
SCREENING DI POTENZIALI AGENTI CARDIOPROTETTIVI SU ... · 3.2 Procedura di isolamento dei...
116
UNIVERSITÀ DI PISA DIPARTIMENTO DI FARMACIA Corso di Laurea Magistrale in Farmacia Tesi di Laurea SCREENING DI POTENZIALI AGENTI CARDIOPROTETTIVI SU PREPARAZIONI DI MITOCONDRI CARDIACI Relatori: Prof. Vincenzo Calderone Dott.ssa Lara Testai Candidato: Alessandra Paglietti Anno Accademico 2014/2015
Transcript of SCREENING DI POTENZIALI AGENTI CARDIOPROTETTIVI SU ... · 3.2 Procedura di isolamento dei...

UNIVERSITÀ DI PISA
DIPARTIMENTO DI FARMACIA
Corso di Laurea Magistrale in Farmacia
Tesi di Laurea
SCREENING DI POTENZIALI AGENTI
CARDIOPROTETTIVI SU PREPARAZIONI DI
MITOCONDRI CARDIACI
Relatori:
Prof. Vincenzo Calderone
Dott.ssa Lara Testai
Candidato:
Alessandra Paglietti
Anno Accademico 2014/2015

INDICE
1. Introduzione
1.1 Danno da Ischemia/Riperfusione ............................................................................ 1
1.2 Morte cellulare indotta durante l’ischemia .............................................................. 3
1.3 Morte cellulare indotta durante la riperfusione ....................................................... 5
1.4 Meccanismi di morte cellulare nel danno da I/R ..................................................... 8
1.5 Meccanismi di cardioprotezione............................................................................ 10
1.5.1 Il precondizionamento ischemico (IPreC) ...................................................... 10
1.5.2 Il post condizionamento ischemico (IPostC) .................................................. 12
1.5.3 Il remoto IPreC e IPostC (ReIPreC, ReIPostC) .............................................. 13
1.6 Il precondizionamento farmacologico (Ph-PreC) .................................................. 14
1.6.1 Triggers ........................................................................................................... 16
1.7 Canali del potassio coinvolti nella cardioprotezione ............................................ 20
1.7.1 Canali ............................................................................................. 21
1.7.2 Canali mitocondriali al potassio sensibili al calcio ......................................... 26
1.7.3 Canali mitoKv ................................................................................................. 30
1.7.4 Canali mitoTASK-3 ........................................................................................ 31
1.8 Protein Kinasi ........................................................................................................ 32
1.9 Poro MPTP ........................................................................................................... 45
1.9.1 Poro MPTP come target cardioprotettivo ....................................................... 47
1.9.2 Poro MPTP come target per il condizionamento ischemico........................... 48
1.10 Uniporter mitocondriale del ...................................................................... 52
1.10.1 Regolatori dell’ uniporter mitocondriale del ...................................... 54
1.11 Metodi innovativi di drug delivery mitocondriale............................................... 57
2. Scopo della tesi .......................................................................................................... 59

3. Materiali e Metodi
3.1 Animali .................................................................................................................. 66
3.2 Procedura di isolamento dei mitocondri cardiaci .................................................. 67
3.3 Protocollo sperimentale 1: valutazione delle variazioni del potenziale di
membrana mitocondriale ............................................................................................. 69
3.4 Protocollo sperimentale 2: valutazione delle variazioni nell’uptake di ....... 71
3.5 Protocollo sperimentale 3: valutazione dei movimenti di K⁺, utilizzando una
sonda Tl⁺-sensibile ...................................................................................................... 73
3.6 Protocollo sperimentale 4: valutazione delle variazioni del potenziale di
membrana mitocontriale utilizzando Rhodamina 123 ................................................. 75
3.7 Analisi statistica .................................................................................................... 77
4. Risultati e Discussione............................................................................................... 78
5. Bibliografia ................................................................................................................ 95

1
1. INTRODUZIONE
1.1 DANNO DA ISCHEMIA/RIPERFUSIONE
L’ischemia acuta miocardica è una delle principali cause di malattia nella società
occidentale e nonostante i recenti progressi nella terapia, rappresenta ancora la
principale causa di morte.
Le cardiopatie ischemiche sono correlate a una parziale o totale occlusione delle arterie
coronarie, le quali portano ad una ostruzione dei vasi così da limitare l’afflusso
sanguigno nei vari tessuti o organi. Questo ridotto apporto di sangue causa una
diminuzione dell’ossigenazione e dell’apporto nutritivo di tali aree, per cui questo
flusso non è più sufficiente per la sopravvivenza della cellula, che quindi va incontro a
morte. Singolarmente, il danno da ischemia è aumentato anche nella fase di
riperfusione, ovvero la fase successiva all’ischemia in cui vengono ripristinate le
condizioni di normalità per la vita cellulare (normale apporto di nutrienti e ossigeno): in
questo caso si parla di danno da Ischemia/Riperfusione (I/R).
Nel miocardio, la morte cellulare durante un episodio di ischemia è causato sia dal
processo di necrosi sia da quello di apoptosi. Mentre la necrosi può avvenire sia durante
l’evento ischemico sia quello di riperfusione, l’apoptosi si verifica soprattutto durante il
periodo della riperfusione (Zhao & Vinten-Johansen, 2002).
In particolare, il meccanismo di necrosi, come forma principale della morte cellulare dei
miociti, si instaura generalmente con una certa rapidità che può scatenare una risposta
infiammatoria significativa. Inoltre, si verificano diversi cambiamenti nelle cellule che
includono gravi danni cellulari, rigonfiamento degli organelli, denaturazione e
coagulazione delle proteine citoplasmatiche e la ripartizione di organelli cellulari.
Questi cambiamenti ultrastrutturali sono legati alla mancanza di ossigeno, alla
deplezione di ATP, alla perdita dell'omeostasi del calcio e ai difetti nella permeabilità
della membrana; il tutto può provocare la contrattura della cellula che può condurre alla
rottura della membrana e all'eventuale morte cellulare (Cotran et al., 1999).
Al contrario, il meccanismo di apoptosi consiste in un processo energia-dipendente in
cui la morte cellulare segue una sequenza geneticamente programmata e controllata di
eventi; le sue caratteristiche morfologiche principali includono la perdita asimmetrica di
fosfolipidi dalla membrana cellulare, la condensazione della cromatina, e la formazione

Introduzione
2
di vescicole citoplasmaticiche. Nella fase finale di apoptosi, alcuni frammenti cellulari
formano i corpi apoptotici che sono confinati all’interno della cellula in modo tale da
prevenire l'innesco di una risposta infiammatoria (Haunstetter & Izumo, 2000).
L’esistenza contemporanea di entrambi i tipi di morte cellulare nel miocardio può
rappresentare il grado finale di un letale danno miocardico dopo la comparsa dell’evento
di ischemia e riperfusione (James, 1998).
Quindi, i meccanismi di danno che innescano la morte cellulare durante la fase
ischemica e la fase della riperfusione sono differenti.

Introduzione
3
1.2 MORTE CELLULARE INDOTTA DURANTE L’ISCHEMIA
Abbassamento del pH intracellulare
Durante l’evento ischemico la richiesta energetica delle cellule miocardiche
aumenta, quindi ci sarà un incremento del processo della glicolisi e per questo
motivo, si avrà un aumento metabolico di acido lattico con conseguente
acidificazione del pH intracellulare (Halestrap et al., 1998). L’acidificazione
della cellula è anche dovuta al contributo dato dalla degradazione di ATP in
ADP e AMP, in cui il pH scende fino a 6.2 nei primi 13 minuti (Garlick et al.,
1979).
Diminuzione di ATP
Con l’evento ischemico si ha l’interruzione di apporto di ossigeno e ciò
determina il blocco della fosforilazione ossidativa mitocondriale e il veloce
consumo di ATP, che viene convertito in ADP e in AMP e come tale esce dalla
cellula ed è la causa della riduzione della funzionalità cardiaca (gravi riduzioni
di ATP conducono a danni irreversibili del miocardio) (Sanada et al., 2011).
Modificazioni nelle concentrazioni ioniche
A seguito della diminuzione della concentrazione di ATP si ha l’inibizione della
pompa / ATPasi e quindi, incremento della concentrazione intracellulare
di . L’inibizione di questa pompa, e il conseguente accumulo di , causa
l’inibizione dell’antiporto / il quale fa ulteriormente diminuire il pH
intracellulare. (Testai et al., 2015).
L’antiporto che ha lo scopo di pompare all’esterno della cellula ioni
e far entrare ioni all’interno della cellula, viene inibito o invertito
provocando un aumento di ioni all’interno della cellula. Tale
concentrazione è incrementata dall’inibizione della pompa ATPasi.
(Sanada et al., 2011).
Questi eventi sono responsabili di un sovraccarico di ioni che potrebbero
contribuire all’apertura irreversibile dei canali MPTP (mithocondrial
permeability transition pore).
Introduzione
4
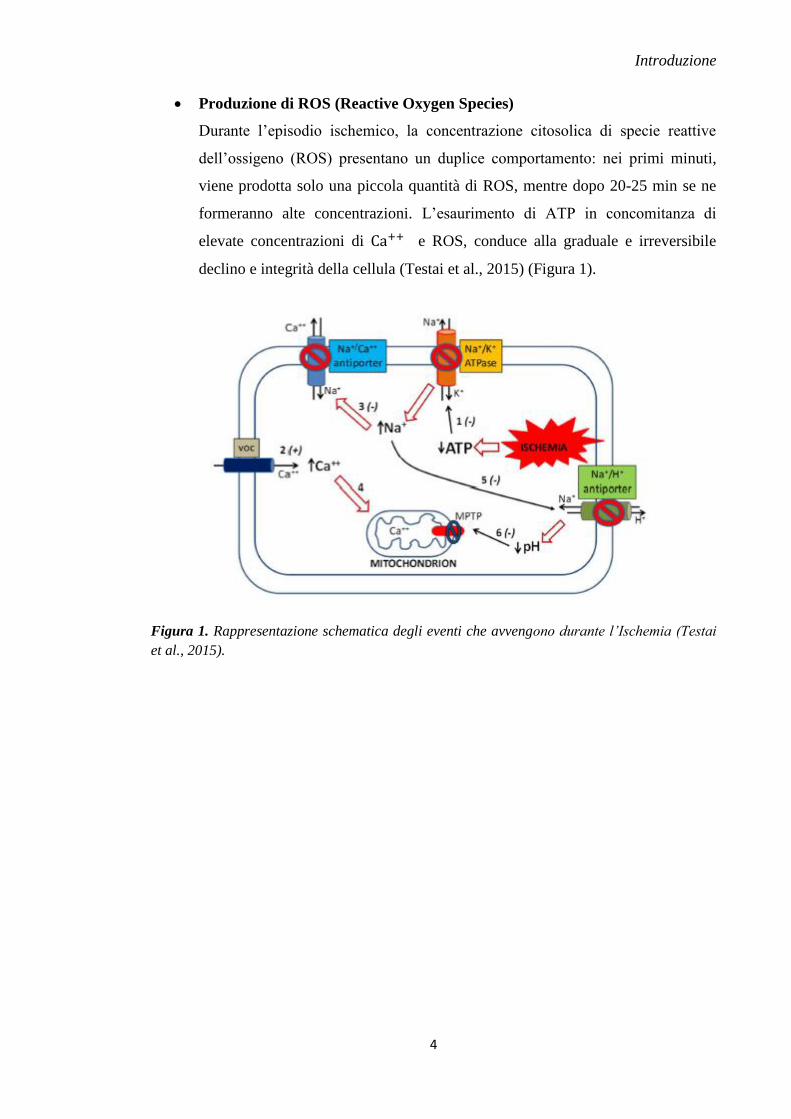
Produzione di ROS (Reactive Oxygen Species)
Durante l’episodio ischemico, la concentrazione citosolica di specie reattive
dell’ossigeno (ROS) presentano un duplice comportamento: nei primi minuti,
viene prodotta solo una piccola quantità di ROS, mentre dopo 20-25 min se ne
formeranno alte concentrazioni. L’esaurimento di ATP in concomitanza di
elevate concentrazioni di e ROS, conduce alla graduale e irreversibile
declino e integrità della cellula (Testai et al., 2015) (Figura 1).
Figura 1. Rappresentazione schematica degli eventi che avvengono durante l’Ischemia (Testai
et al., 2015).

Introduzione
5
1.3 MORTE CELLULARE INDOTTA DURANTE LA RIPERFUSIONE
Stabilizzazione del pH fisiologico
Oltre al ripristino del flusso ematico, dovuto all’inizio della riperfusione,
avviene una rapida normalizzazione del pH extracellulare che determina il
formarsi di un forte gradiente di ioni attraverso la membrana plasmatica.
Principalmente come risultato si ottiene un ulteriore flusso massivo di
all’interno della cellula che permette l’espulsione degli ioni in eccesso
(Schäfer et al., 2001).
Produzione di ROS (Reactive Oxygen Species)
L’inizio della riperfusione è associato ad un’elevata produzione di ROS,
soprattutto dal complesso 1-3 della catena respiratoria, ma anche da altri enzimi
ossidativi mitocondriali tra cui le xantine ossidasi, NAPH ossidasi,
monoammine ossidasi e aconitasi (Kevin, et al., 2003) (Chen & Zweier, 2014).
Quindi, l’improvviso ripristino del metabolismo aerobico, fondamentale per la
ripresa della produzione energetica di ATP, ha come conseguenza un accumulo
di ROS, in particolare di anione superossido ( ). In condizioni fisiologiche il
superossido viene convertito in perossido di idrogeno ( ) dalla superossido
dismutasi (SOD) ed è successivamente inattivato dalla catalasi in e in .
L’evento ischemico potrebbe aver compromesso i meccanismi antiossidanti
della cellula e quindi, se i mitocondri non sono in grado di eliminarli, i ROS
possono andare a danneggiare le strutture cellulari, gli enzimi o le proteine
canale presenti sulla membrana cellulare (Sanada et al., 2011).
Infatti, le proteine mitocondriali sono particolarmente suscettibili al danno
indotto da ROS: questi hanno effetti diretti sulla respirazione e giocano un ruolo
centrale nell’apertura dei canali MPTP.
Accumulo di intracellulare
Gli ioni già accumulati durante l’evento ischemico entrano anche dai
canali voltaggio dipendenti di tipo L (Smart et al., 1997).
Le cellule però, già danneggiate da un lungo periodo ischemico, non sono in
grado di ristabilire istantaneamente l’omeostasi di intracellulare; solo dopo

Introduzione
6
30-60 minuti di riperfusione avviene una progressiva e graduale ripresa
dell’escrezione e il ri-immagazzinamento ATP-dipendente di nel reticolo
sarcoplasmatico (Sanada et al., 2011). L’accumulo di questi ioni nei primi stadi
della riperfusione è responsabile di una serie di fenomeni che accelerano il
danno cellulare: attivazione di lipasi, di nucleasi e di proteasi che vanno a
compromettere la struttura della cellula (viene attivata la Calpaina, proteasi Ca-
dipendente la cui azione è inibita dalla diminuzione di pH); apertura dei canali
MPTP.
Apertura del poro MPTP (Mitochondrial Permeability Transition Pore)
I canali MPTP sono pori di natura proteica ad alta conduttanza, ancorati tra la
membrana esterna e quella interna. Quando sono attivati consentono la
comunicazione tra il citoplasma e la matrice mitocondriale. La loro struttura non
è ancora pienamente caratterizzata, si ipotizza la presenza del canale anionico
voltaggio-dipendente nella membrana mitocondriale esterna (VDAC) e
dell’adenina nucleotide traslocasi (ANT) in quella interna; è stata accertata
anche, la presenza a livello strutturale della proteina traslocatrice TSPO (nella
membrana mitocondriale interna) e della Ciclofillina D (nella matrice
mitocondriale) (Testai et al., 2015). Durante l’evento ischemico i pori MPTP
sono chiusi, inibiti dal pH acido. Invece, durante la riperfusione, la rapida
energizzazione del mitocondrio conduce a entrata massiva di ioni , già
accumulato nel citosol durante l’ischemia, che induce una “distruttiva”
ipercontrazione; questo fattore, in aggiunta all’accumulo di ROS e al ripristino
dei valori di pH fisiologici, promuove l’apertura dei canali MPTP. L’apertura di
un solo poro è sufficiente a provocare un’immediata depolarizzazione che ne
stimola l’attivazione a cascata (Scorrano et al., 1997). Come conseguenza
dell’apertura degli MPTP la membrana mitocondriale è resa permeabile a tutti i
soluti con una dimensione inferiore ai 1.5 kDa che raggiungono un equilibrio
chimico ai lati della membrana. Invece, le molecole con dimensione maggiore
come le proteine, rimangono intrappolate all’interno della matrice creando un
gradiente osmotico che richiama acqua all’interno della membrana. In queste
condizioni si ha il rigonfiamento del mitocondrio e sebbene le creste della
membrana mitocondriale interna riescano ad espandersi senza andare incontro a
rottura, la membrana esterna si rompe e induce alla fuoriuscita di proteine
Introduzione
7
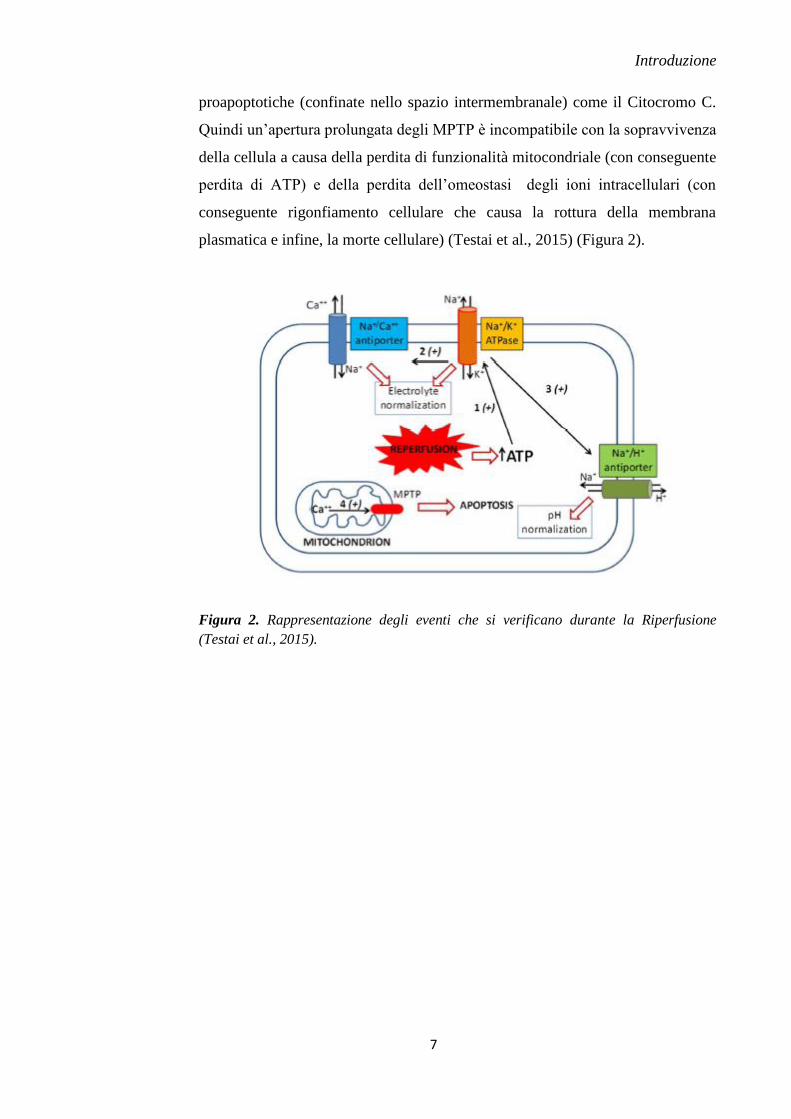
proapoptotiche (confinate nello spazio intermembranale) come il Citocromo C.
Quindi un’apertura prolungata degli MPTP è incompatibile con la sopravvivenza
della cellula a causa della perdita di funzionalità mitocondriale (con conseguente
perdita di ATP) e della perdita dell’omeostasi degli ioni intracellulari (con
conseguente rigonfiamento cellulare che causa la rottura della membrana
plasmatica e infine, la morte cellulare) (Testai et al., 2015) (Figura 2).
Figura 2. Rappresentazione degli eventi che si verificano durante la Riperfusione
(Testai et al., 2015).

Introduzione
8
1.4 MECCANISMI DI MORTE CELLULARE NEL DANNO DA I/R
La morte cellulare causata dal danno da Ischemia/Riperfusione è dovuta all’innesco di
meccanismi di tipo apoptotico, autofagico e necrotico.
Molte ricerche hanno cercato di definire quale forma di morte cellulare avvenga e in che
modo si distribuisca nella zona danneggiata: infatti, alcuni studi hanno proposto che
l’attivazione della morte per necrosi si ha nel momento in cui si ha un abbassamento dei
livelli di mitocondriale e questi raggiungono valori estremamente bassi o si
azzerano del tutto.
Allo stesso modo, sembra che i livelli intracellulari di ATP potrebbero servire anche da
switch molecolare: l’apoptosi si innesca in presenza di alti livelli, invece, a bassi valori
di ATP si avvia il meccanismo di necrosi.
Ulteriori ricerche hanno ipotizzato che dopo l’I/R il destino della cellula sia determinato
dall’estensione dell’apertura degli MPTP nel mitocondrio: nel caso in cui l’apertura sia
minima, la cellula è in grado di ristabilire le normali condizioni fisiologiche; se è di tipo
moderato, la cellula potrebbe generare il meccanismo di morte programmata (apoptosi);
invece, se l’apertura degli MPTP è massiva (severa), la cellula subirà necrosi a causa
dell’insufficiente produzione di energia. Comunque, in tutte e tre le situazioni il destino
cellulare è influenzato dalla risposta del mitocondrio allo stress.
Indipendentemente dalla modalità finale di morte cellulare, è fondamentale che i
meccanismi coinvolti siano tutti interconnessi tra loro.
La morte cellulare indotta da I/R sembra essere un processo attivo che quindi, può
essere inibito con interventi specifici. Studi recenti stanno approfondendo il fatto che i
mitocondri possano essere considerati importanti mediatori e regolatori di tutte le forme
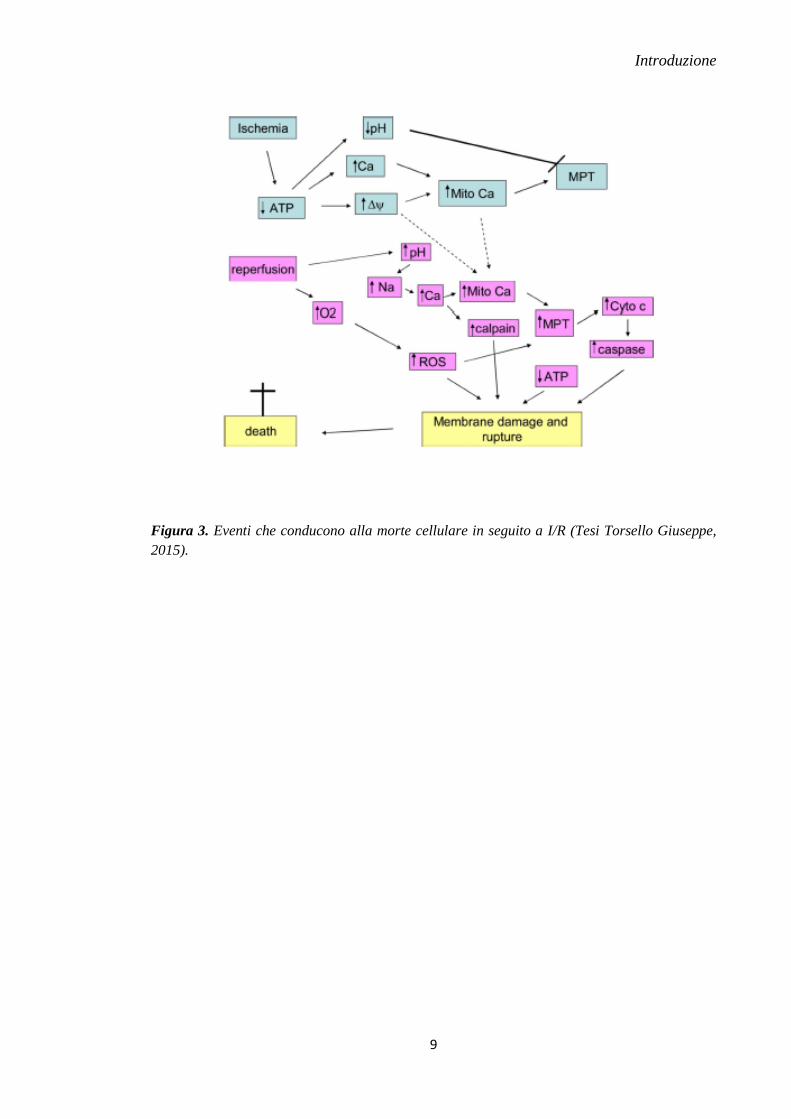
di morte cellulare da I/R. In particolare, i canali MPTP sembrano svolgere il ruolo
principale di regolazione sia della morte apoptotica che necrotica e avere un ruolo
importante nel danno da I/R (Murphy & Steenberger, 2008) (Figura 3).
Introduzione
9
Figura 3. Eventi che conducono alla morte cellulare in seguito a I/R (Tesi Torsello Giuseppe,
2015).
Introduzione
10
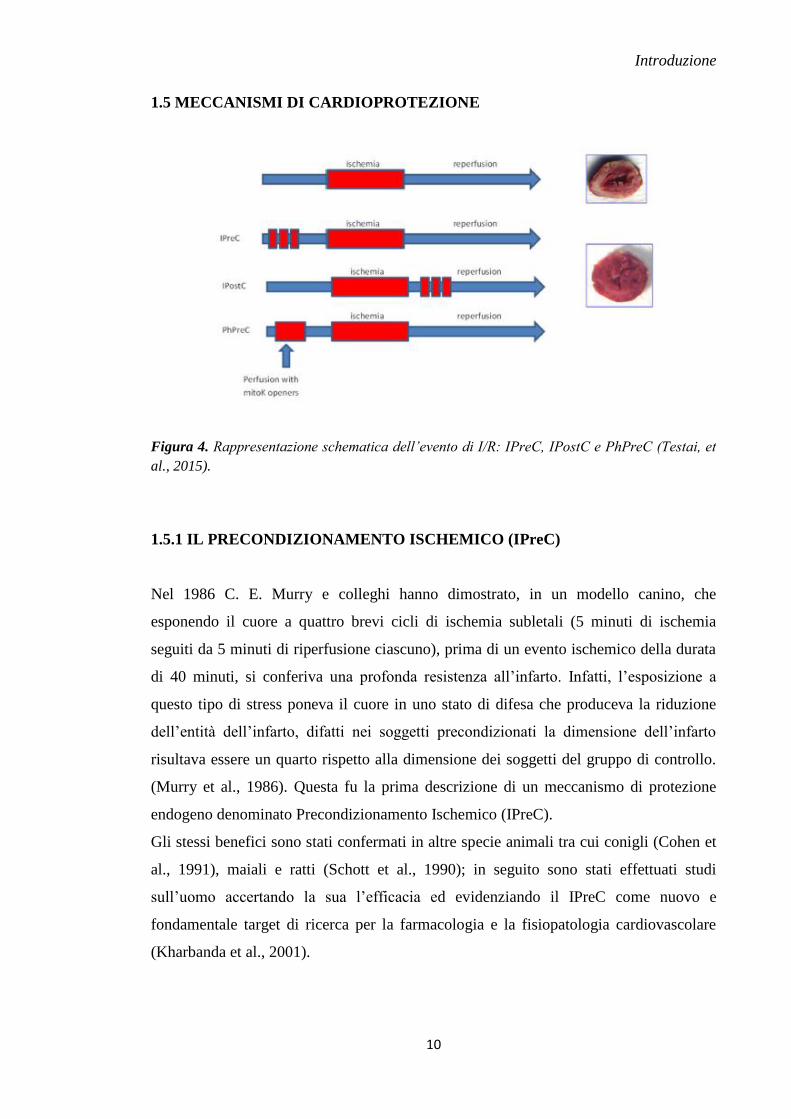
1.5 MECCANISMI DI CARDIOPROTEZIONE
Figura 4. Rappresentazione schematica dell’evento di I/R: IPreC, IPostC e PhPreC (Testai, et
al., 2015).
1.5.1 IL PRECONDIZIONAMENTO ISCHEMICO (IPreC)
Nel 1986 C. E. Murry e colleghi hanno dimostrato, in un modello canino, che
esponendo il cuore a quattro brevi cicli di ischemia subletali (5 minuti di ischemia
seguiti da 5 minuti di riperfusione ciascuno), prima di un evento ischemico della durata
di 40 minuti, si conferiva una profonda resistenza all’infarto. Infatti, l’esposizione a
questo tipo di stress poneva il cuore in uno stato di difesa che produceva la riduzione
dell’entità dell’infarto, difatti nei soggetti precondizionati la dimensione dell’infarto
risultava essere un quarto rispetto alla dimensione dei soggetti del gruppo di controllo.
(Murry et al., 1986). Questa fu la prima descrizione di un meccanismo di protezione
endogeno denominato Precondizionamento Ischemico (IPreC).
Gli stessi benefici sono stati confermati in altre specie animali tra cui conigli (Cohen et
al., 1991), maiali e ratti (Schott et al., 1990); in seguito sono stati effettuati studi
sull’uomo accertando la sua l’efficacia ed evidenziando il IPreC come nuovo e
fondamentale target di ricerca per la farmacologia e la fisiopatologia cardiovascolare
(Kharbanda et al., 2001).

Introduzione
11
Quindi, il IPreC consiste in un breve e transitorio episodio di I/R della durata di 2-5
minuti, che conferisce al miocardio un incremento della resistenza nei confronti di una
successiva sequenza prolungata di episodi di I/R.
Studi successivi, hanno dimostrato che IPreC è distinguibile in due fasi distinte:
- la prima fase, definita “classic IPreC”, dura per le tre ore successive allo stimolo ed è
dovuta dal coinvolgimento di biomolecole già esistenti che agiscono come effettori,
infatti, vanno ad attivare vie di segnalazione cellulare che da un lato avviano un
meccanismo pro-survival e, dall’altro, inibiscono l’azione di segnali pro-morte cellulare
(Yang et al., 2010);
- la seconda fase, più tardiva, chiamata “second window of IPreC”, ha inizio circa dopo
24 ore dall’inizio dello stimolo e si prolunga per i tre giorni successivi, è caratterizzata
da una riprogrammazione genetica della cellula che attiva la trascrizione di geni stress-
responsivi e dalla successiva sintesi di proteine che conferiscono un fenotipo
cardioprotettivo (Hausenloy & Yellon, 2010).
Sia la fase precoce che la tardiva hanno molte caratteristiche in comune: lo stimolo
ischemico precondizionante provoca il rilascio di una serie di sostanze che, legandosi a
recettori sulla superficie cellulare, innescano il meccanismo di protezione, dando inizio
a una cascata di eventi intracellulari. Tuttavia, nelle due fasi sembrano essere coinvolti
meccanismi di reazione differenti: nella fase precoce sono presenti reazioni che possono
essere completate in un breve periodo di tempo come l’attivazione di canali ionici,
l’attivazione tramite fosforilazione di enzimi esistenti, il rapido ricambio o la
traslocazione di sostanze (Yang et al., 2010); invece, la fase tardiva comprende reazioni
che richiedono un tempo maggiore per essere completate tra cui la modulazione
genomica e la sintesi ex novo di svariate proteine tra cui canali proteici, recettori,
enzimi, immunotrasmettitori, ma anche modificazioni post-trasduzionali e traslocazione
di proteine (Hausenloy et al., 2010).

Introduzione
12
1.5.2 IL POST CONDIZIONAMENTO ISCHEMICO (IPostC)
Un certo grado di protezione si può ottenere anche con la riperfusione graduale dopo
l’ischemia (Sato et al., 1997). Tenendo conto dell’estensione del danno provocato
durante la riperfusione e nella ricerca di un metodo di protezione in grado di essere
applicato nel momento preciso in cui si dovrebbe avere l’inizio di questi danni, Zhao e
la sua equipe hanno ipotizzato di intervenire proprio all’inizio della riperfusione. La
loro ricerca ha condotto all’allestimento del protocollo del Postcondizionamento
Ischemico (IPostC). Tramite studi sul modello canino (sotto anestesia) sottoposto ad
ischemia miocardica prolungata, hanno eseguito tre occlusioni di 30 secondi ciascuna a
partire da 30 secondi dopo l’inizio della riperfusione. Le occlusioni erano separate l’una
dall’altra da un periodo di riperfusione della durata di 30 secondi. Attraverso questa
tecnica, gli autori hanno riportato la riduzione dell’estensione dell’infarto, dell’edema
tissutale e della disfunzione endoteliare post-ischemica che favorisce le lesioni da
riperfusione: infatti, si è riscontrata una riduzione del 40% dell’entità dell’infarto
provocato sul cuore canino (Zhao et al., 2003).
Come è stato osservato per IPreC, gli stessi effetti benefici del protocollo di IPostC sono
stati confermati in altre specie animali (roditori come topi e ratti, conigli, maiali, gatti,
scimmie) (Skyschally et al., 2009) tra cui l’uomo (Staat et al., 2005).
Attualmente, i meccanismi coinvolti nel IPostC non sono stati completamente compresi,
ma è stato evidenziato che durante questo evento si verifica una riduzione della
produzione di ROS, del sovraccarico di ioni e del fenomeno infiammatorio (Testai
et al., 2015).

Introduzione
13
1.5.3 IL REMOTO IPreC E IPostC (ReIPreC, ReIPostC)
Schmidt e i suoi colleghi hanno dimostrato che brevi e alternati cicli di ischemia degli
arti offrono una significativa protezione durante un infarto del miocardio, andando a
preservare la funzionalità cardiaca e a ridurre le aritmie tipiche nella riperfusione
(Schmidt et al., 2007).
Allo stesso modo, Li e la sua equipe hanno osservato che brevi periodi di ischemia
dell’arto vanno a ridurre il danno miocardico dopo un periodo ischemico attraverso
l’inibizione dello stress ossidativo (Li et al., 2006).
Inoltre, è stato dimostrato che episodi di ischemia a livello renale, nei primi minuti della
riperfusione, vanno a ridurre l’entità dell’infarto miocardico nel ratto (Kerendi et al.,
2005).
Questi fenomeni sono denominati ReIPreC e ReIPostC: si tratta di brevi episodi di I/R
indotti in un organo distante dal cuore, prima o dopo una prolungata ischemia, e
permettono di produrre un effetto cardioprotettivo.
Benché i meccanismi molecolari coinvolti nel ReIPreC e nel RIPostC non siano ancora
ben compresi (ulteriori studi sono in gran parte di tipo osservazionale), questi fenomeni
sono stati osservati distintamente in molte specie di mammiferi tra cui ratti, conigli e
maiali (Testai et al., 2015).

