
Le Sindromi Mielodisplastiche - AIRTUM · • Con le tecniche convenzionali dovrebbero essere...
57
F.Merli, Ematologia, ASMN RE Le Sindromi Mielodisplastiche Francesco Merli Ematologia, ASMN Reggio Emilia
Transcript of Le Sindromi Mielodisplastiche - AIRTUM · • Con le tecniche convenzionali dovrebbero essere...

F.Merli, Ematologia, ASMN RE
Le Sindromi
Mielodisplastiche
Francesco MerliEmatologia, ASMN
Reggio Emilia

F.Merli, Ematologia, ASMN RE
Definizione
Le Sindromi Mielodisplastiche (MDS) costituiscono un gruppo eterogeneo di disordini clonali della cellula staminale emopoietica caratterizzate, nella maggior
parte dei pazienti, da:
•citopenia periferica che coinvolge una o più linee emopoietiche(eritroide, megacariocitaria o granulocitaria)
•midollo osseo ipercellulare
•displasia di una o più linee cellulari emopoietiche
•rischio variabile (mediamente del 25%) di trasformazione in LMA

F.Merli, Ematologia, ASMN RE
INCIDENZA GLOBALE: • 4-5/100.000 nuovi casi all’anno e aumenta in
modo costante all’aumentare dell’età– 30/100.000 nuovi casi/anno nei soggetti > 70 aa– 0,4/100.000 nuovi casi/anno nei paz. < 30 aa– 0,01/100.000 nuovi casi all’anno nell’età
pediatrica (4% delle neoplasie pediatriche).
Epidemiologia
ETA’ MEDIANA ALLA DIAGNOSI: 76 anniCAUSE DI MORTE:
- 34% dei casi per trasformazione leucemica- 23% dei casi per cause infettive- 53% per altre comorbidità
SOPRAVVIVENZA A LUNGO TERMINE: 35%
SOPRAVVIVENZA MEDIA: 2-3 anni (< 3 mesi – >10 aa)

F.Merli, Ematologia, ASMN RE
AUMENTO DELL’INCIDENZA DELLE MDS CON L’ETA’
0
100
200
300
400
500
600
700
800
<30 30-40 40-50 50-60 60-70 70-80 >80

F.Merli, Ematologia, ASMN RE
Clinica
CITOPENIA:- anemia: presente nel 90% dei casi alla diagnosi; grave nel 60%- neutropenia: presente nel 75% dei casi- piastrinopenia: presente nel 45% dei casi (piùfrequente in caso di t-MDS o MDS ad alto rischio); isolata solo nel 7% dei casi- citopenia bi- o trilineare: nel 60% dei casi all’esordio
EPISODI INFETTIVI O EMORRAGIE MUCO-CUTANEE
MANIFESTAZIONE AUTOIMMUNE: 10% dei casi
EPATO-SPLENOMEGALIA: rara

F.Merli, Ematologia, ASMN RE4,338Nessuna citopenia sulla base dei valori definiti
10,594Totale13Neutropenia + piastrinopenia
761Piastrinopenia isolata220Neutropenia isolata
Altre citopenie senza anemia49,2438Totale
63Anemia grave + neutropenia + piastrinop.67Anemia moderata + neutrop. + piastrinop.
29Anemia lieve + neutrop. + piastrinop.52Anemia grave + piastrinop.25Anemia grave + neutrop.73Anemia moderata isolata + piastrimop.47Anemia moderata + neutrop.51Anemia lieve + piastrinopenia31Anemia lieve + neutropemia
Anemia + altre citopenie36321Totale
87grave isolata
156moderata isolata78 lieve isolata
Anemia isolata senza altre citopenie%N° di casiTipo di citopenia

F.Merli, Ematologia, ASMN RE
Eziologia
MDS primarie MDS secondarie (t-MDS)
• agenti alchilanti
• inibitori della topoisomerasi II
• radiazioni ionizzanti
• TMO autologo
• storia familiare di neoplasia (OR = 1,92)
• fumo di sigaretta (OR = 1,65)
• esposizione a pesticidi (OR = 4,55), solventi o benzene
• polimorfismo genetico
•CLINICA:•età giovane•citopenie più severe•displasia midollare piùmarcata•midollo osseo ipocellulare o con fibrosi•maggiore incidenza di anomalie cariotipiche•prognosi infausta

