
Il management del paziente con Angioedema Ereditario · Bradichinina Callicreina plasmatica HMWK:...
40
LINICAL C PRACTICE I MANUALI DI Il management del paziente con Angioedema Ereditario Definizione e classificazione Manifestazioni cliniche e criteri diagnostici Patogenesi dell’angioedema ereditario Trattamento farmacologico Messaggi per la pratica clinica
Transcript of Il management del paziente con Angioedema Ereditario · Bradichinina Callicreina plasmatica HMWK:...

LINICAL C P R A C T I C EI MANUALI DI
Il management del paziente con Angioedema Ereditario
Definizione e classificazione
Manifestazioni cliniche e criteri diagnostici
Patogenesi dell’angioedema ereditario
Trattamento farmacologico
Messaggi per la pratica clinica

2
© 2011 Effetti Srlvia Gallarate 106 - 20151 Milano
Tutti i diritti di riproduzione, traduzione e adattamento parziale o totale,
con qualunque mezzo sono riservati
Fotografa il QR Code con il tuo telefonino.Scaricherai automaticamente la Pubblicazione in formato pdf

Sommario
3 SOMMARIO
Introduzione Pagina 5
Definizione e classificazione Pagina 7
Manifestazioni cliniche e criteri diagnostici Pagina 14
Patogenesi dell’angioedema ereditario Pagina 22
Trattamento farmacologico Pagina 27
Messaggi per la pratica clinica Pagina 35

4I MANUALI DI CLINICAL PRACTICE
FACULTYMauro Cancian
Clinica Medica I, Dipartimento di Scienze Mediche e Chirurgiche, Università di Padova
Marco CicardiMedicina Interna, Ospedale Luigi Sacco,
Università degli Studi di Milano
Enrico CillariImmunologia Clinica, Unità Operativa di Patologia Clinica,
Azienda Ospedaliera “Cervello” di Palermo
Vincenzo MontinaroDivisione di Nefrologia, Trapianti e Dialisi,
Azienda Ospedaliera Universitaria “Consorziale Policlinico” di Bari
Roberto PerriconeAllergologia e Immunologia Clinica, Università Tor Vergata di Roma
Massimo TriggianiAllergologia e Immunologia Clinica, Università Federico II di Napoli
Andrea ZanichelliDipartimento di Scienze Cliniche, Ospedale Luigi Sacco,
Università degli Studi di Milano

L’angioedema ereditario (AEE) è una malattia ge-netica che colpisce presumibilmente un numerodi persone compreso tra 1:10.000 e 1:50.000.
Rientra, quindi, nella categoria delle malattie rare.Queste sono oltre cinquemila, e complessivamenterappresentano circa il 10% delle patologie umane. Sitratta di affezioni ad andamento cronico, invalidante ocon decorso fatale: è stimato che solamente 1/3 dei pa-zienti potrà essere curato e avere una vita accettabile. Infatti, a causa della loro rarità sono malattie difficilida diagnosticare e da curare, e la loro limitata cono-scenza comporta - per coloro che ne sono affetti e peri loro familiari - notevoli difficoltà nell’individuare iCentri specializzati nella diagnosi e nella cura e,quindi, nell’accesso agli eventuali trattamenti disponi-bili, con il risultato di ritardi inutili nella diagnosi, convisite mediche multiple e prescrizione di farmaci etrattamenti che spesso risultano inappropriati e persinodannosi (Figura 1).Altra criticità è la carenza di informazioni e di stru-menti che aiutino il medico nella formazione e nellapratica clinica: l’identificazione di queste malattie e il
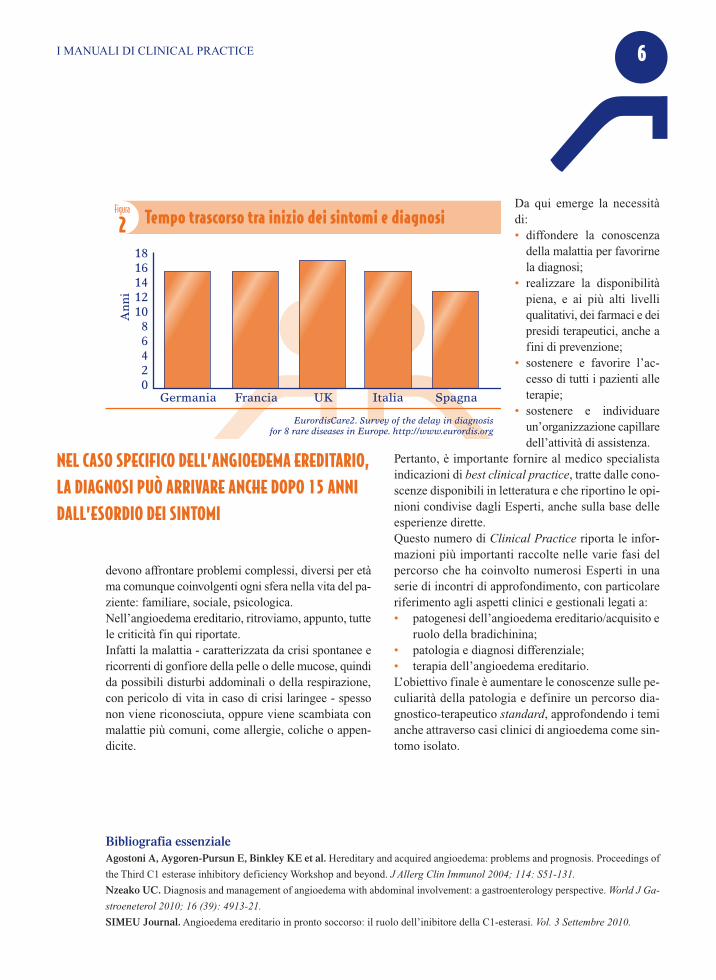
progresso nella ricerca di nuove procedure per la lorodiagnosi e terapia si fondano - né potrebbe essere di-versamente - proprio sulla diffusione delle conoscenzescientifiche già acquisite. Eppure, si stima che media-mente ci vogliano dai 3 ai 5 anni per arrivare a una dia-gnosi di malattia rara e non è raro raggiungere i 7; nelcaso specifico dell’angioedema ereditario, la diagnosipuò arrivare anche dopo 15 anni dall’esordio dei sin-tomi (Figura 2). Appare evidente che, dalla diagnosi all’impostazionedella terapia farmacologica - classificata come orfana,al pari della scarsa assistenza e della gestione dellecomplicanze invalidanti o della disabilità residua - si
I PAZIENTI CON MALATTIE RARE PAGANO RITARDI EANCHE ERRORI DI DIAGNOSI CON LA CONSEGUENZADI RICEVERE SPESSO TRATTAMENTI INAPPROPRIATISE NON DANNOSI
5
Introduzione
INTRODUZIONE
Figura
1 Stato dell’arte malattie rare: EURORDIS survey
Tratto da: The Voice of 12,000 Patients”, EURORDIS, www. Eurordis. org
Il 25% dei pazienti aspetta tra i 5 e i 30 anni per una diagnosi
Il 40% dei pazienti ha una diagnosi errata, con conseguenti interventi medico-chirurgici inappropriati
Al 33% dei pazienti la diagnosi è stata comunicata in termini “non soddisfacenti”; nel 12.5% dei casi in modo “inaccettabile”
Nel 25% dei casi, la natura genetica della malattie non è stata comunicata al paziente o alle famiglie. Nel 50% soltanto, è stato fornito un counselling genetico
Nel 25% dei casi, la diagnosi è stata posta in regioni territoriali diverse dalla propria e il 2% in altro paese
Bibliografia essenzialeAgostoni A, Aygoren-Pursun E, Binkley KE et al. Hereditary and acquired angioedema: problems and prognosis. Proceedings of
the Third C1 esterase inhibitory deficiency Workshop and beyond. J Allerg Clin Immunol 2004; 114: S51-131.
Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Ga-
stroeneterol 2010; 16 (39): 4913-21.
SIMEU Journal.Angioedema ereditario in pronto soccorso: il ruolo dell’inibitore della C1-esterasi. Vol. 3 Settembre 2010.
devono affrontare problemi complessi, diversi per etàma comunque coinvolgenti ogni sfera nella vita del pa-ziente: familiare, sociale, psicologica. Nell’angioedema ereditario, ritroviamo, appunto, tuttele criticità fin qui riportate. Infatti la malattia - caratterizzata da crisi spontanee ericorrenti di gonfiore della pelle o delle mucose, quindida possibili disturbi addominali o della respirazione,con pericolo di vita in caso di crisi laringee - spessonon viene riconosciuta, oppure viene scambiata conmalattie più comuni, come allergie, coliche o appen-dicite.
Da qui emerge la necessitàdi:• diffondere la conoscenzadella malattia per favorirnela diagnosi;
• realizzare la disponibilitàpiena, e ai più alti livelliqualitativi, dei farmaci e deipresidi terapeutici, anche afini di prevenzione;
• sostenere e favorire l’ac-cesso di tutti i pazienti alleterapie;
• sostenere e individuareun’organizzazione capillaredell’attività di assistenza.
Pertanto, è importante fornire al medico specialistaindicazioni di best clinical practice, tratte dalle cono-scenze disponibili in letteratura e che riportino le opi-nioni condivise dagli Esperti, anche sulla base delleesperienze dirette. Questo numero di Clinical Practice riporta le infor-mazioni più importanti raccolte nelle varie fasi delpercorso che ha coinvolto numerosi Esperti in unaserie di incontri di approfondimento, con particolareriferimento agli aspetti clinici e gestionali legati a: • patogenesi dell’angioedema ereditario/acquisito e
ruolo della bradichinina; • patologia e diagnosi differenziale; • terapia dell’angioedema ereditario. L’obiettivo finale è aumentare le conoscenze sulle pe-culiarità della patologia e definire un percorso dia-gnostico-terapeutico standard, approfondendo i temianche attraverso casi clinici di angioedema come sin-tomo isolato.
�EurordisCare2. Survey of the delay in diagnosisfor 8 rare diseases in Europe. http://www.eurordis.org
Figura
2 Tempo trascorso tra inizio dei sintomi e diagnosi
181614121086420
Germania
An
ni
Francia UK Italia Spagna
6I MANUALI DI CLINICAL PRACTICE
NEL CASO SPECIFICO DELL’ANGIOEDEMA EREDITARIO,LA DIAGNOSI PUÒ ARRIVARE ANCHE DOPO 15 ANNIDALL’ESORDIO DEI SINTOMI

L'angioedema ereditario (HAE) è una malattia rarache nella maggior parte dei casi è trasmessa comecarattere autosomico dominante ed è dovuta a una
mutazione del gene che codifica per l’inibitore del C1(C1-INH) sul cromosoma 11. Ad oggi sono state iden-tificate oltre 180 mutazioni del gene; tuttavia circa unquarto dei pazienti non presenta ascendenti affetti,avendo acquisito una mutazione de novo.La mutazione provoca un deficit dell’inibitore fun-zionale del C1, che si estrinseca in un ridotto livellosierico per diminuita sintesi o in una ridotta attivitàdell'inibitore dell'esterasi C1; entrambi le circostanzedeterminano l’attivazione della via classica del com-plemento. L'attivazione incontrollata del comple-mento genera mediatori vasosattivi che induconoedema: il quadro clinico dell'angioedema ereditario è
caratterizzato da edema ricorrente del tessuto sotto-cutaneo - al viso e alle estremità - e delle mucose - piùdi frequente della laringe e dell'intestino (Figura 1). Esistono due tipi principali di angioedema ereditarioda carenza di C1 inibitore: • il tipo I - riscontrato nell’85% dei casi - caratte-
rizzato da concentrazioni marcatamente ridottedell’inibitore (dal 5 al 30% del normale);
• il tipo II - colpisce il 15% dei casi - caratterizzato
7
Definizione eclassificazione
DEFINIZIONE E CLASSIFICAZIONE
Zingale LC, et al. CMAJ 2006
Figura
1 Classificazione dell’angioedema senza orticaria secondo le caratteristiche cliniche o eziopatogenetiche (n=776)
Pazienti Rapporto Età all’esordio, anni Numero % maschi/femmine Mediana Range
Correlato ad un fattore specifico* 124 16 0.51 39 13-76
Malattia/infezione autoimmune 55 7 0.62 49 3-78
ACE-inibitore correlato 85 11 0.93 61 32-84
Da deficit di C1-inibitore 197 25 Ereditario 183 0.88 8 1-34 Acquisito 14 1.8 56.5 42-76
Eziologia non nota (idiopatica) 294 38
Istaminergico 254 0.56 40 7-86
Non-istaminergico 40 1.35 36 8-75
Edema periferico/generalizzato 21 3 0.17 -
*Correlato al cibo, ad un farmaco, a puntura di insetto, ad un allergene ambientale o ad altro stimolo fisico
L'ANGIOEDEMA EREDITARIO COMPARE IN PERSONEPRIVE DELLA CAPACITÀ DI SINTETIZZARE C1 INIBITORE NORMALMENTE FUNZIONANTE

