







Le lingue
Pagine
Legale


ISBN: 978-84-16276-13-4 ISBN: 978-84-16276-00-4
Neurologia e neurochirurgia
Manuale CTOdi Medicina e Chirurgia
Prima edizione


Neurologia e neurochirurgia
TraduttoriSimone MichelineGiuseppe Lucente
AutoreManuel Amosa Delgado
CollaboratoriMaría Elena Capilla Cabezuelo
Fernando Díaz OteroLuis Jiménez Roldán
Isabel Herrera HerreraJaime Herreros Rodríguez
José Vicente Hervás GarcíaLydia López ManzanaresBelén Pilo de la Fuente
Juan Pablo Romero MuñozDolores Vilas Rolán
Manuale CTOdi Medicina e Chirurgia
Prima edizione

I diritti di traduzione, di memorizzazione elettronica, di riproduzione e adattamento totale oparziale, con qualsiasi mezzo (compresi i microfi lm e le copie fotostatiche), sono riservati pertutti i Paesi. Tuttavia il lettore potrà eff ettuare fotocopie per uso personale nei limiti del 15%del presente volume dietro pagamento alla SIAE del compenso previsto dall’art. 68, comma4, della legge 22 aprile 1941 n. 633 ovvero dall’accordo stipulato tra SIAE, AIE, SNS e CNA,CONFARTIGIANATO, CASA, CLAAI, CONFCOMMERCIO, CONFESERCENTI il 18 dicembre2000. Le riproduzioni ad uso diff erente da quello personale potranno avvenire, per un numerodi pagine non superiore al 15% del presente volume, solo a seguito di specifi ca autorizzazionerilasciata da CTO EDITORIAL. S.L.
L’Editore ha compiuto ogni sforzo per ottenere e citare le fonti esatte delle illustrazioni. Qualorain qualche caso non fosse riuscito a reperire gli aventi diritto è a disposizione
© CTO EDITORIAL, S.L. 2015. Tutti i diritti riservati
Editing, design e il layout: CTO Editorial
C/ Francisco Silvela, 106; 28002 MadridTfno.: (0034) 91 782 43 30 - Fax: (0034) 91 782 43 43E-mail: [email protected] Web: www.grupocto.es
ISBN Neurologia e neurochirurgia: 978-84-16276-13-4ISBN Opere complete: 978-84-16276-00-4Deposito legale: M-29262-2014
Stampato in Spagna
NOTA
La medicina è una scienza in continua evoluzione. La ricerca e l’esperienza clinica ampliano costantemente le nostre conoscenze, soprattutto in relazione alle modalità terapeutiche e alla farmacologia. Qualora il testo faccia riferimento al dosaggio o alla posologia di farmaci, il lettore può essere certo che autori, curatori ed editore hanno fatto il possibile per garantire che tali riferimenti siano conformi allo stato delle conoscenze al momento della pubblicazione del libro. Tuttavia, si consiglia il lettore di leggere attentamente i foglietti illustrativi dei farmaci per verifi care personalmente se i dosaggi raccomandati o le controindicazioni specifi cate diff eriscano da quanto indicato nel testo. Ciò è particolarmente importante nel caso di farmaci usati raramente o immessi di recente sul mercato.

Neurologia e neurochirurgia
Manuale CTOdi Medicina e Chirurgia
Prima edizione

VI
01. Introduzione: anatomia, semiologia e fisiologia del sistema nervoso .......................... 1
1.1. Breve ricordo anatomico ................................................................................................. 1
1.2. Alterazioni delle funzioni superiori .......................................................... 2
1.3. Disturbi della funzione motoria ...................................................................... 3
1.4. Alterazioni della sensibilità ........................................................................................ 4
1.5. Alterazioni della coordinazione. Atassia ....................................... 5
1.6. Alterazione dei nervi cranici .................................................................................... 6
1.7. Alterazioni campimetriche e pupillari ............................................... 7
1.8. Sindromi lobari ...................................................................................................................................... 8
1.9. Sindromi troncoencefaliche .................................................................................... 9
1.10. Rifl essi e sindromi midollari .................................................................................... 10
02. Coma. Morte encefalica ............................................................ 12
2.1. Coma ............................................................................................................................................................................ 12
2.2. Segni del valore localizzatore ............................................................................... 12
03. Demenze ........................................................................................................................ 14
3.1. Concetto e classifi cazione .......................................................................................... 14
3.2. Malattia di Alzheimer .............................................................................................................. 15
3.3. Demenza frontotemporale (malattia di Pick) .................... 16
3.4. Demenza vascolare ..................................................................................................................... 16
3.5. Demenza da corpi di Lewy ......................................................................................... 17
04. Malattie cerebrovascolari ..................................................... 18
4.1. Territori vascolari cerebrali .......................................................................................... 18
4.2. Classifi cazione e fattori di rischio ................................................................. 18
4.3. Malattie cerebrovascolari ischemiche ............................................... 19
4.4. Emorragia intraparenchimale .............................................................................. 24
4.5. Malformazioni vascolari .................................................................................................... 25
4.6. Emorragia subaracnoidea ............................................................................................. 25
05. Disturbi del movimento ............................................................ 31
5.1. Tremore .................................................................................................................................................................. 31
5.2. Distonie .................................................................................................................................................................. 32
5.3. Mioclonie ............................................................................................................................................................ 32
5.4. Tic ........................................................................................................................................................................................ 33
5.5. Sindrome delle gambe inquiete .................................................................... 33
5.6. Corea. Malattia di Huntington ............................................................................ 33
5.7. Malattia di Parkinson idiopatica ..................................................................... 33
5.8. Altre sindromi parkinsoniane ............................................................................... 35
06. Malattie da alterazione della mielina ......... 38
6.1. Sclerosi multipla ................................................................................................................................. 38
6.2. Altre malattie demielinizzanti .............................................................................. 41
07. Epilessia ........................................................................................................................... 42
7.1. Classifi cazione ......................................................................................................................................... 42
7.2. Diagnosi ................................................................................................................................................................ 43
7.3. Eziologia ............................................................................................................................................................... 44
7.4. Alcune sindromi epilettiche specifi che .......................................... 44
7.5. Trattamento. Farmaci anticomiziali ......................................................... 45
7.6. Epilessia e gravidanza ............................................................................................................ 46
08. Malattie degenerative del sistema nervoso ........................................................................... 48
8.1. Atassia di Friedrich ........................................................................................................................ 48
8.2. Sclerosi laterale amiotrofi ca ..................................................................................... 48
Ind
ice
Neur
ologia
e ne
uroc
hirur
gia

VII
09. Malattie virale e prioniche del sistema nervoso ........................................................................... 50
9.1. Encefalite erpetica ed altre encefaliti virali ............................. 50
9.2. Leucoencefalopatia multifocale progressiva ..................... 51
9.3. Altre malattie virali del SNC ...................................................................................... 51
9.4. Malattie prioniche .......................................................................................................................... 51
10. Malattie nutrizionali e metaboliche del sistema nervoso ............ 54
10.1. Malattie neurologiche in seguito a defi cit nutrizionali .................................................................................................................. 54
10.2. Encefalopatia anossico-ischemica ............................................................. 55
11. Neuropatie ................................................................................................................. 56
11.1. Considerazioni generali ..................................................................................................... 56
11.2. Sindrome di Guillain-Barré .......................................................................................... 57
11.3. Neuropatia diabetica ............................................................................................................... 58
11.4. Neuropatie dell’infezione da HIV ................................................................ 58
12. Malattie della placca motrice ....................................... 60
12.1. Miastenia gravis ................................................................................................................................ 60
13. Miopatie ........................................................................................................................... 64
13.1. Distrofi e muscolari ........................................................................................................................ 64
14. Cefalee ................................................................................................................................. 67
14.1. Considerazioni generali ..................................................................................................... 67
14.2. Cefalea tensiva ....................................................................................................................................... 67
14.3. Emicrania ............................................................................................................................................................ 67
14.4. Cefalea a grappolo (cluster headache), istaminica o di Horton ........................................................................................................... 69
15. Sindrome da ipertensione endocranica ......................................................................................................... 70
15.1. Fisiopatologia ........................................................................................................................................... 70
15.2. Eziologia ............................................................................................................................................................... 70
15.3. Clinica ......................................................................................................................................................................... 70
15.4. Sindromi da erniazione cerebrale ............................................................... 70
15.5. Diagnosi ................................................................................................................................................................ 71
15.6. Trattamento .................................................................................................................................................. 71
15.7. Sindrome dell’ipertensione intracranica benigna (pseudotumor cerebri) ..................................................................... 71
16. Idrocefalia .................................................................................................................... 73
16.1. Concetto e classifi cazione ............................................................................................ 73
16.2. Eziopatogenesi ...................................................................................................................................... 73
16.3. Clinica ......................................................................................................................................................................... 73
16.4. Trattamento .................................................................................................................................................. 74
16.5. Idrocefalia cronica dell’ adulto ............................................................................ 75
17. Tumori intracranici ................................................................................. 76
17.1. Considerazioni generali ..................................................................................................... 76
17.2. Metastasi cerebrali ........................................................................................................................ 76
17.3. Gliomi ......................................................................................................................................................................... 77
17.4. Medulloblastoma (PNET-Tumore Neuroectodermico Primitivo- infratentoriale) ................ 79
17.5. Meningioma ................................................................................................................................................ 80
17.6. Neurinoma dell’ VIII (schwannoma vestibolare) .......... 80
17.7. Tumori della regione pineale ................................................................................ 81
17.8. Tumori ipofi sari ..................................................................................................................................... 81
17.9. Craniofaringioma ............................................................................................................................. 81
17.10. Linfoma cerebrale primitivo .................................................................................... 82
17.11. Emangioblastoma .......................................................................................................................... 82
17.12. Riassunto delle caratteristiche anatomopatologiche ............................................................................................................... 82
IndiceNeurologia e neurochirurgia

VIII
18. Traumi cranioencefalici .............................................................. 84
18.1. Scala del coma di Glasgow ......................................................................................... 84
18.2. Gestione del TCE in urgenza .................................................................................... 84
18.3. Fratture craniche ............................................................................................................................. 84
18.4. Commozione cerebrale ...................................................................................................... 86
18.5. Ematoma epidurale .................................................................................................................... 86
18.6. Ematoma subdurale .................................................................................................................. 87
18.7. Contusione cerebrale emorragica .............................................................. 87
18.8. Lesione assonale diff usa .................................................................................................. 88
18.9. Complicanze e sequele dei neurotraumatismi dell’ encefalo .................................................... 88
19. Ascesso cerebrale ed empiema subdurale .............................................................. 90
19.1. Ascesso cerebrale ............................................................................................................................ 90
19.2. Empiema subdurale ................................................................................................................... 91
20. Patologia rachimidollare .......................................................... 92
20.1. Dolore lombare .................................................................................................................................... 92
20.2. Lombosciatalgia. Ernia discale lombare ........................................ 93
20.3. Cervicobrachialgia. Ernia discale cervicale ............................. 94
20.4. Stenosi del canale lombare ....................................................................................... 95
20.5. Spondilolistesi ......................................................................................................................................... 96
20.6. Spondilodisciti ........................................................................................................................................ 96
20.7. Tumori intrarachidei .................................................................................................................. 97
20.8. Ascesso epidurale spinale ............................................................................................. 98
20.9. Siringomielia .............................................................................................................................................. 98
21. Anomalie dello sviluppo ....................................................... 101
21.1. Craniosinostosi ...................................................................................................................................... 101
21.2. Malformazioni di Chiari ...................................................................................................... 102
22. Neurochirurgia funzionale .................................................. 103
22.1. Nevralgia del trigemino ..................................................................................................... 103
22.2. Chirurgia del dolore intrattabile ..................................................................... 103
Bibliografia .............................................................................................................................. 105
Indice Neurologia e neurochirurgia



1
Neurologia e neurochirurgia
1.1. Breve ricordo anatomico (Figura 1)
Emisferi cerebrali
In ciascun emisfero si distinguono:
• La corteccia cerebrale o sostanza grigia, di circa 2 o 3 cm di spessore, si
divide in quattro lobi: frontale, parietale, temporale e occipitale.
• La sostanza bianca che forma i sistemi di connessione: capsula interna,
esterna ed estrema.
• Corpo calloso, formato da fi bre che interconnettono entrambi gli emisferi.
Diencefalo
• Talamo. Nucleo della sostanza grigia localizzato nella zona mediale del
cerebro, in entrambi i lati del terzo ventricolo.
• Ipotalamo. Si occupa della regolazione delle funzioni viscerali: omeosta-
si, ciclo sonno-vigilia, controllo endocrino…
Gangli della base
I nuclei grigi del cervello sono formazioni di sostanza grigia situati in prossimi-
tà della base del cervello. Sono il nucleo caudato, putamen e pallido (gli ultimi
due costituiscono insieme il nucleo lenticolare).
Tra questi nuclei si trovano interposte due lamine di sostanza bianca, chiama-
te capsula interna e capsula esterna.
Tronco dell’encefalo
Il tronco dell’encefalo si divide anatomicamente.
• Mesencefalo. In esso si possono incontrare i nuclei dei nervi cranici III
e IV, inoltre dei tubercoli quadrigemini, il nucleo rosso e la substantia
nigra.
01
Midollo spinale
Cervello
CervellettoPonte
Bulbo rachideo
Figura 1. Anatomia del sistema nervoso centrale
• Protuberanza o ponte. Dove si localizzano i nuclei dei nervi cranici V
motore, VI, VII e VIII, e i peduncoli cerebrali medi, che collegano il tronco
dell’encefalo con il cervelletto.
• Bulbo rachideo. Nel quale si possono localizzare i nuclei dei nervi cra-
nici IX, X, XI e XII, così come i centri di controllo delle funzioni cardiaca,
vasocostrittiva e respiratoria e altre attività rifl esse come il vomito.
INTRODUZIONE: ANATOMIA, SEMIOLOGIA E FISIOLOGIA DEL SISTEMA NERVOSO
ORIENTAMENTONegli ultimi anni, ha perso rilievo l’importanza di questo tema nel MIR, tuttavia una lettura attenta è fondamentale poiché aiuterà a capire gli altri argomenti e, dunque, a rispondere alle domande-tipo del caso clinico nelle quali i dati semiologici sono la chiave diagnostica.

201 ∙ I n t roduz ione : anatomia , sem io log ia e f i s i o l og ia de l s i s tema ne rvoso
Manuale CTO di Medicina e Chirurgia, Prima edizione
Cervelletto
Posteriore al tronco dell’encefalo, controlla la via motoria indiretta, coor-
dinando le attività motorie, eff ettuando le correzioni necessarie alla sua
realizzazione e regolando il tono posturale e l’equilibrio.
1.2. Alterazioni delle funzioni superiori
Le alterazioni delle funzioni superiori conseguono a danni della sostanza gri-
gia corticale.
Disturbi del linguaggio
I disturbi del linguaggio sono i seguenti:
• Disartria. È un disturbo specifi co dell’articolazione del linguaggio dove
le basi dello stesso (grammatica, comprensione e scelta della parola)
restano intatte.
• Afasia. È una perdita o compromissione del linguaggio causato da un
danno cerebrale, con integrità delle strutture neuromuscolari che pro-
ducono lo stesso. Consegue a lesioni nell’emisfero dominante, che è il
sinistro nel 99% dei destrimani e nei mancini può essere il sinistro, il
destro o a dominanza bilaterale.
Tipi de afasia
Esistono cinque tipi di afasia, che possono diff erire secondo le caratteristiche
di fl uenza, comprensione, denominazione e ripetizione (Tabella 1).
Fluenza Comprensione Denominazione Ripetizione
Broca No Sí No No
Wernicke Sí No No No
Conduzione Sí Sí No No
Globale No No No No
Transcorticale motoria
No Sí No Sí
Transcorticale sensitiva
Sí No No Sí
Tabella 1. Diagnosi diff erenziale delle afasie
I pazienti con afasia de Broca presentano incapacità di emettere il linguag-
gio, con comprensione conservata. Si produce per lesione nell’area di Broca
nel lobo frontale dominante (area 44 e/o 45 di Brodmann o nerviopercolare
o triangolare) (Figura 2).
L’afasia di Wernicke si produce per lesioni nell’area di Wernicke o parte po-
steriore della circonvoluzione temporale superiore o giro sopramarginale.
I pazienti non comprendono e, a loro volta, presentano un aumento della
fl uenza, tra cui verborrea, con innumerevoli parafasie. Non si rendono bene
conto del loro problema di linguaggio.
L’afasia di conduzione può esserci a seguito di lesioni del fascicolo arcuato,
ma anche in svariate lesioni posteriori. La comprensione rimane conservata,
però il paziente presenta diffi coltà nella denominazione e ripetizione, con lin-
guaggio fl uente e innumerevoli parafasie
Figura 2. Localizzazione anatomica dei principali tipi di afasia
Le afasie transcorticale motoria o sensitiva presentano le stesse caratteri-
stiche delle afasie motoria o sensitiva corrispondenti, però si caratterizzano per
la conservazione della capacità di ripetizione. Si producono a seguito di infarti
estesi nelle zone di vascolarizzazione frontale delle grandi arterie cerebrali.
L‘afasia globale è la forma più grave e frequente di afasia, a seguito di gran-
di lesioni nelle aree anteriori e posteriori del linguaggio. La prognosi di recu-
pero è sfavorevole.
Agnosia
L’agnosia è l’incapacità di riconoscere uno stimolo visivo, tattile o uditivo
quando non esiste alterazione nella comprensione né problemi nelle aree di
sensibilità primaria, sensoriali o uditive.
Rifl ettono un problema a livello corticale.
• Agnosia visiva (lesioni delle aree di associazione visive):
- Prosopoagnosia. Incapacità di riconoscere volti umani preceden-
temente conosciuti o apprenderne di nuovi.
- Simultagnosia. Incapacità di percepire due stimoli visivi in manie-
ra simultanea.
• Agnosia tattile (lesione parietale controlaterale):
- Astereognosia. Incapacità di riconoscere al tatto un oggetto con gli
occhi chiusi, nonostante si descrivano le sue caratteristiche elementari.
- Atopognosia. Impossibilità di localizzare uno stimolo tattile.
- Agrafoestesia. Incapacità di riconoscere una determinata fi gura
tracciata sulla superfi cie corporale.
• Altre (lesioni parietali non dominanti):
- Asomatognosia. Mancanza di riconoscimento delle proprie parti
del corpo.
- Anosognosia. Incapacità di riconoscere la propria malattia.
Aprassia
L’aprassia è l’incapacità di portare a termine operazioni conosciute a se-
guito di un ordine verbale o per imitazione in pazienti con una adeguata
comprensione e senza defi cit motori o sensoriali primari che interferiscano
con lo sviluppo del movimento.
• Aprassia ideomotoria. Incapacità di sviluppare un atto motorio prece-
dentemente acquisito come risposta ad un ordine verbale. È la tipologia
più comune di aprassia.
• Aprassia ideativa. Incapacità di portare a termine una sequenza or-
dinata di atti motori (es. accendere una sigaretta) per perdita del pro-

3
01Neurologia e neurochirurgia
gramma mentale di atti complessi, pur essendo in grado di realizzare
ciascun atto separatamente e correttamente.
• Aprassia costruttiva. Incapacità di disegnare o costruire fi gure semplici.
• Aprassia dell’abbigliamento. Incapacità di vestirsi correttamente
quando vengono consegnate i diversi capi dell’abbigliamento.
• Aprassia della marcia. Incapacità di iniziare la deambulazione in posi-
zione eretta a seguito della perdita dei programmi motori acquisiti per
camminare, mantenendo la capacità motoria in decubito. Caratteristi-
camente, si riscontra nella idrocefalia normotensiva (insieme ad inconti-
nenza urinaria e demenza) e in lesioni frontali bilaterali.
• Aprassia bucolinguofacciale. Incapacità di aprire o chiudere la boc-
ca o gli occhi quando viene richiesto dall’esaminatore, nonostante lo si
possa fare spontaneamente.
1.3. Disturbi della funzione motoria
Fisiologia della funzione motoria
Sistema piramidale
I neuroni dello strato corticale V della corteccia motoria primaria (area 4 di
Brodmann) utilizzano i propri assoni per formare il primo tratto del sistema
piramidale che è composto da due neuroni motori: il primo motoneurone,
che si origina nella corteccia e le cui fi bre discendono attraverso la capsu-
la interna, fi no al corno grigio anteriore del midollo o sino i nuclei motori
dei nervi cranici rispettivamente, ove ha sede il secondo motoneurone la
cui fi bra nervosa, attraverso radici e nervi, si va a porre in sinapsi (giunzione
neuro-muscolare) con la fi bra muscolare.
• Fascio genicolato. Si occupa del controllo volontario della muscolatu-
ra innervata dai nervi cranici.
• Fascio corticospinale. Possono diff erenziarsi due fasci a partire dal bulbo:
- Fascio corticospinale laterale (FCSL). È incrociato e scorre attra-
verso il cordone laterale del midollo.
- Fascio corticospinale anteriore o ventrale (FCSV). E’ ipsilaterale e di-
scende attraverso il cordone anteriore. Innerva la muscolatura assiale.
Fisiologia della placca motrice (Figura 3)
L’acetilcolina è il neurotrasmettitore impiegato nella placca motrice della
giunzione neuromuscolare, dal momento che questa è un modello di sinapsi
chimica. L’acetilcolina si sintetizza nelle vescicole terminali presinaptiche del
secondo motoneurone, si libera nello spazio intersinaptico e va a collocarsi sui
recettori postsinaptici della placca motoria del muscolo . Quando il potenziale
di azione riverberato dal secondo motoneurone percorre l’assone e raggiunge
la terminazione presinaptica, la depolarizza e si aprono canali di calcio voltag-
gio dipendenti. Il calcio attrae vescicole di acetilcolina e provoca la sua esoci-
tosi. L’acetilcolina si unisce ai suoi recettori, situati nella membrana muscolare
sottostante alla terminazione assonica, aprendone i canali ionici.
Dopo l’apertura dei canali, si determina un ingresso massiccio di sodio fa-
vorito del gradiente (all’interno della membrana muscolare, il potenziale è
di circa -80 mV). In questo modo, si ha una inversione locale del potenziale
(passa da -80 a +60 mV), che è il potenziale di placca motrice, che si trasmet-
te alla fi bra muscolare generando un potenziale di azione muscolare e la
contrazione muscolare.
L’acetilcolina scompare velocemente dalla fessura sinaptica per la presenza
dell’enzima acetilcolinesterasi.
Celluladi Schwann
Guaina di mielina
Assone motorio
Placca motrice terminale
Vescicole
Terminazionenervosa
Pieghepostsinaptiche
Fibra muscolare
Figura 3. Fisiologia della placca motrice
Cervelletto e gangli basali
Entrambi formano parte della via motoria indiretta.
Fondamentalmente, il cervelletto aiuta a coordinare le attività motorie e ad
eff ettuare gli aggiustamenti correttivi di queste attività. Inoltre, interviene
nella regolazione della postura e dell’equilibrio.
I gangli basali, a loro volta, contribuiscono a pianifi care e a regolare i processi
complessi del movimento muscolare, attraverso il controllo del tono musco-
lare e dell’intensità relativa, della direzione e della sequenza dei movimenti
necessarie.
Alterazioni motorie
Il defi cit di forza si quantifi ca come mostrato nella Tabella 2.
0 Nessuna contrazione o movimento. Paralisi completa
1 Contrazione che non arriva a determinare nessun movimento
2 Movimento ma non contro gravità
3 Movimento contro gravità però non contro resistenza
4Movimento contro resistenza ma inferiore rispetto al limite massimo del controlaterale o previsto in base all’età
5 Forza normale
Tabella 2. Quantifi cazione del defi cit della forza muscolare
La lesione del primo motoneurone del fascio piramidale, può avere una fase
iniziale chiamata fase di “shock midollare” nella quale sono presenti segni ana-
loghi a quelli della lesione del secondo motoneurone, però quando il danno si
stabilizza, il paziente avrà clinicamente i segni tipici del danno del primo.
Altre alterazioni della funzione motoria, come i disturbi extrapiramidali, le
crisi comiziali motorie, i problemi di coordinazione, così come l’atassia e l’a-
prassia o il disturbo non paralitico del movimento saranno trattati più avanti
in questo capitolo.
Le paralisi possono derivare da:
• Lesioni della via piramidale (primo motoneurone).
- Corteccia e capsula interna: emiparesi facio-brachio-crurale con-
trolaterale alla lesione.
- Tronco encefalico: emiparesi controlaterale con defi cit dei nervi
cranici ipsilaterali.

401 ∙ I n t roduz ione : anatomia , sem io log ia e f i s i o l og ia de l s i s tema ne rvoso
Manuale CTO di Medicina e Chirurgia, Prima edizione
- Le lesioni midollari si presentano con paraparesi o tetraparesi, a
seconda della localizzazione della lesione, e se interessa solo una
via piramidale, la paresi è ispilaterale alla stessa.
• Lesioni del motoneurone del corno anteriore midollare e dei nuclei mo-
tori troncoencefalici (secondo motoneurone).
• Lesioni del nervo periferico.
• Lesioni della placca neuromuscolare (miastenia gravis, sindrome mia-
steniforme paraneoplastica di Eaton-Lambert, botulismo…).
• Miopatie (Figura 4).
Muscolo
1º motoneurone(piramidale)
Decussazione
Via corticospinale mediale (10%)Via corticospinale laterale (90%)
Nervi periferici
Placca motrice
2º motoneurone (corno grigio anteriore)
Figura 4. Vie motore
A livello clinico è di grande importanza la diff erenza tra lesione del primo e
del secondo motoneurone (Tabella 3).
1.ª motoneurone 2.ª motoneurone
Rifl essi osteotendinei
Aumentati Diminuiti o assenti
Risposta cutaneoplantare
Estensione (Babinski) Flessione
Muscolo ∙ Ampi gruppi muscolari
∙ Atrofi a modesta, da non uso
∙ Muscoli isolati o piccoli gruppi
∙ Amiotrofi a precoce ed accentuata
∙ Contratture
∙ Fascicolazioni
Tono Aumentato (paralisi spastica) Diminuito (paralisi fl accida)
Tabella 3. Diagnosi diff erenziale delle lesioni del primo e secondo motoneurone
R I C O R D ABisogna considerare se esistono sintomi associati per orientare la diagnosi, ad esempio: · 1.º motoneurone + afasia = corteccia motoria. · 1.º motoneurone + nervi cranici controlaterali (sindrome alterna)
= tronco dell’encefalo. · 1.º motoneurone isolato = midollo. · 2.º motoneurone + defi cit sensitivo = nervo periferico. · 1.º motoneurone + 2.º motoneurone = sclerosi laterale amiotro-
fi ca (SLA).
1.4. Alterazioni della sensibilità (Figura 5)
Sensibilità vibratoria, posizionalee propriocettiva
Talamo
Nucleigracilis e cuneatus
Cordoniposteriori Ganglio
radicolare
Via spinotalamica
Termoalgesica (dolore e temperatura)
Figura 5. Vie sensitive
Recettori sensoriali
• Recettori sensoriali primari. In questo caso, sono le terminazioni pro-
prie del nervo che operano come sensori.
• Recettori sensoriali secondari. Costituiti da cellule neurali specia-
lizzate o non neurali, che operano come recettori e trasduttori dello
stimolo verso il neurone sensoriale primario, attraverso il meccanismo
sinaptico.
Tra le proprietà dei ricettori sensoriali, hanno rilevanza due principali: la sca-
rica ripetitiva (a maggior intensità e maggior frequenza di scarica) e l’adat-
tabilità o fatica (di fronte ad uno stimolo costante, passato un certo tempo,
la frequenza di scarica è ogni volta è più lenta fi no a che si riduce al minimo
o sparisce).

5
01Neurologia e neurochirurgia
Vie sensitive del SNC
• Sistema colonna dorsale-lemnisco mediale. Conducono informazioni
sensitive chiamate epicritiche o di discriminazione fi ne e vibratoria. Salgo-
no per le colonne posteriori del midollo ipsilaterale, facendo la prima si-
napsi nei nuclei bulbari di Goll e Burdach, e passando, a livello del bulbo, al
lato opposto, formando il lemnisco mediale e fi nendo nel talamo (nucleo
ventrale posterolaterale). È una via di conduzione molto rapida e presenta
un alto grado di collocazione spaziale rispetto all’origine dello stimolo.
• Sistema anterolaterale. La sensibilità che conduce si chiama protopa-
tica, con sottotipi diversi: dolore, temperatura e sensazioni di tatto gros-
solane. Ha la sua prima sinapsi nel corno dorsale della sostanza grigia
midollare; da questa, dopo essere passata al lato opposto del midollo,
sale per le colonne bianche anteriori e laterali (fascio spinotalamico la-
terale), per terminare nei nuclei sensitivi dei nervi cranici ai diff erenti,
successivi livelli del tronco e nel nucleo ventrale posterolaterale del ta-
lamo. È un sistema più lento, con un minor grado di defi nizione spaziale.
Dal talamo, si distribuiscono fi no alla corteccia sensoriale (terzo neurone che
proietta alla corteccia parietale), dove esiste una rappresentazione sensitiva
del corpo, il cosi detto omuncolo sensitivo di Penfi eld.
Clinica
• Sintomi positivi: parestesia (percezione di sensazioni anomale senza
l’applicazione di uno stimolo apparente) e disestesia (sensazione ano-
mala dopo l’applicazione di uno stimolo).
• Sintomi negativi: ipoestesia (diminuzione della percezione) o anestesia
(assenza completa della percezione).
La distribuzione dei defi cit sensoriali è indicativa della localizzazione lesiona-
le nel sistema nervoso (Figura 6).
• Polineuropatia: ipoestesia, disestesia e parestesia a livello distale negli
arti, con distribuzione “a guanto o a calza”.
• Lesioni centromidollari: defi cit in un dermatomo (“livello sospeso”)
per la sensibilità al dolore e termica con conservazione di quella tattile e
propriocettiva, (defi cit dissociato della sensibilità).
• Lesioni midollari: forniscono livelli sensitivi la cui identifi cazione è indi-
cativa del livello lesionale.
- Cordone posteriore: alterazione epicritica ipsilaterale.
- Cordone anteriore: alterazione protopatica controlaterale.
Midollo Nervo Corteccia o talamo
Figura 6. Anatomia delle alterazioni della sensibilità
• Lesioni talamiche: interessano tutte le sensibilità dell’emicorpo con-
trolaterale, incluso quelle della faccia. A volte possono produrre un qua-
dro di dolore o iperpatia nell’emicorpo interessato (sindrome di Déjeri-
ne- Roussy).
• Lesioni corticali parietali: interessano le sensibilità combinate,
con conservazione relativa delle primarie (tatto, dolore e tempe-
ratura).
1.5. Alterazioni della coordinazione.Atassia
Si defi nisce atassia l’alterazione della coordinazione che, senza debolezza
motoria e in assenza di aprassia, altera la direzione e l’ampiezza del movi-
mento volontario, la postura e l’equilibrio. La presenza di atassia implica un
danno in uno dei seguenti sistemi: propriocezione, cervelletto o vestibolare
(Figura 7).
Atassia
Tip
iSensitiva Cerebellare Vestibolare
Emisferica Vermiana Centrale Periferica
Ezio
log
ia
· Neuropatia periférica
· S. Tabetica
· Deg. Subacuta combinata midollare
· Spondilosi cervicale
· Tumorale
· Vascolare
· Postinfettiva
· Droghe (alcohol)
· Farmaci (fenitoina)
· Ictus vertebrobasilare
· Sclerosi multiple
· Tumori angolo pontocerebellare
· Posizionale
· Neuronite
· Labirintite
· Meniere
Marciae membri
Membri Marcia Bipedestazionee marcia
Figura 7. Tipo di atassia ed eziologia
Tipo sindromico di atassia
• Atassia sensitiva. Interessa principalmente l’andatura e gli arti in-
feriori in maniera simmetrica. È caratteristica l’assenza di vertigini,
nistagmo o disartria e si nota un chiaro peggioramento quando il
paziente chiude gli occhi o esercita un movimento in situazioni con
scarsa luminosità. In posizione eretta, con gli occhi aperti, c’è un au-
mento della base di appoggio, e il paziente può cadere se chiude gli
occhi (segno di Romberg). Il segno di Romberg non si riscontra nelle
lesioni cerebellari.
• Atassia cerebellare. L’atassia cerebellare può interessare la stazione
eretta, l’andatura e gli arti e, a diff erenza dell’atassia sensitiva, persiste
anche con il sussidio visivo e non si aggrava maggiormente con la chiu-
sura degli occhi. Si associa ad ipotonia, disartria, tremore cinetico e ni-
stagmo.

601 ∙ I n t roduz ione : anatomia , sem io log ia e f i s i o l og ia de l s i s tema ne rvoso
Manuale CTO di Medicina e Chirurgia, Prima edizione
• Atassia vestibolare. L’atassia o disequilibrio vestibolare si caratterizza
per un’alterazione dell’equilibrio durante la stazione eretta e l’andatura,
senza incoordinazione nei movimenti degli arti quando il paziente vie-
ne esaminato in decubito. La vertigine ed il nistagmo sono tipicamente
associati e non è presente disartria. Il test di Romberg è positivo.
1.6. Alterazione dei nervi cranici (Figura 8)
Paralisi dei nervi cranici oculomotori
Le paralisi o paresi dei nervi cranici oculomotori (nervo oculomotore comune
[III],trocleare o patetico [IV] ed oculomotore esterno [VI]) producono diplopia
binoculare. La diplopia monoculare si osserva nella lussazione del cristallino.
Lesioni del III nervo cranico (nucleo oculomotore comune)
Si presenta con debolezza dei muscoli innervati (costrittore pupillare, retto
superiore, inferiore ed interno, obliquo inferiore) e ptosi (elevatore della pal-
pebra ipsilaterale), producendo diplopia verticale o obliqua binoculare. La
causa più frequente è la mononeuropatia diabetica.
• Le lesioni compressive si caratterizzano inizialmente per midriasi areat-
tiva della pupilla, seguita da paresi della muscolatura extraoculare. Le
lesioni ischemiche non coinvolgono la pupilla, dal momento che sono
confi nate nella porzione centrale del nervo, e le fi bre motorie si localiz-
zano perifericamente.
• Nel seno cavernoso, la lesione del III nervo solitamente si associa a le-
sioni di altri nervi cranici (IV e VI con oftalmoplegia completa; primo e
secondo ramo del trigemino).
• Per la fessura orbitaria su-
periore passano i nervi III, IV
e VI ed il primo ramo del V
(oftalmico) e la vena oftalmi-
ca. Le lesioni a questo livello
non interessano il secondo
ramo del trigemino.
Lesioni del IV nervo cranico (nucleo trocleare)
Il nucleo del IV nervo si localizza nel mesencefalo. La paralisi del IV nervo pro-
duce clinica di diplopia verticale che aumenta con lo sguardo verso basso e
verso il lato opposto alla lesione. I pazienti presentano come caratteristiche
una deviazione della testa verso il lato opposta alla lesione dal momento che
l’inclinazione verso lo stesso lato aumenta la diplopia (test dell’inclinazione
cefalica di Bielschowsky).
La causa più frequente della lesione sono i traumi cranici, seguiti dalla neu-
ropatia ischemica.
Lesioni del VI nervo cranico (nucleo oculomotore esterno)
Il nucleo del VI nervo si localizza nella protuberanza. Da questo si origina
il fascicolo longitudinale mediale, inter neuroni che attraversano la linea
media e salgono per produrre sinapsi nel subnucleo del retto interno del III
nervo controlaterale, permettendo in questo modo lo sguardo coniugato
sul piano orizzontale.
La paresi del VI nervo cranico provoca una limitazione dell’abduzione dell’oc-
chio, producendo diplopia binoculare orizzontale che aumenta quando il
paziente guarda verso il lato della lesione.
• La lesione del fascicolo longitudinale mediale produce la così detta of-
talmoplegia internucleare (paralisi dell’adduzione di un occhio con
nistagmo nell’occhio abducente). Le sue cause più frequenti sono la
sclerosi multipla e le lesioni vascolari.
• La lesione a livello della punta della squama del temporale produce la
sindrome di Gradenigo (paresi del VI nervo, dolore facciale ipsilaterale
per lesione del trigemino e sordità).
R I C O R D AOftalmoplegia internucleare: lesione del fascio longitudinale me-diale (nei giovani c’è il sospetto di malattia demielinizzante mentre, negli anziani, ischemia nel tronco dell’encefalo).
Mesencefalo
Protuberanza
Bulbo
Localizzazione Clinica della lesione
III NC
VI NC
VII NC
XII NC
V NC
Mesencéfalo
Protuberanza
Ponte
Bulbo
Nucleo principale (ponte)Nucleo spinale (protuberanza e bulbo)
Alterazione del movimento orizzontale(gli occhi deviano verso il lato contrario alla lesione)
Paralisi facciale (la bocca devia verso il lato sano)se periferica è completa: superiore + inferiore
- se centrale rispetta la porzione superiore
Paresi, amiotrofia e fascicolazioni della linguache devia verso il lato della lesione
Emi-ipoestesia facciale ipsilaterale
Midriasi areattivaAlterazione dei movimenti verticali
Figura 8. Localizzazione dei nervi cranici nel tronco dell’encefalo
R I C O R D APtosis: · Miastenia gravis · Síndrome de Horner · Lesión del III PC

7
01Neurologia e neurochirurgia
R I C O R D AIl IV nervo cranico è il più lungo e sottile, inoltre nasce dalla faccia posteriore del tronco encefalico. La causa più frequente della sua le-sione sono i traumi cranioencefalici. Il VI nervo cranico realizza un lungo viaggio attraverso lo spazio su-baracnoideo, quindi è suscettibile a lesioni in seguito ad aumenti della pressione intracranica.
Lesione del nervo trigemino o V nervo cranico
Il nervo trigemino innerva i muscoli della masticazione e raccoglie la sensi-
bilità dell’emivolto ipsilaterale. È formato da tre rami: oftalmico, mascellare
e mandibolare. La manifestazione clinica più frequente è il dolore nell’emi-
volto ipsilaterale. Può anche presentarsi con ipoestesia dell’emivolto ipsila-
terale, deviazione della mandibola verso il lato malato con debolezza nella
masticazione ed assenza del rifl esso corneale.
Lesione del nervo facciale o VII nervo cranico
Il nervo facciale innerva i muscoli della mimica facciale di tutta la metà viso
corrispondete, le ghiandole lacrimali, le salivatorie sottomandibolari e su-
blinguali, e la sensibilità gustativa dei due terzi anteriori ella lingua. La lesio-
ne periferica o nucleare produce paresi o paralisi dei muscoli dell’emivolto
ipsilaterale, in modo tale che cercando di alzare entrambe le commissure,
la bocca viene deviata verso il lato sano, il paziente presenta fronte liscia e
diffi coltà nel chiudere la palpebra ipsilaterale.
La lesione sopranucleare (corticale) produce paralisi unicamente della parte
inferiore dell’emivolto controlaterale (paralisi di tipo centrale:l’innervazione
della parte inferiore è controlaterale, mentre l’innervazione della parte supe-
riore è bilaterale e, quindi, rimase preservata).
Lesione del nervo stato-acustico o VIII nervo cranico
A sua volta, è formato da due nervi, il cocleare ed il vestibolare. Il nervo cocleare
è sensoriale e trasmette gli stimoli uditivi. Il nervo vestibolare interviene nella re-
golazione dell’equilibrio e nell’orientamento nello spazio. La lesione del nervo co-
cleare produce tinnito o acufene, così come la diminuzione dell’acutezza uditiva.
Lesione del nervo glossofaringeo o IX nervo cranico
Innerva i muscoli costrittore superiore della faringe e stilofaringeo, la sensibi-
lità del terzo posteriore della lingua e dell’orofaringe. Una sua lesione produ-
ce una lieve disfagia, perdita della sensibilità del terzo posteriore della lingua,
perdita del rifl esso faringeo e deviazione della parte posteriore verso il lato
sano (segno della tendina di Vernet). Raramente si ha una sua lesione isolata.
Lesione del nervo vago o X nervo cranico
Una sua lesione intracranica produce disfagia, disartria, disfonia e anestesia
laringea. Raramente si ha una sua lesione isolata.
Lesione del nervo spinale o XI nervo cranico
È un nervo motore puro che innerva i muscoli sternocleidomastoideo e trape-
zio. Una sua lesione produce debolezza muscolare ipsilaterale a questo livello.
Lesione del nervo ipoglosso o XII nervo cranico
È un nervo motore che innerva l’emilingua controlaterale (muscolo genio-
glosso). Una sua lesione produce emiatrofi a ipsilaterale della lingua e devia-
zione della stessa verso il lato della lesione.
1.7. Alterazioni campimetriche e pupillari
Difetti campimetrici
Si veda la Sessione di Oftalmologia; riassumendo:
• Lesioni alla retina e al nervo ottico portano all’apparizione di scotomi
o amaurosi.
• Lesioni al chiasma producono generalmente emianopsia eteronima
bitemporale.
• Lesioni del tratto (bendelletta) ottica danno luogo ad emianopsia
omonima controlaterale incongruente (diff erenze tra il difetto campi-
metrico di ciascun occhio). Di solito si accompagna al difetto pupillare
aff erente.
• Le lesioni delle radiazioni ottiche inferiori (temporali) producono
una quadrantopsia omonima controlaterale di predominio superiore.
• Le lesioni delle radiazioni ottiche superiori (parietali) producono
una quadrantopsia omonima controlaterale di predominio superiore.
• Lesioni ad entrambe le radiazioni ottiche producono un’emianopsia
omonima controlaterale che, a diff erenza di quelle della bendelletta,
sarà più congruente e senza danneggiare la funzione della pupilla.
• Lesioni ad entrambe le radiazioni ottiche producono Lesioni alla cortec-
cia visiva primaria producono emianopsia omonima controlaterale con
la caratteristica che la macula, zona di miglior visione, rimanga intatta.
R I C O R D A La quadrantopsia bitemporale superiore si produce per compressio-ne delle fi bre inferiori del chiasma e una delle sue cause è di solito il tumore ipofi sario. Al contrario, i craniofaringiomi, che comprimono innanzitutto le fi bre superiori, provocano una quadrantopsia bi-temporale inferiore.
Alterazioni pupillari
• Anisocoria essenziale. Un 15-30% della popolazione normale presen-
ta una diff erenza nella dimensione pupillare di 0,4-1 mm con una reat-
tività normale alla luce.
• Difetto pupillare aff erente relativo. Consiste in una diminuzione della
risposta pupillare costrittoria di fronte ad uno stimolo luminoso diretto,
con una risposta normale se si stimola l’occhio controlaterale (risposta
consensuale normale), ed indica una lesione del nervo ottico.
• Sindrome di Horner. Si produce per lesione alle fi bre pupillari simpati-
che. L’innervazione simpatica che dilata a pupilla si origina a livello ipota-
lamico e discende per il segmento laterale troncoencefalico fi no al nucleo
intermedio laterale del midollo nei segmenti C8-D2. Da qui passa al gan-
glio cervicale superiore della catena simpatica paravertebrale e sale con
il plesso pericarotideo, per incorporarsi al ramo oftalmico del trigemino e
raggiungere la pupilla attraverso i nervi ciliari lunghi. Una lesione a qual-
siasi livello di questi può produrre la sindrome di Horner che si presenta
con la triade di restringimento della rima palpebrale, miosi ed enoftalmo.
A volte si aggiunge un’anidrosi facciale (quest’ultima quando la lesione è
precedente alla biforcazione carotidea; se la lesione è posteriore alla bifor-

801 ∙ I n t roduz ione : anatomia , sem io log ia e f i s i o l og ia de l s i s tema ne rvoso
Manuale CTO di Medicina e Chirurgia, Prima edizione
cazione, non si manifesta anidrosi). La pupilla risponde adeguatamente
alla luce e agli stimoli vicini. L’anisocoria è più evidente con l’oscurità e la
pupilla risponde sia con midriasi che con miosi.
• Lesioni alle fi bre pupillari parasimpatiche o difetto pupillare aff eren-
te (lesione III nervo cranico) produce dilatazione pupillare senza rispo-
sta alla luce. Quando la dilatazione pupillare areattiva si accompagna
ad una relativa preservazione della motilità oculare, l’eziologia di solito
risulta compresa nello spazio subaracnoideo. Lesioni ischemiche del III
nervo cranico inizialmente lasciano intatta la pupilla (dal momento che
l’ischemia di solito interessa le fi bre interne e, come si è già visto, le pa-
rasimpatiche si collocano della porzione esterna del III nervo).
• Pupilla tonica di Adie. Si produce a seguito di una lesione del ganglio
ciliare per cause locali (infi ammazione, infezione o trauma) o come par-
te di una neuropatia periferica o autonoma (sindrome di Guillain-Barré,
sindrome di Fisher, sindrome di Shy-Drager, amiloidosi, neuropatia sen-
sitiva ereditaria, sindrome di Charcot-Marie-Tooth, diabete, alcolismo o
sindrome paraneoplastica). È una pupilla midriatica, generalmente uni-
laterale, che non risponde alla luce, e la cui risposta nel vedere da vicino
è lenta e tonica. L’anisocoria è ancor più evidente in condizioni di lumi-
nosità. Risponde sia al midriatico che al miotico. Può accompagnarsi a
movimenti vermiformi dei margini dell’iride.
• Pupilla di Argyll-Robertson. È una lesione pupillare bilaterale con
pupille piccole e irregolari che rispondono scarsamente alla luce, però
mantengono l’accomodazione per la visione da vicino (dissociazione
luce-accomodazione). Presenta una risposta adeguata a miotico e scar-
sa a midriatico. Pare sia conseguente ad una lesione mesencefalica ro-
strale e si osserva in pazienti con neurosifi lide.
1.8. Sindromi lobari (Figura 9)
Lobo frontale
• Le aree motoria e premotoria sono collegate nello specifi co con i mo-
vimenti volontari e una loro lesione produce paralisi spatica controlate-
rale (primo motoneurone). Le aree motorie primarie, come le sensitive,
si organizzano somatotopicamente in maniera che le aree corticali si
relazionino con le aree corporali specifi che (Figura 10).
• Nel lobo frontale, si trova il centro dello sguardo coniugato. Una sua le-
sione produce una deviazione cefalica e coniugata oculare verso il lato
della lesione. Al contrario, una sua irritazione (crisi comiziale) devia gli
occhi e la testa verso il lato opposto.
• Lesioni dell’area motoria supplementare dell’emisfero dominante ini-
zialmente provoca mutismo, per successivamente trasformarsi in afasia
motoria transcorticale. Quando interessa l’area di Broca, si sviluppa l’afa-
sia motoria o non fl uente. Lesioni più ampie in questa zona provocano
agrafi a ed aprassia bucolinguofacciale.
Figura 10. Organizzazione somatotopica delle aree corticali motorie e sensitive
• La lesione bilaterale delle aree frontali mediali parasagittali provocano
un quadro di aprassia della marcia ed incontinenza urinaria
• Le aree prefrontali hanno una funzione meno specifi ca. Una sua lesione
si relaziona con la mancanza d’iniziativa e spontaneità (stato apatico o
abulico), diminuzione delle relazioni interpersonali, cambi di persona-
lità (a volte con marcata disinibizione sociale, instabilità ed impulsività,
specialmente a seguito di lesioni frontali basali) ed un leggero deterio-
Lobo frontale Lobo temporale Lobo parietale Lobo occipitale
Area motoria e premotoria(paralisi spastica controlaterale)
Area di Broca(afasia motoria)
Corteccia somatosensoriale (ipo-estesia controlaterale)
Radiazioni ottiche superiori(quadrantopsia
inferiore controlaterale)
Corteccia visiva primaria(emianopsia omonimacontrolaterale con rispettomaculare; cecitá corticale)
Corteccia uditiva(allucinazioni uditive;
sordità corticale)
Radiazioni ottiche inferiori(quadrantopsia superiore
controlaterale)
Area di Wernicke(afasia sensitiva)
Centro dello sguardo coniugato(deviazione verso la lesione)
Corteccia prefrontale(mutismo abulia morìa
riflessi arcaici)
Figura 9. Alterazioni delle funzioni superiori e sindromi dei lobi

9
01Neurologia e neurochirurgia
ramento intellettivo, con mancanza di attenzione e concentrazione, in-
capacità di analizzare i problemi e perseverazioni.
Lobo parietale
• Le alterazioni sensoriali che si sviluppano come conseguenza di una le-
sione del lobo parietale sono state descritte precedentemente (si veda
il paragrafo Sindromi sensoriali e agnosia) ed includono l’astereognosia,
l’atopognosia, la perdita di discriminazione tra due punti, l’estinzione
parietale, l’anosognosia e l’asomatognosia.
• Il difetto campimetrico per lesione parietale è un’emianopsia omonima
controlaterale congruente, con un netto predominio nei campi inferiori
(quadrantopsia omonima inferiore per lesione delle radiazioni ottiche
superiori).
• L’aprassia costruttiva e quella dell’abbigliamento, così come l’anoso-
gnosia e l’eminegligenza spaziale (asomatognosia), si osservano più
frequentemente in lesioni parietali destre, anche se possono trovarsi
anche in quelle sinistre.
• La lesione del lobo parietale dominante porta allo sviluppo dell’alessia,
sindrome di Gerstmann (agrafi a, alessia, acalculia, agnosia digitale e di-
sorientamento destra-sinistra), stereognosia bimanuale (agnosia tattile)
e aprassia ideatoria e ideomotoria (possono anche apparire a seguito di
lesioni frontali).
Lobo temporale
• Lesioni del lobo temporale dominante pro-
ducono quadrantopsia omonima superiore
per lesione delle radiazioni ottiche inferiori,
afasia di Wernicke o fl uente, amusia (incapa-
cità di leggere e scrivere musica) e alterazio-
ne dell’apprendimento del materiale verbale
presentato attraverso la via auditiva.
• Lesioni del lobo temporale non dominante
producono lo stesso difetto campimetrico,
alterazione nelle relazioni spaziali, deterio-
ramento nell’apprendimento del materiale
non verbale presentato visivamente e un’in-
capacità nel riconoscere le melodie.
• Lesioni in qualsiasi lobo temporale può dar
luogo ad allucinazioni ed illusioni uditive e
comportamenti psicotici con aggressività.
• La lesione temporale bilaterale può provo-
care la sindrome amnesica di Korsakoff , la
sindrome di Klüver-Bucy (apatia, placidezza,
incremento dell’attività sessuale e mancanza
di riconoscimento degli oggetti commestibi-
li) e sordità corticale.
Lobo occipitale
• Una lesione unilaterale produce un’emianop-
sia omonima controlaterale congruente con
conservazione della visione maculare e con
presenza eventuale di allucinazioni visive.
• Una lesione occipitale bilaterale provoca: 1)
cecità corticale dovuta a lesione delle aree vi-
sive primarie (scissura calcarina). Pazienti con
lesioni occipitali mediali estese di carattere
acuto e bilaterale con cecità corticale possono
portare alla negazione della cecità stessa (anosognosia visiva) e confabu-
lazioni in merito a ciò che stanno guardando: è la sindrome di Anton-
Babinski; 2) prosopagnosia; 3) simultanagnosia; 4) sindrome di Balint, che
implica aprassia ottica (errore nel dirigere la mirata verso una direzione a
seguito di un ordine, potendolo invece fare spontaneamente).
R I C O R D A Le rappresentazioni corticali visive ed uditive sono bilaterali. Per questo anche se possono verifi carsi paralisi ed ipoestesia di un solo emisfero, perché possa esserci cecità o sordità complete di origine corticale, è necessario che vi sia una lesione in entrambi gli emisferi.
1.9. Sindromi troncoencefaliche
Generalmente, bisogna pensare che una lesione a livello del tronco dell’en-
cefalo risulta sempre associata a lesioni dei nervi cranici ipsilaterali con “vie
lunghe” (motorie o sensitive) controlaterali. I nervi cranici indicano il livello
della lesione (Figura 11).
III parLM
NR
VP
ET
VI parVII par
V par
ETLM
VP
XII par V par
ET
VP
LM
Mesencefalo
Ponte
Bulbo
Sindrome di Parinaud
Sindrome di Claude
Sindrome di Benedikt
Sindrome di Weber
Sindromedi Millard-Gubler
Sindromedi Wallenberg
Sindrome bulbare mediale
· Paralisi dello sguardo coniugato verso l’alto· Difficoltà per la convergenza e l’accomodazione· Anisocoria e midriasi
· XII nervo ipsilaterale· Emiplegia controlaterale che rispetta il volto (VP)· Atassia sensitiva controlaterale (LM)
III nervoipsilaterale
+
· Atassia controlaterale (NR)· Mov. anormali controlaterali (corea, tremore e ballismo) (NR)
· III nervo ipsilaterale· Emiparesi controlaterale (VP)
· Emiplegia controlaterale rispettando il viso (VP)· Paresi del VI e VII nervi ipsilaterali
· Emi-ipoestesia facciale ipsilaterale (V Nervo)· Emi-ipoestesia corporale controlaterale (ET) (sd sensitiva alterna)
Inoltre: sindrome vertiginosa, disartria e disfagia,diplopía, sindrome Horner ipsilaterale ed atassiacerebellare ipsilaterale
Figura 11. Sindromi del tronco encefalico (VP: via piramidale; LM: lemnisco mediale; ET: via spinotalamica; HR: nucleo rosso)

