Genetica Clinica - sunhope.it · Malattie da mutazioni di cellule somaticheà Sono mutazioni che...
48
Leonardo Aurino 1 Genetica Clinica Classificazione delle malattie genetiche Malattie monogeniche (mendeliane) à Possono essere ereditate in modo AD, AR, X-linked Sono malattie caratterizzate dalle seguenti condizioni: • Mutazione di un singolo gene • Sono malattie rare • Hanno un alto rischio di ricorrenza familiare • Hanno un pattern di ereditarietà facilmente riconoscibile • Per molte di esse è disponibile una tecnica di diagnosi molecolare e prenatale Tra queste malattie si annoverano: talassemie, emofilia A, fibrosi cistica, distrofia muscolare di Duchenne, Neurofibromatosi 1, anemia falcimorme, deficit di G6PD e molte altre. Malattie cromosomicheà secondarie ad anomalie di struttura del/dei cromosomi o ad alterazioni de numero (stati di aneuploidia). Sono malattie caratterizzate da: • Deficienza o eccesso di interi cromosomi (quindi non di un singolo gene ma di molti geni) • Rare, ad eccezione di alcune (s. di Down) • Frequentemente sono abortive • Frequentemente determinano ritardo mentale, bassa statura e segni di dismorfismo Tra queste malattie si annoverano: sindrome di Down, s. di Edwars, s. di Pateau, s. di Turner, s. di Klineferter ecc. Malattie poligenicheà sono malattie determinate da un numero elevato di geni. Sono un gruppo di malattie: • Molto frequenti • Hanno una modalità di trasmissione difficilmente riconoscibile • Sono spesso influenzate da circostanze ambientali Esempi di malattie poligeniche sono il diabete mellito, autismo, coronaropatie ecc. Malattie da mutazioni di cellule somaticheà Sono mutazioni che insorgono de novo in una cellula somatica. In una certa percentuale di casi sono secondarie ad una predisposizione ereditata attraverso mutazioni di cellule germinali. Su questa predisposizione insorgono nuove mutazioni, che causano la malattia (teoria del doppio hit di Knudson). Tale schema è alquanto ricorrente nelle mutazioni di geni oncogeni (mutazioni GOF) e di geni oncosoppressori (mutazioni LOF) che causano lo sviluppo di tumori. Malattie da mutazioni del genoma mitocondrialeà Il corredo mitocondriale cellulare ha sempre origine materna (in quanto solo l’ovocita possiede mitocondri, mentre lo spermatozoo ne è privo). L’eredità di tali malattie è pertanto matrilineare. Tra queste malattie si annoverano epilessia miotonica e atrofia ottica di Leibner. WWW.SUNHOPE.IT
Transcript of Genetica Clinica - sunhope.it · Malattie da mutazioni di cellule somaticheà Sono mutazioni che...

Leonardo Aurino
1
GeneticaClinicaClassificazionedellemalattiegenetiche
Malattie monogeniche (mendeliane) à Possono essere ereditate in modo AD, AR, X-linked Sono malattie caratterizzate dalle seguenti condizioni:
• Mutazione di un singolo gene• Sono malattie rare• Hanno un alto rischio di ricorrenza familiare• Hanno un pattern di ereditarietà facilmente riconoscibile• Per molte di esse è disponibile una tecnica di diagnosi molecolare e prenatale
Tra queste malattie si annoverano: talassemie, emofilia A, fibrosi cistica, distrofia muscolare di Duchenne, Neurofibromatosi 1, anemia falcimorme, deficit di G6PD e molte altre.
Malattie cromosomicheà secondarie ad anomalie di struttura del/dei cromosomi o ad alterazioni de numero (stati di aneuploidia). Sono malattie caratterizzate da:
• Deficienza o eccesso di interi cromosomi (quindi non di un singolo gene ma di molti geni)• Rare, ad eccezione di alcune (s. di Down)• Frequentemente sono abortive• Frequentemente determinano ritardo mentale, bassa statura e segni di dismorfismo
Tra queste malattie si annoverano: sindrome di Down, s. di Edwars, s. di Pateau, s. di Turner, s. di Klineferter ecc.
Malattie poligenicheà sono malattie determinate da un numero elevato di geni. Sono un gruppo di malattie:
• Molto frequenti• Hanno una modalità di trasmissione difficilmente riconoscibile• Sono spesso influenzate da circostanze ambientali
Esempi di malattie poligeniche sono il diabete mellito, autismo, coronaropatie ecc.
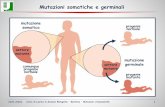
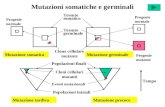
Malattie da mutazioni di cellule somaticheà Sono mutazioni che insorgono de novo in una cellula somatica. In una certa percentuale di casi sono secondarie ad una predisposizione ereditata attraverso mutazioni di cellule germinali. Su questa predisposizione insorgono nuove mutazioni, che causano la malattia (teoria del doppio hit di Knudson). Tale schema è alquanto ricorrente nelle mutazioni di geni oncogeni (mutazioni GOF) e di geni oncosoppressori (mutazioni LOF) che causano lo sviluppo di tumori.
Malattie da mutazioni del genoma mitocondrialeà Il corredo mitocondriale cellulare ha sempre origine materna (in quanto solo l’ovocita possiede mitocondri, mentre lo spermatozoo ne è privo). L’eredità di tali malattie è pertanto matrilineare. Tra queste malattie si annoverano epilessia miotonica e atrofia ottica di Leibner.
WWW.SUNHOPE.IT

Leonardo Aurino
2
TerminologiaLocus: designa la posizione di un gene o di un'altra sequenza significativa all'interno di un cromosoma. Allele: si definiscono alleli le due o più forme alternative dello stesso gene che si trovano nella stessa posizione su ciascun cromosoma omologo (locus genico). Gli alleli controllano lo stesso carattere ma possono portare a prodotti quantitativamente o qualitativamente diversi. Ogni individuo ha due alleli per ogni gene, di cui uno di derivazione materna e l’altro di derivazione paterna (fatta eccezione per i geni posti sul cromosoma X e Y, cioè gli eterocromosomi, del soggetto di genere maschile). Aplotipo: è la successione di alleli di loci vicini su un cromosoma. Tali alleli sono di norma in un rapporto di linkage disequilibrium tra di loro, e pertanto vengono segregati insieme durante i processi di formazione dei gameti. Polimorfismo: si definisce polimorfismo una variabilità allelica presente in almeno l’1% della popolazione generale Mutazione: Si definisce mutazione una variante allelica di un gene presente in meno dell’1% della popolazione generale. Le mutazioni possono essere distinte in:
1. Puntiformi: mutazione di una o di poche bp (paia di basi azotate). Possono realizzarsi per: a) Transizione: sostituzione di una coppia di basi azotate puriniche con un’altra coppia di basi puriniche o di una coppia di basi pirimidiniche con un’altra coppia di basi pirimidiniche. b) Trasversione: sostituzione di una coppia Pu/Pi con una Pi/Pu Sia nelle transizioni che nelle trasversioni, l’alterazione della sequenza di basi azotate può alterare la conformazione del codone originario, causando: - mutazioni missenso: il neocodone formatosi codifiche per un amminoacido diverso da quello originario - mutazioni nonsenso: il neocodone formatosi è un codone di stop, che arresta quindi la traduzione da parte del ribosoma. - mutazione neutra: il neocodone formatosi codifica per un amminoacido diverso ma di struttura simile a quello originario - mutazione silente: non genera variazioni amminoacidiche (per il fenomeno della “ridondanza”) c) Inserzioni/Delezioni: in cui vi è l’aggiunta o la perdita di una o di poche basi azotate all’interno di una tratto di un gene. Tale fenomeno determina: - se il numero di basi inserite o delete è multiplo di 3: non vi è spostamento della “cornice di lettura”. Tali mutazioni sono quindi da considerarsi in frame. - se il numero di basi inserite o delete non è multiplo di 3: vi è spostamento della “cornice di lettura”. Tali mutazioni, definite mutazioni frameshift, sono di tipo out of frame.
2. Riarrangiamento genomico: - traslocazioni: errato scambio di parti di cromosomi non omologhi durante il riarrangiamento cromosomico. Si distinguono due tipi principali di traslocazioni: intracromosomica, cioè all'interno di uno stesso cromosoma, o intercromosomica, cioè tra due cromosomi diversi. Nelle traslocazioni intercromosomiche distinguiamo le traslocazioni non reciproche, cioè spostamento di un segmento di un cromosoma a un altro, o reciproche, cioè avviene uno spostamento reciproco di parti tra due cromosomi, uno dà un pezzo all'altro; un particolare tipo di traslocazione intercromosomica è la traslocazione robertsoniana (cioè la traslocazione tra cromosomi acrocentrici, ossia i cromosomi 13,14,15, 21 e 22) - inversioni: consistono nella rottura del filamento di DNA in due punti; il frammento così ottenuto viene reincorporato, grazie alla riparazione ad opera di specifici enzimi (come le DNA ligasi), nel cromosoma, ma viene invertito di orientamento.
WWW.SUNHOPE.IT

Leonardo Aurino
3
- delezioni: consiste nell'assenza di un tratto di un cromosoma, con conseguente perdita di materiale genetico e non è reversibile.
3. Mutazioni in corso di splicing, cioè durante la processazione dell’mRNA nascente, in cui vengono rimossi gli introni e uniti gli esoni. È questo il caso della progeria, in cui vi è un errore di splicing a carico dell’mRNA derivante dal gene della lamina A.
Omozigote: individuo con alleli identici di un locus specifico Eterozigote: individuo con alleli diversi di un locus specifico Emizigote: individuo con una singola copia di un gene. È questo il caso dei geni localizzati sul cromoscoma X e Y del soggetto di sesso maschile. Tali cromosomi, non avendo i propri omologhi, definiscono lo stato di emizigosi. Anche nella monosomia Turner X0, i geni espressi nell’unico cromosoma X sono in stato di emizigosi. Dominante: genotipo che si manifesta nel fenotipo anche allo stato di eterozigote Recessivo: genotipo che si manifesta nel fenotipo solo allo stato di omozigote Penetranza: è la frequenza con cui un genotipo (dominante o recessivo) si manifesta nel fenotipo di una popolazione. La penetranza può essere:
- completa (100%), quando tutti i soggetti portatori di una specifica mutazioni sono malati - incompleta (< 100%), quando, a parità di genotipo, non tutti i soggetti mostrano il
medesimo fenotipo. Espressività: è il grado di espressione fenotipica di un medesimo genotipo. Un genotipo può determinare, ad esempio, una malattia di una certa gravità di un individuo, mentre può causare la medesima malattia con un gravità molto più sfumata in un altro. Tale variabilità dipende dall’espressività del genotipo. In base a questo principio, i genotipi possono avere espressività costante o espressività variabile. Mosaicismo: condizione per cui in un individuo sono presenti duo o più linee cellulari geneticamente diverse. Tale condizione deriva da mutazioni che occorrono molto precocemente a carico delle zigote, nell’atto delle sue prime divisioni mitotiche. Imprinting: L'imprinting genomico indica l'espressione differenziata di materiale genetico a seconda dell'origine parentale. Si tratta di un meccanismo di regolazione genica che riguarda 156 di geni conosciuti, la maggior parte di questi sono posti sul cromosoma 11 e 15. Nella spermatogenesi parliamo di imprinting paterno, mentre nell'oogenesi di tipo materno. L’imprinting consiste in una differente metilazione di un determinato locus genico che costituisce una sorta di "impronta", la quale impone l'espressione di uno solo dei due alleli di quel determinato locus genico, ossia quello della madre o quello del padre. Viene definito “imprinted” la copia del gene che non viene espressa (materno se è di orgine ovocitaria, paterno se è di origine spermatocitica). Esistono, riassumendo, circa 156 geni che subiscono il processo dell’imprinting (materno/paterno). Per tali geni, quindi, l’individuo possiede una sola copia funzionante (in quanto l’altra è imprinted). Una eventuale delezione del gene non imprinted determina la comparsa di una “nullisomia” per quel dato gene, determinando la comparsa di malattia. Le principali malattie derivanti dal fenomeno dell’imprinting sono:
1. Sindrome di Prader-Willi: Causata da delezione sul cromosoma 15 paterno di una regione (15q11-q13), con cromosoma 15 materno imprinted (inattivo poiché metilato). La sindrome è caratterizzata da facies peculiare (occhi a mandarla, strabismo, capelli chiari e ipopigmentazione cutanea), bassa statura e ritardo mentale medio, criptorchidismo (nel maschio). Caratteristica è l’iperfagia che porta ad obesità già prima del compimento dei 2-3 anni.
2. Sindrome di Angelmam:è l’analogo della sindrome di Prader-Willi, causata questa volta da delezione sul cromosoma 15 materno di una regione (sempre la 15q11-q13), con cromosoma 15 paterno imprinted. La sindrome è caratterizzata da facies tipica (facies “allegra”, bocca larga, lingua protrusa, mandibola prominente). Vi è un grave ritardo mentale con assenza di parola. Il bambino ha
WWW.SUNHOPE.IT

Leonardo Aurino
4
un comportamento estremamente allegro e spesso ride anche al di fuori delle situazioni in cui è previsto (happy puppet syndrome)
3. Sindrome di Silver-Russel: : Causata da delezione sul cromosoma 11 paterno di una regione, con cromosoma 11 materno imprinted (e dunque inattivo poiché metilato). La sindrome è caratterizzata da nanismo disarmonico e faccia triangolare.
Malattieautosomichedominanti Nelle malattie autosomiche dominanti, il fenotipo degli eterozigoti è indistinguibile da quello degli omozigoti affetti. Le malattie autosomiche dominanti, come tutte le malattie autosomiche, non hanno preferenza di sesso, in quanto colpiscono sia maschi che femmine. La trasmissione è di tipo verticale, in quanto non ci sono salti generazionali. Se un singolo genitore è affetto da una malattia AD (R*r), mentre l’altro genitore è sano (rr), allora la progenie sarà per il 50% malata (R*r) e per il 50% sana (rr). Se entrambi i genitori sono affetti da una malattia AD (entrambi R*r), allora il 75% dei figli sarà malato (in quanto sarà R*R* 50% o R*r 25%). solo il 25% dei figli (quelli rr) saranno sani. Per motivi statistici, è molto più probabile che sia solo un genitore ad essere affetto da una malattia AD, pertanto il genotipo dei soggetti affetti da malattie AD è spesso eterozigote. Nelle malattie AD un individuo affetto ha spesso uno dei due genitori che risulta affetto dalla stessa malattia. Esempi di malattie autosomiche dominanti Acondroplasia: è una malattia genetica, ereditata con modalità autosomica dominante, caratterizzata da una mutazione specifica (G380R, cioè sostituzione di glicina con arginina) all’interno del gene FGFR3, posto sul cromosoma 4. Il gene FGFR3 codifica per un recettore espresso dal condrocita che, se attivato, ne inibisce la crescita. La mutazione G380R del gene FGFR3 determina l’attivazione costitutiva del recettore. La malattia ha una penetranza di quasi il 100%, mentre l’espressività è variabile. L’acondroplasia è la più frequente causa di nanismo disamormonico conosciuta, con un’incidenza di 1:4000. La malattia è causa di bassa statura (130 cm per i maschi e 125 cm per le femmine), con particolare riferimento agli arti (la grandezza di testa e tronco è relativamente normale). L’acondroplasico ha, inoltre, una facies caratteristica, caratterizzata da bozze frontali, fronte olimpica e naso appiattito. Il quoziente intellettivo è, in una buona parte dei casi, del tutto normale. Nonostante la malattia sia AD, circa l’80% dei soggetti affetti non sono figli di acondroplasici, in quanto, in questi casi, la malattia è frutto di mutazioni “de novo”. Neurofibromatosi di tipo I o Morbo di Von Recklinghausen: è una malattia neurocutanea ereditata con modalità AD, causata dalla mutazione del gene oncosoppressore NF1, posto sul cromosoma 17. Il ruolo del prodotto proteico del gene NF1 è quello di inibire l’oncogene p21. La malattia ha un’incidenza di circa 1:3000. La penetranza è pari a quasi il 100%, mentre l’espressività è molto variabile in virtù della grandezza del gene NF1 (più un gene è grande più una sua mutazione ha espressività variabile). Da un punto di vista clinico, la NF1 è caratterizzata da lesioni cutanee tipiche, le “macchie
WWW.SUNHOPE.IT

Leonardo Aurino
5
caffelatte”, macchie piatte, di color marrone chiaro, di dimensione e forme variabili. Sono presenti fin dalla nascita e tendono ad ingrandirsi e ad aumentare di numero nei primi anni di vita. Altre lesioni che si associano sono gli amartomi pigmentati dell’iride o noduli di Lisch. Altre tipiche lesioni i neurofibromi, tumori benigni che originano dalla superficie dei nervi periferici, composti da cellule di Schwann e fibroblasti che, nella variante plessiforme, tendono a localizzarsi al volto e hanno una crescita molto pronunciata, potendo raggiungere dimensioni enormi e rappresentare la causa di evidentissime deformità. I neurofibromi hanno inoltre una data tendenza all’evoluzione maligna. Nel 15% dei pazienti con NF-1 si assiste alla formazione di tumori di strutture nervose, come astorocitomi, meningiomi, ependimomi e gliomi dell’ottico. Anche neoplasie non interessanti il sistema nervoso sono frequenti nei soggetti affetti da NF-1 (leucemie, neoplasie gastriche, intestinali, feocromocitomi). Tumori ereditari: alcune mutazioni germinali ereditate in modalità AD di geni oncosoppressori possono rendersi causa di “tumori ereditari”, come nel caso delle mutazioni dei geni BRCA 1 e 2, responsabili di tumori mammari e ovarici, del gene APC, responsabile della FAP (associata a CCR nel 100% dei casi) e nelle mutazioni germinale di alcuni geni coinvolti nel riparo degli errori di accoppiamento delle basi azotate (funzione definita Mismatch Repair System, come hMSH2, hMLH1, hPMS1, hPMS2), come avviene nella sindrome di Lynch, associata al CCR.
MalattieautosomicherecessiveNelle malattie autosomiche recessive, il fenotipo degli eterozigoti è indistinguibile da quello degli omozigoti sani. La trasmissione non dipende dal sesso, come per tutte le malattie autosomiche. La trasmissione di una malattia AR è di tipo trasversale, cioè interessa più individui nella stessa fratria. Normalmente, un individuo affetto da una malattia autosomica recessiva (AR) ha entrambi i genitori sani ma portatori della mutazione, capaci pertanto di trasmetterla al 25% dei figli. In questo caso, il quadrato di Punnet sarà Rr* x Rr*, che darà luogo a figli sani nel 25% dei casi, figli portatori nel 50% e figli affetti nel 25%. Le malattie AR sono molto più frequenti in caso di unioni tra consanguinei, come avviene in alcune culture e religioni (quaccheri, mormoni, anabattisti) o in alcune regioni insulari. Esempi di malattie autosomiche recessive Fibrosi cistica: anche nota come mucoviscidosi, è la malattia autosomica recessiva letale più comune nella razza caucasica. Ha una incidenza di 1:2500 nati vivi, mentre la frequenza di portatori sani (non sintomatici) è addirittura di 1:25. Nelle aree ad alta endogamia, come la Gran Bretagna, l’incidenza della malattia è molto maggiore, di circa 1:337 nati vivi. La fibrosi cistica è causata da una mutazione a carico di un gene posto sul cromosoma 7, definito CFTR, il cui prodotto proteico funge da canale per il Cl- nelle membrane delle cellule epiteliali. Le mutazioni del gene CFTR attualmente conosciute sono oltre 1900. La mutazione più frequente è senza dubbio una delezione di tra paia di basi che comporta la perdita di una fenilalanina in posizione 508 del tradotto proteico. Tale mutazione è quindi definita ΔF508. Le mutazioni del gene CFTR sono state suddivise in 5 classi:
1. Classe I, difetto di produzione: in cui manca la sintesi della proteina 2. Classe II: difetto di maturazione della proteina, cioè la proteina non raggiunge la membrana
(in questa classe figura la ΔF508) 3. Classe III: difetto di regolazione, in cui la proteina si forma, raggiunge la membrana, ma
non risponde ai segnali attivatori
WWW.SUNHOPE.IT

