02.glucocorticoidi
20
RECETTORI INTRACELLULARI I recettori intracellulari sono fattori di trascrizione attivati da ligando. In seguito all’attivazione sono in grado di modulare la sintesi proteica in senso positivo o negativo. Essendo recettori intracellulari il ligando riconosciuto deve essere liposolubile, in grado, cioè, di attraversare le membrane cellulari. Il tempo di attivazione è di decine di minuti o addirittura ore. I recettori intracellulari possono essere suddivisi in due famiglie principali (recettori citosolici e recettori nucleari), ognuna delle quali contiene altre sottoclassi recettoriali. I recettori citosolici sono localizzati a livello del citosol, accoppiati a strutture proteiche (hsp) che mantengono il recettore in una forma inattiva. I recettori nucleari sono presenti a livello del nucleo cellulare, anch'essi in forma inattiva. Sono suddivisibili in tre classi: CLASSE 1) Recettore per gli ormoni tiroidei, recettore per la vitamina D3 CLASSE 2) Recettore per l’acido retinoico CLASSE 3) Recettori per gli ormoni steroidei N.B. Nella classe 3 i recettori per gli estrogeni (ER) sono nucleari, mentre i recettori per i glucocorticoidi (GR), l’aldosterone (MR), il progesterone (PR) e gli androgeni (AR) sono citoplasmatici. Tutti i recettori per gli steroidi formano omodimeri. I recettori intracellulari si assomigliano molto sia dal punto di vista strutturale, della sequenza di amminoacidi e nel meccanismo d' azione. Ogni recettore è costituito da 3 regioni funzionali: - porzione carbossiterminale: contiene il dominio LBD o HBD (Ligand/o hormone binding domain), sito di legame per l’ormone, responsabile della dimerizzazione, in assenza del ligando è occupato dalle hsp90 e hsp56. - porzione centrale: contiene il dominio DBD (DNA binding domain), sequenza costituita da circa 70aa, in prevalenza basici con struttura a dita di zinco (con due atomi di Zn coordinati ognuno con quattro cisteine) per favorire il legame con il DNA. Le diversità, sebbene lievi, di tale sequenza, sono la ragione della
-
Upload
michele-scilla-fresiello -
Category
Documents
-
view
547 -
download
0
Transcript of 02.glucocorticoidi

RECETTORI INTRACELLULARI
I recettori intracellulari sono fattori di trascrizione attivati da ligando. In seguito all’attivazione sono in
grado di modulare la sintesi proteica in senso positivo o negativo. Essendo recettori intracellulari il ligando
riconosciuto deve essere liposolubile, in grado, cioè, di attraversare le membrane cellulari.
Il tempo di attivazione è di decine di minuti o addirittura ore.
I recettori intracellulari possono essere suddivisi in due famiglie principali (recettori
citosolici e recettori nucleari), ognuna delle quali contiene altre sottoclassi recettoriali. I
recettori citosolici sono localizzati a livello del citosol, accoppiati a strutture proteiche
(hsp) che mantengono il recettore in una forma inattiva. I recettori nucleari sono presenti
a livello del nucleo cellulare, anch'essi in forma inattiva.
Sono suddivisibili in tre classi:
CLASSE 1) Recettore per gli ormoni tiroidei, recettore per la vitamina D3
CLASSE 2) Recettore per l’acido retinoico
CLASSE 3) Recettori per gli ormoni steroidei
N.B. Nella classe 3 i recettori per gli estrogeni (ER) sono nucleari, mentre i recettori per i glucocorticoidi
(GR), l’aldosterone (MR), il progesterone (PR) e gli androgeni (AR) sono citoplasmatici. Tutti i recettori per
gli steroidi formano omodimeri.
I recettori intracellulari si assomigliano molto sia dal punto di vista strutturale, della sequenza di
amminoacidi e nel meccanismo d' azione.
Ogni recettore è costituito da 3 regioni funzionali:
- porzione carbossiterminale: contiene il dominio LBD o HBD (Ligand/o hormone binding domain), sito di
legame per l’ormone, responsabile della dimerizzazione, in assenza del ligando è occupato dalle hsp90 e
hsp56.
- porzione centrale: contiene il dominio DBD (DNA binding domain), sequenza costituita da circa 70aa, in
prevalenza basici con struttura a dita di zinco (con due atomi di Zn coordinati ognuno con quattro cisteine)
per favorire il legame con il DNA. Le diversità, sebbene lievi, di tale sequenza, sono la ragione della

specificità di legame con le sequenze HRE (Hormone Responsive Elements, in italiano: sequenze che
rispondono agli ormoni).
- porzione amminoterminale: corrisponde al dominio A/B, contenente siti di legame per coattivatori o
corepressori.
La specificità di azione del recettore è definita da diversi fattori: la variabilità del dominio A/B, e quindi la
differente interazione con co-attivatori e co-repressori, la differente espressione della proteina recettoriale
nei vari tessuti, il metabolismo tessuto-specifico degli ormoni.
I recettori intracellulari possono essere attivati da:
A. Interazione con il ligando e conseguente dimerizzazione (GR, MR, AR)
B. In assenza del ligando, tramite una fosforilazione in serina e treonina del recettore innescata
dall’attivazione di recettori di membrana per fattori di crescita (ER, PR, TR). Questo tipo di attivazione per
ora è stato dimostrata solamente in vitro.
Il recettore normalmente è presente in una forma non attiva. Il recettore non attivo è legato a strutture
proteiche che hanno la funzione di mantenere il recettore in forma quiescente, in modo tale che non possa
essere espressa la propria azione. Queste proteine che inibiscono il recettore sono definite HSP (dall'
inglese Heat Shock Protein, cioè proteine dello shock termico) seguite da un numero che indica il peso
molecolare.
Normalmente il recettore inattivo è presente in una conformazione ripiegata a 90 gradi e legato alle HSPs.
In seguito al legame con l'ormone a livello della sezione carbossi-terminale, il recettore passa da 90 a 180
gradi e stacca la HSP. Si scopre così la porzione centrale del recettore, quella ad affinità per il DNA. Prima di
migrare nel nucleo, il recettore attivato si lega con un altro recettore, e ,così dimerizzato, si dirige verso il
nucleo, vi penetra ed interagisce con specifiche sequenze di DNA. L' azione che si può ottenere può essere
attivatoria o inibitoria sull'espressione genica.
L’interazione del dimero attivato con il promotore favorisce la formazione di complessi macroproteici che
stimolano o reprimono la trascrizione, per mezzo di modificazioni cromatiniche che espongono o
nascondono i geni da trascrivere. Queste modificazioni sono: l’acetilazione delle lisine degli istoni (ad opera
di HAT o istone-acetil-transferasi) con srotolamento della cromatina e aumento dell’espressione genica, e la
deacetilazione istonica (da parte di HDAC o istone-deacetilasi) con compattazione della cromatina e
riduzione dell’espressione genica.
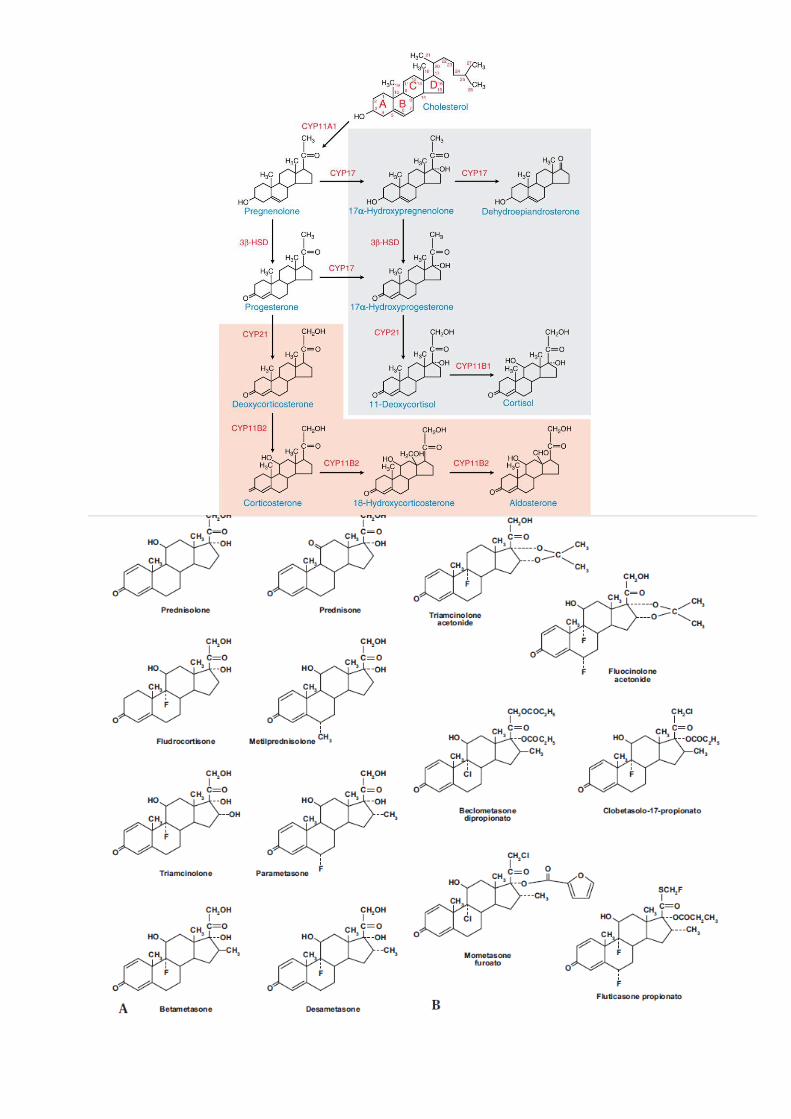
GLUCOCORTICOIDI
1. Cenni storici
Intorno al 1930 Philip Hench, un medico americano responsabile del reparto di reumatologia della Mayo
Clinic di Rochester nel Minnesota osservò che alcune sue pazienti, affette da artrite reumatoide, avevano
una remissione della sintomatologia durante la gravidanza e, poiché ciò era accompagnato da un
ingrandimento della ghiandola surrenalica, sospettò che ciò fosse dovuto ad un ormone corticosteroideo,
che egli definì sostanza X (inizialmente si supponeva che la sost.X fosse un ormone estrogenico ma non lo
era). Tra il 1930-1940 furono isolati 26 steroidi surrenalici da Edward Kendall, un collaboratore di Hench a
Rochester, e da Tadeus Reichstein a Basilea in Svizzera. Fu utilizzata una tonnellata di surrene animale per