1001 ∙ I n t roduz ione : anatomia , sem io log ia e f i s i o l og ia de l s i s tema ne rvoso
Manuale CTO di Medicina e Chirurgia, Prima edizione
Sindrome bulbare laterale o sindrome di Wallenberg
È conseguente ad una occlusione dell’arteria vertebrale o cerebellare poste-
roinferiore (PICA).
Clinicamente, si caratterizza per: 1) sindrome vertiginosa con nausea e vomi-
to per lesione dei nuclei vestibolari; 2) disartria e disfagia per paresi della cor-
da vocale, faringe e velopendulo del palato ipsilaterale, come conseguenza
di una lesione del nucleo ambiguo; 3) diplopia, probabilmente secondaria
all’estensione della lesione al ponte, parte inferiore, dove si trova il VI nervo
cranico; 4) ipoestesia facciale ipsilaterale conseguente a lesione del nucleo
trigeminale; 5) ipoestesia somatica controlaterale per lesione del tratto spi-
notalamico; 6) sindrome di Horner ipsilaterale; 7) atassia cerebellare ipsilate-
rale secondaria alla lesione del peduncolo cerebellare inferiore e cervelletto.
R I C O R D A I nervi cranici ci informano sul livello della lesione. Non bisogna di-menticare la regola 2-2-4-4: i due primi nervi “non arrivano al tron-co”, il III ed il IV arrivano al mesencefalo; i nervi V, VI, VII e VIII al ponte, e gli ultimi quattro al bulbo.
1.10. Rifl essi e sindromi midollari
I principali rifl essi midollari sono i seguenti:
• Rifl esso miotatico e di stiramento muscolare (Figura 12). La stimo-
lazione dei fusi (all’aumentare della lunghezza della fi bra muscolare)
produce una contrazione rifl essa delle grandi fi bre scheletriche che
stanno intorno. Questo rifl esso si produce per una via monosinaptica
(non partecipano gli interneuroni). La quantifi cazione dei rifl essi viene
esposta nella Tabella 4.
Figura 12. Rifl esso miotatico
Arefl essia 0
Iporefl essia +
Rifl essi normali ++
Iperrefl essia +++
Clono ++++
Tabella 4. Quantifi cazione dei rifl essi osteotendinei
• Rifl esso tendineo. Un aumento della tensione muscolare inibisce di-
rettamente il muscolo, senza danneggiare i muscoli adiacenti.
• Rifl esso fl essorio o di retrazione (Figura 13). Davanti ad uno stimolo
sensoriale cutaneo di qualsiasi tipo, ma soprattutto doloroso (per que-
sto è stato chiamato anche rifl esso nocicettivo o del dolore), si produce
una contrazione dei muscoli fl essori dell’estremità e un rilassamento
degli estensori.
• Rifl essi midollari che producono uno spasmo muscolare. In parti-
colare si produce per una frattura ossea e per irritazione del peritoneo
parietale in una peritonite.
• Rifl essi autonomi. Comprendono svariate funzioni, come cambi nel
tono vascolare a seconda della temperatura locale, sudorazione, rifl essi
intestinali e vescicali. Questi tipi di rifl essi di solito sono segmentari, però
occasionalmente vengono attivati in maniera simultanea, in grandi por-
zioni del midollo, di fronte ad uno stimolo nocicettivo forte o la reple-
zione eccessiva viscerale.
Bisogna avere ben presenti le principali vie che percorrono il midollo (Figu-
ra 14 e Tabella 5) per poter riconoscere le sindromi cliniche.
Riflesso flessore Riflesso estensore incrociatoInibizionereciproca
Inibita
Inibita
Eccitata
Eccitata
Circuito polisinaptico
Figura 13. Rifl esso fl essorio
Viaspinotalamica
Cordone posteriore Via corticospinalelaterale
Figura 14. Principali vie motorie e sensitive del midollo spinale

11
01Neurologia e neurochirurgia
Sindromi midollari
Eziologia Clinica
Mielopatia trasversale
Idiopatica (meccanismo inmunoallergico), virale, sclerosi multipla, LES, Sjögren
∙ Defi cit motorio. Paraplegia o tetraplegia inizialmente fl accida e arefl essica (shock midollare); successivamente appaiono segni di lesione del primo motoneurone. Rifl essi osteotendinei esaltati al di sotto della lesione.
∙ Defi cit sensoriale. Interessate tutte le sensibilità.
∙ Disturbi autonomici. Malfunzionamento sfi nterico vescicale (urgenza minzionale) e rettale (stitichezza).
∙ Altri sintomi autonomici sono l’anidrosi, i cambiamenti cutanei trofi ci e la disfunzione sessuale (impotenza).
Emisezione midollare (Sindrome di Brown-Séquard)
Traumi penetranti, lesioni extramidollari compressive
∙ Perdita della sensibilità dolorifi ca e termica controlaterale (lesione del tratto spinotalamico incrociato).
∙ Perdita della sensibilità propriocettiva ipsilaterale con atassia sensoriale (interruzione dei cordoni posteriori)
∙ Paralisi spastica ipsilaterale (lesione della via piramidale incrociata)
Sindrome midollare centrale
Siringomielia, idromielia e tumori centromidollari
Defi cit sensoriale sospeso bilaterale con conservazione della sensibilità tattile (defi cit sensoriale dissociato)
Lesione delle colonne posterolaterali
Degenerazione subacuta combinata del midollo (defi cit di B
12), mielopatia
vacuolare associata all’AIDS, compressione midollare
∙ Atassia sensoriale con perdita di sensibilità propriocettiva e conservazione della sensibilità del dolore e termica
∙ La disfunzione corticospinale bilaterale produce spasticità, iperrefl essia agli arti inferiori e risposta cutaneoplantare in estensione (lesione del primo motoneurone)
Sindrome cordonale posteriore
Neurosifi lide ∙ Atassia sensoriale
∙ Implica dolori lancinanti alle gambe, incontinenza urinaria e arefl essia rotulea ed achillea.
∙ La disfunzione dei cordoni posteriori nella regione cervicale produce una sensazione di “scarica elettrica” discendente con la fl essione del collo (segno di Lhermitte)
Sindrome dell’arteria spinale anteriore
Dissecazione aortica, aterosclerosi, chirurgia dell’aorta addominale
∙ Paraplegia o tetraplegia acuta con disfunzione vescicale e intestinale e anestesia disfunción vesical e intestinal y anestesia del dolore e termica y térmica infralesionale.
∙ Non è presente perdita delle sensibilità propriocettive
Tabella 5. Principali sindromi midollari
I d e e c h i ave L’associazione tra aprassia della marcia, incontinenza urinaria e de-menza appare caratteristicamente nella idrocefalo normoteso e nelle lesioni frontali bilaterali.
Il fascio corticospinale ed il fascio corticonucleare (fascio genicolato) sono i due tratti principali che formano il sistema piramidale. In que-sto sistema esiste un primo motoneurone la cui lesione interessa ampi gruppi muscolari, rifl essi osteotendinei aumentati, segno di Babinski e ipertonia “lama di rasoio” con assenza di fascicolazioni e spasmi. La le-sione del secondo motoneurone (nel corno anteriore del midollo spi-nale) è caratterizzata da una paresi o paralisi di muscoli isolati o in pic-coli gruppi, rifl essi miotatici diminuiti o assenti, spasmi e fascicolazioni.
Fondamentalmente esistono due vie sensoriali: il sistema colonna dorsale lemnisco mediale (sensibilità epicritica, si decussa a livello del bulbo e le sue fi bre fanno le sinapsi nei nuclei di Goll e Burdach) ed il sistema anterolaterale (sensibilità protopatica: dolorifi ca, ter-mica e tattile protopatica; le fi bre si incrociano portandosi nel lato opposto ,al livello di ingresso nel midollo).
Il livello del defi cit sensitivo è indicativo per la localizzazione della lesione.
Defi cit sensitivo“sospeso” per le sensibilità dolorifi ca e termica, con conservazione di quella tattile e propriocettiva (defi cit dissociato della sensibilità), si osserva in lesioni centromidollari, come la sirin-gomielia.
Nella sindrome cerebellare vermiana, si manifestano atassia della marcia e scarsa o nulla atassia degli arti: nella sindrome cerebella-re emisferica, si trovano atassia degli arti e, con maggior frequenza che nel precedente, dismetria, dissingergia, adiadococinesia e di-smetria.
Bisogna sottolineare che lesioni nel tronco encefalico appaiono sempre caratterizzate da lesioni di nervi cranici ipsilaterali con “vie lunghe” (motorie o sensoriali) controlaterali. I nervi cranici indicano il livello della lesione.
Le sindromi disartria-mano goff a e atassia-emiparesi si possono avere sia con lesioni della capsula interna controlaterale sia in quel-le pontine.
Nelle lesioni bulbari si distinguono due sindromi: laterale o di Wal-lenberg (occlusione dell’arteria vertebrale o cerebellare posteroin-feriore) e mediale (occlusione dell’arteria spinale anteriore o verte-brale).

1212
Neurologia e neurochirurgia
ORIENTAMENTO
Questo tema è poco importante per il MIR. Bisogna prestare attenzione ai concetti messi in evidenza nei “Idee chiave” e ripassare i segni con valore localizzatore della lesione, specialmente la esplorazione delle pupille ed i riflessi troncoencefalici.
2.1. Coma
Defi nizioni
• Vigilanza: stato di coscienza di se stessi e dell’ambiente che lo circonda.
• Sonnolenza/obnubilazione: tendenza al sonno, di fronte a stimoli
lievi il paziente risponde, però in poco tempo ritorna al suo stato ini-
ziale.
• Stupor: solo stimoli precisi e molto importanti per raggiungere una ri-
presa parziale della coscienza, ed il paziente arriva a rispondere parzial-
mente momentaneamente.
• Coma: stato patologico di incoscienza resistente a stimoli esterni im-
portanti.
Fisiopatologia
Il livello normale di coscienza dipende dall’attivazione degli emisferi ce-
rebrali ad opera di gruppi neuronali localizzati nel sistema reticolare atti-
vatore (SRA) del tronco encefalico, compreso tra la porzione rostrale del
ponte e la parte caudale del diencefalo, e riveste un’importanza fonda-
mentale per il mantenimento dello stato di vigilanza.
Lesioni emisferiche possono causare il coma a seguito di alcuni dei se-
guenti meccanismi: 1) lesioni strutturali generalizzate o bilaterali, 2) lesioni
unilaterali che comprimono l’emisfero controlaterale, e 3) compressione
troncoencefalica conseguente ad erniazione.
I disturbi metabolici sono la causa più frequente di coma senza segni di fo-
calità con funzione troncoencefalica intatta.
02
2.2. Segni del valore localizzatore
I segni con valore localizzatore nei pazienti in coma sono: il modello respi-
ratorio (Figura 15), le alterazioni pupillari, i movimenti oculari rifl essi e le
posture rifl esse.
R I C O R D AIl pattern respiratorio di Cheyne-Stokes (periodi di iperventila-zione con pause di apnea) può manifestarsi anche nell’uremia e nell’insuffi cienza cardiaca congestiva. Il pattern di Kussmaul (iperventilazione ritmica con respiri profondi o batipnea) si ma-nifesta negli stati di acidosi. Entrambi i pattern possono manife-starsi nell’ipossia.
R I C O R D A Pupille midriatiche areattive: lesione mesencefalica. Pupille pun-tiformi reattive: lesione pontina. Alterazione pupillare unilaterale: lesione strutturale.
R I C O R D A Le posture rifl esse di decorticazione e di decerebrazione corrispon-dono a dei punteggi che vanno da 3 a 2, rispettivamente, nella va-lutazione della risposta motoria nella scala del coma di Glasgow (in questa scala, la risposta motoria è, a sua volta, il parametro più importante).
Stato di pseudocoma
• Mancanza di risposta psicogena. Il paziente sembra senza risposta,
però fi siologicamente è sveglio. L’esame neurologico clinico è normale
e la risposta oculovestibolare è intatta.
• Mutismo acinetico. Stato di vigilia senza possibilità di elaborazione
di risposta. Può essere dovuto al danno cerebrale bilaterale (quadro
apallico), lesione in porzione superiore del mesencefalo e diencefalo o
idrocefalia acuta.
COMA. MORTE ENCEFALICA

13
02Neurologia e neurochirurgia
Emisferi cerebrali
Patron respiratorio
Altre cause Rifl essi
troncoencefaliciDeviazione
dello sguardoPupille
Diencefalo (talamo e ipotalamo)
Cheyne-Stokes ∙ Uremia
∙ Anossia
∙ ICC
Miotiche areattive
Mesencefalo Hperventilazione neurogena centrale
∙ Chetoacidosi diabetica
∙ Acidosi lattica (Kussmaul)
∙ Ipossiemia Midriatiche areattive
Oculocefalici anormali
=
Non ci sono gli “occhi di bambola”
Ponte Apneustica
Bobbing oculare
Puntiformi reattive
Rifl esso corneale abolito
Bulbo rachideo ∙ Cluster
∙ Atassica de Biot(agonica)
Rifl esso nasale abolito
Posture riflesse Decorticazione Decerebrazione
Figura 15. Segni del valore localizzatore nei pazienti in coma
I d e e c h i ave Il coma è il grado più profondo di diminuzione del livello di coscienza.
La causa più frequente del coma sono i disturbi metabolici.
Il livello di coscienza si valuta nell’esplorazione neurologica attra-verso la scala internazionale di Glasgow (si veda il Capitolo 18. Trau-mi cranioencefalici).
La presenza dei rifl essi oculocefalici (movimento coniugato degli occhi in direzione opposta alla rotazione della testa) indica l’integri-tà funzionale del tronco encefalico.

141414
Neurologia e neurochirurgia
Cause di demenza progressiva
∙ Sindrome di Alzheimer (50-90%)
∙ Infarti cerebrali multipli (5-10%)
∙ Alcool (5-10%)
∙ Disturbi endocrinometabolici:
- Ipotiroidismo
- Defi cit di vitamina B12
∙ Neoplasie intracraniche
∙ Ematoma subdurale cronico
∙ Idrocefalia a pressione normale
∙ Altre malattie degenerative:
- Malattia di Pick
- Sindrome di Parkinson
- Malattia di Huntington
- Paralisi sopranucleare progressiva
∙ Infezioni del SNC:
- HIV
- Sifi lide
- Creutzfeldt-Jakob
Tabella 6. Cause piú frequenti di demenza
Diagnosi delle demenze
La diagnosi delle demenze è clinica: una storia clinica dettagliata è fon-
damentale. Lo sforzo prioritario deve tradursi nell’identifi cazione di quel
gruppo di demenze trattabili. Per questo, un primo approccio deve
contemplare un’analitica completa tra cui ematologia, elettroliti sierici,
biochimica sanguinea, prove di funzione renale, epatica e tiroidea (TSH),
livelli delle vitamine B12
, TC craniale e sierologia, almeno per l’HIV e la
sifi lide.
Tra gli studi neuropsicologici, il più adeguato è il mini mental test, che per-
mette di studiare rapidamente la memoria, l’orientamento spaziotemporale,
il linguaggio, la scrittura, la lettura, il calcolo e la praxis visuo-spaziale e ideo-
motoria. Si da un punteggio da 0 a 30 punti, considerandosi normale da 27
a 30 punti, deterioramento cognitivo lieve da 24 a 27 e demenza sotto i 24
punti (Tabella 7).
ORIENTAMENTO
E fondamentale conoscere la differenza tra i diversi tipi di demenza e le caratteristiche della demenza più frequente, l’Alzheimer.
3.1. Concetto e classifi cazione
La demenza costituisce la principale causa di incapacità a lungo termine
nella terza età. Interessa il 2% della popolazione tra i 65-70 anni di età ed il
20% dei maggiori di 80 anni.
Si defi nisce come un deterioramento cronico delle funzioni superiori, acqui-
sito (a diff erenza del ritardo mentale) ed in presenza di un livello di coscienza
e di attenzione normali (a diff erenza del delirio).
R I C O R D ALa principale diff erenza tra demenza e delirio è che, in quest’ultimo, è minore il livello di coscienza e la memoria immediata risulta alte-rata (dipendente dall’attenzione).
Le cause più frequenti di demenza progressiva si mostrano nella Tabel-
la 6.
Anche se la maggior parte delle demenze sono irreversibili (70%) e non
hanno trattamento, ad eccezione del sintomatico, è importante identifi care
quelle che potenzialmente potrebbero essere trattate.
R I C O R D A
Le pseudodemenze sono deterioramenti cognitivi reversibili che possono apparire nei disturbi depressivi. A diff erenza delle demen-ze, migliorano con la agripnia o privazione del sonno.
03DEMENZE

15
03Neurologia e neurochirurgia
Negli ultimi anni, sono state applicate le tecniche radiologiche alla diagnosi
delle demenze; fondamentalmente sono stati realizzati degli studi con ri-
sonanza magnetica e SPECT/PET. La caratteristica è l’atrofi a temporale e le
disfunzioni temporoparietali nella fase iniziale della malattia d’Alzheimer, o
l’atrofi a e disfunzione frontale nella demenza frontotemporale.
TestPunteggio massimo
Orientazione Che anno, mese, giorno, data e stazione dell’anno è? Dove ci troviamo, in quale piano, in quale città,
provincia e nazione?
5
5
Ripetizione Nominare tre oggetti (uno per volta) e chiedere
al paziente di ripeterli successivamente 3
Attenzione e calcolo Deve dire lettera per lettera una parola di cinque
lettere al contrario (es. la parola “Mondo”) o sottrarre il numero 3 di volta in volta, partendo dal 30 e arrivando al 18
5
Memoria Chiedere i tre oggetti nominati precedentemente 3
Linguaggio ∙ Indicare una matita. Il paziente deve dare il nome a
questo oggetto. ∙ Il paziente deve ripetere parole semplici come: “no”,
“sempre”, “quando” o “ma” ∙ Dare al paziente i seguenti ordini: ∙ “Prenda questo foglio con la mano destra” “Pieghi il
foglio a metà” “Metta il foglio sul pavimento” ∙ Il paziente deve scrivere una frase che vuole ma che
abbia un senso ∙ Il paziente deve copiare, inserendo tutti gli angoli
e l’intersezione, due pentagoni disegnati
2
13
1
1
1
Totale 30
Tabella 7. Mini mental test
Si possono distinguere due tipi di demenze, in funzione della localizzazione
delle lesioni: corticali e subcorticali (Tabella 8).
Corticale Subcorticale
Anatomia patologica
∙ Corteccia dei lobi frontali, parietali e temporali
∙ Ippocampo
Nuclei grigi profondi dell’encefalo
Clinica ∙ Afasia
∙ Aprassia
∙ Agnosia
∙ Acalculia
∙ Ritardo psicomotorio
∙ Movimenti anormali
∙ Disartria
∙ Alterazioni posturali
∙ Depressione
Esempi ∙ Alzheimer
∙ Pick
∙ Creutzfeldt-Jakob
∙ Meningoencefalite
∙ Ipossia
∙ Vascolare
∙ Neoplasie
∙ Post-trauma
∙ Huntington
∙ Parkinson e Parkinson plus
∙ Wilson
∙ HIV
∙ Vascolare
∙ Neoplasie
∙ Post-trauma
Tabella 8. Correlazione anatomo-clinica nelle demenze
3.2. Malattia di Alzheimer
Epidemiologia
La malattia di Alzheimer è la causa più frequente di demenza in Occidente.
L’età d’inizio dei sintomi è intorno ai 65 anni, e la prevalenza si duplica ogni
5 anni.
Anatomia patologica
Si caratterizza per una degenerazione progressiva e selettiva della popola-
zione neuronale nella corteccia entorinale, nell’ippocampo, nelle aree corti-
cali di associazione temporali, frontali e parietali, nei nuclei subcorticali e del
tronco (locus coeruleus e nuclei del rafe) (Figura 16).
Figura 16. TC di un paziente con malattia di Alzheimer. Mostra un aumento marcato del sistema ventricolare e dei solchi. La scissura di Silvio ed i corni temporali dei ventricoli laterali sono i più gravemente colpiti
A livello macroscopico, la perdita di neuroni si traduce in un’atrofi a gene-
ralizzata, più grave nei lobi temporali, che si accompagna alla dilatazione
secondaria del sistema ventricolare.
R I C O R D A Le lesioni istologiche tipiche della malattia di Alzheimer sono i depositi intracellulari di tau () iperfosforilata e le placche della -amiloide. Il deposito di quest’ultima si produce anche nei cer-velli anziani e nelle altre patologie come la sindrome di Down, l’angiopatia congofi la e la miosite da corpi inclusi.
Istologicamente, possono trovarsi grovigli o matasse neurofi brillari e plac-
che di amiloide (placche senili o neuritiche) che, anche se non sono pato-
gnomoniche, nell’Alzheimer sono specialmente frequenti nell’ippocampo e
nel lobo temporale.
La somatostatina è il neurotrasmettitore che con più frequenza sembra ri-
dotto, anche se la acetilcolina è quello che sembra più relazionato con il
grado di deterioramento cognitivo.
Genetica e fattori di rischio
L’età è il principale fattore di rischio per lo sviluppo della malattia di Alzhei-
mer. Approssimatamente in un 25% dei casi la storia clinica presenta antece-
denti con un’età di inizio precoce.

1603 ∙ Demenze
Manuale CTO di Medicina e Chirurgia, Prima edizione
Sono stati implicati tre locus cromosomici le cui mutazioni sono state asso-
ciate alla malattia di Alzheimer con inizio precoce:
• Gene della proteina precorritrice amiloide nel cromosoma 21.
• Gene della presenilina1 nel cromosoma 14. È nel locus più fre-
quentemente implicato nei casi di Alzheimer ad inizio precoce
(70%).
• Gene della presenilina2 nel cromosoma 1, con un’incidenza molto
bassa.
I fattori di rischio associati alla malattia di Alzheimer sporadica sono:
• Vulnerabilità genetica. La presenza dell’allele E4 dell’apolipoproteine E.
• Età.
• Sesso. Più frequente nelle donne.
• Storia di trauma cranico precedente.
Clinica
Si tratta di una malattia ad inizio insidioso e con progressione lenta, con un’e-
voluzione media di circa 8-10 anni dall’inizio fi no alla morte.
1. Stadio preclinico: errori puntuali di memoria, senza che esistano altri
defi cit. È in questa fase iniziale che il paziente può soff rire di depressio-
ne.
2. Fase de stato: alterazione della memoria recente e della capacità di
apprendimento. Inizialmente la memoria remota si mantiene intatta.
Progressivamente si associano alterazioni di altre funzioni corticali che
vanno aumentando di gravità: afasia, aprassia, agnosia, alterazione
nell’astrazione e nell’ideazione, mancanza d’iniziativa…
3. Fase fi nale: tutti i defi cit precedenti sono molto gravi. Solo nelle fasi
molto evolute possono comparire segni extrapiramidali, come anda-
tura impacciata, postura incurvata, bradicinesia generalizzata e rigidità.
Generalmente, la causa della morte è una malattia intercorrente, come
infezioni.
Trattamento farmacologico
I potenziali obiettivi del trattamento farmacologico sono: 1) miglioramen-
to cognitivo; 2) rallentamento della progressione; 3) ritardo nella comparsa
della malattia.
• Inibitori dell’acetilcolinesterasi. Indicati nelle fasi lieve e moderata
della malattia; non modifi cano a lungo termine la progressione della
malattia, però provocano un miglioramento delle funzioni cognitive du-
rante i primi mesi di trattamento.
• Memantina. È un antagonista non competitivo dei recettori dell’ N-me-
til-D-aspartato (NMDA) del glutammato, indicato nelle fasi moderate ed
avanzate della malattia d’Alzheimer.
3.3. Demenza frontotemporale(malattia di Pick)
È una malattia degenerativa caratterizzata da una marcata perdita asimme-
trica di neuroni nelle regioni anteriori dei lobi frontali e temporali, con nor-
malità nel resto del cervello (Figura 17).
Istologicamente ci sono due dati caratteristici: 1) neuroni di Pick, sono neu-
roni tumefatti, pallidi, che non si contaminano con le macchie solite e lo-
calizzate nei lobi frontali; 2) corpi di Pick, sono inclusioni citoplasmatiche
localizzate nelle regioni temporali anteriori. Non si osservano matasse neu-
rofi brillari nè placche senili.
Figura 17. Malattia di Pick
R I C O R D A La demenza di Pick si diff erenzia dall’Alzheimer perché si sviluppa nelle persone più giovani: non si presenta con amnesia, né aprassia né agnosia, pur essendo una demenza corticale; le alterazioni com-portamentali e del linguaggio sono più precoci, e non appaiono matasse né placche neuritiche.
Interessa pazienti di media età, essendo una delle demenze più frequenti
tra i 45 e i 65 anni. Si manifesta come una demenza lentamente progres-
siva, dove i cambiamenti di personalità sono i sintomi più evidenti: dif-
fi coltà nelle relazioni sociali, nelle emozioni, nell’insight, accompagnata
dalla perdita delle capacità esecutive. Con l’avanzamento della malattia,
l’apatia e l’abulia dominano in quadro. Insieme a questi sintomi, l’insuf-
fi cienza della memoria recente e della capacità di apprendimento sono
molto frequenti. Il linguaggio viene colpito già nella fase iniziale, poten-
dosi presentare come il primo sintomo (la così chiamata afasia prima-
ria progressiva). Non appaiono alterazioni tipo agnosia o aprassia, come
nell’Alzheimer.
3.4. Demenza vascolare
Demenza multinfartuale
È quella che si produce come sequela di multiple aree di infarto cerebrale.
Si ha il sospetto della malattia quando la demenza inizia bruscamente, e
soprattutto se esistono antecedenti di qualsiasi tipo di malattia vascolare
cerebrale e si accompagna con segni di focalità neurologica. La causa più
frequente è l’embolia cerebrale bilaterale recidivante.
Malattia di Binswanger
Chiamata anche encefalopatia subcorticale arterosclerotica, è una forma di
demenza vascolare associata a ipertensione arteriosa e aterosclerosi, conse-
guente a malattia dei piccoli vasi (arterie perforanti). Si caratterizza per una
demielinizzazione diff usa della sostanza bianca subcorticale con aumento
delle dimensioni dei ventricoli sottostanti (Figura 18). Si presenta come una
demenza subcorticale, con andatura tipica a piccoli passi e base di appoggio
allargata, paralisi pseudobulbare e segni corticospinali.

17
03Neurologia e neurochirurgia
Figura 18. RM cerebrale. Paziente con malattia di Binswanger. Si nota una iperdensità diff usa periventricolare corrispondente al concetto di leucoaraiosi
R I C O R D ALe demenze con causa vascolare sono le seconde a livello di inci-denza. Possono essere corticali o subcorticali, e si caratterizzano da una comparsa brusca, con focalità neurologica, e un decorso clinico fl uttuante, “a gradini”.
La leucoaraiosi è un termine neuro-radiologico che descrive le aree ipodense
nella tomografi a computerizzata (TC) o iperintense nella risonanza magneti-
ca (RM), a distribuzione periventricolare e in centro semiovale, che rifl ettono
la demielinizzazione. È tipica di questa malattia, però non è patognomonica.
3.5. Demenza da corpi di Lewy
È la terza causa di demenza nell’anziano, dopo l’Alzheimer e la demenza
vascolare. Lo studio anatomopatologico rivela un predominio dei corpi di
Lewy a livello neocorticale. I pazienti presentano un deterioramento cogni-
tivo lentamente progressivo di tipo frontosubcorticale.
Le fl uttuazioni cognitive sono molto frequenti, con variazioni rilevanti nell’at-
tenzione e nello stato di allerta. Le allucinazioni visive o presenziali sono carat-
teristiche, così come le alterazioni nel sonno REM (nella fase di atonia musco-
lare, la presenza di un’attività fi sica incessante aiuta a confermare la diagnosi).
Si accompagna abitualmente ad un parkinsonismo che ha un predomi-
nio della clinica rigido-acinetica, con scarso tremore e scarsa risposta alla
L-dopa, può essere indiff erenziabile dalla malattia del Parkinson . È fre-
quente l’elevata suscettibilità ai neurolettici, con peggioramento motorio
e cognitivo.
I d e e c h i ave Si defi nisce demenza il deterioramento progressivo delle funzioni superiori, acquisito e con preservazione a livello dello stato di co-scienza. La prevalenza della demenza aumenta con l’età
La demenza si classifi ca in irreversibile (la maggior parte) o rever-sibile, e in corticale o subcorticale (si vedano le tabelle del capito-lo).
La malattia di Alzheimer è la causa più frequente di demenza in Occidente. È una demenza corticale, di predominio temporopa-rietale. Il suo inizio è insidioso e la progressione lenta, l’età avan-zata è il principale fattore a rischio per il suo sviluppo. Per il suo
trattamento si impiegano inibitori dell’acetilcolinesterasi (done-pezil, rivastigmina, galantamina) nelle fasi lieve e moderata, e antagonisti non competitivi dei ricettori glutammatergici NMDA (memantina) nelle fasi avanzate.
La demenza frontotemporale o di Pick è anch’essa una demenza corticale. Si presenta con afasia, apatia, abulia e altre alterazioni comportamentali, però senza amnesia, né aprassia ne agnosia.
La demenza vascolare è la seconda, per prevalenza. Rilevanti sono la demenza multinfartuale per embolia bilaterale recidivante (ini-zio brusco e con focalità neurologica) e la malattia di Binswanger o encefalopatia aterosclerotica subcorticale, nella quale è tipica la leucoaraiosi o demielinizzazione periventricolare.
C a s i c l i n i c i Di fronte ad una storia progressiva di 8 anni d’evoluzione, a par-tire dai 60 anni, di deterioramento intellettivo, errori inspiegabili nell’attività quotidiana, scarsa attenzione all’igiene personale, che porta il malato ad una dipendenza assoluta ai suoi familiari, con immobilità totale, incontinenza degli sfi nteri, perdita di peso, con-vulsioni, mioclonia e morte, è possibile stabilire una diagnosi di:
1) Malattia di Parkinson.2) Degenerazione epatolenticolare o malattia di Wilson.3) Encefalopatia spongiforme di Creutzfeldt-Jacob da “proteina prione”.4) Demenza vascolare.5) Demenzia del tipo Alzheimer.
RC: 5

181818
Neurologia e neurochirurgia
L’incidenza globale in Spagna delle malattie cerebrovascolari non si cono-
sce con precisione, si stima intorno ai 150-250 casi su 100.000 abitanti/anno.
Il tasso di mortalità è di 95 per 100.000 abitanti/anno, costituendo la terza
causa di morte dopo le cardiopatie e il cancro.
4.1. Territori vascolari cerebrali
I territori vascolari cerebrali vengono descritti nella Figura 19.
04
4.2. Classifi cazione e fattori di rischio
Si distinguono due grandi gruppi di lesioni vascolari: ischemici ed emorragici.
• Le lesioni ischemiche rappresentano l’80-85% dei casi. Possono esse-
re focali (per ostruzione arteriosa o venosa) o diff use (arresto cardiaco,
anossia o ipoperfusione). Si possono anche classifi care in trombotiche
o tromboemboliche.
• L’emorragia intracranica rappresenta approssimativamente un 15-20%
di tutti gli accidenti cerebrovascolari, e l’ipertensione arteriosa costitui-
MALATTIE CEREBROVASCOLARI
ORIENTAMENTO
Questo tema è il più importante di tutta la neurologia, e per tanto va studiato approfonditamente. All’interno dello stesso, è fondamentale conoscere i tipi di accidenti cerebrovascolari (ACV), le diverse eziologie e fattori di rischio, la clinica in funzione dei territori vascolari interessati, i metodi diagnostici, il trattamento in fase acuta e la profilassi. È necessario studiare i “Idee chiave” di questo tema e non dimenticarsi i riquadri a cui prestare attenzione all’interno del testo stesso.
A. cerebrale anteriore
A. corioidea anteriore
A. cerebrale media
A. cerebrale posteriore
A. cerebrale anteriore
A. comunicanteanteriore
A. cerebellareanterosuperiore
A. cerebellareanteroinferiore
A. spinale anteriore
A. cerebellarepostero-inferiore
A. carotide interna
A. cerebrale media
A. corioidea anteriore
A. comunicante posteriore
A. cerebrale posteriore
A. basilare
A. cerebellarepostero-inferiore
Figura 19. Territori vascolari cerebrali

19
04Neurologia e neurochirurgia
sce il principale fattore associato (50-70% dei casi). La maggior parte di
queste emorragie sono localizzate in profondità negli emisferi cerebrali.
Malattie cerebrovascolari e cardiovascolari hanno in comune gli stessi fattori
di rischio ma i primi riconoscono nell’ipertensione arteriosa il principale fat-
tore eziologico.
R I C O R D A L’ipertensione arteriosa è un fattore di rischio sia per le malattie ce-rebrovascolari sia per la cardiopatia ischemica, però il rischio relati-vo è maggiore nella prima. Inoltre, il trattamento dell’ipertensione arteriosa ha dimostrato un maggior eff etto protettivo nella preven-zione delle malattie cerebrovascolari che in quella della cardiopatia ischemica.
4.3. Malattie cerebrovascolari ischemiche
Classifi cazione
Le malattie cerebrovascolari ischemiche si classifi cano in:
• Attacco ischemico transitorio (TIA). Defi cit neurologico con durata
minore di 24 ore.
• Ictus o stroke. Defi cit neurologico che dura più di 24 ore, causato dalla
diminuzione del fl usso sanguigno in un territorio cerebrale.
• Ictus progressivo. È un defi cit neurologico d’inizio improvviso che pro-
gredisce o presenta fl uttuazioni mentre il paziente è sotto osservazione.
Può essere dovuto ad un evento emorragico, a stenosi trombotica pro-
gressiva di un’arteria, edema cerebrale, chiusura dei collaterali o ipoten-
sione arteriosa.
R I C O R D A Un ictus maligno è un ictus del territorio dell’arteria cerebrale me-dia o della carotide che subisce una complicazione in presenza di edema cerebrale con spostamento della linea media (swelling) e diminuzione del livello di coscienza, compromettendo la vita del paziente, associato a segni di focalità neurologica propria degli in-farti. In questi casi può essere utile la craniotomia decompressiva per evitare l’erniazione cerebrale. In particolare è specialmente utile in pazienti giovani con infarti nell’emisfero non dominante.
Eziologia
• Infarto aterotrombotico. L’aterosclerosi dei grandi vasi extracrani-
ci, soprattutto della carotide, è la principale causa di ictus ischemico
(Figura 20).
• Infarto cardioembolico. I quadri embolici si presentano con defi cit
completo dall’inizio. Costituiscono approssimativamente un 20% degli
accidenti di tipo ischemico, essendo la fi brillazione atriale la causa più
frequente.
Altri fattori di rischio sono i seguenti:
- Trombi murali. Aree discinetiche dopo l’infarto acuto del miocar-
dio o miocardiopatie, soprattutto la dilatativa.
- Malattia valvolare. Specialmente frequente in pazienti con fi -
brillazione atriale e stenosi mitralica. Altra causa è l’endocardite
infettiva o non infettiva, quest’ultima associata a processi tumo-
rali di base.
• Altre. Aumento della dimensione del ventricolo sinistro, forame ovale
pervio ed aneurismi ventricolari.
Figura 20. Angiografi a carotidea dove si nota una lesione arteriosclerotica grave all’origine dell’arteria carotide interna
R I C O R D A Gli accidenti cerebrovascolari (ACV) ischemici embolici di solito si producono nel territorio dell’arteria cerebrale anteriore. Si presen-tano con un defi cit completo sin dall’inizio ed hanno un maggior rischio di trasformazione in emorragici (soprattutto a seguito della riperfusione, per danno endoteliale).
• Infarto lacunare. Gli accidenti cerebrovascolari (ACV) ischemici embo-
lici di solito si producono nel territorio dell’arteria cerebrale anteriore. Si
presentano con un defi cit completo sin dall’inizio ed hanno un maggior
rischio di trasformazione in emorragici (soprattutto a seguito della riper-
fusione, per danno endoteliale) (Figura 21).
Braccio anteriore della capsula interna(atassia-emiparesi, disartria-mano torpe)
Braccio posteriore della capsula interna(ictus motorio puro)
Nucleo ventrale postero-lateraledel talamo (ictus sensitivo puro)
Base della protuberanza(atassia-emiparesi,
disartria-mano torpe,ictus motorio puro)
Figura 21. Localizzazione delle sindromi lacunari più frequenti
• Infarti con eziologia poco frequente:
- Cause ematologiche:
Emoglobinopatie. Anemia falciforme.
Sindrome di iperviscosità. Policitemia, trombocitosi, leuce-
mia, macroglobulinemia, mieloma…
Sindrome di ipercoagulabilità, tumori, mutazione fattore V
Leyden…

2004 ∙ Ma la t t i e ce reb rovasco la r i
Manuale CTO di Medicina e Chirurgia, Prima edizione
In associazione ad anticorpi antifosfolipidici o anticardio-
lipinici. Si dovrebbe sospettare in pazienti con aborti ripetuti
e storia di trombosi venosa.
- Arteriopatia non arteriosclerotica. Dissecazioni, malattia di
moyamoya, displasia fi bromuscolare.
- Malattie sistemiche. Connettivopatie, sindromi mieloproliferative,
malattie del metabolismo.
- Trombosi venosa cerebrale.
• Infarto con eziologia sconosciuta. Dopo un approfondito studio diagno-
stico, non si è trovato il meccanismo eziopatogenetico che ne sta alla base.
Sindromi vascolari
La localizzazione delle sindromi vascolari più frequenti è rappresentata nella
Figura 22.
Arteria carotide interna
In molti casi i sintomi possono simulare quelli della lesione all’arteria cere-
brale media. La clinica tipica è l’amaurosi fugax per occlusione dell’arteria
oftalmica, che consiste in una perdita indolore unilaterale della vista, che si
verifi ca in 10-15 s e dura pochi minuti. Nel fondo oculare si possono osserva-
re talvolta emboli di colesterolo nei vasi retinici.
L’associazione di amaurosi fugax, dolore cervicale e sindrome di Horner è
tipica della dissezione dell’arteria carotide.
R I C O R D A Durante l’esplorazione del fondo oculare nell’amaurosi fugax si pos-sono osservare cristalli di colesterolo nei vasi retinici. Inoltre, all’e-splorazione del fondo dell’occhio durante un’occlusione dell’arteria centrale della retina, sono caratteristici il pallore retinico e la “mac-chia rosso ciliegia” a livello maculare.
Arteria cerebrale anteriore
La causa più probabile è un embolia d’origine cardiaca, non l’aterotrom-
bosi.
Non bisogna dimenticare però che se si forma un embolo cardiaco, l’ar-
teria che viene interessata più frequentemente è l’arteria cerebrale media
(ACM), non l’arteria cerebrale anteriore (ACA). L’occlusione distale dell’arte-
ria comunicante anteriore produce:
• Emiparesi ed emi-ipoestesia controlaterale di predominio crurale.
• Diminuzione dell’attività psicomotoria e del linguaggio spontaneo
come conseguenza delle lesioni nelle aree prefrontali.
• Rifl esso di prensione, suzione e rigidità paratonica a seguito di lesione
delle aree motorie supplementari frontali.
• Aprassia della marcia e, a volte, incontinenza urinaria per lesione del
lobo frontale parasagittale (nelle lesioni bilaterali).
R I C O R D A Nelle lesioni frontali bilaterali, può apparire la triade di Hakim Adams (aprassia della marcia, incontinenza urinaria e deteriora-mento cognitivo), caratteristica dell’idrocefalia normotensiva.
Arteria cerebrale media
È la sindrome vascolare più frequente. Si presenta con:
• Emiparesi ed emi-ipoestesia controlaterali, di predominio faciobra-
chiale.
• Emianopsia omonima controlaterale.
• Deviazione oculocefalica verso il lato della lesione, rimangono pre-
servati i rifl essi oculocefalici ed oculovestibolari.
• Afasia di Broca, Wernicke o globale, a seconda della localizzazione e
dell’estensione della lesione (in lesioni nell’emisfero dominante).
• Possibile asomatognosia (eminegligenza spaziale), anosognosia e diso-
rientamento spaziale in lesioni dell’emisfero non dominante.
ACA: arteria cerebrale anteriore ACM arteria cerebrale media ACP arteria cerebrale posteriore
Area motoria e premotoria MMII(paralisi spastica controlaterale)
Area motoria e premotoria MMSSe volto (paralisi spastica controlaterale)
Centro della mirata coniugata(deviazione verso la lesione)
Corteccia prefrontale(mutismo, abulia, memoria,
riflessi arcaici)
Area di Broca(afasia motoria)
Corteccia auditiva(allucinazioni auditive,
sordità corticale)
Area di Wernicke(afasia sensitiva)
Radiazioni ottiche(emianopsia omonima
controlaterale)
Talamo (síndrome talamica)
Corteccia visuale primaria(emianopsia omonima
controlaterale con rispettomaculare; cecità corticale)
Corteccia somatosensorialeMMSS e volto
(ipoestetica controlaterale)
Corteccia somatosensorialeMMII (ipoestesia controlaterale)
VOLTO
Figura 22. Localizzazione delle sindromi vascolari più frequenti

21
04Neurologia e neurochirurgia
R I C O R D A L’afasia è caratteristica dell’ischemia a livello corticale nel territorio vascolare dell’arteria cerebrale media dell’emisfero dominate.
Arteria coroidea anteriore
Origina dalla porzione sopraclinoidea dell’arteria carotide interna, si presen-
ta con emiparesi ed emi-ipoestesia controlaterali, volto incluso, ed a volte
emianopsia controlaterale omonima. A livello clinico, la diagnosi diff erenzia-
le in presenza di lesioni dell’arteria cerebrale media di solito risulta diffi cile.
Arteria cerebrale posteriore
In seguito ad una lesione occipitale, può dar origine a emianopsia controla-
terale che di solito lascia intatta la visione maculare. I rifl essi pupillari riman-
gono conservati. A volte si riscontrano alessia ed acalculia.
R I C O R D A Se siamo in presenza di alessia con agrafi a, la lesione vascolare si trova nel territorio della cerebrale media; al contrario, se c’è alessia senza agrafi a, il territorio interessato è quello della cerebrale poste-riore.
Se si è in presenza di una lesione della circolazione prossimale, si avrà una
sindrome talamica (emianestesia controlaterale per tutti i tipi di sensibilità,
iperpatia o dolore nell’emicorpo interessato, mano con movimenti pseudo-
atetoidi).
R I C O R D A Una maniera facile d’identifi care il territorio arterioso interessato nella malattia ischemica cerebrale è valutare se esiste emiparesi ed emianopsia: la sindrome dell’arteria cerebrale anteriore si presen-ta con emiparesi crurale controlaterale senza emianopsia. Quello della cerebrale media (più frequente) si presenta con emiparesi fa-ciobrachiale ed emianopsia omonima controlaterale; quella della cerebrale posteriore si presenta senza emiparesi, però con emianop-sia omonima congruente controlaterale e preservazione maculare.
Sistema vertebrobasilare
Sono meno frequenti di quelli localizzati nella circolazione anteriore. I pro-
cessi ischemici a questo livello producono le cosidette “sindromi crociate”o
“alterne”, caratterizzate da alterazioni delle vie lunghe controlaterali (emipa-
resi, emi-ipoestesia) e segni ipsilaterali cerebellari o dei nervi cranici.
R I C O R D A La patologia vascolare del sistema vertebrobasilare produce le sin-dromi crociate: emiparesi e emi-ipoestesia controlaterale e lesione ipsilaterale dei nervi cranici (il nervo cranico determina il lato della lesione).
L’ischemia vertebrobasilare può produrre una perdita brusca della coscienza,
con o senza recupero posteriore, preceduta da sintomi di disfunzione tron-
coencefalica (diplopia, vertigini, atassia…). Le sindromi più importanti sono
state descritte precedentemente nel Capitolo 1, nel paragrafo delle Sindromi
mesencefaliche, pontine e bulbari.
Infarti lacunari
Le sindromi lacunari più frequenti sono:
• Ictus motorio puro. Localizzato nel braccio posteriore della capsula
interna, anche se può localizzarsi anche nella porzione anteriore del
ponte.
• Ictus sensoriale puro. Come causa di un infarto lacunare a livello del
nucleo ventrale posterolaterale del talamo.
• Atassia-emiparesi. Infarto lacunare localizzato nel braccio anteriore
della capsula interna o nella ponte.
• Disartria-mano goff a. Nel braccio anteriore o nel ginocchio della cap-
sula interna controlaterale all’emicorpo interessato, anche se può pro-
dursi anche a seguito di lesioni nel ponte.
Studio diagnostico
La prova diagnostica iniziale in qualsiasi dei diversi tipi di infarto cerebrale
è la TC cranica, per stabilire se la natura dell’ictus è ischemica o emorragica,
così come per scartare cause che possano presentarsi come un processo
vascolare (tumori, sanguinamento, metastasi…) e ci informa in merito all’e-
stensione della lesione ischemica. Durante le prime 24-72 ore, possono non
osservarsi lesioni ischemiche, anche se è possibile individuare segni indiretti,
come asimmetrie dei solchi corticali per edema, spostamento delle strutture
o aumento della densità dell’arteria cerebrale media, nel suo tragitto basale.
È di scarsa utilità per la visualizzazione degli infarti vertebrobasilari dovuti
agli artefatti ossei che si generano nella fossa posteriore. La TC rispetto alla
RM individua meglio i sanguinamenti, anche se quest’ultima è più sensibile
nella visualizzazione delle lesioni della fossa posteriore (criterio di scelta).
Negli infarti lacunari, la prova iniziale di scelta in fase acuta è sempre la TC
cranica (per la diagnosi diff erenziale ischemico-emorragica), però per un
suo studio successivo è necessario realizzare una RM cranica (Figura 23),
dato che la TC non individua gli infarti minori di 5 mm e quelli situati nella
fossa posteriore.
Figura 23. RM cerebrale. Si osserva un piccolo infarto lacunare profondo sinistro indicato dalla freccia
Per riassumere, la Figura 24 espone lo studio diagnostico e terapeutico de-
gli accidenti cerebrovascolari.

