Tutte le deficienze in anticorpi sono associate ad un ... · catena leggera delle Ig e inattiva...
40
Immunodeficienze primarie B Le immunodeficienze primarie B comprendono un gruppo eterogeneo di disordini accumunati dalla marcata riduzione o l’assenza di immunoglobuline nel siero. Tutte le deficienze in anticorpi sono associate ad un aumentata suscettibilità alle infezioni da parte di batteri capsulati (Streptococcus pneumonie, Haemophilus influenza) che causano bronchiti e polmoniti. Il trattamento dei pazienti con difetto degli anticorpi consiste nella somministrazione di immunoglobuline. Tale somministrazione avviene Le deficienze anticorpali possono essere raggruppate in 3 grandi categorie: i) difetti nello sviluppo dei linfociti B; ii) sindromi da IperIgM; iii) immunodeficienza comune variabile.
Transcript of Tutte le deficienze in anticorpi sono associate ad un ... · catena leggera delle Ig e inattiva...

ImmunodeficienzeprimarieB
Le immunodeficienze primarie B comprendono un gruppo eterogeneo di disordiniaccumunatidallamarcatariduzioneol’assenzadiimmunoglobulinenelsiero.Tutteledeficienzeinanticorpisonoassociateadunaumentatasuscettibilitàalleinfezionidaparte di batteri capsulati (Streptococcus pneumonie, Haemophilus influenza) che causanobronchitiepolmoniti. Il trattamentodei pazienticondifettodeglianticorpiconsistenellasomministrazionediimmunoglobuline.TalesomministrazioneavvieneLedeficienzeanticorpalipossonoessereraggruppatein3grandicategorie:i)difettinellosviluppodei linfocitiB; ii)sindromidaIperIgM;iii) immunodeficienzacomunevariabile.

ImmunodeficienzecausatedadifettinellosviluppodeilinfocitiB
IdifettidellosviluppodeilinfocitiB sono caratterizzati da unaprofondaipogammaglobulinemia,una forte riduzione o assenza dilinfocitiBnel sangueperifericoeinfezioni batteriche ricorrenti neiprimi5annidivita.Il blocco delllo sviluppo deilinfociti B avviene prima dellae s p r e s s i o n e d e l l eImmunoglobuline complete dimembrana.Mutazioni nella tyrosin chinasiBtk sono responsabili dell’85%delle agammaglobulinemie. Circala metà dei restanti casi sonodovuti a mutazioni in geni checodificano per componenti delpre-BCRodelBCR.

StadidisviluppodeilinfocitiB
I principali eventi che si verificanodurante lamaturazionedei linfociti Bs o n o i l r i a r r a n g i a m e n t o el ’ e sp re s s i one de i g en i de l l eimmunoglobuline. Gli stadi dellosviluppo dei linfociti possono essereidentificati in base allo stato delriarrangiamento dei geni codificati lecatene dei recettori dell’antigene(BCReTCR). I linfocitiBsi sviluppanonel midol lo osseo e i l pr imoprecursoreorientatoversolosviluppoBprendeilnomedipro-B.

I linfociti pro-B possonoessere distinti in baseall’espressione di CD10 eCD19. Nella fase pro-Biniziano ad essere espressiRAG1eRAG2 responsabilidella ricombinazione deisegmenti genici D, J dellacatenapesanteµ.DopolaricombinazioneD-Jun segmento genico Vricombina con il segmentogenicoDJ.L ’ e n z i m a t e r m i n a ld e o s s i n u c l e o t i d i ltransferasi(TdT)chemedial’aggiuntadinucleotidialleg i u n z i o n i V / D / J èaltamente espresso inquestostadio.

Lo stadio successivo al pro-Bè rappresentatodallecellulepre-B chesonocaratterizzatedalcompleto riarrangiamento della catenapesante delle Ig che viene espressa sullamembrana cellulare e in associazione allecatenesostitutiveV-preBe λ5.Talerecettoresi associa alle molecole Igα e Igβ chetrasducono il segnale costituisce il recettorepre-B.I linfociti pre-B vanno incontro a una elevataproliferazione.L’espressionedelpre-BCRoltread indurre la proliferazione delle cellule chehannoriarrangiatoproduttivamente lacatenaIgµ invia i segnali che mediano l’esclusioneallelica.InoltrestimolailriarrangiamentodellacatenaleggeradelleIgeinattival’espressionedelle catene sostitutive. In seguito alriarrangiamento della catena leggera, questasostituirà le catene λ5 e V-preB sullamembrana cellulare e i linfociti esprimenti laIgM completa sono definiti linfociti Bimmaturi.
SviluppodeilinfocitiB

X-linkedagammaglobulinemia(XLA)L’X-linked agammaglobulinemia è stata una fra le prime immunodeficienzedescritte.Nel1952Bruton riporta il casodiunbambinodi8anni con ricorrenti infezionibattericheedassenzadellafrazionedelleglobulinenelsiero.Questaosservazionefufattasullabasediduenuoveacquisizioniscientifiche:-L’applicazione della tecnica dell’elettroforesi sulle proteine seriche insiemeall’identificazionecheglianticorpicorrispondevanoallafrazionedelleglobuline.-L’utilizzodegliantibiotici(1940)Il bambino descritto da Bruton fu trattato con somministrazione sottocutaneamensiledigammaglobuline.-L’analisi di altri casi di bambini affetti da agammaglobulinemia avevanodimostrato che questa colpiva maggiormente i maschi e aveva un pattern ditrasmissioneX-linked.

Nel 1970 è stato dimostrato che i pazienti affetti da X-linkedagammaglobulinemiamostravano una forte riduzione nel numero di linfociti Bcircolanti. Nel 1993 due gruppi contemporaneamente riportaronol’identificazionedelgene responsabiledellaXLA rivelandocheessocodificavaperunachinasicitoplasmaticaederalocalizzatosulcromosomaX.QuestachinasifudenominataBruton’sTyrosinkinase(BTK)Btk appartiene alla famiglia di tirosin chinasi citoplasmatiche chiamate TecchinasicheincludeTec,Itk,RlkeBmx.LeTecchinasisonoespresseprincipalmentenellecellulediorigineematopietica.LecelluleBelepiastrineesprimonoBtkeTec;lecelluleTesprimonoItkRlkeTeclecellulemieloidiinclusiimastocitiesprimonoBtk,TecItkeRlk.
MutazionidellaBruton’styrosinekinasecausanoXLA

Btk presenta nella regione NH-terminale il dominio PH (omologo della plecstrina)che rende possibile l’associazione tra BTK e il fosfatidil inositolo-3, 4, 5-trifosfato(PIP3)generatodallaPI3K.IdominiTH,SH2eSH3diinterazioneconaltreproteineNellaregioneCOOHterminaleildominiochinasico.
Espressionedelletecchinasi