da quantità normali o elevate di una proteina im-munologicamente cross-reagente, ma funzional-mente inattiva.
Essi sono indistinguibili dal punto di vista clinico, madeterminati da mutazioni con effetti differenti. Neltipo I, la mutazione impedisce la sintesi di un prodottoproteico ritrovabile nel palsma, per cui è caratteriz-zato da un basso livello sia antigenico che funzionaledell’inibitore del C1. La mutazione che provoca il tipoII, quasi sempre localizzata nell’esone 8 a livello o invicinanza del sito attivo, consente la produzione diuna proteina misurabile nel plasma ma alterata nellasua funzione: i livelli del C1 inibitore antigenico sononormali o talvolta aumentati, ma i livelli di quello fun-zionale sono ridotti. E’ stato descritto, inoltre, un terzo tipo di angioedemafamiliare (HAE di tipo III), nel quale tuttavia risul-tano normali sia i livelli dell’inibitore antigenico chequelli funzionali. Nei primi casi descritti, la patologia era presente solo
nelle donne e correlava con variazioni fisiologiche ofarmacologicamente indotte dei livelli di estrogeni. Successivamente sono state comunque riconosciute fa-miglie in cui la malattia era presente anche in soggettimaschi. In alcune famiglie la malattia segrega con mu-tazioni del gene per il fattore XII sul cromosoma 5.Pertanto, ad oggi si distinguono due categorie princi-pali di angioedema ereditario:• HAE da deficit genetico del C1 inibitore, che in-
clude il tipo I e II;• HAE con livelli normali di C1 inibitore, tipo III,
che include:- variante con mutazioni del gene che codifica peril fattore XII,- variante idiopatica.
Oltre all’angioedema ereditario, sono state descrittedue forme acquisite, sintomatologicamente indistin-guibili dalla forma ereditaria: una caratterizzata da ca-renza di C1 inibitore, l’altra da alterato catabolismo dipeptidi vasoattivi.
ANGIOEDEMA DA CARENZA ACQUISITA DI C1 INIBITORE (AAE)L’angioedema da carenza acquisita di C1 inibitore,spesso identificato semplicemente con angioedemaacquisito, è una condizione ancora più rara dell’an-gioedema ereditario ed è associata frequentemente a
ESISTONO DUE TIPI DI ANGIOEDEMA EREDITARIO,DETERMINATI DA DIFFERENTI MUTAZIONI DEL GENECHE CODIFICA PER L’INIBITORE C1
8I MANUALI DI CLINICAL PRACTICE
Bas M. et al. Allergy 2007
Arg Arg Val Tyr Ile His Pro Phe His Leu
Arg Arg Val Tyr Ile His Pro Phe
Arg Pro Pro Gly Phe Ser Pro Phe Arg
Phe ArgArg Pro Pro Gly Phe Ser Pro
Figura
2 Ruolo dell’enzima di conversione dell’angiotensina (ACE) nella regolazione dei sistemi renina-angiotensina e callicreina-chinina
Angiotensina I
Angiotensina II
ACE
Recettore AT1
Recettore AT2 HMWK
Bradichinina
Callicreina plasmatica
HMWK: Chininogeno ad alto peso molecolare

malattie linfoproliferative e/o anticorpi anti-C1 inibi-tore, piu raramente è stata descritta nel contesto dimalattie autoimmuni, infezioni, neoplasie. In questi casi la carenza di C1 inibitore è dovuta adaumentato consumo come ben documentato in pre-senza di anticorpi diretti contro il C1 inibitore. La pre-sentazione clinica è simile a quella dell’angioedemaereditario, ma per la trasmissione manca la familia-rità, e generalmente si manifesta dopo la quarta de-cade di vita.
ANGIOEDEMA ACQUISITO FARMACO-INDOTTOAumentate concentrazioni di bradichinina possonoderivare anche da alterazioni del metabolismo dellabradichinina stessa. E’ il caso dell’angioedema daACE-inibitori (ACE-i). Infatti, l’enzima di conver-sione dell’angiotensina (ACE) svolge due funzioni: a) converte l’angiotensina I in angiotensina II, b) inattiva la bradichinina (Figura 2). Pertanto, l’ini-
bizione farmacologica dell’ACE riduce il catabo-lismo della bradichinina.
L’incidenza di angioedema da ACE-i è piuttostobassa: 0.1-1% in pazienti trattati con ACE-i. La po-polazione africana e il sesso femminile sono i più col-
piti. L’insorgenza della sintomatologia non ha un an-damento costante: è più frequente dopo il primo mesedi trattamento, ma sono descritti casi anche dopo 10anni di trattamento. Negli ultimi anni si è comunqueriscontrato un aumento di angioedema da utilizzo diACE-i: circa il 30% dei pazienti visitati al Pronto Soc-corso per angioedema ha sviluppato sintomi legati al-l’utilizzo di ACE-i. Di eziologia diversa è, invece, l’angioedema associatoall’utilizzo di anti-infiammatori non steroidei (FANS)o di salicilati, dovuto all’eccessiva produzione di pro-staglandine, e spesso associato a orticaria.Infine, esistono alcune forme di angioedema indi-stinguibili sintomatologicamente dall’AAE, tuttaviasenza deficit di C1-INH o pregresso utilizzo di far-maci, e non responsivi agli anti-istaminici; per questimotivi vengono indicati con il termine di angioedemaidiopatico non istaminergico (AEI).
9 DEFINIZIONE E CLASSIFICAZIONE
OLTRE ALLE FORME DI ANGIOEDEMA EREDITARIO,SONO STATE DESCRITTE ANCHE FORME ACQUISITECOME QUELLA INDOTTA DA ACE-INIBITORI
Come accade per altri sistemi operanti nell'organismo,quali quello della coagulazione e quello delle chinine,l’attivazione del complemento avviene con un mecca-nismo detto a cascata, per attivazione sequenziale deivari componenti complementari che circolano informa inattiva. L’attivazione della via classica dipende dalla intera-zione di tre proteine del complemento, C1, C4 e C2,con il complesso antigene-anticorpo. La reazione inizia con il legame del C1 alle immuno-globuline di tipo IgG1, IgG3 e IgM fissate a un anti-gene multivalente. Il primo componente delcomplemento (C1) è costituito da tre sub componenti,C1q, C1r e C1s.
Il C1q è composto da sei catene disposte radialmentea ombrello e svolge un'azione di ricognizione legan-dosi specificamente alla regione Fc delle immunoglo-buline. Il C1r e il C1s svolgono, invece, un’azioneenzimatica: il legame di due o più catene di C1q alleimmunoglobuline attiva il C1r, che a sua volta attiva ilC1s. C1r e C1s scindono enzimaticamente le componentiC4 e C2 della cascata complementare - il C4 in duefrazioni: il C4a e C2b che rimangono in circolo e ilC4b e C2a che si legano covalentemente alla mem-brana cellulare. C1s attiva il C2 in due frazioni: la C2ae la C2b. La C2b rimane in fase fluida mentre la C2asi lega al C4b, dando luogo al complesso C4b-C2a.
C1- inibitore e l’attivazione del complemento

Questo complesso costituisce l'enzima C3 convertasidella via classica, capace di legarsi al C3 e di scindereC3 in C3a (anafilotossina) e in C3b, che si lega allamembrana cellulare, formando la C5 convertasi. Tale cascata è regolata da molecole inibitrici della viaclassica: il C1 inibitore (C1-INH), il C4 binding pro-tein (C4bp), e la serina-proteasi, detta fattore I. Il C1-INH è una SERPINa - acronimo di SERine Pro-tease INhibitor, ossia un inibitore delle proteasi seri-niche, e agisce bloccando le attività enzimatiche delcomplesso C1s e C1r; inoltre, è il più importante ini-bitore fisiologico del sistema di contatto della coagu-lazione. Il C4bp inibisce la via classica del complemento pro-vocando la dissociazione del dimero C4b,2a e facili-tando l’azione inibitrice del fattore I sul C4b, da cuideriva il C4bi (inattivato).Le restanti componenti della cascata complementare
(C5, C6, C7, C8, C9) si legano in successione allamembrana cellulare, inserendosi nel contesto dellostrato lipidico della parete cellulare. L'ultimo compo-nente della cascata, il C9, ha la capacità di polimeriz-zare nel punto in cui si è inserito, formando dei porinella membrana cellulare, attraverso i quali acqua eioni hanno libero accesso all'interno della cellula, de-terminandone dapprima il rigonfiamento osmotico e,poi, la lisi.Ancora, la proteolisi dei componenti C3, C4 e C5 ge-nera i relativi frammenti, che si assemblano tra loronelle varie fasi di attivazione del complemento e iframmenti C3a, C4a e C5a, ad attività anafilotossini-nica, che inducono una risposta infiammatoria fissan-dosi ai relativi recettori di membrana presentiprevalentemente sui mastociti, sui neutrofili, sulle cel-lule endoteliali.Il C1 inibitore regola tre sistemi a cascata: la via clas-
�BL Zuraw. N Engl J Med 2008
+
C2
Figura
1 Ruolo del C1 inibitore
Via classica del complemento Via d’attivazione del contatto
Via fibrinolitica
Proteasi C1 del
complemento
Attivazione
AssemblaggioC3 convertasi
C1INH
C1qC1r C1r
C1sC1s
C1qC1r C1r
C4bC2a
C4C1sC1s
C1qC1r C1r
C1sC1s
C1INH
C1INH
C1INH
Icatibant
C1INH
+
+ +
+
Fattore XI
Chininogeno ad altopeso molecolare
FattoreXIa
Fibrina
Degradazionedella fibrina
Chininogeno ad altopeso molecolare
Chininogeno ad altopeso molecolare
FattoreXII
Precallicreina
Chininogeno ad altopeso molecolare
Callicreina
Chininogeno ad altopeso molecolare
Recettore B2della bradichinina
Membranacellulare
Callicreina
Plasmina
Plasminogeno
C1INH
C1INH eecallantide
C1INH eecallantide
Bradichinina
+
+
FattoreXIIa
++
+
10I MANUALI DI CLINICAL PRACTICE

11 DEFINIZIONE E CLASSIFICAZIONE
sica del complemento; il sistema callicreina-chininadel sistema di contatto; la via fibrinolitica. Nell’attivazione della via classica del complemento,la proteasi C1 è attivata e, quindi, assembla la C3 con-vertasi. Il C1 inibitore svolge un ruolo primario nell'attiva-zione della via classica del complemento, nella for-mazione di chinina del sistema di contatto e nellafibrinolisi (figura 1); la disfunzione causa perdita del-l'omeostasi con disegolazione del complemento e con-seguente attivazione del sistema di contatto. In assenza della funzionalità di C1-INH, l'attivazionedi C1 porta a un'incontrollata attività C1, con esauri-mento di C4 e C2 e rilascio di un peptide vasoattivo -la bradichinina - da C2.
L'edema episodico, localizzato, che non conserva l'im-pronta, deriva infatti dagli effetti vasodilatatori dellabradichinina sulle venule postcapillari.Nella via di attivazione da contatto, tracce di fattoreXIIa attivano altro fattore XII e la precallicreina. Il fat-tore XIIa attivato a sua volta attiva il fattore XI in fat-tore XIa, portando ad aumento della formazione difibrina. Il fattore XIIa attivato e la callicreina si attivano reci-procamente e, quindi, la callicreina plasmatica cliva ilchininogeno ad alto peso molecolare con rilascio dibradichinina.Lungo la via fibrinolitica, infine, il plasminogeno at-tivato a plasmina (una serina-proteasi) dal fattore XII,determina la lisi della fibrina.
• Donna di 37 anni affetta da crisi di angioedemacutaneo e delle mucose del volto, scarsamenteresponsive a steroidi e anti-istaminici, e scatenateda microtraumi cutanei: il lavarsi il viso o i ca-pelli o lo struccarsi comportano la comparsa diangioedemi dolorosi e urenti.
• Familiarità per angioedema assente. La sintoma-tologia è tale da compromettere severamente lecapacità relazionali: la paziente non si lava per ti-more di angioedemi, non esce di casa, ha fotofo-bia con conseguente incupimento del tonodell’umore.
• Gli esami di laboratorio rivelano valori anticor-pali alterati [IgG1mg/dl = 261 (vn.580-700);IgG2=258 (vn.290-350); IgG3 = 13 (vn.68-82);IgG4 = 9 (vn.29-35); sIgA = 1.82 (5-20)], con al-terato rapporto CD4/CD8 e, a sorpresa, valoridell’angiotensin converting enzyme (ACE) au-mentati [(ACE = 29.8 (vn.<20)]. Tuttavia la pa-ziente, che è, inoltre, intollerante al glutine e aiderivati del latte, mostra valori di C1 inibitorefunzionale e quantitativo normali, eccetto un mo-desto consumo del complemento nelle fasi acutedell’attacco.
CASO CLINICOQualità della vita e patologie associate ad angioedema ereditario
Figura
1 Ruolo dell’enzima di conversione dell’angiotensina (ACE)
Induce la conversione di angiotensinogeno in angiotensina I e II
Induce la degradazione della bradichinina in peptidi inattivi
Gli ACE-inibitori riducono chemotassi neutrofila polmonare in evidenze sperimentali (di stress respiratorio acuto, fibrosi polmonare)
Gli ACE-inibitori riducono l’infiltrazione delle cellule T helper CD4+ esprimenti IL-17 (Th17) nelle malattie autoimmuni, come l’encefalomielite autoimmune (EAE) e la gastrite autoimmune

CommentoLe orticarie fisiche costituiscono il 15-20% di tuttele forme di orticarie/angioedema ricorrenti. Hanno incomune la caratteristica di essere scatenate da unacausa fisica nota: comparsa dopo esercizio fisico; osfregamento cutaneo (dermografismo sintomatico);o dopo esposizione della cute al caldo, al freddo, allaluce solare, a vibrazioni o pressioni; o dopo applica-zioni di acqua sulla pelle. Si ritiene che alla basedelle manifestazioni suddette siano coinvolte le ma-stcellule con liberazione di istamina (Figura 1). A riguardo, le citochine sembrano svolgere un ruolochiave: l’espressione di interleuchina 17 (IL-17) èstata associata a molte malattie autoimmuni.
Essa è stata riscontrata nel siero e negli organi bersa-glio di pazienti affetti da artrite reumatoide, sclerosimultipla, malattie infiammatorie croniche intestinali(MICI) e lupus eritematoso sistemico (LES), oltreche nel liquido sinoviale di pazienti affetti da artritedi Lyme. Nel morbo di Crohn, cellule producenti solo IL-17 oIL-17 e IFN-a, sono state identificate nel sangue pe-riferico e nella mucosa intestinale. Anche nella granulomatosi di Wegener è presenteuna risposta TH17 aumentata. Infine sono state regi-strate alte concentrazioni di IL-17 e IL-23 nel siero,nella saliva e nelle ghiandole salivari dei pazienti af-fetti da sindrome di Sjogren.
12I MANUALI DI CLINICAL PRACTICE
Bibliografia essenzialeAbbas AK, Lichtman AH, Pober JS. Immunologia cellulare e molecolare. Padova, Piccin, 2002.
Agostoni A, Aygoren-Pursun E, Binkley KE et al. Hereditary and acquired angioedema: problems and prognosis: proceedings of
the third C1 esterase inhibitory deficiency workshop and beyond. J Allerg Clin Immunol 2004; 114: S51-131.
Banerji A, Clark S, Blanda M et al.Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioe-
dema who present to the emergency department. Ann Allergy Asthma Immunol. 2008; 100(4): 327-32.
Bas M, Adams V, Suvorava T et al. Nonallergic angioedema: role of bradykinin. Allergy 2007; 62: 842-856.
Binkley KE, Davis A. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioe-
dema. J Allergy Clin Immunol. 2000; 106/3: 546-550.
Brickman CM, Tsokos GC, Balow JE et al. Immunoregulatory disorders associated with hereditary angioedema. I. Clinical ma-
nifestations of autoimmune disease. J Allergy Clin Immunol 1986; 77(5): 749-57.
Bork K, Barnstedt SE, Koch P, et al. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet 2000; 356(9225):
213-7.
Bork K.Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy, Asthma & Clinical Immunology 2010;
6: 15.

