

![Ematuria [modalit compatibilit ] - Prof. Cesare Selli - … · 2009-07-30 · Definizione • Più di 5 globuli rossi per campo microscopico ad alto ingrandimento nel sedimento urinario](https://static.fdocumenti.com/doc/165x107/5b754e867f8b9a924c8d31d4/ematuria-modalit-compatibilit-prof-cesare-selli-2009-07-30-definizione.jpg)


![LEZIONE2BIS MOD [modalit compatibilit ]) ematuria sindrome nefrosica e nefritica.pdf · LA SINDROME NEFROSICA Entità clinica che riconosce molte cause, caratterizzata da un aumento](https://static.fdocumenti.com/doc/165x107/5c6a9f5709d3f20f7f8cef11/lezione2bis-mod-modalit-compatibilit-ematuria-sindrome-nefrosica-e-nefriticapdf.jpg)


Le lingue
Pagine
Legale


- Omeostasi idro-elettrolitica
- Approccio al bambino con ematuria
- Fisiopatologia dell’edema nelle patologie nefrologiche
- Glomerulonefriti
- Sindrome Nefrosica
- Approccio al bambino con disturbi minzionali (poliuria, enuresi)
- Le infezioni delle vie urinarie
- Insufficienza Renale nel bambino (IRA e IRC)

Omeostasi idro-elettrolitica
Fede C, La Mazza A, Vitale A, Crisafulli RM, Salpietro V, Fede C
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
L’acqua è l’elemento più abbondante del corpo umano,il suo contenuto varia in rapporto al sesso,
l’età e la costituzione del soggetto: nel neonato il contenuto di acqua è il 75% del peso corporeo,nel
giovane adulto di età > o = di 25 anni è il 60%,nella donna di pari età il 50%, nell’uomo di età > o =
di 80 anni è il 50% e nelle donna della stessa età il 45 %.
L’acqua corporea totale si divide in 2 compartimenti:acqua intracellulare e acqua extracellulare.
La prima rappresenta il 67% del peso corporeo ed è indice della massa cellulare corporea.
L’acqua extracellulare raggiunge in media il 33% del peso corporeo suddividendosi in acqua del
plasma,della linfa interstiziale,dei tessuti di sostegno e acqua transcellulare.
L’acqua in noi è una traccia del mare primordiale in cui siamo nati; rappresenta la sostanza
fondamentale di ogni cellula e partecipa a tutti i processi biologici che in essa si svolgono.
Talete di Mileto,filosofo greco antico vissuto tra il VII-VI sec.A.C. affermava “l’acqua è l’origine
di tutte le cose….”. Per i Sumeri, popolazione vissuta nella Mesopotamia meridionale tra il 4000 e
il 1500 A.C. nelle terre tra il Tigri e l’Eufrate, è medico “colui che capisce d’acqua”. Oggi, la
medicina quantistica molecolare riconosce le cause delle malattie nell’alterata polarità dell’acqua.
I reni, pur rappresentando appena di 1/200 del peso corporeo svolgono un ruolo fondamentale nel
mantenimento dell’omeostasi dell’acqua corporea infatti,sono in grado di filtrare ogni giorno
duecento litri d’acqua e riassorbire il 99,9%. Questa capacità dei reni di concentrare e di diluire le
urine ha avuto inizio, in tempi lontani, quando gli organismi viventi si sono trasferiti dal mare alla
terra ferma ed hanno dovuto risparmiare acqua, per sopravvivere alle temperature del nuovo
ambiente.
Oggi, il rene umano è capace di una escrezione di 20 l/die di urine con una osmolarità minima di 50
mOsm/l o di una escrezione di appena 0,5 l/die di urine con una osmolarità massima di 1200
mOsm/l.
L’acqua è sicuramente tra tutti gli elementi costituenti il corpo umano quello più soggetto a
movimento esterno e interno. La perdita giornaliera di acqua nell’adulto corrisponde a circa il 4%
della massa corporea e deve quindi essere riassunta con la dieta. Nei bambini questa percentuale è
molto più elevata circa il 15% ed essi sono quindi più soggetti a disidratazione. Il bilancio
dell’acqua è determinato dall’equilibrio tra il volume di acqua assunta e quello di acqua eliminata
dall’organismo Tale equilibrio è regolato dal centro ipotalamico della sete, che modula l’assunzione

di acqua e dall’ormone antidiuretico (ADH o vasopressina) secreto dall’ipofisi posteriore che
aumenta il riassorbimento di acqua nel rene.
La regolazione del ricambio idro-salino avviene tramite meccanismi nervosi (centrali e periferici) e
ormonale, che esplicano la loro azione sul rene,organo cardine della omeostasi idro-elettrolitica.
La conoscenza dei fabbisogni idro-elettrolitici è importante per conoscere le potenziali perdite di
liquidi ed elettroliti, per esempio a causa del vomito o diarrea. Per i bambini si raccomanda un
apporto di acqua pari a 1,5 ml/kcal di energia spesa, ovvero 35-50 ml/kg o 1000-1500ml/mq di s.c..
Momenti critici per un corretto equilibrio idro-elettrolitico sono quelli che si osservano in corso di
malattie acute critiche,quando diventa evidente il sequestro di acqua nel compartimento
extracellulare con conseguente edema. In questi casi una non corretta gestione del bilancio idro-
elettrolitico può condurre a un deficit circolatorio e all’ipoperfusione tissutale,per cui devono essere
somministrati liquidi (soluzioni saline 0,9 % o ringer lattato o soluzioni colloidi) per mantenere il
volume intravascolare e un’adeguata circolazione onde evitare uno stato di ipoperfusione e
un’insufficienza pre-renale.
Altri momenti clinici critici che meritano particolare attenzione sono i casi di grave malnutrizione in
cui le cellule si impoveriscono a poco a poco di tutti i loro componenti. In questi casi si può
determinare la sindrome da rialimentazione (refeeding sindrome). Per evitarla occorre evitare
eccessivi apporti di sodio e acqua, in quanto il rene fatica ad eliminarli con possibile loro
accumulo,ma essere generosi nella somministrazione di potassio,fosfati e magnesio,onde evitare
una pericolosa diminuzione della loro concentrazione ematica.
Bibliografia
1. Rocca G.: Medicina quantistica molecolare.La dinamica della vita. Tecniche nuove, Milano 2008
2. Hahnemann S.: Organon of medicine. Kessinger Publishing,2007

Approccio al bambino con ematuria
Conti G, Vitale A, Crisafulli RM, Salpietro V, Fede C
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
Epidemiologia
L’ematuria è un sintomo importante nella nefro-urologia, essendo presente nella maggior parte delle
malattie del parenchima renale e delle vie escretrici.
Il riscontro di microematuria può conseguire a screening oppure ad esami occasionali. In Giappone,
mediante uno screening scolastico, è stata rilevata una microematuria isolata nello 0.25% della
popolazione di bambini di 6-12 anni. Negli USA la prevalenza di ematuria microscopica
asintomatica è risultata in alcuni Stati dello 0.05-2%.
Fisiopatologia
Molto interessante è lo studio delle lesioni elementari e delle basi molecolari che sottendono una
ematuria glomerulare. L’ipotesi più accreditata, documentata solo eccezionalmente in microscopia
elettronica, è la formazione di soluzioni di continuo, “breaks o gap” della membrana basale
capillare glomerulare che permettono il passaggio nello spazio urinifero delle emazie. La recente
introduzione della microscopia a scansione ha consentito una visualizzazione del tutto convincente
del fenomeno ed ha dato la dimostrazione del ruolo giocato dal complemento nell’induzione di
ematuria.
In vari modelli sperimentali di glomerulonefrite, indotte con somministrazione di immunocomplessi
preformati si è osservato che compariva ematuria solo se veniva attivato e fissato il complemento,
ed era possibile evitare l’ematuria in animali depleti di complemento. L’attivazione della via
comune C5-C9, porta alla costituzione di una molecola detta complesso di attacco di membrana
(C5b-C9 o MAC) ricco di fosfolipidi. Questo si fissa alla membrana ed assume una posizione di
transmembrana, favorendo in situ la produzione di specie reattive dell’O2 molecolare. La
perossidazione dei fospolipidi di membrana che consegue è il meccanismo principale alla base
della formazione delle soluzioni di continuo della parete dei capillari adiacenti al punto di
inserzione della macromolecola del C5-C9.
È peraltro interessante rilevare che, accanto al meccanismo principale di fissazione del
complemento attivato nell’induzione dell’ematuria glomerulare, questa possa essere clinicamente
evidente per alterazioni non infiammatorie della membrana basale, conseguenti ad anomalie della
composizione molecolare del componente Collageno IV, quali la porzione non collagenosica di vari
tipi di catena alpha (come accade nella ematuria da Sindrome di Alport) o per alterazioni

biochimiche ancora non note, ma che ne condizionano un minore spessore ed una maggiore fragilità
(come nella malattia da membrane basali sottili).
Sedimento urinario: distinzione ematuria glomerulare e non glomerulare
Si considera normale un sedimento urinario con rari globuli rossi (GR) sulle prime urine emesse al
mattino e ottenuto dopo una centrifugazione standard (x3000 giri/minuto) e lettura ad
ingrandimento x400 del sedimento.
La conta può essere normalizzata a GR/mm3 o con la conta di Addis in urine delle 24 ore a GR/min,
ma il sedimento a fresco, se eseguito correttamente, consente di ottenere più informazioni di metodi
più complicati. Una microematuria è significativa quando sono presenti più di 5 GR per campo
microscopico (pcm) ad ingrandimento di x 400, valore che corrisponde a >5 GR/mm3 o >5000
GR/minuto nella conta di Addis
La valutazione microscopica del sedimento è essenziale anche per distinguere un’ematuria da
colorazioni rosse delle urine da pigmenti esogeni (alimentari o farmacologici) o endogeni come in
caso di mioglobinuria (da rabdomiolisi) o emoglobinuria (da anemia emolitica).
Inoltre l’analisi del sedimento permette di valutare la morfologia delle emazie. La presenza di
emazie dismorfiche nelle urine è considerata indicativa di una nefropatia glomerulare. Il
dismorfismo sarebbe prodotto dall’azione della pressione intracapillare sulle emazie che, spinte
attraverso brecce (o gap) della membrana basale glomerulare, ne vengono modificate
definitivamente nella forma. Si ritiene pertanto che quando più del 30-50% delle emazie sono
dismorfiche e pallide la diagnosi più probabile sia di sanguinamento glomerulare. La corretta
interpretazione del dato morfologico deve tenere conto del ruolo dell’osmolarità delle urine e
dell’entità dell’ematuria (che è espressione della velocità di passaggio dei GR attraverso le
soluzioni di continuo della parete capillare glomerulare). L’associazione di emazie dismorfiche con
ematuria glomerulare e emazie ben conservate con ematuria urologica non è infatti assoluta ed
emazie ben conservate possono caratterizzare una macroematuria glomerulare con estrema
riduzione del tempo di passaggio delle emazie attraverso un filtro glomerulare alterato, mentre
l’ipertonicità delle urine può dare immagini difficili da interpretare correttamente. Tutto il
sedimento è invece più informativo, poiché la ricchezza in detriti cellulari e in cilindri, la presenza
di acantociti conferma il reperto di emazie dismorfiche nel sospetto di una microematuria
glomerulare. Allo stesso modo la presenza di coaguli ematici nelle urine e/o di cristalli e/o batteri
nel sedimento può rafforzare l’ipotesi di un’ematuria di origine urologica.
Lo studio del sedimento, quindi, si presenta fondamentale per formulare una prima ipotesi sulla
causa di ematuria ed avviare un procedimento diagnostico diversificato.