Introduzione
14
1.6 IL PRECONDIZIONAMENTO FARMACOLOGICO (Ph-PreC)
Una procedura clinica di IPreC, che va ad anticipare un infarto miocardico, è quasi
impossibile da applicare poiché non si può predire l’inizio dell’evento ischemico.
Al contrario, una strategia clinica di cardioprotezione che si basa sul IPostC
concettualmente è applicabile in pazienti con infarto acuto del miocardio, ma ciò
conduce a molte difficoltà, rischi e svantaggi.
La definizione delle vie di segnalazione di IPreC e di IPostC pone le basi per lo
sviluppo di strategie farmacologiche che vadano a mimare l’azione di protezione
attraverso l’uso di sostanze in grado di innescare la stessa catena di reazioni
mitocondriali. Entrambi i due protocolli, nei quali vengono reclutate vie di segnalazioni
analoghe, sono mediati da numerosi fattori endogeni tra cui adenosina, acetilcolina,
bradichinina e oppioidi, e anche molecole gassose come NO e S. Infatti, durante la
fase di precondizionamento si ha il rilascio di una serie di sostanze che, legandosi a
recettori sulla superficie cellulare, innescano il meccanismo di protezione dando inizio a
una cascata di eventi intracellulari. Queste “sostanze-innesco” vengono denominate
Triggers.
Durante IPreC e IPostC è coinvolta l’attivazione della Proteina Chinasi C (PKC); si
tratta di isoenzimi che spesso esercitano ruoli opposti sia in condizioni normali sia negli
stati non fisiologici. Sono coinvolti anche altre forme di chinasi come RISK
(reperfusion injury salvage kinase), GSK3B (glycogen synthase kinase 3 beta) e STAT3
(signaling traducer and activator of trascription 3) (Testai et al., 2015).
All’interno della membrana mitocondriale sono presenti diverse tipologie di canali al
potassio che si possono ritrovare anche nella membrana plasmatica cellulare. È stato
ipotizzato che questi canali possano essere considerati fattori scatenanti ed effettori
finali nel processo di cardioprotezione.
È stata dimostrata la presenza di canali al potassio ATP-sensibili e Ca-attivati voltaggio
operati (Kv7.4) ed è stato avvalorato che questi sono coinvolti nella regolazione del
volume mitocondriale, del potenziale di membrana, della regolazione di pH e
dell’apoptosi cellulare.
Inoltre, è stato dimostrato che la Connessina 43, una proteina trasmembranale che
consente la comunicazione diretta tra il citoplasma di cellule adiacenti attraverso delle
giunzioni, sia presente a livello del mitocondrio e a quel livello partecipa al IPreC. È
interessante notare che, la Connessina 43 mitocondriale va a contribuire

Introduzione
15
all’assorbimento di calcio andando a formare strutture di semicanali o andando a
modulare trasportatori ionici già esistenti (Boengler et al., 2005). È stato ipotizzato che
questa proteina possa essere coinvolta nella cardioprotezione indotta dal diazossido, un
attivatore , poiché è stato osservato che nei topi con deficienza di Connessina 43,
questo attivatore risultava privo dei suoi effetti benefici di cardioprotezione (Heinzel et
al., 2005).

Introduzione
16
1.6.1 TRIGGERS
Adenosina
È stato il primo attivatore individuato nel processo di IPreC attraverso studi
effettuati su tessuto cardiaco di coniglio; infatti, è stato osservato che i valori di
questa sostanza incrementano durante brevi periodi di ischemia. Di
conseguenza, è stato osservato che questo aumento provoca la riduzione
dell’inotropismo cardiaco e della dilatazione delle resistenza periferiche e
quindi, come risultato si ottiene un bilanciamento della richiesta/domanda di
ossigeno (Liu et al., 1991).
Quando questa viene liberata dal miocardio ischemico, sembra che induca
l’apertura dei canali attraverso l’attivazione/traslocazione della
Proteina Chinasi C (PKC); infatti, l’adenosina va a legarsi con il proprio
recettore A1, accoppiato a proteina G, e ciò provoca l’attivazione della
Fosforilasi C (PLC) che va a catalizzare l’idrolisi del Fosfatidilinositolo 4,5-
difosfato (PIP2) in Inositolo 1,4,5-trifosfato (IP3) e Diarcilglicerolo (DAG),
stimolando due diverse isoforme della PKC (α e δ), le quali a loro volta
andranno a fosforilare i canali MitoK ATP (Figura 5).
Figura 5. Schema di attivazione della protezione miocardica esercitata dalla
liberazione di adenosina (Rastaldo et al., 2006)

Introduzione
17
Bradichinina
È stato dimostrato che la bradichinina viene rilasciata dalle cellule cardiache
durante i brevi periodi di ischemia (Goto et al., 1995). La bradichinina è un
importante peptide endogeno che media il precondizionamento ischemico
indotto al fine di ottenere cardioprotezione, mediante l'attivazione di una cascata
di trasduzione di segnale univoco che rapidamente produce resistenza/tolleranza
del miocardio a condizioni ischemiche sostenute. Il precondizionamento indotto
da questo peptide esercita effetti cardioprotettivi contro la disfunzione contrattile
postischemica simile a quello che si osserva con il IPreC, in seguito
all’attivazione del proprio recettore B2. La bradichinina esercita questi effetti
mediante l’attivazione del pathway di segnalazione di PI3K/Akt/iNOS e la
regolazione dello stato di ossido-riduzione attraverso il rilascio di NO. Infatti,
questo peptide sembra mimare il precondizionamento ischemico attraverso
molteplici mediatori precondizionanti tra cui i ROS, NO, guanil monofofostato
ciclasi (cGMP), la proteina chinasi G (PKG) e i canali . Diversi studi
hanno descritto che il trattamento con diversi antagonisti della bradichinina, tra
cui HOE-140, selettivo per il recettore B2, hanno completamente annullato
l’effetto protettivo del miocardio, evidenziando quindi il ruolo di questo peptide
come mediatore nella protezione nel precondizionameto ischemico (Sharma et
al., 2015) (Figura 6).
Figura 6. Schema di attivazione del Precondizionamento Ischemico indotto dalla
Bradichinina (Sharma et al., 2015).
Introduzione
18
Oppioidi
Tra le sostanze attivate durante il precondizionamento ischemico si hanno anche
i peptidi oppioidi endogeni; infatti, è stato dimostrato che a seguito di brevi
episodi di ischemia si ha un incremento dei livelli di encefalina nel tessuto
miocardico. Inoltre, è stata confermata la presenza nel miocardio dei recettori
oppioidi μ, δ e κ, i quali sono accoppiati a proteina G. Sostanze agoniste di
questi recettori vanno a mimare l’effetto cardioprotettivo sviluppato durante il
precondizionamento (Maslov et al., 2014).
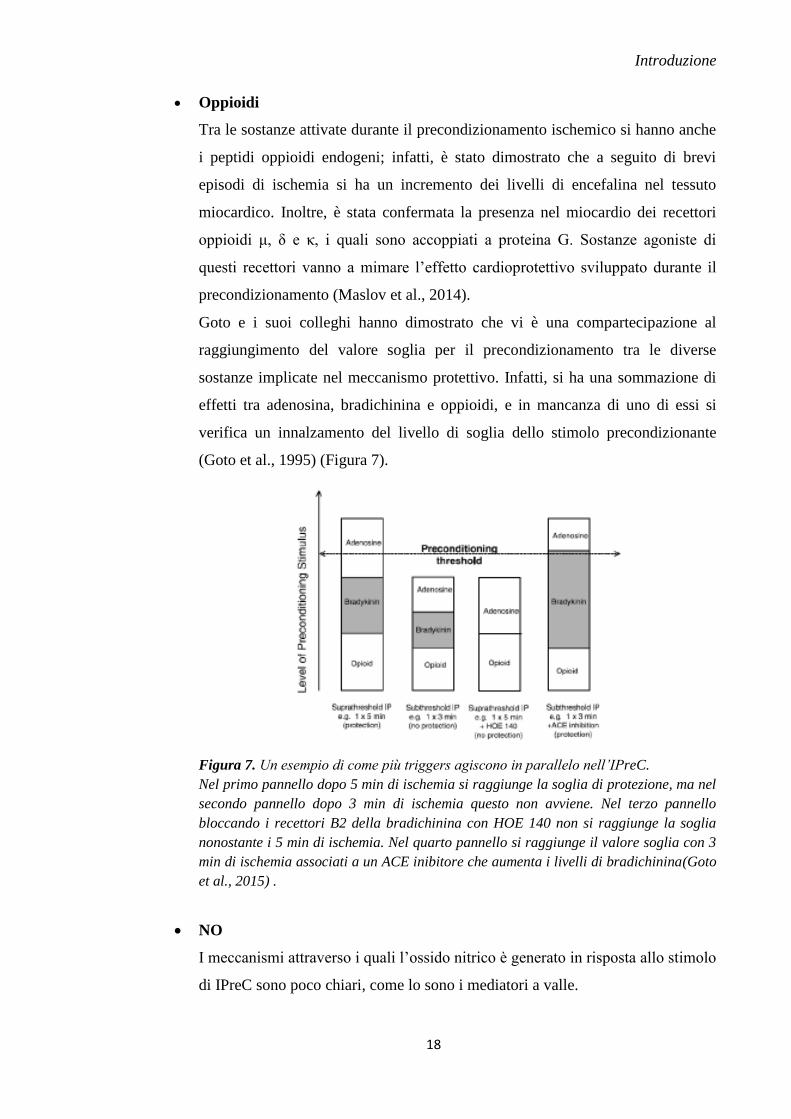
Goto e i suoi colleghi hanno dimostrato che vi è una compartecipazione al
raggiungimento del valore soglia per il precondizionamento tra le diverse
sostanze implicate nel meccanismo protettivo. Infatti, si ha una sommazione di
effetti tra adenosina, bradichinina e oppioidi, e in mancanza di uno di essi si
verifica un innalzamento del livello di soglia dello stimolo precondizionante
(Goto et al., 1995) (Figura 7).
Figura 7. Un esempio di come più triggers agiscono in parallelo nell’IPreC.
Nel primo pannello dopo 5 min di ischemia si raggiunge la soglia di protezione, ma nel
secondo pannello dopo 3 min di ischemia questo non avviene. Nel terzo pannello
bloccando i recettori B2 della bradichinina con HOE 140 non si raggiunge la soglia
nonostante i 5 min di ischemia. Nel quarto pannello si raggiunge il valore soglia con 3
min di ischemia associati a un ACE inibitore che aumenta i livelli di bradichinina(Goto
et al., 2015) .
NO
I meccanismi attraverso i quali l’ossido nitrico è generato in risposta allo stimolo
di IPreC sono poco chiari, come lo sono i mediatori a valle.

Introduzione
19
S
Il solfuro di idrogeno è un neurotrasmettitore endogeno gassoso prodotto
principalmente dagli enzimi cistationina β-sintetasi (CBS) e cistationina γ-liasi
(CGL), sono stati recentemente suggeriti come mediatori della fase tardiva del
IPreC; infatti, è stato dimostrato che su cardiomiociti di ratto, il trattamento con
il solfuro di idrogeno conferiva cardioprotezione ritardata (16-28 ore dopo lo
stimolo precondizionante) e che l’inibizione della biosintesi di questa molecola
andava ad abolire questa cardioprotezione. Ciò ha confermato il ruolo chiave di
questo neurotrasmettitore durante la fase tardiva del precondizionamento (Pan et
al., 2006).
Introduzione
20
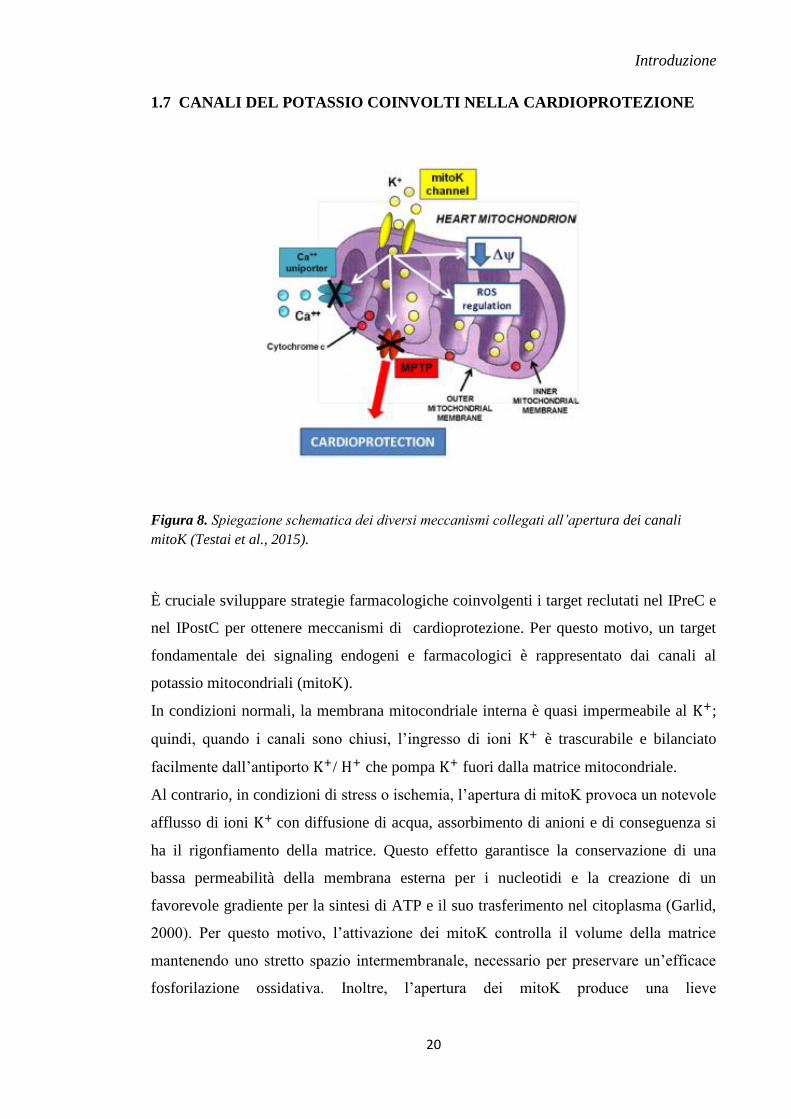
1.7 CANALI DEL POTASSIO COINVOLTI NELLA CARDIOPROTEZIONE
Figura 8. Spiegazione schematica dei diversi meccanismi collegati all’apertura dei canali
mitoK (Testai et al., 2015).
È cruciale sviluppare strategie farmacologiche coinvolgenti i target reclutati nel IPreC e
nel IPostC per ottenere meccanismi di cardioprotezione. Per questo motivo, un target
fondamentale dei signaling endogeni e farmacologici è rappresentato dai canali al
potassio mitocondriali (mitoK).
In condizioni normali, la membrana mitocondriale interna è quasi impermeabile al ;
quindi, quando i canali sono chiusi, l’ingresso di ioni è trascurabile e bilanciato
facilmente dall’antiporto / che pompa fuori dalla matrice mitocondriale.
Al contrario, in condizioni di stress o ischemia, l’apertura di mitoK provoca un notevole
afflusso di ioni con diffusione di acqua, assorbimento di anioni e di conseguenza si
ha il rigonfiamento della matrice. Questo effetto garantisce la conservazione di una
bassa permeabilità della membrana esterna per i nucleotidi e la creazione di un
favorevole gradiente per la sintesi di ATP e il suo trasferimento nel citoplasma (Garlid,
2000). Per questo motivo, l’attivazione dei mitoK controlla il volume della matrice
mantenendo uno stretto spazio intermembranale, necessario per preservare un’efficace
fosforilazione ossidativa. Inoltre, l’apertura dei mitoK produce una lieve

Introduzione
21
depolarizzazione del potenziale di membrana, responsabile dell’assorbimento ridotto di
nella matrice, preservandola da un dannoso sovraccarico e dalla successiva
apertura degli MPTP (Hausenloy et al., 2005). Quindi, l’apertura dei mitoK come target
dell’IPreC previene la formazione e l’apertura degli MPTP, andando a ridurre il rilascio
di fattori pro-apoptotici durante la riperfusione, preservando l’integrità della membrana
mitocondriale.
1.7.1 CANALI
Il canale mitocondriale al potassio sensibile all’ATP è stato il primo canale mitoK
riconosciuto come un target efficace per il precondizionamento farmacologico. Sostanze
che inducono l’apertura di questi canali, come Bimakalim e Cromakalim, sono state
indicate in quanto sono in grado di produrre effetti cardioprotettivi a dosi tali da non
influenzare la durata del potenziale d’azione, suggerendo l’esistenza di un sito di azione
intracellulare indipendente dai canali del sarcolemma (Yao et al., 1994) (Grover et al.,
1995).
Successivamente, per poter confermare il ruolo dei canali sono stati effettuati
studi sui mitocondri di cuore bovino in cui sono stati osservati gli effetti del Diazossido,
agente attivate dell’apertura di questi canali, ad una concentrazione nettamente inferiore
a quella necessaria per l’apertura del canale del sarcolemma (Garlid et al., 1997).
Anche se per diversi anni ci sono stati dibattiti su quale tipo di canale
(sarcolemmatico o mitocondriale) fosse coinvolto nella cardioprotezione, attualmente è
stato accettato che i mitocondri giocano un ruolo cruciale nell’evento I/R ed è probabile
che il ruolo del canale sia prevalente a questo livello (Garlid & Halestrap,
2012).
Infatti, numerosi studi a sostegno di questa ipotesi sono stati raccolti negli ultimi anni, a
dimostrazione che IPreC e più recentemente anche IPostC, può essere riprodotto
attraverso l’uso di diverse sostanze che agiscono come attivatori selettivi dei canali
.
Fino ad oggi, non è ancora stata assegnata al nessuna identità molecolare ben
definita; tuttavia, è stata proposta una somiglianza con quello del sarcolemma. Infatti, è
probabile che il canale sia un complesso etero-ottamerico, contente le

Introduzione
22
subunità 6.1 o 6.2 e subunità recettoriali della sulfonilurea (SUR2), mentre la
subunità SUR1 sembra che non sia presente. Recentemente, è stata descritta una
variante SUR2 specifica del mitocondrio (mitoSUR2), generato dallo splicing
intraesonico della classica subunità SUR2; è importante considerare che in topi in cui
non è espresso SUR2, e in cui è rimasta espressa la forma mitoSUR2, la strategia IPreC
è ancora efficace nel ridurre il danno ischemico (Ye et al., 2009).
Inoltre, lo scambio farmacologico tra i canali e il complesso mitocondriale
respiratorio II (deidrogenasi succinato) ha portato ad ipotizzare che il complesso II
potrebbe essere un ulteriore componente di questo canale mitocondriale (Ardehali et al.,
2005).
È interessante osservare che il gruppo seguito da O’Rourke è riuscito a fare passi in
avanti nella comprensione della struttura molecolare di questo canale: infatti, hanno
dimostrato che il canale ROMK (canale al potassio della midollare esterna), che
normalmente è espresso sulla membrana superficiale delle cellule renali, è localizzato
sulla membrana interna dei mitocondri cardiaci e ha la funzione di mediare il flusso di
ioni ATP-sensibile, così da indurre la protezione contro stimoli di morte cellulare.
Questa osservazione ha portato all’ipotesi che il canale mitoROMK possa formare il
poro nella subunità del canale (Foster et al, 2012).
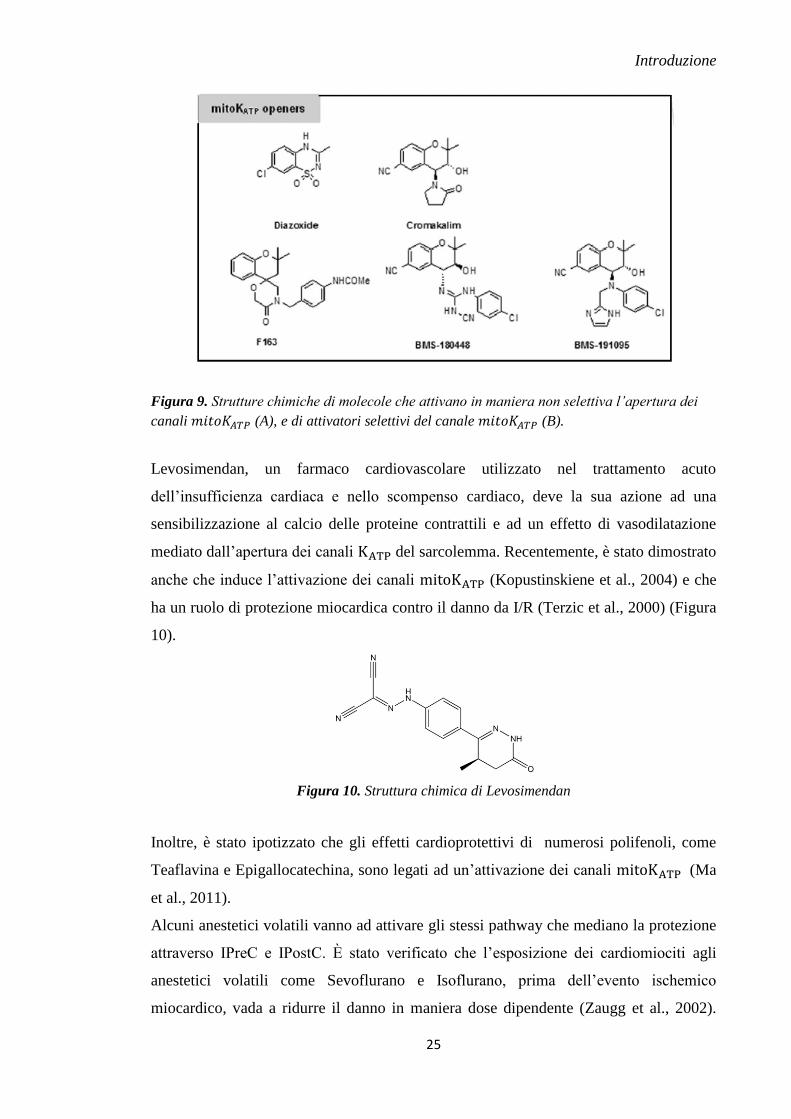
1.7.1.1 ATTIVATORI DEI CANALI
I primi esperimenti fatti per confermare le proprietà cardioprotettive degli attivatori del
canale sono stati effettuati con Cromakalim o suoi analoghi, in cui il nucleo
di base è costituito dal benzopirano. Il Cromakalim è completamente privo di selettività
per i canali , infatti causa l’attivazione dei canali del sarcolemma
presente nelle cellule muscolari lisce vascolari e nei cardiomiociti. Il nucleo
benzopirano ha rappresentato l’elemento maggiormente sottoposto a manipolazione
chimica per lo sviluppo di nuovi attivatori del canale (Mannhold, 2004)
(Figura 9).
Attraverso vari studi è stato evidenziato che il Diazossido, un prototipo della classe
delle benzotiazine, ha la proprietà di riuscire ad aprire con una certa selettività, sebbene
non assoluta, i canali ; infatti, a dose moderate può attivare questi canali
producendo una fase di precondizionamento farmacologico, legate all’attivazione dei
(Garlid et al., 1996) (D’hahan et al., 1999) (Figura 9).

Introduzione
23
Sebbene il ruolo di ROS nel IPreC sia ancora controverso, Heusch e i suoi colleghi
hanno dimostrato che il diazossido conferisce una protezione attraverso la generazione
di radicali liberi (Pain et al., 2000). È stato ipotizzato inoltre, che esistano dei
meccanismi indipendenti dai canali e un possibile bersaglio molecolare è
probabilmente la succinato deidrogenasi mitocondriale (Rodrigo et al., 2004).
Inoltre, è stato dimostrato che il complesso II sia un importante regolatore o un
componente del , poiché il diazossido è in grado di attivare questo canale, ma
anche di inibire il complesso II. Al contrario, l’uso di inibitore del complesso II, come
l’aptenin A5, fa sì che si abbia l’apertura del canale anche a concentrazioni
minori (Wojtovich & Brooks, 2009). Tuttavia, il diazossido provoca effetti aspecifici,
quali vasodilatazione e iperglicemia, legati all’attivazione dei canali
sarcolemmatici, difficili da gestire per poter sviluppare target farmacologici nella
cardioprotezione.
La Bristol Mayer Squibb Company nel tentativo di sviluppare molecole che abbiano
selettività verso i canali , ha sintetizzato nuovi ibridi attraverso la
coniugazione della parte benzopiranica del Cromakalim con un nucleo
cianoguanidinico. Questa ricerca ha condotto allo sviluppo di due interessanti composti:
- BMS 180448: molecola dotata di una buona potenza cardioprotettiva e con una bassa
azione vaso rilasciante.
- BMS 191095: un 4-N-aril-benzopirano sostituto, circa 30 volte più selettivo rispetto al
BMS 180448. La sua azione farmacologica è antagonizzata dall’acido 5-idrossi-
decanoico (5-HD), un bloccante selettivo del canale . Sfortunatamente, questa
molecola non può essere utilizzata in clinica poiché induce tossicità neuronale, per cui
può essere sfruttata solo per scopi di ricerca (Grover & Atwal, 2002) (Figura 9).
Successivamente, sono state sviluppate nuove molecole ottenute mediante l’inserimento
in posizione 4 del benzopirano di un nucleo 2’-carbossialchil-indolo o 2’-carbossialchil-
indolinico. In particolare, la seconda molecola ha mostrato una migliore azione
cardioprotettiva sia in vivo che in vitro, e una debole attività vasorilasciante. Si suppone
che questi nuovi composti possano andare a influenzare il canale visto che i
loro effetti cardioprotettivi vengono completamente bloccati dal 5-HD (Lee et al.,
2003). Inoltre, Jung e i suoi colleghi hanno osservato che un analogo del benzopiranil-
indolo, KR-31466, può ridurre il danno ipossico in cellule H9c2 cardiache attraverso
l’apertura dei canali . È stato ipotizzato che questa protezione da morte

Introduzione
24
indotta da ipossia si verifica anche per il coinvolgimento di meccanismi di attivazione
della PKC (Jung et al, 2003).
Recentemente, Breschi e colleghi hanno sintetizzato una nuova serie di derivati
benzopiranici sostituiti con gruppi spiromorfolinici e spiromorfolonici. L’inserimento di
uno spirociclo sostituito sul carbonio 4 al gruppo benzopirano ha conferito una certa
rigidità alla struttura in questa porzione della molecola tale da rendere migliore la
propria selettività nei confronti dei canali . Alcuni di questi composti hanno
mostrato un buon profilo cardioprotettivo nei modelli di ex vivo di I/R e modesti effetti
a livello della muscolatura liscia vascolare. In particolare, il derivato N-acetil-
spiromorfolone (F163) è stato sottoposto ad ulteriori studi, in quanto è risultato il
migliore della serie di questi composti. È stata dimostrata la sua enantioselettività nel
profilo della cardioprotezione, infatti l’enantiomero levogiro è l’unico responsabile della
sua azione (Rapposelli et al., 2009). In particolare, sono stati confermati gli effetti
cardioprotettivi di F163 attraverso studi più approfonditi effettuati sia su colture di
cardiomioblasti sottoposti ad anossia/riperfusione sia in vivo nel modello di infarto
acuto del miocardio; inoltre, è stato dimostrato il suo target mitocondriale (Calderone et
al, 2010).
Attualmente, è stato osservato che il nuovo composto è capace di produrre tutti gli
effetti tipici dovuti all’apertura del canale : ovvero provoca una debole
depolarizzazione, una riduzione della ricaptazione di ioni , un aumento nel rilascio
di ioni e un debole rigonfiamento mitocondriale. Infatti, è stata dimostrata
l’esistenza di una stretta correlazione tra l’induzione di depolarizzazione e il rilascio di
ioni dalla matrice mitocondriale; in particolare, è stato osservato che è sufficiente
una piccola depolarizzazione per influenzare notevolmente il rilascio di ioni
accumulati nella matrice mitocondriale. Questi studi hanno suggerito che il nuovo
composto, F163, possa rappresentare un interessante prototipo per lo sviluppo di
innovativi attivatori del canale , evidenziando un’azione molto più potente
rispetto al diazossido e priva di effetti significativi a livello della pressione sanguigna e
del metabolismo del glucosio (Calderone et al., 2010)(Figura 9).
Introduzione
25
Figura 9. Strutture chimiche di molecole che attivano in maniera non selettiva l’apertura dei
canali (A), e di attivatori selettivi del canale (B).
Levosimendan, un farmaco cardiovascolare utilizzato nel trattamento acuto
dell’insufficienza cardiaca e nello scompenso cardiaco, deve la sua azione ad una
sensibilizzazione al calcio delle proteine contrattili e ad un effetto di vasodilatazione
mediato dall’apertura dei canali del sarcolemma. Recentemente, è stato dimostrato
anche che induce l’attivazione dei canali (Kopustinskiene et al., 2004) e che
ha un ruolo di protezione miocardica contro il danno da I/R (Terzic et al., 2000) (Figura
10).
Figura 10. Struttura chimica di Levosimendan
Inoltre, è stato ipotizzato che gli effetti cardioprotettivi di numerosi polifenoli, come
Teaflavina e Epigallocatechina, sono legati ad un’attivazione dei canali (Ma
et al., 2011).
Alcuni anestetici volatili vanno ad attivare gli stessi pathway che mediano la protezione
attraverso IPreC e IPostC. È stato verificato che l’esposizione dei cardiomiociti agli
anestetici volatili come Sevoflurano e Isoflurano, prima dell’evento ischemico
miocardico, vada a ridurre il danno in maniera dose dipendente (Zaugg et al., 2002).

Introduzione
26
Questi effetti sono antagonizzati dal 5-HD, a dimostrazione del coinvolgimento dei
canali (Van Allen et al., 2012).
Attualmente, nessuno studio specifico ha tentato di identificare il sito di legame degli
attivatori sui canali ; tuttavia, in analogia con i canali del sarcolemma,
l’interazione con la subunità SUR sembra essere l’ipotesi più probabile.
1.7.2 CANALI MITOCONDRIALI AL POTASSIO SENSIBILI AL CALCIO
Canali e loro attivatori
Nel 2002, O’Rourke e la sua equipe hanno dimostrato l’esistenza di canali del
potassio a livello della membrana interna mitocondriale in cui la conduttanza era
influenzata dall’ingresso di ioni ( ) attraverso esperimenti
effettuati su cuore di cavia (Xu et al., 2002). Precedentemente, era stata descritta
la presenza di questo canale nei mitocondri delle cellule del glioma, ma senza
assegnare loro alcuna funzione specifica (Siemen et al., 1999).
Lo studio condotto da O’Rourke e colleghi consisteva nell’impiegare mitoplasti
patch-clamp mediante i quali sono riusciti a identificare il passaggio di correnti
potassiche attraverso questo canale. Inoltre, hanno evidenziato il ruolo svolto dal
canale nella protezione contro il danno da I/R. Infatti, l’uso di
attivatori di questo canale, come la molecola NS1619 (Figura 11), inducono una
riduzione delle dimensioni dell’infarto in seguito a un periodo di I/R. Questi
effetti, inoltre, sono antagonizzati dall’ uso di un bloccante selettivo, come la
Paxillina (Xu et al., 2002) (Ardeali & O’Rourke, 2005). Ulteriori studi, hanno
confermato che l’apertura farmacologica dei canali viene mimata nel
precondizionamento precoce e ritardato (Cao et al., 2005), così come nel
postcondizionamento (Jin et al., 2012).
Tuttavia, concentrazioni micromolari di NS1619 possono provocare anche
effetti anomali a livello mitocondriale, tra cui una diminuzione del controllo
respiratorio, una insensibilità ai bloccanti selettivi del canale , e una
profonda caduta del potenziale di membrana anche in assenza di ioni
(Bednarczyk et al., 2008). Da osservare che, alte concentrazioni di questa
molecola provocano l’inibizione dei canali al calcio di tipo L, delle correnti al

Introduzione
27
cloro attivate dal calcio, dei canali al calcio voltaggio-gated, e canali al potassio
e al sodio. Questi effetti aspecifici rendono difficile la comprensione e
l’interpretazione del ruolo dei canali svolto nella cardioprotezione
(Park et al., 2007).
Degno di attenzione è il composto NS11021 (Figura 11), un nuovo attivatore dei
canali , chimicamente non correlato al precedente, più potente e più
specifico rispetto al composto NS1619 (Bantzen et al., 2007). Questa molecola
esercita effetti protettivi attraverso l’attivazione dei canali , visto che
questi effetti sono completamente antagonizzati dalla Paxillina. La
somministrazione di concentrazioni nanomolecolari di NS11021 induce un
miglioramento della performance bioenergetica del mitocondrio cardiaco,
attraverso un incremento dell’assorbimento di ioni e un debole
rigonfiamento della membrana, senza ingenti cambiamenti nei valori del
potenziale di membrana mitocondriale (ΔΨ).
Oltre alla sintesi dei vari composti della serie NS, è stato sviluppato un nuovo
composto, il CGS7184 (etil-1-[(4-clorofenil)-amino]osso]-2-idrossi-6-
trifluorometil-1H-indolo-3-carbossilato) (Figura 11); questo provoca la
riduzione della produzione di ROS nel mitocondrio isolato dal cervello di ratto
(Kulawiak et al., 2008) e la depolarizzazione del potenziale di membrana
mitocondriale nella linea cellulare EAhy926 (Wrzosek et al., 2009). Questi
effetti, che sembrano prevedere eventuali proprietà di protezione, dovrebbero
essere correlati alla modulazione dell’omeostasi degli ioni , probabilmente
mediante l’interazione con il reticolo sarcoplasmatico (Wrzosek et al., 2012).
Attualmente, è stato ipotizzato che la struttura del canale espresso a livello
mitocondriale e sarcolemmatico sono conservate, dunque il canale
potrebbe essere composto da quattro subunità alfa, che formano il poro, e da
quattro subunità beta, le quali hanno il compito di modulare la sensibilità al
calcio e l’attività (Meera et al., 1996).
È stata descritta una subunità beta-1 ausiliaria, necessaria per IPreC e rilevabile
anche a livello del sarcolemma (Wang et al., 2008); tuttavia, alcune varianti di
splicing a livello mitocondriale non possono essere escluse.
Nel 2013, Toro e la sue equipe hanno dimostrato l’esistenza di KCNMA1
(Slo1), un gene a livello mitondriale, che codifica per il canale . La
loro ipotesi è stata supportata mediante esperimenti effettuati su topi in cui

Introduzione
28
questo gene non era espresso e dove NS1619 non riusciva a svolgere la sua
funzione cardioprotettiva (Singh et al., 2013).
Figura 11. Struttura chimica di molecole che inducono l’apertura del canale
.
Recentemente, è stato dimostrato che Naringenina, un flavanone che si ritrova in
abbondanza nel genere Citrus, possiede un’attività cardioprotettiva mediata
dall’apertura dei canali (Figura 12). Infatti, il trattamento effettuato
con questa molecola prima dell’inizio dell’attacco ischemico acuto in vivo o
durante l’intero evento ischemico produce il dimezzamento della dimensione del
danno da I/R. Inoltre, su mitocondri di cuore isolato, Naringenina è in grado di
produrre gli stessi effetti tipici degli attivatori del canale tra cui
aumento dell’entrata di ioni potassio all’interno del mitocondrio, una debole
depolarizzazione mitocondriale e una riduzione dell’assorbimento di ioni calcio.
Il target d’azione è stato dimostrato con l’uso di bloccanti selettivi come
Paxillina e Iberiotossina (Testai et al., 2013). Recentemente, è stato osservato
che Naringina, il glicoside della Naringenina, abbia effetti protettivi in cellule
cardiache H9c2 danneggiate da alti livelli di glucosio, attraverso il pathway
MAPK (Chen & Zweier, 2014); questa ipotesi, se confermata anche in modelli
in vivo in cui naringina viene idrolizzata nell’aglicone, potrebbe significare che
il reclutamento di MAPK sia un evento a monte dell’attivazione del canale
mitoBK.