13 DEFINIZIONE E CLASSIFICAZIONE
Bowen T, Cicardi M, Bork K et al. Hereditary angioedema: a current state of the art review, Vii: Canadian Hungarian 2007 Inter-
national Consensus algorithm for the diagnosis, therapy and management of hereditaru angioedema. Ann Allergy Asthma Immunol
2008; 100: S30-40.
Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor- associated angioedema. Immunol Allergy Clin North Am.
2006; 26: 725-737.
Canessa C, Vierucci A, Azzari C. Cellule TH17 nella patologia umana: buone o cattive? Rivista di Immunologia e Allergologia Pe-
diatrica 2010; 1: 19-26. www.riap.it.
Crepaldi G, Baritussio A. Trattato di medicina interna. Piccin 2002; 3: 200-210.
Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin N Am 2006; 26: 739-751.
HAEdb-C1 inhibitor gene mutation database: http://hae.enzim.hu/stat.php
Gompels MM, Lock RJ, Abinun M et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol 2005; 139: 379-394.
Grigoriadou S et al. Clinical Immunology Review Series: An approach to the patient with angioedema. Clin Exp Immunol. 2009;
155(3): 367-77. Review.
Nielsen EW, Gran JT, Straume B et al. Angioedema ereditario: nuove osservazioni cliniche e lo screening autoimmune, integrare
e analisi callicreina-chinina. J Intern. 1996; 239(2): 119-30.
Platten M, Youssef S, Eun Mi Hur et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modula-
tes TH1- and TH17-mediated autoimmunity. PNAS 2009; 106: 35
Polati E, Sette P, Parolini M. Sepsi: miriadi di risposte, aspetti controversi, soluzioni possibili. Di A Galli. Springer 2004: 55-70
Stegbauer J, Lee DH, Seubert S, Ellrichmann G et al. Role of the renin-angiotensin system in autoimmune inflammation of the
central nervous system. Proc Natl Acad Sci USA. 2009; 106(35): 14942-7. Epub 2009 Aug 19
Zingale LC, Beltram L, Zanichelli A et al.Angioedema without urticaria: a large clinical survey. CMAJ. 2006; 175(9): 1065-70.
Zuraw BL. Hereditary Angioedema. N Engl J Med 2008; 359: 1027-1036.

Attualmente in Italia circa 1500 persone sono af-fette da angioedema ereditario, ma meno del 50%di queste sa di essere malata, e spesso la diagnosi
viene effettuata a distanza, anche di 15 anni, dal-l’esordio della sintomatologia. Le sedi maggiormente colpite sono la cute del viso odelle estremità, la mucosa gastroenterica e le primevie aeree (Figura 1).L’angioedema è provocato da un edema profondo deltessuto sottocutaneo o della mucosa dovuto a un au-
mento transitorio di permeabilità dei vasi sanguignidel derma profondo; l’edema pruriginoso della cute- tipico dell’orticaria - è dovuto, invece, a un aumentotransitorio di permeabilità dei piccoli vasi sanguignidel derma superficiale (Figura 2). L’angioedema ereditario ha un andamento cronico, ein almeno il 50% dei casi le manifestazioni clinichecompaiono in età pediatrica - raramente prima dei 3anni - si aggravano in età puberale e persistono pertutta la vita, con importanti ricadute sulla qualità divita del malato. I pazienti non trattati hanno in mediaun attacco ogni 7-14 giorni, ma la frequenza è moltovariabile nei diversi casi.La forma cutanea ha tipicamente un andamentolento: la deformazione del distretto colpito aumentaprogressivamente fino a raggiungere l’estensionemassima in 12-36 ore, per poi regredire completa-mente in un tempo che varia tra 2 e 4 giorni.
14
Manifestazioni cliniche e criteri diagnostici
I MANUALI DI CLINICAL PRACTICE
L’ANGIOEDEMA INTERESSA SOPRATTUTTO LA CUTE DEL VISO E DELLE ESTREMITÀ, LA MUCOSA GASTROENTERICA E LE PRIME VIE AEREE
Bork K, et al. Am J Med 2006
Figura
1 Sede e percentuale di frequenza di angioedema nei vari organi e distretti
100
80
60
40
20
0
% p
azie
nti (
n=20
1)
Cute
Gastroente
rico
Laringe
Ugola
Lingua
Encefa
lo
Vescica
Torace
Musc
oli
Artico
lazioni
Rene
Esofa
go
Analisi per paziente100% 97%
54%
22%9% 8% 5% 5% 4% 4% 4% 2%

Più spesso viene interessato un singolo distretto, manon è infrequente una localizzazione multipla o unandamento migrante.L'edema cutaneo (Figura 3) non è accompagnato daprurito, discromia, arrossamento o orticaria - mal-grado fugaci rash eritematosi, di solito con aspetto acarta geografica (eritema marginato) possano prece-dere la comparsa del vero angioedema - e spesso èsenza grave dolore. L’edema della parete intestinale (Figura 4) può cau-sare forti crampi addominali, talvolta associati a vo-mito e/o diarrea. Il quadro clinico, in assenza diedema del sottocutaneo, è difficilmente distinguibileda un attacco di addome acuto, e può accompagnarsia leucocitosi e a versamento peritoneale visibile al-l’esame ecografico, tale da esporre il paziente a inu-tili interventi chirurgici. L’evento più temuto è, tuttavia, l'edema della glottide(Figura 5), spesso indistinguibile da un attacco ana-filattico e, peraltro, non responder alla comune tera-pia farmacologica antiallergica. Tutte le localizzazioni orali e faringee, nonché quelleesterne del collo, devono essere considerate ad altorischio per evoluzione in edema della glottide e,quindi, in insufficienza respiratoria acuta. Queste localizzazioni hanno, come tutte le altre, unaprogressione lenta nell’arco di alcune ore, ma il pas-saggio dai sintomi generici di edema di faringe/cavoorale (disfagia, senso di corpo estraneo, alterazioninel timbro della voce) all’insufficienza respiratoriaacuta può essere difficilmente prevedibile ed estre-mamente repentino. Si stima che più della metà deisoggetti abbia sviluppato un episodio di edema la-
15
L’EDEMA CUTANEO È DISTINGUIBILE DALL’ORTICARIA, MENTRE L’EDEMA DELLA PARETE INTESTINALE PUÒ MIMARE UN ATTACCO DI ADDOME ACUTO ED ESPORRE IL PAZIENTE A INUTILI INTERVENTI CHIRURGICI
Figura
2 Struttura della cute
Angioedema
Orticaria
Figura
3 Differenza tra angioedema e orticaria
Figura
4 Angioedema della mucosa intestinale
Wong RC et al. Gastroenterology 1999
MANIFESTAZIONI CLINICHE E CRITERI DIAGNOSTICI

ringeo, e in circa il 30% dei casi non diagnosticati èstato causa di morte per asfissia. Infine, nell’1-2%dei casi, possono essere presenti sintomi neurologicio essere coinvolte altre sedi: l’apparato urinario, l’ap-parato muscolo scheletrico, le sierose toraciche ol’apparato genitale. Spesso gli attacchi presentano undecorso prevedibile: sono preceduti da prodromi -sensazione di formicolio - e l’edema si aggrava len-tamente ma inesorabilmente nel corso delle prime 24ore per poi gradualmente ridursi e scomparire nelle48-72 ore seguenti; inoltre, possono verificarsi in se-guito a traumi o piccoli interventi chirurgici, dopo unesercizio fisico intenso, durante le mestruazioni o peruno stress emotivo. Pertanto, sulla base della sola sintomatologia edemi-gena possono essere incluse condizioni eterogenee e
16I MANUALI DI CLINICAL PRACTICE
L’EDEMA DELLA GLOTTIDE, SPESSO INDISTINGUIBILE DA UN ATTACCO ANAFILATTICO, È NON RESPONDER ALLA TERAPIA ANTIALLERGICA
Cicardi M, Zanichelli A. Intern Emerg Med Published online 22 May 2010, DOI: 10.1007/s11739-010-0408-3
Figura
6 Angioedema
Angioedema
Con orticaria Senza orticariaDovuto a cause specifiche
(farmaci, cibi, ecc)
Risoluzione dopo profilassi con antistaminico
Non risoluzione dopo profilassi con antistaminico
Deficit di C1 inibitore
Dovuto ad ACE-inibitori
Idiopaticonon-istaminergicoAngioedema idiopatico
istaminergico
Dovuto aparassiti, infezioni,
malattie autoimmuni
Figura
5 Laringoscopia durante e dopo angioedema dell’epiglottide
Tsunoda K et al. Laryngoscope 2000
TUTTE LE LOCALIZZAZIONI ORALI E FARINGEE DEVONO ESSERE CONSIDERATEAD ALTO RISCHIO PER EVOLUZIONE IN EDEMA DELLA GLOTTIDE E, QUINDI, IN INSUFFICIENZA RESPIRATORIA ACUTA

con patogenesi molto diversa. Tuttavia alcune sem-plici caratteristiche saranno utili a orientare la dia-gnosi differenziale (Figura 6):• insorgenza acuta/cronica• con/senza orticaria• con/senza fattori scatenanti.L’angioedema con orticaria è una condizione acutadi frequente riscontro nella popolazione generale, tal-volta recrudescente, e sempre direttamente correlataa un evento scatenante. Il mediatore principale delle manifestazioni è l’ista-mina, in grado di determinare angioedema con orti-caria, e nei casi più gravi anafilassi. L’angioedema senza orticaria è più frequentementeuna condizione cronica, e raccoglie un gruppo etero-geneo di malattie, nelle quali tuttavia la patogenesinon è istaminergica, ma ricollegabile all’azione dellabradichinina.L’eterogeneità clinica e la facile sovrapposizionedella sintomatologia con fenomeni più propriamenteallergici, fa sì che in Italia la condizione non abbiauno specialista medico di riferimento, venendo ge-stita prevalentemente dai medici del Pronto Soccorso,e solo in piccola percentuale da dermatologi, aller-gologi e internisti. Questo si discosta da quanto avviene nel resto d’Eu-ropa, in particolare al Nord, dove sono maggiormentecoinvolti i dermatologi. Nel contempo, la scarsa specificità dei sintomi fa sìche la condizione possa a lungo non essere inqua-drata come entità nosologica specifica, e rimanere
come interrogativo diagnostico-terapeutico, ovvia-mente a rischio di gravi conseguenze, soprattutto incondizioni di emergenza. Da qui l’importanza di unadiagnosi tempestiva e corretta di angioedema eredi-tario (Figura 6).
CRITERI DIAGNOSTICI La diagnosi di angioedema ereditario si avvale di unaaccurata anamnesi familiare, di criteri clinici e di cri-teri di laboratorio:• storia familiare conclamata di angioedema da ca-
renza di C1 inibitore;• angioedema sottocutaneo, non pruriginoso, non
eritematoso, autolimitante, solitamente ricorrentee di lunga durata (più di 12 ore), con nessuna oscarsa orticaria, talvolta preceduto da eruzionecutanea a tipo eritema marginato;
• dolori addominali ricorrenti (spesso con vomitoe/o diarrea), senza altra causa, a risoluzione spon-tanea in 12-72 ore;
• edemi ricorrenti laringei;• livelli antigenici di C1 inibitore <50% del nor-
17
LA DIAGNOSI DI ANGIOEDEMA EREDITARIO SI AVVALE DI UNA ACCURATA ANAMNESI FAMILIARE,DI CRITERI CLINICI E DI CRITERI DI LABORATORIO
Cicardi M, Zanichelli A. Intern Emerg Med Published online 22 May 2010, DOI: 10.1007/s11739-010-0408-3
Figura
7 Diagnosi biochimica di angioedema dovuto a deficit ereditario (HAE) o acquisito (AAE) di C1 inibitore
Tipo di C1-INH C1-INH C4 C1q Anticorpiangioedema quantitativo funzionale quantitativo quantitativo anti C1-INH
HAE di tipo I Basso Basso Basso Normale Negativi
HAE di tipo II >50% Basso Basso Normale Negativi
AAE Basso/normale Basso Basso Basso Alto titolo nel 70% nel 70% dei pazienti dei pazienti
Livelli bassi= al di sotto del 50% dei valori normali
MANIFESTAZIONI CLINICHE E CRITERI DIAGNOSTICI

