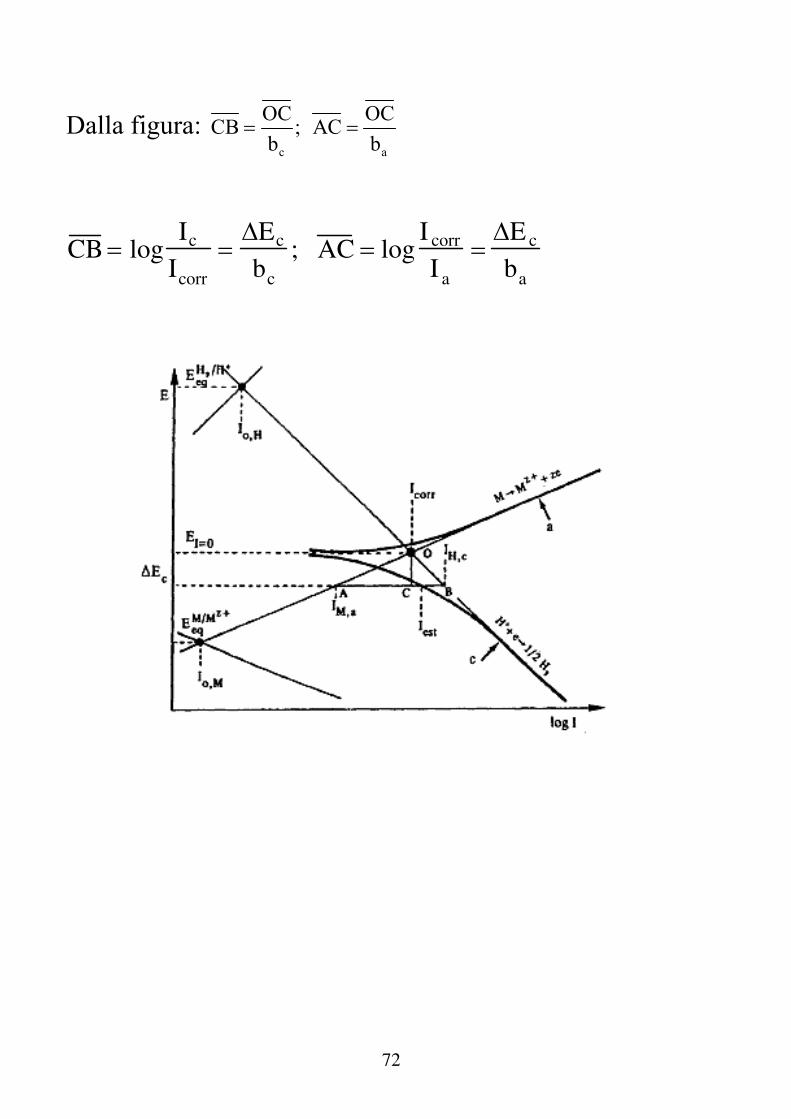
72
Dalla figura: c a
OC OCCB ; AC
b b
CB logIc
Icorr
Ec
bc
; AC logI corr
I a
E c
ba

73
I c Icorr 10Ec bc ; Ia Icorr 10Ec ba Sostituendo nell'espressione della Iest: I est I c Ia I corr 10Ec bc 10Ec ba Nell'ipotesi che ΔEc sia piccolo (nel tratto lineare della curva I Vs. E) e quindi trascurabile rispetto a ba e bc, nello sviluppo in serie ci si può fermare al secondo termine:
c c
2
E b c c
c c
E E10 1 ln10 ln10 ....
b 2b
da cui:
I est I corr 1 2.3E c
bc
1 2.3Ec
ba

74
I corr 1
2.3babc
ba bc
Iest
Ec
1
2.3ba bc
ba b c
1
R p
che permette di definire la Resistenza di Polarizzazione:
R p E c
Iest
In pratica il campo di applicazione di questa equazione è per polarizzazioni di 5 ÷ 20 mV. Il valore di norma più usato è 10 mV.