Btkèespressadurantelosviluppo delle cellule Bdallo stadio pro-B finoallostadiomaturo.In seguito ad attivazioneB t k t r a s l o c a a l l amembrana plasmaticaattraverso l’interazionefra il dominio TH con ilPIP3.
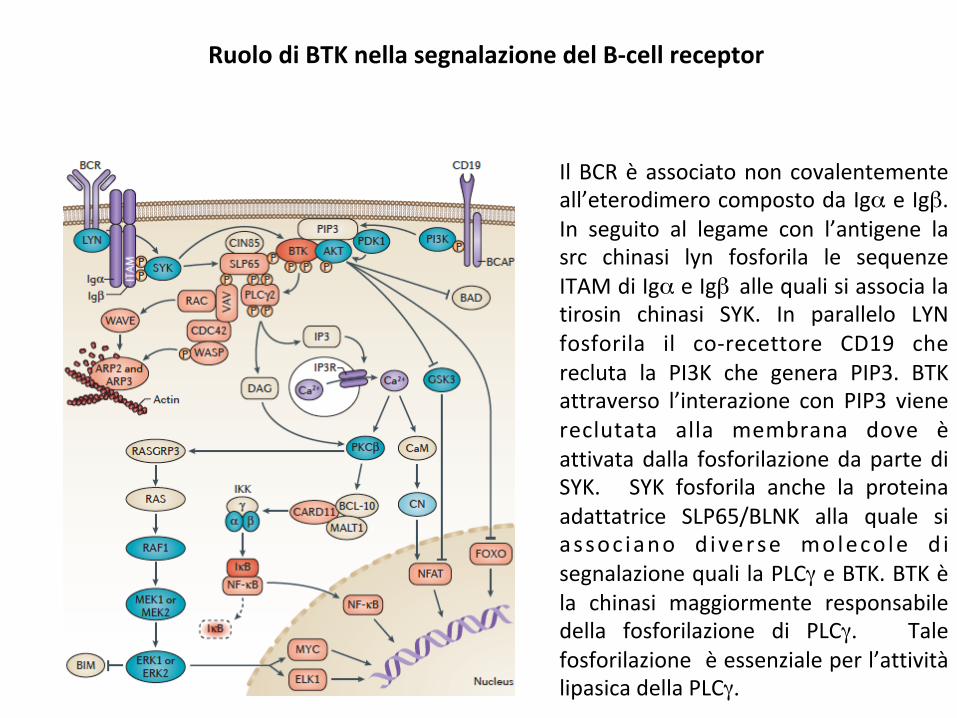
RuolodiBTKnellasegnalazionedelB-cellreceptor
IlBCRèassociatononcovalentementeall’eterodimerocompostodaIgαeIgβ.In seguito al legame con l’antigene lasrc chinasi lyn fosforila le sequenzeITAMdiIgαeIgβ allequalisiassocialatirosin chinasi SYK. In parallelo LYNfosforila il co-recettore CD19 cherecluta la PI3K che genera PIP3. BTKattraverso l’interazione con PIP3 vienereclutata alla membrana dove èattivatadalla fosforilazionedapartediSYK. SYK fosforila anche la proteinaadattatrice SLP65/BLNK alla quale siassoc iano d iverse moleco le d isegnalazionequalilaPLCγeBTK.BTKèla chinasi maggiormente responsabiledella fosforilazione di PLCγ. Talefosforilazioneèessenzialeperl’attivitàlipasicadellaPLCγ.

FrequenzerelativedicelluleaidiversistadidisviluppoB
Lostudiodeiprecursoridel le cel lule B nelmidollo dei pazientia f f e t t i d a X LA hadimostrato una forteriduzione delle cellulepre-B e delle cellulee s p r i m e n t i l e I gcomplete associato adun aumen to de l l ecellule pro-B (CD19+,TdT+ , CD34+ ) . C iòdetermina un aumentode l rapporto de l lecellule proB/ preB. Incircolo i pazienti XLAp r e s e n t a n o p o c h ilinfocitiB.
Neibambininormalifrail10%eil25%dellecelluleCD19+delmidollosonorappresentatedalinfocitiproBmentreil35-60%èrappresentatodacellulepre-B.
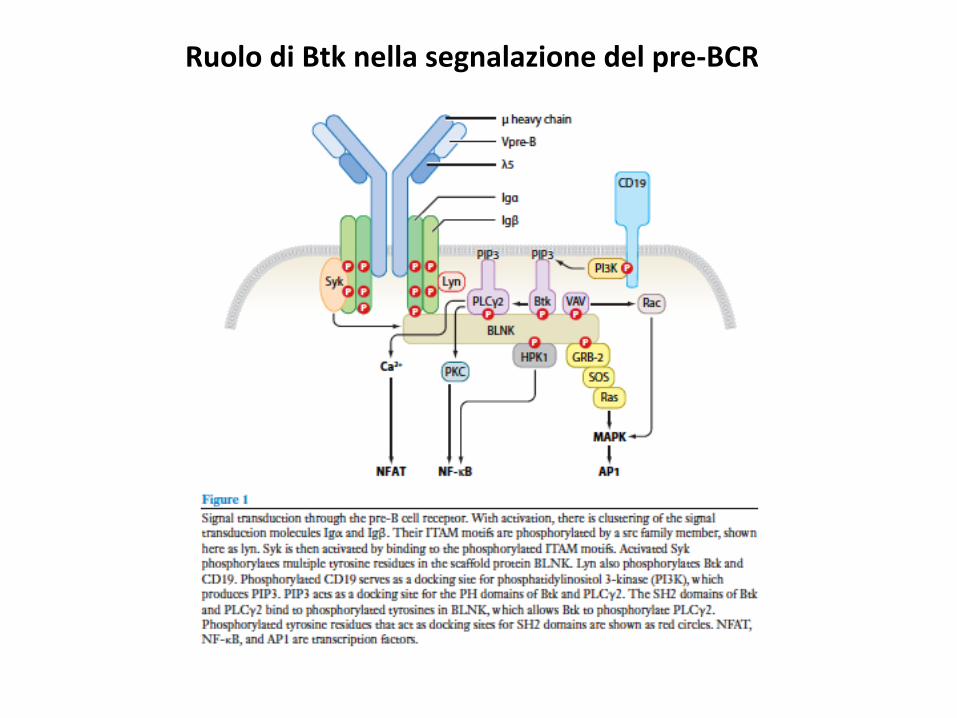
RuolodiBtknellasegnalazionedelpre-BCR
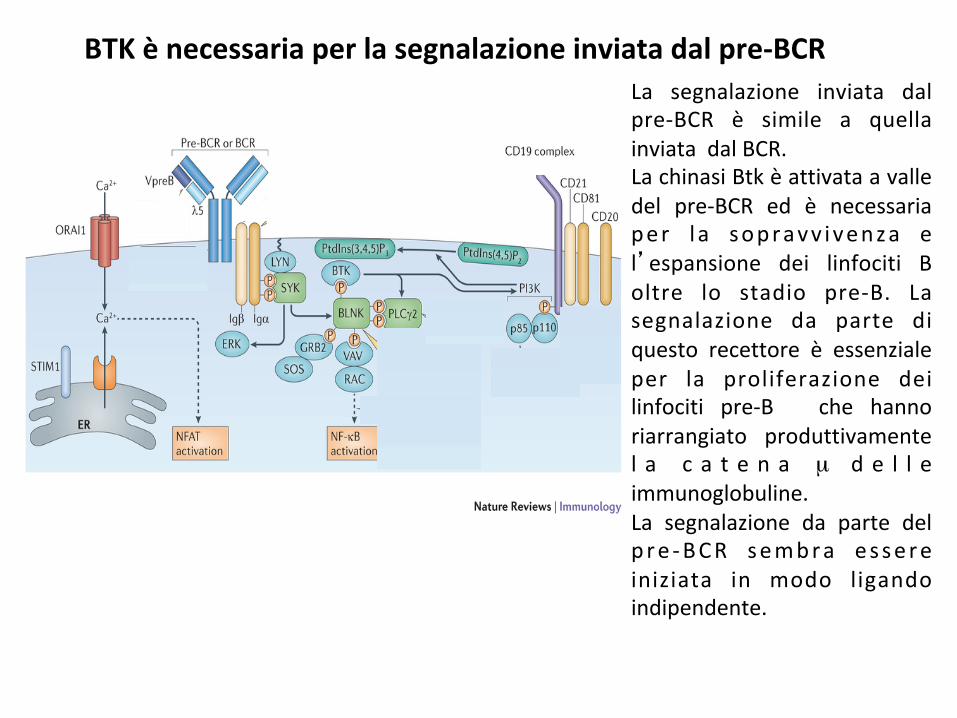
La segnalazione inviata dalpre-BCR è simile a quellainviatadalBCR.LachinasiBtkèattivataavalledel pre-BCR ed è necessariaper l a sopravv ivenza el’espansione dei linfociti Boltre lo stadio pre-B. Lasegnalazione da parte diquesto recettore è essenzialeper la proliferazione deilinfociti pre-B che hannoriarrangiato produttivamentel a c a t e n a µ d e l l eimmunoglobuline.La segnalazione da parte delp re -BCR sembra es sereiniziata in modo ligandoindipendente.
BTKènecessariaperlasegnalazioneinviatadalpre-BCR