F.Merli, Ematologia, ASMN RE
Patogenesi
APOPTOSI
MITOCONDRI
AUTOIMMUNITA’
CELLULE NK
MICROAMBIENTE
ANGIOGENESI
TELOMERI
ALTERAZIONI MOLECOLARI
ANOMALIE EPIGENETICHE
ALTERAZIONI DELLA CELL. STAMINALE
EMOPOIETICA
FENOTIPO MIELODISPLASTICO
ALTERAZIONI CITOGENETICHE
MITOCONDRI

F.Merli, Ematologia, ASMN RE
Diagnosi
1. Morfologica
2. Immunofenotipica
3. Citogenetica
4. Molecolare

F.Merli, Ematologia, ASMN RE
Criteri diagnostici minimi nelle MDS
(A)Prerequisiti (devono essere entrambi presenti)
- Citopenia costante in una o più delle seguenti linee cellulari: eritroide (emoglobina <11 g dL−1); neutrofila (ANC < 1500_L−1) o megacariocitica (piastrine <100,000_L−1)
- Esclusione di tutte le malattie emopoietiche e non che possono essere la causa primitiva della citopenia/displasia.
Valent P. et al, Leuk Res 2007; 31:727-736.

F.Merli, Ematologia, ASMN RE
Criteri diagnostici minimi nelle MDS
(B) Criteri decisivi MDS-relati (almeno uno deveessere presente)
- Displasia in almeno il 10% di tutte le cellule di una delle seguenti linee cellulari nello striscio di sangue midollare: eritroide, neutrofila o megacariocitica
o >15% di sideroblasti ad anello- 5–19% di blasti nello striscio di sangue midollare- anomalie cromosomiche tipiche (evidenziate con
tecniche di citogenetica convenzionale o con FISH)Valent P. et al, Leuk Res 2007; 31:727-736.

F.Merli, Ematologia, ASMN RE
Criteri diagnostici minimi nelle MDS
(C) Co-criteri (per pazienti in cui siano verificati i cirteri ‘A’ ma non i ‘B’, e per quelli che mostrano le tipiche caratteristiche cliniche delle MDS, ad es. una anemia macrocitica trasfusione-dipendente)
- Fenotipo anomalo delle cellule midollari, chiaramente indicativo della presenza di una popolazione clonale di cellule eritroidi e/o mieloidi, individuato tramite citometria a flusso
- Evidenza di una popolazione clonale mediante indagini molecolari, test HUMARA, gene chip profiling, o analisi delle mutazioni puntiformi (ad es. mutazioni di RAS)
- Riduzione marcata e persistente della formazione di colonie (± formazione di cluster) da parte dei progenitori emopoietici midollari e/o circolanti (test CFU).
Valent P. et al, Leuk Res 2007; 31:727-736.

F.Merli, Ematologia, ASMN RE
Diagnosi
MORFOLOGIA
Sono necessari per una diagnosi accurata:
1.Striscio di sangue midollare
2.Striscio di sangue periferico
3.Biopsia osteomidollare
ICUS (idiopathic cytopenia of uncertain significance): presenza in una o più linee mieloidi di citopenia costante (di durata ≥ 6 mesi) che non rispetti i criteri minimi per una diagnosi di MDS e che non sia sostenuta da altra patologia.

F.Merli, Ematologia, ASMN RE
IMMUNOFENOTIPO
• Scopo diagnostico: analisi quantitativa e qualitativa di cellule CD34+, monociti, cellule mieloidi maturanti, cellule eritroidi
• Scopo prognostico: espressione di CD7 sui blasti mieloidi
• Monitorare la risposta al trattamento: riduzione della displasia

F.Merli, Ematologia, ASMN RE
CITOGENETICA• Le indagini di citogenetica andrebbero sempre eseguite su
sangue midollare • Con le tecniche convenzionali dovrebbero essere analizzate
almeno 20-25 metafasi• La presenza di un clone viene definita quando sono individuabili
due cellule midollari che mostrano la stessa anomalia strutturale o la stessa acquisizione di materiale genetico, oppure tre cellule midollari che mostrano la stessa perdita di materiale cromosomico.
• Nei casi dubbi (ad es. scarso numero di metafasi, diagnosi differenziale tra MDS a basso rischio e ICUS) è consigliabile utilizzare la tecnica FISH
• Sub-clone e cariotipo complesso• La presenza di anomalie cromosomiche clonali è riscontrabile
alla diagnosi nel 40-70% dei pazienti con MDS de novo nel 95% dei casi di MDS secondaria

