Chimica Elettroanalitica Avanzata Modulo A - users.unimi.itusers.unimi.it/ECEA/Spettoelettrochimica...
35
1 Spettroelettrochimica Prof. Prof. Patrizia R. Patrizia R. Mussini Mussini Dipartimento di Chimica Fisica ed Elettrochimica Via Golgi 19, 20133 Milano [email protected] Corso di Laurea Magistrale in Scienze Chimiche Chimica Chimica Elettroanalitica Elettroanalitica Avanzata Avanzata Modulo A Modulo A Combinando l’elettrochimica con una o più tecniche spettroscopiche si ottengono preziose informazioni per il riconoscimento delle specie che si formano per effetto del trasferimento di carica. Inoltre la risposta A vs t di una specie generata o consumata nel processo può fornire informazioni quantitative sul meccanismo e sulla cinetica della reazione. La gamma delle tecniche spettroscopiche combinabili con elettrochimica in situ è vastissima. Ne esamineremo solo alcuni esempi.
Transcript of Chimica Elettroanalitica Avanzata Modulo A - users.unimi.itusers.unimi.it/ECEA/Spettoelettrochimica...

1
Spettroelettrochimica
Prof.Prof. Patrizia R. Patrizia R. MussiniMussiniDipartimento di Chimica Fisica ed Elettrochimica
Via Golgi 19, 20133 [email protected]
Corso di Laurea Magistrale in Scienze Chimiche
Chimica Chimica ElettroanaliticaElettroanalitica AvanzataAvanzataModulo AModulo A
Combinando l’elettrochimica con una o più tecniche spettroscopiche si ottengono preziose informazioni per il riconoscimento delle specie che si formano per effetto del
trasferimento di carica.
Inoltre la risposta A vs t di una specie generata o consumata nel processo può fornire informazioni quantitative sul
meccanismo e sulla cinetica della reazione.
La gamma delle tecniche spettroscopiche combinabili con elettrochimica in situ
è vastissima. Ne esamineremo solo alcuni esempi.
2
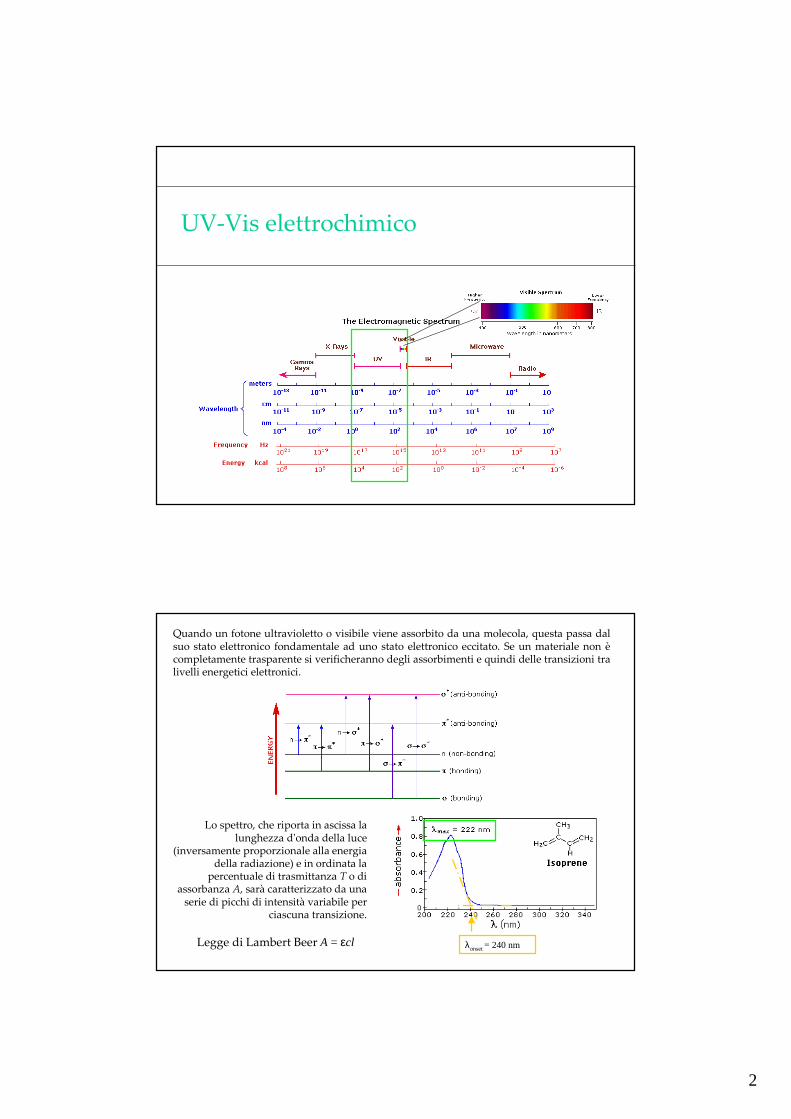
UV-Vis elettrochimico
Quando un fotone ultravioletto o visibile viene assorbito da una molecola, questa passa dal suo stato elettronico fondamentale ad uno stato elettronico eccitato. Se un materiale non è completamente trasparente si verificheranno degli assorbimenti e quindi delle transizioni tra livelli energetici elettronici.
Lo spettro, che riporta in ascissa la lunghezza d'onda della luce
(inversamente proporzionale alla energia della radiazione) e in ordinata la
percentuale di trasmittanza T o di assorbanza A, sarà caratterizzato da una
serie di picchi di intensità variabile per ciascuna transizione.
Legge di Lambert Beer A = εcl λonset = 240 nm

3
Efficienza di coniugazione e λ di assorbimento in sistemi π coniugati
λ cresce → shift batocromico λ cala → shift ipsocromico
ε cresce → shift ipercromico ε cala → shift ipocromico
All’aumentare della coniugazione λ cresce(cala l’energia per la transizione tra HOMO e LUMO)
h (J s-1) × c(m s-1)/(λλλλonset(m) × qe(C))
λ di assorbimentoed energia del gap tra HOMO e LUMO
h (J s-1) × c(m s-1)/(λλλλmax(m) × qe(C))
Criterio del massimo del picco di assorbimento
Criterio dell’onsetdel picco di assorbimento
Si riferisce alla conformazionepiù popolata
Si riferisce alla conformazione di maggior coniugazione efficace
Spettroscopicamente si può stimare solo il gap, mentre elettrochimicamente (con la voltammetria)
si possono stimare sia il gap sia i singoli livelli HOMO e LUMO