2204 ∙ Ma la t t i e ce reb rovasco la r i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Profi lassi e trattamento
Trattamento in fase acuta
• Norme generali. Evitare ipertermie, iperglicemia ed aumento eccessi-
vo della pressione arteriosa, così come diminuzioni brusche di quest’ul-
tima.
• Fibrinolisi con rt-PA. Indicata la somministrazione di rt-PA in pazienti
con:
- Ictus ischemico con meno di 4-5 ore dall’inizio dei sintomi.
- L’età non è in sé criterio di esclusione (nonostante ciò, negli anziani
con età superiore agli 80 anni si raccomanda non superare le 3 ore
di fi nestra terapeutica ed agire con cautela seguendo le indicazio-
ni).
- Punteggio della scala NIHSS (scala internazionale di gravità clinica
dell’ictus) inferiore ai 25 punti.
- Assenza di tutti i criteri di esclusione che appaiono nella Tabella 9.
• Antiaggregante. In pazienti che non compiono i criteri di fi brinolisi.
L’uso dell’acido acetilsalicilico (AAS), 300 mg nelle prime 48 ore a segui-
to dell’ictus ischemico, riduce il rischio di recidiva ed il tasso di mortalità
a medio termine.
Criteri di esclusione
∙ Presenza di emorragia nella TC cranica precedente alla somministrazione del farmaco
∙ Presentazione clinica suggestiva di emorragia subaracnoidea (ESA), anche con TC normale
∙ Defi cit neurologico scarso o sintomi che migliorano rapidamente ∙ Scala NIHSS > 25 punti ∙ Convulsioni all’inizio dell’ictus ∙ Presenza di diatesi emorragica (trombopenia < 100.000, trattamento
attuale con anticoagulanti, o trattamento con eparina durante 48 h precedenti e TTPA aumentato)
∙ Pressione arteriosa > 185/110 o necessità di intervento aggressivo per ridurla
∙ Glucosio sanguineo > 400 mg/dl o < 50 mg/dl ∙ Storia di ictus previo e DM concomitante ∙ Ictus negli ultimi tre mesi ∙ Emorragia grave o manifesta o recente ∙ Precedenti di lesione del SNC: emorragia, neoplasia, aneurisma,
chirurgia… ∙ Patologia grave concomitante (endocarditi, pancreatiti, gastropatia
ulcerativa recente, aneurisma, neoplasia con rischio emorragico, epatopatia grave)
∙ Chirurgia maggiore o trauma importante negli ultimi tre mesi
Tabella 9. Criteri di esclusione per la fi brinolisi endovenosa
Trat
tam
ento
Pro
ve c
om
ple
men
tari
(dia
gn
osi
ezi
olo
gic
a)Fa
tto
ri
di r
isch
ioEz
iolo
gia
Cla
ssifi
cazi
on
eClinica sospetta ACV
Ictus (> 24 ore)
TIA (< 24 ore)
Normale, però clinica evidente:ripetere in 72 ore
· Pressione arteriosa· Analitica
· ECG· Rx torace
TC cranica: elezione in fase acuta.Si farà RM se clinica della fossaposteriore o sindrome lacunareo sospetto di trombosi venosa
ScartareACV
Patologico
Emorragico (20%)Iperdensità
Ischemico (80%)
< 24 oreCancellazione strutture
Effetto massa
> 24 oreIpodensità focale
Trombotico Embolico Infarto lacunare Trombosi venosa Altro
· Processi settici· Gravidanza e puerperio· Disidratazione (anziani)· Anticoncettivi orali· Traumi cranici· Processi ematologici
· Arterite: temporale,· Takayasu...· Dissezione arteriale· Malattia moyamoya (nuvole di fumo)· Stati di ipercoagulazione· Malattia di Binswanger· Displasia fibromuscolare· Vasospasmi· Anticoncettivi orali
Aterosclerosibiforcazione carotidea
(ITA tabacco colesterolo)
Fibrillazione auricolareTrombo murale (IAM)
Valvolopatia (SM)Di solito vanno alla cerebrale media (80%)
Ipertensione arteriosa(lipoialinosi) RM
Angiografia
· Eco-doppler carotideo· Angiografia· Ecografia transesofagica· ECG
Valutare fibrinolisia seconda del tempo
d’evoluzione
· Antiaggregazione con AAS (ticlopidina o clopidogrel di 2° scelta)· Stenosi carotidea
Sintomatica Asintomatica> 70 % Tromboendoarteriectomia50-70% ??< 50% AAS
Anticoagulazione(differita se infarto esteso) Controllo stretto
della PA AAS
Segno della vuotain TC con contrasto
Anticoagulazione
Figura 24. Algoritmo diagnostico e terapeutico degli accidenti cerebrovascolari

23
04Neurologia e neurochirurgia
La Figura 25 mostra il protocollo di gestione e trattamento dell’ictus in fase
acuta.
Clinica suggerente di ictus
TC cranica Altra causa Trattamentospecifico
Emorragia Ischemiao normalità
Gestionedell’emorragia
intracranica
Controindicazionidi fibrinolisi?
Si No
Antiagreggazione· AAS· Clopidogrel
Tempo d’evoluzione
Aterosclerotico· Lacunare· Idiopatico Fibrinolisi i.v. TrombectomiaFibrinolisi
intrarterialeAnticoagulazione
(controindicatoin fase acuta se
grande area d’infarto)TC cranica di controllo 24-48 h
(confermare che non ci sia sanguinamento)
· FA· Trombosi venosa cerebrale· Dissezione carotidea/vertebrale· Ictus progressivo· Trombo murale cardiaco
Antiaggregazione/anticoagulazionesecondo eziologia
Figura 25. Schema d’intervento sull’ictus in fase acuta
Trattamento endovascolare
• Fibrinosi intrarteriosa: applicabile fi no a 6 ore dall’inizio dei sintomi.
• Trombectomia meccanica: applicabile fi no a 8 ore dall’inizio dei sintomi.
Prevenzione primaria
• Trattamento dell’ipertensione arteriosa: una diminuzione della pres-
sione arteriosa diastolica di 5-6 mmHg riduce il rischio del 42% circa.
• A seguito dell’IAM: anticoagulanti orali (INR 2-3, se si associa FA) e trat-
tamento con statina anche con valori di colesterolo nella norma.
• FA: anticoagulazione orale, soprattutto se si associa patologia valvolare.
R I C O R D A Le indicazioni all’anticoagulazione in fase acuta sono: fi brillazione atriale, dissezione carotidea, ictus ischemico progressivo o trombosi dei seni venosi durali. In fase cronica, si anticoagula se si compiono due dei seguenti criteri: età superiore ai 65 anni, fi brillazione atria-le, presenza di fattori di rischio cardiovascolari o dimostrazione di trombi intracardiaci.
Prevenzione secondaria
• Patologia vascolare cerebrale con origine nel territorio carotideo o
vertebrobasilare. La prevenzione secondaria nella carotide sintomati-
ca viene riassunta nello schema della Figura 26.
Con stenosi carotidea asintomatica si raccomanda l’antiaggrega-
zione. Tuttavia, quando la stenosi è emodinamicamente signifi ca-
tiva (>70%) ed evolutiva nel tempo, può essere di benefi cio l’en-
darterectomia carotidea, sempre che la morbilità operatoria non
superi il 5%.
Stenosi carotideasintomatica
< 50%(non significativa)
> 70%(significativa)50-70%
Antiaggregazione Individualizzazionedel caso
Endarterectomia
Figura 26. Prevenzione secondaria della stenosi carotidea sintomatica
R I C O R D A Se la stenosi sintomatica della carotide è del 50-69%, si opta per l’endarterectomia carotidea, al posto dell’antiaggregazione, sola-mente se si tratta di maschi giovani, non diabetici, con speranza di vita superiore ai 5 anni e con un rischio chirurgico inferiore al 6%. Se la stenosi sintomatica della carotide è completa (100%), non si realizza chirurgia, ma solo antiaggregazione.
• Patologia vascolare cardioembolica. La profi lassi secondaria di scelta
è l’anticoagulazione orale. Quando l’area d’ischemia cerebrale aumenta,
non si raccomanda l’anticoagulazione in fase acuta, dato l’alto rischio
di trasformazione emorragica dell’infarto. In questi casi, si consiglia di
realizzare un’anticoagulazione diff erita nel tempo.
Trombosi venosa
Nel 25-40% dei casi non si conosce la causa. Sono state descritte associazioni
con processi septici sistemici o locali (meningiti) in approssimatamente un
15% delle trombosi venose. Altre eziologie associate vengono illustrate nella
Tabella 10.
La clinica è molto vasta, dalle forme asintomatiche a quelle che si presentano
con cefalea o coma. Di solito inizia con una sindrome di ipertensione intracra-
nica, essendo la cefalea il sintomo più frequente. Può susseguirsi un quadro di
focalità neurologica con crisi focali o generalizzate, emiparesi, lesione dei nervi
cranici. All’esame obiettivo, è possibile osservare edema della papilla.
Comuni Poco frequenti
∙ Infezioni
∙ Gravidanza-puerperio
∙ Disidratazione (negli anziani)
∙ Contraccettivi orali
∙ Coagulopatia (trombocitosi), Ematologico (anemia cel. falciformi, emoglobinuria parossistica notturna)
∙ Tumori (invasione locale, es. meningiomi)
∙ Traumi
∙ Malattia infiammatoria intestinale
∙ Sindrome di Behçet
∙ Lupus anticoagulante
∙ Abuso di droghe
∙ Sindromi paraneoplastiche
Tabella 10. Cause di trombosi venosa cerebrale

2404 ∙ Ma la t t i e ce reb rovasco la r i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Per quanto riguarda le prove complementari, sono utili:
• Liquido cefalorachideo (LCR). Un aumento de pressione.
• TC. Potrebbe essere normale o mostrare dei dati indiretti di edema ce-
rebrale. Il segno della “” vuota (Figura 27) è caratteristico, e consiste in
un raff orzamento delle pareti del seno trombizzato attorno ad una zona
centrale isodensa che, teoricamente, corrisponde al trombo.
• RM. È la tecnica di scelta, anche se non si esclude la realizzazione di
un’angiografi a cerebrale.
Figura 27. TC cranica di un paziente con trombosi del seno longitudinale superiore, dove si osserva indicato dalla freccia il segno della “ vuota”, che mostra la presenza di un trombo all’interno del seno longitudianle superiore.
• Angiografi a cerebrale. È la tecnica diagnostica che permette di evi-
denziare l’esistenza di un’ostruzione venosa, anche se la RM ha dimo-
strato una buona correlazione con l’immagine angiografi ca e una gran-
de affi dabilità diagnostica (Figura 28).
Figura 28. TC (immagine a sinistra) e RM (immagine a destra) che mostrano una trombosi del seno longitudinale superiore con segno della “d vuota” in pazienti con diagnosi di otite media
4.4. Emorragia intraparenchimale
I processi vascolari emorragici rappresentano approssimativamente il
20% degli ACV. Dato che l’emorragia nello spazio subaracnoideo o nel
parenchima cerebrale produce un minor danno tissutale che l’ischemia,
i pazienti che sopravvivono alla fase acuta possono mostrare un marcato
recupero funzionale.
La Figura 29 e la Tabella 11 mostrano le cause più frequenti di emorragia
intracranica spontanea. In questo paragrafo si tratterà solamente l’emorra-
gia intraparenchimale.
TC cranica: iperdensità
ACV emorragico(20%)
Emorragiasubaracnoidea
Emorragia intraparenchimatosa
Emorragiaipertensiva
Emorragia lobarespontanea
• Rottura microaneurismi di Charcot Bouchard• Deficit neurologico focale con inizio brusco e decorso progressivo• Clinica:
Sostanza bianca subcorticale
Anziano Giovane Altro
Si veda lo schemadel coma
Putamen
Talamo
Ponte
Cervelletto
Emiparesi ed emi-ipoestesiacontrolateraliDeviazione degli occhiverso il lato della lesioneDiminuzione del livellodi coscienza
Sd. talamica+ emiplegia controlaterali
Coscienza preservatainizialeCefalea occipitale, atassia,VomitoIdrocefalia per compr. IV ventricolo
ANGIOPATIAAMILOIDE
MALFORMAZIONEVASCOLARE
Causa piùfrequentedi em. Noipertensionearteriosanell’anzianoRecidivante
· Angioma venoso: la più freq. asintomatica· Malformazione arteriovenosa: la più freq. sintomatica· Angioma capillare (telangiectasia)· Angioma cavernoso
Bisogna sospettarlonei pazienti giovanicon emorragiain una zona atipicae storia di cefaleaunilaterale e pulsatilecon convulsioni.In questi casi bisognafare un’angiografia.Cerebellare (>3 cm di diametro)
Erniazione
· Trattamento: controllo pressione arteriosa (evitare cambi bruschi)
- Trattamento dell’IEC
- Chirurgia sì
Figura 29. Emorragia intraparenchimatosa. Algoritmo diagnostico
Eziologia
∙ Emorragia ipertensiva ∙ Aneurisma ∙ Malformazioni arterovenose ∙ Vasculopatie (amiloide, moyamoya, vasculite) ∙ Coagulopatie ∙ Emorragia intratumorale ∙ Abuso di droghe (cocaina, simpaticomimetici, amfetamine) ∙ Conseguente ad infarto venoso
Tabella 11. Cause dell’ematoma intraparenchimatoso cerebrale
L’emorragia intraparenchimale o intracerebrale è di solito causata dalla rot-
tura delle arterie situate nella parte profonda del cervello.

25
04Neurologia e neurochirurgia
A diff erenza dell’ictus schemico, d’inizio improvviso, quelli emorragici di
solito evolvono nel corso di alcuni minuti, e si accompagnano a cefalea,
nausea e vomito. La sintomatologia neurologica dipenderà dalla localizza-
zione e dalla dimensione dell’emorragia.
La prova diagnostica di preferenza quando si sospetta un’emorragia è la TC
cranica (Figura 30).
Figura 30. TC cranica dove viene mostrato un ematoma intraparenchimale profondo
• Emorragia intracerebrale focale ipertensiva. Le localizzazioni anato-
miche più frequenti sono profonde: putamen, talamo, ponte e cervel-
letto (Tabella 12).
Putamen (35-50%)
Emiparesi e emi-ipoestesia controlaterali, deterioramento del livello di coscienza, deviazione oculocefalica verso il lato dell’emorragia con mantenimento dei rifl essi del tronco
Talamo (10-15%)
Deterioramento del livello di coscienza, sindrome talamica ed emiplegia controlaterali
Cervelletto (10-30%)
Mantenimento iniziale del livello di coscienza, cefalea occipitale, atassia, vomito, idrocefalia ostruttiva (per compressione del IV ventricolo)
Ponte (10-15%)
Stato di coma, prognosi infausta
Tabella 12. Clinica e localizzazione dell’emorragia intracerebrale
• Malformazioni vascolari. Si devono sospettare fondamentalmente in
pazienti giovani non ipertesi con emorragie superfi ciali.
• Angiopatia amiloide o congofi la. È la causa più frequente di emorra-
gia spontanea non ipertensiva in pazienti anziani, di solito con localiz-
zazione lobare subcorticale. Clinicamente si presentano come ematomi
spontanei ricorrenti.
R I C O R D A Di fronte ad un’emorragia lobare spontanea e ricorrente in un an-ziano non iperteso, bisogna sospettare un’angiopatia congofi la. In questa, come nell’Alzheimer, si deposita -amiloide.
• Altre cause di sanguinamento cerebrale focale. Coagulopatie, trat-
tamento con anticoagulanti e trombolitici, tumori (metastasi, GBM, me-
dulloblastoma), droghe (amfetamine e cocaina), trasformazione emor-
ragica da ictus ischemico…).
Trattamento
Il trattamento medico si basa nel controllo della pressione arteriosa e nell’u-
tilizzo di manitolo ed altri agenti osmotici per la riduzione della pressione
intracranica.
L’indicazione chirurgica degli ematomi intraparenchimali è un tema
enormemente dibattuto nella letteratura.
In generale, si accetta che la chirurgia non è indicata nel caso di emato-
mi profondi (gangli della base e tronco encefalico), e si raccomanda in
pazienti con emorragia cerebellare acuta di 3-4 cm o più di diametro con
deterioramento del livello di coscienza (se il paziente rimane in stato di
allerta e l’ematoma è di piccole dimensioni, può non essere necessaria la
chirurgia) e segni radiologici di erniazione transtentoriale inversa.
Non esiste consenso nel resto dei casi, anche se pare che la chirurgia
assumerà un ruolo significativo nel trattamento in pazienti giovani con
emorragia lobare sintomatica di dimensione moderata, con marcato ef-
fetto massa, e produzione di deterioramento progressivo del livello di
coscienza.
R I C O R D A L’ematoma cerebellare è l’unico ematoma profondo intracranco che si può asportare chirurgicamente (solo se è superiore ai 3 cm e produce un deterioramento clinico o erniazione).
Occasionalmente, gli ematomi profondi (putaminali e talamici) e quelli cere-
bellari possono drenare nel sistema ventricolare, producendo un’emorragia
intraventricolare e un’idrocefalia acuta che richiede la collocazione di un dre-
naggio ventricolare esterno.
4.5. Malformazioni vascolari
Questo termine ingloba diversi tipi di lesione vascolare non neoplastica del
sistema nervoso centrale. Queste malformazioni vascolari vengono riassun-
te nella Tabella 13.
4.6. Emorragia subaracnoidea
Si defi nisce come la presenza di sangue nello spazio subaracnoideo o nel
sistema ventricolare, dove normalmente è presente solo il liquido cefalo-
rachideo.
Epidemiologia
Ha un’incidenza di circa 10/100.000 abitanti. L’80% si manifesta tra i 40-65
anni. È più comune nelle donne che negli uomini (3:2), specialmente duran-
te la gravidanza.
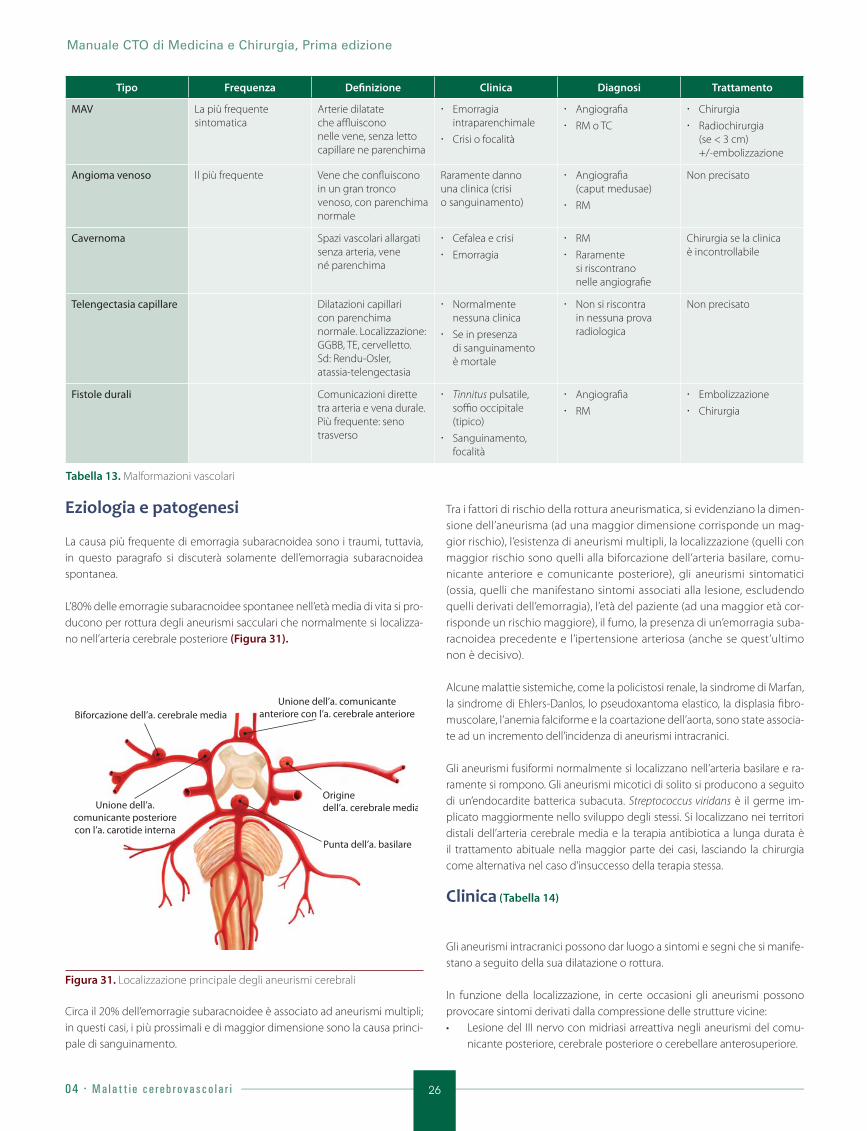
2604 ∙ Ma la t t i e ce reb rovasco la r i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Eziologia e patogenesi
La causa più frequente di emorragia subaracnoidea sono i traumi, tuttavia,
in questo paragrafo si discuterà solamente dell’emorragia subaracnoidea
spontanea.
L’80% delle emorragie subaracnoidee spontanee nell’età media di vita si pro-
ducono per rottura degli aneurismi sacculari che normalmente si localizza-
no nell’arteria cerebrale posteriore (Figura 31).
Biforcazione dell’a. cerebrale media
Unione dell’a. comunicanteanteriore con l’a. cerebrale anteriore
Unione dell’a.comunicante posteriorecon l’a. carotide interna
Punta dell’a. basilare
Originedell’a. cerebrale media
Figura 31. Localizzazione principale degli aneurismi cerebrali
Circa il 20% dell’emorragie subaracnoidee è associato ad aneurismi multipli;
in questi casi, i più prossimali e di maggior dimensione sono la causa princi-
pale di sanguinamento.
Tra i fattori di rischio della rottura aneurismatica, si evidenziano la dimen-
sione dell’aneurisma (ad una maggior dimensione corrisponde un mag-
gior rischio), l’esistenza di aneurismi multipli, la localizzazione (quelli con
maggior rischio sono quelli alla biforcazione dell’arteria basilare, comu-
nicante anteriore e comunicante posteriore), gli aneurismi sintomatici
(ossia, quelli che manifestano sintomi associati alla lesione, escludendo
quelli derivati dell’emorragia), l’età del paziente (ad una maggior età cor-
risponde un rischio maggiore), il fumo, la presenza di un’emorragia suba-
racnoidea precedente e l’ipertensione arteriosa (anche se quest’ultimo
non è decisivo).
Alcune malattie sistemiche, come la policistosi renale, la sindrome di Marfan,
la sindrome di Ehlers-Danlos, lo pseudoxantoma elastico, la displasia fi bro-
muscolare, l’anemia falciforme e la coartazione dell’aorta, sono state associa-
te ad un incremento dell’incidenza di aneurismi intracranici.
Gli aneurismi fusiformi normalmente si localizzano nell’arteria basilare e ra-
ramente si rompono. Gli aneurismi micotici di solito si producono a seguito
di un’endocardite batterica subacuta. Streptococcus viridans è il germe im-
plicato maggiormente nello sviluppo degli stessi. Si localizzano nei territori
distali dell’arteria cerebrale media e la terapia antibiotica a lunga durata è
il trattamento abituale nella maggior parte dei casi, lasciando la chirurgia
come alternativa nel caso d’insuccesso della terapia stessa.
Clinica (Tabella 14)
Gli aneurismi intracranici possono dar luogo a sintomi e segni che si manife-
stano a seguito della sua dilatazione o rottura.
In funzione della localizzazione, in certe occasioni gli aneurismi possono
provocare sintomi derivati dalla compressione delle strutture vicine:
• Lesione del III nervo con midriasi arreattiva negli aneurismi del comu-
nicante posteriore, cerebrale posteriore o cerebellare anterosuperiore.
Tipo Frequenza Defi nizione Clinica Diagnosi Trattamento
MAV La più frequente sintomatica
Arterie dilatate che affl uiscono nelle vene, senza letto capillare ne parenchima
∙ Emorragia intraparenchimale
∙ Crisi o focalità
∙ Angiografi a
∙ RM o TC
∙ Chirurgia
∙ Radiochirurgia (se < 3 cm) +/-embolizzazione
Angioma venoso Il più frequente Vene che confl uiscono in un gran tronco venoso, con parenchima normale
Raramente danno una clinica (crisi o sanguinamento)
∙ Angiografi a (caput medusae)
∙ RM
Non precisato
Cavernoma Spazi vascolari allargati senza arteria, vene né parenchima
∙ Cefalea e crisi
∙ Emorragia
∙ RM
∙ Raramente si riscontrano nelle angiografi e
Chirurgia se la clinica è incontrollabile
Telengectasia capillare Dilatazioni capillari con parenchima normale. Localizzazione: GGBB, TE, cervelletto. Sd: Rendu-Osler, atassia-telengectasia
∙ Normalmente nessuna clinica
∙ Se in presenza di sanguinamento è mortale
∙ Non si riscontra in nessuna prova radiologica
Non precisato
Fistole durali Comunicazioni dirette tra arteria e vena durale. Più frequente: seno trasverso
∙ Tinnitus pulsatile, soffi o occipitale (tipico)
∙ Sanguinamento, focalità
∙ Angiografi a
∙ RM
∙ Embolizzazione
∙ Chirurgia
Tabella 13. Malformazioni vascolari

27
04Neurologia e neurochirurgia
• Oftalmoplegia, lesione del ramo oftalmico del V nervo e cefalea retro-
oculare negli aneurismi del seno cavernoso.
• Lesione del campo visivo negli aneurismi della porzione sopraclinoidea
dell’arteria carotide interna.
Gradi Criteri
Grado 0 Aneurisma intatto
Grado I ∙ Asintomatico o minima cefalea
∙ Rigidità nucale
Grado IA Senza reazione meningea o cerebrale ma con defi cit neurologici
Grado II ∙ Cefalea moderata o grave
∙ Rigidità nucale
∙ Senza defi cit neurologici gravi ad eccezione della lesione dei nervi cranici
Grado III Sonnolenza, confusione o defi cit focali lievi
Grado IV ∙ Stupor
∙ Emiparesi moderata o grave
∙ Alterazioni vegetative e possibile rigidità da decerebrazione precoce
Grado V ∙ Coma profondo.
∙ Rigidità da decerebrazione, stato moribondo
Tabella 14. Classifi cazione di Hunt e Hess dell’emorragia subaracnoidea
R I C O R D A Di fronte ad una midriasi non reattiva in una persona adulta, bisogna scartare un aneurisma nell’arteria comunicante posteriore.
Sfortunatamente, la clinica più evidente della presenza di un aneurisma è
quella conseguente alla rottura dello stesso. Il paziente riporta una cefalea
intensa (“la peggior cefalea della sua vita”), rigidità nucale, nausea e vomito.
Sono altrettanto comuni la fotofobia e la letargia. Nel momento della rottura,
circa la metà dei pazienti perde provvisoriamente la coscienza, con aumento
acuto della pressione intracranica che può raggiungere o superare i livelli
della pressione arteriosa. L’aumento della pressione intracranica può causare
paresi del VI nervo cranico. Nel fondo oculare si possono osservare papille-
dema ed emorragie subialoidee.
Nella metà dei casi, può esserci una clinica da “cefalea sentinella” i giorni pre-
cedenti la rottura dell’aneurisma, dovuta a piccoli sanguinamenti subarac-
noidei o all’interno della parete della malformazione.
R I C O R D A Di fronte ad un paziente che arriva al Pronto Soccorso con una bru-sca ed intensa cefalea, bisogna scartare che non si tratti di un’emor-ragia subaracnoidea.
Esistono una serie di scale per classifi care l’emorragia subaracnoidea in fun-
zione della situazione neurologica inziale del paziente, che assume un valore
prognostico. La più utilizzata al momento è la World Federation of Neurologic
Surgeons (WFNS), che fornisce una classifi cazione del paziente in funzione
del suo livello di coscienza, misurato attraverso la scala di Glasgow, e la pre-
senza o meno di focalità neurologica (Tabella 15).
Grado HSALivello di coscienza (Scala di Glasgow)
Focalità neurologica maggiore
1 15 -
2 13-14 -
3 13-14 +
4 7-12 +/-
5 3-6 +/-
Focalità neurologica maggiore: emiparesi/emiplegia o afasia
Tabella 15. Scala di classifi cazione dell’emorragia subaracnoidea della WFNS
Prognosi
Circa la metà dei pazienti malati di aneurisma muoiono a seguito della rottu-
ra dello stesso. La mortalità nei primi giorni è del 10%.
Inoltre, possiede una morbilità molto alta, dato che circa un terzo dei pa-
zienti che sopravvivono presenta sequele neurologiche moderate o gravi. I
pazienti con età superiore ai 70 anni hanno una prognosi peggiore. Il fattore
più importante al momento di stabilire la prognosi è la situazione neurolo-
gica iniziale.
Diagnosi
L’algoritmo diagnostico dell’emorragia subaracnoidea è rappresentato nella
Figura 32.
Clinica
Diagnosi ESA
Diagnosi eziologica
· Cefalea brusca ed intensa· Rigidità della nuca· Vomito · Fotofobia
Punzionelombare
TC cerebralesenza contrasto
Angiografia cerebrale 4 vasi
Figura 32. Algoritmo diagnostico dell’emorragia subaracnoidea spontanea
R I C O R D A La TC cerebrale è la prova iniziale nel momento in cui si sospetta un’emorragia subaracnoidea. Tuttavia, la prova più sensibile è la puntura lombare.
Diagnosi eziologica
Angiografi a di quattro vasi. I suoi obiettivi sono defi nire la localizzazione e
la morfologia dell’aneurisma, identifi care altri possibili aneurismi intatti, deli-
neare i vasi adiacenti all’aneurisma e valutare il grado di vasospasmo.
Se l’angiografi a non mostra nessun aneurisma, andrà ripetuta in 2-3 settima-
ne (Figura 33).

2804 ∙ Ma la t t i e ce reb rovasco la r i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Figura 33. Arteriografi a cerebrale con aneurisma sacculare
Complicanze
Medica
L’iponatremia rappresenta la complicanza medica più frequente. A causa
dell’eccessiva stimolazione simpatica, possono prodursi aritmie cardiache in
quasi tutti i pazienti (essendo la tachicardia sinusale la più frequente). Si pro-
duce anche ischemia subendocardica ed aree di necrosi miocardica focale,
con i conseguenti cambi elettrocardiografi ci, deterioramento della funzione
cardiaca ed edema polmonare. L’ipertensione arteriosa si può controllare
con -bloccanti, che riducono anche il rischio di aritmie.
Neurologica
• Idrocefalia. Può svilupparsi in forma acuta nelle prime 24 ore, dovuto
al fatto che il sangue all’interno delle cisterne basali o nel sistema ven-
tricolare impedisce la normale circolazione di liquido cefalorachidiano.
In questi casi è consigliata la collocazione di un drenaggio ventricola-
re esterno. L’idrocefalia può anche svilupparsi alcune settimane dopo il
sanguinamento. Si tratta di un’idrocefalia comunicante che si manife-
sta clinicamente con deterioramento cognitivo, incontinenza urinaria e
disturbi del movimento. Il trattamento in questo caso è la derivazione
ventricoloperitoneale.
• Risanguinamento. Esistono due picchi di incidenza di risanguina-
mento, che avvengono nelle prime 24-48 ore (nelle prime 24 ore, può
sanguinare un 4% degli aneurismi) e dopo una settimana. Il risangui-
namento presenta una mortalità del 75%, e la clinica è la stessa che nel
primo episodio, anche se possono apparire nuovi defi cit neurologici.
Il miglior modo di evitare il risanguinamento è escludere l’aneurisma
dalla circolazione generale per via endovascolare (embolizzazione) o
mediante la chirurgia.
• Vasospasmo. È la principale causa di morbimortalità in pazienti che
hanno avuto un’emorragia.
Si sviluppa tra il 4.º-12.º giorno post-sanguinamento (con massima in-
cidenza tra il 6.º ed 8.º giorno) e la clinica corrisponde ad un defi cit del
territorio vascolare interessato (per ischemia) o peggioramento neuro-
logico non spiegabile per altre cause. La quantità di sangue nella TC si
correla con la gravità del vasospasmo. Nella profi lassi del vasospasmo si
usa un calcioantagonista (nimodipina).
Una volta accertato il vasospasmo, la principale linea di trattamento è
la cosidetta terapia “tripla” (emodiluizione-ipervolemia-ipertensione). Il
principale inconveniente di questo trattamento è che aumenta il rischio
di risanguinamento dell’aneurisma, se questo non è stato escluso dalla
circolazione.
R I C O R D A La profi lassi del vasospasmo si realizza con la nimodipina; tuttavia successivamente bisogna applicare la terapia “tripla”.
Trattamento
Nel momento in cui bisogna trattare questi pazienti, si deve distinguere il
trattamento dell’emorragia subaracnoidea con quello della sua causa sotto-
stante (generalmente un aneurisma).
Emorragia subaracnoidea
Gli obiettivi principali del trattamento dell’emorragia subaracnoidea sono la
prevenzione del risanguinamento e del vasospasmo. Per quanto riguarda
il primo, il paziente deve essere collocato in una stanza tranquilla, in riposo
assoluto, con la testa alzata di 30º, per facilitare il drenaggio venoso intracra-
nico. Bisogna sempre tenere sotto controllo la pressione arteriosa (né troppo
alta né troppo bassa). Bisogna evitare che il paziente soff ra di stitichezza e
vomito. Bisogna somministrargli un analgesico dal momento che il dolore
comporta una scarica simpatica che aumenta la pressione arteriosa. Qualora
fosse necessario, si può sedare il paziente con del diazepam. Se si presen-
tano crisi, il farmaco che va utilizzato è la fenitoina (non abbassa il livello di
coscienza). L’utilità del desametasone in questi casi è dibattuta, anche se di
solito si utilizza per ridurre la sintomatologia dolorosa. Bisogna associare la
nimodipina per realizzare la profi lassi del vasospasmo cerebrale. Prestare at-
tenzione alla funzione polmonare (per evitare atelettasie e polmonite).
Aneurisma
Attualmente, esistono due procedimenti il cui obiettivo è quello di isolare
l’aneurisma dalla circolazione cerebrale: l’embolizzazione per via endovasco-
lare e la craniotomia con clipping chirurgico (Figura 34).
Figura 34. Clipping di un aneurisma cerebrale

29
04Neurologia e neurochirurgia
Nei pazienti che presentano un buono stato neurologico (gradi I-III), gli aneu-
rismi di solito vanno trattati in maniera precoce (nei primi 4 giorni); invece
se la situazione neurologica è sfavorevole (gradi IV-V), a volte è consigliabile
trattarlo in maniera diff erente (a partire dal decimo giorno). Il trattamento
precoce dell’aneurisma, con qualsiasi tipo di metodica, permette la gestio-
ne delle complicanze, soprattutto del vasospasmo, diminuendo il rischio di
risanguinamento con la terapia “tripla”. In linea generale, sia l’embolizzazione
sia il clipping hanno una prognosi simile a breve termine. In questi ultimi
anni, la collocazione dello stent e del palloncino stanno facilitando l’embo-
lizzazione aneurismatica.
I d e e c h i ave Gli accidenti cerebrovascolari (ACV) possono essere ischemici (80-85% dei casi) o emorragici (15-20% dei casi). I fattori di rischio sono per buona parte comuni a quelli della patologia ischemica cardiaca. Risulta l’ipertensione arteriosa il principale fattore di rischio negli ACV così come per gli aterosclerotici e gli emorragici, e la fi brillazio-ne atriale, nel caso degli embolici.
Gli ACV embolici producono un defi cit completo già dall’inizio e con maggior tendenza alla trasformazione emorragica.
Gli ACV ischemici per lesione dell’arteria carotide interna sono do-vuti principalmente all’aterosclerosi. La sua clinica tipica è l’amau-rosi fugax. Se questa si accompagna a dolore cervicale e sindrome di Horner ipsilaterali, bisogna sospettare una dissezione carotidea.
Gli ACV ischemici per lesione dell’arteria cerebrale anteriore sono poco frequenti. La sua eziologia di solito è embolica, e la sua clinica tipica consiste nell’emiparesi e nell’emi-ipoestesia controlaterali di predominio crurale, rifl essi arcaici e disinibizione a livello compor-tamentale.
Gli ACV ischemici per lesione dell’arteria cerebrale media sono i più frequenti. La sua clinica tipica consiste nell’emiparesi e nell’emi-ipoestesia controlaterali di predominio faciobrachiale, emianopsia omonima controlaterale, afasia (se si lesiona l’emisfero dominante), agnosia, alessia con agrafi a e deviazione oculare ipsilaterale.
Gli ACV ischemici per lesione dell’arteria cerebrale posteriore pos-sono essere distali (emianopsia omonima controlaterale con ma-cula intatta, alessia ed acalculia) o prossimali (sindrome talamica: emianestesia globale controlaterale, iperpatia nell’emicorpo inte-ressato dalla lesione e defi cit nei movimenti).
Gli ACV ischemici per lesione vertebrobasilare producono le sin-dromi crociate: emiparesi ed emi-ipoestesia controlaterali e lesione ipsilaterale dei nervi cranici (il nervo cranico determina il lato della lesione).
Le sindromi disartria-mano goff a ed atassia-emiparesi si possono verifi care sia in lesioni della capsula interna controlaterale sia in quelle del ponte.
Tra le sindromi lacunari, bisogna sapere che si producono per lesio-ne dei piccoli vasi (lipoialinosi) e che il più frequente è l’ictus moto-rio puro (per lesione del braccio posteriore della capsula interna o del ponte).
La trombosi venosa durale può essere causata a livello sistemico e locale (si veda la Tabella 11) e di solito ha inizio con ipertensione intracranica. È tipico il segno della delta vuota alla TC.
La prima prova diagnostica che si realizza in presenza di un ACV è la TC cranica per valutare la presenza di emorragie (la presenza d’i-schemia potrebbe non essere visualizzabile nelle prime 24-72 ore).
Il trattamento in fase acuta degli ACV ischemici consiste nell’adot-tare misure di supporto (con particolar controllo costante della pressione arteriosa), fi brinolisi con rt-PA (salvo controindicazioni), antiaggregazione e anticoagulazione (se con fi brillazione atriale, dissezione carotidea).
La profi lassi ed il trattamento in fase cronica degli ACV ischemici consistono nel controllo dei fattori di rischio cardiovascolare (so-prattutto dell’ipertensione arteriosa), antiaggregazione, ipolipe-mizzanti (incluso nei normocolesterolemici), anticoagulazione (fi brillazione atriale o determinate cardiopatie come la miocardio-patia dilatativa) ed endarterectomia carotidea, se la stenosi è signi-fi cativa.
Le emorragie intraparenchimali producono una clinica di iperten-sione intracranica e deterioramento del livello di coscienza. Si clas-sifi cano in emorragie ipertensive (profonde) ed emorragie lobari spontanee (causa più frequente negli anziani per angiopatia ami-loide, e nei giovani per malformazioni vascolari).
Le sindromi disartria-mano goff a ed atassia-emiparesi si possono trovare sia in lesioni della capsula interna controlaterale sia in quel-le del ponte.
C a s i c l i n i c i Uomo di 85 anni, con precedenti di emorragia cerebrale da due anni, viene ricoverato per un quadro acuto di emiparesi destra e sonnolenza. Nella TC fatta in urgenza, si riscontra un grande ematoma intracerebra-le lobare frontoparietale sinistro. Il paziente non è iperteso. Quale tra le seguenti è l’eziologia più probabile dell’emorragia del paziente?
1) Metastasi.2) Aneurisma.3) Trauma.4) Fumo/Droghe o farmaci.5) Angiopatia amiloide.
RC: 5
Un uomo di 62 anni si presenta al Pronto Soccorso con instabilità e senso di stordimento sopraggiunti bruscamente. All’esame obiettivo si trova un nistagmo orizzontale, una sindrome di Horner destra, una perdita della sensibilità del dolore nell’emivolto destro e brachiocrurale sinistra, atassia degli arti destri e disfagia. Quale sarà la diagnosi sospettata?
1) Infarto dell’arteria basilare.2) Infarto del ponte.3) Infarto dell’arteria vertebrale sinistra.4) Infarto dell’arteria cerebrale destra.5) Infarto laterale bulbare destro.
RC: 5
Un uomo di 58 anni, fumatore di 2 pacchetti di sigarette al giorno, bevitore abituale, iperteso senza essere sotto controllo costante, ha

3004 ∙ Ma la t t i e ce reb rovasco la r i
Manuale CTO di Medicina e Chirurgia, Prima edizione
riportato negli ultimi giorni due episodi bruschi, di 15 e 45 minuti di durata, con visione off uscata nell’occhio sinistro e parestesia della mano destra. L’esame neurologico è normale. Tra i seguenti, qual è la diagnosi più probabile?
1) Emicrania con aura.2) Crisi parziali complesse.3) Neuropatia ottica alcolica/tabagica.4) Ischemia cerebrale transitoria nel territorio carotideo.5) Attacco di una malattia demielinizzante ricorrente-remittente.
RC: 4
Un paziente di 65 anni, con storia d’ipertensione arteriosa ed iper-colesterolemia, presenta un episodio ischemico transitorio nel terri-torio carotideo destro. La valutazione clinica e l’ECG non mostrano evidenza di cardiopatia. Si realizza un’ arteriografi a cerebrale, che mostra stenosi dell’arteria carotide interna destra del 30%. Che te-rapia potrebbe essere indicata per questo paziente?
1) Anticoagulazione.2) Chirurgia carotidea.3) Angioplastica carotidea.4) Antiaggreganti piastrinici.5) Nessuna.
RC: 4
Uomo di 50 anni, con episodi ripetuti di ischemia cerebrale transito-ria consistenti in perdita di forza e paresi del braccio e della gamba destre, ed amaurosi fugax nell’occhio sinistro. Presenta stenosi del 75% all’inizio della carotide interna sinistra. Qual è l’intervento cor-retto?
1) Anticoagulazione con dicumarinici, 6-12 mesi.2) Anticoagulazione con eparina, 1 settimana.3) Anticoagulazione con eparina ed antiaggreganti per le piastrine.4) Endarterectomia della carotide interna sinistra.5) Bypass aortocarotideo con vena safena autologa.
RC: 4
Paziente di 30 anni che arriva al Pronto Soccorso con amaurosi tran-sitoria dell’occhio destro in forma acuta e cefalea con dolori nella regione cervicale destra. All’esame obiettivo , si riscontra una sin-drome di Horner destra. Qual è la diagnosi più probabile, tra le se-guenti?
1) Stenosi carotidea destra.2) Ematoma subdurale traumatico. 3) Dissezione carotidea destra.4) Trombosi dell’arteria centrale della retina.5) Sindrome di Horton.
RC: 3

31
Neurologia e neurochirurgia
I disturbi del movimento hanno il loro substrato patologico principalmente
nei gangli basali (Figura 35).
Pallido laterale
Pallido mediale
Caudato
Talamo
Subtalamo
Putamen
Figura 35. Anatomia dei gangli basali
Non è possibile identifi care un tipo specifi co di movimento prodotto dai
gangli basali.
I disturbi extrapiramidali si dividono in ipercinesie (tremore, distonia, corea,
atetosi, ballismo, mioclonus, acatisia, gambe inquiete…) ed ipocinesia (par-
kinsonismo).
5.1. Tremore
Il tremore si defi nisce come la presenza di oscillazioni ritmiche di una parte
del corpo, conseguente a contrazioni alternate o sincroniche di gruppi mu-
scolari opposti.
05
Classifi cazione
Sulla base della situazione funzionale nella quale si presenta, il tremore si
può classifi care in:
• Tremore a riposo. Si produce in assenza di attività muscolare volonta-
ria. L’esempio tipico è il tremore osservato nella malattia di Parkinson.
• Tremore d’azione. Si produce con la contrazione muscolare volonta-
ria e si suddivide in tremore posturale e cinetico o di movimento. Il pri-
mo è causato dal mantenimento della posizione e sono degli esempi
il tremore fi siologico, il tremore fi siologico accentuato, il tremore es-
senziale ed il tremore posturale che possono presentarsi nella malattia
di Parkinson e negli altri disturbi del movimento. Sono esempi tipici
del tremore posturale quello che si produce bevendo, mangiando, al-
lacciarsi un bottone o scrivere. Il tremore cinetico appare con qualsiasi
forma di movimento e può manifestarsi all’inizio (tremore iniziale), du-
rante (tremore di transizione) o alla fi ne (tremore terminale o inten-
zionale). Il tremore cinetico è caratteristico della patologia cerebellare
o troncoencefalica (sclerosi multipla, vascolare, tumorale e patologia
degenerativa).
R I C O R D A Il tremore tipico della malattia di Parkinson è a riposo, ma si può trovare frequentemente anche il tremore posturale.
Clinica (Tabella 16)
• Tremore fi siologico accentuato (8-12 Hz), però con maggiore ampiez-
za. È assente a riposo e presente con il mantenimento della posizione. E’
il risultato di un incremento dell’attività periferica b-adrenergica associa-
ta ad un aumento del livello di catecolamine circolanti. È comune negli
stati di agitazione stato di ansietà e in quei disturbi metabolici che com-
portano un’iperattività -adrenergica (tireotossicosi, feocromocitoma,
ipoglicemia). Si presenta anche con l’assunzione di alcuni farmaci (cate-
colamine, metilxantine) o con la sospensione di altri (-bloccanti, mor-
DISTURBI DEL MOVIMENTO
ORIENTAMENTOSi tratta di un tema importante che include due malattie molto richieste in sede d’esame; il Parkinson e la corea di Huntington. Bisogna saper riconoscere le loro caratteristiche e saperle differenziare dagli altri quadri extrapiramidali.

3205 ∙ D i s tu rb i de l mov imento
Manuale CTO di Medicina e Chirurgia, Prima edizione
fi na e alcool). Può essere controllato adeguatamente con i -bloccanti
(propranololo).
Tipo de tremore Riposo Posturale Cinetico
Fisiologico accentuato - ++ +
Essenziale - (+) ++++ + (+)
Parkinsoniano ++++ ++ +
Mesencefalico ++ +++ +++
Cerebellare - + +++
Tabella 16. Correlazione clinico-eziologica del tremore
• Tremore essenziale (Tabella 17). È la forma più comune di tremore sin-
tomatico e il disturbo del movimento più frequente. Si eredita con carat-
tere autosomico dominante ad alta penetranza. Si trova in un 30% delle
anamnesi familiari dei pazienti, anche se a volte può essere in forma spo-
radica. Può cominciare a qualsiasi età e persiste per tutta la vita. All’inizio
è unilaterale ed intermittente, però successivamente diventa bilaterale ed
asimmetrico, potendo colpire qualsiasi parte del corpo. Tipicamente pro-
duce oscillazioni fl esso-estensorie a livello del polso o di avvicinamento-
separazione delle dita quando le braccia sono tese. La sua frequenza è di
4-12 Hz, e si può associare a compiti specifi ci (scrivere, tenere un oggetto
in una posizione determinata…). Si aggrava con lo stress, con l’ansia e la
fatica. Caratteristicamente migliora con l’assunzione di alcool.
Quasi il 50% dei pazienti con tremore essenziale presenta alcune for-
me di distonia associata. Sono delle varianti il tremore cefalico isolato,
il tremore della voce, della lingua, ortostatico… Non ci sono dati di pa-
tologia extrapiramidale o cerebellare (anche se la presenza di rigidità a
“ruota dentata” associata al tremore non è un criterio di esclusione).
Il trattamento si realizza con propranololo o primidone, ed è stata utiliz-
zata in casi rari la tossina botulinica. In casi eccezionali si può utilizzare
la stimolazione talamica.
Criteri d’inclusione
∙ Presenza di tremore posturale visibile e persistente, che interessa le mani o gli avambracci, può essere accompagnato da tremore cinetico. Può essere asimmetrico e interessare altre parti del corpo
∙ Durata prolungata (più di5 anni)
Criteri d’esclusione
∙ Presenza diì alterazioni neurologiche associate, ad eccezione della rigidità a “ruota dentata” (segno di Froment)
∙ Esistenza di altre cause di tremore fi siologico accentuato (es. ipertiroidismo)
∙ Esposizione a farmaci tremorigeni o sospensione di farmaci antitremorigeni
∙ Antecedente di trauma cranico nei 3 mesi precedenti all’inizio dei sintomi
∙ Evidenza clinica di tremore psicogeno
∙ Inizio improvviso
Tabella 17. Criteri diagnostici del tremore essenziale
R I C O R D A Il tremore essenziale è il più frequente dei tremori sintomatici. Può essere asimmetrico ed associarsi a rigidità a “ruota dentata” in for-ma analoga al tremore di un paziente con Parkinson.
R I C O R D A Il tremore fi siologico ed essenziale vengono trattati con b-bloccan-ti; tuttavia, il tremore a riposo del Parkinson si tratta con anticoli-nergici.
5.2. Distonie
Concetto
Sono movimenti involontari sostenuti che producono una deviazione o tor-
sione di un’area corporea. Non cessa con la volontà e si possono innescare a
seguito di movimenti o azioni specifi che.
È possibile che i sistemi dopaminergici e noradrenergici giochino un ruolo
importante nella patogenesi della distonia primaria.
Classifi cazione
• Distonia focale.
• Distonia segmentaria. Si presentano movimenti distonici in aree del
corpo contigue. Include la sindrome di Meige, che si presenta con ble-
farospasmo e distonia oromandibolare.
• Distonia multifocale. Interessa i muscoli di più regioni non conti-
gue.
• Emidistonia. Si presenta con lesioni strutturali nei gangli basali contro-
laterali, in modo particolare nel putamen.
• Distonia generalizzata.
Trattamento
Nel trattamento sintomatico della distonia lieve si utilizzano le benzodiaze-
pine (diazepam, clonazepam, lorazepam) ed altri miorilassanti, come il ba-
clofen o la tizanidina. Nei casi di distonia moderata o grave si utilizzano gli
anticolinergici (triesifenidile, biperidene).
La tossina botulinica è il trattamento di prima scelta della distonia fo-
cale.
R I C O R D A Sono farmaci di seconda scelta il baclofen, la carbamazepina ed anche il valproato.
5.3. Mioclonie
Sono movimenti involontari, improvvisi e di scarsa durata, causati da una
contrazione muscolare attiva. Si distinguono dall’asterissi (fl apping tre-
mors) perché quest’ultima è un movimento rapido e aritmico, con pause
brevi dell’attività muscolare e causa perdita del tono posturale (silenzio
elettrico nell’elettromiogramma [EMG]).
Nel trattamento sintomatico delle mioclonie, risultano molto effi caci far-
maci come il clonazepam, il valproato, il piracetam, primidone e 5-idrossi-
triptofano.

33
05Neurologia e neurochirurgia
5.4. Tic
Sono movimenti stereotipati, privi di fi nalità, che si ripetono in maniera irre-
golare.
Si caratterizzano perché si possono bloccare con la propria volontà ed au-
mentano con lo stress. Possono persistere durante il sonno.
Sindrome di Gilles de la Tourette
È la forma più grave di tic multipli.
Sono criteri diagnostici:
1. Multipli tic motori e uno o più tic fonici.
2. I tic si presentano molte volte al giorno, quasi tutti i giorni durante più
di un anno.
3. Il tipo, la gravità e la complessità dei tic cambiano con il tempo.
4. Inizio precedente ai 21 anni.
5. I movimenti involontari e rumori non possono essere giustifi cati da altri
mezzi.
6. Si associano ad ecolalia e coprolalia.
È caratteristica l’associazione con disturbi ossessivo-compulsivi e defi cit dell’
attenzione.
Il trattamento si realizza con neurolettici (aloperidolo, pimozide), clonidina
ed altri dopaminergici.
5.5. Sindrome delle gambe inquiete
Disturbo del movimento che si caratterizza per disestesia predominante agli
arti inferiori, che si presenta di solito a riposo e che si allevia con il movimen-
to. Si può associare a movimenti periodici durante il sonno.
L’eziologia più frequente è idiopatica, dovendo scartare la polineuropatia
sensoriale (uremica, diabetica…), anemia sideropenica o la coesistenza di
altre patologie. Il trattamento delle forme idiopatiche si basa sull’uso di ago-
nisti dopaminergici o levodopa, così come benzodiazepine o oppiacei.
5.6. Corea. Malattia di Huntington
Il termine corea (“ballo”) fa riferimento ai movimenti aritmici, rapidi, irrego-
lari, scoordinati ed incessanti che possono colpire qualsiasi parte del corpo
(Figura 36).
La malattia di Huntington (HD) è la forma più comune di corea ereditaria. Può
avere inizio a qualsiasi età, anche se la maggior incidenza si colloca tra la 4.ª e
la 5.ª decade, evolvendo lentamente fi no alla morta in un periodo che va dai
10 ai 25 anni.
Si eredita con carattere autosomico dominante ed è il risultato dell’espansio-
ne della tripletta CAG nel braccio corto del cromosoma 4.
È caratteristica l’atrofi a del nucleo caudato, con dilatazione secondaria dei
corni anteriori dei ventricoli laterali.
Figura 36. TC cranica di un paziente con malattia di Huntington, nella quale si osserva la marcata atrofi a dell’encefalo in entrambi i nuclei caudati, che provoca la dilatazione corni anteriori dei ventricoli laterali
Clinica
È caratterizzata dalla triade: disturbo del movimento, deterioramento cogni-
tivo e clinica psichiatrica.
• Disturbo del movimento. Il disturbo del movimento più caratteristi-
co è la corea. Le alterazioni dei movimenti oculari a volte sono i segni
più precoci (perdita dei movimenti saccadici).
• Deterioramento cognitivo. Demenza subcorticale.
• Disturbi psichiatrici e del comportamento. La manifestazione più
frequente sono i disturbi aff ettivi, e la depressione unipolare o bipolare,
che interessa un 50% dei casi. Il rischio di suicidio è maggiore rispetto
alla popolazione generale. Disturbi psicotici come la schizofrenia si ma-
nifestano in un 5-10% dei casi.
Diagnosi
Può essere diagnosticata con la storia clinica, l’esame obiettivo e l’anam-
nesi familiare, o attraverso l’analisi genetica che evidenzia un numero ec-
cessivo di ripetizioni della tripletta CAG (più di 40 ripetizioni) nel cromo-
soma 4.
Trattamento
Dal momento in cui non esiste nessun trattamento patogenetico effi cace,
normalmente si realizza un trattamento sintomatico.
5.7. Malattia di Parkinson idiopatica
È la sindrome parkinsoniana più comune. Interessa più frequentemente gli
uomini, con un’età media d’esordio di 55 anni. Solo un 5-10% inizia prima
dei 40 anni. La media di incidenza annuale varia tra i 7-19 casi ogni 100.000
abitanti, e la sua prevalenza è ampiamente variabile in funzione dell’età e
dell’area geografi ca.

3405 ∙ D i s tu rb i de l mov imento
Manuale CTO di Medicina e Chirurgia, Prima edizione
Patogenesi
È sconosciuta. Il parkinsonismo è più comune nell’anziano, e l’età avanzata
è il fattore di rischio più importante nell’eziologia di questa malattia. Sono
stati presi in considerazione altri fattori di rischio con meccanismo patoge-
netico: genetici (normalmente sporadici; sono state descritte forme AD con
esordio precoce); ambientali (MPTP, il manganese, l’alluminio, l’arsenico, il
mercurio, lo zinco, i pesticida o gli erbicidi), traumi…
Anatomia patologica
Nella malattia di Parkinson, avviene una perdita neuronale nella porzione
compatta della sostanza nera, anche se possono essere colpiti altri nuclei
come il locus coeruleus, i nuclei del rafe, il nucleo basale di Meynert, le co-
lonne mediolaterali del midollo ed i gangli simpatici e parasimpatici (Fi-
gura 37).
Il marker anatomopatologico più caratteristico è rappresentato dai corpi di
Lewy.
Figura 37. Corpi di Lewy in un paziente con malattia di Parkinson
Clinica
È una sindrome clinica caratterizzata da tremore a riposo, bradicinesia,
rigidità ed instabilità posturale. I primi due sono i più tipici (Figura 38).
Il tremore a riposo è un movimento oscillatorio distale a 4-6 Hz che
normalmente interessa le mani, però può anche interessare le labbra, la
lingua, la mandibola e gli arti inferiori. Raramente colpisce la testa o le
corde vocali.
Tipicamente, all’inizio è asimmetrico. Costituisce la forma di presentazione
più frequente (60-70% dei pazienti) e può risultare l’unica manifestazione
della malattia per alcuni anni. Il tremore posturale si presenta circa nel 60%
dei pazienti, a volte associato a tremore a riposo.
R I C O R D A Il tremore a riposo come l’atto di “contare moneta” è tipicamente asimmetrico all’inizio della malattia.
Bradicinesia Andatura festinante Instabilità posturale
Limitazionenella sopraelevazione
della mirata
Rigidità(fenomeno
“ruota dentata”)
Ipomimia facciale
Figura 38. Paziente con malattia di Parkinson
La bradicinesia consiste in un rallentamento generalizzato dei movi-
menti. È la manifestazione più incapacitante della malattia. Risulta dalla
perdita dei meccanismi dopaminergici inibitori dello striato e ipoattività
dei neuroni del globo pallido esterno. Si manifestano con ipomimia fac-
ciale, diminuzione della frequenza del battito delle ciglia, linguaggio mo-
notono ed ipofonico suscettibile a fatica, micrografi a, diffi coltà ad alzarsi
dalla sedia e rigirarsi nel letto. L’andatura è tipica, con fl essione anteriore
del tronco, piccoli passi, trascinando i piedi e con perdita del movimento
oscillatorio delle braccia (marcia festinante).
La rigidità è un incremento della resistenza alla mobilizzazione passi-
va che predomina nella muscolatura fl essoria. È costante durante tutto
il movimento (rigidità plastica), anche se si produce il fenomeno della
rigidità a “ruota dentata”, che viene considerata come l’interferenza del
tremore nella rigidità plastica durante la mobilizzazione passiva dell’arto
(si tratta di una spiegazione parziale, data la possibilità di riscontro della
“ruota dentata” in pazienti senza tremore a riposo). Si produce per disi-
nibizione pallidale con incremento dell’attivazione soprasegmentale dei
meccanismi rifl essi spinali normali e, quindi, con un aumento della scarica
degli -motoneuroni.
R I C O R D A Nelle forme secondarie di parkinsonismo di solito predomina la rigi-dità e non il tremore.
L’instabilità posturale si può manifestare come propulsione (tendenza a
spostarsi in avanti) o retropulsione (spostamento all’indietro).
Le manifestazioni a livello oculare includono limitazioni nell’ elevazione dello
sguardo e rifl esso glabellare inesauribile.

