
tesi triennale
46
UNIVERSIT ` A DEGLI STUDI DI MODENA E REGGIO EMILIA Dipartimento di Scienze Fisiche, Informatiche e Matematiche Corso di Laurea in FISICA Tesi di Laurea Modello tight-binding per il grafene. Relatore: Prof. Franca Manghi Laureando: Lorenzo Rossi Anno Accademico 2014-2015
-
Upload
mipiacemrgoldstein -
Category
Documents
-
view
22 -
download
0
description
modello tight binding del grafene, accenno alle proprietà topologiche
Transcript of tesi triennale

UNIVERSITA DEGLI STUDI DI MODENA E REGGIO EMILIA
Dipartimento di
Scienze Fisiche, Informatiche e MatematicheCorso di Laurea in FISICA
Tesi di Laurea
Modello tight-binding per il grafene.
Relatore:
Prof. Franca ManghiLaureando:
Lorenzo Rossi
Anno Accademico 2014-2015

Capitolo 1
Introduzione
Nelle seguenti pagine verranno trattate le proprieta elettroniche di cristalli esagonali bidimensio-nali. Il sistema cristallino in esame verra idealizzato descrivendolo come una matrice periodica diatomi perfettamente localizzati, trascurando quindi le vibrazioni reticolari ed eventuali imperfezio-ni. Infine l’intera trattazione si basera su una descrizione ad elettrone singolo andando a definirele bande energetiche del solido tramite un modello ad elettroni non interagenti in cui la mutuarepulsione verra tenuta in considerazione solamente tramite un campo medio.
Il cristallo reale a cui faremo riferimento sara il grafene, materiale costituito da un singolo fogliodi grafite avente notevoli proprieta elettroniche e meccaniche1, tuttavia non tutte le proprieta cheverranno introdotte all’interno del modello avranno un’effettiva corrispondenza nelle proprieta spe-rimentalmente misurate del grafene; esso rappresenta quindi piu che altro un punto di riferimentoda cui partire per studiare le proprieta elettroniche di reticoli bidimensionali esagonali.
Il calcolo delle bande elettroniche verra realizzato tramite il metodo tight-binding per cui svilup-peremo inizialmente tale metodo in forma generale nel capitolo 2; nel capitolo 3 i risultati generaliverranno applicati al caso di reticolo esagonale semplice, introducendo opportune ipotesi che ver-ranno mantenute anche nei capitoli successivi. Nel capitolo 4 viene inserita all’interno del modellouna dimerizzazione e successivamente l’interazione spin orbita; quest’ultima permette di introdurreil concetto di isolante topologico ed enunciare il rapporto tra l’invariante topologico Z2 e le bandeenergetiche di un sistema unidimensionale ottenuto dal precedente riducendone di una dimensionela periodicita. Infine nel capitolo 5 si ripropone l’analisi effettuata nel capitolo 4 per un sistemaleggermente diverso caratterizzato da una, cosiddetta, distorsione Kekule.
1Si veda ad esempio A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov e A. K. Geim, The electronicproperties of graphene, Rev. Mod. Phys., 81, 2009, pp.109-162.
1

2

Capitolo 2
Metodo tight-binding
Il metodo tight-binding sfrutta una combinazione lineare di autostati atomici per descrivere gliautostati dell’intero cristallo. Esso fornisce buoni risultati quando la sovrapposizione tra autosta-ti atomici di atomi vicini e ragionevolmente piccola, permettendo di considerare gli elettroni delcristallo localizzati in regioni prossime all’atomo di appartenenza; esso prende forma quindi da unpunto di partenza diametralmente opposto a quello su cui si basa il modello ad elettrone quasilibero.
Nello sviluppare il metodo prendiamo in considerazione un reticolo di Bravais con base. Suppo-nendo di aver definito i vettori di base del reticolo diretto indichiamo con ~R una loro combinazionelineare intera in modo che ogni cella del reticolo sia univocamente individuata da uno di tali vettori;siano poi ~τi, con i ∈ I insieme finito di indici, i vettori della base, ovvero i vettori posizione deidiversi siti appartenenti ad una stessa cella rispetto all’origine della cella stessa. In questo modoogni atomo del cristallo e univocamente identificabile tramite la coppia (~R, i).
Per ogni coppia (~R, i) e possibile scrivere:
H(~r, ~p) = H~R,i(~r, ~p) + ∆U~R,i(~r) (2.1)
dove:
• H(~r, ~p) e l’Hamiltoniana cristallina (a elettrone singolo)
• H~R,i(~r, ~p) e l’Hamiltoniana atomica (a elettrone singolo) del sito (~R, i)
• ∆U~R,i(~r) e la correzione che deve essere apportata al potenziale dell’Hamiltoniana atomica
H~R,i(~r, ~p) per ottenere l’Hamiltoniana cristallina H(~r, ~p) (tale termine include quindi l’intera-zione con gli altri nuclei e un’interazione media con gli elettroni appartenenti agli altri atomidel cristallo).
Data la simmetria traslazionale del reticolo la forma funzionale di H~R,i(~r, ~p) e ∆U~R,i(~r) deve risul-
tare indipendente da ~R mentre ci aspettiamo che dipende da i se il cristallo non e monoatomico.
Supponiamo poi di conoscere gli autostati delle Hamiltoniane atomiche; sia ad esempio |~R, i, α〉 unautostato di H~R,i associato all’autovalore E~R,i,α:
3

2 – Metodo tight-binding
H~R,i |~R, i, α〉 = E~R,i,α |~R, i, α〉 (2.2)
data la simmetria traslazionale del reticolo ci aspettiamo che anche l’insieme degli autovalori{E~Riα} sia indipendente da ~R mentre possa dipendere da i se il cristallo non e monoatomi-co; analogamente ci aspettiamo che valgano le stesse proprieta anche per la forma funzionaledi φ~R,i,α(~r) = 〈~r |~R, i, α〉, dove con |~r〉 si e indicato un autostato dell’operatore posizione.
Nell’ipotesi in cui ∆U~R,i(~r) sia grande solo nelle regioni di spazio in cui gli autostati di H~R,i(~r, ~p)
siano prossimi ad annullarsi possiamo supporre che gli stati |~R, i, α〉 forniscano gia una prima
approssimazione di autostati dell’Hamiltoniana cristallina localizzati sul sito i della cella ~R. Talistati tuttavia non soddisfano la forma prevista dal teorema di Bloch per cui anziche sviluppareil ragionamento con essi sfruttiamo delle loro combinazioni lineari aventi la forma opportuna,definiamo cosı:
|~k, i, α〉 =∑~R
ei~k·(~R+~τi) |~R, i, α〉 (2.3)
per ogni vettore ~k permesso dalle condizioni periodiche al contorno all’interno della prima zonadi Brillouin1 (essendo la cardinalita dell’insieme dei ~k considerati pari alla cardinalita dell’insieme
dei vettori ~R il numero di stati di Bloch cosı definiti e pari al numero di stati localizzati trovati inprecedenza2).
Per migliorare il grado di accuratezza possiamo ora, invece di utilizzare un singolo stato |~k, i, α〉per approssimare un autostato dell’Hamiltoniana cristallina avente la forma prevista dal teoremadi Bloch, sfruttare delle combinazioni lineari quali:
|n,~k〉 =∑~R
∑i
c(n)iα |~k, i, α〉 (2.4)
ricercando i coefficienti c(n)iα in modo tale soddisfare al meglio la relazione:
H |n,~k〉 = εn(~k) |n,~k〉 (2.5)
Per trovare l’espressione corretta per i coefficienti c(n)iα possiamo proiettare la precedente equazione
su |~0, i, α〉 e suddividendo opportunamente l’Hamiltoniana otteniamo:
〈~0, i, α| (H~0,i + ∆U~0,i) |n,~k〉 = εn(~k) 〈~0, i, α|n,~k〉
da cui, sfruttando la definizione di autostato atomico, si deriva:
E~0,i,α 〈~0, i, α|n,~k〉+ 〈~0, i, α|∆U~0,i) |n,~k〉 = εn(~k) 〈~0, i, α|n,~k〉
espandendo quindi lo stato |n,~k〉 si trova:
1Si noti che anche supponendo gli stati |~R, i, α〉 normalizzati e ortogonali per α differenti, gli stati |~k, i, α〉 nonrisultano in generale normalizzati, ma di cio se ne dovra tenere conto solo al termine della discussione, quandosi vorranno definire autostati normalizzati dell’Hamiltoniana cristallina; inoltre a tale punto saranno gia stateintrodotte opportune ipotesi semplificatrici che permetteranno di definire agevolmente le costanti di normalizzazione
2Si veda ad esempio Ashcroft, Mermin Solid state physics pag. 178
4

(εn(~k)− E~0,i,α) 〈~0, i, α|
∑j
∑β
c(n)iβ
∑~R
ei~k·(~R+~τj) |~R, j, β〉
−〈~0, i, α|∆U~0,i
∑j
∑β
c(n)iβ
∑~R
ei~k·(~R+~τj) |~R, j, β〉
= 0
e ponendo
• S(~0, i, α; ~R, j, β) = 〈~0, i, α|~R, j, β〉 integrale di sovrapposizione
• ∆U(~0, i, α; ~R, j, β) = 〈~0, i, α|∆U~0,i |~R, j, β〉 correzione energetica
si ottiene:
(εn(~k)− E~0,i,α)
∑j
∑β
c(n)iβ
∑~R
ei~k·(~R+~τj)S(~0, i, α; ~R, j, β)
−∑
j
∑β
c(n)iβ
∑~R
ei~k·(~R+~τj)∆U(~0, i, α; ~R, j, β)
= 0
da cui:
(εn(~k)− E~0,i,α)
∑j
∑β
c(n)iβ Z(~k; i, α; j, β)
−∑
j
∑β
c(n)iβ W (~k; i, α; j, β)
= 0
avendo posto
• Z(~k; i, α; j, β) =∑
~R ei~k·(~R+~τj)S(~0, i, α; ~R, j, β)
• W (~k; i, α; j, β) =∑
~R ei~k·(~R+~τj)∆U(~0, i, α; ~R, j, β)
Ci si riconduce cosı all’equazione lineare omogenea:∑j
∑β
c(n)iβ
{(εn(~k)− E~0,i,α)Z(~k; i, α; j, β)−W (~k; i, α; j, β)
}= 0 (2.6)
e ripetendo il ragionamento, proiettando su tutti i possibili |~0, i, α〉 al variare di i e α, otteniamo
un sistema di |{i}| · |{α}| equazioni lineari omogenee nelle |{j}| · |{β}| incognite c(n)jβ . Richiedendo
la solubilita di tale sistema imponendo la nullita del determinante della matrice dei coefficienti, si
ottengono quindi |{j}| · |{β}| autovalori εn(~k) e, a partire dai coefficienti c(n)jβ trovati, i corrispettivi
autostati |n,~k〉.
5

6

Capitolo 3
Reticolo esagonale semplice
Applichiamo ora il modello tight-binding ad un reticolo bidimensionale esagonale quale quello delgrafene. Per poter proseguire e necessaria la conoscenza esplicita delle proprieta cristallografichedel reticolo, definiamo quindi nel piano una base ortonormale {~e1, ~e2} ed un’origine O del sistemadi riferimento. Poiche un reticolo esagonale non puo essere descritto con un semplice reticolo diBravais ma e necessaria l’introduzione di una base biatomica1 definiamo, oltre ai vettori del reticolodiretto, anche due opportuni vettori per individuare la posizione dei due siti atomici all’internodella stessa cella elementare (ricordiamo tuttavia che questi due siti sono della stessa natura es-sendo entrambi atomi di carbonio).
O
~a1
~a2
1
2
Figura 3.1: Reticolo diretto; in rosso la cella di Wigner Seitz
Definiamo quindi i vettori di base del reticolo diretto come:
~a1 = a(
12
√32
){~e1,~e2}
~a2 = a(
12 −
√32
){~e1,~e2}
(3.1)
dove a = |~a1| = |~a2| e la distanza a secondi vicini, che nel caso del grafene risulta essere a ≈ 2,46A,e i vettori posizione dei siti atomici come:
~τ1 =a
2
(0 1√
3
){~e1,~e2}
~τ2 =a
2
(0 − 1√
3
){~e1,~e2}
(3.2)
La distanza a primi vicini risulta quindi d = |~τ1 − ~τ2| =a√3≈ 1,42A.
1Si veda ad esempio Ashcroft, Mermin Solid state physics pag. 75
7