Iter diagnostico e cause dell’ematuria
Le cause dell’ematuria nel bambino sono molteplici (Tab. I).
L’approccio ad un bambino con ematuria avviene secondo elementi classici: anamnesi, esame
obiettivo, test laboratoristici a partire da quelli meno complicati ed eseguibili ambulatorialmente a
quelli più complessi e invasivi che richiedono ospedalizzazione
Nella raccolta della storia clinica familiare l’identificazione di malattie renali ereditarie merita
particolare attenzione sia per porre il sospetto di una nefrolitiasi sia di una nefropatia familiare
(sindrome di Alport, malattie delle membrane sottili, ematuria familiare benigna, nefropatia e
depositi IgA).
Nell’anamnesi personale del bambino bisogna considerare particolarmente l’epoca di comparsa
dell’ematuria in rapporto a precedenti episodi infettivi (faringotonsilliti, granulomi o ascessi dentari,
impetigine, ascessi superficiali e profondi) o traumi addominali recenti o alla possibile assunzione
di farmaci (analgesici, FANS, anticoagulanti).
Un punto importante è l’attenta considerazione delle condizioni cliniche nel cui contesto si è
rilevata l’ematuria. La presenza di subedemi palpebrali e malleolari possono far ipotizzare una
sindrome nefritica acuta. Il riscontro di porpora agli arti inferiori e ai glutei, che si accompagna
spesso a manifestazioni addominali (dolore colico di tipo ischemico, melena o rettorragia) e/o ad
artralgie permettono di diagnosticare una sindrome di Schoenlein-Henoch.
Gli esami diagnostici inizialmente sono molto semplici ed includono oltre all’analisi del sedimento
urinario, il dosaggio della proteinuria molto utile nel rilevare modificazioni della permselettività
glomerulare.
Gli esami ematochimici comprendono la valutazione della filtrazione glomerulare (clearance della
creatinina), indici di flogosi (emocromo, VES, PCR), assetto immunologico (protidemia e quadro
elettroforetico, immunoglobuline), complementemia. Una riduzione del complemento sierico
caratterizza alcune forme di glomerulonefriti primitive come la glomerulonefrite acuta postinfettiva,
la glomerulonefrite membrano-proliferativa e alcune forme secondarie, soprattutto quelle in corso di
lupus eritematoso sistemico ed in corso di crioglobulinemie di accompagnamento ad infezione da
HCV, molto rare in età pediatrica.
Nei casi di ematuria ricorrente che si accompagnano a sordità del bambino o di un familiare è
consigliabile effettuare un esame audiometrico che se rileva un difetto neurogenico bilaterale per i
toni medio-alti, può far ipotizzare una sindrome di Alport da confermare successivamente alla
biopsia.

In tutti i pazienti con ematuria è utile l’esecuzione di un’ecotomografia reno-vescicale, anche per
individuare, il più precocemente possibile, patologie tumorali come il tumore di Wilms o il
rabdomiosarcoma vescicale
Nel sospetto di una microematuria non glomerulare è utile l’esecuzione di un’urinocoltura ed il
dosaggio della calciuria. È ancora oggi difficile stabilire con certezza il significato patologico del
riscontro di ipercalciuria nei bambini ed i rapporti precisi con l’ematuria. Screening scolastici hanno
infatti dimostrato la relativa frequenza di ipercalciuria asintomatica, osservata nel 3-9% dei bambini
sani, di cui solo una piccola parte, circa il 20%, sviluppa nel tempo microematuria e calcolosi. Nei
bambini in cui, a dieta libera, è riscontrabile un’ipercalciuria, è utile la ripetizione dell’esame dopo
una settimana di dieta ipocalcica a contenuto controllato di sodio (300 mg/1.73 m2 di calcio e 2
g/1.73 m2 di sodio al giorno). Un rapporto calciuria/creatininuria >0,2 sulle urine raccolte a digiuno
dopo il periodo di dieta, rileva un’ipercalciuria renale. È comunque sempre importante rilevare se
l’ipercalciuria è asintomatica o se è associata a microematuria; nel primo caso assume rilevanza
quando osservata in un contesto di familiarità per calcolosi e/o microematuria, nel secondo caso
rappresenta sempre una condizione da monitorare.
Le indagini strumentali più approfondite (cistografia, urografia, cistoscopia) sono riservate ad un
momento successivo per la conferma di una litiasi o di una malformazione delle vie urinarie.
Biopsia renale
L’iter descritto permette una diagnosi nel 50-60% dei casi. Nella restante percentuale dei casi la
natura della microematuria isolata rimane imprecisata. In questi casi il problema principale è
rappresentato dalla scelta di procedere o meno ad esami più invasivi, come la biopsia renale.
Il ricorso alla biopsia renale viene limitato ad un numero ristretto di casi: episodi di macroematuria
recidivante, microematuria importante persistente con alterati segni clinici o umorali (creatininemia
aumentata, ANA e/o n-DNA positivi) o urinari (proteinuria significativa). In queste condizioni è più
probabile il riscontro bioptico di una glomerulopatia, che deve essere seguita nel tempo con un
follow-up ambulatoriale scrupoloso.
Più difficile decidere se e quando fare l’accertamento istologico nelle microematuria di entità
discreta senza proteinuria o nelle macroematurie recidivanti con sedimento negativo nei periodi
intervallari. In questi casi è fondamentale escludere le cause urologiche ed attendere un follow up
più lungo che può arrivare a 1-2 anni in cui si valuta anche la costanza del reperto (microematuria
persistente) e l’eventuale comparsa di proteinuria nel tempo.

La decisione di non procedere ad accertamento bioptico renale deve sempre essere considerata
temporanea, poiché non può essere valida sempre nel futuro di un paziente. Infatti il profilo clinico
di una nefrite è dinamico e può indurre a rapidi cambiamenti decisionali.
La “finestra pediatrica” del Registro delle biopsie renali Italiane, un’iniziativa del Gruppo di
Immunopatologia Renale Italiano della Società Italiana di Nefrologia Pediatrica, ha censito le
biopsie renali pediatriche eseguite in Italia durante 3 anni (1992-1994) ed ha permesso di valutare
le associazioni fra quadro clinico d’esordio e biopsia renale su un numero notevole di nefropatici
pediatrici, 432 pazienti minori di 15 anni.
Nei bambini in cui l’indicazione alla biopsia renale era microematuria associata a proteinuria
dosabile ma non nefrosica (4 <50 mg/Kg/die) la nefrite più comunemente riscontrata era stata la
Nefropatia a depositi IgA (IgAN,30,4%). Frequente anche la GN in corso di Schönlein–Henoch
(23%), mentre meno frequenti erano la GN membrano-proliferativa (5,2%) e la GN in sindrome di
Alport (4,4%). Nei bambini in cui la biopsia renale era stata eseguita per microematuria isolata
associata a proteinuria in tracce minime o assenti, il 35% presentava una IgAN, il 25% una malattia
a membrane basali sottili. Molto meno rappresentate erano altre forme, quali GN membrano–
proliferative o in corso di sindrome di Alport.

Tabella I: Cause di ematuria in età pediatrica
Cause nefrologiche
- Glomerulonefrite acuta
- Glomerulonefriti (GN) croniche (nefriti a depositi IgA, GN membrano proliferative,
GN rapidamente progressive, GN proliferativa mesangiale, etc)
- Sindrome di Schoenlein-Henoch
- Sindrome emolitico-uremica
- Collagenopatie (LES, vasculiti, etc)
- Sindrome di Alport e nefriti familiari
- Malattia da membrana basale sottile
Cause urologiche
- Tumori renali (nefroblastoma, adenocarcinoma a cellule chiare)
- Tumori vescicali (rabdomiosarcoma)
- Litiasi renale
- Uropatie malformative (giunto pielo-ureterale, valvole dell’uretra o ureterocele)
- Malattia policistica
- Traumi
Altre cause
- Trombosi vene renali o infarto renale
- Litiasi microscopica (ipercalciuria, iperossaluria)
- Difetti della coagulazione
- Nefriti interstiziali immuno-allergiche (farmacologiche)
- Ipertensione arteriosa e malformazioni vascolari
- Sindrome dolore lombare-ematuria

Bibliografia
1. Fede C, Conti G, Il bambino con ematuria. Messina Medica 1994; 11: 14-17
2. Ward JF, Kaplan GW, Mevorach R, Stock JA, Cilento BG Refined microscopic urinalysis
for red blood cell morphology in the evaluation of asymptomatic microscopic hematuria in a
pediatric population J Urol, 1998; 160: 1492-1495
3. Coppo R, Amore A, Peruzzi L, Conti G. Ematuria. Nefrologia Pediatrica a cura di Rosanna
Gusmano, Ed UTET Periodici, pag 69-74
4. Bergstein J, Leiser J, Andreoli S. The clinical significance of asymptomatic gross and
microscopic hematuria in children. Arch Pediatr Adolesc Med. 2005; 159: 353-355.
5. Fogazzi GB, Edefonti A, Garigali G, Giani M, Zolin A, Raimondi S, Mihatsch MJ, Messa
P. Urine erythrocyte morphology in patients with microscopic haematuria caused by a
glomerulopathy. Pediatr Nephrol 2008: 23:1093–1100
6. Hoppe B, Kemper MJ. Diagnostic examination of the child with urolithiasis or
nephrocalcinosis. Pediatr Nephrol. 2010; 25: 403-413.
7. Coppo R, Gianoglio B, Porcellini MG, Maringhini S. Frequency of renal diseases and
clinical indications for renal biopsy in children (report of the Italian National Registry of
Renal Biopsies in Children). Nephrol Dial Transplant. 1998; 13: 293-297.