Leonardo Aurino
6
4. Classe IV: difetto di trasporto del Cl-, cioè il canale del cloro si forma, ma è poco efficiente. 5. Classe V: ridotta sintesi. La proteina si forma normalmente ed è wild-type, ma è sintetizzata
in quantità fortemente ridotta rispetto al solito. La funzione del tradotto proteico del gene CFTR è, come detto, un canale del Cl- ATP-dipendente. La funzione specifica della proteina non è ancora del tutto nota, ma si è visto che CFTR inibisce l’attività del canale del sodio ENaC. In assenza di CFTR vi è un esaltata funzione del canale epiteliale ENaC, che determina sequestro di Na e acqua dai secreti cellulari, rendendoli densi e viscosi. Da un punto di vista clinico, la fibrosi cistica è caratterizzata da un interessamento di tutti gli organi e apparati in grado di produrre secreti (apparato GI, ghiandole sudoripare, apparato respiratorio). I secreti di questi apparati sono particolarmente densi e viscosi e hanno una estrema difficoltà alla progressione e all’allontanamento. Pertanto, la FC si manifesta con:
1. Insufficienza pancreatica esocrina: nell’85% dei casi è presente già alla nascita. La steatorrea e il malassorbimento sono i caratteri più evidenti. Il malassorbimento è causa di meteorismo, flatulenza, alvo frequente con feci abbondanti, dolori addominali.
2. Ileo da meconio: è uno dei segni che più lascia supporre una fibrosi cistica. 3. Sterilità maschile per occlusione dei dotti efferenti dell’epididimo. 4. Frequenti infezioni respiratorie (prima causa di morte per FC). Una patologia infettiva
broncopolmonare compare praticamente in ogni paziente affetto da FC. Meno del 5% dei pazienti ha un interessamento respiratorio nullo o lieve.
La diagnosi di fibrosi cistica può avvenire seguendo tre percorsi:
- Tramite esami di screening eseguiti in tutti i neonati, atti a ricercare soggetti potenzialmente malati, dosando la tripsina immunoreattiva (IRT) in una goccia di sangue al 3° giorno di vita.
- Tramite accertamenti diagnostici motivati da sintomi sospetti (steatorrea cronica, ileo da meconio, scarso accrescimento stato-ponderale, disidratazione da perdita di sali)
- Tramite accertamenti effettuati in parenti di pazienti con diagnosi accertata di FC. In caso di un test screening positivo per elevati livelli di tripsina immunoreattiva, il neonato viene ulteriormente valutato tramite:
1. Test del sudore: consiste nella misurazione della quantità di NaCl presente nel sudore. Nella FC i livelli di NaCl sono superiori alla norma. Il gold standard per la diagnosi di FC resta comunque la misurazione della quantità di Cl dopo iontoforesi pilocarpinica. Vengono considerati patologici valori di Cloro nel sudore superiori a 60 mEq/l. Valori compresi tra 40 e 60 mEq/l sono da considerarsi dubbi.
2. La diagnosi genetica tramite sequanziamento del gene CFTR, alla ricerca di eventuali delezioni o mutazioni puntiformi.
La terapia della FC si basa su:
- Terapia nutrizionale: si avvale si integrazione vitaminica (sopratutto del gruppo ADEK, visto il malassorbimento lipidico secondario all’insufficienza pancreatica esocrina) e la sostizuione enzimatica pancreatica (lipasi, amilasi).
- Terapia respiratoria: utilizzo di ventilazione a pressione esoiratoria positiva (PEPmask) e terapia antibiotica al bisogno.
WWW.SUNHOPE.IT

Leonardo Aurino
7
Ad oggi esiste un farmaco, l’ivacaftor, utilizzabile esclusivamente nella FC secondaria a mutazione G541D. Tale farmaco è in grado di aumentare le performance della proteina CFTR, ripristinando un flusso ottimale di sali e fluidi sulla superficie polmonare.
MalattieX-linked(ereditarietàdiaginica)Si distinguono:
- Malattie X-linked Dominanti: Non può mai essere trasmessa da maschio a maschio (in quanto il figlio maschio eredita dal padre il cromosoma Y, mentre il cromosoma X è materno). Nella maggior parte dei casi, tali malattie sono letali per il sesso maschile, quindi risulta maggiormente affetto il sesso femminile. Tra queste malattie si annovera la Sindrome di Rett.
- Malattie X-linked Recessive: Anche in questo caso la trasmissione padre-figlio maschio è impossibile. Le malattie XL-R interessano quasi esclusivamente il sesso maschile, in quanto possessore di una sola X in stato di emizigosi. Le figlie femmine di un maschio malato sono portatrici al 100%. La femmina portatrice (Xx*) ha la possibilità del 50% di generare maschi affetti e del 50% di generare femmine portatrici. La possibile di una figlia femmina malata è, sebbene raramente, possibile. La figlia femmina può essere malata nel momento in cui sia affetta da monosomia x*0, nel momento in cui si sia realizzata una forte lyonizzazione del cromosoma X sano, e nel momento in cui la madre portatrice Xx* si sia unita con un maschio malato x*Y à 50% possibilità di figlia femmina malata.
Esempi di malattie X-linked Recessive Emofilia: è un disordine ereditario X-linked recessivo causa di un difetto della coagulazione. Si conoscono 3 tipi di emofilia:
- Emofilia A: mutazione del gene codificante per il fattore VIII della coagulazione, che ne causa carenza totale o parziale. L’incidenza della malattia è di 20:100.000 nati vivi. Come detto, la malattia è causata dalla mutazione del gene codificante per il fattore VIII della coagulazione, che può essere una mutazione puntiforme, una delezione o la formazione di un codone di stop. Il principale hot-spot del gene è la sequenza TCGA, dove più frequentemente si localizzano le mutazioni. Si conoscono più di 80 mutazioni responsabili dello sviluppo della malattia. L’emofilia può essere la risultante di un difetto quantitativo (95% dei casi) o qualitativo (5%) del fattore VIII. La diagnosi prenatale di emofilia è possibile tramite villocentesi. La gravità dell’emofilia A è variabile in base all’entità del deficit quantitativo/qualitativo del fattore VIII. L’attività coagulante del fattore VIII in un individuo sano va dal 50% al 200%. In base alla riduzione dell’attività coagulante del fattore VIII si definisce: a) emofilia grave se < o = a 1% b) emofilia moderata se compresa tra 1 e 5% c) emofilia lieve se compresa tra 5-25% Clinicamente l’emofilia A si manifesta con ematomi, emartri, cisti ossee siero-ematiche, ematuria. Eventi drammatici in corso di emofilia A sono le emorragia cerebrali, intra o extraparenchimali. La terapia dell’emofilia A è essenzialmente sostitutiva, basta sull’uso di pasla fresco congelato o crioprecipitati, contenenti F VIII. Ad oggi è possibile utilizzare anche FVIII umano ricombinante. La desmopressina si rivelata capace di aumentare di 2-3 volte i livelli di fattore VIII della coagulazione.
WWW.SUNHOPE.IT

Leonardo Aurino
8
- Emofilia B: mutazione del gene codificante per il fattore IX della coagulazione, che ne causa carenza totale o parziale. La clinica è del tutto simile all’emofilia A.
- Emofilia C: mutazione del gene codificante per il fattore XI della coagulazione, che ne causa carenza totale o parziale
Distrofia muscolare di Duchenne: vedi distrofie
Malattiecromosomiche Per cariotipo si intende il patrimonio cromosomico di una cellula di un individuo. Normalmente è composto da 22 coppie di autosomi (cromosomi non sessuali) e da due eterosomi (cromosomi sessuali), XX nella femmina e XY nel maschio. Lo studio del cariotipo è strutturato nel seguente metodo:
• Prelievo di sangue periferico, da cui si ottengono linfociti. In alternativa è possibile utilizzare colture cellulari (come nel caso dell’amniocentesi).
• Le cellule prelevate vengono stimolate per 72 ore con fitoemoagglutinina (PHA), un mitogeno per i linfociti
• Le cellule stimolate vengono bloccate in metafase tramite l’uso della colchicina • Le cellule bloccate in metafase vengono rigonfiate tramite l’utilizzo di una soluzione
ipotonica a base di KCl per ottenere una lisi osmotica. • Le cromosomi liberati vengono colorati per ottenere il bandeggio. La colorazione più usata
è quella di Giemsa, a base di Blu di Metilene, eosina e Azure A-B-C. I bandeggi esistenti sono diversi (bandeggio G, Q, C, R).
Nello studio dei cromosomi bisogna indicare:
• Braccio lungo (q) e braccio corto (p) • Posizione del centromero, per la quale si distinguono:
- cromosomi metacentrici, cioè con centromero centrale - cromosomi acrocentrici (13, 14, 15, 21, 22) - cromosomi submetacentrici (come X e Y)
Si definiscono aneuploidie le anomalie di numero dei cromosomi. Le aneuploidie sono distinte in autosomiche ed eterocromosomiche. Aneuploidie autosomiche Trisomia del 21 (s. di Down)à cariotipo: 47, XY, +21 (nel maschio) o 47, XX, +21 (nella femmina). Nel 70% dei casi tale aneuploidia porta ad aborto. La trisomia del 21 è la trisomia autosomica più frequente, con una incidenza di 1/2500 nati vivi. La malattia è causata nel 93% da una trisomia libera del 21, una condizione derivante da una non disgiunzione meiotica durante la gametogenesi (spermatogenesi o ovogenesi, anche se nel 75% l’errore è nell’ovogenesi ed è correlato all’età della madre). Il rischio di non disgiunzione nell’ovogenesi cresce con l’età della madre: è di 1:1500 se l’età è inferiore a 30 anni, 1:350 con età pari a 35 anni e di 1:20-45 con età pari o superiore a 45 anni. A seguito di questo processo, uno dei due gameti presenta una copia soprannumeraria del cromosoma 21, con un totale di 24 cromosomi (anziché 23). Tale gamete, dopo fecondazione, porta alla formazione di uno zigote con 47 cromosomi (+21). Altra condizione genetica che può determinare la trisomia del 21 è la traslocazione robertsoniana, responsabile del 4-5% delle sindromi di Down. Il cromosoma 21 è un cromosoma acrocentico, cioè
WWW.SUNHOPE.IT

Leonardo Aurino
9
con centromero disposto all’estremità del cromosoma. I cromosomi acrocentrici sono gli unici che possono essere interessati dalla traslocazione robertsoniana. In questo caso, il braccio lungo del cromosoma 21 si fonde a un altro cromosoma acrocentrico spesso il cromosoma 14 [45,XX o XY,t(14;21)]. Questa condizione, a differenza della trisomia libera, non è influenza dall’età materna. Il 2,5% delle sindromi di Down è determinata da un fenome di mosaicismo, ossia un errore durante le prime divisioni mitotiche dello zigote. Alcune cellule avranno, pertanto, un patrimonio genico normale, mentre altre un corredo 47, XX o XY, + 21. Da un punto di vista clinico, la sindrome di Down è causa di ritardo mentale moderato, bassa statura (70% dei casi), facies caratteristica con epicanto, brachicefalia, lingua protrusa, displasia auricolare, naso appiattito, mano scimmiesca, talvolta pterigium colli, lassità dei legamenti. L'incidenza delle cardiopatie congenite nei neonati con sindrome di Down arriva fino al 50% e il 7% di tutti i bambini con una cardiopatia risulta affetto dalla condizione. Un difetto interventricolare è la forma più comune, con il 40% dei pazienti affetti. A seguire il difetto interatriale, riscontrabile nell'8%, la pervietà del dotto di Botallo, nel 7%, e infine l'1% dei nati con sindrome di Down presenta la tetralogia di Fallot. Anche se l'incidenza generale di neoplasie tra gli individui con sindrome di Down è la stessa che nel resto della popolazione, vi è una maggior probabilità di sviluppare alcuni tumori maligni, come le leucemie e il cancro del testicolo. Trisomia del 18 (s. di Edwards) à L’anomalia determina aborto in quasi il 98% dei casi. È una malattia estremamente grave, che determina, nel 2% dei bambini che nascono vivi, la morte entra 1 anno di vita (nel 90% dei casi). La malattia è causata nel 90% da una non disgiunzione materna, è correlata dunque all’età materna. Alla nascita si osserva peso inferiore per età gestazionale, ipertonia, facies triangolare, occipite prominente, epicanto, labio-palatoschisi, padiglioni auricolari da “fauno”, caratteristica è la contrattura del 2° dito sul 3° e del 4° sul 5°. Nel 90% dei casi vi sono cardiopatie congenite. Possibile la microcefalia e la spina bifida. Trisomia del 13 (S. di Patau) à nel 97,5% dei casi la gravidanza non giunge a termine (abortiva). Solo il 9% dei bambini supera l’anno di vita. La malattia è causata nel 75% da una trisomia libera secondaria a non disgiunzione materna, nel 20% ad una traslocazione robertsoniana e nel 5% dei casi a mosaicismo. Alla nascita il neonato è ipotonico, presenta difetti dello scalpo (aplasia cutis), microftalmia, microcefalia, labio-palatoschisi, polidattilia. Nell’80% dei casi vi è una grave cardiopatia congenita, mente nel 50% dei casi vi è cecità e sordita. Possibile è la fusione degli occhi (monoculo). Aneuploidie eterocromosomiche Si ricordi che il sesso genetico è determinato dalla presenza o assenza del cromosoma Y. Il cromosoma Y è un cromosoma molto piccolo, contenente solo 48 geni, di cui il più importante è SRY, necessario all’acquisizione del fenotipo maschile. Nel sesso femminile, invece, la presenza di due cromosomi X viene compensata tramite il fenomeno della lyonizzazione, per il quale uno dei due cromosomi X viene reso silenziato per processi di metilazione (eterocromatizzazione), con formazione del corpo di Barr. Tale processo si avvera già allo stadio di blastocisti. In realtà l’inattivazione del cromosoma X è subtotale, in quanto due regioni vengono salvaguardate dal processo. Tali regioni, note come pseudoautosomiche, sono le regioni PAR1 e PAR2. La regione PAR1 contiene il gene SHOX (SHOrt stature homeoboX) , la cui aploinsufficienza determina arresto della crescita e bassa statura (questo è il motivo per il quale nella monosomia XO o sindrome di Turner vi è bassa statura, mentre nella trisomia X e nella sindrome di Klinefelter vi è alta statura).
WWW.SUNHOPE.IT

Leonardo Aurino
10
Sindrome di Klinefelterà La malattia ha un’incidenza di 1/1000 maschi nati vivi. Solo il 50% delle gravidanze giunge a termine. I difetti cromosomici nella sindrome di Klinefelter possono essere vari:
• 47 XXY (95% dei casi) per una non disgiunzione paterna • 48 XXXY • 49 XXXXY • 46 XX con traslocazione del gene SRY • Mosaicismo
Data la presenza del cromosoma Y o comunque del gene SRY, il fenotipo sarà sempre maschile, anche se vi sarà sempre la presenza del corpo di Barr. Prima della pubertà non si osservano, di regola, sintomi che rivelino l’affezione. La diagnosi viene posta di regola in età adulta per infertilità e ipogonadismo. In età puberale, il soggetto con sindrome di Klinefelter presenta aspetto longilineo, eunucoide, con muscolatura ipotrofica. La virilizzazione è spesso subnormale, con peli pubici e ascellari scarsi, testicoli ipotrofia con diffusa sclerosi tubulare alla biopsia (pertanto vi è azoospermiaàinfertilità). È di frequente riscontro una ginecomastia sia per iperplasia ghiandolare che per abbondanza dei depositi di grasso (lipomastia). Da un punto di vista laboratoristico vi è una netta riduzione degli androgeni e un aumento di FSH e LH. La malattia viene trattata con testosterone, la cui infusione deve essere iniziata solo in pubertà per impedire la saldatura delle cartilagini d’accrescimento. Sindrome di Turner à è una monosomia XO. Ha una incidenza di 1/2500 nati vivi. Gli embrioni XO hanno una scarsa vitalità, si suppone che la sindrome di Turner sia causa del 7-10% degli aborti spontanei. La sindrome di Turner è, comunque, l’unica monosomia vitale. Nel 45% dei casi la sindrome è determinata da una monosomia vera XO, per un errore nella spermatogenesi non correlato all’età del padre. In caso di monosomia vera il corpo di Barr è assente. Nel 50% dei casi la sindrome è determinata da mosaicismo. Una certa popolazione cellulare avrà un corredo XO (con corpo di Barr o cromatina sessuale assente) mentre altre avranno cariotipo normale 46, XX con cromatina sessuale presente. Nel 5% dei casi la sindrome è causata da una perdita subtotale di uno dei cromosomi X. Da un punto di vista clinico, la sindrome di Turner è caratterizzata da:
• Alterazioni somatiche: bassa statura, con altezza massima raggiungibile compresa tra 130 e 145 cm, pterigium colli, viso triangolare con impianto basso delle orecchie, torace a scudo con capezzoli ipoplasici, valgismo del gomito e/o del ginocchio. Di frequente risconto sono le cardiopatie congenite, in particolare la coartazione aortica e la valvola aortica bicuspide.
• Alterazioni gonadiche: Le ovaie si mostrano atresiche, fibrotiche e allungate (streaks ovarium). Ne deriva una produzione di estrogeni pressoché assente, con conseguente ipogonadismo ipergonadotropo. Ciò si traduce in un quadro clinico di mancato sviluppo puberale, con assenza del menarca (amenorrea primaria), ipoplasia di utero/vulva/vagina, sterilità, iposviluppo mammario. Solo nei casi di mosaicismo è possibile uno sviluppo puberale subnormale e una possibile fertilità.
• Alterazioni neuropsichiche: nel 40-50% vi è un ritardo mentale lieve-moderato.
WWW.SUNHOPE.IT

Leonardo Aurino
11
Distrofiemuscolariprogressive Le DMP sono un gruppo di malattie genetiche caratterizzate da necrosi del tessuto muscolare (parametro fondamentale, altrimenti si parlerebbe di semplice miopatia) e progressiva sostituzione fibro/adiposa del tessuto muscolare. Le DMP sono distinte, secondo la classificazione di Walton/Natrass, in:
• Duchenne, XL • Emery-Dreyfuss, XL • Distrofia dei cingoli, AD o AR • Distrofia facio-scapolo-omerale di Landouzy-Dejerine, AD • Distrofie distali: distrofia scandinava ad esordio tardivo, la miopatia distale di Miyoshi e la
miopatia a corpi inclusi, AD o AR • Distrofia oculo faringea, AD (comparsa in V decade di ptosi palpebrale e disfagia) • Distrofie miotoniche
Una classificazione più recente e aggiornata è quella patogenetica:
• DMP da difetti delle proteine nucleari: emerinopatie e laminopatie A/C (distrofia di Emery-Dreyfuss)
• DMP da difetti delle proteine citoplasmatiche: calpainopatie (distrofia dei cingoli) e fukutinopatie (cingoli)
• DMP da difetti delle proteine della EMC: merosinopatie e lamininopatie • DMP da difetti delle proteine del sarcolemma: caveolinopatie e disferlinopatie • DMP da difetti del complesso distrofina-glicani: distrofinopatie e glicanopatie
DistrofinopatieIl termine distrofinopatie ha, ad oggi, sostituito quello di distrofia di Duchenne-Becker, indicando in maniera omnicomprensiva tutte le patologie connesse alle mutazioni del gene per la distrofina. Il gene della distrofina è localizzato sul cromosoma X in sede p21. È il gene più grande dell’intero genoma, essendo composto da 2.400.000 bp, con 80 esoni, 8 promotori e 5 isoforme. Il prodotto proteico del gene è la distrofina, una proteina con struttura quaternaria composta da 4 domini chiave:
1. Dominio N terminale legante l’actina 2. Dominio C-terminale legante la ECM 3. Dominio cistein-rich 4. Dominio centrale rod-domain
Il ruolo della distrofina è quello di creare un’impalcatura proteica tesa tra i filamenti contrattili di actina e la membrana sarcolemmatica. L’interazione con il sarcolemma è mediata da proteine come il distroglicano α/β e i sarcoglicani. La funzione propria della distrofina è quella di fungere da “ammortizzatore”, dissipare cioè l’energia cinetica prodotta dalla contrazione muscolare. Si distinguono, in base alla gravità, più forme di distrofinopatie:
• Distrofinopatia grave (ex Distrofia muscolare di Duchenne): totale assenza della distrofina • Distrofinopatia benigna (ex Distrofia muscolare di Becker): riduzione ma non totale assenza
della distrofina oppure ipofunzione della distrofina espressa in quantità noramali. • Distrofinopatie nelle portatrici sane
Le distrofinopatie sono malattie genetiche ereditate con modalità X-linked recessiva, con incidenza di 1/3500 maschi nati vivi. La patogenesi della malattia è da ricercare in una mutazione del gene della distrofina. Tali mutazioni sono:
WWW.SUNHOPE.IT