35
05Neurologia e neurochirurgia
La disfunzione autonomica si manifesta con scialorrea, disfagia, stitichezza,
tendenza all’ipotensione, ipersudorazione, nicturia ed urgenza minzionale.
La nicturia è il sintomo più precoce e frequente della clinica urinaria.
R I C O R D A La paralisi verticale dello sguardo è tipica del Parkinson e la limitazio-ne alla infraversione è tipica della paralisi sopranucleare progressiva.
I disturbi non motori nella malattia di Parkinson includono cambi della per-
sonalità, deterioramento delle funzioni superiori (maggiore nelle fasi avanza-
te della malattia, al contrario di ciò che succede nei parkinsonismi secondari,
dove è più frequente una sua comparsa precoce), depressione e disturbi del
sonno.
Diagnosi
La diagnosi della malattia di Parkinson è clinica. I criteri diagnostici vengono
illustrati nella Tabella 18.
Criteri diagnostici
1. Due dei seguenti segni o sintomi
∙ Tremore a riposo ∙ Rigidità ∙ Bradicinesia ∙ Instabilità posturale
2. Miglioramento signifi cativo con L-dopa
3. Scartare i parkinsonismi secondari
4. Assenza di segni incompatibili con la malattia di Parkinson
∙ Oftalmoplegia sopranucleare con paralisi nella infraversione dello sguardo
∙ Lesione corticospinale ∙ Lesione del corno anteriore ∙ Segni cerebellari ∙ Polineuropatie ∙ Mioclonie ∙ Crisi oculogire
Tabella 18. Criteri diagnostici della malattia di Parkinson
Trattamento
Farmacologico
• Levodopa (L-dopa). Associata ad un inibitore della dopadecarbossilasi
periferica (cardidopa-benserazide) continua ad essere il trattamento di
prima linea, quasi tutti i pazienti che inizialmente migliorano perdono
la risposta alla levodopa in 3-8 anni, con la conseguente comparsa di
fl uttuazioni motorie (fenomeno wearing off o fi ne dose, ipercinesia da
inizio dose, distonia da fi ne dose, discinesia bifasica, fenomeno on-off ,
errori delle dosi), e discinesie (picco di dosi, bifasicache). L’associazione
di levodopa con agonisti dopaminergici permette un controllo parziale
delle stesse e la riduzione delle dosi di levodopa.
R I C O R D A È propria dei parkinsonismi secondari la scarsa risposta a levo-dopa.
• Inibitori della COMT. Gli inibitori della catecol-O-metil-transferasi (en-
tacapone, tolcapone) aumentano anche la biodisponibilità della levo-
dopa, inibendo il suo catabolismo, per tanto si possono somministrare
associati alla levodopa. Esistono teorie secondo le quali si possa associa-
re levodopa + carbidopa + entacapone.
• Agonisti dopaminergici. Si utilizzano nella monoterapia quando c’è
una lesione lieve-moderata, specialmente in pazienti giovani o associati
alla levodopa nelle fasi avanzate.
• Anticolinergici (triesifenidile, biperidene). Sono utili per il trattamento
dei pazienti giovani con predominio clinico di tremore a riposo.
R I C O R D A Il tremore a riposo viene trattato con anticolinergici. Il tremore po-sturale con propranololo.
• Deprenil o selegilina e rasagilina. Sono inibitori selettivi della MAO-B;
è stato dimostrato un eff etto neuroprotettore.
• Amantadina. È scarsamente effi cace per il controllo dei sintomi.
Attualmente si preferisce nelle forme iniziali e lievi del Parkinson utilizzare gli
agonisti dopaminergici combinandoli occasionalmente con IMAO-B, men-
tre nelle forme sintomatiche (moderate-gravi), si utilizza come trattamento
prioritario la levodopa.
Chirurgico
R I C O R D A Nel trattamento chirurgico, sono preferibili le tecniche di stimola-zione piuttosto che l’ablativa. La stimolazione bilaterale del nucleo subtalamico off re il miglior risultato.
Il trattamento chirurgico si può pianifi care in pazienti relativamente giova-
ni, con sintomatologia incapacitante, che non rispondano al trattamento
o che presentino intolleranze o sequele importanti allo stesso che ne limi-
tino l’uso.
Attualmente, si può dire che la tecnica chirurgica prioritaria è la stimolazione
bilaterale del nucleo subtalamico.
5.8. Altre sindromi parkinsoniane
Paralisi sopranucleare progressiva (malattia di Steele-Richardson-Olszewski)
Si tratta di un’entità clinica che interessa gli anziani nello stesso periodo di
età che la malattia di Parkinson, e che è caratterizzata da:
• Sindrome parkinsoniana con bradicinesia, rigidità, scarso tremore ed
instabilità posturale, con frequenti cadute, specialmente all’indietro. La ri-
gidità è più evidente nella muscolatura assiale che nei membri e porta ad
una postura in estensione cervicale. L’andatura è rigida e con base ampia.
Scarsa o nessuna risposta al trattamento con levodopa.
• Distonia che interessa principalmente il collo (dato caratteristico).
Un’altra forma di distonia è il blefarospasmo.

3605 ∙ D i s tu rb i de l mov imento
Manuale CTO di Medicina e Chirurgia, Prima edizione
• Disfunzione corticobulbare o corticospinale con aumento dei rifl essi
miotatici, segno di Babinski, disartria, disfagia e labilità emotiva (sindro-
me pseudobulbare).
• Paralisi dello sguardo coniugato sul piano verticale, specialmente
della infraversione dello sgurado nelle fasi iniziali. Si considera il segno
clinico più caratteristico di questa malattia.
• Demenza.
R I C O R D A Il paziente con malattia di Parkinson tende a cadere in avanti, mentre nella PSP, le cadute sono all’indietro e più frequenti.
Va considerata la diagnosi in pazienti anziani con frequenti cadute, segni
extrapiramidali, rigidità cervicale e paralisi dello sguardo verticale . I farmaci
antiparkinsoniani producono scarsi benefi ci.
Atrofi e multisistemiche
Con questa denominazione si fa riferimento ad un gruppo eterogeneo di
patologie degenerative del sistema nervoso.
Clinicamente possono presentarsi con una combinazione di:
• Clinica parkinsoniana.
• Sindrome piramidale.
• Segni e sintomi cerebellari.
• Segni e sintomi autonomici.
R I C O R D A Non sono proprie dell’atrofi a multisistemica la demenza e le crisi comiziali.
Il quadro clinico può variare nello stesso paziente durante il tempo della
sua evoluzione.
Quando predomina la clinica parkinsoniana, si parla di degenerazione
striato-nigrica; quando predomina la clinica autonomica, di sindrome
di Shy-Drager e quando si tratta di atassia e sindrome piramidale si par-
la propriamente di atrofia olivopontocerebellare (OPCA) (forma spora-
dica).
A livello anatomopatologico, le AMS presentano perdita neuronale e gliosi
(senza corpi di Lewy) che possono interessare le seguenti strutture: sostan-
za nera, caudato e putamen, pallido, olive inferiori, protuberanza, cervellet-
to e colonne mediolaterali midollari.
Per quanto riguarda il trattamento, gli agonisti dopaminergici non sono i
più effi caci della levodopa anche se alcuni pazienti li tollerano meglio.
R I C O R D A Di fronte ad un parkinsonismo con clinica disautonomica precoce e nessuna risposta alla L-dopa, bisogna pensare alla sindrome di Shy-Drager.
I d e e c h i ave Il tremore essenziale è il più frequente dei tremori sintomatici. Pre-senta le stesse caratteristiche dei tremori del Parkinson: velocità in-termedia, asimmetrico, coesistenza con altri disturbi del movimen-to (distonie, rigidità a “ruota dentata”) però tipicamente è posturale, presenta precedenti familiari e migliora con l’assunzione di alcool.
Le distonie possono essere trattate con le benzodiazepine, altri rilassanti muscolari ed anticolinergici, a seconda della gravità.
La tossina botulinica è il trattamento di prima scelta delle disto-nie focali.
I tic si estinguono con la volontà ed aumentano con lo stress. La forma più grave di tic multipli è la sindrome di Gilles de la Touret-te, che si manifesta con ecolalia e coprolalia. Viene trattata con neurolettici.
La corea di Huntington comporta: disturbi del movimento, de-terioramento cognitivo e disturbi psichiatrici. Si eredita in forma
autosomica dominante e presenta il fenomeno dell’anticipazio-ne (per espansione della tripletta di nucleotidi CAG). È caratte-ristica l’atrofi a del nucleo caudato e il trattamento sintomatico prevede l’assunzione di neurolettici.
Il tremore a riposo, la bradicinesia, la rigidità, l’instabilità po-sturale caratterizzano la sindrome parkinsoniana. La malattia di Parkinson idiopatica è la forma più comune di sindrome par-kinsoniana. In questa predomina la clinica del tremore (forma di presentazione più frequente) e la bradicinesia (manifestazione più incapacitante della malattia). il miglioramento con la L-dopa è signifi cativo.
La paralisi sopranucleare progressiva è una delle diagnosi diff e-renziali del Parkinson idiopatico. Comporta distonie, sindrome piramidale, paralisi della infraversione dello sguardo e demenza.
Le atrofi e multisistemiche sono caratterizzate da una clinica par-kinsoniana con predominio della rigidità e lesione a carico di al-tre strutture nervose, come la via piramidale, il cervelletto o il sistema vegetativo, a partire dagli stadi iniziali della malattia. La demenza non forma parte del quadro.

37
05Neurologia e neurochirurgia
C a s i c l i n i c i Un paziente di 60 anni riferisce che da anni gli tremano le mani quando impugna il cucchiaio, il bicchiere o la penna, soprattutto se è nervoso o aff aticato, e questi sintomi migliorano con l’assunzio-ne di piccole quantità di vino. Suo padre morì presentando tremore nelle mani e alla testa. L’esplorazione neurologica mostra tremore simmetrico in entrambe le mani. Questo quadro clinico è probabil-mente una sequela di:
1) Un ipotiroidismo familiare.2) Malattia di Parkinson incipiente.3) Sintomi di deprivazione alcolica.4) Un tremore essenziale.5) Una nevrosi da ansia organica familiare.
RC: 4
Un uomo di 70 anni,presenta tremore a riposo di 4 Hz e diffi coltà nei movimenti dell’estremità superiore destra da un anno. Quando cam-mina l’oscillazione delle braccia è ridotto sul lato destro. Ha iniziato un trattamento con 750 milligrammi di levodopa e 75 milligrammi di carbidopa al giorno, con scomparsa dei sintomi. Quale malattia e quale evoluzione sono le più probabili?
1) Corea di Huntington con deterioramento cognitivo progressivo.2) Paralisi sopranucleare progressiva con comparsa tardiva di una limi-
tazione dello sguardo sul piano verticale, sia superiore sia inferiore.3) Malattia di Parkison con estensione del tremore alla gamba destra.4) Malattia di Creutzfeldt-Jakob con rapido deterioramento cognitivo
e frequenti mioclonie.5) Tremore essenziale familiare con comparsa di tremore nell’estremi-
tà superiore sinistra e testa.
RC: 3
Un uomo di 45 anni di età soff re di deterioramento cognitivo pro-gressivo. All’esame obiettivo ambulatoriale il paziente presenta mo-vimenti involontari irregolari delle estremità. Il padre del paziente morì a 60 anni in un centro psichiatrico e anch’egli presentava questi movimenti involontari delle estremità. Qual è la diagnosi più proba-bile?
1) Corea di Sydenham.2) Malattia di Huntington.3) Malattia di Lafora.4) Malattia di Hallervorden-Spatz.5) Paralisi sopranucleare progressiva.
RC: 2

383838
Neurologia e neurochirurgia
ORIENTAMENTO
Tema importante. Bisogna focalizzare l’attenzione sulla sclerosi multipla, apprendendo le sue diverse forme evolutive ed il trattamento adeguato per ciascuna di esse.
Le malattie demielinizzanti sono in insieme di patologie neurologiche che
tendono a colpire i giovani adulti. Si caratterizzano da un’infi ammazione e
distruzione selettiva della mielina del sistema nervoso centrale, senza attac-
care il sistema nervoso periferico (Tabella 19).
Malattie demielinizzanti
acquisite
∙ Sclerosi multipla
∙ Sindrome di Devic
∙ Malattia di Balo
∙ Malattia di Marchiafava-Bignami
∙ Mielinolisi centrale pontina
∙ Encefalomielite disseminata acuta
∙ Encefalomielite emorragica necrotizzante acuta
Malattie demielinizzanti
ereditarie
∙ Leucodistrofia metacromatica. Alterazione funzionale dell’arilsulfatasi A. Ereditarietà autosomica recessiva
∙ Leucodistrofia sudanofila
∙ Adrenoleucodistrofia. Eredità correlata al cromosoma X
∙ Malattia di Pelizaeus-Merzbacher
∙ Eredità recessiva del cromosoma X
∙ Leucodistrofia con cellule globoidi o malattia di Krabbe
∙ Deficit di galactocerebrosidasi
Tabella 19. Malattie da alterazione della mielina
6.1. Sclerosi multipla
Epidemiologia
La sclerosi multipla costituisce la forma più frequente di malattia da altera-
zione della mielina nel sistema nervoso centrale. Interessa prevalentemen-
te i pazienti tra i 20-45 anni, più frequentemente le donne (60% dei casi).
06La prevalenza aumenta allontanandosi dalla latitudine equatoriale nei due
emisferi (l’incidenza nelle zone geografi che con clima temperato è mag-
giore a quella che si osserva nelle zone tropicali) e predomina nella popola-
zione bianca. Diversi studi sostengono la correlazione tra fattori ambientali
e lo sviluppo della malattia.
R I C O R D A La sclerosi multipla è molto più frequente nei paesi nordici e furono i vichinghi che facilitarono il suo sviluppo.
Genetica
Esiste una chiara suscettibilità genetica per lo sviluppo della sclerosi multi-
pla, l’associazione tra HLA-DR2 e HLA-DQ, con una maggior frequenza nei
gemelli monozigoti.
Anatomia patologica
Comparsa di aree o placche di demielinizzazione nella sostanza bianca del
SNC. In queste s’infi ltrano le cellule T (CD4+) ed i macrofagi, con assenza dei
linfociti B e delle cellule plasmatiche. Quando la placca diventa cronica, si
osservano come linfociti predominanti quelli B e T con fenotipo soppressore
(CD8+). Viene così colpito l’oligodendrocita.
Non c’è nessuna correlazione tra il numero di placche, la loro dimensione ed
i sintomi clinici.
R I C O R D A Non viene mai viene colpito il sistema nervoso periferico, perché la cellula che forma la mielina a questo livello è la cellula di Schwann, e non l’oligodendrocita.
Decorso clinico
Le manifestazioni cliniche sono molto variabili, e si distinguono in quattro
forme (Figura 39).
• Forma remittente (remittente-ricorrente o RR). É la forma tradiziona-
le, e costituisce l’85% dei casi.
MALATTIE DA ALTERAZIONE DELLA MIELINA

39
06Neurologia e neurochirurgia
• Forma secondariamente progressiva (SP).
• Forma primaria progressiva (PP). Il 10% dei pazienti presentano un
decorso progressivo dall’inizio della malattia, senza attacchi.
• Forma progressiva ricorrente (PR). Il 5% dei pazienti presentano un
deterioramento progressivo dall’inizio però nel corso della malattia si
verifi cano attacchi.
Forma primariamente progressiva (PP)
Corso clínico
Formaremittente-ricorrente (RR)
Forma secondariamenteprogressiva (SP)
Forma progressiva ricorrente (PR)
Figura 39. Forme cliniche della sclerosi multipla
Si considerano fattori prognostici negativi quelli descritti nella Tabella 20.
Fattori prognostici negativi
∙ Paziente maschio
∙ Inizio in età avanzata
∙ Malattia progressiva dall’inizio della presentazione dei sintomi
∙ All’inizio segni motori e cerebellari
∙ Scarso recupero da un attacco
∙ Breve intervallo tra i due primi attacchi
∙ Lesioni multiple nella RM all’esordio
Tabella 20. Marcatori prognostici che prevedono un’evoluzione più grave della sclerosi multipla
Sintomi
La Tabella 21 mostra i sintomi neurologici più frequenti dall’inizio della malattia.
Per quanto riguarda la neurite ottica (NO), è più frequente la neurite retro-
bulbare (fondo oculare normale) che la papillite (tumefazione papillare del
fondo oculare).
R I C O R D A Lo scotoma centrocecale ed il dolore alla mobilizzazione oculare in-dicano neurite ottica. Nei giovani adulti è indicativo di SM.
Sintomi neurologici
∙ Sintomi sensitivi (61%), peggiorano con il caldo
- Ipoestesia (37%)
- Parestesia (24%)
∙ Visione offuscata per neurite ottica (36%)
∙ Debolezza ed altri sintomi motori (35%)
∙ Diplopia (15%)
∙ Atassia (11%)
Tabella 21. Clinica di presentazione della SM
È frequente la lesione della via piramidale con clinica corrispondente ad
una malattia da primo motoneurone. Nelle lesioni midollari è frequente
l’associazione con urgenza minzionale, impotenza e perdita delle sensibi-
lità del cordone posteriore ( profonde ed epicritiche) che provoca atassia
sensitiva e segno di Romberg. Se la lesione cordonale è a livello cervicale
può apparire una sorta di scarica elettrica discendente al fl ettere il collo: è il
segno di Lhermitte.
R I C O R D A In un anziano con segno di Lhermitte, bisogna scartare una spondilo-si cervicale più che SM.
È frequente in questi pazienti la diplopia, generalmente secondaria ad una
lesione del fascicolo longitudinale mediale che provoca un’oftalmoplegia
internucleare.
Il coinvolgimento del cervelletto o delle sue vie di connessione a livello tron-
coencefalico provocano l’apparizione di atassia, disartria cerebellare (parola
scandita), nistagmo e tremore cinetico.
L’alterazione cognitiva è comune nei casi avanzati, essendo la perdita della
memoria la manifestazione più frequente.
La depressione appare come reazione alla conoscenza della propria malattia
o con l’evoluzione.
In fase avanzata è caratteristica la sintomatologia frontale con euforia e com-
portamento disinibito.
Altri sintomi sono la fatica intensa con la marcia o esercizio moderato, e sin-
tomi parossistici come crisi comiziali (1-4%), distonia, vertigini, acufeni o ne-
vralgia del trigemino.
Diagnosi
La base per la diagnosi continua ad essere clinica.
La diagnosi clinica di SM richiede l’esistenza di criteri di disseminazione spa-
ziale (sintomi e segni che indicano per lo meno due lesioni indipendenti nel
SNC) e temporale (due o più episodi di defi cit neurologico separati tra di loro
da almeno un mese senza nuovi sintomi).
Prove complementari
• LCR. Si osserva un lieve aumento dei linfociti e delle proteine totali nel
40% dei pazienti; aumento delle gamma globuline nel 70%; aumento

4006 ∙ Ma la t t i e da a l te raz ione de l l a m ie l i na
Manuale CTO di Medicina e Chirurgia, Prima edizione
delle IGG nell’80%, e bande oligoclonali in poco più del 90%, anche se
nessuno di questi dati è patognomonico. Le bande oligoclonali indi-
cano l’esistenza di attività immunologica primarie nel SNC, e possono
apparire in altre malattie che si presentano con infi ammazione del SNC
come neurolue, AIDS o panencefalite subacuta sclerosante.
• Potenziali evocati. Osservare un rallentamento nella conduzione di
una via sensoriale suggerisce una lesione demielinizzante, anche in as-
senza di clinica. Attualmente si utilizzano quasi esclusivamente i poten-
ziali visivi evocati.
R I C O R D A Le bande oligoclonali di IGG nel LCR si osservano frequentemente in varie malattie (SM, panencefalite subacuta sclerosante, neurolue, AIDS).
• Neuroimaging. La RM è la prova più sensibile nella SM (Figura 40).
Permette di determinare con un solo studio la disseminazione spaziale
(dimostrando diverse lesioni) e temporale (dimostrando lesioni acute
[che captano contrasto] e croniche). La somministrazione di gadolinio
permette di valutare le lesioni captanti come recenti.
Figura 40. RM cerebrale (A) e cervicale (B), dove si osservano placche di demielinizzazione che interessano la sostanza bianca tipiche della SM
R I C O R D A La RM è la prova diagnostica migliore, dal momento in cui può og-gettivare le disseminazioni spaziale e temporale (le lesioni acute captano contrasto).
Trattamento
Non esiste attualmente alcun trattamento in grado di curare la malattia (Fi-
gura 41).
• Trattamento sintomatico dell’attacco. Gli attacchi d’intensità elevata
richiedono cortisone ad alte dosi per via endovenosa per 3-7 giorni, gli
attacchi d’intensità lieve (sintomatologia esclusivamente sensitiva) pos-
sono essere trattati con cortisone per via orale, con riduzione progressi-
va della dose nell’arco di un mese. Quando un paziente con SM soff re di
un deterioramento brusco si deve valutare se è un nuovo attacco con
nuova attività della malattia o se si tratta di un peggioramento transito-
rio (pseudoattacco).
• Trattamento per modifi care il corso della malattia. I farmaci di prima
linea.
- Glatiramer acetato. La sua somministrazione subcutanea giorna-
liera riduce il numero di attacchi nelle forme RR di un 30%.
- Interferone (1a e 1b). Riducono anch’essi di un 30% il numero di
attacchi nei pazienti con SM di tipo RR.
I criteri per il trattamento farmacologico di prima linea sono:
1. SM remittente ricorrente con due attacchi negli ultimi tre anni.
2. SM secondariamente progressiva con persistenza di attacchi in pa-
zienti che possano ancora deambulare.
3. Successiva ad un attacco tipico di SM con dati clinici radiologici o
del LCR che indicano un alto rischio di sviluppare SM.
SM RR
Sintomaneurológico acuto
Trattamentodi base
Attacco Pseudoattacco
Moderatograve
Lieve(sensitivo)
Attacchifrequenti
Attacchisporadici
Intereferone o acetato
di glatiramero
Osservazione
Corticoterapiaintravenosa
Corticoterapiaorale
Buonarisposta
Intolleranzao nessuna
risposta
Continuareil trattamento
Cambiareil trattamento
Figura 41. Algoritmo terapeutico della SM
I farmaci approvati attualmente come trattamento di seconda linea
sono due:
- Natalizumab. È il farmaco più effi cace per il trattamento della SM
al giorno d’oggi. Il suo eff etto collaterale principale è la comparsa di
leucoencefalopatia multifocale progressiva.
- Fingolimod. La sua effi cacia è superiore al trattamento con i far-
maci di prima linea però inferiore a quella del natalizumab.
I criteri standard per il trattamento di seconda linea sono:
1. SM RR che non risponde al trattamento di prima linea.
2. SM RR molto grave che evolve rapidamente.
Altri farmaci che possono essere utili sono azatioprina, mitoxantrone e
metotrexate.
• Il trattamento sintomatico delle sequele viene esposto nella Tabella 22.

41
06Neurologia e neurochirurgia
Sequele Farmaco
Spasticità ∙ Baclofen
∙ Benzodiazepine
Fatica ∙ Amantadina
∙ Pemolina
Sintomi parossistici (dolore, distonia, tremore)
∙ Carbamazepina
∙ Gabapentina
Disfunzione erettile Sildenafil
Iperreflessia vescicale (urgenza minzionale, incontinenza)
Anticolinergici (oxibutina, tolterodina)
Atonia vescicale (ritenzione)
Colinomimetici (betanecol)
Depressione Inibitori della recaptazione di serotonina
Tabella 22. Trattamento sintomatico delle sequele della SM
Sclerosi multipla e gravidanza
Le pazienti in gravidanza subiscono un minor numero di attacchi durante
la gestazione ed uno maggiore durante i primi tre mesi post-parto. Questo
peggioramento si attribuisce ai livelli alti di prolattina che possono generare
una stimolazione del sistema immunitario.
6.2. Altre malattie demielinizzanti
Mielinolisi centrale pontina. È una malattia demielinizzante del tronco ce-
rebrale caratterizzata da segni di paralisi pseudobulbare (disartria e disfagia),
paraparesi o tetraparesi, che conserva però il battito delle ciglia ed i movi-
menti oculari verticali. Generalmente si manifesta 2-6 giorni dopo la corre-
zione rapida di un’iponatremia, però è stata anche descritta in associazione
ad alcolismo cronico e trapianto epatico. Presenta una prognosi infausta e
non esiste un trattamento effi cace.
I d e e c h i ave La sclerosi multipla (SM) colpisce prevalentemente giovani donne essendo la causa più frequente di disabilità neurologica in giovani adulti.
Si produce una reazione immunologica contro gli oligodendrociti, cellule produttrici della mielina a livello del SNC. Il SNP non è mai colpito.
Si osservano bande oligoclonali IgG nel LCR in praticamente tutti i pazienti con SM anche se non è un segno specifi co (si osservano anche in neurolue, infezioni per morbillo…).
La forma evolutiva RR che si presenta con attacchi acuti è la forma più frequente e quella che risponde meglio al trattamento immu-nomodulatore.
Il trattamento immunomodulatore che diminuisce il numero di at-tacchi e l’interferone ed il glatiramer acetato.
Il trattamento sintomatico dell’attacco si realizza con cortisone via orale (se l’attacco è lieve) o via endovenosa (se moderato o grave).
I sintomi sensitivi (ipoestesia e parestesia) costituiscono la presen-tazione clinica più frequente, seguiti dalla neurite ottica.
Il segno di Lhermitte e l’oftalmoplegia internucleare in un giovane adulto deve far pensare ad una SM.
La diagnosi di SM è clinica e richiede l’esistenza di criteri di dissemi-nazioni temporale (episodi di defi cit neurologici separati nel tem-po) e spaziale (lesioni in zone distinte del SNC); entrambi i criteri possono essere valutati attraverso un’unica metodica: la RM.
La correzione rapida di un’iponatremia è la causa più frequente di mielinolisi pontina centrale.

424242
Neurologia e neurochirurgia
Una convulsione o crisi epilettica è un fenomeno parossistico originato a
seguito di un’attività normale, eccessiva e sincronica di un gruppo di neuroni
del SNC che può presentarsi clinicamente secondo diverse forme.
L’epilessia è defi nita dalla presenza di crisi epilettiche ricorrenti dovute ad
un processo cronico sottostante. L’esistenza di una convulsione isolata o di
crisi ricorrenti dovute a fattori correggibili o evitabili, non è necessariamente
un’epilessia.
Una sindrome epilettica è un’epilessia con un insieme di sintomi e segni
che normalmente si presentano contemporaneamente, evidenziando un
meccanismo sottostante comune.
Si parla di stato epilettico quando una crisi dura più di 30 minuti o quando
esistono crisi ripetute, tra le quali il paziente non recupera conoscenza.
7.1. Classifi cazione (Tabella 23)
Le convulsioni parziali (focali) sono quelle in cui l’attività elettrica rimane
circoscritta ad un’area concreta della corteccia cerebrale, con la caratteri-
stica che durante la crisi la coscienza rimane conservata (parziale semplice)
o alterata (parziale complessa). La sintomatologia con la quale si presenta
la crisi dipenderà dall’area corticale dove si situano i neuroni responsabili
della stessa.
• Le crisi parziali semplici possono produrre sintomi motori, sensitivi,
autonomi (sudorazione, piloerezione), visivi (allucinazioni semplici o
complesse), uditivi (suoni semplici o elaborati), olfattivi (odori inten-
si o poco abituali) o psichici (paura, depersonalizzazione, déjà vu). Le
crisi motorie possono cominciare in un’area molto ristretta ed esten-
dersi gradualmente (in secondi o minuti) ad un’area emicorporale più
estesa (progressione jacksoniana). A volte, dopo una crisi motoria, può
persistere una debolezza dell’area colpita (paralisi di Todd), limitata a
minuti o ore.
07
Crisi parziali
∙ Semplici (con sintomi motori, sensitivi, autonomi o psichici)
∙ Complesse
∙ Con generalizzazione secondaria
Crisi generalizzate
∙ Assenze
∙ Tonico-cloniche
∙ Toniche
∙ Atoniche
∙ Miocloniche
Crisi non classificate
∙ Convulsioni neonatali
∙ Spasmi infantili
Tabella 23. Classifi cazione delle crisi epilettiche (Lega internazionale dell’Epilessia, 1981)
• Durante le crisi parziali complesse (Tabella 24), il paziente mostra
diffi coltà nel mantenere un contatto normale con l’ambiente, unito
ad alterazioni del comportamento che possono andare dall’immobili-
tà o automatismi basici (suzione, deglutizione), a comportamenti più
elaborati; dopo la crisi, si presenta caratteristicamente un periodo di
confusione.
R I C O R D A Le crisi parziali complesse sono più frequenti negli adulti, di solito si sperimentano sensazioni strane e ciò è dovuto all’attivazione del lobo temporale, nella maggior parte dei casi.
• Le crisi generalizzate originano simultaneamente in entrambi gli emi-
sferi, anche se è diffi cile scartare completamente l’esistenza di un’attività
focale iniziale che si propaghi con rapidità e che, occasionalmente, è
riconoscibile per l’esistenza di sintomi focali precedenti alla perdita di
conoscenza (aura).
EPILESSIA
ORIENTAMENTO
È raccomandabile conoscere la classificazione dell’epilessia e capire le differenze tra crisi parziali (semplici e complesse) e generalizzate. Esistono alcune caratteristiche tipiche come lo stato di assenza che è stata chiesta molto spesso. Bisogna tener presente l’eziologia più frequente delle crisi, a seconda dei gruppi di età e delle indicazioni ed effetti secondari dei principali anticonvulsivi.

43
07Neurologia e neurochirurgia
• Le assenze (piccoli mali) si comportano come brevi episodi di perdita
brusca del livello di coscienza, senza alterazione del controllo postuale;
caratteristicamente, durano secondi e possono ripetersi molte volte al
giorno, di solito si accompagnano da piccoli segni motori bilaterali (bat-
tito di ciglia, masticazione) e si recupera la coscienza in modo altrettan-
to brusco, senza confusione successiva né ricordo dell’episodio.
L’età d’inizio di solito si situa tra i 4 anni ed il principio dell’adolescenza,
essendo la causa più frequente di crisi epilettiche crisi in questo pe-
riodo; non si accompagnano ad altri problemi neurologici, rispondono
positivamente al trattamento farmacologico ed in un 60-70% dei casi si
attenuano durante l’adolescenza. I risultati mostrati nell’EEG sono tipi-
camente delle scariche generalizzate e simmetriche con una punta-on-
da a 3 Hz che coincide con la crisi, anche se nell’EEG intercritico esistono
più periodi di attività anormale che quelli visibili clinicamente. L’iperven-
tilazione aumenta la frequenza del tracciato anomalo.
Assenza Crisi parziale complessa
Generalizzata Parziale
Senza aura Può presentarsi aura
< 14 anni Adulti
Secondi Minuti
Varie durante il giorno e durante i minuti
Variabile
Scarsi automatismi Automatismi frequenti
Nessun periodo postcritico Periodo postcritico
∙ Valproato
∙ Etosuccimide
Carbamazepina
Tabella 24. Crisi parziale complessa versus assenza tipica
R I C O R D A La perdita di conoscenza e gli automatismi possono apparire nelle crisi parziali complesse e nelle assenze. Clinicamente si diff erenzia-no nella presenza di un periodo confusionale dopo la crisi in quelle parziali complesse e non in quelle di assenza.
• Le convulsioni tonico-cloniche (grandi mali) di solito cominciano bru-
scamente, senza un avviso previo, anche se alcuni pazienti riferiscono
sintomi poco defi niti nelle ore precedenti, che non devono essere con-
fusi con le auree causate da un’origine focale della crisi. La fase inizia-
le è una contrazione tonica generalizzata, accompagnata da cianosi,
aumento di frequenza cardiaca e della pressione arteriosa, e midriasi.
In 10-20 secondi normalmente incomincia la fase clonica, con durata
variabile. Nel postcritico, esiste un’assenza di risposta agli stimoli esterni,
fl accidità muscolare ed ipersalivazione che può compromettere le vie
aeree, seguita da una fase di lento recupero del livello di coscienza (mi-
nuti-ore) accompagnata da confusione. Il paziente riferisce stanchezza,
cefalea e mialgie durante varie ore dopo la crisi.
L’EEG mostra diversi tracciati durante il periodo della crisi: esiste un’at-
tività rapida a basso voltaggio, con scariche generalizzate e polipun-
te ad alto voltaggio nella fase tonica; nella fase clonica appare una
punta-onda a bassa frequenta: e nel postcritico, si evidenzia un rallen-
tamento globale che si risolve insieme ad un recupero del livello di
coscienza. Sono il tipo di crisi più frequente nel contesto dei disturbi
metabolici.
• Le crisi atoniche si caratterizzano per la repentina perdita del tono mu-
scolare di pochi secondi di durata, ed un’alterazione breve del livello di
coscienza, senza presentare confusione posteriore. Di solito si presenta-
no nel contesto di sindromi epilettiche conosciute.
• Le mioclonie sono contrazioni brevi dei muscoli che possono essere
originate a diversi livelli (corticale, subcorticale, midollare). Quando esi-
ste un’origine corticale, sono considerati fenomeni epilettici, mostrando
nell’EEG delle scariche di punta-onda bilaterali e sincroniche. Di solito si
presentano con altri tipi di crisi anche se sono la principale manifesta-
zione di alcune sindromi epilettiche.
7.2. Diagnosi
Il primo passo è distinguere le crisi da altri sintomi transitori. La sincope e le
pseudocrisi sono le entità più frequenti che vengono confuse con l’epilessia
(Tabella 25).
R I C O R D A Una delle diagnosi diff erenziali più diffi cili è caratterizzata dalle crisi neurotiche o conversive che di solito vengono scatenate da un’emo-zione intensa.
• Elettroencefalogramma. Non è di per sé una prova che permetta di
diagnosticare o escludere l’epilessia.
• Studi neuroradiologici. La TC e la RM sono le tecniche d’elezione, es-
sendo la RM più sensibile nel riscontrare alterazioni strutturali del siste-
ma nervoso centrale.
Caratteristiche Crisi epilettica Sincope
Fattori scatenanti immediati
Abitualmente no Stress, manovra di Valsalva bipedestazione
Sintomi previ Nessuno o aura Sudorazione, nausea…
Postura all’inizio Indiff erente Più frequente in bipedestazione
Passaggio alla perdita di conoscenza
Brusco Più frequente progressivo
Durata dello stato di incoscienza
Minuti Più frequente secondi
Durata dei movimenti tonico-clonici
30-60 secondi Meno di 15 secondi
Aspetto facciale Schiuma dalla bocca e cianosi
Pallore
Basso livello di coscienza successivo
Da minuti a ore Pochi minuti
Dolore muscolare posteriore
Frequente Poco frequente
Morsicatura della lingua
A volte Poco frequente
Incontinenza A volte Poco frequente
Tabella 25. Diagnosi diff erenziale dell’epilessia

4407 ∙ Ep i l ess ia
Manuale CTO di Medicina e Chirurgia, Prima edizione
7.3. Eziologia
Nella Tabella 26 sono mostrate le cause più frequenti di crisi epilettiche, a
seconda dell’età in cui appaiono.
• Febbre. Le crisi febbrili sono un processo tipico dell’età infantile (tra i 3
mesi ed i 5 anni), che si associa più frequentemente con l’aumento della
temperatura, indipendentemente dall’origine della stessa.
Le crisi febbrili semplici generalizzate durano meno di 15 minuti, pre-
sentano un buon recupero posteriore ed i risultati nel periodo inter-
critico sono normali o negativi; frequentemente esistono antecedenti
familiari di crisi febbrili od epilessia; sono ricorrenti in un terzo dei casi
(anche se solo il 10% dei pazienti soff rono più di un episodio) ed è più
probabile che la crisi si manifesti nel primo anno di vita; non vengono
associate ad un rischio maggiore di manifestare epilessia.
Le crisi febbrili complesse sono quelle che presentano segni focali, una
durata superiore ai 15 minuti, o si ripetono nel corso dello stesso episo-
dio febbrile; si associano ad un 2-5% d’incremento del rischio di manife-
stare epilessia posteriormente.
Le crisi febbrili possono trattarsi con diazepam via rettale o intravenosa,
anche se dato che scompaiono spontaneamente, la gestione adeguata
è il controllo della temperatura, preferibilmente con la somministrazio-
ne di paracetamolo. In pazienti con crisi febbrili tipiche ricorrenti, può
somministrarsi diazepam orale o rettale nel caso di aumento della tem-
peratura. Non è indicato il trattamento continuato con anticomiziali
come profi lassi delle crisi febbrili.
Età Eziologia
Neonati (< 1 mesi)
∙ Ipossia perinatale
∙ Emorragia intracranica
∙ Infezioni del SNC
∙ Disturbi metabolici
∙ Astinenza da droghe
∙ Alterazioni genetiche
∙ Alterazioni dello sviluppo
Lattanti e bambini (1 mese-12 anni)
∙ Crisi febbrili
∙ Alterazioni genetiche
∙ Infezioni del SNC
∙ Alterazioni dello sviluppo
∙ Traumi
∙ Idiopatiche
Adolescenti (12-18 anni)
∙ Traumi
∙ Idiopatiche
∙ Alterazioni genetiche
∙ Tumori
∙ Consumo di droghe
Adulti giovani (18-35 anni)
∙ Traumi
∙ Astinenza da alcol
∙ Consumo di droghe
∙ Tumori
∙ Idiopatiche
Adulti (> 35 anni)
∙ Malattie cerebrovascolari
∙ Tumori
∙ Astinenza da alcol
∙ Disturbi metabolici
∙ Malattie degenerative del SNC
∙ Idiopatiche
Tabella 26. Eziologia delle crisi epilettiche a seconda dell’età d’inizio
• Trauma cranioencefalico (TCE). Le crisi che appaiono nella prima ora
dopo il TCE (immediate) di solito non comportano un rischio di epi-
lessia a lungo termine e si tratta normalmente di crisi generalizzate. Le
crisi precoci (tra la prima ora ed il settimo giorno dopo il TCE) di solito
è più frequente nei bambini, vengono associate a lesioni traumatiche
signifi cative ed, a diff erenza di quelle immediate, comportano il rischio
di epilessia tardiva e si tratta dunque di crisi parziali; si è dimostrato che
l’utilizzo di farmaci antiepilettici è utile come prevenzione primaria di
queste crisi.
Infi ne, le crisi tardive (quelle che appaiono dopo la prima settimana)
sono più frequenti negli adulti e si tratta di crisi parziali con tendenza
alla generalizzazione; non si raccomanda l’utilizzo di farmaci antiepilet-
tici per prevenire una sua comparsa.
• Patologia cerebrovascolare. È la responsabile del 50% dei casi nuovi
di epilessia nelle persone con età maggiore dei 64 anni.
R I C O R D A In generale, le crisi parziali indicano una lesione strutturale del pa-renchima nervoso, essendo recidivanti e diffi cili da controllare. Le alterazioni metaboliche di solito si manifestano come crisi genera-lizzate.
7.4. Alcune sindromi epilettiche specifi che
Epilessie parziali benigne dell’infanzia
Si caratterizzano dall’essere dipendenti dall’età, con inizio dopo i 18 mesi,
con crisi generalmente poco frequenti, senza deterioramento neurologi-
co associato e prove complementari normali, ad eccezione dell’EEG che
presenta un’attività di fondo normale con complessi focali, localizzati più
frequentemente nella regione temporale media. Più della metà dei pa-
zienti con epilessia parziale benigna dell’infanzia manifestano un’epilessia
rolandica.
L’epilessia rolandica comincia soprattutto tra i 7-10 anni manifestandosi
nel 98% dei casi intorno ai 14 anni. L’80% delle crisi appare durante il sonno
e di solito presentano focalità facciali. Di solito non richiede trattamento,
data la sua risoluzione spontanea.
Epilessia dell’infanzia con scarsa risposta al trattamento
La sindrome di West appare durante il primo anno di vita, più frequente-
mente tra il 4.º e 7.º mese, ed è più frequente nei maschi (1,5:1). La triade che
defi nisce la sindrome è formata da:
• Spasmi infantili. Contrazioni muscolari brevi (1-3 s), più frequente-
mente generalizzate e di predominio della muscolatura fl essoria, che
appare al risveglio, non persistendo durante il sonno.
• Ritardo dello sviluppo psicomotorio.
• Ipsaritmia intercritica. E’ un criterio imprescindibile per la diagnosi
della sindrome di West. Si tratta di un’attività basale disorganizzata, con
onde lente ad alto voltaggio, intercalate da onde acute. Durante le crisi
appaiono diversi tipi di onde, seguite da un’attenuazione del voltaggio.

45
07Neurologia e neurochirurgia
R I C O R D A L’ipsaritmia è intercritica, ossia tra le varie crisi, ed è il criterio impre-scindibile per la diagnosi di West.
La sindrome di Lennox-Gastaut presenta un’età d’inizio variabile tra 1 e 7
anni, con un picco massimo tra i 2-4 anni. È caratterizzata dalla triade di mol-
teplici tipi di convulsione, interessamento psicomotorio ed alterazioni
nell’EEG.
R I C O R D A La stessa lesione cerebrale nelle diversi età si manifesta secondo forme diverse (nei < 1 anno con sindrome di West e nei > 1 anno con sindrome di Lennox-Gastaut), a causa dei cambi maturativi del sistema nervoso centrale.
Epilessie generalizzate nell’adulto
L’epilessia mioclonica giovanile è il prototipo di epilessia generalizzata idio-
patica. Rappresenta il 10% di tutte le epilessie, ed è l’epilessia mioclonica più
frequente. L’età d’inizio è tra gli 8 ed i 25 anni. La maggior parte dei pazienti
presenta diversi tipi di crisi, soprattutto nelle miocloniche: un 90% presenta
crisi tonico-cloniche e nel 30% assenze tipiche.
Le crisi si presentano come spasmi muscolari brevi, di solito negli arti supe-
riori, al risveglio, favorite dalla privazione precedente del sonno e dal consu-
mo di alcool. Il livello di coscienza è mantenuto, tranne nelle crisi gravi.
R I C O R D A La presenza di mioclonie ed altri tipi di crisi in un adolescente, dopo essere stato ad una festa (consumo di alcol e privazione del sonno) è tipica dell’epilessia mioclonica giovanile.
L’EEG mostra un’attività parossistica punta-onda nella maggior parte dei casi,
ed sempre è patologica durante il sonno. Un terzo dei pazienti presenta atti-
vità parossistica fotosensibile, anche se non è frequente che ci sia una corre-
lazione clinica. Il trattamento come nelle crisi generalizzate è l’acido valproico.
Sclerosi mesiale temporale
La sclerosi mesiale temporale (SMT) interessa entrambi i sessi e costituisce
la sindrome epilettica più frequente (20%), è caratterizzata dalla perdita dei
neuroni e gliosi (sclerosi) nella parte mediale del lobo temporale, interessan-
do l’ippocampo ed il giro dentato. Nella maggior parte dei casi esiste un pre-
cedente di danno cerebrale nei primi 5 anni di vita (meningoencefalite, TCE,
crisi febbrili, complicanze perinatali…). Di solito si presenta tra i 5-15 anni,
inizialmente di tipo parziale semplice e con buona risposta al trattamento.
Successivamente si converte in parziale complessa e poi generalizzata, ogni
volta con una peggior risposta al trattamento medico, fi no a farsi refrattaria
intorno ai 10 anni dalla sua diagnosi.
Non esistono segni specifi ci della SMT, tuttavia la clinica è molto caratteri-
stica. Inizia con aura epigastrica consistente in fastidi epigastrici che salgo-
no fi no alla regione cervicale. Altre aure possibili, anche se meno frequenti,
sono la sensazione di paura, aure psichiche, olfattive o gustative. Successi-
vamente segue un intervallo di rallentamento e/o disconnessione parziale
dall’ambiente, con risposta inconsistente agli stimoli esterni, alternarnando
periodi di buona risposta agli ordini e alle domande verbali con altri nei quali
ciò non è possibile, ed accompagnandosi di solito da automatismi oroali-
mentari o manuali.
Si può manifestare con disturbi autonomici, tra cui quelli del ritmo cardiaco.
È tipica l’associazione con il disturbo della memoria anterograda.
Le prove di studio complementari mostrano una lesione a livello della parte
mediale del lobo temporale, essendo molto tipiche le iperintensità dell’ip-
pocampo nelle sequenze della risonanza magnetica.
Solamente il 10-30% delle SMT è controllabile attraverso un trattamento
medico, essendo questa entità il paradigma dell’epilessia farmacoresistente,
con buona risposta alla chirurgia. In questi casi la realizzazione di un’amigda-
loippocampectomia è la tecnica di elezione.
7.5. Trattamento. Farmaci anticomiziali
Inizio del trattamento
L’indicazione per iniziare un trattamento antiepilettico dopo la diagnosi di
epilessia prevede 2 crisi senza causa chiara ed identifi cabile. Lo schema del
trattamento viene mostrato nella Figura 42.
Trattamentoin monoterapia
Buon controllo (70%) Controlloinsoddisfacente (30%)
Politerapia2-3 farmaci
Buon controllo (15%) Controlloinsoddisfacente (15%)
Totalmente refrattarieal trattamento (10%)
Chirurgiadell’epilessia (5%)
Figura 42. Controllo delle crisi ad inizio recente
In un 70% dei bambini e in un 60% degli adulti epilettici con un buon control-
lo terapeutico, si può sospendere il trattamento dopo un periodo senza crisi.
Non esistono dei criteri comuni però si considerano segni di buona effi cacia
terapeutica: assenza di crisi dopo la sospensione del trattamento, presenza
di un EEG normale, un’esame neurologico senza alterazioni, un solo tipo di
crisi ed un periodo da 1 a 5 anni senza crisi. La maggior parte delle recidive
avviene nei primi 3 mesi dopo la sospensione del trattamento.
Meccanismo d’azione dei farmaci anticomiziali • Inibizione dei canali di Na+: fenitoina, carbamazepina, topiramato.
• Inibizione dei canali di Ca2+: fenitoina, valproato, etosuccimide.

4607 ∙ Ep i l ess ia
Manuale CTO di Medicina e Chirurgia, Prima edizione
Trattamento chirurgico dell’epilessia
L’intervento chirurgico verrà pianifi cato in quei pazienti con crisi poco con-
trollate nonostante l’assunzione di una corretta terapia nell’arco di almeno
un anno.
Sono state utilizzate diverse tecniche chirurgiche:
• Epilessia lesionale:
- Epilessia del lobo temporale: resezione standard del lobo tem-
porale.
- Epilessia extratemporale: resezione della lesione.
• Chirurgia di disconnessione: transezioni subpiali multiple (epilessie
parziali il cui fuoco si localizza nelle aree del linguaggio) e callostomia
totale o subtotale (multipli fuochi non resecabili, crisi tonico-cloniche
secondariamente generalizzate, convulsioni toniche o atoniche, con
ricadute).
• Stimolazione del nervo vago: convulsioni parziali non trattabili.
• Stimolazione cerebrale profonda: nelle diverse parti del cervello.
• Emisferectomia: sindromi panemisferiche con convulsioni non trat-
tabili.
7.6. Epilessia e gravidanza
Durante la gravidanza delle gestanti epilettiche, si mantiene la frequenza
delle crisi nel 50% con miglioramenti nel 20% ed un peggioramento nel
30%, che non è prevedibile dal momento che non dipende dal tipo di epi-
lessia, né dal numero di crisi negli ultimi mesi, né dal comportamento nelle
gravidanze precedenti..
Dato che non esiste un farmaco d’elezione durante la gravidanza, bisogna
mantenere il trattamento in corso alle minime dosi effi caci.
R I C O R D A Di fronte ad una donna incinta controllata con un farmaco anticon-vulsivo, non bisogna realizzare modifi che nel trattamento dal mo-mento che una crisi convulsiva può essere letale per il feto.
Bisogna iniziare un trattamento con l’acido folico per prevenire i difetti di
chiusura del tubo neurale (spina bifi da). Nel 50% dei nati recentemente da
madri in trattamento con farmaci antiepilettici induttori enzimatici, come la
fenitoina, si produce un defi cit transitorio e reversibile dei fattori di coagula-
zione della vitamina K-dipendenti, per tanto bisogna attuare una profi lassi
nelle ultime settimane di gravidanza e nel nascituro.
• Diminuzione della liberazione di glutammato: lamotrigina.
• Potenziamento della funzione dei recettori GABA: benzodiazepine,
barbiturici.
• Aumento della disponibilità del GABA: gabapentina, tiagabina, viga-
batrina.
Farmaci antiepilettici secondo il tipo di crisi
(Tabella 27 e Tabella 28)
Esiste un farmaco antiepilettico ad uso recente che è il levetiracetam,
usato ampiamente nella pratica clinica grazie alla sua effi cacia nel control-
lo delle crisi parziali e generalizzate, dato il suo profi lo farmacodinamico
con poche interazioni ed eff etti collaterali. Inoltre è disponibile per via en-
dovenosa.
Tipi di crisi Trattamento
Tonico-Cloniche generalizzate
Indistintamente: fenitoina, carbamazepina, fenobarbital o valproato
Parziali 1.º Carbamazepina
2.º Fenitoina
Miocloniche 1.º Clonazepam
2.º Valproato
Assenze 1.º Etosuccimide (tipiche)
2.º Valproato (atipiche)
Stato epilettico
1.º Diazepam i.v., fenitoina i.v., fenobarbital i.v.
2.º Anestesia con propofol e midazolam
Tabella 27. Trattamento dell’epilessia
Farmaco Eff etti secondari
Difenilidantoina Irsutismo, iperplasia gengivale
Carbamazepina Epatici ed ematologici
Topiramato Sonnolenza. Litiasi renale
Etosuccimide Sindrome parkinsoniana ed eff etti ematologici
Fenobarbital Sedazione. Bambini: iperattività
Benzodiazepine Si veda la Sessione di Psichiatria
Acido valproico Epatici, ematologici, pancreatite, alopecia
Lamotrigina Esantema
Vigabatrina Disturbi del comportamento e del campo visivo
Gabapentina Intolleranza gastrointestinale
Felbamato Ematologici
Tabella 28. Eff etti secondari dei principali antiepilettici
I d e e c h i ave L’EEG permette di diff erenziare le crisi parziali da quelle generaliz-zate: le prime sono quelle che attivano una sola regione concreta della corteccia, mentre le ultime producono un’attività elettrica si-multanea in entrambi gli emisferi, attivando tutta la corteccia.
Le crisi parziali semplici non alterano la coscienza mentre le crisi complesse sì.
Le crisi parziali semplici possono produrre sintomi motori, sensitivi, autonomi, sensoriali o psichici, a seconda dell’area corticale inte-ressata.
Le crisi parziali complesse producono un’alterazione del livello di coscienza ed automatismi con un periodo di confusione tra le crisi.
Le crisi generalizzate più importanti sono le assenze, o piccolo male, le crisi tonico-cloniche, o grande male.