4

5
Assorbimenti in complessi ottaedrici di metalli d. Da sinistra:
metal centred transition (MC), ligand to metal charge transfer (LMCT), metal to ligand charge transfer (MLCT)
ligand-ligand transition (L-L)
Transizioni elettroniche in complessi di metalli di transizione
Si possono distinguere con il solvatocromismo (le loro λ di assorbimento sono molto più sensibili al solvente delle altre transizioni)
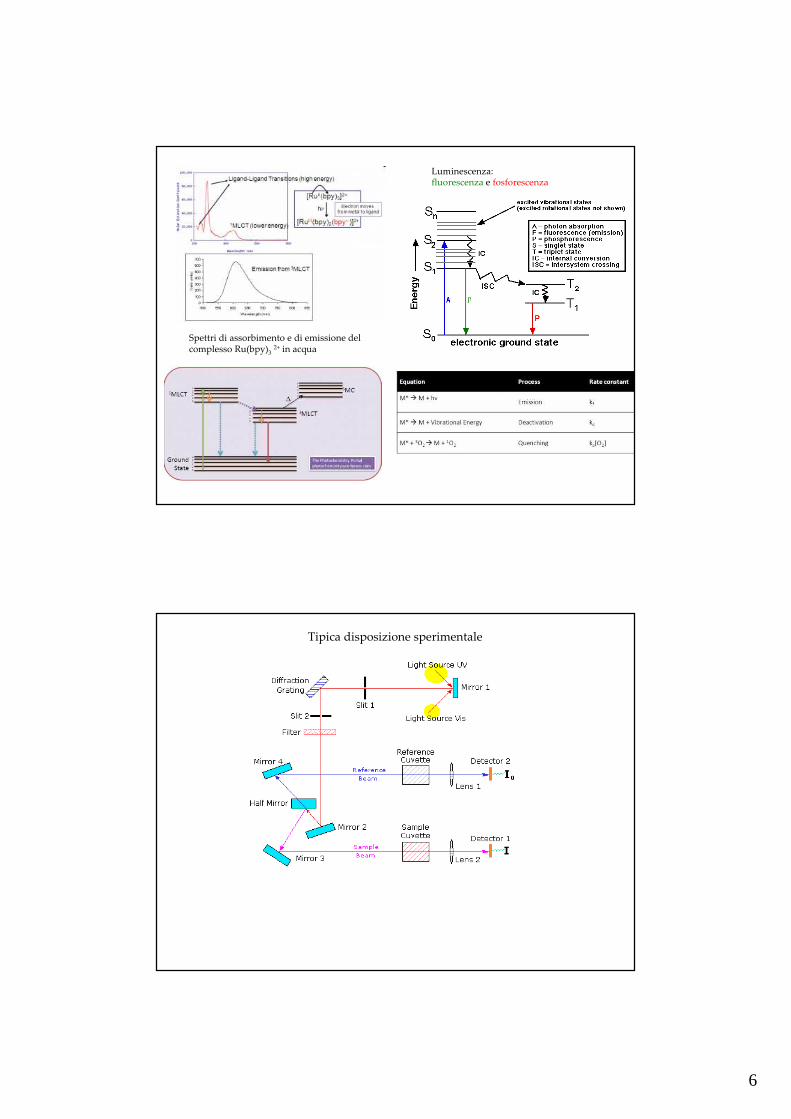
6
Spettri di assorbimento e di emissione del complesso Ru(bpy)3
2+ in acqua
Luminescenza:fluorescenza e fosforescenza
Tipica disposizione sperimentale

7
Le misure con elettrochimica in situ possono essere fatte in assorbanza o in riflettanza (se l’elettrodo non è trasparente o è ricoperto con un film opaco). La celletta, che deve essere alloggiata nello scompartimento dello spettrometro, è ricavabile facilmente in una cuvetta di quarzo per spettroscopia UV-Vis; ne esistono anche di commerciali.
Con cammino ottico molto stretto (per
esaltare il processo
elettrodico rispetto al
bulk.
Nella cella vengono alloggiati l’elettrodo di lavoro (solitamente vetro conduttore ITO o FTO, oppure una retina di oro o platino), l’elettrodo di riferimento (AgCl o calomelano) e il controelettrodo (in genere filo di Pt)

8
E.Kaya, A. Balan, D. Baran, A. Cirpan, L. Toppare
Organic Electronics, 12(1), 2011, 202-209
Un esempio di studio spettroelettrochimico di polimeri elettrocromici
Electrochemical p-type doping electronic absorption spectra of P1between 0.0 and 1.3 V with 0.1 V potential intervals.

9
(A) UV–vis–NIR spectra of P2 in film form (left) and in toluene (right). (a) 0.5, (b) 0.8, (c)1.0, (d) 1.2, and (e) −2.16 V and (a′) 0%, (b′) 5%, (c′) 10%, (d′) 20%, (e′) 30%, (f′) 40% (g′) 50% TFA (v:v) and (h′) dedoped form with 1 mL TEA. (B) UV–vis–NIR spectra of P3 in film form (left) and in toluene (right). (a) 0.5, (b) 0.8, (c) 1.0, (d) 1.2, (e) −2.0, and (f) −2.10 V and (a′) 0%, (b′) 5%, (c′) 10%, (d′) 20%, and (e′) 30% TFA (v:v).
EPR (o ESR) elettrochimico

10
Electron paramagnetic resonance (EPR) o Electron spin resonance (ESR)
La spettroscopia di electron paramagnetic resonance (EPR) o electron spin resonance (ESR) è una tecnica che studia specie chimiche che hanno uno o più elettroni spaiati, come radicali liberi organici o inorganici oppure complessi inorganici con uno ione metallico di transizione.
I concetti della spettroscopia EPR sono analoghi a quelli della spettroscopia risonanza magnetica nucleare (nuclear magnetic resonance, NMR), ma in essa vengono eccitati spin elettronici invece chespin di nuclei atomici. L’analisi della curva di uno spettro EPR o ESR spectrum dà informazioni sulla struttura del materiale.Per un radicale con M nuclei equivalenti, ciascuno con spin I, il numero di righe EPR atteso é 2MI + 1. (ad es. il radicale metile CH3ha tre 1H nuclei equivalenti con I = 1/2 e quindi si osservano 2MI + 1 = 2(3)(1/2) + 1 = 4 righe.
Per un radicale con M1 nuclei equivalenti di spin I1 e M2 nuclei equivalenti di I2 il numero di righe atteso è (2M1I1 + 1) (2M2I2 + 1) (ad es. il radicale metossimetilico H2C(OCH3) ha due 1H nuclei tra loro equivalenti con I = 1/2 e altri tre nuclei 1H tra loro equivalenti con I = ½ e quindi si osservano (2M1I1 + 1) (2M2I2 + 1) = [2(2)(1/2) + 1][2(3)(1/2) + 1] = [3][4] = 12 righe.
N@C60
endofullerene d’azoto radicale
CH3
radicale CH2OCH3
In un esperimento spettroelettrochimico in situ, in cui avvengono uno o più trasferimenti elettronici, si monitora con EPR in diretta la comparsa di specie paramagnetiche, e analizzandone lo spettro si ottengono informazioni sulla loro localizzazione e/o su cambiamenti strutturali della molecola.
Romanovski, Stela Maris de M. et al..J. Braz. Chem. Soc.2010, 21 (5)
Combinazione diSpettrometro ESR,
Potentiostato GalvanostatoSpettrometro UvVis Diode array
Cella elettrochimica nella cavità dello spettrometro ESR
Cella spettroelettrochimica per EPR