Leonardo Aurino
12
• Nel 75% delezioni più o meno ampie (nel caso della distrofina, minore è l’estensione della delezione maggiore è il danno arrecato alla proteina) del gene, che normalmente insorgono negli hot spot, localizzati all’esone 2 e 11 (per l’estremo 5’ del gene) e dell’esone 44 (posto all’esone 3’ del gene). Tali delezioni determinando una perdita di coppie di basi azotate, possono determinare: a) per le delezioni di un numero di coppie di basi multiplo di 3: mutazioni in frameà la proteina sarà anomala (più corta) ma ancora grossolanamente funzionante à fenotipo Becker b) per le delezioni di un numero di coppie di basi non multiplo di 3: mutazioni out frame o mutazioni con formazioni di codone di stopà la proteina sarà, nel caso delle formazioni di un codone di stop, del tutto assente, oppure estremamente modificata e del tutto non funzionanteà fenotipo Duchenne.
• Nel 25% il gene è interessato da mutazioni puntiformi. La conseguenza del danno genico si riversa sul tradotto proteico, sicché la distrofina non può più agire da ponte (e dissipatore di energia cinetica) tra la F-actina e il β-destroglicano. Pertanto, durante la contrazione muscolare, si realizzano danni alla membrana sarcolemmatica che causano ingresso di ioni Ca+2 e attivazione di caspasi/proteasi che causano necrosi muscolare. Clinica della distrofinopatia grave (fenotipo Duchenne) Data l’ereditarietà X-linked recessiva, si manifesta quasi esclusivamente nel sesso maschile. In 1/3 dei casi la mutazione avviene come evento spontaneo, mentre nei 2/3 dei casi vi è una chiara ereditarietà. Si ricorda che la malattia può interessare, sebbene raramente, anche il genere femminile. Ciò avviene nelle donne portatrici “sane” di malattia nel momento in cui il fenomeno di lyonizzazione interessi prevalentemente il cromosoma X sano. In questi rari casi è possibile visualizzare segni laboratoristici o, a volte, perfino clinici di malattia. L’esordio della malattia è precoce. Alla nascita i muscoli scheletrici sono perfettamente normali. Intorno ad 1 anno di età vi è uno stadio subclinico di malattia, caratterizzato da una aumento delle transaminasi, di verosimile origine muscolare. La capacità di deambulare viene appresa molto tardivamente, di norma dopo i 2 anni di età. Si osserva, già a questa età, una pseudoipertrofia dei gastrocnemi, secondaria a sostituzione fibro-adiposa del tessuto muscolare. I deficit motori si fanno evidenti all’età di 2-3 anni, con incapacità nel compiere movimenti e mantenere la stazione eretta in maniera idonea, con conseguenti frequenti cadute. Nel momento di alzarsi il paziente fa forza con le braccia (segno di Grower) Le deambulazione è anserina e nel 50% dei casi vi è un ritardo mentale. La sintomatologia peggiora con i mesi estivi e con il caldo in generale, in quanto l’elevata temperatura determina un “allargamento” delle maglie del sarcolemma, cagionando un maggior ingresso di ioni Ca+2. L’età in cui viene persa totalmente la capacità di deambulare è assai variabile (soprattutto in base alla localizzazione geografica) e va dai 6 ai 12 anni. Il dolore, come in tutte le distrofinopatie è assente. Tuttavia, poiché la miocitolisi interessa prevalentemente i muscoli estensori, può esservi un iperfunzionamento dei muscoli flessori, causa di retrazioni tendinee di segmenti muscolari, con blocchi articolari funzionali. Il dolore, in questo caso, compare ed è di tipo posturale. La prima causa di morte in tutti i pazienti con distrofinopatia grave è la cardiomiopatia di Duchenne o cardiomiopatia distrofinopatica. Interessa soggetti di età compresa tra i 20 e 30 anni. La gravità del coinvolgimento cardiaco è indipendente dal grado di coinvolgimento muscolo-scheletrico. La cardiomiopatia è presente, nel 25% dei casi, già a 6 anni, anche se asintomatica. Il decorso della cardiomiopatia di Duchenne è distinto in 8 stadi:
1. Stadio preclinico: caratterizzato da anomalie all’ECG e all’ECOcardio. All’ECG si mostra una depressione del segmento PQ, un allungamento dell’intervallo QT e un aumento
WWW.SUNHOPE.IT

Leonardo Aurino
13
dell’indice cardiomiopatico, calcolato con !"/!" che è > 4,6. All’ECOcardio si notano discinesie regionali, fibrosi regionale e ipertrofia settale.
2. Stadio ipertrofico: All’ECG segni di ipertrofia ventricolare sinistra (QRS ampi, somma della S in V1 e della R in V6 che supera i 35 mm). All’ECOcardio persiste ipertrofia settale.
3. Stadio aritmogeno: all’ECG o meglio all’Holter-ECG compaiono extrasistole, anche bigemine e run di TV. ). All’ECOcardio persiste ipertrofia settale.
4. Stadio della fibrosi parcellare: ECG: abbassamento dell’onda P e del complesso QRS (da perdita di miociti). All’ECO aumento dello spessore settale e di parete libera per fibrosi.
5. Stadio della firosi diffusa: peggioramento dei succitati parametri. 6. Stadio della cardiomiopatia dilatativa: All’ECOcardio: dilatazione delle 4 camere, FE <
45% e segni di insufficienza valvolare. 7. Stadio dello scompenso cardiaco refrattario alla terapia 8. Morte, a meno di trapianto cardiaco (molto difficile per gli affetti da malattie genetiche)
La seconda causa di morte del soggetto con distrofinopatia grave è l’insufficienza respiratoria, causata da una progressiva compromissione dei muscoli respiratori e dall’evoluzione della scoliosi. Ciò che indica la progressione della limitazione respiratoria è la riduzione della capacità vitale (CV), cioè la massima quantità di aria mobilizzata in un atto respiratorio massimale (v.n. 2,5-5,5 litri). La CV, fisiologicamente, aumenta fino al raggiungimento dei 18 anni, per poi iniziare a declinare gradualmente con l’età. L’aumento della CV è correlato alla deambulazione. Finchè il paziente con DMD deambula, la sua CV aumenta. Al momento in cui la capacità di deambulare viene persa, la CV declina significativamente, dopo un periodo di plateau, al ritmo di 200 ml/anno e con essa l’aspettativa di vita. Inoltre, nella DMD, la CV è fortemente ridotta dalla curvatura cifoscolitica del rachide, che determina un tipico pattern restrittivo alla spirometria. Clinica della distrofinopatia benigna (fenotipo Becker) Da un punto di vista clinico, il soggetto con distrofinopatia benigna presenta sintomi simili alla DMD, ma notevolmente più attenuati. I sintomi tendono a comparire dopo i 10 anni, talvolta in età adulta. La perdita della deambulazione avviene tra la V e la VI decade. Caratteristicamente, l’ipertrofia dei gastrocnemi è molto più accentuata. La cardiomiopatia di Becker, se compare, lo fa in maniera più grave, anche perché il paziente affetto cammina e dunque ha un maggior consumo di ossigeno. Per questo motivo può essere consigliata la cessazione della deambulazione. In questi soggetti il trapianto cardiaco è più facilmente approvato. L’insufficienza respiratoria, intesa come perdita progressiva della CV, è molto più sfumata rispetto alla DMD, in quanto strettamente dipendente dalla capacità a deambulare. Distrofinopatia nelle portatrici sane (carriers) Si tenga presente che, siccome 1/3 delle DMD deriva da una mutazione sporadica “de novo”, non tutte le madri di soggetti affetti da DMD sonoportatrici. Nel 10% vi è comparsa di sintomatologia (manifesting carriers). In questo gruppo di pazienti il coinvolgimento cardiaco è molto superiore rispetto a quello muscolare. La progressione a scompenso cardiaco è comunque molto tardiva, ma può richiedere trapianto. Il 10% delle portatrici sono invece silenti, cioè non manifestano sintomi. In alcune di esse può essere comunque presente ipertrofia dei gastrocnemi. Nel restante 80% dei casi può essere un incremento della CPK sierica, soprattutto della forma CPK-MM.
WWW.SUNHOPE.IT

Leonardo Aurino
14
Diagnosi di distrofinopatia: La diagnosi di distrofinopatia si basa, innanzitutto, sul sospetto clinico-anamnestico. Esami di laboratorio utili sono:
1. Dosaggio transaminasi (nella fase preclinica) 2. Dosaggio CPK-MM, che mostra un incremento dei valori anche di 100 volte 3. Dosaggio di LDH e aldolasi, che risultano aumentate.
Il gold standard per la diagnosi di distrofinopatia è la PCR. Nel maschio la PCR determina amplificazione di tutti gli esoni eccetto quelli deleti, mentre nella femmina può esservi solo riduzione del segnale poiché vi è disponibilità anche dell’allele sano. La biopsia muscolare si esegue solo in caso di test genetico non diagnostico. Essa mostra infiltrazione macrofagica e sostituzione fibroadiposa muscolare. All’immunoistochimica vi è una riduzione/assenza del segnale derivante dalla distrofina. All’EMG si realizza un quadro di danno miopatico, con riduzione dell’intensità e del numero dei potenziali d’azione motori. Terapia: le distrofinopatie, anche quelle gravi (DMD), sono malattie inguaribili ma non incurabili, potendo intervenire per migliorare la qualità e la quantità della vita. Ad oggi, la maggior parte dei pazienti, anche quelli affetti da DMD, supera i 30 anni. Negli anni ’70 la terapia si basava solo sulla fisioterapia e sull’uso di ortesi per prevenire le anchilosi in flessione e la cifoscoliosi. Negli anni ’80 la terapia consisteva in:
1. Precoce trattamento chirurgico delle retrazioni tendinee, causa di anchilosi in flessione, tramite tenotomie multiple entro 6 mesi dalla comparsa di retrazione. - tenotomia Achillea à previene il piede equino - tenotomia del tendine del semimebranoso/semitendinoso à previene il ginocchio varo e flesso - tenotomia del tendine del tensore di fascia lata à previene la flessione delle cosce sul bacino.
2. Precoce trattamento della scoliosi, per ridurre la perdita della CV. Negli anni ’90 si interveniva con un precoce trattamento della insufficienza respiratoria a mezzo di PEP mask da utilizzare durante la notte. La terapia farmacologica consiste nell’utilizzo di corticosteroidi (deflazacort alla dose di 0,75mg/kg/die alle ore 8 del mattino), la cui azione sembrerebbe essere quella di stabilizzare la membrana sarcolemmatica. Negli anni 2000 la terapia consiste in:
• Microtracheotomia (naso artificiale di Rideau): si attua quando la CV è ancora buona, ma vi è una perdita > 50 ml/anno. Tale tecnica permette di preservare la funzione fonatoria e di utilizzare la ventilazione tracheotomia di notte (mentre di giorno si chiude la stomia)
• Terapia farmacologica con gentamicina in pazienti con specifiche mutazioni puntiformi determinanti la formazione di un codone di stop. L’unica mutazione che permette l’utilizzo di gentamicina è quella in cui vi è una tripletta UGA (codone di stop definito Opale) seguita da una C (tale mutazione è responsabile solo dell’1% dei casi di distrofia muscolare grave). Effetti avversi dell’uso di amminoglucosidi (gentamicina) sono la nefrotossicità (che in questo caso è trascurabile in quanto si fa una singola simmonistrazione settimanale) e la ototossicità (solo per i portatori di una specifica mutazione del mt-DNA)
• Terapia genica: Il trattamento del futuro si basa sulla manipolazione genetica. La prospettiva terapeutica considerata migliore si basa sull’utilizzo di oligonucleotidi antisenso. Tali oligonucleotici, iniettati sottocute, inibiscono l’espressione genica ibridizzandosi con l’mRNA ottenuto dalla trascrizione del gene della distrofina, in particolare legandosi alla sequenza mutata o deleta. L’esclusione di tali sequenze mutate determinerebbe la formazione di una proteina troncata, più corta, ma parzialmente funzionante. Oggi questa
WWW.SUNHOPE.IT

Leonardo Aurino
15
tecnica, definita exon skipping, è limitata solo alle mutazioni dell’esone 53 (risultati poco soddisfacenti).
DistrofiamuscolarediEmery-DreifussÈ una DMP dovuta a mutazione del gene localizzato sul cromosoma Xq28 codificante per l’emerina, proteina deputata al mantenimento della forma e dell’integrità del nucleo delle fibrocellule muscolari scheletriche e cardiache. Trattasi dunque di una emerinopatia X-linked recessiva. Esiste una forma di distrofia di Emery-Dreifuss AR legata a mutazione del gene lamina A/C localizzato sul cromosoma 1. L’esordio della malattia avviene in genere nella prima decade di vita e si manifesta in genere con ipotrofia e ipostenia muscolare a localizzazione omero-tibio-peroneale.. Tipicamente la distrofia interessa anche i muscoli nucali (rigidità nucale). In questa sindrome è peculiare l’interessamento cardiaco (quasi costante), che si manifesta con blocchi della conduzione dell’impulso cardiaco e cardiomiopatia dilatativa. La possibilità di una morte cardiaca improvvisa impone un costante monitoraggio cardiologico e l’impianto di un pacemaker/defibrillatore cardiaco. La diagnosi si basa sulla biopsia muscolare nei difetti di emerina e sull’analisi genetica nei difetti di lamina A/C. Si ricordi che il difetto di lamina A/C può associarsi ad una polineuropatia congenita mista ad interessamento distale, la sindrome di Charcot-Marie-Tooth. La terapia prevede la correzione delle contratture muscolari e l’impianto di pace-maker/ICD. Distrofiafacio-scapolo-omeralediLandouzy-DejerineÈ una delle forme più frequenti di distrofia muscolare. Ha una trasmissione AD. Si riconoscono due forme di malattia:
• Forma classica (90% dei casi) causata da una delezione subtelomerica del cromosoma 4 in sede q35. Esordisce tra i 7 e 15 anni con sintomi quali ipostenia dell’orbicolare dell’occhio e della bocca, con difficoltà a serrare con forza le palpebre e a fischiare o gonfiare le gote. L’ipostenia interessa di norma, sebbene più tardivamente, anche il cingolo scapolo-omerale (da cui il nome di distrofia facio-scapolo-omerale) con impossibilità nel sollevare le braccia in abduzione oltre un dato limite. Durante questa manovra si assiste anche alla protrusione posteriore con intrarotazione delle scapole. La malattia in fase avanzata può interessare anche i muscoli prossimali degli arti inferiori e il cingolo pelvico.
• Forma infantile (10% dei casi), con esordio intorno ai 2 anni, con distribuzione dell’ipostenia che ricalca quella dell’adulto ma con severità maggiore. La compromissione dei muscoli facciali da luogo ad una completa amimia. La perdita della deambulazione avviene intorno ai 10 anni d’età.
La diagnosi di distrofia di Landouzy-Dejerine si basa sull’anamnesi genetica (positiva), sul dosaggio della CK/aldolasi/LDH (che sono aumentati) e sull’EMG (pattern miopatico). La biopsia muscolare dimostra polidimensionalità delle fibre e lieve fibrosi con infiltrazione leucocitaria (spt macrofagi). Distrofiemuscolarideicingoli(LGMD-Limb-Girdlemuscolardystrophies)Sono distrofie che interessano prevalentemente la muscolatura prossimale scapolo-omerale e pelvi-femorale (cingoli) Si conoscono di tale malattie forme AD e AR.
WWW.SUNHOPE.IT

Leonardo Aurino
16
Forme autosomiche dominanti: Sono le forme più benigne e più rare (meno del 10% di tutte le LGMD). Tra queste si annoverano:
• LGMD 1A: dovuta a mutazione della miotilina • LGMD 1B: dovuta a mutazione del gene codificante per la lamina A/C. Trattasi dunque di
una laminopatia. L’età di esordio è compresa tra 9-65 anni. La clinica è variabile potendosi riscontrare distrofia isolata dei cingoli, da sola o associata a cardiomiopatia dilatativa, iperCKemia asintomatica.
• LGMD 1C: dovuto a mutazioni del gene codificante per la caveolina • LGMD 1D: mutazione non ancora conosciuta • LGMD 1E: mutazione non ancora conosciuta • LGMD 1F: mutazione non ancora conosciuta • LGMD1G: mutazione non ancora conosciuta
Forme autosomiche recessive:
• LGMD 2A: dovute a mutazioni del gene della calpaina. Trattasi dunque di una calpainopatia. È la forma più frequente di LMGD, soprattutto nel Sud Italia. L’età d’esordio è compresa tra 15 e 50 anni. Si riscontra evidente ipotonotrofia dei due cingoli, con evidente scollamento delle scapole nell’atto di abduzione e sollevamento delle braccia. L’evoluzione della malattia è più grave nei maschi. L’iperCKemia è solitamente molto alta. Il coinvolgimento cardiaco manca in quasi il 70% dei casi, mentre il coinvolgimento respiratorio è graduale, con deterioramento della CV nel 30% dei casi.
• LGMD 2B: dovuta a mutazioni della disferlina. Trattasi di una disferlinopatia. Insorge tipicamente in pazienti che praticano sport a livello agonistico. Il coinvolgimento dei cingoli e le scapole alate non sono molto importanti, mentre è più indicativa una ipotonotrofia dei muscoli distali, con atrofia dei muscoli della loggia posteriore della gamba. Vi è significativa iperCKemia. L’interessamento cardiaco, con aritmie, è presente nel 20% dei casi.
• LGMD 2C, 2D, 2E, 2F: sono tutte dovute a mutazione dei sarcoglicani, rispettivamente α, β, γ, δ. Trattasi di sarcoglicanopatie. Nel sud Italia prevale la forma 2C. La forma più frequente è la 2D. Vi è notevole iperCKemia. L’interessamento cardiaco è presente soprattutto nelle forme 2C e 2F, mentre l’interessamento respiratorio è presente in tutte e quattro le forme.
• LGMD 2G: dovuta a mutazioni del gene codificante per la telethonina. Vi interessamento muscolare prossimale e distale con cardiomiopatia.
• LGMD 2H: dovuta a mutazioni per il gene codificante per TRIM32. I ¾ delle persone affette sono maschi. La malattia è tipica di alcune comunità chiuse con endogamia (quaccheri, mormoni, anabattisti). Nel bambino è tipica la pseudoipertrofia dei gastrocnemi, negli adulti l’atrofia dei muscoli del volto (à amimia). Il coinvolgimento cardiaco conduce nella maggior parte dei casi a BBD, specie in età avanzata.
• LGMD 2I: dovuta a mutazioni del gene codificante per la fukutin-related protein. Si conoscono due mutazioni di questo gene, la R54W (con evoluzione più rapida e clinica più grave) e la L276I (più benigna). Il rapporto M:F è di 11:4. L’iperCKemia è di solito molto frequente. Non vi è mai coinvolgimento cardiaco, mentre il coinvolgimento respiratorio è grave e debilitante e rappresenta la prima causa di morte nei soggetti affetti. Gli individui affetti spesso necessitano di ventilazione meccanica assistita.
• LGMD 2J: conseguente a mutazioni del gene codificante per la titina.
WWW.SUNHOPE.IT

Leonardo Aurino
17
DistrofiamiotonicaLe distrofie miotoniche sono malattie multisistemiche, che interessano il muscolo scheletrico e in varia misura il muscolo cardiaco (àblocchi di conduzione e cardiomiopatia dilatativa), il corpo vitreo dell’occhio (àcataratta) , il SNC (à ritardo intellettivo) , le ghiandole sessuali (àipotrofia /atrofia gonadica e sterilità), le ghiandole endocrine (àdiabete e ipotiroidismo) e gli annessi cutanei (àcalvizie precoce). Sono state identificate due forme di distrofia miotonica:
• TIPO I: definita distrofia di Steinert, causata da mutazioni del gene della miotonina protein chinasi (DMPK)
• TIPO II: definita PROMM, caratteristica per l’interessamento miotonico prossimale, secondaria a mutazione del gene ZNF9.
Entrambe le malattie sono ereditate con meccanismo autosomico dominante E sono caratterizzate dall’espansione di nucleotidi ( tipletta CTG per la distrofia di Stienert e quadripletta CCTG nella PROMM). Clinicamente la distrofia di Steinert, più frequente, si caratterizza per il coinvolgimento della muscolatura distale e dei muscoli mimici. Vi è comunque ipostenia muscolare generalizzata. Nella PROMM, malattia molto rara, è invece tipico l’interessamento prossimale. Entrambe le forme sono tipicamente associate al fenomeno miotonico (da cui il nome di distrofie miotoniche), ossia l’incapacità di decontrarre un muscolo subito dopo l’utilizzo. Da un punto di vista elettromiografico il fenomeno miotonico si caratterizza per la persistenza di un potenziale d’azione anche dopo la sospensione del movimento volontario. Esempi di fenomeni miotonici sono la difficoltà di rilasciare la mano dopo averla stretta a pugno o dopo aver stetto una mano in forma di saluto, nella difficoltà ad aprire gli occhi dopo averli serrati, o nella difficoltà a deglutire, per contrazione dei muscoli oro-faringei, durante la masticazione (soprattutto se il cibo in questione è particolarmente freddo, come un gelato). Il fenomeno miotonico peggiora sensibilmente con le temperature fredde, con l’emotività e con l’assunzione di alcuni farmaci, mentre si riduce con il persistere dell’attività volontaria. Il fenomeno miotonico è secondario ad un difetto dei canali ionici, e migliora sensibilmente con l’utilizzo di farmaci attivi sui canali, come il chinino e gli antiaritmici procainamide e mexiletina. La prognosi è infausta, la morte avviene in prossimità del compimento dei 50 anni d’età. La morte nella tipologia 1 avviene spesso per aritmie fatali.
Atrofiamuscolarespinale(SMA)La SMA rientra fra le distrofie muscolari. In realtà si tratta di un insieme di entità nosologiche, tutte caratterizzata da una progressiva degenerazione del II moneurone, siti a livello delle corna grigie anteriori del midollo, con conseguente danno neurotrofico muscolare. Le SMA sono malattie genetiche ereditate in modo AR (ad eccezione della sindrome di Kennedy che è X-linked recessiva). La principale mutazione delle SMA è a carico del gene SMN1 (95% dei casi) sito sul cromosoma 5 in posizione 5q13. Come detto, col termine SMA si intendo diverse entità clinico-nosologiche, in particolare:
• SMA tipo I (malattia di Werding-Hoffmann): Si manifesta nei primissimi giorni di vita o addirittura già in utero, con una riduzione dei movimenti fetali spontanei. Il neonato si presenta con ipotonia generalizzata (aspetto definito come floppy infant), paralisi dei muscoli prossimali del tronco che causano una tipica respirazione prettamente addominale. I muscoli del viso sono risparmiati dalla malattia e l’aspetto del neonato è attento e vivace. La malattia ha un decorso rapido a causa della paralisi dei muscoli respiratori. L’interessamento dei muscoli deglutitori è causa di scialorrea. A causa della disfagia (su base organica),
WWW.SUNHOPE.IT