47
07Neurologia e neurochirurgia
C a s i c l i n i c i Un episodio caratterizzato da sensazione epigastrica che si estende verso il torace, con diffi coltà a relazionarsi con l’ambiente, movimenti di masticazione, distonia di una mano e mancanza di risposta, di un minuto di durata, con amnesia postcritica è una crisi:
1) Parziale semplice.2) Parziale secondariamente generalizzata.3) Parziale complessa.4) Assenza tipica.5) Assenza atipica.
RC: 3
Un paziente di 40 anni, senza precedenti di rilievo, viene portato al Pronto Soccorso per aver manifestato deviazione della testa verso la sinistra, convulsioni che iniziarono agli arti di sinistra e si generaliz-zarono subito ai quattro arti, con perdita di coscienza, incontinenza vescicale e stato confusionale di una mezz’ora di durata. Indipen-dentemente dai risultati dell’esame obiettivo e delle analisi di labo-ratorio di routine, dovrebbe realizzarsi al più presto possibile, come prima misura:
1) TC cerebrale.2) Determinazione dell’ alcolemia.3) Determinazione dei livelli di oppiacei nel sangue e nell’urina.4) Elettroencefalogramma.5) Puntura lombare.
RC: 1
Bambino di 3 anni che inizia a presentare sintomi catarrali e, un’ora successiva, manifesta un episodio di perdita di conoscenza, movi-menti tonico-clonici delle estremità e revulsione oculare, con una durata approssimativa di due minuti. All’esame obiettivo presenta una temperatura di 39 ºC, esame neurologico normale, tranne ten-denza al sonno, faringe molto congesta con amigdale ipertrofi che e timpani iperemici. Quale comportamento tra i seguenti bisognereb-be adottare in questo momento?
1) Iniziare trattamento con antitermici e controllo successivo.2) Realizzare una puntura lombare per analizzare il liquido cefalora-
chideo.3) Richiedere un EEG urgente.4) Iniziare trattamento con diazepam intravenoso.5) Richiedere una TC cranica.
RC: 1
Sono tipici delle assenze gli episodi bruschi e ripetitivi di disconnes-sione dall’ambiente, con scariche generalizzate e simmetriche pun-ta-onda a 3 Hz nell’EEG. Compaiono nei bambini, risolvendosi spes-so nell’adolescenza e si controllano adeguatamente con i farmaci.
Le crisi tonico-cloniche sono il tipo di crisi più frequente nei distur-bi metabolici.
La causa più comune di convulsioni a seconda dell’età: neonati, en-cefalopatia ipossica; lattanti e bambini, crisi febbrili; adolescenti ed adulti giovani, traumi; anziani, malattie cerebrovascolari. I tumori sarebbero la causa più frequente negli adulti con età media.
La sindrome di West compare nel primo anno di vita e si caratteriz-za per la triade: spasmi infantili, alterazione dello sviluppo psico-motorio ed ipsaritmia.
La sindrome di Lennox-Gastaut è l’evoluzione che presentano i pa-zienti con West verso i 2-4 anni.
L’acido valproico (valproato) è il farmaco l’anticonvulsivo più utiliz-zato dato il suo ampio spettro d’azione. Produce alopecia ed epa-totossicità.
La carbamazepina è indicata nelle crisi parziali, però non va som-ministrata durante gli stati di assenza. Produce epatotossicità, ane-mia aplastica e sindrome di Stevens-Johnson.
L’ ACTH è indicata nel trattamento della sindrome di West.

484848
Neurologia e neurochirurgia
ORIENTAMENTO
Tema difficile nel quale bisogna concentrarsi sullo studio delle malattie dei motoneuroni. È raccomandabile capire bene il caso clinico, conoscendo perfettamente la clinica della SLA.
8.1. Atassia di Friedrich
È il tipo più frequente di atassia ereditaria. Si eredita in forma autosomica
recessiva ed il gene anomalo si localizza nel braccio corto del cromosoma 9.
Anatomia patologica
Il dato più caratteristico è la perdita neuronale nei gangli delle radici dorsali.
In secondo luogo, può presentarsi una degenerazione retrograda delle fi bre
nervose dei cordoni posteriori, tratto spinocerebellare e nervi periferici. La
maggior parte dei casi implica anche una degenerazione della via piramida-
le (corticospinale).
Il midollo spinale è atrofi co. A livello sistemico, spiccano le alterazioni cardia-
che, con fi brosi interstiziale cronica ed ipertrofi a ventricolare.
Clinica
Si presenta nell’infanzia o nell’adolescenza con atassia progressiva della
marcia. Segue atassia degli arti e disartria cerebellare. È molto caratteristi-
ca l’associazione di iporefl essia miotatica con risposte plantari in estensione
(segno di Babinski). La perdita della capacità di deambulazione si manifesta
approssimatamente 15 anni dopo.
R I C O R D A È tipica l’associazione di iporefl essia (dato dal 2.ª motoneurone) con Babinski (dato dal 1.ª motoneurone), e dell’atassia.
La lesione dei cordoni posteriori giustifi ca la perdita della sensibilità profon-
da (artrocinetica, posizionale e vibratoria). In alcuni casi si verifi ca amiotrofi a
distale ed ipoestesia a guanto o a calza per lesione del nervo periferico. Può
08esserci nistagmo, sordità e atrofi a ottica. Non c’è lesione delle funzioni su-
periori. Si associano anche cifoscoliosi e pié cavo. Un 10-20% dei pazienti
presenta un diabete mellito.
R I C O R D A È frequente l’associazione di cifoscoliosi, piedi cavi, diabete mellito e miocardiopatia ipertrofi ca.
La principale causa di morte è l’insuffi cienza cardiaca secondaria a miocar-
diopatia ipertrofi ca. Non esiste un trattamento effi cace.
Diagnosi
È fondamentalmente clinica. Nell’EMG si evidenziano segni di denervazione
dei muscoli distali. Il LCR è normale. Nella TC si può osservare una moderata
atrofi a cerebellare nelle fasi avanzate. È importante scartare in questi pazien-
ti un defi cit sottostante di vitamina E.
R I C O R D A Il defi cit di vitamina E può provocare una degenerazione nervosa con atassia.
8.2. Sclerosi laterale amiotrofi ca
La sclerosi laterale amiotrofi ca (SLA) è la forma più frequente di malattia pro-
gressiva del motoneurone, essendo la sua incidenza globale di 1-3 casi per
ogni 100.000 abitanti/anno. Di solito è più frequente negli uomini (1,5:1). Si
distinguono fondamentalmente due forme: familiare (10% con inizio pre-
coce) e sporadica (90%, con inizio sopra i 50 anni).
Anatomia patologica
Processo degenerativo che interessa, con gradi diversi, il primo ed il secondo
motoneurone, producendo secondariamente atrofi a delle fi bre muscolari. A
volte è possibile trovare pazienti con lesione selettiva del primo motoneuro-
ne (sclerosi laterale primaria), dei neuroni dei nuclei troncoencefalici (paralisi
MALATTIE DEGENERATIVE DEL SISTEMA NERVOSO

49
08Neurologia e neurochirurgia
Sclerosi laterale amiotrofica primaria
Paralisi pseudobulbare
Paralisi bulbare
Atrofia muscolare spinale
Figura 43. Forme di SLA
I d e e c h i ave Tra le atassie degenerative ereditarie, si trova l’atassia di Friedreich (AF), che è la più frequente.
L’AF si caratterizza per degenerazione neuronale dei gangli dorsali midollari (atassia della marcia) e cerebellare, associata a cifoscoliosi, piedi cavi, diabete mellito e miocardiopatia ipertrofi ca.
Problemi cardiaci sono le principali cause di morte in questi pazienti.
La SLA può essere ereditaria (10%) o sporadica (90%) e si caratte-rizza per degenerazione selettiva del primo e del secondo moto-neurone, con sensibilità, capacità cognitiva e funzione autonomica conservate.
La clinica è una combinazione di sintomi dovuti all’interessamento del primo e del secondo motoneurone, con un inizio asimmetrico ed una sopravvivenza media di 3 anni.
L’unico trattamento disponibile è il riluzolo (blocca i recettori NMDA del glutammato) con un benefi cio discreto.
bulbare progressiva) o secondo motoneurone (atrofi a muscolare spinale)
(Figura 43).
Clinica
Si presenta con debolezza muscolare lentamente progressiva con segni di
lesione del primo e/o secondo motoneurone, sia delle estremità sia dei nervi
cranici (disartria e disfagia).
Non si presentano defi cit sensitivi, ne disfunzioni vescicali, deterioramento
intellettivo o disfunzioni sessuali. La media di sopravvivenza è di 3 anni dall’i-
nizio dei sintomi.
R I C O R D A Debolezza muscolare progressiva, con inizio asimmetrico e distale, con lesione dei nervi cranici bassi e scarsa lesione della muscolatu-ra, è tipica della SLA.
Trattamento
Attualmente si sta commercializzando solo il riluzolo come trattamento
per questa malattia. Sembra che abbia un benefi cio discreto sull’aumento
della sopravivenza, specialmente in pazienti con inizio bulbare della clinica.
C a s i c l i n i c i Donna di 64 anni che si presenta con una clinica progressiva, negli ultimi 4 mesi, di debolezza alla gamba destra. All’esame obiettivo si evidenzia una paresi con amiotrofi a dell’arto inferiore destro ed una iperrefl essia miotatica dello stesso. Qual è la sua diagnosi?
1) Ernia al discale lombare.2) Sindrome di Guillain-Barré.3) SLA.4) Neuropatia per malattia di Lyme.5) SM.
RC: 3
Paziente di 50 anni che presenta in maniera insidiosa debolez-za e spasmi all’arto superiore destro. All’esame obiettivo neu-rologico si individua spasticità nell’emicorpo destro, atrofia del primo muscolo interosseo della mano destra, con riflessi osteo-tendinei presenti e senza deficit sensitivi. Qual è la diagnosi più probabile in questo paziente, tra le seguenti?
1) Siringomielia.2) Tumore a livello del forame magno.3) SLA.4) Mielopatia cervicale di origine vascolare.5) Astrocitoma midollare cervicale.
RC: 3

505050
Neurologia e neurochirurgia
9.1. Encefalite erpetica ed altre encefaliti virali
Nelle meningiti, il processo infettivo si limita solo alle meningi; invece
nell’encefalite viene interessato anche il parenchima cerebrale.
Eziologia
Sono causa frequente d’encefalite epidemica gli arbovirus e l’enterovirus.
Il virus herpes simplex tipo I è l’agente eziologico più frequente dell’encefa-
lite sporadica.
Clinica
Si presenta con febbre, cefalea, segni meningei ed il segno più caratteristico
dell’encefalite: il deterioramento dello stato di coscienza con segni di focalità
neurologica corticale.
Si possono manifestare allucinazioni, stato di agitazione, cambi del compor-
tamento e anche una clinica psicotica. Fino a un 50% dei pazienti presenta
crisi focali o generalizzate.
R I C O R D A Meningismo con deterioramento cognitivo e focalità neurologica sono suggestivi di encefalite.
Prove complementari
• LCR. Caratteristicamente esiste pleiocitosi linfocitaria (> 95% dei casi)
con iperproteinorrachia e glucosio normale.
09
R I C O R D A La presenza di emazie abbondanti nel LCR è suggestiva di un’ence-falite necroemorragica secondaria ad herpes simplex.
• Coltura del LCR. In generale è di scarso rendimento.
• Sierologia. È una diagnosi retrospettiva però possiede uno scarso valo-
re nella diagnosi e nel trattamento in fase acuta.
• TC, RM e EEG. L’esistenza di focalità a livello frontotemporale nell’EEG è
suggestiva di encefalite erpetica. Nella TC, l’encefalite erpetica può dar
luogo a zone d’ipodensità ed eff etto massa nelle regioni frontotempo-
rali che possono captare contrasto. La RM è la tecnica radiologica per
eccellenza per rilevare cambiamenti del segnale nel parenchima cere-
brale nell’encefalite (Figura 44).
Figura 44. RM che mostra una lesione della regione temporale destra, che in un contesto di febbre e meningismo è altamente suggestiva di encefalite erpetica
MALATTIE VIRALE E PRIONICHE DEL SISTEMA NERVOSO
ORIENTAMENTO
È un tema di poca importanza però può servire per ripassare l’encefalite erpetica richiesta anche nelle Malattie Infettive. Le altre malattie infettive virali presentano un quadro molto caratteristico, tra queste è necessario conoscere solamente la più tipica. La malattia di Creutzfeldt-Jacob (ECJ) è la malattia prionica per eccelenza.

51
09Neurologia e neurochirurgia
R I C O R D A L’encefalite erpetica coinvolge maggiormente la zona frontotem-porale.
• Biopsia cerebrale. Ha una sensibilità di più del 95% ed una specifi cità
prossima al 100%. Tuttavia la sua applicazione è diminuita dopo l’intro-
duzione dell’aciclovir come trattamento dell’encefalite erpetica (ampia
effi cacia con scarsi eff etti secondari). Si indica in casi dubbi o con scarsa
riposta al trattamento per confermare la diagnosi.
• Reazione a catena della polimerasi (PCR). La PCR del DNA del virus
herpes simplex ha sostituito la biopsia cerebrale come tecnica diagno-
stica d’elezione nell’encefalite erpetica.
Trattamento
L’aciclovir è il farmaco d’elezione delle encefaliti da herpes simplex, inoltre è
effi cace per il trattamento del virus di Epstein-Barr e della varicella-zoster. Si
deve somministrare in caso di minimo sospetto clinico, senza ritardi.
R I C O R D A La PCR del LCR è la tecnica diagnostica d’elezione nelle encefaliti er-petiche.
Prognosi
La mortalità nell’encefalite erpetica raggiunge un 20% e fi no ad un 40% dei
pazienti che sopravvivono presenta una disabilità grave (in relazione diret-
ta con l’età del paziente, il ritardo del trattamento e lo stato di coscienza).
9.2. Leucoencefalopatia multifocale progressiva
Anatomia patologica
La leucoencefalopatia multifocale progressiva (LMP) colpisce pazienti im-
munodepressi e si caratterizza per una demielinizzazione multifocale che
colpisce il SNC, con lesioni di dimensioni variabili. Gli oligodendrociti con-
tengono inclusioni cristalline che sono particelle del virus JC (papovavirus).
Clinica
I pazienti abitualmente mostrano alterazioni visive, generalmente emianopsia
omonima e deterioramento delle funzioni superiori con demenza ed alterazio-
ni della personalità. Durante l’evoluzione è tipica la presenza di defi cit motori.
Prove complementari
Alla TC si osservano lesioni ipodense nella sostanza bianca che non captano
contrasto e non presentano edema. La RM è più sensibile per evidenziare
queste lesioni.
L’EEG mostra un rallentamento focale o diff uso. Il LCR è tipicamente norma-
le con iperproteinorrachia modesta ed occasionalmente leucocitosi (< 25
cellule/μl).
La diagnosi defi nitiva necessita dell’osservazione di cambiamenti anatomo-
patologici tipici alla biopsia. Dato che un 80-90% della popolazione adulta
normale è sieropositiva per il virus JC, l’identifi cazione dell’antigene o geno-
ma del virus non è diagnostica di LMP se non è accompagnata da cambia-
menti anatomopatologici tipici. La reazione a catena della polimerasi (PCR)
non è utile ai fi ni diagnostici in questa malattia.
R I C O R D A In un paziente con AIDS e clinica neurologica si deve scartare la toxoplasmosi. Se nella TC le lesioni non captano contrasto e non presentano eff etto massa bisogna sospettare una LMP come pri-ma opzione.
Prognosi
La regola è la morte in pochi mesi. Non esiste trattamento efficace, an-
che se alcuni casi migliorano con la ricostituzione del sistema immuni-
tario.
9.3. Altre malattie virali del SNC
Sono riassunte Tabella 29.
Malattie Virus associato Clinica caratteristica
Paraparesi spastica
tropicale
HTLV-1 Iperefl essia e spasticità
progressiva nelle estremità
inferiori
LMP Virus JC Disturbi visivi (emianopsia
omonima), demenza e alterazioni
della personalità
Panencefalite
subacuta
sclerosante
Morbillo ∙ Cattivo rendimento scolastico
e alterazioni della personalità
∙ Successivamente
deterioramento neurologico
e tetraparesi spastica
Tabella 29. Malattie del SNC di origine virale
9.4. Malattie prioniche (Tabella 30 e Figura 45)
Con questo termine si classifi cano un insieme di malattie il cui agente pato-
geno è una proteina infettiva carente dell’acido nucleico chiamata prione.
Le malattie prioniche umane (si veda la Tabella 30) includono la malattia
di Creutzfeldt-Jakob (MCJ), kuru, malattia di Gerstmann-Sträussler-Scheinker
(GSS) e l’insonnia familiare fatale (IFF).
R I C O R D A Le bande oligoclonali nel LCR sono presenti in molte patologie ed indicano solamente l’attivazione di un piccolo pool di linfociti B nel SNC.

5209 ∙ Ma la t t i e v i ra l e e p r i on i che de l s i s tema ne rvoso
Manuale CTO di Medicina e Chirurgia, Prima edizione
Malattie prioniche infettive
Kuru Cannibalismo
MCJ iatrogeno
∙ Trapianto di cornea
∙ Innesto della duramadre
∙ Estratti ipofi sari
∙ Procedimenti neurochirurgici
Malattie prioniche ereditarie
MCJ Mutazioni nel gene della PrP nel cromosoma 20
GSS Mutazioni nel gene della PrP nel cromosoma 20
IFF Mutazioni nel gene della PrP nel cromosoma 20
Malattie prioniche sporadiche
MCJ (85%) ∙ Mutazione somatica
∙ Conversione spontanea di PrPc a PrPsc
∙ Suscettibilità a fattori ambientali
Tabella 30. Categorie eziologiche delle malattie prioniche
Forma normale(PrPc)
Forma patológicao prione (PrPc)
Figura 45. Confi gurazione spaziale di una proteina prionica
Malattia di Creutzfeldt-Jakob (MCJ)
Epidemiologia
La maggior parte dei casi è sporadica però nel 5-15% è stato dimostrato
un pattern d’ereditarietà autosomico dominante, in relazione alla mutazione
nel gene della PrP nel cromosoma 20, ed un tipo di trasmissione da persona
a persona in caso d’interventi neurochirurgici con materiale non decontami-
nato, trapianti di cornea o innesti di duramadre umana ed attraverso estratti
ipofi sari di ormoni della crescita, ottenuti da cadaveri colpiti dalla malattia. I
casi sporadici non sono ancora ben spiegabili.
Clinica
È caratterizzata da una demenza rapidamente progressiva che associa mio-
clonie nel 90% dei pazienti ed atassia cerebellare. Altri sintomi extrapirami-
dali includono tremore, coreoatetosi e parkinsonismo (60%). Nel 50% sono
presenti segni di coinvolgimento piramidale. L’evoluzione è invariabilmente
fatale. Recentemente è stata descritta la nuova variante di questa malat-
tia che si associa epidemiologicamente alla diff usione della encefalopatia
spongiforme bovina (“mucca pazza”). Inizia nei pazienti più giovani (16-40
anni), ha un decorso lento e non si osservano mutazioni nel cromosoma
20. La clinica inizia con alterazioni psichiatricche, parestesie nelle estremità
inferiori ed atassia. La sopravvivenza media è di 15 mesi, comparata con i 5
mesi della MCJ sporadica.
R I C O R D A La variante associata alla malattia della mucca pazza si distingue dalla forma classica di MCJ perché appare in pazienti più giovani e predominano i sintomi psichiatrici e l’atassia sulle demenze e sulle mioclonie.
Prove complementari
• LCR. Generalmente normale o con lieve aumento delle proteine. E’ sug-
gestiva la positività della proteina 14-3-3, anche se presenta falsi positivi
e negativi.
• TC e RM. Possono mostrare atrofi a corticale generalizzata però d’inten-
sità minore rispetto a ciò che ci aspettiamo dal grado di demenza.
• EEG. Un pattern tipico può suggerire la diagnosi. Su un’attività di base
lenta, è frequente l’apparizione di scariche di onde acute bilaterali e sin-
crone (ogni 0,5-2,5 s e con una durata di 200-600 ms). Questo pattern si
osserva nella maggior parte dei casi anche se può non essere presente
nelle fasi inziali o molto avanzate. L’EEG generalmente è normale nella
nuova variante.
R I C O R D A La MCJ classica possiede un pattern tipico nell’EEG, mentre la nuova variante mostra un EEG normale.
• Studio anatomopatologico. È il metodo diagnostico d’elezione (la
biopsia o l’autopsia). È caratterizzata da una degenerazione spongifor-
me massiva nella corteccia cerebrale, però prominente nei nuclei della
base, nel talamo e nel cervelletto. Abitualmente non sono coinvolti
né il tronco encefalico né il midollo spinale. L’evidenza di placche di
amiloide composte da PrPsc è diagnostica.
I d e e c h i ave Le encefaliti epidemiche sono causate da arbovirus ed enterovirus.
L’encefalite sporadica più frequente è quella secondaria al virus her-pes simplex tipo 1.
La clinica dell’encefalite è simile a quella della meningite (febbre, cefalea, meningismo), con alterazione cognitiva e focalità neu-rologica.
Nel LCR si osserveranno le caratteristiche della meningite virale: iper-proteinorrachia, pleiocitosi linfocitaria e glucosio normale.

53
09Neurologia e neurochirurgia
L’encefalite erpetica mostra focalità frontotemporale, essendo la RM la prova d’immagine d’elezione.
La PCR del DNA del virus herpes nel LCR è la tecnica diagnostica d’elezione sostituendo la biopsia cerebrale.
Il trattamento con aciclovir è sicuro ed effi cace; per questo si deve instaurare rapidamente di fronte al sospetto d’encefalite.
La paraparesi spastica tropicale è stata messa in relazione con l’infe-zione per HTLV-1 in zone tropicali. Può simulare una sclerosi multi-pla con presenza di bande oligoclonali nel LCR e placche di demie-linizzazione nella RM.
La LMP è caratterizzata da demenza ed emianopsia in pazienti con AIDS. È causata dall’infezione per il papovavirus JC che provoca de-
mielinizzazione simile alla sclerosi multipla. La diagnosi richiede la biopsia cerebrale.
La panencefalite subacuta sclerosante si presenta nei bambini con età compresa tra i 5-15 anni con storia di morbillo nei pri-mi anni di vita. L’EEG mostra un pattern caratteristico, e nel LCR si osservano bande oligoclonali ed un aumento del titolo di Ac antimorbillo.
Le malattie prioniche sono un’insieme di malattie che hanno in co-mune la presenza di una proteina come agente patogeno. La MCJ classica si caratterizza per demenza rapidamente progressiva, mio-clonie ed atassia. L’EEG mostra un pattern tipico anche se la diagno-si è anatomopatologica.
C a s i c l i n i c i Paziente di 60 anni senza precedenti d’interesse che in un periodo di 6 mesi sviluppa una clinica di deterioramento cognitivo intenso. All’esame obiettivo si osserva, a parte l’esistenza di una sindrome rigido-acinetica, un’atassia della marcia e mioclonie. Di fronte a que-sto quadro, quale diagnosi tra le seguenti siamo obbligati a valutare in primo luogo?
1) Una LMP.2) Una panencefalite sclerosante subacuta.3) Una forma sporadica di Creutzfeldt-Jakob.4) Un’atrofi a multisistemica.5) Un’encefalopatia di Wernicke.
RC: 3

545454
Neurologia e neurochirurgia
ORIENTAMENTO
Tema d’importanza discreta. Si raccomanda di studiare bene l’encefalopatia di Wernicke e la degenerazione subacuta combinata del midollo.
10.1. Malattie neurologiche in seguito a defi cit nutrizionali
Encefalopatia di Wernicke
Eziologia e patogenesi
Si presenta in pazienti alcolisti e malnutriti (per iperemesi, cancro, inanizio-
ne…) a causa di un defi cit della tiamina o della vitamina B1. Il defi cit della
tiamina produce un disturbo del metabolismo cerebrale del glucosio.
Clinica
Si caratterizza dalla triade oftalmoparesi, atassia e stato confusionale.
Prove complementari
Il LCR è normale o mostra un minimo aumento delle proteine. Nell’EEG si
riscontra un’attività lenta in forma diff usa. Le prove della funzione vestibo-
lare risultano alterate in tutti i casi, in maniera bilaterale e simmetrica nelle
fasi iniziali. Nei casi non trattati, c’è un aumento del piruvato sierico con una
diminuzione della transchetolasi.
Evoluzione
Anche se tutti questi sintomi possono comparire simultaneamente, di solito l’of-
talmoplegia e/o l’atassia precedono il quadro confusionale di giorni o settimane.
Dopo aver somministrato il trattamento vitaminico, il recupero continua in
maniera tipica. Innanzitutto migliora l’oftalmoparesi e, successivamente, l’a-
10
tassia. È possibile che rimangano come sequele un nistagmo orizzontale, un
aumento della base d’appoggio e marcia instabile.
Nell’ultima fase migliora il quadro confusionale e può comparire un disturbo
amnesico con incapacità nel conservare le nuove informazioni; è la sindro-
me di Korsakoff , la cui prognosi di recupero varia; la maggior parte conserva
un defi cit di memoria variabile e meno del 20% dei pazienti manifesta un
recupero completo.
Un 15-20% dei pazienti con malattia di Wernicke muore, generalmente, a
causa di infezioni intercorrenti o per insuffi cienza epatica.
Trattamento
Va somministrata la vitamina B1.
Per evitare il suo sviluppo, bisogna somministrare tiamina prima delle solu-
zioni glucosate a pazienti con rischio di denutrizione.
Degenerazione subacuta combinata del midollo
Si instaura per un defi cit di vitamina B12
.
• Clinica. A livello ematologico, produce un’anemia megaloblastica. A li-
vello midollare produce una sindrome posterolaterale (viene colpita la
sensibilità epicritica associata a sintomatologia del primo motoneuro-
ne). A livello mentale determina un quadro di deterioramento cognitivo
simile ad una demenza.
• Diagnosi. Si conferma mediante la determinazione dei livelli di vitami-
na B12
ed il test di Schilling.
• Trattamento. Somministrazione della vitamina B12
parenterale (intra-
muscolare).
Pellagra
È una malattia nutrizionale dovuta a defi cit di niacina o acido nicotinico. Le
sue manifestazioni sono dermatite, diarrea e demenza. La dermatite è bila-
terale, simmetrica e compare in zone esposte alla luce solare a causa della
fotosensibilità.
MALATTIE NUTRIZIONALI E METABOLICHE DEL SISTEMA NERVOSO

55
10Neurologia e neurochirurgia
C a s i c l i n i c i Uomo di 59 anni, ex bevitore e sottoposto a gastrectomia per emor-ragia digestiva 15 anni prima. Senza trattamento in corso. Si presenta per un quadro insidioso di diffi coltà a camminare che peggiora nell’o-scurità. All’esame obiettivo si riscontra un’atassia della marcia, il pa-ziente cade al suolo durante la prova di Romberg, risposte plantari in estensione e mantenimento della sensibilità algesica, abolite la vibra-toria e la posizionale. Segnali tra le seguenti l’aff ermazione corretta:
1) La diagnosi è degenerazione cerebellare alcolica.2) Si tratta di una degenerazione combinata subacuta del midollo spi-
nale.3) Bisogna scartare una lesione centromidollare.4) La diagnosi è SM in forma primaria progressiva.5) Il trattamento preferenziale è tiamina intravenosa.
RC: 2
R I C O R D A Il defi cit di niacina produce la pellagra, la malattia delle 3 “D”: diarrea, demenza e dermatite.
Le manifestazioni neurologiche della pellagra sono diverse, predominando
la clinica dell’encefalopatia; altre manifestazioni sono la mielopatia e la neu-
ropatia periferica.
10.2. Encefalopatia anossico-ischemica (Figura 46)
Clinica
Se l’anossia dura più di 3-5 minuti, si produce un danno cerebrale irrever-
sibile. Le aree cerebrali più sensibili all’anossia sono i gangli basali, il cervel-
letto, l’ippocampo e le regioni fronto-parieto-occipitali. Questo condiziona
le possibili sequele di un’encefalopatia anossica; 1) clinica extrapiramidale,
fondamentalmente mioclonie; 2) atassia cerebellare; 3) sindromi amnesiche
tipo Korsakoff , e 4) agnosia visuale.
Trattamento
Si basa sul ripristino delle funzioni cardiorespiratorie, in maniera urgente.
Figura 46. Encefalopatia anossica per arresto cardiorespiratorio
I d e e c h i ave L’encefalopatia di Wernicke si manifesta principalmente negli al-colisti, per iperemesi e malnutrizione.
L’encefalopatia di Wernicke è caratterizzata dalla triade oftalmo-paresi (soprattutto bilaterale ed asimmetrica del VI nervo crani-co), atassia e sindrome confusionale che appaiono e scompaio-no con il trattamento. La sindrome confusionale evolve in una sindrome amnesica anterograda: sindrome di Korsakoff .
Il trattamento dell’encefalopatia di Wernicke è la vitamina B1.
La degenerazione subacuta combinata del midollo si produce per un defi cit di vitamina B
12, e viene diagnosticata attraverso la
determinazione dei livelli della stessa, così come mediante il test de Schilling.
La degenerazione subacuta combinata del midollo si presen-ta con perdita della sensibilità vibratoria e propriocettiva, e clinica del primo motoneurone per lesione dei cordoni poste-riori e laterali midollari, rispettivamente. Può anche produrre anemia megaloblastica. Il suo trattamento è la vitamina B
12
parenterale.
La pellagra si produce per defi cit di niacina. È la malattia delle 3 “D”: dermatite, diarrea e demenza.
Le aree cerebrali più sensibili all’anossia sono i gangli basali, il cer-velletto, l’ippocampo e le regioni fronto-parieto-occipitali.

565656
Neurologia e neurochirurgia
ORIENTAMENTO
Tema d’importanza intermedia. Bisogna focalizzarsi sulle considerazioni generale e sulla sindrome di Guillain-Barré.
11.1. Considerazioni generali
Si utilizza il termine generico di neuropatia periferica per defi nire quei distur-
bi dei nervi periferici, a prescindere dalla sua causa (Figura 47).
Si parla di polineuropatie per riferirsi a patologie con esordio graduale
che interessano i multipli tronchi nervosi e che si caratterizzano per es-
sere simmetrici e generalizzati, con lesioni normalmente distali. La mo-
noneurite multipla è una lesione simultanea o consecutiva dei tronchi
nervosi non contigui, con evoluzione di giorni o anni. A volte presentano
un carattere confl uente, con diffi cile diagnosi diff erenziale con le poli-
neuopatie.
Le mononeuropatie sono lesioni focali di un unico tronco nervoso. Vengo-
no studiate dettagliatamente nella Sessione di Ortopedia.
11Le radiculopatie sono disturbi che interessano le radici nervose. Il termine si
riferisce alla lesione di radici nervose multiple in forma consecutiva.
Le plessopatie sono disturbi che interessano il plesso nervoso, di origine
traumatica, compressiva o autoimmune.
Clinica
• Disturbi sensitivi. Di solito è la prima manifestazione clinica.
• Disturbi motori. Caratteristicamente il paziente avrà dei segni di lesio-
ne del secondo motoneurone.
La presenza di atrofi a muscolare dipende dal tipo di neuropatia, essen-
do molto importante nelle lesioni assonali e poco frequente nelle ma-
lattie demielinizzanti.
• Disturbi autonomici. I sintomi autonomici includono soprattutto
ipotensione ortostatica, ritenzione urinaria, stitichezza, diarrea, impo-
tenza.
Diagnosi
• Bisogna valutare la presenza di pregresse infezioni virali, malattie
sistemiche (diabete mellito, uremia, porfi ria, defi cit vitaminici di B1, B
6,
acido folico, acido pantotenico, B12
, epatopatia cronica, amiloidosi, ipo-
NEUROPATIE
Assonale
Velocità di conduzione normaleLatenze distali normaliAmpiezza del potenziale ridotta
Demielinizzante
Diminuzione della velocità di conduzione Aumento delle latenze distaliSe non è omogenea: - Dispersione - Blocchi
Ampiezza normale
Figura 47. Fisiopatologia delle neuropatie

57
11Neurologia e neurochirurgia
tiroidismo, pneumopatia cronica, acromegalia, malassorbimento, carci-
nomi, linfomi, policitemia vera, mieloma, gammopatia monoclonale…),
utilizzo di farmaci (amiodarone, cisplatino, dapsone, idralazina, isonia-
zide, metronidazolo, difenilidantoina, piridossina, talidomide, vincristi-
na, nitrofurantoina), esposizione a sostanze tossiche (solventi, pestici-
di, metalli pesanti o alcool).
• Palpazione dei tronchi nervosi. Il segno di Tinnel (sensazione elettrica
con la percussione del nervo) è caratteristico delle neuropatie compres-
sive.
• Neurofi siologia. I disturbi assonali si caratterizzano da una perdita
dell’ampiezza del potenziale di azione, mentre quelle demieliniz-
zanti presentano un aumento della latenza, una diminuzione della
velocità di conduzione, con la possibile comparsa di fenomeni di
blocco della conduzione e della dispersione del potenziale nervoso
(Figura 48).
• Biopsia del nervo. Si utilizza generalmente il nervo surale. È indicato
nei casi di mononeuriti multiple e nella diagnosi di alcuni disturbi infan-
tili determinati geneticamente (leucodistrofi a metacromatica, malattia
di Krabbe…).
Neurofisiologia Demielinizzante
Diminuzione della velocità di conduzioneAumento delle latenze distali
Se demie-linizzazione non omogenea
Dispersione del potenzialeBlocchi di conduzione
Guillain-BarréGenetiche:
AssonaleVelocità di conduzione normaleDiminuzione dell’ampiezza del potenziale
Metaboliche e tossiche (ad eccezione di quelle incluse nelle miste)Charcot-Marie-Tooth tipo II
Miste DiabeticaAssociata a linfomaAssociata a mieloma
- Charcot-Marie- Tooth-tipo I- Déjerine-Sottas- Refsum
Figura 48. Classifi cazione delle polineuropatie secondo lo studio neurofi siologico
11.2. Sindrome di Guillain-Barré
Si tratta di una poliradiculoneuropatia demielinizzante acuta d’origine im-
munologica. Di solito interessa i giovani maschi.
R I C O R D A C. jejuni si tratta con eritromicina e può generare un quadro clinico e istologico simile alle malattie infi ammatorie intestinali.
In più dei due terzi dei casi si osservano pregresse infezioni virali respiratorie
e gastrointestinali. I virus più frequentemente implicati sono quelli del grup-
po herpes (citomegalovirus, virus di Epstein-Barr).
Più recentemente, Campylobacter jejuni è stato descritto in pazienti con Guil-
lain-Barré e storia di pregressa gastroenterite. È stato anche associato con
precedenti interventi chirurgici, linfomi e lupus eritematoso sistemico. Viene
considerata una malattia autoimmunitaria mediata da linfociti ed anticorpi
anti-GM1.
L’anatomia patologica mostra aree di demielinizzazione limitata al sistema
nervoso periferico.
Clinica
Si presenta con un quadro di tetraparesi fl accida ed arefl essica con
scarsi sintomi sensitivi. Di solito non interessa gli sfi nteri. Nella maggior
parte dei casi la debolezza inizia dagli arti inferiori e sale progressiva-
mente fi no ad interessare tutto il corpo.
La progressione della paresi è molto variabile, in casi gravi si può arrivare
alla plegia completa con incapacità respiratoria, per debolezza della mu-
scolatura diaframmatica o intercostale, diffi coltà a parlare e nella deglu-
tizione, a causa di una debolezza della muscolatura faringea. Le lesioni
sono simmetriche e l’atrofi a infrequente.
Può presentarsi paresi facciale bilaterale nel 50% dei casi; altri nervi cra-
nici che possono essere colpiti sono quelli che innervano la lingua e la
muscolatura deputata alla deglutizione, però non interessa gli oculo-
motori.
A livello sensitivo può presentarsi parestesia distale specialmente all’inizio
del quadro, però non esiste un defi cit della sensibilità marcato. È frequente
la presenza del dolore nella zona lombare o gli arti inferiori all’esordio della
malattia.
I sintomi autonomici includono tachicardia, ipotensione posturale, iperten-
sione e sintomi vasomotori. La Tabella 31 mostra i risultati che mettono in
dubbio o scartano la diagnosi
Risultati che mettono
in dubbio la diagnosi
∙ Debolezza asimmetrica in forma marcata e persistente
∙ Livello sensoriale non ben defi nito
∙ Disfunzione intestinale o vescicale all’esordio
∙ Disfunzione intestinale o vescicale persistente
∙ Pleocitosi mononucleare > a 50 cell/mm3
∙ Pleocitosi di polimorfonucleari
Risultati che scartano la diagnosi
∙ Sindrome sensitiva pura
∙ Storia recente di contatti con solventi (esacarboni)
∙ Metabolismo anormale delle porfi rine
∙ Infezione difterica recente
∙ Evidenza d’intossicazione per piombo
∙ Diagnosi defi nitiva di:
- Poliomielite
- Botulismo
- Neuropatia tossica (dapsone, organofosforadi)
Tabella 31. Sintomi e segni non compatibili con sindrome di Guillain-Barré
Decorso
È caratteristica la rapida progressione della debolezza, che raggiunge l’acme
in 4 settimane nel 90% dei casi. Il recupero di solito inizia dopo 2-4 settimane
e può durare mesi.
Prognosi
Anche se la maggior parte dei pazienti mostra un eccellente recupero fun-
zionale, esiste un 5% di mortalità e nel 50% restano alcune sequele.

5811 ∙ Neuropat i e
Manuale CTO di Medicina e Chirurgia, Prima edizione
Prove complementari
• LCR. È tipica la dissociazione albuminocitologica (proteine alte senza
cellule) che inizia dopo la prima settimana e si mantiene così per mesi,
anche a seguito della risoluzione del quadro clinico.
R I C O R D A Se il LCR presenta una pleocitosi importante, bisogna pensare ad una sindrome di Guillain-Barré associata ad infezione per HIV.
• Studi neurofi siologici. Nelle fasi inziali, le velocità di conduzione mo-
torie distali di solito sono normali ed è proporzionalmente maggiore
l’abolizione dell’onda F, che valuta la conduzione motoria prossimale,
essendo il primo segno diagnostico. Nell’80% dei pazienti si osserva un
rallentamento della velocità di conduzione e aumento delle latenze di-
stali (demielinizzazione). Non coinvolge tutti i nervi essendo la demieli-
nizzazione non uniforme.
R I C O R D A Nella miastenia gravis si utilizza l’elettromiografi a; nella sindrome di Guillan Barre l’elettroneurografi a.
Diagnosi
I criteri diagnostici sono elencati di seguito.
• Necessari:
- Debolezza progressiva in una o più estremità per neuropatia.
- Arefl essia.
- Durata della malattia inferiore a 4 settimane.
- Esclusioni di altre cause.
• Suggestivi::
- Debolezza simmetrica relativa.
- Minimo coinvolgimento sensitivo.
- Alterazione di un qualsiasi nervo cranico.
- Assenza di febbre.
- Evidenza elettrofi siologica di demielinizzazione.
Si devono scartare le entità elencate nella Tabella 32.
Diagnosi diff erenziale con…
∙ Paralisi ipopotassiemica
∙ Mielite acuta
∙ Botulismo
∙ Poliomielite
∙ Porfi rie
∙ Difterite
∙ Neuropatie tossiche (dapsone, tallio, nitrofurantoina)
∙ Neuroborrellia o malattia di Lyme
Tabella 32. Diagnosi diff erenziale della sindrome di Guillain-Barré
Trattamento
Consiste nel supporto della funzione cardiorespiratoria y prevenzione delle
infezioni intercorrenti.
Il trattamento con plasmaferesi o la somministrazione di immunoglobuli-
ne intravenose rappresentano i trattamenti d’elezione per quei pazienti che
hanno perso la capacità di deambulare autonomamente, la combinazione
di entrambi i trattamenti non sembra essere superiore alla somministrazione
di uno solo dei due.
11.3. Neuropatia diabetica
Il diabete mellito si può complicare con una vasta gamma di malattie del
sistema nervoso periferico che in generale si possono classifi care in 2 tipi:
polineuropatie simmetriche ed asimmetriche, anche se abitualmente i pa-
zienti presentano manifestazioni cliniche miste. La presenza di dolore è ca-
ratteristica nella maggior parte di esse
Polineuropatie simmetriche
È caratteristica la combinazione di degenerazione assonale (abitualmente
distale) e demielinizzazione segmentaria.
• Polineuropatia sensitiva distale. È la forma più frequente tra le poli-
neuropatie diabetiche.
R I C O R D A Le neuropatie simmetriche sono più frequenti in pazienti diabetici con un cattivo controllo metabolico.
Polineuropatie asimmetriche
Sono meno comuni, si manifestano frequentemente negli anziani e posso-
no apparire prima delle polineuropatie simmetriche durante il decorso della
malattia. La loro eziologia è normalmente vascolare.
• Neuropatie craniche. Possono essere la prima manifestazione di un
diabete. Il III nervo cranico è quello più frequentemente aff etto.
• Neuropatie da intrappolamento.
• Neuropati del tronco.
R I C O R D A La neuropatia diabetica del III nervo cranico preserva la motilità pupil-lare. Le forme compressive invece, a questo livello, non la risparmiano.
Trattamento
Il trattamento della neuropatia diabetica è poco effi cace. Include un buon con-
trollo metabolico e il trattamento sintomatico del dolore con analgesici abituali
e, se non c’è risposta, con carbamazepina, amitriptilina, fenitoina o clonazepam.
Le neuropatie da intrappolamento, come la sindrome del tunnel carpale,
possono richiedere decompressione chirurgica.
11.4. Neuropatie dell’infezione da HIV
La neuropatia periferica è molto frequente durante l’infezione da HIV. Può
apparire in tutte le fasi dell’infezione e in varie occasioni in forma subclinica.
Si può presentare sotto varie forme che sono riassunte nella Tabella 33.

59
11Neurologia e neurochirurgia
I d e e c h i ave Le polineuropatie interessano multipli tronchi nervosi, le mono-neuriti multiple tronchi nervosi non adiacenti e le mononeuropa-tie interessano focalmente un unico tronco nervoso.
Nella sindrome di Guillain-Barré, i due terzi dei pazienti pre-sentano una pregressa infezione respiratoria o gastrointesti-nale. È caratterizzata da una tetraparesi flaccida ed assenza di riflessi, simmetrica ed ascendente. Si può associare ad una pa-ralisi facciale bilaterale nella metà dei casi e presenta sintomi autonomici.
Nella sindrome di Guillain-Barré, è tipico un LCR con dissociazio-ne albuminocitologica. Il trattamento è il supporto della funzione cardiorespiratoria. Si possono anche utilizzare la plasmaferesi o le immunoglobuline endovenose. Gli steroidi non sono utili.
La neuropatia diabetica può essere simmetrica (sensitiva distale, autonomica, acuta dolorosa e amiotrofi ca diabetica) o asimmetri-ca (cranica, essendo il III nervo il più frequentemente interessato, per intrappolamento e del tronco).
La forma più frequente di polineuropatia diabetica è la sensitiva distale.
La causa più frequente di disautonomia è la polineuropatia diabetica.
Nelle infezioni dovute all’HIV possono comparire le seguenti neu-ropatie: simmetrica distale e mononeurite multipla (nelle fasi avanzate), polineuropatie demielinizzanti acute o croniche (nelle fasi precoci dell’infezione), o poliradiculite (probabilmente secon-dario a CMV).
La forma più frequente di neuropatia in pazienti con AIDS è la sim-metrica distale.
C a s i c l i n i c i Un paziente diabetico, di 69 anni, si presenta per comparsa brusca di dolore oculare destro e visione doppia. All’esame obiettivo si no-tano una ptosi destra e una paralisi di tutti i movimenti di questo occhio, ad eccezione dell’abduzione. Le pupille sono normali, cosi come l’acutezza visiva. La diagnosi più probabile è:
1) Aneurisma dell’arteria comunicante posteriore.2) Oftalmite fungina diabetica.3) Mononeuropatia diabetica del III nervo.4) Processo espansivo del seno cavernoso.5) Oftalmoplegia internucleare.
RC: 3
Un paziente di 28 anni si presenta con un quadro iniziato da 48 ore, di dolore lombare e parestesie della muscolatura delle gambe. All’esame obiettivo si riscontra paralisi degli arti inferiori e debo-lezza prossimale degli arti superiori. All’esame obiettivo sensorio e nervi cranici risultano normali. Rifl essi miotatici e risposte plantari assenti. Non vengono riferiti antecedenti di interesse, ad eccezione di una gastroenterite acuta 15 giorni fa. Segnali tra le seguenti il comportamento più importante nella gestione di questo paziente:
1) Vigilanza stretta della funzione respiratoria e ventilazione meccani-ca in caso di deterioramento.
2) Decompressione chirurgica immediata del midollo cervicale.3) Risonanza magnetica della colonna cervicale dal C3 verso il basso.4) Puntura lombare immediata per scartare iperproteinorachia.5) Trattamento con 1 mg/kg/giorno di prednisone, della durata di una
settimana.
RC: 1
Neuropatia simmetrica distale Mononeuriti multiple PolirradiculitisNeuropatie demielinizzanti
infiammatorie
∙ HIV
∙ Citomegalovirus
∙ ddI y ddC (antiretrovirali)
∙ Isoniazide
∙ Alcaloidi della vinca
∙ Defi cit di vitamina B12
∙ HIV
∙ Citomegalovirus
∙ Citomegalovirus
∙ HIV
∙ Varicella zoster
∙ Tuberculois
∙ Sifi lide
∙ Acute (tipo Guillain-Barré)
∙ Croniche
Tabella 33. Cause di alterazioni del nervo periferico nell’infezione da HIV secondo la manifestazione

606060
Neurologia e neurochirurgia
Le malattie della placca motrice includono fondamentalmente la miastenia
gravis (disordine post-sinaptico) (Figura 49), la sindrome miastenica di Lam-
bert-Eaton ed il botulismo (disordini pre-sinaptici) (Tabella 34).
Placca motrice
Assone
Muscolo
Normale Miastenia gravis
Rivestimentomielinico
Sito di liberazione
Recettori dell’Ach
Figura 49. (A) Giunzione neuromuscolare normale. (B) Miastenia gravis
12.1. Miastenia gravis
Si tratta di un disordine autoimmunitario che si manifesta con debolezza
e aff aticabilità della muscolatura scheletrica. Globalmente colpisce più fre-
12
quentemente le donne in qualsiasi fascia di età, con un picco d’ inci-
denza tra la 2ª e 3ª decade di vita, leggermente più tardi i maschi (4.ª-5.ª
decade).
E’ la malattia autoimmunitaria meglio caratterizzata. In un 85-90% dei casi
esistono anticorpi diretti contro i recettori nicotinici dell’Acetilcolina (Ach).
Per cui, anche se la quantità di Ach liberata è normale, non è effi cace nel
legarsi al recettore. Pare che il timo giochi un ruolo importante nella genesi
della risposta autoimmunitaria, dato che è colpito nel 75% dei pazienti (nel
65% è iperplastico e nel 10% si trova un timoma).
Clinica
Si manifesta con debolezza e aff aticabilità muscolare con distribuzione
tipica, senza alterazione delle altre funzioni neurologiche e con 3 caratteri-
stiche chiave:
• Carattere fl uttuante della debolezza, con peggioramento dopo l’eser-
cizio e miglioramento dopo il riposo o il sonno. I pazienti lamentano
maggiore debolezza durante il pomeriggio.
• Colpisce la muscolatura cranica, preferenzialmente la oculare estrin-
seca, con ptosi e diplopía. Può simulare un’oftalmoplegia internucleare.
Nella maggior parte dei pazienti (85%) la debolezza si generalizza alla
muscolatura degli arti , essendo di carattere prossimale e simmetrica,
con conservazione dei rifl essi miotatici e senza amiotrofi a. Non ci sono
alterazioni sensitive, autonomiche o pupillari.
• Risposta clínica ai farmaci colinergici (anticolinesterasici).
Forme cliniche
• Miastenia oculare: esiste unicamente debolezza della muscolatura
oculare.
• Miastenia generalizzata: colpisce tutta la muscolatura non limitando-
si a quella oculare.
• Crisi miastenica: la debolezza muscolare respiratoria produce insuf-
fi cienza respiratoria o la debolezza bulbare impedisce la deglutizione,
con necessità di inserire una sonda per l’alimentazione per il rischio di
aspirazione.
MALATTIE DELLA PLACCA MOTRICE
ORIENTAMENTOTema d’importanza intermedia nel MIR ma relativamente facile. E’ raccomandabile incentrarsi soprattutto sulla miastenia gravis e sapersi orientare nella diagnosi differenziale con la síndrome di Lambert-Eaton ed il botulismo.

61
12Neurologia e neurochirurgia
R I C O R D A Nella miastenia gravis, i rifl essi osteotendinei (ROT), le pupille ed il SNA sono intatti, a diff erenza del botulismo e della síndrome di Lambert-Eaton.
Diagnosi
Per stabilire la diagnosi defi nitiva, si utilizzano le seguenti prove complementari:
• Test del Tensilon® (edrofonio). L’ edrofonio è un fármaco che inibisce
l’acetilcolinesterasi, la cui somministrazione produce un miglioramento
immediato ma transitorio.
• Dimostrazione degli anticorpi antirecettori dell’acetilcolina.
Questi appaiono in un 85-90% dei pazienti con miastenia genera-
lizzata e nel 50% di quelli con miastenia oculare. La loro presenza è
diagnostica, però la loro assenza non esclude la diagnosi; non sono
patognomonici di miastenia gravis. La loro titolazione inoltre, non
corrisponde direttamente alla gravità della malattia, ma serve alla
monitorizzazione dell’evoluzione e della risposta al trattamento in
pazienti isolati.
• Studi neurofi siologici. Le velocità di conduzione nervosa sono norma-
li. L’ampiezza del potenziale d’azione prima di uno stimolo unico, è nor-
male. Inoltre, una stimolazione nervosa ripetuta produce una risposta
decrementale (Figura 50).
Figura 50. Risposta decrementale in paziente con miastenia gravis durante la stimolazione nervosa ripetitiva a 3 Hz
Miastenia gravis Sindrome Lambert-Eaton Botulismo
Patogenesi ∙ Ac-antirecettore del Ach-autoimmmunitaria 75% (post-sinaptico)
∙ Alt. Timiche : 65% iperplasia, 10% timoma
∙ Ac-anticanale del calcio (pre-sinaptico)
∙ Paraneoplastico (Ca. bronchiale)
∙ Blocco della liberazione Ach (pre-sinaptico)
∙ Tossina botulinica
∙ Cl. botulinum
Sesso ed età ∙ Prevalenza: Femmine
∙ Qualsiasi età: Picco 20-30 anni Picco 50-60 anni
Prevalenza: Maschi > 40 anni ∙ Sesso indiff erente
∙ Qualsiasi età
∙ Forma più frequente nei lattanti
Debolezza ∙ Muscolatura extraoculare
∙ Prossimale Arti inf. (85%)
∙ Asimmetrica
∙ Prossimale Arti inf.
∙ Extraoculare (70%)
∙ Bulbare (m. Extraoculare)
∙ Discendente Simmetrica
Refl essi miotatici Normali
Pupille Normali Risposta diminuita
Disautonomia No Si Si
Migliora Riposo, sonno
Anticolinesterasici (test al Tensilon®)
Esercizio Non migliora dopo esercizio
Peggiora ∙ Esercizio
∙ Stress
∙ Gravidanza
∙ Tubocurarina
∙ Esametonio
Primo potenziale (stimolo unico)
Normale
(aumento del jitter in singola fi bra)
Stimolazione ripetitiv 2-3 Hz
Stimolazione ripetitiva > 10 Hz
Trattamento ∙ Anticolinesterasici:
- Piridostigmina
- Neostigmina
∙ Corticosteroidi
∙ Timectomia
∙ Plasmaferesi
Trattamento del tumore subiacente ∙ Antitossina equina (non utile nella forma infantile)
∙ Supporto vitale
Diagnosi ∙ Test del Tensilon®
∙ Dosaggio di Ac antirecettore dell’ acetilcolina
∙ TC torace per ricerca timoma
∙ Ac contro il canale del calcio(prova più sensibile)
∙ TC torace per ricerca tumore
Tabella 34. Diagnosi diff erenziale delle sindromi miasteniche

6212 ∙ Ma la t t i e de l l a p lacca mot r i ce
Manuale CTO di Medicina e Chirurgia, Prima edizione
L’elettromiografi a a singola fi bra, mostra un incremento del jitter. Il jitter
rappresenta la variabilità dell’intervallo interpotenziale, ovvero, la varia-
bilità nella latenza tra due fi bre muscolari appartenenti alla medesima
unità motoria. Nella miastenia gravis, la stimolazione ripetuta ad alte
frequenze, incrementa il jitter (Figura 51), mentre, nella síndrome di
Lambert-Eaton e nel botulismo, il jitter si incrementa a basse frequenze
e diminuisce ad alte frequenze.
• Radiologia. Si deve eff ettuare una TC o RM toracica per scoprire even-
tuali alterazioni timiche (iperplasia o timoma).
Figura 51. Elettromiografi a a singola fi bra di un paziente con miastenia gravis che mostra un prolungamento del jitter o variabilità dell’intervallo interpotenziale
Si dovrebbe eff ettuare la diagnosi diff erenziale con la sindrome di Lambert-
Eaton ed il botulismo (si veda la Tabella 34 e Figura 52).
Paralisi bulbare discendente simmetrica
Ipotonia (diminuzione del tono muscolare)
Bambini
Adulti
Alimenti
Tossina
FeriteAlimenti
Figura 52. Fisiopatologia e clinica del botulismo
Trattamento
Lo schema di trattamento è riassunto nella Figura 53.
Si ricordi che la timectomia è indicata nei casi di timoma ed in tutte le forme
generalizzate in pazienti tra la pubertà e 55 anni. Nelle forme oculari pure
non è stata dimostrata l’effi cacia della timectomia.
Trattamento della miastenia gravis
Formageneralizzata
Crisimiastenica
Trattamento di supporto(ventilazione, liquidi)
Anticolinesterasici(piridostigmina)
Anticolinesterasici(piridostigmina)
Insufficiente
Indicazioni a timectomia: Timoma Forma generalizzata
Rischio chirurgicoMigliora
Non miglioraBasso Alto
Plasmaferesio immunoglobina i.v.
Timectomia
Immunosoppressione(prednisone azatioprinaciclosporina)
Forma oculareesclusiva
Valutazione dello statoclinico e se il paziente
ne ha bisogno
Plasmaferesio immunoglobuline ev
Figura 53. Trattamento delle forme distinte di miastenia gravis
R I C O R D A Gli anticolinesterasici in monoterapia si utilizzano solo nelle forme oculari pure; negli altri casi è necessario aggiungere altri approcci terapeutici.
I d e e c h i ave La miastenia gravis è dovuta al blocco dei recettori colinergici post-
sinaptici da parte di anticorpi diretti contro di essi ( anche se tali an-ticorpi non si trovano in tutti i pazienti). Nel 75% dei pazienti vi è associata un’alterazione del timo (iperplasia o timoma).
La miastenia gravis è caratterizzata da debolezza muscolare dei m. estrinseci degli occhi e prossimali degli arti, che migliora con il ripo-so. Non ci sono alterazioni sensitive, autonomiche, pupillari nè dei ROT.
La diagnosi di miastenia gravis si eff ettua con il test all’ Edrofonio e la determinazione degli anticorpi antirecettore dell’Ach (la prova più specifi ca per la diagnosi)
Il trattamento basilare della miastenia gravis è costituito dagli anti-colinesterasici. La timectomia si eff ettuerà nelle forme generalizzate in pazienti tra la pubertà ed i 55 anni.
La sindrome di Lambert-Eaton ed il botulismo sono disordini presi-naptici che si manifestano con ROT e risposta pupillare diminuiti e con disautonomia, a diff erenza della miastenia gravis.

