
Spettroscopia Strutturale Sommario -...
32
Spettroscopia Strutturale Sommario ❖Cristallografia a raggi X ๏ Principi delle tecniche diffrattive ๏ Diffrazione di raggi X su cristalli ๏ Diffrazione di raggi X su fibra ๏ Diffrazione di raggi X da soluzioni diluite a largo angolo ๏ Cenni alla diffrazione di neutroni ๏ Il Protein Data Bank ❖Cenni a software per visualizzazione di strutture ❖(Crio) microscopia elettronica (e database di mappe elettroniche a bassa risoluzione) ❖NRM strutturale Materiale didattico scaricabile da http://homepage.sns.it/tozzini/didattica.html
Transcript of Spettroscopia Strutturale Sommario -...

Spettroscopia Strutturale
Sommario❖Cristallografia a raggi X
๏Principi delle tecniche diffrattive๏Diffrazione di raggi X su cristalli๏Diffrazione di raggi X su fibra๏Diffrazione di raggi X da soluzioni diluite a largo angolo๏Cenni alla diffrazione di neutroni๏Il Protein Data Bank
❖Cenni a software per visualizzazione di strutture ❖(Crio) microscopia elettronica (e database di mappe elettroniche a bassa risoluzione)❖NRM strutturale
Materiale didattico scaricabile dahttp://homepage.sns.it/tozzini/didattica.html
Spettroscopia Strutturale
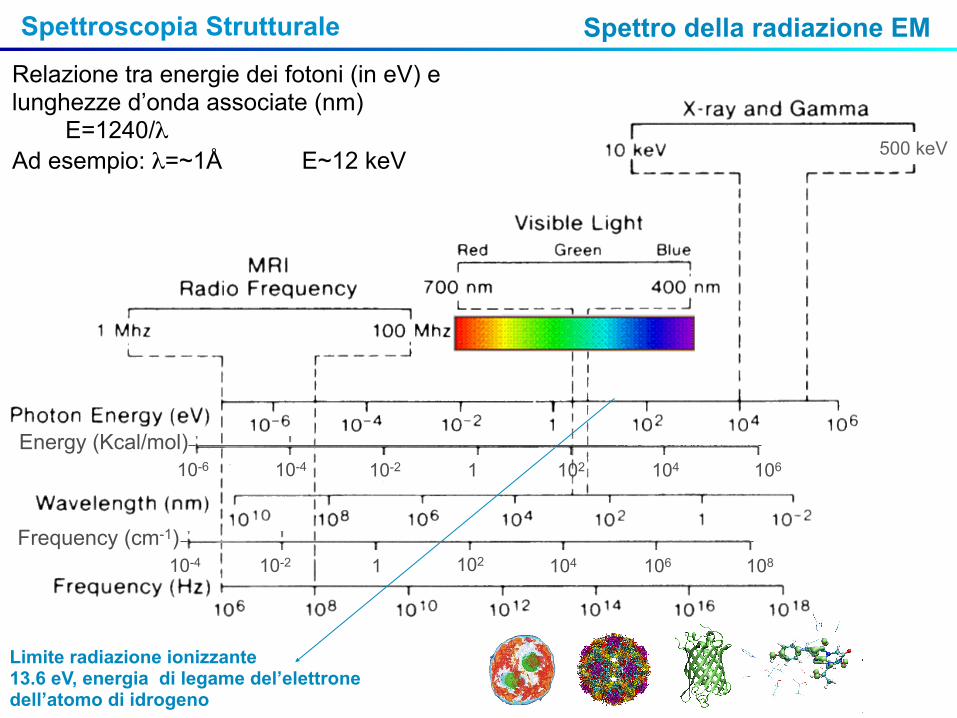
Relazione tra energie dei fotoni (in eV) e lunghezze d’onda associate (nm) E=1240/λ Ad esempio: λ=~1Å E~12 keV
Limite radiazione ionizzante13.6 eV, energia di legame del’elettrone dell’atomo di idrogeno
Spettro della radiazione EM
1 102 104 10610-210-410-6
Energy (Kcal/mol)
102 104 106 108 1 10-210-4
Frequency (cm-1)
500 keV
Spettroscopia Strutturale
E =m0c2
�1− v2
c2
λ =h
m0v
�1− v2
c2
Spettroscopia Strutturale
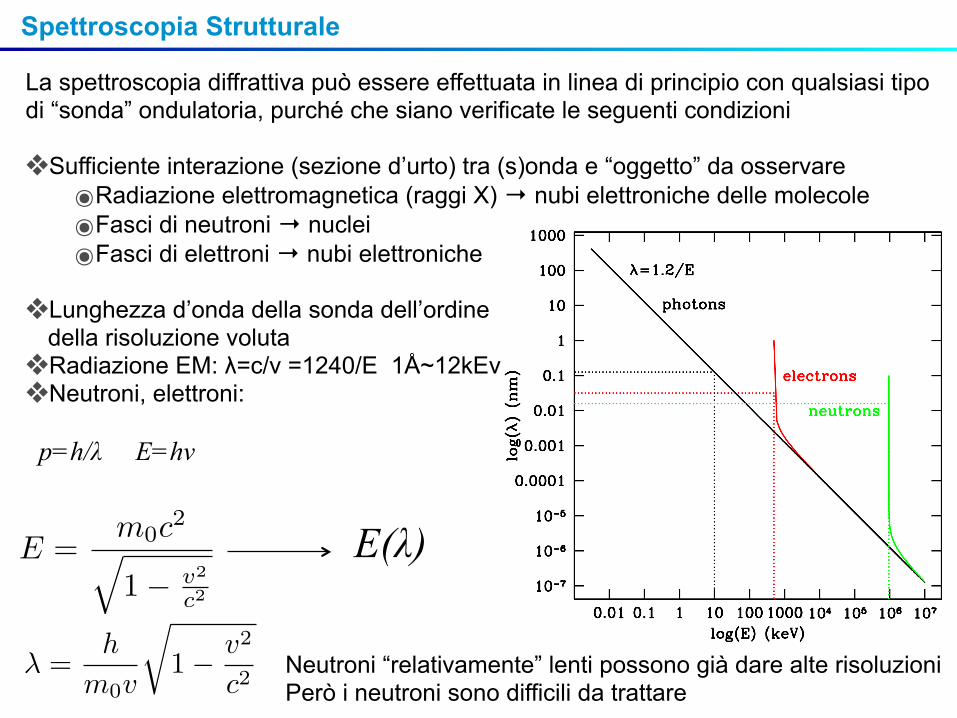
La spettroscopia diffrattiva può essere effettuata in linea di principio con qualsiasi tipo di “sonda” ondulatoria, purché che siano verificate le seguenti condizioni
❖Sufficiente interazione (sezione d’urto) tra (s)onda e “oggetto” da osservare๏Radiazione elettromagnetica (raggi X) → nubi elettroniche delle molecole๏Fasci di neutroni → nuclei๏Fasci di elettroni → nubi elettroniche
❖Lunghezza d’onda della sonda dell’ordine della risoluzione voluta ❖Radiazione EM: λ=c/ν =1240/E 1Å~12kEv❖Neutroni, elettroni:
p=h/λ E=hν
E(λ)
Neutroni “relativamente” lenti possono già dare alte risoluzioniPerò i neutroni sono difficili da trattare
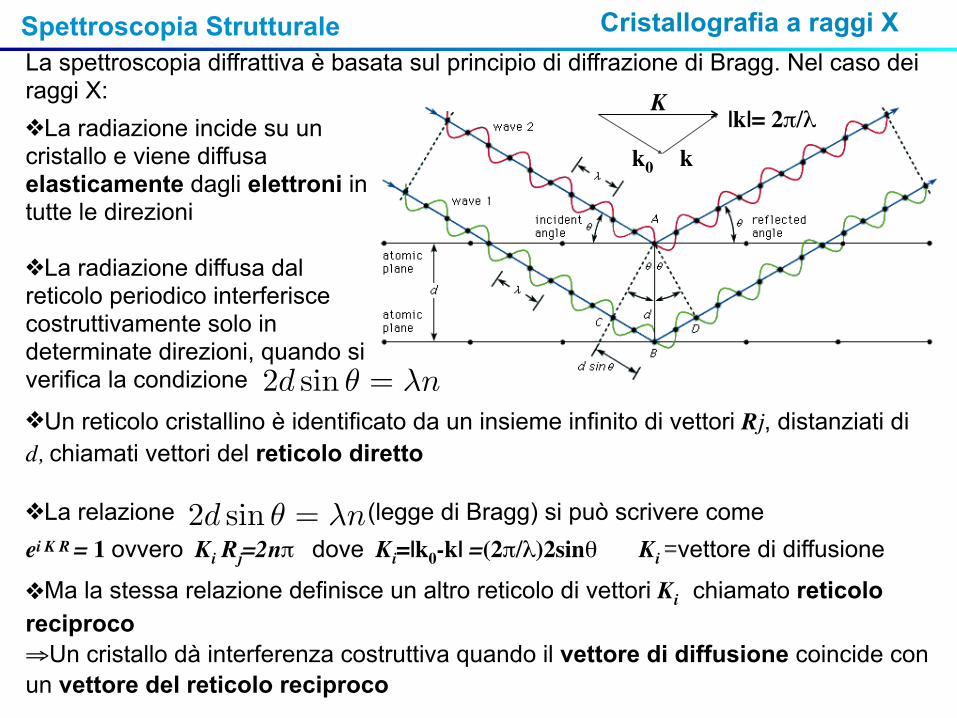
Spettroscopia Strutturale Cristallografia a raggi X La spettroscopia diffrattiva è basata sul principio di diffrazione di Bragg. Nel caso dei raggi X:❖La radiazione incide su un cristallo e viene diffusa elasticamente dagli elettroni in tutte le direzioni
❖La radiazione diffusa dal reticolo periodico interferisce costruttivamente solo in determinate direzioni, quando si verifica la condizione
❖Un reticolo cristallino è identificato da un insieme infinito di vettori Rj, distanziati di d, chiamati vettori del reticolo diretto
❖La relazione (legge di Bragg) si può scrivere come ei K R = 1 ovvero Ki Rj=2nπ dove Ki=|k0-k| =(2π/λ)2sinθ Ki =vettore di diffusione