Leonardo Aurino
18
l’alimentazione spesso diviene di tipo artificiale, a mezzo di SNG o di PEG. La morte avviene di norme entro 6-8 mesi, molto raramente i bambini superano l’anno di vita.
• SMA tipo II: definita anche forma intermedia di SMA, esordisce dopo i primi 6 mesi di vita, durante i quali il bambino si muove in modo apparentemente normale. Dopo il compimento del 6° mese il bambino mostra notevole difficoltà alla stazione eretta, mentre quella seduta, una volta ottenuta, viene mantenuta. L’atrofia e la paralisi muscolare sono particolarmente evidenti a livello dei muscoli del bacino e degli arti inferiori, motivo per il quale vi è la difficoltà a reggersi in piedi e a deambulare. I muscoli del tronco e quelli respiratori sono relativamente risparmiati. Il decorso della malattia è cronico e l’aspettativa di vita va oltre l’età adulta in molti casi. La principale caratteristica della SMA di tipo II è la cifoscoliosi che ha esordio molto precoce (primi anni di vita) che può determinare un deficit respiratorio restrittivo che può richiedere ventilazione meccanica.
• SMA tipo III (sindrome di Kugelberd-Welander): forma definita lieve di SMA, si manifesta ad un’età molto variabile. Spesso la malattia compare nei primissimi anni di vita, quando il bambino è già in grado di camminare , con un difficoltà nella corsa, nel salire le scale e nel rialzarsi da terra. Non raramente gli stessi segni possono essere osservati anche in adolescenza o nel giovane adulto. Nella forma SMA III i muscoli del tronco e quelli respiratori sono poco interessati, per cui la scoliosi e le difficoltà respiratorie ad essa connesse non sono di solito presenti.
• SMA tipo IV: definita anche SMA dell’adulto, è una forma che colpisce, appunto, gli adulti, in quanto i sintomi si presentano di norma oltre i 35 anni. Ha un inizio insidioso ed una lenta progressione.
• SMA X-linked (sindrome di Kennedy): anche nota come atrofia muscolare bulbo-spinale si manifesta solo nei maschi, in quanto ereditata in modo eterocromosico recessivo (X-linked recessivo). Questa forma di SMA è associata ad una mutazione del gene che codifica per il recettore per gli androgeni, posto sul cromosoma X. La mutazione consiste in una espasione di triplette CAG, che vengono ripetute da 38 a 62 volte (valori normali di triplette CAG nel gene sono compresi tra 9-36). La malattia determina una condizione di ipogonadismo, con atrofia testicolare e ginecomastia, oltre ad un condizione di distrofia muscolare che interessa prevalentemente i muscoli facciali e la lingua. Il decorso della SMA X-linked è variabile, solitamente tende comunque a progredire molto lentamente.
Da un punto di vista genetico, come detto, le SMA (ad eccezione della sindrome di Kennedy) sono malattie AR legate a mutazioni del gene SMN1 posto sul 5q13. In realtà, in tale locus si trovano due geni, SMN1 e SMN2. Solo la contemporanea mutazione su entrambi i cromosomi 5 di SMN1, ma non di SMN2, porta alla malattia. Il gene SMN1 (SMN sta per survival motor neuron) è un fattore di sopravvivenza dei motoneuroni di II ordine. Le mutazioni del gene SMN2, posto sempre sul 5q13, a differenza di SMN1, non sono causa di malattie, nonostante i geni siano quasi identici (cambiano solo per 2 singoli nucleotidi, uno all’esone 7 e uno all’esone 8). Sia il gene SMN1 che SMN2 producono la proteina SMN, ma quella prodotta da SMN2 è molto meno funzionante e quindi insufficiente. I pazienti con SMA sono, quindi, in grado di produrre una certa quota di SMN, che risulta però insufficiente a causa della delezione del gene SMN1. La diagnosi di SMA è soprattutto clinica ed è confermata dall’indagine genetica. I genitori di soggetti affetti da SMA sono quasi sempre portatori sani (in quanto la malattia è AR) e, in quanto tali, hanno il rischio del 25% di generare un altro figlio affetto da SMA ad ogni nuova gravidanza. Solo meno del 2% dei casi di SMA insorge, infatti, come mutazione “de novo”. È disponibile un test quantitativo che mette in evidenza il numero di geni SMN1 per definire lo stato di portatore sano di SMA. Il test ha una affidabilità del 95%. La diagnosi prenatale di SMA si basa sull’analisi genetica delle cellule ottenute mediante villocentesi.
WWW.SUNHOPE.IT

Leonardo Aurino
19
CardiomiopatiegeneticheLe cardiomiopatie genetiche sono un gruppo di patologie genetiche che colpiscono primitivamente il muscolo cardiaco. Le CM vengono ad oggi classificate dalla WHO nel seguente modo: 1.CM genetiche
• CM ipertrofica • CM dilatativa • CM restrittiva • CM aritmogena del VD
2.CM specifiche: cardiomiopatia distrofinopatica (da distrofinopatie) o neuromuscolare (da atassia di Friederich) 3.CM secondarie: a patologie infiammatorie (miocarditi), a patologie metaboliche, valvolari, ischemiche, ipertensive. 4.CM non classificate
• Fibroelastosi endocardica • CM mitocondriali • Mildly-Dilated CM • CM con miocardio non compatto
CardiomiopatiaipertroficaÈ una cardiomiopatia determinante una ipertrofia asimmetrica del SIV, soprattutto nella sua variante subaortica, con prevalente interessamento del VS (raramente interessa anche il VD). La modalità di trasmissione della malattia è AD nel 70%, con penetranza incompleta e espressività variabile. Nella sua variante “giapponese” l’ipertrofia interessa soprattutto il setto apicale. Sono state individuate più di 100 mutazioni in almeno 9 geni diversi, tutti codificanti per proteine sarcomeriche. Tra questi geni si annoverano:
1. Miosina cardiaca legante la proteina C (responsabile del 30% dei casi di CM ipertrofica) 2. Catena pesante della β-miosina (20% dei casi) 3. TnT cardiaca (5% dei casi) 4. TnI cardiaca 5. Catena leggera essenziale della β-miosina 6. Catena leggera regolatrice della β-miosina 7. Α-tropomiosina 8. Titina
Si noti che il 40% dei soggetti affetti da CM ipertrofica ha delle mutazioni in geni non ancora individuati. Da un punto di vista patogenetico, il disordine strutturale sarcomerico è causa di ingresso di ioni Ca+2, con conseguente morte cellulare e sostituzione, dopo una fase di compenso, con tessuto fibroso. L’eccesso di calcio determina inoltre anche un certo grado di disfunzione mitocondriale, con ridotta capacità da parte della fibrocellula muscolare cardiaca di produrre energia. Da un punto di vista anatomo-patologico, un cuore affetto da CM ipertensiva mostra:
• Aumento del peso (anche del 100-200%) • Aumento di spessore del SIV (90%) e della parete libera del VS + SIV (10% dei casi) • Riduzione del volume endocavitario del VS
WWW.SUNHOPE.IT

Leonardo Aurino
20
• Evidente ipertrofia del miocita • Perdita della normale disposizione spaziale in parallelo delle fibre (miocardial disarray) • Fibrosi
Da un punto di vista fisiopatologico, le modificazioni che occorrono a carico del VS determinano, col tempo, uno stato di progressiva ipertrofica concentrica del VS, con riduzione del volume endocavitario del VS e conseguente deficit diastolico. Il deficit diastolico deriva tanto dall’aumento della massa muscolare cardiaca che dalla fibrosi endocardica, in quanto entrambi le condizioni determinano un aumento del tempo necessario al rilasciamento telesistolico, con conseguente riduzione del tempo totale di diastole. Inoltre, la riduzione del volume endocavitario del VS determina una riduzione del volume telediastolico. Come detto, nella maggior parte dei casi esiste inoltre una spiccata ipertrofia del SIV, soprattutto nella sua porzione sottovalvolare, configurando un quadro di stenosi aortica sottovalvolare, causa di una riduzione della gittata sistolica. L’accelerazione che il sangue subisce al momento in cui transita attraverso la stenosi sottovalvolare determina, per effetto Venturi, un SAM (systolic anterior movement), cioè un movimento della cuspide antero-mediale della valvola mitralica, determinando una insufficienza mitralica funzionale. La presenza o l’assenza dell’ostruzione all’efflusso ventricolare sx permette di distinguere, inoltre, le cardiomiopatie ipertrofiche in:
1. CM ipertrofica forma ostruttiva (con ostruzione subaortica o medioventricolare) 2. CM ipertrofica non ostruttiva
Da un punto di vista clinico, il paziente con CM ipertrofica ha spesso una storia familiare positiva per morte improvvisa cardiaca, oltre ad una storia personale di episodi di sincopi e dolori anginosi, astenia e dispnea. Le principali manifestazioni della CM ipertrofica sono:
1. Angina e CAD 2. Sincope 3. Dispnea 4. Morte improvvisa cardiaca 5. Aritmie 6. Scompenso cardiaco (prima diastolico, poi, nell’end-stage della malattia, di tipo sistolico).
All’e.o. si registra un itto palpabile, sdoppiamento del secondo tono T2 (da ritardo di chiusura della valvola aortica), soffio sistolico aspro, eiettivo, di conformazione a diamante sul focolaio di Erb da stenosi valvolare aortica sottovalvolare, più un eventuale soffio olosistolico, dolce e aspirativo al 4° s.i.s. da insufficienza mitralica funzionale. All’Holter-ECG delle 24-48-72 ore si nota:
• Riduzione del PR + BBD • Segni di ipertrofia ventricolare sinistra, distinta in 4 gradi:
I) : somma della S più profonda in V1 + somma della R più alta in V6 > 35 mm (indici di Sokolov) II) : punto I a cui si aggiunge un’area T < 2/3 dell’area del QRS III ) punto I a cui si aggiunge una T piatta IV) punto I a cui si aggiunge una T invertita
All’ECOcardiografia si nota: • Dimensioni delle camere e volumi ventricolari ridotti con FE che può essere normale o
lievemente ridotta (come da ipertrofia concentrica) • Indice cardiaco, ossia spessore SIV/spessore parete libera VS > 1.3 (indice di ipertrofia
asimmetrica) • Visualizzazione del SAM
WWW.SUNHOPE.IT

Leonardo Aurino
21
• Valutazione del grado di impedenza all’efflusso VS, tramite misurazione dell’AVA e del gradiente medio/picco transaortico.
La Cardio-RM permette di valutare lo stadio della fibrosi cardiaca e le eventuali alterazioni del microcircolo. Da un punto di vista prognostico, la CM ipertrofica col tempo tende a virare in una CM dilatativa, per progressivo sfiancamento del muscolo cardiaco e assottigliamento delle pareti ventricolari. Tale condizione configura l’end stage della CM ipertrofica e può essere favorita dall’uso di inotropi negativi (β-bloccanti e CCB non diidropiridinici). La prognosi della CM ipertrofica è strettamente connessa alla comparsa di morte improvvisa cardiaca. La morte cardiaca improvvisa per CM ipertrofica è nella maggior parte dei casi secondaria a complicanze aritmiche ipercinetiche ventricolari. In alcuni casi, un arresto cardiaco può essere determinato, tipicamente in corso di sforzo, quando vi è una ostruzione acuta all’efflusso VS. Fattori prognostici negativi sono:
• Presenza di run di TV all’holter-ECG • Esordio in età pediatrica/giovanile • Familiarità per morte cardiaca improvvisa • Presenza di una forma ostruttiva (dimostrata da un elevato gradiente pressorio transvalvolare
all’ECOcardio) • Presenza di alcune mutazioni specifiche
CardiomiopatiadilatativaMalattia del miocardio caratterizzata da una riduzione della funzione sistolica del VS o di entrambi i ventricoli, associata a dilatazione cardiaca. La malattia mostra nel 50% una forte familiarità. I geni che, se mutati, sono responsabili dello sviluppo della malattia sono 10, tutti codificanti per proteine sarcomeriche (come nel caso delle distrofinopatie X-linked), del citoscheletro, proteine nucleari (come nel caso dell’emerina) o proteine transmembrana. In alcuni soggetti la cardiomiopatia dilatativa può essere secondaria anche a danno autoimmune diretto contro antigeni miocardici, come le proteine contrattili (miosina) o le chaperonine HSP-60. Da un punto di visa anatomo-patologico, macroscopicamente si osserva:
• Cuore globoso, dal peso aumentato (cor bovinum, anche se l’aumento del peso è inferiore rispetto alla CM ipertrofica)
• Dilatazione delle cavità ventricolari e dunque degli osti valvolari atrio-ventricolari (insufficienza mitralica/tricuspidalica secondaria)
• Formazione di trombi in recessi ventricolari e/o atriali per la stasi di sangue conseguente alla riduzione della funzione sistolica
Microscopicamente, invece, si nota: • Ispessimento fibrotico/fibroelastico, prima parcellare poi diffuso • Infiltrati linfomonocitari • Fibrosi interstiziale pericellulare e perivasale • Ipertrofia dei cardiomiociti con aumento della lunghezza
Da un punto di vista fisiopatologico, la CM dilatativa è causa di un aumento del volume endocavitario del VS con assottigliamento della parete ventricolare. Il miocardio mostra inoltre una scarsa contrattilità in quanto distesamente fibrotico. Tale condizione determina una riduzione della gittata sistolica, con aumento del residuo telesistolico. Pertanto, vi è una netta riduzione della FE, fino allo scompenso di tipo sistolico. I pazienti con scompenso sono distinti in base alle classi NYHA:
WWW.SUNHOPE.IT

Leonardo Aurino
22
• I classe NYHA: l’ordinaria attività non provoca sintomatologia (né angina, né dispena né cardiopalmo)
• II classe NYHA: lieve limitazione dell’attività fisica. Sintomi assenti a riposo ma compaiono con sforzi prolungati
• III classe NYHA: sintomi compaiono anche per sforzi lievi e abituali • IV classe NYHA: sintomi presenti anche a riposo
La ridotta gittata sistolica determina, inoltre, una riduzione della perfusione di organi come il SNC (possibili sincopi/lipotimie e stroke ischemici), rene (riduzione della VFG). L’incapacità dell’atrio sinistro di svuotarsi in un ventricolo sinistro con deficit sistolico (e quindi con residuo telesistolico aumentato) determina una condizione di stasi polmonare, che può esitare in edema polmonare, dapprima interstiziale dunque alveolare. Frequente è inoltre il riscontro di un versamento pleurico trasudatizio nei recessi costo-frenici o costo-mediastinici di sinistra. All’esame obiettivo, la CM dilatativa mostra i segni di uno scompenso cardiaco congestizio, con itto apicale ampio, toni parafonici, soffi da insufficienza atrio-ventricolari (soffi olosistolici “a getto di vapore”), rantoli polmonari bilaterali da edema, congestione venosa centrale e periferica (edemi declivi, turgore giugulare, fegato da stasi con polso epato-giugulare). All’ECG:
• Scarsa progressione dell’onda R nelle precordiali • Onde Q seguite da onde R basse (definiti complessi QS) nelle precordiali • Alterazioni della ripolarizzazione (come BBS, inversione delle onde T) • Tachiaritmie, specialmente FA
All’RX-torace: • Cardiomegalia, con indice cardiotoracico > 0.55 • Accentuazione arco inferiore di sx (corrispondente al VS) • Congestione ilare e possibile obliterazione seno costo-frenico sx (da edema polmonare
cronico) All’ECOcardiogramma:
• Aumento del volume endocavitario del VS e del VD • FE < 40% • Assottigliamento parete libera e SIV • Insufficienze valvolari atrio-ventricolari
La prognosi della cardiomiopatia dilatativa è infausta. Nel momento in cui compare scompenso cardiaco, il 30% muore entro 1 anno, il 65% entro 5 anni. Fattori prognostici negativi, di avvenuto scompenso, sono:
• Indice cardiotoracico all’Rx > 0.55 • Insufficienza valvolare atrioventricolare • BBS e FA • Aumento del peptide natriuretico atriale (BNP) • Classe NYHA IV
CardiomiopatiaaritmogenadelVDConosciuta storicamente anche cardiomiopatia aritmogena del ventricolo destro, è ad oggi definita semplicemente cardiomiopatia aritmogena in quanto è ormai riconosciuto l’interessamento biventricolare. La malattia è caratterizzata da una sostituzione del tessuto miocardico con un tessuto fibro-adiposo (adipositas cordis), responsabile di alcune forme di aritmia ipercinetica. La malattia è trasmessa in modo autosomico dominante con penetranza variabile. Le mutazioni interessano 8 loci genici, disposti su 4 cromosomi diversi. I geni mutati sono definiti ARVD 1-2-3-
WWW.SUNHOPE.IT

Leonardo Aurino
23
4-5-6-7-8, codificanti per il recettore rianodinico cardiaco (un recettore-canale per il Ca) e per alcune proteine strutturali come la placofillina e la desmoplachina). Si conoscono due varianti cliniche e genetiche che sono invece ereditata con carattere autosomico recessivo (variante Naxos e Carvajal). La variante Naxos, tipica dell’isola greca di Naxos (la più grande delle Cicladi), è caratterizzata da cardiomiopatia aritmogena in soggetti con capelli chiari e lanosi, lentiggini e ipercheratosi palmo-plantare. La proteina mutata nella forma Naxos è la placoglobina. Siccome i geni mutati sono quelli che codificano per canali per il calcio e per proteine strutturali delle giunzioni intercellulari, è facile comprendere il nesso con lo sviluppo di aritmie ipercinetiche. La sostituzione fibroadiposa non ha invece una sua specifica spiegazione, secondo alcuni Autori potrebbe essere un reliquato di una miocardite. Morfologicamente si definiscono due varianti di cardiomiopatia aritmogena:
1. Variante infiltrativa: in questa forma vi è una infiltrazione retiforme di tessuto adiposo. La sostituzione adiposa è più spesso limitata solo al ventricolo destro. Il tessuto adiposo è causa di formazione di aritmie da rientro e eventuale morte improvvisa.
2. Variante cardiomiopatica: vi è sostituzione massica del mio cardio ventricolare con tessuto fibroadiposo. Il processo interessa entrambi i ventricoli, di solito risparmiando il setto interventricolare. La riduzione quantitativa di unità contrattili è causa di insufficienza di pompa, pertanto si instaura uno scompenso diastolico che necessita spesso di trapianto cardiaco.
CanalopatieLe malattie causate da mutazioni nei geni che codificano per le subunità dei canali ionici sono indicate come "canalopatie". Canalopatie dei canali del Na+ Sindrome di Brugadaà è una malattia trasmessa in modalità AD, secondaria a mutazione missenso (con LOF) o nonsenso del gene SCN5A, posto sul cromosoma 3. In realtà, tale anomalia è stata riscontrata in non più del 20% dei soggetti affetti. Il cuore dei soggetti affetti è apparentemente sano, ma vi è un consistente rischio di aritmie ipercinetiche ventricolari (soprattutto FV) e di morte cardiaca improvvisa, specie nei soggetti affetti di sesso M di età compresa tra 30 e 40 anni. La diagnosi di sindrome di Brugada si basa, innanzitutto, su alcuni rilievi ECGrafici. Esistono 3 varianti:
1. Sopraslivellamento ST >/= 2 mm nelle derivazioni precordiali di destra (V1-2-3) con seguente onda T negativa (coved type)
2. Sopraslivellamento ST >/= 2 mm nelle precordiali destre con seguente onda T positiva o bifasica (saddle-back pattern).
3. Sopraslivellamento del tratto ST < 2 mm ed un'onda T positiva. Il tipo 3 non è affatto infrequente in soggetti sani.
Sia il tipo 2 che il tipo 3 non sono ritenuti diagnostici se non si convertono in tipo 1 dopo l’esecuzione di un test di provocazione alla flecainide Sindrome di Lev-Lenegre (AD)à associata a mutazione del gene SCN5A, tipica dell’anziano. Canalopatie dei canali del K+ Sindrome del QT lungo (AD o AR)à gruppo di patologie aritmogene legate ad un allungamento della fase di ripolarizzazione ventricolare, con aumentato rischio di FV e torsioni di punta, per mutazioni con LOF di LQT1, 2, 5, 6, 7. La diagnosi si basa sul riscontro di QT lungo in pazienti
WWW.SUNHOPE.IT

