
Sequenziamento Esomico. Maria Valentini (CRS4), Cagliari, 18 Novembre 2015
33
Sequenziamento esomico: analisi dati e casi di studio Maria Valentini 18 Novembre 2015 1
-
Upload
crs4-research-center-in-sardinia -
Category
Science
-
view
858 -
download
1
Transcript of Sequenziamento Esomico. Maria Valentini (CRS4), Cagliari, 18 Novembre 2015

Sequenziamento esomico: analisi dati e casi di studio
Maria Valentini 18 Novembre 2015
1

• Cosa e’ l’esoma, come si studia e perche’.
• Cosa e’ una variante, quante ce ne sono e il genoma di riferimento
• Next Generation Sequencing e analisi dei dati generati.
• Caso studio 1
• Caso studio 2
• Conclusioni
2
Introduzione
3
Cosa e’ l’esoma
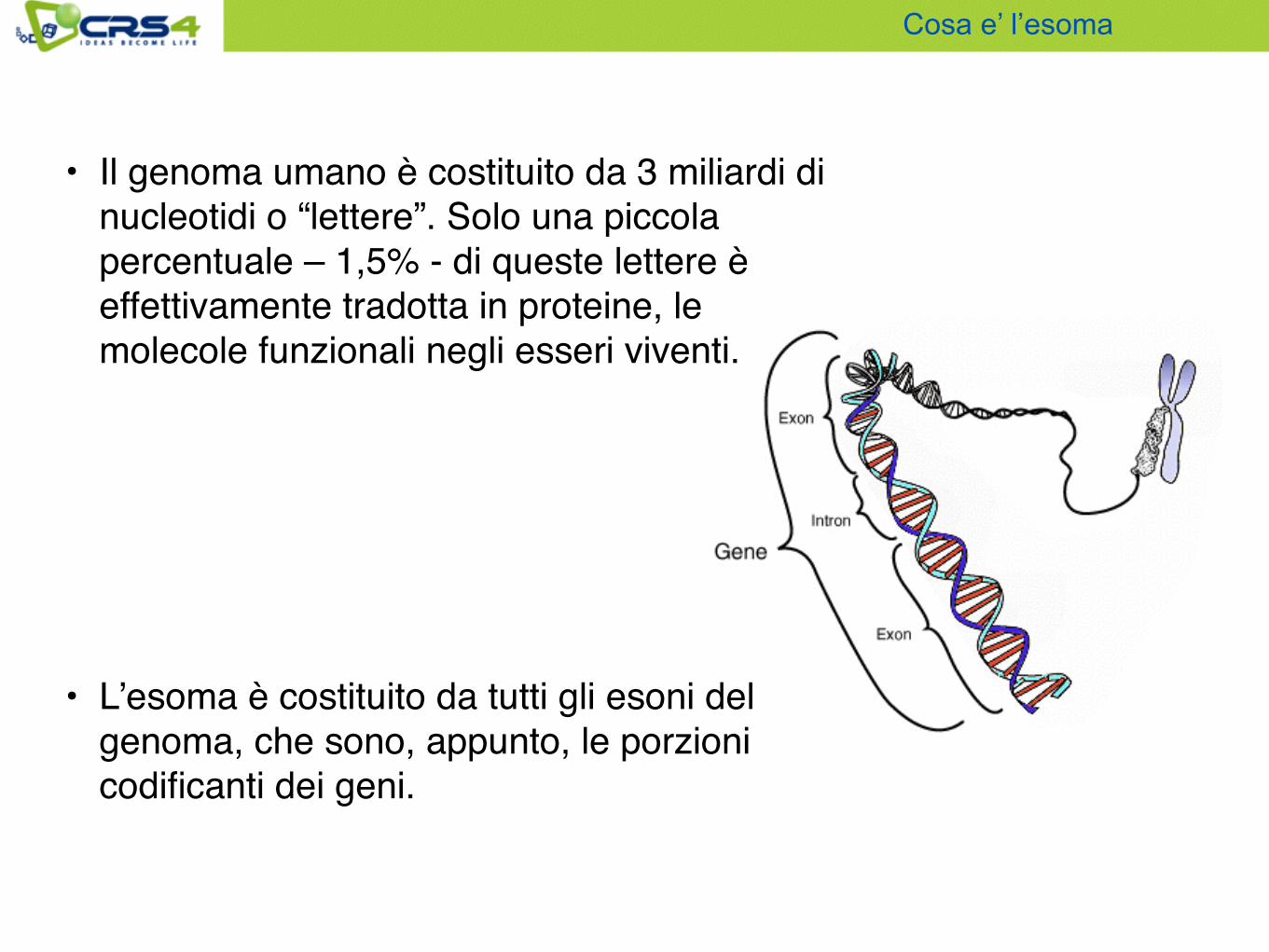
• Il genoma umano è costituito da 3 miliardi di nucleotidi o “lettere”. Solo una piccola percentuale – 1,5% - di queste lettere è effettivamente tradotta in proteine, le molecole funzionali negli esseri viventi.
• L’esoma è costituito da tutti gli esoni del genoma, che sono, appunto, le porzioni codificanti dei geni.

4
Cosa e’ l’exome sequencing
Exome sequencing (o Whole-Exome Sequencing - WES) è il sequenziamento dell'intera regione codificante del genoma di un individuo.
Il termine esone deriva da “regione espressa” (o ‘exon’, EXpressed regiON), poiché sono queste le regioni che vengono tradotte, o espresse come proteine. L’esoma e’ la porzione di genoma che codifica le proteine.