3 – Reticolo esagonale semplice
Γ
~b1
~b2
M ′K
MK ′
Figura 3.2: Reticolo reciproco; in blu la prima zona di Brillouin
Definiti i vettori di base del reticolo diretto possiamo ricavare i vettori di base del reticolo reciproco{~b1,~b2} dalle relazioni: {
~b1 · ~a1 = 2π~b1 · ~a2 = 0
{~b2 · ~a1 = 0~b2 · ~a2 = 2π
(3.3)
e si trova:
~b1 =2π
a
(1 1√
3
){~e1,~e2}
~b2 =2π
a
(1 − 1√
3
){~e1,~e2}
(3.4)
Possiamo infine definire i punti di alta simmetria del reticolo reciproco come:
• Γ =(0 0
){~e1,~e2}
• K ′ = 2πa
(23 0
){~e1,~e2}
• M = 2πa
(12
12√3
){~e1,~e2}
• K = 2πa
(13
1√3
){~e1,~e2}
• M ′ = 2πa
(0 1√
3
){~e1,~e2}
Possiamo ora ripartire da (2.6) introducendo ipotesi opportune in modo da rendere tale equazione,e le altre costituenti il sistema, piu maneggevoli.
Dobbiamo innanzi tutto specificare quali orbitali atomici sia sensato utilizzare per approssimaregli autostati dell’Hamiltoniana cristallina, e infatti conveniente considerare solo quegli autostatiassociati ad energie prossime all’energia di Fermi del cristallo poiche sono essi a fornire il principalecontributo. Nel caso specifico del grafene e sufficiente prendere in considerazione un solo orbitaleper sito, la configurazione elettronica del carbonio in tale reticolo prevede infatti due elettronifortemente localizzati nell’orbitale 1s, tre elettroni impegnati nel realizzare forti legami σ in or-bitali ibridi sp2 disposti nel piano xy a 120◦ l’uno dall’altro e infine un elettrone maggiormentedelocalizzato disposto in un orbitale pz e realizzante legami π con gli atomi vicini. Solo quest’ul-timo orbitale fornira quindi un contributo significativo nella definizione degli autostati cristalliniassociati ad energie prossime all’energia di Fermi.
In quest’ipotesi la (2.6) si riduce, tenendo anche conto del numero di siti per cella, a:
8

2∑j=1
c(n)i
{(εn(~k)− E~0,i)Z(~k; i; j)−W (~k; i, ; j)
}= 0 (3.5)
dove:
• Z(~k; i; j) =∑
~R ei~k·(~R+~τj)S(~0, i; ~R, j)
• W (~k; i; j) =∑
~R ei~k·(~R+~τj)∆U(~0, i; ~R, j)
• E~0,i e l’energia dell’unico orbitale coinvolto del sito i; dato pero che nel sistema in esame idue atomi della base sono identici possiamo porre E0 = E~0,1 = E~0,2
Introduciamo quindi ipotesi semplificatrici anche sugli integrali di sovrapposizione S(~0, i; ~R, j) =
〈~0, i, α; ~R, j, α〉 e sulle correzioni energetiche ∆U(~0, i; ~R, j) = 〈~0, i, α|∆U~0,i |~R, j, α〉 supponendoche tutti gli integrali di sovrapposizione tra autostati atomici localizzati su siti differenti sianotrascurabili:
S(~0, i; ~R, j) = δ~0~Rδij
e che le uniche correzioni energetiche significative siano solo a primi vicini:
∆U(~0, i; ~R, j) /= 0 ⇐⇒ (~0, i) (~R, j) coincidenti o primi vicini
In queste ipotesi2 otteniamo:
Z(~k; i; j) =∑~R
ei~k·(~R+~τj)δ~0~Rδij = ei
~k·~τjδij
e:
W (~k; 1; 1) =∑~R
ei~k·(~R+~τ1)∆U(~0,1; ~R,1)
= ei~k·~τ1∆U(~0,1;~0,1)
W (~k; 2; 2) =∑~R
ei~k·(~R+~τ2)∆U(~0,2; ~R,2)
= ei~k·~τ2∆U(~0,2;~0,2)
W (~k; 1; 2) =∑~R
ei~k·(~R+~τ2)∆U(~0,1; ~R,2)
= ei~k·~τ2 [∆U(~0,1;~0,2) + ei
~k·~a1∆U(~0,1;~a1,2) + e−i~k·~a2∆U(~0,1;−~a2,2)]
W (~k; 2; 1) =∑~R
ei~k·(~R+~τ1)∆U(~0,2; ~R,1)
= ei~k·~τ1 [∆U(~0,2;~0,1) + e−i
~k·~a1∆U(~0,2;−~a1,1) + ei~k·~a2∆U(~0,2;~a2,1)]
2Nell’ipotesi fatta relativamente agli integrali di sovrapposizione risulta diretto verificare che la costante dinormalizzazione da applicare agli stati (2.3), supponendo gli autostati atomici normalizzati, e pari a 1√
|{~R}|
9

3 – Reticolo esagonale semplice
Data l’identica natura dei siti atomici coinvolti dobbiamo poi supporre che:
∆U(~0,1;~0,1) = ∆U(~0,2;~0,2)
e che:
∆U(~0,1;~0,2) = ∆U(~0,1;~a1,2) = ∆U(~0,1;−~a2,2) =
= ∆U(~0,2;~0,1) = ∆U(~0,2;−~a1,1) = ∆U(~0,2;~a2,1)
pertanto per semplicita indichiamo i primi con t0 ed i secondi con t e riscriviamo i coefficientiW (~k; i; j) come3:
W (~k; 1; 1) = t0ei~k·~τ1
W (~k; 2; 2) = t0ei~k·~τ2
W (~k; 1; 2) = tei~k·~τ2ϕ(~k)
W (~k; 2; 1) = tei~k·~τ1ϕ∗(~k)
dove ϕ(~k) = [1 + ei~k·~a1 + e−i
~k·~a2 ]. In questo modo il sistema di equazioni lineari che dobbiamorisolvere si riduce a:(
(εn(~k)− E0 − t0)ei~k·~τ1 −tei~k·~τ2ϕ(~k)
−tei~k·~τ1ϕ∗(~k) (εn(~k)− E0 − t0)ei~k·~τ2
)(c(n)1
c(n)2
)=
(00
)(3.6)
Moltiplicando quindi la prima equazione per e−i~k·~τ1 e la seconda per e−i
~k·~τ2 ci possiamo poiricondurre all’usuale forma di un problema agli autovalori:(
0 tei~k·(~τ2−~τ1)ϕ(~k)
te−i~k·(~τ2−~τ1)ϕ∗(~k) 0
)(c(n)1
c(n)2
)= (εn(~k)− E0 − t0)
(c(n)1
c(n)2
)(3.7)
che prevede come soluzioni:
ε±(~k)− E0 − t0 = ±|t|√|ϕ(~k)|2 (3.8)
dove:
|ϕ(~k)|2 = 3 + 2 cos(~k · ~a1) + 2 cos(~k · ~a2) + 2 cos(~k · (~a1 + ~a2))
Ridefinendo lo zero dell’energia come E0 + t0 possiamo affermare che le due bande trovate sonosimmetriche rispetto ad esso e si annullano se e solo se |ϕ(~k)|2 = 0.
Gli unici punti in cui cio accade sono4
3Senza perdita di generalita possiamo supporre t, t0 ∈ R, cosı come i termini ad essi analoghi che introdurremoin seguito; il potenziale e infatti scrivibile tramite una funzione reale del vettore posizione e gli stati possono esserescelti in modo da soddisfare alla stessa condizione; in generale inoltre ci aspettiamo che tali coefficienti siano negativi
4Il risultato e noto tuttavia una dimostrazione di questo risultato e presente anche nel capitolo successivo
10

K ′ =2π
a
(43 0
){~e1,~e2}
K =2π
a
(13
1√3
){~e1,~e2}
e tutti i punti da essi raggiungibili tramite combinazioni lineari intere di vettori di base del reticoloreciproco. In un qualunque intorno di tali punti si trova poi che:
|ϕ(~k)|2 =3a2
4(δx
2 + δy2) + o(δx
2, δy2) ≈ 3a2
4|~δ|2
dove ~δ e il vettore distanza (nel reticolo reciproco) da essi, e quindi la relazione di dispersione puoessere ivi approssimata tramite un andamento lineare nel modulo della distanza. Nei punti K eK ′ si vengono cosı a delineare i cosiddetti coni di Dirac, che rappresentano la peculiarita dellebande energetiche del grafene (cfr. Figura 3.3); una legge di dispersione lineare permette infattidi trattare gli elettroni di conduzione del grafene analogamente a particelle relativistiche prive dimassa, prevedendo quindi un’elevata conducibilita per questo materiale.
Figura 3.3: Bande energetiche del grafene; ~k ∈ [− 2πa, 2πa
] × [− 85πa, 85πa
], a = 1,|t| = 1
Ognuna delle due bande energetiche trovata secondo questo modello e infatti degenere in spinper cui, dovendo allocare due elettroni per cella, arriviamo a concludere che, nello stato di minimaenergia, la banda inferiore risulta completamente occupata mentre la banda superiore risulta vuota.Essendoci pero un gap nullo tra le due bande la conduzione e possibile e si e pertanto soliticlassificare il grafene come un semi-metallo.
11

12

Capitolo 4
Reticolo esagonale condimerizzazione
Partendo da un sistema analogo al precedente introduciamo una leggera variazione ipotizzandola presenza di una piccola anisotropia nel sistema che non altera apprezzabilmente la strutturageometrica ma influisce sui coefficienti di interazione1. Consideriamo quindi nuovamente un reticoloesagonale descritto dagli stessi vettori di base del reticolo diretto introdotti in (3.1):
O
~a1
~a2
1
2
Figura 4.1: Reticolo diretto; in rosso la cella di Wigner Seitz
~a1 = a(
12
√32
){~e1,~e2}
~a2 = a(
12 −
√32
){~e1,~e2}
(4.1)
e dagli stessi vettori posizione dei siti atomici definiti in (3.2):
~τ1 =a
2
(0 1√
3
){~e1,~e2}
~τ2 =a
2
(0 − 1√
3
){~e1,~e2}
(4.2)
e di conseguenza otteniamo anche gli stesi vettori di base del reticolo reciproco trovati in (3.4):
~b1 =2π
a
(1 1√
3
){~e1,~e2}
~b2 =2π
a
(1 − 1√
3
){~e1,~e2}
(4.3)
1Cosı come proposto da T. C. Lang, A. M. Essein, V. Gurarie and S. Wessel, Z2 topological invariants in twodimaensions from quantum Monte Carlo Phys. Rev. B 87, 205101 (2013)
13

4 – Reticolo esagonale con dimerizzazione
Γ
~b1
~b2
M ′K
MK ′
Figura 4.2: Reticolo reciproco; in blu la prima zona di Brillouin
Supponiamo pero ora che l’interazione tra i due siti all’interno di una stessa cella sia leggermentediversa dall’interazione che essi hanno con gli atomi di celle differenti, ad esempio in seguito ad unaleggera trazione o compressione effettuata nella direzione individuata dal vettore ~e2; nelle stesseipotesi introdotte in precedenza notiamo che questo corrisponde alla definizione di due differentitipologie di interazione a primi vicini, come in precedenza possiamo infatti porre:
t0 = ∆U(~0,1;~0,1) = ∆U(~0,2;~0,2)
mentre differenziamo le correzioni energetiche tra siti vicini nel seguente modo:
t′ = ∆U(~0,1;~0,2) = ∆U(~0,2;~0,1)
t = ∆U(~0,1;~a1,2) = ∆U(~0,1;−~a2,2) = ∆U(~0,2;~a2,1) = ∆U(~0,2;−~a1,1)
Ripercorrendo gli stessi passi compiuti in precedenza giungiamo quindi al problema agli autovalori:
(0 ei
~k·(~τ2−~τ1)f(t, t′,~k)
e−i~k·(~τ2−~τ1)f∗(t, t′,~k) 0
)(c(n)1
c(n)2
)= (εn(t, t′,~k)− E0 − t0)
(c(n)1
c(n)2
)(4.4)
dove pero:
f(t, t′,~k) = t′ + t(ei~k·~a1 + e−i
~k·~a2) (4.5)
Gli autovalori risultano dunque:
ε±(t, t′,~k)− E0 − t0 = ±√|f(t, t′,~k)|2
dove:
|f(t, t′,~k)|2 = |t|2(
2 + q2 + 2q cos(~k · ~a1) + 2q cos(~k · ~a2) + 2 cos(~k · (~a1 + ~a2))
(4.6)
avendo posto q = |t′||t| .
Sfruttando i valori espliciti di ~a1 e ~a2 possiamo riscrivere la (4.6) come:
|f(t, t′,~k)|2
|t|2= q2 + 4q cos
(kxa
2
)cos
(ky√
3a
2
)+ 4 cos2
(kxa
2
)(4.7)
14