Fisiopatologia dell’edema nelle patologie nefrologiche
Giovanni Conti, Agata Vitale, Lorena Silipigni, Antonella La Mazza, Vincenzo Salpietro, Carmelo Fede
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
Il movimento di fluidi fra il comparto vascolare e quello interstiziale è regolato dalla legge di
Starling dove le variabili principali sono la differenza di pressione idrostatica e di pressione
oncotica fra i due comparti. Secondo l’Equilibrio di Gibbs-Donnan: “il prodotto delle
concentrazioni di ciascun paio di cationi e anioni diffusibili posti ad un lato della membrana è
uguale al prodotto dello stesso paio di ioni posti all’altro lato”. A livello plasmatico, la pressione
osmotica è maggiore perché vi sono le proteine (che hanno un peso maggiore) ed un maggior
numero di ioni diffusibili.
L'edema in corso di patologie renali può originare secondo due meccanismi patogenetici diversi:
quello dell' "underfill" (sottoriempimento) e quello dell' "overfill" (sovrariempimento).
Secondo la teoria “underfill”, in particolare durante una sindrome nefrosica, si può avere una
riduzione della pressione oncotica da massiva proteinuria e conseguente ipoalbuminemia.
Dall’altro, secondo la teoria “overfill”, in corso di glomerulonefrite si può determinare un aumento
della pressione idrostatica da ritenzione di sodio ed acqua. Le due teorie hanno meccanismi
patogenetici opposti e richiederebbero anche interventi terapeutici differenti.
Nella sindrome nefrosica, particolarmente in età pediatrica, si ha una perdita massiva e persistente
di proteine per alterata permselettività del filtro glomerulare (Fig1). L'intensità dell’ipoalbuminemia
è variabile e dipende dalla capacità di sintesi epatica, dallo stato nutrizionale del soggetto e dalla
eventuale presenza di patologie associate. L'ipoalbuminemia determina una riduzione della
pressione oncotica nel compartimento plasmatico con diminuzione del gradiente plasma-interstizio,
cui consegue una maggior fuoriuscita di liquidi verso l'interstizio. Il rene attua dei meccanismi di
compenso che tendono a ristabilire l'equilibrio: aumento del flusso linfatico e vasocostrizione
capillare che tentano di ristabilire il gradiente fra pressione oncotica plasmatica e interstiziale.
Inoltre la scarsa distensibilità dell'interstizio con rapido aumento della pressione idrostatica
interstiziale frena la fuoriuscita di liquidi dal comparto vascolare. Caratteristiche fondamentali
dell’edema da “underfill”, soprattutto ai fini terapeutici, sono l'ipovolemia, l'attivazione del sistema
renina-angiotensina-aldosterone, la vasocostrizione periferica. La funzione renale è per lo più
conservata.
Invece, secondo la teoria "overfill" (Fig.2), il danno glomerulare indurrebbe una ritenzione
primitiva di sodio con aumento della volemia e fuoriuscita di liquidi dal comparto vascolare per
aumento della pressione idrostatica plasmatica. In questa situazione c'è un aumento della volemia

ed espansione del volume interstiziale, il sistema renina-angiotensina-aldosterone è soppresso e
sono presenti ipertensione e contrazione della funzione renale
I due meccanismi, però, possono essere presenti in fasi diverse della stessa malattia. L'edema da
"underfill" si verifica più frequentemente nella sindrome nefrosica in età pediatrica e poiché la
caratteristica principale è la contrazione del volume plasmatico sono presenti sintomi da ipotensione
e da ipovolemia. Emoglobinemia ed ematocrito sono aumentati. La causa più frequente è la
glomerulonefrite a lesioni minime.
L'edema da "overfill", a volume plasmatico espanso, è più frequente nella sindrome nefrosica
dell’adulto e nelle glomerulonefriti ed è caratterizzato da ipertensione e sintomi di ipervolemia.
Emoglobinemia e ematocrito sono normali.
Terapia
I due meccanismi possono essere presenti in diverse fasi della stessa malattia; tuttavia è importante
per la terapia riconoscere quale dei due distinti meccanismi è presente o prevalente.
L'edema a volume plasmatico contratto non richiede una correzione urgente e frequentemente la
terapia "causale" ne consente il controllo. Qualora l'ipovolemia sia sintomatica l'approccio più
razionale è quello di espandere il volume plasmatico con albumina (1 g/kg/die nel bambino; 40
g/die nell'adulto) e talora con mannitolo al 18% (10 ml/kg fino a un massimo di 200 ml, per non
incorrere nel rischio di tubulotossicità). Raramente si utilizzano plasma expanders e plasma.
L'edema con volume espanso, da sovraccarico, richiede invece un trattamento più urgente perchè
può evolvere in edema polmonare o ipertensione grave.
L'obiettivo terapeutico principale è ridurre la volemia, negativizzando il bilancio del sodio.
Pertanto è necessario limitare l'introduzione di sodio e utilizzare i diuretici.
Se la funzione renale è normale con edemi modesti i diuretici di prima scelta sono i tiazidici:
idroclorotiazide 1mg/kg/die.
Negli edemi refrattari o in presenza di contrazione della funzione renale è necessario l'uso dei
diuretici dell'ansa, tenendo conto dell'ototossicità, in particolare in caso di ipoalbuminemia grave.
La dose di furosemide parte da 1 mg/kg/die e può arrivare fino a 250-500 mg/die nell'adulto.
Talora è utile l'associazione con i tiazidici.
I diuretici risparmiatori di potassio hanno un utilizzo razionale nelle situazioni di attivazione del
sistema renina-angiotensina-aldosterone e in associazione a diuretici dell'ansa per evitare
l'ipopotassiemia. Lo spironolattone si usa a dosi di 1-3 mg/kg/die.

Fig 1: Patogenesi dell’edema in nefrologia: teoria “underfill”
Fig 2: Patogenesi dell’edema in nefrologia: teoria “overfill”

Bibliografia
1. Favia I, Garisto C, Rossi E, Picardo S, Ricci Z. Fluid management in pediatric intensive
care. Contrib Nephrol. 2010;164:217-226.
2. Dhir V, Arya V, Malav IC, Suryanarayanan BS, Gupta R, Dey AB. Idiopathic systemic
capillary leak syndrome (SCLS): case report and systematic review of cases reported in the
last 16 years. Intern Med. 2007; 46: 899-904.
3. Ulinski T, Aoun B. Pediatric idiopathic nephrotic syndrome: treatment strategies in steroid
dependent and steroid resistant forms. Curr Med Chem. 2010; 17: 847-53.

Glomerulonefriti
Chimenz R.,Catena MA., Crisafulli, RM., Privitera C., Fede C.
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
Le glomerulonefriti sono processi infiammatori a carico del glomerulo del Malpighi su base
immunologica, idiopatica o causata da agenti infettivi o non infettivi, e con uno spettro di
manifestazioni cliniche estremamente vario, che va da forme asintomatiche a forme gravi, evolventi
in modo acuto o cronico verso l’insufficienza renale. L’infiammazione del glomerulo con
proliferazione cellulare con essudazione di polimorfonucleati ed alterazioni vascolari con microressi
ed alterazione della permeabilità vascolare, é responsabile delle sindromi "nefritiche" e
"nefrosiche”.
NEFRITI
La sindrome nefritica acuta si caratterizza clinicamente per la presenza di sintomi e segni quali :
- Ematuria
- Oliguria
- Edemi di lieve entità
- Ipertensione
- Proteinuria, Insufficienza renale acuta, anemia
I principali quadri di glomerulo nefrite in età pediatrica sono rappresentati da:
- Glomerulonefrite acuta post-infettiva
- Nefropatia a depositi di IgA (s. di Berger)
- Glomerulonefrite da SSH
- Sindrome di Alport
- Glomerulonefrite membranosa
- Glomerulonefrite membrano-proliferativa
Glomerulonefrite acuta post-infettiva: la grande maggioranza di tali glomerulo nefriti conseguono a
infezioni acute da ceppi nefritogeni di streptococco beta emolitico di gruppo A, ma altri agenti
eziologici possono essere rappresentati anche da altri batteri (stafilococco, meningococco,
pneumococco, salmonelle, treponema p, leptospira), virus (HBV, CMV, EBV, varicella zoster,
echovirus, coxsackie B4, virus di influenza, parotite e rosolia), toxoplasma, plasmodium m.
Patogeneticamente alla base della malattia vi è un’attivazione della cascata della via alterna del
complemento, molto probabilmente innescata dalla presenza di immunocomplessi circolanti ( ruolo
di antigene M, nephritis strain-associated protein, endostreptosina, proteasi) ciò determina il
richiamo dei PMN ed un conseguente danno di membrana transitorio. Clinicamente, dopo 10-15

giorni dal superamento di un’infezione, classicamente faringite o dopo 21 giorni da un’infezione
della cute, compaiono dapprima sintomi aspecifici (malessere generale, pallore, astenia, anoressia,
dolenzia lombare) seguiti poi dai sintomi principali della patologia, ovvero edemi, macroematuria,
oliguria ed ipertensione arteriosa. Meno frequentemente il quadro clinico può essere rappresentato
da forme paucisintomatiche, encefalopatia ipertensiva, sindrome nefrosica o scompenso cardiaco.
Per avere conferma diagnostica, in caso di sospetto di glomerulo nefrite acuta postinfettiva,
andremo ad eseguire innanzitutto un esame delle urine per la ricerca di proteinuria, ematuria e dei
caratteristici cilindri eritrocitari, ematuria e proteinuria.
• Parlando di ematuria, ovvero presenza di elementi corpuscolati del sangue nelle urine,
dobbiamo distinguere tra ematuria macroscopica e microscopica e tra ematuria asintomatica
o sintomatica ( associata a disuria, dolori addominali, patologie sistemiche…), precisando
che, nonostante l’ematuria possa essere la manifestazione di molti disordini del rene o delle
vie urinarie, il suo semplice riscontro non è necessariamente indice di malattia. L’ematuria
macroscopica non è comune in età pediatrica; può derivare dal rene (ed in tal caso le urine
possono essere di colore francamente rosso, rosa a lavatura di carne, color marsala o
marrone) o da vescica o uretra ( in tal caso le urine possono essere di colore dal rosa tipo
lavatura di carne al rosso acceso). Si parla di ematuria microscopica quando la ricerca di
emoglobina con Hema –combistick sia positiva e quando nel sedimento si riscontrino più di
5 GR per campo a forte ingrandimento(x400) della centrifugazione di 10 mL di urina. Le
cause di ematuria possono essere distinte in glomerulari( glomerulo nefriti acute e croniche,
nefropatie ereditarie, sindr.emoliticouremica, porpora di Schonlein Henoch,vasculiti, diabete
mellito, amiloidosi..) e non glomerulari (cistiti, pielonefriti, calcolosi, trauma).
Tornando a parlare di glomerulo nefrite acuta post infettiva, oltre all’esame urine, andremo
ad eseguire esami di laboratorio quali complementemia ( ↓ C3 nel 90% dei casi), TAS,
ricerche virologihe, indici di funzionalità renale, ionogramma urinario; la diagnosi
differenziale va sempre posta con la nefropatia da IgA, sindrome di Alport e glomerulo
nefrite membranoproliferativa. La terapia è semplicemente dietetica e sintomatica, senza
alcuna indicazione all’avvio di terapia antibiotica.
Nefropatia da IgA( malattia di Berger): nefropatia ad eziologia immunitaria, con quadro
istologico( evidenziabile con biopsia renale) caratteristico con depositi granulari mesangiali
di Ig, essenzialmente IgA. Clinicamente i pzt presentano macroematuria ricorrente in
coincidenza di episodi febbrili e microematuria persistente con proteinuria. La diagnosi è
essenzialmente clinica e bioptica e la terapia prevede l’uso di ACE inibitori, cortisone e
immunosoppressori.