Introduzione
29
Figura 12 . Struttura chimica di Naringenina
Canali
Oltre ai canali , è stata confermata l’esistenza di un canale al potassio
sensibile al calcio ad intermedia conduttanza, o 3.1. Questo canale è
stato rilevato nel mitocondrio purificato dalle cellule del carcinoma del colon
(De Marchi et al., 2009), nelle cellule HeLa e nei fibroblasti embrionali (Sassi et
al., 2010). Il canale risulta avere lo stesso peso molecolare del canale
localizzato nella membrana plasmatica, e ciò suggerisce che possano avere
equivalenti proprietà biofisiche e farmacologiche. Il ruolo farmacologico di
questo canale non è ancora stato approfondito, ma l’uso di un inibitore
come TRAM-34, provoca iperpolarizzazione della membrana mitocondriale, in
accordo con il tipico profilo degli attivatori del canale al potassio mitocondriale
(Sassi et al., 2010).
Attualmente, il canale non è stato rilevato a livello cardiaco.
Canali
Recentemente, il canale , un canale al potassio sensibile al calcio a
debole conduttanza, è stato identificato, localizzato mediante microscopia
elettronica, e purificato a livello della membrana interna del mitocondrio
cardiaco di cavia e a livello del mitocondrio delle cellule neuronali (Dolga et al.,
2013). Inoltre, è stato dimostrato che la loro attivazione mimi il
precondizionamento farmacologico e quindi conferisca cardioprotezione, come è
mostrato dal miglioramento funzionale e metabolico registrato durante la
riperfusione su cuori di cavia isolati e perfusi. In più, l’attivazione del canale
sembra che induca, similmente all’apertura dei canali

Introduzione
30
, un controllo sulla produzione intramitocondriale di ROS (Stowe et
al., 2013).
Tra i composti sintetizzati come attivatori del canale , DCEBIO
(Figura 13) che ha dimostrato di indurre cardioprotezione. La sua azione è
antagonizzata da l’uso sia di NS8593, un antagonista dell’isoforma del canale
, sia da TBAP, uno scavenger del perossinitrile.
DCEBIO
Figura 13. Struttura chimica di un attivatore del canale .
1.7.3 CANALI mitoKv
Nel 2005, Szabò e la sua equipe hanno evidenziato la presenza di canali al potassio
voltage-gated 1.3 (Kv1.3) all’interno della membrana del mitocondrio dei linfociti T
(Szabò et al., 2005). Successivamente, questi canali sono stati localizzati anche nei
mitocondri dei macrofagi (Vicente et al, 2006) e dei neuroni dell’ippocampo
(Bednarczyk et al., 2010). Sebbene i canali Kv1.3 descritti nei mitocondri dei neuroni
dell’ippocampo presentino una diversa conduttanza rispetto ai canali Kv1.3 localizzati
nei linfociti, entrambi i canali evidenziano una sensibilità alla margatossina. È
interessante notare che un’iperpolarizzazione del potenziale di membrana mitocondriale
può essere registrato durante l’inibizione del canale mitoKv1.3, indicando che nei
mitocondri eccitati questi canali sono normalmente attivati.
Inoltre, ulteriori studi hanno evidenziato che nei mitocondri dei macrofagi J774 sono
espressi i canali Kv1.3 e Kv1.5 e che l’inibizione di questi canali potrebbe
efficacemente indurre apoptosi in questa linea cellulare di macrofagi. Pertanto, i risultati
ottenuti indicano che il meccanismo proposto per i canali mitoKv1.3 può essere esteso
ad altri canali Kv localizzati nei macrofagi, importanti componenti del sistema
immunitario (Leanza et al., 2012). Recentemente, è stato dimostrato che l’inibizione del
canale mitoKv1.3 induce un effetto drastico sul processo di apoptosi delle cellule

Introduzione
31
tumorali mediante un pathway Bax/Bak indipendente (Leanza et al., 2012).
Attualmente, i canali mitoKv1.3 non sono stati riconosciuti a livello cardiaco.
Canali mitoKv7.4
Il canale mitoKv7.4 è di recente scoperta, infatti, è stato per la prima volta
localizzato, mediante studi effettuati con Western blot, microscopia elettronica e
analisi immunocitochimiche, a livello del mitocondrio dei cardiomiociti di ratto.
Inoltre, attraverso ulteriori studi di funzionalità mitocondriale, è stato dimostrato
che l’attivazione farmacologica di questo canale induce depolarizzazione del
potenziale di membrana mitocondriale, provocando la riduzione
dell’assorbimento di mitocondriale e quindi, inducendo la riduzione dei
danni indotti dal danno I/R (Testai et al, 2016).
1.7.4 CANALI mitoTASK-3
Il canale mitoTASK-3 è stato localizzato a livello del mitocondrio del melanoma, delle
cellule cheratinocitiche (Szewczyk et al., 2009) e dell’ippocampo di embrioni di ratto
(Kajma & Szewczyk, 2012). Tuttavia, le sue proprietà funzionali devono ancora essere
approfondite.
Szewczyk e la sua equipe hanno dimostrato che questo canale svolge un ruolo protettivo
nei cheratinociti umani sottoposti a radiazioni UVB (Toczylowska-Maminska et al.,
2014).
È importante osservare che a oggi non è stata riconosciuta la presenza del canale
mitoTASK-3 nel cuore.

Introduzione
32
1.8 PROTEIN KINASI
La cascata di eventi mediata dai complessi chinasici rappresenta il principale crocevia di
pathway mitocondriali indotti dal danno miocardico da I/R. La prima chinasi ad essere
stata scoperta nel coinvolgimento di IPreC e IPostC è la PKC; tuttavia, ad oggi stanno
emergendo numerose chinasi coinvolte in queste fasi.
Proteina Chinasi C (PKC)
Nel 1994 Ytrehus e la sua equipe sono stati i primi a descrivere il ruolo della
PKC nel precondizionamento ischemico; essi hanno evidenziato che inibitori
selettivi della PKC tra cui staurosporina e polimixina B impediscono l’effetto
cardioprotettivo del precondizionamento nei conigli. Allo stesso modo, l’uso di
attivatori della PKC, come gli esteri del forbolo, inducono il cuore in uno stato
protetto (Ytrehus et al., 1994).
La PKC è una serina/treonina chinasi attivata da cofattori lipidici che derivano
dalla degradazione dei fosfolipidi di membrana da parte della Fosfolipasi C. Nel
cuore è presente in diverse isoforme, che possono essere classificate in isoforme
classiche: α, β e γ che dipendono sia dal DAG sia dal , e in isoforme nuove:
ε, δ e η che sono -indipendenti e sono attivate solo dal DAG; inoltre, sono
presenti isoforme atipiche come la ζ che non richiede né DAG né calcio. Le
isoforme della PKC attivate possiedono un’alta specificità per il legame con una
proteina nominata Recettore attivato da Chinasi C (RACK); questi recettori si
trovano solo a livello di alcuni organelli all’interno della cellula dove hanno il
compito di traslocare l’isoforma di PKC in prossimità di una specifica proteina
substrato e il loro legame con la PKC completa l’attivazione dell’isoforma e
causa la fosforilazione del substrato nelle vicinanze (Johnson et al, 1996).
Quindi, l’attivazione della PKC richiede la traslocazione fisica dell’enzima dal
citosol al sito di legame nella membrana del sarcolemma e questo sembra essere
un evento chiave del precondizionamento ischemico (Liu et al., 1994).
Esaminando le varie isoforme della PKC, Gregory e i suoi colleghi hanno
dimostrato il ruolo della PKC-ε nell’IPreC: infatti, attraverso studi su cuore di
topo fosfolambano-deficiente e con livelli ridotti di questa isoforma, hanno
evidenziato un’insufficienza nel recupero contrattile durante la riperfusione
seguente i 40 minuti di ischemia. L’accoppiamento di questi topi con altri che

Introduzione
33
esprimevano la ε-RACK ha portato a dimostrare che nei transgeni risultanti si
aveva una maggiore traslocazione di PKC-ε e un miglioramento nella
contrattilità cardiaca durante la riperfusione (Gregory, et al, 2004).
Recentemente, è stato dimostrato che la PKC-ε va ad attivare l’enzima
mitocondriale Aldeide Deidrogenasi 2 (ALDH2), la quale ha il compito di
rimuovere i prodotti della perossidazione lipidica per garantire una protezione
delle funzioni mitocondriali (Chen et al., 2008). Inoltre, Baines e la sua equipe
hanno osservato che la traslocazione di PCK-ε può essere considerata un
meccanismo aggiuntivo per la cardioprotezione: la diretta fosforilazione dei
componenti MPTP inibisce l’apertura del poro (Baines et al., 2002).
Per quanto riguarda l’isoforma δ, il suo ruolo è controverso infatti, sembrerebbe
che la sua inibizione durante la riperfusione abbia azione cardioprotettiva.
Churchill e il suo gruppo hanno dimostrato che il precondizionamento
ischemico, in un modello isolato di cuore di ratto perfuso, causa una
diminuzione della PKC-δ nella frazione mitocondriale, contrastata da un
aumento di PKC-ε (Churchill et al., 2010). Questi recenti studi hanno indicato
che la PKC-δ non ha un ruolo protettivo nel cuore, contribuendo a sottolineare i
ruoli divergenti delle isoforme ε e δ nella cardioprotezione (Simkhovich et al.,
2013).
Attualmente, è stato dimostrato che l’attivazione dell’isoforma δ innesca la
piruvato deidrogenasi chinasi mitocondriale e ciò provoca l’inibizione sia della
piruvato deidrogenasi sia della rigenerazione di ATP. Inoltre, l’attivazione di
questa isoforma induce una perfusione compromessa dei miociti dopo l’evento
ischemico, determinando in tal modo ulteriori lesioni tissutali (Testai et al.,
2015).
La Proteina Chinasi C può essere attivata attraverso la via della proteina G
(Fosfolipasi C e DAG) oppure in seguito alla modulazione di ROS, che avviene
durante la fase di riperfusione. Il suo meccanismo d’azione e i suoi target non
sono ancora del tutto definiti, ma sicuramente sono stati fatti passi in avanti nella
comprensione del suo ruolo nella cardioprotezione.
In particolare, è stato osservato che attraverso la via della proteina G, la PKG va
a mediare l’attivazione della PKC- ε1 e PKC- ε2. La PKC-ε1, situata nei pressi
della membrana mitocondriale interna, fosforila e apre i canali con
conseguente aumento di produzione di ROS. Invece, la PKC-ε2 ha il compito di

Introduzione
34
inibire l’apertura degli MPTP e quindi di protezione del cuore (Costa & Garlid,
2008).
L’azione della PKC provoca inoltre, un innalzamento del pH intracellulare
agendo sullo scambiatore / (Simkhovich et al., 2013).
Per quanto riguarda la PKC-α, sembra che questa attivi direttamente la 5’-
nucleotidasi, un enzima che genera un aumento dei livelli di adenosina a partire
dall’adenosina monofosfato (AMP) (Kitakaze et al., 1993). È stato dimostrato,
ulteriormente, che la PKC può aumentare l’affinità del recettore A2b per
l’adenosina attraverso la fosforilazione dello stesso recettore o l’accoppiamento
a proteine; sembra possibile, infatti, che questo recettore possa rispondere
all’adenosina endogena solo dopo la sensibilizzazione della PKC, che ne
abbassa la soglia di sensibilità (Kuno et al., 2007) (Yang et al., 2010).
Altri ruoli riconducibili alla PKC sono: la riduzione della morte cellulare per
apoptosi (riconducibile soprattutto all’isoforma ε) e la riduzione del volume
dell’infarto (Saurin et al., 2002); inoltre, sembra che la PKC contribuisca alla
fosforilazione e all’attivazione della Connessina-43, una proteina fondamentale
per la formazione delle gap-junctions intercellulari (Schultz et al., 2003) (Figura
14).
Figura 14. PKC Signaling (Simkhovic, et al., 2013).

Introduzione
35
Proteina Chinasi G (PKG)
Si tratta di una proteina serina/treonina chinasi cGMP-dipendente. È stata
implicata per la prima volta nell’IPreC in studi in cui è stato dimostrato
l’aumento dei livelli di cGMP in cuori precondizionati (Iliodromitis et al., 1996).
Quando viene attivata dalla cascata del Fosfatidilinositolo-3-chinasi (PIK3) che
conduce a un incremento dei valori di cGMP, la PKG va a mediare l’apertura dei
canali MitoKATP attraverso l’attivazione della PKC-ε (Costa et al., 2005). È
stato dimostrato attraverso studi su mitocondri di topo, che cGMP e PKG sono
in grado di inibire l’apertura degli MPTP (Takuma et al., 2001). Hausenloy e il
suo gruppo hanno ipotizzato che l’attivazione della PKG possa mediare
l’inibizione degli MPTP che si verifica al momento della riperfusione
miocardica (Hausenloy & Yellon, 2006). È stato suggerito, inoltre, che
l’attivazione della PKG possa andare a diminuire la comunicazione tra le gap-
junction intercellulari (Kwak et al., 1995) (Figura 15).
Figura 15. PKG signaling (Costa et al., 2005).
Proteina Chinasi A (PKA)
Sanada e la sua equipe hanno dimostrato che durante la fase di
precondizionamento si verifica l’attivazione della PKA (Sanada et al., 2004).
È interessante notare (in comune con p38 MAPK) come la PKA potrebbe avere
un duplice ruolo nel precondizionamento: la sua attivazione durante l’ischemia
sembrerebbe essere dannosa, mentre l’attivazione durante la fase di
precondizionamento porterebbe a protezione (Makaula et al., 2005).

Introduzione
36
Durante il precondizionamento si ha quindi, l’attivazione della PKA correlata
alla generazione di cAMP indotta dall’ischemia. Secondo alcuni studi è stato
suggerito che la mediazione della PKA nel processo di IPreC provochi una
protezione nei miociti dalla loro distruzione mediata dalla Calpaina al momento
della riperfusione (Inserte et al., 2004).
Tra i meccanismi d’azione più avvalorati è stato riscontrato quello su modelli di
cuore canino in cui si osserva che l’attivazione transitoria della PKA durante la
fase di precondizionamento, conferisce protezione al miocardio provocando
l’inibizione della Rho GTP-asi e la Rho Chinasi. Gli effettori di questa cascata
di reazioni non sono ancora del tutto chiari (Sanada et al, 2004).
Tirosin Chinasi (TK)
Possono essere classificati in: recettori tirosin-chinasici che provocano
l’attivazione della PKC e quindi, possono essere definiti anche come trigger
dell’IPreC; oppure, recettori citosolici che possono agire come mediatori nella
fase di precondizionamento andando ad agire a valle o in parallelo con la PKC.
Queste molecole sono state identificate da Maulik e dalla sua equipe, i quali
dimostrarono che l’isoflavone Genisteina, antagonista tirosin-chinasico, poteva
andare a bloccare la protezione indotta dal precondizionamento nel cuore di topo
(Maulik et al., 1998).
Studi successivi hanno suggerito che la TK in congiunzione con il recettore del
fattore di crescita epidermica possa essere richiesta per attivare il pathway PI3K-
Akt, implicato nella protezione indotta dalla IPreC (Krieg et al., 2002). Benché
il meccanismo attraverso il quale il recettore della TK induce protezione non sia
del tutto chiaro, sembrerebbe però, che possa esserci una correlazione con
l’attivazione delle Proteine Chinasi Mitogeno-Attivate (MAPK) (Hausenloy &
Yellon, 2006).
Le MAPK esistono sotto quattro isoforme: la 42 e 44 kDa Extracellular Signal-
regulated Kinase (ErK 1/2), la 38 kDa (p38MAPK), la c-Jun N-terminal Kinase
(JNK) e la Big MAP Kinase 1 (BMK1 o Erk5). Esse trasmettono i segnali ai
loro bersagli mediante l’attivazione di varie vie di segnalazione intracellulare
(Widmann et al.,1999) (Figura 16).

Introduzione
37
Figura 16. Schema di attivazione della famiglia delle proteine MAPK. La loro
attivazione prevede una cascata di fosforilazione a partire dalla MAPK Kinase Kinase
(MKKK) che fosforila la MPAK Kinase (MKK) che attiva la MAPK (Widmann et al.,
1999).
Erk 1/2
Si tratta di una serina/treonina proteina chinasi ed è stata decritta per la prima
volta nel 1990. La sua azione come mediatore nella fase di precondizionamento
è controversa, però molti studi hanno sostenuto l’idea che questa possa
contribuire al meccanismo protettivo.
Durante il precondizionamento, questa proteina chinasi viene attivata dalla
PKC-ε (Ping et al., 1999); altri studi hanno dimostrato che i ROS, generati in
risposta allo stimolo precondizionante, sono tra gli attivatori di Erk 1/2
(Samavati et al., 2002).
Studi più recenti sembra abbiano dimostrato l’ipotesi secondo cui l’attivazione
di Erk 1/2 andrebbe a ridurre la morte cellulare provocata dal danno da I/R
attraverso meccanismi anti-apoptotici (Hausenloy et al., 2004).
Attraverso studi condotti su miociti di topo che sovraesprimono la PKC-ε è stata
osservata un’interazione tra questa proteina chinasi e Erk 1/2 a livello
mitocondriale. È stato dimostrato che, mentre la PKC-ε può legarsi a
componenti degli MPTP e inibendoli, Erk 1/2 attraverso la p90RSK, può andare
a fosforilare e inibire la proteina GSK-3β, provocando l’inibizione dell’apertura
di MPTP e di conseguenza si ha un effetto cardioprotettivo (Juhaszova et al.,

Introduzione
38
2004). Gao e i suoi colleghi hanno dimostrato la traslocazione di Erk 1/2 verso
le gap-junctions. Un’ipotesi accreditata sembra essere la riduzione di queste
comunicazioni come meccanismo cardioprotettivo nel precondizionamento (Gao
et al., 2004).
Studi più recenti suggeriscono che l’IPreC induca 2 fasi di attivazione di Erk
1/2: la prima si verifica subito dopo lo stimolo precondizionate, mentre la
seconda si ha al momento della riperfusione miocardica. È interessante notare
che andando ad inibire la prima fase di attivazione si va a inibire anche la
seconda; quindi, la prima fase di attivazione di Erk 1/2 è necessaria per poter
eseguire la seconda fase, e forse può avvenire attraverso la traslocazione della
chinasi al suo sito d’azione (Hausenloy et al., 2005).
p38 MAPK
La p38 MAPK è composta da 4 isoforme principali: p38α, p38β, p38γ e p38δ
(Widmann et al., 1999) e quelle che hanno evidenziato avere un interesse a
livello della cardioprotezione sono la α e la β. Infatti, sembra che la p38α vada a
mediare processi di morte cellulare, invece, la p38β sembra che contribuisca alla
sopravvivenza della cellula (Nemoto et al., 1998). Studi successivi, hanno
suggerito che i livelli di p38α vanno ad aumentare durante l’evento ischemico, e
il precondizionamento sembra proteggere i miociti andando a ridurre
l’attivazione di questa isoforma (Saurin et al., 2000). Allo stesso tempo però,
altri studi hanno dimostrato un aumento dell’attività della p38β MAPK nel cuore
precondizionato di suino (Schulz et al., 2003).
Il ruolo di questa proteina nella cardioprotezione non è ancora del tutto chiaro, in
quanto alcuni studi hanno dimostrato che la sua inibizione durante il danno da
I/R fosse protettiva. Recentemente, è stato osservato che l’IPreC media
l’attivazione transitoria di p38 MAPK e riduce la sua attivazione durante la
prolungata fase ischemica (Marais et a., 2001) (Hausenloy et al., 2006).
Durante l’IPreC, la PKC e la TK sembrano risultare potenziali attivatori della
p38 MAPK.
Benché sia stato ipotizzato che questa proteina possa essere attivata da parte dei
ROS prodotti in risposta all’apertura dei , ciò non è ancora stato
dimostrato direttamente (Hausenloy et al., 2006).

Introduzione
39
Per quanto riguarda i target della p38 MAPK, sono stati osservati 2 principali
substrati: la MAPK-activating protein kinase 2 (MAPKAPK 2), una chinasi in
grado di fosforilare piccole proteine come la HSP27 che conferisce stabilità al
citoscheletro (studi hanno evidenziato la traslocazione p38 MAPK-dipendente
dal citosol al citoscheletro); la αB cristallina la cui fosforilazione e attivazione
conduce alla protezione contro il danno da I/R (Armstrong et al., 1999) (Eaton et
al., 2001).
Baines e la sua equipe, mediante studi su cuori di topo che sovraesprimevano la
PKC-ε, hanno evidenziato un’interazione nel signaling tra p38 MAPK e la stessa
PKC (Baines et al., 2002). Comunque, per poter capire il ruolo della p38 MAPK
nella cardioprotezione sono necessari ulteriori studi e approfondimenti.
Schulz e la sua equipe hanno evidenziato che l’attivazione della Connessina-43
da parte della p38 MAPK (in particolare, si ha l’incremento dell’isoforma β)
porterebbe a protezione nell’IPreC andando a ridurre la conduttanza delle gap-
junctions (Schulz et al., 2003).
JNK
Questa proteina è stata scoperta nel 1991 come appartenente alla famiglia delle
MAPK; questa differiva però, da Erk 1/2 in quanto sembrava essere attivata a
seguito di stress cellulare come calore, shock osmotico, raggi UV, endotossine e
citochine e per questo motivo è stata nominata anche Stress-Actived Protein
Kinase (SAPK) (Kyriakis et al., 1994).
Alcuni studi hanno confermato l’attivazione di JNK in risposta a uno stimolo
precondizionante e il suo ruolo come mediatore nella protezione indotta da
IPreC (Sato et al., 2000). Invece, altre ricerche hanno dimostrato la sua
attivazione a seguito di uno stimolo di precondizionamento, ma hanno smentito
il suo contributo alla protezione (Iliodromitis et al., 2002). Addirittura, studi
effettuati su modelli di ischemia cerebrale su topo hanno evidenziato che l’IPreC
inibisce l’attivazione di JNK all’inizio della fase ischemica, suggerendo un ruolo
dannoso nella fase del precondizionamento (Gu et al., 2000).
Nell’IPreC probabili attivatori della proteina JNK sono i ROS, che sembrano
mediarne l’attivazione indotta dalla riperfusione. Come effettivamente i ROS
generati da uno stimolo precondizionante, attivino JNK è ancora sconosciuto e
da dimostrare (Knight & Buxton, 1996).

Introduzione
40
Inoltre, altri esperimenti su miociti di coniglio hanno dimostrato un’attivazione
della JNK a monte da parte della PKC-ε (Ping et al., 1999).
Come per le altre proteine appartenenti alla famiglia delle MAPK anche la
proteina JNK interviene nel signaling attivato dalla PKC a livello dei
mitocondri, evidenziando questa proteina come il target mitocondriale più
accreditato (Baines et al., 2002).
BMK1
Si tratta di una proteina descritta recentemente; appartiene alla famiglia delle
MAPK ed è chiamata Big MAP Kinase 1 (BMK1 o Erk5) ed è stata individuata
attraverso studi che ne hanno dimostrato un aumento di attività in cuori
precondizionati di suino (Takeishi et al., 1999).
Tra i suoi potenziali effetti cardioprotettivi ci sono: la riduzione della
conduttanza delle gap-junctions e quindi un probabile coinvolgimento della
Connessina 43 (Cameron et al., 2004); la fosforilazione e inibizione del fattore
pro-apoptotico BAD (Pi et al., 2004).
PI3K e Akt
La Fosfatidilinositolo-3-chinasi (PI3K) è una famiglia di enzimi coinvolti in
complessi meccanismi cellulari; ha il compito di fosforilare l’idrossile in
posizione 3 dell’anello inositolico del Fosfatidilinositolo bifosfato (PIP2) e di
trasformarlo in Fosfatidilinositolo trifosfato (PIP3), il quale andrà ad attivare (tra
le varie vie di segnalazione) la proteina serina/treonina chinasi Akt anche
denominata PKB, la quale è implicata anche nella sopravvivenza cellulare
(Cantley, 2002).
Wick e la sua equipe hanno dimostrato che Phosphoinositide-dependent protein
kinase 1 (PDK1) è un mediatore dell’attivazione di Akt (Wick et al., 2000).
Ulteriori studi hanno dimostrato il coinvolgimento della cascata PI3K-Akt nella
protezione da precondizionamento ischemico (Tong et al., 2000).
La TK è un attivatore noto di PI3K-Akt; studi hanno avvalorato che la Src TK
vada ad innescare la cascata nel pathway di PI3K-Akt nell’IPreC (Cantley,
2002). Altre ricerche hanno rilevato che, oltre ad essere un attivatore a monte dei
, PI3K-Akt possono essere attivati a loro volta a valle dei ,

Introduzione
41
probabilmente dai ROS generati dall’apertura dei canali (Hausenloy et al.,
2006).
Tong e la sua equipe hanno dimostrato che questa chinasi svolge il ruolo di
mediatore nel precondizionamento ischemico, in quanto induce in maniera
diretta la produzione di NO e PKC-ε (Tong et al., 2000).
Target di Akt sono la Caspasi 9, la fosforilazione e inibizione dei fattori pro-
apoptotici BAX e BAD, la fosforilazione e attivazione del fattore anti-apoptotico
Bcl-2, la Glycogen synthase kinase GSK-3β.
In particolare, la fosforilazione e l’inattivazione di GSK-3β provoca inibizione
dell’apertura degli MPTP, che è uno dei principali contributi della cascata di
PI3K-Akt nella protezione da IPreC (Juhaszova et al., 2004).
Recentemente, Davidson e la sua equipe hanno provato che in cellule
sovraesprimenti Akt e sottoposte a stress ossidativo si ha la mancata apertura dei
canali MPTP (Davidson et al., 2006).
Come nel caso di Erk 1/2, alcuni studi hanno suggerito che anche Akt possa
avere 2 fasi di attivazione a seguito di uno stimolo precondizionante, in cui la
seconda fase di attivazione, che si verifica alla riperfusione, è necessaria per la
protezione da IPreC (Hausenloy et al., 2005) (Figura 17).
Figura 17. PI3K-Akt signaling

Introduzione
42
GSK-3β
La Glycogen synthase kinase 3β è una proteina implicata nel
precondizionamento ischemico. Infatti, la sua fosforilazione (e inibizione) è il
target di diversi mediatori tra cui Akt e p70S6K, e probabilmente anche di PKC
e Erk 1/2 (Tong et al., 2002).
Murphy e i suoi colleghi hanno dimostrato che la fosforilazione e l’inibizione di
questa proteina ritarda e inibisce la formazione degli MPTP (infatti, sembra che
sia il target cardioprotettivo principale di questa via di signaling), però i
meccanismi sono ancora sconosciuti (Murphy & Steenbergen, 2005).
Un altro target fondamentale nella fase di precondizionamento è la
fosforilazione del fattore pro-apoptotico BAX, quindi l’inibizione di GSK-3β a
questi livello, fa sì che si abbia la riduzione dell’attivazione di questo fattore.
mTOR
La Mammalian Target of Rapamycin (mTOR) è una proteina chinasi, in
particolare un’atipica serina/treonina chinasi, che svolge le sue principali
funzioni cellulari mediante l’interazione con specifiche proteine per formare 2
differenti complessi: mTOR Complex 1 (mTORC1) e mTOR Complex 2
(mTORC2). Il primo ha il compito di regolare la sintesi proteica, la crescita e la
proliferazione cellulare, l’autofagia e le risposte agli stress cellulari; mentre il
secondo sembra essere coinvolto nella regolazione della sopravvivenza cellulare.
Tra i target più avvalorati attraverso il quale mTOR esercita il suo effetto
cardioprotettivo nell’IPreC si ha l’attivazione, tramite fosforilazione sulla
treonina 389, della p70 ribosomal S6 Kinase (p70S6K), ovvero una proteina che
media la fosforilazione e l’inibizione del fattore pro-apoptotico BAD,
l’inibizione di GSK-3β e la regolazione positiva dell’autofagia, meccanismo
importante nel danno da I/R.
L’uso di un inibitore specifico di mTOR, la Rapamicina, ha dimostrato bloccare
gli effetti protettivi del precondizionamento ischemico mediante, l’inibizione
della fosforilazione di p70S6K (Murphy & Steenberger, 2008).

Introduzione
43
Sfingosina Chinasi (SK)
La Sfingosina 1-fosfato (S1P) è una proteina attivata dalla catalisi della
Sfingosina chinasi (SK), la quale fosforila la Sfingosina.
L’attivazione/inibizione di questo pathway è importante per la motilità della
cellula, per l’organizzazione del citoscheletro, per la crescita cellulare e può
influenzare il destino della cellula andando ad alterare l’equilibrio tra Ceramide
(molecola pro-apoptotica) e S1P (molecola che media l’attivazione di processi
per la salvaguardia della cellula) (Karliner, 2013).
La SK esiste in due isoforme, SK1 e SK2; l’isoforma SK1 sembra svolgere un
ruolo più significativo nella cardioprotezione. In particolare, Jin e la sua equipe
hanno effettuati studi su cardiomiociti di topo ed hanno definito la via di
signaling della SK, innescata dall’azione del Monoganglioside GM1. Questo
fosforila ed attiva la PKC-ε, e in seguito la SK, la quale fosforila la S1P (Jin et
al., 2004). Così, la S1P può contrastare l’azione pro-apoptotica della Ceramide,
oppure può legarsi con i suoi recettori mediante una traslocazione extracellulare.
Nel momento in cui si lega con i suoi recettori, soprattutto il recettore ,
accoppiati a proteine G, favorisce l’attivazione del pathway della proteina Akt
che fosforila e inibisce la GSK-3β, con la conseguente inibizione di apertura
degli MPTP, e il fattore pro-apototico BAD (Karliner, 2013).
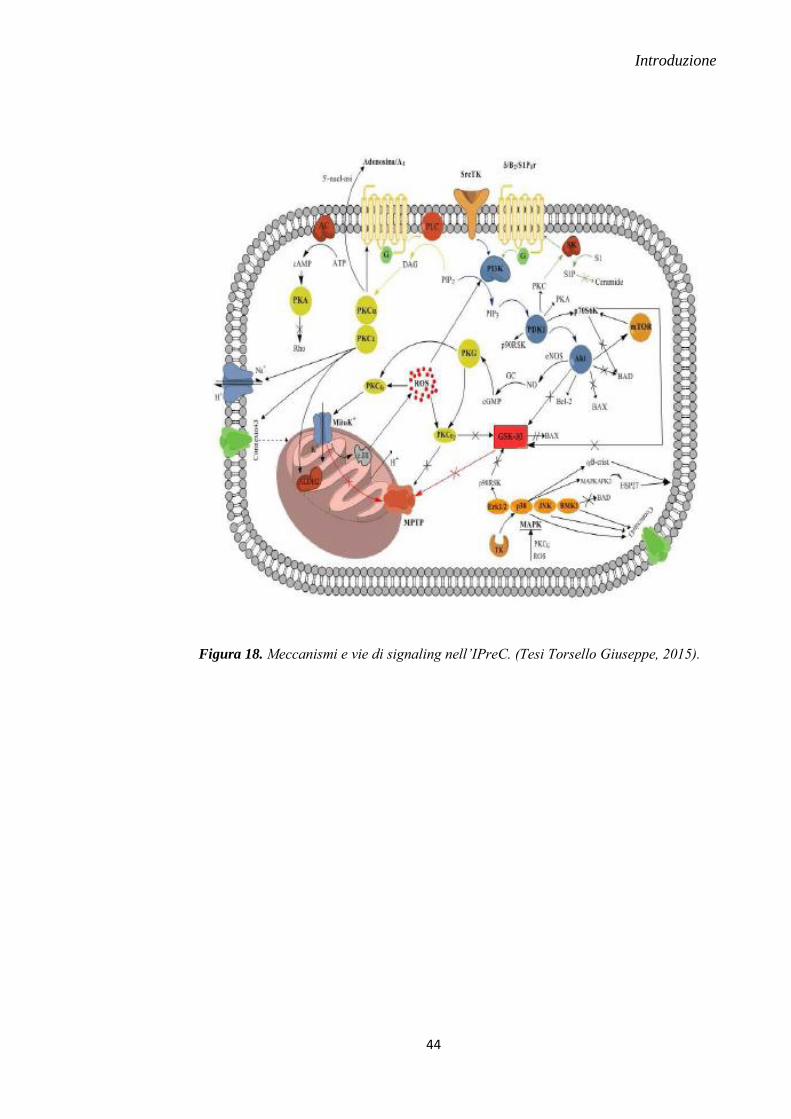
Introduzione
44
Figura 18. Meccanismi e vie di signaling nell’IPreC. (Tesi Torsello Giuseppe, 2015).

Introduzione
45
1.9 PORO MPTP
A seguito di accumulo di intramitocondriale si forma il poro MPTP, con
conseguente incremento della permeabilità dei soluti con massa inferiore a 1.5 kDa
(Bernardi, 1999). La formazione del poro è modulata dal , ma anche da altri fattori,
quali anossia, ROS, pH e potenziale di membrana mitocondriale.
Questo poro è composto da 3 subunità principali:
- canale anionico voltaggio-dipendente (VDAC) localizzato sulla membrana esterna del
mitocondrio; composto da un polipeptide (Shoshan-Barmatz et al., 2006) ed interagisce
con un gran numero di proteine della membrana esterna, come il recettore
benzodiazepinico periferico, Bax, Bcl-2, CK e esochinasi.
Ne esistono 3 isoforme: VDAC1 è la più espressa e svolge un ruolo importante nella
morte cellulare poiché induce apoptosi (Zaid et al., 2005); VDAC2 al contrario, blocca
il processo apoptotico, e quindi la mancata presenza di quest’ultima rende più probabile
la morte cellulare (Cheng et al., 2003), ed infine VDAC3, di cui però non si conosce il
ruolo preciso nel processo di morte celulare.
L’attivazione di VDAC dipende dal voltaggio e dal potenziale di membrana; questo
canale svolge un ruolo chiave nella regolazione del flusso metabolico ed energetico
attraverso la membrana mitocondriale esterna. Inoltre, è coinvolto nel trasporto di ATP,
ADP, metaboliti ed infine, è un importante regolatore dell’omeostasi di
(Schoshan-Barmatz et al., 2003).
- Adenina nucleotide traslocasi (ANT): componente centrale dell’MPTP e proteina
maggiormente presente a livello del mitocondrio (Halestrap et al., 2003). Il ruolo
prevalente di ANT è di trasportare ATP fuori dal mitocondrio e ADP all’interno. Anche
di questa struttura proteica ne esistono 3 isoforme: ANT1, presente a livello del cuore e
del muscolo cardiaco, interagisce con VDAC e CyP-D per formare l’MPTP; ANT2
espresso solo in cellule tumorali; ANT3 presente in tutti gli altri tessuti.
Esistono due conformazioni: “c” nella quale la porzione ANT si lega al CyP-D e
sensibilizza MPTP al ; “m” nella quale invece, si lega con ADP e ATP
desensibilizzando il poro al (Halestrap et al., 1998).
- Ciclofillina D (CyP-D): può legarsi alla porzione ANT oppure al complesso
ANT/VDAC. Quando i livelli di nella matrice sono elevati, questa porzione
proteica modifica la conformazione di ANT, promuovendo la formazione dell’MPTP.
Una riduzione di CyP-D aumenta la capacità di trattenre con maggiore resistenza

Introduzione
46
al rigonfiamento e alla formazione del poro (Schinzel et al., 2005), prevenendo la morte
cellulare indotta da I/R ed eccessiva presenza di nel mitocondrio (Baines et al.,
2005). Una maggiore espressione invece, porta alla formazione del MPTP (Li et al.,
2004) e causa rigonfiamento della matrice e morte cellulare (Baines et al., 2005). Questa
subunità controlla quindi direttamente la formazione di MPTP e la sua apertura.
In condizioni fisiologiche la membrana mitocondriale è impermeabile a tutti i metaboliti
e ioni, l’MPTP quindi non si forma; ciò è fondamentale per il mantenimento delle
normali funzioni mitocondriali.
In condizioni di stress, come a causa di un aumento di causato dal danno I/R si ha
un cambiamento nella conformazione delle subunità che costituiscono il poro e ciò ne
causa la successiva formazione. L’apertura dell’MPTP ha come immediata conseguenza
l’idrolisi di ATP, una caduta drastica del potenziale di membrana a causa della
permeabilità di soluti con massa inferiore a 1.5 kDa, accumulo di acqua all’interno della
matrice con successivo swelling mitocondriale, rottura della membrana esterna e
rilascio di fattori pro-apoptotici quali il Citocromo C (Weiss et al., 2003) (Figura 19).
Figura 19. Struttura molecolare e localizzazione dell’MPTP
Quindi, l’MPTP si tratta di un mediatore critico del danno acuto da I/R, ed è perciò, un
importante target per la cardioprotezione. Ricerche effettuate su modelli animali hanno
mostrato che un’inibizione farmacologica dell’apertura degli MPTP conduce ad una
diminuzione dell’entità dell’infarto miocardico.