11
Particolarmente vantaggiosa è la combinazione ternaria ESR UV EC
Spettri UV-Vis di un film di polianilina ciclando il potenziale tra -50 mV e 350 mV. Vi è sovrapposizione degli assorbimenti del materiale di partenza e dei prodotti formati nel ciclo redox. Non si riesce ad assegnare in modo non ambiguo gli assorbimenti delle specie cariche
Monitorando però in contemporanea con EPR la natura e concentrazione delle specie paramagnetiche si riesce a scomporre il segnale UV nelle componenti delle specie paramagnetiche e non:
Spettro UV-Vis puro del polarone generato elettrochimicamente, ottenuto per analisi dello spettro globale supportata da EPR
Spettro UV-Vis puro del bipolarone (privo di spin) generato elettrochimicamente, , ottenuto per analisi dello spettro globale supportata da EPR
NMR elettrochimico
12
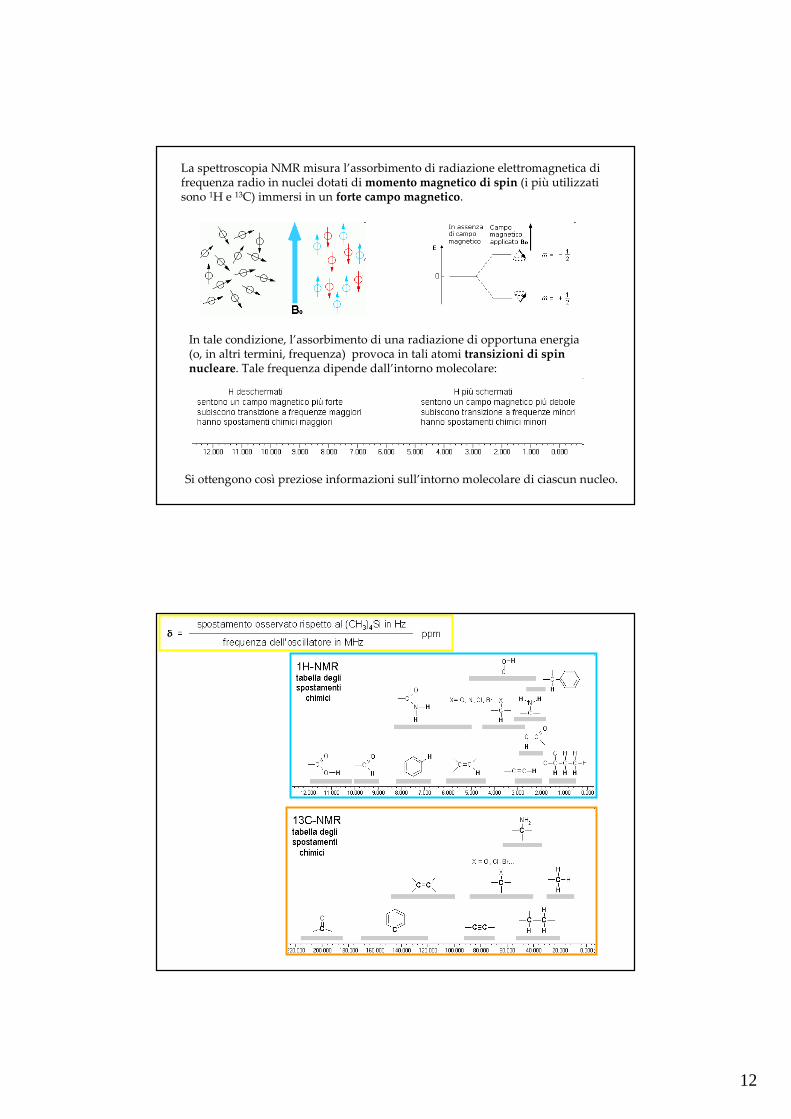
La spettroscopia NMR misura l’assorbimento di radiazione elettromagnetica di frequenza radio in nuclei dotati di momento magnetico di spin (i più utilizzati sono 1H e 13C) immersi in un forte campo magnetico.
In tale condizione, l’assorbimento di una radiazione di opportuna energia (o, in altri termini, frequenza) provoca in tali atomi transizioni di spin nucleare. Tale frequenza dipende dall’intorno molecolare:
Si ottengono così preziose informazioni sull’intorno molecolare di ciascun nucleo.

13
Inoltre fenomeni di accoppiamento tra nuclei adiacenti danno luogo ad uno splitting dei segnali che dipende dalla molteplicità di tali nuclei, e quindi dà informazioni su di essa.
La combinazione della spettroscopia NMR (in particolare 1H) con l’elettrochimica in situ fornisce un metodo per seguire cambiamenti strutturali delle molecole redox attive durante i processi di trasferimento di carica.
Riduzione reversibile del p-Benzochinone al corrispondente idrochinone seguita in situ mediante spettroelettrochimica NMR
Questa tecnica può anche essere un utile complemento all’ EPR elettrochimicoperché fornisce informazioni sulle specie non paramagnetiche

14
spettrometro NMR combinato con potentiostato
Un problema nella costruzione della cella è che non deve contenere metalli che possano essere attratti dal forte campo magnetico. La cella sotto rappresentata (IFW Dresda) è pressoché priva di metalli, essendo basata su elettrodi a fibra lunga di carbonio. Grazie ai progressi nelle apparecchiature NMR non è più richiesta la rotazione del campione per raggiungere alta risoluzione nelle misure, e quindi questa cella, di facile ed economica preparazione, si può usare senza problemi con un’ampia gamma di spettrometri NMR e alle frequenze caratteristiche di diversi nuclei.
R.D. Webster, Analytical Chemistry 2004, 76, 1603-1610

15
R.D. Webster, Analytical Chemistry 2004, 76, 1603-1610
IR elettrochimico

16
Lo spettro IR rappresenta in ascissa il numero d'onda (più spesso utilizzata rispetto alla frequenza) e in ordinata la percentuale di radiazione trasmessa. Si può suddividere in tre diverse zone:
1. zona dei gruppi funzionali, che si estende da 3800 a 1300 cm-1 e comprende bande dovute sia a stiramenti che a deformazioni di gruppi funzionali (per esempio legami N-H, O-H, C-H, C=C, C=O, N=O, ecc.), con questi ultimi compresi tra 1600 e 1300 cm-1. È da notare che i legami con l'idrogeno si trovano a frequenze molto alte per via della massa molto ridotta di quest'atomo;
2. zona delle fingerprints, impronte digitali, da 1300 a 650 cm-1 e che deve il suo nome alla presenza di bande strettamente caratteristiche di ciascuna singola molecola in quanto originate da vibrazioni corali dell'intero scheletro molecolare;
3. zona del lontano IR, che si estende da 650 a 200 cm-1 e presenta bande dovute a stiramenti di atomi pesanti, deformazioni di gruppi privi di idrogeno e vibrazioni di scheletro.
symmetrical stretching
asymmetrical stretching scissoring rocking twisting wagging
Quando un fotone infrarosso viene assorbito da una molecola, questa passa dal suo stato vibrazionale fondamentale ad uno stato vibrazionale eccitato.
Modi di vibrazione

17
Da Bard e Faulkner
Disposizione strumentale tipica
Cella per IR elettrochimico

18
La spettroelettrochimica IR è stata molto applicata per studiare specie adsorbite
(reagenti, intermedi, prodotti), per esaminare specie prodotte nel sottile strato di soluzione tra elettrodo e finestra, e per
sondare il doppio strato.
Questo approccio è stato particolarmente efficace con
specie che si distinguono bene in IR quali CO e CN−.
In casi favorevoli si riesce a capire com’è orientata la molecola e la dipendenza
dell’orientamento dal potenziale.
Si veda ad esempio gli spettri differenziali (tra 0.2 V e 0.4 V) riportati a fianco per diverse zone dello spettro. I picchi
negativi corrispondono a modi di vibrazione a 0.4 V (molecola adsorbita) e quelli positivi a 0.2
V (molecola in soluzione).
Raman elettrochimico