❖Ma la stessa relazione definisce un altro reticolo di vettori Ki chiamato reticolo reciproco⇒Un cristallo dà interferenza costruttiva quando il vettore di diffusione coincide con un vettore del reticolo reciproco
k0 k|k|= 2π/λ
K
2d sin θ = λn
2d sin θ = λn
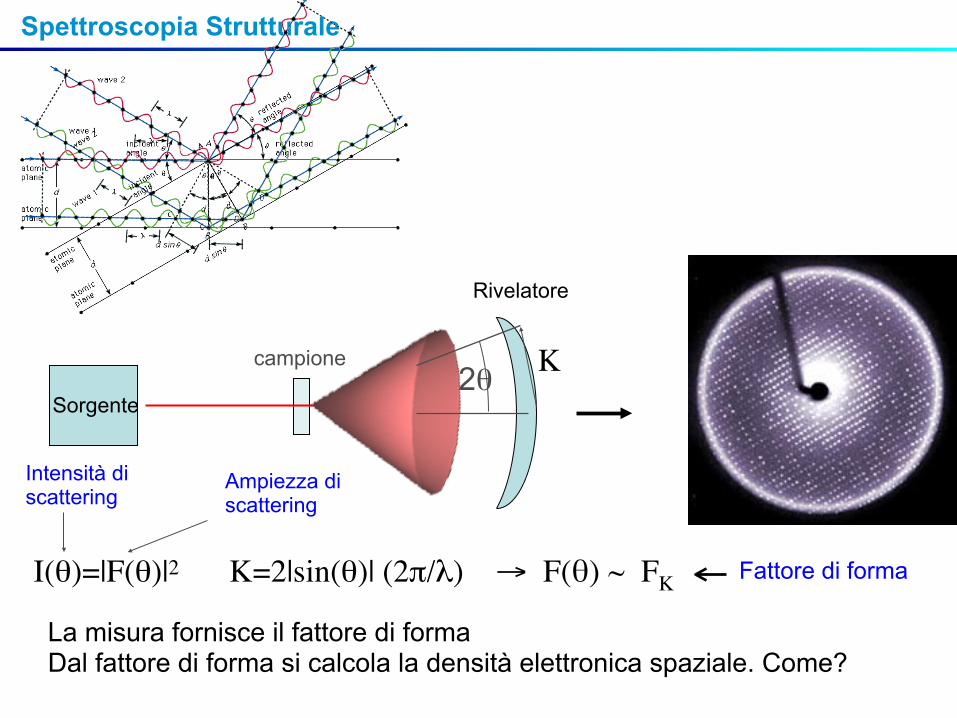
Spettroscopia Strutturale
Intensità di scattering
Ampiezza di scattering
I(θ)=|F(θ)|2 K=2|sin(θ)| (2π/λ) → F(θ) ~ FK Fattore di forma
La misura fornisce il fattore di formaDal fattore di forma si calcola la densità elettronica spaziale. Come?
Sorgente
campione
Rivelatore
2θ K
F (k) =�
dr(2π)3
f(r)e−ikr = 1
FT [ρ] =
=�
i
F (k)e−ikr =�
i
e−ikr =�
j
δk,Kj = FKj
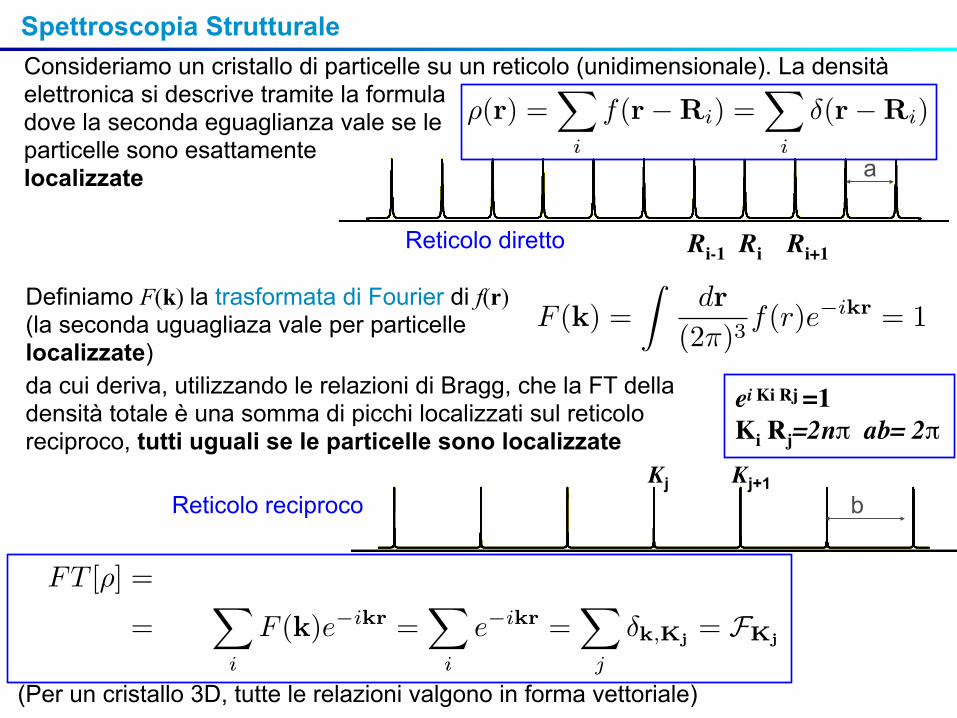
Spettroscopia StrutturaleConsideriamo un cristallo di particelle su un reticolo (unidimensionale). La densità elettronica si descrive tramite la formuladove la seconda eguaglianza vale se le particelle sono esattamentelocalizzate
ei Ki Rj =1 Ki Rj=2nπ ab= 2π
Ri Ri+1Ri-1Reticolo diretto
Reticolo reciprocoKj Kj+1
a
b
(Per un cristallo 3D, tutte le relazioni valgono in forma vettoriale)
Definiamo F(k) la trasformata di Fourier di f(r)(la seconda uguagliaza vale per particelle localizzate)da cui deriva, utilizzando le relazioni di Bragg, che la FT della densità totale è una somma di picchi localizzati sul reticolo reciproco, tutti uguali se le particelle sono localizzate
ρ(r) =�
i
f(r−Ri) =�
i
δ(r−Ri)

f(r) =�
Kj
FKjeiKjr
Spettroscopia Strutturale
Ma gli elettroni sono delocalizzati intorno ai siti reticolari,→ f(r) ~ gaussiana Ri
La FT di una densità di particelle localizzate su siti di un reticolo diretto è identicamente uguale alla somma di delta localizzate sui siti del reticlo reciprocoMa che collegamento c’è tra FK (fattore di forma misurato) e ? FK =
In pratica, per ogni fissato vettore di diffusione (cioè fissata la direzione di diffusione) viene selezionata una differenza di fase per la quale si ha interferenza costruttiva e quindi intensità diversa da zero. Quindi lo scanning sulla direzione di diffusione (θ o equivalentemente k) equivale a fare una trasformata di Fourier dei punti da cui proviene la radiazione diffusa
→ Il fattore di forma è proporzionale alla trasformata di Fourier della densità spaziale di carica elettronica
€
FK j
Kj
con F(k) ~ gaussiana
⇒Misurando FK, si ricava f(r) e quindi ρ(r) con una trasformata di Fourier inversa
FK FK
ρ(r) =�
i
f(r −Ri)
FT [ρ] = FKj =�
j
F (k)δk,Kj

f(r) =�
i
f i(r − ri)
Spettroscopia Strutturale
In un cristallo di proteine, in ogni cella (sito reticolare) si trova una proteina→f(r) = somma di densità elettroniche dovute ai singoli atomi
⇒ Il fattore di forma strutturale è la somma di fattori di forma atomici pesati con un fattore di fase dipendente dalla posizione dell’atomo all’interno della cella
⇒ Dalla misura di FK, noti (tabulati) i fattori di forma atomici, si ricavano le posizioni atomiche