Introduzione
47
1.9.1 PORO MPTP COME TARGET CARDIOPROTETTIVO
L’importante scoperta effettuata nel 1988 da Crompton e la sua equipe, in cui l'apertura
dell’MPTP potrebbe essere inibita dall’immunosoppressore, Ciclosporina-A, ha
facilitato l'indagine sull’MPTP come mediatore del danno da I/R come un obiettivo di
cardioprotezione
In primo luogo, hanno scoperto che il trattamento di cardiomiciti ventricolari di ratto
adulto con Ciclosporina A induceva una protezione contro la morte cellulare indotta
dalla simulazione di un danno acuto da I/R; avendo scoperto questo meccanismo,
Crompton e i suoi colleghi sono stati i primi ad usare questa molecola per fare ulteriori
studi per comprendere il ruolo degli MPTP come target nella cardioprotezione
(Nazareth et al., 1991).
Successivamente, Griffiths e Halestrap hanno evidenziato gli effetti prodotti mediante
pre-trattamento con Ciclosporina A su cuori isolati di ratto perfusi, osservando un
migliore recupero funzionale miocardico e una certa quantità di ATP salvaguardata in
seguito a un acuto infarto miocardico (Griffiths & Halestrap, 1993).
Nonostante la presenza di induttori dell’MPTP come il calcio, il fosfato, lo stress
ossidativo e la deplezione di ATP, le condizioni acide intracellulari durante l’evento
ischemico inibiscono l’apertura dell’MPTP, facendo supporre che queste condizioni
estreme inibiscano MPTP durante l’ischemia. Invece, il rapido ristabilimento del pH
fisiologico nei primi minuti della riperfusione fa sì che provochi l’apertura di questi
canali. Ad avvalorare questa affermazione, sono stati effettuati diversi studi per
confermare che durante il periodo di riperfusione si abbia l’apertura del canale MPTP
(Di Lisa et al., 2001) (Murata et al., 2001) (Matsumoto-Ida et al., 2006).
Successivamente, è stato confermato che l’apertura di MPTP si verifica, in particolare,
al momento della comparsa della riperfusione, infatti, è stato dimostrato, su cuori di
ratto perfusi, che si ha una riduzione dell’entità dell’infarto miocardico nel momento in
cui viene somministrata Ciclosporina A solo alla comparsa della ripefusione (Hausenloy
& Yellon, 2003). Invece, gli effetti cardioprotettivi indotti dall’inibizione di MPTP
vengono completamente persi se molecole inibitrici di MPTP vengono somministrate
dopo 15 minuti dall’inizio della riperfusione miocardica. Quindi, è evidente che è
importante intervenire nei primi minuti della riperfusione per poter avere effetti
cardioprotettivi.
Il ruolo fondamentale svolto dal poro MPTP come mediatore del danno da I/R è stato
confermato mediante studi genetici in cui sono stati utilizzati topi con deficienza di

Introduzione
48
CYpD mitocondriale, un componente regolatorio di questo poro, che presentano una
ridotta entità dell’infarto miocardico rispetto ai topi wild-type (Basso et al., 2005).
Il fatto che l’apertura del poro MPTP si verifichi nei primi minuti della riperfusione, ha
definito una finestra di tempo critico per poter utilizzare molecole inibitorie di questo
canale come strategia cardioprotettiva.
Pertanto, qualsiasi strategia terapeutica che è stata progettata come target del danno da
I/R deve essere somministrata prima o al momento della comparsa immediata della
riperfusione per evitare l’apertura dell’MPTP. La tempistica critica dell’intervento
terapeutico ha avuto importanti implicazioni per lo sviluppo di nuove terapie
cardioprotettive in ambito clinico (Hausenloy et al., 2010) (Hausenloy et al., 2013).
1.9.2 PORO MPTP COME TARGET PER IL CONDIZIONAMENTO
ISCHEMICO
Diversi studi effettuati hanno dimostrato che l’apertura di MPTP è inibita dalla
riperfusione in cuori soggetti sia a IPreC sia a IPostC e anche a RePreC.
Attualmente, i meccanismi attraverso cui il condizionamento ischemico prevengono
l’apertura dell’MPTP al momento della riperfusione non sono ancora chiari, sebbene
siano stati proposti due pathway, in cui l’uno non esclude l’altro (Hausenloy et al.,
2009). Nella prima ipotesi, definita “inibizione indiretta dell’MPTP”, il
condizionamento ischemico andrebbe a modulare diversi fattori tra cui lo stress
ossidativo, la concentrazione di calcio mitocondriale, l’accumulo di fosfato, i livelli di
ADP/ATP e il pH intracellulare, che sono tutti elementi noti che influiscono
sull’apertura del canale nel momento della riperfusione.
Al contrario, la seconda ipotesi, denominata “inibizione diretta dell’MPTP”, presuppone
che l’attivazione dei noti mediatori di segnalazione di condizionamento ischemico sia in
grado di modulare l’apertura dell’MPTP in seguito all’interazione diretta con i
componenti del canale.
Inibizione indiretta di MPTP
Dato il ruolo importante svolto dal sovraccarico mitocondriale di ioni calcio
nell’indurre l’apertura di MPTP al momento della comparsa della riperfusione,
sono state effettuate diverse ricerche che hanno cercato di capire se l’effetto del
condizionamento ischemico potesse andare ad attenuare il sovraccarico di
mitocondriale e citosolico prodotto durante il danno da I/R. I primi studi

Introduzione
49
effettuati hanno suggerito che durante il precondizionamento ischemico,
mediante l’apertura di canali mitocondriali al potassio ATP-dipendenti
( ), si potrebbe avere una riduzione nel sovraccarico di ioni calcio
mitocondriale che può provocare una parziale depolarizzazione del potenziale di
membrana del mitocondrio (Wang et al., 2001). Tuttavia, è ancora motivo di
discussione il fatto che l’apertura del canale possa effettivamente
indurre una sufficiente depolarizzazione della membrana mitocondriale tale da
ridurre l’accumulo di ioni nel mitocondrio (Murata et al., 2001). Per di
più, il contributo del canale nella protezione indotta da IPreC è ancora
controversa.
Recentemente, diversi studi hanno suggerito che lo stress ossidativo in seguito
alla riattivazione della catena di trasporto degli elettroni nei primi minuti della
riperfusione miocardica e il ripristino del pH fisiologico potrebbero essere tra i
fattori più importanti che vanno ad indurre l’apertura dell’MPTP durante il
danno da I/R. Inoltre, l’ablazione genetica dell’uniporter mitocondriale del
di recente scoperta, potrebbe indurre una riduzione della sensibilità all’apertura
dell’MPTP calcio-indotta (Pan et al., 2013).
Per di più, è stato ipotizzato che la conservazione dei livelli di ATP
mitocondriale possa essere un fattore critico nella cardioprotezione esercitata da
IPreC. Studi successivi hanno dimostrato che IPreC induce una protezione
mediante la riduzione di ATP consumato durante l’ischemia miocardica e
attraverso la preservazione della produzione di energia mitocondriale durante
l’attacco acuto da I/R (Kobara et al., 1996).
Il rapido ripristino del pH fisiologico intracellulare nei primi minuti della
riperfusione miocardica, da un pH acido indotto dall’ischemia, si pensa che
induca l’apertura dell’MPTP. Studi recenti hanno ipotizzato che IPreC e IPostC
possano inibire l’apertura di MPTP al momento della comparsa della
riperfusione andando a ritardare il ripristino del pH fisiologico, sebbene il
meccanismo non è ancora del tutto chiaro (Cohen et al., 2007).
Inofatti, alcuni studi hanno riportato che IPreC inibisce l’attività dello
scambiatore / nel momento della riperfusione attraverso un meccanismo
di rallentamento del ripristino del normale pH e andando a ridurre la
concentrazione di sodio intracellulare e l’accumulo di calcio (Xiao et al., 2000),
anche se al momento questo risultato è stata controverso. A questa evidenza si

Introduzione
50
aggiunge quella che vede il coinvolgimento di Akt nell’inibizione dello
scambiatore / , suggerendo che questa proteina chinasi interviene nel
rallentare il ritorno del pH valori fisiologici.
Inibizione diretta di MPTP
È dimostrato che sia IPreC che IPostC svolgono un ruolo cardioprotettivo
mediante l’attivazione di una varietà di specifiche cascate di segnalazioni molte
delle quali terminano al livello dei mitocondri, dove mediano l'inibizione
dell'apertura MPTP al momento della riperfusione.
È ben noto, che l'attivazione acuta di un pathway di proteine chinasi
cardioprotettive, come il RISK e il Survival Activation Factor Enhancement
(SAFE) al momento della comparsa della riperfusione miocardica, possono
limitare l’entità dell’infarto miocardico (Hausenloy et al., 2011).
In particolare, è stato dimostrato che IPreC e IPostC provocano l’attivazione di
questo pathway, andando ad inibire l’inibizione dell’apertura del canale MPTP
(Davidson et al., 2006).
Il meccanismo attraverso cui l’attivazione del pathway RISK va a mediare
l’effetto inibitorio sull’apertura di MPTP non è ancora ben definita, potrebbe
fare ciò attraverso l’attivazione di mediatori a valle come PKG, GSK-3β, o
esochinasi II, oppure potrebbe avvenire attraverso la modificazione di fattori di
induzione dell’MPTP come stress ossidativo, calcio o modifiche di pH.
Diversi esperimenti hanno evidenziato che le chinasi di salvataggio del pathway
RISK come Akt (Bijur & Jope, 2003), Erk 1/2 (Baines et al., 2002), e PKG
(Costa et al., 2005) possono trasferirsi nei mitocondri e in alcuni casi la
traslocazione mitocondriale è stata associata a inibitori MPTP, anche se il
meccanismo reale attraverso cui queste chinasi citosoliche sono in grado di
accedere ai componenti interni della membrana MPTP non sono ancora noti.
Clarke e la sue equipe hanno messo in discussione il legame che mette in
relazione queste chinasi cardioprotettive che compiono traslocazione nei
mitocondri e l'inibizione degli MPTP riportando gli effetti indiretti di IPreC e
IPostC svolti nell’attenuazione dello stress ossidativo come il meccanismo di
inibizione MPTP (Clarke et al., 2008). Potenziali bersagli a valle dei componenti
di Akt e Erk 1/2 del pathway RISK che sono stati collegati all’inibizione di
MPTP includono PKG, GSK-3β, e esochinasi II.

Introduzione
51
Dal momento che i mitocondri sono strutture dinamiche capaci di cambiare la
loro morfologia (Ong et al., 2013), recentemente, è stato ipotizzato che il
mitocondrio vada incontro a fissione e ad apertura del canale MPTP in risposta
al danno acuto da I/R; è stato riportato che un’ inibizione genetica o
farmacologica della fissione del mitocondrio inibisca l’apertura dell’MPTP e
quindi riduca la morte cellulare (Ong et al., 2010). Queste scoperte hanno
suggerito che ci possa essere un collegamento tra la morfologia del mitocondrio
e la suscettibilità all’apertura del poro, in quanto l’inibizione della fissione
mitocondriale indotta dal danno acuto I/R sembra possa prevenire l’apertura di
questi canali al momento della riperfusione.

Introduzione
52
1.10 UNIPORTER MITOCONDRIALE DEL
Figura 20. Schema molecolare e localizzazione uniporter mitocondriale del (Kamer et
al., 2015).
Il Calcio è fondamentale per la funzione mitocondriale e delle cellule, e la sua
segnalazione è altamente localizzata nella cellula. In condizioni patologiche, un grande
aumento nei livelli di calcio mitocondriale è stato ipotizzato che possa provocare
l’attivazione dell’MPTP, con conseguente morte cellulare. La proteina responsabile
dell’assorbimento mitocondriale di calcio, l’uniporter mitocondriale del , è stato
identificato nel 2011, e la sua identificazione molecolare ha stimolato e rinvigorito la
ricerca in questo settore.
L’uniporter mitocondriale del mostra multeplici funzioni nella regolazione della
produzione di ATP, nell’apertura dei canali MPTP e successivamente nel processo di
apoptosi (Testai, 2015).
Il uniporter è formato da due domini transmembranali, ed è localizzato nella
membrana interna del mitocondrio, oligomerizzato a formare un complesso con più
ampio peso molecolare (Figura 20). Le due subunità che costituiscono questo uniporter
sono:
- MICU (Mitochondrial Calcium Uptake 1), una proteina mitocondriale di 54kDa che si
lega al calcio; quando questa viene silenziata si ha l’inibizione dell’assorbimento di
mitocondriale (Baughman et al., 2011).
- MICU2 (Mitochondrial Calcium Uptake 2), una proteina di 45kDa analoga a MICU1,
presente all’interno della membrana mitocondriale. Questa proteina sembra inibire la
funzione del uniporter nel momento in cui sono minori i livelli di calcio citosolico.

Introduzione
53
Quando MICU2 è silenziato, è stato evidenziato un aumento dei livelli di calcio
mitocondriale. Nel caso in cui questa proteina è sovraespressa, si verifica una debole,
ma statisticamente significativa diminuzione della concentrazione di calcio
mitocondriale.
Un breve tratto di aminoacidi che si affacciano nello spazio intermembranale del
mitocondrio sembra influenzare il trasporto di calcio: il Rosso Rutenio ed il relativo
composto Ru360 sono noti potenti inibitori di questo uniporter; ma, mutazioni su residui
in questo tratto e nel terminale carbossilico hanno conferito una marcata resistenza sia
nell'assorbimento di calcio sia nell'effetto inibitorio della Ru360, andando a suggerire
che questa è l'unità che forma il poro dell’uniporter (Baughman et al., 2011).
La forza trainante che influisce sull'assorbimento del calcio mediante il uniporter è
stato ipotizzato essere il potenziale di membrana negativo, che viene stabilito dalla
catena respiratoria (Rizzuto et al., 2012). Inoltre, ulteriori ricerche basate sulle
misurazioni in mitoplasti hanno dimostrato che il uniporter è un canale ionico
specifico al calcio (Baughman et al., 2011).
Poiché l’apertura dell’MPTP e la successiva morte delle cellule è generalmente avviata
da un grande afflusso di calcio nei mitocondri attraverso l’uniporter al calcio, c'è un
grande interesse nel fatto che topi in cui non è espresso questo canale, risultano protetti
dall’apertura MPTP e dalla morte cellulare risultante a seguito di ischemia e di
riperfusione. (Moore, 1971).
Diversi studi effettuati su modelli di cuore perfuso di topo in cui non è espresso
l’uniporter al calcio che sono soggetti a danno I/R, hanno mostrato che non vi è nessuna
differenza nell’entità dell’infarto o nel recupero della funzionalità contrattile rispetto ai
topi wild type. Inoltre, è stato osservato che, benché non avvenisse nessuna apertura del
poro MPTP nei mitocondri in cui non è espresso il uniporter che sono stati esposti
a grandi quantità di calcio, ciò non preclude l’apertura di MPTP indotta da ROS,
indipendenti dalle concentrazioni di calcio, in cellule dopo l’evento di ischemia e
riperfusione. Quindi, è possibile che il poro MPTP possa essere attivato nei cuori in cui
non è espresso il uniporter in maniera indipendente dalle concentrazioni di calcio
mitocondriale; questo sarebbe coerente con i suggerimenti che ROS è l'attivatore
primario dell’MPTP in vivo (Zorov et al., 2014).
Se l'attivazione del canale MPTP si verifica nel cuore in cui non è espresso l’uniporter
del calcio, allora ci si aspetterebbe che la Ciclosporina A, un inibitore del canale MPTP,
dovesse fornire una protezione nei cuori di animali che non esprimono questo uniporter.

Introduzione
54
Questo aspetto è stato studiato da Pan e la sua equipe su modelli di topi in cui questo
uniporter non è espresso: hanno scoperto che in seguito all’evento di ischemia e
riperfusione a cui sono stati soggetti i cuori di topi wild type, la somministrazione di
Ciclosporina A, svolgeva effetti protettivi; invece, in cuori di topo in cui non era
espresso il uniporter non li svolgeva.
Alcuni studi hanno evidenziato che vari flavonoidi naturali, tra cui il Camferolo,
attivano direttamente l’uniporter mitocondriale del , mostrando multeplici
funzioni nella regolazione della produzione di ATP, nell’apertura dei canali MPTP e
successivamente nel processo di apoptosi, a concentrazioni compatibili con l’assunzione
quotidiana di alimenti ricchi di flavonoidi. Tali evidenze suggeriscono che si possa
ottenere in vivo una modulazione nell’assorbimento di mitocondriale a seconda
della dieta alimentare (Montero et al., 2004).
1.10.1 REGOLATORI DELL’ UNIPORTER MITOCONDRIALE DEL
MCUb
MCUb è una proteina di 33kDa che sembra molto simile al uniporter nella
struttura e nell’orientamento, ma che agisce in maniera contraria a questo canale.
Infatti, MCUb non consente il passaggio degli ioni calcio attraverso le
membrane mitocondriali interne, e sembra ridurre il movimento del calcio nei
mitocondri quando oligomerizza con uniporter. Inoltre, questo canale ha
una differente regolazione dell’assorbimento di calcio mitocondriale a seconda
del tessuto coinvolto, infatti nei cardiomiociti non permette l’entrata di calcio
nel mitocondrio (Plovanich et al., 2013).
EMRE
Nel 2013 Mootha e la sue equipe hanno identificato un regolatore
transmembranale ad unico transito che sembra sia in funzione del
uniporter, denominata Essential Mitocondrial Calcium Uniporte Regulator
(EMRE). Studi su modelli di cellule e organelli isolati di topo, in cui è stato
silenziato EMRE, hanno evidenziato una completa inefficienza del
uniporter. Quindi, ricerche successive hanno suggerito che MICU1 e

Introduzione
55
MICU2 sono importanti regolatori dell’uniporter, ma che non sono essenziali
per la sua funzionalità: infatti, EMRE rappresenta il primo regolatore essenziale
per l’attività del uniporter.
Ad oggi l’orientamento di questo regolatore non è ancora conosciuto, ma è stato
ipotizzato che possa ad andare ad interagire sia con MICU sia MICU2 al livello
della membrana interna del mitocondrio e che possa legarsi a sua volta anche
con il uniporter (Sancak et al., 2013).
SLC25A23
Si tratta di una tasca che contiene il carrier mitocondriale dello scambiatore Mg-
ATP/Pi. Studi in cui è stato silenziato questo regolatore, hanno evidenziato una
riduzione ma una totale eliminazione, dell’uptake di calcio mitocondriale.
Cellule Hela in cui non era espresso questo regolatore hanno mostrato una
debole riduzione del potenziale di membrana mitocondriale in seguito all’uptkae
di calcio mitocondriale. Per questo motivo, è stato ipotizzato che SLC25A23
potrebbe regolare l’azione del uniporter mediante l’attivazione di un
trasportatore di anioni fosfato, ma che possa anche interagire direttamente con il
canale. Quindi, il silenziamento di questo regolatore sembra possa avere una
potenziale azione protettiva nei confronti della morte cellulare (Hoffman et al.,
2014).
Inoltre, Amigo e la sue equipe hanno riportato che una diminuzione di
SLC25A23 va a ridurre la capacità di ritenzione di calcio nel mitocondrio,
coerente alla maggiore apertura del canale MPTP (Amigo et al.,2013).
MCUR1
Mitochondrial Calcium Uniporter Regulator 1, o MCUR1, è una proteina di
40kDa formata da due domini trasmembranali e da una regione coiled-coil, che,
come nel caso del uniporter, ha N e C-terminazioni che si affacciano nello
spazio intermembranale. Le cellule che sono prive di questa proteina sembrano
simili alle cellule in cui non è espresso l’uniporter, e per questo motivo,
ovviamente, non si verifica l’assorbimento di calcio. Inoltre, questa tipologia di
cellule mostra un incremento nell’attività autofagica e un aumento nella

Introduzione
56
resistenza alla morte cellulare per apoptosi e necrosi (Mallilankaraman et al.,
2012).
Nel caso di una sovraespressione di MCUR1 è stato evidenziato un incremento
nell’uptake di calcio mitocondriale, ma solo nel caso in cui era espresso il
canale. Questo ha suggerito che il calcio deve essere necessariamente trasportato
mediante uniporter.
ALTRI REGOLATORI
Sono coinvolte diverse proteine tra cui lo scambiatore mitocondriale sodio-
calcio (NCLX), proteine non accoppiate (UPC) 2 e 3, la tasca leucina-EF-hand
che contiene la proteina trasmembranale 1 (LETM1), EPR57, TRCP3. Queste
proteine sono importanti per la fisiologia del calcio mitocondriale, ma i loro
ruoli esatti, sia fisici e biochimici, non sono ancora stati completamente chiariti
(Rizzuto et al., 2012).

Introduzione
57
1.11 METODI INNOVATIVI DI DRUG DELIVERY MITOCONDRIALE
Le caratteristiche che deve presentare una molecola ideale che deve agire come
cardioprotettiva deve avere la capacità di penetrare facilmente nella cellula ed essere
efficacemente accumulata a livello del proprio target mitocondriale. La mancanza di
queste proprietà farmacocinetiche è un serio limite nell’applicazione clinica.
Per questo motivo, sono state proposte strategie di drug delivery innovative e stimolanti
al fine di migliorare l’ingresso selettivo di molecole nei mitocondri e minimizzare
eventuali effetti aspecifici e indesiderati.
Quindi, per ottenere farmaci anti-ischemici altamente selettivi e abili nell’essere
selettivamente trasportati all’interno del mitocondrio, la strategia più sfruttata è quella di
combinare la singola molecola selezionata con il catione “mitocondrio specifico”,
Trifenilfosfonio ( ), mediante un linker alifatico appropriato. Generalmente, la
lunghezza dello spaziatore influenza il grado di idrofobicità di questi agenti
mitocondriotropici e di conseguenza, ciò influisce sulla loro efficacia sia in modelli in
vivo sia in vitro.
Quindi, la strategia consiste nello sviluppare un derivato coniugato con il , che
rapidamente e ampiamente si possa accumulare a livello dei mitocondri, guidato dal
potenziale di membrana mitocondriale (circa di -180mV) (Porteous et al., 2010).
Il maggiore vantaggio nell’utilizzo di cationi lipofili è la loro bassa reattività e debole
interazione con le componenti cellulari.
Per esempio, al fine di indirizzare il Resveratrolo all’interno del mitocondrio, questo
polifenolo è stato combinato con il .
Lo stesso destino è stato riservato a molte sostanze antiossidanti, tra cui MitoVitE,
MitoQ e MitoL, coniugate con il . Ad oggi, il derivato MitoQ ha evidenziato
effetti cardioprotettivi nei modelli di danno da I/R (Smith & Murphy, 2010) e in più
recentemente, Mito-Tempol, un nitrossido-piperidina coniugata con il TPP+, ha
mostrato effetti cardioprotettivi in modelli in vivo su ratto (Dickey et al., 2013).
Oltre ai cationi lipofili, anche alcuni peptidi sono stati utilizzati per veicolare molecole a
livello mitocondriale; in particolare, i peptidi Szeto-Schiller (SS) sono in grado di
legarsi selettivamente alla membrana interna mitocondriale, indipendentemente dal
valore del potenziale. Questa caratteristica è un vantaggio poiché queste molecole
possono penetrare anche dentro organelli danneggiati, in cui è presente un ridotto del
potenziale (Szeto & Schiller, 2011).

Introduzione
58
Un importante differenza tra i cationi lipofili e i peptidi SS è che quest’ultimi
possiedono un’attività intrinseca nel mitocondrio. In particolare, i peptidi SS-02 e SS-31
sono in grado di comportarsi da scavenger verso il perossido di idrogeno, i radicali
idrossilici, e il perossinitrile in modelli in vivo e ridurre il danno da I/R in modelli ex
vivo. Secondo ricerche effettuate in vivo è stata evidenziata la loro capacità di andare a
promuovere la respirazione mitocondriale e la sintesi di ATP, con conseguente
riduzione nella produzione di ROS, e l’inibizione del rigonfiamento mitocondriale
(Szeto, 2008) (Figura 21).
Figura 21. Strutture chimiche di molecole mito-targeted: (A) molecole coniugate con TPP+,
(B) molecole coniugate con il peptide SS.

59
2. SCOPO DELLA TESI
Le malattie cardiovascolari, in particolare le cardiopatie ischemiche, sono una della
maggiori cause di morte nei paesi industrializzati. Questa patologia è dovuta ad una
parziale o totale occlusione delle arterie coronarie, che porta ad una ostruzione dei vasi
così da limitare l’afflusso sanguigno nei vari tessuti. Come conseguenza si ha una
diminuzione dell’ossigenazione e dell’apporto nutritivo di tali aree, fino al
raggiungimento di una condizione tale che non permette la sopravvivenza della cellula,
che quindi va incontro a morte. Paradossalmente, il danno da ischemia è aumentato
anche nella fase della riperfusione, ovvero la fase successiva all’ischemia in cui
vengono ripristinate le condizioni di normalità per la vita cellulare.
La presenza di particolari canali al potassio sulla membrana interna di mitocondri
conferisce a quest’organello un ruolo fondamentale nella cardioprotezione. Infatti, in
seguito all’ischemia e alla riperfusione, la cellula va incontro ad apoptosi che avviene
come conseguenza della generazione di ROS, dell’attivazione dello scambiatore
Na /H , di una risposta infiammatoria e dell’apertura di un poro localizzato a livello
della membrana del mitocondrio, poro di transizione di permeabilità mitocondriale
(MPTP), promossa dall’ingresso massivo di ioni calcio all’interno del mitocondrio.
L’apertura di questo poro è prevenuta dall’attivazione dei canali al potassio
mitocondriali. L’attivazione di questi canali, attraverso una parziale depolarizzazione
della membrana mitocondriale, determina un ridotto sovraccarico di ioni calcio nella
matrice del mitocondrio. Questa riduzione di concentrazione, a sua volta previene al
momento della riperfusione la sostenuta apertura del MPTP, inducendo
cardioprotezione. Pertanto, si comprende quanto sia critico il ruolo del mitocondrio nel
fenomeno di cardioprotezione e l’importanza di sviluppare farmaci in grado di agire su
questi target mitocondriali fondamentali.
Nell’ottica di selezionare composti potenzialmente attivi nella protezione miocardica
del danno da I/R, questa tesi di Laurea si è posta l’obiettivo di valutare la capacità dei
composti in esame di influenzare alcuni importanti parametri di funzionalità
mitocondriale, quali le variazioni del potenziale di membrana mitocondriale (ΔΨm), i
movimenti dello ione calcio attraverso la membrana mitocondriale in termini di uptake
di calcio e i flussi potassici. Altro obiettivo di questa tesi di Laurea è stato indagare il