Leonardo Aurino
24
con storia di TV sostenuta o familiarità per morte cardiaca improvvisa. I valori normali dell'intervallo QT sono compresi fra 0.30 e 0.44 (0.45 per le donne) secondi. La normalità dell'intervallo QT può essere riconosciuta anche con una stima grossolana, ovvero quando il QT è inferiore a metà dell'intervallo RR. In alcuni soggetti affetti il QT può essere normale, ma si presenta una onda T “matched”, ossia con gobba sulla branca ascendente. La terapia si basa sull’uso di supplementi di potassio (KCl) e β-bloccanti. Nei casi refrattari si impianta un ICD. Il soggetto affetto deve evitare sport agonistici, in quanto l’attività fisica aumenta il rischio di sviluppo di aritmie. Bisogna inoltre evitare l’assunzione di β2-agonisti, antidepressivi, neurolettici e alcuni antibiotici capaci di determinare un allungamento del QT (macrolidi). Sindrome di Jervell-Lange-Nielsenà è una variante della sindrome del QT lungo associata a sordità neurosensoriale congenita. Sindrome del QT corto (AD)à gruppo di patologie aritmogene caratterizzate da una riduzione del periodo refrattario ventricolare ( QT corto), con aumentato rischio di tachiaritmie ventricolari. Le mutazioni responsabili sono di tipo GOF a carico dei geni SOT1, 2, 3. La diagnosi si basa sul riscontro di QT permanentemente < 300msec seguito da onde T alte ed aguzze. La terapia si basa sull’uso della chinidina e sull’impianto di ICD. Canalopatie dei canali del Ca+2
Sindrome di Timothy (AD o AR)à La sindrome di Timothy è una malattia genetica rara che si manifesta con malformazioni corporee, disfunzioni cardiache e disordini psichiatrici. I bambini affetti dalla sindrome di Timothy hanno un aspetto fisico tipico, caratterizzato da anomalie facciali quali sella nasale infossata, sviluppo incompleto della mascella superiore, labbro superiore sottile, orecchie a bassa attaccatura e viso arrotondato. Sono inoltre affetti da sindattilia (fusione delle dita) delle mani e dei piedi, disfunzioni cardiache congenite (QT-lungo e tachicardia ventricolare ), ritardo mentale e disturbo dello spettro autistico. Esistono due forme di sindrome di Timothy (TS1 e TS2). La TS2 è la più grave perché coinvolge maggiormente cuore e cervello rispetto alla TS1. L’esito della malattia è generalmente infausto: la maggior parte dei bambini affetti muore intorno ai 2-3 anni, di solito a causa di un arresto cardiaco, infezioni o altre complicazioni. Chi sopravvive sviluppa forme di ritardo mentale e disturbi dello spettro autistico. La sindrome è associata a mutazioni nel gene CACNA1C, che codifica per una proteina chiamata Cav1.2, responsabile del trasporto dello ione calcio. Tachicardia ventricolare polimorfa catecolaminergica (AR o AD)à malattia causa di un abnorme afflusso di Ca+2 nel miocardio, conseguentemente a stimoli catecolaminergici (sforzo fisico/stress), con predisposizione a gravi aritmie. La malattia tende a presentarsi già entro la prima decade di vita. Le mutazioni responsabili della sindrome sono quelle a carico del gene codificante per la calsequestrina (AR) o per il recettore rianodinico (AD).
WWW.SUNHOPE.IT

Leonardo Aurino
25
Genetica:NEFROLOGIA RenePolicisticoIl rene policistico è una condizione ereditaria, caratterizzata dal progressivo sviluppo, all’interno del rene, di numerosi cisti che, sostituendosi al parenchima, determinano una riduzione della funzione renale, fino a IRC. Si conoscono due forme di questa malattia: una giovanile (pediatrica) e una dell’adulto. La forma giovanile o pediatrica è una malattia ereditata con meccanismo autosomico recessivo. Il gene mutato si trova sul cromosoma 6 e codifica per la fibrocistina, una proteina prodotta dalle cellule epiteliali dei dotti collettori. In questa forma il rene mantiene la sua forma, non è particolarmente ingrandito, ma nella corticale sono presenti numerose cisti di diametro prossimo ai 4 cm. La malattia coinvolge entrambi i reni ed causa di IRC. La forma dell’adulto è ereditata con meccanismo autosomico dominante, ha una penetranza del 100%. I geni coinvolti si trovano sul cromosoma 16 e 4 e codificano per la policistina o PDK1 e PDK2. Le mutazioni di questi geni impediscono la maturazione del glomerulo e dei tubuli convoluti prossimali e distali con conseguente formazione di lesioni aberranti (cisti) che si ingrandiscono nel tempo, raggiungendo la massima grandezza verso i 20-40 anni. In un certo numero di casi, la malattia può essere asintomatica. Quando la malattia è sintomatica, invece, i reni appaiono molto ingranditi, con peso di ciascuno prossimo ai 4 kg e con diametro di 20-30 cm (normalmente è di 10-12 cm). Le cisti hanno localizzazione corticale ma spesso debordano nella midollare. L’evoluzione verso l’IRC e la dialisi è inevitabile e si avvera di norma entro i 54 anni nelle mutazioni di PDK1 e verso i 74 anni nelle mutazioni interessanti PDK2. Sintomo comune è l’ematuria, dovuta ad emorragia intracistica. Evento drammatico è la rottura di cisti, che può determinare abbondante ematuria se il sangue si riversa negli ureteri, oppure, se la rottura avviene nello spazio retroperitoneale, un quadro di peritonite (con dolore e febbre). Può esservi ipertensione, parallelamente all’aumento di dimensioni del rene. Nella forma dell’adulto, nel 30% dei casi, delle cisti si localizzano anche a livello epatico e/o splenico e/o pancreatico. tali cisti possono essere sede di infenzione, possono andare incontro a rottura o possono causare compressione delle strutture limitrofe. Sia nella forma giovanile che in quella adulta, la diagnosi si basa sull’ecografia renale e sulla TAC (nei casi dubbi). La terapia consiste nella prevenzione delle infezioni e della formazione di calcoli, sulla dialisi (o trapianto renale) e sul controllo dell’ipertensione arteriosa. SindromediAlport(X-linkedoAR)È una condizione genetica caratterizzata dalla progressiva perdita della funzione renale e uditiva. Talvolta può esservi anche interessamento oculare. La presenza di ematuria è quasi sempre riscontrabile. La sindrome di Alport è causata da mutazione dei geni COL4A3, COL4A4 e COL4A5, codificanti tutti per catene di collagene. La mutazione più frequente, nell’ 85% dei casi, è quella di COL4A5, localizzato sul cromosoma X (la malattia in questo caso è X-linked e interessa quasi inevitabilmente solo il sesso maschile) . nelle femmine portatrici di tale mutazione solitamente vi è solo emauria, senza progressione verso l’IRC. I geni COL4A3 e COL4A4 sono invece localizzati sul cromosoma 2 e vengono ereditati in modo AR. La mutazione di tali geni impedisce la corretta produzione di collagene di tipo IV, costituente essenziale delle membrane basali a livello renale e auricolare. La perdita di struttura delle MB renali rende impossibile la filtrazione glomerulare, con conseguente proteinuria ed ematuria. La malattia evolve, inevitabilmente, verso l’IRC.
WWW.SUNHOPE.IT

Leonardo Aurino
26
Per fare diagnosi di sindrome di Alport è necessario che si presentino almeno 4 criteri su 10: • Storia familiare di ematuria inspiegata associata a nefrite • Ematuria persistente • Sordità neurosensoriale bilaterale (la sordità si completa in prossimità dei 25 anni) • Mutazione del gene COL4 • Evidenza immunoistochimica dell’epitopo di Alport a livello delle membrane basli
glomerulari • Lesioni oculari (lenticono anterione, cataratta subcapsulare) • IRC terminale in almeno due membri della famiglia • Estese anomalie all’ultrastruttura delle membrane basali
SindromenefrosicacongenitaLa sindrome nefrosica, in generale, è definita da:
• Proteinuria che supera il range nefrosico ( > 3g/24h) • Ipoalbuminemia • Edemi diffusi • Iperlipidemia e iperTGemia • Anoressia, astenia • Anemia • Ipocalcemia/iperfosfatemia e conseguente iperPTH
Tra le sindromi nefrosiche congenite si annoverano:
1. Sindrome nefrosica congenita di tipo finnico: Nefropatia AR dovuta a mutazione del gene nefrina posta sul cromosoma 19. La malattia è presente fin dalla nascita. Pochi sopravvivono oltre l’anno di vita. Morfologicamente il rene mostra cisti corticali dei tubuli convoluti prossimali, ipertrofia delle cellule epiteliali, con espansione del mesangio e ispessimento della parete dei capillari. La diagnosi si basa sulle seguenti condizioni: a) parto prematuro b) placenta ingrandita c)elevati livelli di α-fetoproina nel liquido amniotico (come espressione di proteinuria)
2. Sclerosi mesangiale diffusa: la malattia è idiopatica. È associata a pseudoermafroditismo (sindrome di Dash) o a microencefalia (sindrome di Galloway-Mowat). Si manifesta con ipertensione arteriosa, proteinuria e sindrome nefrosica entro il primo anno di vita. Vi è una progressione rapida verso l’IRC entro i 4 anni.
NefroblastomaoTumorediWilmsÈ il tumore maligno renale più frequente nel paziente pediatrico. Il tumore origina dalle cellule del blastema nefrogenico. La neoplasia è monolaterale nel 95% dei casi, è voluminosa, con margini netti, circoscritta da capsula fibrosa molto soffice e incline alla rottura. È un tumore trifasico, formato cioè da tre componenti cellulari:
1. Componente epiteliale, che abbozza strutture glomerulari e tubulari 2. Componente mesenchimale differenziata, che può formare qualsiasi tipologia di tessuto
(muscolare liscio o striato, adiposo, osseo o cartilagineo) 3. Componente mesenchimale indifferenziato, definito blastema
il nefroblastoma è diagnosticano spesso nei primi 3 anni di vita. La massa, espandendosi, determina compressione dei surreni, dell’intestino, del fegato, della colonna vertebrale e dei vasi
WWW.SUNHOPE.IT

Leonardo Aurino
27
renali. La sintomatologia classica è rappresentata da massa addominale palpabile e manifestazioni ostruttive intestinali, algie addominali e talvolta ematuria. In caso di nefroblastoma si prevede di norma una chemioterapia neoadiuvante che precede l’intervento chirurgico, pertanto la biopsia prechirurgica è indispensabile. Da un punto di vista patogenetico la maggior parte della letteratura scientifica concorda sul fatto che il tumore di Wilms si sviluppi in seguito ad una mutazione genetica. Il gene implicato è denominato WT1 (da "Wilms Tumor"). Si tratta di un gene oncosoppressore, cioè di un gene che, se mutato in entrambi gli alleli, provoca l'insorgenza di un tumore. Nelle forme ereditarie la mutazione a livello di uno degli alleli viene ereditata da un genitore, mentre la mutazione sull'altro allele viene acquisita successivamente. Nelle forme sporadiche (non ereditarie), al contrario, entrambe le mutazioni sono acquisite. Il gene WT1 è stato localizzato sul braccio corto del cromosoma 11 in posizione 11p13 e codifica per una proteina implicata nelle prime fasi dello sviluppo embriologico del rene (differenziazione dei nefroblasti). Altra mutazione possibile è quella del gene WT2, posto sempre sul cromosoma 11.
Genetica:NEUROLOGIA MorbodiHuntington(AD)La malattia di Huntington, detta anche corea di Huntington è una malattia neurodegenerativa genetica, trasmessa con carattere autosomico dominante. La malattia è caratterizzata, oltre che dai tipici movimenti coreici, da demenza e disturbi della personalità. Nella maggior parte dei casi i primi sintomi della malattia iniziano verso i 40-50 anni, anche se si contano casi ad esordio più precoce, intorno ai 20 anni (Morbo di Huntington giovanile). Da un punto di vista genetico, la malattia è caratterizzata da una mutazione del gene IT15, definito huntingtina, localizzato sul braccio corto del cromosoma 4 in posizione 16.3 (4p16.3). La mutazione del gene deriva da un’espansione di triplette patologiche, brevi sequenze nucleotidiche del tipo CAG. La sequenza CAG è ripetuta dalle 6 alle 26 volte nel cromosoma 4p16.3 normale, mente nei soggetti con corea di Huntington tale ripetizione supera le 40 volte. La maggior parte delle ripetizioni CAG avviene durante il processo di meiosi-spermatogenesi, sicché la malattia viene di norma trasmessa dal padre. Tale espansione tende ad aumentare di generazione in generazione, ed essendo la gravità della patologia correlata al numero di espansione, di norma i figli presentano sintomatologie più drammatiche rispetto ai genitori stessi. Ciò spiega l’osservazione che la malattia di Huntington del paziente di 15-40 anni sia di gran lunga prognosticamente peggiore rispetto alla stessa del paziente di 50 anni. Il prodotto del gene IT15 è una proteina nota come huntingtina, la cui distribuzione è sia nervosa che extranervosa e il cui ruolo è quello di acceleratore del complesso della Dineina. La ripetizione CAG, codificante per una glutammina (Gln), determina la comparsa di un numero estremamente elevato di residui glutamminici all’interno della huntingtina. Da un punto di vista neuropatologico, le lesioni tipiche della malattia di Huntington sono localizzate allo striato (caudato + putamen) e alla neocorteccia fronto-temporale (sono interessati esclusivamente i neuroni spinosi dello striato e i neuroni degli strati III, IV e V della neocorteccia). Siccome la proteina huntingtina è rappresentata anche in altri tessuti ma nei pazienti con malattia di Huntington non si verificano altre anomalie sistemiche, è lecito supporre che il danno cerebrale sia da ricondurre all’interazione esiste tra huntingtina e qualche non meglio precisata proteina cerebro-specifica.
WWW.SUNHOPE.IT

Leonardo Aurino
28
Clinica Le tre manifestazioni tipiche della corea di Huntington sono:
1. Disturbi del movimento di tipo coreico 2. Disturbi della personalità 3. Deterioramento intellettivo
In più del 50% dei casi i primi segni indicativi della patologia sono dei cambiamenti di personalità rilevati dai familiari. Il soggetto diviene aggressivo, sociopatico o talvolta depresso. Non sono rari i comportamenti francamente psicotici. I disturbi intellettivi sono sempre presenti. Il paziente, nel divenire sociopatico, si estranea e inizia a trascurare se stesso, compresa l’igiene personale. Si assiste ad un calo dell’attenzione e della capacità critica, i disturbi del sonno spesso coesistono. Le alterazioni del movimento sono la caratteristica più tipica della malattia. I movimenti coreici sono afinalistici, ampi e irregolari, tendono a interessare principalmente le sezioni distali degli arti. All’inizio i movimenti sono meno evidenti e si localizzano al volto e alle mani, sicché il paziente per un certo lasso di tempo viene etichettato come “nervoso”. Col tempo tali movimenti coreici interessano tutto il soma, il paziente non riesce a mantenere una stazione di riposo se non per pochi secondi, essendo scosso di continuo da movimenti involontari. Anche la motilità oculare saccadica coniugata viene costantemente alterata. Compare infatti impersistenza dello sguardo, in quanto il paziente non è in grado di compiere movimenti saccadici diretti verso uno stimolo distraente. I movimenti coreici sono intensificati dallo stresso e dagli stimoli emotivi, mentre tendono a scomparire nel sonno. La malattia risparmia i nervi cranici e qualsiasi tipo di sensibilità. Col progredire della malattia, i disturbi del movimento e i deficit cognitivi sempre più evidenti portano il paziente in uno stato vegetativo. Il paziente perde qualsiasi autonomia e lo stato nutrizionale è scadentissimo. Di norma una polmonite ab ingestis è la causa del decesso, che avviene regolarmente tra i 10 e i 20 anni dalla diagnosi. Altra causa di decesso può essere il suicidio. Diagnosi Gli esami ematochimici e liquorali sono normali. L’EEG mostra anomalie diffuse ma non patognomoniche. La TAC mette in evidenza una dilatazione del III ventricolo “a farfalla”, per l’atrofia dello striato (che forma una parete del ventricolo). La PET con fluorodeossiglucosio mostra un evidente ipometabolismo nelle aree corrispondenti al caudato e al putamen. La malattia di Huntington è diagnosticata facilmente grazie alla peculiarità della sintomatologia associata alla presenza di una storia familiare positiva per malattia di Huntington. La diagnosi definitiva si ottiene con PCR su genoma di linfociti ematici. Con tale metodica è possibile riconoscere il numero di ripetizioni CAG del gene IT15 del cromosoma 4p. se il numero di triplette è > di 35 è possibile confermare la diagnosi. La disponibilità di un test così affidabile permette di effettuare diagnosi prenatale, con tutti i problemi etici ad essa connessi. Terapia Nonostante i farmaci neurolettici convenzionali, come l’aloperidolo siano moderatamente efficaci sulle manifestazioni coreiche, sono in grado di determinare diversi effetti collaterali, tra cui distonie/discinesie tardive, parkinsonismo, sindrome maligna da neurolettici. Si utilizzano pertanto neurolettici atipici, tra cui i più tollerati e efficaci sono l’olanzapina e la quetiapina. La depressione del tono dell’umore risponde bene alle comuni terapie antidepressive (SSRI, triciclici). La terapia con alcuni antidepressivi può aumentare il rischio di suicidio, già particolarmente elevato nei pazienti con malattia di Huntington.
WWW.SUNHOPE.IT

Leonardo Aurino
29
AtassiadiFriederich(AR)È la forma più frequente, rappresentando il 50% delle ARCA (atassie cerebellari autosomiche recessive). La malattia è legata alla mutazione del gene FRDA, posto sul cromosoma 9, che codifica per una proteina mitocondriale, la fratassina, espressa ubiquitariamente, ma con alti livelli nel cuore e nel SNC. La mutazione consiste in una espansione della tripletta GAA del gene, da 67 a 1700 ripetizioni (normalmente sono 6-36), con perdita di funzione della proteina, che conduce ad alterazioni della catena respiratoria mitocondriale e del metabolismo del ferro, causando stress ossidativo e danno cellulare. Le modificazione neuropatologiche della AF coinvolgono primariamente le grandi cellule T dei gangli spinali con degenerazione e atrofia dei cordoni posteriori del midollo e delle radici spinali posteriori. Si associa degenerazione del tratto spino-cerebellare dorsale e ventrale, accentuata nel tratto cervicale, e la degenerazione dei fasci piramidali che colpisce il tratto lombare maggiormente. Vi sono lievi alterazioni del cervelletto con perdita di cellule del Purkinje, di neuroni del nucleo dentato e atrofia dei peduncoli cerebellari medio e superiore. Il SN periferico è colpito da perdita di fibre sensitive mieliniche di grosso calibro. L’età di esordio è prima dei 20 anni, in media tra gli 8 e i 15. Il sintomo di esordio è l’atassia della marcia (solo raramente la scoliosi, la disartria e cardiomiopatia possono precederla), conseguente alla degenerazione dei cordoni posteriori e dei gangli dorsali, pertanto è di tipo misto tabeto-cerebellare con peggioramento alla chiusura degli occhi. Tipica è l’assenza dei riflessi degli arti inferiori (che manca se la patologia è ad esordio tardivo); è presente segno di Babinski e ipostenia progressiva degli arti inferiori per interessamento piramidale; l’alterazione più caratteristica dei movimenti oculari è l’instabilità della fissità dello sguardo, cui si aggiunge il nistagmo. Altre caratteristiche cliniche sono la disartria cerebellare, la riduzione del visus per atrofia del nervo ottica, l’ipoacusia neurosensoriale, la disfagia, i disturbi sfinterici, piede cavo, diabete mellito e cifoscoliosi. In genere lo sviluppo cognitivo è normale. Nel 40% dei pz è presente cardiomiopatia ipertrofica concentrica, causa di morte. In fase avanzata compare amiotrofia distale degli arti superiori e inferiori (diminuzione progressiva del normale trofismo dei muscoli e delle connesse capacità funzionali) e deficit di forza progressivo, che costringono il pz alla sedia a rotelle dopo 10-15 anni dall’esordio. L’età media dell’exitus è 40-50 anni. Per la diagnosi, è consigliata la ricerca genetica dell’AF e successiva valutazione alla RMN cervicale, che evidenzia atrofia nella parte superiore del midollo, mentre la RMN encefalo evidenzia raramente atrofia del cervelletto (colpiti sono invece i nuclei cerebellari). Non esiste terapia capace di arrestare la patologia, ma sono stati proposti farmaci antiossidanti (vitamina E e analoghi del CoQ come l’idebenone.), chelanti del ferro (deferiprone) e farmaci capaci di incrementare l’espressione della fratassina (Epo). Il trattamento delle complicanze della cardiopatia migliora l’aspettativa di vita e la riabilitazione è un aspetto fondamentale. Neurofibromatosi(AD)È una sindrome neurocutanea ereditata con modalità AD. Se ne distinguono due tipi:
• Neurofibromatosi di tipo I o Morbo di Von Recklinghausen: è una malattia neurocutanea ereditata con modalità AD, causata dalla mutazione (traslocazione) del gene oncosoppressore NF1, posto sul cromosoma 17. Il ruolo del prodotto proteico del gene NF1 è quello di inibire l’oncogene p21. La malattia ha un’incidenza di circa 1:3000. La penetranza è pari a quasi il 100%, mentre l’espressività è molto variabile in virtù della grandezza del gene NF1 (più un gene è grande
WWW.SUNHOPE.IT