In realtà quasi mai si procede alla trasformazione di Fourier per ricavare la f(r), ma piuttosto si parte da un modello di proteina che viene raffinato tramite iterazioni successive fino a che le FK sperimentale e calcolata dal modello coincidono
O
ClCl-
K+
Fattori di forma atomici fi(k=0) = = carica elettronica totale
FK =�
i
f iKeiKri
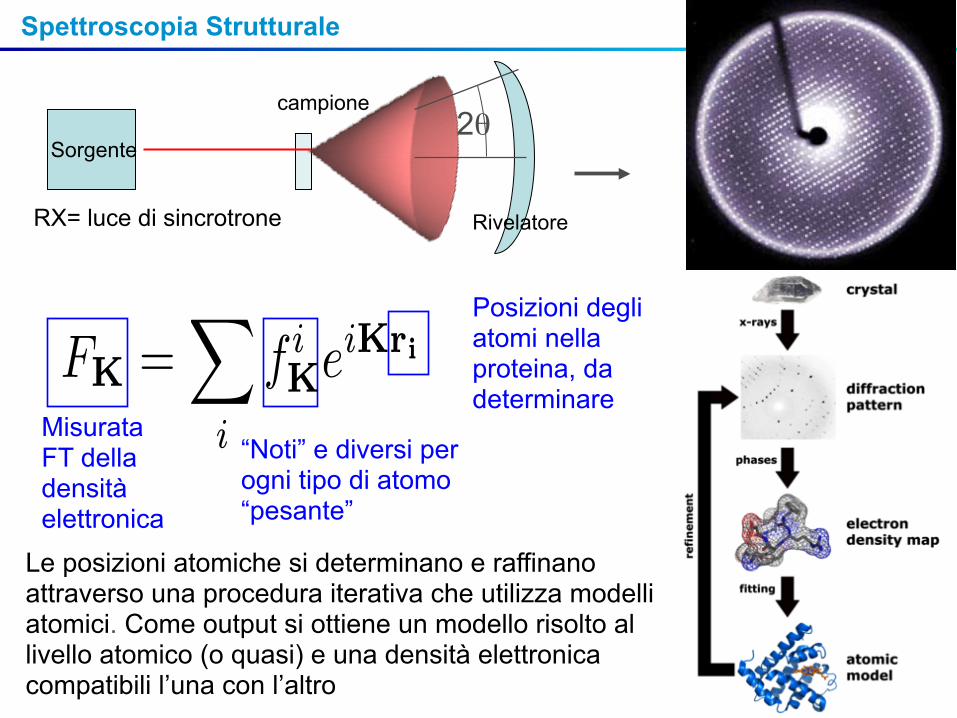
Spettroscopia Strutturale
Sorgente
campione
Rivelatore
2θ
MisurataFT della densitàelettronica
“Noti” e diversi per ogni tipo di atomo “pesante”
Posizioni degli atomi nella proteina, da determinare
Le posizioni atomiche si determinano e raffinano attraverso una procedura iterativa che utilizza modelli atomici. Come output si ottiene un modello risolto al livello atomico (o quasi) e una densità elettronica compatibili l’una con l’altro
RX= luce di sincrotrone
FK =�
i
f iKeiKri

Spettroscopia StrutturaleRisultati finali:❖Mappa elettronica e modello atomico della molecola a risoluzione di pochi Å❖La risoluzione dipende dalla purezza del cristallo e dalla temperatura: la posizione degli atomi è determinata entro le fluttuazioni termiche
Fattore di temperatura fattore di Debye-Waller
Problemi collegati alla tecnica1. La posizione degli atomi di H non è risolta perché questi non hanno sufficiente densità elettronica2. Le molecole vanno cristallizzate❖non tutte le molecole possono essere cristallizzate (ad es. le prot di membrana)❖le molecole cristallizzate hanno strutture “non naturali” e bloccate in una singola conformazione, dipendente dalle condizioni di cristallizzazione3. Problema dello scattering secondario dovuto all’interazione del fascio diffuso con ulteriori siti cristallini dopo il primo. Questo è un problema lieve con i raggi X che interagiscono debolmente con la materia, ma è un problema più grosso con gli elettroni4. Problema della fase: Il pattern di diffrazione in realtà fornisce solo il “modulo” del numero complesso F(k) (che si ricava dalla radice di I(θ)) con conseguente perdita di informazione. Se si adotta la procedura iterativa partendo da una struttura approssimata, questa perdita di informazione è compensata dalla conoscenza a priori sul sistema. Altrimenti, se si parte da zero, l’informazione sulla fase va recuperata con specifiche tecniche (sostituzione con “atomi pesanti”)
B =8π2
3�u2� fi(r) ∝ e−(r−ri)
2/2�u2�
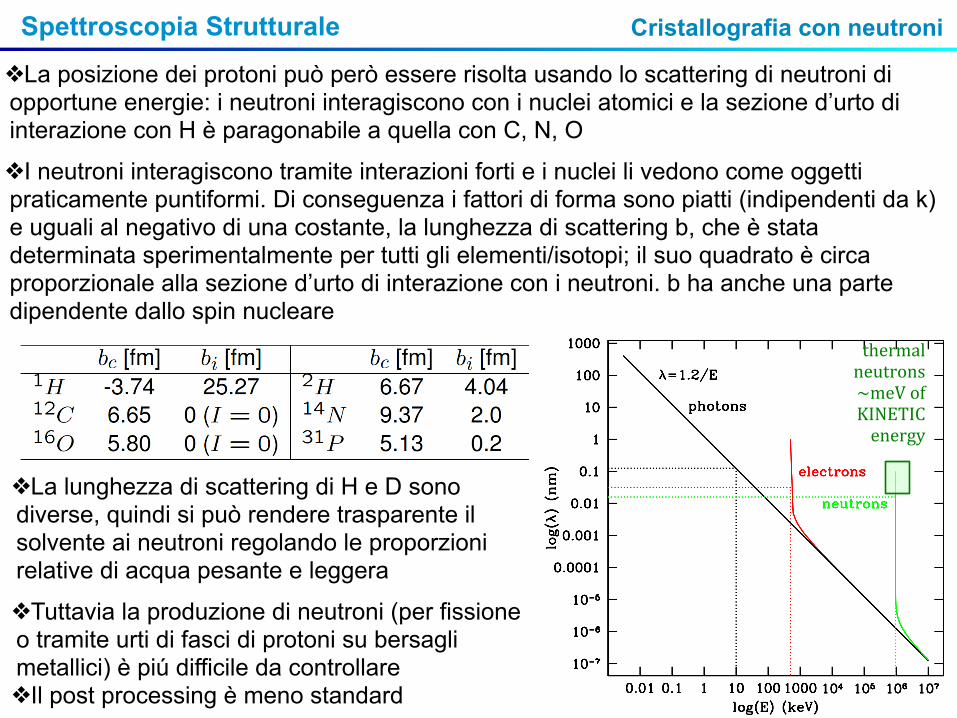
Spettroscopia Strutturale
❖La posizione dei protoni può però essere risolta usando lo scattering di neutroni di opportune energie: i neutroni interagiscono con i nuclei atomici e la sezione d’urto di interazione con H è paragonabile a quella con C, N, O
❖I neutroni interagiscono tramite interazioni forti e i nuclei li vedono come oggetti praticamente puntiformi. Di conseguenza i fattori di forma sono piatti (indipendenti da k) e uguali al negativo di una costante, la lunghezza di scattering b, che è stata determinata sperimentalmente per tutti gli elementi/isotopi; il suo quadrato è circa proporzionale alla sezione d’urto di interazione con i neutroni. b ha anche una parte dipendente dallo spin nucleare