Scopo della tesi
60
meccanismo mitocondriale responsabile di un’eventuale azione “protettiva” per
selezionati composti.
In quest’ottica, i composti testati appartengono a due gruppi selezionati:
Composti analizzati in relazione ad analogie strutturali con Naringenina
(Extrasynthese), flavonone del genere Citrus:
Flavonoidi che si ritrovano in abbondanza nel genere Citrus.:
- Eriodictiolo (Extrasynthese)
- Tangeretina (Extrasynthese)
- Esperetina (Extrasynthese) e il suo glicoside Esperidina (Extrasynthese)
Flavonoidi che presentano un’analogia con Naringenina per la presenza di un
gruppo idrossilico in posizione 5 e/o 7 sull’anello biciclico:
- Crisina (Extrasynthese)
- Apigenina (Extrasynthese)
- Luteolina (Indena)
Flavonoidi monoidrossilati, che quindi presentano una struttura chimica più
semplice:
- 4’-idrossi-flavanone (Sigma-Aldrich)
- 5-idrossi-flavone (Sigma-Aldrich)
- 6-idrossi-flavanone (Sigma-Aldrich)
- 7-idrossi-flavone (Sigma-Aldrich)
Composti in cui la struttura flavonoidica classica è complicata con numerose
sostituzioni di gruppi idrossilici sull’anello ciclico e biciclico (isolati e
purificati da materiale vegetale, gentilmente forniti dal laboratorio della
Prof.ssa Braca):
- Myricetina
- Kaempferolo
Proantocianidine più o meno complesse strutturalmente (isolati e purificati
da materiale vegetale, gentilmente forniti dal laboratorio della Prof.ssa
Braca):
- Isoginkgetina
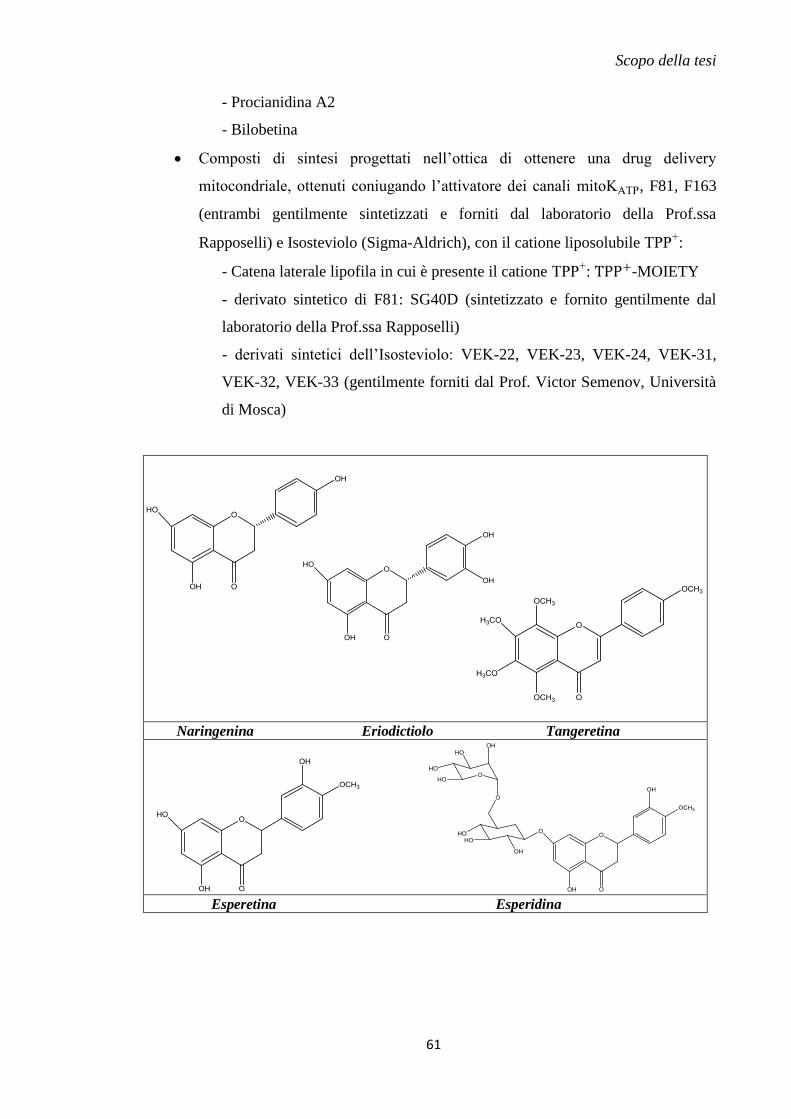
Scopo della tesi
61
- Procianidina A2
- Bilobetina
Composti di sintesi progettati nell’ottica di ottenere una drug delivery
mitocondriale, ottenuti coniugando l’attivatore dei canali mitoKATP, F81, F163
(entrambi gentilmente sintetizzati e forniti dal laboratorio della Prof.ssa
Rapposelli) e Isosteviolo (Sigma-Aldrich), con il catione liposolubile TPP :
- Catena laterale lipofila in cui è presente il catione TPP : -MOIETY
- derivato sintetico di F81: SG40D (sintetizzato e fornito gentilmente dal
laboratorio della Prof.ssa Rapposelli)
- derivati sintetici dell’Isosteviolo: VEK-22, VEK-23, VEK-24, VEK-31,
VEK-32, VEK-33 (gentilmente forniti dal Prof. Victor Semenov, Università
di Mosca)
Naringenina Eriodictiolo Tangeretina
Esperetina Esperidina
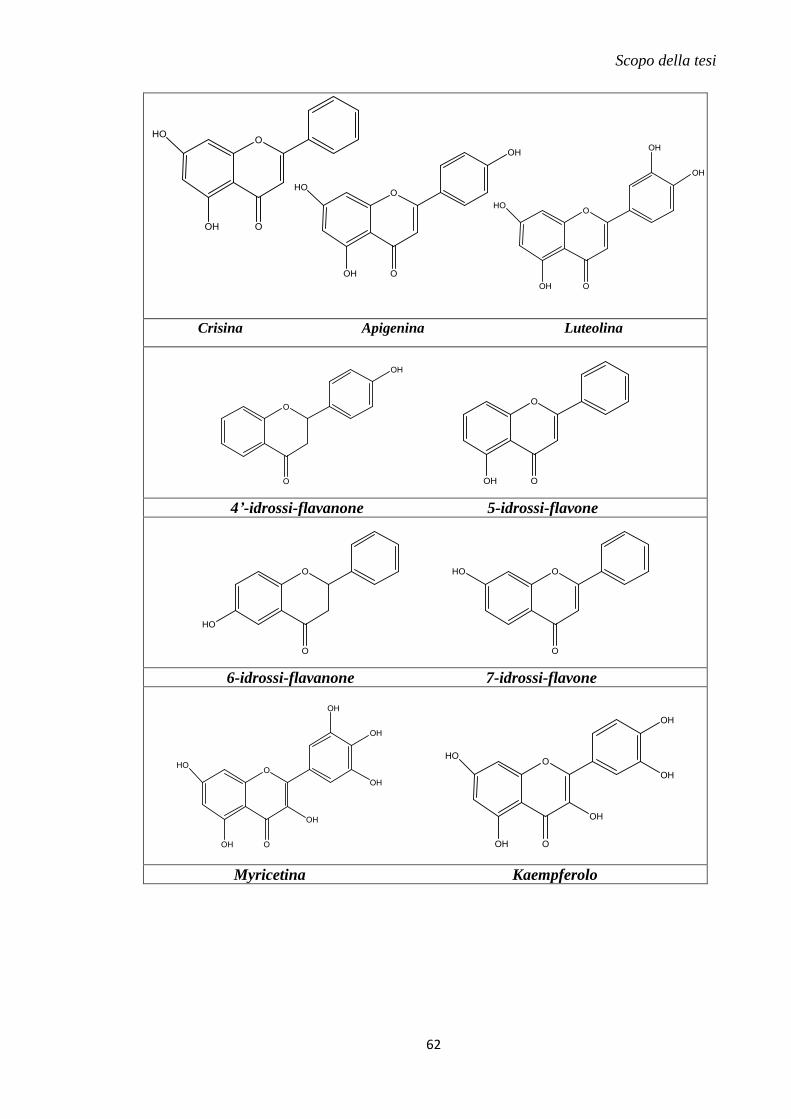
Scopo della tesi
62
Crisina Apigenina Luteolina
4’-idrossi-flavanone 5-idrossi-flavone
6-idrossi-flavanone 7-idrossi-flavone
Myricetina Kaempferolo
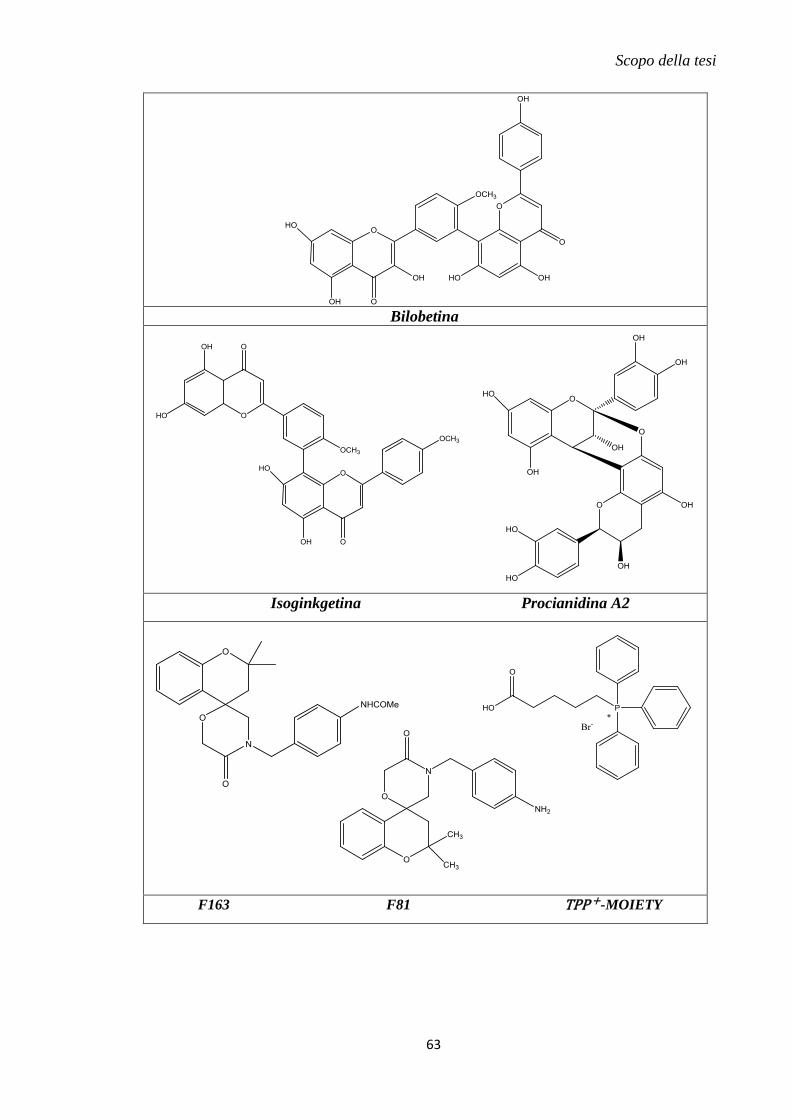
Scopo della tesi
63
Bilobetina
Isoginkgetina Procianidina A2
F163 F81 -MOIETY
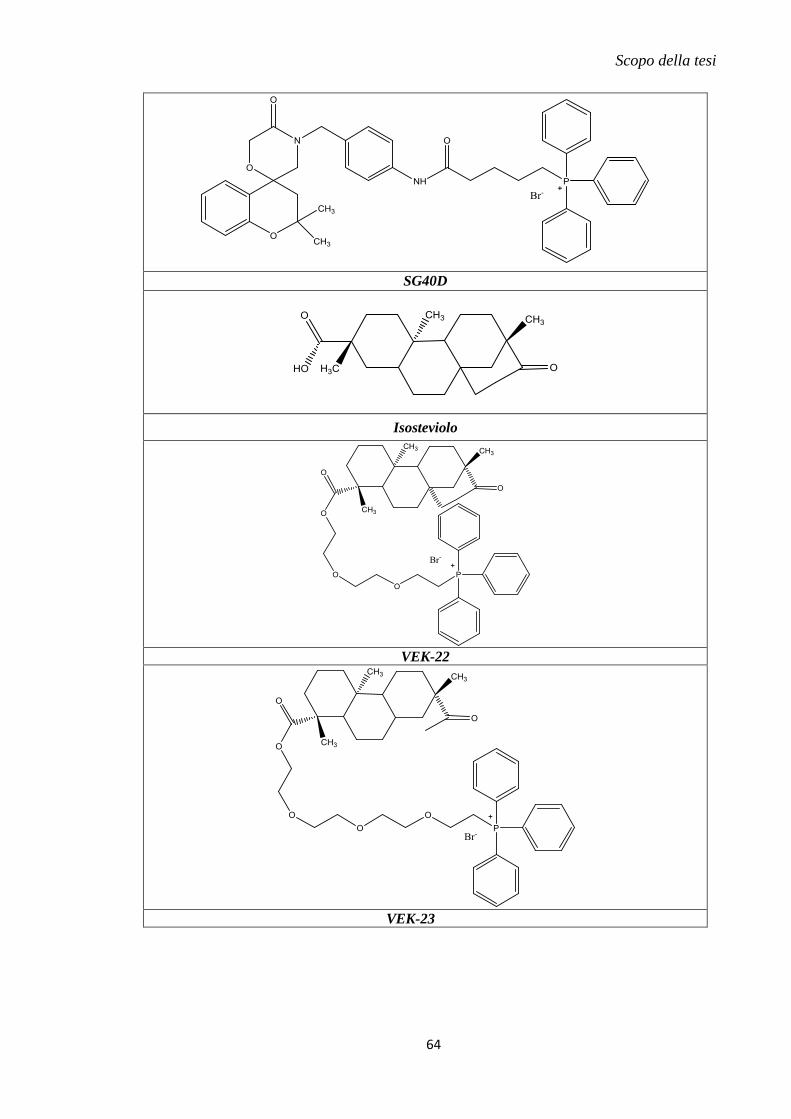
Scopo della tesi
64
SG40D
Isosteviolo
VEK-22
VEK-23
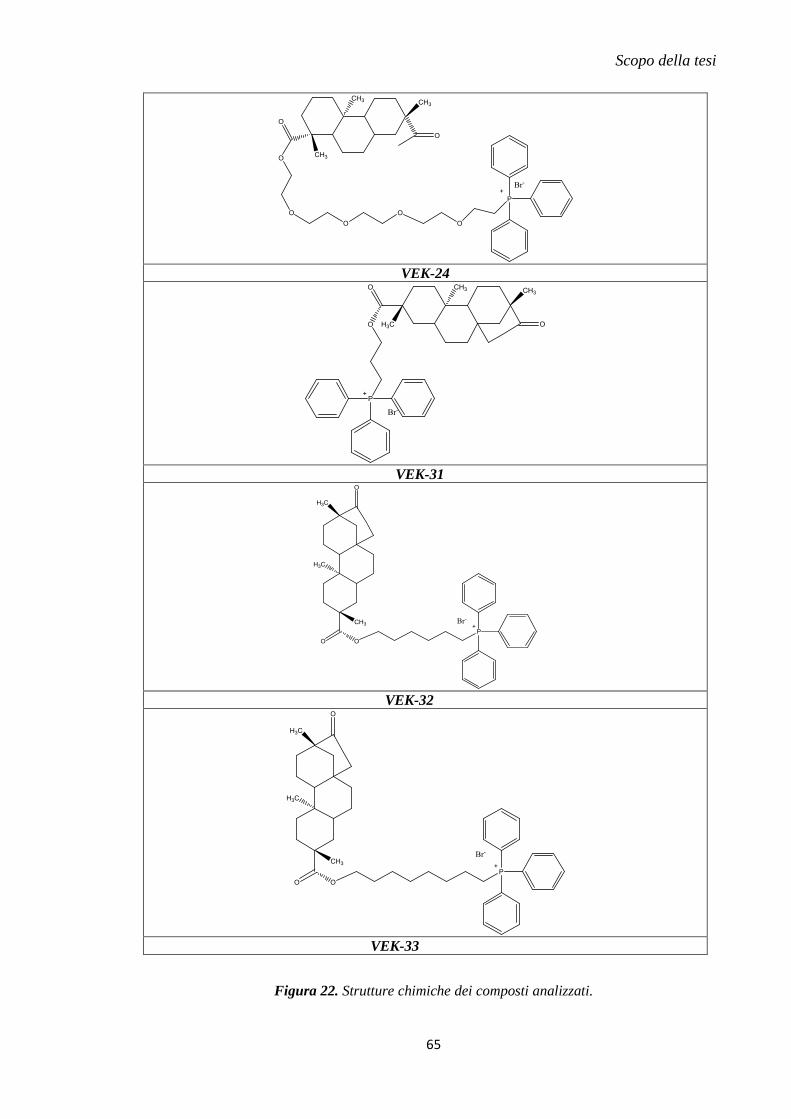
Scopo della tesi
65
VEK-24
VEK-31
VEK-32
VEK-33
Figura 22. Strutture chimiche dei composti analizzati.

66
3. MATERIALI E METODI
3.1 ANIMALI
La sperimentazione è stata condotta su ratti albini del ceppo Wistar, di sesso maschile
con un peso compreso tra 300 e 400g.
Gli animali sono stati allevati in gabbie di dimensioni appropriate al peso e al numero di
individui, adeguatamente rifornite di cibo e acqua, esposti a cicli di luce-buio di 12 ore
in conformità con la normativa comunitaria (2010/63/UE) e italiana (D.L. del 4/03/2014
n°26).
Il giorno dell’esperimento gli animali sono stati anestetizzati con una iniezione
intraperitoneale di Pentobarbitale sodico (100 mg/Kg).
Figura 23. Ratto Wistar

Materiali e Metodi
67
3.2 PROCEDURA DI ISOLAMENTO DEI MITOCONDRI CARDIACI
Dopo il dissanguamento ascellare dell’animale, una sternotomia e una pericardiotomia,
il cuore viene espiantato e immediatamente posto in buffer di isolamento denominato
STE [composizione: Saccarosio (Sigma-Aldrich) 250 mM; TRIS (Sigma-Aldrich) 5
mM; EGTA (Sigma-Aldrich) 1 mM, portando il pH a 7.4 con aggiunta di HCl quando la
soluzione ha raggiunto una T di circa 4°C] mantenuto in ghiaccio.
Quindi, il cuore subisce un’operazione di sminuzzamento e lavaggio dal sangue residuo
con STE, utilizzando forbici chirurgiche. Questa operazione viene effettuata più volte
per cercare di allontanare il più possibile il sangue residuo.
Una volta completato lo sminuzzamento, le parti del cuore vengono trasferite in un tubo
da centrifuga, sospese in circa 10 ml di STE, e il tutto viene omogeneizzato mediante
l’uso di Ultra-Turrax (IKA, T-18 basic).
L’omogeneizzazione viene eseguita a cicli, per evitare un eccessivo surriscaldamento
del composto, e mantenendo il tutto in ghiaccio.
Successivamente, dopo aver aggiunto ulteriori 30ml di STE all’omogenato ottenuto,
viene effettuata una prima centrifugazione a 1075 giri per 3 minuti a 4°C, al termine
della quale il surnatante viene trasferito in un nuovo tubo da centrifuga e messo in
ghiaccio; invece, al pellet vengono aggiunti circa 10 ml di STE e, trasferito in un Potter
con pestello, viene risospeso. La sospensione ottenuta viene, quindi, portato a un
volume di circa 20 ml con STE e nuovamente centrifugato a 1075 giri per 3 minuti a
4°C. Al termine delle centrifugazione, il pellet viene scartato, mentre il surnatante viene
trasferito in un tubo da centrifuga pulito.
I surnatanti ottenuti dalle prime due centrifughe vengono nuovamente centrifugati a
11950 giri per 10 minuti a 4°C. Al completamento di questa centrifuga, vengono scartati
i surnatanti ottenuti, invece, viene aggiunto a ciascun pellet circa 10 ml di STE e viene
di nuovo sospeso.
A questo punto, dopo aver portato a volume di circa 20 ml con STE, viene effettuata
una successiva centrifugazione a 11950 giri per 10 minuti a 4°C e al termine della
quale, con sistema analogo a quello scritto sopra, si risospende il pellet utilizzando un
secondo buffer di isolamento denominato ST [composizione: Saccarosio (Sigma-
Aldrich) 250 mM; TRIS (Sigma-Aldrich) 5 mM, con T di circa 4°C e pH a 7.4 ottenuto
con l’utilizzo di HCl].

Materiali e Metodi
68
Il surnatante viene scartato, mentre il pellet viene risospeso con 400μl di ST e
conservato in una eppendorf posta in ghiaccio per tutta la durata dell’esperimento, fino
ad un massimo di 2h.
La concentrazione di proteine mitocondriali viene determinata utilizzando la reazione di
Bradford.
La validità della procedura di isolamento è stata confermata da esperimenti preliminari
con la misurazione della funzionalità respiratoria attraverso il saggio di bioluminescenza
dell’ATP, in accordo con il metodo Drew e Leenwenburg.
I mitocondri (1mg/ml) sono stati sospesi in ST con l’aggiunta di Succinato 20 mM,
30 mM, e ADP 5’-difosfato 200μM. La reazione è iniziata dall’aggiunta del
reagente luciferina-luciferasi. Il bianco era costituito dalla stessa preparazione, ma con
la sostituzione dell’ADP 5’-difosfato con acqua bidistillata. Dopo 3 minuti dall’inizio
della reazione, è stata effettuata la misurazione con un luminometro. Nelle preparazioni
testate il saggio ha permesso di determinare la luce prodotta dall’ossidazione ATP-
dipendente della luciferina attraverso l’enzima luciferasi. Nei preparati a cui è stato
aggiunto l’inibitore della ATP-sintetasi oligomicina (2μg/ml) o disaccoppianti della
fosforilazione ossidativa, 2,4-dinitrofenolo (DNP) (100μM), non c’è stata produzione di
ATP e quindi luce.

Materiali e Metodi
69
3.3 PROTOCOLLO SPERIMENTALE 1: VALUTAZIONE DELLE VARIAZIONI
DEL POTENZIALE DI MEMBRANA MITOCONDRIALE
Per determinare il potenziale di membrana ( ) dei mitocondri isolati, sono stati
utilizzati cationi liposolubili. La liposolubilità è importante poiché solo in questo modo
gli ioni possono attraversare liberamente le membrane biologiche e distribuirsi fra
matrice e il mezzo di isolamento extramitocondriale. La concentrazione del mezzo può
essere determinata con un elettrodo iono-selettivo che ci permette di controllare
l’accumulo dello ione e quindi di valutare il potenziale di membrana.
Il catione lipofilo utilizzato è il (tetrafenil-fosfonio) (Sigma-Aldrich).
I cambiamenti di concentrazione del nel mezzo di sospensione vengono misurati
con un mini-elettrodo sensibile al (WPI TipTPP), accoppiato ad un elettrodo di
riferimento, utilizzando un sistema di acquisizione dati (BIOPAC Systems inc.).
I mezzi di sospensione che sono stati utilizzati sono due: uno denominato Swelling
Buffer [composizione: KCl (Carlo Erba) 120 mM; (Sigma-Aldrich) 5 mM;
Hepes (Sigma-Aldrich) 10 mM; Acido Succinico (Sigma-Aldrich) 10 mM;
(Sigma-Aldrich) 2 mM, portando il pH a 7.4 con aggiunta di NaOH]; l’altro, detto
Mannitol Buffer, in cui il gradiente osmotico del potassio è sostituito dal mannitolo
[composizione: Mannitolo (Sigma-Aldrich) 240 mM; (J.T.Baker) 5 mM;
Hepes (Sigma-Aldrich) 10 mM; Acido Succinico (Sigma-Aldrich) 10 mM;
(Carlo Erba) 2 mM, portando il pH a 7.4 con NaOH].
Il buffer di sospensione scelto viene messo all’interno di un piccolo becker, nel quale
vengono immersi i due elettrodi sopra citati; viene aggiunta una concentrazione nota di
(10μM), di EGTA (1mM) (agente chelante dello ione calcio) e successivamente i
mitocondri (in concentrazione 1 mg/ml), mantenendo la soluzione in continua
agitazione magnetica. Si attende che il catione venga accumulato nei mitocondri per
avere una stima del .
Raggiunto il plateau, valutiamo le variazioni di potenziale di membrana costruendo
curve cumulative delle sostanze da analizzare con concentrazioni che variano da 1μM a
300μM. In esperimenti preliminari è stata valutata la capacità dei veicoli (DMSO all’1%
e al 5%), di influenzare il ΔΨ.
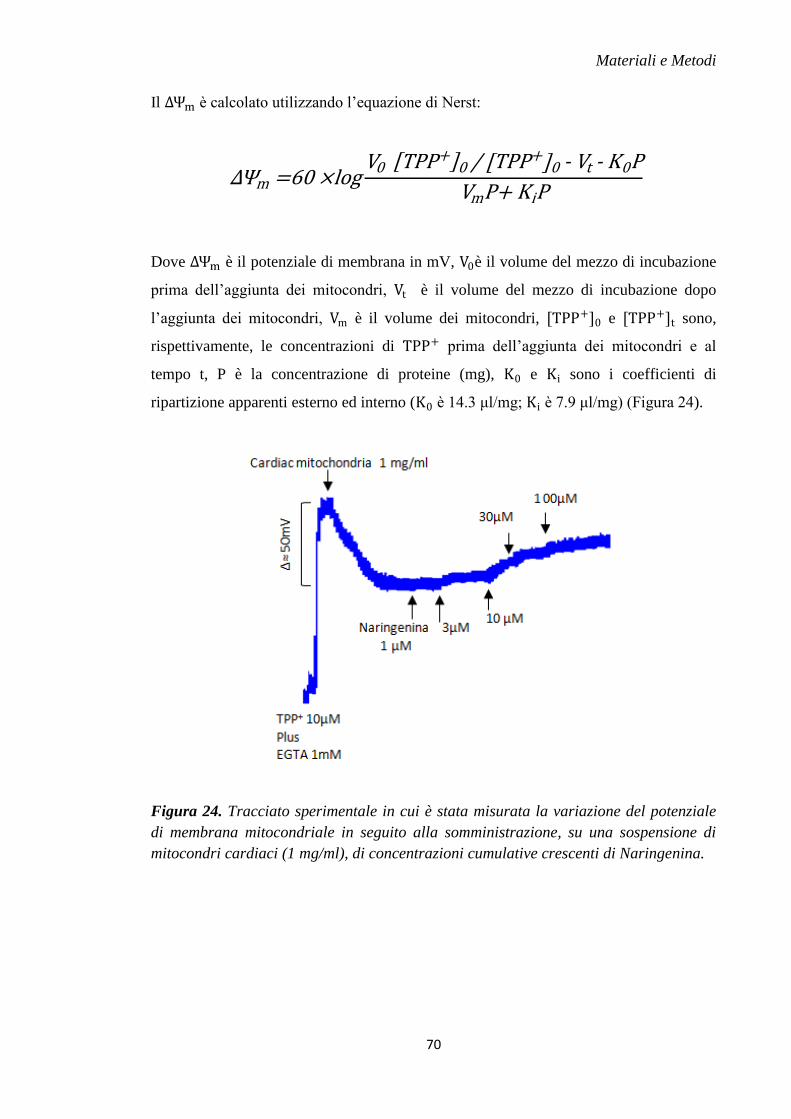
Materiali e Metodi
70
Il è calcolato utilizzando l’equazione di Nerst:
Dove è il potenziale di membrana in mV, è il volume del mezzo di incubazione
prima dell’aggiunta dei mitocondri, è il volume del mezzo di incubazione dopo
l’aggiunta dei mitocondri, è il volume dei mitocondri, e sono,
rispettivamente, le concentrazioni di prima dell’aggiunta dei mitocondri e al
tempo t, P è la concentrazione di proteine (mg), e sono i coefficienti di
ripartizione apparenti esterno ed interno ( è 14.3 μl/mg; è 7.9 μl/mg) (Figura 24).
Figura 24. Tracciato sperimentale in cui è stata misurata la variazione del potenziale
di membrana mitocondriale in seguito alla somministrazione, su una sospensione di
mitocondri cardiaci (1 mg/ml), di concentrazioni cumulative crescenti di Naringenina.

Materiali e Metodi
71
3.4 PROTOCOLLO SPERIMENTALE 2: VALUTAZIONE DELLE VARIAZIONI
NELL’UPTAKE DI
I cambiamenti delle concentrazioni di nel mezzo di sospensione vengono
continuamente misurati con un mini-elettrodo sensibile al (WPI TipCa)
accoppiato ad un elettrodo di riferimento, utilizzando un sistema di acquisizione dati
(BIOPAC Systems inc.).
La selettività dell’elettrodo al rispetto agli altri cationi è maggiore di .
Per correlare la misurazione potenziometrica in mV con la concentrazione di
corrispondente in soluzione, prima di ogni esperimento è stata fatta una curva di
calibrazione utilizzando concentrazioni note di ( - - 3 - M).
Il buffer di sospensione, denominato Swelling Buffer, privo della presenza di ioni calcio
[composizione: KCl (Carlo Erba) 120 mM; (Sigma-Aldrich) 5 mM; Hepes
(Sigma-Aldrich) 10 mM; Acido Succinico (Sigma-Aldrich) 10 mM; (Sigma-
Aldrich) 2 mM, portando il pH a 7.4 con aggiunta di NaOH] viene messo all’interno di
un piccolo becker, nel quale vengono immersi i due elettrodi e sottoposto ad agitazione
magnetica continua. A questa soluzione si aggiunge (100μM) e le sostanze in
esame e il loro veicolo (DMSO all’1%). Quindi, si aggiungono i mitocondri (1mg/ml) e
si attende che il si accumuli all’interno di questi ultimi (ciò è valutabile grazie alla
variazione in mV che si registra).
Misurando la scomparsa del dal mezzo, si risale alla quantità accumulata
all’interno della matrice mitocondriale (Figura 25).
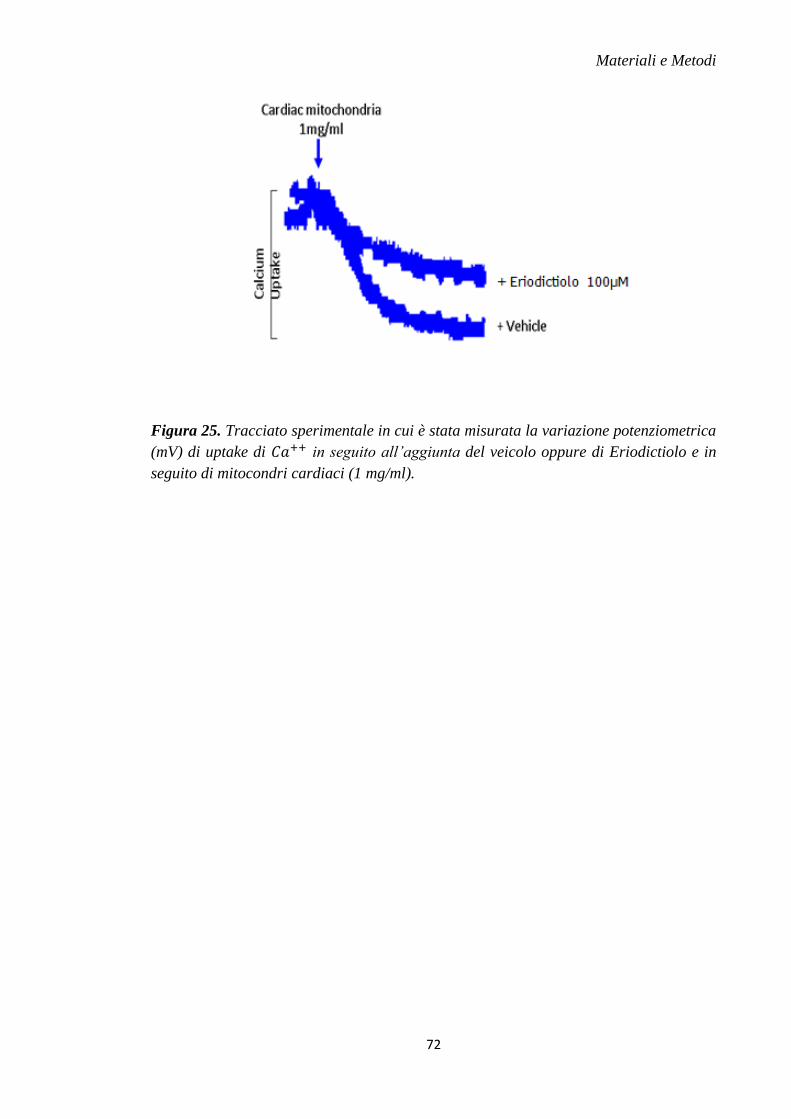
Materiali e Metodi
72
Figura 25. Tracciato sperimentale in cui è stata misurata la variazione potenziometrica
(mV) di uptake di in seguito all’aggiunta del veicolo oppure di Eriodictiolo e in
seguito di mitocondri cardiaci (1 mg/ml).

Materiali e Metodi
73
3.5 PROTOCOLLO SPERIMENTALE 3: VALUTAZIONE DEI MOVIMENTI DI
, UTILIZZANDO UNA SONDA - SENSIBILE
Si sfrutta il fatto che il entra nelle cellule e nelle strutture subcellulari attraverso i
canali del potassio.
L’apertura dei canali mitoK permette il passaggio del all’interno del mitocondrio.
Questi movimenti sono analizzati utilizzando una sonda fluorescente che legandosi al
emette fluorescenza, seguendo la procedura riportata nello specifico kit, FluxOR™
Potassium Ion Channel (Invitrogen™).
Il protocollo di isolamento dei mitocondri cardiaci è lo stesso che è stato descritto in
precedenza. Al termine dell’ultima centrifugazione a 11950 giri per 10 minuti a 4°C, il
pellet viene risospeso in 2ml di Loading Buffer e posto ad agitazione per circa 10 minuti
al buio, così che la sonda viene caricata all’interno del mitocondrio, grazie alle
caratteristiche chimiche che gli permettono di essere permeabile alla membrana.
All’interno la sonda non genera fluorescenza, quindi si eseguono dei lavaggi in ST per
allontanare l’eccesso della sonda e, alla fine, si recupera il pellet e lo si sospende in
400μl di ST.
La concentrazione di proteine mitocondriali viene determinata mediante l’uso della
reazione di Bradford.
Quindi, si diluiscono 0.5 mg/ml di mitocondri con un buffer “potassium-free”,
denominato Mannitol Buffer, dove il gradiente osmotico del è sostituito dal
mannitolo [composizione: Mannitolo (Sigma-Aldrich) 240 mM; (J.T.Baker)
5 mM; Hepes (Sigma-Aldrich) 10 mM; Acido Succinico (Sigma-Aldrich) 10 mM;
(Carlo Erba) 2 mM, portando il pH a 7.4 con NaOH] e ATP 200μM.
80 µl della sospensione mitocondriale vengono messi in ogni well di una piastra da 96
pozzetti e viene misurato il baseline utilizzando uno spettrofluorimetro (EnSpire, Perkin
Elmer) (λ eccitazione: 488 nm; λ emissione: 525nm).
Successivamente, si aggiungono Valinomicina (2µM) (ionoforo dello ione potassio),
DMSO, Eriodictiolo, Isosteviolo, SG40D e il loro veicolo (DMSO al 3%) (sostanze che
sono state precedentemente diluite a concentrazione desiderata con Stimulus Buffer, il
buffer che contiene lo ione ) e si effettuano 12 letture fino al raggiungimento del
plateau.

Materiali e Metodi
74
Quando richiesto dal protocollo sperimentale, l’agente bloccante del canale mitoK,
come l’acido 5-idrossi–decanoato (5-HD, bloccante selettivo dei ), lo si fa
incubare per 5 minuti prima dell’aggiunta dei composti attivatori mitoK.
Il saggio consiste nel misurare la fluorescenza emessa dopo che lo ione entra nella
matrice mitocondriale attraverso i canali del e si lega con la sonda.

Materiali e Metodi
75
3.6 PROTOCOLLO SPERIMENTALE 4: VALUTAZIONE DELLE VARIAZIONI
DEL POTENZIALE DI MEMBRANA MITOCONTRIALE UTILIZZANDO
RHODAMINA 123
Rhodamina 123 è un catione lipofilo fluorescente che viene accumulato nei mitocondri
in proporzione al potenziale di membrana mitocondriale; perciò, rappresenta una valida
alternativa alla misurazione del ΔΨ qualora non sia possibile utilizzare l’approccio
potenziometrico. Quando questa sonda migra all’interno del mitocondrio, l’organello si
comporta da quencher e si assiste ad una scomparsa della fluorescenza, che si misura
allo spettrofluorimetro (Enspire, Perkin Elmer) (λeccitazione: 490nm; λemissione:
535nm, alla temperatura di 37°).
Il protocollo di isolamento dei mitocondri cardiaci è lo stesso descritto in precedenza.
Anche in questo caso, la concentrazione di proteine mitocondriali viene determinata
mediante l’uso della reazione di Bradford.
La messa a punto di questo protocollo è stato un risultato di questa tesi di Laurea,
poiché non è mai stato effettuato in questo laboratorio.
Quindi, 80µl della sospensione mitocondriale (1 mg/ml) vengono messi in ogni well di
una piastra da 96 pozzetti e quindi, si procede con i trattamenti farmacologici e il loro
veicolo (DMSO al 3%) e con lo spettrofluorimetro si procede alla misurazione del
baseline.
Successivamente, si aggiungono 10μl di Rhodamina 123 (5μM) al buio e si effettuano
diverse letture ripetute ogni 15 secondi per 6 minuti attraverso EnSpire.
Infine, viene misurato l’incremento della fluorescenza che si registra in risposta alla
depolarizzazione del potenziale di membrana mitocondriale quando la sonda fuoriesce
dal mitocondrio.

Materiali e Metodi
76
SOSTANZE:
Ciascuna sostanza analizzata è stata solubilizzata in DMSO, preparando la soluzione-
madre di concentrazione - M.
Le diluizioni successive sono state realizzate utilizzando i diversi buffer a seconda del
protocollo realizzato.
, EGTA, sono stati solubilizzati in acqua bidistillata per ottenere una
concentrazione di - M per i primi due, per una concentrazione di - M; le
successive diluizioni sono state effettuate in buffer adeguati al tipo di protocollo
eseguito.

Materiali e Metodi
77
3.7 ANALISI STATISTICA
I dati ottenuti sono stati analizzati applicando il t test di Student o ANOVA two/way.
Un valore di P<0.05 è stato considerato indice di differenza statisticamente
significativa.
Per quanto riguarda le curve concentrazione-risposta depolarizzante ottenute con i
composti progettati per migliorare la delivery mitocondriale (VEK-22, VEK-23, VEK-
24, VEK-31, VEK-32, VEK-33, SG40D), il valore di potenza è espresso come ,
corrispondente al logaritmo negativo della concentrazione alla quale si ha il 50%
dell’effetto massimale; il valore di efficacia massima è invece espresso come ,
corrispondente all’effetto depolarizzante ottenuto alla massima concentrazione del
composto in esame.