Sindrome di Alport: è una forma di glomerulo nefrite ereditaria ( trasmissione X linked,
oppure autosomico dominante o autosomico recessiva) caratterizzata da un difetto del
collagene di tipo IV che determina un danno delle membrane basali, soprattutto quelle
glomerulari. Clinicamente si caratterizza per macroematuria e microematuria persistente con
proteinuria ,Sordità neurosensoriale ed Insufficienza renale cronica. La diagnosi è bioptica e
genetica e, anche in questo caso, la terapia prevede l’uso di ACE inibitori, cortisone e
immunosoppressori.
Bibliografia
1) E. Avner et al.: “Pediatric Nephrology” Lippincot Williams e Wilkins
2) K.K. Kher et al.: « Clinical Pediatric Nephrology » Informa healthcare

Sindrome Nefrosica
Chimenz R.,Catena MA., Crisafulli, RM., Privitera C., Fede C.
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
La sindrome nefrosica è caratterizzata da alterata permeabilità della membrana basale del glomerulo
renale cui conseguono proteinuria massiva (>50 mg/Kg/die), ipodisprotidemia, edemi
(ipoalbuminemia ↓ pressione oncotica plasmatica), iperlipidemia (conseguenza dell’aumentata
sintesi epatica di LPA dovuta all’edema)
Le specifiche alterazioni istologiche definiscono i 4 principali tipi di sindrome nefrosica, ovvero la
Sindrome nefrosica a lesioni minime (forma + frequente nel bambino), la glomerulosclerosi focale,
la glomerulonefrite membranoproliferativa e la glomerulonefrite membranosa.
All’esame urine: proteinuria massiva (selettiva nella SNLM, non selettiva nella GSF, membranosa e
MP), ematuria (non presente nella SNLM), cilindri ialino-granulosi,lipiduria.
Agli esami bioumorali: ipoproteinemia, aumento lipidi totali, diminuzione calcemia, piastrinosi,
aumento di VES iperfibrinogemia, C3 e C4 normali nella SNLM, nella membranosa e
membranoproliferativa.
La normalizzazione dei parametri bioumorali (ipodisprotidemia, ipocalcemia, piastrinosi ed
iperlipidemia) avverrà in seguito alla risoluzione della fase acuta con negativizzazione della
proteinuria
La terapia sarà dietetica (dieta iposodica e normoproteica), sintomatica (infusione di albumina e
furosemide), patogenetica (cortisone, ciclofosfamide e ciclosporina). La terapia steoridea all’esordio
di sindrome nefrosica viene eseguita con deltacortene al dosaggio di 60 mg/m2/die per quattro
settimane, seguite da quattro settimane a giorni alterni a 40 mg/m2/die alterni secondo schema
ISKDC.
Il follow-up clinico laboratoristico prevede controlli della proteinuria con albustick per
monitorizzare precocemente eventuali recidive.
La terapia delle recidive prevede la ripresa della terapia steroidea a dosaggio piena a 60 mg/m2/die
fino alla negativizzazione della proteinuria per tre giorni non consecutivi con successiva riduzione
dello steroide a 40 mg/m2/die alterni per quattro settimane.

Bibliografia
1. “Corticosteroid therapy for nephrotic syndrome in children”. Elisabeth M Hodson, Narelle S
Willis, Jonathan C Craig. The Cochrane library 2010, Isuue 4
2. “Interventions for idiopathic steroid-resistant nephrotic syndrome in children” Elisabeth M
Hodson, Doaa Habashy, Jonathan C Craig. The Cochrane library 2010, Isuue 4
3. “Early age at debut is a predictor of steroid-dependent and frequent relapsing nephritic
syndrome” R. Frydensbjerg et al. Pediatric Nephrology (2010) 25: 1299-1304

Approccio al bambino con disturbi minzionali
Silipigni L, Privitera C, Catena MA, Crisafulli RM, Fede C
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
Poliuria
Definizione
Il termine “poliuria” indica un’emissione eccessiva di urine in un tempo stabilito, ovvero oltre i
2L/m2/24 ore (adulti e bambini > 40 ml/Kg/die; lattanti 0-2 anni > 100 ml/Kg/die).
Eziologia
La poliuria è un sintomo frequente di patologia renale; essa deve essere distinta dalla “pollachiuria”,
cioè dall’emissione frequente di una quantità totale normale di urina. La poliuria si associa spesso a
polidipsia e nicturia, mentre la pollachiuria no.
Di fronte ad un bambino con poliuria, un elemento importante per la diagnosi differenziale è
rappresentato dalla osmolarità urinaria da cui dipende anche il peso specifico (PS). In base a tali
valori è possibile distinguere l’ipostenuria (urine ipotoniche ovvero a basso PS) dall’isostenuria
(urine isotoniche o leggermente ipertoniche). L’ipostenuria può essere determinata dalla mancanza
di ormone antidiuretico (diabete insipido primario), mancata risposta delle cellule tubulari
all’ormone antidiuretico (diabete insipido nefrogenico) o assenza di stimoli alla secrezione di
ormone antidiuretico (polidipsia psicogena). Altre condizioni possono essere date dall’ipercalcemia
o dalla deficienza cronica di potassio. L’isostenuria compare, invece, in pazienti con diuresi
osmotica e con responsività all’ormone antidiuretico (diabete mellito, fase poliurica
dell’insufficienza renale, somministrazione di diuretici).
Segni e sintomi
Nel bambino piccolo la diagnosi di poliuria e polidipsia può essere difficile. I primi segni da non
sottovalutare nel lattante sono la suzione vigorosa, il vomito ripetuto, la letargia, il pannolino
eccessivamente bagnato di urine e la stipsi ostinata. Il bambino più grande, con senso della sete
conservato, chiederà insistentemente di bere e, se non vi sono restrizioni idriche, può anche
bilanciare le perdite urinarie. Spesso sono presenti irritabilità (conseguenza della disidratazione),
deficit di sviluppo (scarso introito calorico a causa della sazietà legata alla sola introduzione di
liquidi), nicturia, polidipsia notturna e febbre intermittente senza causa apparente. E’ molto
importante l’esame obiettivo teso ad evidenziare eventuali segni di disidratazione (secchezza delle
mucose, diminuito turgore cutaneo, fontanella depressa, ridotta lacrimazione, tachicardia, pallore,
ipotensione, tempo di refill > 2”). Il riconoscimento precoce della poliuria è molto importante al fine
di prevenire anche le complicanze più gravi della disidratazione e diselettrolitemia quali shock
ipovolemico, convulsioni, ipossia e conseguente ritardo mentale e/o exitus.
Iter diagnostico
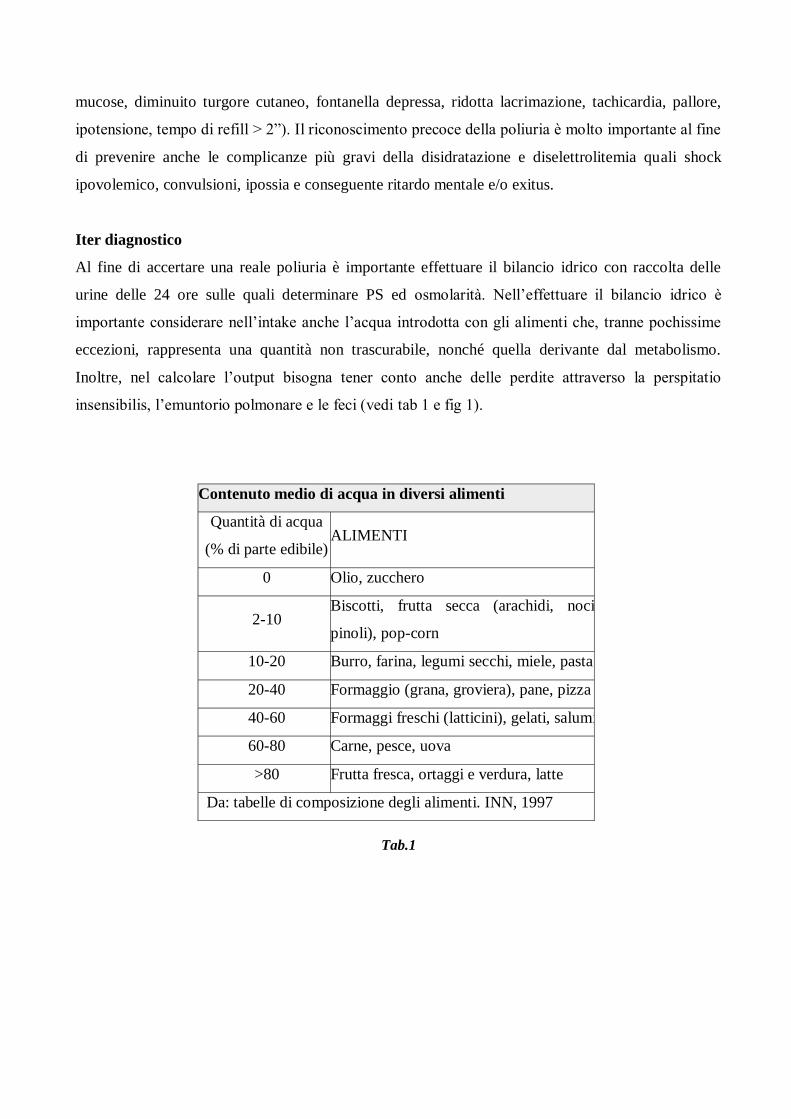
Al fine di accertare una reale poliuria è importante effettuare il bilancio idrico con raccolta delle
urine delle 24 ore sulle quali determinare PS ed osmolarità. Nell’effettuare il bilancio idrico è
importante considerare nell’intake anche l’acqua introdotta con gli alimenti che, tranne pochissime
eccezioni, rappresenta una quantità non trascurabile, nonché quella derivante dal metabolismo.
Inoltre, nel calcolare l’output bisogna tener conto anche delle perdite attraverso la perspitatio
insensibilis, l’emuntorio polmonare e le feci (vedi tab 1 e fig 1).
Tab.1
Contenuto medio di acqua in diversi alimenti
Quantità di acqua
(% di parte edibile) ALIMENTI
0 Olio, zucchero
2-10 Biscotti, frutta secca (arachidi, noci,
pinoli), pop-corn
10-20 Burro, farina, legumi secchi, miele, pasta
20-40 Formaggio (grana, groviera), pane, pizza
40-60 Formaggi freschi (latticini), gelati, salumi
60-80 Carne, pesce, uova
>80 Frutta fresca, ortaggi e verdura, latte
Da: tabelle di composizione degli alimenti. INN, 1997