e sommando e sottraendo 4q cos(kx2
)possiamo arrivare a scrivere:
|f(t, t′,~k)|2
|t|2=
(q − 2 cos
(kxa
2
))2
+ 4q cos
(kxa
2
)(1 + cos
(ky√
3a
2
))(4.8)
Tale forma e utile per ricercare i valori di ~k in cui |f(t, t′,~k)|2 si annulla: essa e infatti scritta comela somma di due termini:
•(q − 2 cos
(kxa2
))2che e sempre non negativo
• 4q cos(kxa2
) (1 + cos
(ky√3a
2
))che e invece scritto tramite il prodotto di due fattori di cui il
secondo e sempre non negativo mentre il primo ha segno dipendente da kx.
Limitandoci pero a considerare −πa ≤ kx ≤πa , scelta peraltro non restrittiva, anche il primo fattore
del secondo termine risulta non negativo e quindi, affinche |f(t, t′,~k)|2 si annulli, entrambi i terminisi devono annullare.
Affinche il secondo termine si annulli non e possibile che il primo dei due fattori sia nullo altrimentiil primo termine risulterebbe diverso da zero, pertanto si deve avere:
1 + cos
(ky√
3a
2
)= 0 ⇐⇒ ky =
2π√3a
(e repliche periodiche)
mentre il primo termine si annulla se:
q − 2 cos
(kxa
2
)= 0 ⇐⇒ cos
(kxa
2
)=q
2
Notiamo cosı che:
• per q > 2 non si ha soluzione
• per q = 2 si ha come soluzione kx = 0
• per 1 < q < 2 si ha come soluzione 0 < kx <2π3a (e opposto)
• per q = 1 si ha come soluzione kx = 2π3a (e opposto)
• per 0 < q < 1 si ha come soluzione 2π3a < kx <
πa (e opposto)
• per q = 0 si ha come soluzione kx = πa (e opposto)
Possiamo dunque concludere che la modulazione dell’interazione all’interno della cella comportauna deformazione delle bande, ed in particolar modo dei punti di contatto tra esse, fino a giungere,per q > 2, all’apertura di un gap (cfr. Figura(4.3), Figura(4.4) e Figura(4.5)).
Per q = 1, ovvero nel caso di reticolo non dimerizzato, ritroviamo il risultato riportato nella sezioneprecedente: gli unici punti in cui le bande assumono il valore dell’energia di Fermi, arrivandosi a
toccare, sono i punti K = 2πa
(13
1√3
){~e1,~e2}
e K ′ = 2πa
(− 1
31√3
){~e1,~e2}
(e tutti i punti raggiun-
gibili da essi tramite combinazioni lineari intere di vettori di base del reticolo reciproco e quindiad essi equivalenti).
15
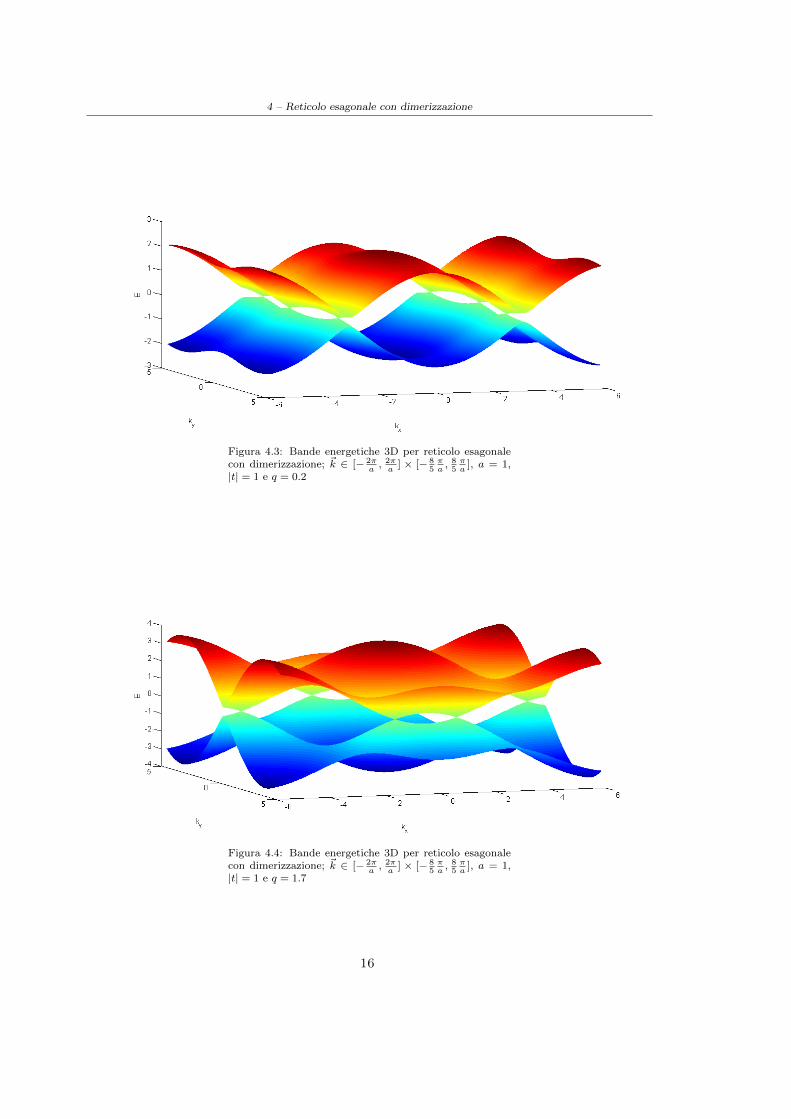
4 – Reticolo esagonale con dimerizzazione
Figura 4.3: Bande energetiche 3D per reticolo esagonalecon dimerizzazione; ~k ∈ [− 2π
a, 2πa
] × [− 85πa, 85πa
], a = 1,|t| = 1 e q = 0.2
Figura 4.4: Bande energetiche 3D per reticolo esagonalecon dimerizzazione; ~k ∈ [− 2π
a, 2πa
] × [− 85πa, 85πa
], a = 1,|t| = 1 e q = 1.7
16
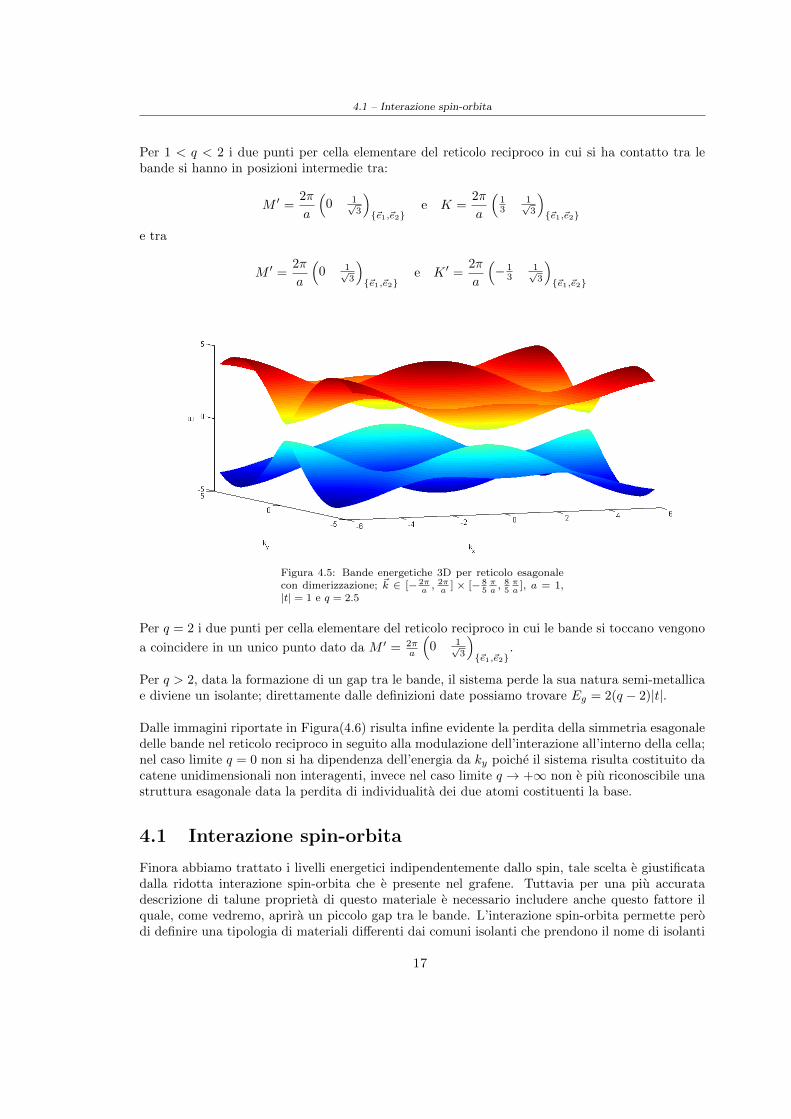
4.1 – Interazione spin-orbita
Per 1 < q < 2 i due punti per cella elementare del reticolo reciproco in cui si ha contatto tra lebande si hanno in posizioni intermedie tra:
M ′ =2π
a
(0 1√
3
){~e1,~e2}
e K =2π
a
(13
1√3
){~e1,~e2}
e tra
M ′ =2π
a
(0 1√
3
){~e1,~e2}
e K ′ =2π
a
(− 1
31√3
){~e1,~e2}
Figura 4.5: Bande energetiche 3D per reticolo esagonalecon dimerizzazione; ~k ∈ [− 2π
a, 2πa
] × [− 85πa, 85πa
], a = 1,|t| = 1 e q = 2.5
Per q = 2 i due punti per cella elementare del reticolo reciproco in cui le bande si toccano vengono
a coincidere in un unico punto dato da M ′ = 2πa
(0 1√
3
){~e1,~e2}
.
Per q > 2, data la formazione di un gap tra le bande, il sistema perde la sua natura semi-metallicae diviene un isolante; direttamente dalle definizioni date possiamo trovare Eg = 2(q − 2)|t|.
Dalle immagini riportate in Figura(4.6) risulta infine evidente la perdita della simmetria esagonaledelle bande nel reticolo reciproco in seguito alla modulazione dell’interazione all’interno della cella;nel caso limite q = 0 non si ha dipendenza dell’energia da ky poiche il sistema risulta costituito dacatene unidimensionali non interagenti, invece nel caso limite q → +∞ non e piu riconoscibile unastruttura esagonale data la perdita di individualita dei due atomi costituenti la base.
4.1 Interazione spin-orbita
Finora abbiamo trattato i livelli energetici indipendentemente dallo spin, tale scelta e giustificatadalla ridotta interazione spin-orbita che e presente nel grafene. Tuttavia per una piu accuratadescrizione di talune proprieta di questo materiale e necessario includere anche questo fattore ilquale, come vedremo, aprira un piccolo gap tra le bande. L’interazione spin-orbita permette perodi definire una tipologia di materiali differenti dai comuni isolanti che prendono il nome di isolanti
17
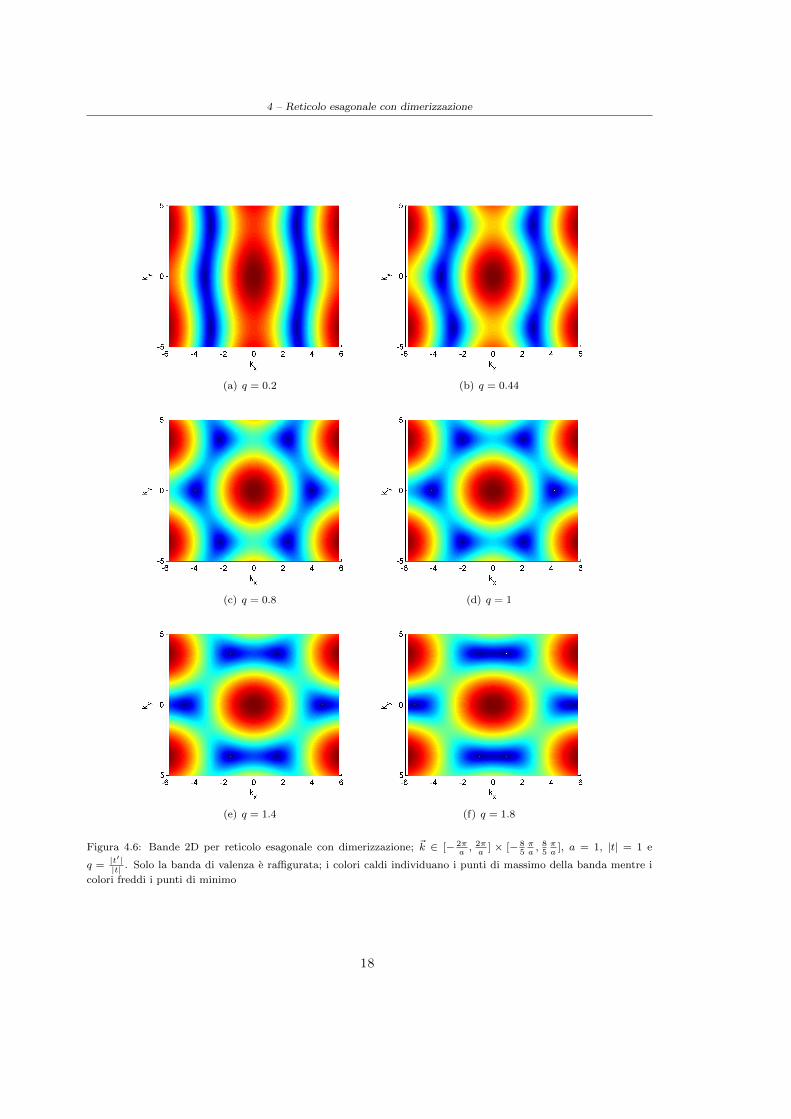
4 – Reticolo esagonale con dimerizzazione
(a) q = 0.2 (b) q = 0.44
(c) q = 0.8 (d) q = 1
(e) q = 1.4 (f) q = 1.8
Figura 4.6: Bande 2D per reticolo esagonale con dimerizzazione; ~k ∈ [− 2πa, 2πa
] × [− 85πa, 85πa
], a = 1, |t| = 1 e
q =|t′||t| . Solo la banda di valenza e raffigurata; i colori caldi individuano i punti di massimo della banda mentre i
colori freddi i punti di minimo
18

