La “riforma” dell’IVA 2010. Il VAT PACKAGE Lineamenti generali 2.
Upload
truongkienCategory
view
216download
0
Progettazione di primer per PCR. Verifica della specificità
La PCR (Polymerase Chain Reaction) ha progressivamente assunto importanza tra le tecnologie ricombinanti in quanto in diversi protocolli applicativi rappresenta un mezzo potente per raggiungere i più disparati obiettivi. Per ottenere buoni risultati con la PCR è essenziale progettare in modo accurato gli oligonucleotidi (primer) più adatti allo scopo che vogliamo raggiungere e che serviranno da innesco alla DNA polimerasi durante la reazione di amplificazione. Nella fase di progettazione dei primer è fondamentali considerare una serie di fattori:
- specificità di appaiamento - lunghezza (18-25 bp) - contenuto in GC (circa 60%); empiricamente, Tm = 2x(nAT)+4x(nGC) - Tm: è opportuno che siano molto vicine (differenze superiori a 2°C possono determinare amplificazioni aspecifiche) - verificare ed evitare la possibile formazione di strutture secondarie - l'estremità 3' del primer deve terminare con una GC ma non averne più di 3 consecutive.
Inoltre, per un’eventuale ottimizzazione delle condizioni, è utile realizzare reazioni di PCR a varie temperature ed in condizioni variabili di primer e di Mg++. L'aggiunta di altri fattori, quali sali e denaturanti o detergenti, può influire sulla stringenza e sulla specificità di appaiamento; le contaminazioni possono essere combattute mediante aggiunta di uracil-N-glicosilasi (UNG), che digerisce i prodotti della PCR precedente.
Sebbene queste considerazioni possano apparire fuori dal campo d'interesse prettamente bioinformatico, sono riportate in quanto non ha senso acquisire competenza negli strumenti di progettazione senza conoscere almeno i principi base della tecnica. Anzi, proprio dalla conoscenza dei fattori critici per la PCR sono nati i migliori programmi di analisi, che consentono a loro volta di valutare con un'accuratezza impensabile per calcoli "manuali" rischi e vantaggi della scelta di uno specifico primer o di condizioni di PCR.
Consideriamo un esempio in cui si voglia lavorare alla temperatura di massima stringenza di appaiamento primer-target. In particolare, si vuole amplificare un frammento interno della sequenza O49339 presente nel database UniProtKB, per verificarne l’espressione, la correttezza della sequenza depositata nel database, la tessuto specificità o altro. La prima cosa da fare è rintracciare la sequenza nucleotidica codificante (coding sequence). Per le successive fasi di analisi sono disponibili vari programmi bioinformatici, pubblici e commerciali. Tra questi ultimi, si riportano esempi di uso di un software commerciale, il pacchetto LASERGENE© della ditta DNASTAR:

In programmi come LASERGENE© è necessario che le sequenze abbiano un formato specifico; a tal fine, nell'esempio riportato, si usa il programma EditSeq©:
EditSeq© consente anche di invertire la sequenza o, cosa più importante per la progettazione dei primer, di ottenere la sequenza complementare inversa (reverse complement), che serve per l'individuazione del reverse primer:
Inoltre, EditSeq© consente di tradurre la sequenza nucleotidica in aminoacidica:
Può essere molto utile anche creare un documento con word processor, dove poter annotare tutte le informazioni del caso. Nell’ esempio: sequenza codificante, codoni di START e STOP (evidenziati in rosso), posizione e sequenza dei primer, che sono evidenziati in rosso:

Per analizzarne le eventuali strutture secondarie dei primer si può usare un altro programma del software package LASERGENE©, chiamato Primer Select© (o qualsiasi analogo prodotto commerciale o pubblico). Primer Select© è organizzato nelle solite sezioni File, Edit ed Aiuto ed in quelle specifiche Conditions, Locate, Log, Report ed Options. Ovviamente, tali sezioni sono riportate solo a titolo d'esempio e possono differire tra programmi; tuttavia esse rispecchiano la possibilità di modificare numerosi parametri:
Primer Select© richiede in primo luogo che siano impostate le condizioni iniziali di amplificazione e quelle dei primer (di seguito sono mostrate le opzioni preimpostate, modificabili):
Si possono anche impostare le condizioni di mispriming, ovvero di appaiamento errato:

A questo punto si possono inserire le sequenze di ciascun primer:
per poi valutare eventuali appaiamenti dannosi ai fini dell'amplificazione, ad es. tra forward primer:
Si può notare che il programma mostra tutti i possibili appaiamenti; per ciascuno di essi indica (in rosso) le basi complementari e riporta per ciascuna coppia il valore di deltaG. Ecco invece la schermata che mostra i possibili appaiamenti tra reverse primer:

Nell'esempio precedente tutte le coppie avevano medesimi valori di deltaG; nell'appaiamento tra reverse primer, invece, alcune combinazioni risultano più stabili. Ecco, infine, la scheramata dei possibili appaiamenti tra forward e reverse primer:
È importante che non ci sia (o almeno sia ridotto il più possibile) appaiamento al 3’ fra i primer, in modo da evitare l’eventuale allungamento da parte della polimerasi. Ciò infatti è oltremodo dannoso in quanto, al ciclo successivo di PCR, avremo in soluzione qualcosa diverso dal primer iniziale. Per quanto riguarda le regioni 5’, piccole zone di appaiamento sono tollerabili, poiché comunque la polimerasi non è in grado di allungare la sequenza dell’oligonucleotide (alterandolo in modo irreversibile). Zone complementari devono essere intervallate da “bolle di repulsione” che destabilizzano le strutture, la presenza di sequenze non complementari all’estremità dei primer aiuta a sciogliere eventuali complessi formatesi in soluzione grazie all’agitazione termica durante lo step di

appaiamento. Anche l’esistenza di forcine (hairpin) può creare problemi, in questo caso si possono fare le stesse considerazioni sopra elencate oltre a tenere presente che tanto più stretto è il loop della forcina tanto più questa è instabile. Il programma è in grado di predire anche le forcine:
Infine è importante verificare, mediante Primer-BLAST che i primer selezionati mostrino specificità di appaiamento con il bersaglio (target) e non con altre sequenze indesiderate. Ecco i risultati:

che mostrano allineamento tra primer forward e 3 sequenze, identiche al 100%: un mRNA predetto, una sequenza genomica ed un mRNA identificato. E' importante controllare i rapporti tra sequenze identificate, poichè un primer che risulti allinearsi con più sequenze potrebbe apparire erroneamente aspecifico. Infatti, proprio in questo esempio, la seconda sequenza riconosciuta è quella genomica, ma la prima è la corrispondente predizione del messaggero e la terza è lo stesso messaggero, identificato e sequenziato. Scorrendo nella finestra con i risultati di BLAST verso le sequenze con score più bassi, si osservano allineamenti parziali del forward primer con regioni genomiche differenti:
ciò indica il rischio di amplificazione spuria e la necessità, quindi, di badare alla stringenza, adottando una temperatura di amplificazione non troppo più bassa di quella di fusione (Tm) del primer (forward o reverse) che mostra la Tm più bassa.
Progettazione di costrutti ricombinanti e ricerca dei siti di restrizione
Quando si clona in un vettore di espressione per ottenere un prodotto proteico chimerico, è necessario mantenere il corretto registro di lettura delle sequenze codificanti. Per studiare la localizzazione subcellulare di una proteina in vivo si può clonarne la sequenza codificante a valle di quella codificante una proteina come la GFP (o una sua variante); ecco un vettore:

ECFP rappresenta la proteina che si colora (tag) e MCS è invece il sito di policlonaggio, posto al 3’ rispetto al tag e caratterizzato dal fatto di avere siti di restrizione unici all’interno dell’intero vettore:
La progettazione dei primer per clonare l’intera sequenza codificante ha vincoli di posizione: si deve includere il codone di STOP originale e variare il meno possibile a livello del codone di START:
A questo punto, sulla sequenza clonata va eseguita un’analisi di restrizione in silico, ad es. usando ancora un'applicazione di LASERGENE ©, ovvero MapDraw ©. Questo e programmi analoghi permettono di ottenere una mini-mappa del frammento considerato:

elencando anche i siti di taglio unici:
e fornire un elenco di enzimi di restrizione che non tagliano all’interno della sequenza:

Le possibilità indicate dal programma servono ad individuare il sito od i siti più adeguati al vettore, all'inserto ed alle esigenze di clonaggio. Immaginando di aver scelto Bam HI, diventa necessario ridisegnare i primer di in modo da "aggiungere" alla sequenza dell'amplificato "code" rappresentanti i siti di riconoscimento per Bam HI; in tal modo, dopo la PCR, l'amplificato potrà essere digerito e inserito nel sito Bam HI del vettore. Tuttavia sarà necessario anche fare in modo che, dopo la ligazione, la sequenza nucleotidica della proteina d’interesse sia in registro con il tag (nell'esempio illustrato, ECFP). Vediamo cosa succede digerendo il vettore e l’amplicone con Bam HI e simulando la ligazione, nel caso in cui i primer con code non siano stati disegnati in modo corretto:
Prima del taglio:
Dopo il taglio:
Post-ligazione:
Le lettere evidenziate in verde rappresentano l’ultima posizione di ogni codone nel registro di lettura (frame) corrispondente a quello del tag, in rosso è evidenziato il sito di riconoscimento e taglio per Bam HI. Come si vede, usando i primer in figura viene perso il registro di lettura e l'ATG iniziale non è tradotto in metionina, ottenendo un prodotto tronco o comunque tutt’altro che l’originale. Quanto sopra è rapidamente determinabile tramite traduzione dinamica in sequenza aminoacidica della sequenza di DNA dei possibili costrutti con code, mediata da semplici tools bioinformatici, inclusi in software commerciali o disponibili on line presso molti server europei ed americani quali, ad esempio, quelli presenti nella sezione DNA-->protein del server Expasy di Ginevra di cui un esempio è stato riportato nella lezione sulla pagina di proteomic tools di Expasy.
Analisi di cromatogrammi da sequenziamento
Numerosi programmi consentono la visualizzazione e correzione dei cromatogrammi derivanti da sequenziatori automatici e la loro trasformazione in sequenza letterale. Ad esempio EditView consente di impostare i parametri di visualizzazione sia per il cromatogramma che per la sequenza:

Ecco come si presentano il cromatogramma di una sequenza di DNA (visione parziale):
e la sua rappresentazione letterale dell'alfabeto:
Si può notare che a ciascun picco corrisponde, automaticamente, una base: tuttavia quando ci si sposta verso l'estremità 3' della sequenza, i picchi sono meno netti e distanziati e si possono generare errori:
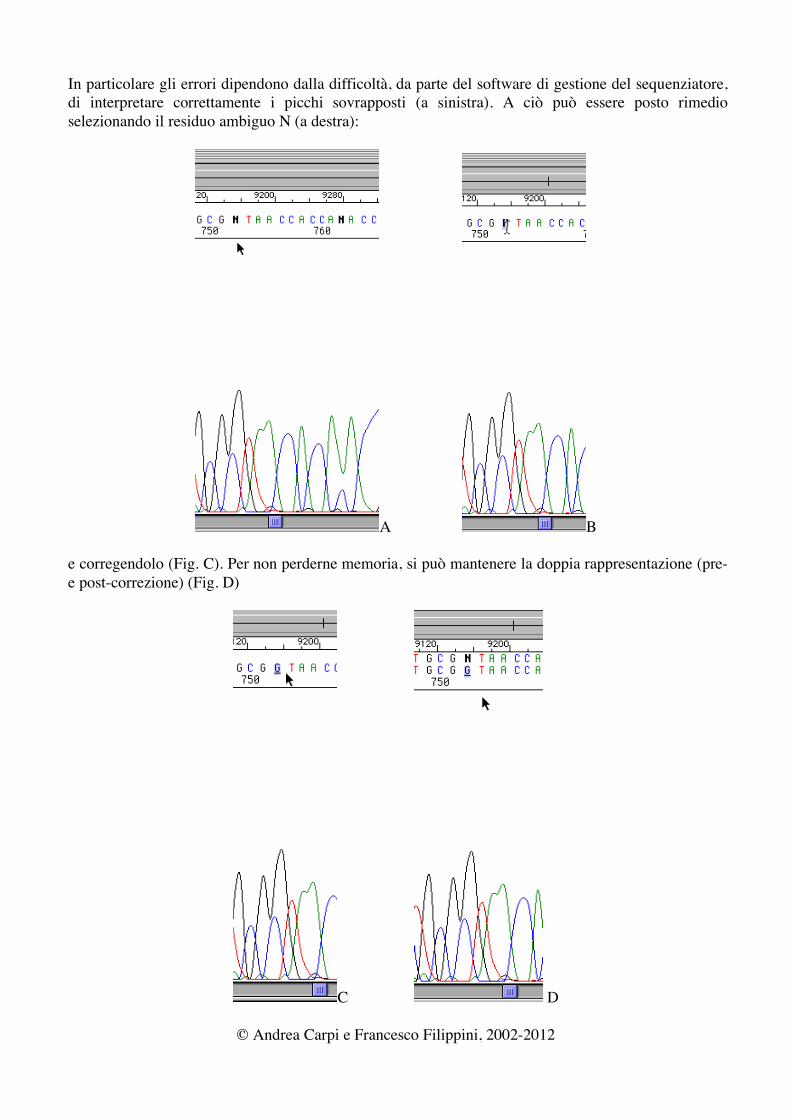
In particolare gli errori dipendono dalla difficoltà, da parte del software di gestione del sequenziatore, di interpretare correttamente i picchi sovrapposti (a sinistra). A ciò può essere posto rimedio selezionando il residuo ambiguo N (a destra):
A B
e corregendolo (Fig. C). Per non perderne memoria, si può mantenere la doppia rappresentazione (pre- e post-correzione) (Fig. D)
C D
© Andrea Carpi e Francesco Filippini, 2002-2012