5
Perche’ l’exome sequencing
Ciò è di estremo interesse sia per la dignostica di routine che per la ricerca scientifica. Infatti, anche se la regione codificante rappresenta soltanto l'1% di tutto il genoma, si stima che fino all'85% di tutte le mutazioni patogene siano contenute in questa regione.
Identificando le varianti di un gene possiamo capire se la proteina e’ “mal funzionante” e fare delle ipotesi sui meccanismi biologici che provocano la malattia!
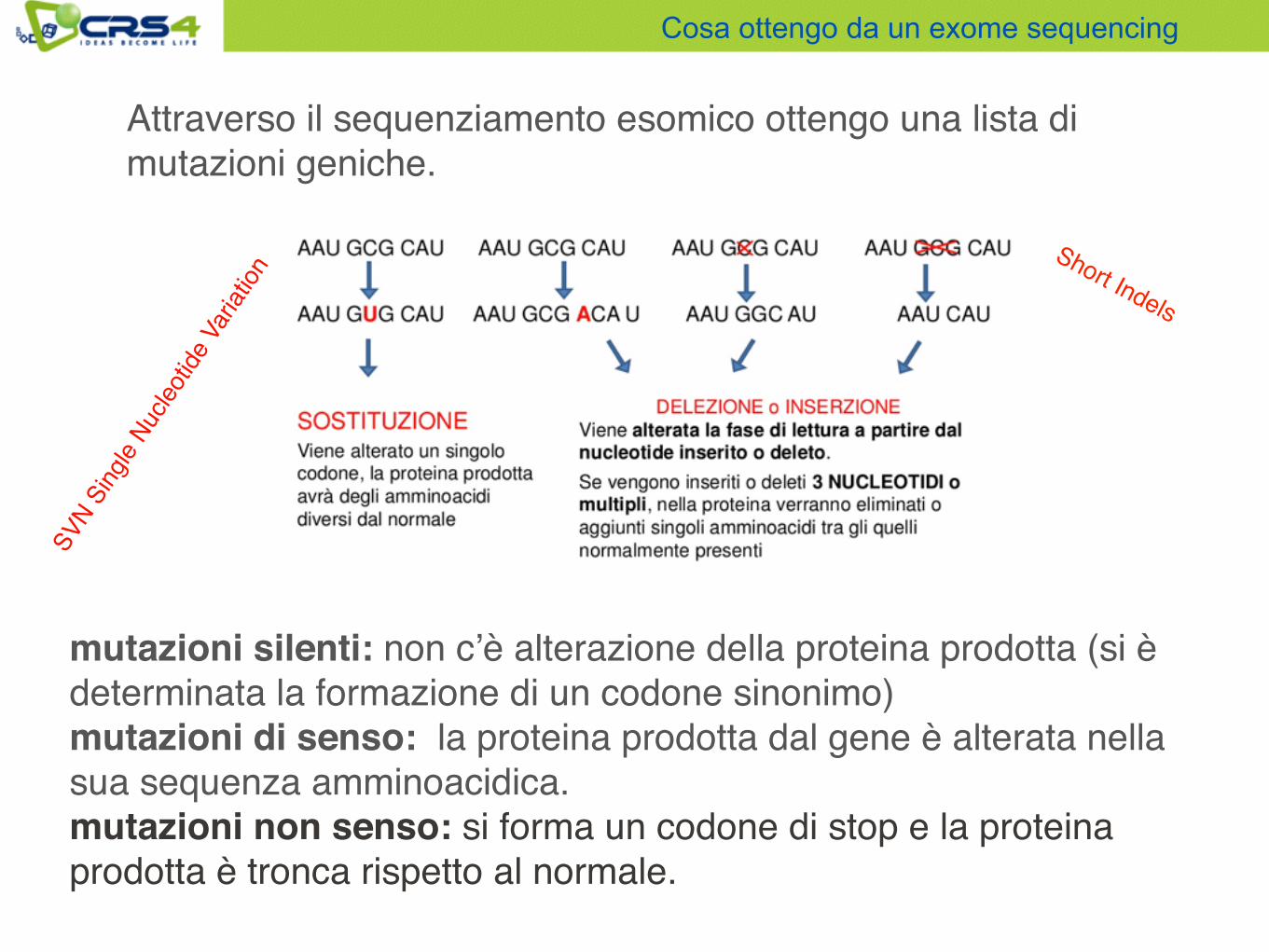
Attraverso il sequenziamento esomico ottengo una lista di mutazioni geniche.
6
Cosa ottengo da un exome sequencing
mutazioni silenti: non c’è alterazione della proteina prodotta (si è determinata la formazione di un codone sinonimo)mutazioni di senso: la proteina prodotta dal gene è alterata nella sua sequenza amminoacidica.mutazioni non senso: si forma un codone di stop e la proteina prodotta è tronca rispetto al normale.
SVN
Single
Nuc
leotid
e Vari
ation
Short Indels
7
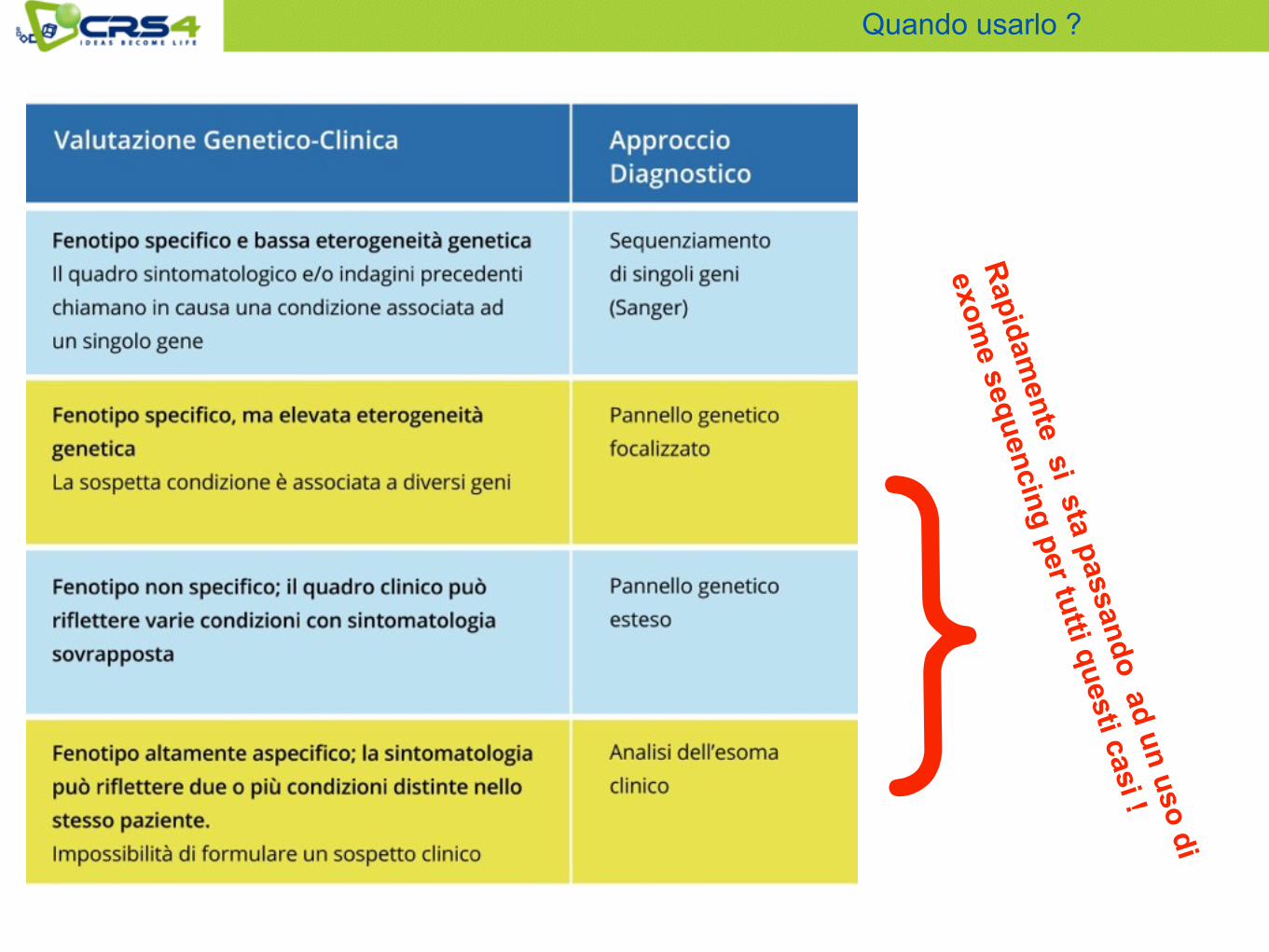
Quando usarlo ?
}R
apidamente si sta passando ad un uso di
exome sequencing per tutti questi casi !

8
Vi sono situazioni cliniche in cui la valutazione dell’intero esoma è praticamente l’unica strada percorribile! • Condizioni patologiche in cui è probabile un’elevata eterogeneità
genetica e variabilità fenotipica (numerosi geni candidati e/o sintomatologia riconducibile a una tra diverse condizioni);
• Quadro sintomatologico compatibile con la presenza di più condizioni genetiche distinte nello stesso paziente;
• Situazioni compatibili con la presenza di mutazioni de novo o comunque non ereditate attraverso la linea germinale parentale (assenza di altri familiari affetti, assenza di specifiche mutazioni patogeniche nei genitori, ecc);
Quando e’ inevitabile usarlo?