4.1 – Interazione spin-orbita
topologici.
Dobbiamo innanzi tutto definire un’opportuna forma per l’interazione spin-orbita ed un modellomolto utilizzato, introdotto da C. L. Kane and E. J. Mele2, schematizza lo spin-orbita tramiteun’interazione a secondi vicini che, nella rappresentazione dell’Hamiltoniana tramite stati di Blochdefiniti in (2.3), comporta, supponendo il sito individuato dal vettore posizione ~rl secondo vicinocon il sito individuato da ~rj e avente il comune primo vicino in ~rm, la comparsa di un fattoreaggiuntivo:
itkm(−1)12−σ
(~rm − ~rl)× (~rj − ~rm)
|(~rm − ~rl)× (~rj − ~rm)|ei~k(~rj−~rl) (4.9)
nell’elemento di matrice H(l, j), dove i e l’unita immaginaria, tkm e un parametro reale carat-teristico dell’intensita dell’interazione e σ = ± 1
2 e la componente z dello spin. Notiamo quindiche, dovendo rappresentare l’interazione tra il momento angolare di spin ed il momento angolareorbitale, l’interazione e direzionale, essendo il suo segno dipendente dal senso, orario o antiora-rio, tramite cui viene raggiunto un secondo vicino. Il segno dipende inoltre dalla componente zdello spin per cui gli stati di base dovrebbero essere scritti tramite spinori a due componenti ela matrice hamiltoniana risulterebbe conseguentemente di rango 4; tuttavia operando in tal modosi trovano solamente due bande distinte, entrambe doppiamente degeneri. E quindi sufficientestudiare studiare il problema per una data componente dello spin, avendo la consapevolezza cheper la componente opposta si otterrebbe un risultato analogo in cui le bande sono semplicementescambiate tra loro. Scegliamo quindi di prendere (−1)
12−σ = 1 e continuiamo a lavorare con stati
di base ad una sola componente.
Con l’introduzione di questi termini il problema agli autovalori definito in (4.4) diviene:
(g(tkm,~k) ei
~k·(~τ2−~τ1)f(t, t′,~k)
e−i~k·(~τ2−~τ1)f∗(t, t′,~k) −g(tkm,~k)
)(c(n)1
c(n)2
)= (εn(t, t′, tkm,~k)−E0 − t0)
(c(n)1
c(n)2
)(4.10)
Infatti i secondi vicini dei siti della tipologia j sono solo siti della tipologia j e quindi i terminifuori dalla diagonale rimangono inalterati mentre i termini sulla diagonale, nulli in assenza dispin-orbita, divengono:
H(1,1) = itkm
[+ei
~k·~a1 + ei~k·~a2 + e−i
~k·(~a1+~a2) − e−i~k·~a1 − e−i~k·~a2 − ei~k·(~a1+~a2)]
H(2,2) = itkm
[−ei~k·~a1 − ei~k·~a2 − e−i~k·(~a1+~a2) + e−i
~k·~a1 + e−i~k·~a2 + ei
~k·(~a1+~a2)]
ovvero:
H(1,1) = g(tkm,~k) = 2tkm
[sin(~k · (~a1 + ~a2)
)− sin(~k · ~a1)− sin(~k · ~a2)
]H(2,2) = −g(tkm,~k) = −2tkm
[sin(~k · (~a1 + ~a2)
)− sin(~k · ~a1)− sin(~k · ~a2)
]
2C. L. Kane and E. J. Mele, Quantum Spin Hall Effect in Graphene, Phys. Rev. Lett., 95, 226801 (2005)
19
4 – Reticolo esagonale con dimerizzazione
Conseguentemente, risolvendo il problema agli autovalori, si trova:
ε±(t, t′, tkm,~k)− E0 − t0 = ±√g2(tkm,~k) + |f(t, t′,~k)|2
Conoscendo il comportamento di |f(t, t′,~k)|2, e sufficiente studiare l’andamento di g(tkm,~k) pervalutare l’effetto dell’interazione spin-orbita sulle bande energetiche del sistema e conseguentementesulla sua natura semi-metallica o isolante. Per studiare il comportamento di g(tkm,~k) inseriamo ivalori espliciti di ~a1 e ~a2 ottenendo:
g(tkm,~k)
tkm= 4 sin
(kxa
2
)[cos
(kxa
2
)− cos
(ky√
3a
2
)]
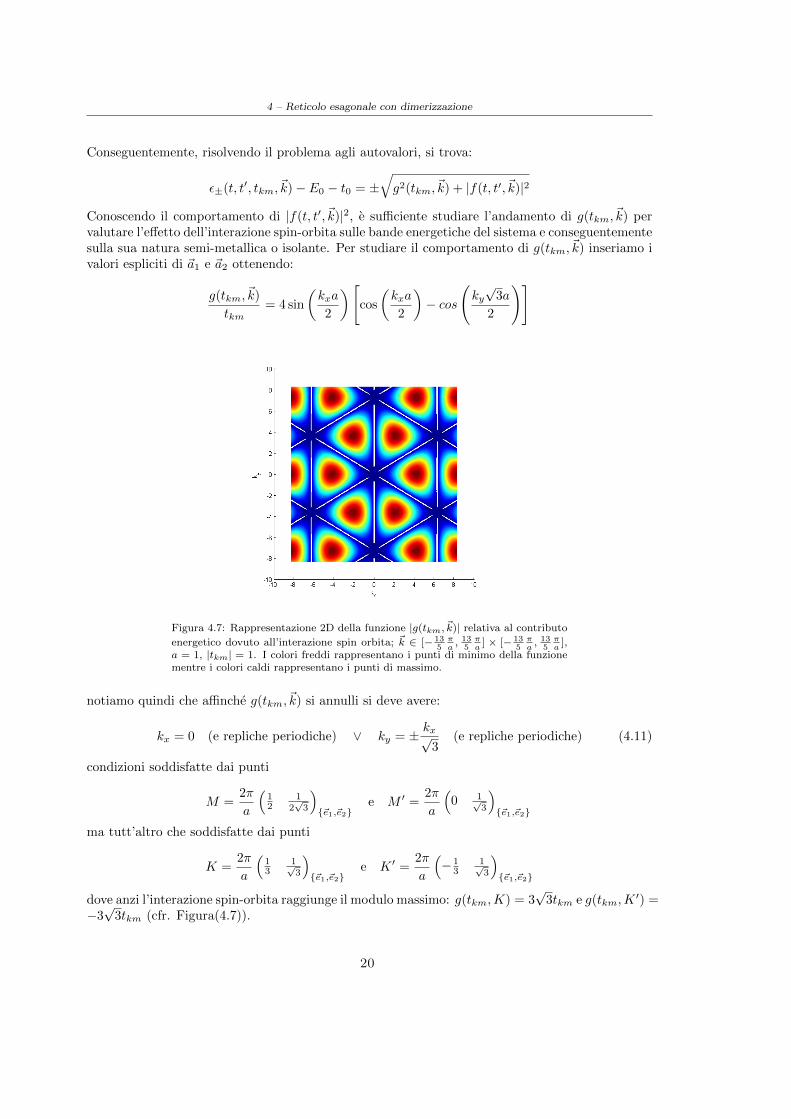
Figura 4.7: Rappresentazione 2D della funzione |g(tkm, ~k)| relativa al contributo
energetico dovuto all’interazione spin orbita; ~k ∈ [− 135πa, 13
5πa
] × [− 135πa, 13
5πa
],a = 1, |tkm| = 1. I colori freddi rappresentano i punti di minimo della funzionementre i colori caldi rappresentano i punti di massimo.
notiamo quindi che affinche g(tkm,~k) si annulli si deve avere:
kx = 0 (e repliche periodiche) ∨ ky = ± kx√3
(e repliche periodiche) (4.11)
condizioni soddisfatte dai punti
M =2π
a
(12
12√3
){~e1,~e2}
e M ′ =2π
a
(0 1√
3
){~e1,~e2}
ma tutt’altro che soddisfatte dai punti
K =2π
a
(13
1√3
){~e1,~e2}
e K ′ =2π
a
(− 1
31√3
){~e1,~e2}
dove anzi l’interazione spin-orbita raggiunge il modulo massimo: g(tkm,K) = 3√
3tkm e g(tkm,K′) =
−3√
3tkm (cfr. Figura(4.7)).
20
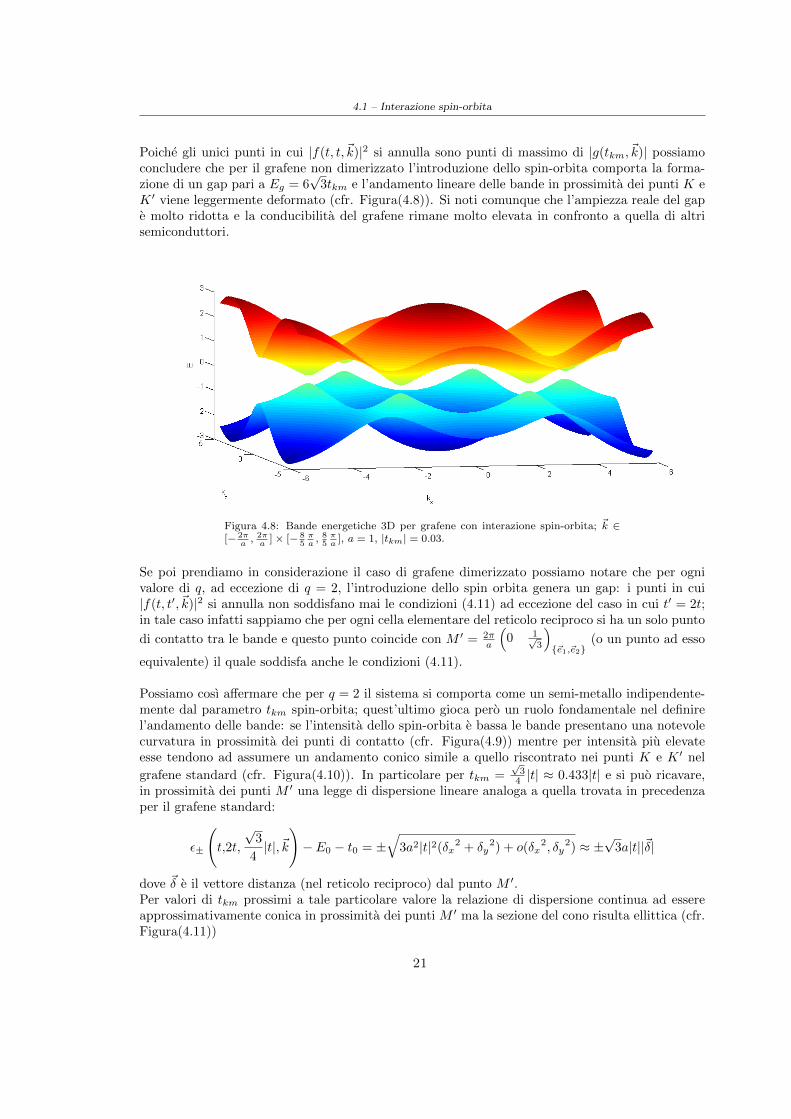
4.1 – Interazione spin-orbita
Poiche gli unici punti in cui |f(t, t,~k)|2 si annulla sono punti di massimo di |g(tkm,~k)| possiamoconcludere che per il grafene non dimerizzato l’introduzione dello spin-orbita comporta la forma-zione di un gap pari a Eg = 6
√3tkm e l’andamento lineare delle bande in prossimita dei punti K e
K ′ viene leggermente deformato (cfr. Figura(4.8)). Si noti comunque che l’ampiezza reale del gape molto ridotta e la conducibilita del grafene rimane molto elevata in confronto a quella di altrisemiconduttori.
Figura 4.8: Bande energetiche 3D per grafene con interazione spin-orbita; ~k ∈[− 2π
a, 2πa
]× [− 85πa, 85πa
], a = 1, |tkm| = 0.03.
Se poi prendiamo in considerazione il caso di grafene dimerizzato possiamo notare che per ognivalore di q, ad eccezione di q = 2, l’introduzione dello spin orbita genera un gap: i punti in cui|f(t, t′,~k)|2 si annulla non soddisfano mai le condizioni (4.11) ad eccezione del caso in cui t′ = 2t;in tale caso infatti sappiamo che per ogni cella elementare del reticolo reciproco si ha un solo punto
di contatto tra le bande e questo punto coincide con M ′ = 2πa
(0 1√
3
){~e1,~e2}
(o un punto ad esso
equivalente) il quale soddisfa anche le condizioni (4.11).
Possiamo cosı affermare che per q = 2 il sistema si comporta come un semi-metallo indipendente-mente dal parametro tkm spin-orbita; quest’ultimo gioca pero un ruolo fondamentale nel definirel’andamento delle bande: se l’intensita dello spin-orbita e bassa le bande presentano una notevolecurvatura in prossimita dei punti di contatto (cfr. Figura(4.9)) mentre per intensita piu elevateesse tendono ad assumere un andamento conico simile a quello riscontrato nei punti K e K ′ nel
grafene standard (cfr. Figura(4.10)). In particolare per tkm =√34 |t| ≈ 0.433|t| e si puo ricavare,
in prossimita dei punti M ′ una legge di dispersione lineare analoga a quella trovata in precedenzaper il grafene standard:
ε±
(t,2t,
√3
4|t|,~k
)− E0 − t0 = ±
√3a2|t|2(δx
2 + δy2) + o(δx
2, δy2) ≈ ±
√3a|t||~δ|
dove ~δ e il vettore distanza (nel reticolo reciproco) dal punto M ′.Per valori di tkm prossimi a tale particolare valore la relazione di dispersione continua ad essereapprossimativamente conica in prossimita dei punti M ′ ma la sezione del cono risulta ellittica (cfr.Figura(4.11))
21
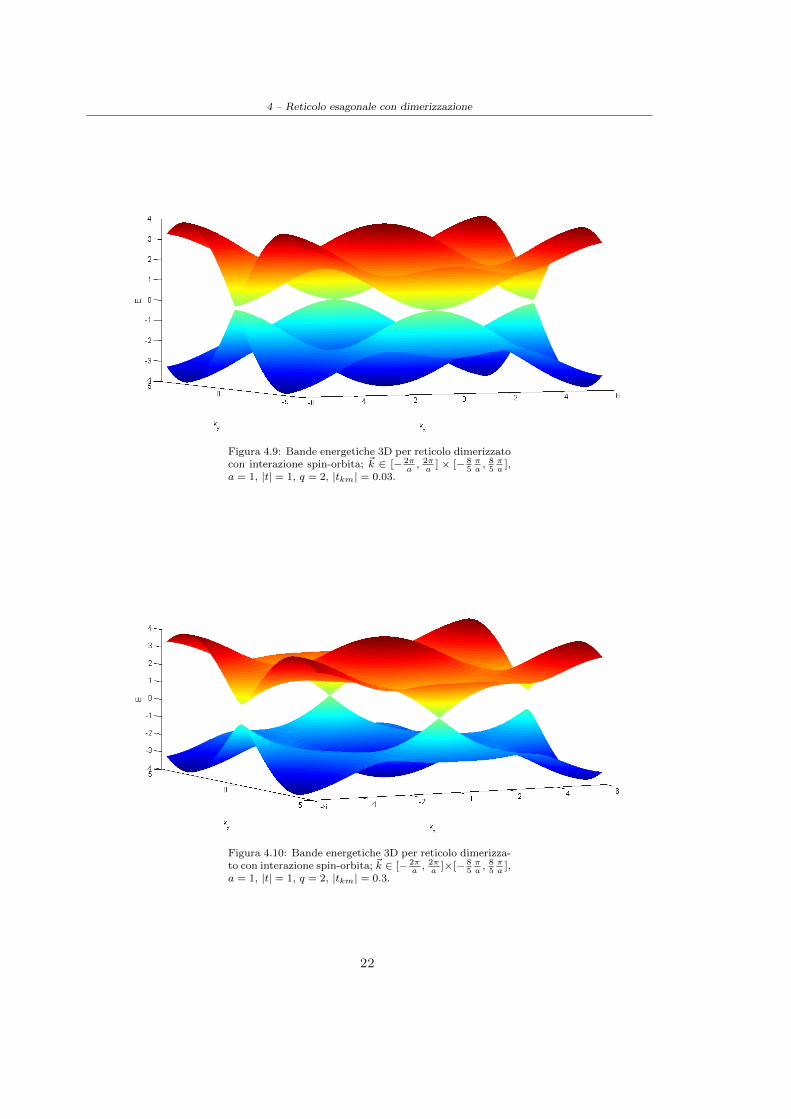
4 – Reticolo esagonale con dimerizzazione
Figura 4.9: Bande energetiche 3D per reticolo dimerizzatocon interazione spin-orbita; ~k ∈ [− 2π
a, 2πa
] × [− 85πa, 85πa
],a = 1, |t| = 1, q = 2, |tkm| = 0.03.
Figura 4.10: Bande energetiche 3D per reticolo dimerizza-to con interazione spin-orbita; ~k ∈ [− 2π
a, 2πa
]×[− 85πa, 85πa
],a = 1, |t| = 1, q = 2, |tkm| = 0.3.
22
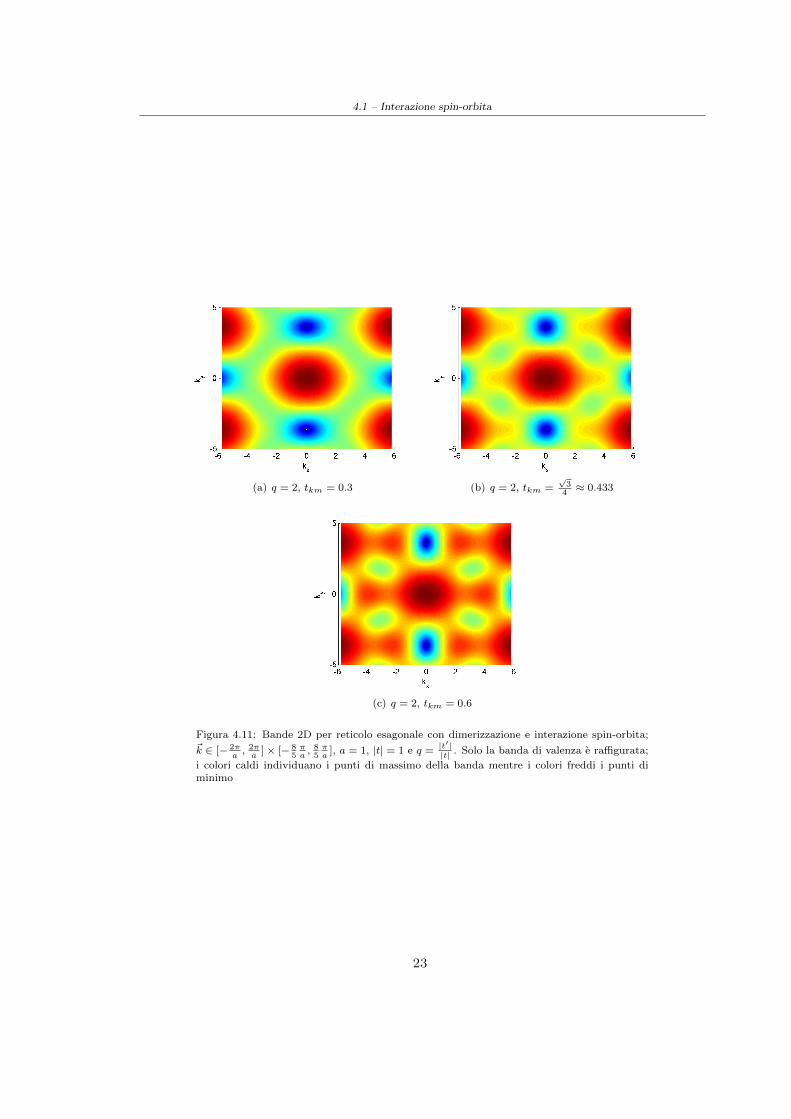
4.1 – Interazione spin-orbita
(a) q = 2, tkm = 0.3 (b) q = 2, tkm =√34≈ 0.433
(c) q = 2, tkm = 0.6
Figura 4.11: Bande 2D per reticolo esagonale con dimerizzazione e interazione spin-orbita;~k ∈ [− 2π
a, 2πa
]× [− 85πa, 85πa
], a = 1, |t| = 1 e q =|t′||t| . Solo la banda di valenza e raffigurata;
i colori caldi individuano i punti di massimo della banda mentre i colori freddi i punti diminimo
23