75
- -
76
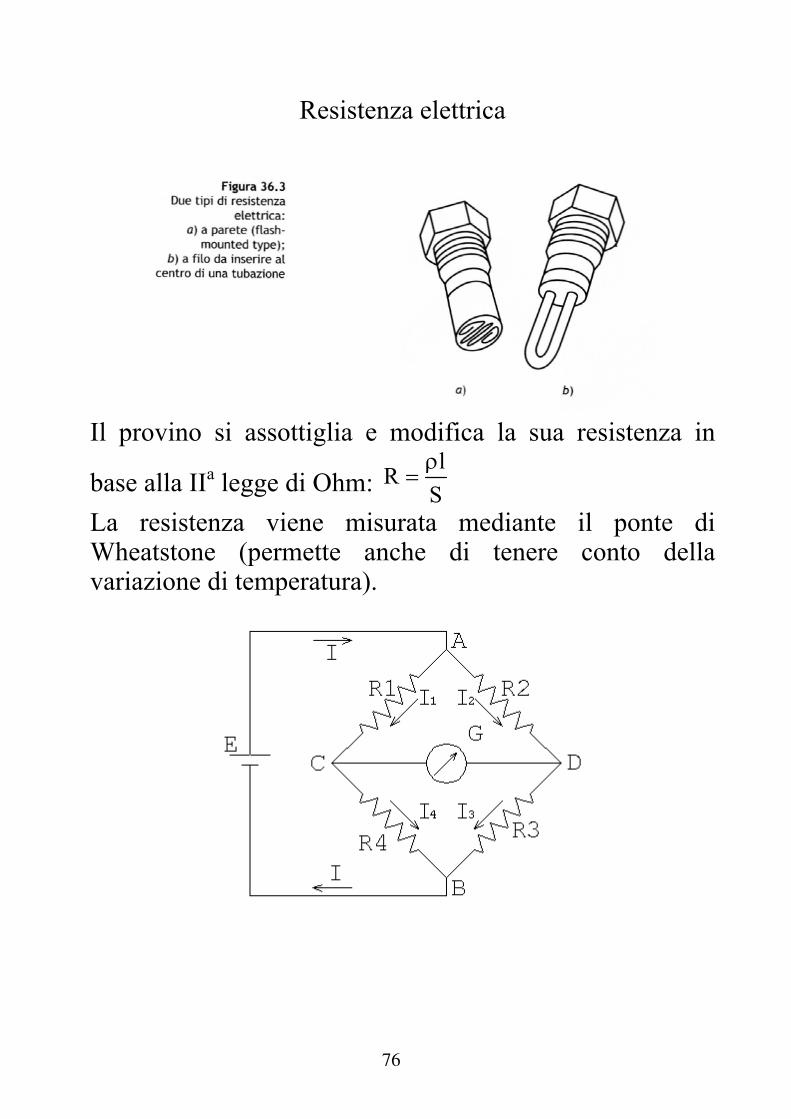
Resistenza elettrica
Il provino si assottiglia e modifica la sua resistenza in
base alla IIa legge di Ohm: l
RS
La resistenza viene misurata mediante il ponte di Wheatstone (permette anche di tenere conto della variazione di temperatura).