FunzionidelrecettorepreB
Le cellule preB Cµ+SLC+(surrogate light chain)+nei pazienti XLA sono dip i c c o l e d imen s i o n isuggerendo che Btk èn e c e s s a r i a p e rl ’ e s p a n s i o n e e l asopravv i venza de l l ecellulepreB.

Mutazioni in Btk
Delle più di 600 mutazioni descritte in Btk, più del 90% sono dovute a sostituzioni di una singola coppia di basi e inserzioni o delezioni di meno di 5 coppie di basi.
In base alla età did i a g n o s i , l aconcentrazione di IgM nel plasma e ilnumerodicelluleBincircololemutazionidiB t k s o n o s t a t eclassificate in gravi emoderate. Questeultime permettonola produzione di unaminima quantità dienzima.

AgammaglobulinemieautosomicherecessiveNon tutti i pazienti con profondaipogammaglobulinemia, riduzione oassenza di linfociti B e infezioni nei primimesidivitasonomaschicondifetti inBtk.Giàneglianni‘70erastatodimostratochecirca un 10%di femmine presentavano unfenotipo XLA. Tali pazienti manifestavanoprecocemente la malattia, presentavanomenodell’1%deinormalivaloridicelluleBi n c i r c o l o e u n a p r o f o n d aagammaglobulinemia.Mutazioni nella catena µ delle Ig, di Igα,B L N K ( S L P 6 5 ) e λ 5 c a u s a n oagammaglobulinemie.DifettinellefasiprecocidellosviluppodeilinfocitiBsonoassociatia:• infezionibatterichericorrentineiprimi5annidivita• profondaipogammaglobulinemia• forteriduzioneoassenzadilinfocitiBcircolantii

Agammaglobulinemierecessiveautosomiche
L’85% dei difetti del losviluppo B sono dovuti amutazioni in Btk, 5% hannomutazioninellacatenaµ,unaminoranza mutazioni in λ5Igα o BLNK. I pazienti conmutazioni della catena µ delle Ig tendonoadavereunfenot ipo più grave deipazienti con mutazioni inBtk.

TrattamentodelleXLA
Primadella identificazionedapartedi Brutondella XLA i pazienti affetti daquestaimmunodeficienza morivano di infezioni croniche o acute. Fra gli anni ‘50-’70l’introduzione della somministrazione intramuscolare di gammaglobuline cheassicurava una bassa concentrazione di IgG faceva si che la maggior parte deipazientimorivaprimadell’etàadultaacausadi infezioniacute,polmoniti, infezionidaenterovirus.Apartiredaglianni‘80laterapiadelleXLAvieneeffettuatamediantetrasferimento delle IgG per via endovenosa o sottocutanea associata asomministrazione di antibiotici. Questo trattamento ha notevolmente aumentatol’etàmediadisopravvivenzadiquestipazienti.
DalpuntodivistaclinicoipazienticonagammaglobulinemiaautosomicarecessivanonsonodistinguibilidaipazientiXLA.Ancheinquestocasoesisteunanotevolevariabilitànella gravità delle infezioni. Nei pazienti con mutazioni nella catena pesante delleimmunoglobuline, Igα e BLNK presentano infezioni da Pseudomonas e Stafilococco,neutropenia suggerendo che l’ipogammaglobulinemia è la causa dell’aumentatasuscettibilitàalleinfezioni.La terapia in questi pazienti è la somministrazione di immunoglobuline associata atrattamentoantibiotico.

ImmunodeficienzeanticorpalidadifettifunzionalidellecelluleB
Q u e s t o g r u p p o d iimmunodeficienze sono causatedadifettinellaattivazioneenellaproduzione di anticorpi da parted e i l i n f o c i t i B . Q u e s t eimmunodeficienzeincludono:i) la sindrome da IperIgM,ii)l’immunodeficienza comunevariabile.

SindromedaiperIgM
Sindrome da iperIgM= questa sindrome include un gruppo eterogeneo diimmunodeficienzeprimariecaratterizzatedalivellisericibassioassentidiIgG,IgA,IgE,manormalioelevati livellidiIgM.Ipazientiaffettidaquestasindromesonosuscettibiliadinfezionipolmonarieopportunistiche.L’iperIgMècausatadadifettinellacommutazionediclassedelleimmunoglobulinedaIgM/IgDaIgG,IgAoIgE.