male in 2 determinazioni separate e dopo il primoanno di vita;
• livelli di attività funzionale di C1 inibitore <50%del normale in 2 determinazioni separate e dopoil primo anno di vita;
• mutazione del gene di C1 inibitore che altera lasintesi e/o la funzionalità della proteina.
Quest’ultima metodica va riservata solo ai casi di as-senza di familiarità: lo screening genetico è piutto-sto complesso, infatti in mancanza di regionispecifiche o di mutazioni specifiche responsabili deldeficit di C1-INH l’intero gene deve essere sequen-ziato per arrivare alla diagnosi.
La diagnosi viene stabilita in presenza di almeno uncriterio clinico e di almeno un criterio di laboratorio,mentre la presenza di storia familiare conclamata diangioedema da carenza di C1 inibitore associata a uncriterio di laboratorio permette di definire il pazienteportatore asintomatico.Inoltre, va ricordato che, in assenza della funzionalitàdi C1-INH, l'attivazione di C1 porta a un'incontrol-lata attività C1, con esaurimento delle frazioni C4 eC2. Pertanto il dosaggio del C4 è un utile parametroindiretto di carenza di C1-INH (Figura 7). Per contro, nel caso di episodi di anafilassi istamino-mediata, i valori della triptasi sierica rimangono ele-vati per circa 6 ore dopo l’episodio acuto e testimo-niano l’attivazione (degranulazione) dei mastocititissutali, le cellule effettrici primarie delle reazioniallergiche. Infine, l’analisi genetica per l’individuazione di mu-tazioni nel gene che codifica per FXII può essered’ausilio per la diagnosi e nel definire la trasmissibi-lità dell’angioedema di tipo III con C1-INH normale.
18I MANUALI DI CLINICAL PRACTICE
L’anafilassi sistemica rappresenta l’evento più dram-matico e potenzialmente letale delle manifestazioniallergiche; è una reazione immunologica di tipo Imediata dall’interazione tra anticorpi specifici dellaclasse IgE e un antigene. L’interazione tra le IgE - presenti sui recettori(FceRI) delle cellule effettrici primarie delle rea-zioni allergiche (mastociti tissutali e basofili circo-lanti) - e l’antigene (allergene) determina una seriecomplessa di eventi biochimici che inducono il rila-scio di mediatori chimici vasoattivi, principalmenteistamina, triptasi, leucotrieni, prostaglandine, plate-let activating factor. Anche meccanismi non IgE-mediati possono in-durre l’anafilassi: i meccanismi alla base di questereazioni includono l’attivazione diretta di mastocitie basofili non mediata da anticorpi di classe E, l’at-tivazione del complemento o di proteine plasmati-che della coagulazione, o del sistema
callicreina-chinina e meccanismi neuro-psicogeni.L’effetto del rilascio di istamina e di altri mediatorivasoattivi liberati dai mastociti tessutali e dai baso-fili circolanti determina un rapido passaggio di li-quidi - fino al 50% del volume circolante in 10minuti - dallo spazio intravascolare a quello extra-vascolare, tale da indurre grave ipotensione e col-lasso, associato a possibili aritmie e ischemiamiocardica, oppure compromissione della funzioneventilatoria per broncocostrizione delle vie aeree in-feriori o edema a carico di quelle superiori. Clinicamente si distingue:• una sintomatologia cutanea caratterizzata da
prurito, eritema, orticaria e angioedema, concoinvolgimento delle mucose adiacenti: pruritodel palato, prurito e parestesia del faringe, pru-rito ed edema delle mucose genitali;
• una sintomatologia a carico dell’apparato respi-ratorio: rinorrea, disfonia, raucedine, edema la-
LA DIAGNOSI DI ANGIOEDEMA EREDITARIO È STABILITA IN PRESENZA DI ALMENO UN CRITERIOCLINICO E DI ALMENO UN CRITERIO DI LABORATORIO
Anafilassi: patogenesi e sintomatologia

19 MANIFESTAZIONI CLINICHE E CRITERI DIAGNOSTICI
• Una donna di 46 anni viene visitata in ProntoSoccorso per un episodio di edema del volto, concoinvolgimento delle palpebre e della lingua, in-sorto da più di 24 ore e refrattario al trattamentodomiciliare con corticosteroidi per via parente-rale.
• In passato la paziente era stata più volte ricove-rata per episodi dolorosi addominali, e in una cir-costanza aveva effettuato una laparotomiaesplorativa per sospetta endometriosi, peraltronon riscontrata.
• L’anamnesi familiare viene ritenuta non signifi-cativa. Al Pronto Soccorso i valori dell’emogasa-nalisi risultano nella norma. Viene instaurata unaterapia a base di cortisonici e prometazina. A di-stanza di circa un’ora la sintomatologia appareleggermente migliorata, tuttavia si procede a unricovero presso il reparto di medicina/dermatolo-gia. Poco dopo il ricovero la donna comincia emanifestare segni di stress respiratorio che diven-tano presto chiaramente indicativi di edema dellaglottide; viene subito somministrata adrenalina,ma senza alcun beneficio. La paziente entra incoma che rimane irreversibile.
CommentoQuesto caso è esemplificativo dell’andamento dellasintomatologia e della necessità di un tempestivo in-quadramento diagnostico, tanto più in presenza diuna sintomatologia subdola, che può tuttavia rapida-mente evolvere verso situazioni di emergenza - neisoggetti con angioedema ereditario l’edema dellaglottide, per quanto in genere molto meno repentinoche nell’anafilassi, può essere rapidamente progres-sivo soprattutto nel caso siano interessate regioniadiacenti (la paziente aveva una edema della lingua). Viene evidenziato l’iter diagnostico classico dell’an-gioedema, dove spesso tra l’esordio della malattia ela diagnosi intercorre diverso tempo, con il rischio diinterventi chirurgici inutili. Va rimarcato, che, qua-lora in corso di angioedema periferico dovesse insor-gere l’edema della laringe, bisogna mettersiimmediatamente nelle condizioni di intervenireanche in modo invasivo con intubazione o tracheoto-mia. Nel caso citato, la sintomatologia non è dipesadal rilascio di istamina, per cui la terapia antianafi-lattica non ha alcun effetto (vedi box anafilassi). Da qui l’imperativo della diagnosi corretta e della di-sponibilità di farmaci salva-vita.
CASI CLINICI1. Valutazione del paziente affetto da angioedema: dalla sintomatologia all’emergenza
• Il caso tratta di un soggetto di sesso maschile di38 anni, affetto da angioedema ereditario e confamiliarità per la condizione, trasportato d’ur-genza al Pronto Soccorso in preda a nausea, vo-mito, dolori addominali trafittivi e refrattari aterapia sintomatica.
• Gli esami di laboratorio effettuati all’ingresso
mostrano: emoconcentrazione, leucocitosi neutrofila, proteina C-reattiva (PCR) aumentata, funzionalità epato-pancreatica nella norma.
• Durante il ricovero al Pronto Soccorso la sinto-matologia edematosa peggiora con la comparsadi edema della glottide.
2. Urgenza chirurgica o angioedema addominale?
ringeo, tosse, ostruzione laringea, broncospa-smo, fino all’arresto respiratorio;
• un interessamento dell’apparato gastrointesti-nale: crampi addominali, nausea, vomito, incon-tinenza fecale, diarrea, con o senza incontinenza
urinaria e crampi uterini;• un coinvolgimento dell’apparato cardiovascolare
con sintomi e segni di diversa gravità: tachicar-dia, alterazioni pressorie, aritmia, shock, fino al-l’arresto cardiaco.
20I MANUALI DI CLINICAL PRACTICE
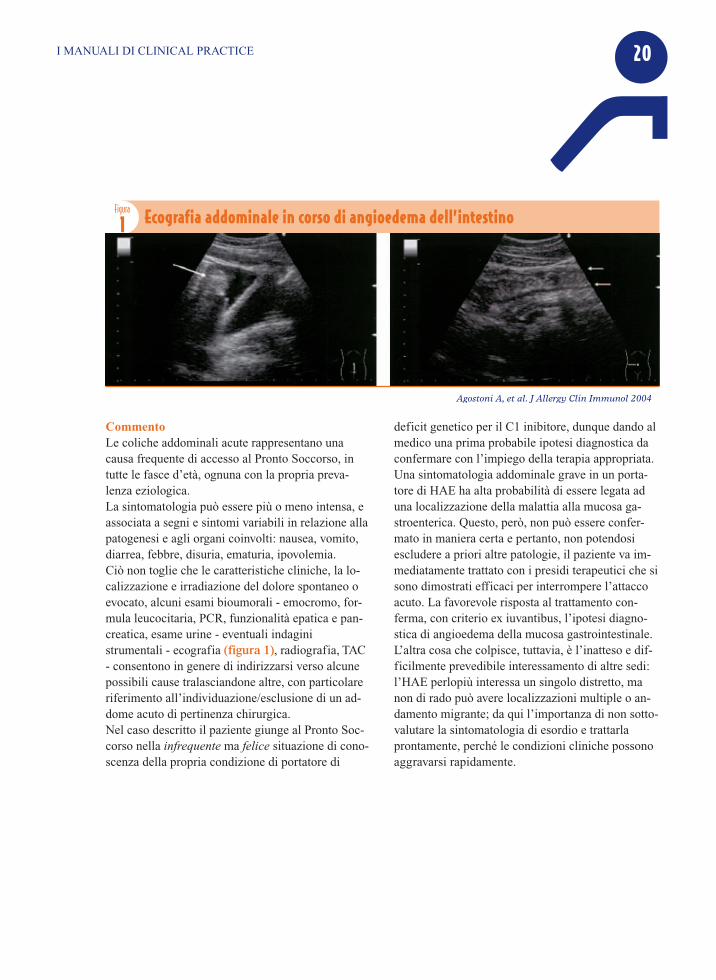
CommentoLe coliche addominali acute rappresentano unacausa frequente di accesso al Pronto Soccorso, intutte le fasce d’età, ognuna con la propria preva-lenza eziologica. La sintomatologia può essere più o meno intensa, eassociata a segni e sintomi variabili in relazione allapatogenesi e agli organi coinvolti: nausea, vomito,diarrea, febbre, disuria, ematuria, ipovolemia. Ciò non toglie che le caratteristiche cliniche, la lo-calizzazione e irradiazione del dolore spontaneo oevocato, alcuni esami bioumorali - emocromo, for-mula leucocitaria, PCR, funzionalità epatica e pan-creatica, esame urine - eventuali indaginistrumentali - ecografia (figura 1), radiografia, TAC- consentono in genere di indirizzarsi verso alcunepossibili cause tralasciandone altre, con particolareriferimento all’individuazione/esclusione di un ad-dome acuto di pertinenza chirurgica. Nel caso descritto il paziente giunge al Pronto Soc-corso nella infrequente ma felice situazione di cono-scenza della propria condizione di portatore di
deficit genetico per il C1 inibitore, dunque dando almedico una prima probabile ipotesi diagnostica daconfermare con l’impiego della terapia appropriata.Una sintomatologia addominale grave in un porta-tore di HAE ha alta probabilità di essere legata aduna localizzazione della malattia alla mucosa ga-stroenterica. Questo, però, non può essere confer-mato in maniera certa e pertanto, non potendosiescludere a priori altre patologie, il paziente va im-mediatamente trattato con i presidi terapeutici che sisono dimostrati efficaci per interrompere l’attaccoacuto. La favorevole risposta al trattamento con-ferma, con criterio ex iuvantibus, l’ipotesi diagno-stica di angioedema della mucosa gastrointestinale.L’altra cosa che colpisce, tuttavia, è l’inatteso e dif-ficilmente prevedibile interessamento di altre sedi:l’HAE perlopiù interessa un singolo distretto, manon di rado può avere localizzazioni multiple o an-damento migrante; da qui l’importanza di non sotto-valutare la sintomatologia di esordio e trattarlaprontamente, perché le condizioni cliniche possonoaggravarsi rapidamente.
Figura
1 Ecografia addominale in corso di angioedema dell’intestino
Agostoni A, et al. J Allergy Clin Immunol 2004

21 MANIFESTAZIONI CLINICHE E CRITERI DIAGNOSTICI
Bibliografia essenzialeAgostoni A, Aygoren-Pursun E, Binkley KE et al. Hereditary and acquired angioedema: problems and prognosis. Proceedings of
the Third C1 esterase inhibitory deficiency Workshop and beyond. J Allerg Clin Immunol 2004; 114: S51-131.
Bowen, T, Cicardi M, Farkas H et al. 2010 international consensus algorithm for the diagnosis, therapy and management of here-
ditary angioedema. Allergy Asthma Clin Immunol. 2010; 6(1): 24.
Cicardi M, Zingale LC, Zanichelli A et al.Angioedema ereditario da carenza di C1 inibitore. Consensus Document italiano per la
diagnosi e la terapia. Giornale It di Allergologia ed Immunol Clin 2008; 18(4): 114-122.
Cugno M, Zanichelli A, Foieni F et al. C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress.
Trends Mol Med 2009; 15(2): 69-78.
de Crescenzo G. Trattamento farmacologico delle reazioni anafilattiche ed anafilattoidi in Reazioni anafilattiche ed anafilattoidi.
G. Marone. Eds Springer 1997: 115-131.
Farkas H. Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma Clin Immunol 2010; 6: 2-10.
Kaplan AP, Greaves MW.Angioedema. J Am Acad Dermatol 2005; 53: 373-88.
Marone G. “Epidemiologia e rilevanza delle reazioni anafilattiche” in “Reazioni anafilattiche ed anafilattoidi. Patogenesi, preven-
zione, diagnosi e terapia” Eds Springer 1997: 1-12.
Marone G. Human Basophils and Mast Cells: Biological Aspects. Karger, Basel 1995: 28.
Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Ga-
stroeneterol 2010; 16(39): 4913-21.
Rook A, Wilkinson DS, Ebling FJB, Champion RH, Burton JL. Textbook of Dermatology. Blackwell Scientific Publications.
Fourth edition.
Temiño VM, Stokes Peebles R. The Spectrum and Treatment of Angioedema. The American Journal of Medicine 2008; 121: 282-
286.
Tsunoda K, Hozaki F, Aikawa J. Angioedema for the epiglottis associated with enalapril. Laryngoscope 2000; 110(12): 2147-8.
Wong RC, Phan TG, Adelstein S et al. Image of the month. Gastroenterology 1999; 514: 77.
Zingale LC, Castelli R, Zanichelli A et al. Acquired deficiency of the inhibitor of the first complement component: presentation,
diagnosis, course, and conventional management. Immunol Allergy Clin North Am 2006; 26: 669-690.