4 – Reticolo esagonale con dimerizzazione
4.2 Caratterizzazione topologica
Nella precedente sezione e stata introdotta l’interazione spin-orbita e si e constatato come questa,a meno di casi limite particolari, generi un gap tra le bande energetiche del grafene, sia esso di-merizzato o non. Tuttavia il cristallo che si viene a descrivere in questo modo non e un sempliceisolante, bensı un isolante topologico.
Questa particolare classe di materiali, attualmente alquanto studiata3, differisce dai comuni iso-lanti poiche, sebbene non siano presenti stati di conduzione all’interno del cristallo (quando questoe trattato come un reticolo tridimensionale infinito), se si riduce la periodicita del reticolo di unadimensione si vengono a formare stati di conduzioni localizzati sulle superfici. Analogamente sel’isolante topologico in questione e costituito da un reticolo bidimensionale infinito esso non con-duce, ma riducendo di una dimensione la periodicita del sistema, ottenendo quindi un nastro, sivengono a definire stati di conduzione localizzati lungo i bordi.
Un isolante viene quindi classificato come isolante (topologicamente) triviale o isolante topologicoa seconda del valore assunto da un invariante topologico, solitamente indicato come Z2, dipendentedalla natura della struttura a bande del cristallo. Se Z2 ≡ 0 (mod 2) l’isolante e triviale, se inveceZ2 ≡ 1 (mod 2) l’isolante e non triviale. Il calcolo di questo invariante puo essere eseguito inmodi differenti4, ma in particolare, nel caso di un’Hamiltoniana non interagente con simmetria diinversione spaziale e temporale, quale quella definita in (4.10), esso puo essere calcolato tramite larelazione:
(−1)Z2 =∏
~k∈TRIM
α−(~k) (4.12)
ovvero tramite il prodotto degli autovalori α−(~k), associati agli autostati occupati dell’Hamil-toniana, dell’operatore di inversione spaziale (detto anche operatore parita), calcolati nei punti
invarianti sotto l’operazione di inversione temporale, ossia quei punti ~k tali da differire da −~k perun vettore del reticolo reciproco; i punti suddetti (spesso indicati come punti TRIM, dall’ingleseTime Reversal Invariant Momenta) per il sistema in esame sono:
• Γ =(0 0
){~e1,~e2}
• M = 2πa
(12
12√3
){~e1,~e2}
• M ′ = 2πa
(0 1√
3
){~e1,~e2}
• M ′′ = 2πa
(12 − 1
2√3
){~e1,~e2}
mentre l’operatore parita, rappresentato nella base degli stati di Bloch definiti in (2.3), diviene:
3Si veda ad esempio M. Z. Hasan and C. L. Kane, Colloquium: Topological insulators, Rev. Mod. Phys., 82(2010) 3045-3067
4Si veda ad esempio F. Grandi, Effetti di correlazione elettronica negli isolanti topologici, Tesi di laurea magistralediscussa nel Dipartimento di Scienze Fisiche, Informatiche e Matematiche, Universita di Modena e Reggio Emilia,A.A. 2013-2014
24