PatologiamolecolaredelleIperIgM
All’inizio degli anni ‘60 diversi gruppi hannodescritto pazienti con infezioni ricorrenti e elevatilivelli di IgMma ridotti livelli di IgG. Tali pazientii n i z i a l m e n t e d e f i n i t i a f f e t t i d adisgammaglobulinemiaeranoper lamaggiorpartemaschi.Iltermineiper-IgMèstatoutilizzatoperlaprima volta nel 1974. Attualmente essendoevidente che la caratteristica comune di questipazientièildifettonellacommutazionediclasseèstatopropostocheildifettodellacommutazionediclasse definisce un gruppo di pazienti chepresentanoinfezionibatterichericorrentienellorosieronormalioelevatilivellidiIgMeridottilivellidiIgG,IgA,IgE.
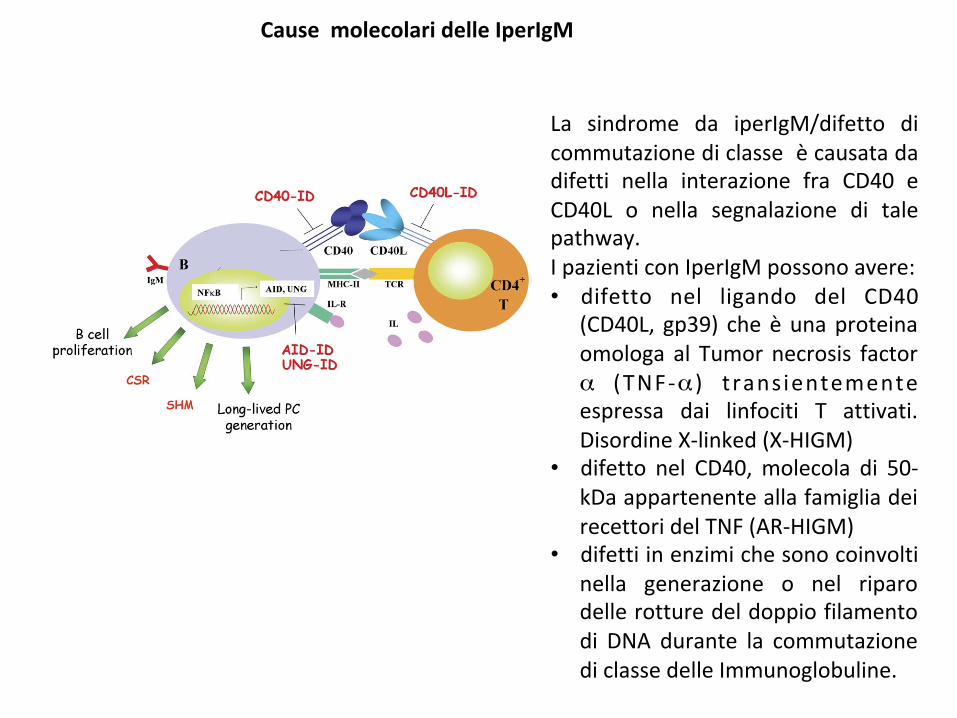
La sindrome da iperIgM/difetto dicommutazionediclasseècausatadadifetti nella interazione fra CD40 eCD40L o nella segnalazione di talepathway.IpazienticonIperIgMpossonoavere:• difetto nel ligando del CD40
(CD40L, gp39) cheèunaproteinaomologa al Tumornecrosis factorα (TNF-α ) transientementeespressa dai linfociti T attivati.DisordineX-linked(X-HIGM)
• difetto nel CD40,molecola di 50-kDaappartenenteallafamigliadeirecettoridelTNF(AR-HIGM)
• difettiinenzimichesonocoinvoltinella generazione o nel riparodellerotturedeldoppiofilamentodi DNA durante la commutazionediclassedelleImmunoglobuline.
CausemolecolaridelleIperIgM
and SHM (Fig 2). On the other hand, these defects alsocompromise monocyte–dendritic cell activation and henceaffect T-cell priming. This explains why the immunologicand clinical features of defects of CD40-mediated signal-ing are broader and more severe than pure antibody defi-ciencies.
X-linked immunodeficiency with hyper-IgM(CD40L deficiency)
Inherited as an X-linked trait, CD40L deficiency wasthe first form of hyper-IgM syndrome for which themolecular basis was identified.
Most patients present in infancy with recurrent bacterialor opportunistic infections, including Pneumocystisjiroveci–induced pneumonia and diarrhea caused byCryptosporidium species infection.7,13 Neutropenia is acommon finding and might further contribute to suscepti-bility to bacterial infections. Severe liver–biliary tract dis-ease, progressing to sclerosing cholangitis, occurs in asignificant number of patients (with some discrepancy be-tween the European and the United States registries) and isoften associated with Cryptosporidium species infection.A high proportion of patients (50% in the European series)die before the fourth decade of life, despite substitutiontreatment with Igs. This has prompted more aggressiveforms of treatment on the basis of hematopoietic stemcell transplantation. However, a retrospective analysis of38 European patients treated with hematopoietic stemcell transplantation has shown that only 22 (58%) ofthem were cured.14
Patients with CD40L deficiency have markedly de-creased serum IgG and IgA levels and normal to increasedIgM levels. Occasionally, IgA levels might be normal.
This is likely due to a CD40L-CD40–independent CSRmechanism, in which B cells are activated by B-lympho-cyte stimulator/B-cell activating factor (BLyS/BAFF) anda profileration-inducing ligand (APRIL) molecules (ex-pressed by dendritic cells activated by IFN-a, IFN-g, orLPS), along with signals provided by cytokines (TGF-b)and B-cell receptor engagement.15
Flow cytometry represents the most important screen-ing assay for diagnosis of CD40L deficiency. In mostpatients, in vitro activation of T lymphocytes with phorbolesters and ionomycin fails to induce expression of theCD40L molecule, as revealed by staining with CD40L-specific mAbs or a CD40-Ig chimeric construct. In anycase final confirmation of mutations requires molecularanalysis. Genomic abnormalities are scattered along theCD40L gene (Fig 3).16 In a minority of patients, mildermutations that allow binding of CD40 (albeit at reducedintensity) are associated with a less severe clinical course,which in a few cases was characterized uniquely by parvo-virus B19–related anemia.
B lymphocytes from the patients coexpress surface IgM(sIgM) and sIgD; in most cases the proportion of memory(CD271) B lymphocytes and the frequency of SHM arereduced. Importantly, defects of CSR and SHM reflect ab-normalities of the T-cell compartment, as indicated by thefact that the patients’ B cells can be induced to undergoCSR in vitro on activation with CD40 agonists and cyto-kines.17
In addition, lymph nodes from CD40L-deficient pa-tients show primary follicles but are devoid of germinalcenter formation, indicating the importance of CD40L-CD40 interaction for maturation of secondary lymphoidorgans.
FIG 2. Schematic representation of CD40L-CD40 interaction and its effects on CSR and SHM. Activated CD41
T cells in the lymph nodes interact with CD40-expressing B cells and secrete ILs that interact with cytokine-specific B-cell receptors (IL-R). CD40-mediated signaling activates the NF-kB signaling pathway and ultimatelyresults in the expression of AID and UNG. Mutations that affect CD40-mediated B-cell activation result in someof the forms of hyper-IgM syndrome. MHC-II, Major histocompatibility complex of class II; TCR, T-cell recep-tor; CD40L-ID, X-linked immunodeficiency caused by CD40L defect; CD40-ID, immunodeficiency caused byCD40 deficiency; XL-EDA-ID, X-linked immunodeficiency with ectodermal dystrophy; AID-ID, immunodefi-ciency caused by AID deficiency; UNG-ID, immunodeficiency caused by UNG deficiency; PC, plasma cell.
J ALLERGY CLIN IMMUNOL
APRIL 2006858 Notarangelo et al
Basic
and
clinica
limm
unolo
gy
and SHM (Fig 2). On the other hand, these defects alsocompromise monocyte–dendritic cell activation and henceaffect T-cell priming. This explains why the immunologicand clinical features of defects of CD40-mediated signal-ing are broader and more severe than pure antibody defi-ciencies.
X-linked immunodeficiency with hyper-IgM(CD40L deficiency)
Inherited as an X-linked trait, CD40L deficiency wasthe first form of hyper-IgM syndrome for which themolecular basis was identified.
Most patients present in infancy with recurrent bacterialor opportunistic infections, including Pneumocystisjiroveci–induced pneumonia and diarrhea caused byCryptosporidium species infection.7,13 Neutropenia is acommon finding and might further contribute to suscepti-bility to bacterial infections. Severe liver–biliary tract dis-ease, progressing to sclerosing cholangitis, occurs in asignificant number of patients (with some discrepancy be-tween the European and the United States registries) and isoften associated with Cryptosporidium species infection.A high proportion of patients (50% in the European series)die before the fourth decade of life, despite substitutiontreatment with Igs. This has prompted more aggressiveforms of treatment on the basis of hematopoietic stemcell transplantation. However, a retrospective analysis of38 European patients treated with hematopoietic stemcell transplantation has shown that only 22 (58%) ofthem were cured.14
Patients with CD40L deficiency have markedly de-creased serum IgG and IgA levels and normal to increasedIgM levels. Occasionally, IgA levels might be normal.