77
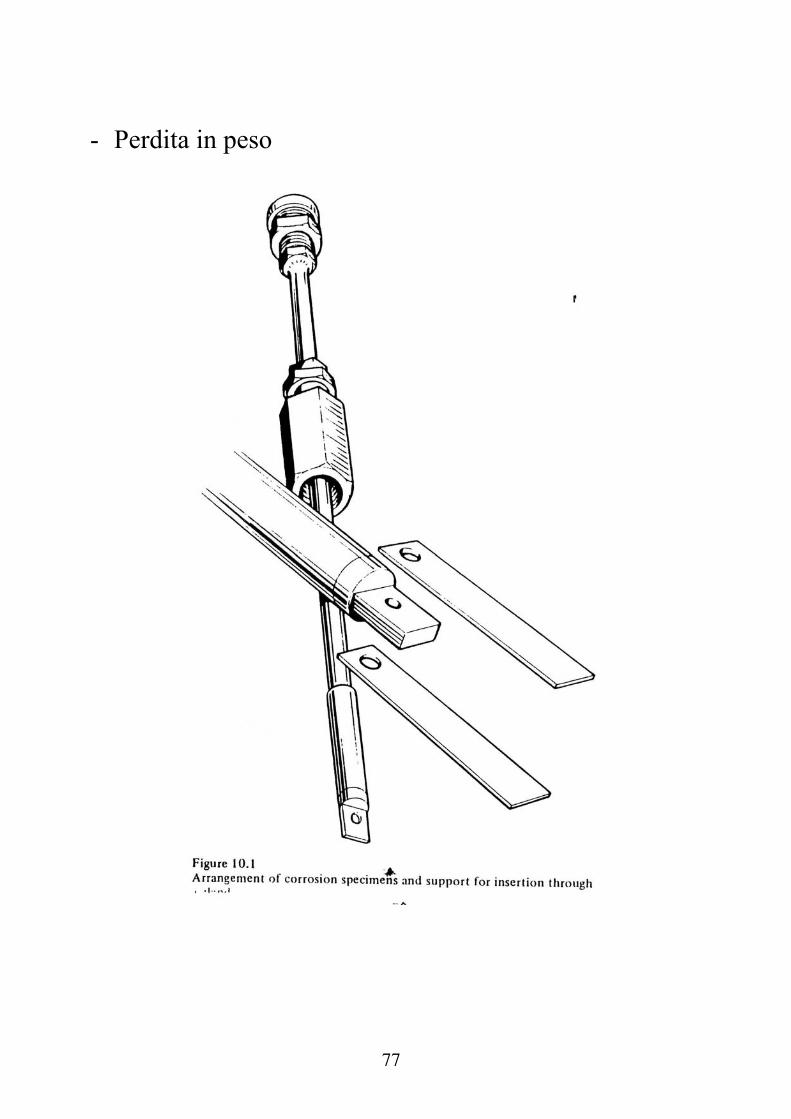
- Perdita in peso
78
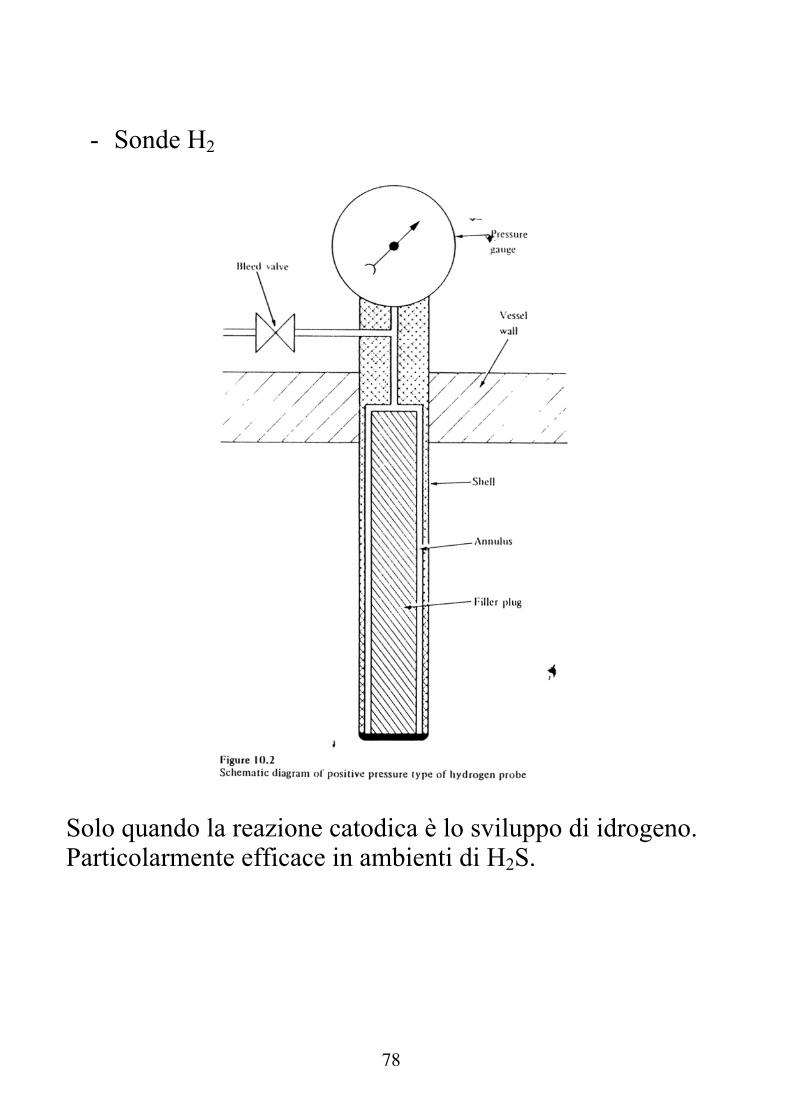
- Sonde H2
Solo quando la reazione catodica è lo