19
La spettroscopia Raman è il metodo di elezione per analizzare la struttura di ogni tipo di materiale carbonioso, come grafite, grafene, nanotubi di carbonio e fullereni. Dall’analisi dei modi vibrazionali si ottengono informazioni strutturali quali ad esempio il grado di cristallinità, di funzionalizzazione, e così via.
Un altro esempio di applicazione é lo studio dello stato e qualità dei materiali nanostrutturati per batterie.
Condizione necessaria affinché si verifichi la risonanza con la radiazione elettromagnetica è che la molecola sia anisotropicamente polarizzabile nel caso di transizione rotazionale, ovvero la vibrazione deve implicare una variazione della polarizzabilità nel caso di transizione vibrazionale.“Regola di esclusione“: se una molecola possiede un centro di simmetria nessun modo vibrazionale può essere contemporaneamente Raman-attivo e attivo all'infrarosso: ad esempio, nel caso della molecola CO2 le transizioni Raman sono legate allo stretching simmetrico del legame C-O, mentre tale modo, non producendo variazione del momento di dipolo molecolare, non è invece attivo all'infrarosso.
•Stokes, che possiede energia minore rispetto alla radiazione originaria incidente, visto che una parte di tale energia è utilizzata per promuovere una transizione a un livello superiore
•anti-Stokes, che riceve invece un contributo energetico dallo stato eccitato quando passa a un livello inferiore, per cui è caratterizzata da maggiore energia;
•Rayleigh (scattering elastico), che mantiene la stessa energia della radiazione incidente.
La spettroscopia Raman (dal cognome del fisico indiano scopritore di tale effetto, premio Nobel 1930) è complementare a quella infrarossa nello studio di modi a bassa frequenza, quali in particolare vibrazioni e translazioni.
Si utilizza tipicamente una luce laser nel campo visibile, del vicino infrarosso o nel vicino ultravioletto. Trattandosi di una spettroscopia di scattering si fa incidere sul campione la radiazione elettromagnetica monocromatica di intensità e frequenza nota e si misura la radiazione diffusa tramite rivelatore posto a 90º rispetto al campione. La radiazione può essere diffusa in tre modi:
Il Raman è come noto molto meno esigente dell’IR per quanto riguarda forma e
dimensioni del campione.
Cella per Raman elettrochimico

20
Szunerits et al., Langmuir 2008, 24, 6327−6333Funzionalizzazione della grafite vetrosa con sali di diazonio in liquidi ionici
La spettroscopia Raman con elettrochimica in situ viene qui impiegata per discriminare tra la riduzione di NO2 a
NH2 e quella di N=N a NH−NH
Le bande Raman di NO2 a 1103 cm-1 e 1336 cm-1 vengono conservate fino a un potenziale di circa –1.1 V(SCE); dopo, il segnale viene interamente perduto. Le bande N=N decrescono significativamente a potenziali più negativi di –1.1 V(SCE), per la riduzione dell’azobenzene al suo radical anione e poi dianione. Si riesce così a determinare il potenziale ottimale per la conversione selettiva di NO2 a NH2
SERS (Surface-enhanced Raman Spectroscopy)

21
Un ostacolo all’applicazione della spettroscopia Raman a molti problemi è stata la sua scarsa sensibilità, legata al fatto che è basato su transizioni proibite.
Tuttavia, la sensibilità del Raman può essere aumentata se si registra il segnale avendo l’analita assorbito su una superficie resa rugosa, ad esempio per cicli redox del metallo, oppure funzionata con nanoparticelle, di un metallo opportuno, come argento, rame e oro. Questo effetto, scoperto da
Fleischmann per caso, si chiama perciò Surface Enhanced Raman Spectroscopy (SERS)
Non riguarda però tutti i modi vibrazionali indiscriminatamente, ma in particolare quelli coinvolti nell’interazione con la superficie; e in ogni caso lo spettro può cambiare significativamente rispetto a quello Raman, se cambia la simmetria.
Spettro Raman del 2-mercaptoethanol liquido (sotto) e spettro SERS di un monostrato di
2-mercaptoetanolo su argento rugoso (sopra). Spectra are
scaled and shifted for clarity. Si vede una differenza nelle regole
di selezione: alcune bande appaiono solo nello spettro
Raman o solo in quello SERS.

22
L’effetto di esaltazione del segnale da parte della superficie è attribuito a due effetti:•elettromagnetico: le microstrutture della superficie rugosa o nanoparticellare causano forti amplificazioni locali dei campi elettrici delle radiazioni incidente e riflessa, con formazione di onde plasmoniche superficiali (onde elettromagnetiche che si propagano lungo la superficie parallelamente all’interfase)•chimico: la molecola adsorbita interagisce con la superficie metallico portando a trasferimenti di carica; l’effetto di amplificazione è paragonabile a quello alla base della spettroscopia Raman di Risonanza.
Entrambi gli effetti valgono solo su piccole distanze, e quindi la SERS è specifica per molecole adsorbite sulla superficie.
L’applicazione della SERS ai sistemi elettrochimici è che deve essere fatta specificamente su Ag, Au o Cu. Però, essendo la parte elettromagnetica dell’efftto mantenuta fino a diversi nanometri, si può rivestire il metallo SERS attivo con uno strato sottile di quello che interessa (ad es. Pd su Au), e ottenere ancora l’amplificazione.
L’amplificazione è normalmente dell’ordine di 101010105555−−−−106 ma può arrivare fino a un fattore 1010101015151515−−−−1014, fino a permettere la rilevazione anche di singole molecole adsorbite, in particolare con nanoparticelle e/o con la tecnica SERRS, che combina l’espediente della amplificazione superficiale con quello della spettroscopia Raman di risonanza (in cui la lunghezza d’onda di eccitazione corrisponde a una transizione elettronica nella molecola),
Dicroismo circolare elettrochimicoe una sua applicazione per lo studio di polimeri conduttori chirali

23
Circular dichroism for chiral conducting polymer monitoring I
Symmetrical LCP + RCP→ plane polarized light.
At a λ where light absorption takes place, εLCP ≠ εRCP → the resulting light becomes helliptically polarized
In a chiral medium LCP and RCP propagate at different speeds→ the PL plane is rotated(rotation angle measured in polarimetry)
Two perpendicular
plane-polarized light
components having a 90°
phase shift →left and right
circularlypolarized light (LCP and RCP)
LCP
RCP
Circular dichroism for chiral conducting polymer monitoring II
Enantiomer molecules exhibit mirror imaged CD spectra of opposite sign
cl
AA RCPLCPRCPLCP εε
−=−=∆ ε
ϑ hellipticity
CD spectra are a powerful tool for probing macromolecular and supramolecular chirality in 3D space (e.g. nucleic acids, proteins etc.)
Increasingly defined 3D order of the macromolecule constituted by chiralsubunits→ increasingly sharp and bisignate signals (Cotton effect)
random coilloose helix
(no H bonds)
β sheet
α helix