This is likely due to a CD40L-CD40–independent CSRmechanism, in which B cells are activated by B-lympho-cyte stimulator/B-cell activating factor (BLyS/BAFF) anda profileration-inducing ligand (APRIL) molecules (ex-pressed by dendritic cells activated by IFN-a, IFN-g, orLPS), along with signals provided by cytokines (TGF-b)and B-cell receptor engagement.15
Flow cytometry represents the most important screen-ing assay for diagnosis of CD40L deficiency. In mostpatients, in vitro activation of T lymphocytes with phorbolesters and ionomycin fails to induce expression of theCD40L molecule, as revealed by staining with CD40L-specific mAbs or a CD40-Ig chimeric construct. In anycase final confirmation of mutations requires molecularanalysis. Genomic abnormalities are scattered along theCD40L gene (Fig 3).16 In a minority of patients, mildermutations that allow binding of CD40 (albeit at reducedintensity) are associated with a less severe clinical course,which in a few cases was characterized uniquely by parvo-virus B19–related anemia.
B lymphocytes from the patients coexpress surface IgM(sIgM) and sIgD; in most cases the proportion of memory(CD271) B lymphocytes and the frequency of SHM arereduced. Importantly, defects of CSR and SHM reflect ab-normalities of the T-cell compartment, as indicated by thefact that the patients’ B cells can be induced to undergoCSR in vitro on activation with CD40 agonists and cyto-kines.17
In addition, lymph nodes from CD40L-deficient pa-tients show primary follicles but are devoid of germinalcenter formation, indicating the importance of CD40L-CD40 interaction for maturation of secondary lymphoidorgans.
FIG 2. Schematic representation of CD40L-CD40 interaction and its effects on CSR and SHM. Activated CD41
T cells in the lymph nodes interact with CD40-expressing B cells and secrete ILs that interact with cytokine-specific B-cell receptors (IL-R). CD40-mediated signaling activates the NF-kB signaling pathway and ultimatelyresults in the expression of AID and UNG. Mutations that affect CD40-mediated B-cell activation result in someof the forms of hyper-IgM syndrome. MHC-II, Major histocompatibility complex of class II; TCR, T-cell recep-tor; CD40L-ID, X-linked immunodeficiency caused by CD40L defect; CD40-ID, immunodeficiency caused byCD40 deficiency; XL-EDA-ID, X-linked immunodeficiency with ectodermal dystrophy; AID-ID, immunodefi-ciency caused by AID deficiency; UNG-ID, immunodeficiency caused by UNG deficiency; PC, plasma cell.
J ALLERGY CLIN IMMUNOL
APRIL 2006858 Notarangelo et al
Basic
and
clinica
limm
unolo
gy
and SHM (Fig 2). On the other hand, these defects alsocompromise monocyte–dendritic cell activation and henceaffect T-cell priming. This explains why the immunologicand clinical features of defects of CD40-mediated signal-ing are broader and more severe than pure antibody defi-ciencies.
X-linked immunodeficiency with hyper-IgM(CD40L deficiency)
Inherited as an X-linked trait, CD40L deficiency wasthe first form of hyper-IgM syndrome for which themolecular basis was identified.
Most patients present in infancy with recurrent bacterialor opportunistic infections, including Pneumocystisjiroveci–induced pneumonia and diarrhea caused byCryptosporidium species infection.7,13 Neutropenia is acommon finding and might further contribute to suscepti-bility to bacterial infections. Severe liver–biliary tract dis-ease, progressing to sclerosing cholangitis, occurs in asignificant number of patients (with some discrepancy be-tween the European and the United States registries) and isoften associated with Cryptosporidium species infection.A high proportion of patients (50% in the European series)die before the fourth decade of life, despite substitutiontreatment with Igs. This has prompted more aggressiveforms of treatment on the basis of hematopoietic stemcell transplantation. However, a retrospective analysis of38 European patients treated with hematopoietic stemcell transplantation has shown that only 22 (58%) ofthem were cured.14
Patients with CD40L deficiency have markedly de-creased serum IgG and IgA levels and normal to increasedIgM levels. Occasionally, IgA levels might be normal.
This is likely due to a CD40L-CD40–independent CSRmechanism, in which B cells are activated by B-lympho-cyte stimulator/B-cell activating factor (BLyS/BAFF) anda profileration-inducing ligand (APRIL) molecules (ex-pressed by dendritic cells activated by IFN-a, IFN-g, orLPS), along with signals provided by cytokines (TGF-b)and B-cell receptor engagement.15
Flow cytometry represents the most important screen-ing assay for diagnosis of CD40L deficiency. In mostpatients, in vitro activation of T lymphocytes with phorbolesters and ionomycin fails to induce expression of theCD40L molecule, as revealed by staining with CD40L-specific mAbs or a CD40-Ig chimeric construct. In anycase final confirmation of mutations requires molecularanalysis. Genomic abnormalities are scattered along theCD40L gene (Fig 3).16 In a minority of patients, mildermutations that allow binding of CD40 (albeit at reducedintensity) are associated with a less severe clinical course,which in a few cases was characterized uniquely by parvo-virus B19–related anemia.
B lymphocytes from the patients coexpress surface IgM(sIgM) and sIgD; in most cases the proportion of memory(CD271) B lymphocytes and the frequency of SHM arereduced. Importantly, defects of CSR and SHM reflect ab-normalities of the T-cell compartment, as indicated by thefact that the patients’ B cells can be induced to undergoCSR in vitro on activation with CD40 agonists and cyto-kines.17
In addition, lymph nodes from CD40L-deficient pa-tients show primary follicles but are devoid of germinalcenter formation, indicating the importance of CD40L-CD40 interaction for maturation of secondary lymphoidorgans.
FIG 2. Schematic representation of CD40L-CD40 interaction and its effects on CSR and SHM. Activated CD41
T cells in the lymph nodes interact with CD40-expressing B cells and secrete ILs that interact with cytokine-specific B-cell receptors (IL-R). CD40-mediated signaling activates the NF-kB signaling pathway and ultimatelyresults in the expression of AID and UNG. Mutations that affect CD40-mediated B-cell activation result in someof the forms of hyper-IgM syndrome. MHC-II, Major histocompatibility complex of class II; TCR, T-cell recep-tor; CD40L-ID, X-linked immunodeficiency caused by CD40L defect; CD40-ID, immunodeficiency caused byCD40 deficiency; XL-EDA-ID, X-linked immunodeficiency with ectodermal dystrophy; AID-ID, immunodefi-ciency caused by AID deficiency; UNG-ID, immunodeficiency caused by UNG deficiency; PC, plasma cell.
J ALLERGY CLIN IMMUNOL
APRIL 2006858 Notarangelo et al
Basic
and
clin
icalim
munolo
gy
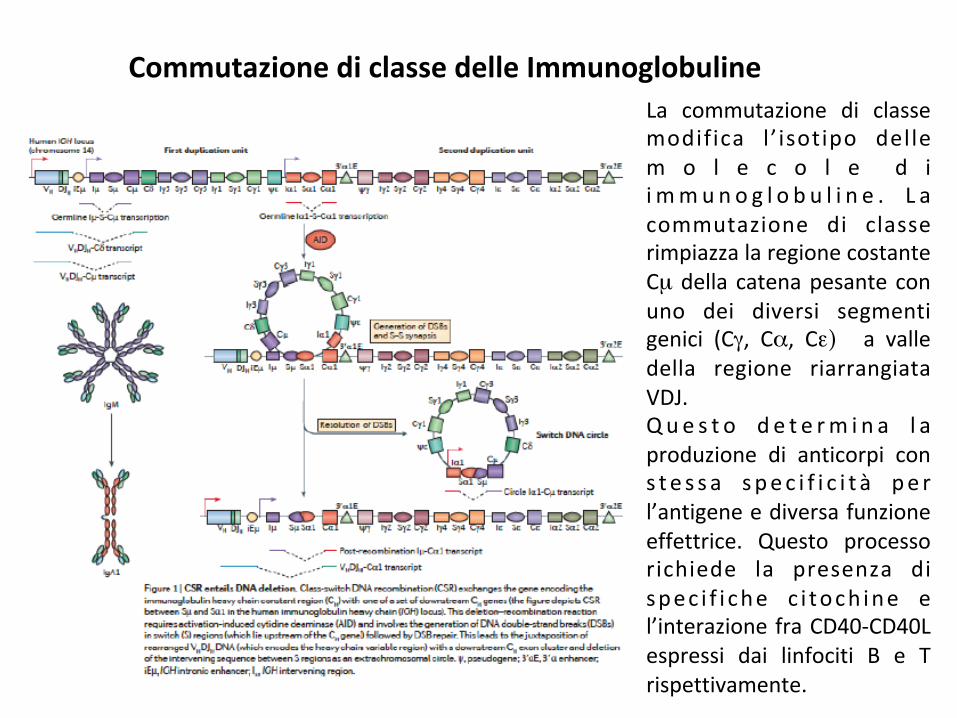
CommutazionediclassedelleImmunoglobulineLa commutazione di classemodifica l ’ isotipo dellem o l e c o l e d ii mmun o g l o b u l i n e . L acommutazione di classerimpiazzalaregionecostanteCµdella catenapesanteconuno dei diversi segmentigenici (Cγ, Cα, Cε) a valledella regione riarrangiataVDJ.Q u e s t o d e t e rm i n a l aproduzione di anticorpi cons t e s s a spec i f i c i t à pe rl’antigeneediversafunzioneeffettrice. Questo processorichiede la presenza dispec i f i che c i toch ine el’interazionefraCD40-CD40Lespressi dai linfociti B e Trispettivamente.