Leonardo Aurino
30
più una sua mutazione ha espressività variabile). Da un punto di vista clinico, la NF1 è caratterizzata da lesioni cutanee tipiche, le “macchie caffelatte”, macchie piatte, di color marrone chiaro, di dimensione e forme variabili. Sono presenti fin dalla nascita e tendono ad ingrandirsi e ad aumentare di numero nei primi anni di vita. Altre lesioni che si associano sono gli amartomi pigmentati dell’iride o noduli di Lisch. Altre tipiche lesioni i neurofibromi, tumori benigni che originano dalla superficie dei nervi periferici, composti da cellule di Schwann e fibroblasti che, nella variante plessiforme, tendono a localizzarsi al volto e hanno una crescita molto pronunciata, potendo raggiungere dimensioni enormi e rappresentare la causa di evidentissime deformità. I neurofibromi hanno inoltre una data tendenza all’evoluzione maligna. Nel 15% dei pazienti con NF-1 si assiste alla formazione di tumori di strutture nervose, come astorocitomi, meningiomi, ependimomi e gliomi dell’ottico. Anche neoplasie non interessanti il sistema nervoso sono frequenti nei soggetti affetti da NF-1 (leucemie, neoplasie gastriche, intestinali, feocromocitomi).
• Neurofibromatosi di tipo 2: molto più rara della precedente, ereditata anch’essa in modo AD. Sono costanti i neurinomi di uno o di entrambi i nervi acustici (il neurinoma bilaterale dell’acustico è patognomonico della NF2). Vi è quindi, spesso, sordità neurosensoriale retrococleare uni/bilaterale. La mutazione responsabile è quella gene della merlina un oncosoppressore, posto sul cromosoma 22.
Sindromedell’X-FragileosindromediMartin-Bell(X-linked) Causa genetica più frequente di ritardo mentale nei maschi e seconda causa in generale (la prima è la s. di Down, mentre la sindrome di Rett è la più frequente causa nelle femmine). Ha prevalenza di 1 maschio/ 6000 . Il gene (FMR1) è localizzato sulla porzione terminale del braccio lungo del cromosoma X formato da 38 Kb con 17 esoni, è una malattia da triplette poiché legata all'espansione di una tripletta nucleotidica (CGG) che provoca una fragilità del cromosoma X. L’espansione determina, inoltre, una metilazione estrema delle citosine a livello del promotore del gene FMR1, con conseguente silenziamento genico. Il gene interessato ha una funzione regolativa che partecipa ad importanti processi neuronali come il trasporto dei messaggeri e la regolazione della traduzione nelle sinapsi. Sulla base della lunghezza della regione amplificata distinguiamo 4 tipi di alleli:
1. Normali → 5-50 ripetizioni → non si espandono in seguito a trasmissione � 2. Intermedio → 50-55 ripetizioni → sono destinate ad espandersi, a differenza di quelle
normali, durante la trasmissione alla generazione successiva e portano a premutazione � 3. Premutazione → 55-200 ripetizioni → si pensava in passato che questi pazienti non
mostrassero nessun segno clinico ma poi si è riscontrato che sono possibili differenti fenotipi associati alla premutazione ◦ Manifestazione in forma lieve dei segni clinici della sindrome come lassità legamentosa, �tendenza alla depressione e qualche tratto dismorfico ◦ Menopausa precoce ◦ Sindrome con atassia e tremore associata all'X-fragile (FXTAS) �
4. Mutazione piena → oltre 200 ripetizioni �Importante sottolineare che esistono differenze a seconda del genitore che trasmette l'anomalia infatti: � a) Se si trasmette dalla madre → questa ha un cromosoma X normale e uno con premutazione per cui la probabilità che essa trasmetta l'uno o l'altro è la stessa, se trasmette il cromosoma con mutazione ad un maschio questo presenterà la malattia a patto che la madre abbia una premutazione, mentre se ha un genotipo intermedio il figlio sarà normale ma portatore della premutazione. Se invece la trasmissione avviene ad una figlia femmina nel caso in cui si tratti di una mutazione piena i sintomi saranno più lievi perchè ella presenta l'altro X sano e può produrre metà della proteina FMR normale � b) Se si trasmette dal padre → può trasmettere la mutazione piena solo alle figlie femmine .
WWW.SUNHOPE.IT

Leonardo Aurino
31
Diagnosi
Su test genetici eseguiti su tutti i pazienti con ritardo mentale o comportamento autistico e non sul quadro clinico � Quadro clinico � Nei maschi → esordisce nell'infanzia con ritardo dello sviluppo motorio e del linguaggio. �Otiti, sinusiti, epilessia. Deficit cognitivo lieve da disturbi dell'apprendimento a ritardo mentale grave. Comportamenti autistici come il battere le mani, tendenza a mordersi le mani, hand flapping (sfarfallamento delle mani presente anche del disturbo di Rett), scarso contatto oculare nel tentativo di evitare lo sguardo fisso (evitamento dello sguardo diverso da quello presente nei bambini autistici infatti i bimbi con X-fragile sono socialmente molto responsivi e spesso affettuosi). Sono presenti disturbi dell'umore, d'ansia e aggressività. Macroorchidismo � Nelle femmine → disturbi cognitivi e del comportamento lievi e consistono in disturbi emotivi e dell'apprendimento, timide o ansiose, problemi di attenzione � Entrambi i sessi → faccia stretta ed allungata, orecchie e fronte prominenti, piedi piatti. Scrivere è molto difficile per loro, non hanno il controllo delle proprie mani e dei muscoli delle dita per scarso tono muscolare.
CollagenopatieSindromediMarfan(AD)Patologia autosomica dominante che colpisce il tessuto connettivo. Le manifestazioni della sindrome di Marfan interessano prevalentemente:
• apparato osteo-arto-muscolare: l'altezza dei pazienti, che sono più alti della media dei coetanei, e spesso sviluppano (soprattutto durante l'adolescenza) segni di magrezza eccessiva (intesa non soltanto come un semplice sottopeso, ma anche come un habitus in generale eccessivamente affusolato e dinoccolato). Un'altra caratteristica è la lunghezza e forma affusolata delle dita, che viene chiamata aracnodattilia. L'aracnodattilia, che tende a causare un'ipermobilità delle dita, è legata alla lassità generalizzata dei legamenti.
• Cuore: insufficienza della valvola mitrale e della tricuspide, talora dovute al prolasso dei lembi, talora alla rottura delle corde tendinee.
• occhi: tipica è la sublussazione della lente o cristallino, causa di visi di rifrazione (spt miopia)
• grossi vasi: non è rara la dissezione aortica. Nella maggior parte dei casi, la sindrome di Marfan è causata dalle mutazioni del gene FBN1 (15q21) che codifica per la fibrillina-1, una proteina essenziale del tessuto connettivo. SindromediEhlers-DanlosTrattasi in realtà di un gruppo di patologie ereditarie, contraddistinte da lassità legamentosa e iperelasticità cutanea. Tale sindrome è determinata da una anomalie di un collagene. Si conoscono diversi tipi di sindrome di Ehlers-Danlos:
• classica (distinta in gravis e mitis): mutazione del gene COL5A1 e COL5A2, codificanti per collageno di tipo V. si manifesta con lassità legamentosa e iperelasticità cutanea,
WWW.SUNHOPE.IT

Leonardo Aurino
32
pseudotumori mollusciformi, lividi anche a seguito di traumi minimi, facili prolassi ed ernie viscerali. 1/3 dei pazienti presenta una dilatazione della radice aortica.
• Tipo “ipermobile”: la mutazione interessa il collagene ma il gene non è stato ancora individuato. I sintomi sono lassità legamentosa e iperelasticità cutanea, con frequenti lussazioni articolari, con dolore cronico soprattutto ai cingoli. Frequente è il prolasso mitralico e la dilatazione della radice aortica. Vi è assenza del frenulo labiale.
• Tipo “vascolare”: difetto del collagene III per mutazione del gene COL3A1. I sintomi sono soprattutto quelli di fragilità delle parete dei vasi, con frequenti emorragie epatiche, intestinali, uterine, lividi frequenti e varici venose.
• Tipo “cifoscoliosi”: si manifesta per mancanza del gene PLOD1, codificante per la lisil-idrossilasi, un enzima necessario ai processi di idrossilazione dei residui di lisina del collagene. I pazienti affetti mostrano ipotonia muscolare grave in età prepuberale, causa di curvature cifoscoliotiche gravi del rachide. Vi è iperlassità legamentosa e fragilità dei vasi
• Tipo “artroclasia”: imputabile ad una mancanza di una catene del collageno di tipo I, dovuta al passaggio dell’esone 6 nel gene COL1A1 o COL1A2. Si manfesta con iperlassità legamentosa grave e generalizzata con multiple lussazioni. Vi è lussazione congenita delle anche.
• Tipo “dermatosparassi”: malattia estremamente rara, con nessun caso in Italia e solo 10 casi al mondo. È causata dalla mancanza dell’enzima procollagene 1 N-term peptidase, codificato dal gene ADMST2. L’enzima è necessario per tagliare l’estremità N-terminale del procollagene I e trasfrormarlo in collagene I. I sintomi sono iperelasticità cutanea, con pelle in eccesso che si mostra molle e soffice, frequenti sono i lividi.
La diagnosi di sindrome di Ehlers-Danlos si basa su:
1. Storia clinica e anamnestica 2. Test genetici: Possono rilevare alterazioni nei geni ADAMTS2, COL1A1, COL1A2,
COL3A1, COL5A1, COL5A2, PLOD1 e TNXB. Si rivelano un utile strumento per identificare, in particolar modo, i tipi “Vascolare” e “Artoclasia”.
3. Esame delle urine: È disponibile un test che aiuta ad identificare il tipo “Cifoscoliosi”. Il test misura il livello di un enzima prodotto dalla mutazione di un gene.
Test diagnostici prenatali. Sono utili per rilevare mutazioni di geni associati nel feto, se un parente stretto presenta la malattia. Il tipo “Cifoscoliosi” e “Vascolare” possono essere rilevati tramite amniocentesi, prelevando un campione di liquido amniotico, per valutare i livelli di attività enzimatica. Al momento non esistono cure efficaci. Esistono però delle precauzioni da adottare, come:
• Evitare di sollevare e trasportare oggetti pesanti • Utilizzare colle adesive o strisce al posto di punti di sutura in caso di ferite • Alte dosi di acido ascorbico (essenziale cofattore nella sintesi di collagene) • Follow-up cardiologico ed oftalmologico (vi è possibilità di lussazione della lente a causa
della lassità dei legamenti zonulare dello Zinn)
SindromediBethlemÈ una collagenopatia causata da mutazioni in una delle tre subunità del collagene di tipo VI, per mutazioni del gene COL6A1, COL6A2 e COL6A3 (più frequente). Viene anche definita come miopatia prossimale benigna progressiva, in quanto vi è un certo grado di miopatia. La sindrome di Bethlem si manifesta nell'adulto, benché sia spesso documentabile un'ipotonia neonatale, ridotti movimenti, torcicollo e ritardo della deambulazione. Nell'adulto sono
WWW.SUNHOPE.IT

Leonardo Aurino
33
caratteristiche le contratture in flessione dei polsi, dei gomiti, delle ginocchia e delle dita, accompagnate spesso a debolezza lentamente progressiva. Queste manifestazioni sono responsabili di difficoltà di deambulazione in più dei 2/3 dei soggetti con più di 50 anni affetti da questa miopatia. La progressione della miopatia può coinvolgere il diaframma, rendendo difficoltosa la respirazione notturna. Altre manifestazioni sono:
• Lassità articolare delle mani e dei piedi • Scoliosi o cifosi • Pectus excavatum • Tendenza alla formazione di cheloidi
SindromediUllrichLa distrofia muscolare di Ullrich è una distrofia muscolare grave caratterizza da debolezza muscolare a insorgenza precoce associata a contratture delle articolazioni prossimali e iperelasticità di quelle distali. È una patologia associata a difetti del collagene VI che si trasmette per modalità autosomica recessiva, benché in alcuni casi si possa dimostrare una modalità di trasmissione del tipo autosomico dominante.
Le mutazioni recessive caratteristiche della sindrome di Ullrich provocano un accorciamento della porzione terminale (estremità carbossiterminale) delle proteine collageniche COL6A2 e meno frequentemente delle proteine COL6A3 e COL6A1. Tali mutazioni provocano un'aberrazione dell'appaiamento del proto-collagene e conseguente alterata disposizione delle fibrille. Nel caso di trasmissione dominante la mutazione è simile a quelle riscontrate nella sindrome di Bethlem. La UCMD è caratterizza da debolezza muscolare di entità grave; i soggetti non riesco a camminare o camminano solo per brevi periodo. Alla rigidità della colonna vertebrale può associarsi scoliosi e dislocazione congenita dell'anca. I problemi respiratori portano alla morte nella prima o nella seconda decade di vita. È inoltre possibile riscontrare:
• Tendenza alla formazione di cheloidi • Ipercheratosi follicolare • Cute vellutata dei piedi e delle mani
GlicogenosiSono malattie del metabolismo del glicogeno nelle quali le cellule accumulano tale omopolimero per incapacità di mobilizzarlo, a causa di un deficit enzimatico geneticamente determinato. Le sedi dove maggiormente si accumula glicogeno quelle in cui il metabolismo di tale sostanza di riserva è particolarmente attivo, cioè fegato, muscolo, miocardio e rene. All’esame istologico dei tessuti, le cellule si mostrano generalmente ingrandite e contengono glicogeno disposto, ad eccezione della glicogenosi di tipo II, a mo’ di rosetta in seda citoplasmatica (per fegato e rene) e in sede interfibrillare nel muscolo cardiaco e scheletrico. La classificazione delle glicogenosi si fonda sulla natura dei rispettivi deficit enzimatici.
• Glicogenosi di tipo I (malattia di von Gierke):la trasmissione è AR. è caratterizzata da deposizione di glicogeno in fegato e rene per un deficit di glucosio-6-fosfatasi (secondario a mutazione del gene per la G6P posto sul cromosoma 17) enzima presente solo in questi due tessuti, che sono pertanto gli unici in grado di aumentare la glicemia riversando in circolo il glucosio ottenuto dalla glicogenolisi. Si genera pertanto una condizione di ipoglicemia, a sua volta responsabile di una riduzione dei livelli plasmatici di insulina. La riduzione dei livelli di insulina è causa di lipolisi (in quanto il glicerolo ottenibile dagli acidi grassi virrà utilizzato per la neoglucogenesi) e dismissione in circolo di acidi grassi dal tessuto adiposo. Una certa quota di questi acidi grassi raggiunge il fegato e determina steatosi epatica. I sintomi che caratterizzano questo tipo di glicogenosi sono: ipoglicemia a digiuno,
WWW.SUNHOPE.IT

Leonardo Aurino
34
epatomegalia (dovuta ad un aumento del glicogeno nel fegato e a steatosi), ritardata crescita nell'adolescenza, iperlipidemia , aumento dei corpi chetonici e un aumento degli acidi grassi in circolo (perché l'ipoglicemia induce formazione di corpi chetonici, prodotti a partire dagli acidi grassi mobilizzati dal tessuto adiposo).
• Glicogenosi di tipo II (malattia di Pompe): è una malattia genetica a trasmissione AR caratterizzata da mancanza dell’enzima maltasi acida (α-1, 4-glicosidasi lisosomiale), enzima presente in ogni tessuto. L’enzima fa parte del processo metabolico della glicogenolisi. In mancanza di questo enzima, il glicogeno si accumula in tutti gli organi, segregato in vacuoli intracitoplasmatici. In realtà la patologia è una malattia lisosomiale ereditaria. Tra i tessuti che più risentono dell’accumulo di glicogeno nella malattia di Pompe vi sono i muscoli, tanto che si assiste ad una progressiva miopatia, soprattutto dei muscoli addominali e in special modo del diaframma. In base al decorso clinico, si distinguono: A) Infantile Onset, si manifesta in età neonatale ed è e caratterizzata da un predominante interessamento cardiaco con coinvolgimento muscolare scheletrico (tipica assenza di tono muscolare alla nascita: “floppy baby”). In assenza di terapia, il decorso della malattia è fatale e avviene solitamente entro il primo anno di vita B) Late Onset, si manifesta con esordio variabile (dopo il primo anno di vita fino ai 60/70 anni), ed è caratterizzata da un'ampia variabilità di espressione clinica, ovvero: - Progressiva debolezza della muscolatura prossimale, in particolare degli arti inferiori (è il quadro clinico più comune); - Insufficienza respiratoria, causata da debolezza del diaframma e dei muscoli respiratori accessori: può presentarsi come ortopnea e dispnea e con occasionale cefalea mattutina; - Aumento dei livelli di creatin-chinasi (CK) da 1,5 a 15 volte i limiti superiori della norma.
• Glicogenosi di tipo III: in cui il deficit enzimatico è quello dell’enzima deramificante α-1, 6- glicosidasi. In questa malattia l’accumulo di glicogeno interessa il fegato in maniera quasi comparabile al tipo I, mentre il muscolo scheletrico e cardiaco sono poco coinvolti. A livello epatico l’accumulo di glicogeno è causa di epatomegalia e innalzamento delle transaminasi seriche. Caratteristica è una debolezza muscolare che si mostra soprattutto con l’esercizio fisico intenso. L’aspettativa di vita è quasi normale.
• Glicogenosi di tipo IV (amilopectinosi o malattia di Anderson): è una forma rara e gravissima di glicogenosi, determinata dall’assenza dell’enzima ramificante. La mancanza di tale enzima determina la formazione di catene di glicogeno dotate di poche ramificazioni, ma in compenso molto lunghe. Tale conformazione sterica del glicogeno lo rende simile all’amilopectina (da cui il nome di amilopectinosi) e notevolmente non idrosolubile. L’accumulo di glicogeno non è notevolissimo, ma la sua completa insolubilità determina fenomeni necrotici a carico del fegato, con conseguente cirrosi e ipertensione portale. La morte avviene di norma al 2-3 anno di vita.
• Glicogenosi di tipo V (malattia di McArdle): è una glicogenosi poco grave, determinata dalla mancanza di fosforilasi muscolare, enzima muscolare indispensabile per l’utilizzo di glicogeno ai fini ergogenici. Vi è solo una limitata intolleranza all’esercizio fisico, che se intenso può determinare un aumento dell’aldolasi, dell’LDH e della CPK.
WWW.SUNHOPE.IT

Leonardo Aurino
35
GangliosidosiSono un gruppo di malattie metaboliche geneticamente determinate contraddistinte dall'accumulo di gangliosidi (dei glicolipidi particolarmente abbondanti nel Sistema Nervoso) nei lisosomi a causa di difetti enzimatici lisosomiali. Si conoscono due gangliosidosi principali, la GM1 e la GM2. GM1 (AR) La gangliosidosi GM1 è una rara malattia neurodegenerativa caratterizzata dall’accumulo all’interno dei lisosomi di gangliosidi GM1. Si conoscono tre forme di gangliosidosi GM1:
• Forma infantile: fa il suo esordio prima dei 3 mesi di vita con encefalopatia progressiva e amaurosi. È presente epato-splenomegalia e facies caratteristica, con deformazioni scheletriche (cifoscoliosi). Durante i primi 6 mesi di vita vi è un arresto dello sviluppo, seguito da deterioramento neurologico progressivo. I soggetti affetti non superano i due anni di vita.
• Forma giovanile: esordisce tra 1 e 5 anni di vita con atassia e crisi epilettiche. Il coinvolgimento viscerale è lieve. La morte sopravviene entro 20 anni dalla diagnosi.
• Forma dell’adulto: definita anche gangliosidosi GM1 cronica viene diagnosticata in età adulta. Le manifestazioni cliniche sono simili a quelle del Parkinson giovanile e alla miotonia. La prognosi è molto variabile
Alla base delle gangliosidosi GM1 vi il deficit dell’enzima β-galattosidasi, responsabile del metabolismo dei gangliosidi M1. L’accumulo di gangliosidi determinerebbe l’innesco del processo apoptotico neuronale. GM2 (AR) O malattia di Tay-Sachs, è caratterizzata dall'accumulo di gangliosidi GM2 da deficit dell'enzima esoaminossidasi A. Anche per la GM2 si differenziano forme ad esordio infantile, giovanile e dell’età adulta. La clinica è pressochè uguale alla gangliosidosi GM1.
Sfingolipidosi Gli sfingolipidi sono lipidi di membrana costituiti prevalentemente da sfingosina. Appartengono al gruppo degli sfingolipidi i ceramidi e la sfingomielina (costituente essenziale della mielina). Tra le sfingolipidosi si annoverano: Malattia di Gaucher (AR) È una patologia da accumulo lisosomiale secondaria al deficit dell’enzima glucocerebrosidasi. La malattia è caratterizzata dall’accumulo di glucocerebrosidi a livello epatico, splenico e del midollo osseo. La prevalenza della malattia è di 1:100.000. Le manifestazioni cliniche sono molto variabili, potendo definire:
• Tipo I: definita forma non neurologica, corrisponde al 95% del totale. Vi è epatosplenomegalia, patologia scheletrica (infarti ossei con dolore, osteonecrosi) e pancitopenia.
• Tipo II: forma neurologica acuta, con disfunzione del tronco encefalico ad esordio precoce (entro 1 anno di vita) e organomegalia.
WWW.SUNHOPE.IT