24
Particularly dealing with polypyrroles or polythiophenes, chirality is frequently introduced in the polymer by attaching chiral pendants to the electroactive backbone through suitable linkers.
Chiral monomers for the preparation of chiral polymers
the presence of carbon stereocentersinvariably characterizes the chiral
substituents
Experimental conditions affect the chirality manifestations of the polymers designed according to this strategy.
Chiral pendants: regioregular polymerization
S
OR
S
OR
( )n
S
OR
Br Br
S
OR
Br H S
OR
CH3
CH3CH3
R =
1) FeCl3
2) NH2-NH2
poly 12eq NBS
1) 1eq n-BuLi2) H2O
1) LDA
2) MgBr 2Et2O3) Ni(dppp)Cl 2
( )n
poly 2
G. Bidan, S. Guillerez and V. Sorokin, Adv. Mater., 1996, 8, 157
Poly 1
Poly 2
CD
A good CD signal is only obtained with the
regioregular polymer prepared along the more
laborious synthetic route.
25
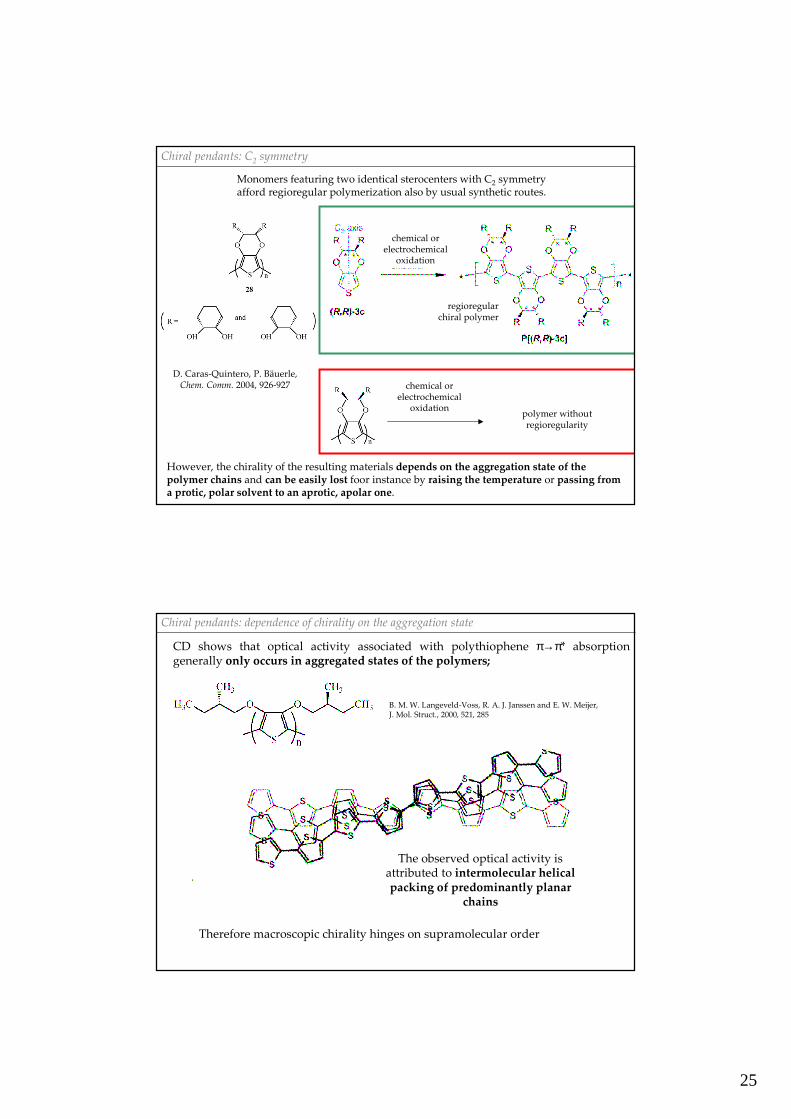
Chiral pendants: C2 symmetry
D. Caras-Quintero, P. Bäuerle,Chem. Comm. 2004, 926-927
Monomers featuring two identical sterocenters with C2 symmetry afford regioregular polymerization also by usual synthetic routes.
chemical or electrochemical
oxidation
regioregularchiral polymer
chemical or electrochemical
oxidationpolymer withoutregioregularity
However, the chirality of the resulting materials depends on the aggregation state of the polymer chains and can be easily lost foor instance by raising the temperature or passing froma protic, polar solvent to an aprotic, apolar one.
Chiral pendants: dependence of chirality on the aggregation state
CD shows that optical activity associated with polythiophene π→π* absorptiongenerally only occurs in aggregated states of the polymers;
B. M. W. Langeveld-Voss, R. A. J. Janssen and E. W. Meijer, J. Mol. Struct., 2000, 521, 285
The observed optical activity isattributed to intermolecular helicalpacking of predominantly planar
chains
Therefore macroscopic chirality hinges on supramolecular order
26
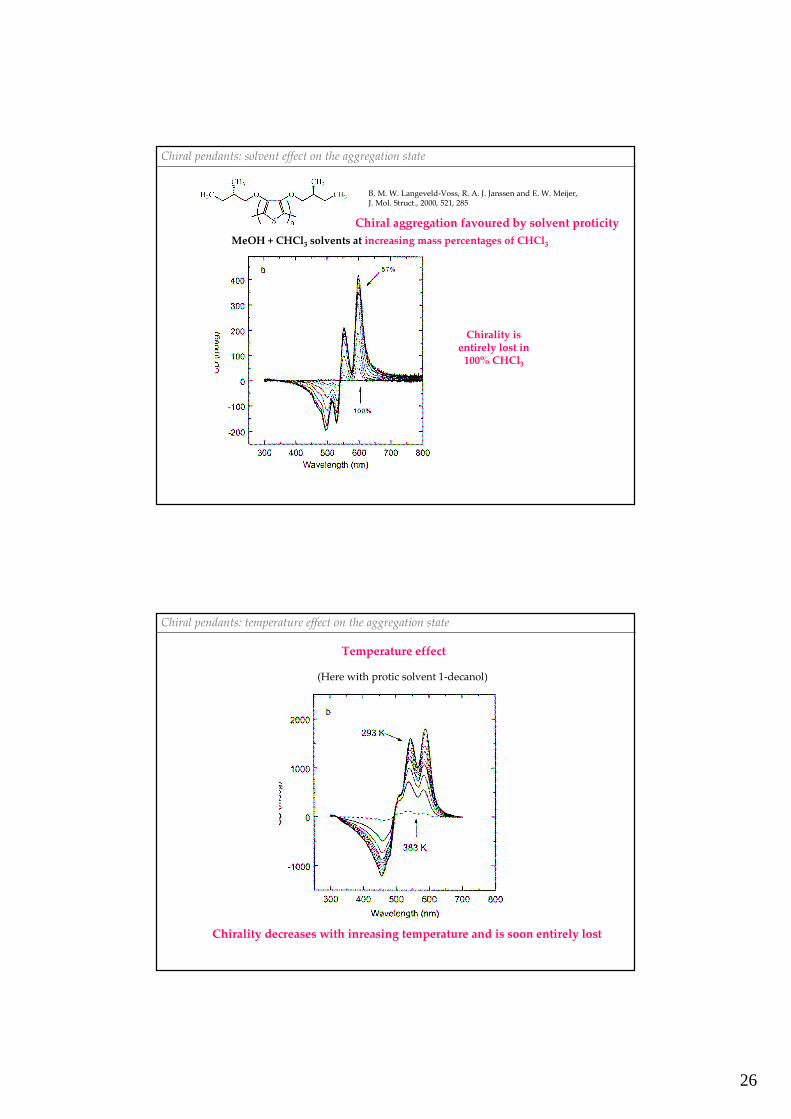
Chiral pendants: solvent effect on the aggregation state
B. M. W. Langeveld-Voss, R. A. J. Janssen and E. W. Meijer, J. Mol. Struct., 2000, 521, 285
MeOH + CHCl3 solvents at increasing mass percentages of CHCl3
Chirality isentirely lost in
100% CHCl3
Chiral aggregation favoured by solvent proticity
Chiral pendants: temperature effect on the aggregation state
(Here with protic solvent 1-decanol)
Chirality decreases with inreasing temperature and is soon entirely lost
Temperature effect
27
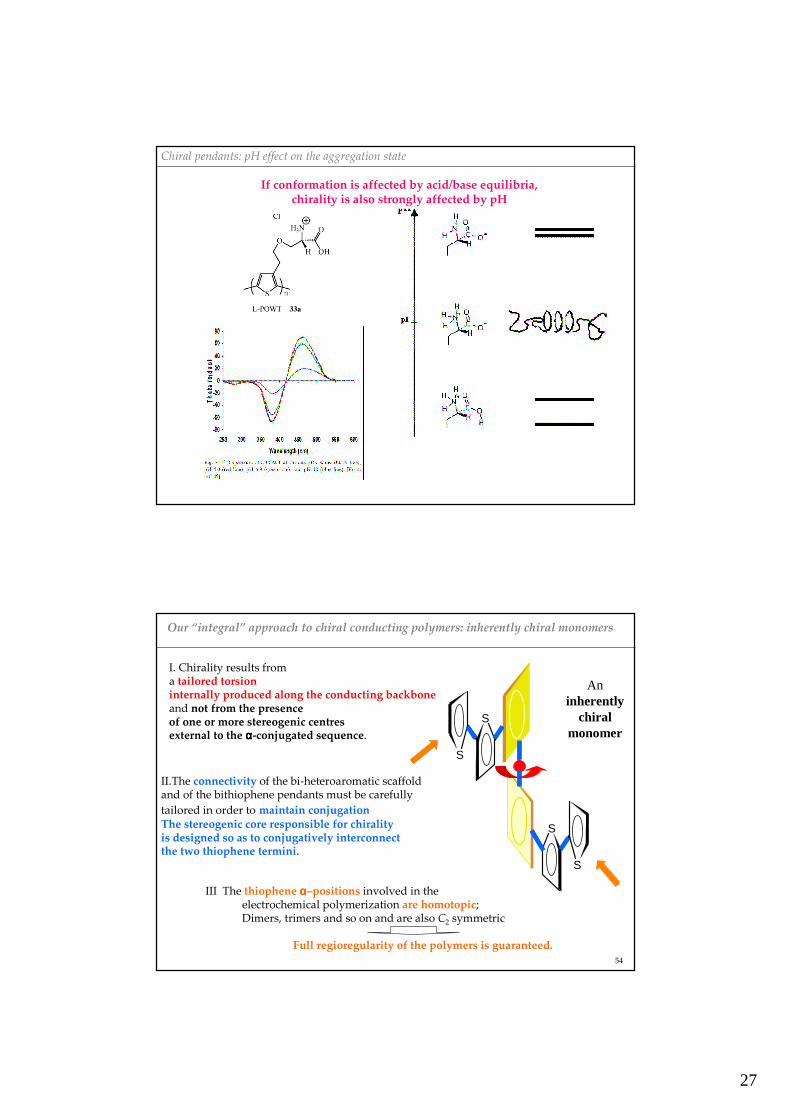
Chiral pendants: pH effect on the aggregation state
If conformation is affected by acid/base equilibria, chirality is also strongly affected by pH
III The thiophene αααα–positions involved in the electrochemical polymerization are homotopic;Dimers, trimers and so on and are also C2 symmetric
Full regioregularity of the polymers is guaranteed.
Our “integral” approach to chiral conducting polymers: inherently chiral monomers
S
S
S
S
II.The connectivity of the bi-heteroaromatic scaffold and of the bithiophene pendants must be carefullytailored in order to maintain conjugationThe stereogenic core responsible for chiralityis designed so as to conjugatively interconnect the two thiophene termini.
I. Chirality results from a tailored torsioninternally produced along the conducting backboneand not from the presenceof one or more stereogenic centres external to the αααα-conjugated sequence.
Aninherently
chiralmonomer
54