Una volta confermata la poliuria, oltre alla valutazione della osmolarità urinaria, del PS e degli
elettroliti urinari, bisogna determinare l’osmolarità plasmatica, la glicemia e gli elettroliti sierici.
L’iperglicemia e la glicosuria sono indicativi di diabete mellito. Invece, si pone diagnosi di diabete
insipido (DI) se l’osmolarità sierica risulta maggiore di 300 mOsm/Kg e quella urinaria inferiore a
300 mOsm/Kg per cui il rapporto osmolarità urinaria/osmolarità plasmatica è <1 talvolta associata
ad ipernatriemia (Na+ > 144 mmol/L). In caso di polidipsia psicogena i valori plasmatici sono
tendenzialmente normali o ridotti. Attraverso il test dell’assetamento è possibile fare diagnosi
differenziale tra DI e polidipsia psicogena: nel primo caso il rapporto osmolarità urinaria/osmolarità
plasmatica rimarrà inferiore a 1 (valore normale >1,5) per la mancata concentrazione delle urine;
diversamente l’aumento dell’osmolarità urinaria fino a valori superiori a quella plasmatica indica
una condizione di polidipsia psicogena. Un ulteriore test dato dalla prova con desmopressina
(DDAVP) permette di differenziare il DI centrale da quello nefrogenico: la somministrazione
endonasale di DDAVP determina una riduzione della polidipsia e contrazione della diuresi nel caso
di diabete insipido centrale; nessuna modifica si osserva nella forma nefrogenica.
Diabete insipido centrale
Il DI centrale dipende da un difetto di sintesi e/o rilascio di ormone antidiuretico o
argininvasopressina (AVP) da parte dei nuclei ipotalamici paraventricolari e sopraottici in seguito
alla loro distruzione o degenerazione. Esso può derivare da cause diverse (Tab2): mutazioni geniche
del gene della vasopressina (rari casi con ereditarietà autosomica dominante, recessiva o X-linked)
trauma (accidentale o chirurgico) dei neuroni produttori della vasopressina, malformazioni congenite
ipotalamiche o ipofisarie, tumori (germinoma o craniofaringioma), patologie infiltrative (istiocitosi a
cellule di Langerhans, ipofisite linfocitica, sarcoidosi), autoimmuni, vascolari ed infettive dei
neuroni produttori di vasopressina o del peduncolo e un incremento del metabolismo della
vasopressina. Nella maggioranza dei casi si tratta di una condizione acquisita; in circa il 10 % dei

bambini, l’eziologia rimane idiopatica. Possono essere presenti deficit di altri ormoni ipofisari. Nelle
forme tumorali, poliuria e polidipsia insorgono generalmente dopo i 5 anni di età. I germinomi
possono essere molto piccoli e non identificabili con la risonanza magnetica nucleare (RMN) per
parecchi anni dopo l’inizio della poliuria, pertanto è importante, altresì, il dosaggio della subunità β
della gonadotropina corionica umana (hCG), spesso secreta da germinomi e pinealomi. Anche
neoplasie ematologiche, come la leucemia mielocitica acuta, possono provocare DI attraverso una
infiltrazione del peduncolo ipofisario e della sella. Le forme familiari sono le più precoci, ma
possono farsi evidenti anche solo durante l’età infantile. La forma familiare autosomica dominante è
secondaria ad una mutazione del gene che codifica per l’AVP-neurofisina II, precursore dell’AVP
localizzato a livello del cromosoma 20p13. Anche la Sindrome di Wolfram (che comprende DI,
diabete mellito, atrofia ottica e sordità) deriva da deficit di vasopressina. Si è identificato il gene di
questo disturbo, ma la sua funzione è ancora sconosciuta. Varie anomalie cerebrali congenite come
la displasia setto-ottica con agenesia del corpo calloso, l’ipoplasia ipofisaria familiare con assenza
del peduncolo, possono essere associate a DI centrale e a difetti nella percezione della sete. Anche la
sindrome della “sella vuota”, forse da infarto ipofisario non riconosciuto, nei bambini si può
associare a DI.
Tab.2

Diagnosi eziologica DI centrale
Al fine di determinare la causa scatenante il DI centrale, in alcuni casi, è necessario utilizzare mezzi
diagnostici complementari, finalizzati alla individualizzazione o alla esclusione di una eventuale
condizione acquisita: Rx o scintigrafia ossea nel sospetto di istiocitosi a cellule di Langerhans;
esame citologico del liquor e dosaggio hCG nel sospetto di germinoma; Rx torace, dosaggio ACE
nel plasma e liquor nel sospetto di sarcoidosi. Un esame strumentale fondamentale per la diagnosi
differenziale e la RMN. L’importanza di questa indagine radiologica deriva dalla dimostrazione
dell’immagine d’iperintensità della neuroipofisi nel 95-100% dei soggetti normali e dalla sua
mancata identificazione nei pazienti con diabete insipido centrale, suggestiva di lesione occulta a
carico del sistema nervoso centrale. In pazienti con DI autosomico dominante familiare, nonché, in
rari casi di DI idiopatico, tale iperintensità può essere presente anche in assenza di livelli circolanti
di vasopressina; questo è da attribuirsi a un difetto di rilascio di vasopressina-neurofisina-II mutato,
il cui accumulo potrebbe essere responsabile di una degenerazione retrograda di natura tossica dei
nuclei ipotalamici. Un altro riscontro radiologico importante è rappresentato dall’identificazione di
un ispessimento del peduncolo ipofisario associato alla mancata visualizzazione della neuroipofisi,
dimostrato in corso di DI secondario a istiocitosi a cellule di Langerhans, germinoma, DI idiopatico,
post infettivo, infiammatorio cronico. L’identificazione radiologica di anomalie della linea mediana
(oloprosencefalia, agenesia del setto pellucido ecc.) suggerisce un’origine mal formativa.
Follow up DI centrale
Nei pazienti con DI centrale è importante effettuare un attento follow up clinico, radiologico ed
endocrinologico. La comparsa di deficit ipofisari anteriori ed in particolar modo il deficit di ormone
della crescita è piuttosto frequente; inoltre ulteriori deficit ormonali possono sopraggiungere anche a
distanza di lungo tempo. È fondamentale un follow up radiologico, anche in presenza di peduncolo
ipofisario nella norma, in quanto non è prevedibile una sua evoluzione verso il germinoma.
La dimensione del peduncolo ispessito può variare e andare incontro ad una risoluzione spontanea
fino alla normalizzazione, alla parziale atrofia, all’ispessimento ulteriore o alla persistenza cronica
dell’ispessimento. In particolare, quando lo spessore del peduncolo supera i 6,5 mm, vi è
l’indicazione ad eseguire la biopsia soprattutto se le dimensioni dell’adenoipofisi sono aumentate o
vi è un coinvolgimento del terzo ventricolo. La risonanza magnetica con mezzo di contrasto può
essere utile per identificare quei casi di DI centrale con normali dimensioni del peduncolo ipofisario
ed alterazione del flusso sanguigno nell’ipofisi posteriore. La cadenza del follow up dipende dalle
alterazioni riscontrate nel singolo paziente. Di seguito è riportato un algoritmo per la diagnosi e il
follow up orientativo per una possibile linea di comportamento.

Diabete insipido nefrogenico (DIN)
Il DI nefrogenico (vasopressina-insensibile), condizione più rara rispetto al DI centrale, insorge per
cause genetiche o acquisite. Le cause genetiche sono meno comuni ma più gravi di quelle acquisite
con insorgenza della sintomatologia già nelle prime settimane di vita e maggiore evidenza dopo lo
svezzamento o quando si istaurano lunghi periodi di sonno. Molti neonati, inizialmente, manifestano
febbre, vomito e disidratazione. L’ingestione e l’escrezione protratte di notevoli volumi di acqua
può portare ad idronefrosi non ostruttiva, idrouretere e megavescica. Il DIN congenito associato a X
deriva da mutazioni inattivanti del recettore V2 della vasopressina. Il DIN congenito autosomico
recessivo dipende da difetti del gene dell’acquaporina-2. Il DIN congenito autosomico dominante è
associato a mutazioni del processing del gene dell’acquaporina-2.
Le cause acquisite possono essere date da ipercalcemia, ipokaliemia o dall’utilizzo di alcuni farmaci
(litio, demeclociclina, foscarnet, clozapina, amfotericina, meticillina, rifampicina). La capacità
renale di concentrare le urine può essere compromessa anche in presenza di ostruzione ureterale,
insufficienza renale cronica, malattia policistica del rene, malattia midollare cistica, sindrome di
Sjogren ed anemia a cellule falciformi.

Cenni di terapia
In primo luogo il trattamento deve essere rivolto alla risoluzione del problema eziologico che ha
portato al DI qualora questo sia possibile. Nei casi di DI centrale il farmaco di prima scelta per il
trattamento della poliuria è dato dalla desmopressina 0 1- desamino-8-D-arginina vasopressina
(DDAVP=minirin) alla posologia di :
5-20 microgrammi (1-4 spruzzi) ogni 12 ore per la forma nasale
0,1-0,3 mg ogni 8 ore per la forma orale.
Durante il trattamento bisogna monitorare attentamente l’andamento clinico ed aggiustare la
posologia in base alla risposta individuale di ciascun paziente, al fine di evitare sovradosaggi e
limitare il rischio di intossicazione d’acqua (primo segno di allarme la cefalea) ed iponatriemia.
Nel diabete insipido nefrogenico, laddove non è possibile intervenire sulla causa scatenante, la
terapia si avvale dei diuretici diazidici come la clorotiazide e il clortalidone.
Bibliografia
1. Nelson Textbook of Pediatrics. XVIII edizione.
2. Bartolozzi G e Guglielmi M. Pediatria. Principi e pratica clinica. II edizione.
3. Stefano Ghirardello, Maria-Luisa Garrè, Andrea Rossi and Mohamad Maghnie. The Diagnosis
of Children with Central Diabetes Insipidus. Journal of Pediatric Endocrinology & Metabolism
2007; 20: 359-375.
4. Maghnie M, Cosi G, Genovese E, et al. Central diabetes insipidus in children and young aduts.
N Engl J Med 2000; 343: 998-1007.