Leonardo Aurino
36
• Tipo III: forma neurologica subacuta, con disfunzioni neurologiche (atassia, epilessia e aprassia oculo-motoria) che insorgono in età adolescenziale.
Malattia di Niemann-Pick (AR) È una malattia lisosomiale a trasmissione AR dovuta al deficit dell’enzima sfingomielinasi acida, l’enzima che trasforma la sfingomielina in ceramide. Si genera, pertanto, in virtù del deficit enzimatico, un accumulo di sfingomielina all’interno delle cellule. In base al quadro clinico si definiscono due tipi di malattie:
• Tipo A: esordio nel primo anno di vita con epatosplenomegalia grave e interessamento neurologico (ipotonia generalizzata, ritardo dello sviluppo). Il deterioramento neurologico porta rapidamente a morte.
• Tipo B: esordio in età variabile (anche adulta) con il segno più tipico che è l’epatosplenomegalia, talvolta associata a segni polmonari.
Malattia di Fabry (X-linked) La malattia di Fabry (FD) è una malattia da deposito lisosomiale multisistemica, progressiva, ereditaria, caratterizzata da specifici segni neurologici, cutanei, renali, cardiovascolari, cocleo-vestibolari e cerebrovasculari. La malattia di Fabry è un difetto del metabolismo dei glicosfingolipidi, dovuto alla riduzione o assenza di attività dell'enzima lisosomiale alfa-galattosidasi A, da mutazione del gene GLA (Xq21.3-q22), che codifica per l'enzima. La diminuzione dell'attività provoca l'accumulo di globoside Gb3 all'interno dei lisosomi, che a sua volta scatena una cascata di eventi cellulari. Il quadro clinico comprende un ampio spettro di sintomi, che varia dalle forme lievi nelle femmine eterozigoti, ai casi gravi nei maschi emizigoti con le forme classiche, che non mostrano alcuna attività residua dell'alfa-galattosidasi A. Questi pazienti possono presentare tutti i segni tipici della malattia a livello neurologico (dolore), cutaneo (angiocheratoma), renale (proteinuria, insufficienza renale), cardiovascolare (cardiomiopatia, aritmia), cocleo-vestibolare e cerebrovascolare (ictus, episodi ischemici transitori). Le femmine possono mostrare sintomi lievi-gravi. Il dolore è un sintomo comune precoce (dolore cronico caratterizzato da parestesia con bruciore e prurito e rare crisi episodiche caratterizzate da dolore acuto con senso di bruciore). Il dolore può risolversi nell'età adulta. Possono insorgere anidrosi o ipoidrosi, che causano intolleranza al calore e all'esercizio. Altri segni clinici sono l'angiocheratoma, le alterazioni della cornea, il tinnito, l'affaticamento cronico, le anomalie cardiache e cerebrovascolari (ipertrofia ventricolare sinistra, aritmia, angina), la dispnea e la nefropatia.
MucopolisaccaridosiLe mucopolisaccaridosi sono malattie geneticamente determinate dovute ad accumulo di mucopolisaccaridi (cioè glucosamminoglicani o GAG), a causa di deficit enzimatici lisosomiali. L’importanza di alcuni mucopolisaccaridi, come il GAG acido ialuronico in distretti come cartilagini, articolazioni e ossa spiega come si realizzino spiccatissime malformazioni. Tipicamente, nelle mucopolisaccaridosi vi è sclerosi delle capsule articolari, delle valvole cardiache e del connettivo in ogni distretto anatomico. I mucopolisaccaridi, liberati dalle cellule andate in contro a necrosi dopo averli accumulati in sede intracitoplasmatica (in quanto non possono degradarli per l’assenza di enzimi lisosomiali) vanno a localizzarsi, tramite il torrente ematico, a livello delle cellule endoteliali, determinandone rigonfiamento. Tale condizione porta il nome di pseudoateromatosi. L’eccesso di mucopolisaccaridi raggiunge le urine, dove può essere dosato.
WWW.SUNHOPE.IT

Leonardo Aurino
37
Si conoscono almeno 7 tipi di mucopolisaccaridosi. Per ogni tipo di MPS vi è un enzima lisosomiale deficitario e un GAG accumulato (i principali GAG che si accumulano sono il dermatan solfato, eparan solfato, cheratan solfato, condroitin solfato)
Genetica:OCULISTICA RetinoblastomaTumore oculare, rappresenta, dopo il neuroblastoma, il tumore maligno più frequente in età pediatrica. Insorge a livello retinico, unilaterlamente o talvolta bilateralmente. Si differenziano due tipi di retinoblastoma:
• Retinoblastoma ereditario: 40% dei casi. È dovuto all’eredità AD di un allele mutato del gene codificante per la proteina del Retinoblastoma (RB1), posto sul cromosoma 13, da uno dei due genitori. In questi individui, affinchè si manifesti il tumore, è necessaria una seconda mutazione dell’allele sano (teoria del double hit di Knudson). Nelle forme ereditarie il retinoblastoma può essere bilaterale e ha una evoluzione molto veloce.
• Retinoblastoma sporadico 60% dei casi. Non è influenzato da nessuna componente ereditaria. Trattasi quindi di mutazioni somatiche (e non germinali) della del gene codificante per la proteina RB1. Di norma queste forme sono meno gravi e spesso unilaterali.
La sintomatologia si presenta di norma entro i primi 5 anni di vita. Il principale segno è la leucocoria, cioè un riflesso biancastro del fondo oculare dovuto alla presenza del tumore che invade il vitreo posteriore. Spesso vi è strabismo. RetinitepigmentosaMalattia eredo-degenerativa della retina periferica. La malattia è definita “pigmentosa” per la caratteristica deposizione di pigmenti nella periferia retinica, con disposizione definito “ad osteoblasti”, espressione della degenerazione dei fotorecettori e dell’epitelio pigmentato stesso. La malattia può essere trasmessa in modo:
• ADà forma più benigna • AR • X-linkedà forma più severa con riduzione estrema del visus già a 20 anni. • Mitocondriale
Clinicamente, la RP si manifesta con : • Nictalopia à cioè cecità notturna (per riduzione del numero di bastoncelli) • Alterazione della percezione cromatica (nelle fasi più avanzate) • Riduzione della acutezza visiva centrale (la retinite pigmentosa inizia alla periferia della
retina ma ha uno sviluppo centripeto, fino a raggiungere la macula. In quel momento la visione diviene “a cannocchiale”).
• Fotopsie: i pazienti hanno allucinazioni visive elementari.
WWW.SUNHOPE.IT

Leonardo Aurino
38
Genetica:EMATOLOGIA Emofilia(X-linkedR)è un disordine ereditario X-linked recessivo causa di un difetto della coagulazione. Si conoscono 3 tipi di emofilia:
- Emofilia A: mutazione del gene codificante per il fattore VIII della coagulazione, che ne causa carenza totale o parziale. L’incidenza della malattia è di 20:100.000 nati vivi. Come detto, la malattia è causata dalla mutazione del gene codificante per il fattore VIII della coagulazione, che può essere una mutazione puntiforme, una delezione o la formazione di un codone di stop. Il principale hot-spot del gene è la sequenza TCGA, dove più frequentemente si localizzano le mutazioni. Si conoscono più di 80 mutazioni responsabili dello sviluppo della malattia. L’emofilia può essere la risultante di un difetto quantitativo (95% dei casi) o qualitativo (5%) del fattore VIII. La diagnosi prenatale di emofilia è possibile tramite villocentesi. La gravità dell’emofilia A è variabile in base all’entità del deficit quantitativo/qualitativo del fattore VIII. L’attività coagulante del fattore VIII in un individuo sano va dal 50% al 200%. In base alla riduzione dell’attività coagulante del fattore VIII si definisce: a) emofilia grave se < o = a 1% b) emofilia moderata se compresa tra 1 e 5% c) emofilia lieve se compresa tra 5-25% Clinicamente l’emofilia A si manifesta con ematomi, emartri, cisti ossee siero-ematiche, ematuria. Eventi drammatici in corso di emofilia A sono le emorragia cerebrali, intra o extraparenchimali. La terapia dell’emofilia A è essenzialmente sostitutiva, basta sull’uso di pasla fresco congelato o crioprecipitati, contenenti F VIII. Ad oggi è possibile utilizzare anche FVIII umano ricombinante. La desmopressina si rivelata capace di aumentare di 2-3 volte i livelli di fattore VIII della coagulazione.
- Emofilia B: mutazione del gene codificante per il fattore IX della coagulazione, che ne causa carenza totale o parziale. La clinica è del tutto simile all’emofilia A.
- Emofilia C: mutazione del gene codificante per il fattore XI della coagulazione, che ne causa carenza totale o parziale
DeficitGlucosio6-fosfatodeidrogenasi(FAVISMO)(X-linked)È una forma di anemia da causa mista, poiché il globulo rosso ha una condizione di enzimopenia (G6PD), ma l’emolisi non si presenta se non vi è esposizione ad alcuni farmaci (primachina, un antimalarico, fluorochinoloni di I generazione, cotrimossazolo, doxorubicina) o a prodotti derivanti dal consumo di fave (le fave contengono grosse quantità di L-Dopo, convertita in dopachinone, il quale converte il glutatione ridotto, GSH, in glutatione ossidato, GSSG). Il gene che codifica per l’enzima G6PD è posto sul cromosoma X. La malattia è trasmessa, quindi, secondo il sesso. I maschi o sono sani o sono malati, mentre le femmine possono essere sane, malate o portatrici. La malattia nelle carriers sane può comunque presentarsi, sebbene in misura misura molto più sfumata rispetta alla forma classica, qualora vi sia una lyonizzazione massimale del cromosoma X con l’allele wild-type. La mutazione del gene codificante per la G6PD è molto spesso una mutazione puntiforme. L’enzima G6PD è il primo enzima della via dei pentoso-fosfati, una via che produce NADPH utile per il processo di conversione del glutatione ossidato GSSG in glutatione ridotto GSH. Il deficit di NAPDH che si genera negli individui affetti è causa di una riduzione dei livelli di glutatione ridotto, con conseguente suscettibilità a tutti gli stimoli ossidativi. Il globulo rosso, in particolare, utilizza continuamente glutatione GSH per prevenire l’ossidazione dei gruppi tiolici (-SH) dell’emoglobina e della membrana. In carenza di GSHm l’emoglobina si
WWW.SUNHOPE.IT

Leonardo Aurino
39
denatura e precipita sulla membrana dell’emazia, formando il corpo di Heinz, danneggiandola e determinando emolisi intravascolare. Clinica: la malattia, per quanto detto, si manifesta quindi con crisi di emolisi intravascolare, con comparsa acuta di segni di anemia emolitica, quali:
1. aumento della bilirubina non coniugata e comparsa di ittero (l’aumento della bilirubina determina soventemente litiasi biliare)
2. possibile IRA da NTA da emoglobinuria, causa anche di dolori lombari, oltre che di oligo-anuria, aumento dell’azotemia e della creatininemia.
3. Anemia normocromica/normocitica e colorazione pallida di cute e mucose (su fondo giallo dato dall’ittero)
4. Feci e urine ipercromiche per aumento dei bilinogeni fecali e urinari. Anemiafalciforme(AR)Anche definita drepanocitosi, è una emoglobinopatia qualitativa, caratterizzata da una mutazione puntiforme di un singolo amminoacido della catena β dell’emoglobina. Più specificamente si ha una sostituzione, in posizione 6, di un residuo di Acido Glutammico con una Valina. Tale modificazione della struttura primaria della proteina determina un cambiamento morfo-strutturale dell’Hb. La nuova Hb prodotta è definita HbS. A causa della mutazione, quando l’HbS passa da uno stato Ossi ad uno Deossi, cioè nel sangue venoso dove vi è bassa pO2, l’emoglobina polimerizza formando dei cristalli birinfrangenti all’interno dell’eritrocita, deformandolo e determinando l’acquisizione della tipica conformazione dell’emazie a falce. Tali emazie anomale, vengono prematuramente captate dalle cellule del reticolo endotelio (soprattutto milza e in parte fegato e macrofagi) e avviate verso l’emocateresi. Inoltre, gli eritrociti a falce hanno elevata tendenza ad agglutinarsi nel torrente circolatorio e causare ostruzioni e infarti. Ciò spiega la duplice sintomatologia dell’anemia drepanocitica, l’anemia emolitica cronica e le crisi dolorose (crisi falcemiche) da infarti venosi ossei e polidistrettuali. L’emoglobina S è causa di malattia conclamata solo in condizioni di omozigosi (cioè tutta l’Hb è HbS), mentre la condizione di eterozigosi si definisce “trait falcemico”. Nei soggetti con trait falcemico solo il 30% o meno dell’Hb è HbS. Le manifestazioni cliniche dell’anemia drepanocitica si evidenziano solo dopo il 6° mese di vita, quando la maggior parte dell’HbF è sostituita con HbS. La clinica, come detto, consiste in:
1. Anemia emolitica cronica: causa di ritardo nell’accrescimento, astenia e pallore cutaneo, litiasi biliare (da aumento della bilirubinaà formazione di calcoli bruni di bilirubinato di calcio), ittero, crisi aplastiche da consumo di vitamine emoattive (B9 e B12), feci e urine ipercromiche
2. Fenomeni vaso-occlusivi: la loro insorgenza è più frequente in caso di disidratazione e di esposizione al freddo. Frequenti sono gli infarti oseei, splenici, e talvolta anche cerebrali, polmonari, miocardici, retinici e renali. Possibile anche il priapismo doloroso per occlusione del circolo a livello dei corpi cavernosi.
La diagnosi si basa sull’elettroforesi dell’Hb (HbS migra più lentamente della HbA). TalassemiaCon il termine talassemia si identificano una serie di disordini genetici caratterizzati da una alterazione della sintesi emoglobinica, causa di eritropoiesi eritrocitaria ed emolisi, con conseguente anemia. Le principali alterazioni emoglobiniche quantitative nella pratica clinica sono:
• Talassemia α • Talassemia β
WWW.SUNHOPE.IT

Leonardo Aurino
40
Talassemia β (AR) Le β-talassemie presentano un modello di ereditarietà di tipo mendeliano semplice autosomico recessivo. Il difetto coinvolge il cromosoma 11 a livello del gene codificante per le catene β dell’Hb. In letteratura sono state descritte più di 200 mutazioni in grado di abolire o di ridurre fortemente la sintesi della β-globina. Il difetto più comune è una mutazione puntiforme del gene in un “hot spot” del processo di trascrizione in RNA o di traduzione in proteina (alterazioni del promoter, delle zone di splicing ecc.). I bambini affetti dalla forma più grave di β-talassemia (morbo di Cooley) sono omozigoti per l’allele mutato, presentando una ridottissima o assente produzione di catene β. Il quadro clinico delle sindromi β-talassemiche è estremamente variabile, in rapporto al difetto molecolare e, soprattutto, al genotipo del soggetto. Si riconoscono: Talassemia Maior: la β-talassemia maior o Morbo di Coleey) è una condizione determinata da un genotipo che può essere: A) omozigote recessivo per gli alleli β0 B) doppia eterozigosi β0/β-tal+ C) omozigosi per Hb Lepore (condizione data da un ineguale appaiamento di due cromosomi omologhi con formazione di un gene di fusione δβ. I soggetti affetti in forma omozigote producono solo Hb Lepore, osseia α2-(δβ)2 ). La mutazione porta fondamentalmente alla ridotta o addirittura assente sintesi di catene β e quindi di HbA, con una conseguente gravissima anemia dovuta alla precipitazione di catene α libere che sedimentando all’interno delle emazie e ne stimolano l’emolisi. Si ottiene così una lisi degli eritroblasti in sede endomidollare e una emolisi delle emazie mature in sede periferica. L’eccesso di catene α va coniugandosi con le rimanenze di catene γ, formando HbF che in effetti è aumentata in questi pazienti. Il morbo di Cooley è dunque una grave anemia congenita, refrattaria ad ogni trattamento terapeutico, con grave ittero pre-epatico da emolisi e epatosplenomegalia. La grandezza di milza e fegato è dovuta ad una aumentata emocateresi dei globuli rossi e alla funzione emopoietica vicariante del fegato. L’imponente grado di anemia stimola la formazione di EPO che agendo a livello midollare ne stimola l’iperplasia, fino alla deformazione del contenitore osseo, facilmente visibile soprattutto nelle ossa del neurocranio (“cranio a spazzola”). La facies degli individui affetti assume un aspetto “asiatico” con taglio mongoloide degli occhi e prominenti mascellari con denti radi. La lisi delle emazie si accompagna ad un aumento della sideremia fino all’emosiderosi che danneggia fegato, ipofisi e cuore. L’anemia è molto grave (3-6g/dl) con un ridotto numero di globuli rossi (2 milioni/mm3). Le emazie sono fortemente ipocromiche e hanno forma bizzarra (poichilocitosi) e aspetto a “target cells”. La diagnosi si pone tramite elettroforesi dell’Hb, che mostra un aumento considerevole dei livelli di HbF. Nei soggetti con genotipo β0-β-Tal+ è dosabile una certa quota di HbA, mentre nei soggetti omozigoti per β0 l’HbA non è dosabile. La vita media delle emazie è ridotta a circa 20 giorni e la bilirubinemia, soprattutto indiretta ottenuta dal catabolismo dell’eme, sale a valori alti 6-8mg/dl (valori normali 0.2-1mg/dl). La terapia trasfusionale è d’obbligo spesso accompagnata da splenectomia. Al fine di evitare l’accumulo tissutale di ferro (emosiderosi) che danneggia soprattutto il cuore (à scompenso cardiaco refrattario di tipo dilatativo). Si utilizzano, pertanto, farmaci chelanti ferro.
• Talassemia Intermedia. Condizione determinata dai seguenti genotipi: A) omozigosi per geni β-talassemici lievi: β-tal+-β-tal+ B) contemporanea presenza di β-talassemia Maior e α-talassemia. E’ una talassemia caratterizzata da anemia che, a differenza della forma Maior, non necessita, se non occasionalmente, di emotrasfusioni. In questi soggetti vi è una certa quota di eritropoiesi inefficace, pertanto vi è un esaltato assorbimento di Fe, dato dall’iperplasia dell’eritrone. Pertanto, sebbene molto più tardivamente dei soggetti trasfusione-dipendente, possono sviluppare una emosiderosi, e necessitare pertanto di una terapia chelante il Fe. Può
WWW.SUNHOPE.IT

Leonardo Aurino
41
essere necessaria una splenectomia qualora la milza divenga megalica, con conseguente ipersplenismo e peggioramento dell’anemia.
• Talassemia Minor, condizione caratterizzata da una eterozigosi di tipo: A) β/β-tal+ B) β/β-tal0 E’ una condizione caratterizzata da assente o lieve anemia e senza sintomatologia clinica, diagnosticabile solo laboratoristicamente, con l’esame emocromocitometrico e morfologico del sangue e con l’elettroforesi dell’Hb. In Italia si contano più di 3 milioni di talassemici eterozigoti. La finalità della diagnosi di β-talassemia minor è principalmente eugenica, consentendo di informare il portatore che la sua unione con un altro β-talassemico eterozigote può portare nel 25% dei casi ad un figlio gravemente malato (morbo di Cooley). L’eterozigosi per la β-talassemia comporta una modestissima riduzione della sintesi di catene β, e quindi dell’HbA. Il lieve eccesso di catene α va a complessarsi con catene δ, formando HbA2 (α2δ2). La diagnosi si basa, pertanto, proprio sul riscontro di livelli lievemente maggiori di HbA2 (normalmente non supera il 3% del totale). Inoltre, tipicamente il soggetto con talassemia minor ha un elevato numero di GR (in rapporto ai livelli di Hb) microcitici e ipocromici, con aumento dell’RDW.
Talassemia α La sintesi delle catene α è regolata da due loci genici, posti sui cromosomi 16. Vi sono quindi, in totale, 4 geni codificanti per catene α. La gravita dell’α –talassemia dipende dal numero di geni “α-talassemici”, o meglio, mancanti. Infatti, a differenza delle β-talassemia, il principale difetto genico delle α-talassemia è la delezione di uno o più geni α. In base al numero di alleli α mancanti si distinguono:
• Genotipo --/-- : Anche definita α-talassemia 0 omozigote. In questo caso vi una delezione di tutti i geni codificanti per catene α in entrambi i cromosomi 16. Vi è completa assenza di geni codificanti per catene α. Ciò determina l’impossibilità di sintetizzare catene α, condizione non compatibile con la vita. In questa condizione, durante la vita fetale, le catene γ che si producono non trovano catene α cone le quali appaiarsi, precipitando sottoforma di tetramero γ4 (Hb Bart). L’Hb Bart ha un’affinità per l’O2 più di 10 volte superiore alle Hb normali e perciò non cede ossigeno ai tessuti. Ciò realizza il quadro clinico dell’anasarca, con morte del feto in utero (con macerazione) o subito dopo la nascita.
• Genotipo --/αα: Anche definita α-talassemia 0 eterozigote (tratto α-talassemico). In questo caso, vi è delezione dei geni codificanti per catene α di un singolo cromosoma 16. La produzione di catene α è quindi lievemente ridotta, consentendo la formazione di, durante la vita fetale e per i primi 6 mesi di vita di Hb Bart (tetramero γ4). Dopo i 6 mesi di vita, il passaggio ad una forma di HbA è possibile in quanto vi è una sufficiente quota di catene α. Clinicamente, il soggetto affetto da α-talassemia 0 eterozigote ha un quadro ematologico simile a quello della β-talassemia minor, con modesta anemia (asintomatica o subclinica) e aumentato numero di GR circolanti, piccoli e ipocromoci. La diagnosi nell’adulto può essere raggiunta solo per esclusione o per studi familiari.
• Genotipo –α/-- : definita anche malattia da HbH, è una condizione di doppia eterozigosi per l’α-talassemia 0 e l’α-talassemia+ . Un cromosoma 16 è totalmente mancante di geni codificanti per catene α, mentre l’altro cromosoma 16 (omologo) possiede una singola copia genica (α-talassemia+). In definita, quindi, il paziente possiede un singolo gene α, sufficiente a produrre una quota di catene α-globiniche che risulta però insufficiente. L’HbA è quindi presente, sebbene in concentrazioni ridotte. Vi sarà pertanto una forma di anemia che varia da severa a moderata, di tipo ipocromico/microcitico. Durante la vita fetale, la ridotta concentrazione di catene α spiega la formazione di HbH(γ4).
WWW.SUNHOPE.IT