78
4. RISULTATI E DISCUSSIONE
Il mitocondrio gioca un ruolo fondamentale nella morte cellulare e nella protezione dal
danno da Ischemia/Riperfusione (I/R), in particolar modo per la presenza di particolari
canali al potassio sulla membrana interna. Infatti, in seguito al danno da I/R, la cellula
va incontro a un processo apoptotico come conseguenza della generazione di ROS,
dell’attivazione dello scambiatore Na /H , di una risposta infiammatoria e dell’apertura
di un poro localizzato a livello della membrana del mitocondrio, poro di transizione di
permeabilità mitocondriale (MPTP), promossa dall’ingresso massivo di ioni calcio
all’interno del mitocondrio. L’apertura di questo poro è prevenuta dall’attivazione dei
canali al potassio mitocondriali; l’attivazione di questi canali, attraverso una parziale
depolarizzazione della membrana mitocondriale, determina un ridotto sovraccarico di
ioni calcio nella matrice del mitocondrio. Questa riduzione di concentrazione, a sua
volta previene al momento della riperfusione la sostenuta apertura del MPTP,
inducendo cardioprotezione.
Pertanto, si comprende quanto sia critico il ruolo del mitocondrio nel fenomeno di
cardioprotezione e quanto sia importante sviluppare farmaci in grado di agire su questi
target mitocondriali fondamentali. Per questo motivo, la conoscenza di target
farmacologici risulta essere un importante campo di ricerca nell’ambito delle patologie
cardiovascolari, che ancora oggi rappresentano la principale causa di morte.
Nell’ottica di selezionare composti potenzialmente attivi nella protezione miocardica
del danno da I/R, questa tesi di Laurea si è posta l’obiettivo di valutare la capacità dei
composti in esame di influenzare alcuni importanti parametri di funzionalità
mitocondriale, quali ΔΨ, uptake di calcio e flussi potassici. Altro obiettivo di questa tesi
di Laurea è stato indagare il meccanismo mitocondriale responsabile di un’eventuale
azione “protettiva” per selezionati composti.
In prima analisi, sulla base degli studi ottenuti nel laboratorio dove ho svolto la mia tesi
di Laurea, per poter effettuare uno screening di potenziali molecole cardioprotettive e
quindi, per predire gli output coinvolti nel meccanismo di azione cardioproettiva
esplicati a livello del mitocondrio, abbiamo effettuato degli studi di tipo
potenziometrico utilizzando il modello sperimentale dei mitocondri cardiaci isolati.
Il flavanone Naringenina, appartenete al genere Citrus, è stato il nostro flavonoide di
riferimento per lo studio, in quanto in precedenti studi ha mostrato avere parziale attività
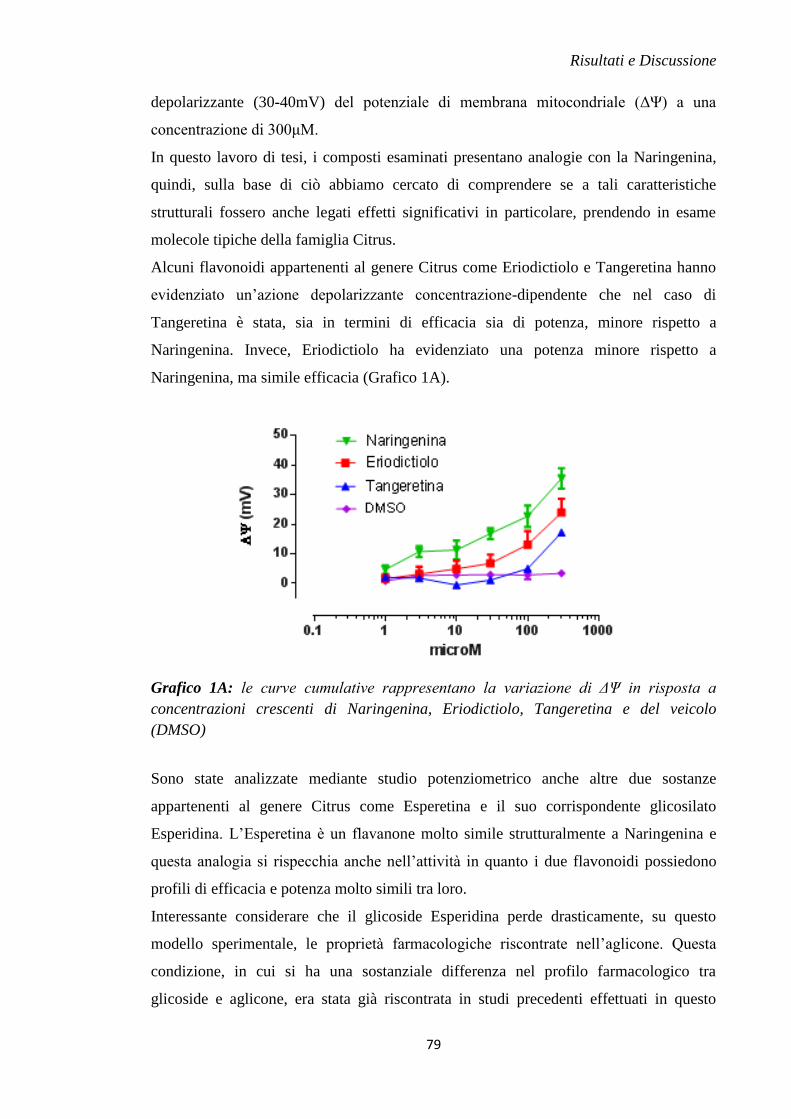
Risultati e Discussione
79
depolarizzante (30-40mV) del potenziale di membrana mitocondriale (ΔΨ) a una
concentrazione di 300μM.
In questo lavoro di tesi, i composti esaminati presentano analogie con la Naringenina,
quindi, sulla base di ciò abbiamo cercato di comprendere se a tali caratteristiche
strutturali fossero anche legati effetti significativi in particolare, prendendo in esame
molecole tipiche della famiglia Citrus.
Alcuni flavonoidi appartenenti al genere Citrus come Eriodictiolo e Tangeretina hanno
evidenziato un’azione depolarizzante concentrazione-dipendente che nel caso di
Tangeretina è stata, sia in termini di efficacia sia di potenza, minore rispetto a
Naringenina. Invece, Eriodictiolo ha evidenziato una potenza minore rispetto a
Naringenina, ma simile efficacia (Grafico 1A).
Grafico 1A: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Naringenina, Eriodictiolo, Tangeretina e del veicolo
(DMSO)
Sono state analizzate mediante studio potenziometrico anche altre due sostanze
appartenenti al genere Citrus come Esperetina e il suo corrispondente glicosilato
Esperidina. L’Esperetina è un flavanone molto simile strutturalmente a Naringenina e
questa analogia si rispecchia anche nell’attività in quanto i due flavonoidi possiedono
profili di efficacia e potenza molto simili tra loro.
Interessante considerare che il glicoside Esperidina perde drasticamente, su questo
modello sperimentale, le proprietà farmacologiche riscontrate nell’aglicone. Questa
condizione, in cui si ha una sostanziale differenza nel profilo farmacologico tra
glicoside e aglicone, era stata già riscontrata in studi precedenti effettuati in questo
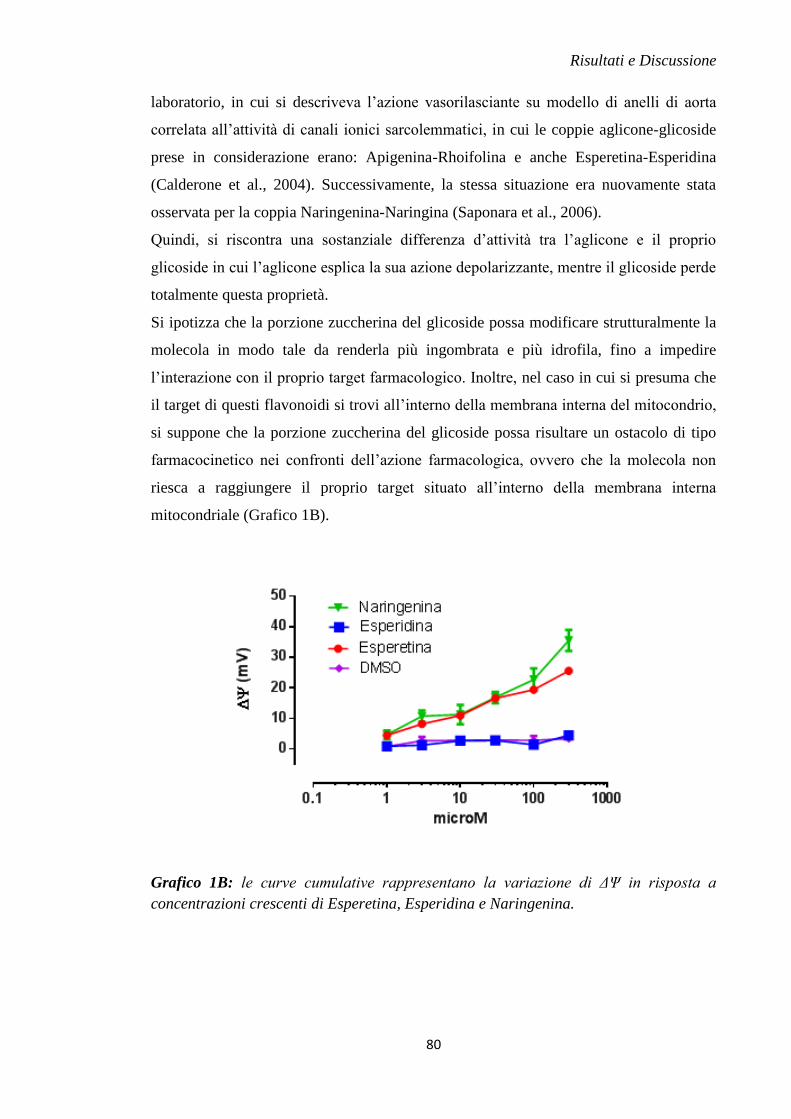
Risultati e Discussione
80
laboratorio, in cui si descriveva l’azione vasorilasciante su modello di anelli di aorta
correlata all’attività di canali ionici sarcolemmatici, in cui le coppie aglicone-glicoside
prese in considerazione erano: Apigenina-Rhoifolina e anche Esperetina-Esperidina
(Calderone et al., 2004). Successivamente, la stessa situazione era nuovamente stata
osservata per la coppia Naringenina-Naringina (Saponara et al., 2006).
Quindi, si riscontra una sostanziale differenza d’attività tra l’aglicone e il proprio
glicoside in cui l’aglicone esplica la sua azione depolarizzante, mentre il glicoside perde
totalmente questa proprietà.
Si ipotizza che la porzione zuccherina del glicoside possa modificare strutturalmente la
molecola in modo tale da renderla più ingombrata e più idrofila, fino a impedire
l’interazione con il proprio target farmacologico. Inoltre, nel caso in cui si presuma che
il target di questi flavonoidi si trovi all’interno della membrana interna del mitocondrio,
si suppone che la porzione zuccherina del glicoside possa risultare un ostacolo di tipo
farmacocinetico nei confronti dell’azione farmacologica, ovvero che la molecola non
riesca a raggiungere il proprio target situato all’interno della membrana interna
mitocondriale (Grafico 1B).
Grafico 1B: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Esperetina, Esperidina e Naringenina.
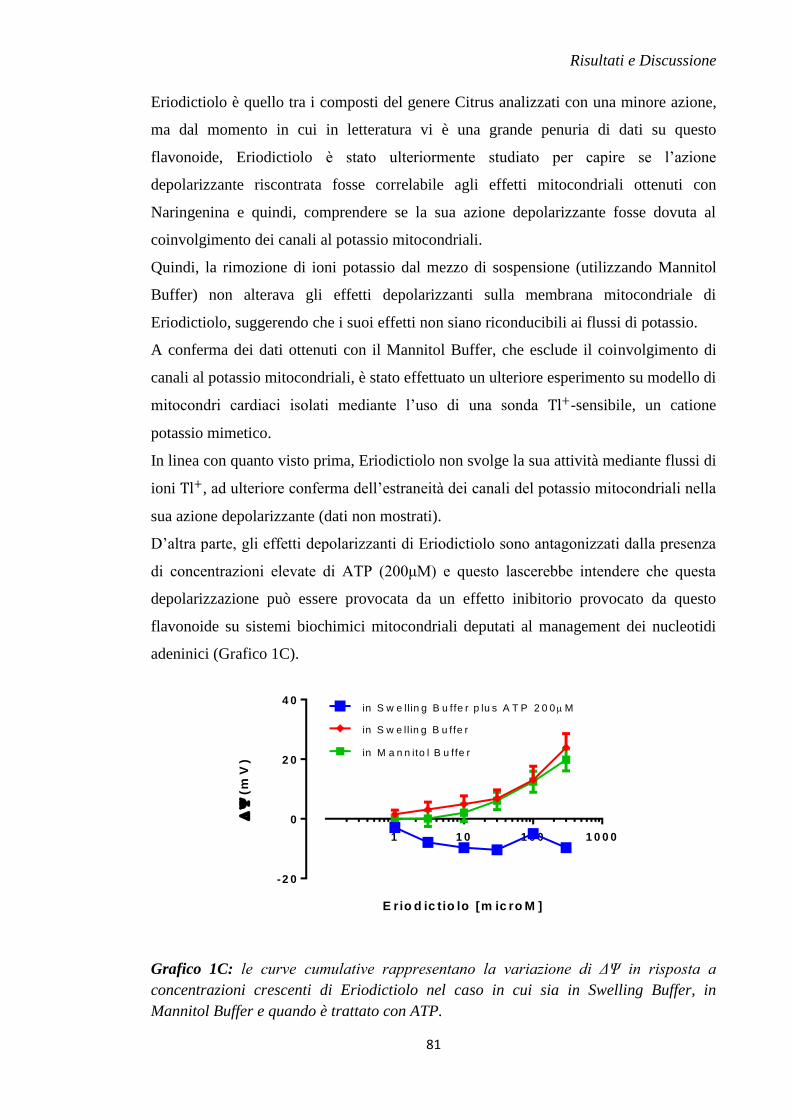
Risultati e Discussione
81
Eriodictiolo è quello tra i composti del genere Citrus analizzati con una minore azione,
ma dal momento in cui in letteratura vi è una grande penuria di dati su questo
flavonoide, Eriodictiolo è stato ulteriormente studiato per capire se l’azione
depolarizzante riscontrata fosse correlabile agli effetti mitocondriali ottenuti con
Naringenina e quindi, comprendere se la sua azione depolarizzante fosse dovuta al
coinvolgimento dei canali al potassio mitocondriali.
Quindi, la rimozione di ioni potassio dal mezzo di sospensione (utilizzando Mannitol
Buffer) non alterava gli effetti depolarizzanti sulla membrana mitocondriale di
Eriodictiolo, suggerendo che i suoi effetti non siano riconducibili ai flussi di potassio.
A conferma dei dati ottenuti con il Mannitol Buffer, che esclude il coinvolgimento di
canali al potassio mitocondriali, è stato effettuato un ulteriore esperimento su modello di
mitocondri cardiaci isolati mediante l’uso di una sonda -sensibile, un catione
potassio mimetico.
In linea con quanto visto prima, Eriodictiolo non svolge la sua attività mediante flussi di
ioni , ad ulteriore conferma dell’estraneità dei canali del potassio mitocondriali nella
sua azione depolarizzante (dati non mostrati).
D’altra parte, gli effetti depolarizzanti di Eriodictiolo sono antagonizzati dalla presenza
di concentrazioni elevate di ATP (200μM) e questo lascerebbe intendere che questa
depolarizzazione può essere provocata da un effetto inibitorio provocato da questo
flavonoide su sistemi biochimici mitocondriali deputati al management dei nucleotidi
adeninici (Grafico 1C).
(mV
)
1 1 0 1 0 0 1 0 0 0
-2 0
0
2 0
4 0
in M a n n ito l B u ffe r
in S w e llin g B u ffe r p lu s A T P 2 0 0M
in S w e ll in g B u ffe r
E rio d ic tio lo [m ic ro M ]
Grafico 1C: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Eriodictiolo nel caso in cui sia in Swelling Buffer, in
Mannitol Buffer e quando è trattato con ATP.
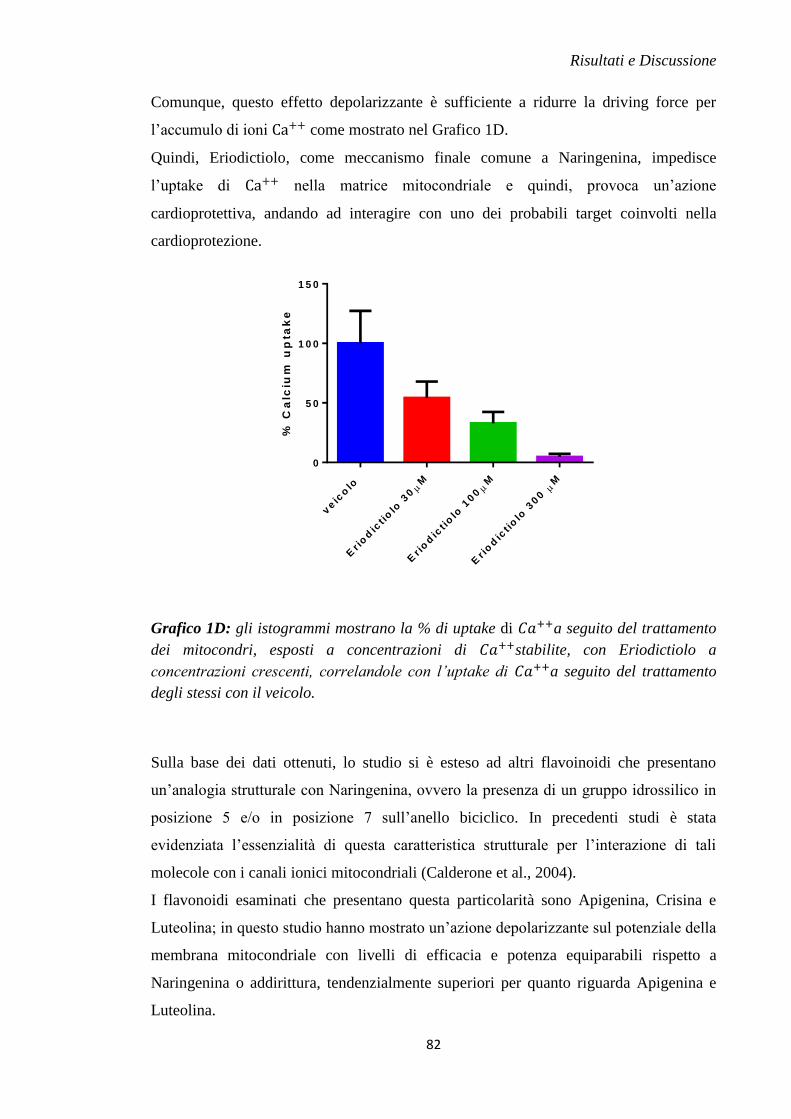
Risultati e Discussione
82
Comunque, questo effetto depolarizzante è sufficiente a ridurre la driving force per
l’accumulo di ioni come mostrato nel Grafico 1D.
Quindi, Eriodictiolo, come meccanismo finale comune a Naringenina, impedisce
l’uptake di nella matrice mitocondriale e quindi, provoca un’azione
cardioprotettiva, andando ad interagire con uno dei probabili target coinvolti nella
cardioprotezione.
veic
olo
Er i
od
ict i
olo
30M
Er i
od
ict i
olo
100M
Er i
od
ict i
olo
300
M
0
5 0
1 0 0
1 5 0%
Ca
lciu
m u
pta
ke
Grafico 1D: gli istogrammi mostrano la % di uptake di a seguito del trattamento
dei mitocondri, esposti a concentrazioni di stabilite, con Eriodictiolo a
concentrazioni crescenti, correlandole con l’uptake di a seguito del trattamento
degli stessi con il veicolo.
Sulla base dei dati ottenuti, lo studio si è esteso ad altri flavoinoidi che presentano
un’analogia strutturale con Naringenina, ovvero la presenza di un gruppo idrossilico in
posizione 5 e/o in posizione 7 sull’anello biciclico. In precedenti studi è stata
evidenziata l’essenzialità di questa caratteristica strutturale per l’interazione di tali
molecole con i canali ionici mitocondriali (Calderone et al., 2004).
I flavonoidi esaminati che presentano questa particolarità sono Apigenina, Crisina e
Luteolina; in questo studio hanno mostrato un’azione depolarizzante sul potenziale della
membrana mitocondriale con livelli di efficacia e potenza equiparabili rispetto a
Naringenina o addirittura, tendenzialmente superiori per quanto riguarda Apigenina e
Luteolina.
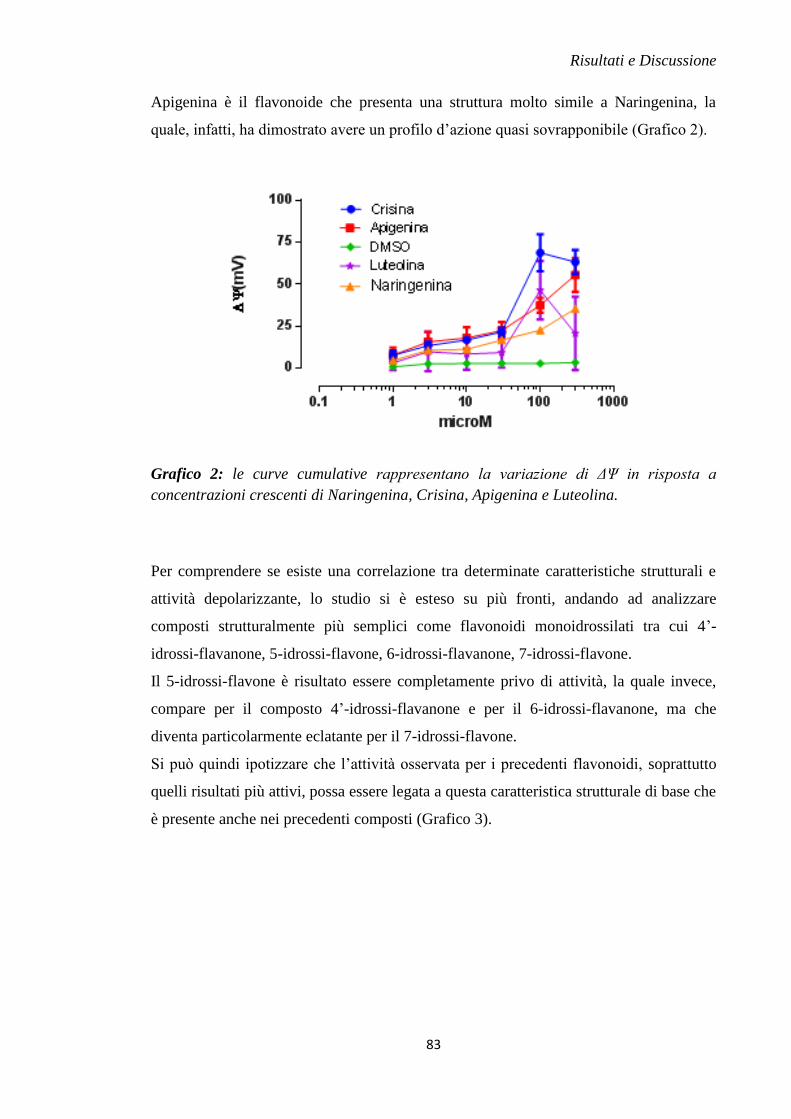
Risultati e Discussione
83
Apigenina è il flavonoide che presenta una struttura molto simile a Naringenina, la
quale, infatti, ha dimostrato avere un profilo d’azione quasi sovrapponibile (Grafico 2).
Grafico 2: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Naringenina, Crisina, Apigenina e Luteolina.
Per comprendere se esiste una correlazione tra determinate caratteristiche strutturali e
attività depolarizzante, lo studio si è esteso su più fronti, andando ad analizzare
composti strutturalmente più semplici come flavonoidi monoidrossilati tra cui 4’-
idrossi-flavanone, 5-idrossi-flavone, 6-idrossi-flavanone, 7-idrossi-flavone.
Il 5-idrossi-flavone è risultato essere completamente privo di attività, la quale invece,
compare per il composto 4’-idrossi-flavanone e per il 6-idrossi-flavanone, ma che
diventa particolarmente eclatante per il 7-idrossi-flavone.
Si può quindi ipotizzare che l’attività osservata per i precedenti flavonoidi, soprattutto
quelli risultati più attivi, possa essere legata a questa caratteristica strutturale di base che
è presente anche nei precedenti composti (Grafico 3).
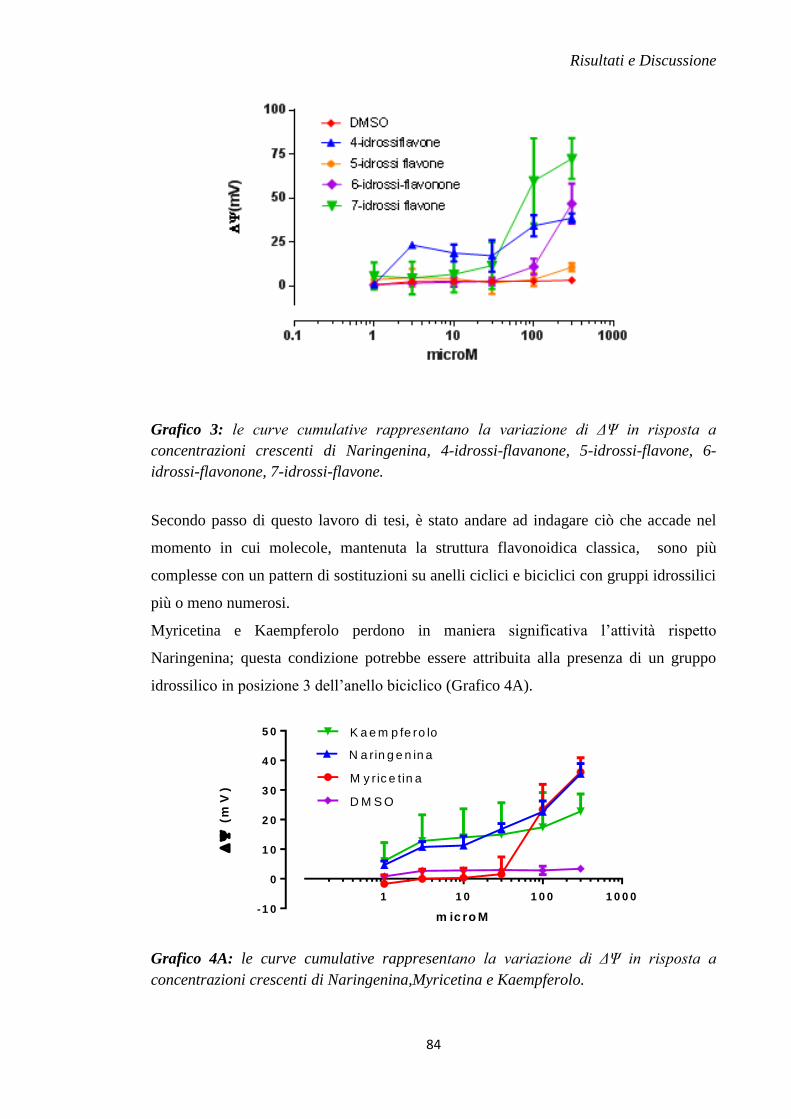
Risultati e Discussione
84
Grafico 3: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Naringenina, 4-idrossi-flavanone, 5-idrossi-flavone, 6-
idrossi-flavonone, 7-idrossi-flavone.
Secondo passo di questo lavoro di tesi, è stato andare ad indagare ciò che accade nel
momento in cui molecole, mantenuta la struttura flavonoidica classica, sono più
complesse con un pattern di sostituzioni su anelli ciclici e biciclici con gruppi idrossilici
più o meno numerosi.
Myricetina e Kaempferolo perdono in maniera significativa l’attività rispetto
Naringenina; questa condizione potrebbe essere attribuita alla presenza di un gruppo
idrossilico in posizione 3 dell’anello biciclico (Grafico 4A).
m ic ro M
(m
V)
1 1 0 1 0 0 1 0 0 0-1 0
0
1 0
2 0
3 0
4 0
5 0
D M S O
M y r ic e tin a
K a e m p fe ro lo
N a rin g e n in a
Grafico 4A: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Naringenina,Myricetina e Kaempferolo.
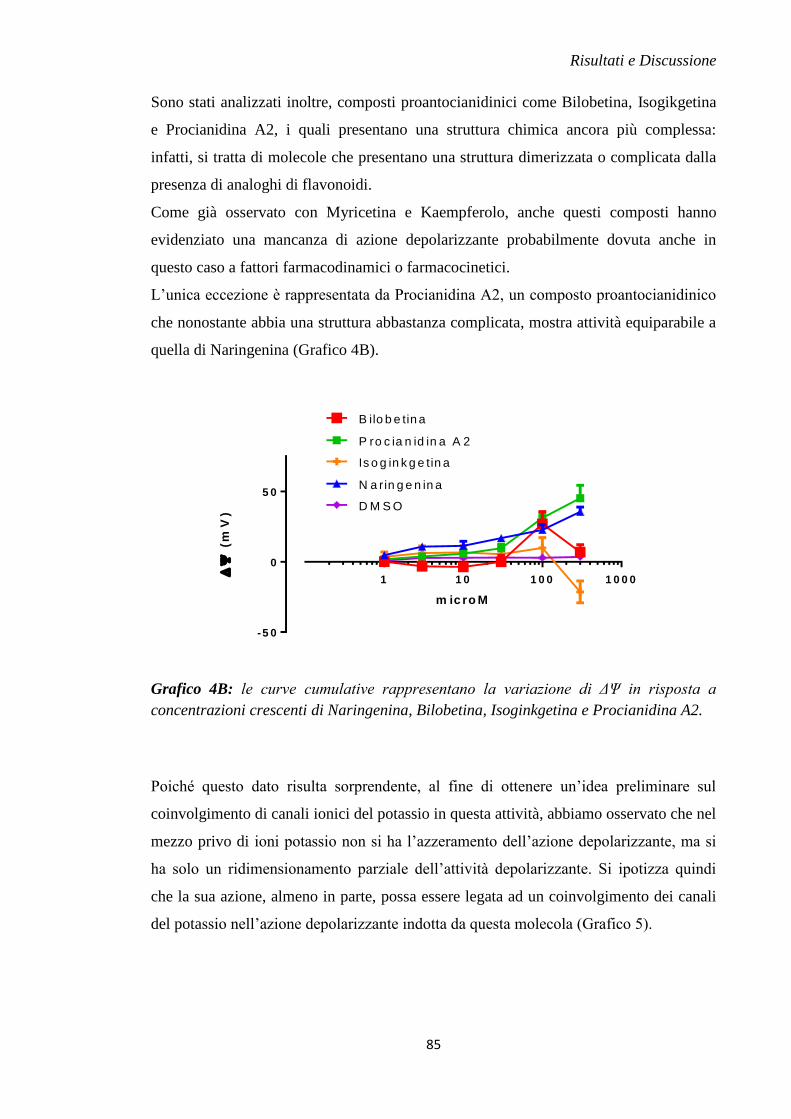
Risultati e Discussione
85
Sono stati analizzati inoltre, composti proantocianidinici come Bilobetina, Isogikgetina
e Procianidina A2, i quali presentano una struttura chimica ancora più complessa:
infatti, si tratta di molecole che presentano una struttura dimerizzata o complicata dalla
presenza di analoghi di flavonoidi.
Come già osservato con Myricetina e Kaempferolo, anche questi composti hanno
evidenziato una mancanza di azione depolarizzante probabilmente dovuta anche in
questo caso a fattori farmacodinamici o farmacocinetici.
L’unica eccezione è rappresentata da Procianidina A2, un composto proantocianidinico
che nonostante abbia una struttura abbastanza complicata, mostra attività equiparabile a
quella di Naringenina (Grafico 4B).
m ic ro M
(m
V)
1 1 0 1 0 0 1 0 0 0
-5 0
0
5 0
D M S O
Is o g in k g e tin a
P ro c ia n id in a A 2
B ilo b e tin a
N a rin g e n in a
Grafico 4B: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Naringenina, Bilobetina, Isoginkgetina e Procianidina A2.
Poiché questo dato risulta sorprendente, al fine di ottenere un’idea preliminare sul
coinvolgimento di canali ionici del potassio in questa attività, abbiamo osservato che nel
mezzo privo di ioni potassio non si ha l’azzeramento dell’azione depolarizzante, ma si
ha solo un ridimensionamento parziale dell’attività depolarizzante. Si ipotizza quindi
che la sua azione, almeno in parte, possa essere legata ad un coinvolgimento dei canali
del potassio nell’azione depolarizzante indotta da questa molecola (Grafico 5).
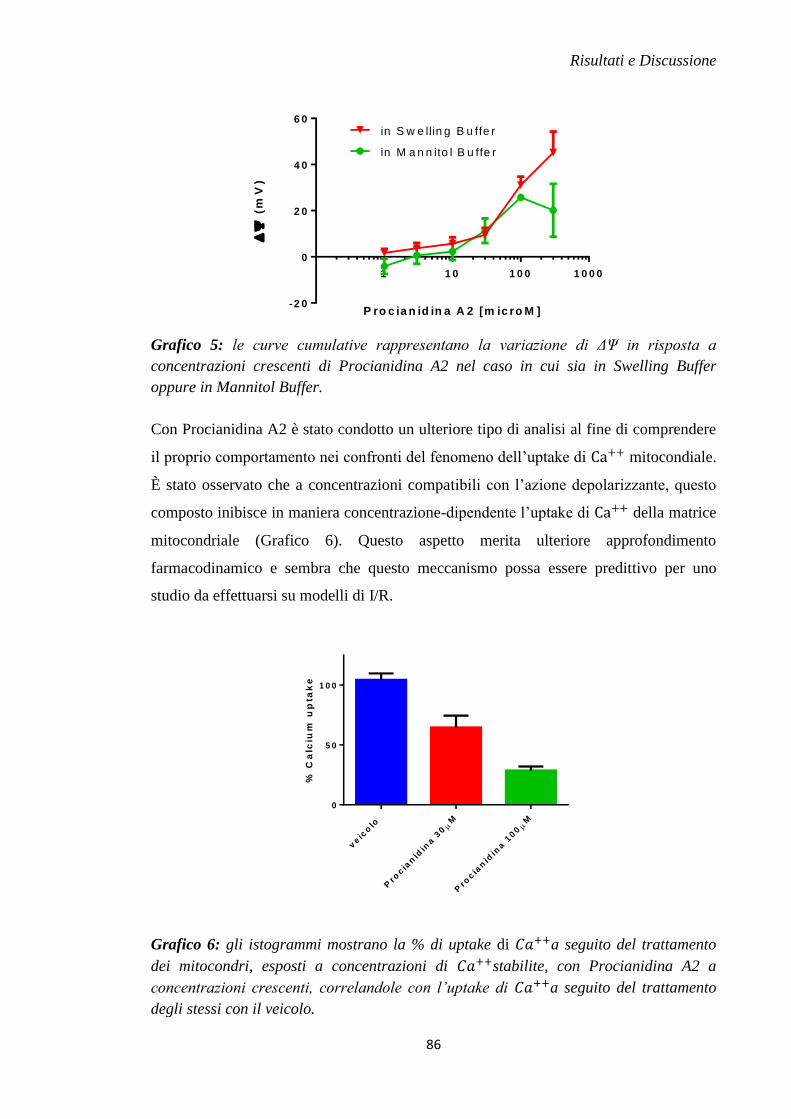
Risultati e Discussione
86
P ro c ia n id in a A 2 [m ic ro M ]
(m
V)
1 1 0 1 0 0 1 0 0 0
-2 0
0
2 0
4 0
6 0
in M a n n ito l B u ffe r
in S w e llin g B u ffe r
Grafico 5: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Procianidina A2 nel caso in cui sia in Swelling Buffer
oppure in Mannitol Buffer.
Con Procianidina A2 è stato condotto un ulteriore tipo di analisi al fine di comprendere
il proprio comportamento nei confronti del fenomeno dell’uptake di mitocondiale.
È stato osservato che a concentrazioni compatibili con l’azione depolarizzante, questo
composto inibisce in maniera concentrazione-dipendente l’uptake di della matrice
mitocondriale (Grafico 6). Questo aspetto merita ulteriore approfondimento
farmacodinamico e sembra che questo meccanismo possa essere predittivo per uno
studio da effettuarsi su modelli di I/R.
veic
olo
Pro
cia
nid
ina 3
0M
Pro
cia
nid
ina 1
00M
0
5 0
1 0 0
% C
alc
ium
up
tak
e
Grafico 6: gli istogrammi mostrano la % di uptake di a seguito del trattamento
dei mitocondri, esposti a concentrazioni di stabilite, con Procianidina A2 a
concentrazioni crescenti, correlandole con l’uptake di a seguito del trattamento
degli stessi con il veicolo.

Risultati e Discussione
87
Una seconda parte dello studio si è invece, dedicata ad un altro aspetto di tipo
farmacocinetico in cui per determinati composti di sintesi si immagina poter sviluppare
e applicare una delivery mitocondriale. In particolare, lo studio si è dedicato alla
valutazione di effetti mitocondriali indotti da molecole attivatrici di canali ionici
mitocondriali, nonché l’analisi di un composto naturale di natura terpenica come
Isosteviolo, riportato in letteratura come possibile attivatore del canale del potassio
mitocondriale di tipo (Xu et al., 2007), sebbene la struttura chimica sembra
discostarsi dai modelli farmacofori già visti precedentemente.
Inoltre, i composti su cui è stato effettuato questo tipo di studio sono F163 e F81, due
composti spirobenzopirani; per questi due composti sono stati già effettuati nel nostro
laboratorio studi che hanno dimostrato il loro coinvolgimento nella cardioprotezione
mediata da canali (Breschi et al., 2006) (Calderone et al., 2010).
Isosteviolo sulla base di evidenze sperimentali non del tutto esaurienti, è considerato un
attivatore del canale di tipo funzionale, quindi per questo composto si è reso
necessario compiere una prima analisi per capire la sua azione a livello del mitocondrio
cardiaco isolato.
È stato osservato che Isosteviolo causa una depolarizzazione concentrazione-dipendente
e questo risultato è in linea con l’azione cardioprotettiva riscontrata in letteratura.
Il comportamento è analogo a quello di Eriodictiolo, ovvero la sua depolarizzazione non
è antagonizzata dal Mannitol Buffer, ma lo è dall’ATP. Questo dato in modo
ragionevole fa escludere il coinvolgimento dei canali al potassio e il fatto che gli effetti
depolarizzanti di Isosteviolo sono antagonizzati dalla presenza di concentrazioni elevate
di ATP fa ipotizzare che questa depolarizzazione può essere legata ad interazioni con i
sistemi biochimici mitocondriali deputati al management dei nucleotidi adeninici
(Grafico 7).
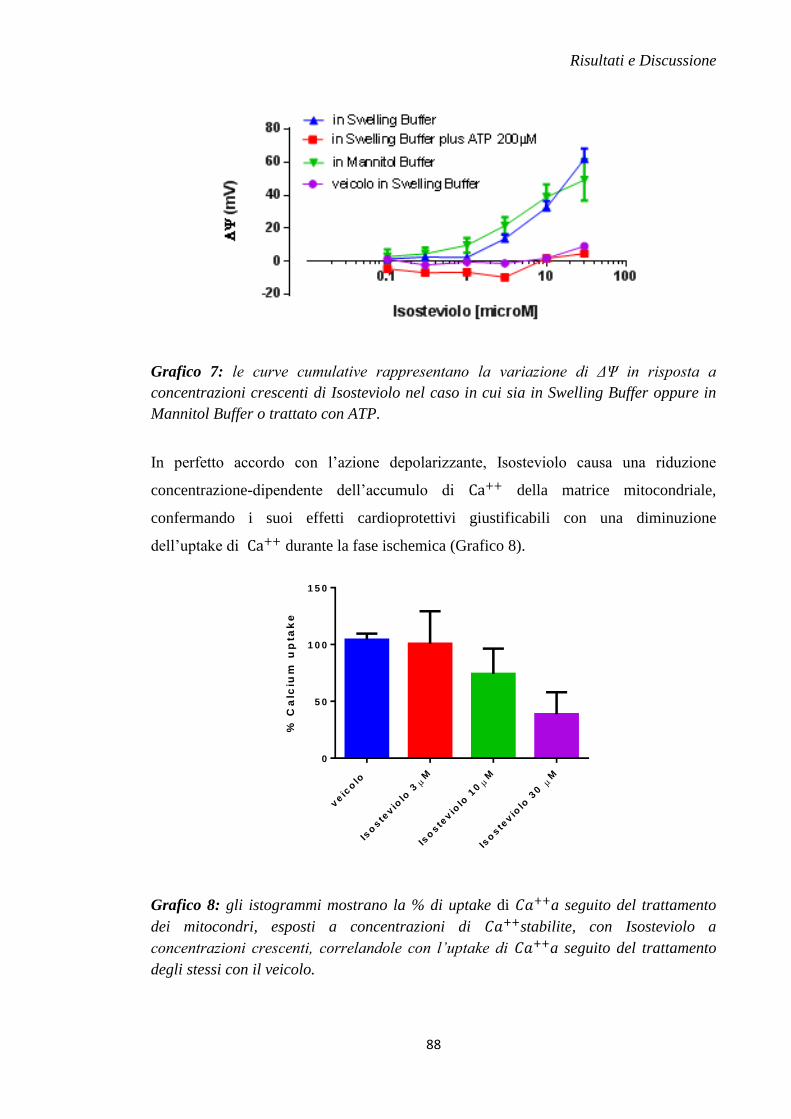
Risultati e Discussione
88
Grafico 7: le curve cumulative rappresentano la variazione di ΔΨ in risposta a
concentrazioni crescenti di Isosteviolo nel caso in cui sia in Swelling Buffer oppure in
Mannitol Buffer o trattato con ATP.
In perfetto accordo con l’azione depolarizzante, Isosteviolo causa una riduzione
concentrazione-dipendente dell’accumulo di della matrice mitocondriale,
confermando i suoi effetti cardioprotettivi giustificabili con una diminuzione
dell’uptake di durante la fase ischemica (Grafico 8).
veic
olo
Iso
ste
vio
lo 3M
Iso
ste
vio
lo 1
0M
Iso
ste
vio
lo 3
0
M
0
5 0
1 0 0
1 5 0
% C
alc
ium
up
tak
e
Grafico 8: gli istogrammi mostrano la % di uptake di a seguito del trattamento
dei mitocondri, esposti a concentrazioni di stabilite, con Isosteviolo a
concentrazioni crescenti, correlandole con l’uptake di a seguito del trattamento
degli stessi con il veicolo.