Il medico tedesco Heinrich Irenaeus Quincke fu ilprimo a descrivere, nel 1882, un quadro clinico cheegli indicò con il proprio nome, e che a distanza di
pochi anni il dott William Osler chiamerà edema an-gioneurotico ereditario. Da allora numerosi progressi sono stati compiuti nellacomprensione dei meccanismi patogenetici dell’an-gioedema bradichinina-mediato, in particolare delruolo del C1 inibitore (C1-INH). La carenza di C1-INH provoca instabilità nei diversisistemi controllati da questa proteina: complemento,fase di contatto, coagulazione e fibrinolisi (Figura 1),svolgendo un ruolo primario nell’omeostasi dei di-
versi sistemi proteolitici; tuttavia, il mediatore del-l’aumento della permeabilità vascolare è la bradichi-nina, che svolge questa funzione legandosi alrecettore B2 localizzato sulle cellule dell’endoteliovascolare.Infatti, l’aumentata attività della bradichinina è cen-trale nella patogenesi di tre delle cinque forme di an-gioedema non allergico: angioedema ereditario(HAE), angioedema acquisito (AAE) da carenza diC1 inibitore, e angioedema indotto dagli inibitori delsistema renina-angiotensina. La bradichinina è un nanopeptide (Figura 2) prodottoa partire dal chininogeno ad alto peso molecolare,grazie all’azione proteolitica della calleicreina. Il peptide, fisiologicamente attivo, appartiene algruppo delle proteine dette chinine. I suoi effetti com-prendono vasodilatazione, aumento della permeabi-lità vascolare, contrazione delle cellule muscolarilisce non vascolari e dolore. Finora sono stati identificati due tipi di recettori dellabradichinina, distinti in B1 e B2; nella patogenesi del-l’angioedema è di rilievo solo il recettore B2.
22
Patogenesi dell’angioedema ereditario
I MANUALI DI CLINICAL PRACTICE
LA CARENZA DI C1-INH PROVOCA INSTABILITÀ NEI DIVERSI SISTEMI CONTROLLATI DALLA BRADICHININA: COMPLEMENTO, FASE DI CONTATTO,COAGULAZIONE E FIBRINOLISI
Davis AE. Ann Allergy Asthma Immunol 2008
Figura
1 Ruolo del C1 inibitore nell’inibizione proteasica nel plasma
Via Proteasi % capacità inibitoria del C1 inibitore
Via del contatto Callicreina 42-48% Fattore XIIa 47% Fattore XI 90%
Via del complemento C1r 100% C1s 100% MASP2 n.d.
Via fibrinolitica Attivatore del plasminogeno - Plasmina -

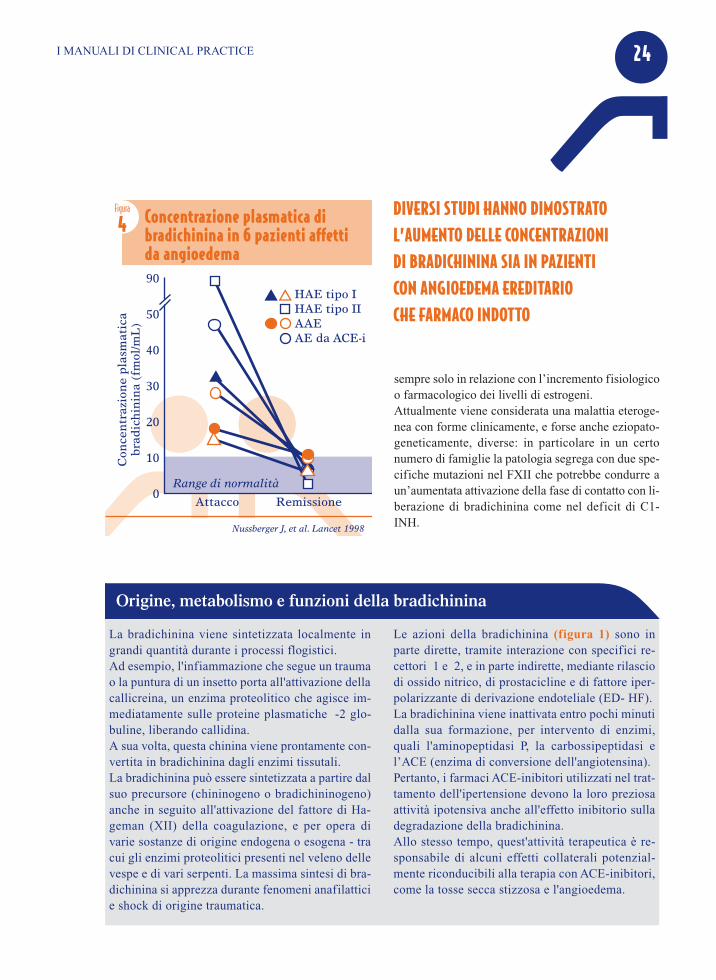
La bradichinina si lega ai recettori B2, costitutiva-mente espressi sulla superficie delle cellule endote-liali, stimolando la produzione di ossido nitrico;questo fa contrarre l’actina intracellulare con aper-tura delle giunzioni interendoteliali, facilitando il pas-saggio di fluidi dall’interno all’esterno dei vasi, e laformazione dell’edema (Figura 3).A questo proposito, Massimo Cugno e coll. hanno va-lutato con il metodo radioimmunologico dopo sepa-razione con cromotografia liquida ad alta prestazionei dosaggi di bradichinina durante e dopo la remissionedell’attacco acuto di angioedema:• nei pazienti con deficit genetico di C1-INH i do-
saggi di bradichinia sono risultati molto aumentatidurante l’attacco, tra 18.0 e 90.0 pM (valori nor-mali 0.2-7.1 pM);
• nei pazienti con storia di angioedema acquisito daACE-i, i valori di bradichinina sono risultati par-ticolarmente alti durante la somministrazione delfarmaco, mentre in un paziente i valori sono di-minuiti del 93% con la sospensione dell’ACE-i;
• in un paziente con angioedema idiopatico, il do-saggio di bradichinina nel sangue refluo dal brac-cio edematoso presentava alti valori di bradichina(20.0 pM) rispetto al braccio controlaterale nonedematoso (6.6 pM);
• 4 pazienti con angioedema istamino-mediato mo-stravano valori normali/bassi di bradichinina mal-grado l’attacco acuto.
Precedentemente risultati analoghi erano stati ottenutida Nussberger e coll, con una sperimentazione simile
su 6 pazienti affetti da forme diverse di angioedema(Figura 4).Le conoscenze sull’eziopatogenesi dell’HAE di tipoIII con C1-INH normale sono, invece, più limitate.Venne descritto per la prima volta dieci anni fa e neiprimi casi le caratteristiche distintive, oltre alla man-canza del difetto biochimico, erano costituite dallapresenza di sintomi nel solo sesso femminile, e quasi
23 PATOGENESI DELL’ANGIOEDEMA EREDITARIO
NELL’ANGIOEDEMA EREDITARIO SONO COINVOLTI IRECETTORI B2 DELLA BRADICHININA, ESPRESSISULLA SUPERFICIE DELLE CELLULE ENDOTELIALI
Figura
2 Bradichinina
Peso molecolare: 1060.21 DaltonFormula: C50H73N15O11
Figura
3 Struttura del vaso in condizioni normali (a) e in seguito al legame della bradichinina al recettore B2 (b)
BradichininaRecettore B2
della bradichinina
Cellulaendoteliale
Giunzioni interendoteliali
serrate
Bradichinina
Recettore B2della bradichinina
Giunzioni interendoteliali
aperte
A B
sempre solo in relazione con l’incremento fisiologicoo farmacologico dei livelli di estrogeni. Attualmente viene considerata una malattia eteroge-nea con forme clinicamente, e forse anche eziopato-geneticamente, diverse: in particolare in un certonumero di famiglie la patologia segrega con due spe-cifiche mutazioni nel FXII che potrebbe condurre aun’aumentata attivazione della fase di contatto con li-berazione di bradichinina come nel deficit di C1-INH.
24I MANUALI DI CLINICAL PRACTICE
DIVERSI STUDI HANNO DIMOSTRATO L’AUMENTO DELLE CONCENTRAZIONI DI BRADICHININA SIA IN PAZIENTI CON ANGIOEDEMA EREDITARIO CHE FARMACO INDOTTO
Nussberger J, et al. Lancet 1998
Figura
4 Concentrazione plasmatica di bradichinina in 6 pazienti affetti da angioedema90
50
40
30
20
10
0Attacco Remissione
Con
cen
traz
ion
e p
lasm
atic
ab
rad
ich
inin
a (f
mol
/mL
)
HAE tipo IHAE tipo IIAAEAE da ACE-i
Range di normalità
La bradichinina viene sintetizzata localmente ingrandi quantità durante i processi flogistici. Ad esempio, l'infiammazione che segue un traumao la puntura di un insetto porta all'attivazione dellacallicreina, un enzima proteolitico che agisce im-mediatamente sulle proteine plasmatiche �-2 glo-buline, liberando callidina. A sua volta, questa chinina viene prontamente con-vertita in bradichinina dagli enzimi tissutali.La bradichinina può essere sintetizzata a partire dalsuo precursore (chininogeno o bradichininogeno)anche in seguito all'attivazione del fattore di Ha-geman (XII) della coagulazione, e per opera divarie sostanze di origine endogena o esogena - tracui gli enzimi proteolitici presenti nel veleno dellevespe e di vari serpenti. La massima sintesi di bra-dichinina si apprezza durante fenomeni anafilatticie shock di origine traumatica.
Le azioni della bradichinina (figura 1) sono inparte dirette, tramite interazione con specifici re-cettori �1 e �2, e in parte indirette, mediante rilasciodi ossido nitrico, di prostacicline e di fattore iper-polarizzante di derivazione endoteliale (ED- HF).La bradichinina viene inattivata entro pochi minutidalla sua formazione, per intervento di enzimi,quali l'aminopeptidasi P, la carbossipeptidasi el’ACE (enzima di conversione dell'angiotensina). Pertanto, i farmaci ACE-inibitori utilizzati nel trat-tamento dell'ipertensione devono la loro preziosaattività ipotensiva anche all'effetto inibitorio sulladegradazione della bradichinina. Allo stesso tempo, quest'attività terapeutica è re-sponsabile di alcuni effetti collaterali potenzial-mente riconducibili alla terapia con ACE-inibitori,come la tosse secca stizzosa e l'angioedema.
Origine, metabolismo e funzioni della bradichinina
25 PATOGENESI DELL’ANGIOEDEMA EREDITARIO
Skidgel RA, Erdös EG. The Pharmacological Basis of Therapeutics 10th ed. 2005
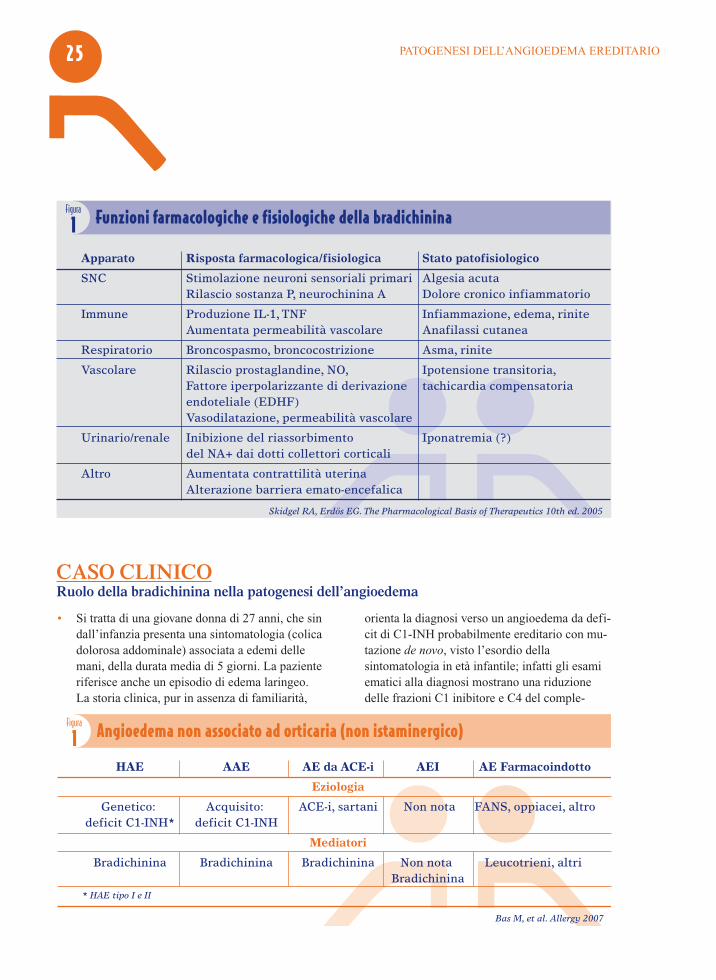
Apparato Risposta farmacologica/fisiologica Stato patofisiologico
SNC Stimolazione neuroni sensoriali primari Algesia acuta Rilascio sostanza P, neurochinina A Dolore cronico infiammatorio
Immune Produzione IL-1, TNF Infiammazione, edema, rinite Aumentata permeabilità vascolare Anafilassi cutanea
Respiratorio Broncospasmo, broncocostrizione Asma, rinite
Vascolare Rilascio prostaglandine, NO, Ipotensione transitoria, Fattore iperpolarizzante di derivazione tachicardia compensatoria endoteliale (EDHF) Vasodilatazione, permeabilità vascolare
Urinario/renale Inibizione del riassorbimento Iponatremia (?) del NA+ dai dotti collettori corticali
Altro Aumentata contrattilità uterina Alterazione barriera emato-encefalica
Figura
1 Funzioni farmacologiche e fisiologiche della bradichinina
• Si tratta di una giovane donna di 27 anni, che sindall’infanzia presenta una sintomatologia (colicadolorosa addominale) associata a edemi dellemani, della durata media di 5 giorni. La pazienteriferisce anche un episodio di edema laringeo.La storia clinica, pur in assenza di familiarità,
orienta la diagnosi verso un angioedema da defi-cit di C1-INH probabilmente ereditario con mu-tazione de novo, visto l’esordio dellasintomatologia in età infantile; infatti gli esamiematici alla diagnosi mostrano una riduzionedelle frazioni C1 inibitore e C4 del comple-
CASO CLINICORuolo della bradichinina nella patogenesi dell’angioedema
Bas M, et al. Allergy 2007
Figura
1 Angioedema non associato ad orticaria (non istaminergico)
HAE AAE AE da ACE-i AEI AE Farmacoindotto
Eziologia
Genetico: Acquisito: ACE-i, sartani Non nota FANS, oppiacei, altro deficit C1-INH* deficit C1-INH
Mediatori
Bradichinina Bradichinina Bradichinina Non nota Leucotrieni, altri Bradichinina* HAE tipo I e II