avere un grammo di corticosteroidi. Dopo che la sostanza X fu abbastanza purificata, nel 1948 fu effettuata
la prima somministrazione di cortisone in 9 pazienti affetti da artrite reumatoide. Sulla base delle
conoscenze del tempo gli effetti antinfiammatori del cortisone erano del tutto sorprendenti ed inaspettati,
fino ad allora erano utilizzati nella cura della artrite reumatoide solo aspirina e salicilati e come cura
palliativa del dolore veniva al massimo somministrato l’oppio. L’impatto sul mondo medico di queste
osservazioni, che aprirono la strada alla terapia di patologie fino allora incurabili, fu tale che nel 1950
Hench, Kendall e Reichsten ricevettero il premio Nobel per la Medicina. Dal momento della scoperta degli
effetti antinfiammatori circa trenta principi attivi ad azione glucocorticoide sono stati sintetizzati
dall’industria farmaceutica e largamente impiegati nella terapia di numerose patologie
immunoinfiammatorie ed attualmente i glucocorticoidi sono tra i farmaci più potenti ed usati per il
trattamento di tali patologie.
2. Fisiologia
La parte corticale della ghiandola surrenale produce una serie di ormoni steroidei distinguibili in:
corticosteroidi a 21 atomi di carbonio ed androgeni a 19 atomi di carbonio. I corticosteroidi vengono distinti
secondo la loro attività principale in glucocorticoidi (attivi sul metabolismo dei carboidrati) e
mineralcorticoidi (attivi sul metabolismo degli elettroliti). I principali glucocorticoidi naturali sono: cortisolo
(idrocortisone), cortisone, corticosterone, prodotti nella zona fascicolata. I principali mineralcorticoidi sono:
aldosterone, desossicorticosterone, prodotti nella zona glomerulare. I principali androgeni sono:
testosterone, androstenedione, deidroepiandrosterone, prodotti nella zona reticolare.

Il cortisolo (o idrocortisone) è quello maggiormente prodotto: nelle 24h ne vengono secreti 8-25 mg,
mentre di aldosterone ne vengono secreti 0,15 mg/die. I corticosteroidi non sono immagazzinati nelle
ghiandole surrenali ma vengono continuamente prodotti, pertanto la quantità sintetizzata è equivalente
alla quantità secreta. La produzione non è costante, ma presenta delle variazioni circadiane. La
concentrazione plasmatica di cortisolo è massima nelle prime ore della mattina (tra le 8 e le 9 del mattino)
per poi ridursi e raggiungere il valore minimo durante il pomeriggio.
La biosintesi degli ormoni steroidei avviene a partire dal colesterolo ed è regolata dalla
adrenocorticotropina (ACTH) polipeptide a catena singola di 39 aminoacidi prodotto dalla adenoipofisi
derivante dalla pro-opiomelanocortina (POMC), la cui sintesi è a sua volta stimolata dall’ormone
ipotalamico, corticotrophin releasing hormone (CRH). L’ACTH attiva l’enzima adenilciclasi con conseguente
aumento intracellulare di cAMP ed attivazione di specifiche chinasi di tipo A (protein kinase A, PKA). A loro
volta le PKA attivano diversi enzimi per fosforilazione, tra cui una esterasi che libera il colesterolo, il
precursore dei corticosteroidi, dai depositi intracellulari dove è in forma esterificata, rendendolo
disponibile per la successiva metabolizzazione. Il colesterolo libero viene trasportato sul lato interno della
membrana mitocondriale, dove avviene la prima reazione della catena biosintetica dei corticosteroidi:
l’ossidazione del colesterolo a pregnenolone da parte del citocromo P-450. Il fattore limitante (the rate
limiting step) nella steroidogenesi è quindi la disponibilità di colesterolo libero per l’ossidazione da parte
dell’enzima mitocondriale.
L’asse ipotalamo-ipofisi-surrene che regola la sintesi dei glucocorticoidi si attiva in condizioni di stress.
Molecole proinfiammatorie (Prostaglandine, IL1/IL6,TNFα) stimolano la produzione di CRH ipotalamico. Il
risultato finale è un aumento di sintesi dei glucocorticoidi endogeni che tendono a ristabilire l’equilibrio
perturbato. L’aumento dei glucocorticoidi circolanti inoltre inibisce la sintesi e gli effetti di CRH e di ACTH,
mediante un meccanismo a feedback negativo, sia diretto (su ipotalamo e ipofisi) sia indiretto (bloccando la
sintesi delle molecole proinfiammatorie).

L’inibizione prolungata è causata da vari meccanismi di depressione genica sia a livello ipotalamico sia
ipofisario: inibizione della sintesi di CRH, riduzione dei recettori ipofisari per il CRH, inibizione della
produzione di cAMP stimolata da CRH, diminuzione dei livelli di RNA messaggero di ACTH e della pro-
opiomelanocortina. È stata infatti dimostrata la presenza di GRE negativi (vedi meccanismo di azione)nei
promotori dei geni codificanti per CRH e POMC.
3. Meccanismo d’azione molecolare
Il cortisolo, principale glucocorticoide naturale regola l’espressione di 2000-3000 proteine enzimatiche
coinvolte nel metabolismo intermedio, è provvisto sia di effetti sul metabolismo glicidico ed elettrolitico sia
di attività antinfiammatoria, ed è il termine di paragone per l’attività dei glucocorticoidi sintetici. Le
modificazioni chimiche sulla molecola del cortisolo hanno prodotto composti di sintesi con elevata attività
antinfiammatoria e trascurabile azione mineralcorticoide. D’altra parte non è stato finora possibile separare
l’attività antinfiammatoria dagli effetti sul metabolismo di carboidrati, proteine e lipidi, probabilmente
poiché sono mediati dallo stesso tipo di recettore. Pertanto i glucocorticoidi di sintesi più potenti nel
sopprimere le reazioni infiammatorie sono anche quelli che causano i maggiori effetti collaterali metabolici.
La struttura chimica di base dei glucocorticoidi è rappresentata dalla
formula del ciclopentanoperidrofenantrene a 21 atomi di carbonio.
La posizione dei radicali sostituiti ai vari atomi di carbonio rispetto al
piano della molecola steroidea è importante per l’attività biologica.
Sono designati α e β i gruppi rispettivamente al di sotto ed al di sopra
del piano dello steroide. Le caratteristiche essenziali per l’attività
biologica sono: (a)Doppio legame C4-C5 e gruppo chetonico in C3
necessari per attività glucocorticoide e mineralcorticoide; (b) Gruppo
OH in posizione beta al C11; (c) Catena laterale di due atomi di C ed
OH in posizione alfa al C17; (d) Gruppi metilici in C18 e C19 tutte
necessarie per attività glucocorticoide.
Le modificazioni chimiche della molecola di cortisolo hanno dato origine a derivati sintetici con attività glucocorticoide
e mineralcorticoide maggiormente distinte. Inoltre, queste modificazioni hanno generato derivati con potenze
maggiori e più lunga durata d’azione. Per nessuno dei derivati disponibili è possibile separare gli effetti
antiinfiammatori da quelli sul metabolismo intermedio o dagli effetti soppressivi sull’asse ipotalamo-ipofisi-surrene.
In aggiunta alla sintesi corticale i livelli tissutali di cortisolo sono regolati localmente dall’attività dell’enzima
11-beta deidrogenasi che interconverte il cortisolo a cortisone (inattivo).
Esistono due forme di 11-beta deidrogenasi (11β-HSD 1 e 11β-
HSD2). Il cortisolo per essere attivo deve avere un gruppo OH in
posizione 11, l’isoforma enzimatica 2 trasforma l’OH in un gruppo
chetonico (=O), dando origine al cortisone che è inattivo; l’isoforma
1 catalizza la reazione inversa trasformando il cortisone in cortisolo
attivo. Il bilancio tra questi due enzimi regola la concentrazione di cortisolo a livello tissutale. In particolare
questa interconversione, che inibisce il legame del cortisolo al recettore mineralcorticoide, inattivandolo a
cortisone, avviene nel fegato, nel rene e nel colon dove è maggiormente espresso l’isozima 2.
I glucocorticoidi interagiscono con specifici recettori citosolici regolando sia positivamente che
negativamente la sintesi di proteine nelle cellule bersaglio. I glucocorticoidi entrano nelle cellule bersaglio
per diffusione passiva e si legano con alta affinità a specifici recettori citosolici (glucocorticoid receptors,
GR). Il complesso glucocorticoide-recettore forma degli omodimeri che traslocano nel nucleo dove