28
SS
SS
S
S
Each one is approximately planar and thus of high effective conjugation
Intrinsic regioregularity in
polymerization: Only two sites available for
polymerization, reciprocally far away,
symmetrical,
and easily accessible.
Intrinsic regioregularity in
polymerization: Only two sites available for
polymerization, reciprocally far away,
symmetrical,
and easily accessible.
Structural peculiarities of the TBTX monomer2,2’-bis(2,2’-bithiophene-5-yl)-3,3’-bi-1-benzothiophene
SS
SS
S
S
Inherent dissimmetryIn spite of including no
stereogenic center, the whole molecule is chiral, exhibiting a C2 symmetry axis. The energy
barrier is sufficiently high(about 50 kcal mol-1) to yield
stable enantiomers that can be separated and stored.
Inherent dissimmetryIn spite of including no
stereogenic center, the whole molecule is chiral, exhibiting a C2 symmetry axis. The energy
barrier is sufficiently high(about 50 kcal mol-1) to yield
stable enantiomers that can be separated and stored.
(R) -TBTX (S) -TBTX
Intrinsic 3D character: bulky substituents on both β positions of the central thiophene rings, induce a 70o node between the two
moietes which does not totally impair conjugation
Intrinsic 3D character: bulky substituents on both β positions of the central thiophene rings, induce a 70o node between the two
moietes which does not totally impair conjugation
-
70o
Connectivity allows double bond conjugation along the whole backboneConnectivity allows double bond conjugation along the whole backbone
55
-3.0 -2.5 -2.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 1.5
E / V(Fc+|Fc)
i/cS
v0.
5 /
(A m
ol-1
dm
3 cm
-2 V
-0.5 s
0.5 )
-0.5
0
0.5
1
1.5
2
2.5
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7
E / V(Fc+|Fc)
i/cSv
0.5 /
(A
mol
-1 d
m3 c
m-2 V
-0.5 s
0.5)
0.2 V/s
0.5 V/s
0.1 V/s
0.2 V/s
0.5 mV/s
1 V/s
2 V/s
Two nearlyequivalentfirst oxidationsites
Eg, onset = 2.93eV
Eg, max = 3.20 eV
Energy gapsintermediate between
linear terthiophene and tetrathiophene
Electrochemical activity of the TBTX monomer as a racemate
Some residualinteraction
between the twomoieties through
the 70o node
Deposition of anelectroactive
product
Ep nearly constant withscan rate → rather fast
electron transfer
56