F.Merli, Ematologia, ASMN RE
ALTERAZIONI CITOGENETICHE E MOLECOLARI
90103
90
t-MDS -5/del(5q) o – 7/del(7q) +8t(11q23)Cariotipi complessi
10-2010
5-101075
5-610-20
MDS de novo -5/del(5q)+8-7/del(7q)-Y17p-del(20q)t(11q23)Cariotipi complessi
Anomalia Incidenza (%)
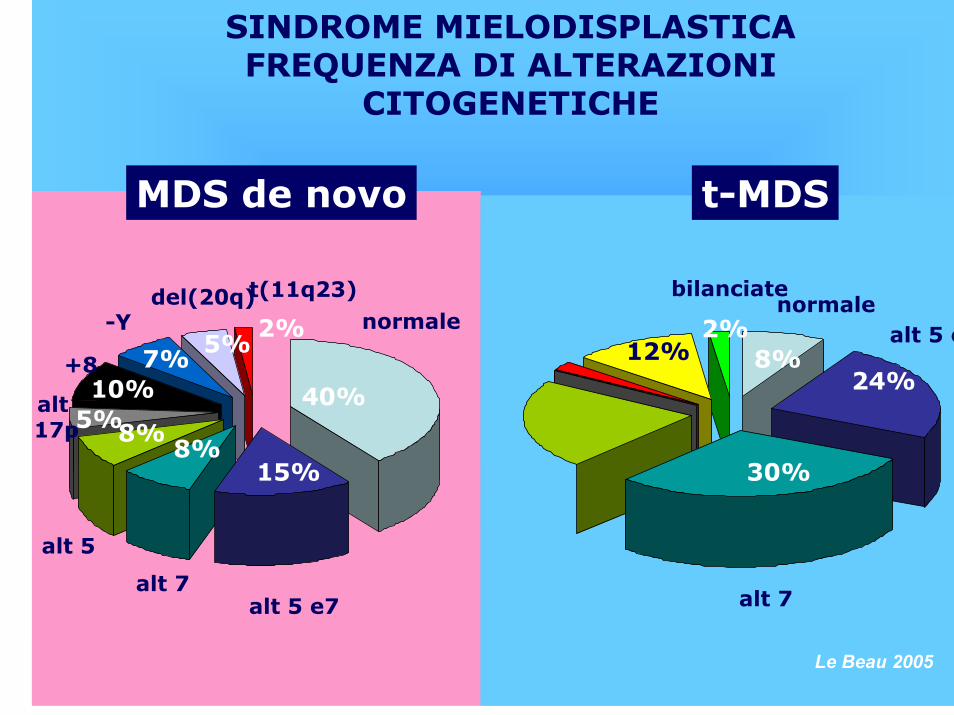
F.Merli, Ematologia, ASMN RE
SINDROME MIELODISPLASTICAFREQUENZA DI ALTERAZIONI
CITOGENETICHE
10%
8%15%
40%22%
normale-Y altre
alt 5 e7
MDS de novo
8%5%
7%5%
2%
alt 7
alt 5
alt 17p
+8
del(20q)t(11q23)
alt 5
2%
t(11q23)
30%
24%8%
normale
alt 7
t-MDS
alt 5 e712%
2%
bilanciate
Le Beau 2005

F.Merli, Ematologia, ASMN RE
–delezione isolata di 5q;–maggiore incidenza nel sesso
femminile e nella fascia di etàtra 60 e 65 anni;
–decorso indolente (sopravvivenza mediana: 80 mesi);
–bassa incidenza di complicanzeinfettive o emorragiche;
–splenomegalia nel 20% dei casi;
–evoluzione in LMA in circa il 10% dei casi;
–presenza di grave anemia macrocitica refrattaria, conta piastrinica normale o elevata, modesta leucopenia;
–percentuale di blasti midollari <5% e presenza di megacariociti ipolobulati nel MO
Sindrome 5q-

F.Merli, Ematologia, ASMN RE
Monosomia 7
Clinica:- età mediana inferiore rispettoai pazienti con “sindrome 5q-“;- citopenia refrattaria severa con tendenza a sviluppare episodi infettivi gravi;- prognosi infausta (sopr. mediana di 14 m.)
Terapia:-TMO allogenico -Agenti ipometilanti (AZA)
Biologia molecolare:Associazione tra questa anomalia citogenetica e mutazioni del gene RAS, mutazioni di AML1 e ipometilazione di p15INK4b
25% dei casi mutati nell’adulto,
50% dei casi pediatrici