interagiscono con specifiche sequenze di DNA (glucocorticoid responsive elements, GRE) situate nelle
regioni regolatrici di specifici geni. Tale interazione altera la velocità di trascrizione, causando induzione o
repressione genica.
I recettori dei glucocorticoidi, che fanno parte di una superfamiglia genica che include i recettori degli
ormoni steroidei, degli ormoni tiroidei, della vitamina D e dell’acido retinoico, sono stati identificati in quasi
tutti i tessuti ed hanno una
densità variabile da 3000 a
10000 per cellula. L’entità della
risposta biologica dipende dal
numero di recettori che
interagiscono con il ligando.
Sono state identificate due
isoforme del recettore
glucocorticoide umano derivate da splicing alternativo dell’RNA messaggero. L’isoforma α (GRα) consiste di
777 aminoacidi e rappresenta la proteina capace di legare il glucocorticoide e di interagire con i GRE
modulando l’espressione genica mentre l’isoforma β (GRβ) consiste di 742 aminoacidi, con un HBD
accorciato. Le due isoforme sono identiche nei primi 727 aminoacidi, ma poi differiscono al
carbossiterminale in quanto GRα presenta altri 50 residui mentre GRβ presenta 15 aminoacidi non
omologhi. L’isoforma recettoriale funzionante è l’α (N.B.: riguardo a β il professore a lezione ha detto che il
recettore β lega l’ormone, ma non è in grado di dimerizzare e quindi di trascrivere, sul suo libro però c’è
scritto che GRβ, poiché ha un HBD più corto, non è in grado di interagire con il ligando, ma compete per il
sito di legame sui GRE pur essendo inattivo sulla trascrizione, pertanto una elevata espressione di GRβ
potrebbe contribuire al fenomeno della resistenza tissutale ai glucocorticoidi).

La regolazione dell’espressione genica da parte dei glucocorticoidi dipende dallo stato di acetilazione
degli istoni dipendente a sua volta dalla attività di enzimi specifici come le istone acetiltransferasi (HATs)
e le istone deacetilasi (HDACs).
1) In assenza dell’ormone GRα forma un complesso eteromerico insieme a due molecole di heat shock
protein hsp90 ed una molecola di hsp56 (step 1 della figura).L’associazione con hsp90 mantiene il
recettore in una conformazione adatta a legare l’ormone, mentre impedisce il legame al DNA.
2) In seguito al legame dell’ormone all’HBD del recettore si verifica la dissociazione delle molecole hsp
e la liberazione di un monomero ormone-recettore (step 2).
3) Il monomero ormone-recettore dimerizza formando omodimeri che traslocano nel nucleo (step 4).
4) Il dimero nel nucleo interagisce, per mezzo del dominio DBD con sequenze nucleotidiche specifiche
dette GRE positive, presenti a monte della TATAbox nei promotori di geni di cui causano un
aumento della loro espressione, reclutando funzioni acetilasiche, istone-acetil-transferasi HAT che
portano a uno srotolamento della cromatina (step 5).
5) È stato dimostrato d’altra parte che il GRα, sotto forma di monomero, può legarsi a sequenze
nucleotidiche differenti, dette GRE negative nel promotore di geni specifici, la cui espressione
risulta inibita tramite il reclutamento di deacetilasi istoniche HDAC (step 6) (ad es. il meccanismo
del feedback negativo è mediato da sequenze GRE negative presenti nei promotori di CRH e
POMC).
Quindi se il promotore di un gene presenta una GRE positiva il dimero GRα ne induce un aumento della sua
espressione, se presenta una GRE negativa il monomero GRα ne induce una repressione trascrizionale.
L’azione antiinfiammatoria e immunosoppressiva dei glucocorticoidi, però, non avviene attraverso
l’interazione con sequenze GRE negative, infatti la maggior parte dei geni codificanti per proteine
proinfiammatorie non presenta tali sequenze nel promotore, bensì avviene attraverso l’interazione con altri
fattori di trascrizione, ovvero bloccando l’attività transattivante di fattori come AP-1 (jun/fos) e NF-κB
(p50/p65) che nel corso della risposta infiammatoria attivano la trascrizione di geni per citochine,
chemochine, proteine d’adesione, cox, lipossigenasi, fosfolipasi A2, iNOs.
L’inibizione di AP-1 e NF-κB può avvenire attraverso tre diversi meccanismi:
a) A livello citosolico, interazione diretta proteina-proteina tra GRα monomerico da un lato e AP-1 o
NF-κB dall’altro(step 3).
b) A livello nucleare, interazione del dimero con AP-1 e NF-kB quando questi sono legati alle loro
sequenze responsive, reclutando funzioni deacetilasiche e interferendo dunque con la loro attività
transattivante (step 7)
c) Induzione da parte del dimero GRα di IkBα, che mantiene l’NF-κB in forma non attiva nel
citoplasma.
Quindi i geni proinfiammatori risultano downregolati dai glucocorticoidi indipendentemente dalla
presenza di sequenze GRE negative, ma dall’interferenza diretta o indiretta con l’attività transattivante di
NF-kB e AP-1 con effetto immunosoppressivo ed antinfiammatorio. Inoltre a dosi più alte i glucocorticoidi
sono anche in grado di indurre l’espressione di geni antiinfiammatori, nel cui promoter sono presenti GRE
positive.