A l i v e l l o m o l e c o l a r e l acommutazione di classe è unareazione di ricombinazionedelezione fra le sequenze diswitch localizzate al 5’ di ognigene codificante la regionecostante della catena pesantedelleIg.Questo meccanismo procedeattraverso la generazione dirotture del doppio filamento diDNA nelle regioni di switchseguito dalla unione delleestremità con conseguentesostituzione della nuova regionecostante della immunoglobulinaavalledelsegmentogenicoVDJ.Questo processo richiede latrascrizione delle sequenze I-S-Cche avviene in presenza dis p e c i f i c h e c i t o c h i n e el ’espress ione del la c i t id ind e a m i n a s i i n d o t t adall’attivazione (AID: activationinduceddeaminase).
Meccanismomolecolaredellecommutazionediclasse

Il processo dellacommutazione diclasse è iniziatodalla trascrizionedelle sequenze I=i n t e r v e n i n g ,S = s w i t c h e i lcluster di esoni Cc h e g e n e r a itrascritti definitigerminali o sterili.Questo libera deifilamenti singoli diD N A c h erappresentano isubstratidiAID.
LacommutazionediclasserichiedelatrascrizionedelleregioniI-S-C

TrascrittigermlineLa trascrizione della regione S èessenziale per la commutazione diclassediunparticolareisotipo.In ciascuno dei geni CH è presenteun promotore inducibile dallecitochineunesoneI(intervening)laregioneSintronicaelaregioneCH.Nel corso della risposta immunel’ambiente citochinico attiva latrascrizione germline in specificigeniCHmediandolacommutazionediclassedispecificiisotipi.Es: le cellule spleniche di topo inpresenza di IL-4 e CD40 attivano latrascrizione germline di Cγ e Cεpromuovendo lo switch isotipicoversoleIgG1eleIgE.

IntroduzionedellerotturedeldoppiofilamentodelDNAdapartediAIDItrascrittigerminalisiappaianoalfilamentodiDNAcodificantelasciandol’altrofilamentodiDNAlibero.Activationinduceddeaminasi(AID)convertelacitosinainuracile.L’uracil-N-glicosilasicreasitiabasiciL’endonucleasiApegeneradeitaglinelDNA.I tagli del doppio filamento di DNAsono riparati dagli stessi sistemi diriparo attivati da agenti che causanorotture del doppio filamento di DNA(es: radiazioni ionizzanti) cheincudono il non homologous endjoining)
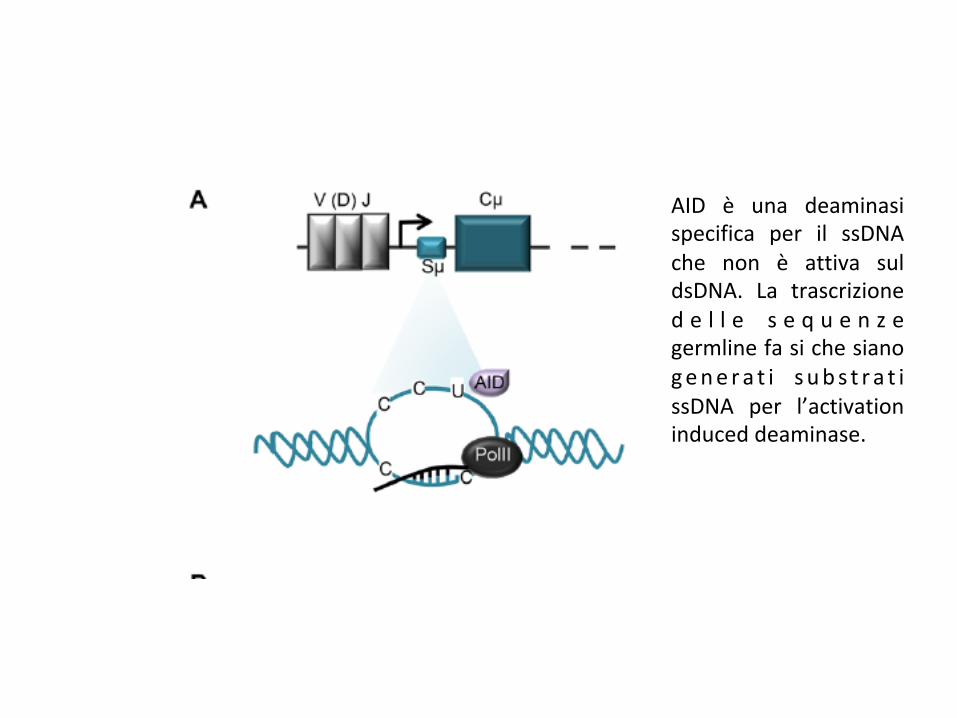
AID è una deaminasispecifica per il ssDNAche non è attiva suldsDNA. La trascrizioned e l l e s e q u e n z egermlinefasichesianogene ra t i s ubs t r a t issDNA per l’activationinduceddeaminase.