26I MANUALI DI CLINICAL PRACTICE
mento: C1 inibitore funzionale 11% (v.n. 64-131%); C1 inibitore antigene 4% (v.n. 70-115%); C4 12 mg/dL (v.n. 20-50).
• All’età di 14 anni, essendo le manifestazionimolto frequenti, inizia un trattamento con un anti-fibrinolitico, che tuttavia a lungo andare si rivelainsufficiente sia per una profilassi a lungo ter-mine, sia per controllare gli eventi in acuto.
• All’età di 18 anni, con l’inizio degli studi univer-sitari, si ha una ulteriore esacerbazione della sin-tomatologia che viene quindi trattata con un C1inibitore plasmatico in acuto, e successivamentecon androgeni- che determinano immediata alte-razione del ciclo mestruale senza un significativomiglioramento della sintomatologia.
La paziente quindi sospende gli androgeni e cominciail trattamento in acuto con un antagonista selettivodella bradichinina - icatibant - che rispetto al C1 ini-bitore può più agevolmente essere somministrato a
domicilio dal medico curante. Col trattamento degliattacchi in fase precoce la paziente riesce a ridurremarcatamente la durata degli angioedemi, ottenendouna qualità di vita quasi normale.L'angioedema ereditario (figura 1) compare in per-sone geneticamente prive della capacità di sintetiz-zare C1 inibitore normalmente funzionante. Lemutazioni responsabili dell'angioedema ereditariosono eterogenee e, a causa di mutazioni spontanee,nel 20-25% dei pazienti manca una storia familiarenegli ascendenti. Tuttavia, i valori bioumorali permettono un inqua-dramento diagnostico e orientano la terapia. Malgrado le procedure di profilassi instaurate, ilcontinuo ricorrere degli attacchi causati dall’elevataconcentrazione di bradichinina circolante a causadel deficit del C1-INH, rende ragione dell’utilizzomirato degli antagonisti recettoriali della bradichi-nina.
Bibliografia essenzialeBas M, Adams V, Suvorava T et al. Nonallergic angioedema: role of bradykinin. Allergy 2007; 62: 842-856.
Binkley KE, Davis A 3rd. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of an-
gioedema. J Allergy Clin Immunol 2000; 106(3): 546-50.
Cicardi M, Banerji A, Bracho F et al. Icatibant, a New Bradykinin-Receptor Antagonist, in Hereditary Angioedema. N Engl J Med
2010; 363: 532-541.
Cugno M, Nussberger J, Cicardi M et al. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol 2003; 3: 311-
317.
Davis AE 3rd. Hereditary angioedema: a current state-of-the-art review III: mechanisms of hereditary angioedema. Ann Allergy
Asthma Immund 2008; 100: S7-12.
Dewald G, Bork K.Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with nor-
mal C1 inhibitor. Biochem Biophys Res Commun 2006; 343(4): 1286.
Donaldson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C' 1-
esterase. Am J Med 1963; 35: 37-44.
Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Im-
munol 2010; 126(5): 918-25.
Kaplan AP, Joseph K.The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol 2010;
104(3): 193-204.
Quincke H. Über akutes umschriebenes Hautödem. Monatsh Prakt Derm 1882; 1: 129-131.
Nussberger J, Cugno M, Amstutz C et al. Plasma bradykinin in angio-oedema. Lancet 1998; 351: 1693-1697.
Osler W. Hereditary angio-neurotic oedema. Am J Med Sci 1888; 95(2): 362-67.
Skidgel RA, Erdös EG. Histamine, Bradykinin and Their Antagonists. Chapter 24 Goodman & Gilman The Pharmacological Basis
of Therapeutics 10th ed. 2005.

In seguito alle migliorate conoscenze eziopatogene-tiche, oggi è possibile impostare un percorso dia-gnostico più preciso e un trattamento specifico per
l’angioedema eredtario da carenza di C1 inibitore(C1-INH). Ciò non toglie che la persistente associazione del sin-tomo angioedema a un fatto propriamente allergicoe il conseguente trattamento con anti-istaminici, cor-tisone e adrenalina, continua a esporre il pazienteportatore di deficit da C1-INH a gravi rischi per lasalute nonché a interventi strumentali/chirurgici inu-tili. Quindi, di fronte a un soggetto con attacco acuto diedema - tranne nei casi in cui è necessario mantenerela pervietà della vie aeree - va posta particolare at-tenzione a raccogliere un’accurata anamnesi, con re-lativa analisi dei sintomi e diagnosi differenziale(vedi figura 6 pagina 16) per instaurare prontamentela terapia più appropriata. La terapia dell’angioedema da deficit di C1-INH pre-vede tre diversi momenti:• trattamento dell’attacco acuto• profilassi a breve termine degli attacchi• profilassi a lungo termine degli attacchi.Per questo tipo di patologia sono oggi disponibili inItalia quattro farmaci con finalità d’uso diverse (fi-gura 1): l’inibitore umano della C1-esterasi, l’anta-gonista dei recettori B2 della bradichinina icatibant,il danazolo e l’acido tranexamico. In particolare, l’utilizzo del danazolo e dell’acido tra-nexamico è frutto di dati clinici derivanti dai primianni Sessanta e della lunga esperienza dei clinici; tut-tavia, devono essere somministrati continuativa-mente, pertanto il beneficio clinico va soppesatocontro il rischio di effetti collaterali a lungo termine(vedi di seguito il caso clinico), particolarmente ele-vato per i derivati del testosterone, non indicati prima
dei 18 anni e nelle donne in gravidanza.Gli obiettivi del trattamento dell’evento acuto sonodi ridurre la mortalità e le complicanze dell’attacco,evitando comportamenti e trattamenti che ne favori-scono l’insorgenza. L’inibitore umano della C1-esterasi è entrato in usoper il trattamento in fase acuta già a partire dal 1980.Diversi preparati si sono resi disponibili; quello re-gistrato attualmente in Europa e in Italia è il concen-trato plasmatico pastorizzato, da somministrare pervia endovenosa alla dose di 20 U/kg. La rapidità d’azione - già apprezzabile entro 30 mi-nuti dalla somministrazione - lo rende utilizzabileanche nelle situazioni di emergenza come l’edemadella glottide. Tuttavia, il ruolo chiave svolto dalla bradichininanella patogenesi dell’angioedema da carenza di C1-INH pone il suddetto mediatore come punto di par-tenza cruciale per lo sviluppo di terapie efficaci.
ICATIBANTIcatibant è il primo antagonista competitivo selettivoper i recettori B2 della bradichinina, indicato per iltrattamento sintomatico degli attacchi acuti di an-gioedema ereditario negli adulti con carenza di C1-
27
Trattamentofarmacologico
TRATTAMENTO FARMACOLOGICO
ICATIBANT È IL PRIMO ANTAGONISTA COMPETITIVO SELETTIVO PER I RECETTORI B2 DELLA BRADICHININA INDICATO PER IL TRATTAMENTODEGLI ATTACCHI ACUTI DI ANGIOEDEMA EREDITARIODA CARENZA DI C1-INIBITORE

esterasi inibitore. Le proprietà farmacocinetiche in-dicano che, dopo una somministrazione sottocutanea,la biodisponibilità assoluta di icatibant è del 97%; iltempo per raggiungere la massima concentrazione èdi 0,5 ore, con volume di distribuzione di circa 20-25L; l'emivita terminale è di circa 1.2 ore. Icatibant è ampiamente metabolizzato da enzimi pro-teolitici in metaboliti inattivi, che sono in gran parteeliminati nelle urine. Gli studi in vitro hanno confermato che icatibant nonviene degradato tramite vie metaboliche ossidative;non è un inibitore dei principali isoenzimi del cito-cromo P450; non è un induttore dei citocromi CYP1A2 e 3A4.
Gli studi FASTIcatibant è stato di recente valutato in due studi difase III - denominati FAST (For Angioedema Subcu-taneous Treatment 1) - dal disegno identico: doppiocieco, randomizzati e multicentrici, in pazienti chenei sei mesi precedenti avevano avuto più di 7 attac-chi di angioedema ereditario cutaneo o addominale
con sintomi moderati/severi.Nello studio FAST-1, 56 pazienti sono stati rando-mizzati a ricevere icatibant oppure placebo, mentrenello studio europeo, FAST-2, 74 soggetti sono statitrattati con icatibant o acido tranexamico per viaorale, alla dose di 3 g al giorno per 2 giorni. Icatibant è stato somministrato una sola volta per viasottocutanea alla dose di 30 mg. In entrambi gli studi, alla fase controllata è seguitauna fase in aperto durante la quale gli altri episodi diangioedema sono stati trattati con icatibant con pos-sibilità di utilizzare sino a 3 dosi da 30 mg nelle 24ore (Figura 2).
Endpoint• Sulla base del sintomo principale - dolore addo-
minale, dolore cutaneo, edema cutaneo - l’en-dpoint principale è stato il tempo dall’inizio dellarisoluzione dei sintomi del 30% (TOR30+) ri-portato dal paziente usando la scala di valuta-zione visual analogue scale (VAS): - rappresentato da una riduzione del punteggiodella VAS di ≥30 mm per una VAS basale di 100mm e di ≥21 mm per una VAS basale di 30 mm- calcolato retrospettivamente come il tempo nelquale la risoluzione dei sintomi è stato documen-tata da 3 misurazioni consecutive sulla VAS.
• Tempo all’iniziale miglioramento dei sintomi se-condo il paziente e il medico
• Tempo alla quasi completa risoluzione dei sin-tomi (TOR90+): risoluzione del 90% dei sintomi
28I MANUALI DI CLINICAL PRACTICE
ICATIBANT È STATO VALUTATO IN DUE STUDI MULTICENTRICI IN DOPPIO CIECO, RANDOMIZZATIVERSO PLACEBO (STUDI FAST 1 E 2) OPPURE ACIDO TRANEXAMICO
SIMEU Journal 2010
Figura
1 Trattamenti farmacologici per l’angioedema ereditario da deficit di C1-inibitore
Principio attivo Indicazioni Via di somministrazione Dosaggio
Inibitore umano Attacco acuto Endovena 20 U/kg della C1-esterasi Profilassi a breve termine
Icatibant Attacco acuto Sottocutanea 30 mg
Danazolo Profilassi a lungo termine Orale 50-200 mg/die* Profilassi a breve termine
Acido tranexamico Profilassi a lungo termine Orale 2-3 g/die*
* Dose minima efficace