4.2 – Caratterizzazione topologica
P (~k) =
(0 e−2i
~k·~τ1
e−2i~k·~τ2 0
)(4.13)
Dobbiamo quindi innanzi tutto calcolare gli autostati dell’Hamiltoniana nei punti TRIM. Diret-tamente dai risultati trovati nella sezione precedente possiamo notare che g(tkm,~k) si annulla incorrispondenza di tali punti e quindi l’Hamiltoniana assume la forma:
H(t, t′,~k) =
(0 ei
~k·(~τ2−~τ1)f(t, t′,~k)
e−i~k·(~τ2−~τ1)f∗(t, t′,~k) 0
)(4.14)
e di conseguenza gli autovettori, corrispondenti rispettivamente a ε+(t, t′,~k) e ε−(t, t′,~k) risultano:
(c(+)1 (t, t′,~k)
c(+)2 (t, t′,~k)
)=
1√2
(ei~k·(~τ2−~τ1)f(t,t′,~k)
|ei~k·(~τ2−~τ1)f(t,t′,~k)|1
)(4.15)
e:
(c(−)1 (t, t′,~k)
c(−)2 (t, t′,~k)
)=
1√2
(− ei
~k·(~τ2−~τ1)f(t,t′,~k)
|ei~k·(~τ2−~τ1)f(t,t′,~k)|1
)(4.16)
Calcolando poi tali autovettori nei punti TRIM si ottiene:
(c(±)1 (t, t′,Γ)
c(±)2 (t, t′,Γ)
)=
1√2
(± t′+2t|t′+2t|1
)⇒
(c(±)1 (Γ)
c(±)2 (Γ)
)=
1√2
(∓11
)(c(±)1 (t, t′,M)
c(±)2 (t, t′,M)
)=
1√2
(± e−i
π3 t′
|e−iπ3 t′|
1
)⇒
(c(±)1 (M)
c(±)2 (M)
)=
1√2
(∓e−iπ3
1
)(c(±)1 (t, t′,M ′′)
c(±)2 (t, t′,M ′′)
)=
1√2
(± ei
π3 t′
|eiπ3 t′|
1
)⇒
(c(±)1 (M ′′)
c(±)2 (M ′′)
)=
1√2
(∓eiπ3
1
)(c(±)1 (t, t′,M ′)
c(±)2 (t, t′,M ′)
)=
1√2
± ei2π3 (t′−2t)
|ei2π3 (t′−2t)|1
⇒
(c(±)1 (t, t′,M ′)
c(±)2 (t, t′,M ′)
)=
1√2
(±eiπ3 s
1
)
dove s = sgn(|t′| − 2|t|).
Trovati gli autovettori della matrice hamiltoniana nei punti TRIM possiamo calcolare gli autovaloridell’operatore parita a cui appartengono; in particolare, essendo sufficiente il calcolo degli autovaloriassociati ai soli stati occupati, possiamo concentrarci sugli autovettori appartenenti a ε− ottenendo:
25

4 – Reticolo esagonale con dimerizzazione
P (Γ) =
(0 11 0
)⇒ P (Γ)
(c(−)1 (Γ)
c(−)2 (Γ)
)=
(0 11 0
)1√2
(11
)=
1√2
(11
)
= (1)
(c(−)1 (Γ)
c(−)2 (Γ)
)
P (M) =
(0 e−i
π3
eiπ3 0
)⇒ P (M)
(c(−)1 (M)
c(−)2 (M)
)=
(0 e−i
π3
eiπ3 0
)1√2
(e−i
π3
1
)=
1√2
(e−i
π3
1
)
= (1)
(c(−)1 (M)
c(−)2 (M)
)
P (M ′′) =
(0 ei
π3
e−iπ3 0
)⇒ P (M ′′)
(c(−)1 (M ′′)
c(−)2 (M ′′)
)=
(0 ei
π3
e−iπ3 0
)1√2
(eiπ3
1
)=
1√2
(eiπ3
1
)
= (1)
(c(−)1 (M ′′)
c(−)2 (M ′′)
)
P (M ′) =
(0 −e−iπ3−eiπ3 0
)⇒ P (M ′)
(c(−)1 (M ′)
c(−)2 (M ′)
)=
(0 −e−iπ3−eiπ3 0
)1√2
(−e−iπ3 s
1
)= (s)
1√2
(−e−iπ3
1
)
= (s)
(c(−)1 (M ′)
c(−)2 (M ′)
)
Possiamo cosı concludere che che: ∏~k∈TRIM
α−(~k) = (1)(1)(1)(s)
pertanto: {s = +1 ⇐⇒ |t′| > 2|t| ⇒ (−1)Z2 = 1 ⇐⇒ Z2 ≡ 0 (mod 2)
s = −1 ⇐⇒ |t′| < 2|t| ⇒ (−1)Z2 = −1 ⇐⇒ Z2 ≡ 1 (mod 2)(4.17)
Troviamo quindi che q = |t′||t| = 2 rappresenta il valore limite che sancisce il passaggio da isolante
topologico (q < 2) ad isolante triviale (q > 2) indipendentemente dal valore assunto da tkm;ricordiamo pero che e necessario che quest’ultimo valore sia diverso da zero affinche il sistema siaisolante e quindi anche un isolante topologico.
4.3 Nastri
Sappiamo che la proprieta peculiare degli isolanti topologici bidimensionali e la formazione di staticonduttivi sui bordi in seguito alla riduzione di periodicita del cristallo in una dimensione. An-diamo dunque a definire un sistema con le stesse proprieta del reticolo esagonale bidimensionale
26

4.3 – Nastri
considerato in precedenza, questa volta pero illimitato nella direzione x ma limitato nella direzioney. Un sistema di questo tipo e generalmente chiamato nastro e nel caso di un reticolo esagonale,quale nel caso del grafene, vengono solitamente definite due tipologie di nastri (dette ad armchairo a zigzag) a seconda della terminazione che viene realizzata (cfr. Figura(4.12)).
Figura 4.12: (a) nastro a zig zag, (b) nastro ad armchair
Possiamo estendere la formulazione del modello tight-binding definita in precedenza anche a que-sto sistema non illimitato prendendo un’unita ripetitiva estesa su tutto lo spessore del nastro etrattando quindi quest’ultimo come un sistema unidimensionale.
~a
Figura 4.13: Nastro con dimerizzazione; evidenziata da una linea tratteggiatarossa l’unita ripetitiva
Nel caso di nastro a zig zag, l’unico che prenderemo in esame, il vettore di base del reticolo direttodiviene:
~a = a~e1 (4.18)
dove a e la distanza a secondi vicini, e conseguentemente il vettore di base del reticolo reciprocodiviene:
~b =2π
a~e1 (4.19)
la prima zona di Brillouin risulta quindi data dall’intervallo unidimensionale [M ′,M ] = [−πa ,πa ].
Supponendo di considerare un nastro di spessore tale da contenere n siti nell’unita ripetitiva elavorando nelle stesse ipotesi considerate sino ad ora, si arriva a dover diagonalizzare una matrice
27
4 – Reticolo esagonale con dimerizzazione
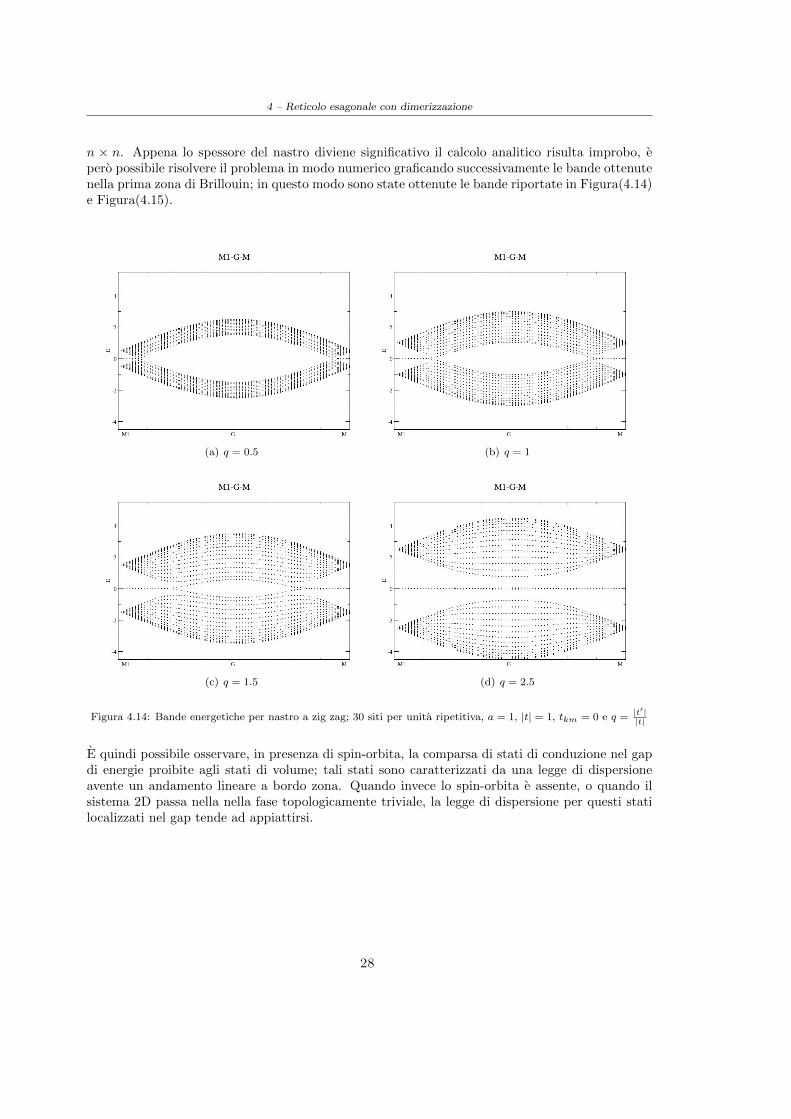
n × n. Appena lo spessore del nastro diviene significativo il calcolo analitico risulta improbo, epero possibile risolvere il problema in modo numerico graficando successivamente le bande ottenutenella prima zona di Brillouin; in questo modo sono state ottenute le bande riportate in Figura(4.14)e Figura(4.15).
(a) q = 0.5 (b) q = 1
(c) q = 1.5 (d) q = 2.5
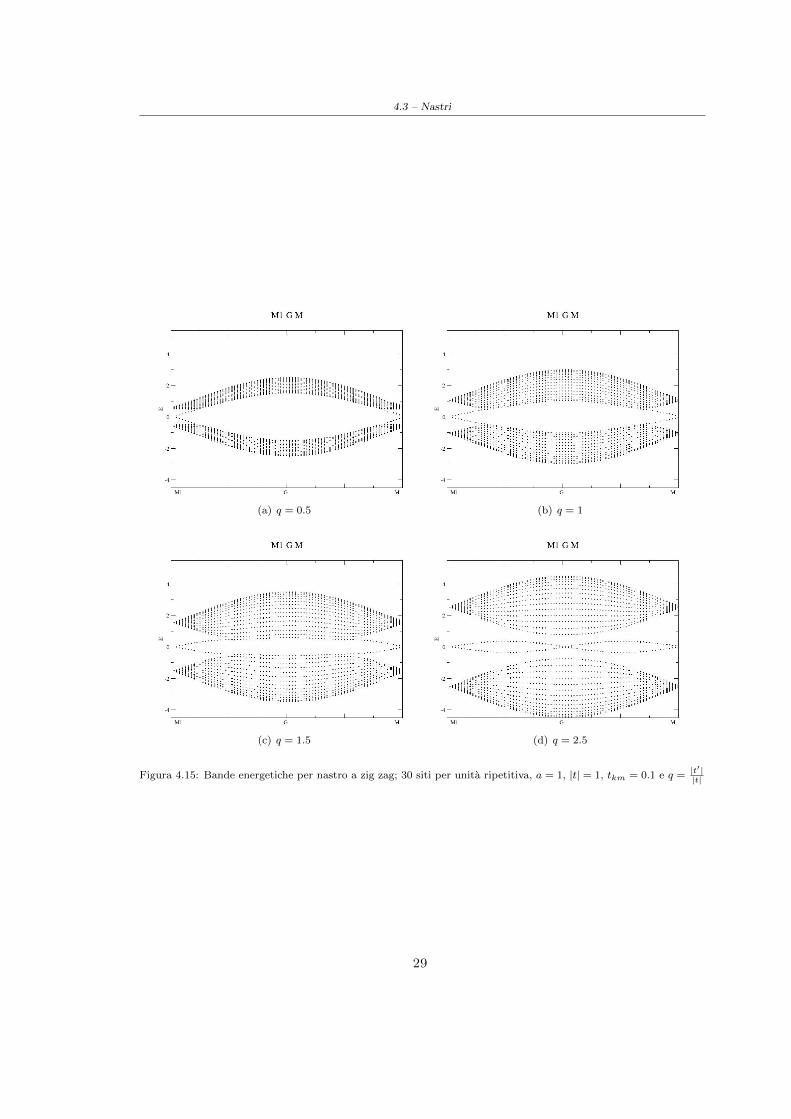
Figura 4.14: Bande energetiche per nastro a zig zag; 30 siti per unita ripetitiva, a = 1, |t| = 1, tkm = 0 e q =|t′||t|
E quindi possibile osservare, in presenza di spin-orbita, la comparsa di stati di conduzione nel gapdi energie proibite agli stati di volume; tali stati sono caratterizzati da una legge di dispersioneavente un andamento lineare a bordo zona. Quando invece lo spin-orbita e assente, o quando ilsistema 2D passa nella nella fase topologicamente triviale, la legge di dispersione per questi statilocalizzati nel gap tende ad appiattirsi.
28
4.3 – Nastri
(a) q = 0.5 (b) q = 1
(c) q = 1.5 (d) q = 2.5
Figura 4.15: Bande energetiche per nastro a zig zag; 30 siti per unita ripetitiva, a = 1, |t| = 1, tkm = 0.1 e q =|t′||t|
29

