
Legami deboli o interazioni deboli - TIM 2009.pdf · grazie ai moti termici in grado di provocare...
55
Legami deboli o interazioni deboli La forza di un legame chimico viene stabilita in base alla energia del legame (corrispondente all'energia necessaria per spezzarlo): Tipo di legame energia di legame (kj/mole) energia di legame (kcal/mole) Legami forti (ionici o covalenti) 100 ÷ 1000 24 ÷ 240 Legami deboli 0.1 ÷ 30 0.024 ÷ 7.1 ALCUNE ENERGIE DI LEGAME Tipo di legame Energia (kj/mole) Energia (kcal/mole) C-H 415 99 C-C (singolo 1.54 Å) 347 83 C-C (doppio 1.34 Å) 610 145 C-C (triplo 1.20 Å) 836 200 C-C (benzene 1.39Å) 518 124 C-F 485 116 C-Cl 328 78 C-Br 276 66 C-I 240 57 C-N 293 70 C-O 358 85 H-H 435 104 H-Cl 431 103 O-H 463 110 N-H 389 93 Tipi di interazioni deboli
Transcript of Legami deboli o interazioni deboli - TIM 2009.pdf · grazie ai moti termici in grado di provocare...

Legami deboli o interazioni deboli
La forza di un legame chimico viene stabilita in base alla energiadel legame (corrispondente all'energia necessaria per spezzarlo):Tipo di legame energia di legame (kj/mole) energia dilegame (kcal/mole) Legami forti (ionici o covalenti) 100 ÷ 1000 24 ÷ 240Legami deboli 0.1 ÷ 30 0.024 ÷ 7.1
ALCUNE ENERGIE DI LEGAMETipo di legame Energia (kj/mole) Energia (kcal/mole) C-H 415 99C-C (singolo 1.54 Å) 347 83C-C (doppio 1.34 Å) 610 145C-C (triplo 1.20 Å) 836 200C-C (benzene 1.39Å) 518 124C-F 485 116C-Cl 328 78C-Br 276 66C-I 240 57C-N 293 70C-O 358 85H-H 435 104H-Cl 431 103O-H 463 110N-H 389 93
Tipi di interazioni deboli

Le interazioni deboli, essenzialmente di natura elettrostatica, sirealizzano "tra" le molecole o specie chimiche già costituite conlegami di tipo covalente (forze intermolecolari), oppure "tra" partidiverse di una singola molecola generalmente piuttosto grande. Learee interessate alle forze di attrazione devono essere,tendenzialmente, cariche di segno opposto.Le molecole si possono attrarre vicendevolmente solo a distanzaravvicinata. Tali forze attrattive, dette genericamente “forze di vander Waals” sono meno intense di quelle che si realizzano neilegami covalenti e, già a temperature relativamente basse, questedeboli interazioni di legame persistono per tempi molto brevi.Per quanto le forze intermolecolari siano dotate di scarsa energia, laloro esistenza è fondamentale nel determinare gli stati diaggregazione delle sostanze e sono anche responsabili di altrenotevoli proprietà dei composti molecolari, come ad esempio la lorostruttura cristallina, il punto di fusione, il punto di ebollizione, icalori di fusione e di vaporizzazione, la tensione superficiale e ladensità.Le forze intermolecolari, in aggiunta a quanto detto, giocano unruolo importante nello stabilizzare le forme tridimensionali dellemacromolecole biologiche (enzimi e proteine). Lo "shape" di talimacromolecole è spesso indispensabile per una corretta esplicazionedell'attività fisiologica.Le forze intermolecolari possono essere PERMANENTI oTRANSITORIE. Le prime sono dovute all’interazione di speciechimiche contenenti gruppi permanentemente poveri di elettroni ealtrettante zone elettron-ricche (molecole dipolari): in questacategoria rientra il legame idrogeno. Le seconde si presentanograzie ai moti termici in grado di provocare rapide oscillazioni dellenuvole elettroniche che determinano la formazione di dipolimomentanei.Possibili disposizioni nell'interazione attrattiva tra dipoli permanenti

Interazioni DIPOLO - DIPOLO
Tutte le molecole tra le quali esiste questo tipo di interazione sonopolari (Dipoli Permanenti) e tendono ad orientarsi disponendositesta-coda con il risultato di una forza attrattiva (esempi: H2O*,H2S, HCl, SO2). Allo stato gassoso, quando le molecole siavvicinano a sufficienza, l'interazione dipolare tende a manifestarsi,ma spesso non è in grado di legare permanentemente le molecoleperchè queste sono dotate, a quella temperatura, di una quantità dimoto sufficiente a prevenire la formazione di agglomerati stabili(condensa).
Interazioni DIPOLO - DIPOLO INDOTTO
Si instaurano tra molecole polari e altre apolari ma che risultanofacilmente polarizzabili per induzione da parte delle molecolepolari. L'intensità del legame dipende dalla polarizzabilità dellemolecole (vedi oltre).
Interazioni DIPOLO ISTANTANEO - DIPOLO ISTANTANEO(forze di dispersione di London)
Si instaturano tra molecole apolari. Le forze di legame sono dovutealla distorsione momentanea della nuvola elettronica che si propagaper induzione alle molecole circostanti. Nella figura a destravengono illustrati, molto sommariamente, tre brevi istanti insuccessione per raffigurare la formazione del dipolo istantaneo-dipolo istantaneo indotto nel caso di una molecola biatomicaomonucleare. La formazione dei dipoli e di quelli indotti persisteper tempi dell'ordine di 10-14 o 10-15 secondi ma sufficiente perprodurre una netta forza di natura attrattiva.

Come già detto per le forze dipolo-dipolo indotto, anche l'entità diquesta interazione dipende dalla polarizzabilità delle molecole. Ildato semiquantitativo indica che la polarizzabilità, e quindil'intensità dell'interazione, cresce nell'ordine He, H2, F2, Cl2, Br2,I2.Alle molecole molto simmetriche e rigide (dette "hard"), comequelle monoatomiche dei gas nobili, é associata una energia diattrazione elettrostatica, dovuta alle forze di dispersione di London,di appena pochi decimi di kilocaloria per mole, con il risultato diuna interazione veramente debole (a temperatura ambiente sononello stato aeriforme). Se raffreddati a sufficienza, i gas nobilidiventano liquidi: la loro temperatura di ebollizione aumentascendendo lungo il gruppo perchè in tal senso aumentano le forze didispersione di London.Nel caso degli alogeni, la variazione graduale del loro stato diaggregazione, da uno stato meno condensato ad uno più condensato,è in accordo con l'incremento dei valori di polarizzabilità che sihanno scendendo lungo il gruppo.

Cloro (gas)
Bromo (liquido e vapori)
Iodio (solido e vapori)
Polarizzabilità
La polarizzabilità di un atomo o di una molecola si riferisceprincipalmente alla facilità con cui gli elettroni possono esserespostati dalla loro posizione media. Gli elettroni interessati sonoquelli di valenza, meno fortemente legati ai nuclei. Se siconsiderano gli elementi di un gruppo di non metalli, lapolarizzabilità aumenta scendendo lungo il gruppo a causa dellamaggiore distanza dal nucleo e all'opera di schermo degli elettroni

nei gusci più interni. Questo concetto si estende, nella maggioranzadei casi, anche alle dimensioni delle molecole, nel senso che lemolecole di maggiori dimensioni presentano maggiorepolarizzabilità
Il LEGAME A PONTE IDROGENO
Evidenza del Legame Idrogeno
Si considerino gli idruri più semplici degli elementi del quartogruppo. Se si costruisce un grafico nel quale in ascissa si pongono lesostanze in ordine sequenziale di peso molecolare crescente e inordinata il relativo punto di ebollizione, si osserva un gradualeaumento di questa proprietà.
L'aumento del punto di ebollizione è spiegato considerando che lemolecole aumentano di dimensione e hanno un numero di elettronimaggiore; questo fatto determina un aumento delle forze didispersione a causa della maggiore polarizzabilità.
Se adesso ripetiamo l'operazione con gli idruri degli elementi delquinto, sesto e settimo gruppo, noteremo qualcosa di diverso.
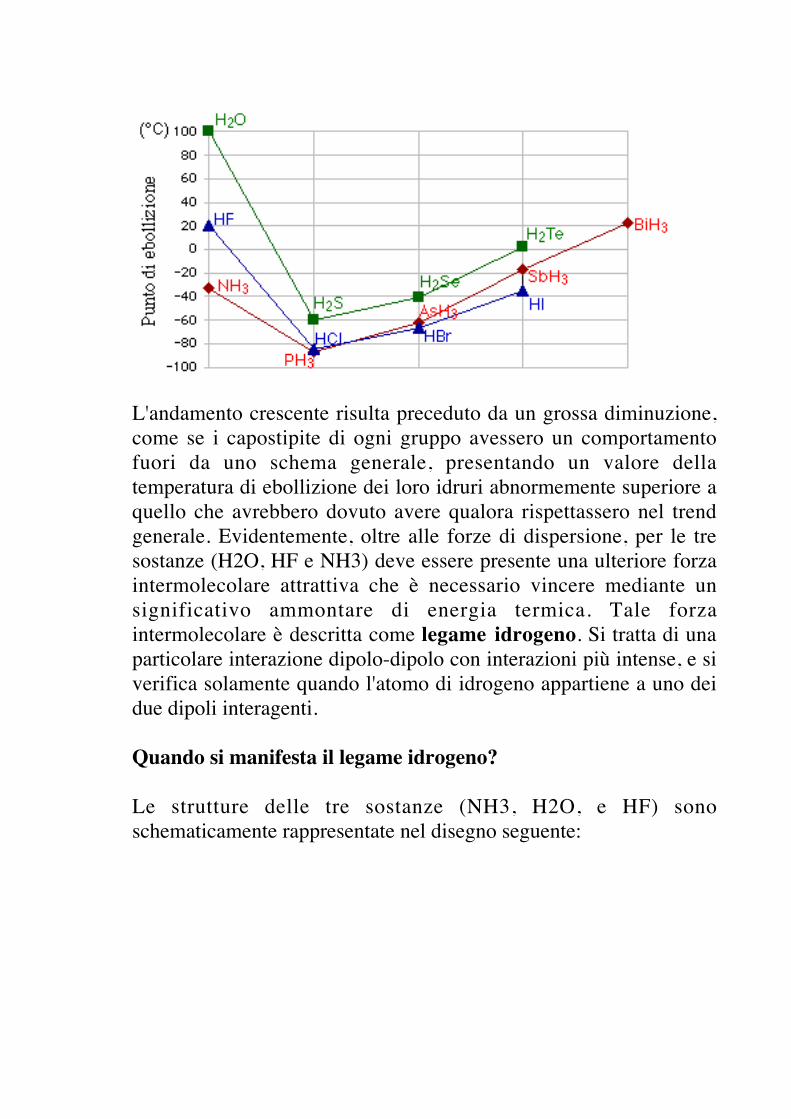
L'andamento crescente risulta preceduto da un grossa diminuzione,come se i capostipite di ogni gruppo avessero un comportamentofuori da uno schema generale, presentando un valore dellatemperatura di ebollizione dei loro idruri abnormemente superiore aquello che avrebbero dovuto avere qualora rispettassero nel trendgenerale. Evidentemente, oltre alle forze di dispersione, per le tresostanze (H2O, HF e NH3) deve essere presente una ulteriore forzaintermolecolare attrattiva che è necessario vincere mediante unsignificativo ammontare di energia termica. Tale forzaintermolecolare è descritta come legame idrogeno. Si tratta di unaparticolare interazione dipolo-dipolo con interazioni più intense, e siverifica solamente quando l'atomo di idrogeno appartiene a uno deidue dipoli interagenti.
Quando si manifesta il legame idrogeno?
Le strutture delle tre sostanze (NH3, H2O, e HF) sonoschematicamente rappresentate nel disegno seguente:

Per esse si possono evidenziare le seguenti condizioni:
* L'atomo di idrogeno è legato, mediante legame covalente, adun elemento molto elettronegativo, pertanto il legame risultafortemente polarizzato e l'idrogeno acquista una carica positivarelativamente grande* Su ciascun elemento cui è legato l'idrogeno è presente almenouna coppia di eletroni non impegnata in legame (coppia solitaria olone pair
Se due molecole dello stesso tipo (es. due molecole di acqua o duemolecole di ammoniaca) si ritrovano vicine e disposte in manieratale che l'idrogeno positivizzato rientri nella sfera di azione di unlone pair dell'elemento elettronegativo, l'idrogeno stesso (d+) verràattratto con la formazione di un legame più intenso rispetto ad unasemplice interazione dipolo-dipolo. Qualcuno si sbilanciaaffermando che si può pensare alla "tendenziale" formazione di unlegame dativo.Il legame idrogeno viene indicato usualmente con una lineatratteggiata:

Il legame idrogeno che si instaura nell'acqua ha una forza di circaquindici volte minore rispetto ad un normale legame covalente H-O(7 kcal contro 110) e una distanza di legame media notevolmentemaggiore.Nell'acqua liquida i legami idrogeno si formano e si distruggonocontinuamente ma, mediamente, la loro influenza sullo stato diaggregazione è di fatto fondamentale.Ogni molecola di acqua può formare due legami idrogeno. Nelghiaccio, quindi a bassa temperatura e bassa agitazione termica, lastruttura tridimensionale tende ad essere tetraedrica e ciascunamolecola di acqua forma due legami idrogeno stabili (fitti tratteggiazzurri):
Il risultato più evidente, derivante dalla struttura solida dell'acqua, eche le molecole si dispongono lasciando una maggiore quantità dispazio libero rispetto alla struttura dinamica in fase liquida. In virtùdi questo fatto, la densità del ghiaccio risulta minore rispetto a

quella dell'acqua liquida, e quindi il solido (H2O(s)) tende agalleggiare sul liquido (H2O(l))
In NH3 il legame idrogeno è più debole perchè l'elettronegativitàdell'azoto è minore rispetto a quella dell'ossigeno. Molto forterisulta invece il legame idrogeno in HF, ma questa sostanza bolle auna temperatura inferiore a quella dell'acqua sia perchè può formareun solo legame idrogeno ma anche a causa della formazione diaggregati stabili persistenti in fase gassosa.
Importanza del legame idrogeno
* Se non ci fosse il legame idrogeno l'acqua bollirebbe a circa -100°C, ben 200 gradi di differenza rispetto alla realtà. In altritermini l'acqua non sarebbe quella dispensatrice di vita che noiconosciamo.* Interviene anche in moltissime sostanze di natura organica (es.alcoli e ammine).* La struttura secondaria tipo a-elica è stabilizzata dallaformazione di legami a idrogeno tra l'idrogeno ammidico di unlegame peptidico e l'ossigeno carbossilico che lo sovrasta. Lastruttura terziaria di alcune proteine ed enzimi viene mantenutaanche con il contributo di questo tipo di legame.* L'accoppiamento delle basi, nel DNA, è ottenuto e in partemantenuto, da un certo numero di opportuni legami idrogeno che siinstaurano tra le coppie Adenina-Timina (2 legami idrogeno) eGuanina-Citosina (tre legami idrogeno).
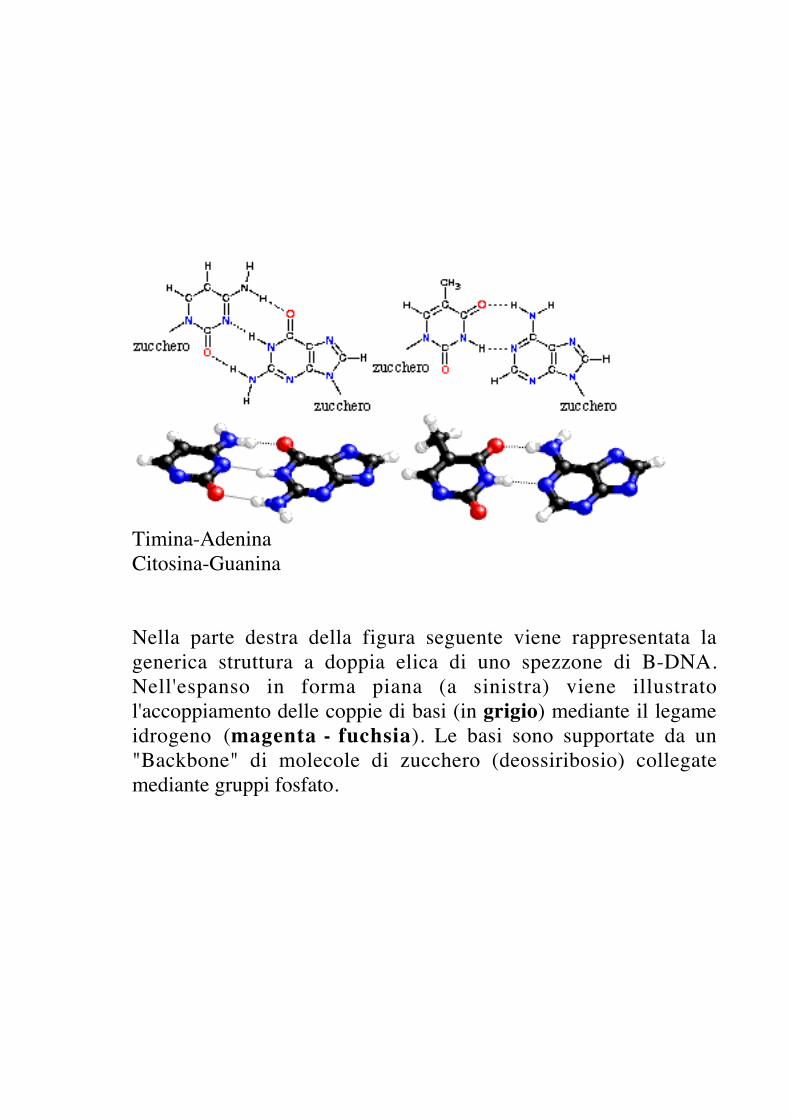
Timina-AdeninaCitosina-Guanina
Nella parte destra della figura seguente viene rappresentata lagenerica struttura a doppia elica di uno spezzone di B-DNA.Nell'espanso in forma piana (a sinistra) viene illustratol'accoppiamento delle coppie di basi (in grigio) mediante il legameidrogeno (magenta - fuchsia). Le basi sono supportate da un"Backbone" di molecole di zucchero (deossiribosio) collegatemediante gruppi fosfato.
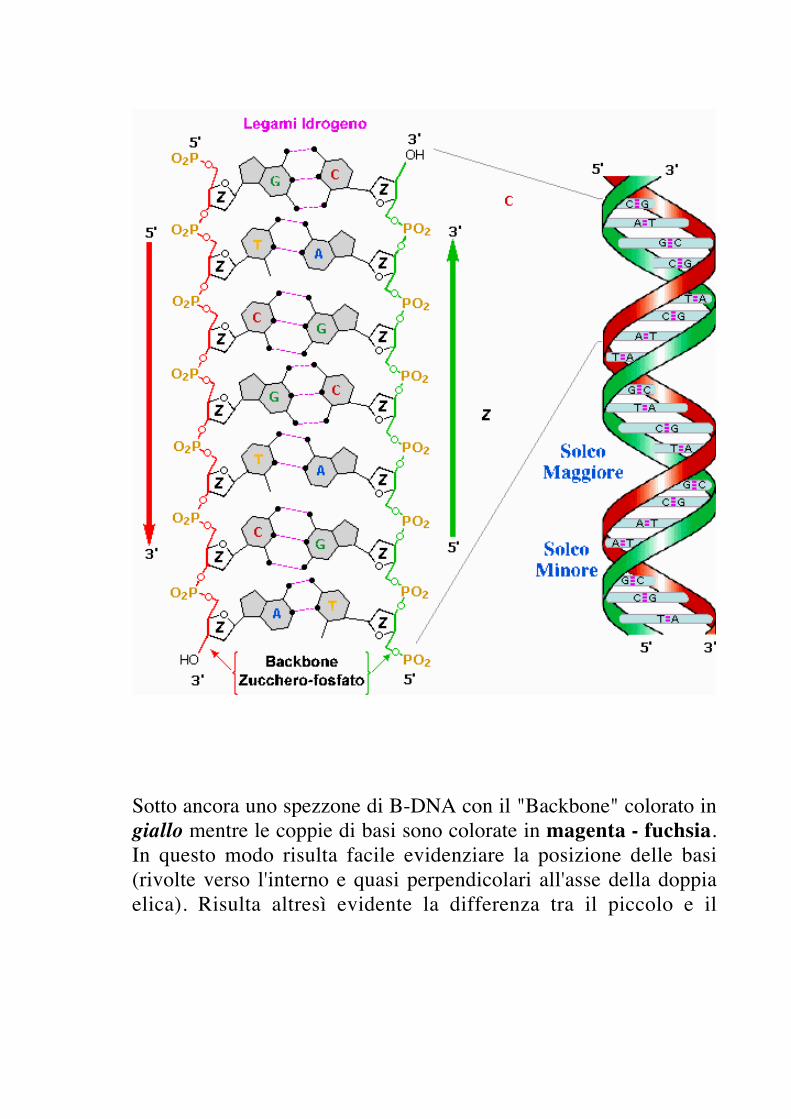
Sotto ancora uno spezzone di B-DNA con il "Backbone" colorato ingiallo mentre le coppie di basi sono colorate in magenta - fuchsia.In questo modo risulta facile evidenziare la posizione delle basi(rivolte verso l'interno e quasi perpendicolari all'asse della doppiaelica). Risulta altresì evidente la differenza tra il piccolo e il

g rande solco (in quest'ultimo le basisono più facilmente accessibili ad agenti esterni).