F.Merli, Ematologia, ASMN RE
Cariotipo complesso
• 3 anomalie cariotipiche indipendenti• 30% dei casi mutati• Processo multistep• Pregressa terapia con alchilanti• TMO allogenico• Recentemente sono state osservate risposte
citogenetiche in pazienti ad alto rischio trattati con decitabina o con lenalidomide

F.Merli, Ematologia, ASMN RE
Il cariotipo è il più importante parametro prognostico: analisi di 2124 pazienti
Anomalie clonali:49,8% dei paz.con SMD de novo 68,5% dei paz. SMD secondaria
Validità prognostica delle categorie citogeneticheidentificate dall’IPSS. Mediana di sopravvivenza: paz. a citogenetica favorevole: 54 mesipaz. a citogenetica intermedia: 31 mesipaz. a citogenetica sfavorevole: 11 mesi
Mediana di sopravvivenza:•53.4 mesi: paz. a cariotipo normale•24 mesi: paz. con 1 o 2 anomalie cromosomiche•8.7 mesi: paz. a cariotipo complesso
Haase et al, Blood, 2007; 110:4385

F.Merli, Ematologia, ASMN RE
Impatto prognostico delle anomalie più comuni
5q-: mediana di sopravvivenza 80 mesi se presente come singola anomalia, di 47 mesi se presente insieme ad un’altra anomalia e di 7 mesi se parte di un cariotipo complesso. Se il 5q coinvolto in una traslocazionemediana di soli 4.4 mesi -7: mediana di sopravvivenza 14 mesi se presente come singola anomalia o insieme ad un’altra anomalia e di 8 mesi se parte di un cariotipo complesso7q-: prognosi migliore rispetto ai pazienti con -7, ma differenza non statisticamente significativa+8: mediana di sopravvivenza 22 mesi se presente come singola anomalia, di 44 mesi se presente insieme ad un’altra anomalia e di 17 mesi se parte di un cariotipo complesso
Haase et al, Blood, 2007; 110:4385

F.Merli, Ematologia, ASMN RE
Decisioni terapeutiche nelle SMD
Anomalia citogenetica
Cariotipo complessoNormale/Singola an.
Azacitidina/Decitabina
Trapianto Allogenico
5q- Lenalidomide
+8 ATG
N Eritropoietina
- 7 Azacitidina

F.Merli, Ematologia, ASMN RE
Classificazione
Principali modifiche apportate dalla classificazione WHO:
1. Riduzione della quota di blasti per la diagnosi di LMA
2. RMCD
3. Sindrome 5q-
4. RAEB-1 e RAEB-2
5. CMML
1982: classificazione FAB
1999: classificazione WHO
Catenacci DVT, Schiller GJ, Blood Reviews2005; 19: 301-319.

F.Merli, Ematologia, ASMN RE
SMD: CLASSIFICAZIONE WHO (1)
SINDROMI MIELODISPLASTICHE (blasti< 20%)
Anemia refrattaria con sideroblasti ad anello (RARS)
Anemia refrattaria senza sideroblasti ad anello (RA)(criteri come in classif. F.A.B. ma solo anemia)
Citopenia refrattaria con displasia multilineare (RCMD)(almeno 2 citop. e morfol. displastica in almeno 2 linee)
Citopenia refrattaria con displasia multilineare e sideroblasti ad anello (RCMD-RS)
(come sopra + sideroblasti > 15%)

F.Merli, Ematologia, ASMN RE
SMD CLASSIFICAZIONE WHO (1)
SINDROMI MIELODISPLASTICHE (blasti< 20%)
Anemia refrattaria con eccesso di blasti tipo 1 (RAEB1)(blasti mid. 5 – 9 %; blasti perifer. < 5%)
Anemia refrattaria con eccesso di blasti tipo 2 (RAEB 2)(blasti midollari 10 - 19% o periferici > 5%)
Sindrome 5q-(criterio citogenetico e morfologico)
SMD non classificabili(neutropenia o piastrinopenia isolata; SMD con fibrosi)

F.Merli, Ematologia, ASMN RE
SMD: CLASSIFICAZIONE WHO (2)
Disordini mielodisplastici /mieloproliferativi
• leucemia mielomonocitica cronica (LMMC)(segni di displasia + monociti > 1 x 103 /µl:tipo 1 se blasti < 5% nel sangue e < 10% nel midollo;tipo 2 se blasti ≥ 5% nel sangue o ≥ 10% nel midollo)
• leucemia mieloide cronica atipica• leucemia mielomonocitica giovanile• RARS con piastrinosi (entità provvisoria)• forme non classificabili