29
Pt electrode,CH2Cl2+0.1 M TBAPF6,
0.2 V s-1
-1.5
-1
-0.5
0
0.5
1
1.5
2
2.5
0 0.25 0.5 0.75 1 1.25 1.5
E / V(SCE)
i/c
/ (
A m
ol -1
dm3 c
m-2
)
I
VI
XII
XVIII
XXIV
XXXI
XXXV
0.0005 M TX
Pt foil electrode, CH2Cl2 + 0.1 M
TBAPF6, 0.2 V s-1
Electropolymerization of the TBTX monomer as a racemate: EQCM monitoring
In all operatingconditions
fast and regular electropolymerizationeven at low monomer
concentration -0.10
-0.05
0.00
0.05
0.10
0.15
0.20
0.25
-1 -0.75 -0.5 -0.25 0 0.25 0.5 0.75 1 1.25
E /V (Fc+/Fc)
I /
mA
-1100
-1000
-900
-800
-700
-600
-500
-400
-300
-200
-100
0
ηf / H
z
0
0.0000005
0.000001
0.0000015
0.000002
0.0000025
0.000003
0.0000035
0.000004
0 25 50 75 100 125 150 175 200 225
t /s
ηm/g
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
mo
no
me
r un
its nm -2
Au electrode (EQCM), CH2Cl2 + 0.1 M TBAPF6, 0.2 V s-1
0.001 M TX
Regular increa
se of the
polymer film
Counteranioningress/egress
57
Maldi spectra indicate that the material isconstitued by oligomers, from dimers topentamers
They are stable upon anodic potential cycling(provided that a suitable counter ion is used) and exhibit a partial charge trapping effect
CV and UVvis spectroscopy point tosignificantly improved conjugation
0.0000000
0.0000002
0.0000004
0.0000006
0.0000008
0.0000010
0.0000012
0 20 40 60 80 100 120
t /s
∆∆ ∆∆m
/g
0
0.5
1
1.5
2
Counter anions nm
-2
-600
-400
-200
0
200
400
600
800
1000
1200
1400
-0.3 -0.1 0.1 0.3 0.5 0.7 0.9
E / V(Fc+/Fc)
I/ m
A
-300
-250
-200
-150
-100
-50
0
∆f / H
z
TBAPF6(0.1 M in CH2Cl2; same
as in electropolymerization)
EQCM: stability of the electrodeposited film as a function of the counteranion
-600
-400
-200
0
200
400
600
800
1000
1200
1400
-0.3 -0.1 0.1 0.3 0.5 0.7 0.9
E /V(Fc+/Fc)
I / m
A
-300
-250
-200
-150
-100
-50
0
∆f / H
z
0.0000000
0.0000002
0.0000004
0.0000006
0.0000008
0.0000010
0.0000012
0 20 40 60 80 100 120
t /s
∆∆ ∆∆m
/g
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
Cou
nter anions nm-2
TBABF4(0.1 M in CH2Cl2)
very stable upon potential cycling in a monomer-free solution with the polymerization
counteranion or a smaller one
-600
-400
-200
0
200
400
600
800
1000
1200
1400
-0.3 -0.1 0.1 0.3 0.5 0.7 0.9
E / V(Fc+/Fc)
I /
mA
-300
-250
-200
-150
-100
-50
0
∆f / H
z
0
0.0000002
0.0000004
0.0000006
0.0000008
0.000001
0.0000012
0 20 40 60 80 100 120
t /s
∆∆ ∆∆m
/g
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
Counter anions nm
-2
TBAPTS(0.1 M in CH2Cl2)
Much more difficult is the ingress/egress of a bulkier and very different anion; possible trapping of
the counterion and/or polymer degradation 58
at IPC PAS, Warsaw
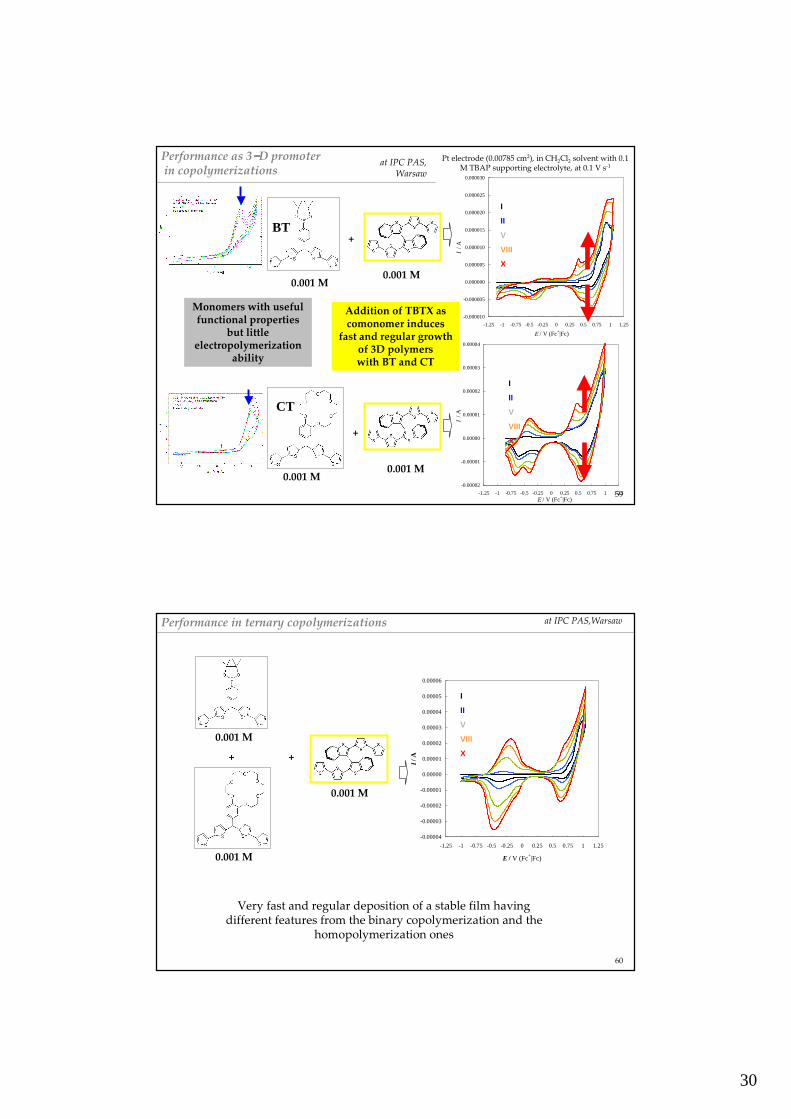
30
Performance as 3−−−−D promoterin copolymerizations
Pt electrode (0.00785 cm2), in CH2Cl2 solvent with 0.1 M TBAP supporting electrolyte, at 0.1 V s-1
-0.000010
-0.000005
0.000000
0.000005
0.000010
0.000015
0.000020
0.000025
0.000030
-1.25 -1 -0.75 -0.5 -0.25 0 0.25 0.5 0.75 1 1.25
E / V (Fc+|Fc)
I /
A
I
II
V
VIII
X
-0.00002
-0.00001
0.00000
0.00001
0.00002
0.00003
0.00004
-1.25 -1 -0.75 -0.5 -0.25 0 0.25 0.5 0.75 1 1.25E / V (Fc+|Fc)
I /
A
I
II
V
VIII
X
Addition of TBTX as comonomer induces
fast and regular growthof 3D polymers with BT and CT
0.001 M
+
0.001 M
+
0.001 M
0.001 M
Monomers with usefulfunctional properties
but little electropolymerization
ability
BT
CT
59
at IPC PAS, Warsaw
-0.00004
-0.00003
-0.00002
-0.00001
0.00000
0.00001
0.00002
0.00003
0.00004
0.00005
0.00006
-1.25 -1 -0.75 -0.5 -0.25 0 0.25 0.5 0.75 1 1.25
E / V (Fc+|Fc)
I/ A
I
II
V
VIII
X
Performance in ternary copolymerizations
0.001 M
0.001 M
0.001 M
++
Very fast and regular deposition of a stable film havingdifferent features from the binary copolymerization and the
homopolymerization ones
60
at IPC PAS,Warsaw

31
n
P-
F F
F
FF F
FF
F
F
F
FF F
F
F
FF
P +
NN
N
NH3+
NH3+
H3N+
P-F F
F
FF F
FF
F
F
F
FF F
F
F
FF
P+
S
S
S
S
SS
OO O
OOO
SSS
S
S
SS
S
S
S
S
S
S
OO
O
OO
O
S
S
SS
S
S
S S
O
O
OO
O
O
S
S
S
P-
F
F
FF
FF F F
FFF
FF
F
F
F
F F
P+
O
O-F
F F
OO-
FF
F
O
O-
FF
F
The copolymers are stableand fully retain the functional properties of the comonomer
Analytical Chemistry (2009)
A MT + TBTX copolymer molecularly imprinted with melamine has given an excellent performance as the recognition element of a selective piezomicrogravimetric chemosensor,
in terms of linear concentration range, detection limit, and selectivity
Performance in a molecularly imprinted polymer
61
IPC PAS,Warsaw
S
S
S
BrS
S
Br
Br
SS
SS
S
S
1) n-BuLi
2) CuCl 2
T = - 90 °C
Br2
CH2Cl2
SS
SnBu 3
(Ph3P)4Pd
toluene
+_( )
Synthesis and enantiomer separation
TBTX is an easily accessible product and its synthesis involves commercially available starting materials.
4 6 8 10 12
0
200
400
600
800
1000
1200
-2000
-1500
-1000
-500
0
-200
0
200
400
(+)
Elution time (min)
Sig
nal (
µV)
(-)
The racemateseparation into
enantiomers was successfully achieved at a semi-preparative scale level by hplc on
a chiral stationary phase.