4. Effetti antinfiammatori ed immunosoppressivi dei glucocorticoidi
L’infiammazione è una risposta dell’organismo a stimoli lesivi endogeni o esogeni sostenuta dalla
liberazione di numerosi mediatori, tra cui hanno particolare importanza citochine ed eicosanoidi, e dal
coinvolgimento di numerose cellule sia facenti parte del tessuto infiammato, sia ivi migrate dal sangue.
La reazione infiammatoria acuta è una risposta positiva che tende ad eliminare la causa scatenante ed a
ristabilire l’equilibrio perturbato. Talvolta, per fenomeni ancora non del tutto chiari che portano anche
all’attivazione del sistema immunitario, l’infiammazione si cronicizza con produzione di mediatori e
migrazione cellulare incontrollate che possono a loro volta causare un grave danno tissutale ed organico.
Tra le cause e gli effetti della attivazione del sistema immunitario è di particolare rilievo la produzione da
parte di numerose cellule infiammatorie di citochine come IL-1, TNF-α, IL-6, IL-8. Queste proteine, oltre a
indurre la propria sintesi, hanno numerose azioni di amplificazione del processo infiammatorio come
stimolazione della sintesi dei mediatori lipidici, chemiotassi, attivazione e proliferazione cellulare.
I glucocorticoidi inibiscono la sintesi e/o gli effetti dei mediatori lipidici e delle citochine. Inoltre bloccano
attivazione, migrazione e proliferazione di tutte le cellule coinvolte nella risposta infiammatoria. Tali effetti
dipendono sia dall’inibizione della sintesi di proteine proinfiammatorie e immunostimolanti sia
dall’induzione della sintesi di proteine antiinfiammatorie e immunosoppressive (vedi tabella
sotto).
Grazie a tali meccanismi i glucocorticoidi sono i più potenti farmaci antinfiammatori ed immunosoppressori
attualmente disponibili. Sono farmaci antiinfiammatori principalmente perché inibiscono la sintesi di IL-1 e

TNFα e di enzimi quali la Fosfolipasi A2 (perciò è anche un potente antiasmatico). Sono farmaci
immunosoppressivi perché sono in grado di indurre l’apoptosi linfocitaria.
a) Inibizione della sintesi di proteine proinfiammatorie e immunostimolanti
I glucocorticoidi inibiscono la sintesi di citochine coinvolte nella risposta infiammatoria, tale inibizione
dipende in gran parte dagli effetti del GRα attivato su altri fattori di trascrizione come AP-1 e NF-κB. È noto
infatti che sostanze come i lipopolisaccaridi (LPS) batterici o altri stimoli mitogeni inducono la sintesi di
citochine attivando fattori di trascrizione come AP-1 ed NF- κB . Grazie a questi meccanismi, i
glucocorticoidi sono in grado di inibire la sintesi delle citochine e gli effetti delle citochine stesse sulla
induzione dell’espressione genica. A questo riguardo assume particolare importanza per l’effetto
antiflogistico dei glucocorticoidi la loro capacità di inibire l’induzione da parte di IL-1 e TNF-α di enzimi
come fosfolipasi A2, ciclossigenasi e nitrossidosintasi inducibili. Il risultato finale è il blocco della sintesi e
del rilascio dei mediatori come prostaglandine e nitrossido, blocco che media, almeno in parte, gli effetti
dei glucocorticoidi sulla componente vascolare dell’infiammazione: inibizione della vasodilatazione e
dell’aumento della permeabilità capillare.
Oltre agli aspetti vascolari, una caratteristica fondamentale della risposta infiammatoria è, come sopra
discusso, la migrazione leucocitaria. Questa è provocata da sostanze chemiotattiche come leucotrieni, PAF,
chemochine e da una serie di molecole di adesione che insieme mediano la marginazione e poi l’attacco dei
leucociti circolanti all’endotelio capillare e la migrazione leucocitaria attraverso le giunzioni tra le cellule
endoteliali. I glucocorticoidi inibiscono fortemente la migrazione leucocitaria bloccando la formazione di
mediatori lipidici, chemochine e molecole di adesione. Gli effetti antinfiammatori finora citati dipendono da
inibizione della sintesi di proteine direttamente coinvolte nel processo infiammatorio: citochine, molecole
di adesione, enzimi biosintetici di mediatori.
Grazie all’inibizione, indiretta, della fosfolipasi A2, i glucocorticoidi inibiscono sia la produzione di
prostanoidi sia di leucotrieni per cui gli effetti sono sia sulla fase vascolare, che su quella cellulare
dell’infiammazione. Inoltre poiché non esiste un inibitore selettivo per le lipoossigenasi, i glucocorticoidi
sono gli unici farmaci in grado di inibire la produzione dei leucotrieni broncocostrittori agendo più in alto
nella cascata a livello dell’acido arachidonico. (importante farmaco antiasmatico).
STUDI SULL’ATTIVITA’ INIBENTE IL METABOLISMO DELL’ACIDO ARACHIDONICO
ESPERIMENTO N.1: EDEMA DA CARRAGENINA vs. PREPARAZIONI ENZIMATICHE SUBCELLULARI
Nel 1972 immediatamente dopo la scoperta del meccanismo d’azione dei FANS sulla sintesi delle
prostaglandine ci si chiese quale fosse l’effetto dei glucocorticoidi sul metabolismo dell’acido arachidonico,
in quanto gli effetti clinici erano gli stessi ma quelli dei glucococorticoidi molto più potenti.
Si effettuarono test in vitro e in vivo. Per i test in vitro si valutò l’attività inibente sulla prostaglandina
sintetasi. Per i test in vivo si utilizzò la metodica dell’edema indotto da carragenina (modello
infiammatorio sperimentale in cui nelle due zampe posteriori di un ratto si inietta un agente flogogeno, la
carragenina, un polisaccaride solforato, che induce infiammazione nella zampa, che aumenta di volume a
causa dell’edema. L’edema che si produce deve essere valutato: una volta si sacrificava l’animale, si
amputavano gli arti e si pesavano; oggi si usa il pletismometro ad acqua, la zampa si introduce in
un’ampolla e lo spostamento del volume di acqua, pari a quello della zampa che ne ha indotto lo
spostamento, viene misurato dall’apparecchio).
Per i FANS l’attività inibente in vitro della prostaglandina sintetasi corrisponde a una buona attività in vivo
antiinfiammatoria.
Differentemente il desometasone e gli altri glucocorticoidi presentano un’attività in vivo notevolmente
maggiore rispetto a quella dei FANS, ma non presentano alcuna attività inibente l’enzima in vitro. I
glucocorticoidi, infatti, non hanno effetto su preparazioni enzimatiche subcellulari, in quanto necessitano
della cellula intera per esplicare la loro azione che si basa sulla regolazione genica.