Risultati e Discussione
89
F163, F81 e Isosteviolo sono molecole idonee per la coniugazione molecolare con il
catione lipofilo trifenilfosfonio ( ) in modo tale da consentire a questi composti un
migliore raggiungimento del proprio target nella membrana mitocondriale.
Sfortunatamente, F163 sebbene migliore di F81, in considerazione dei precedenti studi
(Breschi et al., 2008), non presenta caratteristiche idonee alla coniugazione con questo
catione, quindi è stato usato solo come farmaco di riferimento.
F81 è stato coniugato con legato a una piccola catena alifatica (C4) ( -
MOIETY) mediante un legame ammidico a ottenere il composto denominato SG40D.
Per questo studio di delivery mitocondriale, non è stato possibile utilizzare il metodo
potenziometrico per la presenza di interferenze intrinseche al metodo stesso, quindi è
stato utilizzato un approccio di tipo spettrofluorimetrico usando come sonda
fluorescente la Rhodamina 123, in cui siamo andati ad osservare l’effluso di questa
sonda, precedentemente incamerata dal mitocondrio, in relazione ai diversi trattamenti
farmacologici. Si tratta di un modello efficace sebbene meno sensibile rispetto al
modello potenziometrico.
La messa a punto di questo metodo è stato uno dei risultati di questa tesi, poiché non era
mai stato effettuato prima in questo laboratorio e per fare ciò, si è utilizzato come
trattamento le diverse concentrazioni (0.01 μM-1μM) di m-cloro-carbonilcianuro-
fenilidrazone (CCCP) un agente disaccoppiante con effetti depolarizzanti evidenti.
Quindi, come già noto, F81 si rivela avere azione depolarizzante del potenziale di
membrana mitocondriale meno efficace rispetto a F163, in quanto la depolarizzazione
indotta da F163 a una concentrazione di 300μM è raggiunta ed eguagliata da F81 alla
concentrazione di 1mM, mentre, concentrazioni minori di F81 non hanno mostrato
depolarizzazione rilevabili.
Però, F81 coniugato con la -MOIETY, ovvero il composto SG40D, ha provocato
una depolarizzazione concentrazione-dipendente che alla concentrazione di 100μM è
già massimale: quindi, con una concentrazione 3 volte minore rispetto a F163 e 10 volte
minore a F81, porta un effetto depolarizzante che è incrementato in efficacia di circa 2-3
volte; infatti, SG40D ha evidenziato avere un valore di potenza espresso come pEC50 di
4.42 ± 0.12 e un valore di efficacia massima espressa come Emax (%) di 90 ± 8
(Grafico 9B).
Di per sé -MOIETY, anche a concentrazioni molto elevate, non evoca effetti
depolarizzanti, quindi, realmente la coniugazione di un composto di per sé non ottimo
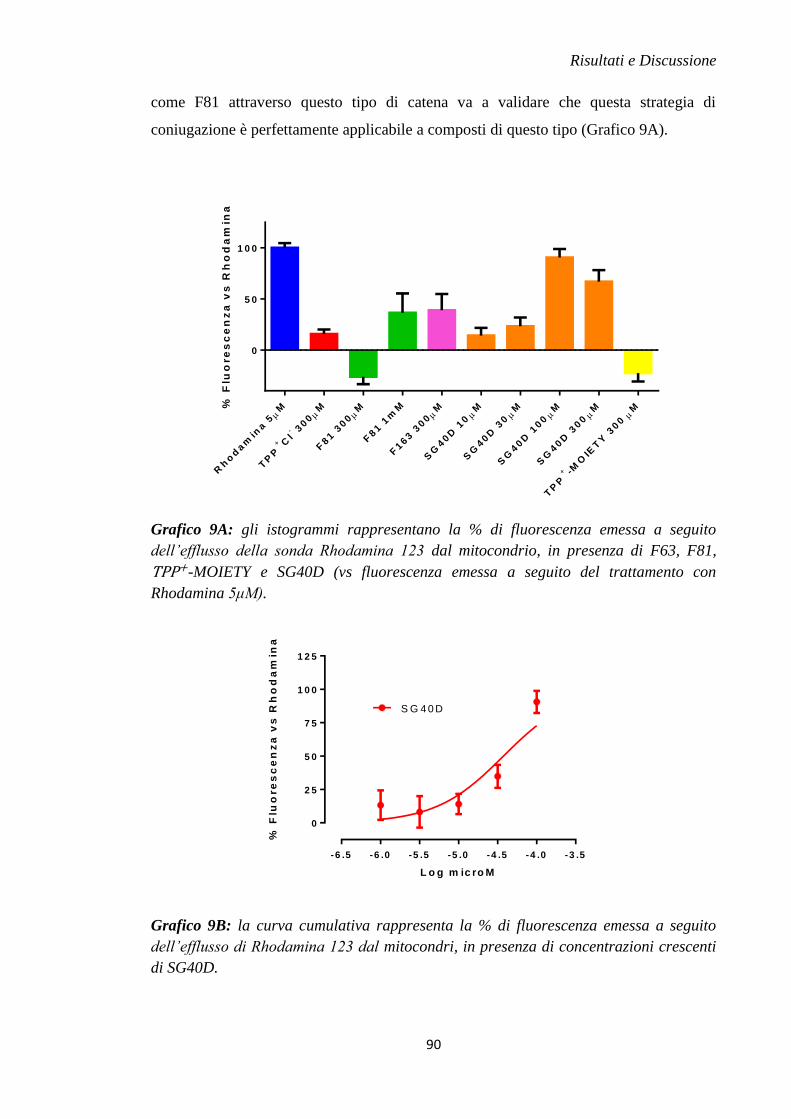
Risultati e Discussione
90
come F81 attraverso questo tipo di catena va a validare che questa strategia di
coniugazione è perfettamente applicabile a composti di questo tipo (Grafico 9A).
% F
luo
re
sc
en
za
vs
Rh
od
am
ina
Rh
od
am
ina 5M
TP
P+ C
l- 3
00
M
F81 3
00
M
F81 1
mM
F163 3
00
M
SG
40D
10M
SG
40D
30M
SG
40D
100M
SG
40D
300M
TP
P+ -M
OIE
TY
300M
0
5 0
1 0 0
Grafico 9A: gli istogrammi rappresentano la % di fluorescenza emessa a seguito
dell’efflusso della sonda Rhodamina 123 dal mitocondrio, in presenza di F63, F81,
-MOIETY e SG40D (vs fluorescenza emessa a seguito del trattamento con
Rhodamina 5μM).
-6 .5 -6 .0 -5 .5 -5 .0 -4 .5 -4 .0 -3 .5
0
2 5
5 0
7 5
1 0 0
1 2 5
S G 4 0 D
L o g m ic ro M
% F
luo
re
sc
en
za
vs
Rh
od
am
ina
Grafico 9B: la curva cumulativa rappresenta la % di fluorescenza emessa a seguito
dell’efflusso di Rhodamina 123 dal mitocondri, in presenza di concentrazioni crescenti
di SG40D.

Risultati e Discussione
91
Successivamente, abbiamo applicato questa strategia di delivery mitocondrilale
all’Isosteviolo coniugandolo al catione lipofilo mediante 6 catene diverse le quali
sono costituite da catene alchiliche di lunghezza crescente oppure da gruppi
polietilenglicoli (PEG) con lunghezza crescente come indicato in tabella 1.
Composto Struttura chimica N° spaziatori tra
Isosteviolo e
Isosteviolo (R)
-MOIETY
VEK-22
3
VEK-23
4
VEK-24
5
VEK-31
3
VEK-32
5
VEK-33
7
Tabella 1: struttura chimica dei derivati dell’Isosteviolo (VEK-22, VEK-23, VEK-24,
VEK-31, VEK.32, VEK-33).
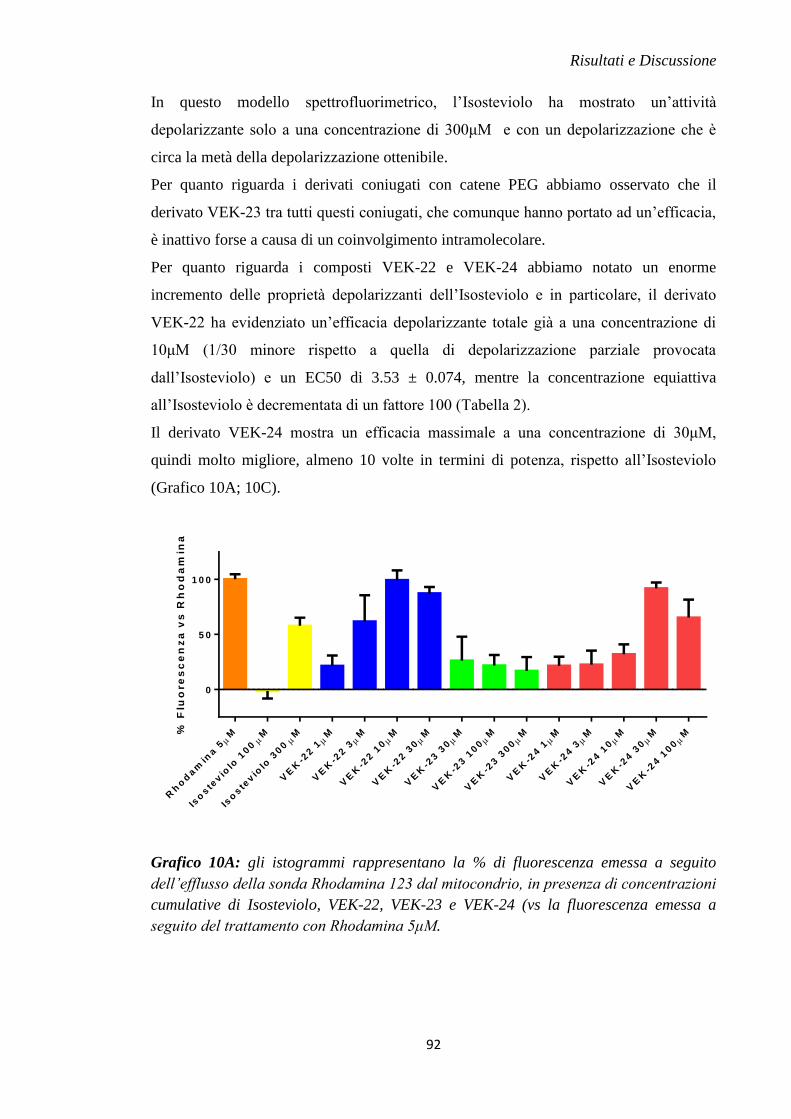
Risultati e Discussione
92
In questo modello spettrofluorimetrico, l’Isosteviolo ha mostrato un’attività
depolarizzante solo a una concentrazione di 300μM e con un depolarizzazione che è
circa la metà della depolarizzazione ottenibile.
Per quanto riguarda i derivati coniugati con catene PEG abbiamo osservato che il
derivato VEK-23 tra tutti questi coniugati, che comunque hanno portato ad un’efficacia,
è inattivo forse a causa di un coinvolgimento intramolecolare.
Per quanto riguarda i composti VEK-22 e VEK-24 abbiamo notato un enorme
incremento delle proprietà depolarizzanti dell’Isosteviolo e in particolare, il derivato
VEK-22 ha evidenziato un’efficacia depolarizzante totale già a una concentrazione di
10μM (1/30 minore rispetto a quella di depolarizzazione parziale provocata
dall’Isosteviolo) e un EC50 di 3.53 ± 0.074, mentre la concentrazione equiattiva
all’Isosteviolo è decrementata di un fattore 100 (Tabella 2).
Il derivato VEK-24 mostra un efficacia massimale a una concentrazione di 30μM,
quindi molto migliore, almeno 10 volte in termini di potenza, rispetto all’Isosteviolo
(Grafico 10A; 10C).
% F
luo
re
sc
en
za
vs
Rh
od
am
ina
Rh
od
am
ina 5M
Iso
ste
vio
lo 1
00M
Iso
ste
vio
lo 3
00M
VE
K-2
2 1M
VE
K-2
2 3M
VE
K-2
2 1
0 M
VE
K-2
2 3
0 M
VE
K-2
3 3
0 M
VE
K-2
3 1
00
M
VE
K-2
3 3
00
M
VE
K-2
4 1M
VE
K-2
4 3M
VE
K-2
4 1
0 M
VE
K-2
4 3
0 M
VE
K-2
4 1
00
M
0
5 0
1 0 0
Grafico 10A: gli istogrammi rappresentano la % di fluorescenza emessa a seguito
dell’efflusso della sonda Rhodamina 123 dal mitocondrio, in presenza di concentrazioni
cumulative di Isosteviolo, VEK-22, VEK-23 e VEK-24 (vs la fluorescenza emessa a
seguito del trattamento con Rhodamina 5μM.
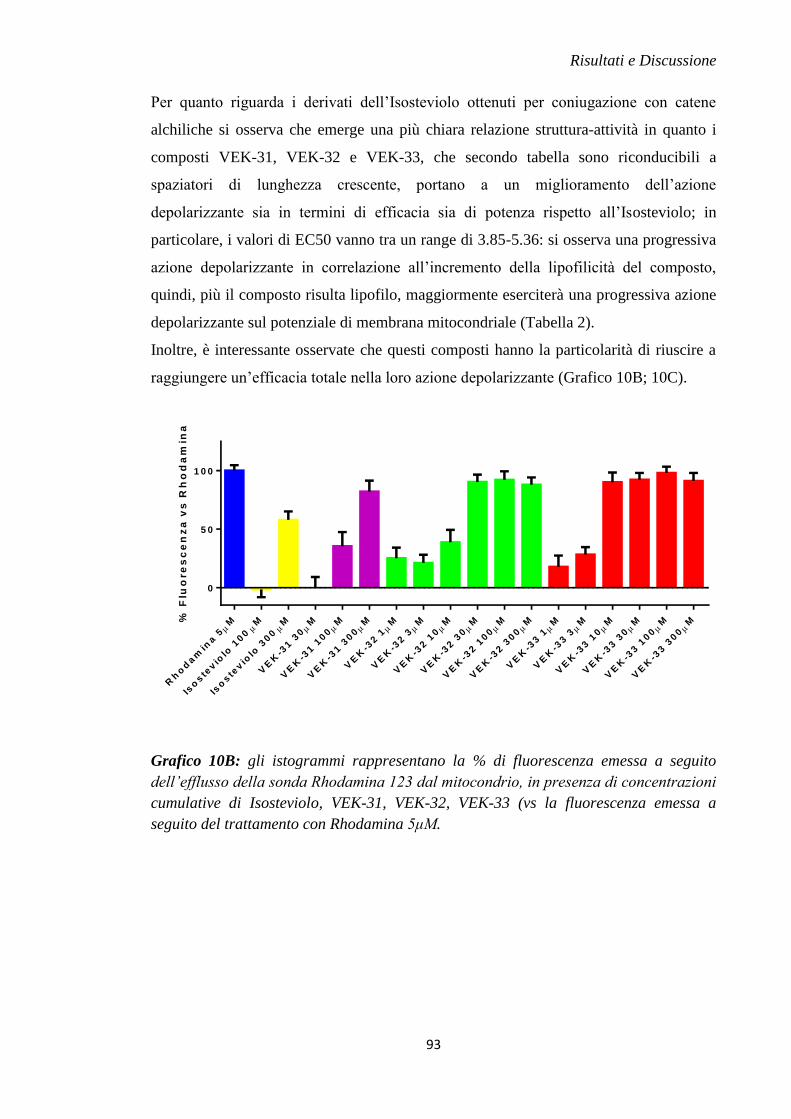
Risultati e Discussione
93
Per quanto riguarda i derivati dell’Isosteviolo ottenuti per coniugazione con catene
alchiliche si osserva che emerge una più chiara relazione struttura-attività in quanto i
composti VEK-31, VEK-32 e VEK-33, che secondo tabella sono riconducibili a
spaziatori di lunghezza crescente, portano a un miglioramento dell’azione
depolarizzante sia in termini di efficacia sia di potenza rispetto all’Isosteviolo; in
particolare, i valori di EC50 vanno tra un range di 3.85-5.36: si osserva una progressiva
azione depolarizzante in correlazione all’incremento della lipofilicità del composto,
quindi, più il composto risulta lipofilo, maggiormente eserciterà una progressiva azione
depolarizzante sul potenziale di membrana mitocondriale (Tabella 2).
Inoltre, è interessante osservate che questi composti hanno la particolarità di riuscire a
raggiungere un’efficacia totale nella loro azione depolarizzante (Grafico 10B; 10C).
% F
luo
re
sc
en
za
vs
Rh
od
am
ina
Rh
od
am
ina 5M
Iso
ste
vio
lo 1
00M
Iso
ste
vio
lo 3
00M
VE
K-3
1 3
0 M
VE
K-3
1 1
00
M
VE
K-3
1 3
00
M
VE
K-3
2 1M
VE
K-3
2 3M
VE
K-3
2 1
0 M
VE
K-3
2 3
0 M
VE
K-3
2 1
00
M
VE
K-3
2 3
00
M
VE
K-3
3 1M
VE
K-3
3 3M
VE
K-3
3 1
0 M
VE
K-3
3 3
0 M
VE
K-3
3 1
00
M
VE
K-3
3 3
00
M
0
5 0
1 0 0
Grafico 10B: gli istogrammi rappresentano la % di fluorescenza emessa a seguito
dell’efflusso della sonda Rhodamina 123 dal mitocondrio, in presenza di concentrazioni
cumulative di Isosteviolo, VEK-31, VEK-32, VEK-33 (vs la fluorescenza emessa a
seguito del trattamento con Rhodamina 5μM.
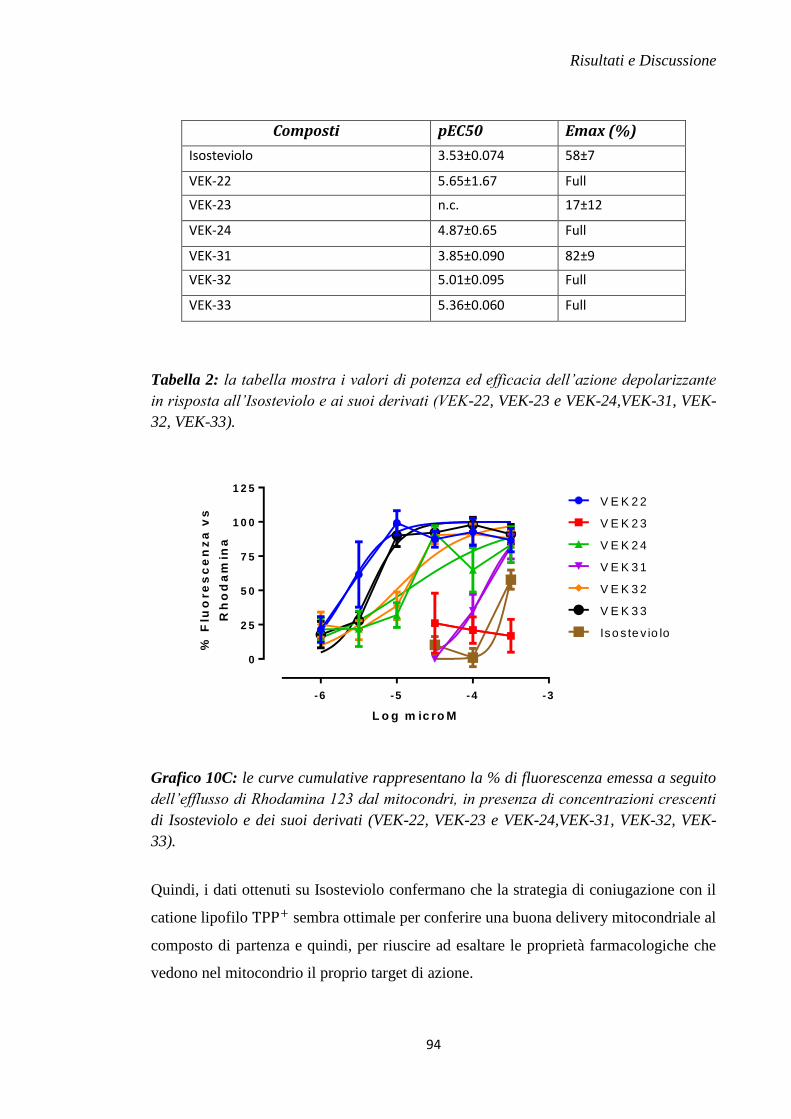
Risultati e Discussione
94
Composti pEC50 Emax (%)
Isosteviolo 3.53±0.074 58±7
VEK-22 5.65±1.67 Full
VEK-23 n.c. 17±12
VEK-24 4.87±0.65 Full
VEK-31 3.85±0.090 82±9
VEK-32 5.01±0.095 Full
VEK-33 5.36±0.060 Full
Tabella 2: la tabella mostra i valori di potenza ed efficacia dell’azione depolarizzante
in risposta all’Isosteviolo e ai suoi derivati (VEK-22, VEK-23 e VEK-24,VEK-31, VEK-
32, VEK-33).
L o g m ic ro M
-6 -5 -4 -3
0
2 5
5 0
7 5
1 0 0
1 2 5
V E K 2 2
V E K 2 3
V E K 2 4
V E K 3 1
V E K 3 2
V E K 3 3
% F
luo
re
sc
en
za
vs
Rh
od
am
ina
Iso s te v io lo
Grafico 10C: le curve cumulative rappresentano la % di fluorescenza emessa a seguito
dell’efflusso di Rhodamina 123 dal mitocondri, in presenza di concentrazioni crescenti
di Isosteviolo e dei suoi derivati (VEK-22, VEK-23 e VEK-24,VEK-31, VEK-32, VEK-
33).
Quindi, i dati ottenuti su Isosteviolo confermano che la strategia di coniugazione con il
catione lipofilo sembra ottimale per conferire una buona delivery mitocondriale al
composto di partenza e quindi, per riuscire ad esaltare le proprietà farmacologiche che
vedono nel mitocondrio il proprio target di azione.

95
5. BIBLIOGRAFIA
Amigo I, Traba J, González-Barroso MM, Rueda CB, Fernández M, Rial E, Sánchez A,
Satrústegui J, Del Arco A, Glucagon regulation of oxidative phosphorylation requires
an increase in matrix adenine nucleotide content through Ca2+ activation of the
mitochondrial ATP-Mg/Pi carrier SCaMC-3, J Biol Chem, 2013; 288(11):7791-802
Ardeali H, O’Rourke B, Mitochondrial channels in cell survival and death, J Mol
Cell Cardiol, 2005; 39:7-16
Armstrong SC, Delacey M, Ganote CE, Phosphorylation state of hsp27 and p38 mapk
during preconditioning and protein phosphatase inhibitor protection of rabbit
cardiomyocytes, Journal of molecular and cellular cardiology, 1999; 31:555-567
Arts IC, Hollman PC, Polyphenols and disease risk in epidemiologic studies, Am J Clin
Nutr, 2005; 81(1 Suppl):317S-325S
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill
EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, Loss of cyclophilin
D reveals a critical role for mitochondrial permeability transition in cell death, Nature,
2005; 434(7033):658-662
Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P,
Mitochondrial PCK epsilon and MAPK form signaling modules in the murine heart.
Enhanced mitochondrial PKC-epsilon MAPK interactions and differential MAPK
activation in PKC epsilon-induced cardioprotection, Circ Res, 2002; 90:390-397
Bantzen BH, Nardi A, Calloe K, Madsen LS, Olesen SP, Grunnet M, The small
molecule NS11021 is a potent and specific activator of -activated big-conductance
channels, Mol Pharmacol, 2007; 72:1033-1044
Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P, Properties of the
permeability transition pore in mitochondria devoid of Cyclophilin D, J Biol Chem,
2005; 280:18558-18561
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y,
Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK,
Integrative genomics identifies MCU as an essential component of the mitochondrial
calcium uniporter, Nature, 2011; 476(7360):341-345
http://www.ncbi.nlm.nih.gov/pubmed/?term=Baughman%20JM%5BAuthor%5D&cauthor=true&cauthor_uid=21685886

Bibliografia
96
Bednarczyk P, Barker GD, Halestrap AP, Determination of the rate of K(+) movement
through potassium channels in isolated rat heart and liver mitochondria, Biochim
Biophys Acta, 2008; 1777:540-548
Bednarczyk P, Kowalczyk JE, Beresewicz M, Dołowy K, Szewczyk A, Zabłocka B,
Identification of a voltage-gated potassium channel in gerbil hippocampal
mitochondria, Biochem Biophys Res Commun, 2010; 397:614-620
Bernardi P, Mitochondrial transport of cations: channels, exchangers, and permeability
transition, Physiol Rev, 1999; 79(4):1127-1155
Bijur GN, Jope RS, Rapid accumulation of Akt in mitochondria following
phosphatidylinositol 3-kinase activation, J Neurochem, 2003; 87(6):1427-1435
Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres
P, Konietzka I, Lopez-Iglesias C, Garcia-Dorado D, Di Lisa F, Heusch G, Schulz R,
Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic
preconditioning, Cardiovasc Res, 2005; 67:234-244
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R, Inhibition of permeability
transition pore opening by mitochondrial STAT3 and its role in myocardial
ischemia/reperfusion, Basic Res Cardiol, 2010; 105(6):771-785
Breschi MC, Calderone V, Martelli A, Minutolo F, Rapposelli S, Testai L, Tonelli F,
Balsamo A, New Benzopyran-Based Openers of the Mitochondrial ATP-Sensitive
Potassium Channel with Potent Anti-Ischemic Properties, J Med Chem, 2006; 49:7600-
7602
Breschi MC, Calderone V, Digiacomo M, Manganaro M, Martelli A, Minutolo F,
Rapposelli S, Testai L, Tonelli F, Balsamo A, Spirocyclic benzopyran-based derivatives
as new anti-ischemic activators of mitochondrial ATP-sensitive potassium channel, J
Med Chem, 2008; 51(21):6945-6954
Calderone V, Chericoni S, Martinelli C, Testai L, Nardi A, Morelli I, Breschi MC,
Martinotti E, Vasorelaxing effects of flavonoids: investigation on the possible
involvement of potassium channels, Naunyn Schmiedebergs Arch Pharmacol, 2004;
370(4):290-298
Calderone V, Testai L, Martelli A, Rapposelli S, Digiacomo M, Balsamo A, Breschi
MC, Anti-ischemic properties of a new spiro-cyclic benzopyran activator of the cardiac
channel, Biochemical Pharmacology, 2010; 79:39-47
http://www.ncbi.nlm.nih.gov/pubmed/?term=Konietzka%20I%5BAuthor%5D&cauthor=true&cauthor_uid=15919068

Bibliografia
97
Cameron SJ, Itoh S, Baines CP, Zhang C, Ohta S, Che W, Glassman M, Lee JD, Yan C,
Yang J, Abe J, Activation of big map kinase 1 (bmk1/erk5) inhibits cardiac injury after
myocardial ischemia and reperfusion, FEBS letters, 2004; 566:255-260
Cantley LC, The phosphoinositide 3-kinase pathway, Science, 2002; 296:1655-1657
Cao CM, Xia Q, Gao Q, Chen M, Wong TM, Calcium-activated potassium channel
triggers cardioprotection of ischemic preconditioning, J Pharmacol Exp Ther, 2005;
312:644-650
Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D,
Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart, Science,
2008; 321:1493-1495
Chen J, Guo R, Yan H, Tian L, You Q, Li S, Huang R, Wu K, Naringin inhibits ROS-
activated MAPK pathway in high glucose-induced injuries in H9c2 cardiac cells, Basic
Clin Pharmacol Toxicol, 2014; 114:293-304
Chen YR, Zweier JL, Cardiac mitochondria and reactive oxygen species generation,
Circ Res, 2014; 114:524-537
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ, VDAC2 inhibits BAK
activation and mitochondrial apoptosis, Science, 2003; 301(5632):513-517
Churchill EN, Ferreira JC, Brum PC, Szweda LI, Mochly-Rosen D, Ischaemic
preconditioning improves proteasomal activity and increases the degradation of
deltapkc during reperfusion, Cardiovascular research, 2010; 85:385-394
Clarke SJ, Khaliulin I, Das M, Parker JE, Heesom KJ, Halestrap AP, Inhibition of
mitochondrial permeability transition pore opening by ischemic preconditioning is
probably mediated by reduction of oxidative stress rather than mitochondrial protein
phosphorylation, Circ Res, 2008; 102(9):1082-1090
Cohen MV, Liu GS, Downey JM, Preconditioning causes improved wall motion as well
as smaller infarcts after transient coronary occlusion in rabbits, Circulation, 1991;
84:341-349
Cohen MV, Yang XM, Downey JM, The pH hypothesis of postconditioning: staccato
reperfusion reintroduces oxygen and perpetuates myocardial acidosis, Circulation,
2007; 115(14):1895-1903
Cotran RS, Kumar V, Collins T, Cellular pathology I: cell injury and cell death, In:
Robbins pathologic basis of disease, Philadeplhia, PA: WB Saunderss, 1999, pp. 1-29

Bibliografia
98
Costa AD, Garlid KD, Intramitochondrial signaling: Interactions among mitokatp,
pkcepsilon, ros, and mpt, American journal of physiology. Heart and circulatory
physiology, 2008; 295:H874-882
Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, Critz SD,
Protein kinase g transmits the cardioprotective signal from cytosol to mitochondria,
Circulation research, 2005; 97:329-336
Davidson SM, Hausenloy D, Duchen MR, Yellon DM, Signalling via the reperfusion
injury signalling kinase (risk) pathway links closure of the mitochondrial permeability
transition pore to cardioprotection, The international journal of biochemistry & cell
biology, 2006; 38:414-419
De Marchi U, Sassi N, Fioretti B, Catacuzzeno L, Cereghetti GM, Szabò I, Zoratti M,
Intermediate conductance - activated potassium channel (KCa3.1) in the
innermitochondrial membrane of human colon cancer cells, Call Calcium, 2009;
45:509-516
D’hahan N, Moreau C, Prost AL, Jacquet H, Alekseev AE, Terzic A, Vivaudou M,
Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward
diazoxide revealed by ADP, Proc Natl Acad Sci USA, 1999; 96:12162-12167
Dickey JS, Gonzalez Y, Aryal B, Mog S, Nakamura AJ, Redon CE, Baxa U, Rosen
E, Cheng G, Zielonka J, Parekh P, Mason KP, Joseph J, Kalyanaraman B, Bonner
W, Herman E, Shacter E, Rao VA, Mito-tempol and dexrazoxane exhibit
cardioprotective and chemotherapeutic effects through specific protein oxidation and
autophagy in a syngeneic breast tumor preclinical model, PLoS One, 2013; 8:e70575
Di Lisa F, Menabo R, Canton M, Barile M, Berardi P, Opening of the mitochondrial
permability transition pore causes depletivo of mitochondrial and cytosolic NAD + and
it is causative event in the death of myocytes in postischemic reperefusion of the heart, J
Biol Chem, 2001; 276: 2571-2575
Dolga AM, Netter MF, Perocchi F, Doti N, Meissner L, Tobaben S, Grohm J, Zischka
H, Plesnila N, Decher N, Culmsee C, Mitochondrial small conductance SK2 channels
prevent glutamate-induced oxytosis and mitochondrial dysfunction, J Biol Chem, 2013;
288:10792-10804
Eaton P, Fuller W, Bell JR, Shattock MJ, Alphab crystallin translocation and
phosphorylation: Signal transduction pathways and preconditioning in the isolated rat
heart, Journal of molecular and cellular cardiology, 2001; 33:1659-1671

Bibliografia
99
Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, Garlid KD, O’Rourke B,
Mitochondrial ROMK channel is a molecular component of mitoK (ATP), Circ Res,
2012; 111:446-454
Gao Y, Shan YQ, Pan MX, Wang Y, Tang LJ, Li H, Zhang Z, Protein kinase c-
dependent activation of p44/42 mitogen-activated protein kinase and heat shock protein
70 in signal transduction during hepatocyte ischemic preconditioning, World journal of
Gastroenterology, 2004; 10:1019-1027
Garlick PB, Radda GK, Seeley PJ, Studies of acidosis in the ischaemic heart by
phosphorus nuclear magnetic resonance, The Biochemical journal, 1979; 184:547-554
Garlid KD, Opening mitochondrial k(atp) in the heart--what happens, and what does
not Happen, Basic research in cardiology, 2000; 95:275-279
Garlid KD, Halestrap AP, the mitochondrial channel. Fact or function?, Mol Cell
Cardiol, 2012; 52:578-583
Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ,
Lodge NJ, Smith MA, Grover GJ, Cardioprotective effect of diazoxide and its
interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of
cardiprotection, Circ Res, 1997; 81:1072-1082
Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA, The mitochondrial
channel as a receptor for potassium channel openers, J Biol Chem, 1996;
271:8796-8799
Goto M, Liu Y, Yang XM, Ardell JL, Cohen MV, Downey JM, Role of bradykinin in
protection of ischemic preconditioning in rabbit hearts, Circulation research, 1995;
77:611-621
Gregory KN, Hahn H, Haghighi K, Marreez Y, Odley A, Dorn GW, 2nd, Kranias EG,
Increased particulate partitioning of pkc epsilon reverses susceptibility of
phospholamban knockout hearts to ischemic injury, Journal of molecular and cellular
cardiology, 2004; 36:313-318
Griffiths EJ, Halestrap AP, Protection by Cyclosporin A of ischemia/reperfusion-
induced damage in isolated rat hearts, J Mol Cell Cardiol, 1993; 25:1461-1469
Grover GJ, Atwal KS, Pharmacologic profile of the selective mitochondrial-K(ATP)
opener BMS-191095 for treatment of acute myocardical ischemia, Cardiovasc Drug
Rev, 2002; 20:121-136