Enuresi
Definizione
Si definisce “enuresi” una minzione completa involontaria che avviene durante il sonno oltre l’età
in cui il controllo vescicale dovrebbe essere acquisito. Secondo una definizione del DSM-IV.
effettuata in base alla frequenza degli episodi, enuretico è “chi bagna il letto almeno 2 volte la
settimana per 3 mesi consecutivi”.
L’enuresi è un disturbo molto diffuso in Italia ed è piuttosto frequente tra la popolazione pediatrica.
Infatti circa 1 milione di bambini tra i 6 e i 14 anni bagna il letto la notte.
Il limite temporale entro cui viene raggiunto tale controllo è il 5° anno per le femmine ed il 6° anno
per i maschi.
L’incidenza dell’enuresi varia in base all’età. All’età di 5-6 anni bagna il letto oltre il 10-15% dei
bambini. All’età di 10 anni tale disturbo è presente nel 5% dei bambini. All’età di 15 anni l’enuresi
persiste ancora nel 1-2 % della popolazione pediatrica.
Prevale nel sesso maschile con un’incidenza di 3:2 , ma tale rapporto si pareggia nell’adolescenza.
L’enuresi rivela la presenza di fattori favorenti multigenici; l’anamnesi familiare, infatti, risulta
spesso positiva per enuresi, con un rischio di trasmissione che raggiunge il 77% se i due genitori
sono enuretici; del 44% se solo uno dei genitori è stato enuretico. L’anamnesi familiare risulta,
invece, negativa nel 15% dei pz.
L’importanza dei fattori genetici emerge anche dalla prevalenza del disturbo nel 68% dei gemelli
monozigoti e nel 36% dei gemelli dizigoti. Ed infatti è stato localizzato in un tratto del cromosoma
13 il possibile locus del gene responsabile dell’enuresi familiare: ENUR 1. Tale gene sembrerebbe
correlato con l’enuresi monosintomatica. L’enuresi con disturbi diurni sembrerebbe esser legata,
invece, al gene presente sul cromosoma 12, mentre una correlazione indefinita esiste fra enuresi e il
gene presente sul cromosoma 8.
Non esistono elementi predittivi sull’epoca della risoluzione spontanea del disturbo, ma dai dati
emerge che il 98-99% dei soggetti diventano asciutti entro i 15 anni.
Classificazione
L’enuresi si definisce primaria quando non è stato mai raggiunto un periodo asciutto continuativo
di almeno 6 mesi; e. secondaria, presente nel 18-25% dei casi, quando, invece, compare dopo un
periodo asciutto di almeno 6 mesi.
L’enuresi, come accennato sopra, si distingue in monosintomatica e sintomatica, quest’ultima
caratterizzata dall’associazione con segni di disfunzione minzionale diurna.

Patogenesi
L’enuresi primaria e l’enuresi secondaria sono state generalmente considerate come entità separate
con differente patogenesi.
L’e. primaria sembra dipendere da:
1) disturbi del risveglio per cui i bambini con enuresi non vengono risvegliati dalla
sovradistensione vescicale. Molti genitori di bambini affetti da enuresi riferiscono che i loro figli
hanno un sonno molto profondo e che difficilmente si svegliano. L’enuresi notturna si presenta in
qualsiasi fase del sonno e non è collegata ad una maggiore o minore profondità del sonno.
2) poliuria notturna legata a una carente increzione notturna di vasopressina. Norgaard per primo
ha dimostrato che in una quota di soggetti con enuresi notturna primaria viene persa la pulsatilità
del ritmo circadiano dell'ADH, evidenziando un appiattimento del fisiologico picco notturno. In tal
modo, vengono a mancare l’incremento dell’osmolarità urinaria notturna e la contrazione della
diuresi, per cui la produzione di urine durante la notte dovrebbe essere pari al 50% della
produzione diurna.
3) capacità vescicale notturna ridotta nei bambini con enuresi non–monosintomatica con maggior
instabilità vescicale notturna rispetto alle ore diurne
4) combinazione di questi fattori.
L’e. secondaria prevede come cause patogenetiche la presenza di:
1) stress psicologici come la nascita di un fratellino, un trasferimento, preoccupazioni scolastiche, la
morte di un nonno o di un genitore; 2) infezioni delle vie urinarie; 3) stipsi; 4) disfunzioni
minzionali; 5) diabete mellito tipo I .
Diagnosi
La valutazione diagnostica prevede innanzitutto un inquadramento clinico con l’analisi di alcuni
punti fondamentali: 1) familiarità per enuresi; 2) età di acquisizione del controllo degli sfinteri e
delle tappe dello sviluppo psicomotorio; 3) anamnesi per IVU; 4) anamnesi minzionale nelle ore
diurne con valutazione del mitto urinario, eventuale presenza di mutandine bagnate, fughe di urina o
urgenza minzionale; 5) l’eventuale presenza di incontinenza fecale, di stipsi o parassitosi
intestinale; 6) l’esame della regione lombosacrale per evidenziare la presenza di anomalie che
possano evidenziare una spina bifida occulta.
Nell’enuresi notturna gli esami da effettuare comprendono l’esame delle urine e, nei casi associati a
disturbi minzionali diurni, esplorazione morfologica e funzionale della vescica. Nel sospetto di
lesioni neurologiche, mai monosintomatiche, l’inquadramento diagnostico dovrà essere completato
da esame neurologico ed eventuale RMN del rachide ed elettromiografia sfinterica.

Terapia
Partendo dal presupposto che non si deve curare l’enuresi, ma il bambino enuretico, si comprende
facilmente come l’inizio del trattamento sia strettamente correlato alla richiesta effettiva della
famiglia e del bambino di risolvere il problema. E’ doveroso intervenire quando l’enuresi comincia
ad avere delle ripercussioni psicologiche sul bambino che teme di essere giudicato, si sente diverso
dagli altri, riduce l’autostima, si isola non partecipando alla vita sociale, rinunciando alle gite con
gli amici e ai momenti di comunità. In poche parole non vive più, per colpa dell’enuresi, con
serenità e gioia momenti importanti per la sua formazione e per il raggiungimento di un adeguato
equilibrio psico-fisico-intellettivo. L’approccio è impegnativo poiché prevede il coinvolgimento di
tutto il nucleo familiare e non solo la partecipazione del bambino.
Un fattore determinante, quindi, per iniziare il trattamento è la motivazione del bambino e della
sua famiglia a risolvere il disturbo. È importante che la famiglia sappia che il disturbo talvolta
richiede tempi lunghi per risolversi, che comunque nella maggior parte dei casi si risolverà
spontaneamente e che perciò il bambino non deve essere colpevolizzato, deriso o addirittura punito.
Due diversi approcci vengono proposti per il trattamento dell'enuresi notturna: l'uno
comportamentale, l'altro farmacologico.
L'approccio comportamentale, basato su tecniche di rieducazione vescico-sfinteriale, prevede
innanzi tutto l'osservazione di alcune norme generali quali la riduzione dell'introduzione di liquidi
nelle ore serali, lo svuotamento della vescica prima di dormire, la proscrizione del pannolone
durante le ore notturne perché, anche se comodo, spinge il bambino a rifugiarsi in comportamenti
infantili e l'incentivazione con premi da parte dei genitori al conseguimento dei primi risultati.
La terapia comportamentale dell'enuresi notturna sembra dare i risultati migliori con alti tassi di
guarigione e basse percentuali di ricadute.
L’approccio farmacologico prevede la somministrazione di ormone antidiuretico nell’ipotesi di una
sua carente produzione endogena durante le ore notturne. La desmopressina agisce in maniera
selettiva sui recettori renali aumentando il riassorbimento dell’acqua e dei soluti a livello dei tubuli
distali. Gli effetti collaterali comprendono la cefalea, l’intossicazione da acqua e l’iponatremia. Per
i disturbi minzionali diurni trova, invece, indicazione l’ossibutinina, farmaco ad azione
anticolinergica che agisce riducendo il numero e l’entità delle contrazioni vescicali e di conseguenza
riduce l’instabilità vescicale. Gli effetti collaterali possono essere dati da secchezza delle fauci, rash
cutanei, diarrea o stipsi, cefalea, vertigini. Viene anche descritta la comparsa di episodi di pavor
nocturnus che richiedono la sospensione della terapia. Il trattamento farmacologico dovrà essere
affiancato dal training vescicale utile al fine di aumentare la capacita vescicale e correggere le
abitudini vescicale sbagliate. L’enuresi notturna secondaria dovrebbe essere valutata e trattata come

la forma primaria. Nei casi di enuresi secondaria, in cui sia stata esclusa una eziologia organica, può
essere proposto lo stesso approccio considerando anche un eventuale supporto psicologico qualora
venga individuata una causa scatenante.
Bibliografia
1. Nelson “Trattato di Pediatria” Edizioni Minerva Medica
2. Schwarz Tiene “Manuale di Pediatria”Casa Editrice Ambrosiana Milano
3. Neveus T, Stendberg A, Lachgreen G et al “Il sonno dei bambini con enuresi: uno studio in
polisonnografia” Pediatrics 1999; 3:321-5
4. Katherine M. Graham, MPAS, Jay B. Levi, MD “Enuresi” Pediatrics in Review- Maggio
2009