Il legame ionico.
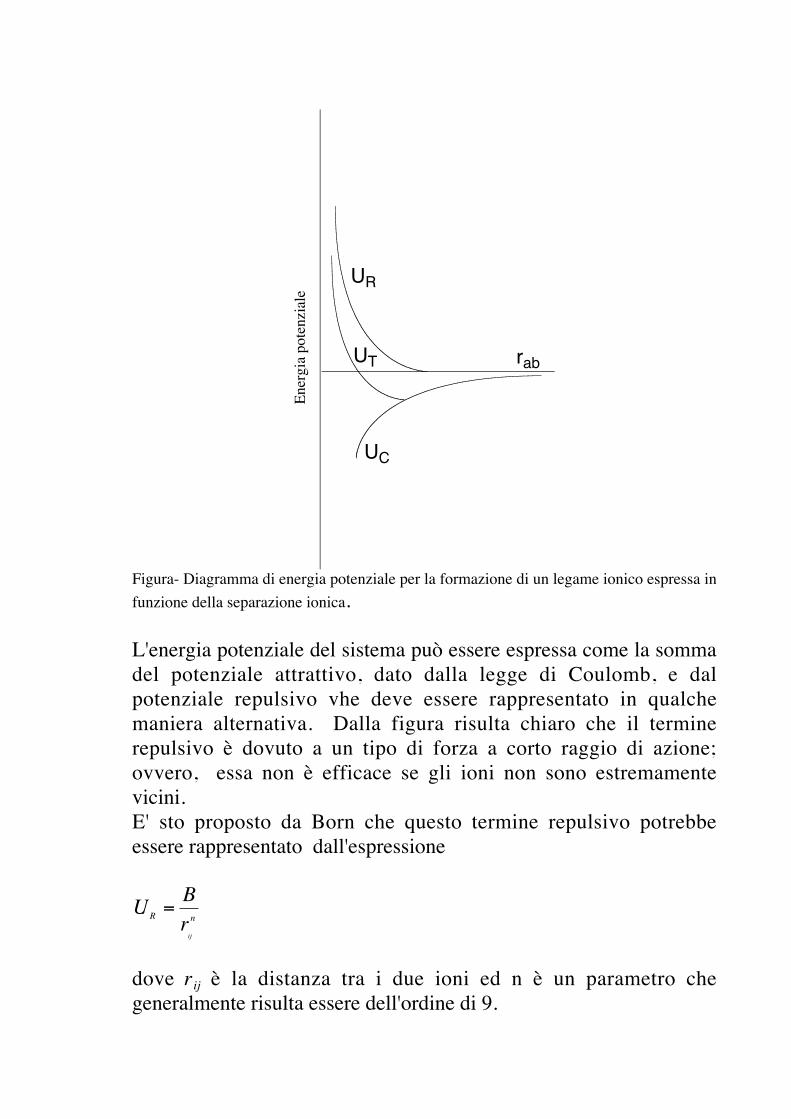
Da un punto di vista matematico, il più semplice tipo di legamechimico è quello che può essere considerato di carattere puramenteelettrostatico. Tale trattamento è risultato pienamente soddisfacenteper gli alogenuri dei metalli alcalini dove il legame implica ilcatione di un atomo fortemente elettropositivo e l'anione di unatomo fortemente elettronegativo. Sebbene sia possibile prendere inconsiderazione il fatto che un legame possa essere parzialmenteionico e parzialmente covalente, il grado di carattere ionico dellegame dipende in misura sostanziale dalla diferenza dielettronegatività degli atomi che si combinano. Nel caso deglialogenuri dei metalli alcalini, dovrebbe essere sicuro considerare illegame quasi esclusivamente ionico. Tuttavia la prova di questopostulato poggia sul successo che si ha nella valutazionequantitativa delle varie proprietà del composto risultante. Ingenerale, si può definire un legame come puramente ionico quandoviene descritto in modo efficace dal modello elettrostatico.Come primo test per la valutazione de modello elettrostatico,dovrebbe essere possibile calcolare l'energia del reticolo cristallino.In prima approssimazione, possiamo rappresentare gli ioni comecariche puntiformi e, per la formazione di una singola molecola uni-univalente, possiamo considerare una carica puntiforme positiva euna negativa poste a distanza infinita e quindi permettere che siavvicinino l'una all'altra. Da quanto sappiamo di elettrostatica,siamo consapevoli che vi sarà una attrazione tra i due ionidescrivibile nei termini della legge di Coulomb. Nello stesso tempo,sappiamo pure che i due ioni non si avvicinano al punto dasovrapporsi. Di conseguenza, va preso in considerazione un terminerepulsivo che entra in gioco in qualche zona apparentementepiuttosto vicina alla distanza di equilibrio. Tale situazione puòessere descritta dal diagramma di energia potenziale mostrato nellaseguente figura
Ener
gia p
oten
zial
e
rab
UR
UT
UC
Figura- Diagramma di energia potenziale per la formazione di un legame ionico espressa infunzione della separazione ionica.
L'energia potenziale del sistema può essere espressa come la sommadel potenziale attrattivo, dato dalla legge di Coulomb, e dalpotenziale repulsivo vhe deve essere rappresentato in qualchemaniera alternativa. Dalla figura risulta chiaro che il terminerepulsivo è dovuto a un tipo di forza a corto raggio di azione;ovvero, essa non è efficace se gli ioni non sono estremamentevicini.E' sto proposto da Born che questo termine repulsivo potrebbeessere rappresentato dall'espressione
€
UR =Brij
n
dove rij è la distanza tra i due ioni ed n è un parametro chegeneralmente risulta essere dell'ordine di 9.

Quindi, l'espressione matematica per l'energia potenziale è:
€
UT =UC +UR = −ziz je
2
r+Brij9
Per i composti uni-univalenti zi = z j=1 e perviò possono essereignorati. Per questa ragione zi e zj= verranno omessi nello sviluppodell'equazione che li riguarda, per essere poi reintrodottinell'espressione finale che sarà valida in tutti i casi.E' possibile valutare il coefficiente b se si considera che la pendenzadella curva dell'energia potenziale è zero alla distanza di equilibriotra gli ioni r0. Di conseguenza, possiamo dire che
€
∂UT
∂r
r= r0
=e2
r02−Bnr0
n−1
r0n .r0
n= 0
e2
r02−nBr0n+1
= 0
Risolvendo l'equazione rispetto a B, si ottiene;
€
B =e2r0
n−1
n
per cui l'espressione dell'energia potenziale può essere data nellaforma:
€
UT = −e2
ro+e2ro
n−1
nr0n
ovvero

€
UT = −e2
r01− 1
n
Se ora si considera l'energia di un intero cristallo, dobbiamomoltiplicare l'espressione di UT per il numero di ioni presenti , e, inaggiunta, bisogna prendere in considerazione i termini attrattivi erepulsivi degli ioni che nel cristallo si trovano a distanze via viacrescenti (i primi vicini, i secondi vicini e così via). Il primo fattorepuò facimente essere determinato considerando una mole sia dianioni che di cationi. Il secondo fattore risulta essere dipendente dalparticolare tipo di struttura cristallina. Se quindi si introducono itermini corrispondenti ai sopraesposti fattori nella equazioneprecedente, e se si generalizza l'espressione per una coppia di ioni dicarica di carica rispettivamente zi e z j si ottiene l'espressionegenerale per l'energia reticolar edi un cristallo
€
U = −ziz j ANe
2
r01− 1
n
dove A è la costante di Madelung e N è il nmero di Avogadro.Infine il parametro n può essere valutato dala compressibilità delcristallo in virtù della relazione
€
κ =18r0
4
Ae2 (n−1)
dove
€
κ è la compressibilità.
In definitiva, per la valutazione dell'energia molecolare risulta difondamentale importanza il valore della costante di Madelung che,come abbiamo detto, dipende dalle carettaristiche geometriche delreticolo cristallino
Sebbene le energie reticolari calcolate nei termini dei questomodello risultino nel complesso piuttosto soddisfacenti, tuttavia i

loro valori possono essere affinati prendendo in considerazione duefattori addizionali: la repulsione che ha origine dalle interazionielettrone-elettrone e l'energia di punto zero degli ioni.Questi termini sono stati incorporati in vario modo da Mayer, Borned Helmotz facendo uso di forme differenti di termini repulsivi
€
U = −ziz j ANe
2
r+ NBe−kr − NC
r 6+ NE0
dove k e C sono due nuovi parametri ed E0 è l'energia di punto zero.

La costante di Madelung.
La figura successiva mostra la cella elementare del cloruro di sodio. Si considerino leinterazioni elettrostatiche esercitate sul sodio centrale da tutti gli ioni presenti nella cella, esuccessivamente, da tutti gli altri presenti nel reticolo.
Dunque uno ione Na+ si trova ad essere circondato da sei ioni Cl- posti in manieraequivalente alla distanza r. Lo ione Na+ subisce quindi una interazione attrattiva da partedei primi vicini pari a
re
E2
6−=
Come secondi vicini, vi sono dodici ioni Na+ posti alla distanza 2r che contribuisconoall’attrazione complessiva come nell’espressione seguente:
2126
22
r
ere
E +−=
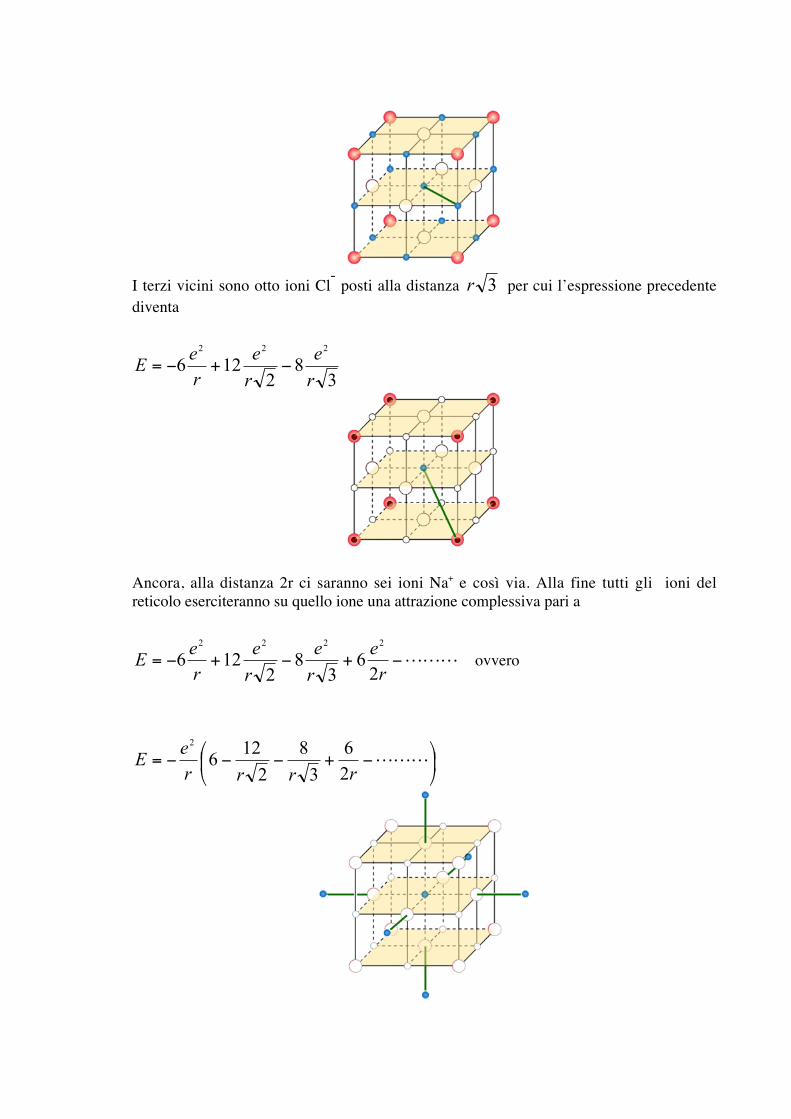
I terzi vicini sono otto ioni Cl- posti alla distanza 3r per cui l’espressione precedentediventa
38
2126
222
r
e
r
ere
E −+−=
Ancora, alla distanza 2r ci saranno sei ioni Na+ e così via. Alla fine tutti gli ioni delreticolo eserciteranno su quello ione una attrazione complessiva pari a
LLL−+−+−=re
r
e
r
ere
E26
38
2126
2222
ovvero
−+−−−= LLLrrrr
eE
26
3
8
2
126
2