F.Merli, Ematologia, ASMN RE
DIFFERENZE WHO E FAB
Scomparsa della categoria AREB-T – più del 20 % dei blasti nel midollo fanno porre diagnosi di LAM (with multilineage dysplasia)
Migliore definizione delle categorie Refractory Anemia (RA) e Refractory Anemia with Ringed Sideroblasts (RARS)
Introduzione di una nuova categoria Refractory Cytopenia with Multilineage Myelodisplasia and Ringed Sideroblasts (RCMD-RS)

F.Merli, Ematologia, ASMN RE
DIFFERENZE WHO E FAB
Suddivisione della categoria RAEB in due sottogruppi, RAEB-1 e RAEB-2, a seconda del numero di blasti nel midollo o nel sangue periferico (5-9% vs 10-19 %)
Definizione di una specifica categoria MDS associata alla del (5q) isolata
Eliminazione della leucemia mielomonocitica cronica dalle categorie incluse nella MDS, e suo inserimento nel gruppo delle MDS/MPD (Myelodisplastic/Myeloproliferative diseases)

F.Merli, Ematologia, ASMN RE
Prognosi
2/30/1citopenia
sfavorevoleintermediofavorevolecariotipo
21-3011-20-5-10<5% blasti midollari
2,01,51,00,50Variabile
Punteggio
favorevole intermedio sfavorevole
Normale -7/del(7q)
-Y * 1 o 2 anomalie complesso
del(5q) *
del(20q) *
* unica anomalia
1997: InternationalPrognosticScoringSystem (IPSS)
Greenberg et al, Blood 1997; 87:2079

F.Merli, Ematologia, ASMN RE
VALORE PROGNOSTICO DEL CARIOTIPO IPSS
Greenberg et Blood 1997; 87:2079

F.Merli, Ematologia, ASMN RE
Stratificazione delle MDS secondo l’IPSS
Basso rischio: 0
Rischio intermedio 1: 0,5 - 1,0
Rischio intermedio 2: 1,5 - 2
Alto rischio: ≥ 2,5
Greenberg et al Blood1997;87:20790.30.4High
0.951.2Int-2
2.73.5Int-1
5.75.7Low
Leukemia free survival (years)
Overall survival(years)
IPSS score

F.Merli, Ematologia, ASMN RE
LIMITI DEL IPSS
• l’IPSS è stato elaborato basandosi sulla classificazione FAB
• include nel gruppo citogenetico a rischio intermedio i pazienti con alterazioni rare e con duplice anomalia
• lo score IPSS attribuisce molta importanza ai fattori predittivi di trasformazione leucemica attribuendo un punteggio di solo 0.5 ai pazienti con 2/3 citopenie considerando alla stessa stregua tutte le citopenie indipendentemente dalla loro severità
MODIFICHE PROPOSTE
• M-IPSS (β2-microglobulina, cariotipo, % di blasti midollari, citopenia)
• LDH• WPSS

F.Merli, Ematologia, ASMN RE
Score prognostico delle MDS basato sulla classificazione WHO (WPSS)
——RegolareNoFabbisogno trasfusionale**
—SfavorevoleIntermedioFavorevoleCariotipo*
RAEB-2
RAEB-1RCMD, RCMD-RS
RA, RARS, 5q–
Categoria WHO
3210Variabile
Sono stati identificati i seguenti gruppi di rischio: molto basso (punteggio = 0), basso (punteggio = 1), intermedio (punteggio = 2), alto (punteggio = 3-4), e molto alto (punteggio = 5-6).
* I diversi cariotipi erano così suddivisi: favorevole: normale, –Y, del(5q), del(20q); sfavorevole: complesso (3 anomalie), anomalie del cromosoma 7; e intermedio: altre anomalie.
**La dipendenza dalle trasfusioni di eritrociti era definita come la necessità di trasfondere almeno una unità di EC ogni 8 settimane per un periodo di 4 mesi.
Malcovati L. et al, J Clin Oncol 2007; 25: 3503-3510.