63
12Neurologia e neurochirurgia
C a s i c l i n i c i Una paziente di 22 anni presenta, da una settimana prima, ptosi pal-pebrale sinistra, senza dolore, con diplopia dello sguardo laterale sinistro. All’esplorazione fi sica si obiettiva l’esistenza di ptosi sini-stra, ed una paresi dell’abduzione dell’occhio sinistro, con pupille isocoriche e normoreagenti alla luce. Che malattia è più probabile che possa aver colpito la paziente?
1) Una neurite ottica sinistra in relazione a sclerosi multipla.2) Una sindrome di Horner.3) Una miastenia gravis.4) Una paralisi del III paio sinistro .5) Una miopatia ipertiroidea con coinvolgimento della muscolatura
extraoculare.
RC: 3
La miastenia gravis è caratterizzata da:
1) Decremento dell’ attivita elettrica presinaptica.2) Blocco dei recettori colinergici nicotinici.
3) Diminuzione della sintesi di acetilcolina.4) Presenza di anticorpi contro i recettori colinergici.5) Migrazione dei recettori fuori dalla fessura sinaptica.
RC: 4
Uomo di 55 anni, aff etto da circa 3 mesi, da debolezza muscolare a livello prossimale delle estremità, secchezza della bocca, dolori muscolari e parestesie ad i 4 arti. Durante l’esplorazione física, si ri-scontra debolezza dei muscoli prossimali. La sensibilità è conserva-ta ed i rifl essi osteotendinei sono ridotti a livello degli arti superiori ed aboliti negli inferiori. Che prove complementari, tra i seguenti, ci aiuterà nello stabilire la diagnosi?
1) Studio del LCR.2) Biopsia del nervo aff etto.3) Biopsia del muscolo aff etto.4) Rx torace.5) RM del midollo spinale.
RC: 4

646464
Neurologia e neurochirurgia
ORIENTAMENTO
Tema totalmente prescindibile per il MIR, poiché negli ultimi anni ci sono state solo due domande. E’ consigliabile ricordare il tipico paziente con distrofia miotonica di Steinert.
13.1. Distrofi e muscolari
I tipi di distrofi a muscolare sono elencati nella Tabella 35.
Tipi di distrofi a muscolare
∙ Distrofi nopate:
- Distrofi a muscolare di Duchenne
- Distrofi a muscolare di Becker
∙ Distrofi a miotonica di Steinert
Tabella 35. Distrofi e muscolari
Distrofi nopatie
Sono malattie recessive legate al cromosoma X, originano dall’alterazione
della distrofi na (proteina codifi cata sul Xp21, necessaria per la contrazione
muscolare). Colpiscono quasi esclusivamente i maschi, poichè le femmine
sono portatrici-trasmittrici.
• Distrofi a muscolare di Duchenne (DMD). La clinica ha esordio intor-
no ai 3-5 anni, con disturbi della marcia e debolezza progressiva della
muscolatura prossimale degli arti e fl essoria del collo, essendo gli arti
inferiori più gravemente colpiti rispetto ai superiori. E’ caratteristica la
pseudoipertrofi a dei polpacci, dovuta alla sostituzione del tessuto mu-
scolare con grasso e tessuto connettivo. Se il paziente prova ad alzarsi
da terra, attua la manovra di Gowers (si arrampica su se stesso) (Figura
54).
Frequentemente si associa a deterioramento intellettivo non progressi-
vo e scoliosi progressiva. Dai 12 anni, la maggior parte degli individui af-
fetti, utilizza la sedia a rotelle. I pazienti muoiono nella seconda decade
di vita a causa di infezioni polmonari intercorrenti. Sono poco comuni
le cause di morte di natura cardiaca, nonostante la miocardiopatia sia
presente in quasi tutti i casi.
13
Figura 54. Manovra di Gowers
C’è un aumento importante, fi n dalla nascita, della CPK sierica e la sua
determinazione è fondamentale per il riconoscimento delle portatrici (il
50% di loro hanno una CPK aumentata).
L’ elettromiografi a (EMG) evidenzia caratteristiche miopatiche con atti-
vità spontanea e potenziali polifasici brevi e di bassa ampiezza.
La biopsia muscolare stabilisce la diagnosi defi nitiva; essa mostra una
totale assenza di distrofi na (Figura 55). Il trattamento con prednisone
può ritardare il corso della malattia.
Figura 55. Patrón di immunocolorazione normale con anticorpo antidistrofi na (sinistra)
• Distrofi a muscolare di Becker. E’ una variante allelica della DMD, ad
inizio più tardivo, evoluzione più benigna e frequenza minore.
La distribuzione del coinvolgimento muscolare è la stessa della DMD,
trovando anche in questo caso un riscontro precoce e prominente di
pseudoipertrofi a muscolare, in particolare gemellare.
MIOPATIE

65
13Neurologia e neurochirurgia
L’età del debutto della malattia è tra i 5-15 anni con deambulazione con-
servata anche dopo i 15 anni. L’aspettativa di vita si attesta tra la 4ª e 5ª
decade ed è meno frequente l’associazione con il ritardo mentale.
La CPK e l’EMG sono analoghi a quelli della DMD. La biopsia mostra una
distrofi na in scarsa quantità e di minor grandezza. Non si conosce anco-
ra adeguatamente il risultato del trattamento con prednisone.
Distrofi a miotonica di Steinert
E’ una malattia con eredità autosomica dominante, trasmessa attraverso un
gene anomalo localizzato nel braccio lungo del cromosoma 19 che codi-
fi ca la miotonina protein-chinasi. Il difetto genetico è la ripetizione di un
trinucleotide (CTG) in questo cromosoma, per il quale si produce il fenome-
no dell’anticipazione.
La mutazione è instabile, ciò spiega l’ampio range di gravità clinica della di-
strofi a; alcuni pazienti sono gravemente colpiti fi n dalla nascita, soprattutto
quando discendono da madre aff etta; altri sono minimamente colpiti dalla
vita adulta.
R I C O R D A Altre malattie che presentano il fenomeno dell’anticipazione sono la Corea di Huntington (tripletta CAG) e la sindrome dell’X fragile. (tripletta CGG)
Clinica
La malattia ha il suo debutto nella 2ª o 3ª decade, con debolezza della mu-
scolatura facciale, fl essoria del collo e degli arti, e atrofi a della muscolatura
facciale, massetere e muscolo temporale. Il coinvolgimento della lingua, fa-
ringe e palato molle, porta a voce nasale e disfagia.
E’ caratteristico il fenomeno miotonico a livello delle mani, palpebre e lingua,
con diffi coltà al rilassamento muscolare che tipicamente migliora con l’eser-
cizio ripetuto e peggiora con il freddo.
Si associa a deterioramento intellettivo, ipersonnia, calvizie frontale, cataratta
subcapsulare, atrofi a gonadica, insuffi cienza respiratoria per debolezza della
muscolatura respiratoria, resistenza all’insulina, alterazioni gastrointestinali e
cardiopatia, con alterazione del sistema di conduzione (blocco AV) (Figura 56).
Prove complementarie
La CPK può essere normale o discretamente aumentata e l’EMG mostra sca-
riche miotoniche (Figura 57).
La clinica peggioracon il freddo
Calvizie frontale
Catarattesubcapsulari
Atrofiamuscolare facciale
Blocco AV
Resistenzaall’insulina
Fenomenomiotonico
Alterazionigastrointestinali Debolezza muscolare
distale degli arti
Figura 56. Distrofi a miotónica de Steinert
Figura 57. Scarica miotonica
La biopsia mostra atrofi a muscolare, preferenzialmente delle fi bre di tipo I,
ma, a diff erenza delle altre distrofi e muscolari, non esiste necorsi delle fi bre
stesse.
Trattamento
Il trattamento di elezione della miotonia è la fenitoina. I disturbi della condu-
zione possono richiedere il l’impianto di pacemaker.
I d e e c h i ave La distrofi a muscolare di Duchenne ha una eredità recessiva, legata al cromosoma X. E’ caratteristica la pseudoipertrofi a dei polpacci e la manovra di Gowers (il paziente si arrampica su se stesso per alzarsi da terra)
La distrofi a miotonica di Steinert ha una eredità autosomica dominante. E’ caratteristico il blocco AV, la calvizie frontale, le cataratte subcapsulari, la resistenza all’insulina ed il fenomeno miotonico a livello delle mani, lingua e palpebre (diffi coltà al ri-lassamento muscolare).

6613 ∙ M iopat i e
Manuale CTO di Medicina e Chirurgia, Prima edizione
C a s i c l i n i c i In una visita routinaria di un paziente di 34 anni di età, si riscontra una glicemia di 160 mg/dL, una CPK di 429 U/l, GTP 62 U/l, GOT 43 U/l e GGT 32 U/l. All’elettrocardiogramma é presente un blocco AV di primo grado. All’esame obiettivo, si apprezzano opacità corneali incipienti e diffi coltà a rilassare un muscolo dopo contrazione intensa, in maniera evidente a livello della mano. Da quale malattia è aff etto il paziente?
1) Una miopatia mitocondriale.2) Una distrofi a muscolare dei cingoli.3) Una distrofi a muscolare di Duchenne.4) Una distrofi a muscolare di Steinert.5) Una distrofi a muscolare di Becker.
RC: 4

67
Neurologia e neurochirurgia
14.1. Considerazioni generali
Il primo obiettivo di fronte ad un paziente con cefalea, è scartare patologie
gravi sottostanti ma trattabili, per cui bisogna riconoscere i criteri di gravità di
una cefalea, che sono raccolti nella Tabella 36.
Criteri di gravità di una cefalea
∙ Cefalea intensa con inizio improvviso
∙ Peggioramento recente di una cefalea cronica
∙ Cefalea di frequenza e/o intensità crescente
∙ Localizzazione unilaterale, sempre dallo stesso lato (eccetto cefalea a grappolo, emicrania parossistica o continua, nevralgia occipitale, del trigemino ed altre cefalee primarie unilaterali)
∙ Manifestazioni concomitanti:
- Alterazione psichica progressiva
- Crisi epilettica
- Alterazioni neurologiche focali
- Papilledema
- Febbre
- Nausea e vomito non spiegabili con una cefalea primaria o con una patologia sistemica
- Presenza di segni meningei
∙ Cefalea precipitata da sforzo fi sico, tosse o cambio di postura
Tabella 36. Manifestazioni d’allarme in una cefalea
Patologie gravi che si manifestano con una cefalea sono:
• Emorragia subaracnoidea (cefalea intensa con inizio improvviso).
• Meningiti (febbre, rigidità nucale…).
• Tumori (cefalea progressiva, di maggiore intensità notturna, con sintomi
neurologici “a focolaio e/o di ipertensione endocranica).
• Arterite temporale (pazienti anziani, con claudicatio mandibolare, au-
mento della VES…).
14
14.2. Cefalea tensiva
E’ il tipo di cefalea più frequente ed è più frequente nelle donne. Si distin-
guono tre forme di cefalea tensiva: episodica sporadica, episodica frequente
e cronica.
Come criteri diagnostici, spiccano episodi di cefalea che durano tra i 30 mi-
nuti ed i 7 giorni, di tipo gravativo, intensità lieve o moderata, localizzazione
bilaterale, non aggravata da sforzi fi sici o non associata a nausea o vomito.
Il trattamento del dolore si realizza con FANS, paracetamolo o analgesici co-
muni. Il trattamento preventivo, a seconda della frequenza, durata ed inten-
sità del dolore, si basa sull’uso di antidepressivi triciclici ed inibitori selettivi
della ricaptazione della serotonina.
14.3. Emicrania
La maggior parte dei pazienti presenta il primo episodio di emicrania tra i 10
ed i 30 anni e nel 60-75% dei casi sono di sesso femminile. Esiste una pre-
disposizione ereditaria; la trasmissione del tratto genetico è probabilmente
autosomica recessiva a penetranza incompleta.
Fisiopatologia
La patogenesi certa dell’emicrania non è nota. Esistono diff erenti ipotesi e
vengono proposte tre fasi o meccanismi:
1. Genesi troncoencefalica con possibile partecipazione dei nuclei del rafe
mediano, soprattutto Nucleo Dorsale del Rafe del mesencefalo, a pro-
iezione ascendente, serotoninergici. Alla base dell’attacco emicranico
vi sarebbe una instabilità della neuromodulazione centrale del dolore.
2. Attivazione vasomotoria con vasocostrizione iniziale, che giustifi che-
rebbe la focalità neurologica dell’emicrania con “aura”, seguita da una
seconda fase di vasodilatazione extracranica responsabile del dolore; è
CEFALEE
ORIENTAMENTOTema d’importanza intermedio-bassa nel MIR, ma che si è presentato costantemente nelle ultime prove. E’ consigliabile incentrarsi sull’emicrania, specialmente su trattamento e profilassi e sulla cefalea a grappolo. Il resto è prescindibile.

6814 ∙ Cefa lee
Manuale CTO di Medicina e Chirurgia, Prima edizione
la classica ipotesi “vasomotoria”, con ”instabilità vascolare”, che si tende
però oggi a considerare epifenomeno e non causa.
3. Attivazione dei neuroni del nucleo trigeminale a livello bulbare, con
successiva liberazione di neuropeptidi vasoattivi nelle terminazioni va-
scolari. Questa fase porta a rigonfi amento tissutale così come l’aumento
di pressione nei vasi sanguigni durante l’episodio di emicrania.
Sottotipi clinici
La Tabella 37 raccoglie le caratteristiche ed i criteri diagnostici di questa
patologia.
Emicrania senza aura
Almeno 5 episodi, obbediendo ad i seguenti criteri:
∙ Durata dell’episodio di 4-72 ore (senza trattamento o trattata senza risultato)
∙ Almeno 2 dei seguenti dati:
- Unilaterale (30-40% sono bilaterali)
- Pulsante (50% dei casi non sono pulsanti)
- Moderata-grave (interferisce con la vita quotidiana)
- Aggravata dal movimento (camminare o salire le scale)
∙ Almeno un sintomo associato:
- Nausea o vomito
- Fotofobia
- Fonofobia
∙ Il dolore non può essere attribuito ad altre patologie
Emicraniacon aura
Ai criteri descritti anteriormente si aggiungono i seguenti:
∙ Uno o più sintomi neurologici focali transitori (90% visivi) prima o durante la cefalea
∙ Durata dell’aura tra 5 e 60 minuti
∙ La cefalea accompagna o segue l’aura entro i 60 minuti
∙ Il dolore non si associa ad altre patologie
Tabella 37. Criteri diagnostici semplifi cati per l’emicrania con e senza aura
Tipo di trattamento
Farmaci Commenti
Attacchi lievi-moderati
FANS (ASA, naprossene o ibuprofene) ∙ Si devono somministrare immediatamente dopo l’inizio della cefalea, ripetendo la dose ogni 4-6 ore
∙ La somministrazione di metoclopramide o domperidone non solo migliora la nausea ed il vomito ma facilita l’assorbimento degli analgesici. Oltre al trattamento analgesico c’è bisogno di riposo, se possibile, in un luogo oscuro e silenzioso.
Atacchi moderato-gravi
Triptani (sumatriptan, nartriptan, zolmitriptan, rizatriptan, almotriptan, eletriptan e frovatriptan)
∙ Sono agonisti dei recettori serotoninergici (5-HT1B e 1D) con azione di vasocostrizione e di riduzione dell’infi ammazione intorno ai vasi
∙ Controindicazioni: cardiopatia ischemica o claudicatio intermittens
Preventivo (se la frequenza è superior a 2 episodi al mese)
-bloccanti (propranololo) Il meccanismo attraverso il quale esercitano la loro azione profi lattica è sconosciuto, anche se si suppone un eff etto bloccante sui recettori serotoninergici 5-HT2
Calcioantagonisti (fl unarizina, cinarizina, verapamil)
I primi due si devono utilizzare con precauzione in pazienti con morbo di Parkinson, sindrome depressiva pregressa, o disturbi extrapiramidali di altro tipo
Antidepressivi triciclici (amtriptillina, nortriptillina)
Specialmente indicati in pazienti con emicrania associata a cefalea tensiva. La loro azione sembra essere indipendente dalla loro attività antidepressiva.
Antagonisti della serotonina (ciproeptadina, pizotifene, metisergide)
Molto effi caci; devono essere somministrati con precauzione a causa dei loro eff etti collaterali importanti, comunque reversibili, a seguito di un uso prolungato: fi brosi pleurica, pericardica e retroperitoneale
Neuromodulatori (valproato, topiramato, lamotrigina)
Si utilizzano principalmente nelle emicranie con aura e nell’aura emicranica senza cefalea
Tossina botulinica E’ indicata unicamente nell’emicrania cronica refrattaria
Tabella 38. Principali opzioni di trattamento del’emicrania
• Emicrania con aura o emicrania classica. Rappresenta il 20% delle
emicranie. E’ una cefalea ricorrente, di predominio emicraniale e caratte-
re pulsante, che può accompagnarsi a nausea, vomito, fotofobia e fono-
fobia, che dura tra le 4 e le 72h. Viene preceduta da una clinica di focalità
neurologica (aura), in cui le manifestazioni visive sono le più frequenti
(scotomi scintillanti, visione off uscata, difetti emianopsici, spettro di for-
tifi cazione...) anche se si possono avere anche sintomi motori o sensitivi.
Precedono la cefalea di 15-30 minuti ed abitualmente scompaiono po-
chi minuti prima dell’inizio della cefalea.
• Emicrania senza aura o emicrania comune. Rappresenta il 75% dei
casi di emicrania. Consiste in cefalee di caratteristiche analoghe a quelle
descritte nell’emicrania con aura ma senza clinica di focalità neurologica
che precedono o accompagnano la cefalea.
Entrambi i tipi di emicrania possono colpire lo stesso paziente. Tra gli episodi
di emicrania il paziente può presentare cefalea tensiva.
Le crisi possono essere scatenate da diversi fattori dietetici, ambientali, psi-
cologici, ormonali o farmacologici.
Complicanze
• Emicrania cronica: quando si presentano più di 15 episodi al mese, per
un periodo superiore ai 3 mesi.
• Stato di male emicranico: più di 72 ore di durata nonostante il tratta-
mento.
• Infarto emicranico o emicrania complicata: quando i sintomi dell’aura
persistono oltre la durata della cefalea e si associano ad una lesione ische-
mica cerebrale dello stesso territorio vascolare, dimostrata attraverso le
neuroimmagini.
Trattamento
Le principali opzioni terapeutiche delle emicranie sono riassunte nella Ta-
bella 38.

69
14Neurologia e neurochirurgia
14.4. Cefalea a grappolo (cluster headache), istaminica o di Horton
Più frequente nei maschi (10:1), può esordire a qualunque età, anche se pre-
ferenzialmente tra i 20 e i 50 anni. Si distingue una forma episodica da un’al-
tra cronica (quando c’è assenza di fasi di remissione per un anno o più o con
remissioni che durano meno di un mese).
E’ caratterizzata da episodi giornalieri di cefalea unilaterale, localizzata
maggiormente a livello perioculare, con irradiazione alla fronte o alla man-
dibola, di alta intensità, la cui durata può variare tra i 15-180 minuti, da
una fi no a 8 volte al giorno. Appare caratteristicamente durante la notte,
approssimativamente un’ora dopo l’inizio del sonno, e può ripresentarsi
durante il giorno, spesso alla stessa ora. In molti casi si accompagna a la-
crimazione, rinorrea, congestione oculare e ostruzione nasale ipsilaterale
al dolore, sudorazione frontale e facciale, edema palpebrale ipsilaterale,
miosi-ptosi ipsilaterale e senso d’ inquietudine motoria. Nel 25% dei casi si
accompagna ad una sindrome di Horner che può occasionalmente persi-
stere.
La cefalea appare quotidianamente in periodi (cluster) che oscillano tra 1-4
mesi, rimanendo poi asintomatica per periodi piuttosto lunghi (1-2 anni).
Non si accompagna ad aura, nausea o storia familiare.
• Trattamento preventivo. Evitare fattori scatenanti, se esistono, come
alcool o altri vasodilatatori.
• Trattamento sintomatico. La terapia d’elezione è il sumatriptan subcu-
taneo, per la sua rapidità ed effi cacia. La seconda linea di trattamento è
l’inalazione di ossigeno ad alto fl usso.
• Trattamento profi lattico. Si considera il verapamil come farmaco d’e-
lezione. Se non c’è risposta, si può provare un ciclo breve di corticoste-
roidi, topiramato, ergotamina in dose unica notturna o il litio.
E’ necessaria la diagnosi diff erenziale con la emicrania cronica parossistica:
più frequente nelle donne, con inizio nell’età adulta. Si tratta di una cefalea
trigemino-autonomica, con episodi di dolore simili alla cefalea a grappolo,
ma di minor durata (2-30 min), e con frequenza maggiore (5-30 episodi al
giorno). La buona risposta all’indometacina è un criterio diagnostico.
I d e e c h i ave Il trattamento degli attacchi di emicrania lieve-moderata è costitu-ito dai FANS. Nel caso di attacchi moderato-gravi si usano i triptani. L’ergotamina è un’alternativa, ma il suo utilizzo cronico può produr-re una cefalea ergotamina-dipendente.
Per la prevenzione degli attacchi di emicrania si utilizzano i -bloccanti, calcioantagonisti, antidepressivi triciclici, antiepilettici e antagonisti della serotonina. Ricorda che la metisirgide (antago-nista della serotonina) può provocare fi brosi pleurica, pericardica e retroperitoneale. L’alcool può scatenare la cefalea a grappolo.
La cefalea a grappolo si presenta nei maschi tra 20-50 anni. Si ma-nifesta con episodi di cefalea unilaterale perioculare, tipicamente notturna. Può causare lacrimazione, rinorrea, senso d’inquietudine e tra tutti, la sindrome di Horner.
Il trattamento d’elezione della cefalea a grappolo è il sumatriptan subcutaneo. La seconda linea è l’ossigeno ad alto fl usso. Come pro-fi lassi, si usa il verapamil.
L’arterite temporale è una causa di cefalea che va sospettata di fron-te a pazienti di più di 50 anni, con emicrania di recente insorgenza, associata a claudicatio mandibolare ed un’arteria temporale senza polso.
C a s i c l i n i c i Donna di 34 anni, con diagnosi di emicrania senz’aura, giunge in vi-sita per i suoi abituali episodi di cefalea in numero di 4-5 al mese. Quale trattamento NON sarebbe indicato?
1) Prendere triptani durante tutti gli attacchi.2) Utilizzare dosi basse giornaliere di ergotamina .3) Somministrare come profi lassi propranololo.4) Trattare tutti gli attacchi acuti con naprossene.5) Utilizzare come profi lssi fl unarizina.
RC: 2
Un paziente di 54 anni riferisce, da circa 10 giorni, una o due crisi di dolore a livello dell’occhio destro, con lacrimazione, nervosismo, che lo sveglia durante la notte e lo obbliga ad alzarsi dal letto, con durata di circa 2 ore. Quale dei seguenti trattamenti pensi che sia più effi cace per calmare il dolore?
1) Ossigeno intranasale.2) Sumatriptan subcutaneo.3) Ibuprofene orale.4) Tramadolo orale.5) Metamizolo intramuscolare.
RC: 2

707070
Neurologia e neurochirurgia
15.1. Fisiopatologia
L’ ipotesi di Monro-Kellie stabilisce che il volume totale del contenuto endo-
cranico (parenchima, sangue e liquido cefalorachidiano) deve essere costan-
te. Premesso che questi si trovano all’interno di una cavità non estensibile,
come il cranio, un incremento del volume di uno di questi componenti (ad
es. Un tumore cerebrale, un ematoma epidurale, subdurale o un’idrocefalia)
indurrà, in maniera compensatoria, una diminuzione del volume delle altre
componenti. Se i meccanismi di compenso si saturano, si produce un au-
mento della pressione endocranica (i cui valori normali nell’adulto oscillano
tra i 5 e i 15mmHg).
15.2. Eziologia
Le cause più frequenti di aumento della Pressione Intracranica (PIC) sono
raccolti nella Tabella 39.
15.3. Clinica
La clinica caratteristica della sindrome da ipertensione endocranica si mani-
festa con:
• Cefalea. Più grave durante la notte. Può svegliare il paziente e peggiora
di mattina o con la manovra di Valsalva.
• Vomito. Di predominio mattutino, tipicamente a “getto”.
• Edema della papilla. E’ il segno obiettivo che denota l’esistenza d’iper-
tensione endocranica.
• Appare inoltre frequentemente diplopia, in generale secondaria a le-
sione del VI paio di nervi cranici.
• Alterazione del livello di coscienza.
15
Eziologia
Traumatismo cranioencefalico
∙ Ematoma epidurale
∙ Ematoma subdurale
∙ Contusione emorragica
∙ Swelling
Idrocefalia
Tumori
Infezioni ∙ Ascesso cerebrale
∙ Empiema subdurale
Processi vascolari ∙ Infarto cerebrale
∙ Trombosi venosa
∙ Ematoma intraparenchimale
Encefalopatie che possono manifestarsi con edema cerebrale
∙ Ipercapnica
∙ Epatica
∙ Sindrome da disequilibrio (dialisi)
Tabella 39. Cause più frequenti di aumento della pressione intracranica
Nelle fasi d’ipertensione endocranica moderata o avanzata, si può osservare
la triade di Cushing: ipertensione arteriosa (il segno più costante), bradicar-
dia e alterazioni del ritmo respiratorio, anche se solo nel 30% dei pazienti si
osserva la triade completa.
15.4. Sindromi da erniazione cerebrale (Figura 58)
• Erniazione uncale. Più frequente nelle lesioni temporali. L’uncus del
lobo temporale ernia attraverso la fessura tentoriale e può comprimere
il III paio dei nervi cranici, dando midriasi ipsilaterale come segno più
precoce, ed il mesencefalo, con emiplegia controlaterale e diminuzione
progressiva del livello di coscienza.
SINDROME DA IPERTENSIONE ENDOCRANICA
ORIENTAMENTOAll’interno di questo tema si deve conoscere molto bene lo pseudotumor cerebri, specialmente i criteri diagnostici e le condizioni associate a questa patologia. Inoltre, è importante conoscere la clinica dell’erniazione uncale e il trattamento generale dell’ipertensione endocranica.

71
15Neurologia e neurochirurgia
• Erniazione subfalcale. Dislocazione del parenchima cerebrale al di
sotto della falce cerabrale. Può comprimere l’arteria cerebrale anteriore.
Può essere un segno precoce di erniazione transtentoriale.
• Erniazione centrale, transtentoriale o tentoriale. Si produce una
dislocazione verso il basso degli emisferi cerebrali e dei gangli basali,
comprimendo successivamente: diencefalo, mesencefalo, protuberan-
za e bulbo.
• Erniazione transtentoriale inversa. Strutture della fossa posteriore er-
niano verso l’alto attraverso la fessura tentoriale.
R I C O R D A Prima di eff ettuare una puntura lombare, bisogna accertare , me-diante una TC cerebrale, che il paziente non abbia nessuna causa che provochi un aumento della pressione intracranica, che generi un gradiente tra il cranio e la regione lombare.
• Erniazione cerebello-amigdalare. Le amigdale cerebellari erniano
attraverso il forame magno, dando compressione bulbare e portando
rapidamente a morte.
Erniazioneuncale
Erniazione subfalciale
Erniazioneamigdalare
Figura 58. Erniazioni del sistema nervoso centrale
15.5. Diagnosi
La diagnosi di certezza si stabilisce mediante la misurazione della Pressione
Intracranica. La esecuzione di esami di neuroimaging sono utili per la dia-
gnosi eziologica.
15.6. Trattamento
Bisogna trattare il problema primario responsabile dell’ipertensione endo-
cranica, laddove fosse possibile.
In quanto al trattamento generale dell’ipertensione endocranica, indipen-
dentemente da quale sia la causa che l’ha provocata, sono utili i seguenti
rimedi (Tabella 40).
Misure di primo livello
∙ Elevazione della testa a 30º
∙ Sedazione e rilassamento
∙ Drenaggio ventricolare esterno
∙ Mannitolo al 20%
∙ Soluzione ipertonica
∙ Iperventilazione
Misure di secondo livello
∙ Craniotomía decompressiva
∙ Coma barbiturico
∙ Ipotermia
Tabella 40. Trattamiento generale dell’ipertensione intracranica
15.7. Sindrome dell’ipertensione intracranica benigna (pseudotumor cerebri)
Si defi nisce con la presenza di clinica d’ipertensione endocranica (la ce-
falea è il sintomo più frequente ed il papilledema è il segno più costan-
te), senza diminuzione del livello di coscienza e senza focalità neurologica
(salvo la diplopia per interessamento del VI paio). Negli esami di neuroi-
maging che si eff ettuano (RM e angio-RM in fase venosa) non si evidenzia
una causa giustifi cabile del quadro. La puntura lombare presenta, in forma
invariabile, un incremeto di pressione del LCR con studio analitico all’inter-
no del range di normalità, eccetto occasionalmente per una diminuzione
delle proteine.
Tra le varie ipotesi, in associazione all’ ipertensione intracranica benigna,
sono stati confermati solo: sesso femminile, età riproduttiva, disordini me-
struali, obesità o aumento recente di peso.
Si tratta di una malattia generalmente autolimitantesi ma ricorrente, il cui
rischio principale, è la perdita della vista per edema della papilla. Il tratta-
mento ha come obiettivo la prevenzione dei defi cit visivi ed il controllo della
cefalea. Lo schema terapeutico viene indicato nella Figura 59.
Figura 59. Gestione terapeutica generale dello pseudotumor cerebri

7215 ∙ S ind rome da i pe r tens ione endocran ica
Manuale CTO di Medicina e Chirurgia, Prima edizione
I d e e c h i ave I fattori che infl uiscono sulla pressione intracranica sono il sangue, il parenchima e il liquido cefalorachidiano. L’ ipotesi di Monro-Kellie stabilisce che la somma dei volumi tra sangue, liquido cefalorachi-diano e parenchima deve rimanere costante.
L’iperventilazione , che implica ipocapnia, provoca una vasocostri-zione, che porta ad una diminuzione della pressione intracranica. Senza dubbio può produrre anche ischemia cerebrale.
L’ipertensione intracranica ,superiore a 15 mmHg nell’adulto, può pro-dursi per aumento del volume del parenchima, del sangue o del LCR.
Le erniazioni cerebrali sono dislocazioni del cervello da un sito a mag-gior ad uno a minore pressione. L’erniazione uncale, che clinicamente si manifesta con una triade consistente in midriasi areattiva in un oc-chio, emiparesia controlaterale alla midriasi e diminuzione del livello di coscienza, costituisce un’emergenza neurochirurgica. L’erniazione cerebello-amigdalare può essere provocata da una puntura lombare.
Nel trattamento dell’ipertensione intracranica, bisogna iniziare con rimedi di primo livello ( che cercano fondamentalmente di diminui-re il volume di sangue, parenchima o LCR) e, nel caso di insuffi cien-za di questi, rimedi di secondo livello che implicano un alto tasso di complicanze.
Esiste una serie dei fattori assocati allo pseudotumor cerebri, tra cui spiccano i farmaci e determinate metabolopatie. Ricorda che il sesso femminile, l’obesità, l’età fertile, ed i disturbi mestruali sono quelli che hanno conferma epidemiologica..
I criteri diagnostici dello pseudotumor cerebri sono 6: clinica d’iper-tensione endocranica, assenza di focalità neurologica, livello di co-scienza normale, studi di neuroimaging normali, pressione del LCR elevata ed analisi del liquor normali.
La gestione terapeutica fondamentale nello pseudotumor cerebri è: eliminare i fattori di rischio, acetazolamide, derivazione lombo-peritoneale e decompressione del nervo ottico.
C a s i c l i n i c i Una donna di 34 anni, obesa, presenta da varie settimane, cefalea ed episodi transitori di perdita della visione binoculare, in particola-re ad alzarsi dal letto. All’esame obiettivo, ha come unico segno un papilledema bilaterale. Una risonanza magnetica cranica ed un’an-gioRM cerebrale risultano normali. Che ulteriore prova richiedereb-be?
1) Doppler dei tronchi suvraaortici.2) EEG e studio del sonno.3) Puntura lombare e misura della pressione del LCR.4) Inizierebbe trattamento antiaggregante prima di realizzare altre
prove.5) Potenziali evocati visivi.
RC: 3
Donna di 24 anni che, negli ultimi 2 mesi, presenta episodi mat-tutini di cefalea accompagnata da nausea e visione off uscata; nell’ultimo episodio ha presentato anche diplopia. All’esame obiettivo si riesce a notare solo papilledema bilaterale e obesità. La risonanza magnetica cerebrale è normale, ad eccezione di un aumento di pressione. Quale dei seguenti interventi terapeutici NON è indicata nel corso della malattia di questa paziente?
1) Punture lombari ripetute.2) Acetazolamide.3) Derivazione lomboperitoneale del LCR.4) Steroidi.5) Indometacina.
RC: 5

73
Neurologia e neurochirurgia
16.1. Concetto e classifi cazione
Il liquido cefalorachidiano (LCR) viene prodotto nei plessi coroidei, fonda-
mentalmente a livello dei ventricoli laterali e quarto ventricolo, in quantità di
circa 500 ml al giorno.
Dai ventricoli laterali, raggiunge il terzo ventricolo attraverso i forami di Mon-
ro, e attraverso l’acquedotto di Silvio arriva al quarto ventricolo nella fossa
posteriore, per riemergere a livello delle cisterne dello spazio subaracnoideo
attraverso i forami del Luschka e Magendie.
Successivamente circola nello spazio subaracnoideo e, secondo le teorie più
recenti, sembra che venga riassorbito a livello delle granulazioni aracnoidee
nella convessità durale (Figura 60).
Cisternamagna
Aracnoidi
Duramadre Senolongitudinale
superiore
Plesso corioideodel ventricolo laterale
Spaziosubaracnoideo
Granulomiaracnoidei
Plesso corioideodel terzo ventricolo
Forodel Luschka
Plesso corioideo del quarto ventricolo
Foro di Magendie
Figura 60. Circolazione del liquido cefalorachidiano
16
Classicamente, si distinguono due tipi di idrocefalia:
• Idrocefalia non comunicante o ostruttiva. Esistenza di un ostacolo a
livello del sistema ventricolare.
• Idrocefalia comunicante o non ostruttiva. Il LCR raggiunge lo spazio
subaracnoideo, ma a questo livello trova diffi coltà per la sua circolazio-
ne o riassorbimento.
16.2. Eziopatogenesi
I meccanismi attraverso il quale si può produrre un’idrocefalia sono i se-
guenti:
• Ipersecrezione del LCR. Molto raro.
• Disturbi del transito liquorale. E’ il meccanismo fondamentale.
• Alterazioni del drenaggio venoso intracranico. Ostacolano il riassor-
bimento del LCR verso il torrente sanguigno.
16.3. Clinica
Idrocefalia del lattante
L’idrocefalia nei lattanti si manifesta con un aumento della circonferenza cra-
nica (macrocefalia), dilatazione delle vene epicraniche, rigonfi amento del-
le fontanelle, segno di Macewen (suono tipico alla percussione del cranio
sopra le zone di dilatazione ventricolare) e transilluminazione positiva della
testa. Sono frequenti il pianto e l’irritabilità. All’esame obiettivo può eviden-
ziarsi, nei casi avanzati, occhio a “sole calante” e alterazioni del ritmo respira-
torio (Figura 61).
La causa più frequente di idrocefalia nei neonati è la stenosi congenita
dell’acquedotto del Silvio.
La diagnosi si realizza mediante misurazione della circonferenza cranica
(metodo più sensibile) ed esami d’immagini (ecografi a transfontanellare o
IDROCEFALIA
ORIENTAMENTO L’argomento più importante di questo tema è l’idrocefalia cronica dell’adulto. In particolar modo, bisogna ricordare la triade classica ed i risultati delle diverse prove che si utilizzano per fare diagnosi.
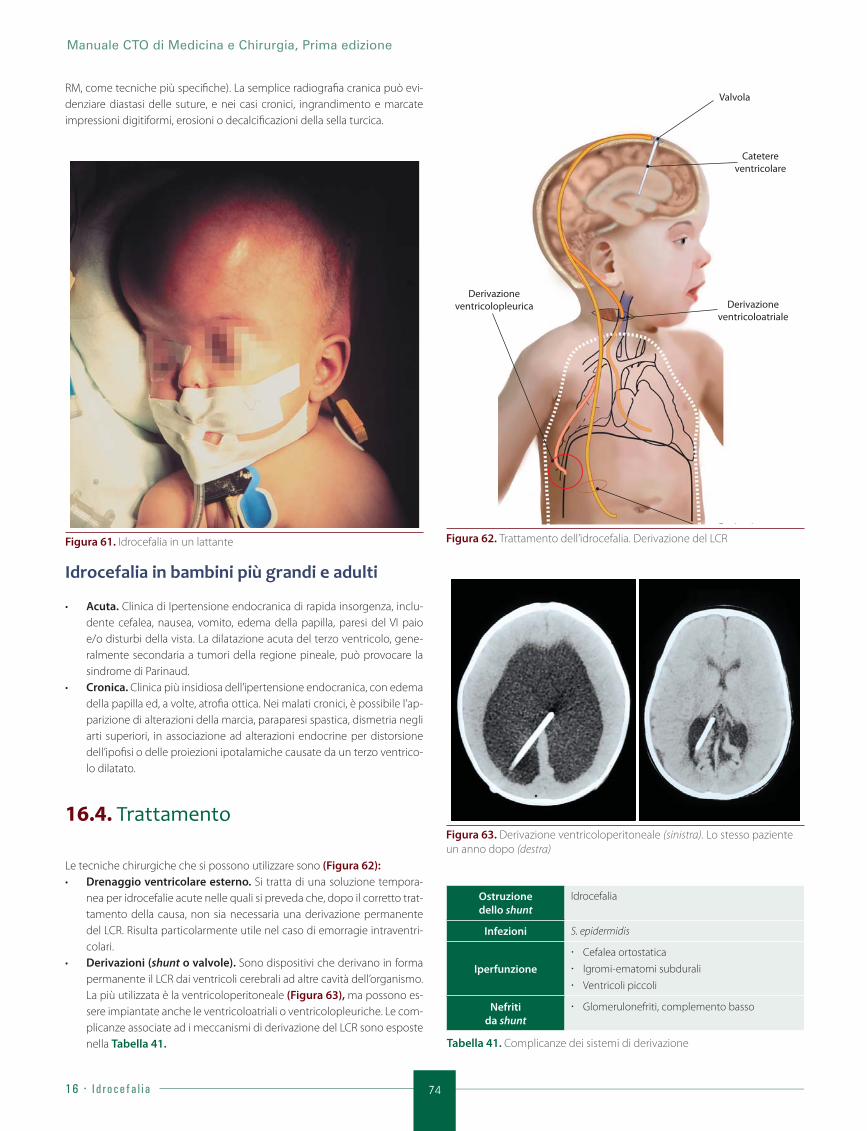
7416 ∙ I d rocefa l i a
Manuale CTO di Medicina e Chirurgia, Prima edizione
RM, come tecniche più specifi che). La semplice radiografi a cranica può evi-
denziare diastasi delle suture, e nei casi cronici, ingrandimento e marcate
impressioni digitiformi, erosioni o decalcifi cazioni della sella turcica.
Figura 61. Idrocefalia in un lattante
Idrocefalia in bambini più grandi e adulti
• Acuta. Clinica di Ipertensione endocranica di rapida insorgenza, inclu-
dente cefalea, nausea, vomito, edema della papilla, paresi del VI paio
e/o disturbi della vista. La dilatazione acuta del terzo ventricolo, gene-
ralmente secondaria a tumori della regione pineale, può provocare la
sindrome di Parinaud.
• Cronica. Clinica più insidiosa dell’ipertensione endocranica, con edema
della papilla ed, a volte, atrofi a ottica. Nei malati cronici, è possibile l’ap-
parizione di alterazioni della marcia, paraparesi spastica, dismetria negli
arti superiori, in associazione ad alterazioni endocrine per distorsione
dell’ipofi si o delle proiezioni ipotalamiche causate da un terzo ventrico-
lo dilatato.
16.4. Trattamento
Le tecniche chirurgiche che si possono utilizzare sono (Figura 62):
• Drenaggio ventricolare esterno. Si tratta di una soluzione tempora-
nea per idrocefalie acute nelle quali si preveda che, dopo il corretto trat-
tamento della causa, non sia necessaria una derivazione permanente
del LCR. Risulta particolarmente utile nel caso di emorragie intraventri-
colari.
• Derivazioni (shunt o valvole). Sono dispositivi che derivano in forma
permanente il LCR dai ventricoli cerebrali ad altre cavità dell’organismo.
La più utilizzata è la ventricoloperitoneale (Figura 63), ma possono es-
sere impiantate anche le ventricoloatriali o ventricolopleuriche. Le com-
plicanze associate ad i meccanismi di derivazione del LCR sono esposte
nella Tabella 41.
Valvola
Catetereventricolare
Derivazioneventricolopleurica Derivazione
ventricoloatriale
D i iFigura 62. Trattamento dell’idrocefalia. Derivazione del LCR
Figura 63. Derivazione ventricoloperitoneale (sinistra). Lo stesso paziente un anno dopo (destra)
Ostruzione dello shunt
Idrocefalia
Infezioni S. epidermidis
Iperfunzione
∙ Cefalea ortostatica
∙ Igromi-ematomi subdurali
∙ Ventricoli piccoli
Nefriti da shunt
∙ Glomerulonefriti, complemento basso
Tabella 41. Complicanze dei sistemi di derivazione

75
16Neurologia e neurochirurgia
• Ventricolostomia premamillare endoscopica. Crea una comunica-
zione diretta tra il terzo ventricolo e lo spazio subaracnoideo. Attual-
mente, si considera la tecnica d’elezione per il trattamento della stenosi
dell’acquedotto del Silvio.
16.5. Idrocefalia cronica dell’ adulto
Anche chiamata idrocefalia normotensiva o idrocefalia a pressione normale.
Da un punto di vista eziologico, si possono trovare forme idiopatiche, (40-
60% dei casi) e secondarie ad altri disordini neurologici come l’emorragia
subaracnoidea (la più frequente in questo gruppo), il traumatismo cranio-
encefalico, postmeningitica o a seguito di tumori. La forma idiopatica è un’i-
drocefalia che si presenta in pazienti di età avanzata (> 60 anni) e colpisce
leggermente di più gli uomini.
La clinica è molto caratteristica, anche se non patognomonica, e si manifesta
con la triade di Hakim-Adams: demenza (è una delle poche cause reversibili
di demenza), incontinenza urinaria e disturbi della marcia, che solitamente
è il segno più precoce e più frequente (la sua assenza deve far dubitare del
quadro). A volte si accompagna a disturbi extrapiramidali (parkinsonismo).
R I C O R D A Domanda MIR frequente: tríade di Hakim-Adams: aprassia della mar-cia, demenza e incontinenza sfi nterica.
La diagnosi si sospetta di fronte ad un riscontro di idrocefalia comunicante
nelle prove d’imaging (TC o RM) in un paziente con clinica compatibile, an-
che se non ci sono dati patognomonici (Figura 64).
Bisogna fare una diagnosi diff erenziale con l’idrocefalia ex vacuo o secon-
daria ad una atrofi a cerebrale, che è un aumento compensatorio della gran-
dezza del sistema ventricolare che appare frequentemente in anziani con
importante atrofi a cerebrale corticosubcorticale e non richiede trattamento.
A diff erenza di quest’ultimo tipo di idrocefalia, nella cronica dell’adulto, esi-
stono segni di riassorbimento transependimale (ipodensità periventricolare
alla TC), rigonfi amento del terzo ventricolo ed assenza di solchi a livello della
convessità.
Attualmente si stanno utilizzando alcuni studi di RM del fl usso del liquido
cefalorachidiano, nei quali si dimostra un aumento di velocità del fl usso a
livello dell’acquedotto di Silvio.
Figura 64. In questa immagine si può osservare una dilatazione del sistema ventricolare in assenza di solchi prominenti nella convessità, il tutto compatibile con idrocefalia normotensiva
La diagnosi si avvale di una monitorizzazione continua della pressione intra-
cranica, nella quale si può osservare un aumento di pressione e/o l’esistenza
di onde patologiche d’ipertensione intracranica. Si può realizzare, inoltre, un
test d’infusione: si introduce della soluzione fi siologica nello spazio intrate-
cale mediante puntura lombare ad una velocità determinata e si registra
il cambiamento di pressione nello spazio subaracnoideo durante il tempo;
con i dati ottenuti, si misura la resistenza all’uscita di LCR, che nel caso dell’i-
drocefalia cronica, si trova aumentata. Un’altra utile manovra diagnostica è
la puntura lombare evacuativa per dimostrare se esiste un miglioramento
clinico signifi cativo dopo l’estrazione di LCR.
Il trattamento d’elezione è la derivazione del LCR (abitualmente ventrico-
loperitoneale).
I d e e c h i ave Il LCR viene prodotto nei plessi coroidei. Da lì circola attraverso i ventricoli nello spazio subaracnoideo e viene riassorbito a livello dei capillari cerebrali (secondo le teorie più recenti).
Classicamente, l’idrocefalia si divide in ostruttiva o non comunican-te, se il problema si localizza a livello ventricolare, e non ostruttiva o comunicante, se l’alterazione della circolazione del LCR è sita a livello dello spazio subaracnoideo.
La causa più frequente di idrocefalia congenita è la stenosi dell’ac-quedotto di Silvio. Il trattamento d’elezione è la ventricolostomia endoscopica.
L’idrocefalia cronica dell’adulto può essere primaria (propria degli anziani) o secondaria ad altri processi, come l’emorragia subarac-noidea, i traumatismi e le meningiti.
Nella diagnosi di idrocefalia cronica dell’adulto, non esiste nessuna prova che mostri dati patognomonici.
La triade clinica caratteristica dell’idrocefalia cronica è: aprassia della marcia, demenza, ed incontinenza sfi nterica. Il disturbo della marcia rappresenta il sintomo più frequente e la clinica d’esordio abituale.
Mediante il test d’ infusione, si registra un aumento della resisten-za all’uscita del LCR, che è il meccanismo eziopatologico implicato nell’idrocefalia cronica.