❖La lunghezza di scattering di H e D sono diverse, quindi si può rendere trasparente il solvente ai neutroni regolando le proporzioni relative di acqua pesante e leggera
❖Tuttavia la produzione di neutroni (per fissione o tramite urti di fasci di protoni su bersagli metallici) è piú difficile da controllare❖Il post processing è meno standard
thermal neutrons~meV of KINETIC energy
Cristallografia con neutroni

Spettroscopia Strutturale Tecniche rX per molecole non cristallizzate
Diffrazione da fibre cristalline: ❖Il campione è composto da microcristalli di fibre allungate allineate lungo l’asse, ma casualmente ruotati lungo l’asse. Questa è la forma tipica con cui si riesce a cristallizzare il DNA (Rosalind Franklin)❖Il pattern di diffrazione è mediato a causa della perdita di ordine rotazionale→proiezione 2D dello spazio reciproco 3D❖ è ancora possibile estrarre informazione sufficiente per determinare le posizioni atomiche soprattutto se c’è sufficiente conoscenza a priori sul sistema
Diffusione “ad angolo largo” da soluzioni diluite: È possibile ottenere informazioni anche da molecole in soluzione, raccogliendo idealmente il segnale diffusivo da singole molecole. In questo caso❖È necessario raccogliere radiazione diffusa su largo angolo perché il segnale è piú debole e piú sparso❖Data l’orientazione casuale delle molecole, il pattern di interferenza è 1D e formato dall’interferenza dalla “singola” molecola❖Il post processing dei dati è piú difficile, procede attraverso trasformate di Fourier con simmetria radiale e si ricavano solo dati a bassa risoluzione; è necessario partire da una struttura già parzialmente nota da ottimizzare…… Ma si possono osservare strutture naturali e cambiamenti conformazionali
Diffrazione da polveri micro-cristalline In questo caso l’orientazione varia nelle tre direzioni spaziali e quindi il pattern di diffrazione va mediato e proiettato su una singola direzione (pattern 1D, simmetrico per rotazione intorno all’asse del fascio incidente) Diffrazione a piccolo angolo, consente di avere dati con esposizone breve e quindi avere misure risolte in tempo. Si ottengono dati a bassissima risoluzione
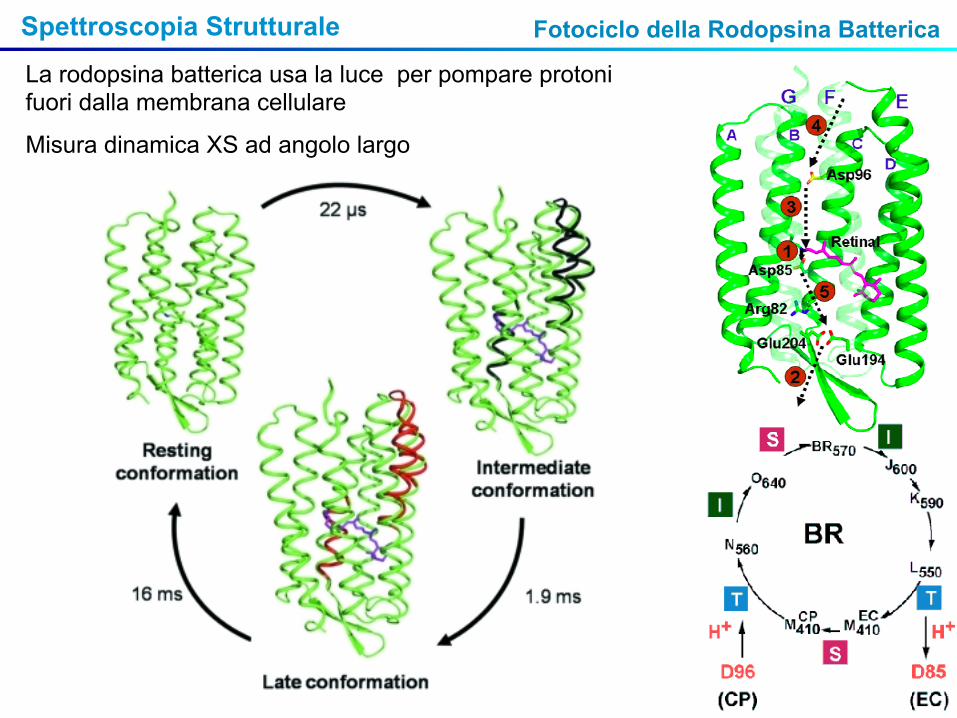
Spettroscopia Strutturale Fotociclo della Rodopsina Batterica
La rodopsina batterica usa la luce per pompare protoni fuori dalla membrana cellulare
Misura dinamica XS ad angolo largo
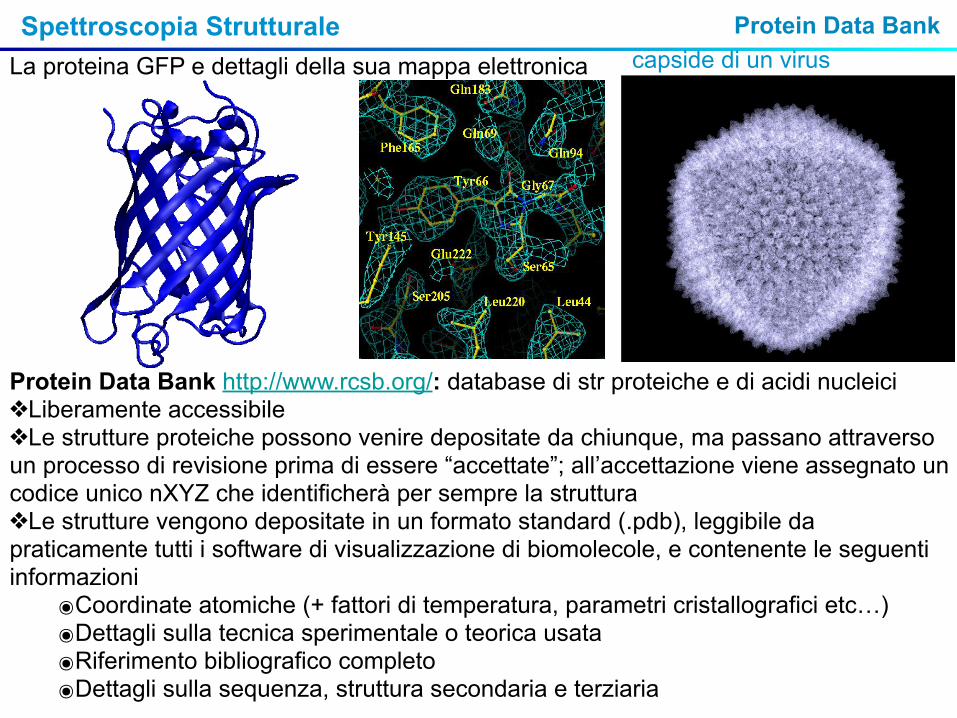
Spettroscopia Strutturale
Protein Data Bank http://www.rcsb.org/: database di str proteiche e di acidi nucleici❖Liberamente accessibile❖Le strutture proteiche possono venire depositate da chiunque, ma passano attraverso un processo di revisione prima di essere “accettate”; all’accettazione viene assegnato un codice unico nXYZ che identificherà per sempre la struttura❖Le strutture vengono depositate in un formato standard (.pdb), leggibile da praticamente tutti i software di visualizzazione di biomolecole, e contenente le seguenti informazioni