Le infezioni delle vie urinarie
Chimenz R, Catena MA, Crisafulli RM, Privitera C, Fede C
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
Per infezione delle vie urinarie si intende “il riscontro di una urocoltura positiva con conta colonie
superiore a 100.000 cfu/mmc”, con presenza di determinati segni clinici e urinari associati.
Epidemiologicamente le IVU rappresentano la seconda causa di morbilità in età pediatrica, dopo le
infezioni delle vie respiratorie, con maggiore incidenza nel primo anno di vita. Dal punto di vista
eziologico, i batteri Gram – della famiglia delle Enterobacteriaceae rappresentano la causa più
frequente di IVU ( E.Coli nell’80-85% dei casi, ma anche Proteus, Klebsiella, Enterobacter sp,
Morganela M);anche i gram +, in misura minore, possono essere agenti eziologici di IVU (PSA,
Enterococcus, S.saprophyticus nelle adolescenti, S.aureus negli ascessi renali, Streptococco di
gruppo B nei neonati).
Le vie seguite dai patogeni per infettare le vie urinarie sono: 1) via ematogena: estremamente rara,
tranne nei neonati in cui l’IVU può instaurarsi nell’ambito di una sepsi; 2) via ascendente: la
colonizzazione della mucosa dei genitali da parte degli enterobatteri determina la diffusione
dell’infezione alla mucosa delle vie urinarie. La predisposizione alle IVU e l’attecchimento
dell’IVU stessa possono essere determinate da anomalie strutturali del tratto urinario (RVU,
anomalie funzionali della vescica quali instabilità vescicale secondaria a dissinergia sfinteriale),
fattori legati all’ospite (per es. la brevità dell’uretra femminile rende le bambine più suscettibili alle
IVU), fattori di virulenza del germe (presenza di adesine e fimbrie, emolisine e citotossine
proteiche, sierotipo, genetica del batterio).
La sintomatologia dell’IVU è multiforme e varia in rapporto a vari fattori, quali l’età, il sesso, la
gravità dell’infezione e la sua localizzazione (alta o bassa). Nel neonato e nel lattante mancano
principalmente i segni locali e si rilevano solo segni generali o a carico di altri organi o apparati:
rifiuto dell’alimento, arresto ponderale, vomito, diarrea, irritabilità, acidosi metabolica, pianto alla
minzione, macroematuria, emissione di urine maleodoranti, febbre sine causa, ipotensione, shock.
Nel bambino di età > 3 anni possono associarsi segni locali: disuria, dolore sovrapubico,
pollachiuria, urgenza minzionale, enuresi, emissione di urine maleodoranti, macroematuria, febbre
sine causa.
Gli scopi della diagnosi in un paziente con IVU sono: 1) conferma della diagnosi, 2) identificazione
del microrganismo responsabile, 3) localizzazione della sede d’infezione (distinzione tra IVU “alta”
e IVU “bassa” ai fini terapeutici e prognostici) , 4) riconoscimento del paziente con malformazione
del tratto urinario. Nel sospetto di IVU va sempre richiesto un esame delle urine, da solo o in

associazione con la coltura delle urine. Per la raccolta delle urine possono essere seguite diverse
metodiche:
- Mitto intermedio nei bambini più grandi, capaci di urinare a comando: le urine
(preferibilmente le prime del mattino) vanno raccolte a metà della minzione, previa
un’accurata pulizia dei genitali esterni. Tale metodica di raccolta delle urine è quella di
elezione in termini di sterilità e non invasività.
- Sacchetto perineale nei bambini più piccoli, non collaboranti. Il sacchetto va applicato ai
genitali previa accurata pulizia e rimosso non appena terminata la minzione; se il bambino
non minge entro 30 minuti dall’applicazione, il sacchetto va sostituito previa nuova pulizia
dei genitali. Tale tecnica di raccolta delle urine rischia di determinare dei falsi positivi e per
tale motivo è opportuno avere tre urocolture positive consecutive per essere certi di essere in
presenza di infezione urinaria.
- Cateterismo vescicale in casi selezionati, data l’invasività della tecnica.
I principali indicatori di infezione all’esame urine sono rappresentati da: esterasi leucocitaria, test ai
nitriti ( in quanto i germi gram- trasformano i nitrati urinari in nitriti; quando siano in gioco altri
germi, tipo cocchi gram pos e Proteus che non producono nitriti, si possono avere risultati
falsamente negativi), sedimento urinario ( riscontro di batteriuria). L’esame colturale delle urine
permette una diagnosi di sicurezza d’infezione: l’urinocoltura è considerata positiva quando
dimostri la crescita di un germe in concentrazione > 100.000 CFU. Per quanto riguarda altri esami
ematici, può essere utile l’esecuzione di un prelievo per emocromo (per la conta dei globuli bianchi)
ed indici di flogosi, anche se, nelle linee guida di riferimento e nella letteratura più recente, non
vengono presi in considerazione tali esami per la distinzione tra IVU alte e basse, in quanto sono
poco correlati con la sede d’infezione; a tale scopo può invece risultare utile il dosaggio della pro
calcitonina.
Una volta posta la diagnosi generica di IVU in un bambino, l’iter diagnostico non deve considerarsi
concluso, soprattutto perché va valutata la presenza di eventuali fattori di rischio quali:
- Ecografia fetale patologica
- Familiarità per Reflusso vescicoureterale
- Scarsa affidabilità del nucleo familiare
Il gold standard per quanto riguarda gli esami strumentali, soprattutto nel sospetto di una IVU
complicata, è rappresentato dall’esecuzione di:
- Cistouretrografia minzionale nel sospetto di un RVU, oggi la metodica ecografica con
mezzo di contrasto che sfrutta il principio delle microbolle (cistosonografia) rappresenta la
tecnica più appropriata.

- Scintigrafia renale con DMSA che permette di valutare anche eventuali esiti a distanza, quali
cicatrici renali.
Terapia delle IVU: andremo a curare un bambino con IVU per risolvere l’infezione acuta, prevenire
una sepsi urinaria, ma anche per ridurre il rischio di danno renale. La scelta dell’antibiotico si basa
sulla sensibilità del microrganismo alla molecola in questione, sulla tollerabilità del paziente
all’antibiotico scelto, sul rispetto del rapporto costo-efficacia.
Ovviamente gli antibiotici scelti per trattare una IVU devono essere metabolizzati a livello
dell’emuntorio renale. La terapia antibiotica va iniziata subito dopo aver raccolto il campione di
urine, in base a:
• sospetto clinico
• positività esame urine al microscopio
• positività stick urine.
Nel caso in cui si tratti di una IVU “complicata” (pzt con febbre alta, settico,disidratato,con vomito
persistente e/o scarsamente compliante) si inizierà con una terapia parenterale, per passare poi dopo
circa 2-4 giorni alla via orale ( per un totale di circa 10 giorni di terapia). In caso invece di IVU non
complicata ( pzt febbrile ma in buone condizioni generali, in grado di assumere liquidi e farmaci per
os, disidratato in modo lieve, e/o ben compliante) si inizierà direttamente una terapia orale. In attesa
dell’antibiogramma si avvia una terapia empirica, scegliendo tra i seguenti farmaci in base alla via
di somministrazione scelta:
- Via iniettiva: ceftriaxone 75 mg/kg ogni 24 h, oppure cefotaxime 150 mg/kg in 3-4 dosi, oppure
amino glicosidi in caso di allergia alle cefalosporine.
- Via orale: cefixime 8 mg/kg ogni 24 h, oppure amoxicillina/clavulanato 50 mg/kg in 2 dosi.
Altri possibili antibiotici da utilizzare sono:gentamicina (7.5 mg/kg/die), tobramicina (5 mg/kg/die),
amikacina (15 mg/kg/die),netilmicina (6-9 mg/kg/die)
La durata ottimale della terapia nei bambini con pielonefrite acuta non è nota.
Le raccomandazioni correnti suggeriscono 7-14 giorni (in media 10 giorni).

Bibliografia
1. “Le infezioni febbrili delle vie urinarie: Raccomandazioni di consenso per la diagnosi, il
trattamento e il follow-up in bambini di età compresa fra 2 mesi e 3 anni”. A cura di un
gruppo di lavoro della Società Italiana di Nefrologia Pediatrica (SINP): Montini G
(Coordinamento), Ammenti A, Cataldi L, Chimenz R, Fanos V, La Manna A, Marra G,
Materassi M, Pecile M, Pennesi M, Pisanello L, Sica F, Toffolo A. Medico e Bambino. 359-
370- 6/09
2. “UTI IN CHILDREN – Urinary tract infection in children: diagnosis, treatment and long-
term management” NHS National Institute for Health and Clinical Excellence – August
2007
3. “Antibiotics for acute pyelonephritis in children” Hodson, Willis, Craig. The Cochrane
Library 2010, Issue 3

Insufficienza Renale nel bambino
Fede C, Vitale A, La Mazza A, Crisafulli RM, Salpietro V, Fede C
UOSD Nefrologia e Reumatologia Pediatrica con Dialisi, AOU Policlinico G Martino, Messina
L’insufficienza renale è una sindrome clinica caratterizzata da una riduzione della funzionalità
renale che può essere improvvisa (IRA) o graduale e progressiva (IRC).
Insufficienza Renale Acuta (IRA)
Nell'insufficienza renale acuta si verifica una riduzione rapida (ore o giorni) della capacità dei reni
di espletare la funzione escretoria e, può essere reversibile. A differenza dell’insufficienza renale
cronica in cui la riduzione della funzione renale si verifica in un arco temporale di mesi o anni ed è
irreversibile. L'incidenza di insufficienza renale acuta varia a seconda delle condizioni ambientali e
socio-economiche. Nei paesi del Terzo Mondo la causa più frequente è la disidratazione da diarrea
profusa. Nei paesi occidentali la causa più frequente sono gli interventi di cardiochirurgia infantile.
Le cause di insufficienza renale acuta possono essere: pre-renali (condizionanti un ridotto apporto
ematico), renali (in cui è lesa l'integrità del parenchima) e post-renali (da ostacolato deflusso
urinario).
L'insufficienza renale acuta pre-renale è espressione di una compromissione funzionale secondaria
ad ipoperfusione acuta del parenchima renale.
Le caratteristiche principali di questo tipo di insufficienza renale acuta sono una riduzione del
volume urinario con urine a basso contenuto sodico ed elevato peso specifico.
L’insufficienza renale acuta parenchimale è conseguenza di un danno acuto a una o più componenti
tissutali del parenchima renale (glomeruli, tubuli, interstizio e vasi).
L’insufficienza renale acuta post-renale è conseguente ad ostruzione acuta delle vie escretrici.
Poichè l'integrità di un solo rene è sufficiente a garantire una adeguata funzione emuntoria questo
tipo di insufficienza renale acuta si instaura quando l'ostruzione interessa entrambe le vie escretrici
(es. l'uretra) o quando ci si trovi in una condizione di rene unico funzionante. L'incidenza
nell'ambito dell'insufficienza renale acuta è variabile a seconda dell'età, nel primo anno di vita
rappresentano circa il 10%.
Le cause più frequenti di insufficienza renale acuta nel primo anno di vita sono rappresentate
dall'ipoperfusione renale, le uropatie ostruttive rimangono importanti nel neonato, come le trombosi
venose. La sindrome emolitico-uremica è particolarmente rilevante nel primo anno di vita e diventa