• Tasso di risposta TOR30+ a 4 ore dopo l’iniziodel trattamento.
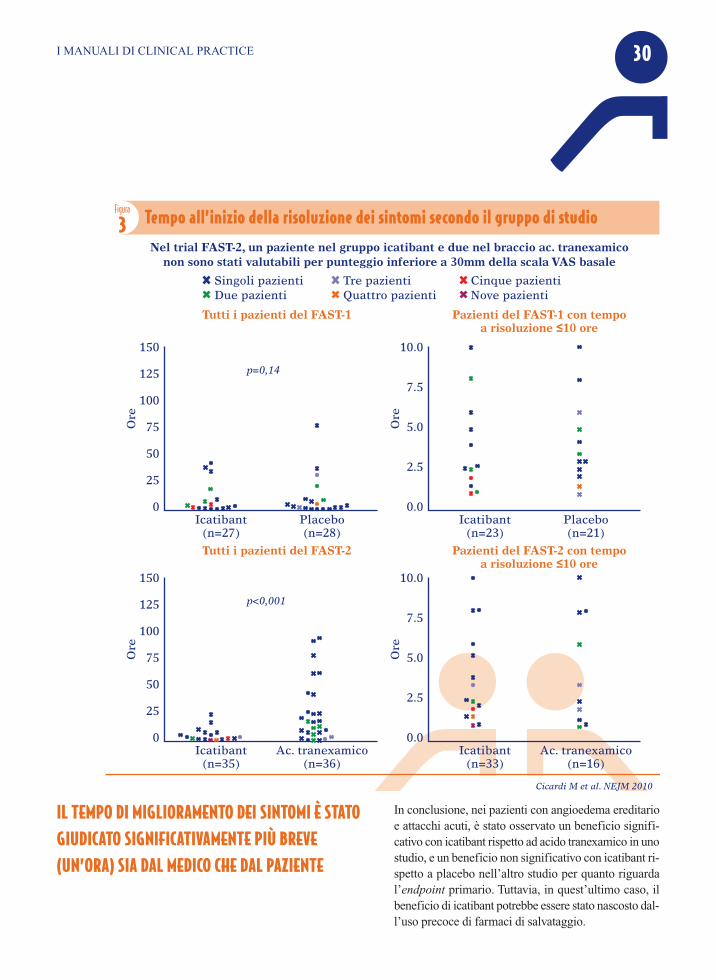
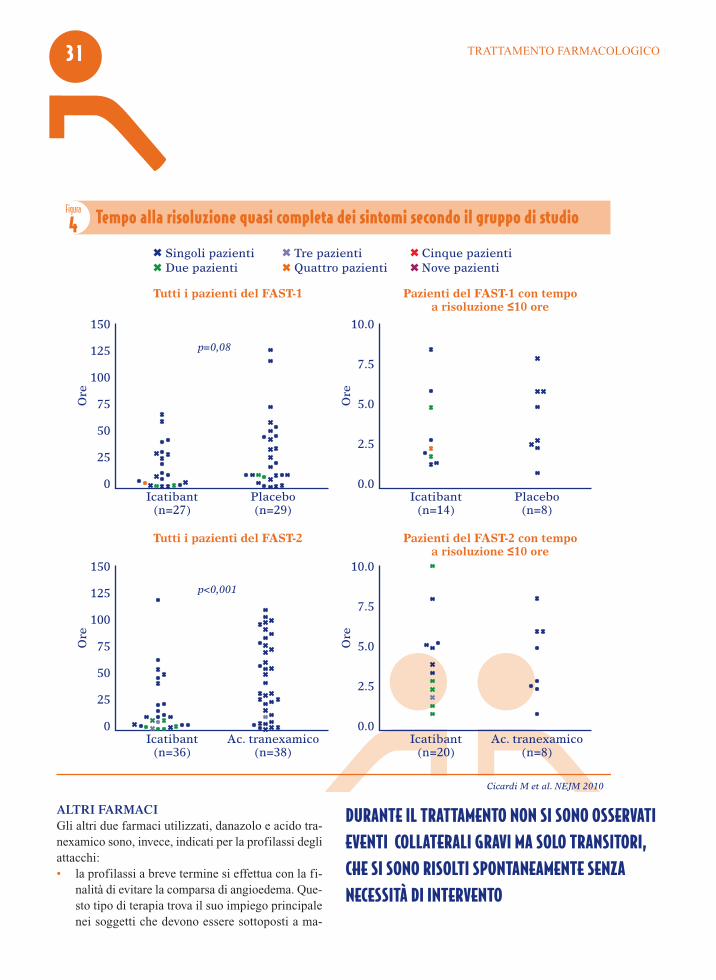
RisultatiL’endpoint è stato raggiunto in 2.5 ore con icatibantversus 4.6 ore con placebo nello studio FAST-1(p=0.14), mentre nel FAST-2, la regressione dei sin-tomi del 30% (TOR30+) è stata raggiunta in 2 oredopo la somministrazione di icatibant rispetto alle 12ore con l’inibitore della fibrinolisi, l’acido tranexa-mico (p<0.001) (Figura 3). Parallelamente, icatibant ha arrestato la sintomatolo-gia (TOR90+) in un tempo altamente significativo ri-spetto all’acido tranexamico (10 ore vs 51 ore;p<0.001) (Figura 4). La percentuale di pazienti con risposta TOR30+ a 4ore dopo l’inizio del trattamento è stata del 67% pericatibant vs 46% per placebo nel FAST-1 e del 80%per icatibant vs 31% per placebo nel FAST-2(P<0.001). Inoltre, in entrambi gli studi il tempo di migliora-mento dei sintomi è stato giudicato sia dal paziente,sia dal medico significativamente più breve - pari ad
un’ora- nel braccio icatibant. In aggiunta, il prose-guimento del trial in aperto ha evidenziato un rapidomiglioramento della sintomatologia con icatibantanche nei pazienti affetti da attacchi di edema dellaglottide. Nello studio FAST-1, 3 pazienti trattati conicatibant e 13 trattati con placebo hanno avuto biso-gno di trattamento con farmaci di salvataggio. Gli effetti collaterali più frequenti nei due trial sonostate reazioni della sede di iniezione, come eritema,gonfiore e, occasionalmente, prurito e dolore; tali sin-tomi sono stati transitori e si sono risolti spontanea-mente, senza alcun intervento. Non è stato osservatonessun evento avverso grave imputabile a icatibant.
29 TRATTAMENTO FARMACOLOGICO
ICATIBANT HA DIMOSTRATO DI RIDURRE O DI ARRESTARE I SINTOMI DELL’ATTACCO ACUTO IN UN TEMPO ALTAMENTE SIGNIFICATIVO RISPETTOALL’ACIDO TRANEXAMICO
Figura
2 Gli studi FAST: pazienti assegnati al braccio di trattamento
Consenso informatoEsami di screening
Primo attacco eleggibile
Attacchi successivi (cutanei, addominali, laringei)
Fase in aperto: estensione in apertoicatibant 30 mg s.c. fino a 3 volte per attacco
Edema cutaneo o addominale Edema laringeo
Trattamento in doppio cieco:icatibant 30 mg s.c. 1x vs comparatore
Trattamento in aperto:icatibant 30 mg s.c. 1x
In conclusione, nei pazienti con angioedema ereditarioe attacchi acuti, è stato osservato un beneficio signifi-cativo con icatibant rispetto ad acido tranexamico in unostudio, e un beneficio non significativo con icatibant ri-spetto a placebo nell’altro studio per quanto riguardal’endpoint primario. Tuttavia, in quest’ultimo caso, ilbeneficio di icatibant potrebbe essere stato nascosto dal-l’uso precoce di farmaci di salvataggio.
30I MANUALI DI CLINICAL PRACTICE
IL TEMPO DI MIGLIORAMENTO DEI SINTOMI È STATOGIUDICATO SIGNIFICATIVAMENTE PIÙ BREVE(UN’ORA) SIA DAL MEDICO CHE DAL PAZIENTE
Cicardi M et al. NEJM 2010
Figura
3 Tempo all’inizio della risoluzione dei sintomi secondo il gruppo di studio
150
125
100
75
50
25
0Icatibant
(n=27)Placebo(n=28)
Ore
Singoli pazientiDue pazienti
Tre pazientiQuattro pazienti
Cinque pazienti Nove pazienti
p=0,14
10.0
7.5
5.0
2.5
0.0Icatibant
(n=23)Placebo(n=21)
Ore
150
125
100
75
50
25
0Icatibant
(n=35)Ac. tranexamico
(n=36)
Ore
p<0,001
10.0
7.5
5.0
2.5
0.0Icatibant
(n=33)Ac. tranexamico
(n=16)
Ore
Nel trial FAST-2, un paziente nel gruppo icatibant e due nel braccio ac. tranexamiconon sono stati valutabili per punteggio inferiore a 30mm della scala VAS basale
Tutti i pazienti del FAST-1 Pazienti del FAST-1 con tempoa risoluzione ≤10 ore
Tutti i pazienti del FAST-2 Pazienti del FAST-2 con tempoa risoluzione ≤10 ore
ALTRI FARMACIGli altri due farmaci utilizzati, danazolo e acido tra-nexamico sono, invece, indicati per la profilassi degliattacchi: • la profilassi a breve termine si effettua con la fi-
nalità di evitare la comparsa di angioedema. Que-sto tipo di terapia trova il suo impiego principalenei soggetti che devono essere sottoposti a ma-
31 TRATTAMENTO FARMACOLOGICO
DURANTE IL TRATTAMENTO NON SI SONO OSSERVATIEVENTI COLLATERALI GRAVI MA SOLO TRANSITORI,CHE SI SONO RISOLTI SPONTANEAMENTE SENZA NECESSITÀ DI INTERVENTO
Tutti i pazienti del FAST-1 Pazienti del FAST-1 con tempoa risoluzione ≤10 ore
Tutti i pazienti del FAST-2 Pazienti del FAST-2 con tempoa risoluzione ≤10 ore
Cicardi M et al. NEJM 2010
Figura
4 Tempo alla risoluzione quasi completa dei sintomi secondo il gruppo di studioFigura
4
150
125
100
75
50
25
0Icatibant
(n=27)Placebo(n=29)
Ore
Singoli pazientiDue pazienti
Tre pazientiQuattro pazienti
Cinque pazienti Nove pazienti
p=0,08
10.0
7.5
5.0
2.5
0.0Icatibant
(n=14)Placebo
(n=8)
Ore
150
125
100
75
50
25
0Icatibant
(n=36)Ac. tranexamico
(n=38)
Ore
p<0,001
10.0
7.5
5.0
2.5
0.0Icatibant
(n=20)Ac. tranexamico
(n=8)
Ore

novre mediche che comportano traumatismi delcavo orale, o delle prime vie aeree, nel caso di in-terventi odontoiatrici, indagini endoscopiche ointubazione tracheale. Traumatismi in tali regionipossono infatti scatenare un edema della regionelaringea, con le ovvie conseguenze. Il concentratoumano della C1-esterasi, somministrato entro le6 ore precedenti l’evento a rischio, rappresenta iltrattamento più diffuso. Qualora l’evento sia programmato, gli androgeni
attenuati somministrati 5 giorni prima dell’inter-vento possono costituire un’alternativa;
• la profilassi a lungo termine ha l’obiettivo di ri-durre gli effetti invalidanti della malattia e di ri-durre la frequenza degli attacchi. In questacircostanza, nei pazienti a elevato rischio di reci-diva o di attacchi di edema laringeo, la terapiaprevede l’impiego di antifibrinolitici o di ormonisessuali maschili.
Oltre all’antagonismo selettivo dei recettori della bra-dichinina, sono in studio diversi prodotti: • i farmaci che sostituiscono il C1 inibitore pla-
smatico (il derivato plasmatico nano filtrato o pa-storizzato, un C1 inibitore ricombinante non diderivazione plasmatica, isolato da latte di coni-glio transgenico)
• i farmaci target specifici, cioè diretti verso altricomponenti del sistema di contatto, e in partico-lare selettivamente contro la callicreina.
32I MANUALI DI CLINICAL PRACTICE
OLTRE ALL’INIBIZIONE SELETTIVA DEI RECETTORIDELLA BRADICHININA OTTENUTA CON ICATIBANT,SONO IN STUDIO DIVERSI ALTRI FARMACI CHE UTILIZZANO MECCANISMI D’AZIONE DIFFERENTI
• Una ragazza di 21 anni lamenta dall’età di 15anni episodi ricorrenti, della durata di 2-5giorni, di gonfiori periferici senza orticaria, diedema della glottide, e di numerose coliche ad-dominali che aumentano di frequenza e intensitàin seguito a traumi fisici o stress psicologici.
• L’anamnesi indica che alcuni componenti dellafamiglia riferiscono sintomi analoghi. Sulla basedi questo quadro clinico-anamnestico, si so-spetta un caso di angioedema ereditario e si ef-fettuano i relativi esami di laboratorio, cheevidenziano: - C1 inibitore funzionale <28% (v.n. 64-131%)- C1 inibitore antigene 7% (v.n. 70-115%)- C4 10 mg/dL (v.n. 20-50)- C1q 97% (v.n. 78-104).
• Viene instaurata la terapia con C1 inibitore emo-derivato al momento degli attacchi acuti addo-minali e delle prime vie aeree, con risoluzionedella sintomatologia nell’arco di alcune ore. Tuttavia, dopo la seconda gravidanza i sintomi
della paziente aumentano in frequenza e inten-sità, e finito l’allattamento si inizia profilassicon danazolo. Il trattamento con androgeni atte-nuati determina una diminuzione della fre-quenza degli attacchi, ma insorgono irregolaritàdel ciclo mestruale, incremento del peso corpo-reo (fino a 10 kg) e variazioni del tono del-l’umore, cosicché è necessario interrompere iltrattamento, con conseguente nuovo incrementodella frequenza degli attacchi.
• La paziente viene, quindi, trattata con icatibantsottocute, che risulta avere un’ottima efficacia euna maggior facilità di somministrazione ri-spetto al C1 inibitore emoderivato.
CommentoIl caso clinico descritto ripropone l’esordio e il de-corso clinico tipico della malattia. Infatti l’esordiodei sintomi avviene in genere in giovane età, e ladiagnosi corretta viene solo molti anni dopo ilprimo episodio - la scarsa conoscenza provoca fra i
CASI CLINICI1. Angioedema: la gestione nelle donne