Fino a pochi anni fa il test genetico per eccellenza consisteva nel sequenziamento di singolo gene (o di un ristretto gruppo di geni) tramite la metodica dell'elettroforesi capillare o sequenziamento Sanger.
9
L’avvento delle nuove tecnologie di sequenziamento (Next Generation Sequencing - NGS o High-Throughput sequencing) ha trasformato lo studio della genetica delle malattie umane portando ad un'epoca di produttività senza precedenti.
Grazie ai costi e ai tempi ridotti, tramite NGS è possibile analizzare un elevato numero di frammenti di DNA in parallelo fino ad ottenere la sequenza dell'intera regione codificante di un individuo o dell’intero genoma.
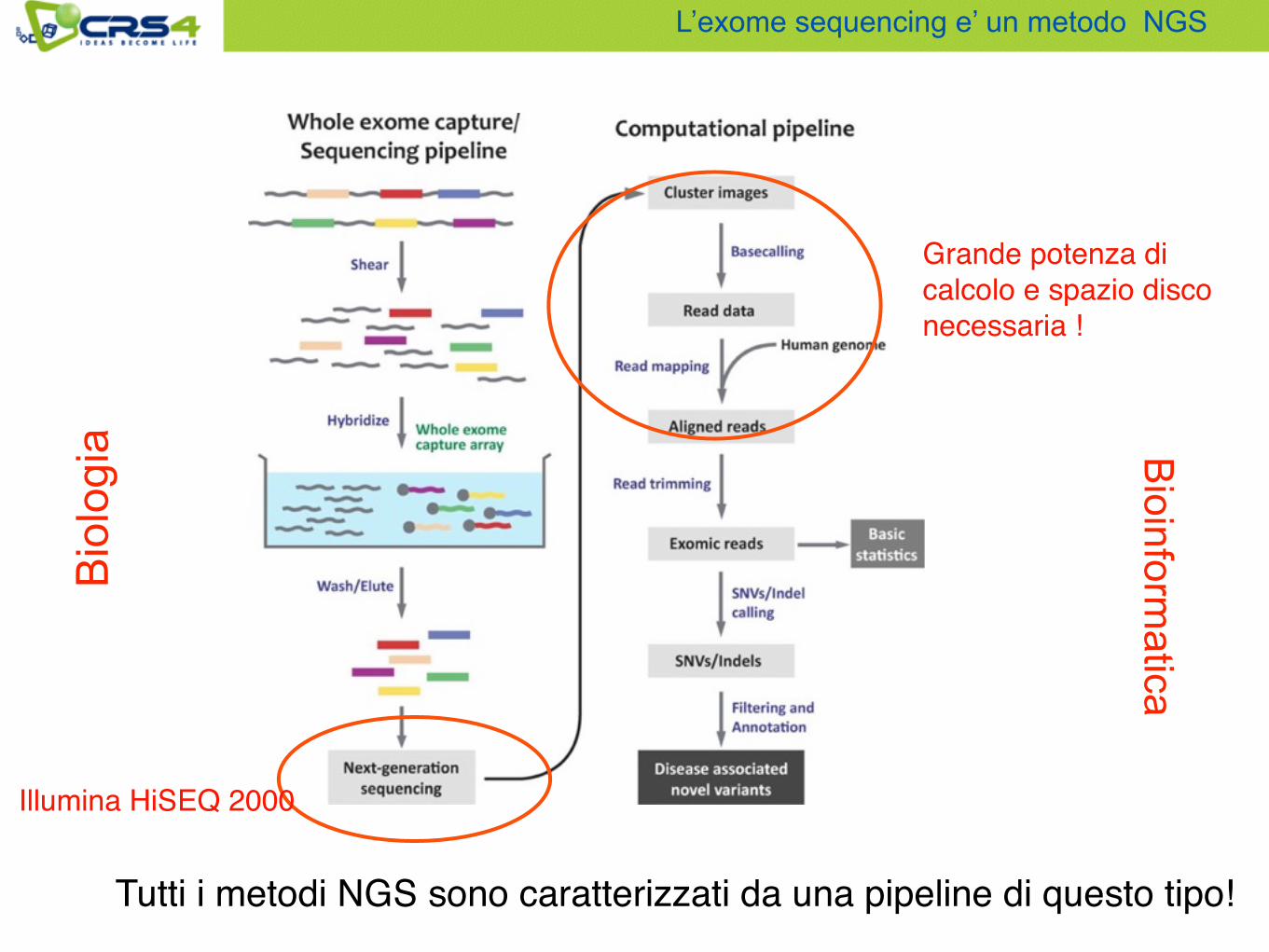
Metodi Next Generation Sequencing
10
L’exome sequencing e’ un metodo NGS
Tutti i metodi NGS sono caratterizzati da una pipeline di questo tipo!
Grande potenza di calcolo e spazio disco necessaria !
Bio
logi
a Bioinform
atica
Illumina HiSEQ 2000
11
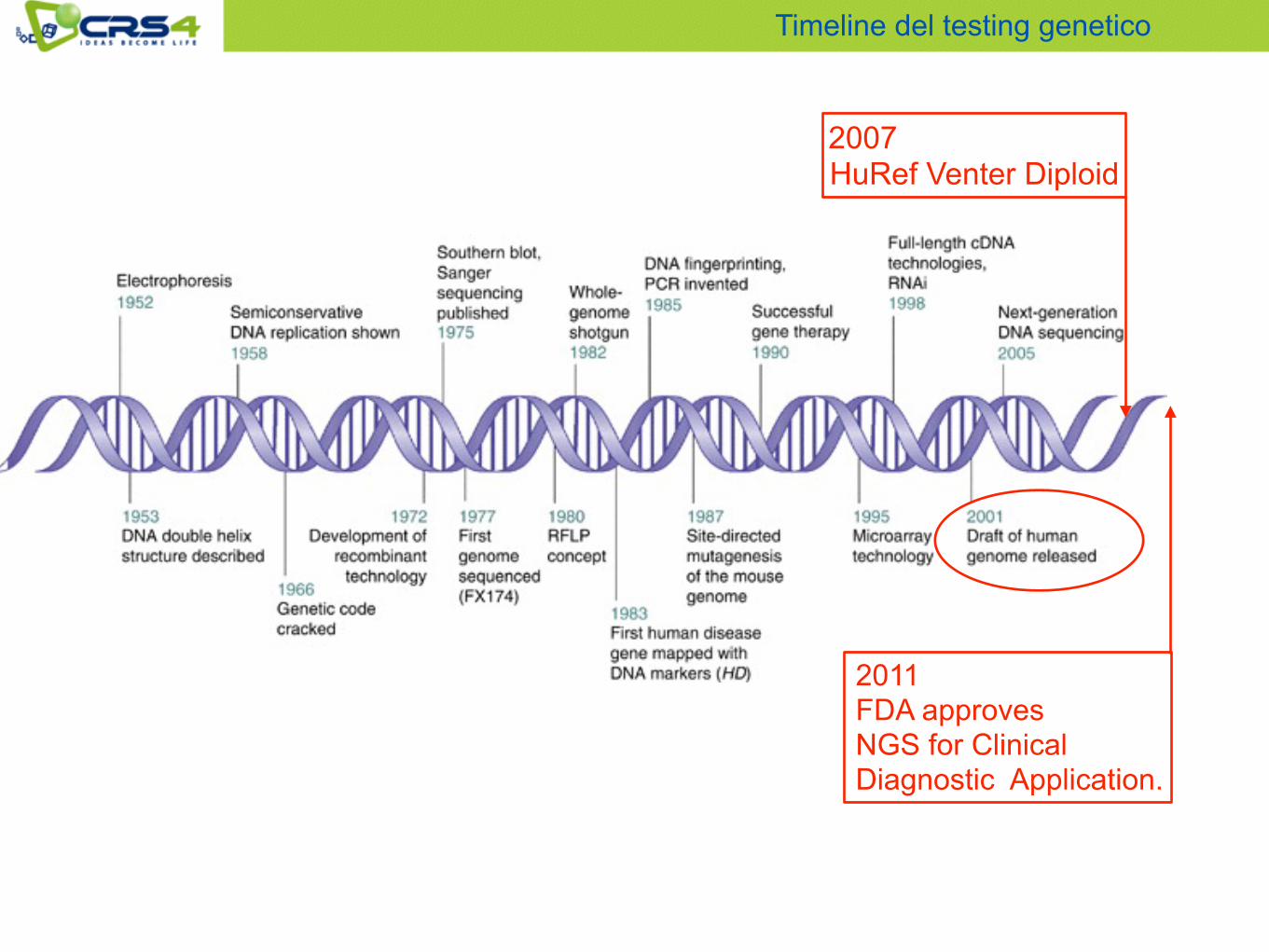
Timeline del testing genetico
2011 FDA approves NGS for Clinical Diagnostic Application.
2007 HuRef Venter Diploid

2015 Cao et al. : De-Novo assembly of a haplotype resolved human genome.
12
Il futuro

• Quanto sono differenti due genomi umani? Più’ due persone sono imparentate piu’ i loro genomi sono simili.
• Si stima che per persone senza legami familiari , due persone prese a caso per strade ad esempio, i genomi differiscono di 1 ogni 1200 o 1500 basi di DNA.
• Quindi ci sono piu’ di tre milioni di differenze tra un genoma umano e un altro. D’altra parte e’ anche vero che siamo simili al 99,9%.
• Ma siamo uguali solo al 99% ai nostri parenti genetici piu’ vicini: gli scimpanze! :)
13
Variazione e Similitudine del genoma

14
Variazione e Similitudine del genoma
• Se ogni genoma umano e’ diverso che cosa vuol dire sequenziare “IL genoma umano”?
• E che cosa vuol dire cercare varianti ? Varianti rispetto a cosa?