F.Merli, Ematologia, ASMN RE
a. terapie di supporto
- trasfusioni di EC/Plt
- gestione degli episodi infettivi
- terapia ferrochelante
b. terapie convenzionali
- fattori di crescita emopoietici
- chemioterapia convenzionale
- trapianto di midollo osseo
Autologo
Allogenico
c. terapie innovative
- farmaci immunomodulanti
- agenti ipometilanti
- terapia immunosoppressiva
d. terapie sperimentali
Terapia

F.Merli, Ematologia, ASMN RE
Terapie di supporto
1. Supporto trasfusionale
2. Terapia ferrochelante
3. Profilassi e tratt. degli episodi infettivi
• Circa il 60% dei pazienti presenta alla diagnosi un’anemia severa (Hb< 10 g/dl);
• nel corso della malattia l’anemia si sviluppa nel 90% dei casi;
• complessivamente circa l’80% dei pazienti prima o poi avrà bisogno di trasfusioni di EC
• le trasfusioni di EC sono l’unica opzione terapeutica in circa il 40% dei pazienti
• valore soglia?
Trasfusioni di Plt: anche a scopo profilattico?

F.Merli, Ematologia, ASMN RE
Terapia FerrochelanteL’emosiderosi secondaria peggiora la sopravvivenza dei pazienti con MDS provocando un aumento del rischio di morte del 40% per ogni aumento di 500 ng/ml della ferritina siericaL’entità del sovraccarico marziale viene in genere determinato con i valori di ferritinemia nella pratica clinica (da mantenere<1000 ng/ml) e con RMN cardiaca ed epaticaTra le cause di morte non leucemica nei pazienti con MDS nel 51% dei casi il decesso avviene per insufficienza cardiaca e nel8% dei casi per cirrosi epaticaLe attuali linee guida raccomandano l’impiego della terapia ferrochelante nei pazienti che:o vengono regolarmente trasfusio hanno MDS a basso rischio e hanno ricevuto 20-50 unità di
EC o hanno MDS a rischio più elevato ma malattia stabileo hanno ferritinemia > 1000 ng/mlo vengono regolarmente trasfusi e sono candidati a TMO
allogenico

F.Merli, Ematologia, ASMN RE
Terapie convenzionali
1. Fattori di crescita emopoietici
2. Chemioterapia convenzionale
3. Trapianto autologo e allogenico

F.Merli, Ematologia, ASMN RE
Fattori di crescita emopoietici• I fattori di crescita granulocitari da soli non sono raccomandati in
profilassi, ma solo in caso di neutropenia febbrile• La rHuEPO da sola è efficace in circa il 20% dei pazienti anemici,
mentre si è osservato un sinergismo in vitro e in vivo con il G-CSF con un incremento dei tassi di risposta fino al 40-50%
• La risposta si osserva in genere dopo 2-3 mesi di trattamento e dura in media 24 mesi
• E’ stato elaborato uno score in grado di predire la risposta al trattamento con EPO e G-CSF (Hellstrom-Lindberg E. et al, Br J Haematol 1997):
Intermedia (23%)
Bassa (7%)
Alta (74%)
Probabilità di risposta
≥ 2 UEC/mese< 2 UEC/meseFabbisogno trasfusionale
> 500 U/L≤ 500 U/LEPO sierica
• Attualmente considerati il gold standard nel trattamento di pazienti con IPSS basso o int-1 con anemia trasfusione-dipendente con bassi livelli endogeni di EPO e fabbisogno trasfusivo

F.Merli, Ematologia, ASMN RE
Chemioterapia convenzionale
• il trattamento chemioterapico con schemi AML-like èriservato a pazienti con IPSS int-2 o alto nel tentativo di modificare la storia naturale della malattia
• Efficace ma insoddisfacente: RC ematologiche del 50-60%, remissioni di breve durata (8 mesi), elevata incidenza di recidiva, sopravvivenza a lungo termine del 14%
• Attualmente raccomandata per pazienti con età <65 anni non candidati a trapianto allogenico e con IPSS int-2 o alto solo se non siano applicabili protocolli sperimentali
• In dubbio il ruolo della chemioterapia prima del trapianto allogenico (le evidenze attuali sono a favore del trattamento chemioterapico precedente)
• NO ARA-C a basse dosi (mielotossica e scarsamente efficace)

F.Merli, Ematologia, ASMN RE
Trapianto autologo
• Ruolo molto controverso nei pazienti con MDS.
• Raccomandato dalle linee guida SIE previo condizionamento mieloablativo per i pazienti che, non disponendo di un donatore HLA-compatibile, raggiungono una remissione completa dopo chemioterapia AML-like, in particolare in quei casi in cui si ottenga una remissione citogenetica (in considerazione dell’elevata incidenza di recidiva di malattia dopo la sola chemioterapia).
• Impiego di staminali periferiche.
• Sopravvivenza libera da malattia a 4 anni di circa il 25%