Leonardo Aurino
42
Quando, dopo il 6° mese di vita, inizia la sostituzione dell’HbF con l’HbA, data la mancanza di catene α sufficienti, le catene β emoglobiniche tendono ad associarsi tra di loro, formando un tetramero β4, definito HbH. L’HbH è una emoglobina fortemente instabile che, precipitando sulla membrana eritrocitaria, ne determina la lisi. L’analisi elettroforetica dell’Hb mostra dei livelli che raggiungono anche il 30% di HbH, la quale migra velocemente oltre l’HbA. La gravita dell’anemia può essere limitata dalla splenectomia.
• Genotipo αα/α- : tale genotipo è definito “portatore α-talassemico”. Il numero di geni codificanti per catene α è del tutto sufficiente, sicché non si realizza anemia né grave né moderata. Spesso vi è solo una anemia lieve ipocromica/microcitica valutabile solo laboratoristicamente. Entro poche settimane dalla nascita può essere dosata una piccola percentuale di Hb Bart.
Consulenzagenetica La CG è l’atto informativo attraverso il quale i pazienti affetti da una malattia geneticamente determinata, o i loro familiari, ricevono informazioni relative a:
• Caratteristiche della malattia • Modalità di trasmissione • Rischio di ricorrenza nella progenie • Possibili terapie
L’individuo che richiede la CG al medico genetista è definito consultando. Il consultando autorizza le indagini, il prelievo, l’archiviazione e l’utilizzo del materiale genico mediante consenso informato. Il consenso informato per consulenze genetiche può essere ampliato o ristretto. Di norma, partecipano alla CG, oltre al medico genetista, anche altre figure professionali, tra cui uno psicologo, un citogenetista (e il laboratorio di biologia molecolare), il ginecologo (in caso il consultando sia una donna in stato di gravidanza) e la nurse di genetica (una figura infermieristica che effettua una selezione di I° livello per inviare pazienti già selezionati al genetista). Aspetti fondamentali di una CG sono:
• Costruzione di un albero genealogico (cosa che può richiedere numerosi incontri) • Verifica della diagnosi
Problemi connessi alla CG sono: • Eterogeneità genetica: situazione per la quale lo stesso fenotipo può essere dovuto a
mutazioni di geni diversi. Ad esempio, un soggetto che necessita di sedie a rotelle può essere affetto da SMA, SLA o distrofia muscolare.
• Fenocopie: condizioni cliniche che simulano malattie genetiche ma che non lo sono (esempio una microcefalia può essere genetica, come nella microcefalia AR, o essere conseguenza di un utero materno settato).
• Casi sporadici: sono mutazioni “de novo”, che insorgono come condizione somatica nello zigote e non come condizione germinale. È indispensabile sapere se una malattia si è presentata come caso sporadico o come difetto ereditario, soprattutto per poter programmare una nuova gravidanza. Ad esempio, una coppia che partorisce un feto con NF1 (malattia AD) ha una possibilità di generare un secondo figlio affetto del 50% se la mutazione è germinale, mentre un secondo figlio affetto come evento sporadico è di 1/10.000.
• Illegittimità: ossia errata attribuzione di paternità, cosa che avviene nel 15% dei casi.
WWW.SUNHOPE.IT

Leonardo Aurino
43
La CG per le cromosomopatie riguarda di solito madri in età avanzata, in quanto il progredire dell’età aumenta notevolmente il rischio di non disgiunzione meiotica a livello dell’ovogenesi, con sensibile aumento del rischio di trisomie cromosomiche. La maggior parte delle trisomie sono incompatibili con la vita, mentre la trisomia del 21, 13 e 18 sono responsabili rispettivamente dalla s. di Down, s. di Patau e s. di Edwards. La CG nella malattie ad ereditarietà mendeliana è necessaria per definire il pattern di ereditarietà AR o AD o X-linked. Le CG possono essere di vario tipo:
• CG prenatale: Un aiuto per decidere cosa sia meglio fare per monitorare lo stato di salute del feto è sicuramente dato dal colloquio con il proprio Genetista, durante il quale si discutono e si chiariscono i seguenti aspetti: -studio sulla possibile malattia genetica per cui viene richiesto l’esame. -Informazioni riguardanti il rischio che il bambino abbia un difetto genetico che si intende ricercare. -Il tipo di analisi più adatta al proprio caso e il tipo di risultati ottenibili (esistono metodiche invasive, come l’amniocentesi-villocentesi-funicolocentesi, e metodiche non invasive come gli screening biochimici+translucenza nucale). -Informazioni sull’affidabilità dell’analisi. -Percentuali del rischio di aborto dovuto alla procedura. -Il tempo necessario per ottenere i risultati dell’analisi e relative modalitàper accedere al risultato degli esami richiesti. -Le possibilità di cura o le alternative possibili se il bambino risulti affettoda una malattia genetica. -Le conseguenze sul piano emotivo per la coppia che esegue l’analisi. Durante la consulenza genetica sarà spiegato nei dettagli in cosa consistono queste procedure, quando e come si effettuano, cosa accade dopo il prelievo e i possibili benefici e rischi correlati.
• CG neonatale: CG richiesta dopo la nascita di un neonato o di un nato morto, al fine di riconoscere l’eventuale patologia genetica/malformativa di base, le conseguenze della malattia, la modalità di trasmissione, il rischio di ulteriori figli affetti e ovviamente le eventuali strategie terapeutiche. Questa consulenza si svolge in collaborazione con diversi specialisti.
• CG dismorfologica: i soggetti con sindromi dismorfico-genetiche vengono valutati sia ai fini diagnostici che di follow-up. Per ogni paziente viene effettuata una anamnesi molto approfondita, raccolta dell'albero genealogico ed esame fisico del paziente che comprende una valutazione dismorfologica (accurata descrizione di tutti i segni dismorfici) e documentazione fotografica. I segni osservati vanno interpretati e categorizzati. E' indispensabile una adeguata preparazione in campo genetico-clinico, una ampia conoscenza della letteratura ed eventualmente l'uso di mezzi diagnostici computerizzati. Viene poi attuata una attività di coordinamento degli interventi di assistenza specialistica multidisciplinare indispensabili sia alla diagnosi che alle cure di tali soggetti. Esistono dei database dismorfologici quali POSSUM (Pictures of Standard Syndromes and Undiagnosed Malformation) e il LDDB (London Dysmorfology DataBase).
• CG riproduttiva: CG utile: a) prima del concepimento, per riconoscere la possibilità di sviluppo di una malattia ereditaria nella gravidanza progettata,
WWW.SUNHOPE.IT

Leonardo Aurino
44
b) prima dell’avvio di programmi di fecondazione assistita (se si hanno malattie genetiche non si può accedere al programma) c) nel caso di una coppia con precedente figlio affetto da patologia genetica d) nei soggetti che sono venuti a conoscenza di una malattia genetica nei propri familiari.
• CG oncologica: il 10-15 % dei tumori ha una sua base genetica. Esempio: ricerca delle mutazioni BRCA1/2 per poter allestire il miglior schema terapeutico possibile prima della comparsa del tumore e stimarne il rischio. Tali test sono quindi devoluti a ottenere una diagnosi precoce tramite screening e follow.up.
• CG richiesta per effetuare test pre-sintomatici: Ossia per richiedere dei test che possano diagnosticare una malattia genetica prima della comparsa dei sintomi. Si esegue solo per malattie curabili (FAP), mentre in caso di malattie non curabili non viene eseguita per le importanti implicazioni psicologiche che possono derivarne.
TestgeneticiI test genetici sono analisi a scopo clinico di DNA, RNA, proteine, cromosomi, metaboliti e altri prodotti, allo scopo di evidenziare mutazioni, genotipi, fenotipi o cariotipi correlati o meno con patologie ereditabili. Questa definizione include i test di screening prenatale, neonatale, sui portatori e sulle famiglie a rischio. I test genetici eseguibili sono:
• Test diagnostici: Sono test eseguibili sia in epoca pre-natale che post-natale, che hanno lo scopo di definire una malattia già esistente e già sintomatica. Esempi sono: valutazione del cariotipo in un soggetto con aneupoloidia, ricerca di muazioni del gene CFTR (fibrosi cistica) in un soggetto con ricorrenti infezioni polmonari.
• Test presintomatici: test eseguiti su soggetti affetti da patologie genetiche con esordio tardivo, quindi prima della comparsa dei sintomi. Hanno lo scopo di predire il futuro clinico del soggetto. Sono utili e vanno eseguiti solo quando vi è la possibilità di una cura efficace, altrimenti andrebbero evitati. Nella maggior parte dei casi, le malattie genetiche ad esordio tardivo sono le AD (vedi malattia di Huntington e rene policistico dell’adulto).
• Test per la valutazione della suscettibilità genetica: Sono test volti a rivelare una eventuale suscettibilità a malattie specifiche geneticamente determinate. È questo il caso della definizione dell’aplotipo HLA (es. HLA-B27 definisce la possibilità di sviluppo di spondilite anchilosante), di una mutazione del gene codificante l’enzima G6PD che può essere responsabile di crisi emolitiche al momento di assunzione di alcuni farmaci/fave, valutazione del deficit di α1-antitripsina che, se associato al fumo, espone all’enfisema panlobulare giovanile, mutazioni del gene codificante per subunità del canale rianodinico per prevenire l’ipertermia maligna secondaria all’anestesia.
• Test per l’identificazione dei portatori eterozigoti: prevalentemente hanno finalità eugeniche. Conoscere di essere portatore, ad esempio di β-talassemia minor, mette in guarda il soggetto dall’unione con un altro soggetto portatore, in quanto tale unione può generare, nel 25% dei casi, un figlio affetto da morbo di Cooley.
• Test con finalità medico legali: Come test per l’attribuzione di paternità e test per identificazione di vittime o indagati (polizia scientifica).
DiagnosiprenatalePer diagnosi prenatale si intende il complesso di indagini, strumantali e laboratoristiche, finalizzate alla diagnosi e al monitoraggio dello stato di salute del concepito. Le indagini sono effettuate
WWW.SUNHOPE.IT

Leonardo Aurino
45
durante tutto l’arco della gravidanza. Si ricordo che la % dei soggetti affetti è, nella pratica, notevolmente inferiore a quella prevedibile teoricamente, in quanto molte anomalie (cromosomiche o genetiche) hanno carattere abortivo. I principali soggetti che richiedono una diagnosi prenatale sono:
1. Madri con età avanzata (> 35 anni) al momento del concepimento 2. Coppie con precedenti figli affetti da cromosomopatie 3. Genitori con mosaicismo/aneuploidie fertili. 4. In caso di gravidanze che abbiano mostrato anomalie specifiche (difetti di accrescimento,
alterazioni di parametri biochimici, anomalie del liquido amniotico, documentata positività al complesso TORCH)
Le indagini eseguibili in diagnostica prenatale sono distinte in: • Indagini del I trimestre à indagini precoci che hanno il compito di suggerire un eventuale
aborto (in Italia è possibile l’interruzione volontaria di gravidanza entro 90 giorni anche in assenza di indicazioni mediche specifiche)
• Indagini del II trimestreà per valutare la possibilità di un aborto tardivo (in Italia, in presenza certa di malattie cromosomiche/citogenetiche è possibile l’aborto fino al 180° giorno. Si ricordi che, comunque, anche nel caso di aborto tardivo, nel momento in cui il feto espulso sia vitale va soccorso).
• Indagini del III trimestre à indagini di preparazione al parto (nel terzo trimestre non è possibile eseguire l’aborto).
Indagini diagnostiche del I e II trimestre Sono distinte in: Test strumentali e non invasivi
• Tritest (sensibilità 70%): è un test biochimico finalizzato a riconoscere patologie specifiche. Si basa sul dosaggio di α-fetoproteina, fβ-HCG, estriolo non coniugato. È indicato in donne di età superiore ai 35 anni al momento del concepimento. a) α-fetoproteina: aumenta nei difetti di chiusura del tubo neurale, nei difetti strutturali della parete addominale (estrofia vescicale e onfalocele), nella labio-palatoschisi, nella s. di Turner. Elevati livelli di AFP sono fisiologici nei parti gemellari. Ridotti livelli di α-fetoproteina sono riscontrabili nella sindrome di Down. b) estriolo non coniugato: si riduce nella s. di Down c) β-HCG: aumenta nella s. di Down. Va eseguito alla 16a settimana.
• Bitest: valuta i livelli di fβ-HCG e PAPP-A. Ha una sensibilità dell’80% per la diagnosi di sindrome di Down (dove la β-HCG libera aumenta e il PAPP-A si riduce) se associato alla valutazione ecografica della traslucenza nucale(v.n. < 2,5-3 cm)
• Quadtest: dosaggio di fβ-HCG, α-fetoproteina, inibina A e estriolo non coniugato. È il test con sensibilità più elevata (90%) ma è eseguibile solo dopo la 15a settimana.
• Ecografia: esame strumentale non invasivo che permette di: a) nel I trimestre: visualizzare la camera gestazionale, definire l’età gestazionale, valutare l’attività cardiaca e calcolare la traslucenza nucale (v.n. < 2,5-3 cm) e riconoscere macroanomalie (come l’anencefalia) b) nel II trimestre: valutare il numero di feti, numero e inserzioni della placenta, entità del liquido amniotico, attività cardiaca, eventuali malformazioni. c) nel III trimestre: indicazioni di tempi, luoghi e modalità del parto. In caso di specifiche malattie genetiche il parto dovrebbe essere eseguito in strutture di III livello, dotate di terapia intensiva neonatale.
• ECOcardiografia fetale: per la valutazione della struttura del cuore, alla ricerca di eventuali cardiopatie congenite.
WWW.SUNHOPE.IT

Leonardo Aurino
46
• Flussimetria doppler dell’arterie uterine e ombelicali: alla ricerca di patologie placentari e sullo studio delle gestosi.
• Isolamento di cellule fetali non invasivo: è una metodica ancora sperimentale tramite isolamente da sangue materno o lavaggio cervicale.
Test invasivi
• Villocentesi: (9-11 w): prelievo sotto guida ecografica per via transaddominale o transcervicale di alcuni grammi di tessuto coriale (villi), che essendo di derivazione trofoblastico, sono di origine fetale. I risultati della villocentesi sono ottenibili in 24 ore poiché i villociti non hanno bisogno di essere messi in coltura (hanno mitosi spontanee). Complicanze sono il sanguinamento cervicale (10% dei casi), corioamniosite, oligoidramnios, sensibilizzazione Rh (se la madre è Rh- bisogna sempre fare profilassi con siero anti-D) e l’aborto (1%)
• Amniocentesi (15-18 w): prelievo sotto guida ecografica per via transaddominale di 20-30 ml di liquido amniotico, contenenti amniociti, cellule di sfaldamento di origine fetale. Il risultato si ottiene dopo circa 3 settimana, in quanto le cellule vanno messe in coltura con fitoemoagglutinina (a volte può essere causa di ritardo diagnostico e superamento della soglia del 180° giorno previsto per l’interruzione di gravidanza tardiva).
• Funicolocentesi: prelievo di sangue fetale tramite il funicolo. È una procedura ecoguidata. È indicata soprattutto per patologie ematologiche.
Terapiagenica L’inserimento di materiale genetico all’interno delle cellule con finalità terapeutiche è definito terapia genica. La procedura di trasferimento del materiale genetico è detto transfezione. Per essere più esatti la terapia genica consiste nel trasferimento di uno o più geni sani in una cellula malata, al fine di curare una patologia causata dall'assenza o dal difetto di uno o più geni (mutati). Dunque, è necessario in primo luogo identificare il singolo gene o i diversi geni responsabili della malattia genetica. La terapia genica può avvalersi di:
• Cellule staminali somatiche • Cellule staminali embrionali (in Italia è vitato) • Trasferimento di singoli o più geni • Modificazioni dell’RNA
Il trasferimento genico può interessare due linee cellulari:
• Cellule germinali: Se si trasferisce un gene ad una linea cellulare germinale (o su cellule staminali totipotenti), la modifica che si apporta al genoma verrà trasmessa alle generazioni successive attraverso le stesse cellule germinali (spermatozoi e ovociti). Attualmente è impossibile controllare completamente le alterazioni genetiche che vengono apportate da un trasferimento genico, di conseguenza si rischia di provocare delle anomalie fisiologiche che verrebbero trasmesse alle generazioni successive; inoltre, l'alterazione di materiale genetico ereditabile pone una serie di interrogativi dal punto di vista etico e morale. Per queste ragioni, la modificazione genetica di linee cellulari germinali non viene considerata una strategia adottabile, e la sua sperimentazione clinica e' vietata in Svizzera come in quasi tutti gli altri paesi del mondo.
• Cellule somatiche: Con il trasferimento genico su linee cellulari somatiche, si modifica il genoma di tessuti (muscoli, polmoni, cervello, ossa, reni, cuore etc.), senza che questa
WWW.SUNHOPE.IT

Leonardo Aurino
47
modificazione venga trasmessa alla generazione successiva; l'alterazione genetica riguarda esclusivamente il paziente su cui è stata realizzata. Ciò non significa che tale terapia sia priva di rischi.
La terapia genica viene principalmente distinta in:
• Ex vivo: Consiste nel prelievo di cellule somatiche della persona interessata. Successivamente queste cellule vengono messe in coltura e trasfettate con il gene d’interesse, inserito tramite un apposito vettore (di norma virale), per poi essere reinfuse e e reimpiantate nel corpo del soggetto. Tale procedura è senza dubbio lunga e onerosa, ma permette di selezionare e amplificare le cellule d’interesse e gode, dunque, di maggior efficienza.
• In vivo: Viene attuata quando le cellule prelevate non possono essere messe in coltura o prelevate e reimpiantate, come nel caso del cervello, del cuore e di molti organi interni. In questo caso, il gene d’interesse viene inserito in un vettore idoneo, il quale viene direttamente immesso nell’organismo per via locale o sistemica. A tale scopo possono essere utilizzati vettori virali e non virali. La coniugazione del DNA da inserire nel vettore non virale può essere di tre tipi: A) lipoplessi: in cui il DNA (carico negativamente) è legato a nanoparticelle lipidiche con cariche positive B) poliplessi: in cui il DNA è legato a nanoparticelle polimeriche (policationi) C) lipopoliplessi: in cui il DNA è legato ad un sistema supramolecolare formato da liposomi e policationi.
Una volta che il gene raggiunge la cellula target, esso può essere integrato nel genoma o rimanerne fuori, in forma episomiale. Nel primo caso, nel momento in cui il gene si integra nel genoma della cellula ospite, esso viene replicato e tradotto, venendo quindi trasmesso a tutte le generazioni cellulari derivanti dalla cellula madre. Nel caso in cui il gene permanga in forma episomiale, al contrario, non avviene la trasmissione alle cellule figlie. È possibile ovviare a questa evenienza inserendo nel gene delle sequenze di replicazione, per permettere la trasmissione dell’episoma alle cellule figlie. Vettori I vettori utilizzabili sono distinti in virali e non virali. Trasferimento non virale: Il trasferimento di DNA senza vettori virali può avvenire per:
• Iniezione di DNA nudo all’interno delle cellule target, adeguatamente prelevate e messe in coltura, a mezzo di specifiche micropipette. Ovviamente, l’operatore dovrà trnafettare le cellule una ad una. Il rendimento di tale tecnica è notevolmente basso, in quanto buona parte dell’acido nucleico introdotto nella cellula viene degradato dai lisosomi.
• Inserimento mediante vettori lipidici (lipoplessi), vettori polimerici (poliplessi) e lipidi/polimerici (lipopoliplessi)
• Bombardamento cellulare a mezzo di gene gun: sono strumenti elettrici o ad alta pressione, detti cannoni genici (gene gun) che permettono di “bombardare” la cellula con nanoparticelle d’oro o di tungsteno ricoperte da DNA.
Trasferimento virale Si basa sull’utilizzo di opportuni virus ricombinanti. I virus hanno una elevata tendenza ad infettare le cellule ospiti e inserirvi il proprio DNA, grazie alla presenza di enzimi come le endonucleasi e le DNA-ligasi. Il genoma può, anche in questo caso,
WWW.SUNHOPE.IT

Leonardo Aurino
48
restare sotto forma episomiale nel nucleo celluare o essere integrato. Il trasferimento con vettori virali ha un’efficienza notevolmente maggiore rispetto al trasferimento non virale. I virus da utilizzare devono, però, essere dotati di alcune caratteristiche:
• Il virus da utilizzare non deve essere in grado di riprodursi, a differenza del virus wild-type. • Il virus non deve essere in grado di causare effetti indesiderati (infezione, attivazione del
sistema immunitario) • Il genoma del virus deve essere abbastanza grande da contenere il gene terapeutico che si
vuole utilizzare (vi è quindi anche un vincolo di dimensione). I virus più utilizzati come vettore sono: retrovirus, lentivirus, adenovirus, herpesvirus, virus adeno-associati. Si tenga conto, però, che in alcuni casi, l’inserzione di materiale genetico all’interno del DNA della cellula ospite (soprattutto quando introdotto tramite virus del tipo retrovirus, lentivirus e virus adeno-associati) ha turbato la normale espressione di protooncogeni umani, determinandone una iperespressione e genesi di tumori. Lo sviluppo di tumore è molto più frequente nei casi in cui si utilizzino virus che hanno una elevata capacità di introdurre il proprio genoma all’interno di quello della cellula ospite, formando un ibridoma. Tra questi virus si annoverano: lentivirus, retrovirus e virus adeno-associati. Al contrario, tale “reazione avversa” è meno frequente con quei virus che hanno dimostrato bassa tendenza all’integrazione nel genoma (e che restano quindi in forma episomiale): adenovirus, herpesvirus.
WWW.SUNHOPE.IT