A l t e r a z i o n i n e l l asegnalazione del CD40 o lamancata interazione con ilCD40L sono alla base dellasindromedaIperIgM.Mutazioni o delezioni nelligandodelCD40causanoX-HIGM.Una forma autosomicarecessiva di IperIgM (AR-HIGM) con un fenotiposimileallaX-HIGMècausatadadifettinelCD40.IpazientiaffettidaX-HIGMeda difetti del CD40 nonp r e s e n t a n o c e n t r igerminativinellamilzaeneilinfonodi e sono soggetti ainfezioni opportunistiche(Pneumocys t i s Car in i i ,Cytomegalovirus)
SindromedaIperIgMcausatadadifettinellainterazioneCD40-CD40L
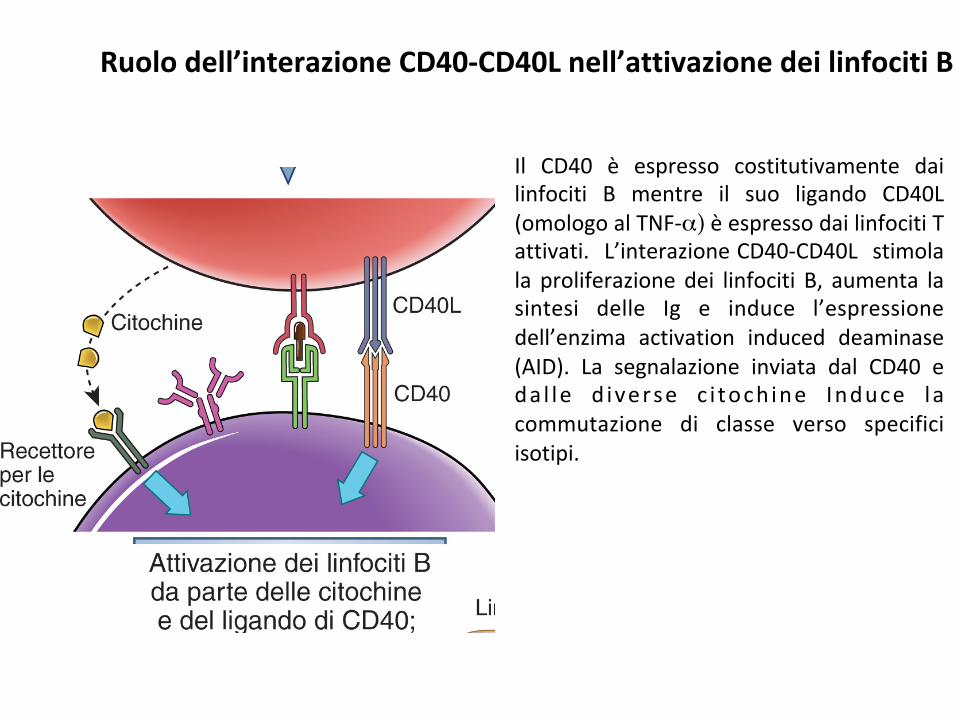
Il CD40 è espresso costitutivamente dailinfociti B mentre il suo ligando CD40L(omologoalTNF-α)èespressodailinfocitiTattivati. L’interazioneCD40-CD40L stimolala proliferazionedei linfociti B, aumenta lasintesi delle Ig e induce l’espressionedell’enzima activation induced deaminase(AID). La segnalazione inviata dal CD40 eda l le d iverse c i toch ine Induce lacommutazione di classe verso specificiisotipi.
Ruolodell’interazioneCD40-CD40Lnell’attivazionedeilinfocitiB
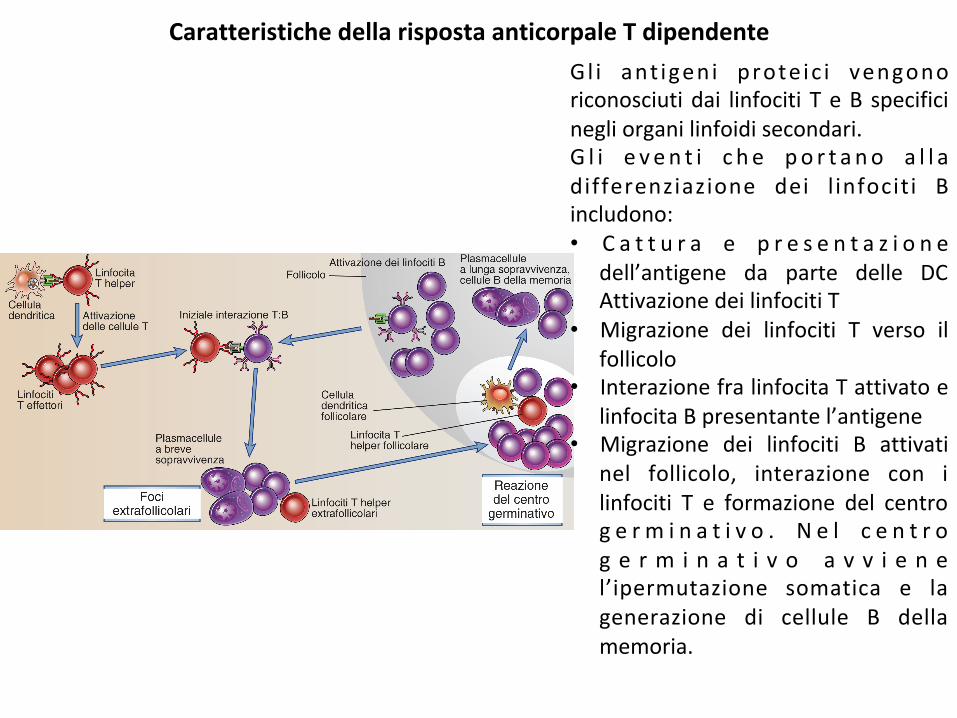
CaratteristichedellarispostaanticorpaleTdipendenteGl i ant igeni prote ic i vengonoriconosciutidai linfociti TeB specificinegliorganilinfoidisecondari.G l i e v en t i c h e po r t a no a l l adifferenziazione dei l infociti Bincludono:• C a t t u r a e p r e s e n t a z i o n e
dell’antigene da parte delle DCAttivazionedeilinfocitiT
• Migrazione dei linfociti T verso ilfollicolo
• InterazionefralinfocitaTattivatoelinfocitaBpresentantel’antigene
• Migrazione dei linfociti B attivatinel follicolo, interazione con ilinfociti T e formazione del centrog e r m i n a t i v o . N e l c e n t r og e r m i n a t i v o a v v i e n el’ipermutazione somatica e lagenerazione di cellule B dellamemoria.