F.Merli, Ematologia, ASMN RE
Trapianto allogenico
• E’ l’unica terapia potenzialmente curativa
• Proponibile a meno del 10% dei pazienti con MDS
• TRM = 27-50%
• Incidenza di recidiva = 23-48%

F.Merli, Ematologia, ASMN RE
•Sovraccarico marziale (ferritina elevata)
•Dipendenza dalle trasfusioni
•Mismatch donatore-ricevente
•Fibrosi midollare
•Eccesso di blasti midollari/stato di malattia al trapianto
•Cariotipo (monosomia 7, anomalie cariotip. multiple)
•IPSS intermedio-2 o alto
•MDS secondaria/LMA
•Comorbidità
•Età avanzata
Fattori prognostici sfavorevoli per l’outcome di un TMO

F.Merli, Ematologia, ASMN RE
Candidati:
Età < 55 anni e MDS ad alto rischio
MDS pediatriche
Età < 55 anni e MDS a basso rischio con cariotipo sfavorevole o non responsivi ad altra terapia
TimingSorgente di cellule staminali: preferibili le staminaliperiferiche
Regimi di condizionamento: standard, RIC
Trapianto aploidentico o da cordone ombelicale

F.Merli, Ematologia, ASMN RE
Terapie innovative
1. Farmaci immunomodulanti
2. Agenti ipometilanti
3. Terapia immunosoppressiva

F.Merli, Ematologia, ASMN RE
Farmaci immunomodulantiTHALIDOMIDE
• riduce la produzione di citochine infiammatorie e pro-apoptotiche, inibisce lo stimolo neoangiogenetico, potenzia la risposta immunitaria antigene-mediata, modula l’adesione cellulare allo stroma midollare, spinge la risposta immune verso il tipo Th2, stimola l’attività dei linfociti T CD8+, aumenta i livelli sierici di EPO e di Hb fetale, riduce la quota di CD4+ e aumenta il numero delle cellule NK
• risposta ematologica nel 25-30% dei casi spt sull’anemia
• risposta dopo 8-12 settimane di terapia, non dose-dipendente
• pazienti con IPSS basso o int-1, anemia isolata, diagnosi recente, età < 65 anni
• 200 mg/die
• 40-50% dei pazienti sospendono il trattamento per tossicità(neuropatia periferica, stipsi, sedazione, astenia)

F.Merli, Ematologia, ASMN RE
Farmaci immunomodulanti
57%16%200-100068Moreno-Aspitiaet al 2006
64%88%200-80047Bouscary et al 2005
67%50%100-80012Bowen et al 2005
37%32%100-30040Musto et al 2002-2004
15%65%100-50034Strupp et al 2002
38%31%100-40083Raza et al 2001
Drop-out
Risposta globale
Dose (mg/die)
N°paz.
Autore

F.Merli, Ematologia, ASMN RE
Farmaci immunomodulanti
LENALIDOMIDE
• Ha maggiore attività immunomodulante (influenza la proliferazione dei linfociti T, la produzione di IL-2 e IFN-γ, e la risposta NK) e antiflogistica, stimola l’eritropoiesi, ha attività anti-proliferativa cariotipo-dipendente
• Mielotossicità
• 83% di risposte eritroidi e 75% di risposte citogenetiche complete nei pazienti con del(5q)
• 36% i remissioni istomorfologiche
• Risposta che compare dopo 4-6 settimane e dura a lungo
• Approvata nel 2005 per i pazienti con MDS con IPSS basso o int-1 e del(5q)
• Ottimi risultati anche nei pazienti con del(5q) associata ad altre anomalie

F.Merli, Ematologia, ASMN RE
Agenti ipometilantiModificazioni epigeneticheMetilazione del DNAGià noti 25 anni fa, oggi impiegati a dosi 10-30 volte minoririspetto alla dose massima tollerataSi integrano nel DNA
AZACITIDINA
•Precedentemente impiegata come chemioterapico
•Approvata nel 2004 per il trattamento di tutti i sottotipi di MDS
•75 mg/mq s.c. per 7 giorni ogni 4 settimane
•Risposta globale nel 40-47% dei casi (10-17% di RC)
•90% delle risposte si osserva dopo il VI ciclo
•Mielotossicità
•Migliori risposte nei pazienti con -7 o +8