La sommatoria in parentesi converge a un valore finito, che indicheremo con α e chechiameremo costante di Madelung.
re
E2
α−=
Come risulta chiaro, il valore della costante d i Madelung dipende dalle caratteristichegeometriche del reticolo cristallino.

Il legame covalente
L'elettrone libero e l'atomo di idrogeno: trattamento secondo lavecchia teoria dei quanti.
Condizione di quantizzazione:"L'azione meccanica estesa a un ciclo deve valere almeno quanto lacostante di Plank o quanto un multiplo intero di questa grandezza
∫ mvds = nh (1)
Elettrone nella scatola.L'elettrone è libero di muoversi in una scatola cubica nella quale siacostante il potenziale a cui è sottoposto. Si consideri, persemplificare, soltanto la componente del moto in una dimensionedella scatola (ovvero, si consideri solamente l'oscillazionedell'elettrone lungo uno spigolo del cubo così che il problema è lostesso del caso di una scatola di potenziale di sezione estremamentestretta ed estesa solo nella direzione della x. In altre parole, stiamoconsiderando il moto di un elettrone in una scatola di potenzialeunidimensionale).
x
a
In un periodo (andata e ritorno da una estremità della scatola) lavariazione della quantità di moto è mvx-(-mvx) = 2mvx , e lacondizione di quantizzazione
∫ mvxdx = 2mvxa = nh (n = 1,2,3,…) (2)

Nell'ipotesi che l'energia posseduta dall'elettrone in questecondizioni sia tutta cinetica, si avrà
mvx =
€
nh2a
; (3)
E = T =
€
12mvx
2= 2
222
8)(
21
mahn
mvm x = (4)
La particella nella scatola può assumere pertanto un numero limitatodi valori discreti di energia, separati in modo discontinuo secondomultipli quadratici interi di una quantità fondamentale o quanto dienergia
€
h2
8ma2. Lo spettro dei valori permessi per l'energia di una
particella nella scatola viene illustrato nella figura seguente
Ci si può servire del modello dell'elettrone nella scatola come di unutile esempio per definire ordini di grandezza per l'energia.

Così l'elettrone in un atomo, considerato come una particella che simuove in una scatola della dimesione dei raggi atomici (10-8 cm)avrà energie dell'ordine del quanto
€
h2
8ma2=
(6.62 ⋅10−27)2
8 ⋅ 9.107 ⋅10−28 ⋅ 4 ⋅10−16=1.6 ⋅10−11erg =10eV
Si tratta di energie dell'ordine dei legami chimici, dei calori direazione, dell'energia dei fotoni visibili e ultravioletti.
Una particella pesante (protone o neutrone) contenuta nel nucleo ècome una particella in una scatola di dimensioni estremamenteridotte (10-12 cm). In queste condizioni il quanto di energia è:
€
h2
8ma2=
(6.62 ⋅10−27)2
8 ⋅1.66 ⋅10−24 ⋅10−24= 3.5 ⋅10−6erg = 2 ⋅106eV = 2MeV
che corrisponde a un ordine di grandezza delle energie tipico deiprocessi nucleari.Infine, una molecola che pesi 100uma, e che si muova in uno spaziodi dimensioni macroscopiche (es. un cubo di un centimetro di lato,ha un quanto di energia dell'ordine di
€
h2
8ma2=
(6.62 ⋅10−27)2
8 ⋅100 ⋅1.6610−21= 3 ⋅10−32erg = 2 ⋅10−20eV
Cioò significa che la stato energetico di un sistema macroscopicopuò corrispondere a una successione grandissima di livelli quanticitalmente vicini da apparire praticamente continui.
Atomo di idrogeno.Si consideri un modelo estremamente semplificato di un atomo diidrogeno, ovvero lo si immagini costituito da un elettrone chepercorre orbite circolari entro un campo a simmetria centralegenerato da un protone

L'equazione del moto per un tale sistema è la seguente
€
mev2
r=e2
r2 (5)
ovvero, la forza centrifuga che agisce su un corpo in moto circolareuniforma eguaglia l'attrazione centripeta esercitata dalla forza diCoulomb.L'espressione dell'energia totale è:
€
E =T +V =12mv 2 − e
2
r= −
12e2
r (6)
Il risultato dell'equazione precedente si giustifica in quanto
€
mev2
r⋅r2
=e2
r2⋅r2 (7)
La condizione di quantizzazione è:
∫ mve ds = nh (n = 1,2,3,…..)
dove l'integrale è esteso a un periodo completo di rotazionedell'elettrone intorno al nucleo. Assumendo in via semplificata cheil moto dell'elettrone attorno al nucleo sia circolare uniforma (r e vcostanti in modulo), la condizione di quantizzazione diventa:
∫ mve ds = mev∫ ds = mev2πr = nh (8)
ovvero

€
mver = n h2π
n=(1,2,3…) (9) 1° postulato di Bohr
Essendo
€
mev2
r⋅r2
=e2
r2⋅r2⇒ m
e
2v 2 =e2
rme ⇒ mev = e me
r (10)
Così la (8) diventa
2πr
€
e me
r = nh (8)
Ne consegue che
€
4π 2r2e2 me
r= n2h2 =>
€
r =n2h2
4π 2me2 (10)
L'elettrone nell'atomo di idrogeno ha perciò a disposizione unospettro di valori permessi quantizzati dell'energia ricavabilidall'espressione:
€
En = −12e2
r= −
2π 2e4me
n2h2= −
RH
n2= −13.59n2
eV = −109677n2
hc (11)
I risultati dela teoria atomica di Borh, ottenuti nella semplice ipotesiche l'elettrone possa percorrere soltanto orbite circolari, vengonogeneralizzati da Sommerfield, il quale conclude correttamente che lasoluzione generale dell'equazione del moto di un corpo in un campoa simmetria centrale è una conica, famiglia di curve delle quali ilcerchio rappresenta solo un caso particolare. Si ammetta dunqueche l'elettrone di un atomo monoelettronico (idrogenoide) percorraun'orbita ellittica. Tutte le ellissi aventi un dato semiasse maggiorea esibiscono la medesima energia di moto orbitalico. Tuttavia,ciascuna delle ellissi caratterizzata da un dato semiasse maggiore,presenta momento della quantità di moto che dipende dal semiasse

minore b . Genericamente si può scrivere una relazione diproporzionalità tra semiasse maggiore a, semiasse minore b e certinumeri interi k ed n tali che
€
ba
=kn
(12)
In 12 n rappresenta il numero quantico postulato da Bohr; k a suavolta quantizza il momento della quantità di moto dell'elettroneorbitante, secondo la relazione|p| =
€
k h2π
Generalmente, il moderno formalismo pone k nella forma
k = l+1 => l = k-1
L'intero l=k-1 prende il nome di numero quantico secondario oazimutale e può assumere i valori 0,1,2,….n-1.
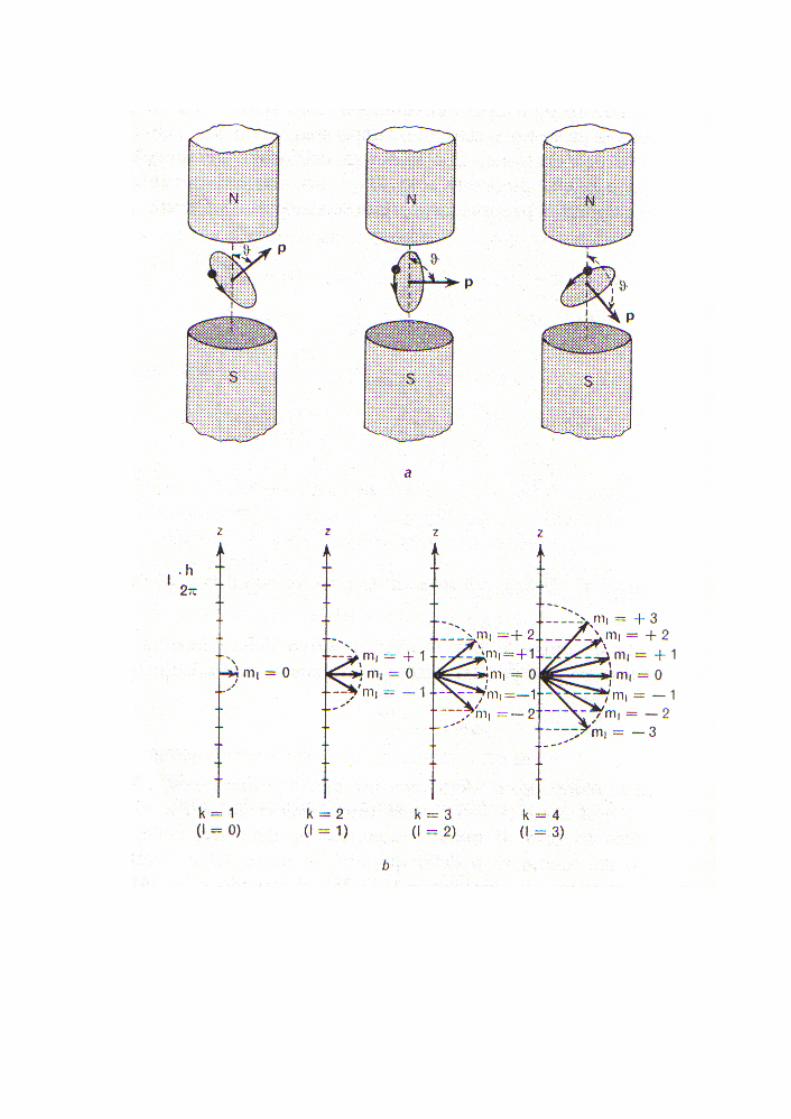
Ogni orbita circolare o ellittica definisce un piano ed una normalead esso, lungo la quale è diretto il vettore p del momento dellaquantità di moto. La meccanica classica non pone alcunalimitazione alle orientazioni che questo vettore può assumererispetto a un a direzione esterna di riferimento (per esempio le linee