RuolodelCD40nellaattivazionedeilinfocitiT
IpazientiaffettidadifettidelCD40 o CD40Lmostrano unaaumentata suscettibilità alleinfezioni opportunistiche( P neumoc y s t i c c a r i n i i ,C r i p t o s po r i d i um ) . T a l iinfezioni sono rare nellei m m u n o d e f i c i e n z estrettamente umorali mapresentiinquelleT.L’interazione CD40-CD40Lattiva le cellule presentantil ’ an t i gene aumentandol ’ e s p r e s s i o n e d i B 7 edell’IL-12.

RuolodelCD40nell’attivazionedeimacrofagi
L’ interazione delCD40 espresso daimacrofagi con i lC D 4 0 L a t t i v a im a c r o f a g ipotenziando la lorocapacitàbattericida.

SindromedaIperIgMcausatedadifettidienzimicoinvoltinellagenerazioneonelriparodellerotturedeldoppiofilamentodiDNAdurantelacommutazionediclasse
delleImmunoglobuline.
Approssimativamente10-15%dei pazienti con difetti nellacommutazione di classe delleIg presentano mutazioninell’enzimaAID.Intalipazientiladiagnosivieneeffettuataneiprimi 5 anni di vita. Questipazienti presentano linfonodigiganti e la sindrome puòe s s e r e s i a a u t o s om i c adominantecherecessiva.Mutazioni nell’enzima UNGsono state documentate in unristretto numero di pazientiaffettidaIperIgMclinicamentesimiliaipazienticonmutazioniin AID. Anche questi pazientimostrano centri germinativimoltosviluppati.

L’interazionefraCD40eCD40LinviasegnalidisopravvivenzaeproliferazioneailinfocitiB
DeficitdiCD40oCD40Ldeterminano l’assenzadeicentrigerminativi.N e i p a z i e n t i c o nmutazioni i AID e UNGl’assenza di switchisotipicosiaccompagnaalla presenza di centrigerminat iv i g igant idimostrando che lasegnazione inviata dalCD40 per indurre laformazione dei centrig e r m i n a t i v i èi n d i p e n d e n t edall’attivazionediAID.

TerapiadelleHIGM
TutteleformediiperIgMrichiedonolasomministrazionediImmunoglobulineperviaintravenosaosottocutaneacheriducelafrequenzaegravitàdelleinfezioni.IltrapiantodicellulestaminalipuòcurarelaX-HIGMelaformacausatadadifettidelCD40.Irisultatidi38trapiantieffettuatiinEuropahannodimostratolaguarigionedel58%deitrapiantatimentreil32%nonsonosopravvissutiacausadiinfezioniodiGVHD.PerquestosistacercandodivalutarealungotermineirischielbeneficideltrapiantodicellulestaminalirispettoallaterapiaconleIg

Immunodeficienzacomunevariabile
Lecommonvariableimmunodeficiency(CVID)includonoungruppoeterogeneodidisordini.Ipazientiaffettidatalemalattiasonoaffettidafrequentiinfezionidopoidieciannidivita.QuestipresentanovalorinormaliobassidiIgMebassilivellidiIgGeIgA.IlnumerodicelluleBnelsangueènormale,mainalcunicasipuòesseremoltobasso.LamaggiorpartedeipazientipresentanounaforteriduzionedellecelluleBdellamemoria.Taledisordinesiaccompagnaamanifestazioniautoimmuni.Lecausegeneticheditalesindromenonsonostatedefinite.LecausediquestodisordinepotrebberoesseredovuteaanomaliedeilinfocitiBoanomaliedeilinfocitiThelper.MutazioniinICOSeCD19sonostateosservateinunaminoranzadiindividuiaffettidaCVID.ICOSènecessarioperlagenerazionedeilinfocitiThelperfollicolari.

Immunodeficienzadell’immunitàinnata:DifettinelburstossidativosonoresponsabilidelChronicgranulomatousdisease
IlCGDèunamalattiageneticararadovuta prevalentemente a difettidei neutrofili. Questa malattia ècausata da mutazioni in uno deigeni che codificano le componentidell’ossidasi fagocitica. Può avereuna t rasmiss ione legato a lcromosoma X o autosomicarecessiva. I pazienti affetti daquestamalattianonsono ingradodi produrre specie reatt ivedell’ossigenoinrispostaainfezionibatteriche o fungine. La maggiorparte delle infezioni in questipazientisonocausatedaS.aureusBurkholderia cepacia complex,A s p e r g i l l u s . L e i n f e z i o n iinteressano la pelle, polmoni,l’intestino.
Iltrattamentoconantibioticieantifunginihaaumentato l’etàmedia di sopravvivenza diquesti pazienti che risulta essere del 50%nellaterzadecadedivita.

IneutrofiliattivatiproduconoROSattraversol’attivazionedellaNADPHossidasi
LamieloperossidasiutilizzaH202+Cl-pergenerareacidiipoalogenati(acidoipocloroso)

TestiperilmodulodiImmunopatologiadelCorsodiPatologiaMolecolareeImmunopatologiaImmunologiacellulareemolecolareA.K.Abbas,A,H.Lichtman,S.PilaiottavaedizioneElsevier
IpersensibilitàimmediataImmunologiadeitrapiantiImmunodeficienzecongeniteMalattiemediatedaanticorpiRisposteanticorpaliagliantigeniTdipendentiComplementoReview:IgEandmastcellsinallergicdisease.GalliSJ,TsaiM.Nat.Med.201218:693-704.Thedevelopmentofallergicinflammation.GalliSJ,TsaiM,PiliponskyAM.2008.454:445-54.Asthmaandallergicinflammation.LocksleyRM.Cell.2010140:777-83.AntibodymediatedorganallograftrejectionR.B.ColvinandR.N.Smith.NatRev.immunol2005;5:807-817LoupyA,LefaucheurC.Antibody-MediatedRejectionofSolid-OrganAllografts.NEnglJMed.2018Sep20;379(12):1150-1160.doi:10.1056/NEJMra1802677.Antibody-mediatedrejectionacrosssolidorgantransplants:manifestations,mechanisms,andtherapies.ValenzuelaNM,ReedEF.JClinInvest.2017;127:2492-2504Moleculardefectsinhumanseverecombinedimmunodeficiencyandapproachestoimmunereconstitution.BuckleyR.H.Annu.Rev.Immunol2004.22:625-655Tcellallorecognitionandtransplantrejectionasummaryandupdate.HeegerPS.Am.J.Transplant2003.3:525-33PrimaryBcellimmunodeficiency:comparisonandcontrasts.ConleyM.E.etal.Ann.Rev.immunol.2009.27:199-277.