b) Induzione della sintesi di proteine antiinfiammatorie e immunosoppressive
I glucocorticoidi esercitano, inoltre, azione antinfiammatoria anche stimolando la sintesi proteica. È stato
infatti dimostrato che questi composti inducono la sintesi ed il rilascio in vitro ed in vivo di una proteina di
37 kDa, la annessina-1, che possiede attività antinfiammatoria ed immunosoppressiva sperimentale simile
a quella dei glucocorticoidi. Il suo promoter include un TATAbox e una sequenza GRE positiva.
Le principali azioni dell’annessina-1 nell’infiammazione sono:
- l’inibizione della attivazione della fosfolipasi A2 con conseguente blocco del rilascio degli eicosanoidi pro-
infiammatori,
- l’inibizione dell’espressione di COX-2 e iNOS,
- l’inibizione della migrazione leucocitaria con meccanismi non del tutto chiari, ma che probabilmente
coinvolgono l’interazione della proteina con molecole di adesione.
- l’induzione del l’apoptosi di cellule infiammatorie, come neutrofili e monociti, fenomeno che può
contribuire alla risoluzione del processo infiammatorio.
Un’altra proteina antinfiammatoria indotta dai glucocorticoidi è la Mitogen Activated Protein Kinase
(MAPK) fosfatasi- 1. Questa fosfatasi inattiva per defosforilazione tutte le proteine appartenenti alla
famiglia delle MAP kinasi, pertanto può inibire l’attività della fosfolipasi A2 citosolica che è attivata dalla
fosforilazione da parte di MAP kinasi. Inoltre la MAPK fosfatasi-1 può defosforilare ed inattivare la Jun N-
terminal kinase con la conseguenza di inibire l’espressione dei geni responsivi ad AP- 1, meccanismo
inibitorio addizionale alla interazione proteina-proteina tra GRα ed AP-1 descritta in precedenza.
Ricercatori italiani hanno dimostrato che i glucocorticoidi inducono la sintesi di una Glucocorticoid-Induced
Leucine Zipper (GILZ) proteina che è in grado di inibire l’attivazione delle MAP kinasi e la trascrizione genica
dipendente da AP-1 e NF-κB .
Un potenziale effetto antinfiammatorio dei glucocorticoidi dipendente da induzione di sintesi proteica
riguarda infine i recettori di IL-1. È noto che la IL-1 produce i suoi numerosi effetti infiammatori ed
immunostimolanti interagendo con due differenti recettori di membrana. Solamente il recettore di tipo I
(80 kDa) è in grado di tradurre il segnale mediando gli effetti della citochina, mentre il recettore di tipo II
(60 kDa) lega la citochina, ma non è in grado di attivare la risposta cellulare. È stato pertanto proposto che il
recettore di tipo II (decoy receptor) agisca da inibitore fisiologico della IL-1. I glucocorticoidi
antinfiammatori inducono potentemente la trascrizione del recettore di tipo II ed inoltre stimolano il
ESPERIMENTO N.2: PERFUSIONE A CASCATA DI DUE POLMONI DI CAVIA
Quando il primo polmone veniva perfuso da desometasaone si liberava qualcosa che agiva sul secondo
polmone riducendo la produzione di mediatori del metabolismo dell’acido arachidonico. Si capì che i
glucocorticoidi stimolavano nel primo polmone la sintesi di un fattore proteico (proteico in quanto veniva
inibito da somministrazione di ciclosimide, un inibitore della sintesi proteica). Questo fattore inibiva il
rilascio dei prodotti del metabolismo dell’acido arachidonico, ma ancora non si capiva a che livello agisse.
Si capì a che livello agiva solo con la somministrazione dello stesso acido arachidonico al sistema. Con la
somministrazione dell’acido arachidonico si bloccava l’effetto dei glucocorticoidi perché questi agiscono a
monte dell’acido arachidonico sulla fosfolipasi a2 e non bloccano le cox e lipossigenasi. Dopo una serie di
ricerche si arrivò alla conclusione che il polipeptide liberato che causava l’effetto antifosfolipasi era
l’annessina-1 (o lipocortina).
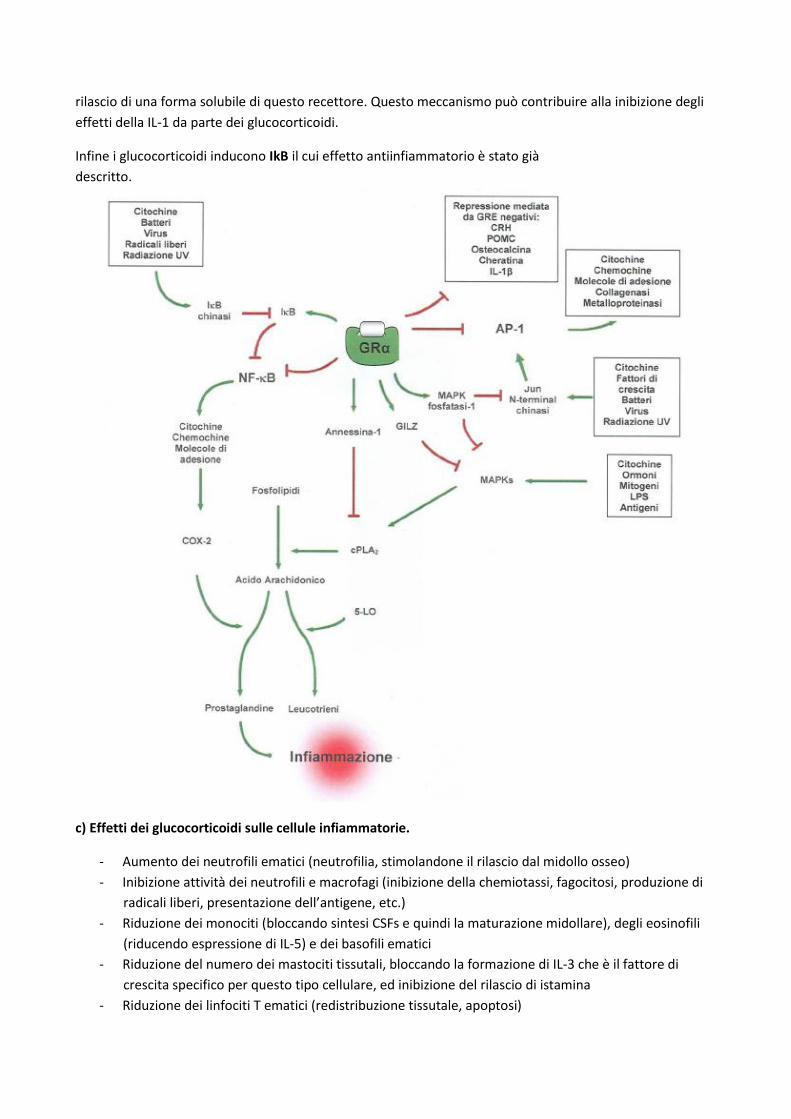
rilascio di una forma solubile di questo recettore. Questo meccanismo può contribuire alla inibizione degli
effetti della IL-1 da parte dei glucocorticoidi.
Infine i glucocorticoidi inducono IkB il cui effetto antiinfiammatorio è stato già
descritto.
c) Effetti dei glucocorticoidi sulle cellule infiammatorie.
- Aumento dei neutrofili ematici (neutrofilia, stimolandone il rilascio dal midollo osseo)
- Inibizione attività dei neutrofili e macrofagi (inibizione della chemiotassi, fagocitosi, produzione di
radicali liberi, presentazione dell’antigene, etc.)
- Riduzione dei monociti (bloccando sintesi CSFs e quindi la maturazione midollare), degli eosinofili
(riducendo espressione di IL-5) e dei basofili ematici
- Riduzione del numero dei mastociti tissutali, bloccando la formazione di IL-3 che è il fattore di
crescita specifico per questo tipo cellulare, ed inibizione del rilascio di istamina
- Riduzione dei linfociti T ematici (redistribuzione tissutale, apoptosi)

- Inibizione della produzione di anticorpi da parte dei linfociti B (anche se il meccanismo non è stato
ancora bene chiarito)
- Inibizione della proliferazione delle cellule endoteliali e dei fibroblasti, con riduzione dei fenomeni
tipici dell’infiammazione cronica (angiogenesi e fibrosi), ma che ritarda anche i meccanismi
riparativi postinfiammatori.
Per quanto riguarda i linfociti, in misura maggiore i linfociti T, i glucocorticoidi sono in grado di indurne
l’apoptosi sia con un meccanismo indiretto mediante il blocco di NFkB e AP1, induttori di proteine
antiapoptotiche, sia con un meccanismo diretto proapoptotico, con l’induzione della sintesi di particolari
proteine (ad es. annessina-1)
5. Indicazioni cliniche dei glucocorticoidi
Gli usi terapeutici dei glucocorticoidi possono essere distinti in due categorie:
- Terapia sostitutiva in stati di deficit corticosurrenalico
- Terapia antiinfiammatoria ed immunosoppressiva
N.B. Con l’eccezione della terapia sostitutiva nelle condizioni di deficit, la somministrazione di
glucocorticoidi è ampiamente EMPIRICA, non cura la malattia: i glucocorticoidi non sono né specifici né
curativi, possiedono piuttosto effetti palliativi in virtù delle loro proprietà antiinfiammatorie ed
immunosoppressive.
(a) Terapia sostitutiva
L’insufficienza surrenalica può essere primaria se causata da lesioni della corteccia surrenalica (per cause
congenite come nel M.di Addison, per fenomeni autoimmuni, a seguito di infezioni o neoplasie) o
secondaria se causata da lesioni dell’ipofisi anteriore o dell’ipotalamo
Insufficienza surrenalica acuta: questa patologia potenzialmente fatale è caratterizzata da sintomi GI
(nausea, vomito, dolori addominali), disidratazione, iponatremia, iperkaliemia, debolezza, letargia e
ipotensione. Essa è solitamente associata a patologie del surrene (insuff.primarie) piuttosto che dell’ipofisi-
ipotalamo, ma talvolta fa seguito a una brusca sospensione della somministrazione di glucocorticoidi ad
alte dosi o per periodi prolungati, che determina una repressione dell’asse ipotalamo-ipofisi-surrene (per
questo motivo è sempre indicata una sospensione graduale della terapia con dosi decrescenti).La terapia
prevede la somministrazione di un bolo da 100 mg di idrocortisone i.v. ogni 8 ore fino alla stabilizzazione,
poi 25 mg i.v. ogni 6-8 ore
Insufficienza surrenalica cronica: è caratterizzata da sintomi simili, ma meno severi. Questi pazienti
richiedono un trattamento giornaliero con corticosteroidi. I regimi di sostituzione prevedono: 25-30 mg di
idrocortisone per os al giorno in 2-3 somministrazioni di cui il 60-70% della dose al mattino.
La somministrazione del solo glucocorticoide è in grado di esplicare anche l’attività sul metabolismo
idrosalino, molti pazienti però in caso di insufficienza surrenalica severa necessitano anche la terapia
sostitutiva con mineralcorticoidi (generalmente si utilizza il fludrocortisone a dosi di 0.05-0.2 mg/die)
Iperplasia surrenalica congenita: questo termine indica un insieme di patologie genetiche in cui è
deficitaria l’attività di uno dei numerosi enzimi necessari per la biosintesi del cortisolo (solitamente il
difetto genico è una mutazione di CYP21, enzima che catalizza la reazione di idrossilazione in posizione
C21). La produzione ridotta di cortisolo e la conseguente mancanza del feedback negativo determinano un