๏Coordinate atomiche (+ fattori di temperatura, parametri cristallografici etc…)๏Dettagli sulla tecnica sperimentale o teorica usata๏Riferimento bibliografico completo๏Dettagli sulla sequenza, struttura secondaria e terziaria
La proteina GFP e dettagli della sua mappa elettronica capside di un virusProtein Data Bank

Spettroscopia Strutturale Esempio: 1GFL
La proteina verde fluorescente (GFP) è stata cristallizzata e risolta alla risoluzione di 1.9Å in forma dimerica nel 1996
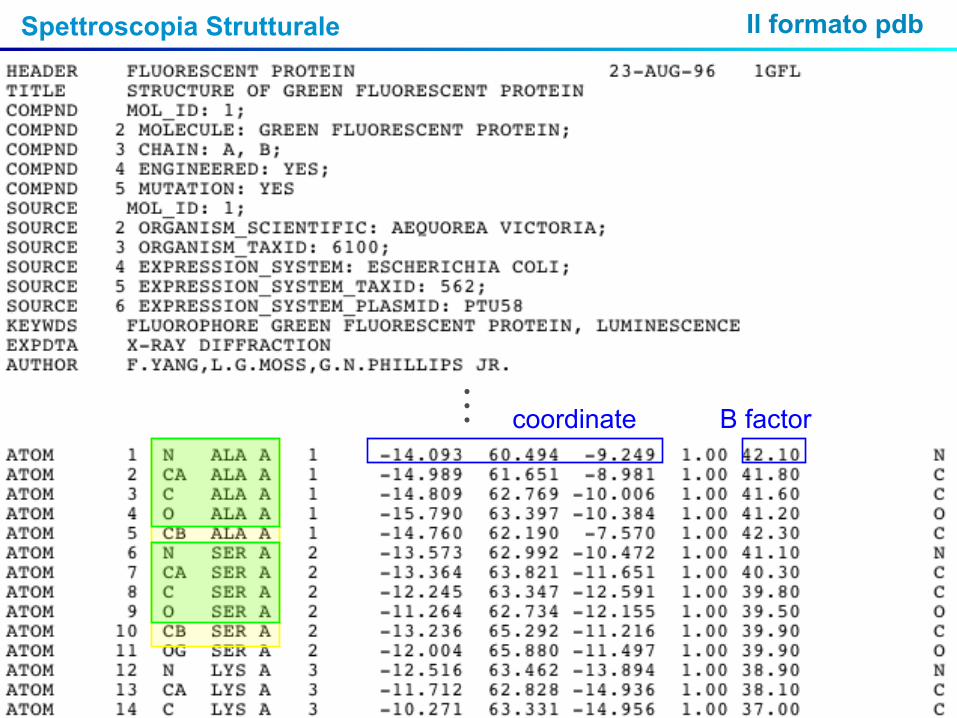
Spettroscopia Strutturale Il formato pdb
…
coordinate B factor
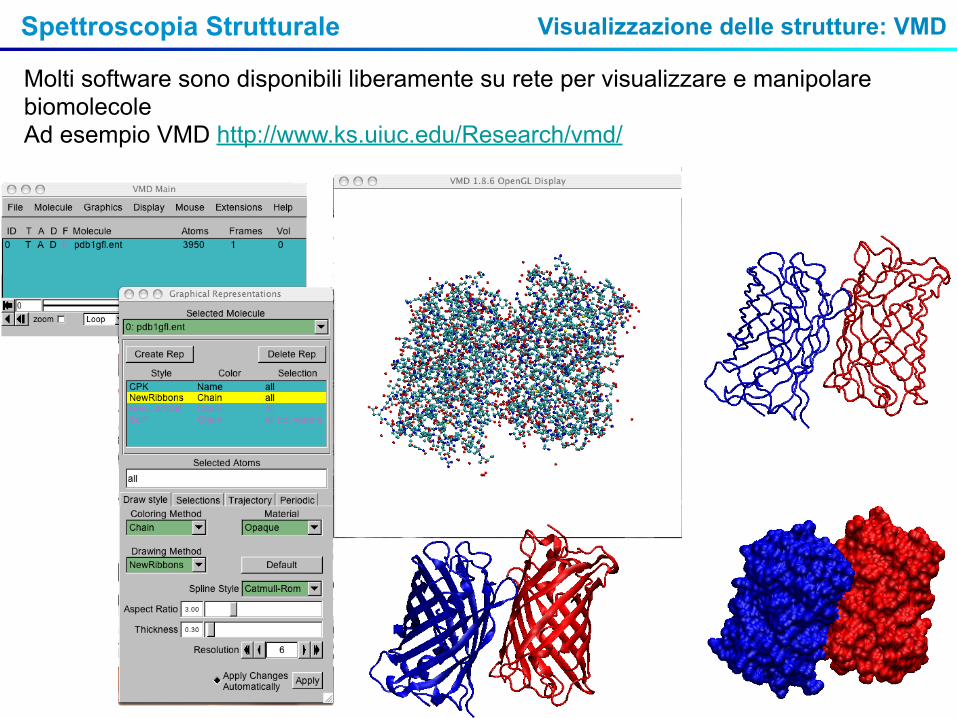
Spettroscopia Strutturale Visualizzazione delle strutture: VMD
Molti software sono disponibili liberamente su rete per visualizzare e manipolare biomolecole Ad esempio VMD http://www.ks.uiuc.edu/Research/vmd/
FK = F (k)�
j
δk,Kj = FT [f ]× FT [{R}]ρ(r) =
�
i
f(r −Ri)
ρ(r) = f(r)
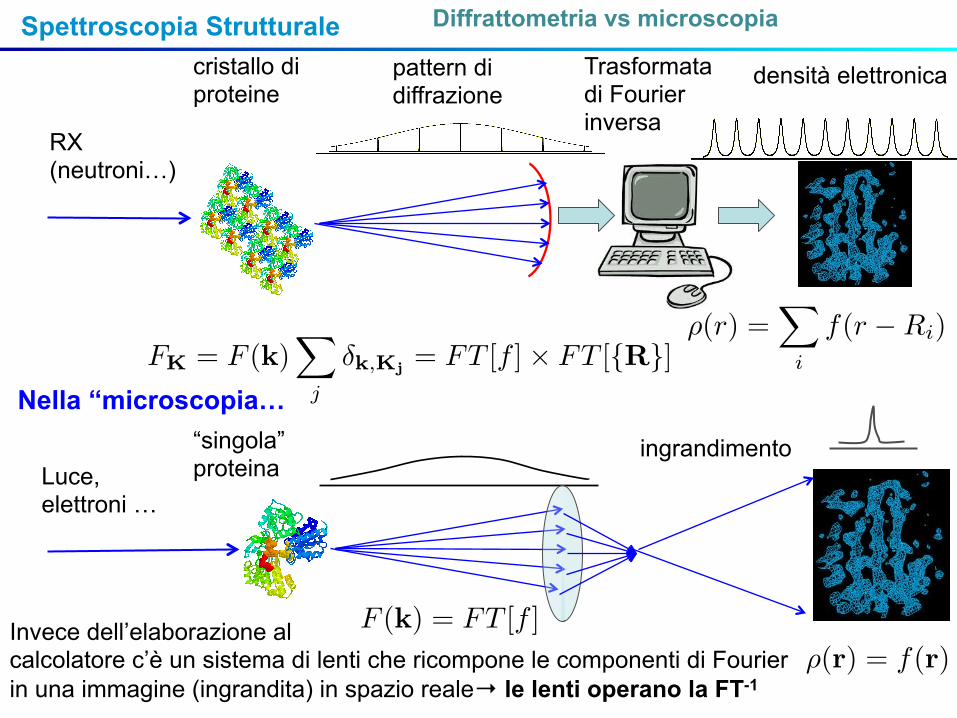
Spettroscopia Strutturale Diffrattometria vs microscopia
RX (neutroni…)
cristallo di proteine
pattern di diffrazione
densità elettronicaTrasformata di Fourier inversa
Luce, elettroni …
“singola” proteina
Nella “microscopia…
ingrandimento
Invece dell’elaborazione alcalcolatore c’è un sistema di lenti che ricompone le componenti di Fourier in una immagine (ingrandita) in spazio reale→ le lenti operano la FT-1
F (k) = FT [f ]
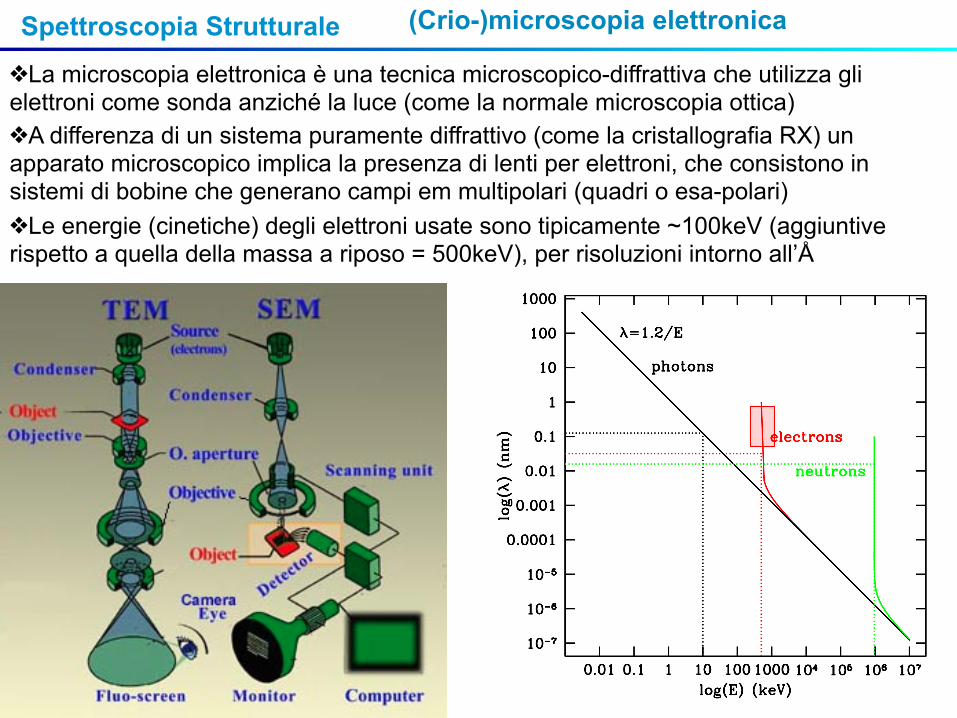
Spettroscopia Strutturale (Crio-)microscopia elettronica
❖La microscopia elettronica è una tecnica microscopico-diffrattiva che utilizza gli elettroni come sonda anziché la luce (come la normale microscopia ottica)❖A differenza di un sistema puramente diffrattivo (come la cristallografia RX) un apparato microscopico implica la presenza di lenti per elettroni, che consistono in sistemi di bobine che generano campi em multipolari (quadri o esa-polari)❖Le energie (cinetiche) degli elettroni usate sono tipicamente ~100keV (aggiuntive rispetto a quella della massa a riposo = 500keV), per risoluzioni intorno all’Å
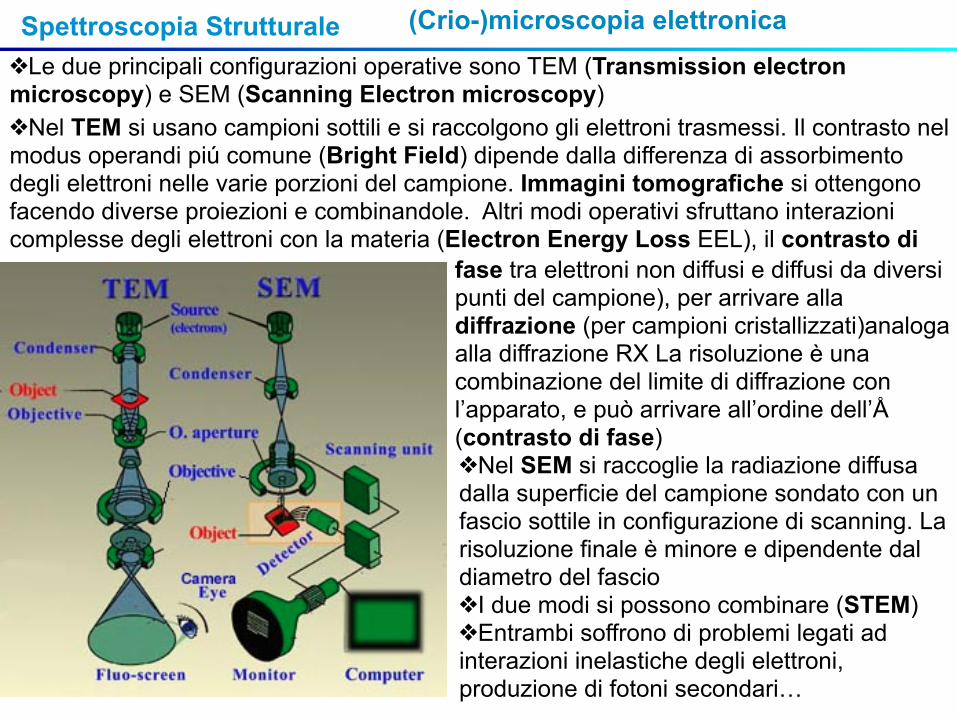
Spettroscopia Strutturale (Crio-)microscopia elettronica❖Le due principali configurazioni operative sono TEM (Transmission electron microscopy) e SEM (Scanning Electron microscopy)❖Nel TEM si usano campioni sottili e si raccolgono gli elettroni trasmessi. Il contrasto nel modus operandi piú comune (Bright Field) dipende dalla differenza di assorbimento degli elettroni nelle varie porzioni del campione. Immagini tomografiche si ottengono facendo diverse proiezioni e combinandole. Altri modi operativi sfruttano interazioni complesse degli elettroni con la materia (Electron Energy Loss EEL), il contrasto di fase tra elettroni non diffusi e diffusi da diversi