30

Capitolo 5
Reticolo esagonale con distorsioneKekule
Prendiamo ora in considerazione un reticolo esagonale in cui, come nel caso trattato nel capitoloprecedente, le correzioni energetiche a primi vicini non sono tutte uguali ma dipendano dal primovicino considerato. Consideriamo in particolare una situazione, rappresentata in Figura(5.1), incui il reticolo bidimensionale puo essere pensato come scomposto in cluster di sei siti interagenti traloro. A differenza del caso precedente in cui la dimerizzazione poteva essere pensata come l’effettodi una trazione o di una compressione del sistema, una rappresentazione di questo tipo puo inve-ce fornire un primo rudimentale approccio all’analisi degli effetti dell’interazione inter-elettronica,modellizzando gli effetti di questa tramite una differenziazione tra le correzioni energetiche intracluster o extra cluster.
O
~a1
~a2
1
23
4
56
Figura 5.1: Reticolo diretto; in rosso la cella di Wigner Seitz
L’approccio con cui affronteremo il problema e il medesimo, andiamo quindi innanzi tutto a definirei vettori di base del reticolo diretto notando pero che in questo caso la cella elementare conterrasei siti atomici; i vettori di base del reticolo diretto divengono quindi:
31

5 – Reticolo esagonale con distorsione Kekule
~a1 = a(
32
√32
){~e1,~e2}
~a2 = a(
32 −
√32
){~e1,~e2}
(5.1)
e i vettori posizione dei siti atomici all’interno di una cella risultano:
~τ1 =a
2
(1 1√
3
){~e1,~e2}
~τ2 =a
2
(1 − 1√
3
){~e1,~e2}
~τ3 =a
2
(0 − 2√
3
){~e1,~e2}
~τ4 =a
2
(−1 − 1√
3
){~e1,~e2}
~τ5 =a
2
(−1 1√
3
){~e1,~e2}
~τ6 =a
2
(0 2√
3
){~e1,~e2}
Mediante le relazioni (3.3) troviamo poi i vettori di base del reticolo reciproco:
~b1 =2π
a
(13
1√3
){~e1,~e2}
~b2 =2π
a
(13 − 1√
3
){~e1,~e2}
(5.2)
Γ
~b1
~b2
M ′
K ′
M ′′′
K ′′
M ′′
Figura 5.2: Reticolo reciproco; in blu la prima zona di Brillouin
e tramite essi possiamo infine definire alcuni dei punti di alta simmetria del reticolo reciproco:
• Γ =(0 0
){~e1,~e2}
• K ′ = 2πa
(13
13√3
){~e1,~e2}
• M ′′′ = 2πa
(13 0
){~e1,~e2}
32

Nelle medesime ipotesi e operando in modo analogo a quanto fatto nella sezione precedente pos-siamo arrivare a scrivere la matrice hamiltoniana nella base degli stati di Bloch (avendo ridefinitoopportunamente lo sero dell’energia in modo d eliminare i contributi costanti E0 + t0) come:
H(t, t′, tkm,~k) =
0 β3(~k)t′ −itkmγ∗(~k) β1(~k)t itkmγ(~k) β5(~k)t′
β∗3(~k)t′ 0 β∗1(~k)t′ −itkmγ∗(~k) β∗5(~k)t itkmγ(~k)
itkmγ(~k) β1(~k)t′ 0 β5(~k)t′ −itkmγ∗(~k) β3(~k)t
β∗1(~k)t itkmγ(~k) β∗5(~k)t′ 0 β∗3(~k)t′ −itkmγ∗(~k)
−itkmγ∗(~k) β5(~k)t itkmγ(~k) β3(~k)t′ 0 β1(~k)t′
β∗5(~k)t′ −itkmγ∗(~k) β∗3(~k)t itkmγ(~k) β∗1(~k)t′ 0
dove:
• β1(~k) = ei~k·τ1
• β3(~k) = ei~k·τ3
• β5(~k) = ei~k·τ5
• γ(~k) = ei~k·(~τ5−~τ1)
[1 + ei
~k·~a1 + ei~k·~a2
]• t rappresenta l’interazione a primi vicini extra cluster
• t′ rappresenta l’interazione a primi vicini intra cluster
• tkm rappresenta l’interazione spin orbita
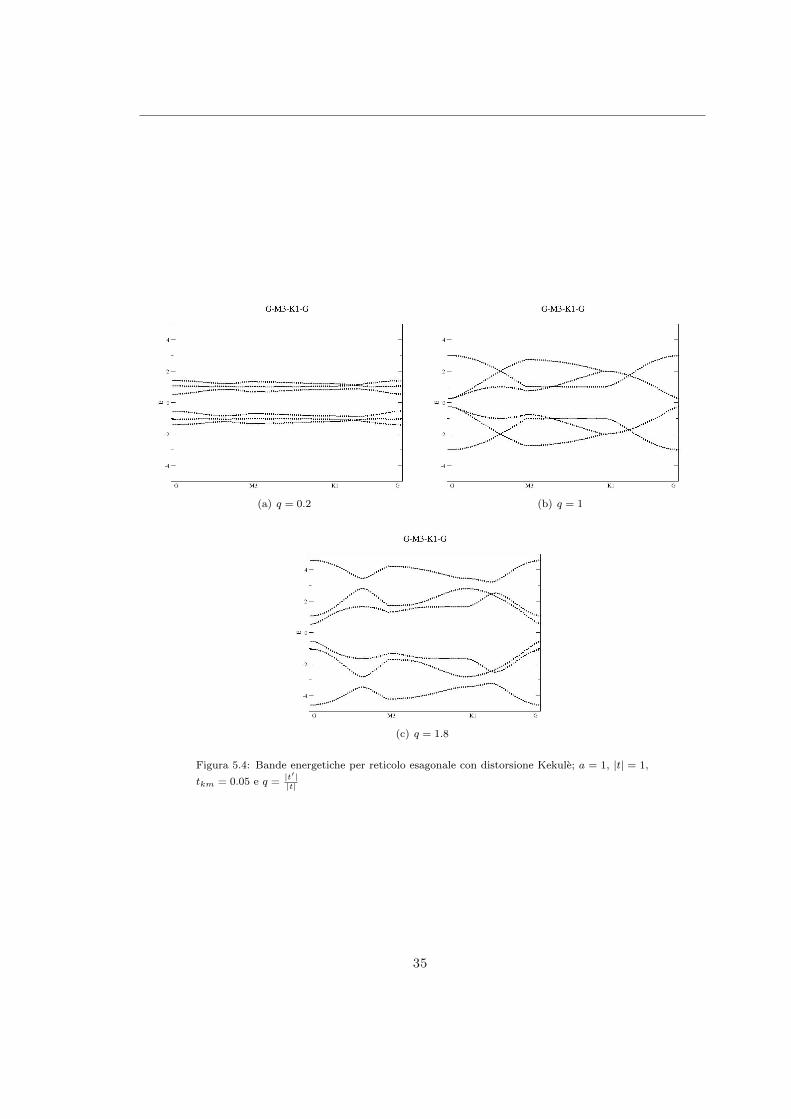
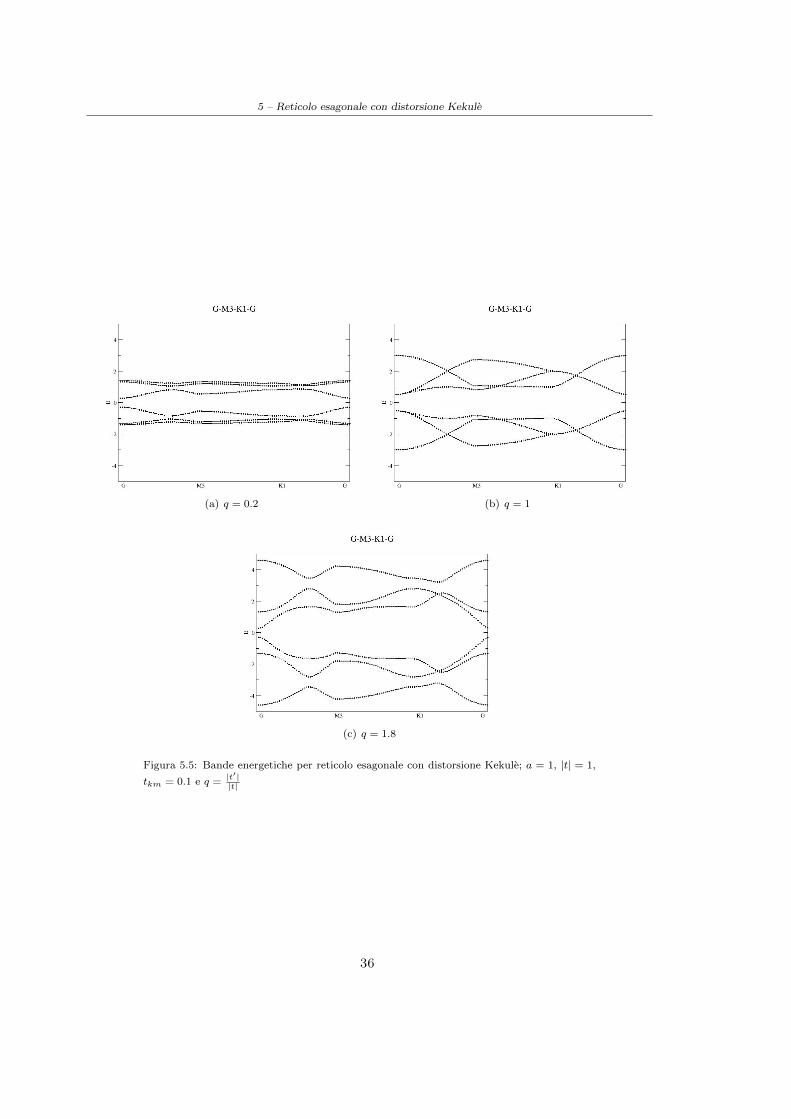
La risoluzione analitica di questo problema agli autovalori 6× 6 e impensabile tuttavia e possibileutilizzare un approccio numerico calcolando le bande energetiche per differenti valori dei parametri
(cfr. Figura(5.3), Figura(5.4), Figura(5.5)). Si noti che per valori di q = |t′|t inferiori a 1 la disper-
sione delle bande e ridotta in quanto il sistema che si sta descrivendo in tale caso viene ad esserecostituito da dimeri isolati poco interagenti tra loro; se invece q > 1 la dispersione e invece favorita.
Notando quindi che il gap minimo si ha sempre in prossimita del punto Γ e possibile restringereun’analisi piu approfondita solo a tale punto dello spazio reciproco, calcolando analiticamenteper quali valori dei parametri i gap si chiude. Calcoliamo quindi la matrice hamiltoniana in Γottenendo:
H(t, t′, tkm,Γ) =
0 t′ −i3tkm t i3tkm t′
t′ 0 t′ −i3tkm t i3tkmi3tkm t′ 0 t′ −i3tkm tt i3tkm t′ 0 t′ −i3tkm
−i3tkm t i3tkm t′ 0 t′
t′ −i3tkm t i3tkm t′ 0
e da essa ricaviamo i seguenti autovalori:
• ε(±1)(Γ) = ±(t+ 2t′)
33