incremento della secrezione di ACTH, con conseguente iperplasia della ghiandola. Ne consegue che altri
steroidi, che precedono il blocco enzimatico nella via biosintetica, vengono prodotti in eccesso, come gli
androgeni. Lo scopo della terapia è pertanto quello di ristabilire livelli fisiologici di ormoni steroidei, di
ridurre la produzione di ACTH e conseguentemente la produzione di androgeni surrenalici. La dose abituale
di cortisolo è di circa 0.6 mg/kg per os al giorno suddivisa in quantità ineguali (due terzi al mattino, un terzo
alla sera). In caso di necessità il mineralcorticoide da somministrare è il fludrocortisone acetato alla dose di
0.05-0.2 mg al giorno.
(b) Terapia antiinfiammatoria e immunosoppressiva
A parte l’uso nella terapia sostitutiva delle insufficienze surrenaliche, i glucocorticoidi sono largamente
utilizzati nella terapia di gravi sindromi a patogenesi immunoinfiammatoria spesso su base
autoimmunitaria. L’uso dei glucocorticoidi in questi casi è palliativo, in quanto le cause iniziali della
malattia, spesso sconosciute, rimangono inalterate. D’altra parte, l’efficace soppressione della sintesi e
degli effetti infiammatori dei vari mediatori da parte dei glucocorticoidi causa forte attenuazione dei
sintomi clinici, che in qualche caso può avere un’azione salvavita. La corretta utilizzazione dei
glucocorticoidi richiede una analisi approfondita della malattia, dello stato del paziente e dei possibili effetti
collaterali. In considerazione del numero e della gravità dei potenziali effetti collaterali, la decisione di
iniziare una terapia con glucocorticoidi richiede sempre un’attenta considerazione dei rischi e dei benefici
per ciascun paziente. Per qualsiasi patologia e per qualsiasi paziente la dose appropriata per ottenere un
effetto terapeutico deve essere individuata per tentativi e riesaminata periodicamente, sia per osservare il
decorso della patologia di base, sia per valutare le eventuali complicanze.
Alcuni criteri per un ottimale uso clinico dei glucocorticoidi in patologie extrasurrenaliche sono:
1. Utilizzare solo in presenza di una diagnosi definita
2. La dose terapeutica va individualizzata per ciascun paziente per successivi tentativi (by trial and error) e
va individuata riducendola gradualmente finchè il peggioramento dei sintomi non indica che si è raggiunta
la dose minima accettabile.
3. Somministrare la dose efficace più bassa per il periodo più breve in un’unica somministrazione alle ore 8.
4. Instaurare quanto prima possibile una somministrazione a giorni alterni
5. Ridurre l’apporto calorico per prevenire l’aumento di peso
6. Ridurre l’assunzione di sodio per prevenire la formazione di edemi, lo sviluppo di ipertensione e la
perdita di potassio. Se necessario aumentare la somministrazione di potassio
7. Laddove possibile, sostituire o integrare con altri farmaci (p.es. antinfiammatori non steroidei) in modo
da ridurre la dose di glucocorticoidi
8. La severità e l’incidenza degli effetti collaterali aumentano con la dose e la durata di somministrazione
9. La sospensione improvvisa di una terapia prolungata con alte dosi di glucocorticoidi può causare una
grave crisi di insufficienza surrenale acuta, che può essere fatale.
10. La somministrazione di alte dosi per brevi periodi (fino ad una settimana), in assenza di
controindicazioni specifiche, causa trascurabili effetti collaterali.
Per quanto riguarda la scelta del farmaco da utilizzare per terapie prolungate la preferenza va ai composti
con durata di azione intermedia non fluorurati come prednisolone e metil-prednisolone. Questi farmaci
hanno minori effetti cutanei e muscolari e permettono una somministrazione orale unica al mattino in
modo da minimizzare gli effetti inibitori sulla produzione ipotalamica di CRH. Infatti al mattino la
cortisolemia endogena è più elevata e di conseguenza è fisiologicamente soppressa la secrezione di CRH.
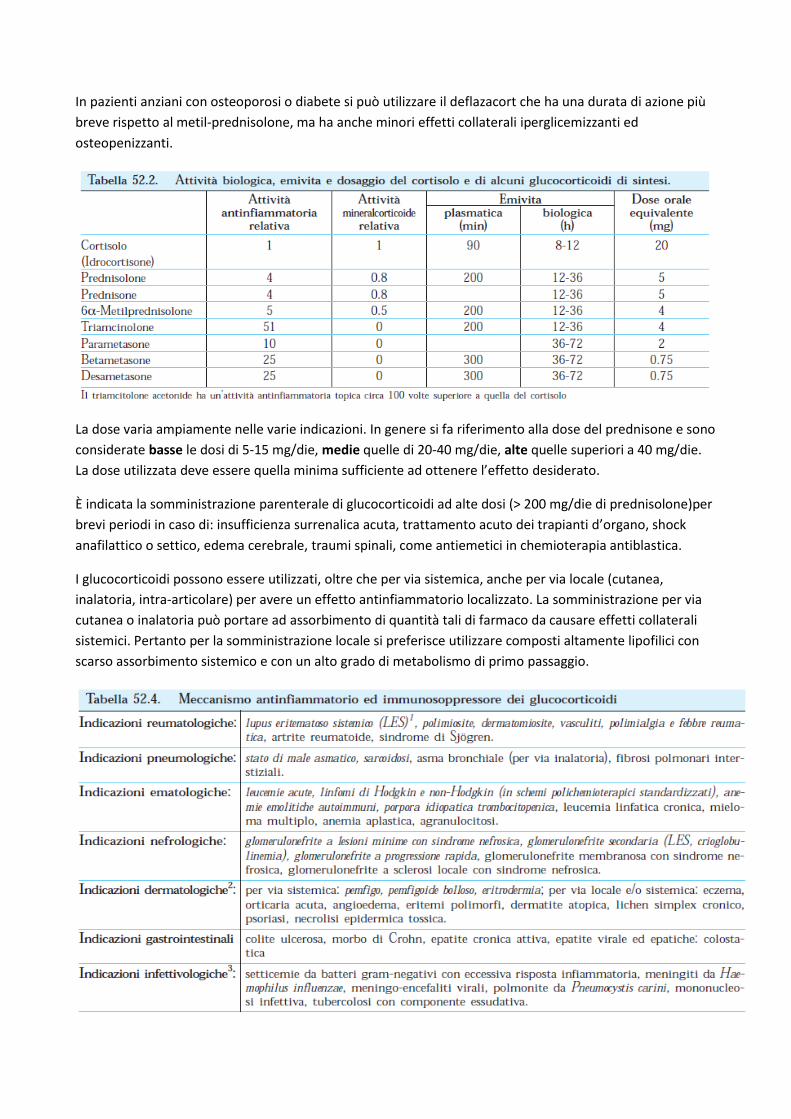
In pazienti anziani con osteoporosi o diabete si può utilizzare il deflazacort che ha una durata di azione più
breve rispetto al metil-prednisolone, ma ha anche minori effetti collaterali iperglicemizzanti ed
osteopenizzanti.
La dose varia ampiamente nelle varie indicazioni. In genere si fa riferimento alla dose del prednisone e sono
considerate basse le dosi di 5-15 mg/die, medie quelle di 20-40 mg/die, alte quelle superiori a 40 mg/die.
La dose utilizzata deve essere quella minima sufficiente ad ottenere l’effetto desiderato.
È indicata la somministrazione parenterale di glucocorticoidi ad alte dosi (> 200 mg/die di prednisolone)per
brevi periodi in caso di: insufficienza surrenalica acuta, trattamento acuto dei trapianti d’organo, shock
anafilattico o settico, edema cerebrale, traumi spinali, come antiemetici in chemioterapia antiblastica.
I glucocorticoidi possono essere utilizzati, oltre che per via sistemica, anche per via locale (cutanea,
inalatoria, intra-articolare) per avere un effetto antinfiammatorio localizzato. La somministrazione per via
cutanea o inalatoria può portare ad assorbimento di quantità tali di farmaco da causare effetti collaterali
sistemici. Pertanto per la somministrazione locale si preferisce utilizzare composti altamente lipofilici con
scarso assorbimento sistemico e con un alto grado di metabolismo di primo passaggio.