• La sequenza completa del genoma umano, sia quella completata nel 2001 che le successive, e’ in effetti una sequenza “rappresentativa” ottenuta dal DNA di diversi individui.
• Ad esempio la versione 19 del genoma umano , GRCh37 the Genome Reference Consortium human genome (build 37), e’ derivata da 13 donatori anonimi di Buffalo, New York.
• Un avviso e’ stato messo sul giornale locale ed i primi 10 maschi e 10 femmine che hanno risposto all’annuncio sono stati invitati ad un incontro con il consigliere genetico del progetto ed a donare il loro DNA.
• Come risultato finale si ha che circa l’80% del genoma di riferimento viene da 8 persone ed in particolare circa il 66% proviene da un maschio designato come RP11.
15
Genoma di riferimento

• In effetti quindi il genoma di riferimento e’ solo una referenza che ci serve per mappare le nostre sequenze.
• Non ci sono assunzioni medico o biologiche sulla sua sequenza ! Quindi anche trovando una variante rispetto al genoma di riferimento bisogna sempre validarla prima di dichiararla mutazione patogena!
16
Genoma di riferimento
Esistono molti database pubblici e metodi di predizione di mutazioni patogene da utilizzare per validare le varianti.
17
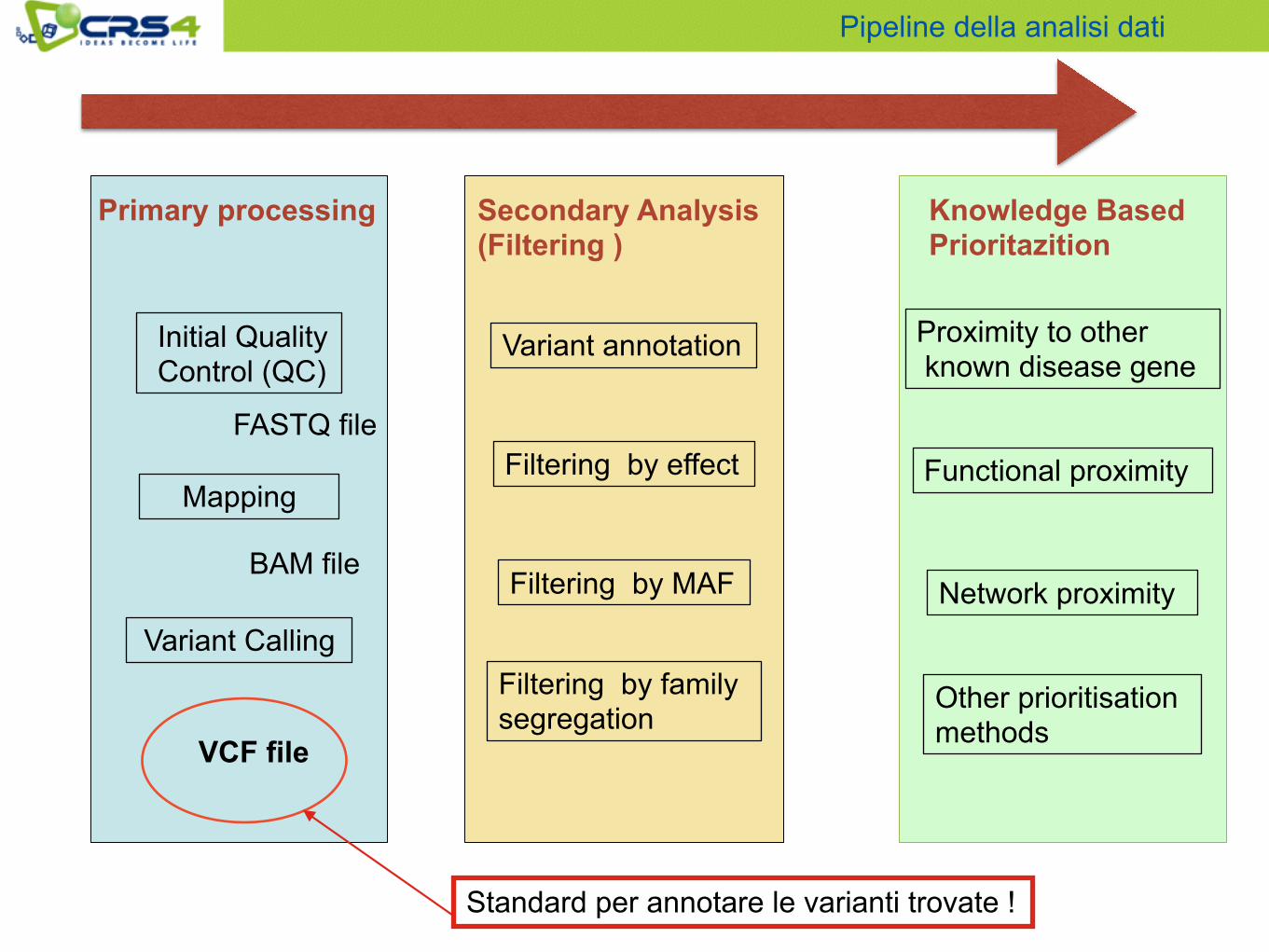
Primary processing
Initial Quality Control (QC)
Mapping
Variant Calling
Secondary Analysis (Filtering )
Variant annotation