di forza di un pur sempre individuabile campo magnetico, debole alpunto da non alterare il moto dell'elettrone). Lo sviluppo della teoriadi Sommerfield giunge alla conclusione che il vettore p puòassumere rispetto a quella direzione soltanto 2l+1 orietazioni.Queste orientazioni permesse sono definite da angoli θ tra il versopositivo della direzione privilegiata e il verso positivo del vettore pnormale al piano dell'orbita , tali che
€
cosϑ =ml
k (13)
dove ml è ancora un numero quantico, detto numero quanticomagnetico che può assumere i valori 0, ±1, ±2, ±3…,±(k-1), ovverotutti i valori compresi tra +l e -l, incluso lo zero. Il numero qunticoml definisce, nelle solite unità h/2π, le proiezioni pz del vettore psulla direzione privilegiata z del campo esterno. Essenso dunque pz= |p| cosθ, e ricordando la 13, si avrà
€
pz = k h2πcosϑ
Questo risultato equivale a dire che la vi è una quantificazionespaziale dell'orbita. La figura successiva illustra bene il concetto

L'elettrone come onda e la meccanica quantistica.
Alla base della meccanica quantistica sta l'idea di definire unafunzione di stato ψ che possa riprodurre e simulare il più fedelmentepossibile il comportamento e le proprietà di un dato sistema fisico.In una tale funzione di stato possono venire soddisfatti tutti irequisiti principali della fisica microscopica: così ad esempio lafunzione che descrive il comportamento di un punto materiale (adesempio un elettrone) nello spazio sarà funzione delle coordinate x,y e z del dato elettrone
ψ = ψ(x,y,z);
tale funzione sarà in grado di rappresentare un punto mareriale,ovvero il corpuscolo.Tuttavia la stessa funzione spaziale potrà venire moltiplicata per unafunzione periodica del tempo, ed assumere quindi le caratteristichedi un fenomeno ondulatorio (ψ infatti è chiamata funzione d'onda,oltre che funzione di stato del sistema esaminato).
Per conoscere le grandezze fisiche Gi associate a un sistemamicroscopico , nel mondo fisico reale si esegue una misuramediante un apparecchio di misura applicato al sistema osservato, el'applicazione dello strumento di misura al sistema osservatoproduce come risultato il valore della grandezza cercata Gi. Si ènotato che spesso in sistema microscopico durante un operazione dimisura viene perturbato in maniera sostanziale,Quando, invece del sistema fisico reale si consideri la funzione distato che ne è il simulacro matematico, allo strumento di misuradella grandezza Gi viene sostituito un operatore matematico Gi , cheriproduce o simula un appareccio di misura, e che applicato allafunzione di stato produce un risultato collegato al valore della stessagrandezza
G ψ
€
→ risultato della misura di G.

Una parte essenziale della mecccanica quantistica è la definizionedegli operatori connessi alle diverse grandezze fisiche. Ledefinizioni di base riguardano gli operatori collegati alla posizionee alla quantità di moto di un corpuscolo.
x → x = x .
y → y = y .
z → z = z .
px = mvx →
€
i h2π
∂∂x
py = mvy →
€
i h2π
∂∂y
pz = mvz →
€
i h2π
∂∂z
Per altre grandezze fisiche, che si possono esprimere come funzionidelle posizioni e delle quantità di moto (o delle velocità), glioperatori risultano delle stesse funzioni degli operatorifondamentali. Così l'operatore della energia potenziale, che come sisa è funzione solo della posizione (V = V(x,y,z) ) sarà ilmoltiplicatore di ciascuna coordinata per la funzione di stato cuil'operatore viene applicato. L'energia cinetica di una particella sipuò esprimere invece in funzione della quantità di moto
€
T =12mv2 =
12m(vx
2 + vy2 + vz
2 ) =12m
(m 2vx2 +m 2vy
2 +m 2vz2 ) =
=12m
(px2 + py
2 + pz2 )

€
T →T =12m
(px.p
x+ p
y.p
y+ p
z.p
z) =
12m
i h2π
∂∂x
i h2π
∂∂x
+ ...
=
=12m
−h2
4π 2
∂ 2
∂x 2− ...
= −
h2
8π 2m∇ 2 ∂ 2
∂x 2+∂ 2
∂y2+∂ 2
∂z 2
= −
h2
8π 2m∇ 2
Una grandezza particolarmente importante è l'energia totale ohamiltoniana H = T+V di un sistema
€
H = T +V = −h2
8π 2m( ∂
2
∂x 2+∂ 2
∂y2+∂ 2
∂z2)+V (x,y,z) =
= −h2
8π 2m∇ 2 +V (x,y,z)
Notiamo che molti operatori sono di derivazione, e quinditrasformano la funzione di stato nella sua derivata, cioè in unadiversa funzione; ciò corrisponde al fatto che le corrispondentimisure fisiche sarebbero tali da alterare sostanzialmente il sistemaesaminato. Per chiarire meglio questo punto, che è strettamentecollegato al principio di indeterminazione, è opportuno considerarequale possa essere il significato fisico diretto di una funzione distato. L'informazione più semplice che possiamo avere su unsistema è se il sistema esista o no (ovvero, in una concezioneprbabilistica, quale probabilità di esistenza possa avere il sistema),e ci potremmo chiedere se il valore numerico della funzione di statopossa da solo rispondere a questo quesito. E' facile però rendersiconto che la funzione di stato non può di per sè avere avere ilsignificato di probabilità di esistenza del sistema, in quanto unaprobabilità è rappresentata da un numero reale , positivo e compresotra 0 e 1, mentre una funzione di stato è sottoposta a pocherestrizioni e assai generali ( per esempio essere continua e

derivabile), e quindi può benissimo assumere valore negativo, ocomplesso, o maggiore di 1. La via più semplice per ottenere unaespressione che possa avere il significato fisico di probabilità diesistenza del sistema è quello di considerare non la funzione di stato ψ ma il suo quadrato ψ2 e, nel caso di ψ complessa, la sua normaψ∗ψ (che son sempre valori reali e positivi), e di normalizzarle,cioè di far sì che il loro integrale
€
ψ *∫ ψdV esteso a tutto lo spazioS in cui può esistere il sistema sia uguale a 1. Nel caso che
€ €
ψ*
S∫ ψdV
ψ* ,ψ( ) = a
la funzione di stato corretta sarà
€
1aψ , per cui è:
€
1aS
∫ ψ* . 1aψdV =
1aψ* . 1
aψ =
1a.a =1
Fisicamente, la normalità di una funzione di stato significa lacertezza di esistenza della particella, certezza che si acquistaripetendo l'operazione di determinazione della probabilità diesistenza in tutti gli elementini di volume dV dello spazio esommando i risultati particolari (infinitesimi) ottenuti. Unprocedimento analogo, di cui non daremo i dettagli, si esegue perconoscere il valor medio di qualunque grandezza osservabile.Qui ci preme rilevare che come uno strumento non deve perturbareuno stato microscopico nel caso della misura di una certa grandezza,allo stesso modo un operatore, che dello strumento è un simulacromatematico, non deve alterare la forma di una funzione di stato; alpiù la deve trasformare in se stessa eventualmente moltiplicata peruna costante
G'ψ = kψ = Gaψ

In tal caso particolare il sistema si dice in autostato per la grandezzaG. La costante k rappresenta il valorte (unico, non soggetto a scattiprobabilistici e quindi esattamente definito) della grandezza G' intale stato e si chiama autovalore Ga, mentre la funzione ψ chedescrive tale stato prende il nome di autofunzione.L'esistenza di autostati e di autovaloriè solo apparentementeun'eccezione al principio di indeterminazione: all'assenza diindeterminazione nel valore di una grandezza fisica (per esempioenergia) in autostato corrisponde indeterminazione infinita nellagrandezza coniugata (tempo):
ΔE x Δt ≥ h ; ΔE→0; Δt→∞
quindi gli autostati dell'energia sono considerati come statistazionari, ossia come stati che non variano nel tempo, per cuil'indeterminazione insita nel tempo diventa irrilevante.
L'equazione di autovalore (G'ψ = kψ = Gaψ) esprime la circostanzache il sistema rappresentato dalla ψ non si altera sotto l'operazionedi misura della G', ed è quindi condizione necessaria e sufficienteper l'esistenza di un autostato. Tra le equazioni di autovalore deltipo G'ψ = kψ = Gaψ la più importante è quella dell'energia in formahamiltoniana, equazione di Schröedinger, che per un singoloelettrone è:
€
Hψ = −h2
8π 2m(∂
2ψ∂x 2
+∂ 2ψ∂y2
+∂ 2ψ∂z 2
)+Vψ = −h2
8π 2m∇ 2ψ+Vψ = Eaψ
Naturalmente un'equazione di autovalore può anche non ammeteresoluzioni, ed allora la grandezza considerata non è in autostato ;oppure l'equazione di autovalore (ed è il caso più comune) ammettesoluzioni solo per alcuni ben determinati e discreti valori dellacostante, ed allora la grandezza G, altre ad essere in autostato,risulta anche automaticamente quantizzata. Benchè gli autostatisiano piuttosto l'eccezione che la regola nei sistemi fisici