F.Merli, Ematologia, ASMN RE
Agenti ipometilanti
DECITABINA• approvata per il trattamento di tutti i sottotipi di MDS • al dosaggio di 15 mg/mq ogni 8 ore per 3 giorni ogni 6 settimane induce una risposta globale di circa il 50%• la dose migliore sembra essere 20 mg/mq/die e.v. per 5 gg ogni 4 sett., con 70% di risposte globali (35% di RC) e un 49% di risposte sulla conta piastrinica• profilo di tossicità accettabile
Quali pazienti?-IPSS int-2 o alto, candidati o meno a TMO- IPSS basso o int-1 con citopenie importanti e mancata risposta a fattori di crescita e thalidomide- monosomia 7 o trisomia 8
Combinazioni con altri farmaci attivi nelle MDS, in particolare inibitori delle istone-deacetilasi, acido valproico o acido retinoico

F.Merli, Ematologia, ASMN RE
Terapia immunosoppressiva
• Nei paz.con MDS si riscontrano numerose anomalie immunologiche
• Si è ipotizzato che almeno in una quota di pazienti con MDS a basso rischio il ruolo patogenetico principale sia svolto dai linfociti T citotossici attivati che aggrediscono i progenitori emopoietici
• I primi studi sulla terapia immunosoppressiva nei pazienti con MDS risalgono agli anni Ottanta.
• Gli steroidi sono stati impiegati a lungo, spt nei pazienti con AR ma attualmente hanno perso di importanza

F.Merli, Ematologia, ASMN RE
Terapia immunosoppressiva
• La CyA è stata impiegata nei pazienti con MDS a basso rischio ottenendo l’indipendenza dalle trasfusioni in circa il 50% dei casi; tuttavia, la risposta è dose-dipendente (maggiore per dosi > 5 mg/kg) e significative sono la tossicità renale e il rischio di infezioni e neoplasie
• L’ATG al dosaggio di 40 mg/kg/die e.v. per 4 giorni consente di ottenere l’indipendenza dalle trasfusioni nel 35-50% dei casi spt in quelli con AR
• Pazienti con maggiori probabilità di risposta sono quelli giovani (età < 60 anni), con un breve periodo di trasfusione-dipendenza, la presenza di HLADRB1 15 (riscontrato in diversi casi di AR) e midollo osseo ipocellulare

F.Merli, Ematologia, ASMN RE
Strategie Generali
di Trattamento

F.Merli, Ematologia, ASMN RE
Rischio IPSS basso o Int-1
Citopenia clinicamente significativa
Terapia di supporto come aggiunta al trattamento
Trombocitopenia,neutropenia
Anemia sintomatica Del(5q) ± altre anomalie
Lenalidomide
Nessuna risposta
Azacitidina,
decitabina,
thalidomide,
EPO-α ± G-CSF,
darbopoietina-α o
trial clinico
Azacitidina,
decitabina o
trial clinico
Nessuna risposta
IST o
Trial clinico
EPO sierica
> 500 mU/mlEPO sierica
≤ 500 mU/ml
EPO-α ± G-CSF,darbopoietina-α
Nessuna risposta
Azacitidina,
decitabina,
thalidomide o
trial clinico
Nessuna risposta
Trial clinico
Candidato per la terapia immumo-soppressiva (IST)
Non candidato per la terapia immumo-soppressiva (IST)
Azacitidina,
decitabina,
thalidomide o
trial clinico
Nessuna risposta
EPO-α ± G-CSF,
darbopoietina-α o
trial clinico
Azacitidina,
decitabina,
thalidomide,
EPO-α ± G-CSF,
darbopoietina-α o
trial clinico
ATG, CyA
Nessuna risposta

F.Merli, Ematologia, ASMN RE
Rischio IPSS Int-2 o alto
Candidato alla terapia intensiva
Non candidato alla terapia intensiva
Donatore disponibile
SI NO
Trapianto di cellule staminali emopoietiche
Terapia ad alta intensitàoazacitidina,
decitabinaotrials clinici
oterapia di
supporto
Azacitidina, decitabinaotrials clinici
oterapia di
supporto

F.Merli, Ematologia, ASMN RE
Conclusioni
Enorme complessità Progressi nella
comprensione di fisiopatologia e basi molecolari
Processo multistep
Nuovi approcci terapeutici
Trattamenti approvati per MDSAssociazioni
di farmaci
Terapia di supporto
Trapianto allogenico

F.Merli, Ematologia, ASMN RE
Ringraziamenti
Dr.ssa Isabella Capodanno borsista Ematologia ASMN
Dr.ssa Caterina Mammidata manager Progetto Aziendale “Linfocare”


