Filtering by effect
Filtering by MAF
Filtering by family segregation
Knowledge Based Prioritazition
Proximity to other known disease gene
Functional proximity
Network proximity
Other prioritisation methods
FASTQ file
BAM file
VCF file
Standard per annotare le varianti trovate !
Pipeline della analisi dati
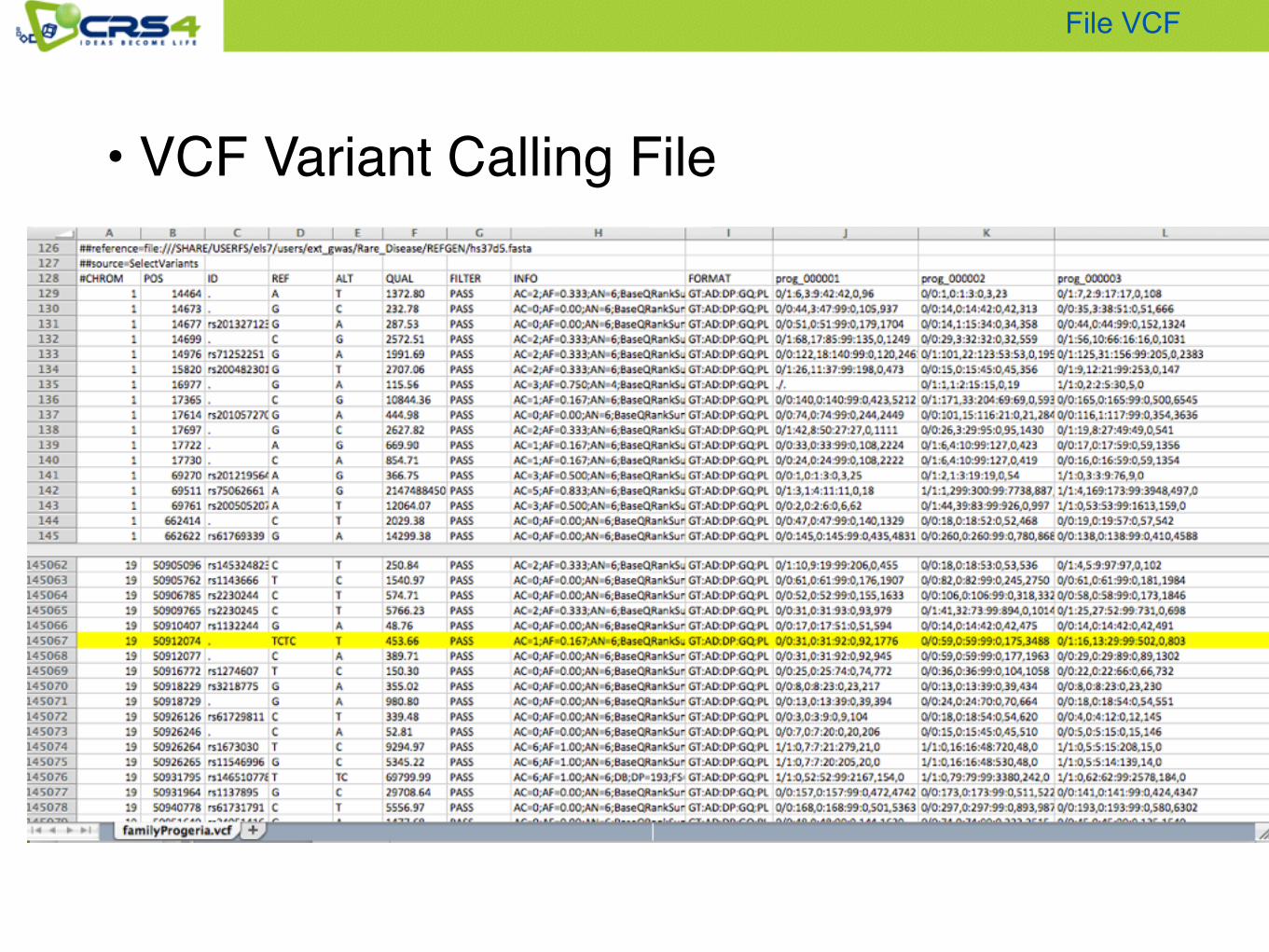
• VCF Variant Calling File
18
File VCF
19
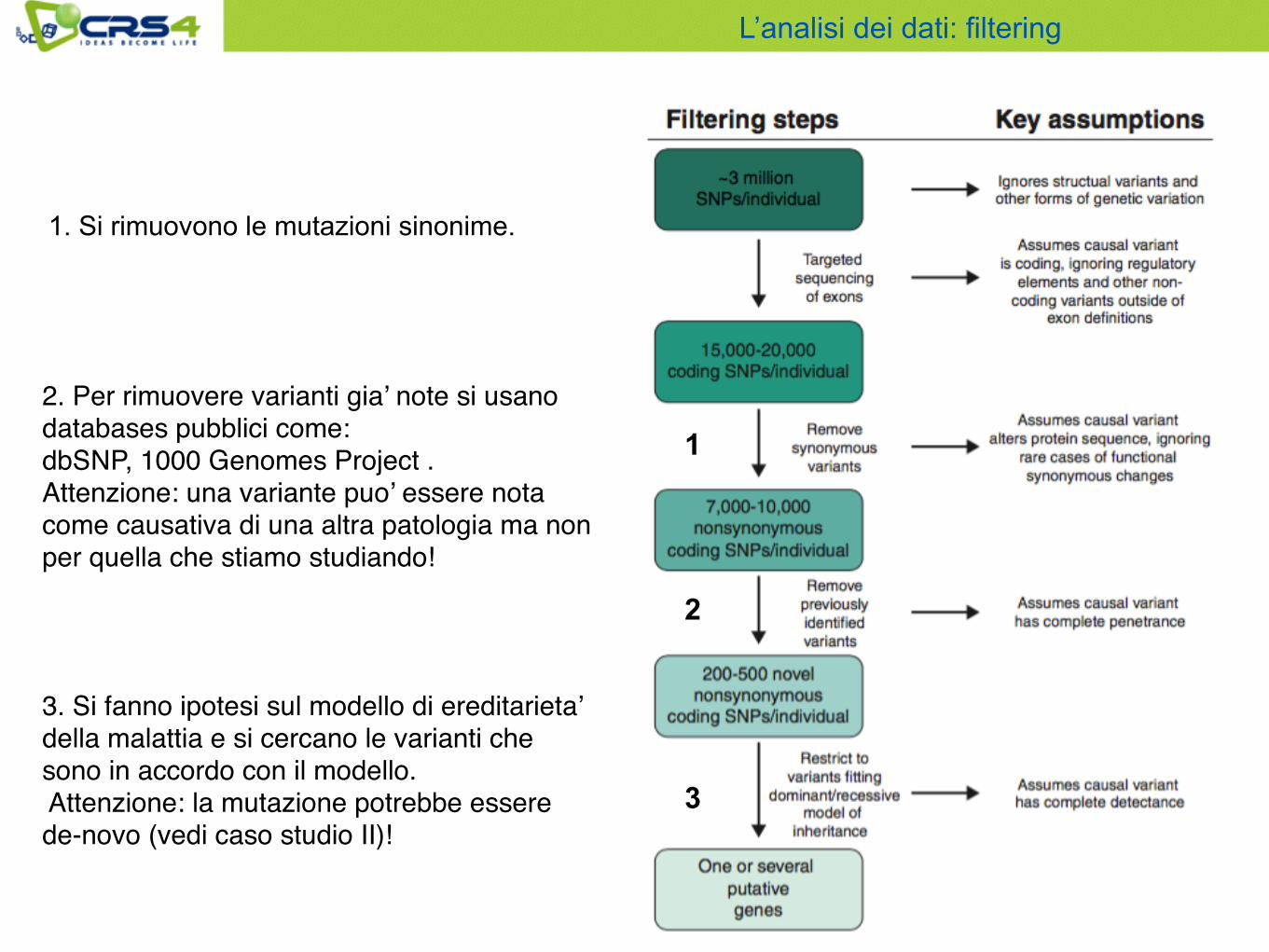
2. Per rimuovere varianti gia’ note si usano databases pubblici come: dbSNP, 1000 Genomes Project . Attenzione: una variante puo’ essere nota come causativa di una altra patologia ma non per quella che stiamo studiando!
1
2
3
1. Si rimuovono le mutazioni sinonime.
3. Si fanno ipotesi sul modello di ereditarieta’ della malattia e si cercano le varianti che sono in accordo con il modello. Attenzione: la mutazione potrebbe essere de-novo (vedi caso studio II)!
L’analisi dei dati: filtering
20
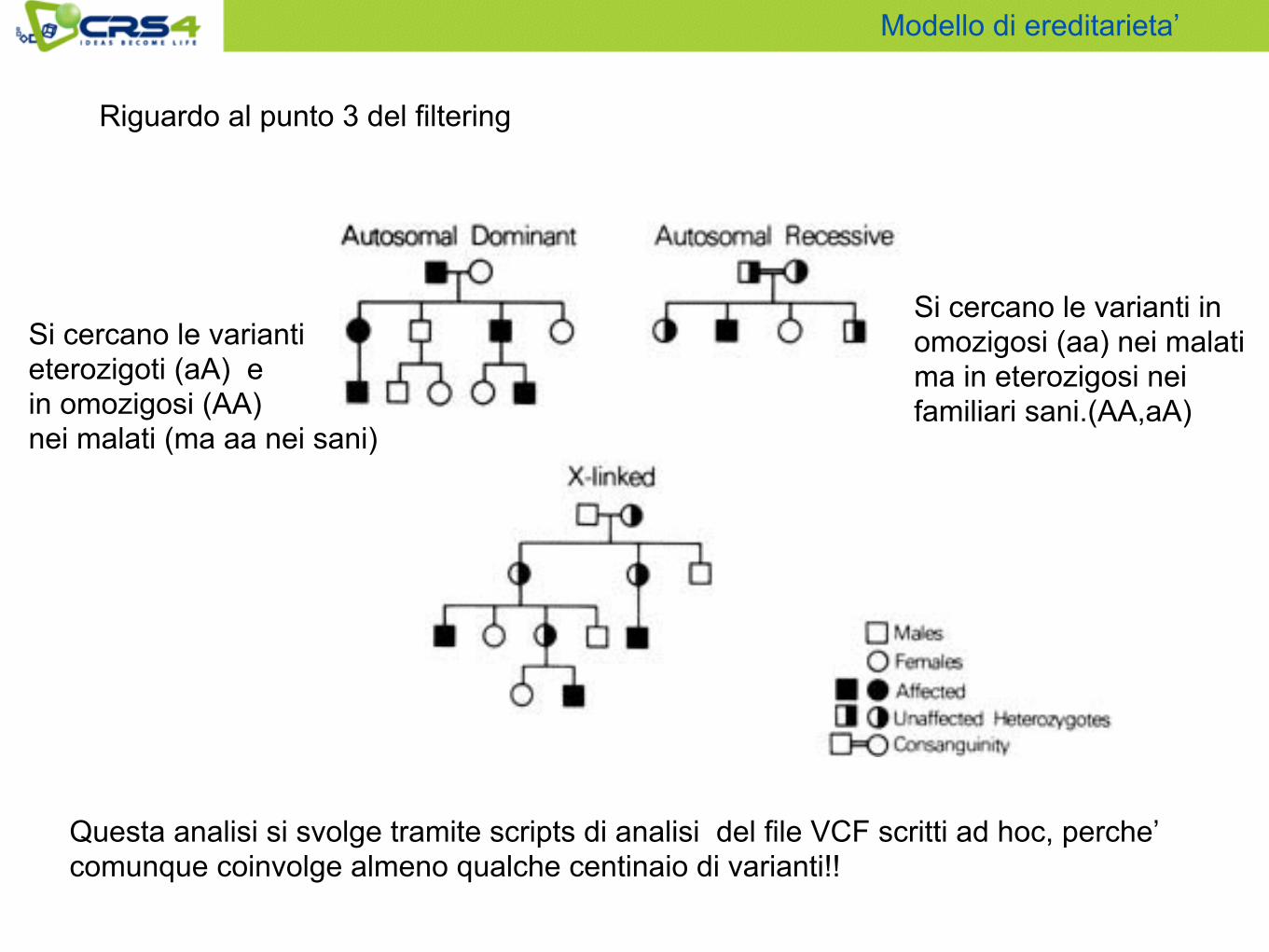
Modello di ereditarieta’
Si cercano le varianti eterozigoti (aA) e in omozigosi (AA) nei malati (ma aa nei sani)
Si cercano le varianti in omozigosi (aa) nei malati ma in eterozigosi nei familiari sani.(AA,aA)
Questa analisi si svolge tramite scripts di analisi del file VCF scritti ad hoc, perche’ comunque coinvolge almeno qualche centinaio di varianti!!
Riguardo al punto 3 del filtering