sviluppo di idrogeno. Particolarmente efficace in ambienti di H2S.

79
Fattori di corrosione • Fattori di corrosione relativi al metallo: natura del metallo composizione chimica superficiale alligazione microstruttura sollecitazioni meccaniche • Fattori di corrosione relativi all'ambiente: pH temperatura moto del fluido composizione chimica potere ossidante

80
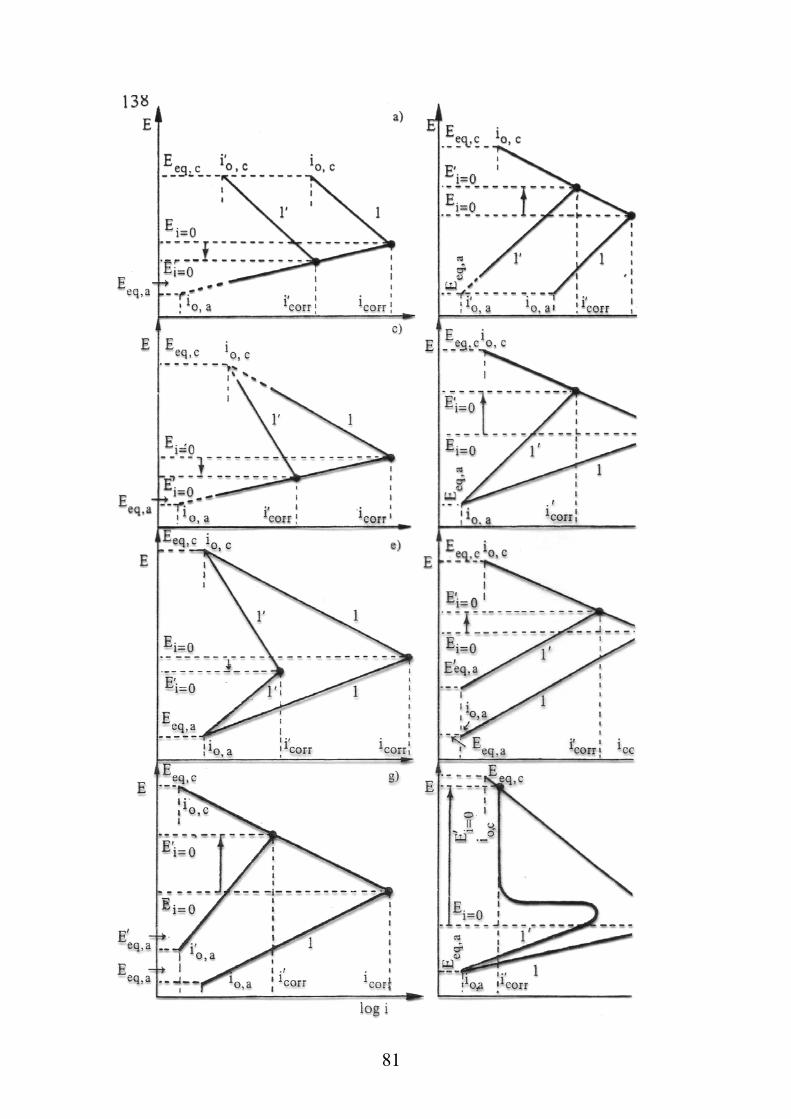
Fattori di corrosione relativi al metallo
81
82
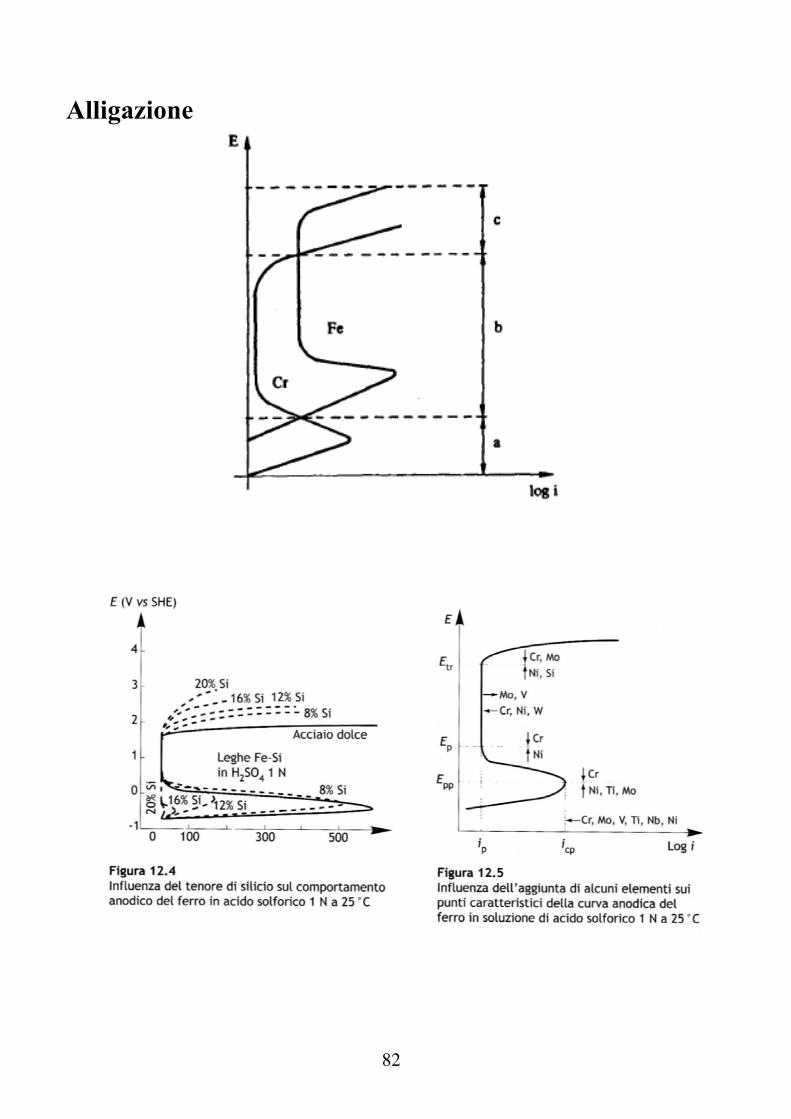