punti del campione), per arrivare alla diffrazione (per campioni cristallizzati)analoga alla diffrazione RX La risoluzione è una combinazione del limite di diffrazione con l’apparato, e può arrivare all’ordine dell’Å (contrasto di fase)❖Nel SEM si raccoglie la radiazione diffusa dalla superficie del campione sondato con un fascio sottile in configurazione di scanning. La risoluzione finale è minore e dipendente dal diametro del fascio❖I due modi si possono combinare (STEM)❖Entrambi soffrono di problemi legati ad interazioni inelastiche degli elettroni, produzione di fotoni secondari…

Spettroscopia Strutturale❖Gli elettroni interagiscono con la densità elettronica della molecola bersaglio; siccome sono facilmente assorbiti (anche dall’aria), il campione va tenuto sotto vuoto❖Alle energie minime necessarie per raggiungere una risoluzione adeguata gli elettroni sono già dannosi per i campioni biologici. Per questo motivo e per aumentare il contrasto e la stabilità i campioni sono talvolta ricoperti da film metallici sottili, o trattati con specifiche sostanze per aumentare il contrasto. Però cosí si altera la loro struttura❖Alternativamente, si usano energie minori e campioni idratati, ma a temperatura criogenica (in azoto (77K) o metano liquido(112K)) ⇒ crio-microscopia elettronica❖La risoluzione può venire aumentata fissando le molecole in matrici bidimensionali su substrati❖Le tecniche di risoluzione dell’immagine e costruzione del modello sono simili a quelle della diffrazione da campioni non cristallinila risoluzione ottenibile è minore (5-10Å), e quindile mappe elettroniche ottenute sono piú grossolane,spesso sufficienti a localizzare solo i Cα o i P del fosfato negli acidi nucleici❖Tecnica adatta per analizzare oggetti molto grandi (da aggregati macromolecolari a virus) in un ambiente quasi fisiologico❖La risoluzione può essere aumentata se esistono modelli ad alta risoluzione delle singole parti da fittare nella mappa elettronica a bassa risoluzione
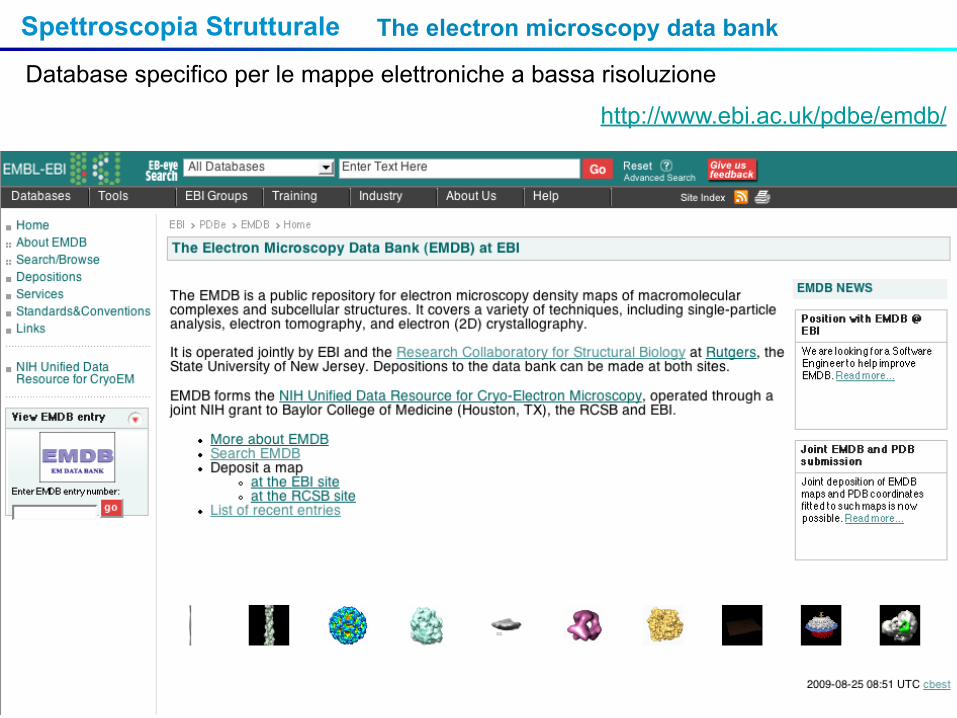
Spettroscopia Strutturale
http://www.ebi.ac.uk/pdbe/emdb/
The electron microscopy data bank
Database specifico per le mappe elettroniche a bassa risoluzione
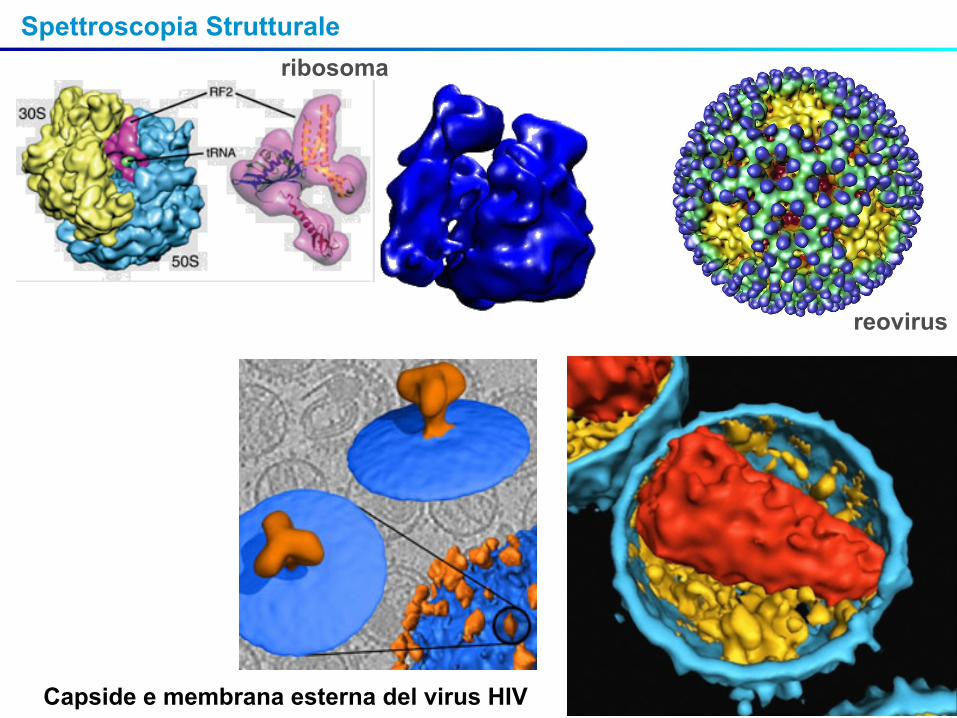
Spettroscopia Strutturaleribosoma
reovirus
Capside e membrana esterna del virus HIV

Spettroscopia Strutturale Spettroscopia strutturale con NMR
❖La spettroscopia a risonanza magnetica nucleare (NMR) non necessita che le molecole siano cristallizzate, e anzi è adatta ad analizzarle in soluzione acquosa❖La tecnica si basa sull’interazione tra il momento magnetico associato ad un nucleo e un campo magnetico esterno ⇒ La molecola deve avere nuclei con spin non nullo (numero di protoni o neutroni dispari)❖I nuclei piú usati sono H, 13C, 15N, 31P, con spin = ½❖Occasionalmente anche 14N (spin 1)❖In presenza di campo magnetico esterno B0 il momento magnetico del nucleo precede alla frequenza di Larmor ωL=B0 γ❖Il rapporto giromagnetico γ=gZe/2Mè il rapporto tra momento magnetico e momento angolare (spin), ed è specifico per ogni nucleo.