Nella tabella sono riportate in corsivo le indicazioni ‘elettive’ nelle quali i glucocorticoidi sono i farmaci di elezione o di prima scelta in assenza di specifiche controindicazioni, le altre indicazioni sono ‘selettive’ in cui i glucocorticoidi non sono farmaci di prima scelta, ma sono utilizzati in casi con complicanze o resistenti ad altre terapie. Nelle infezioni batteriche il trattamento con glucocorticoidi va associato alla somministrazione di chemioantibiotici.
(c) Controindicazioni
Sono controindicazioni assolute alla somministrazione dei glucocorticoidi: l’osteoporosi grave
(schiacciamenti vertebrali), il diabete instabile, le psicosi gravi e gli stati di immunodeficienza. Sono
controindicazioni relative in cui i glucocorticoidi vanno utilizzati con grande cautela e solamente se non
esistono alternative terapeutiche: osteoporosi lieve, diabete stabile, infezioni in atto, ulcera peptica attiva,
ipertensione arteriosa grave, insufficienza renale grave.
6. Farmacocinetica
I glucocorticoidi sono efficacemente assorbiti per via orale essendo liposolubili e scarsamente idrosolubili.
La coniugazione al carbonio 21 con gruppi fosfato ed emisuccinato fa aumentare la idrosolubilità e
permette la somministrazione per via endovenosa . I glucocorticoidi sono assorbiti anche dai siti di
applicazione locale come articolazioni, cute, polmoni. La somministrazione prolungata a livello cutaneo e
polmonare può risultare nell’assorbimento di quantità di farmaco sufficienti a provocare effetti collaterali
sistemici.
Nel plasma cortisolo e prednisolone si legano a due proteine: una globulina, la transcortina, caratterizzata
da alta affinità e bassa capacità totale di legame, e l’albumina, caratterizzata da bassa affinità ed alta
capacità totale di legame.
I glucocorticoidi sono estesamente metabolizzati ed escreti come metaboliti e solo in minima parte
immodificati. Le principali reazioni metaboliche sono: riduzione del doppio legame tra gli atomi di carbonio
4 e 5, che avviene nel fegato ed in tessuti extraepatici, e successiva riduzione del chetone in posizione 3,
che è stata osservata solo nel fegato. I metaboliti ridotti, che sono biologicamente inattivi, vengono
coniugati a livello dell’idrossile in posizione 3 con solfato o acido glicuronico nel fegato ed in piccola parte
anche nel rene. I composti coniugati, più idrosolubili, sono escreti per via renale. La escrezione per via
biliare o fecale è trascurabile nell’uomo. I valori di emivita plasmatica e biologica dimostrano che la
presenza di un doppio legame in posizione 1-2 (prednisolone, metilprednisolone) e di un atomo di fluoro al
carbonio 6 (parametasone) o al carbonio 9 (betametasone, desametasone), riduce la velocità di
metabolismo e fa aumentare la durata di azione del farmaco (Tabella 52.2).
Una via addizionale di metabolismo è la continua interconversione di cortisolo-cortisone e prednisolone-
prednisone catalizzata dall’enzima 11β-idrossisteroidodeidrogenasi. Infatti, gli steroidi cortisone e
prednisone sono biologicamente inattivi avendo un gruppo chetonico all’atomo di carbonio 11 e devono
essere convertiti nei rispettivi cortisolo e prednisolone.
L’eliminazione di questi composti è ridotta nelle gravi insufficienze epatiche e nell’ipotiroidismo, mentre è
aumentata nell’ipertiroidismo, nelle ipoalbuminemie e nelle sindromi nefrotiche.

7. Effetti collaterali
I glucocorticoidi sono farmaci molto efficaci e di grande valore terapeutico, ma potenzialmente molto
pericolosi perché in grado di provocare numerosi e severi effetti collaterali (Tabella 52.5).
I glucocorticoidi di sintesi hanno trascurabile attività mineralcorticoide (il recettore è diverso), ma
conservano l’azione di inibizione sull’asse ipotalamo-ipofisisurrene ed i vari effetti metabolici che sono
responsabili dei gravi effetti collaterali che si verificano in corso della terapia antinfiammatoria con
glucocorticoidi (gli effetti antiinfiammatori e quelli sul metabolismo intermedio sono mediati dallo stesso
recettore).
N.B. Gli effetti collaterali rappresentano manifestazioni eccessive dei rispettivi effetti fisiologici
normalmente esplicati dai glucocorticoidi.
Sindrome di Cushing iatrogena: Sindrome caratterizzata da alterazioni del metabolismo intermedio e da
disturbi endocrini causata dalla somministrazione cronica di dosi farmacologiche di glucocorticoidi
La tossicità dei glucocorticoidi è spesso direttamente proporzionale alla durata della terapia ed alla dose
utilizzata. La somministrazione di dosi farmacologiche per alcuni giorni non causa effetti tossici rilevanti, ma
se la somministrazione si prolunga per alcune settimane o mesi, i seguenti effetti collaterali si manifestano
con maggiore o minore severità nella maggior parte dei pazienti trattati.
Distinguiamo pertanto effetti collaterali DOSE e TEMPO DIPENDENTI, da effetti collaterali INDIPENDENTI
DALLA DOSE e DALLA DURATA DELLA TERAPIA.

Effetti collaterali dose e tempo dipendenti:
a) SOPPRESSIONE DELL’ASSE IPOTALAMO-IPOFISI-SURRENE: 20-30 mg/die di prednisolone
somministrati per un periodo di tempo superiore a 1 settimana, porta SEMPRE all’inibizione della
sintesi del CRH ipotalamico e della POMC ipofisaria, per il meccanismo di feedback negativo
regolato da sequenze GRE negative nei promotori di tali geni. Le manifestazioni cliniche sono
l’atrofia surrenalica con conseguente insufficienza caratteristici della cosiddetta sindrome da
sospensione acuta del trattamento. Per evitare questi effetti in caso di terapia prolungata è buona
norma somministrare il glucocorticoide in dose unica mattutina, cioè quando l’asse ipotalamo-
ipofisi-surrene è già fisiologicamente inibito dal feedback endogeno, e in caso di sospensione del
trattamento farmacologico, questa deve essere graduale.
b) EFFETTO DIABETOGENO: i glucocorticoidi inducono la sintesi a livello epatico di vari enzimi coinvolti
nella gluconeogenesi, e svolgono azione anti-insulinica sia causando un aumento della
concentrazione plasmatica di glucagone, sia a livello periferico riducendo l’espressione delle
proteine trasportatrici del glucosio (GLUT). Il risultato netto è un aumento della glicemia. In questo
modo i glucocorticoidi possono peggiorare il controllo della glicemia in pazienti con diabete
preesistente e possono scatenare una sindrome diabetica in soggetti geneticamente predisposti. In
questi casi è preferenziale l’uso del deflazacort, che, anche se non è noto il motivo, sembra avere
un minor effetto diabetogeno.
c) RITENSIONE IDROSALINA: I glucocorticoidi naturali (cortisolo)causano disturbi del ricambio
elettrolitico con ritenzione di sodio ed aumentata escrezione di potassio (infatti questi presentano
un rapporto attività glucocorticoide/attività mineralcorticoide 1:1, perché legano attivandolo sia il
recettore glucocorticoideo che mineralcorticoideo). I chimici farmaceutici sono però riusciti a
scindere le due attività, producendo glucocorticoidi sintetici che non sono in grado di legare il
recettore mineralcorticoideo (quindi il rapporto attività glucocorticoidea/ mineralcorticoidea per
questi farmaci dovrebbe essere 1:0). In realtà l’attività sul metabolismo idrosalino di questi farmaci
non è mai zero, in quanto è vero che hanno una minore affinità rispetto al cortisolo per il recettore
mineralcorticoide, ma possono causare ugualmente ritenzione di sodio mediante meccanismi di
modulazione genica recentemente identificati. È stato dimostrato che il desametasone stimola, per
la presenza di GRE positivi, l’espressione di una specifica chinasi (Serum- and-
Glucocorticoidregulated Kinase, SGK) che fosforila e attiva gli ENaCs (Canali per il Sodio epiteliali,
che invece sono indotti dal recettore mineralcorticoideo). Questi effetti possono portare a
ritenzione di sodio e ipokaliemia e contribuire allo sviluppo di sindromi ipertensive. In questi casi è
opportuna la riduzione dell’assunzione di sodio, la somministrazione di potassio, e l’aggiustamento
delle dosi.
d) AUMENTATA SENSIBILITÀ ALLE INFEZIONI: per gli effetti immunosoppressivi la terapia con
glucocorticoidi si associa frequentemente sia all’insorgenza di infezioni opportunistiche sia alla
riattivazione di infezioni latenti. È stata anche osservata una riattivazione del citomegalovirus
(CMV) ad esempio in pazienti sottoposti a trapianto d’organo in trattamento con glucocorticoidi.
Tale riattivazione è causata dalla generale immunosoppressione, ma anche dalla diretta
stimolazione della replicazione virale dovuta alla presenza, recentemente dimostrata, di GRE
positivi nel promotore del CMV. In questi casi è di fondamentale importanza la profilassi chemio-
antibiotica.
e) MIOPATIA: debolezza muscolare degli arti e delle articolazioni scapolo-omerale e pelvica a causa
degli effetti catabolici dei glucocorticoidi sul tessuto muscolare. Questa complicazione può
verificarsi anche dopo trattamento di breve durata e con tale severità da impedire la