la causa più importante fra 1 e 4 anni di vita. Nei bambini più grandi le glomerulonefriti acute e le
tubulopatie tossiche acute sono particolarmente importanti.
Quadro clinico
Nella clinica dell'insufficienza renale acuta la diuresi è inferiore a 500ml/1.73 m2/die (nel neonato
inferiore a 1 ml/kg/ora), a volte può essere conservata. Il quadro clinico può essere caratterizzato da
edemi periferici, incremento di peso, emodiluizione, ipertensione arteriosa, in un crescendo clinico
fino allo scompenso cardiaco, all'edema polmonare ed all'edema cerebrale.
La creatininemia é in aumento di 0.2 mg/dl/die nel neonato, di 1 mg/dl/die nel bambino più grande:
non rappresenta un indice di tossicità di per sé, ma marker importante di insufficienza renale.
È possibile che si manifesti un'iperpotassiemia che, quando >7 mEq/l, può comportare gravi
conseguenze cardiache che vanno dalle alterazioni elettrocardiografiche (onda T alta e a punta, QRS
allungato, onda P poco evidente) ad un quadro di aritmia con blocco AV fino alla fibrillazione
ventricolare ed all'arresto cardiaco.
Il decorso clinico passa attraverso una fase d'esordio in cui si verificano i fattori causali e si instaura
la contrazione funzionale, seguita da una fase oligurica in cui si manifestano i sintomi clinici e
biologici. Questa dura da poche ore a più giorni (in media 10-14 giorni). Se la durata è maggiore di
1 mese è probabile che si sia instaurata una necrosi corticale e che l'insufficienza renale sia
irreversibile. Segue infine una fase di recupero con ripresa della diuresi fino alla poliuria.
Inizialmente le orine sono ipotoniche, ricche di sali, e gli indici ritentivi renali ancora elevati. Segue
una graduale normalizzazione del filtrato glomerulare con recupero più lento della funzionalità
tubulare.
Trattamento dell'insufficienza renale acuta
Fin dall'inizio di uno stato di insufficienza renale acuta, prima che questa evolva fino alla necessità
di un trattamento dialitico, é indispensabile mettere in atto i provvedimenti rivolti a controllare
l'iperidratazione, correggere le disionie e l'acidosi e stabilire un'adeguata nutrizione per limitare
l'ipercatabolismo.
La dialisi diventa indispensabile se il bambino è ipercatabolico, in sovraccarico idro-salino e si
presume una oliguria protratta, oppure se esiste un sovraccarico idrosalino che non permette una
terapia infusionale indispensabile per correggere l'acidosi, rialimentare il bambino o istituire altre
terapie infusionali.
Le scelte tecniche prevedono la dialisi peritoneale e l'emodialisi.
La dialisi peritoneale é in generale il trattamento di elezione nell'insufficienza renale acuta
pediatrica, soprattutto nel bambino piccolo in cui i problemi tecnici dell'emodialisi di accesso

vascolare, di volume ematico in circolazione extracorporea e di dialisi iperefficace per lo più
sconsigliano questo tipo di trattamento a meno che non si sovrappongano delle situazioni, quali
ustioni addominali, che ne rendano improponibile l'utilizzo.
L'accesso al peritoneo avviene attraverso un catetere inserito con paracentesi: nell'insufficienza
renale acuta, se si agisce di urgenza e si prevede una durata dell'anuria contenuta si utilizzano
cateteri rigidi, tuttavia per lo più si preferiscono attualmente i cateteri in silastic morbidi tipo
Tencknoff inseriti non tanto a cielo coperto, quanto con tecnica chirurgica.
Gli schemi dialitici più comuni sono di 25-50/ml/kg introdotti in peritoneo e lasciati in sosta per
10-20 min. Generalmente si riesce ad ottenere una disidratazione di 50-80 ml/kg soprattutto in caso
di sovraccarico idrosalino.
L'emodialisi interviene nel trattamento dell'insufficienza renale acuta quando si richiede una rapida
risoluzione di un sovraccarico idrosalino al limite con l'edema polmonare o di correzione di
un'iperpotassiemia a rischio di tossicità cardiaca.
Nell'ultimo decennio é stata utilizzata spesso con successo una tecnica emodialitica peculiare:
l'emofiltrazione continua artero-venosa (CAVH) che ha trovato larga applicazione nel trattamento
di sovraccarichi idrosalini o alterazioni elettrolitiche in bambini in insufficienza renale acuta. La
gittata cardiaca del paziente fornisce il flusso ematico attraverso il circuito e non sono necessarie
pompe per spingere il sangue nel dializzatore. La gamma di filtri molto permeabili é tale da
permettere l'applicazione di questa metodica anche ai prematuri. L'indicazione principale, oltre a
quella tecnica di non avere a disposizione in ambienti particolari, la possibilità di produrre bagno di
dialisi, é rappresentata da situazioni emodinamicamente instabili per cui gli squilibri indotti da una
dialisi tradizionale di poche ore, ad alta efficienza, potrebbero essere problematici.
Insufficienza Renale Cronica (IRC)
L’IRC è una sindrome clinico- metabolica conseguenza della progressiva distruzione dei
nefroni,indipendentemente da quale ne sia la causa,fino alla scomparsa, nella fase terminale (IRT),
della citoarchitettura anatomica dei reni. La riduzione nefronica comporta modificazioni nella
emodinamica renale che determinano aumento del flusso plasmatico e aumento della pressione
idrostatica al fine di permettere ai nefroni residui di compensare quelli mancanti, il prezzo che si
paga è l’ aumentata permeabilità vasale(proteinuria) e della matrice mesangiale, entrambi fattori
favorenti la sclerosi.

Sul piano della funzionalità è presente IRC, quando il filtrato glomerulare è al di sotto di
90ml/min./1.73 da almeno tre-sei mesi. La IRC è stata suddivisa in 5 stadi, che si susseguono senza
soluzione di continuità fino alla fase terminale non più compatibile con la vita in assenza di
trattamento dialitico o trapianto. È possibile nello stadio 1 attraverso campagne di sensibilizzazione
sociale attenzionare la condizione di danno renale ,prevenendo o riducendone i fattori di rischio,in
un periodo in cui la malattia è muta sul piano bioumorale.
Stadio 1: IRC lieve (FG 60-89 % del normale)
(completo compenso biochimico metabolico attraverso la riserva funzionale. Nessun segno di
laboratorio)
Stadio 2: IRC moderata (FG 30-59 % del normale)
(segni iniziali di scompenso funzionale,aumento dei valori di azotemia e creatininemia).
Stadio 3: IRC severa ( FG 15-29 % del normale)
(comparsa di alterazioni biochimico- metaboliche)
Stadio 4: IRC avanzata ( FG 10-15% del normale)
(sintomatologia uremica con interessamento di organi ed apparati)
Stadio 5: IRC terminale (FG < 10ml/min/1.73)
(compromissione multisistemica, marcata ritenzione idrica. Necessità di terapia sostitutiva
(HD,PD,TX)
È possibile nello stadio 1 attraverso campagne di sensibilizzazione sociale attenzionare la
condizione di danno renale, prevenendo o riducendone i fattori di rischio,in un periodo in cui la
malattia è muta sul piano bioumorale.
Entità del problema:
In Italia secondo l’Italkid (registro italiano dei bambini con IRC) l’incidenza di IRC è di 12
bambini/anno per milione di popolazione pediatrica, mentre la necessità di trattamento sostitutivo
dialitico è di circa 3-6 bambini/anno/ per milione di bambini.
Questi dati sono da considerare sottostimati in quanto molti bambini con IRC meno severa
raggiungeranno lo stato di IRT nell’età adulta.
Verosimilmente l’incidenza di IRC in età pediatrica si situa tra 10-30 bambini/anno/milione di
popolazione pediatrica.
Il minor numero di bambini in dialisi rispetto alle altri paesi occidentali

È da ricercare nel fatto che ancor oggi in Italia molti bambini vengono dializzati in centri adulti, per
lo più centri per adulti,vista la anomalia italiana della loro vasta diffusione, con le conseguenze di
una assistenza tecnicamente accurata ma,senza assistenza pediatrica che si fa carico in senso
solistico del bambino.
Le cause di IRC riportate del Registro Pediatrico Italiano dell’Insufficienza Renale Cronica
(Italkid), indicano che le ipodisplasie con presenza o assenza di uropatie malformative sono tra le
cause più frequenti(54-56%) ,seguite dalle malattie ereditarie (15-16%) e dalle malattie cistiche. Il
reflusso vescico-ureterale è la uropatia preminente tra le cause di IRC, seguita dalle valvole
dell’uretra. Le glomerulonefriti nei bambini più piccoli incidono soltanto per il 5-6%, mentre nella
terza infanzia e nell’adolescente le glomeronefriti ed i processi autoimmunitari acquistano
rilevanza.
La sintomatologia dell’IRC, nelle fasi iniziali, può essere vaga e aspecifica (cefalea, astenia,
anoressia, vomito, polidpsia, poliuria, ritardo di crescita), specie nei bambini con danno renale di
tipo malformativo mentre, nei bambini con glomerulonefriti o nefropatie ereditarie sono i segni
della malattia di base che compaiono prima dell’esordio di insufficienza renale che conducono alla
diagnosi.
All’inizio, quindi la ridotta funzione renale è mascherata da un compenso metabolico che rende le
conseguenze cliniche non evidenti per lungo tempo. Con il progredire della perdita di funzione
renale, le capacità adattative del rene divengono insufficienti e si manifesta un quadro clinico di
insufficienza renale, sempre più manifesto ed importante fino ad arrivare a una serie di sintomi
pluriorgano,che diventano sempre più evidenti fino all’uremia, fase di scompenso terminale
incompatibile con la vita.
La gestione dei bambini con IRC richiede un accurato monitoraggio dei parametri clinici (esame
obiettivo, peso, statura, pressione arteriosa) e bioumorali che comprendono emoglobina (anemia),
elettroliti (iponatremia, iperkaliemia, acidosi), azotemia, creatine mia (accumulo di azoto e stadio di
funzionalità renale), livelli di calcio e fosforo e fosfatasi alcalina e (ipocalcemia, iperfosfatemia,
osteodistrofia). Inoltre, è importante il dosaggio periodico, almeno 2 volte l’anno del paratormone e
della densitometria ossea per la valutazione e la prevenzione della osteodistrofia renale. Lo stato
nutrizionale può essere monitorato attraverso il controllo dell’albumina sierica,dello zinco,della
trasferrina, dell’acido folico e della sideremia. La ecocardiografia a cadenza semestrale e la
radiografia del torace a cadenza annuale per valutare la funzionalità cardiaca.
Lo scopo è quello di ritardare la progressione del danno renale e la morbilità del complesso di
organi e apparati che il rene malato cointeressa nel suo progredire verso l’uremia terminale.

Bibliografia
1. Avner E.D.,Harmon W.E.,Niudet P.: Pediatric Nephrology 5th Lippincott.2004
2. De Santo N.,Capasso G.:Nefrologioa Pediatrica ed. Bios Cosenza
3. Textor S.C.:Curr. Opin. Nephrol Hypertension,2010
4. Levey A:S: et al.: Kidney int.,2010,Dec8.
Top Related