5 – Reticolo esagonale con distorsione Kekule
(a) q = 0.2 (b) q = 1
(c) q = 1.8
Figura 5.3: Bande energetiche per reticolo esagonale con distorsione Kekule; a = 1, |t| = 1,
tkm = 0 e q =|t′||t|
34
(a) q = 0.2 (b) q = 1
(c) q = 1.8
Figura 5.4: Bande energetiche per reticolo esagonale con distorsione Kekule; a = 1, |t| = 1,
tkm = 0.05 e q =|t′||t|
35
5 – Reticolo esagonale con distorsione Kekule
(a) q = 0.2 (b) q = 1
(c) q = 1.8
Figura 5.5: Bande energetiche per reticolo esagonale con distorsione Kekule; a = 1, |t| = 1,
tkm = 0.1 e q =|t′||t|
36
• ε(±2)(Γ) = ±(t− t′ + 3√
3tkm)
• ε(±3)(Γ) = ±(t− t′ − 3√
3tkm)
Il gap pertanto si chiude se e solo:
t− t′ + 3√
3tkm = 0 ∨ t− t′ − 3√
3tkm = 0
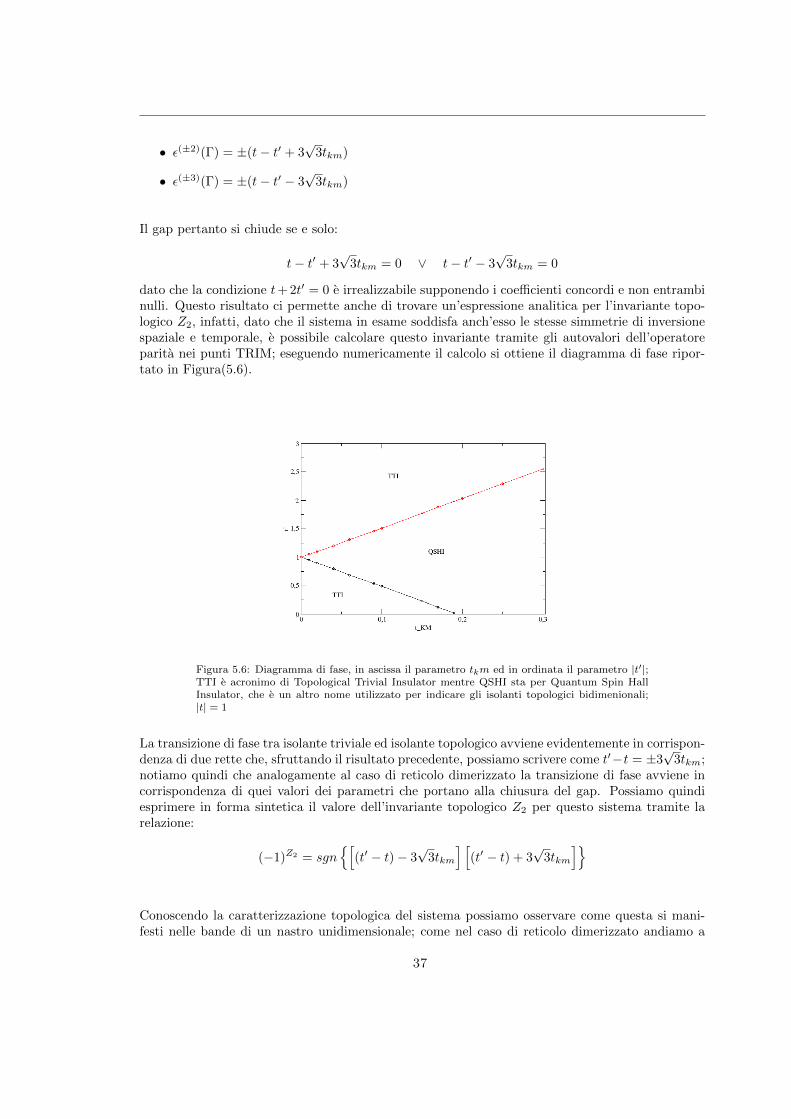
dato che la condizione t+ 2t′ = 0 e irrealizzabile supponendo i coefficienti concordi e non entrambinulli. Questo risultato ci permette anche di trovare un’espressione analitica per l’invariante topo-logico Z2, infatti, dato che il sistema in esame soddisfa anch’esso le stesse simmetrie di inversionespaziale e temporale, e possibile calcolare questo invariante tramite gli autovalori dell’operatoreparita nei punti TRIM; eseguendo numericamente il calcolo si ottiene il diagramma di fase ripor-tato in Figura(5.6).
Figura 5.6: Diagramma di fase, in ascissa il parametro tkm ed in ordinata il parametro |t′|;TTI e acronimo di Topological Trivial Insulator mentre QSHI sta per Quantum Spin HallInsulator, che e un altro nome utilizzato per indicare gli isolanti topologici bidimenionali;|t| = 1
La transizione di fase tra isolante triviale ed isolante topologico avviene evidentemente in corrispon-denza di due rette che, sfruttando il risultato precedente, possiamo scrivere come t′−t = ±3
√3tkm;
notiamo quindi che analogamente al caso di reticolo dimerizzato la transizione di fase avviene incorrispondenza di quei valori dei parametri che portano alla chiusura del gap. Possiamo quindiesprimere in forma sintetica il valore dell’invariante topologico Z2 per questo sistema tramite larelazione:
(−1)Z2 = sgn{[
(t′ − t)− 3√
3tkm
] [(t′ − t) + 3
√3tkm
]}
Conoscendo la caratterizzazione topologica del sistema possiamo osservare come questa si mani-festi nelle bande di un nastro unidimensionale; come nel caso di reticolo dimerizzato andiamo a
37

5 – Reticolo esagonale con distorsione Kekule
~a
Figura 5.7: Nastro a zig zag con distorsione Kekule; l’unita ripetitiva e evidenziata da unalinea tratteggiata rossa
considerare un nastro a zig zag quale quello raffigurato in Figura(5.7).
In questo caso, data la ridotta periodicita del sistema, il vettore di base del reticolo diretto risultapari a:
~a = 3a~e1 (5.3)
conseguentemente il vettore di base del reticolo reciproco diviene:
~b =2π
3a~e1 (5.4)
e la prima zona di Brilluoin viene a coincidere con l’intervallo [M ′,M ] = [−π3 ,π3 ].
Calcolando quindi numericamente le bande energetiche per questo sistema unidimensionale trovia-mo gli andamenti rappresentati in Figura(5.8), Figura(5.9) e Figura(5.10). Tali andamenti sonoanaloghi a quelli trovati per le bande del reticolo con dimerizzazione: quando il sistema e nellafase topologicamente non triviale (ad esempio per (q, tkm) = (1,0.05) e per (q, tkm) = (1,0.1); ilcaso (q, tkm) = (1,0) e un caso limite trovandosi (cfr. Figura(5.6))) si vengono a delineare stati diconduzione con dispersione lineare nel gap di energie proibito agli stati di volume. Quando inveceil sistema passa nella fase topologicamente triviale questi stati di superficie tendono ad avere unadispersione piatta.
38

(a) q = 0.2; TTI (b) q = 1; zona di transizione
(c) q = 1.8 TTI
Figura 5.8: Bande energetiche per nastro a zig zag con distorsione Kekule; 30 siti atomici
per unita ripetitiva, a = 1, |t| = 1, tkm = 0 e q =|t′||t|
39

5 – Reticolo esagonale con distorsione Kekule
(a) q = 0.2; TTI (b) q = 1; QSHI
(c) q = 1.8; TTI
Figura 5.9: Bande energetiche per nastro a zig zag con distorsione Kekule; 30 siti atomici
per unita ripetitiva, a = 1, |t| = 1, tkm = 0.05 e q =|t′||t|
40

(a) q = 0.2; TTI (b) q = 1; QSHI
(c) q = 1.8; TTI
Figura 5.10: Bande energetiche per nastro a zig zag con distorsione Kekule; 30 siti atomici
per unita ripetitiva, a = 1, |t| = 1, tkm = 0.1 e q =|t′||t|
41

42

Capitolo 6
Conclusioni
In questo lavoro, frutto del periodo di tirocinio effettuato presso il Dipartimento di Fisica dell’U-niversita di Modena e Reggio Emilia, sono state studiate le proprieta elettroniche di un reticoloesagonale bidimensionale al variare dei parametri relativi all’interazione a primi vicini e all’intera-zione spin orbita.
Nel sistema caratterizzato da una dimerizzazione si e studiato come questa possa influire sulla for-ma assunta dalla banda di conduzione e di valenza, rendendo il cristallo in esame un semimetalloo un isolante. E stata quindi introdotta l’interazione spin orbita, si e studiato il suo contributoalle bande energetiche ed e stata effettuata la caratterizzazione topologica del sistema per capirequando esso si trova nella fase semimetallica, di isolante triviale o di isolante topologico. Que-sta ha mostrato come le transizioni di fase avvengano per un ben preciso valore del rapportotra l’intensita dei legami dimerizzati e semplici, indipendentemente dall’intensita dell’interazionespin orbita. Quando pero l’interazione spin-orbita e nulla la transizione che avviene e del tiposemimetallo-isolante triviale, mentre in presenza di interazione spin orbita essa e del tipo isolantetopologico-isolante triviale. Una diretta conseguenza delle transizioni di fase e poi stata osservatastudiando le bande energetiche di un nastro unidimensionale.
Nel sistema caratterizzato da distorsione Kekule l’analisi riproposta ha invece mostrato un com-portamento piu complesso: le transizioni di fase avvengono in punti dello spazio dei parametridipendenti sia dal rapporto tra l’intensita dei legami dimerizzati e semplici sia dall’interazione spinorbita, andando a definire un diagramma di fase piu ricco.
Aggiungerei infine alcune riflessioni sulla natura didattica di questa esperienza. Essa mi ha infattiportato per la prima volta ad affrontare seriamente la lettura di riviste specialistiche e di argomentidi cui non era affatto consolidata la padronanza, a lavorare su problemi la cui soluzione, e soprat-tutto il cui metodo risolutivo, non era visibile fin da principio e a prender parte ad un progetto diricerca piu ampio di cui solo una piccola frazione mi era accessibile, con la diretta conseguenza didover partire da lavori gia svolti e risultati gia ottenuti per cercare, tramite piccoli cambiamenti, didare, per quanto possibile, un minimo contributo. Vorrei quindi ringraziare la professoressa FrancaManghi ed il dottor Francesco Grandi per il loro aiuto nell’indicarmi i giusti modi per affrontarequeste difficolta e ancor piu per la grande umanita e cortesia mostrata.
43

44

Bibliografia
• A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov e A. K. Geim, The electronicproperties of graphene, Rev. Mod. Phys., 81, 2009, pp.109-162.
• Ashcroft, Mermin Solid state physics, Saunders College Publishing (1976)
• T. C. Lang, A. M. Essein, V. Gurarie and S. Wessel, Z2 topological invariants in twodimaensions from quantum Monte Carlo Phys. Rev. B 87, 205101 (2013)
• C. L. Kane and E. J. Mele, Quantum Spin Hall Effect in Graphene, Phys. Rev. Lett., 95,226801 (2005)
• M. Z. Hasan and C. L. Kane, Colloquium: Topological insulators, Rev. Mod. Phys., 82(2010) 3045-3067
• F. Grandi, Effetti di correlazione elettronica negli isolanti topologici, Tesi di laurea magistralediscussa nel Dipartimento di Scienze Fisiche, Informatiche e Matematiche, Universita diModena e Reggio Emilia, A.A. 2013-2014
45


