deambulazione, e può pertanto costituire una indicazione per la sospensione del trattamento. Gli
effetti muscolari dipendono in gran parte dall’induzione di geni codificanti per proteine
appartenenti alla famiglia ubiquitina-proteasoma che stimolano la proteolisi muscolare. Questo
effetto collaterale è discusso, la miopatia non è sviluppata da tutti i pazienti, nel caso si verifichi
bisogna limitare la dose al di sotto dei 10 mg/die.
f) RIDISTRIBUZIONE DEL GRASSO CORPOREO: si verificano i caratteristici segni della sindrome
cushingoide iatrogena: gibbo di bufalo (buffalo hump) per accumulo di grasso nella regione
posteriore del collo, faccia a luna piena, obesità del tronco. Occorre la limitazione della posologia <
10 mg/die di prednisolone equivalente
g) OSTEOPOROSI Un grave e debilitante effetto collaterale quasi sempre associato alla terapia di
lunga durata con alte dosi di glucocorticoidi è la osteoporosi, che può portare a fratture spontanee
più frequenti a carico di vertebre e coste. Numerosi effetti contribuiscono all’attività osteoporotica
di questi farmaci. La osteoprotegerina (OPG) è un recettore solubile appartenente alla
superfamiglia dei recettori per il TNF-α che inibisce l’attività osteoclastica interagendo con il
ligando per la osteoprotegerina (OPGL) bloccandone gli effetti biologici. OPG-L è in grado di
stimolare la proliferazione degli osteoclasti e di inibirne l’apoptosi. I glucocorticoidi stimolano
l’espressione di OPG-L mentre inibiscono la sintesi di OPG, con la conseguenza di una aumentata
attività osteoclastica. Inoltre questi composti inibiscono la proliferazione e l’attività degli
osteoblasti riducendo la sintesi di osteocalcina per la presenza di GRE negativi nel promotore
di questa proteina. La osteoporosi (valutatata con la mineralometria ossea) può essere una
indicazione forte per la sospensione del trattamento. Inoltre la possibilità dello sviluppo di
fenomeni osteoporotici deve essere presa in considerazione nell’iniziare un trattamento con
glucocorticoidi nei soggetti anziani e nelle donne in menopausa. È consigliato l’uso preferenziale del
deflazacort, che oltre ad avere un minor effetto diabetogeno ha anche un minor effetto
osteoporotico, associato a una terapia antiosteoporotica(Ca2+, Vit. D, bifosfonati)
h) GASTROLESIVITÀ: non danneggiano la mucosa gastrointestinale come i FANS, ma possono
aggravare un’ulcera preesistente, per aumento della secrezione cloro-peptica e riduzione della
produzione di muco. Controindicati in pazienti con alterazioni gastrointestinali. In caso di ulcera
conclamata sospendere terapia.
i) EFFETTI CUTANEI: i glucocorticoidi riducendo la sintesi di collageno e di KGF (Keratinocyte Growth
Factor) e inibendo la proliferazione di endotelio e fibroblasti, determinano fragilità cutanea e
assottigliamento del tessuto sottocutaneo. I pazienti trattati con alte dosi e per lungo tempo con
glucocorticoidi presentano la comparsa di strie cutanee, ecchimosi e petecchie e una ritardata
guarigione delle ferite. In tal caso è opportuno limitarne la posologia al di sotto di 10 mg/die di
prednisolone equivalente
j) INIBIZIONE DELLA CRESCITA CORPOREA: la somministrazione di glucocorticoidi nei bambini deve
essere attentamente valutata in quanto questi farmaci possono indurre ritardo o nei casi più gravi
arresto della crescita (nanismo), con un meccanismo non ancora ben noto (probabilmente con la
riduzione della sintesi di collageno). È pertanto consigliata una somministrazione unica al mattino,
eventualmente a giorni alterni. Nei trattamenti locali (asma, patologie cutanee) utilizzare i
composti con alto metabolismo di primo passaggio (glucocorticoidi con lunga catena laterale al
C17) per evitare gli effetti sistemici.
k) EFFETTI OCULARI (nei soggetti geneticamente predisposti): formazione di addotti covalenti
steroide-lisine delle proteine del cristallino con formazione di cataratta sottocapsulare posteriore e
glaucoma. La profilassi prevede frequenti esami oftalmologici

l) NECROSI ASETTICA DELLE OSSA (nei soggetti geneticamente predisposti): per un meccanismo
ignoto, pazienti iperuremici ed iperlipidemici. Necrosi a carico di teste omerali e femorali
Effetti collaterali non direttamente collegati alla dose o alla durata del trattamento
a) DISTURBI NEUROPSICHICI: I glucocorticoidi possono provocare varie alterazioni dell’umore e del
comportamento come insonnia, iperattività, agitazione psicomotoria fino a psicosi maniaco-
depressive o schizofreniche. Il meccanismo d’azione è legato all’inibizione dell’espressione dei
recettori 5-HT1A probabilmente bloccando l’attività trascrizionale di elementi responsivi ad NF-κB
presenti nel promotore di questi recettori.
b) PANCREATITE ACUTA
c) REAZIONI ANAFILATTOIDI
GLUCOCORTICOIDI DISSOCIATI
Poiché gli effetti antinfiammatori dipendono in gran parte da inibizione proteica, per interferenza
all’attività transattivante di NfkB e Ap1, mentre gli effetti metabolici sono causati prevalentemente da
stimolazione proteica, recenti studi hanno investigato la possibilità di separare l’attività stimolatoria da
quella di repressione genica. Sono stati pertanto sviluppati alcuni composti, denominati ‘glucocorticoidi
dissociati’ che mantengono l’attività di repressione genica in presenza di una ridotta capacità stimolante.
Tali composti hanno mostrato:
- Attività antinfiammatoria sperimentale simile al prednisolone
- Minori effetti collaterali sul metabolismo del glucosio, tessuti linfoidi ,atrofia cutanea rispetto ai
glucocorticoidi classici
- Effetti simili al prednisolone sull’inibizione dell’asse ipotalamo-ipofisi-surrene
FARMACI ANTI-GLUCOCORTICOIDEI
Sono inibitori della secrezione corticosurrenalica, utili nella terapia dell’iperglicocorticoidismo. Si ricorda che la forma
primaria di questa patologia è definita sindrome di Cushing, in cui vi è un’ipersecrezione autonoma di glucocorticoidi e
in particolare di cortisolo da parte di tumori surrenalici o di iperplasia surrenalica ipersecernente. La forma secondaria
è definita Malattia di Cushing, in cui l’iperproduzione è indotta da un adenoma ipofisario ipersecernente ACTH.
Ketoconazolo è utilizzato principalmente come antimicotico. A dosaggi più elevati, esso inibisce la steroidogenesi
surrenalica attraverso l’inibizione del CYP17 (17α-idrossilasi), che catalizza una delle prime reazioni enzimatiche. A
dosi ancora più elevate blocca anche il CYP11A1 (che catalizza la prima reazione della steroidogenesi) bloccando la
sintesi di tutti gli steroidi. È l’inibitore più efficace e meglio tollerato, il regime terapeutico prevede 600-800 mg al
giorno in due somministrazioni. Tra gli effetti collaterali si annovera l’epatotossicità. Inoltre la potenziale interazione
del ketoconazolo con numerosi enzimi CYP può condurre a un’interazione tra farmaci con gravi conseguenze.
Etomidato, inibitore di CYP11B1 (11β-idrossilasi) l’enzima che converte l’11-deossicortisolo in cortisolo. La dose
raccomandabile è di 0.03-0.1 mg/kg i.v. Ha come vantaggio un rapido effetto in pazienti non trattabili per via orale.
Mitotano è utilizzato nel trattamento del carcinoma surrenalico inoperabile. È convertito dai CYP surrenalici in
composti reattivi citolitici. La dose indicata è di 0.5-3 g per os al giorno in 3 somministrazioni. Può avere effetti
collaterali come disturbi GI, atassia, miastenia.