1. Pazienti con fenotipo di Osteopetrosi ma poi rivalutati a Pycnodysostosis
21
Due casi studio di exome sequencing
2. Un caso di una sindrome molto rara e non ancora geneticamente descritta al momento in cui abbiamo iniziato lo studio.
22
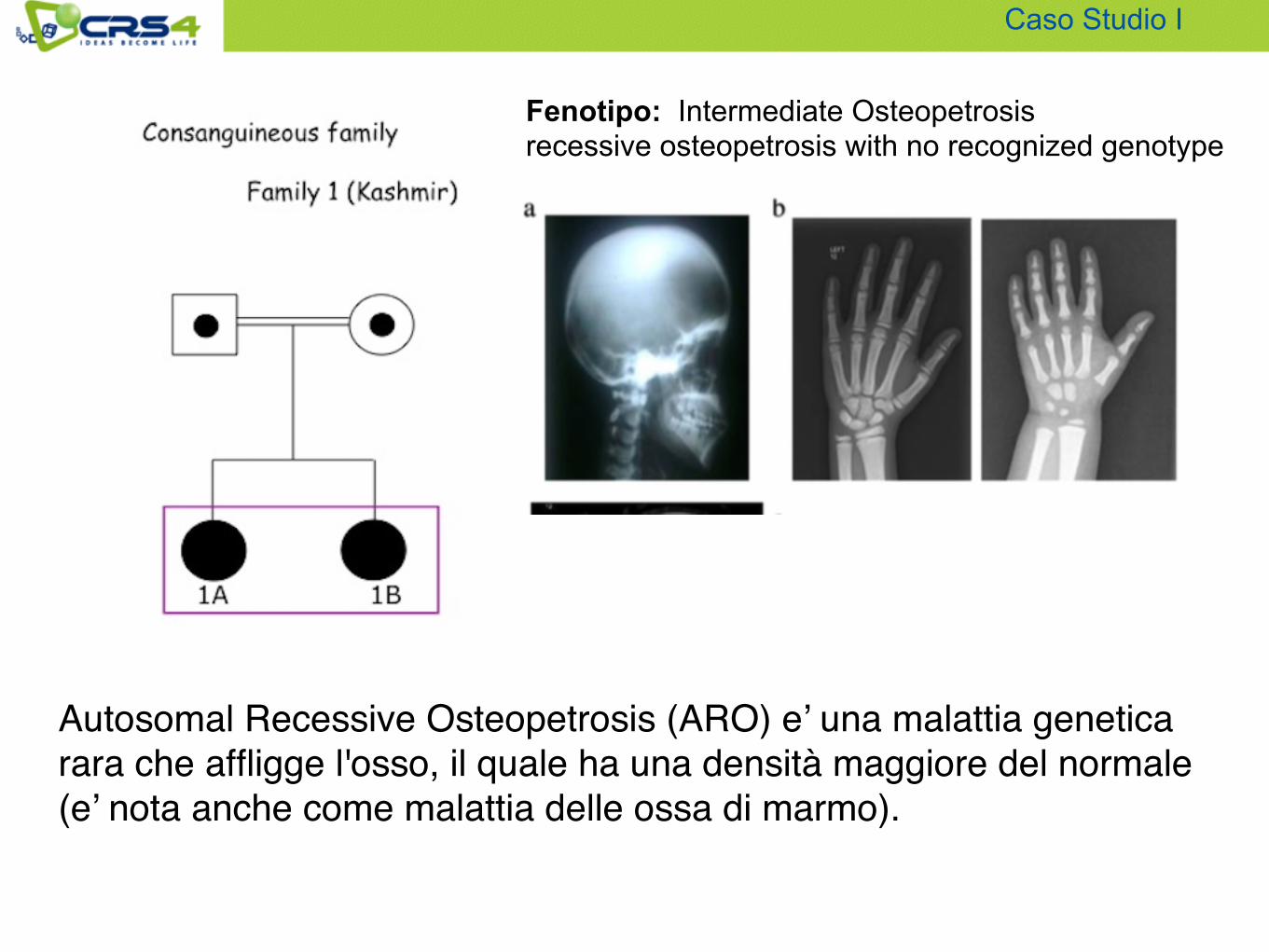
Fenotipo: Intermediate Osteopetrosis recessive osteopetrosis with no recognized genotype
Caso Studio I
Autosomal Recessive Osteopetrosis (ARO) e’ una malattia genetica rara che affligge l'osso, il quale ha una densità maggiore del normale (e’ nota anche come malattia delle ossa di marmo).