Alligazione
83
Fattori di corrosione relativi all'ambiente
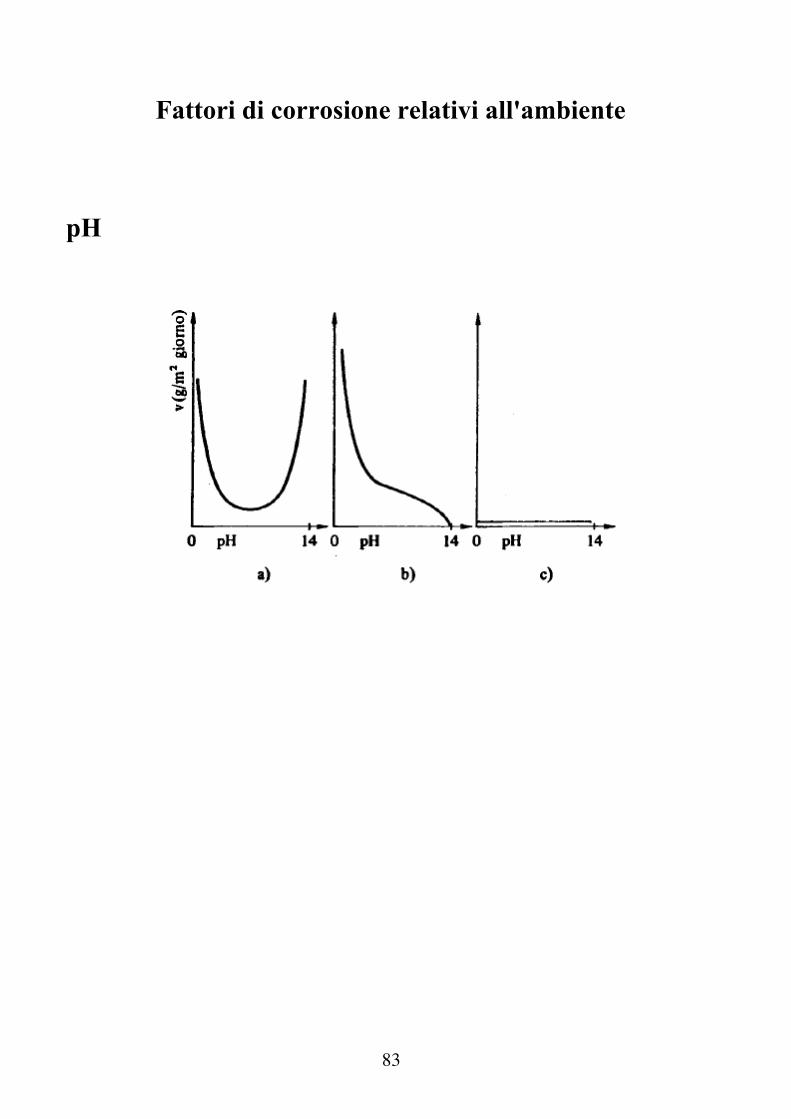
pH
84
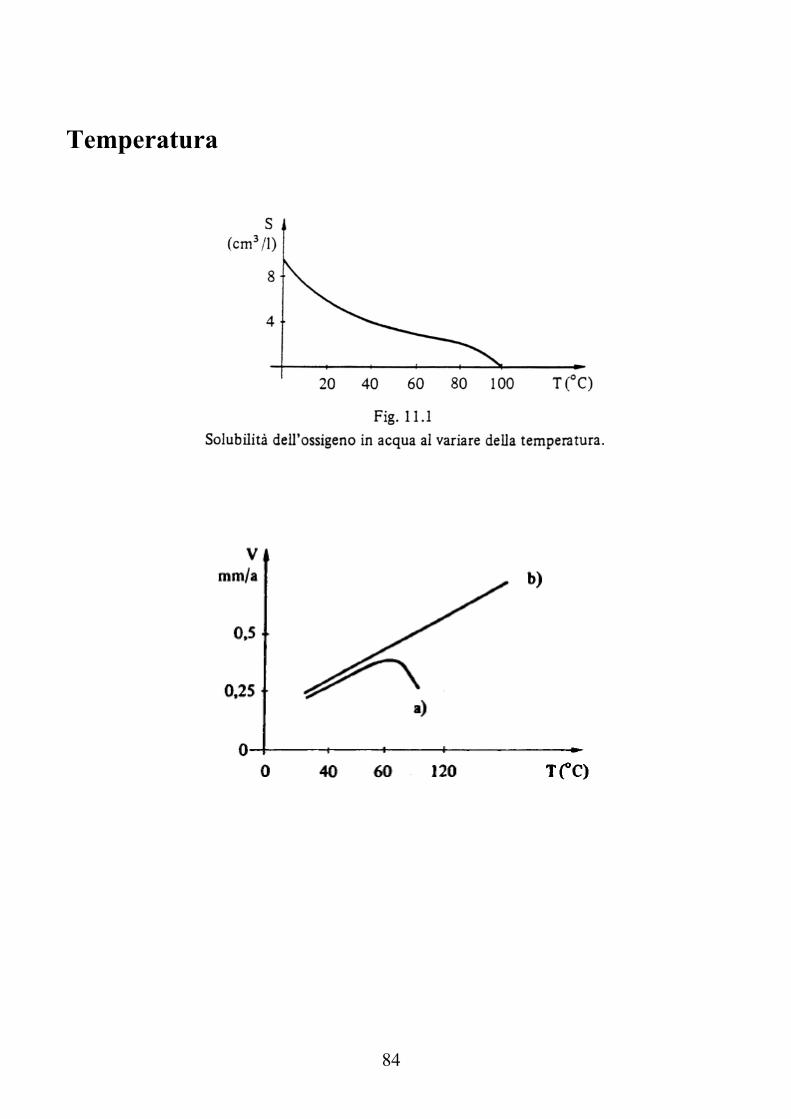