γ (rad/ gauss sec)
ωL0/ 2π
(MHz/T)Sensibilitàrelativa (ad H)e 1.76 104 2.80 104
1H 26.7 42.6 1.000
2H 4.1 6.5 0.0096
13C 6.7 10.7 0.016
14N 1.9 3.07 0.001
31P 10.8 17.2 0.066
g particelle classiche = 1 gelectron~-2 (+correzioni di mecc quantistica dei campi )gproton~5.6 gneutron~ -3.8 …
Spettroscopia Strutturale
zx
y
Bxy
ω
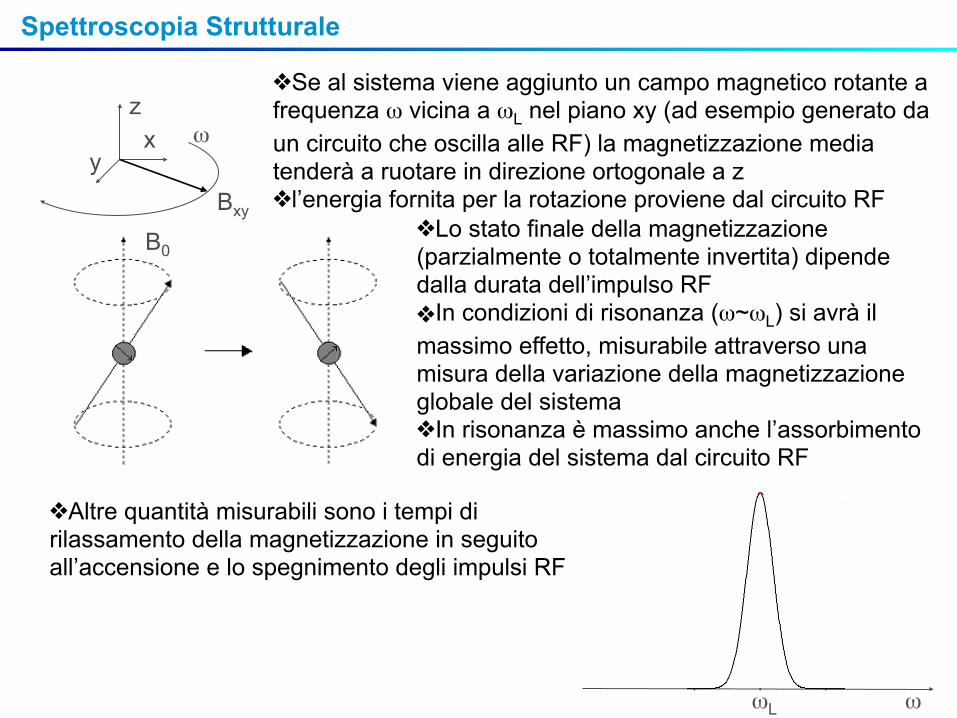
❖Se al sistema viene aggiunto un campo magnetico rotante a frequenza ω vicina a ωL nel piano xy (ad esempio generato da un circuito che oscilla alle RF) la magnetizzazione media tenderà a ruotare in direzione ortogonale a z ❖l’energia fornita per la rotazione proviene dal circuito RF
B0❖Lo stato finale della magnetizzazione (parzialmente o totalmente invertita) dipende dalla durata dell’impulso RF❖In condizioni di risonanza (ω~ωL) si avrà il massimo effetto, misurabile attraverso una misura della variazione della magnetizzazione globale del sistema❖In risonanza è massimo anche l’assorbimento di energia del sistema dal circuito RF
ωL ω
❖Altre quantità misurabili sono i tempi di rilassamento della magnetizzazione in seguito all’accensione e lo spegnimento degli impulsi RF
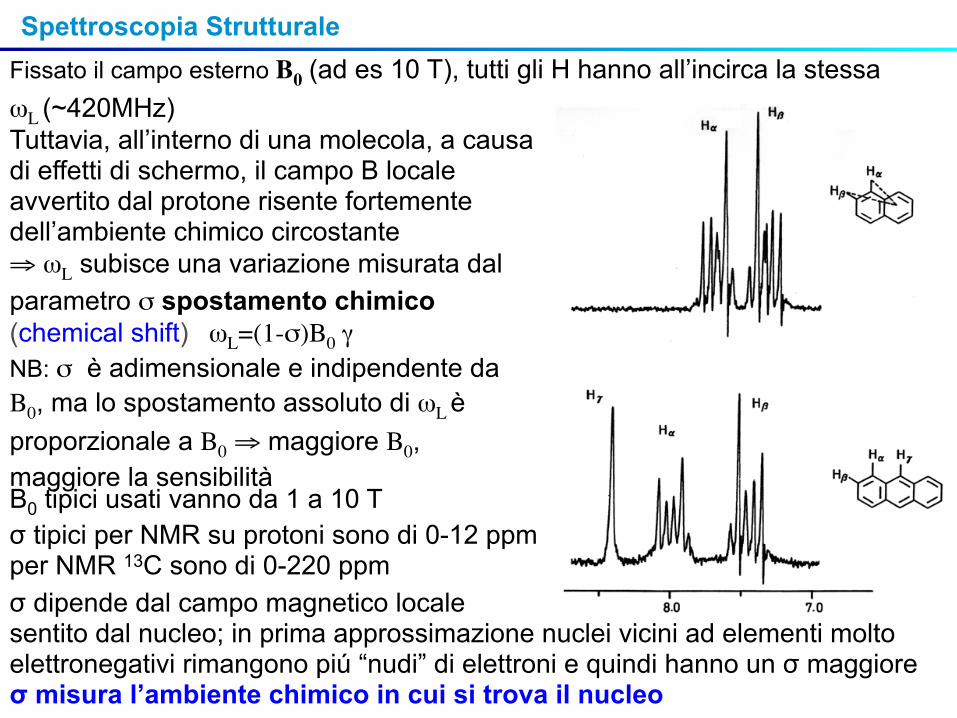
Spettroscopia StrutturaleFissato il campo esterno B0 (ad es 10 T), tutti gli H hanno all’incirca la stessa ωL (~420MHz)
ωL=(1-σ)B0 γNB: σ è adimensionale e indipendente da B0, ma lo spostamento assoluto di ωL è proporzionale a B0 ⇒ maggiore B0, maggiore la sensibilità
Tuttavia, all’interno di una molecola, a causa di effetti di schermo, il campo B locale avvertito dal protone risente fortemente dell’ambiente chimico circostante ⇒ ωL subisce una variazione misurata dal parametro σ spostamento chimico (chemical shift)
B0 tipici usati vanno da 1 a 10 Tσ tipici per NMR su protoni sono di 0-12 ppmper NMR 13C sono di 0-220 ppmσ dipende dal campo magnetico localesentito dal nucleo; in prima approssimazione nuclei vicini ad elementi molto elettronegativi rimangono piú “nudi” di elettroni e quindi hanno un σ maggioreσ misura l’ambiente chimico in cui si trova il nucleo

Spettroscopia Strutturale
Chemical shift dei diversi atomi C nei 20 amminoacidi: il picco principale di ciascuna distribuzione fornisce informazioni grossolane, come il tipo di atomo (Cα o Cβ etc) e a quale amminoacido appartiene
Spettroscopia Strutturale
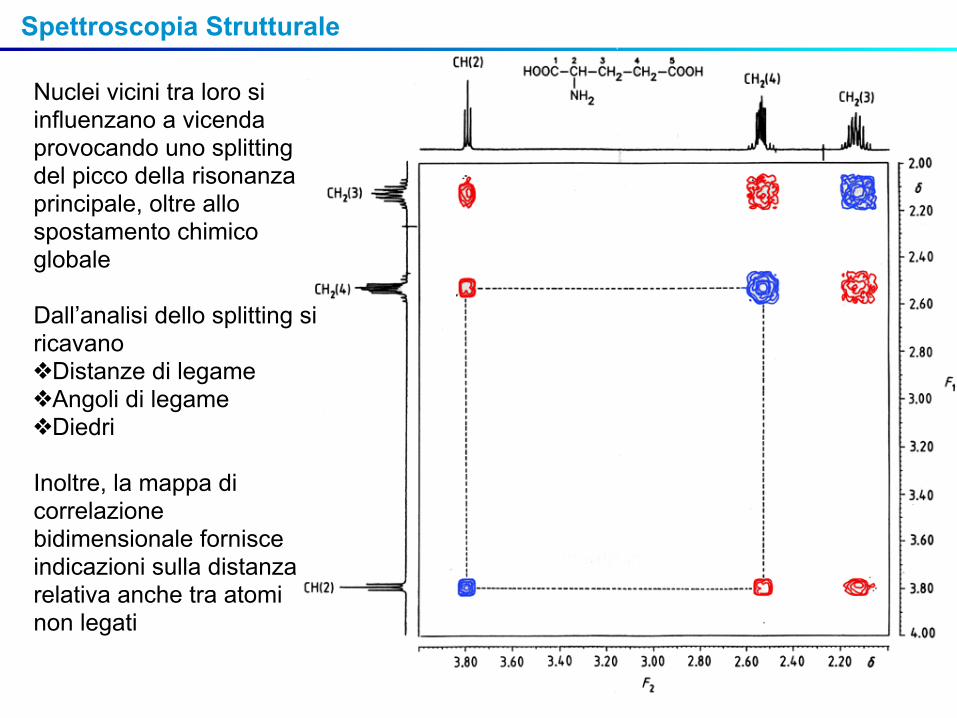
Nuclei vicini tra loro si influenzano a vicenda provocando uno splitting del picco della risonanza principale, oltre allo spostamento chimico globale
Dall’analisi dello splitting si ricavano ❖Distanze di legame❖Angoli di legame❖Diedri
Inoltre, la mappa di correlazione bidimensionale fornisce indicazioni sulla distanza relativa anche tra atomi non legati
Spettroscopia Strutturale
In conclusione