23
Caso Studio I
Abbiamo usato una lista di geni per prioritizzare le varianti passando da 165 a 3. Fondamentale il lavoro dei biologi e medici per ottenere questo risultato !!!
Abbiamo svolto lo studio di sequenziamento esomico nelle 2 sorelle affette e raggiunto per entrambi i campioni un coverage medio di 69x sulle 62 MB di target, raggiungendo un 94% di coverage.
24
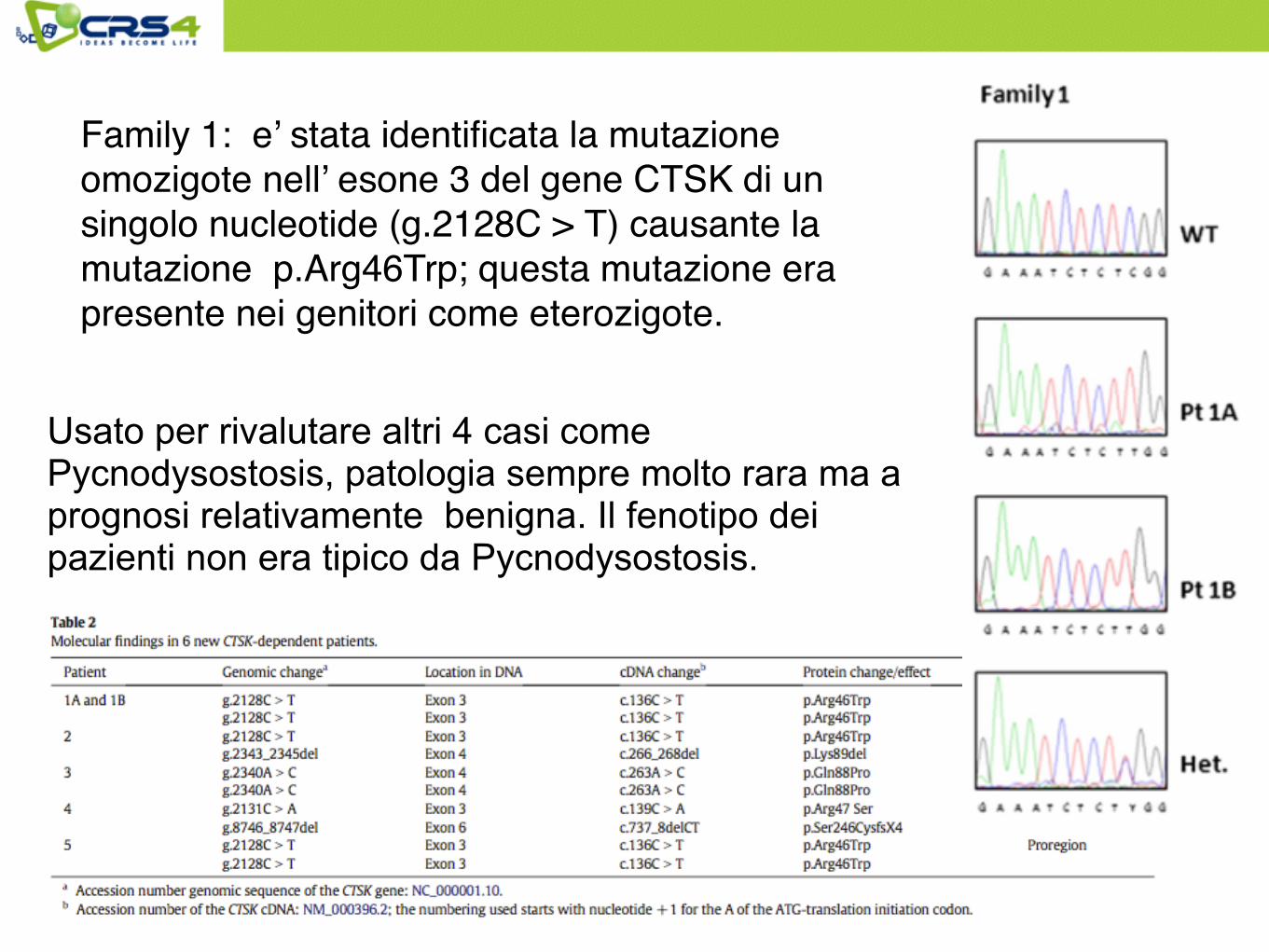
Family 1: e’ stata identificata la mutazione omozigote nell’ esone 3 del gene CTSK di un singolo nucleotide (g.2128C > T) causante la mutazione p.Arg46Trp; questa mutazione era presente nei genitori come eterozigote.
Usato per rivalutare altri 4 casi come Pycnodysostosis, patologia sempre molto rara ma a prognosi relativamente benigna. Il fenotipo dei pazienti non era tipico da Pycnodysostosis.

• Attraverso lo studio e’ stata identificata una mutazione patogena omozigote nel gene CTSK che fino allo studio non si conosceva.
• Importante il fatto che due campioni fossero di due sorelle affette dalla stessa patologia!
• Sono stati quindi testati attraverso sequenziamento Sanger altri 25 pazienti diagnosticati come “recessive osteopetrosis” ed in 4 e’ stata trovata la mutazione nel gene CTSK.
• E’ stata raccomandata l’inclusione del gene CTSK nella lista di geni da controllare per una diagnosi di patologia di accresciuta densita’ ossea.
25
Caso Studio I : Conclusioni

26
Caso Studio II
Progeria o no? Un paziente con una patologia recessiva molto rara.

27
Fenotipo e’ un caso di sindrome progeroide. La ricerca di mutazioni nel gene LMNA non ha dato risultati!!
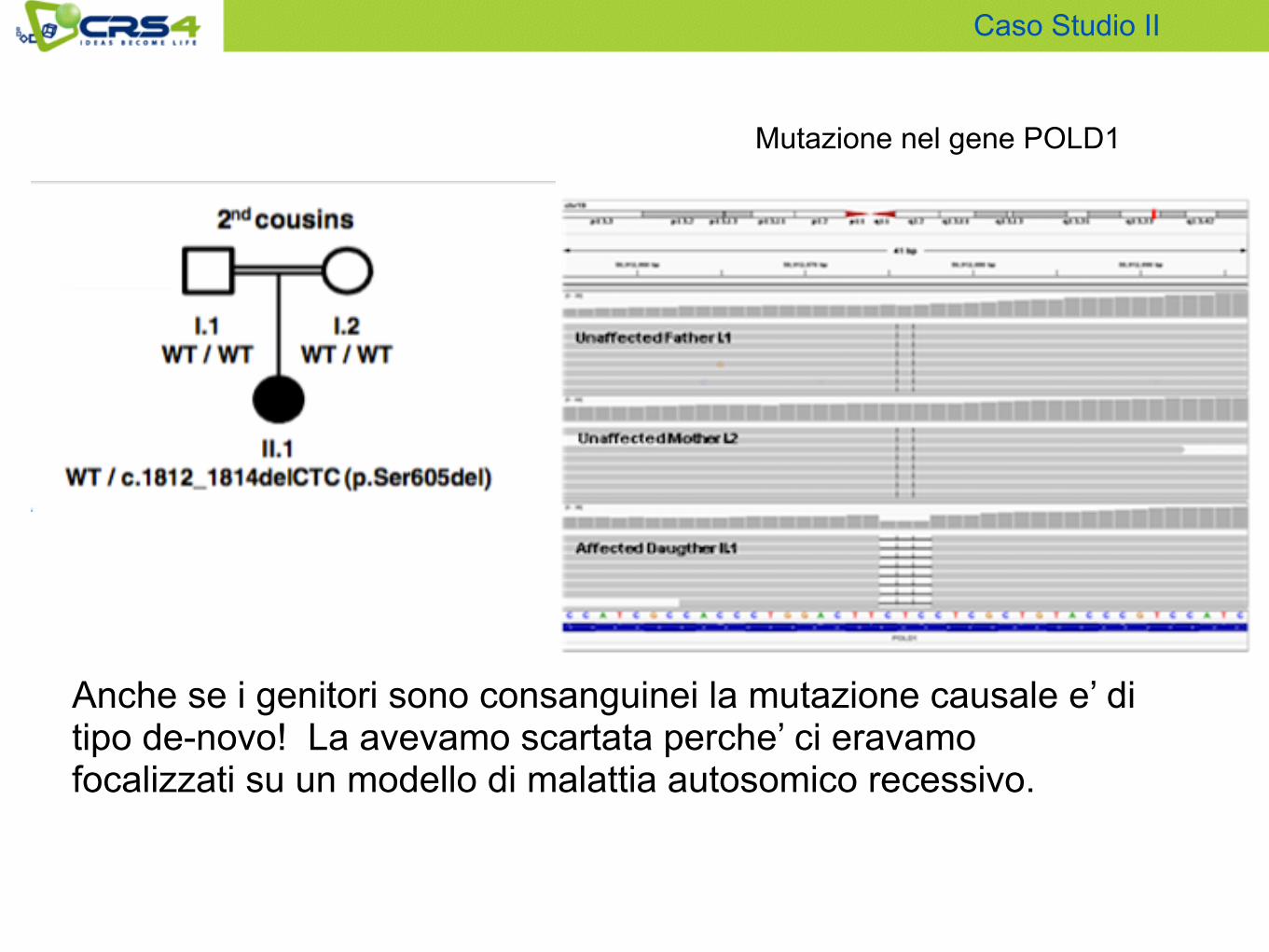
Genitori consanguinei : per questo motivo si sono cercate le varianti omozigoti nella paziente e eterozigoti nei genitori ipotizzando che la mutazione causale fosse ereditata da un antenato comune.
Sono quindi stati analizzati con il sequenziamento esomico i campioni del trio (cioe’ i 2 genitori e la paziente).
Caso Studio II