Bibliografia
100
Grover GJ, D’Alonzo AJ, Parham CS, Darbenzio RB, Cardioprotection with the
opener cromakalim is not correlated with ischemic myocardical action potential
duration, J Cardiovasc Pharmacol, 1995; 26:145-152
Gu Z, Jiang Q, Zhang G, Cui Z, Zhu Z, Diphosphorylation of extracellular signal-
regulated kinases and c-jun n-terminal protein kinases in brain ischemic tolerance in
rat, Brain research, 2000; 860:157-160
Halestrap AP, Brenner C, The adenine nucleotide translocase: a central component of
the mitochondrial permeability transition pore and key player in cell death, Curr Med
Chem, 2003; 10(16):1507-1525
Halestrap AP, Kerr PM, Javadov S, Woodfield KY, Elucidating the molecular
mechanism of the permeability transition pore and its role in reperfusion injury of the
heart, Biochimica et biophysica acta, 1998; 1366:79-94
Haunstetter A, Izumo S, Toward antiapoptosis as a new treatment modality, Circ
Res, 2000; 86(4):371-376
Hausenloy DJ, Baxter G, Bell R, Botker HE, Davidson SM, Downey J, Heusch G,
Kitakaze M, Lecour S, Mentzer R, Mocanu MM, Ovize M, Schulz R, Shannon R,
Walker M, Walkinshaw G, Yellon DM, Translating novel strategies for
cardioprotection: the Hatter Workshop Recommendations, Basic Res Cardiol, 2010;
105:677-686
Hausenloy DJ, Erik BH, Condorelli G, Ferdinandy P, Garcia-Dorado D, Heusch G,
Lecour S, van Laake LW, Madonna R, Ruiz-Meana M, Schulz R, Sluijter JP, Yellon
DM, Ovize M, Translating cardioprotection for patient benefit: position paper from the
Working Group of Cellular Biology of the Heart of the European Society of Cardiology,
Cardiovasc Res, 2013; 98:7-27
Hausenloy DJ, Lecour S, Yellon DM, RISK and SAFE pro-survival signaling pathways
in ischaemic postconditioning: two sides of the same coin, Antioxid Redox Signal, 2011;
14 (5):893-907
Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM, Inhibiting mitochondrial
permeability transition pore opening: a new paradigm for myocardical precondition?,
Cardiovasc Res, 2002; 55:543-543
Hausenloy DJ, Ong SB, Yellon DM, The mitochondrial permeability transition pore as
a target for preconditioning and postconditioning, Basic Res Cardiol, 2009;
104(2):189-202

Bibliografia
101
Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM, Ischemic preconditioning protects
by activating prosurvival kinases at reperfusion, American journal of physiology. Heart
and circulatory physiology, 2005; 288:H971-976
Hausenloy DJ, Yellon DM, Survival kinases in ischemic preconditioning and
postconditioning, Cardiovascular research, 2006; 70:240-253
Hausenloy DJ, Yellon DM, The second window of preconditioning (swop) where are we
now?, Cardiovascular drugs and therapy / sponsored by the International Society of
Cardiovascular Pharmacotherapy, 2010; 24:235-254
Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR, Preconditioning protects by
inhibiting the mitochondrial permeability transition, American journal of physiology.
Heart and circulatory physiology, 2004; 287:H841-849
Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, García-Dorado D, Di Lisa F, Schulz
R, Heusch G, Impairment of diazoxide-induced formation of reactive oxygen species
and loss of cardioprotection in connexin 43 deficient mice, Circ Res, 2005; 97:583-586
Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan
PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, Koch WJ, Cheung JY, Madesh M.,
SLC25A23 augments mitochondrial Ca²⁺ uptake, interacts with MCU, and induces
oxidative stress-mediated cell death, Mol Biol Cell, 2014; 25(6):936-947
Iliodromitis EK, Gaitanaki C, Lazou A, Bofilis E, Karavolias GK, Beis I, Kremastinos
DT, Dissociation of stress-activated protein kinase (p38-mapk and jnks)
phosphorylation from the protective effect of preconditioning in vivo, Journal of
molecular and cellular cardiology, 2002; 34:1019-1028
Iliodromitis EK, Papadopoulos CC, Markianos M, Paraskevaidis IA, Kyriakides ZS,
Kremastinos DT, Alterations in circulating cyclic guanosine monophosphate (c-gmp)
during short and long ischemia in preconditioning, Basic research in cardiology, 1996;
91:234-239
Inserte J, Garcia-Dorado D, Ruiz-Meana M, Agullo L, Pina P, Soler-Soler J, Ischemic
preconditioning attenuates calpain-mediated degradation of structural proteins through
a protein kinase a-dependent mechanism, Cardiovascular research, 2004; 64:105-114
James TN, The variable morphological coexistence of apoptosis and necrosis in human
myocardial infarction: significance for understanding its pathogenesis, clinical course,
diagnosis and prognosis, Coron Artery Dis, 1998; 9(5):291-307
Jin C, Wu J, Watanabe M, Okada T, Iesaki T, Mitochondrial channels are involved
in ischemic postconditioning in rat hearts, J Physiol Sci, 2012; 62:325-332

Bibliografia
102
Jin ZQ, Goetzl EJ, Karliner JS, Sphingosine kinase activation mediates ischemic
preconditioning in murine heart, Circulation, 2004; 110:1980-1989
Johnson JA, Gray MO, Chen CH, Mochly-Rosen D, A protein kinase c translocation
inhibitor as an isozyme-selective antagonist of cardiac function, The Journal of
biological chemistry, 1996; 271:24962-24966
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S,
Ytrehus K, Antos CL, Olson EN, Sollott SJ, Glycogen synthase kinase-3beta mediates
convergence of protection signaling to inhibit the mitochondrial permeability transition
pore, The Journal of clinical investigation, 2004; 113:1535-1549
Jung YS, Kim MY, Kim MH, Lee S, Yi KY, Yoo SE, Lee SH, Baik EJ, Moon CH, Cho
JP, KR-31466, a benzopyranylindiol analog, attenuates hypoxic injury through
mitochondrial K (ATP) channel and protein kinase C activation in heart-derived H9c2
cells, J Pharmacol Sci, 2003; 92:13-18
Kajma A, Szewczyk A, A new pH-sensitive rectifying potassium channel in
mitochondria from the embryonic rat hippocampus, Biochim Biophys Acta, 2012;
1817:1867-1878
Karliner JS, Sphingosine kinase and sphingosine 1-phosphate in the heart: A decade of
progress, Biochimica et biophysica acta, 2013; 1831:203-212
Kerendi F, Kin H, Halkos ME, Jiang R, Zatta AJ, Zhao ZQ, Guyton RA, Vinten-
Johansen J, Remote postconditioning. Brief renal ischemia and reperfusion applied
before coronary artery reperfusion reduces myocardial infarct size via endogenous
activation of adenosine receptors, Basic Res Cardiol, 2005; 100:404-412
Kevin LG, Camara AKS, Riess ML, Novalija E, Stowe DF, Ischemic preconditioning
alters real-time measure of O2-radicals in intact with ischemia and reperfusion, Am J
Physiol, 2003; 284:566-574
Kharbanda RK, Peters M, Walton B, Kattenhorn M, Mullen M, Klein N, Vallance P,
Deanfield J, MacAllister R, Ischemic preconditioning prevents endothelial injury and
systemic neutrophil activation during ischemia-reperfusion in humans in vivo,
Circulation, 2001; 103:1624-1630
Kitakaze M, Hori M, Takashima S, Sato H, Inoue M, Kamada T, Ischemic
preconditioning increases adenosine release and 5'-nucleotidase activity during
myocardial ischemia and reperfusion in dogs. Implications for myocardial salvage,
Circulation, 1993; 87:208-215

Bibliografia
103
Knight RJ, Buxton DB, Stimulation of c-jun kinase and mitogen-activated protein
kinase by ischemia and reperfusion in the perfused rat heart, Biochemical and
biophysical research communications, 1996; 218:83-88
Kobara M, Tatsumi T, Matoba S, Yamahara Y, Nakagawa C, Ohta B, Matsumoto T,
Inoue D, Asayama J, Nakagawa M, Effect of ischemic preconditioning on mitochondrial
oxidative phosphorylation and high energy phosphates in rat hearts, J Mol Cell
Cardiol, 1996; 28(2):417-428
Kopustinskiene DM, Pollesello P, Saris NE, Potassium-specific effects of levosimendan
on heart mitochondria, Biochem Pharmacol, 2004; 68:807-812
Krieg T, Qin Q, McIntosh EC, Cohen MV, Downey JM, Ach and adenosine activate
pi3-kinase in rabbit hearts through transactivation of receptor tyrosine kinases,
American journal of physiology. Heart and circulatory physiology, 2002; 283:H2322-
2330
Kulawiak B, Kudin AP, Szewczyk A, Kunz WS, BK channel openers inhibit ROS
production of isolated rat brain mitochondria, Exp Neuronal, 2008; 212:543-547
Kuno A, Critz SD, Cui L, Solodushko V, Yang XM, Krahn T, Albrecht B, Philipp S,
Cohen MV, Downey JM, Protein kinase c protects preconditioned rabbit hearts by
increasing sensitivity of adenosine a2b-dependent signaling during early reperfusion,
Journal of molecular and cellular cardiology, 2007; 43:262-271
Kwak BR, Saez JC, Wilders R, Chanson M, Fishman GI, Hertzberg EL, Spray DC,
Jongsma HJ, Effects of cgmp-dependent phosphorylation on rat and human connexin43
gap junction channels, Pflugers Archiv: European journal of physiology, 1995;
430:770-778
Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J,
Woodgett JR, The stress-activated protein kinase subfamily of c-jun kinases, Nature,
1994; 369:156-160
Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, Szabò I, Inhibitors of
mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells,
EMBO Mol Med, 2012; 4:577-593
Leanza L, Zoratti M, Gulbins E,Szabò I, Induction of apotosis in macrophages via
Kv1.3 and Kv1.5 potassium channels, Curr Med Chem, 2012; 19:5394-5404
Lecour S, Activation of the protective Survivor Activating Factor Enhancement (SAFE)
pathway against reperfusion injury: Does it go beyond the RISK pathway?, J Mol Cell
Cardiol, 2009; 47(1):32-40

Bibliografia
104
Lee S, Yi KY, Kim SK, Suh J, Kim NJ, Yoo SE, Lee BH, Seo HW, Kim SO, Lim H,
Cardioselective anti-ischemic ATP-sensitive potassium channel ( ) openers:
Benzopyranyl indoline and indole analogues, Eur J Med Chem, 2003; 38:459-471
Li CM, Zhang XH, Ma XJ, Luo M, Limb ischemic postconditioning protects
myocardium from ischemia-reperfusion injury, Scand Cardiovasc J, 2006; 40:312-317
Li Y, Johnson N, Capano M, Edwards M, Crompton M, Cyclophilin-D promotes the
mitochondrial permeability transition but has opposite effects on apoptosis and
necrosis, Biochem J, 2004; 383(Pt 1):101-109
Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM,
Protection against infarction afforded by preconditioning is mediated by a1 adenosine
receptors in rabbit heart, Circulation, 1991; 84:350-356
Liu Y, Ytrehus K, Downey JM, Evidence that translocation of protein kinase c is a key
event during ischemic preconditioning of rabbit myocardium, Journal of molecular and
cellular cardiology, 1994; 26:661-668
Ma H, Huang X, Li Q, Guan Y, Yuan F, Zhang Y, ATP-dependent potassium channels
and mitochondrial permeability transition pores play roles in the cardioprotection of
the theaflavin in young rat, J Physiol Sci, 2011; 61:337-342
Makaula S, Lochner A, Genade S, Sack MN, Awan MM, Opie LH, H-89, a non-specific
inhibitor of protein kinase a, promotes post-ischemic cardiac contractile recovery and
reduces infarct size, Journal of cardiovascular pharmacology, 2005; 45:341-347
Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár
T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman
B, Hajnóczky G, Foskett JK, Madesh M, MCUR1 is an essential component of
mitochondrial Ca2+ uptake that regulates cellular metabolism, Nat Cell Biol, 2012;
14(12):1336-1343
Mannhold R, channel openers: Structure-activity relationships and therapeutic
potential, Med Res Rev, 2004; 24:213-266
Marais E, Genade S, Huisamen B, Strijdom JG, Moolman JA, Lochner A, Activation of
p38 mapk induced by a multi-cycle ischaemic preconditioning protocol is associated
with attenuated p38 mapk activity during sustained ischaemia and reperfusion, Journal
of molecular and cellular cardiology, 2001; 33:769-778
Maslov LN, Naryzhnaia NV, Podoksenov Iu K, Mrochek AG, Gorbunov AS,
Tsibul'nikov S, [opioids--triggers of adaptive phenomenon of ischemic preconditioning
of heart], Rossiiskii fiziologicheskii zhurnal imeni I.M. Sechenova / Rossiiskaia
akademiia nauk, 2014; 100:993-1007

Bibliografia
105
Matsumoto-Ida M, Akao M, Takeda T, Kato M, Kita T, Real-time 2-photon imaging of
mitochondrial function in perfused rat hearts subjected to ischemia/reperfusion,
Circulation, 2006; 114:1497-503
Maulik N, Yoshida T, Zu YL, Sato M, Banerjee A, Das DK, Ischemic preconditioning
triggers tyrosine kinase signaling: A potential role for mapkap kinase 2, The American
journal of physiology, 1998; 275:H1857-1864
Meera P, Wallner M, Jiang Z, Toro L, A calcium switch for the functional coupling
between alpha (hslo) and beta subunits (K, V, Ca beta) of maxi K channel, FEBS Lett,
1996; 382:84-88
Montero M, Lobatón CD, Hernández-Sanmiguel E, Santodomingo J, Vay L, Moreno A,
Alvarez J, Direct activation of the mitochondrial calcium uniporter by natural plant
flavonoids, Biochem J, 2004; 384:19-24
Moore CL, Specific inhibition of mitochondrial Ca++ transport by ruthenium red,
Biochem Biophys Res Commun, 1971; 42(2):298-305
Murata M, Akao M, O’Rourke B, Marban E, Mitochondrial ATP-sensitive potassium
channels attenuate matrix Ca (2+) overload during simulated ischemia and
reperfusion; possible mechanism of cardioprotection, Circ Res, 2001; 89:891-898
Murphy E, Steenbergen C, Inhibition of gsk-3beta as a target for cardioprotection: The
importance of timing, location, duration and degree of inhibition, Expert opinion on
therapeutic targets, 2005; 9:447-456
Murphy E, Steenbergen C, Mechanisms underlying acute protection from cardiac
ischemia reperfusion injury, Physiological reviews, 2008; 88:581-609
Murry CE, Jennings RB, Reimer KA, Preconditioning with ischemia: A delay of lethal
cell injury in ischemic myocardium, Circulation, 1986; 74:1124-1136
Nazareth W, Yafei N, Crompton M, Inhibition of anoxia-induced injury in heart
myocytes by ciclosporin A, J Mol Cell Cardiol, 1991; 23:1351-1354
Nemoto S, Xiang J, Huang S, Lin A, Induction of apoptosis by sb202190 through
inhibition of p38beta mitogen-activated protein kinase, The Journal of biological
chemistry, 1998; 273:16415-16420
Ong SB, Hall AR, Hausenloy DJ, Mitochondrial dynamics in cardiovascular health and
disease, Antioxid Redox Signal, 2013; 19(4):400-414

Bibliografia
106
Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ, The mitochondrial permeability
transition pore and its role in myocardial ischemia reperfusion injury, Journal of
molecular and cellular cardiology, 2015; 78:23-34
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ, Inhibiting
mitochondrial fission protects the heart against ischemia/reperfusion injury,
Circulation, 2010; 121(18):2012-2022
Pain T, Yang X, Critz S, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey
JM, Opening of mitochondrial KATP channels triggers the preconditioned state by
generating free radicals, Circ Res, 2000; 87:460-466
Pan TT, Feng ZN, Lee SW, Moore PK, Bian JS, Endogenous hydrogen sulfide
contributes to the cardioprotection by metabolic inhibition preconditioning in the rat
ventricular myocytes, Journal of molecular and cellular cardiology, 2006; 40:119-113
Park WS, Kang SH, Son YK, Kim N, Ko JH, Kim HK, Ko EA, Kim CD, Han J, The
mitochondrial Ca2+-activated K+channel activator, NS 1619 inhibits L-type Ca2+
channels in rat ventricular myocytes, Biochem Biophys Res Commun, 2007; 362(1):31-
36
Pi X, Yan C, Berk BC, Big mitogen-activated protein kinase (bmk1)/erk5 protects
endothelial cells from apoptosis, Circulation research, 2004; 94:362-369
Ping P, Zhang J, Cao X, Li RC, Kong D, Tang XL, Qiu Y, Manchikalapudi S,
Auchampach JA, Black RG, Bolli R, Pkc-dependent activation of p44/p42 mapks
during myocardial ischemia-reperfusion in conscious rabbits, The American journal of
physiology, 1999; 276:H1468-1481
Ping P, Zhang J, Huang S, Cao X, Tang XL, Li RC, Zheng YT, Qiu Y, Clerk A, Sugden
P, Han J, Bolli R, Pkc-dependent activation of p46/p54 jnks during ischemic
preconditioning in conscious rabbits, The American journal of physiology, 1999;
277:H1771-1785
Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis
HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V,
Mootha VK, MICU2, a paralog of MICU1, resides within the mitochondrial uniporter
complex to regulate calcium handling, PLoS One, 2013; 8(2):e55785
Porteous CM, Logan A, Evans C, Ledgerwood EC, Menon DK, Aigbirhio F, Smith
RA, Murphy MP, Rapid uptake of lipophilic triphenylphosphonium cations by
mitochondria in vivo following intravenous injection: implications for mitochondria-
specific therapies and probes, Biochim Biophys Acta, 2010; 1800:1009-1017
http://www.ncbi.nlm.nih.gov/pubmed/?term=Plovanich%20M%5BAuthor%5D&cauthor=true&cauthor_uid=23409044
http://www.ncbi.nlm.nih.gov/pubmed/?term=Porteous%20CM%5BAuthor%5D&cauthor=true&cauthor_uid=20621583

Bibliografia
107
Rapposelli S, Calderone V, Cirilli R, Digiacomo M, Faggi C, La Torre F, Manganaro
M, Martelli A, Testai L, Enantioselectivity in cardioprotection induced by (S)-(-)-2,2-
dimethyl-N-(4’-acetamidobenzyl)-4-spiromorpholone-chromane, J Med Chem, 2009;
52:1477-1480
Rastaldo R, Penna C, Cappello S, Mancardi D, Pagliaro P, Losano G, Ischemic
postconditioning: An effective strategy of myocardial protection?, Giornale italiano di
Cardiologia, 2006; 7:464-473
Rizzuto R, De Stefani D, Raffaello A, Mammucari C, Mitochondria as sensors and
regulators of calcium signaling, Nat Rev Mol Cell Biol, 2012; 13(9):566-578
Rodrigo GC, Davies NW, Standen NB, Diazoxide causes early activation of cardiac
sarcolemmal channels during metabolic inhibition by an indirect mechanism,
Cardiovasc Res, 2004; 61:570-579
Samavati L, Monick MM, Sanlioglu S, Buettner GR, Oberley LW, Hunninghake GW,
Mitochondrial k(atp) channel openers activate the erk kinase by an oxidant-dependent
mechanism, American journal of physiology. Cell physiology, 2002; 283:C273-281
Sanada S, Asanuma H, Tsukamoto O, Minamino T, Node K, Takashima S, Fukushima
T, Ogai A, Shinozaki Y, Fujita M, Hirata A, Okuda H, Shimokawa H, Tomoike H, Hori
M, Kitakaze M, Protein kinase a as another mediator of ischemic preconditioning
independent of protein kinase c, Circulation, 2004; 110:51-57
Sanada S, Komuro I, Kitakaze M, Pathophysiology of myocardial reperfusion injury:
Preconditioning, postconditioning, and translational aspects of protective measures,
American journal of physiology. Heart and circulatory physiology, 2011, 301:H1723-
1741
Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr
SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK, EMRE
is an essential component of the mitochondrial calcium uniporter complex, Science,
2013; 342(6164):1379-1382
Sánchez G, Fernández C, Montecinos L, Domenech RJ, Donoso P,
Preconditioning tachycardia decreases the activity of the mitochondrial permeability
transition pore in the dog heart, Biochem Biophys Res Commun, 2011; 410(4):916-921
Saponara S, Testai L, Iozzi D, Martinotti E, Martelli A, Chericoni S, Sgaragli G, Fusi F,
Calderone V, (+/-)-Naringenin as large conductance Ca2+-activated K+ (BKCa)
channel opener in vascular smooth muscle cells, British Journal of Pharmacology,
2006; 149: 1013–1021

Bibliografia
108
Sassi N, De Marchi U, Fioretti B, Biasutto L, Gulbins E, Francolini F, Szabò I, Zoratti
M, An investigation of the occurrence and properties of the mitochondrial intermediate
conductance -activated channel mtKCa3.1, Biochem Biophys Acta, 2010;
1797:260-1267
Sato H, Jordan JE, Zhao ZQ, Sarvotham SS, Vinten-Johansen J, Gradual reperfusion
reduces infarct size and endothelial injury but augments neutrophil accumulation, The
Annals of thoracic surgery, 1997; 64:1099-1107
Sato M, Cordis GA, Maulik N, Das DK, Sapks regulation of ischemic preconditioning,
American journal of physiology. Heart and circulatory physiology, 2000; 279:H901-907
Saurin AT, Martin JL, Heads RJ, Foley C, Mockridge JW, Wright MJ, Wang Y, Marber
MS, The role of differential activation of p38-mitogen-activated protein kinase in
preconditioned ventricular myocytes, FASEB journal: official publication of the
Federation of American Societies for Experimental Biology, 2000; 14:2237-2246
Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, Marber MS, Targeted
disruption of the protein kinase c epsilon gene abolishes the infarct size reduction that
follows ischaemic preconditioning of isolated buffer-perfused mouse hearts,
Cardiovascular research, 2002; 55:672-680
Schafer C, Ladilov Y, Inserte J, Schafer M, Haffner S, Garcia-Dorado D, Piper HM,
Role of the reverse mode of the na+/ca2+ exchanger in reoxygenation-induced
cardiomyocyte injury, Cardiovascular research, 2001; 51:241-250
Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial
NN, Moskowitz MA, Korsmeyer SJ, Cyclophilin D is a component of mitochondrial
permeability transition and mediates neuronal cell death after focal cerebral ischemia,
Proc Natl Acad Sci USA, 2005; 102(34):12005-12010
Schmidt MR, Smerup M, Konstantinov IE, Shimizu M, Li J, Cheung M, White
PA, Kristiansen SB, Sorensen K, Dzavik V, Redington AN, Kharbanda RK, Intermittent
peripheral tissue ischemia during coronary ischemia reduces myocardial infarction
through a KATP-dependent mechanism: first demonstration of remote ischemic
preconditioning, Am J Physiol Heart Circ Physiol, 2007; 292:H1883-1890
Schott RJ, Rohmann S, Braun ER, Schaper W, Ischemic preconditioning reduces infarct
size in swine myocardium, Circulation research, 1990; 66:1133-1142
Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G,
Ischemic preconditioning preserves connexin 43 phosphorylation during sustained
ischemia in pig hearts in vivo, FASEB journal: official publication of the Federation of
American Societies for Experimental Biology, 2003; 17:1355-1357

Bibliografia
109
Scorrano L, Petronilli V, Bernardi P, On the voltage dependence of the mitochondrial
permeability transition pore. A critical appraisal, The Journal of biological chemistry,
1997; 272:12295-12299
Sharma R, Randhawa PK, Singh N, Jaggi AS, Bradykinin in ischemic conditioning-
induced tissue protection: Evidences and possible mechanisms, Eur J Pharmacol, 2015;
768:58-70
Shoshan-Barmatz V, Gincel D, The voltage-dependent anion channel: characterization,
modulation, and role in mitochondrial function in cell life and death, Cell Biochem
Biophys, 2003; 39(3):279-292
Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS, The voltage-dependent anion
channel (VDAC): function in intracellular signalling, cell life and cell death, Curr
Pharm Des, 2006; 12(18):2249-2270
Siemen D, Loupatatzis C, Borecky J, Gulbins E, Lang F, -activated K channel of
the -type in the inner mitochondrial pf a human glioma cell line, Biochem Biophys
Res Commun, 1999; 257:549-554
Simkhovich BZ, Przyklenk K, Kloner RA, Role of protein kinase c in ischemic
"conditioning": From first evidence to current perspectives, Journal of cardiovascular
pharmacology and therapeutics, 2013; 18:525-532
Singh H, Lu R, Bopassa JC, Meredith AL, Stefani E, Toro L, MitoBK(Ca) is encoded by
the Kcnma 1 gene, and splicing sequence defines its mitochondrial location, Proc Natl
Acad Sci USA, 2013; 110:10836-10841
Škėmienė K, Jablonskienė G, Liobikas J, Borutaitė V, Protecting the heart against
ischemia/reperfusion-induced necrosis and apoptosis: the effect of anthocyanins,
Medicina (Kaunas), 2013; 49(2):84-88
Skyschally A, van Caster P, Iliodromitis EK, Schulz R, Kremastinos DT, Heusch G,
Ischemic postconditioning: experimental models and protocol algorithms, Basic Res
Cardiol, 2009; 104(5):469-483
Smart SC, Sagar KB, Warltier DC, Differential roles of myocardial ca2+ channels and
na+/ca2+ exchange in myocardial reperfusion injury in open chest dogs: Relative roles
during ischemia and reperfusion, Cardiovascular research, 1997; 36:337-346
Smith RA, Murphy MP, Animal and human studies with the mitochondria-targeted
antioxidant MitoQ, Ann N Y Acad Sci, 2010; 1201:96-103

Bibliografia
110
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, Aupetit JF, Bonnefoy
E, Finet G, André-Fouët X, Ovize M, Postconditioning the human heart,
Circulation, 2005; 112(14):2143-2148
Stowe DF, Gadicherla AK, Zhou Y, Aldakkak M, Cheng Q, Kwok WM, Jiang MT,
Heisner JS, Yang MY, Camara AK, Protection against injury by small -sensitive
channel identified in guinea-pig cardiac inner mitochondrial membrane, Biochim
Biophys Acta, 2013; 828:427-442
Szabò I, Boch J, Jekle A, Soddemann M, Adams C, Lang F, Zoratti M, Gulbins E, A
novel potassium channel in lymphocyte mitochondria, J Biol Chem, 2005; 280:12790-
12798
Szeto HH, Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion
injury, Antioxid Redox Signal, 2008; 10:601-619
Szeto HH, Schiller PW, Novel therapies targeting inner mitochondrial membrane--from
discovery to clinical development, Pharm Res, 2011; 28:2669-2679
Szewczyk A, Jarmuszkiewicz W, Kunz WS, Mitochondrial potassium channels,
IUBMB Life, 2009; 61:134-143
Takeishi Y, Abe J, Lee JD, Kawakatsu H, Walsh RA, Berk BC, Differential regulation
of p90 ribosomal s6 kinase and big mitogen-activated protein kinase 1 by
ischemia/reperfusion and oxidative stress in perfused guinea pig hearts, Circulation
research, 1999; 85:1164-1172
Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T, Anti-apoptotic effect of
cgmp in cultured astrocytes: Inhibition by cgmp-dependent protein kinase of
mitochondrial permeable transition pore, The Journal of biological chemistry, 2001;
276:48093-48099
Terzic A, Dzeja PP, Holmuhamedov EL, Mitochondrial channels: Probing
molecular identity and pharmacology, J Mol Cell Cardiol, 2000; 32:1911-1915
Testai L, Flavonoids and mitochondrial pharmacology: A new paradigm for
cardioprotection, Life Sciences, 2015; 135:68-76
Testai L, Barrese V, Soldovieri MV, Ambrosino P, Martelli A, Vinciguerra I, Miceli F,
Greenwood IA, Curtis MJ, Breschi MC, Sisalli MJ, Scorziello A, Canduela MJ,
Grandes P, Calderone V, Taglialatela M, Expression and function of Kv7.4 channels in
rat cardiac mitochondria: possible targets for cardioprotection, Cardiovasc Res, 2016;
110(1):40-50.

Bibliografia
111
Testai L, Martelli A, Cristofaro M, Breschi MC, Calderone V, Cardioprotective effects
of different flavonoids against myocardial ischaemia/reperfusion injury in Langendorff-
perfused rat hearts, J Pharm Pharmacol, 2013; 65(5):750-756
Testai L, Martelli A, Marino A, D’Antongiovanni V, Ciregia F, Giusti L, Lucacchini A,
Chericoni S, Breschi MC, Calderone V, The activation of mitochondrial BK potassium
channels contribuites to the protective effects of naringenin against myocardial
ischemia/reperfusion injury, Biochem Pharmacol, 2013; 85:1634-1643
Testai L, Raposelli S, Martelli A, Breschi MC, Calderone V, Mitochondrial potassium
channels as pharmacological target for cardioprotective drugs, Medicinal research
reviews, 2015, 35: 520-553
Toczylowska-Maminska R, Olszewska A, Laskowski M, Bednarczyk P, Skowronek K,
Szewczyk A, Potassium channel in the mitochondria of human keratinocytes, J Invest
Dermatol, 2014; 134:764-772
Tong H, Chen W, Steenbergen C, Murphy E, Ischemic preconditioning activates
phosphatidylinositol-3-kinase upstream of protein kinase c, Circulation research, 2000;
87:309-315
Tong H, Imahashi K, Steenbergen C, Murphy E, Phosphorylation of glycogen synthase
kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase-dependent
pathway is cardioprotective, Circulation research, 2002; 90:377-379
Van Allen NR, Krafft PR, Leitzke AS, Applegate RL, Tang J, Zhang JH, The role of
volatile anesthetics in cardioprotection: A systematic review, Med Gas Res, 2012; 2:22
Vicente R, Escalada A, Villalonga N, Texidó L, Roura-Ferrer M, Martín-Satué M,
López-Iglesias C, Soler C, Solsona C, Tamkun MM, Felipe A, Association of Kv1.5 and
Kv1.3 contributes to the major voltage-dependent K+ channel in macrophages, J Biol
Chem, 2006; 281:37675-37685
Wang L, Cherednichenko G, Hernandez L, Halow J, Camacho SA, Figueredo V,
Schaefer S, Preconditioning limits mitochondrial Ca(2+) during ischemia in rat hearts:
role of K(ATP) channels, Am J Physiol Heart Circ Physiol, 2001; 280(5):H2321-8
Wang X, Fisher PW, Xi L, Kukreja RC, Essential role of mitochondrial -activated
and ATP-sensitive channels in sildenafil-induced late cardioprotection, J Mol Cell
Cardiol, 2008; 44:105-113

Bibliografia
112
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek
K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J,
Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett
P, Lesnefsky EJ, Larner AC, Function of mitochondrial Stat3 in cellular respiration,
Science, 2009; 323(5915):793-797
Weiss JN, Korge P, Honda HM, Ping P, Role of the mitochondrial permeability
transition in myocardial disease, Circ Res, 2003; 93(4):292-301
Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F, Mechanism of phosphorylation of
protein kinase b/akt by a constitutively active 3-phosphoinositide-dependent protein
kinase-1, The Journal of biological chemistry, 2000; 275:40400-40406
Widmann C, Gibson S, Jarpe MB, Johnson GL, Mitogen-activated protein kinase:
Conservation of a three-kinase module from yeast to human, Physiological reviews,
1999; 79:143-180
Wojtovich AP, Brooks PS, The complex II inhibitor atpenin A5 protects against cardiac
ischemia-reperfusion injury via activation of mitochondrial channels, Basic Res
Cardiol, 2009; 104:121-129
Wrzosek A, Lukasiak A, Gwozdz P, Malinska D, Kozlovski VI, Szewczyk A, Chlopicki
S, Dolowy K, Large-conductance channel opener CGS7184 as a regulator of
endothelial cell function, Eur J Pharmacol, 2009; 602:105-111
Wrzosek A, Tomaskova Z, Ondrias K, Lukasiak A, Szewczyk A, The potassium
channel opener CGS7184 activates release from the endoplasmic reticulum, Eur J
Pharmacol, 2012; 690:60-67
Xu D, Zhang S, Foster DJ, Wang J, The effects of isosteviol against myocardium injury
induced by ischaemia–reperfusion in the isolated guinea pig heart, Clinical and
experimental pharmacology and physiology, 2007; 34:488–493
Xu WH, Liu YG, Wang S, McDonald T, VanEyk JE, Sidor A, O’Rourke B,
Cytoprotective role of - activated channels in cardiac inner mitochondrial
membrane, Science, 2002; 298:1029-1033
Xuan YT, Tang XL, Qiu Y, Banerjee S, Takano H, Han H, Bolli R, Biphasic response
of cardiac no synthase isoforms to ischemic preconditioning in conscious rabbits,
American journal of physiology. Heart and circulatory physiology, 2000; 279:H2360-
2371

Bibliografia
113
Yang X, Cohen MV, Downey JM, Mechanism of cardioprotection by early ischemic
Preconditioning, Cardiovascular drugs and therapy / sponsored by the International
Society of Cardiovascular Pharmacotherapy, 2010; 24:225-234
Yao Z, Gross GJ, Effects of the channel opener bimakalim on coronary blood
flow, monophasic action potential duration, and infarct size in dogs, Circulation, 1994;
89:1769-1775
Ye B, Kroboth SL, Pu JL, Sims JJ, Aggarwal NT, McNally EM, Makielski JC, Shi NQ,
Molecular identification and functional characterization of a mitochondrial
sulfonylurea receptor 2 splice variant generated by intraexonic splicing, Circ Res,
2009; 105:1083-1093
Ytrehus K, Liu Y, Downey JM, Preconditioning protects ischemic rabbit heart by
protein kinase c activation, The American journal of physiology, 1994;266:H1145-1152
Xiao XH, Allen DG, Activity of the Na(+)/H(+) exchanger is critical to reperfusion
damage and preconditioning in the isolated rat heart, Cardiovasc Res, 2000; 48(2):244-
253
Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V, The voltage-
dependent anion channel-1 modulates apoptotic cell death, Cell Death Differ, 2005;
12(7):751-760
Zaugg M, Lucchinetti E, Spahn DR, Pasch T, Schaub MC, Volatile anesthetics mimic
cardiac preconditioning by priming the activation of mitochondrial K (ATP) channels
via multiple signaling pathways, Anesthesiology, 2002; 97:4-14
Zhao ZQ, Vinten-Johansen J, Myocardical apoptosis and ischemic preconditioning,
Cardiovascular research, 2002, 55:438-455
Zorov DB, Juhaszova M, Sollott SJ, Mitochondrial reactive oxygen species (ROS) and
ROS-induced ROS release, Physiol Rev, 2014; 94(3):909-950


