❖dall’analisi di spettri e mappe NMR si ottengono vincoli su distanze e angoli di legame e distanze tra atomi non legati❖Se questi vincoli sono in numero sufficiente rispetto agli atomi della struttura, è possibile inserirli in un modello che poi viene iterativamente ottimizzato fino a che i vincoli stessi non sono soddisfatti❖Questa procedura non dà come risultato un’unica struttura, ma un insieme di modelli ❖Le differenze strutturali tra i modelli riflettono
๏La reale libertà conformazionale di una molecola in soluzione๏Le fluttuazioni termiche๏L’errore sperimentale
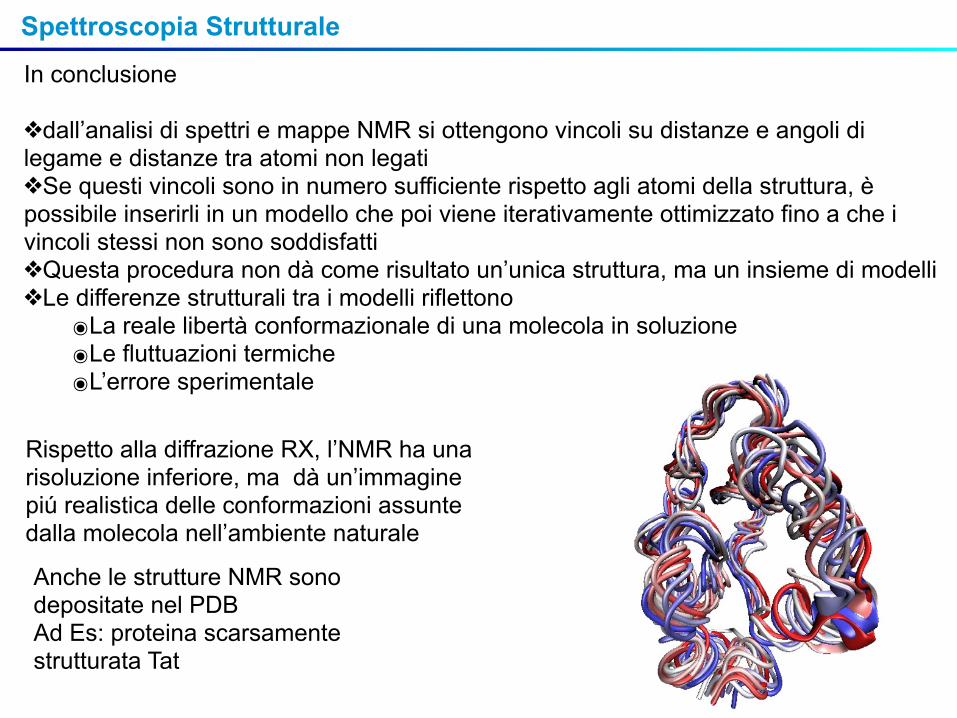
Rispetto alla diffrazione RX, l’NMR ha una risoluzione inferiore, ma dà un’immagine piú realistica delle conformazioni assunte dalla molecola nell’ambiente naturale
Anche le strutture NMR sono depositate nel PDBAd Es: proteina scarsamente strutturata Tat
Spettroscopia Strutturale
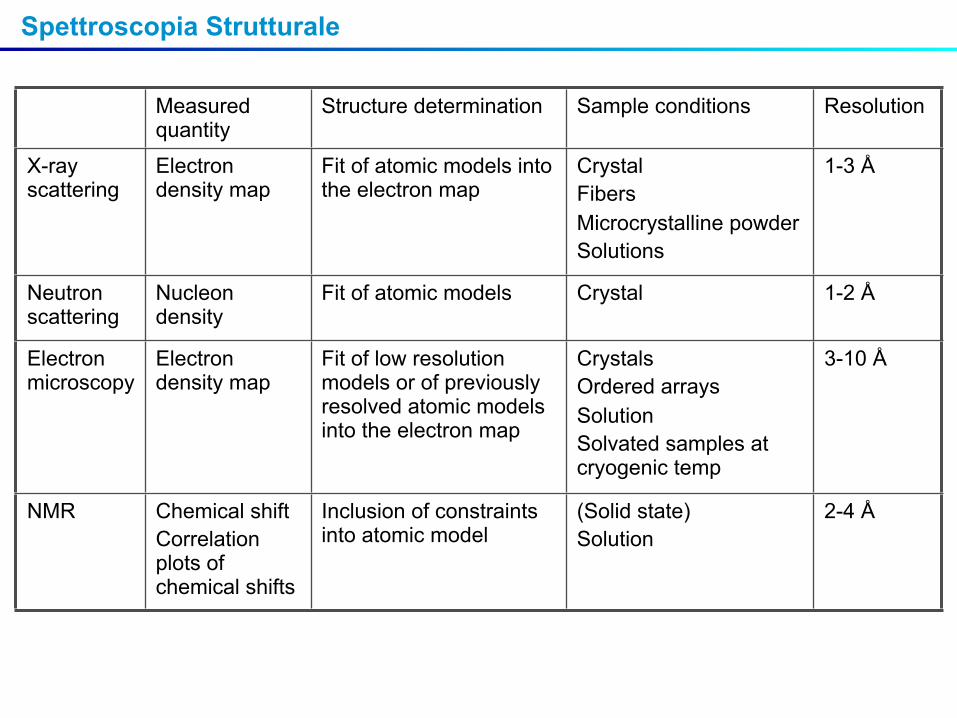
Measured quantity
Structure determination Sample conditions Resolution
X-ray scattering
Electron density map
Fit of atomic models into the electron map
CrystalFibersMicrocrystalline powderSolutions
1-3 Å
Neutron scattering
Nucleon density
Fit of atomic models Crystal 1-2 Å
Electron microscopy
Electron density map
Fit of low resolution models or of previously resolved atomic models into the electron map
CrystalsOrdered arraysSolutionSolvated samples at cryogenic temp
3-10 Å
NMR Chemical shift Correlation plots of chemical shifts
Inclusion of constraints into atomic model
(Solid state)Solution
2-4 Å
Spettroscopia Strutturale
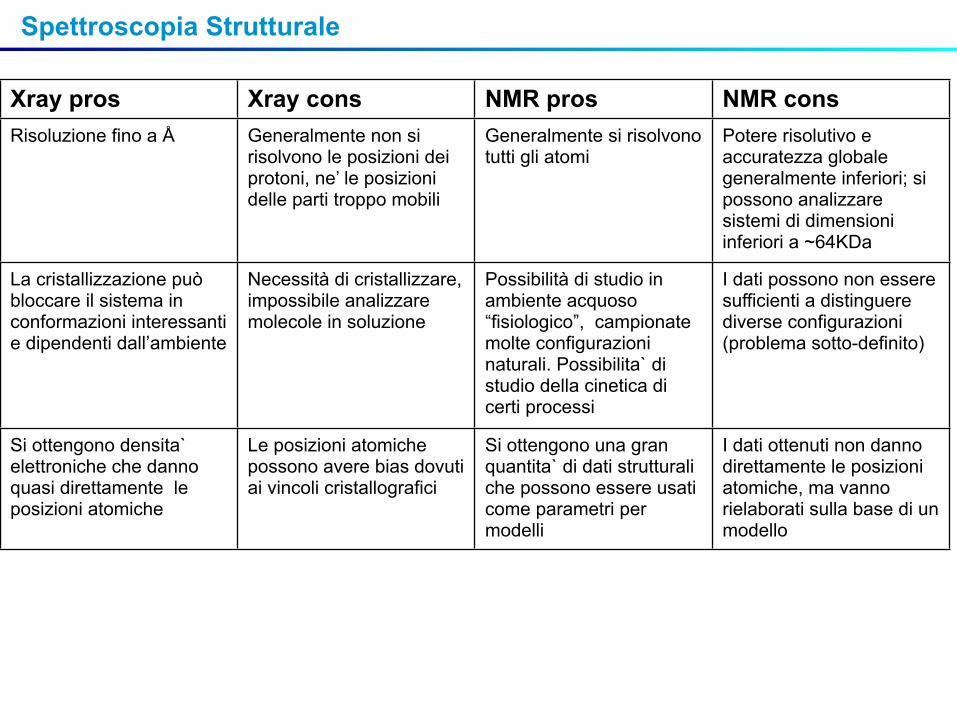
Xray pros Xray cons NMR pros NMR consRisoluzione fino a Å Generalmente non si
risolvono le posizioni dei protoni, ne’ le posizioni delle parti troppo mobili
Generalmente si risolvono tutti gli atomi
Potere risolutivo e accuratezza globale generalmente inferiori; si possono analizzare sistemi di dimensioni inferiori a ~64KDa
La cristallizzazione può bloccare il sistema in conformazioni interessanti e dipendenti dall’ambiente
Necessità di cristallizzare, impossibile analizzare molecole in soluzione
Possibilità di studio in ambiente acquoso “fisiologico”, campionate molte configurazioni naturali. Possibilita` di studio della cinetica di certi processi
I dati possono non essere sufficienti a distinguere diverse configurazioni (problema sotto-definito)
Si ottengono densita` elettroniche che danno quasi direttamente le posizioni atomiche
Le posizioni atomiche possono avere bias dovuti ai vincoli cristallografici
Si ottengono una gran quantita` di dati strutturali che possono essere usati come parametri per modelli
I dati ottenuti non danno direttamente le posizioni atomiche, ma vanno rielaborati sulla base di un modello


