33 TRATTAMENTO FARMACOLOGICO
• Una donna di 65 anni giunge al Pronto Soccorsoin preda a intensi dolori addominali associati anausea; l’esame ecografico mostra versamentolibero in addome e splenomegalia. La storiaanamnestica riporta: ipertensione arteriosa intrattamento con ACE-inibitore; sindrome an-sioso- depressiva, trattata con citalopram; assun-zione di FANS per gonalgia sinistra. Gli esamiematochimici risultano nella norma.
• Sulla base della sintomatologia, la pazienteviene trasferita nel reparto di chirurgia generale;tuttavia, nel giro di tre giorni si assiste alla com-pleta risoluzione della sintomatologia ad ecce-zione della splenomegalia, e viene dimessa conterapia sintomatica.
• Dopo un mese, si ha un nuovo ricovero per sin-tomatologia sovrapponibile, e in questa circo-stanza viene effettuata una videolaparoscopiache mostra - come unico reperto - la presenza diaderenze omento parietali.
• A distanza di un altro mese, si ha un terzo rico-vero per recrudescenza della sintomatologia ad-dominale, con l’aggiunta di edema del volto.
• Allo scopo di un inquadramento diagnostico,vengono effettuati: - esami di immagine gastrointestinali, che tutta-via risultano privi di specificità; - valutazione endocrinologica, che referta una ti-reopatia non autoimmune; - valutazione reumatologica, che evidenzia bassi
livelli di C4 e picco monoclonale IgM, in pre-senza di splenomegalia.
• Si richiedono, quindi, accertamenti ematologicidi approfondimento, che esitano nella diagnosidi linfoma marginale splenico. La pazienteviene, quindi, inquadrata come affetta da an-gioedema intestinale e al volto per deficit di C1-INH secondario a linfoma marginale splenico.
CommentoIl caso presenta diversi punti critici che riguardanola diagnosi, primo fra tutti una differenza con unquadro di addome acuto: gli angioedemi che si loca-lizzano alla mucosa gastrointestinale comportanouna grave sintomatologia dolorosa, spesso con vo-mito e/o diarrea, e sono difficilmente differenziabilida un addome acuto chirurgico. Le caratteristiche cliniche, bioumorali ed eventual-mente strumentali permettono un orientamento dia-gnostico, malgrado ciò non è infrequente il ricorsoalla laparoscopia; esistono poi forme acquisite, indi-stinguibili clinicamente da quelle su base geneticase non per l’insorgenza più tardiva e per l’associa-zione con malattie linfoproliferative che determi-nano un aumentato consumo di C1-INH. In questo caso poi, va posta diagnosi differenzialeanche con l’angioedema farmaco-indotta: gli ACE-inibitori sono i farmaci responsabili del maggior nu-mero di casi di angioedema. Infatti, purmanifestandosi solo nello 0,1-0,5% dei casi trattati,
più alti ritardi diagnostici, tanto che tra i primi sin-tomi e la diagnosi passano in media anche 15 anni.Gli episodi poi sono spesso ricorrenti, a intervalli egravità non preventivabili, con un episodio di edemalaringeo grave, peraltro rilevabile nell’anamnesi, inoltre la metà di tali pazienti nel corso della loro vita.Diversi i fattori scatenanti che possono precipitaregli attacchi: microtraumi, stress, mestruazioni, va-riazioni ormonali, infezioni; la gravidanza e il partosono momenti di particolare pericolosità per la vitadelle pazienti affette da angioedema ereditario. In particolare, è stata notata una associazione tra
fluttuazioni ormonali ed episodi di angioedema:maggior incidenza in corrispondenza del periodopre-mestruale o al termine della gravidanza. - L’aumento delle concentrazioni di progesterone/an-drogeni e la diminuzione dei livelli di estrogenipotrebbe essere alla base della riduzione degli attac-chi che alcune donne registrano in menopausa. Ilcaso evidenzia, inoltre, le pesanti ripercussioni - dalpunto di vista fisico, lavorativo e psicologico - chela malattia, e talvolta la terapia cronica, ha sullaqualità di vita dei pazienti.
2. Un caso di angioedema complicato

34I MANUALI DI CLINICAL PRACTICE
il largo uso di questi farmaci è ritenuto responsabiledell’ospedalizzazione di alcune centinaia di casi al-l'anno per edema della glottide. Le manifestazioniangioedematose cutanee, addominali e laringee as-sociate all'uso degli ACE-inibitori sono simili aquelle che si riscontrano nei pazienti con carenza diC1 inibitore. Un importante problema che si riscontra nell'indivi-duare gli angioedemi legati all'assunzione di ACE-inibitori è dovuto alla possibile mancanza di unrapporto cronologico tra l'assunzione del farmaco ela comparsa dell'angioedema, quindi parecchi pa-zienti continuano ad assumere il farmaco. E' anchepossibile che i soggetti che hanno assunto il far-maco per lungo tempo continuino ad avere per unbreve periodo episodi di angioedema anche dopo lasospensione.
Infatti gli ACE-inibitori certamente non medianol'angioedema attraverso una reazione allergica oidiosincratica, ma sembrano piuttosto facilitarne lacomparsa in individui predisposti.L’angioedema da carenza di C1-INH dovrebbe es-sere sospettato nei pazienti che giungono al ProntoSoccorso con edemi sottocutanei/mucosali o colicheaddominali ricorrenti di eziologia sconosciuta, aprescindere dalla familiarità. L’accurata anamnesi, unitamente alla mancata rispo-sta a terapia antiallergica/sintomatica, o in alterna-tiva al criterio ex iuvantibus della risposta a untrattamento specifico, seguito poi dal ricorso allaconsulenza specialistica e il dosaggio di C4 e C1inibitore, rappresentano le misure più efficaci per ri-durre il ritardo diagnostico e per iniziare una terapiaappropriata.
Bibliografia essenzialeAgostoni A, Cicardi M, Cugno M, Zingale LC, Gioffrè D, Nüssberger J. Angioedema due to angiotensin-converting enzyme in-
hibitors. Immunopharmacology 1999; 44(1-2): 21-5.
Agostoni A, Cicardi M. Drug-induced angioedema without urticaria. Incidence, prevention and management. Drug Safety 2001;
24(8): 599-606.
Bernstein JA. Hereditary angioedema: a current state-of-the-art review, VIII: current status of emerging therapies. Ann Allergy
Asthma Immunol 2008; 100: S41-6.
Bork K, Barnstedt SE.Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary an-
gioedema. Arch Intern Med 2001; 161(5): 714-8.
Cicardi M, Zingale LC, Zanichelli A et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Ex-
pert Opin Pharmacother 2007; 8(18): 3173-81.
Cicardi M, Banerji A, Bracho F et al. Icatibant, a New Bradykinin-Receptor Antagonist, in Hereditary Angioedema. N Engl J Med
2010; 363: 532-41.
Craig TJ, Levy RJ, Wasserman RL et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute he-
reditary angioedema attacks. J Allergy Clin Immunol 2009; 124(4): 801-8.
Cugno M, Zanichelli A, Foieni F et al. C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress.
Trends Mol Med 2009; 15(2): 69-78.
Deeks ED. Icatibant. Drugs 2010; 70(1): 73-81.
Firazyr. Riassunto delle Caratteristiche del Prodotto (RCP).
Reshef A, Leibovich I, Goren A. Hereditary angioedema: new hopes for an orphan disease. Isr Med Assoc J 2008; 10(12): 850-5.
Angioedema ereditario in pronto soccorso: il ruolo dell’inibitore della C1-esterasi. SIMEU Journal. 2010; 3.

35
Messaggi perla pratica clinica
MESSAGGI PER LA PRATICA CLINICA
PATOGENESI• La bradichinina è il mediatore dell’aumentata
permeabilità vascolare ed esplica la sua funzioneattraverso il legame al recettore B2 localizzatosulle cellule dell’endotelio vascolare.
• La bradichinina circolante è elevata per un au-mento della sua sintesi nell’angioedema da ca-renza di C1 inibitore o per una riduzione del suocatabolismo nell’angioedema da ACE-inibitori.
MANIFESTAZIONI CLINICHE• Le manifestazioni cliniche, che insorgono
solitamente entro la seconda decade di vita nelcaso dell’angioedema ereditario e dopo la quartadecade nel caso dell’angioedema acquisito, consistono in angioedemi ricorrenti, autolimi-tantesi, senza orticaria, che durano mediamente
2-4 giorni e possono interessare: il tessuto sottocutaneo del viso o degli arti e la mucosa -più frequentemente - del tratto gastroenterico edelle alte vie respiratorie.
• La sintomatologia non regredisce con terapia sin-tomatica o con la comune terapia anti-allergica.
L’angioedema è definito come edema profondodel tessuto sottocutaneo o della mucosa dovutoad un aumento transitorio di permeabilità dei
vasi sanguigni del derma profondo, a differenza del-l’edema pruriginoso che contraddistingue l’ortica-ria:• Nell’inquadramento della malattia vanno di-
stinte: - forme acute/croniche - con orticaria/senza orticaria - con fattore scatenante/senza fattore scatenante.
• L’angioedema da carenza di C1 inibitore è unacondizione rara che può essere dovuta ad un de-ficit ereditario di C1 inibitore (angioedema ere-ditario, hereditary angioedema, HAE) oppure adun deficit acquisito di C1 inibitore (angioedemaacquisito, acquired angioedema, AAE).
• L’angioedema ereditario è una malattia geneticache si trasmette come carattere autosomico do-minante.
Sono note due varianti fenotipiche della malattia:- tipo 1 caratterizzato da bassi livelli plasmaticidi C1 inibitore sia antigenici che funzionali- tipo 2 caratterizzato da livelli funzionali di C1inibitore ridotti e da livelli antigenici normali oaumentati - è nota una terza variante - tipo 3 - con livellinormali di C1 inibitore. Si tratta di una malattiaeterogenea, con forme clinicamente, e forseanche eziopatogeneticamente, diverse.
• L’angioedema acquisito è una condizione ancorapiù rara dell’angioedema ereditario legato fre-quentemente ad una malattia linfoproliferativae/o alla presenza di autoanticorpi.
• L’angioedema da ACE-inibitori può verificarsidopo ore o anni dalla prima somministrazione erecidivare nel tempo in modo casuale; se la som-ministrazione non viene interrotta aumenta lafrequenza degli attacchi e la gravità dei sintomi.

36I MANUALI DI CLINICAL PRACTICE
TERAPIALe finalità del trattamento dell’angioedema ereditario sono rivolte a: • evitare comportamenti/terapie che favoriscono
l’insorgenza degli attacchi• eliminare la mortalità e ridurre le conseguenze
degli eventi acuti (terapia dell’attacco acuto)• ridurre gli effetti invalidanti della malattia che
derivano dalla frequente ricorrenza di eventiacuti (profilassi a lungo termine)
• prevenire lo scatenamento di attacchi in alcune
condizioni particolari (profilassi a breve ter-mine)
• Icatibant è il primo antagonista competitivo se-let tivo per i recettori B2 della bradichinina, indi-cato per il trattamento sintomatico degli attacchiacuti di angioedema ereditario negli adulti concarenza di C1-esterasi inibitore, ad avere dimo-strato in studi di fase III di ridurre la sintomato-logia più rapidamente rispetto ad acidotranexamico anche nei pazienti più gravi.
DIAGNOSICriteri clinici• Angioedema sottocutaneo, non pruriginoso,
non eritematoso, autolimitante, solitamente ricorrente e di lunga durata (> 12 ore), con nessuna o scarsa orticaria, talvolta prece-duto da eruzione cutanea a tipo eritema margi-nato.
• Dolori addominali ricorrenti (spesso con vomitoe/o diarrea) senza altra causa, a risoluzionespontanea in 12-72 ore.
• Edemi laringei ricorrenti.
• Storia familiare di angioedema da carenza di C1inibitore.
Criteri di laboratorio• Livelli antigenici di C1 inibitore < 50% del nor-
male in 2 determinazioni separate e dopo ilprimo anno di vita.
• Livelli di attivita� funzionale di C1 inibitore < 50% del normale in 2 determinazioni separatee dopo il primo anno di vita.
• Mutazione del gene di C1 inibitore che altera lasintesi e/o la funzione della proteina.

37
supplemento a CLINICAL PRACTICEBimestrale di attualità in Medicina anno VIII - N. 5
Direttore Responsabile: Monica Luciani
Comitato Scientifico: Corrado Blandizzi, Francesco Blasi, Giampiero Carosi, Alessandro Gringeri, Mirco Lusuardi, Sylvie Ménard, Mauro Moroni, Carlo Federico Perno, Giovanni Rosti, Elena Santagostino,
Piercarlo Sarzi Puttini, Mario Scartozzi, Giuseppe Viale, Mauro Viganò, Massimo Volpe
Redazione: Anna Invernizzi, Monica Luciani
Direzione, redazione, amministrazione: via Gallarate 106 - 20151 Milano - Tel.02/3343281
Stampa: La Grafica (Molteno, LC)
Pubblicazione registrata al Tribunale di Milano n.785 del 23-12-2003


LINICAL C P R A C T I C EI MANUALI DI

PROGETTO EDITORIALE CON IL CONTRIBUTO INCONDIZIONATO DI
Riassu
nto de
lle Car
atteris
tiche d
el Prod
otto in
allega
toDep
osito c
/o Ag
enzia I
taliana
del Fa
rmaco
in dat
a 26/
02/2
013
Codice
ZINC:
IIT/HG
/FIR/
11/0
001(1
)







![Intervalli di riferimento nel Tempo di Protrombina, Tempo ... · (FXII), e il cofattore Chininogeno ad alto peso molecolare (HK). [3] L’attivazione degli zimogeni, con passaggio](https://static.fdocumenti.com/doc/165x107/5c658e3109d3f2a86e8cc757/intervalli-di-riferimento-nel-tempo-di-protrombina-tempo-fxii-e-il-cofattore.jpg)