32
Chiroptical properties of the enantiopure antipodes
[αααα]D20 = +1001 (c = 0.1%, CHCl3)
[αααα]D20 = −−−− 991 (c = 0.1%, CHCl3)
Impressive specific rotation, in accordance with the presence of an inherently dissymmetrical chromophore
Absolute configuration was assigned to the enantiomers by comparisonof the experimental CD curves with that calculated for the (R)-enantiomer.
DFT calculations suggested an atropisomerization barrier of about 43 kcal mol-1
MM calculations suggested a dihedral angle of about 70°
High chirality consisten with complementary theoretical computations:
RemarkableCotton effect,
consistent withthe torsion of the whole molecular
backbone
Monomer CV patterns are equal for the racemate and the enantiomers, as expected.
Electropolymerization took placeregularly in all cases; however, at
rigorously constant operatingconditions it was significantly
slower with enantiopure monomersthan with racemate ones
Electrochemical properties of the enantiopure antipodes
After 30 polymerization cycles
racemate
(−−−−) enantiomer
(+) enantiomer
After 50 polymerization cycles
(−−−−) enantiomer
(+) enantiomer
This appearsconsistent with the
lower freedomdegree and higherstereospecificity of
the enantiomer cases.
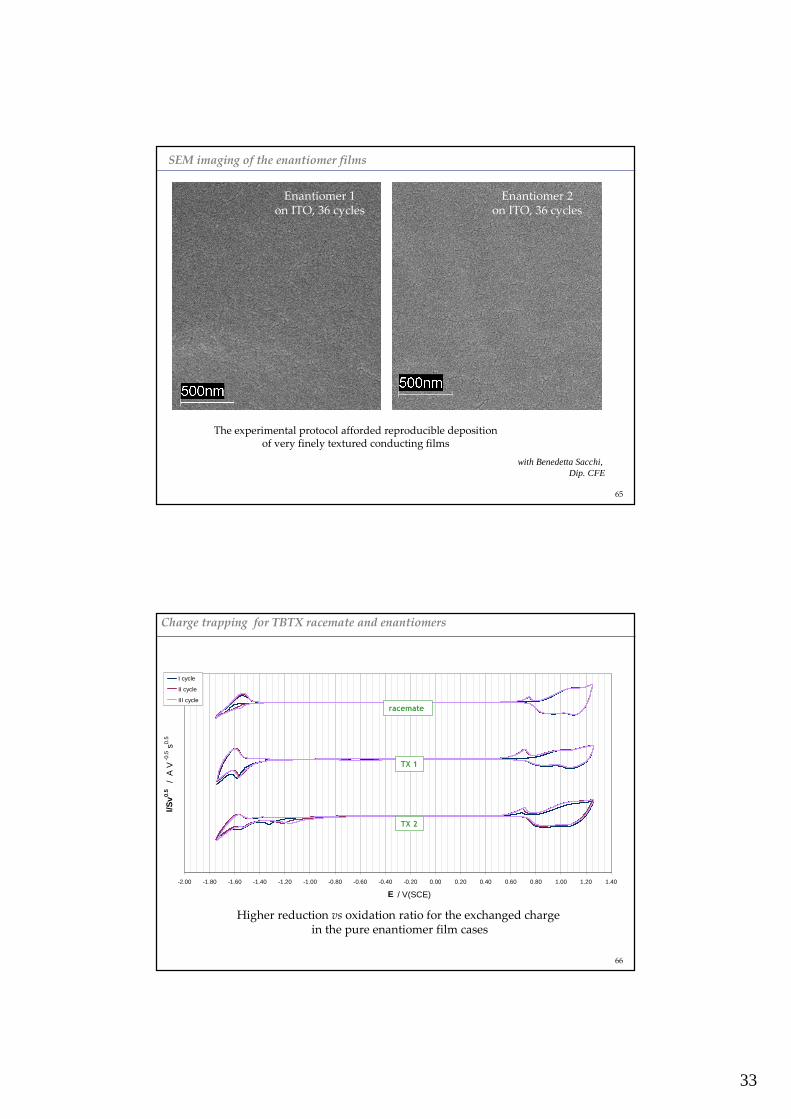
33
SEM imaging of the enantiomer films
Enantiomer 1on ITO, 36 cycles
Enantiomer 2on ITO, 36 cycles
The experimental protocol afforded reproducible depositionof very finely textured conducting films
with Benedetta Sacchi, Dip. CFE
65
-2.00 -1.80 -1.60 -1.40 -1.20 -1.00 -0.80 -0.60 -0.40 -0.20 0.00 0.20 0.40 0.60 0.80 1.00 1.20 1.40
E / V(SCE)
I/Sv0.
5 /
A V
-0.
5 s0.
5
I cycle
II cycle
III cycle
racemate
TX 1
TX 2
Charge trapping for TBTX racemate and enantiomers
Higher reduction vs oxidation ratio for the exchanged chargein the pure enantiomer film cases
66

34
Impedance tests on racemate vs enantiomer (Nyquist plots)
Racemate film,30 cycles on ITO
Enantiomer 1 film,30 cycles on ITO
Racemate film,50 cycles on ITO
Enantiomer 1 film,50 cycles on ITO
p-doped
p-doped
p-doping onset
undoped
undoped
n-doped (charge trapping)
n-doped
67
Circular dichroism on TBTX enantiomers in solution and as electropolymerized films
68
-12
0
12
300 400 500 600 700 800 900nm
mdeg
Enantiopure polymer filmsdeposited on ITO glass electrodes
λ (nm)
CD (mdcg)
Specular bisignate signalsare obtained also for the enantiomer polymer films
Chirality and its CD sign fully transferred from monomers to polymers
→ These non conventional, inherently chiral poly-
thiophenes are remarkably different from traditional
ones, which sometimes exhibit CD manifestations, but only under particular chain aggregation states while are silent under ordinary conditions.
→ These non conventional, inherently chiral poly-
thiophenes are remarkably different from traditional
ones, which sometimes exhibit CD manifestations, but only under particular chain aggregation states while are silent under ordinary conditions.
(-)-(S)
300 350 400 450 500
-60
0
60
md
eg
nm
CD (mdcg)
Enantiopure monomers in solution
(+)-(R) Strongly red shiftedellipticity maxima
Increased conjugation extent gained with polymerization

35
-0.003
-0.002
-0.001
0
0.001
0.002
0.003
0.004
-2 -1.5 -1 -0.5 0 0.5 1 1.5
E / V(SCE)
I/Sv0.
5 /
A V
-0.
5 s0.
5
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900 1000 1100nm
A
UV spectroelectrochemistry
From 0.55 V to 1.15 V → decrease of the absorption band at 450 nm and growth of two new bands at
780 and 1060 nm, attributable to more
conjugated polaronic and
bipolaronic states.
The thianaphthenesystem tends to
reduce the interanular torsion in order to better
delocalize the charges along the
whole chain.
Negligibleschanges
were observed instead
when the potential
was driven toward
negative values.
The original bisignate curve progressively decreases with increasing potential, while a new signal grows at higher wavelengths, as expected on the basis of the UV data.
Interestingly, the new signal exhibits a modest CD activity, indicating a reduction in chirality strength of the doped state.
Holes injection could force the two thianaphthene rings to reduce the interanular torsion in order to gain in electronic delocalization with some loss in the stereogenic efficacy of the atropisomericcore.
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-0.008
-0.006
-0.004
-0.002
0
0.002
0.004
0.006
0.008
0.01
0.012
0 0.2 0.4 0.6 0.8 1 1.2 1.4
E / V(SCE)
I/Sv
/ A
cm
-2 V
-1 s
II
III
IV
V
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-12
0
12
300 400 500 600 700 800 900
nm
mde
g
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900nm
A
-0,2
-0,15
-0,1
-0,05
0
0,05
0,1
300 400 500 600 700 800 900nm
A
CD spectroelectrochemistry
V
The CD signal is fully recovered when the polymer is switched back to neutral.
Therefore the two interconnected heteroaromatic units cannot become coplanarin the heavily doped state (otherwise racemization would occur)