767676
Neurologia e neurochirurgia
17.1. Considerazioni generali
Epidemiologia
I tumori intracranici più frequenti dell’adulto sono le metastasi. Tra i tumori
cerebrali primitivi, spiccano i gliomi (il glioblastoma multiforme è il tumore
cerebrale più frequente nei maggiori di 20 anni).
Le neoplasie intracraniche sono, dopo le leucemie, i processi maligni più fre-
quenti dell’età infantile, e sono la neoplasia solida più frequente in questo
gruppo d’età. La maggior percentuale è costituità dai gliomi (astrocitomi),
seguiti dal medulloblastoma e dal craniofaringioma, essendo eccezionali le
metastasi.
Negli adulti, i tumori cerebrali sono, nella maggior parte, sovratentoriali (80%),
mentre nei bambini si trova una distribuzione piuttosto omogenea tra il com-
partimento sovratentoriale ed infratentoriale, anche se nei primi due anni
sono più frequenti quelli al di sopra del tentorio.
Clinica
• Cefalea. Sintomo più frequente di presentazione, più intensa al matti-
no; può svegliare il paziente e si può associare a sintomi d’ipertensione
endocranica o focalità neurologica.
• Focalità neurologica. Dipende più dalla localizzazione che dal tipo di
tumore (si veda nella pagina seguente la Figura 65).
• Crisi epilettiche (causa più frequente tra i 35-50 anni). Specialmente
epilettogeni sono : oligodendroglioma, gangliocitoma e metastasi.
• Idrocefalia. Soprattutto in tumori infratentoriali o in comunicazione
con i ventricoli.
17.2. Metastasi cerebrali
Le metastasi sono i tumori cerebrali più frequenti nell’adulto, ma sono ec-
cezionali nei bambini. Si localizzano generalmente a livello della giunzione
17
corticosubcorticale degli emisferi cerebrali (80%) e, meno frequentemente,
negli emisferi cerebellari. Rappresentano comunque il tumore più frequente
della fossa posteriore dell’adulto. La maggior percentuale sono di origine
polmonare. Fino ad un 10% sono di origine sconosciuta. Il tumore che ha più
tendenza a metastatizzare a livello cerebrale è il melanoma.
Radiologicamente, si osservano alla TC come lesioni ipodense la cui parete
risulta fortemente captante contrasto (captazione ad anello o immagine a
ciambella) (Figura 66). Abitualmente sono circondati da abbondante ede-
ma vasogenico digitiforme.
Esistono alcune metastasi con particolare tendenza al sanguinamento (per
cui sono iperdense alla TC): coriocarcinoma, melanoma, carcinoma tiroideo,
ipernefroma (rene) e carcinoma broncogeno.
Figura 66. Metastasi cerebrali multiple di un carcinoma polmonare con captazione di contrasto ad anello
Il trattamento d ’elezione per le metastasi cerebrali viene riassunto nella Fi-
gura 67.
TUMORI INTRACRANICI
ORIENTAMENTOConstituisce uno dei temi più importanti in neurochirurgia per il MIR. Può risultare pratico elaborare una lista dove venga mostrato l’aspetto più frequente e caratteristico di ogni tumore, e con quello rispondere, senza gran difficoltà, alla maggior parte delle domande del MIR.
77
17Neurologia e neurochirurgia
RT olocranica
Metastasi
Primitivo non controllato
Non trattare/Rt olocranica palliativa
(*) Radiochirurgia
Primitivocontrollato
Unica Multipla
Chirurgia (*)
Chirurgia (*)
Figura 67. Trattamento d’elezione delle metastasi cerebrali
La sopravvivenza media dei pazienti con metastasi cerebrali trattate è di 6
mesi circa.
17.3. Gliomi
I gliomi sono neoplasie cerebrali che derivano dalle cellule gliali. Sono i tu-
mori primitivi più frequenti del SNC, soprattutto i tumori astrocitari più ag-
gressivi (glioblastoma multiforme). Nella Tabella 42 si riassume la classifi ca-
zione attuale dei gliomi (OMS, 2007).
Tumori astrocitari
∙ Astrocitoma diff uso (fi brillare, protoplasmatico, gemistocitico):
- Astrocitoma di basso grado
- Astrocitoma anaplastico
- Glioblastoma multiforme
› Glioblastoma a cellule giganti
› Gliosarcoma
∙ Astrocitomi localizzati:
- Astrocitoma pilocitico
- Xantoastrocitoma pleiomorfo
- Astrocitoma gigantocellulare subependimale
Tumori oligodendrogliali
∙ Oligodendroglioma
∙ Oligodendroglioma anaplastico
Gliomi misti ∙ Oligoastrocitoma
∙ Oligoastrocitoma anaplastico
Tumori ependimari
∙ Ependimoma
∙ Ependimoma anaplastico
∙ Ependimoma mixopapillare
∙ Subependimoma
Tumori gliali di origine incerta
∙ Gliomatosi cerebri
∙ Astroblastoma
∙ Glioma cordoide del terzo ventricolo
Tabella 42. Classifi cazione dei tumori gliali (OMS, 2007)
Emisferici Crisi + focalitàneurologica
Fossa posteriore
Metastasi Gliomi Linfomi primaridel SNC
Meningioma Cervelletto Tronco encefalicoQuarto
ventricoloAngolo
pontocerebellare
· Più freq da tumori sopradiaframmatici che sottodiaframmatici· Unione corticobubcorticale
· Immunodepressi (HIV) · Multicentrici· Buona risposta a corticosteroidi· RT d’elezione
· Donne· Convessità parasagittale· Iperostosi cranica e blistering· Aumento vascolarizzazione (unico intradurale vascolarizzato dall’art carotide ext. · Ormonodipendente· Se infanzia o multipli (NFM II )
· Cefalea· Idrocefalia· Snd cerebellare
· Gliomi· Bambini
Idrocefalia
· Ependimoma· Il mixopapillare nel filum terminale
· Papilloma dei plessi corioidei· Nei bambini nei ventricoli laterali
Astrocitomaemisferico
Bambini Adulti
· Medulloblastoma vermiano· Possibile disseminazione nel LCR e/o metastasi
Astrocitoma
Miglior prognosi il pilocitico ed il subependimario a cellule giganti (sclerosi tuberosa)
Glioblastoma multiforme
Oligodendroglioma
Quellocon la peggiorprognosi
· Sostanza bianca subcorticale lobo frontale· Crisi e calcificazioni
· Emangioblastoma· Emisferico· Poliglobulia· Von Hippel Lindau
Bambini
Bambini
Adulti
Adulti · Linea mediana (cervelletto, tronco, nervo ottico [NFM I])· Raro negli emisferi
Sostanza bianca subcorticale lobo frontale e temporale
Neurinoma acustico
Meningioma
Colesteatoma (cisti epidermoide o tumore perlato)
Regionechiasmatica
· Adenoma dell’ipofisi più freq è il prolattinoma· Emianopsia bitemporale + clinica endocrinologica
· Craniofaringioma· Stessa clinica campimetrica · Calcificazioni soprasellari
· Coristoma · Neuroipofisi
Regione pineale
· Più freq il germinoma· Idrocefalia + snd di Parinaud· Il più radiosensibile
Figura 65. Diagnosi diff erenziale per localizzazione del tumore

7817 ∙ Tumor i i n t rac ran ic i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Astrocitoma
Gli astrocitomi sono tumori che derivano dagli astrociti. Costituiscono il
gruppo più numeroso dei tumori primitivi del sistema nervoso centrale.
Sono neoplasie che esprimono la proteina gliofi brillare acida (GFAP).
Astrocitomi diff usi o infi ltranti
I tumori che appartengono a questa categoria si caratterizzano per l’aspetto
infi ltrante locale e la loro capacità di metastatizzazione verso siti anatomici
lontani dalla localizzazione iniziale. Inoltre, hanno la capacità di degenerare
in forme più maligne con il passare del tempo. Secondo l’OMS esistono 3 tipi
di astrocitomi diff usi (Tabella 43):
Atipia nucleare
Attività mitotica
Proliferazione endoteliale
Necrosi
Astrocitoma + - - -
Astrocitoma anaplastico
+ + - -
Glioblastoma multifome
+ + + +
Tabella 43. Caratteristiche anatomopatologiche degli astrocitomi diff usi
• Astrocitomi (di basso grado). Corrispondono a tumori di grado II.
Sono tumori che si trovano maggiormente in bambini e giovani adulti.
Alla RM, sono tumori che non captano costrasto o, se lo fanno, in ma-
niera debole. Il trattamento consiste in chirurgia e radioterapia, anche
se, in alcune occasioni, si mantiene un attitudine attendeista d’osserva-
zione, soprattutto se colpisce le aree del linguaggio; la chemioterapia
si riserva alle ricorrenze tumorali. La mediana di sopravvivenza è tra i
5-10 anni.
• Astrocitomi anaplastici (tumori di grado III). Il picco d’incidenza si
trova intorno ai 40 anni. Alla RM non captano solitamente contrasto,
anche se possono farlo maggiormente rispetto agli astrocitomi di grado
II. Il trattamento consiste nella resezione chirurgica, radioterapia e che-
mioterapia, sia sistemica o, negli ultimi anni, locale (carmustina), ovvero
somministrata nel letto chirurgico dopo asportazione del tumore. La
mediana di sopravvivenza si trova tra 2,5 ed i 3 anni.
• Glioblastoma multiforme (tumori di grado IV) (Figura 68). Sono i
tumori primitivi più frequenti degli adulti. L’età media di presentazione
si attesta intorno ai 53 anni. Caratteristicamente, mostrano captazione
ad anello dopo contrasto (Figura 69).
Figura 68. Glioblastoma multiforme temporale destro
Figura 69. Captazione di contrasto ad anello in paziente con glioblastoma multiforme temporale destro
Come l’astrocitoma anaplastico, il trattamento consiste nella chirurgia,
radioterapia e chemioterapia (locale o sistemica). Nonostante la dispo-
nibilità terapeutica, la mediana di sopravvivenza è di un anno. Per que-
sto motivo, in molte occasioni si decide per l’astensione terapeutica per
la possibilità di lasciare sequele al paziente, soprattutto se sono localiz-
zati in prossimità delle aree del linguaggio.
Astrocitomi localizzzati
Sono tumori che si caratterizzano per essere relativamente circoscritti ed
avere una minima capacità di disseminazione all’interno del sistema nervo-
so. Sono più comuni nei bambini e nei giovani adulti. In questa categoria
sono inclusi 3 tipi di tumore:
• Astrocitoma pilocitico (Figura 70 e Figura 71) (grado I della classifi ca-
zione OMS). Costiuisce la neoplasia cerebrale più frequente nei bambi-
ni, con un’incidenza massima nella 2.ª decade di vita. (10-12 anni come
picco d’incidenza). Le fi bre di Rosenthal costituiscono un reperto anato-
mopatologico caratteristico di questi tumori.
Si localizzano soprattutto a livello degli emisferi cerebellari; alla RM si
mostrano come lesioni cistiche con un nodulo captante all’interno. La
resezione chirurgica totale di questo tumore riesce a curare questo tipo
di pazienti, senza necessità di terapie complementari. La prognosi è ec-
cellente nei casi in cui riesca la resezione completa del tumore.
Figura 70. Astrocitoma pilocitico cerebellare

79
17Neurologia e neurochirurgia
• Astrocitoma gigantocellulare subependimale (grado I della classifi -
cazione OMS). E’ un tumore che si associa a sclerosi tuberosa. Si localizza
a livello delle pareti dei ventricoli laterali.
Figura 71. (A) Glioma del tronco. (B) Glioma del nervo ottico derstro
Oligodendroglioma
Rappresenta meno del 10% di tutti i gliomi. La sua caratteristica microsco-
pica più rappresentativa è l’esistenza di cellule rotonde che contengono
nuclei ipercromatici e citoplasma di scarsa capacità tintoriale, con aspetto
a “uovo fritto” (Figura 72). Sono frequenti le cisti, le calcifi cazioni e le emor-
ragie spontanee.
E’ tipico che esordisca con crisi epilettiche, poichè rappresenta il tumore pri-
mitivo più epilettogeno.
Figura 72. Oligodendroglioma con l’immagine tipica delle cellule a “uovo fritto”
R I C O R D A Di fronte ad un tumore del lobo frontale, bisogna pensare fonda-mentalmente a 3 possibilità: metastasi, glioblastoma ed oligoden-droglioma. Quest’ultimo si caratterizza per la presenza di calcifi ca-zioni che i primi 2 non presentano.
La TC evidenza una lesione ipodensa con aree cistiche e calcifi che, che non
capta contrasto. Il trattamento d’elezione è la resezione chirurgica associata
a chemioterapia (PCV: procarbazina, CCNU e vincristina); nel caso degli oligo-
dendrogliomi anaplastici, si può associare radioterapia. La perdita dei bracci
cromosomici 1p e 19q è in relazione ad una miglior risposta alla chemiotera-
pia e ad una miglior sopravvivenza. A volte esistono tumori che sono un mix
tra astrocitomi e oligodendrogliomi, rappresentando gli oligoastrocitomi, tu-
mori a cavallo tra i due.
Ependimoma
La sua caratteristica istologica più tipica sono le formazioni a “rosetta”. Ge-
neralmente sono benigni. A livello intracranico, rappresentano il 5-6% dei
gliomi, e crescono tipicamente sul pavimento del quarto ventricolo (Figura
73), causando idrocefalia. Colpiscono generalmente i bambini.
Figura 73. Ependimoma del quarto ventricolo
Senza dubbio, sono molto più frequenti a livello spinale, dove sono propri
dell’adulto ed hanno una prognosi migliore. La colonna cervicale, costitu-
isce il segmento dove si può trovare questo tipo di tumore con maggior
frequenza. A livello del fi lum terminale, si localizza in maniera specifi ca un
sottotipo di ependimoma, ovvero la variante mixopapillare.
Il trattamento d’elezione è la chirurgia associata a chemioterapia. Si possono
manifestare ripetizioni attraverso il LCR, per le quali si richiede radioterapia
di tutto il neuroasse.
17.4. Medulloblastoma (PNET-Tumore Neuroectodermico Primitivo- infratentoriale)
Si tratta del tumore encefalico più frequente nei bambini minori di 5 anni
e della neoplasia intracranica maligna più frequente dell’età infantile. Isto-
logicamente, sono caratteristiche le formazioni a “rosetta di Homer-Wright”,
anche se non sono patognomoniche perchè possono apparire nel resto dei
tumori embrionari (Figura 74).
Figura 74. Rosette di Homer-Wright, tipiche del medulloblastoma

8017 ∙ Tumor i i n t rac ran ic i
Manuale CTO di Medicina e Chirurgia, Prima edizione
In un terzo dei casi è stata dimostrata una perdita di materiale genetico a
livello del braccio corto del cromosoma 17. Si associa a malattie ereditarie,
come la sindrome carcinoma basocellulare nevoide (sindrome di Gorlin) e la
sindrome di Turcot (poliposi colica e tumore cerebrale).
R I C O R D A Il medulloblastoma origina dal tetto del quarto ventricolo, mentre l’e-pendimoma dal pavimento del quarto ventricolo.
Radiologicamente, si manifesta come un tumore solido della fossa poste-
riore, che capta contrasto in forma omogenea, abitualmente localizzato
sulla linea mediana a livello del verme cerebellare (Figura 75) e tetto del
quarto ventricolo, per cui esordice spesso con sintomi d’ipertensione en-
docranica per una idrocefalia ostruttiva e segni di disfunzione cerebellare
(atassia del tronco). Negli adulti, si localizza più frequentemente a livello
emisferico.
Il trattameento d’elezione è la exeresi chirurgica, seguita da irradiazione
concentrata sul focolaio tumorale e radioterapia preventiva diff usa cranio-
spinale; poi poli-chemioterapia, soprattutto nei bambini tra 18 mesi e 3
anni.
Figura 75. (A) Medulloblastoma nel verme cerebellare. (B) Disseminazione spinale
17.5. Meningioma
Epidemiologia
Il meningioma segue per frequenza i gliomi nei tumori intracranici dell’adul-
to (20%), però è il più frequente dei tumori intracranici extraparenchimali.
Colpisce generalmente donne tra 5.ª e 6.ª decade di vita. Quando si associa
a neurofi bromatosi tipo II appare nell’infanzia e, spesso, in forma di lesioni
multiple.
Anatomia patologica
Crescono a partire dall’aracnoide (leptomeningi), non dalla duramadre. La
loro localizzazione più frequente è a livello della convessità ossea (Figura
76), ma possono apparire dovunque vi siano cellule aracnoidee.
Figura 76. Meningioma del solco olfattivo
Hanno tendenza a calcifi care. I corpi psammomatosi sono un riscontro ana-
tomopatologico caratteristico. La vimentina e l’EMA (antigene epiteliale di
membrana) sono due marcatori istochimici del meningioma.
Diagnosi
Mostrano un aspetto omogeneo circoscritto e ben delimitato alla TC e RM.
Dopo la somministrazione di contrasto, si evidenzia un marcato enhance-
ment ed occasionalmente la cosidetta “coda durale”. Possono presentare
calcifi caizoni (corpi psammomatosi) che sono visibili alla TC e all’RX cranica.
A volte si manifestano con iperostosi e il fenomeno del blistering nella por-
zione ossea adiacente.
Trattamento
La resezione completa può essere curativa. La radioterapia è riservata ai me-
ningiomi maligni o atipici, alle resezioni incomplete o in caso di recidive.
17.6. Neurinoma dell’ VIII (schwannoma vestibolare)
E’ un tumore benigno, a crescita lenta, che origina nel rivestimento mielinico
della porzione intracanalicolare della branca vestibolare dell’ VIII paio di nervi
cranici; è ben delimitato,di forma ovoidale o fusata, di consistenza variabile
da compatta-fi brosa (Antoni tipo A) a molle per presenza di formazioni mi-
crocistiche a contenuto mucoide (Antoni tipo B) (Figura 77). E’ il tumore
più frequente dell’angolo pontocerebellare (il secondo è il meningioma e,
il terzo, l’epidermoide o colesteatoma) ed il tumore primitivo più frequente
della fossa posteriore nell’adulto. Possono essere bilaterali e, in questo caso,
sono patognomonici di neurofi bromatosi tipo II.
Causa ipoacusia neurosensoriale, acufeni e vertigini (lesione dell’ VIII paio).
Durante la sua crescita può comprimere i nervi V e VII, dando luogo a ipoeste-
sia trigeminale con abolizione del rifl esso corneale e paresi facciale. Quando
è molto grande, può arrivare a comprimere il tronco encefalico ed altri nervi
cranici, dando luogo ad atassia, diplopia, compromissione dei nervi cranici
bassi, fi no a disturbi respiratori e coma, se non diagnosticato precocemente.
Il procedimento diagnostico d’elezione è la RM e, in secondo luogo, la TC
con contrasto e studio dell’osso per eventuali modifi cazioni del forame.
Il trattamento può essere chirurgico e/o radiochirurgico (terapia con raggi
gamma da varie fonti [Co-60] circostritto nell’area occupata dal tumore, che
si realizza in un unica sessione). Si descrive più in dettaglio nella sezione di
Otorinolaringoiatria.

81
17Neurologia e neurochirurgia
Figura 77. Neurinoma dell’ VIII paio
17.7. Tumori della regione pineale
I tumori di questa regione (Tabella 44) sono, in generale, più frequenti nei
bambini che negli adulti. Abitualmente portano ad idrocefalia per steno-
si dell’acquedotto di Silvio e danno una clinica d’ipertensione endocranica
senza focalità neurologica.
Il germinoma è il tipo istologico più frequente e rappresenta il tumore più
frequente della regione pineale.
Generalmente, i markers tumorali nel LCR e nel sangue sono negativi, ma
possono presentare un aumento moderato dell’-fetoproteina (-FP), fo-
sfatasi alcalina placentaria o gonadotropina corionica (HCG). Quando sono
positivi, la loro misurazione seriata permette di valutare la risposta al tratta-
mento e diagnosticare precocemente le recidive.
Origine germinale Origine non germinale
∙ Germinoma
∙ Coriocarcinoma
∙ Tumore del seno endodermico
∙ Teratoma
∙ Carcinoma embrionario
∙ Astrocitoma
∙ Pinealoblastoma
∙ Pineocitoma
Tabella 44. Tumori della regione pineale
E’ straordinariamente radiosensibile e può essere trattato con radioterapia;
gli altri richiedono exeresi chirurgia. La prognosi è migliore nel caso del ger-
minoma rispetto ai tumori non germinomatosi.
17.8. Tumori ipofi sari
Adenoma
Sono tumori benigni della porzione anteriore dell’ipofi si (adenoipofi si). Pos-
sono essere funzionanti o secretori, in circa il 70% dei casi (il più frequente è
il prolattinoma), e non funzionanti. A volte sono misti (più frequentemente
produttori di ormone della crescita GH e prolattina). Si classifi cano, in fun-
zione della grandezza, come microadenomi (minori di 1 cm di grandezza)
e macroadenomi (maggiori o uguli ad 1 cm) (Figura 78). La loro incidenza
è simile in entrambi i sessi, e sono più fequenti tra la 3.ª e 4.ª decade di vita.
Il trattamento chirurgico d’elezione è la resezione per via transfenoidale. Tra i
trattamenti medici troviamo la bromocriptina per il prolattinoma e l’octreoti-
de o analoghi, per i tumori secernenti GH. La radioterapia postchirurgica è a
volte molto effi cace nel controllo delle recidive. Una forma rara di presenta-
zione è l’apoplessia ipofi saria, che consiste in un deterioramento neurolo-
gico rapido che si manifesta spesso con cefalea, disturbi visivi (tra cui amau-
rosi improvvisa), oftalmoplegia e riduzione del livello di coscienza, dovuto ad
emorragia, necrosi, o infarto all’interno del tumore o della ghiandola adiacen-
te. Si accompagna a panipopituitarismo, per cui il trattamento consiste in un
trattamento ormonale sostitutivo, soprattutto corticosteroidi, e nella decom-
pressione chirurgica urgente, con l’obiettivo di migliorare la funzione visiva.
Nella sezione di Endocrinologia, metabolismo e nutrizione si aff ronta il tema in
maniera più approfondita.
Figura 78. Macroadenoma dell’ipofi si
17.9. Craniofaringioma
E’ un tumore disembrioblastico che origina a partire dai resti della tasca di
Rathke, che colpisce principalmente bambini e adolescenti, un secondo
picco di frequenza si ha tra i 50 e 60 anni (Figura 79). Da un punto di vi-
sta anatomopatologico (maggiormente negli adulti) solitamente presenta
un’importante componente cistica a contenuto oleoso ed una parete solida
parzialmente calcifi cata (vengono descritte a forma di “parentesi” nell’Rx la-
terale del cranio).
Si manifesta con una clinica di disfunzione neuroendocrina e campimetrica
a causa della compressione del chiasma (emianopsia bitemporale o qua-
drantanopsia inferiore). Può causare statura bassa ed obesità per interessa-
mento ipotalamo-ipofi sario.
Il trattamento d’elezione è la resezione chirurgica che è pero spesso incom-
pleta e diffi coltosa per la frequente marcata adesione alle strutture circostan-
ti ed in particolare alle arterie della base; vengono utilizzate anche tecniche
di evacuazione stereotassica della cisti, terapia intracavitaria con radioisotopi
come l’ittrio ed il fosforo, bleomicina intralesionale, INF-, radioterapia con-
venzionale e radiochirugia.

8217 ∙ Tumor i i n t rac ran ic i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Figura 79. Craniofaringioma
17.10. Linfoma cerebrale primitivo
Abitualmente, si osserva in pazienti con difetti immunologici misti (HIV, con-
nettivopatie), anche se la sua frequenza in individui immunocompetenti sta
aumentando (soprattutto anziani). Si associa ad infezioni da EBV.
Generalmente sono linfomi a cellule B e presentano una distribuzione
perivascolare. Si localizzano con maggior frequenza nei gangli della base,
sostanza bianca periventricolare e corpo calloso. Un dato anatomopa-
tologico caratteristico di questi tumori sono gli infi ltrati perivascolari di
linfociti.
Alla TC cranica captano contrasto omogeneamente, spesso ad anello.
E’ caratteristica l’importante riduzione o la scomparsa delle lesioni alla TC,
dopo un ciclo di varie settimane con corticosteroidi a dosi elevate (tumori
fantasma).
Il trattamento più effi cace è la radioterapia, che attualmente si combina con
chemioterapia con metotrexate.
17.11. Emangioblastoma
E’ un tumore benigno che si presenta con maggior frequenza nella fossa
posteriore, a livello degli emisferi cerebellari. E’ il più frequente tra i tumori
primitivi intrassiali della fossa posteriore nell’adulto. Può essere solido, cisti-
co, con un nodulo murale ipercaptante.
Nonostante la maggior parte di essi siano sporadici, fi no ad un 20% si trova
nella sindrome di Von Hippel-Lindau.
In questi casi, spesso sono multipli e si associano ad emangioblastomi reti-
nici ed altre lesioni viscerali (abitualmente tumori o cisti, soprattutto a livello
pancreatico e del rene).
E’ tipica la relazione di questa malattia con il feocromocitoma. Nella Tabella
45 vengono descritte le associazioni più frequenti tra facomatosi e tumori
del SNC.
R I C O R D A Una cisti con un nodulo captante al suo interno, nel seno di un emi-sfero cerbellare, è un astrocitoma pilocitico se il paziente è un bam-bino, è un emangioblastoma se è un adulto.
Facomatosi Tumori SNC
Sclerosi tuberosa Astrocitoma gigantocellulare subependimale
Neurofi bromatosi tipo I Glioma delle vie ottiche
Neurofi bromatosi tipo II ∙ Neurinoma bilaterale dell’ VIII
∙ Meningiomi
Sturge-Weber Angiomi leptomeningei
Von Hippel-Lindau Emangioblastoma cerebellare
Klippel-Trenaunay Angioma cavernoso del midollo spinale
Tabella 45. Facomatosi e tumori del sistema nervoso centrale
Possono produrre eritropoietina ed è caratteristica la presenza di policitemia
agli esami di laboratorio. Il trattamento d’elezione è la chirurgia (svuotamen-
to della cisti ed exeresi del nodulo murale).
17.12. Riassunto delle caratteristiche anatomopatologiche
Nella Tabella 46 vengono riassunte le caratteristiche anatomopatologiche
più tipiche dei tumori che sono stati trattati in questo capitolo.
Tumori SNCCaratteristiche
anatomopatologiche
Astrocitoma pilocitico Fibre di Rosenthal
Oligodendroglioma Cellule a “uovo fritto”
Tumori embrionari Rosette di Homer-Wright
Meningioma Corpi psammomatosi
Schwannoma vestibolare Fibre di Antoni
Pineocitoma Rosette di Borit
Cisti colloide Materiale PAS(+)
Linfoma Infiltrai linfocitari perivascolari
Cordoma Cellule fisalìfore
Tabella 46. Dati anatomopatoligici caratteristici dei tumori del SNC

83
17Neurologia e neurochirurgia
I d e e c h i ave Negli adulti, i tumori cerebrali più frequenti sono le metastasi ce-rebrali, mentre il primitivo più frequente è il glioblastoma mul-tiforme.
Invece, nei bambini, il tumore più frequente è l’astrocitoma pilo-citico ed il tumore maligno più comune è il medulloblastoma. Le metastasi sono molto rare.
Il sintomo d’esordio più frequente dei tumori cerebrali è la cefa-lea. E’ solitamente più intensa al mattino e può portare il pazien-te a svegliarsi durante la notte.
Le metastasi più frequenti provengono dal carcinoma microciti-co del polmone. Tuttavia, il tumore con maggior tendenza a dare metastasi cerebrali è il melanoma.
Il trattamento d’elezione di una metastasi cerebrale unica, in un sito accessibile e con malattia primitiva controllata, è la chirurgia seguita da radioterapia olocranica.
Gli astrocitomi constituiscono i tumori primitivi più frequenti del SNC.
Gli astrocitomi diff usi si caratterizzano per una tendenza a dege-nerare verso forme più maligne con il passare del tempo.
Il trattamento degli astrocitomi diff usi di alto grado (astrocitoma anaplastico e glioblastoma multiforme) è palliativo.
L’astrocitoma gigantocellulare subependimario si associa a scle-rosi tuberosa
Il tumore primitivo più epilettogeno è l’oligodendroglioma.
I meningiomi sono tumori che hann0 un’infl uenza ormonale evi-dente: compliscono più le donne e sono stati impiegati antago-nisti del progesterone per il loro trattamento.
Il trattamento d’elezione dei meningiomi è l’approccio chirurgi-co. Il grado di resezione infl uisce sulla possibiltà di recidiva del tumore.
La clinica più frequente d’esordio dei tumori della regione pinea-le è l’ipertensione endocranica secondaria ad idrocefalia.
Il tumore più frequente della regione pineale è il germinoma, il cui trattamento d’elezione è la radioterapia.
Gli adenomi ipofi sari possono essere funzionanti o non funzio-nanti. Di solito si manifestano con un’emianopsia bitemporale con prevalenza nei campi superiori, associata con la clinica do-vuta all’iperfunzione ormonale (se sono funzionanti) e/o da ipo-pituitarismo secondario.
Un tumore sellare con cisti e calcifi cazioni orienta la diagnosi ver-so un craniofaringioma .
Un tumore cerebellare associato a policitemia è un emangiobla-stoma.
C a s i c l i n i c i Uomo di 29 anni lamenta nausea ed intorpidimento degli arti di si-nistra, negli ultimi 6 mesi. All’esame obiettivo presenta dismetria e adiadococinesia dell’arto superiore sinistro. La TC cranica mostra una lesione cistica, con nodulo ipercaptante situato nell’emisfero cere-bellare sinistro. Le analisi di laboratori sono normali, ad eccezione di un ematocrito del 58%, ed una TC toracoaddominale ha evidenziato cisti al pancreas e al rene. Più probabilmente, la natura di questa le-sione intracranica sarà:
1) Medulloblastoma.2) Metastasi da carcinoma polmonare.3) Emangioblastoma.4) Astrocitoma pilocitico.5) Glioblastoma multiforme.
RC: 3
Un paziente di 60 anni, con precedenti di cancro polmonare, presen-ta una crisi epilettica. Viene eff ettuata una RM cerebrale, che mo-stra una lesione cistica suggestiva di metastasi. Non c’è evidenza di metastasi extracerebrali. Quali delle seguenti aff ermazioni è FALSA riguardo al trattamento del paziente?
1) Bisogna somministrare trattamento antiepilettico.2) I corticosteroidi sono utili per diminuire l’edema vasogenico.3) Se la lesione è accessibile, può essere indicata la chirurgia.
4) La radioterapia cranica non è indicata.5) La chemioterapia può essere indicata.
RC: 4
In un uomo di 75 anni, dopo l’apparizione di una brusca sindrome neuropsichiatrica, una TC cranio mostra la presenza di una metasta-si cerebrale. Qual’è la localizzazione più probabile del tumore primi-tivo metastatizzante?
1) Gastrica.2) Renale.3) Tiroidea.4) Broncopolmonare.5) Testicolare.
RC: 4
Di fronte ad un bambino di 5 anni con un quadro d’ipertensione en-docranica, alterazioni visive ed ipotalamiche, che presenta in una radiografi a laterale del cranio calcifi cazioni a forma di parentesi a livello soprasellare, quale sarà presumibilmente la diagnosi?
1) Adenoma ipofi sario.2) Glioma del nervo ottico.3) Craniofaringioma.4) Medulloblastoma.5) Pinealoma che causa idrocefalia.
RC: 3

848484
Neurologia e neurochirurgia
I traumi cranioencefalici (TCE) si considerano la prima causa di perdita di coscien-
za (dalla commozione cerebrale fi no ai diff erenti livelli del coma) nella popola-
zione generale, ed il fattore eziologico più frequente di epilessia tra i 18-35 anni.
18.1. Scala del coma di Glasgow
La scala di Glasgow esposta nella Tabella 47.
PunteggioApertura
degli occhiRisposta motoria
Risposta verbale
6 - Obbedisce agli ordini
-
5 - Localizza il dolore Congrua
4 Spontanea Ritira al dolore Confusa
3 Alla voce Flessoria (decorticazione)
Inappropiata
2 Al dolore Estensoria (decerebrazione)
Incomprensibile
1 No No No
Tabella 47. Scala del coma di Glasgow
Il livello di coscienza, stimato secondo il punteggio di questa scala, è il princi-
pale fattore prognostico nel TC. Si defi nisce come TCE lieve un punteggio di
14 o 15; TCE moderato se il punteggio è tra 9-13. Un punteggio totale, uguale
o minore di 8 indica un TCE grave con prognosi grave.
R I C O R D A La risposta motoria è il parametro di maggior valore nella scala di Glasgow.
18
In generale, la prova radiologica d’elezione per la diagnosi delle lesione
intracraniche associate al traumatismo cranioencefalico è la TC cranica.
18.2. Gestione del TCE in urgenza
• Pazienti con rischio basso. Pazienti asintomatici, con cefalea, nausea
o con contusione o abrasione del cuoio capelluto.
In questo gruppo di pazienti si raccomanda l’osservazione domiciliare,
senza indicare nessuna prova d’immagine, sempre che siano accom-
pagnati.
Si raccomanda la realizzazione di una TC cerebrale ai soggetti con:
coagulopatie, alcolisti, abuso di droghe, antecedenti neurochirurgici,
anziani con incapacità ed in quelli con epilessia.
• Pazienti con rischio moderato. Pazienti con amnesia antero e/o re-
trograda postraumatica, perdita di coscienza, convulsioni, pazienti che
hanno presentato vomito, con signifi cativa tumefazione subgaleale,
cefalea progressiva, minori di 2 anni o storia di assunzione di droghe.
In questo tipo di pazienti, si raccomanda l’esecuzione di una TC cere-
brale e, nella maggior parte dei casi, osservazione in ambiente ospe-
daliero per qualche ora.
• Pazienti ad alto rischio. Sono pazienti che presentano un livello di
coscienza depresso, GCS < 14 o pazienti in cui si osserva una dimi-
nuzione progressiva del livello di coscienza, pazienti che presentano
segni di focalità neurologica, TCE penetranti o fratture-infossamento.
In questo gruppo bisogna realizzare una TC cerebrale ed una visita
neurologica.
18.3. Fratture craniche
A seconda del tipo di frattura, si possono classifi care come di seguito:
TRAUMI CRANIOENCEFALICI
ORIENTAMENTO
Gli aspetti più richiesti di questo tema sono l’ematoma epidurale, la scala di Glasgow e le fratture della base del cranio. E’ importante che siano chiari i concetti base di ogni lesione che viene prodotta come conseguenza dei traumi cranioencefalici. Bisogna differenziare un ematoma epidurale da un subdurale acuto, e studiare la scala di Glasgow.

85
18Neurologia e neurochirurgia
Fattura lineare
L’esistenza di una frattura presuppone che il cranio abbia subito un impatto
ad alta energia, ma la prognosi del paziente dipenderà dalla possibile lesione
encefalica sottostante, non dalla frattura. Esiste una scarsa correlazione tra la
lesione ossea ed il danno cerebrale.
Il riscontro di una frattura all’ RX cranica è indicazione ad una TC cranica ur-
gente per valutare le possibili lesioni intracraniche associate (Figura 80).
In generale, non richiede trattamento, ma è necessario tenere il paziente
sotto osservazione.
Figura 80. Frattura lineare frontale
Frattura aperta
Si defi nisce aperta una frattura lineare che è in comunicazione con una lace-
razione della duramadre.
Il rischio d’infezione intracranica con fratture lineari della convessità, è molto
basso, ma aumenta se sono aperte.
Frattura-infossamento
Quando l’energia viene applicata su un’area relativamente piccola, può
prodursi una frattura-infossamento (Figura 81), che è quella nella quale il
tavolato esterno si porta al di sotto del limite anatomico di quello interno.
Il trattamento dipende dal tipo di lesione, però, in generale, richiede un
trattamento chirurgico per sollevare il frammento. In queste fratture c’è un
aumento del rischio di crisi epilettiche postraumatiche.
R I C O R D A Una frattura-infossamento è un fattore di rischio di sviluppo di crisi epilettiche precoci.
Figura 81. Frattura-infossamento parietale
Frattura composta
La frattura composta è una frattura cranica associata ad una ferita del cuoio
capelluto o in continuità con una frattura della parete dei seni paranasali,
celle mastoidee o cavità dell’orecchio medio, con integrità della duramadre.
In questo caso, bisogna eff ettuare una toeletta chirurgica della ferita e som-
ministrare trattamento antibiotico per prevenire una osteomielite o un’infe-
zione del cuoio capelluto (più frequentemente da Staphylococcus aureus).
Frattura diastatica
Sono quelle in cui la linea di frattura coincide con una sutura cranica. Sono
più frequenti in bambini piccoli (minori di 3 anni).
Frattura crescente o evolutiva
Sono tipiche dei bambini e sono caratterizzate dal fatto che la frattura lacera
la duramadre, causando l’erniazione dell’aracnoide attraverso la linea di frat-
tura, facendo in modo che le pulsazioni del LCR amplino progressivamente
la frattura. Vengono chiamate anche cisti leptomeningee postraumatiche e
richiedono la chirurgia per chiudere il difetto meningeo.
Frattura a “ping-pong”
Nei lattanti, a causa della plasticità del cranio, gli infossamenti chiusi di solito
provocano questo tipo di frattura a legno verde. In assenza di danno paren-
chimatoso, la riparazione chirurgica è necessaria solo nelle lesioni frontali
per motivi estetici (in altre localizzazioni, di solito tendono a scomparire con
la crescita) (Figura 82).
Figura 82. Frattura a “ping-pong” in un lattante

8618 ∙ Traumi c ran ioencefa l i c i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Fratture della base del cranio
Le fratture della base del cranio (fratture basilari) si sospettano quando il
paziente ha subito un TCE e presenta alcuni segni obiettivi: emotimpano,
ecchimosi retroauricolare (segno di Battle), ecchimosi periorbitale (occhio
da procione), lesione dei nervi cranici della base (anosmia per lesione del
I nelle fratture frontoetmoidali, lesioni del VII e VIII per le lesioni della rocca
petrosa, e del VI paio in quelle del clivus) e, con minor frequenza , otorrea o
rinorrea liquorale o ematica (Figura 83).
OtorreaSegno di Battle
(ecchimosi retroauricolare)
Ematoma periorbitario bilaterale “occhi da procione”
Figura 83. Segni clinici delle fratture della base del cranio
Le caratteristiche delle fratture della rocca petrosa sono esposte di seguito
nella Tabella 48.
Longitudinali Trasversali
Frequenza 70-90% 12-20%
Perforazione Frequente Rara
Otorrea Frequente Rara
Emotimpano Raro Frequente
Otoliquorrea Frequente Rara
Ipoacusia Trasmissiva Percettiva (cofosi)
Paralisi facciale 20%. Transitoria 50%. Permanente
Vertigini Rare e lievi (posizionali) Frequenti e gravi
Radiologia Schüller Stenvers
Tabella 48. Diagnosi diff erenziale delle fratture della rocca petrosa
Le fratture della base cranica sono diffi cilmente evidenziabili mediante RX,
per cui la prova radiologica d’elezione è la TC cranio con fi nestra ossea.
Un segno indiretto è la presenza di aria intracranica (pneumoencefalo).
Esistono alcune proiezioni radiologiche classiche, ora in disuso, per dia-
gnosticare le fratture della rocca: Schüller per le longitudinali e Stenvers
per le trasversali.
R I C O R D A La TC con fi nestra ossea è la prova radiologica d’elezione delle frat-ture della base del cranio. Il trattamento di queste fratture è conser-vativo nella maggior parte dei casi.
La maggior parte delle fratture della base non necessitano di trattamento.
Tuttavia, possono associarsi a determinate complicanze che richiedono in-
vece un trattamento specifi co: aneurisma carotideo traumatico, fi stola caro-
tido-cavernosa postraumatica, fi stola del LCR, meningiti (anche in assenza di
fi stola del LCR), paralisi del nervo facciale ed altre ancora.
18.4. Commozione cerebrale
E’ la lesione traumatica cerebrale più frequente ma con minori ripercussioni
cliniche. Si defi nisce come un’alterazione del livello di coscienza transitoria
e di durata variabile, senza esiti permanenti, che consegue ad un trauma
non penetrante cerebrale. Si possono produrre diverse alterazioni compor-
tamentali del paziente, amnesia episodica, incoordinazione...che si recupe-
rano in un tempo variabile e generalmente breve. Non è stata dimostrata
alcuna alterazione anatomo-patologica né radiologica dell’encefalo, ma
solo una lieve disfunzione biochimica (diminuzione dell’ATP mitocondriale
o alterazione dei neurotrasmettitori eccitatori). Non necessita di trattamenti
specifi ci.
18.5. Ematoma epidurale
Appare nell’1-3% dei traumi cranici. E’ più comune tra la 2.ª e 3.ª decade
di vita, soprattutto tra i maschi. Gli incidenti stradali sono la causa più fre-
quente. L’85% dei casi sono di origine arteriosa, principalmente per rottura
dell’arteria meningea media dopo una frattura dell’osso temporale o pa-
rietale. Sono seguite, per frequenza, dai traumi del frontale o della fossa
posteriore. La presentazione clinica classica è: perdita di coscienza, seguita
da un periodo di lucidità (intervallo lucido). Successivamente si determina
un deterioramento neurologico con rapida evoluzione, in genere dovuto
ad erniazione uncale per un importante eff etto massa della raccolta ema-
tica. Tuttavia meno del 30% si presenta con la sequenza completa; spesso
non c’è la perdita di coscienza iniziale o c’è, ma senza l’ intervallo lucido
successivo.
La diagnosi si eff ettua mediante TC, che mostra un’immagine iperdensa al di
sotto del tavolato interno, con morfologia a “lente biconvessa”, che compri-
me il parenchima cerebrale sottostante(eff etto massa) (Figura 84).
Figura 84. Emorrragie intracraniche

87
18Neurologia e neurochirurgia
La maggior parte di questi traumi richiede evacuazione chirurgica urgente
con craniotomia. La mortalità, con trattamento precoce, è approssimativa-
mente del 10%.
18.6. Ematoma subdurale (Tabella 49)
E’ la conseguenza di un’emorragia venosa causata dalla rottura delle vene
a ponte corticali, o da una lacerazione del parenchima cerebrale. Si classi-
fi cano, in funzione del tempo di evoluzione dall’impatto, in acuto (i primi
3 giorni dopo il trauma), subacuti (tra i 3 giorni e le 3 settimane), cronico (a
partire dalle 3 settimane).
• Ematoma subdurale acuto. Rappresenta una delle lesioni traumatiche
con maggior morbimortalità (50-90% nonostante la chirurgia). General-
mente la forza dell’impatto è maggiore rispetto all’ematoma epidurale e
solitamente si accompagna a danno parenchimale sottostante, a causa
del quale presenta una peggiore prognosi. Si presenta con deteriora-
mento neurologico a rapida evoluzione. Si diagnostica tramite TC con
un’immagine iperdensa a forma di “semiluna” (Figura 85).
Ematoma epidurale Ematoma subdurale
Origine Sanguinamento arterioso (85%)
Più frequentemente per rottura dell’arteria meningea media
∙ Rottura delle vv. Corticali a ponte
∙ Acuto: prima settimana
∙ Subacuto: 7-10 gg post-TCE
∙ Cronico: TCE minore o non identificato nel 50%. Tipico di anziani ed alcolisti
Clinica Commozione cerebrale
Intervallo lucido
Erniazione uncale (coma a rapida evoluzione)
Anche se < 30% si presentano con la clinica classica
∙ Acuto: clinica d’ erniazione uncale progressiva, a rapida evoluzione
∙ Cronico: cefalea e demenza progressive (simile a ACV ischemico ma fluttuante)
TC ∙ Iperdensità a forma di lente biconvessa
∙ Frequentemente effetto massa
∙ Acuto: iperdensità a forma di “semiluna”
∙ Subacuto: isodenso
∙ Cronico: ipodensità a forma di “semiluna”
Lesione parenchima
In generale, minore e tardiva (per compressione)
∙ In generale, maggiore e dall’inizio (il sangue è in contatto con il parenchima cerebrale)
Mortalità Con diagnosi e trattamento precoce la mortalità è approssimativamente del 10%
∙ Le forme acute hanno una mortalità del 50-90%
Trattamento Evacuazione chirurgica mediante craniotomia
∙ Acuto: evacuazione chirurgica mediante craniotomia
∙ Cronico: evacuazione chirurgica con trapano, con o senza drenaggio subdurale
Tabella 49. Ematoma epidurale ed ematoma subdurale
Si raccomanda l’utilizzo di antiepilettici per il rischio di crisi epilettiche
precoci. Il trattamento richiede evacuazione chirurgica urgente attra-
verso craniotomia..
• Ematoma subdurale subacuto. Sono solitamente isodensi rispetto al
parenchima cerebrale, anche se raramente è utile ricorrere alla RM per
la diagnosi.
• Ematoma subdurale cronico. Si presenta soprattutto in pazienti di età
avanzata o alcolisti cronici, che presentano solitamente, un certo grado
di atrofi a cerebrale (con conseguente aumento dello spazio subdura-
le) e nei pazienti che utilizzano anticoagulanti. Il trauma scatenante è
spesso tanto lieve che né il paziente, né la famiglia lo ricordano.
Figura 85. (A) Ematoma epidurale con forma a lente biconvessa.(B) Ematoma subdurale acuto a forma di “semiluna”. Entambi causano un grande spostamento delle strutture della linea mediana
R I C O R D A L’immagine a “semiluna” è propria dell’ematoma subdurale acuto, e l’immagina a forma di lente “biconvessa” è caratteristica dell’e-matoma epidurale.
I sintomi ed i segni dell’ematoma subdurale cronico sono molto eterogenei
e possono simulare la clinica di altre patologie, come gli accidenti cerebrova-
scolari, tumori, encefalopatie metaboliche, demenza o psicosi. Predominano
le alterazioni dello stato mentale, l’emiparesi, le cadute frequenti e la cefalea.
Alla TC cerebrale sono ipodense (densità dei liquidi) e a forma di “semiluna”.
Se sono sintomatici richiedono evacuazione chirurgica, ma nei casi avanza-
ti, si possono evacuare attraverso alcuni fòri mediante trapano, lasciando o
meno un drenaggio continuo.
18.7. Contusione cerebrale emorragica
Sono lesioni necrotico-emorragicche intraparenchimali traumatiche (iper-
dense alla TC) (Figura 86) la cui localizzazione più fequente è il lobo frontale
(polo e superfi cie orbitaria), la porzione anterobasale del lobo temporale ed il
polo occipitale, zone più sensibili al danno da contraccolpo dovuto al movi-
mento brusco dell’encefalo all’interno della teca cranica per trauma sul bordo
osseo controlaterale. L’indicazione chirurgica dipenderà dalla localizzazione,
dalla grandezza e dallo stato neurologico del paziente. Richiede trattamento
con fenitoina (maggior percentuale di crisi focali precoci o tardive associate).

8818 ∙ Traumi c ran ioencefa l i c i
Manuale CTO di Medicina e Chirurgia, Prima edizione
Figura 86. Contusioni cerebrali emorragiche multiple (frecce)
18.8. Lesione assonale diff usa
E’ una lesione primaria del parenchima cerebrale che si trova in pazienti con
TCE con meccanismo rotazionale di accelerazione-decelerazione. Anatomo-
patologicamente, si riscontrano lesioni diff use degli assoni.Determina un
deterioramento cognitivo precoce e compromissione duratura del livello di
coscienza, senza che si abbia alla TC cerebrale una lesione che giustifi chi il
quadro.
La gravità del danno assonale viene determinata per la localizzazione delle
emorragie puntiformi alla TC cerebrale: grado I se si trovano alla giunzione
corticosubcorticale; grado II, a livello del corpo calloso; grado III,nella porzio-
ne dorsolaterale del tronco encefalico. Sono lesioni che, in genere, presenta-
no una prognosi infausta.
R I C O R D A Importante diminuzione del livello di coscienza dopo TCE; senza riscon-tri rilevanti alla TC; bisogna pensare ad una lesione assonale diff usa.
18.9. Complicanze e sequele dei neurotraumatismi dell’ encefalo
• Infezioni tardive nei traumi aperti (meningite postraumatica ricorren-
te, empiema, ascesso, trombofl ebiti, labirintite purulenta).
• Fistola del LCR.
• Crisi epilettiche postraumatiche.
• Fístola carotido-cavernosa, più frequente nei traumi della base o pe-
netranti. Può apparire inoltre in maniera spontanea. Si determina per
rottura parziale del sifone carotideo all’interno del seno cavernoso e si
manifesta con esoftalmo unilaterale o bilaterale pulsante, soffi o udibile
dal paziente stesso all’interno della testa (di solito è il sintomo iniziale),
ecchimosi congiuntivale importante e, a volte, lesione dei nervi cranici
oculomotori (più frequentemente del VI paio, che è l’unico localizzato
all’interno del seno cavernoso) o dei rami trigeminali del seno caver-
noso (I e II branca). La diagnosi viene confermata con l’angiografi a ed il
trattamento d’elezione è l’embolizzazione che deve essere eff ettuata in
quelle ad alto fl usso o se esistono alterazioni visive.
• Sindrome Soggettiva Postraumatica. Si manifesta tipicamente giorni
o mesi dopo traumi cranioencefalici lievi e si evidenzia con cefalee di
vario tipo, vertigini, irritabilità, ansia, defi cit di concentrazione o sintomi
pseudopsicotici con obiettività neurologica normale.
• Idrocefalia postraumatica. Questo quadro si caratterizza per la triade
di Hakim-Adams. Un 4% dei traumi cranioencefalici gravi possono com-
plicarsi con questo tipo di idrocefalia comunicante. La gestione diagno-
stica e terapeutica è simile alla forma idiopatica vista precedentemente
• Encefalopatia traumatica cronica. E’ una sequela cronica che com-
bina disturbi della personalità, cognitivi (bradipsichia e defi cit della
memoria) e motori (disfunzione cerebellare, parkinsonismo, alterazioni
della via piramidale) .
• Demenza postraumatica.
I d e e c h i ave La scala d Glasgow valuta il livello di coscienza di un paziente trau-matizzato in funzione di tre parametri: risposta verbale, motoria ed oculare. Il punteggio minimo è di 3 punti ed il massimo di 15.
I traumi cranioencefalici (TCE) si dividono in lievi (14-15 punti nella scala di Glasgow), moderati (9 - 13 punti) e gravi (3- 8 punti).
Mediante la storia clinica e l’esame obiettivo i soggetti che hanno subito un TCE devono essere classifi cati come pazienti con basso, moderato o alto rischio di avere una lesione intracranica, poichè la gestione è diversa in ognuno dei 3 gruppi.
Una frattura aperta implica una lacerazione della duramadre e porta ad un rischio elevato d’infezione intracranica. Nelle fratture composte, la duramadre è integra, per cui c’è un rischio basso di infezioni intracraniche.
Nelle fratture della base del cranio, può esistere un coinvolgimento dei nervi cranici, specialmente del I e del II nelle fratture della fossa cranica anteriore, del VII e VIII nelle fratture della rocca petrosa e del VI nelle fratture del clivus.
La fuoriuscita di LCR attraverso l’orecchio o le narici è un riscontro caratteristico, ma poco frequente, delle fratture della base del cra-nio.
L’ematoma epidurale si deve alla rottura dell’ arteria meningea me-dia causata da una frattura ossea. L’ematoma subdurale è di origine venoso, dovuto ad una lesione della corteccia cerebrale.
La clinica più frequente di presentazione di un ematoma epidura-le è quella dovuta ad erniazione uncale; tuttavia, è caratteristica la presenza di un intervallo lucido.
L’età avanzata, l’alcolismo e le alterazioni della coagulazione costi-tuiscono fattori di rischio per l’ematoma subdurale cronico. Solo nella metà dei casi si riconosce un antecedente traumatico.

89
18Neurologia e neurochirurgia
C a s i c l i n i c i Paziente di 25 anni, che ha subìto un TCE ad alta energia, è stato portato in ospedale in coma con un valore nella scala di Glasgow di 5 punti. Si eff ettuano diverse TC encefalo che risultano normali. Una RM eff ettuata a distanza di una settimana dall’incidente, ha evi-denziato una zona di contusione emorragica a livello dello splenio del corpo calloso. A distanza di un mese dal trauma, la situazione persiste immodifi cata, con un punteggio di 5 nella scala di Glasgow, presentando diversi episodi di iperidrosi ed iperpiressia e non evi-denziandosi altre lesioni rispetto a quella citata precedentemente nei successivi controlli radiologici:
1) La causa più frequente di coma continuo in un trauma cranico è lo stato epilettico, e bisognerebbe iniziare un trattamento mirato.
2) Credo che la situazione del paziente è dovuta a cause non neurolo-giche.
3) Credo che il paziente presenti una lesione assonale diff usa. 4) E’ impossibile che un paziente in coma presenti una TC normale.5) Si dovrebbe procedere all’evacuazione chirurgica della lesione del
corpo calloso.
RC: 3
Un bambino di 12 anni presenta una perdita di coscienza breve, dopo una caduta dalla bicicletta. All’arrivo in Pronto Soccorso, è orientato e presenta segni d’impatto nella regione parietale destra.
Due ore più tardi, lamenta cefalea di intensità rapidamente crescen-te, seguita da alterazione del livello di coscienza. La causa più pro-babile del deterioramento cognitivo, tra le seguenti, è:
1) Contusione cerebrale parietal destra.2) Ipertensione intracranica acuta secondaria ad edema cerebrale va-
sogenico.3) Ematoma extradurale acuto.4) Meningite pneumococcica fulminante, per breccia meningea se-
condaria a frattura della base cranica.5) Idrocefalia acuta secondaria a emorragia subaracnoidea postrau-
matica.
RC: 3
Un paziente che ha subìto un trauma cranico, arriva cosciente al Pronto Soccorso. Radiologicamente si apprezza una frattura lineare della volta cranica. A 12 ore dall’incidente, comincia ad avere riduzione progressiva del livello di coscienza ed asimmetria pupillare. Che diagnosi va fatta in prima battuta?
1) Un ematoma subdurale.2) Un ematoma epidurale.3) Una crisi epilettica postraumatica.4) Una meningite.5) Un coma metabolico iatrogeno.
RC: 2

909090
Neurologia e neurochirurgia
ORIENTAMENTOL’aspetto più importante di questo tema è conoscere gli elementi fondamentali dell’ascesso cerebrale.
19.1. Ascesso cerebrale
Si tratta di un processo suppurativo focale all’interno del parenchima cere-
brale. Durante la sua formazione si passa inizialmente per una fase di cerebri-
te nelle vicinanze del focolaio di necrosi con formazione, successivamente,
di una capsula di collagene con gliosi pericapsulare (Figura 87).
Figura 87. Ascessi cerebrali multipli con captazione ad anello
Eziopatogenesi
Si distinguono i seguenti meccanismi patogenetici (Tabella 50):
• Estensione per contiguità da un focolaio infettivo vicino. Si tratta
del meccanismo patogenetico più comune. Generalmente sono ascessi
unici.
19
• Disseminazione ematogena da un focolaio infettivo distante. Abi-
tualmente sono ascessi multipli e, occasionalmente, non si identifi ca il
focolaio emboligeno.
• Post-chirurgico e post-traumatico.
R I C O R D A L’origine più frequente degli ascessi cerebrali a disseminazione ema-tica, è l’ascesso polmonare negli adulti e la tetralogia di Fallot nei bambini.
Estensione per contiguità
∙ Sinusiti ∙ Otiti ∙ Mastoiditi ∙ Osteomieliti
Disseminazione ematica
∙ Polmonare (ascesso, empiema, bronchiectasie)
∙ Cardiopatie con shunt destro-sinistro (T. di Fallot)
∙ Fistole AV polmonari (Rendu-Osler-Weber)
∙ Osteomieliti
∙ Infezioni intraddominali (diverticoliti, colecistiti)
∙ Infezioni dentali
∙ Altre cause di batteriemia
Post-chirurgiche
Post-traumatiche
∙ Traumi penetranti
∙ Fratture della base del cranio
Tabella 50. Eziopatogenesi degli ascessi cerebrali
Microbiologia
In generale, i batteri più frequentemente implicati negli ascessi cerebrali
sono quelli della famiglia degli Streptococchi. Senza dubbio, nel caso degli
ascessi originati a partire da traumi cranioencefalici o da procedimenti chi-
rurgici, il più frequente è lo Staphylococcus aureus.
In un quarto dei casi, la coltura batterica è negativa, mentre in una percen-
tuale molto variabile (può variare da un 10% fi no ad un 90%) possiamo tro-
vare germi multipli.
ASCESSO CEREBRALE ED EMPIEMA SUBDURALE

91
19Neurologia e neurochirurgia
Clinica
L’ascesso cerebrale si manifesta con la tipica triade di: ipertensione endocra-
nica (la cefalea è il sintomo più frequente, riscontrandosi nell’80% dei casi),
febbre ed un quadro di focalità neurologica (sia in forma di defi cit neurologi-
co, che in forma di crisi epilettica). Senza dubbio, in molti casi non si presenta
in forma completa. L’emiparesi e le crisi convulsive si presentano nel 30-50%
delle occasioni, e la febbre, nel 50% dei casi.
Diagnosi
La sensibilità della TC con contrasto è vicina al 100% (lesione ipodensa che
capta contrasto ad anello circondata da un’alone di edema). La RM è più
sensibile della TC in fase di cerebrite.
R I C O R D A Le 5 lesioni che captano contrasto ad anello sono: metastasi, glio-blastoma multiforme, ascesso cerebrale, toxoplasmosi e linfoma.
A livello laboratoristico, si possono evidenziare: leucocitosi, aumento della
VES e della Proteina C Reattiva. La puntura lombare ha un basso valore dia-
gnostico e, in generale, non è indicata per il rischio di erniazione cerebrale.
Trattamento
Il trattamento dell’ascesso cerebrale deve combinare antibioticoterapia e
chirurgia evacuativa. Il trattamento della toxoplasmosi si eff ettua con sulfa-
diazina e pirimetamina.
La chirurgia evacuativa è indicata nelle lesioni uniche, accessibili, con sinto-
matologia d’ipertensione endocranica, eff etto massa o edema importante.
La mortalità è del 10% e le sequele neurologiche frequenti (emiparesi e crisi
epilettiche persistenti).
19.2. Empiema subdurale
E’ un processo suppurativo localizzato nello spazio subdurale cranico. Nel
75% dei casi sono unilaterali e, abitualmente, l’infezione origina per conti-
guità dai seni frontali, etmoidali o dall’orecchio medio (il microrganismo più
frequente è lo Streptococco). E’ rara la disseminazione ematica. Può essere
inoltre post-chirurgico o post-traumatico dovuto principalmente a Staphylo-
coccus aureus.
Si tratta di una malattia grave e progressiva, che, in generale, richiede un
trattamento urgente. Bisogna sospettarlo in pazienti con febbre alta, cefalea,
segni meningei e focalità neurologica unilaterale come nel quadro prece-
dentemente descritto. Possono manifestarsi inoltre, sintomi d’ipertensione
endocranica. Si diagnostica con TC con contrasto.
Il trattamento richiede l’evacuazione chirurgica urgente del materiale puru-
lento mediante craniotomia. La terapia va completata con antibioticotera-
pia. La mortalità si aggira intorno al 20% e sono frequenti le sequele neuro-
logiche.
I d e e c h i ave L’estensione per contiguità costituisce il meccanismo patogene-tico più frequente dell’ascesso cerebrale.
Esiste una triade clinica tipica di presentazione : clinica d’iperten-sionne endocranica, febbre e focalità neurologica.
Alla TC, è una delle lesioni che captano contrasto ad anello.
Il trattameento fondamentale consiste nell’ antibioticoterapia e l’evacuazione chirurgica della lesione.