microscopici, essi rappresentano tuttavia dei casi particolari moltoimportanti e molto significativi; tra le grandezze fisichecaratteristiche dei sistemi microscopici, la più impportante èl'energia, e quindi gran parte delle applicazioni chimiche dellameccanica quantistica tratta della descrizione degli autostatidell'energia negli atomi e nelle molecole, ossia della soluzionedell'equazione di Schröedinger per tali sistemi.Per iniziare vedremo come la meccanica quantistica tratta ilproblema dell'elettrone nella scatola.
Elettrone nella scatola,Per un elettrone che si trovi in una scatola di potenzialeunidimensionale, nella quale non sia soggetto a forze, l'energiapotenziale è costante entro la scatola, e poichè il suo valorenumerico viene determinato dallo zero della scala delle energie cheè arbitrario, possiamo fissare V=0 nell'interno della scatola.L'hamiltoniano consiste quindi di sola enetgia cinetica, e l'equazionedi Schröedinger per l'elettrone entro la scatola diventa allora:
€
Hψ(x ) =Tψ(x ) = −h2
8π 2md 2ψ(x )
dx 2= Eaψ(x )
E' evidente che questa equazione differenziale non solo è risolvibile(il che assicura che l'energia dell'elettrone nella scatola può esisterein autostato) ma è anche di soluzione immediata, in quanto la ψ(x)può essere una qualunque funzione di tipo periodico che contengadue costanti
€
d 2ψ(x )
dx 2= −
8π 2mEa
h2ψ(x ) = −cψ(x )
si vede immediatamente che essa può venire soddisfatta da qualsiasifunzione del tipo
€
ψ(x ) =Asin( cx+α )oppure
€
ψ(x ) =Bcos( cx+β)

oppure
€
ψ(x ) =Ce cxγ
Per precisare ulteriormente la forma sia della ψ(x) che della Eadobbiamo considerare però che la natura fisica del problema imponedelle limitazioni o condizioni al contorno, e che inoltre la ψ(x) deveessere normalizzata. Le condizioni al contorno sono che, nel casopresente, la particella deve avere probabilità finita di esistenza entrola scatola, cioè ψ(x) finito per 0<x<a; ma deve vavere probabilitànulla di trovarsi fuori della scatola o sulle pareti di essa, cioè ψ(x) = 0per X=a e X=0: Prendendo ad esemio la funzione seno, ciò significache la sua fase deve essere zero, e che il suo argonemto
€
x c =πh8mEa deve essere uguale a nπ per x=0 e per x=a, cioè:
€
ca = nπ =πh8mEa
€
n2 =8mEa
h2
la condizione di normalizzazione impone che:
€
1= A sin cx[ ]2
0
a∫ dx = A sin nπ
a
xdx
0
a∫ = A 2 sin2
0
a∫ (nπx
a)dx =
=1 − cos
2nπx
a
2
0
a∫ dx = A 2 1
2x
0
a
+12sin 2nπx
a
0
a
=
=A 2a2
=1
ossia che:
€
A =2a
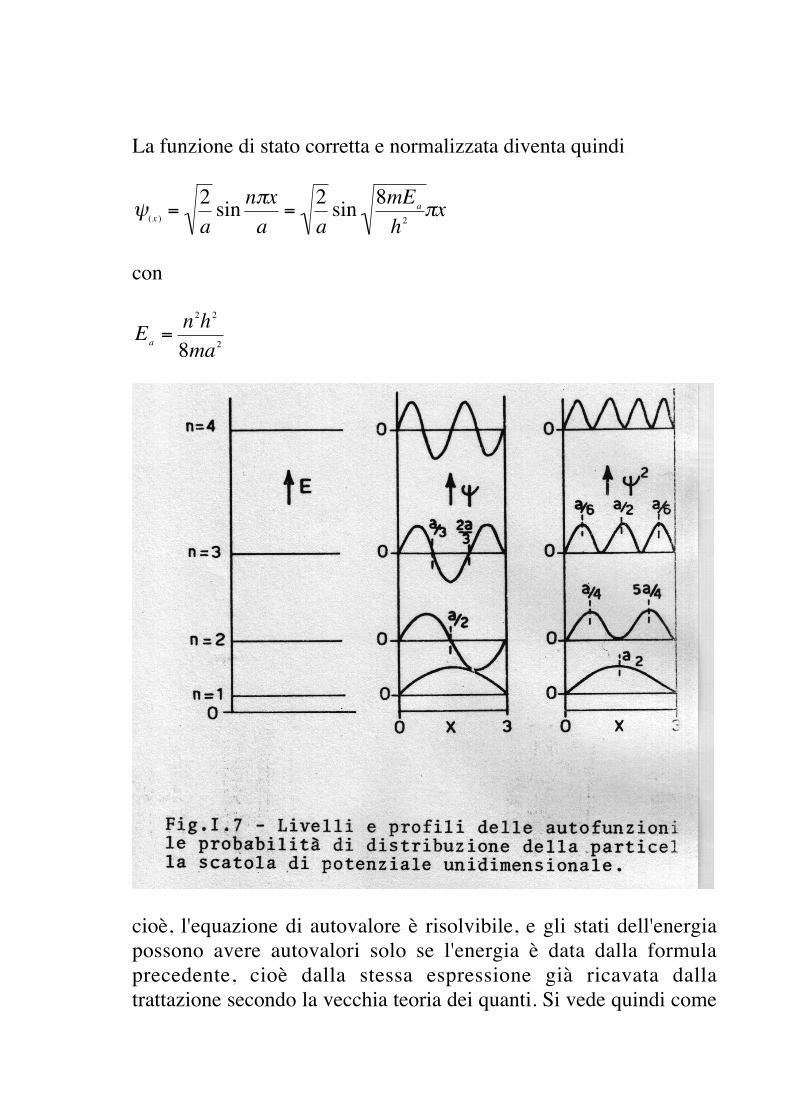
La funzione di stato corretta e normalizzata diventa quindi
€
ψ(x ) =2asin nπx
a=
2asin 8mEa
h2πx
con
€
Ea =n2h2
8ma2
cioè, l'equazione di autovalore è risolvibile, e gli stati dell'energiapossono avere autovalori solo se l'energia è data dalla formulaprecedente, cioè dalla stessa espressione già ricavata dallatrattazione secondo la vecchia teoria dei quanti. Si vede quindi come

il trattamento quantomeccanico porti, per quanto riguarda i valoridell'energia, esattamente alle stesse conclusioni ed agli stessirisultati della vecchia teoria dei quanti, sebbene gli stessi risultatisiano ottenuti in modo più sistematico e corretto; ad esempio, laquantizzazione non ha bisogno di venire imposta, ma è unsottoprodottorichiesto dalle condizioni matematiche di risolvibilitàdell'equazione di autovalore.
Atomo di idrogeno.
L'operatore dell'energia cinetica per l'elettrone nell'atomo diidrogeno è
€
−h2
8π 2m∇ 2
mentre l'energia potenziale è data dall'attrazione elettrostatica per ilnucleo. Perciò l'equazione di Schrödinger è
€
Hψ x,y,z( ) = −h2
8π 2me
ψ x,y,z( )+V x,y,z( )ψ x,y,z( ) =
= −h2
8πme
∂ 2ψ∂x 2
+∂ 2ψ∂y2
+∂ 2ψ∂z 2
−
Ze2
x 2 + y2 + z 2= Eaψ
Risulta più conveniente esprimere la posizoone dell'elettrone in unsistema di coordinate polari anzichè in un sistema di coordinatecartesiane.L'equazione di Schrödinger presenta allora la forma

€
Hψ r,ϑ ,ϕ( ) = −h2
8π 2me
∂ 2ψ∂r 2
+2r∂ψ∂r
+1
r 2 sinϑ∂∂ϑ
sin ∂ψ∂ϑ
+
1r 2 sinϑ
∂ 2ψ∂ϕ 2
−Ze2
rψ = Eaψ
che ha il vantaggio di poter easprimere lal ψ come il prodotto di trefunzioni separate, dipendenti ognuna da una delle tre coordinate r,
€
ϑ e
€
ϕ :
€
ψ r,ϑ ,ϕ( ) = R r( ).Θ ϑ( ).Φ ϕ( )
Ognuna di queste tre funzioni è soluzione di una parte separatadell'equazione di Schrödinger:
€
−h2
8π 2me
∂ 2R∂r 2
+2r∂R∂r
−Ze2
rR −
l l+1( )r 2
R = EA .R
€
1sinϑ
. ∂∂ϑ
sinϑ ∂Θ∂ϑ
−8π 2ml l+1( )
h2−
m 2
sin2ϑ
Θ = 0
€
∂ 2Φ∂ϕ 2
= −m 2Φ
Nella prima di queste equazioni compare solo la variabile r, nellaseconda solo la
€
ϑ e nella terza solo la
€
ϕ . La terza equazione èparticolarmente semplice; essa sembra essere della stessa formadella equazione d'onda per la particella in una scatola. In termini diseno e coseno, la soluzione è:
€
Φm ϕ( ) = A sinmϕ +B cosmϕ
Uno dei requisiti di tale funzione è che deve assere a valore singolo.Per soddisfare questa condizione, la funzione
€
Φm ϕ( ) deve avere lostesso valore per
€
ϕ = 0 e per
€
ϕ = 2π.

Nel caso di
€
ϕ = 0, è
€
Φm 0( ) = A sin 0( )+B cos 0( )
e, quando
€
ϕ = 2π
€
Φm 2π( ) = A sinm2π +B cosm2π
Poichè il valore di
€
Φm ϕ( ) deve essere lo stesso sotto le stessecondizioni, è necessario che
€
B = A sinm2π +B cosm2π
Questa identità può reggere solo se m è zero o se ha un valore interopositivo o negativo. Queste caratteristiche sono quelle che ci sisarebbe aspettato per un numero quantico, e le particolari restrizionia cui è soggetto m sono le stesse riscontrate nel numero quanticomagnetico del modello di Bohr-Sommmerfield.Molto spesso, nel trattamento dell'atomo di idrogeno viene usata lalsoluzione esponenziale dell'equazione
€
Φ:
€
Φm ϕ( ) =Ce± imϕ
Allo scopo di valutare la costante C potrebbe essere convenientescegliere c in modo tale che la funzione
€
Φ ϕ( ) sia una funzioned'onda normalizzabile. Ciò richiede che
€
ΦΦ* dϕ =10
2π∫
e quindi che
€
C 2
0
2π∫ em imϕe± imϕdϕ =1
ovvero

€
C 2
0
2π∫ dϕ = 2πC 2 =1
Alla fine, si vede che il valore della costante C, che dà una funzioned'onda normalizzata, è:
€
Φm ϕ( ) =12π
e± imϕ ,m = ±1,±2,±3....
L'equazione
€
Θ.
Le soluzioni dell'equazione
€
Θ, come di quella radiale,sfortunatamente non sono così semplice come qualla dell'equazione
€
Φ. Tuttavia , capita che l'equazione
€
Θ può essere messa in unaforma che era nota ai matematici sin da parecchio tempo primadell'avvento della meccanica quantistica. Questa particolare forma ènota come equazione di Legendre e fornisce la seguente soluzionenormalizzata:
€
Θ l ,m ϑ( ) =2l+1( ) l− m( )!2 l+ m( )!
Pl
m cosϑ( )
dove
€
Pl
m è la funzione associata di Legendre di grado
€
l ed ordine
€
m . La forma della soluzione è ovviamente piuttosto complicata.Nonostante la natura complicata della soluzione, si possonodesumere alcune importanti considarazioni in merito alla variabilitàdi m e di
€
l: entrambi infatti devono essere numeri interi, poichè nonsono definiti i fattoriali di non interi; si desume inoltre che
€
l , oltrelo zero, può assumere solo valori interi e positivi; si conferema ilfatto che m può valere 0, ±1, ±2, …. , ±
€
l.
L'equazione radiale.
Rimane da risolvere l'equazione R(r) che, come l'equazioneangolare
€
Φ ϕ( ), può essere posta in una forma da lungo tempo notaai matematici come equazione di Laguerre, la cui soluzionenormalizzata è la seguente:

€
Rn ,l r( ) =2Zna0
3 n− l−1( )!2n n+ l( )![ ]
2 e−ρ2ρ lLn+1
2l+1
dove
€
ρ =2Zna0
r,
€
a0 =h2
4π 2µe2, ed
€
Ln+l
2l+1 ρ( ) rappresenta il polinomio
associato di Laguerre. Dalla forma della soluzione vengonoconfermati i limiti dei possibili valori di m ed
€
l ed inoltre si imponeche n possa assumere soltanto valori interi, e che
€
l possa almassimo assumere il valore n-1.
Molecola H2+.
La molecola più semplice è H2+ che è costituita da due nuclei di
idrogeno e da un solo elettrone. Si tratta di una molecola instabile(infatti esiste solo in equilibrio con H e H2 in fase gassosa e non sipuò isolare in composti solidi) ma è abbastanza ben nota nelle sueproprietà chimico-fisiche per via spettroscpica: ha una distanza dilegame di 1.06 Å, energia di legame 64 Kcal/mol e affinitàelettronica 46 Kcal/mol. Avendo un solo elettrone di valenza, essa èabbastanza diversa dalle altre molecole che in genere contengonodue elettroni per ogni legame, ma tuttavia essa è molto iportante inquanto presenta nella forma più semplice il problema di spiegarel'esistenza stabile di una molecola costituita da più atomi, cioè lanatura di un legame chimico. L'equazione di Schröedinger perl'unico elettrone di H2
+ è:

rA rB
HA HB
€
−h2
8π 2m∇ 2ψ −
ZAe2
rAψ −
ZBe2
rBψ = Eψ
Questa equazione può venire risolta in maniera esatta, esprimendola posizione dell'unico elettrone in un sistema di coordinateellittiche, e i risultati così ottenuti permettono di riprodurre congrande precisione le caratteristiche sperimentali della molecola.Tuttavia, l'applicazione dello stesso metodo a una molecola di pocopiù complessa come H2 presenta difficoltà matematicheinsormontabili. Vale quindi la pena affrontare una trattazione piùapprossimata per H2
+ che ovviamente dà risultati meno buoni, mocostituisce un metodo idoneo alla trattazione di molecole piùcomplesse. Discuteremo quindi le proprietà di H2
+ deducibili dallafunzione approssimata arbitraria. Dal punto di vistafenomenologico, si tratta di cercare una funzione di stato capace didescrivere lo stato di moto dell'elettrone attorno ad ambedue i nucleidella molecola. Quando, durante la sua traiettoria, l'elettrone si trovain un punto come p1
HA HBp1 p2
esso è soggetto principalmente all'attrazione del nucleo A che gli èpiù vicino e risente in maniera quasi trascurabile dell'effetto delnucleo B più distante; perciò in tal punto il suo stato può veniredescritto con sufficiente accuratezza da una funzione idrogenoide

centrata sul nucleo A ψa+
, A come ψ1s,A. Analogamente nel punto p2l'elettrone può venire descritto abbastanza bene da un funzionecentrata sul nucleo B ψb
+, B come ψ1s,B. Una formulazione più
generale dello stato dell'elettrone potrà quindi essere rappresentatada una sovrapposizione dei due stati parziali ψ at,A e ψ at,Besprimibile come una combinazione lineare
€
cAψ a
+ + cBψ b
+
dove i coefficienti cA e cB debbono soddisfare la condizione dinormalità. La trattazione matematica del problema così impostatoporta a formlare due funzioni di stato plausibili per lo statofondamentale di H2
+, ψ+ e ψ-, sulla base delle quali si potrà discuterel'energia elettronica e il tipo di distribuzione elettronica.Senza addentrarci, prendiamo atto che esistono per H2
+ dueautovalori dell'energia
€
E ± =11+S
(EH +α ±β)
dove: EH = autovalore dell'energia per l'atomo di idrogeno isolato;S = integrale di sovrapposizione;α = integrale coulombiano;β = intergrale di risonanza.
L'integrale coulombiano α rappresenta l'attrazione dell'elettrone sulnucleo di α (distribuzione di carica ψA*ψA) per il nucleo di B; essoè un termine attrattivo abbastanza piccolo, e spesso lo si consideratrascurabile, particolarmente in molecole neutre.L'integrale β, dal quale dipende la differenziazione energetica tra ψ+
e ψ-, e che quindi è il vero responsabile della formazione e dellastabilità del legame, non ha un significato fisico altrettanto evidentee semplice; esso può tuttavia venir rappresentato come l'attrazionedella densità di carica di sovrapposizione ψAψB (frazione dielettrone che si trova nella zona centrale tra i nuclei dove lasovraposizione tra ψA e ψ B è sensibile) per l'uno o l'altro dei duenuclei. Esso ha segno negativo e importo di alcuni eV , ovvero di

qualche decina di Kcal/mol. Ne consegue che lo stato ψ-, avendo uncontributo negativo di β, ha energia più alta dell'energia EH degliatomi separati, e quindi rappresenta uno stato di instabilità o diantilegame. Il vero stato legante, che rappresenta lo statofondamentale stabile della molecola, è ψ+, con energia E+ più bassadi quella dei singoli atomi separati. Notiamo che all'integrale βcontribuiscono soltanto quegli elementi dello spazio in cui ψA e ψBhanno contemoraneamente valori diversi da zero, cioè β è diversoda zero soltanto quando vi è sovrapposizione, e in valore assolutoesso cresce al crescere della sovrapposizione S, benchè tra i dueintegrali β e S non vi sia una vera e propria proporzionalità. Si puòasserire che l'energia di un legame stabile è in genere tantomaggiore quanto maggiore è la sovrapposizione tra gli orbitaliatomici degli atomi legati.
La densità elettronica in un punto x,y,z dello spazio è data da
€
ψ±
2 x,y,z( ) =1
2±2SψA
2 +ψB2 ±2ψAψB( )
E' particolarmente interessante discutere l'andamento della densitàelettronica nella zona centrale della molecola, cioè lungo il piano disimmetria σ della molecola (luogo dei punti equidistante dai nuclei).
HA HB
σ
ra rb
Poichè in tale piano ra=rb, il valore numerico delle funzioniatomiche di A e di B sarà uguale:
ψA = ψB = ψ(σ)

Sarà quindi
€
ψ+
2 α( ) =1
2±2S2ψσ
2 ±2ψσ
2( )
Essendo
€
ψσ
2 la densità elettronica che si registrerebbe se gli statiatomici fossero rimasti imperturbati , cioè se non fossere intervenutieffetti di legame e di antilegame, notiamo che lo stato legante
€
ψ+presenta un aumento di densità elettronica nella zona in mezzo
tra i nuclei, mentre lo stato antilegante
€
ψ−presenta una diminuizione
nella stessa zona, arrivando anzi ad annullarsi nel centro.Quest'ultimo andamento del resto si può anche prevedere anchesemplicemente dal fatto che il piano di simmetria σ è un pianonodale della funzione
€
ψ−.
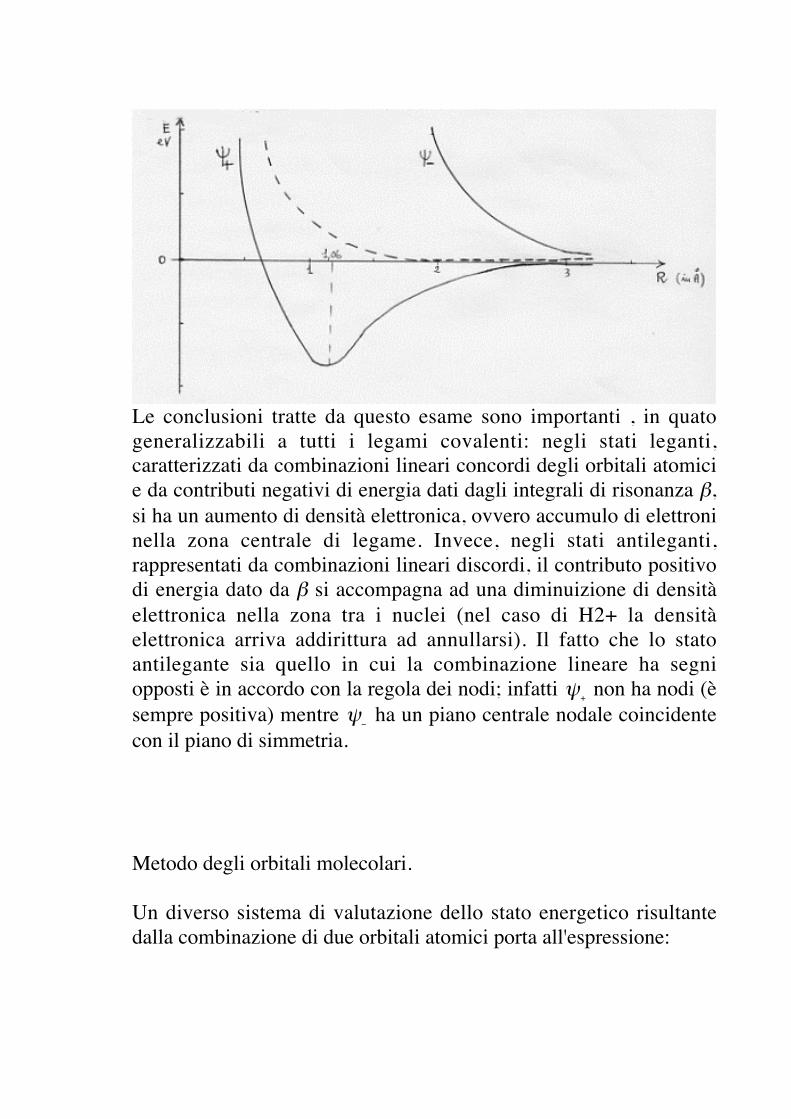
Le conclusioni tratte da questo esame sono importanti , in quatogeneralizzabili a tutti i legami covalenti: negli stati leganti,caratterizzati da combinazioni lineari concordi degli orbitali atomicie da contributi negativi di energia dati dagli integrali di risonanza β,si ha un aumento di densità elettronica, ovvero accumulo di elettroninella zona centrale di legame. Invece, negli stati antileganti,rappresentati da combinazioni lineari discordi, il contributo positivodi energia dato da β si accompagna ad una diminuizione di densitàelettronica nella zona tra i nuclei (nel caso di H2+ la densitàelettronica arriva addirittura ad annullarsi). Il fatto che lo statoantilegante sia quello in cui la combinazione lineare ha segniopposti è in accordo con la regola dei nodi; infatti
€
ψ+ non ha nodi (è
sempre positiva) mentre
€
ψ− ha un piano centrale nodale coincidente
con il piano di simmetria.
Metodo degli orbitali molecolari.
Un diverso sistema di valutazione dello stato energetico risultantedalla combinazione di due orbitali atomici porta all'espressione:

€
E m = Eat +α m β1m SAB
L'energia dell'orbitale molecolare si differenzia dall'energia degliorbitali atomici di partenza per un contributo, sempre positivo, datodall'integrale coulombiano α, e per il contributo positivo o negativodato dall'integrale di risonanza β. L'integrale coulombianorappresenta l'attrazione o repulsione elettrostatica della densità dicarica
€
ψA*∫ ψadV , ossia di un elettrone in un orbitale dell'atomo A,
per la carica effettiva ZB dell'atomo B o viceversa; il suo contributosi può agevolmente trascurare , almeno per molecole che nonabbiano legami fortemente polari. Se si trascura inoltre il valore di Sal denominatore, che è generalmente prossimo a zero, l'espressionedell'energia degli orbitali molecolari si può approssimativamentescrivere come
€
E m = Eat +α m β1m SAB
≈ Eat +α ±β ≈ Eat ±β
La differenza di energia dell'elettrone di valenza tra lo stato atomicodi partenza e lo stato molecolare è quindi espressa quasiesclusivamente dal valore dell'integrale di risonanza β; esso è untermine attrattivo (rappresenta all'incirca l'attrazione della frazionedi carica elettronica che si trova nella zona di sovrapposizione versol'uno o l'altro dei due nuclei); perciò è negativo e lo stato stabile èlal combinazione somma che porta a un livello energeticomolecolere inferiore al livello energetico atomico.La situazione energetica si può illustrare come nel seguente schema:

β
β
ψm=√1/2(ψA+ψB)
ψm=√1/2(ψA-ψB)
ψA ψB
A AB B
E
cioè dalla combinazione di due orbitali atomici della stessa energiasi può formare una combinazione lineare legante e una antilegantecon energie pressochè simmetriche rispetto all'energia degli statiatomici di partenza (a meno del contributo degli integrali α), el'effetto di legame o di antilegame è dato direttamente dal valore diβ. Valutato numericamente, questo integrale è dell'ordine di alcunedecine di Kcal/mol, ovvero dell'ordine di grandezza dell'energia deicomuni legami chimici.Volendo considerare la possibilità di formazione di orbitalimolecolari per combinazione lineare tra autofunzioni atomiche dtipo e di energia diverse nei due atomi di una molecola biatomica,dobbiamo anzitutto tener presente un requisito di simmetria;possono cioè dare combinazioni molecolari efficaci soloautofunzioni atomiche che appartengano allo stesso tipo disimmetria, cioè o ambedue σ o ambedue π.
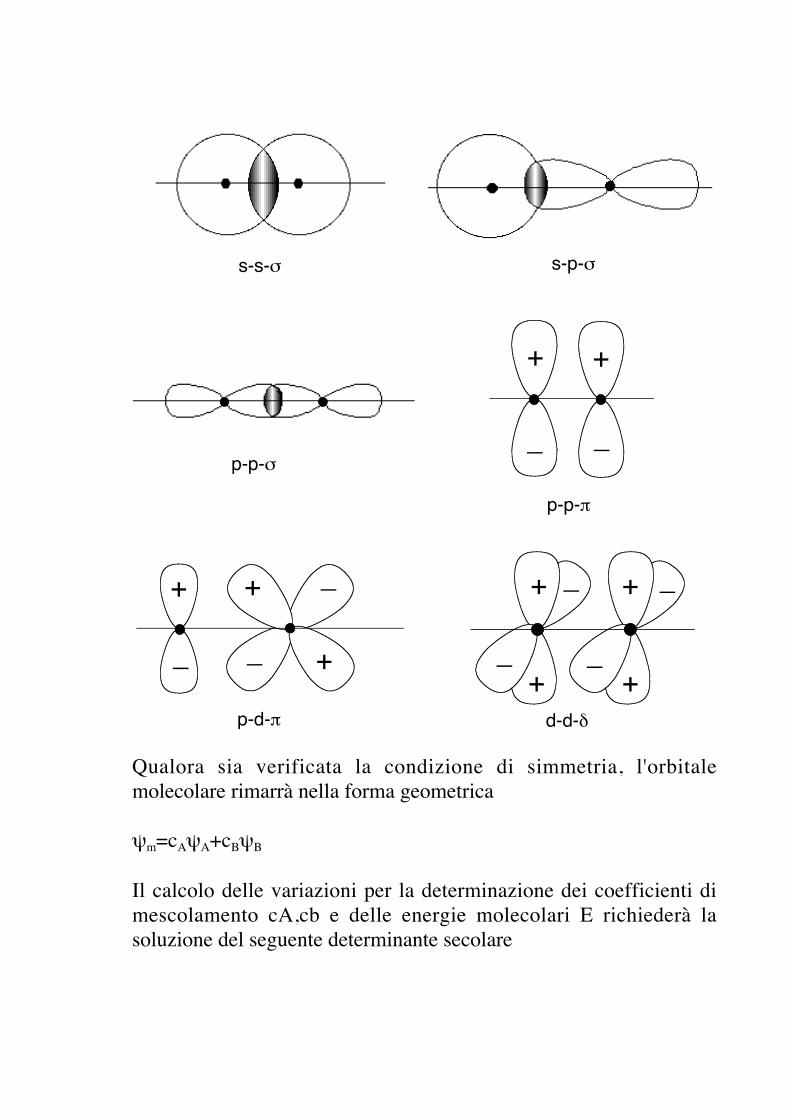
s-s-σ s-p-σ
p-p-σ
+ +
_ _
p-p-π
p-d-π
+
+_
_ _
_
+
+
+ +
+
_
__
d-d-δ
Qualora sia verificata la condizione di simmetria, l'orbitalemolecolare rimarrà nella forma geometrica
ψm=cAψA+cBψB
Il calcolo delle variazioni per la determinazione dei coefficienti dimescolamento cA,cb e delle energie molecolari E richiederà lasoluzione del seguente determinante secolare

Eat,A + αA,B - E
Eat,B + αB,A - E
β − ESAB
β − ESAB
= 0
Trascurando il contributo degli integrali a e ponendo S=0 (il chenon è quantitativamente corretto mam non altera l'aspettoqualitativo dei risultati), il determinante prende la forma:
Eat,A + - E
Eat,B - E
β
β
= 0
e corrisponde all'equazione determinantale
€
E 2 −E(Eat ,A +Eat ,B )+Eat ,A .Eat ,B −β2 = 0
con le radici
€
E1,2 =Eat ,A +Eat ,B
2±
Eat ,A −Eat ,B
2
2
+β 2
E' opportuno considerare due casi limite:a) Se
EA-EB=0 ⇒ E1,2 = Eat ± β
cioè si ritorna alla relazione prima ricavata e l'effetto di legame èdato in pieno dal valore β.

b) Se
€
β <<< EA −Eb
tanto che si possa trascurare b nell'espressione sotto radiceprecedente, che così diventa:
€
E1,2 =EA +EB
2mEA −EB
2EA = cA =1,cB = 0( )EB = cA = 0,cB =1( )
ed i coefficienti di mescolamento diventano 0 e 1. In tal caso (cioèse le energie degli stati atoici di partenza sono molto diverse),l'effetto di legame è trascurabile, ovvero le energie non si sspostandai valori delle energie atomiche, ed in pratica non si hamescolamento tra i due orbitali atomici; gli orbitali molecolaririsultano quindi degenerati in obitali atomici. Nei casi intemedi,ovvero quando le energie degli stati atomici non sono troppodifferenti, dai due orbitali atomici
€
ψA e
€
ψB , a energierispettivamente EA ed E B, si formano quindi per combinazionelineare due orbitali molecolari, uno legante
€
ψm = cAψA + cBψB , e unoantilegante
€
ψm = cAψA − cBψB , con energia rispettivamente più bassadel più basso dei livelli di energia atomica, e più alto del più alto(figura successiva);

E
(cA > cB)
ψA
ψB
ψm=cAψΑ+cBψΒ
ψ∗m=cAψΑ−cBψΒ
EA
EB
l'effetto di legame, ovvero la differenza di energia tra un orbitalemolecolare e quella del più vicino orbitale atomico, è però inassoluto inferiore a
€
β ; questo vuol dire che il peso dell'integraleβ nel determinare gli effetti di legame è tanto maggiore quantominore è la differenza di energia dei due orbitali atomici che sicombinano.
L'orbitale ψm ad energia più bassa (legante) è una combinazionelineare somma, ed in essa è maggiore il coefficiente c relativoall'orbitale atomico di energia più bassa ; l'orbitale molecolare ψm dienergia più alta è una combinazione lineare differenza (e pertanto haun nodo nella zona di legame) ed ha un coefficiente maggiore (cioèuna maggiore partecipazione ) dell'orbitale a energia più alta.Tenendo presente i principi ora illustrati sulla formazione di orbitalimolecolari per combinazione lineare di orbitali atomici, ericordando in particolare che una combinazione lineare risultaefficace ai fini della formazione di legame solo se sono soddisfatti idue requisiti di simmetria (stesso carattere degli orbitali atomici chesi combinano) e di energia (l'energia deve essere il più possibilevicina negli orbitali atomici che si combinano), possiamo passare acostruire il sistemam di livelli energetici per molecole biatomiche

omonucleari generiche, cioè abbiamo più di nu orbitale atomico divalenza capace di entrare in cobinazione molecolare. Da ognicoppia di orbitali atomici corrispondentedsi per simmetria e perenergia prevediamo la formazione di una coppia di orbitalimolecolari, ad energia rispettivament epiù alta e più bassa, cioè diuna coppia molecolare legante e entilegante, con separazioneenergetica data dai valori dell'integrale β , costruito con quel datoorbitale atomico. Poi, una volta stabilito il sistema di orbitalimolecolari, lo riempiremo seguendo il principio di Aufbauperfettamente simile a quallo visto per la struttura elettronica degliatomi, con il numero di elettroni dato dalla somma degli elettronidei due atomi costituenti. Così, per le molecole biatomicheomonuclari degli elementi del primo piccolo periodo, vale ilseguente schema generale di orbitali molecolari:
A AAB
1s1s
2s 2s
2p 2p
k
k*
zσ
yσ∗
xσwπ
vπ∗uσ∗



