• Nel 2013 viene pubblicato un articolo che descrive una sindrome con fenotipo simile in 4 pazienti! La stessa variante in POLD1 e’ presente nel nostro caso.
28
Geni su cui avevamo focalizzato l’attenzione!
29
Anche se i genitori sono consanguinei la mutazione causale e’ di tipo de-novo! La avevamo scartata perche’ ci eravamo focalizzati su un modello di malattia autosomico recessivo.
Mutazione nel gene POLD1
Caso Studio II
30
Caso Studio II
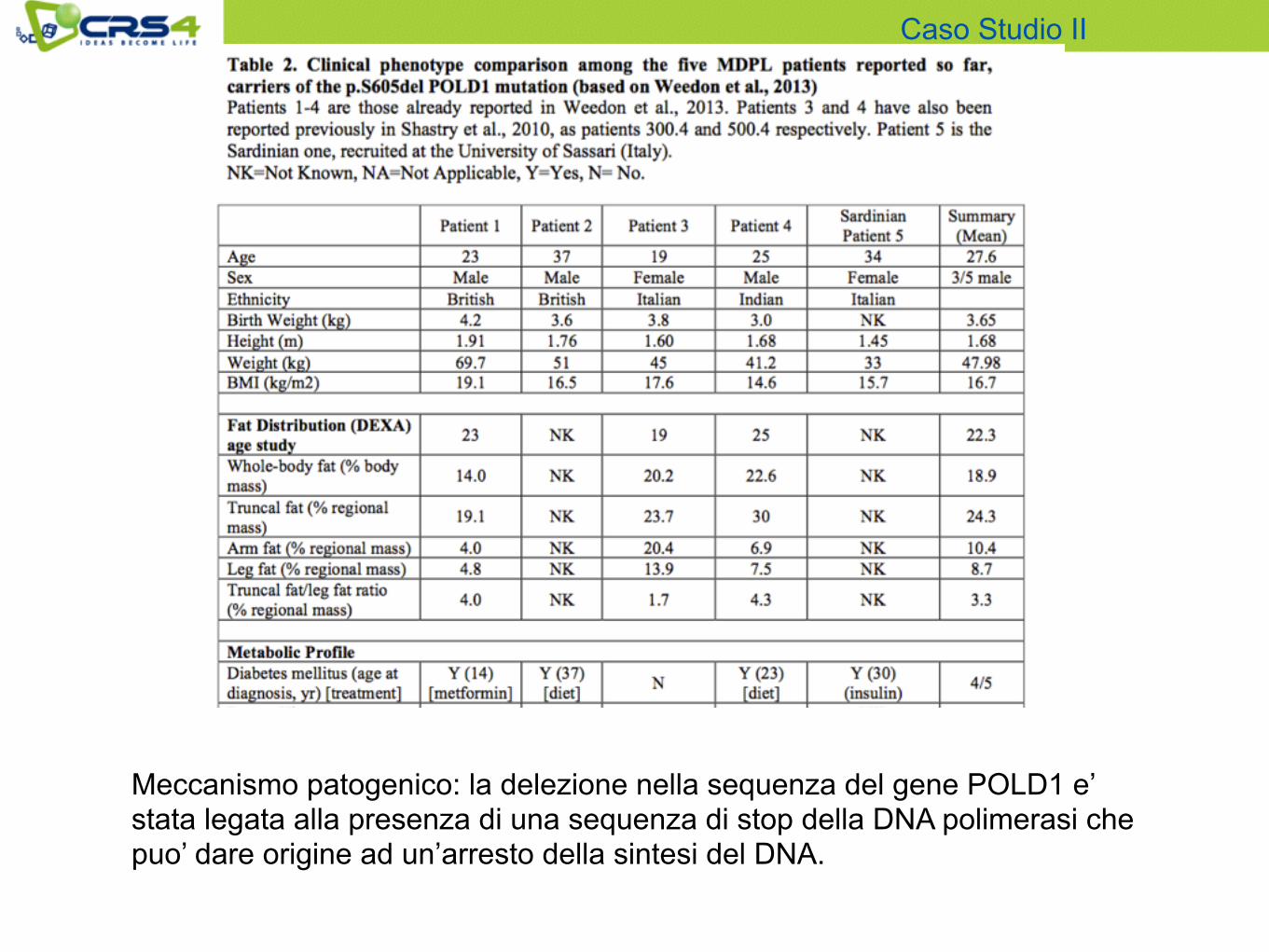
Meccanismo patogenico: la delezione nella sequenza del gene POLD1 e’ stata legata alla presenza di una sequenza di stop della DNA polimerasi che puo’ dare origine ad un’arresto della sintesi del DNA.

• Attraverso lo studio di exome sequencing e’ stato trovato il quinto paziente di MDPL con la stessa mutazione de novo pSer605del nel gene POLD1.
• e’ stata data ulteriore prova genetica che questa e’ una mutazione che provoca la malattia;
• e’ stata ipotizzata una spiegazione del possibile meccanismo per cui insorge la malattia.
• bisogna sempre considerare l’eventualita’ di una mutazione de novo anche se in una malattia molto rara e con genitori consanguinei! Ricordarsi di svolgere di routine la ricerca di mutazioni de novo.
31
Caso Studio II : conclusioni

• Il sequenziamento esomico funziona e permette di guadagnare molto tempo nello studio di patologie di vario tipo.
• I costi stanno velocemente diminuendo e il suo utilizzo diventa sempre maggiore anche in ambito clinico.
• Per uno studio di successo e’ necessario avere una sinergia di competenze (medico+biologo+bioinformatico) durante ogni fase dello studio.
• La mole di dati prodotti (come anche i campioni raccolti per lo studio) deve essere conservata e disponibile per studi futuri. Necessita’ di organizzare biobanche!
32
Conclusioni

• Ringrazio i miei (ex) colleghi del gruppo di bioinformatica del CRS4 , Fred Reinier, Riccardo Berutti, Ilenia Zara.
• Ringrazio i colleghi del CNR -IRGB con cui abbiamo iniziato questo tipo di studi e con cui abbiamo fatto innumerevoli riunioni: Serena Sanna, Laura Crisponi, Carlo Sidore e molti altri…
• Ringrazio i biologi con cui abbiamo lavorato per la pazienza dimostrata con chi come me di biologia conosce ben poco. In particolare Alessandro Puddu, Manuela Oppo, Roberto Cusano.
33
Ringraziamenti


