929292
Neurologia e neurochirurgia
20.1. Dolore lombare
Il dolore al rachide è la causa più frequente d’incapacità in pazienti maggio-
ri di 45 anni. Si classifi ca, in funzione della durata, in dolore lombare acuto
(durata inferiore alle 6 settimane), subacuto (tra le 6 settimane ed i 3 mesi) e
cronico (più di 3 mesi).
La maggior parte delle lombosciatalgie derivano da uno sforzo eccessivo e
sono autolimitanti (lombalgie meccaniche). La valutazione iniziale deve es-
sere indirizzata all’esclusione di quelle eziologie gravi di dolore lombare che,
anche se infrequenti, possono richiedere trattamento immediato (traumi, in-
fezioni, tumori, sidrome della cauda equina). Per questo, bisognerà realizzare
una corretta anamnesi ed esame obiettivo, prestando particolare attenzione
alla presenza di fattori di rischio che facciano sospettare un’origine grave del
dolore, secondo le linee guida europee sulla gestione ambulatoriale delle
lombalgie acute (Tabella 51).
Fattori di rischio di patologia grave
∙ Primo episodio in minori di 20 anni o maggiori di 55 anni
∙ Defi cit neurologico diff uso (incluso la “sindrome della cauda equina”)
∙ Dolore non infl uenzato dalla postura, movimenti o sforzi, che non migliora con il riposo
∙ Dolore esclusivamente dorsale
∙ Deformità strutturale di recente insorgenza
∙ Trattamento prolungato con glucocorticoidi
∙ Antecedenti di trauma recente
∙ Cattivo stato generale
∙ Perdita di peso ingiustifi cata
∙ Diagnosi previa di cancro
∙ Febbre
∙ Storia d’immunosoppressione (trapianto, HIV...)
∙ Consumo di droghe per via parenterale
Tabella 51. Fattori di rischio di eziologia grave del dolore al rachide
20
R I C O R D A Nella lombalgia bisogna scartare tre processi eziologici importanti: infezioni, traumi e tumori.
In assenza di un sospetto fondato di un’eziologia grave del dolore non si
raccomanda la realizzazione di studi complementari.
La maggior parte dei pazienti con dolore al rachide tenderà al miglioramen-
to clinico nel giro di un mese, con o senza trattamento, per cui la gestione
iniziale di un paziente con dolore lombare senza fattori di rischio deve essere
conservativa, con l’obiettivo di determinare un miglioramento della sinto-
matologia (Tabella 52).
Il paziente deve essere correttamente informato sulla natura dei suoi disturbi.
Trattamento del dolore lombare
∙ Dare informazioni positive e tranquillizzare il paziente
∙ Non si raccomanda riposo a letto
∙ Raccomandare ai pazienti che si mantengano attivi e che continuino le normali attività quotidiane, incluso il lavoro.
∙ Farmaci di prima linea, se l’intensità del dolore lo richiede:
- Inizio con paracetamolo
- Se non migliora FANS (massimo 3 mesi)
- Se non migliora, aggiungere un ciclo di miorilassanti (meno di una settimana)
∙ Prescrivere esercizio fi sico a partire da 2-6 settimane
∙ Neurorefl essoterapia nei casi resistenti
Tabella 52. Opzioni terapeutiche nel trattamento del dolore al rachide
Il rimedio principalmente adottato nella fase acuta è stato tradizionalmente
il riposo assoluto a letto; tuttavia, studi recenti hanno dimostrato che il ripo-
PATOLOGIA RACHIMIDOLLARE
ORIENTAMENTO
Si tratta di uno dei temi fondamentali per il MIR, per cui bisogna pianificarne uno studio personalizzato. In particolar modo è necessario prestare molta attenzione alle seguenti parti del tema: attitudine diagnosticoterapeutica da adottare di fronte una lombalgia, lombosciatalgia o cervicobrachialgia, esplorazione delle differenti radici brachiali e crurali (molto importante lo schema del Manuale), la stenosi del canale lombare e la siringomielia.

93
20Neurologia e neurochirurgia
so a letto per più di 2 giorni porta a scarsi risultati. E’ utile la rieducazione po-
sturale, cercando di evitare quelle attività e posture che scatenano il dolore.
Il trattamento farmacologico si basa su farmaci analgesici (paracetamolo),
antinfi ammatori (fondamentalmente FANS) e miorilassanti (questi ultimi,
per non più di una settimana).
Se la sintomatologia persiste per più di 2-6 settimane, nonostante il trattamento
conservativo, o c’è un aumento d’intensità durante lo stesso periodo, è necessario
tornare a valutare il paziente in maniera completa, eff ettuando indagini diagno-
stiche e trattamenti specifi ci, se questo lo richiede. Quando il dolore persiste per
più di 12 settimane (3 mesi) si eff ettua la diagnosi di dolore lombare cronico. In
questi casi, una volta scartata la possibilità di patologie gravi all’esame obiettivo,
non si raccomanda nessuna prova diagnostica aggiuntiva, salvo che si sospetti
una causa specifi ca. In questi pazienti è necessario un programma di riabilitazione
multidisciplinare (unità di terapia del dolore, programmi educativi, esercizi, trat-
tamento psicologico) insieme ad un trattamento analgesico (antidepressivi trici-
clici, capsaicina, oppiacei in caso di fallimento dei trattamenti precedenti). Come
ultima opzione conservativa, si può consigliare neurostimolazione percutanea.
20.2. Lombosciatalgia. Ernia discale lombare
Il termine lombosciatalgia si utilizza per descrivere il dolore lombare irradiato
all’arto inferiore che depone per la compressione di una radice nervosa. La
causa più frequente di lombosciatalgia è l’ernia discale lombare.
Basi anatomiche (Figura 88)
Il disco erniato comprime gli elementi nervosi che scorrono nel canale mi-
dollare e può dar luogo ad una radicolopatia (per compressione della radice
nervosa) o una mielopatia (per compressione del midollo spinale, solo a li-
vello corticodorsale, non a livello lombare basso).
Le radici nervose fuoriescono dal canale vertebrale attraverso i forami di con-
giunzione. Le prime 7 radici cervicali escono dal canale vertebrale passando
al di sopra del margine superiore della vertebra omonima (la radice C6 esce
dal forame tra C5 e C6, quindi al di sopra della vertebra C6). La radice C8 esce
al di sotto della vertebra C7, tra essa e la D1; più in basso le radici dorsali,
lombari e sacrali, fuoriescono dal canale dal forame sottostante alla vertebra
corrispondente (la radice L4 esce perciò dal forame tra L4 ed L5). Ai livelli cer-
vicali, le ernie comprimono direttamente il forame, per cui a questi livelli, col-
picono clinicamente la radice del nome della vertebra sottostante allo spazio.
Nel rachide lombare, le ernie sono generalmente posterolaterali; vanno a
comprimere quindi,schicciandola contro la parete ossea, la radice che si pre-
para ad uscire e non quella che sta già uscendo che resta al di sopra dell’ernia.
Per questo, la radice che si manifesta clinicamente è la radice che “passa”, ovve-
ro quella che uscirà poi nel forame al di sotto; la radice danneggiata è dunque
quella con il nome della vertebra inferiore (un’ernia del disco tra le vertebre L4
ed L5 produce quindi una radicolopatia della L5). Se l’ernia fosse foraminale,
più rara, colpirebbe invece la radice che esce direttamente dal forame, ovvero
la sovrastante (la radice L4 nel caso di ernia del disco L4-L5) (Figura 88).
Clinica
• L’aspetto caratteristico dell’ernia discale lombare è che il dolore si irradia
all’arto inferiore (sciatica), a causa della compressione della radice nervosa.
Nelle radicolopatie compressive, il dolore aumenta tipicamente con
la manovra di Valsalva (tosse, defecazione). Si può riprodurre con varie
manovre esploratorie. La manovra di Lasègue (anche conosciuta come
manovra d’elevazione della gamba tesa dal piano del letto) consiste nel
sollevare la gamba stesa con il paziente in decubito supino, ed è positi-
va se compare dolore con un angolo minore di 60 gradi. La manovra di
Bragard è simile a quella di Lasègue, ma con dolore alla dorsifl essione
passiva del piede. Entrambe le manovre stirano fondamentalmente le
radici L5 ed S1.
• Radicolopatia (Figura 88). Le manfestazioni tipiche dell’interessamen-
to di ogni radice, nel caso dlle ernie posterolaterali, sono riassunte nella
Tabella 53.
Ernia discale
Anello fibroso
Radicecervicale
Nucleo polposo
Vertebra L4
Vertebra L5
Radice L5
Radice L5
Ernia discale posterolaterale L4-5
Ernia discale foraminale
L4-5
· Ernia discale cervicale (A): comprime la radice che esce dallo spazio· Ernie discali lombari (B): le ernie posterolaterali comprimono la radice che esce dallo spazio inferiore· Ernia foraminale: comprime la radice che esce dallo stesso spazio
Figura 88. Ernia discale cervicale (A) ed ernie discali lombari (B)

9420 ∙ Pato log ia rach im ido l l a re
Manuale CTO di Medicina e Chirurgia, Prima edizione
Diagnosi
Le indagini radiologiche devo essere richieste solo in pazienti con sospetto
clinico di una eziologia grave o che non rispondano adeguatamente alla
terapia medica o siano candidati alla chirurgia. L’ indagine d’elezione è la RM
(Figura 89).
Figura 89. RM di un’ernia discale lombare
Per valutare il livello, il grado e, soprattutto,il tipo (aprassia od assonotmesi
delle fi bre nervose) della lesione radicolare si utilizza l’elettromiografi a (EMG).
Trattamento
Il trattamento iniziale dell’ernia discale deve essere conservativo e porta ad
un miglioramento clinico nel 90% dei casi. Nei pazienti con sciatica, il riposo
a letto non si è dimostrato effi cace nel migliorare il dolore o l’impotenza
funzionale.
Quando la prima linea di trattamento non risulta effi cace è indicato il tratta-
mento chirurgico (Tabella 54). La tecnica chirurgica d’elezione è la fl avecto-
mia con asportazione del disco colpito (discectomia o microdiscectomia) .
Indicazioni al trattamento chirurgico
∙ Lesione di una radice che comporta una perdita acuta o progressiva della forza, obiettivabile clinicamente o con EMG (“ernie paralizzanti”). E’ indicazione alla chirurgia urgente.
∙ Segni clinici suggestivi di una sindrome della cauda equina o di lesioni midollari (disfunzione sfi nteriale, anestesia perineale a sella...) E’ indicazione alla chirurgia urgente.
∙ Fallimento del trattamento conservativo, ovvero, dolore invalidante, con caratteristiche radicolari, che non risponde al trattamento medico durante un periodo di 4 settimane.
∙ Invalidità recidivante nonostante il trattamento medico
Tabella 54. Indicazioni alla chirurgia nell’ernia discale lombare
20.3. Cervicobrachialgia. Ernia discale cervicale
Il termine cervicobrachialgia si utilizza per descrivere il dolore cervicale irra-
diato all’arto superiore.
A livello cervicale, le ernie discali si sviluppano preferenzialmente negli spazi
C5-C6 e C6-C7 (ernia cervicale più frequente) e, come le lombari, sono ge-
neralmente localizzate posterolateralmente. La patogenesi è la stessa che
a livello lombare. All’esame obiettivo, la cervicobrachialgia, mostra il segno
di Spurling (l’esaminatore attua una pressione sopra il vertice cranico con la
testa estesa e ruotata verso il lato sintomatico; è positivo se sviluppa dolore).
L’abduzione della spalla (portando la mano sopra la testa) tende ad allevia-
re il dolore. Le manifestazioni di radicolopatia cervicale sono riassunte nella
Tabella 55.
La tecnica d’immagine d’elezione nella patologia cervicale è la RM. L’EMG
può aiutare a stabilire la radice colpita e l’entità del danno.
La RM è indicata nei casi in cui non si abbia miglioramento con il trattamen-
to conservativo, nei casi con defi cit neurologico o nei casi in cui esistano
segni di interessamento mielico.
La maggior parte dei pazienti con cervicobrachialgia (95%) migliora dopo
trattamento conservativo (analgesici, antinfi ammatori e miorilassanti) e non
è necessario eff ettuare indagini radiologiche inizialmente.
Livello dell’ernia discale
L1-L2 L2-L3 L3-L4 L4-L5 L5-S1
Radice abitualmente colpita
L2 L3 L4 L5 S1
Rifl esso alterato Rotuleo A volte, raramente, l’achilleo Achilleo
Defi cit motorio Flessione anca (psoas)
∙ Flessione anca (psoas)
∙ Estensione ginocchio (quadricipite)
∙ Estensione ginocchio (quadricipite)
∙ Dorsifl essione del piede (tibiale anteriore)
∙ Estensione dell’alluce (1° dito )
Flessione plantare del piede (gastrocnemio e soleo)
Defi cit sensitivo Faccia anteriore coscia
Faccia anteriore coscia e ginocchio
∙ Ginocchio e faccia anteromediale della gamba
∙ Malleolo mediale
∙ Bordo mediale del piede
Faccia anterolaterale della gamba. Dorso del piede fi no al 1° dito
∙ Malleolo esterno
∙ Pianta e bordo laterale del piede fi no al 5.º dito
Tabella 53. Esplorazione delle radici nervose del plesso lombosacrale

95
20Neurologia e neurochirurgia
R I C O R D A In uno spazio intervertebrale lombare, emerge la radice della ver-tebra sovrastante, mentre è la radice che fuoriesce dall’interspazio sottostante ad essere generalmente colpita nel caso di un’ernia discale a questo livello; invece in uno spazio intervertebrale cervi-cale, emerge e viene colpita la radice corrispondente alla vertebra sottostante .
Le indicazioni alla chirurgia dell’ernia discale cervicale sono simili alla lomba-
re; si riserva a tutti i casi resistenti al trattamento medico o, precocemente,nei
casi con mielopatia o interessamento radicolare importante che implica un
defi cit motorio. La tecnica chirurgica d’elezione è la discectomia anteriore
con innesto intersomatico osseo o metallico (tecnica di Cloward e di Smith-
Robinson) (Figura 90).
Figura 90. Discectomia cervicale con innesto intersomatico (tecnica di Cloward)
Se esistono segni di mielopatia cervicale, in relazione a spondilosi cervicale
o con uno o più dischi erniati, si può programmare una corporectomia con
innesto e placca cervicale anteriore, una laminoplastica o una laminectomia
posteriore.
20.4. Stenosi del canale lombare
La rachistenosi è una riduzione del diametro anteroposteriore del canale
vertebrale, che può produrre compressione o compromissione vascolare
delle radici della cauda equina. Pertanto, è una diagnosi anatomica che
si stabilisce attraverso le indagini di “imaging” (RM, TC o mielografi a-TC)
(Figura 91).
Figura 91. TC di una rachistenosi lombare
Può essere congenita o acquisita (spondilosi, spondilolistesi, Paget, acrome-
galia, postraumatica…), anche se più frequentemente si tratta di una stenosi
acquisita su una stenosi congenita previa. E’ una patologia la cui incidenza
aumenta con l’età. E’ più frequente a livello dello spazio L4-L5.
La rachistenosi lombare produce tipicamente un dolore lombare, lombo-
sciatalgie (con frequenza e bilaterali), ed è la causa più frequente di claudica-
zione neurogena dell’estremità inferiori, (dolore lombare, alle natiche e alle
gambe, sia camminando sia in posizione fi ssa, che migliora con il riposo).
E’ necessario stabilire la diagnosi diff erenziale con la claudicatio di origine
vascolare (Tabella 56). Il dolore aumenta con l’iperestensione della colonna
e, a diff erenza dell’ernia discale, recede in posizione seduta (con la fl essione
della colonna) ; per questo, i pazienti tendono ad adottare una postura an-
tropoide, anche denominata postura a “carrello del supermercato.
Livello dell’ernia discale
C4-C5 C5-C6 C6-C7 C7-D1
Radice abitualmente colpita C5 C6 C7 C8
Rifl essi alterati Bicipitale Bicipitale
Stiloradiale
Tricipitale Tricipitale
(a volte)
Defi cit motorio Abduzione e fl essione della spalla
∙ Flessione del gomito
∙ Estensione del polso
∙ Estensione del gomito
∙ Flessione del polso
∙ Flessione delle dita
∙ Muscolatura intrinseca della mano
Defi cit sensitivo Spalla e faccia laterale del braccio
Faccia laterale dell’avambraccio fi no al 1° e, meno, 2° dito
Faccia dorsale dell’avambraccio fi no al 3°dito, bordo radiale del 4° dito e mediale del 2°
5.º dito e lato ulnare del 4.º Faccia mediale (ulnare) dello avambraccio
Tabella 55. Esplorazione delle radici nervose del plesso brachiale

9620 ∙ Pato log ia rach im ido l l a re
Manuale CTO di Medicina e Chirurgia, Prima edizione
Il trattamento iniziale è conservativo. La chirurgia è indicata in maniera
urgente nei casi di paresi rilevante bilaterale e nella sindrome della cauda
equina o in maniera programmata, quando la claudicazione persiste per più
di 6 mesi nonostante il trattamento conservativo. La chirurgia consiste nel
decomprimere il canale mediante laminectomia (Figura 92).
Figura 92. Laminectomia lombare
20.5. Spondilolistesi
Si defi nisce come uno scivolamento anteriore della vertebra superiore rispet-
to all’inferiore. In funzione della percentuale di spostamento, è stata classifi -
cata in 5 gradi: grado I, quando lo scivolamento è minore del 25%; grado II,
tra il 25-50%; grado III, 50-75%; grado IV, 75-100%, e grado V o spondiloptosi,
quando la vertebra superiore supera in tutto il suo spessore l’inferiore, e per-
tanto, bascula tendendo alla verticalizzazione. Le spondilolistesi dei gradi I e II
si denominano di basso grado, mentre dal grado III al V sono spondilolistesi di
alto grado. In funzione del meccanismo patogenetico, la spondilolistesi può
essere classifi cata in 5 tipi, mostrati nella Tabella 57.
• Tipo II, istmiche o spondilolisi. Sono prodotte da un’alterazione del-
la pars interarticularis (frattura o allungamento). Sono le spondilolistesi
più frequenti, e si trovano soprattutto a livello L5-S1. La loro incidenza
aumenta con l’età. La clinica consiste in lombalgie e sintomi radicolari.
Nei casi in cui i sintomi non migliorino con il trattamento conservativo
o si noti una progressione della deformità, si procederà a pianifi care un
trattamento chirurgico.
• Tipo III o degenerative. Si devono a processi degenerativi discali e di
altre strutture del segmento vertebrale, come il legamento giallo. Sono
più frequenti nelle donne a partire dai 50 anni. Il punto più colpito è
L4-L5. Da un punto di vista clinico possono provocare claudicazione
neurogena, dovuta alla stenosi prodotta dallo spostamento, lombalgia
meccanica e radicolopatia, per la compressione della radice nervosa a
livello del forame di congiunzione. Se questi sintomi persistono per più
di 3 mesi, nonostante il trattamento medico, o interferiscono con la vita
del paziente o si stabilisce un defi cit neurologico progressivo o il pazien-
te riferisce la comparsa di sintomi agli sfi nteri, è indicato il trattamento
chirurgico, che consiste in una laminectomia decompressiva con fusio-
ne intersomatica.
Tipi di spondilolistesi lombari
Tipo I displastica Deficienza congenita faccetaria
Tipo II ístmica o spondilolisi
Frattura o allungamento della pars interarticularis
Tipo III degenerativa Degenerazione delle strutture della colonna
Tipo IV traumatica Frattura che non colpisce la pars
Tipo V patologica Malattia ossea, che colpisce la pars o il peduncolo
Tabella 57. Spondilolistesi lombare secondo il meccanismo patogenetico
R I C O R D A La spondilolistesi più frequente è la tipo II o spondilolisi o spondi-lolistesi istmica, che colpisce soprattutto L5-S1. La degenerativa (tipo III) colpisce soprattutto L4-L5.
20.6. Spondilodisciti
Si defi nisce spondilodiscite un’infezione del disco e della vertebra adiacente.
La spondilodiscite più comune è quella della regione lombare. Il microrgani-
smo più coinvolto nell’infezione del disco intervertebrale è lo Staphylococcus
aureus.
Il sintomo più frequente d’esordio è il dolore lombare, che aumenta con
qualsiasi movimento, si allevia con il riposo ed abitualmente è ben localizza-
Claudicazione neurogena Claudicazione vascolare
Distribuzione del dolore Territorio di un nervo (dermatomero) Gruppi muscolari con irrorazione comune
Fattori scatenanti ∙ Esercizio d’intensità variabile
∙ Mantenimiento prolungato di una postura
∙ Al mettersi in piedi , prima dell’inizio della marcia
∙ Esercizio con intensità costante, esordio dolore per sforzi via via meno intensi man mano che la malattia avanza
∙ Raro in piedi senza camminare
Distanza da camminare prima dell’apparizione dei sintomi
Variabile Costante
Miglioramento con il riposo ∙ Lento
∙ Dipendente dalla postura (meglio in fl essione della colonna)
∙ Immediato
∙ Non dipende dalla postura
Polsi periferici Conservati Diminuiti o assenti
Pallore cutaneo al sollevare gli AAII No Marcato
Temperatura negli AAII Normale Diminuita
Tabella 56. Diagnosi diff erenziale tra claudicazione neurogena e vascolare

97
20Neurologia e neurochirurgia
to a livello del disco aff etto. La febbre è un sintomo incostante. É raro trovare
anomalie all’esame neurologico. Ci sono scarsi rilievi laboratoristici che indi-
chino un’infezione.
La diagnosi radiologica d’elezione è la RM (Figura 93). La diagnosi defi nitiva
si eff ettua mediante studio microbiologico o istologico del materiale discale,
ottenuto con puntura-aspirazione con ago.
Figura 93. RM tipica di spondilodiscite con coinvolgimento dello spazio intervertebrale ed edema osseo che interessa i corpi vertebrali sovra e sottostanti.
Il trattamento consiste nell’immobilizzazione (riposo a letto e successiva-
mente immobilizzazione con corsetto) ed antibioticoterapia ev. prolun-
gata (4-6 settimane) seguita da un periodo di antibiotici orali di analoga
durata.
20.7. Tumori intrarachidei
Rappresentano un 15% dei tumori primitivi del SNC. Anche se i tumori spinali
più frequenti sono le metastasi, la maggior parte dei tumori primitivi sono
benigni, a diff erenza dei tumori cranici, e tendono a dare una clinica di com-
pressione piuttosto che d’invasione.
I tumori rachidei si classifi cano in 3 gruppi (Tabella 58) (Figura 94):
Extradurali ∙ Metastasi
∙ Cordoma
Intradurali extramidollari ∙ Neurinoma
∙ Meningioma
Intradurali intramidollari ∙ Astrocitoma
∙ Ependimoma
Tabella 58. Tumori intrarachidei
A B
C D
Figura 94. (A) Metastasi epidurale. (B) Neurinoma cervicale. (C) Meningioma dorsale. (D) Astrocitoma cervicale
• Extradurali (i più frequenti, 55%). Crescono nel corpo vertebrale e/o
nello spazio epidurale. Anche se le metastasi si possono trovare in qual-
siasi altro gruppo, di solito hanno localizzazione extradurale.
Un altro tumore inquadrato all’interno di questo gruppo è il cordoma
sacrococcígeo (cellule “fi salífore”, tipiche in anatomía patologica).
• Intradurali extramidollari (40%). In generale, crescono a partire dalle
radici nervose (neurinomi) o dalle leptomeningi (meningiomi).
I neurinomi sono i tumori primitivi intrarachidei più frequenti. Provocano
dolore e defi cit neurologico nel territorio della radice sulla quale cresco-
no.
I meningiomi sono più frequenti nelle donne, più nella regione toraci-
ca. Provocano dolore (rachialgia intercostale) e compressione midollare.
Entrambi si diagnosticano attraverso la RM, ed il trattamento d’elezione
è la chirurgia, che è curativa.
• Intramidollari (5%). Crescono infi ltrando e distruggendo la sostanza
grigia e bianca midollare: astrocitomi (tumore intramidollare più fre-
quente al di fuori del fi lum terminale). In entrambi i casi, il trattamento
è chirurgico (microchirurgia), anche se non sempre è possibile l’aspor-
tazione completa.
R I C O R D A Il trattamento di una metastasi intrarachidea (che sia chirurgico o radioterapico) è palliativo, attraverso una decompressione midollare.
R I C O R D A Il tumore intramidollare più frequente nei bambini è l’astricitoma, mentre negli adulti è l’ependimoma.

9820 ∙ Pato log ia rach im ido l l a re
Manuale CTO di Medicina e Chirurgia, Prima edizione
20.8. Ascesso epidurale spinale
Si tratta di una raccolta purulenta nello spazio epidurale spinale. Si localizza
con maggior frequenza a livello dorsale (50%), seguito dal segmento lomba-
re (35%) e cervicale (15%). Con frequenza si associa a osteomielite o discite.
Il microrganismo più frequentemente implicato nelle forme acute è lo
Staphylococcus aureus; nelle forme croniche, Mycobacterium tuberculosis.
Generalmente si presenta con febbre elevata, dolore e rigidità al rachide.
Si manifesta spesso con sintomi radicolari, ad evoluzione progressiva fi no
ad una compressione midollare con disfunzione degli sfi nteri, paraparesi o
tetraparesi.
La indagine d’immagine d’elezione è la RM (massa epidurale che comprime
il sacco durale) (Figura 95).
Figura 95. RM di un ascesso epidurale cervicale, dove si osserva una massa (segnalata da una freccia bianca) che occupa lo spazio epidurale e cervicale e che comprime il midollo, soprattutto la parte posteriore
R I C O R D A La spondilodiscite e l’ascesso epidurale sono spesso associati. VES e PCR sono utili marcarori per il follow up dell’infezione
Il trattamento consiste nell’immobilizzazione, antibioticoterapia (in maniera
empirica, si raccomanda si cominciare con una cefalosporina di III genera-
zione associata a vancomicina o rifampicina) per 6-8 settimane, e chirurgia
in caso di defi cit neurologico (anche se alcuni autori raccomandano chirur-
gia anche in assenza di defi cit motori). La prognosi è infausta, con elevata
mortalità e sequele neurologiche frequenti (maggiori quanto peggiore è la
situazione neurologica del pazinete al momento della diagnosi).
20.9. Siringomielia
E’ defi nita come l’esistenza di cavità cistiche, anche chiamate “siringhe”, nel
midollo spinale, che possono comunicare o meno con il canale ependimale
centrale. Generalmente si localizzano a livello cervicale e/o dorsale (Figura
96). In alcuni casi si estendono rostralmente fi no a raggiungere il bulbo, de-
nominandosi quindi come siringobulbia.
Figura 96. Siringomielia. RM in cui si apprezza la cavità siringomielica cervicodorsale
Si associa spesso a malformazioni congenite (soprattutto Chiari tipo I), neo-
plasie midollari (fondamentalmente astrocitomi) aracnoiditi e traumi (sirin-
gomielia postraumatica).
Dà luogo ad un quadro clinico tipico, caratterizzato da una sindrome centro-
midollare, con defi cit sospeso e dissociato della sensibilità (abolizione della
sensibilità termodolorifi ca, rispettando le vie sensitive dei cordoni posterio-
ri). La prova diagnostica d’elezione è la RM. Quando è chiaramente sintoma-
tica o il quadro clinico progredisce nei controlli successivi, si può optare per
un trattamento chirurgico.
Nei casi associati a malformazioni di Chiari, il trattamento d’elezione è la cra-
niectomia decompressiva suboccipitale con plastica durale, per aumentare
la grandezza della fossa posteriore. In altri casi si realizzano delle derivazioni
siringosubaracnoidee o siringoperitoneali. L’obiettivo fondamentale del trat-
tamento è evitare la progressione del defi cit neurologico.

99
20Neurologia e neurochirurgia
I d e e c h i ave Di fronte ad un paziente con lombalgia, lombosciatalgia o cervico-brachialgia, senza fattori di rischio per un’eziologia grave, senza de-fi cit motori né sindrome della cauda equina (nel caso di lomboscia-tica) o mielopatia (nel caso di cervicobrachialgia), si deve iniziare un trattamento sintomatico senza realizzare alcuna prova diagnostica aggiuntiva; se dopo 6 settimane il quadro non recede, bisogna pia-nifi care uno studio a fi ni diagnostici.
L’intensità della lombosciatalgia aumenta con le manovre di Valsal-va. Il Lasègue si considera positivo se la lombosciatalgia si manifesta ponendo la gamba a meno di 60° rispetto alla posizione orizzontale.
Il riposo a letto non si è dimostrato utile nel trattamento della lom-balgia e lombosciatalgia.
La radicolopatia L4 colpisce la faccia anteromediale della gamba, il rifl esso rotuleo e l’estensione del ginocchio. La radicolopatia L5 colpisce la faccia anterolaterale della gamba, il dorso del piede e la fl essione dorsale del piede. La radicolopatia S1 colpisce la faccia posteriore della gamba e pianta del piede, il rifl esso achilleo e la fl essione plantare del piede.
Sono indicazioni all’intervento chirurgico di una lombosciatica, il fallimento del trattamento conservativo e le lombosciatiche che si accompagnano a defi cit motorio o sindrome della cauda equina.
La radicolopatia C6 colpisce la faccia laterale dell’avambraccio ed il primo e secondo dito, i rifl essi bicipitale e stiloradiale, la fl essione
del gomito e l’estensione del polso. La radicolopatia C7 colpisce il terzo dito, il rifl esso tricipitale, l’estensione del gomito e la fl essione del polso. La radicolopatia C8 colpisce la faccia mediale dell’avam-braccio, il quarto e quinto dito e la mobilità intrinseca delle dita del-la mano.
Sono indicazioni alla chirurgia di una cervicobrachialgia il fallimen-to del trattamento conservativo e quelle che sono accompagnate da defi cit motorio o mielopatia.
La stenosi del canale lombare è un processo tipico delle persone di età avanzata. Si localizza con maggior frequenza a livello di L4-L5.
La claudicazione neurogena è tipica dele stenosi del canale lomba-re. La sintomatologia aumenta con l’esercizio e recede alla fl essione del tronco.
Il tumore intrarachideo più frequente è la metastasi ed il primitivo più frequente, il neurinoma. Le metastasi costituiscono la causa più frequente di compressione midollare.
S. aureus costituisce il microrganismo più frequentemente isolato dagli ascessi epidurali spinali.
La clinica di un ascesso epidurale spinale è progressiva: inizia con dolore nella regione aff etta, e nel giro di pochi giorni si manifestano i segni neurologici.
La clinica tipica della siringomielia è la dissociazione della sensibili-tà in maniera sospesa, poichè tende a colpire le estremità superiori e rispettare le inferiori.
C a s i c l i n i c i Un uomo di 80 anni riferisce, da circa 9 mesi, dolore intermitten-te alle gambe e parestesie che si manifestano dopo aver cammi-nato 100-200 metri. I sintomi cominciano nelle zone distali delle estremità inferiori, salgono poi a livello gluteo, accompagnati da dolore lombare. Gli episodi sono più frequenti quando cammina in discesa più che in salita, e si alleviano con il sedersi o nell’ap-poggiarsi sui talloni o sporgendosi in avanti mentre cammina. L’e-same neurologico è normale. Quale delle seguenti diagnosi è la più probabile?
1) Disco toracico erniato.2) Stenosi spinale lombare.3) Stenosi dell’ arteria iliaca.4) Miastenia gravis.5) Neuropatia periferica demielinizzante.
RC: 2
Un paziente di 62 anni presenta una storia clinica di cervicalgia irra-diata alle spalle. Da un anno, presenta diffi coltà progressiva a cam-minare, con dolore al braccio destro. All’esame obiettivo, presenta un rifl esso bicipitale abolito ed alcuni rifl essi osteotendinei polici-netici alle gambe.
1) Credo possa avere un tumore midollare, e richiederei una RM cer-vicale.
2) Credo che abbia un’ernia discale con spondilolisi, e richiederei una RM cervicale.
3) Probabilmente presenta una siringomielia e richiederei una RM.4) Credo che abbia una spondilosi cervicale, e richiederei una TC della
colonna cervicale.
5) Credo che uno studio radiologico semplice della colonna ed un trattamento con FANS sia adeguato.
RC: 2
Un muratore subisce un incidente sul lavoro, precipitando da 6 metri di altezza. Presenta un importante dolore a livello lombare e defi cit dell’estensione contro gravità delle dita del piede destro. Bisognerà pensare a:
1) Una lesióne della radice L-3.2) Una lesióne della radice L-4.3) Una lesióne della radice S-1.4) Una lesióne della radice L-5.5) Una lesióne della radice S-2.
RC: 4
Donna di 35 anni, senza precedenti di rilievo, si presenta alla no-stra osservazione per un dolore intenso alla colonna cervicale da 2 giorni, che le impedisce lo svolgimento del suo lavoro di ausiliare amministrativo. Il dolore si irradia verso la spalla destra, aumenta con i movimenti di fl essione e rotazione del collo, e non presenta altre manifestazioni cliniche. L’esame obiettivo generale e neuro-logico risultano normali anche se la valutazione della forza degli arti superiori è stata infi ciata dalla presenza di dolore. Qual’è il comportamento più adeguato da mantenere?
1) Riposo assoluto e collare rigido per 30 giorni.2) RM nucleare urgente e trattare di conseguenza.3) Paracetamolo con codeina, educazione posturale, esercizi lievi, e
seguire l’evoluzione per 7 giorni.4) Radiologia semplice e, nel caso trovassimo rettifi cazione della cur-
vatura fi siologica della colonna, trazione cervicale.

10020 ∙ Pato log ia rach im ido l l a re
Manuale CTO di Medicina e Chirurgia, Prima edizione
5) Radiologia convenzionale e, in assenza di riscontri signifi cativi, ef-fettuare una EMG.
RC: 3
Uomo di 34 anni, senza precedenti di rilievo. Presenta da una setti-mana, dolore nella zona lombare bassa, che non gli ha impedito di svolgere la normale attività lavorativa. Nelle ultime 24 ore, il dolore è aumentato, fi no a diventare severo ed invalidante, rendendogli diffi cile anche la deambulazione o alzarsi dal letto. Il paziente si presenta in Pronto Soccorso, dove si riscontra un normale esame obiettivo generale ed un esame neurologico reso diffi cile dalla pre-senza di dolore, senza alterazioni della sensibilità. Con Lasegue a 60°, Bragard negativo e rifl essi osteotendinei conservati e simme-trici nei 4 arti. Quale comportamento è più adeguato nello studio e nel trattamento di questo paziente?
1) Si realizza un Rx semplice della colonna lombare, che, se normale ci da la diagnosi di lombalgia acuta; si prescrivono analgesici di II livello, miorilassanti, riposo a letto per 2 settimane, e si valuta l’evo-luzione alla fi ne del periodo di riposo.
2) Si fa diagnosi di lombalgia acuta, non si procede a nessuno studio complementare, s’informa il paziente e la sua famiglia sulla pato-logia di cui è aff etto, si prescrivono analgesici di II livello, miorilas-sante, mobilizzazione precoce e si valuta l’evoluzione ad una setti-mana.
3) Si fa diagnosi di ernia discale e si ricovera per il trattamento chirur-gico.
4) Si realizza una TC urgente della colonna lombare, che se normale pone indicazione al ricovero del paziente per completare il quadro con uno studio con RM ed isotopi per sospetta neoplasia o infesio-ne.
5) Si realizza una TC ed una RM urgenti, se risultano normali si consulta uno psichiatra per scartare una componente funzionale.
RC: 2
Un uomo adulto presenta un quadro di dolore e rigidità cervicale, con irradiazione del dolore all’estremità superiore destra, faccia dorsale dell’avambraccio,e terzo dito associato a debolezzza dei fl essori del polso e diminuzione del rifl esso tricipitale. Dopo radiologia conven-zionale e risonanza magnetica, si fa diagnosi di ernia discale cervicale. Il disco erniato si trova a livello di:
1) C1-C2.2) C2-C3.3) C3-C4.4) C4-C5.5) C6-C7.
RC: 5
Paziente di 60 anni che presenta febbre elevata, dolore al rachide e paraparesi. Si realizza una RM della colonna che mostra una massa epidurale che comprime il midollo dorsale. Quale delle seguenti, è la dignosi più probabile?
1) Meningioma dorsale.2) Metastasi epidurale.3) Ematoma epidurale.4) Ascesso epidurale.5) Infarto midollare.
RC: 4

101
Neurologia e neurochirurgia
ORIENTAMENTO
Il tema delle anomalie dello sviluppoha molto poca importanza. Basta solo avere un’idea chiara sui concetti base delle differenti forme di craniosinostosi,e delle malformazioni di Chiari.
21.1. Craniosinostosi
Anche chiamate craniostenosi, sono deformità craniche che si formano dalla
chiusura precoce di una o più suture cartilaginee che separano le ossa mem-
branose del cranio. Si classifi cano a seconda della sutura che si chiude più
precocemente (Figura 97).
• Scafocefalia o dolicocefalia: chiusura precoce della sutura sagittale. E’
la più frequente.
• Brachicefalia o turricefalia: chiusura precoce della sutura coronale
bilaterale. Partecipa frequentemente alle sindromi autosomiche domi-
nanti con dismorfi smi facciali.
• Plagiocefalia anteriore: chiusura precoce della sutura coronale unila-
teralmente.
21 • Plagiocefalia posteriore: chiusura precoce della sutura lambdoidea.
In molte occasioni, la deformità è di origine posturale e non una vera
chiusura precoce.
• Trigonocefalia: chiusura precoce della sutura metopica. E’ in rela-
zione maggiormente con anomalie encefaliche, come la oloprosen-
cefalia.
• Ossicefalia: chiusura precoce di molteplici o tutte le suture craniche
(cranio a torre) provocando ipertensione endocranica.
R I C O R D A La maggior parte delle plagiocefalie posteriori sono posturali. La loro incidenza sta aumentando, dovuta alla raccomandazione di far dormire i neonati in decubito supino per evitare la sindrome del-la morte improvvisa.
La diagnosi si formula alla nascita attraverso l’osservazione della deformità
cranica. Attraverso la palpazione , si può apprezzare una cresta ossea al di so-
pra della sutura precocemente serrata. La chiusura della sutura si conferma
mediante tecniche d’imaging (Rx cranica o TC-3D).
ANOMALIE DELLO SVILUPPO
Figura 97. Craniostenosi

10221 ∙ Anoma l i e de l l o sv i l uppo
Manuale CTO di Medicina e Chirurgia, Prima edizione
I d e e c h i ave La scafocefalia è la craniosinostosi più frequente. La brachicefalia può associarsi a dismorfi smi facciali.
La malformazione di Chiari tipo I è la più frequente. Si tratta di una discesa delle amigdale cerebellari attraverso il forame magno. Può associarsi a siringomielia.
La malformazione di Chiari tipo II si associa a mielomeningocele e ad idrocefalia.
Il trattamento d’elezione è la ricostruzione chirurgica, spesso con la parteci-
pazione congiunta di neurochirurghi e chirurghi maxillofacciali, per la rico-
struzione delle dismorfi e facciali associate. Eccetto che nelle forme più gravi,
in quelle cioè in cui la chiusura di suture multiple può rendere diffi cile la cre-
scita del cervello, la maggior parte delle volte l’indicazione chirurgica è fon-
damentalmente estetica. Alcune forme posturali di plagiocefalia posteriore
possono essere corrette con “caschi” ortopedici che rimodellano il cranio.
21.2. Malformazioni di Chiari
Chiari tipo I
Consistono in una discesa ed allungamento delle amigdale cerebellari al di
sotto del piano del forame magno. E’ frequente l’associazione con siringo-
mielia. Solitamente esordisce in adolescenza e nell’età adulta (età media 40
anni) ed è leggermente più frequente nelle femmine. Il sintomo abituale di
presentazione è la cefalea suboccipitale, che aumenta con la monovra di
Valsalva, seguita talvolta da sintomi d’interessamento troncale, cerebellare
o centromidollare (con maggior frequenza agli arti superiori). Il nistagmo
verticale è un segno tipico.
La tecnica diagnostica d’elezione è la RM. Ultimamente, si stanno impiegan-
do alcuni studi sul fl usso del LCR mediante RM, nei pazienti in cui si riscon-
trano problemi di circolazione a livello del forame magno
Il trattamento d’elezione per i pazienti con Chiari tipo I sintomatici, o asso-
ciati a siringomielia, è la craniectomia decompressiva suboccipitale (che si
può implementare con una laminectomia di C1 e C2) con apertura della
dura madre e collocamento di una plastica durale per ampliare lo spazio
della fossa posteriore.
R I C O R D A Una cefalea suboccipitale nelle persone intorno ai 30 anni, che aumenta con la manovra di Valsalva, è suggestiva di una malformazione Chiari tipo I.
Quando si associa ad idrocefalia , bisogna posizionare una derivazione del
LCR. I pazienti asintomatici devono essere tenuti sotto osservazione nel
tempo ed operati solo nei casi di peggioramento cognitivo.
Chiari tipo II
E’ una discesa del verme cerebellare, quarto ventricolo, protuberanza e bul-
bo, al di sotto del piano del forame magno. Si associa frequentemente con
mielomeningocele ed idrocefalia. Ha esordio generalmente nell’infanzia. Le
manifestazioni cliniche sono dovute a disfunzione del tronco e dei nervi
cranici bassi. Si manifesta con stridore respiratorio, apnea episodica, aspira-
zioni frequenti, retrocolli e/o segni cerebellari. Si diagnostica mediante RM.
E’ necessario posizionare una derivazione ventricoloperitoneale per l’idro-
cefalia e realizzare una decompressione ampia della fossa posteriore. Le dif-
fi coltà respiratorie sono la principale causa di elevata morbimortalità della
malformazione di Chiari tipo II.
Chiari tipo III
Consiste in una discesa delle strutture della fossa posteriore (verme, emisferi
cerebellari e tronco) nel contesto di un encefalomeningocele cervicale alto.
E’ la forma più grave, generalmete incompatibile con la vita.
Chiari tipo IV
E’ un’ipoplasia cerebellare senza erniazione.

103
Neurologia e neurochirurgia
ORIENTAMENTO
In questo tema bisogna studiare approfonditamente la nevralgia del trigemino. Il resto degli argomenti non sono d’importanza fondamentale ai fini dell’esame.
22.1. Nevralgia del trigemino
E’ una sindrome dolorosa del volto, abitualmente unilaterale, d’inizio improv-
viso, carattere lancinante, e localizzazione nel territorio cutaneo di uno o più
rami del trigemino (secondo o terzo con maggioire frequenza). Le crisi dolo-
rose sono di breve durata e di carattere recidivante, con un’intensità tale da
interferire con le normali attività, fi no a spingere, nei casi pù gravi, il paziente
a condotte suicide. Le crisi non svegliano il paziente durante la notte.
Si presentano spontaneamente o dopo stimoli sensoriali, nelle cosidette
“zone grilletto” o “trigger” (sfregamenti sul viso, masticazione, pulizia dei den-
ti, o anche il semplice deglutire o parlare) . Nel caso in cui si abbia un defi cit
neurologico associato al dolore o quando la presentazione non sia di carat-
tere episodico, ma continua, si deve sospettare la possibilità che si stia di
fronte ad un caso di nevralgia secondaria ad altri processi.
Si clasifi cano in:
• Nevralgie essenziali. E’ il gruppo più numeroso. Colpisce solitamente
donne maggiori di 40 anni, con andamento ciclico.
• Nevralgie secondarie ad infi ammazioni, anomalie vascolari, tumo-
ri dell’angolo pontocerebellare, infezioni o patologie demielinizzanti
(sclerosi multipla) che colpiscono il V paio lungo il suo decorso.
Il trattamento iniziale d’elezione è la carbamazepina, a dosi crescenti, ma
con un rigoroso controllo ematologico (rischio di neutropenia). Sono utili,
in seconda linea, la fenitoina, il baclofen, il clonazepam, il gabapentin o la
amitriptillina.
Se non risulta effi cace, può essere indicato il trattamento neurochirurgico. Le
due tecniche più utilizzate sono la rizotomia percutanea (distruzione delle
fi bre nocicettive tramite termocoagulazione a radiofrequenza o creazione di
un vero e proprio danno meccanico attraverso un palloncino gonfi abile) e la
decompressione microchirurgica (separare l’arteria che si trova sul ganglio
di Gasser che, attraverso il battito, irrita il nervo). Altre tecniche che si pos-
22sono utilizzare sono l’alcolizzazione del ganglio di Gasser,la fenolizzazione
delle fi bre sensitive e la rizotomia retrogasseriana.
R I C O R D A Una nevralgia del trigemino con esplorazione patologica e/o con dolore continuo, ovvero non episodica, obbliga a scartare cause se-condarie.
22.2. Chirurgia del dolore intrattabile
Le procedure maggiormente impiegate si classifi cano in 2 grandi gruppi.
Tecniche neuromodulative
• Infusione di farmaci nel sistema nervoso (intratecale o intraventrico-
lare). La principale indicazione per questa terapia è il dolore nocicettivo
(per es. in relazione al cancro) . Tuttavia, si possono utilizzare anche nella
sindrome da fallimento chirurgico alla schiena FBSS (Fayled back surge-
ry sindrome).
• Tcniche di stimolazione:
- Stimolazione del midollo spinale e del nervo periferico. Indi-
cata nel dolore radicolare persistente associato alla sindrome da
fallimento chirurgico alla schiena o alla distrofi a simpatica rifl essa.
- Stimolazione cerebrale profonda del talamo somatosensoria-
le e della sostanza grigia periacqueduttale. Indicata nel dolore
di origene non maligno (sindrome da fallimento chirurgico, dolore
neuropatico dopo lesioni del sistema nervoso centrale o periferico
o dolore trigeminale).
- Stimolazione della corteccia motoria. Sono valide le stesse indi-
cazioni della stimolazione talamica.
Tecniche ablative
Sono solitamente più appropriate per il dolore nocicettivo che per il neuro-
patico. Esistono 3 grandi gruppi:
• Interventi periferici (sul nevo):
- Simpatectomia. Dolore viscerale associato al cancro e a disturbi
vasospastici.
NEUROCHIRURGIA FUNZIONALE

10422 ∙ Neuroch i ru rg ia funz iona le
Manuale CTO di Medicina e Chirurgia, Prima edizione
- Neurectomia. Dolore a seguito di lesione del nervo periferico. (es.
Amputazione di un arto).
- Rizotomia dorsale e ganglioneurectomia. Indicata nel dolore a
livello del tronco o dell’addome associato a neoplasia.
• Interventi spinali:
- Lesioni della zona d’ingresso della radice dorsale. Dolore neu-
ropatico dopo avulsione della radice.
- Cordotomia e mielotomia. Dolore in relazione al cancro.
• Interventi sovraspinali. Come la mesencefalotomia, talamotomia, cin-
golotomia e ipofi sectomia.
I d e e c h i ave La nevralgia del trigemino è caratterizzata da un dolore neuropati-co, episodico e recidivante, che NON SVEGLIA il paziente durante la notte e che si distribuisce lungo i rami del trigemino. Si può scate-nare con alcune manovre o per stimolazione di determinate zone facciali.
I rami più colpiti sono il secondo ed il terzo, ovvero l’infraorbitario ed il mandibolare rispettivamente.
Da un punto de vista eziologico, può essere essenziale o secondaria a diversi processi intracranici, come tumori o sclerosi multipla
Il trattamento d’elezione è la carbamazepina. In caso di fallimento o intol-leranza al trattamento medico, si pianifi cherà un trattamento chirurgico.

105
Neurologia e neurochirurgia Bibliografia
B i b l i o g ra f i aN e u ro l o g i a e n e u ro c h i r u rg i a
Brazis PW, Masdeu JC, Biller J (Eds.). Localization in clinical Neurology, 6th Edition. Wolters Kluwer LWW, 2007.
Daroff RB, Fenichel GM, Jankovic J, Mazziotta JC. Bradley’s Neurology in Clinical Practice, 6th Edition. Elsevier, 2012.
Engel J, Pedley TA. Epilepsy: A comprehensive textbook, 2nd Edition. Lippincott Williams & Wilkins, 2008.
Greenberg MS. Handbook of Neurosurgery, 7th Edition. Thieme Medical Publishers, 2010.
Grupo CTO. Manual CTO de Neurologia y neurocirugía, 8.ª ed. Madrid. CTO Editorial, 2012.
Larner AJ. A Dictionary of Neurological Signs, 3rd Edition. Springer, 2011.
Lindsay KW. Neurology and Neurosurgery Illustrated, 5rd Edition. Churchill Livingstone Elsevier, 2010.
Moore AJ, Newell DW (Eds.). Neurosurgery. Principles and Practice, 2nd Edition. Springer, 2005.
Pahwa R, Lyons KE. Handbook of Parkinson’s Disease, 4th Edition. Informa Healthcare USA, Inc., 2007.
Patten JP. Neurological Diff erential Diagnosis, 2nd Edition. Springer, 1996.
Ropper AH, Samuels M. Adams and Victor’s Principles of Neurology, 8th Edition. McGraw-Hill, 2005.
Rowland LP, Pedley TA. Merrit’s Textbook of Neurology, 12th Edition. Lippincott Williams & Wilkins, 2010.
Top Related