Temperatura
85
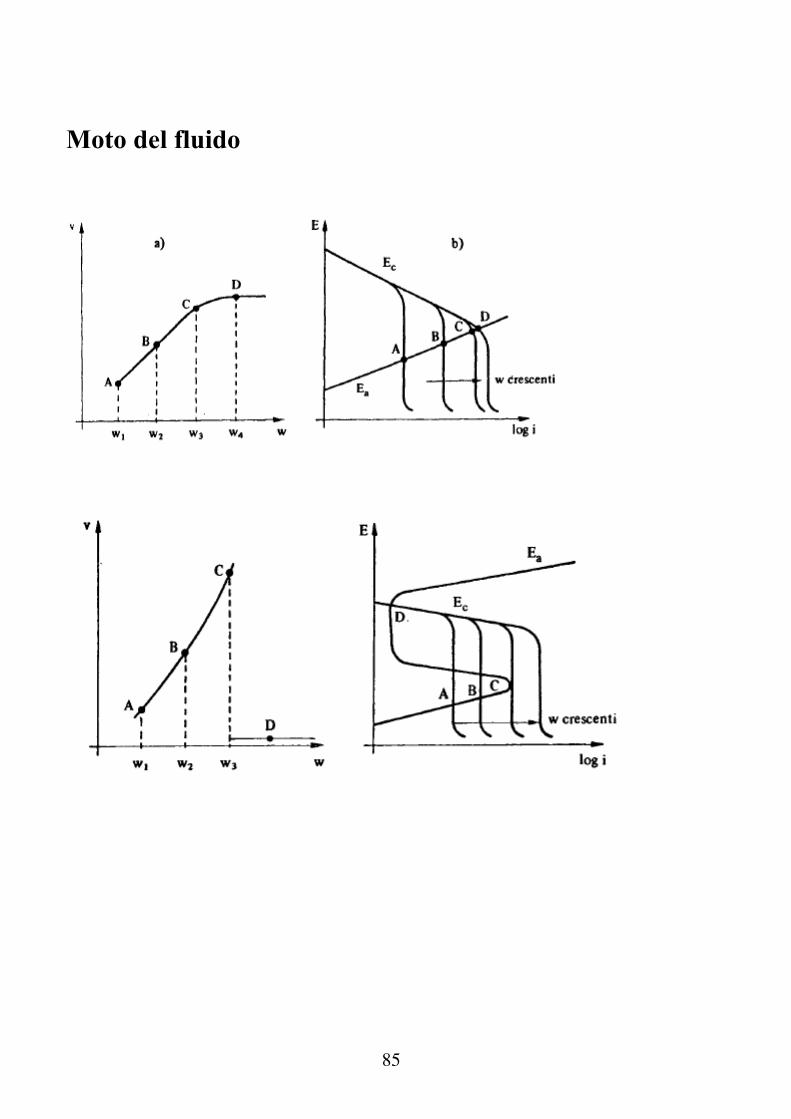
Moto del fluido
86
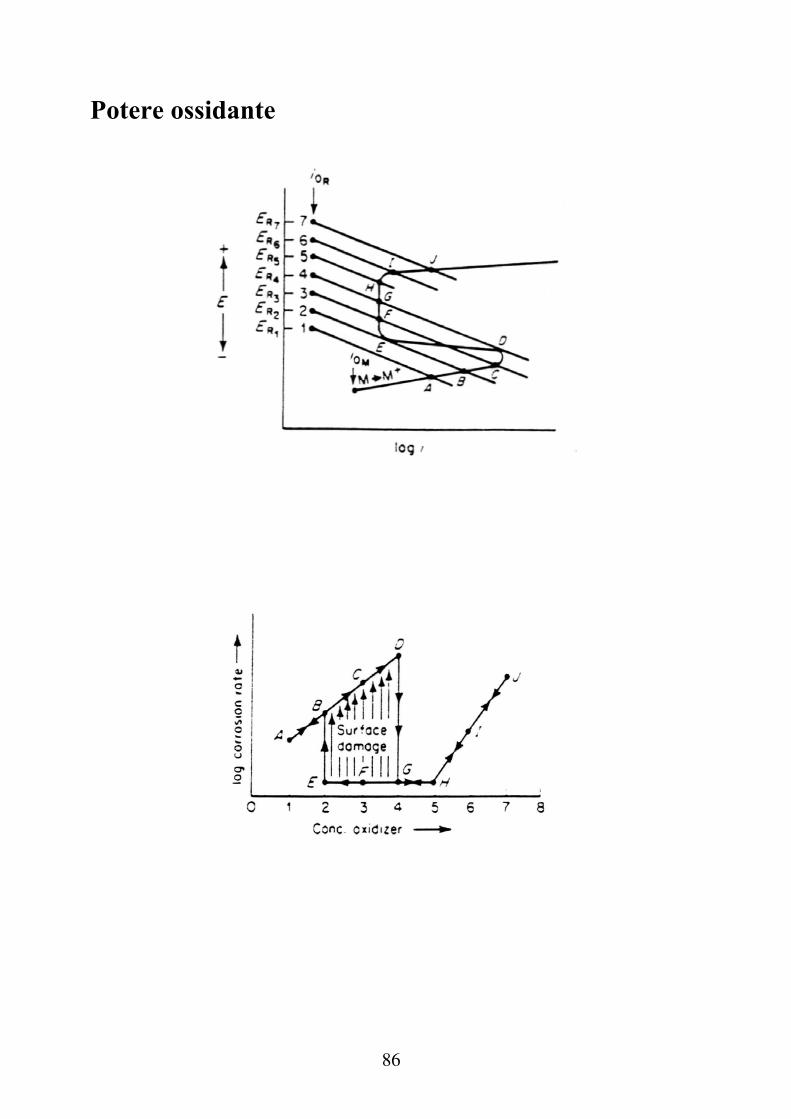
Potere ossidante


















