
DIPARTIMENTO DI MEDICINA SPERIMENTALE TESI DI …padis.uniroma1.it/bitstream/10805/2259/1/TESI di...
41
DIPARTIMENTO DI MEDICINA SPERIMENTALE TESI DI DOTTORATO DI RICERCA IN GENETICA MEDICA - XXVI CICLO - Coordinatore: Prof.ssa Paola Grammatico Caratterizzazione funzionale della proteina SHOC2 e della sua forma miristilata implicata nella sindrome di Mazzanti DOTTORANDA Marialetizia Motta TUTOR Dott. Marco Tartaglia Direttore del reparto di fisiopatologia delle malattie genetiche Dip. EOMM, Istituto Superiore di Sanità, Roma
Transcript of DIPARTIMENTO DI MEDICINA SPERIMENTALE TESI DI …padis.uniroma1.it/bitstream/10805/2259/1/TESI di...

DIPARTIMENTO DI MEDICINA SPERIMENTALE
TESI DI
DOTTORATO DI RICERCA
IN
GENETICA MEDICA - XXVI CICLO -
Coordinatore: Prof.ssa Paola Grammatico
Caratterizzazione funzionale della proteina SHOC2 e
della sua forma miristilata implicata nella sindrome di Mazzanti
DOTTORANDA Marialetizia Motta
TUTOR
Dott. Marco Tartaglia
Direttore del reparto di fisiopatologia delle malattie genetiche
Dip. EOMM, Istituto Superiore di Sanità, Roma

2
INDICE
ABBREVIAZIONI 4
INTRODUZIONE 6
La sindrome di Noonan 6
Caratteristiche cliniche 6
Aspetti genetici 8
Diagnosi 9
Terapia 11
La sindrome di Noonan con “loose anagen hair” o 12
sindrome di Mazzanti
La proteina SHOC2 13
FINALITÀ DELLA RICERCA 16
MATERIALI E METODI 17
Analisi in silico 17
Preparazione dei costrutti 17
Colture cellulari e trasfezione 20
Omogenati cellulari e dosaggio proteico 20
Preparazione di microdomini di membrana (lipid rafts) 21
Elettroforesi e Western Blotting 21
Microscopia confocale a scansione laser (CLSM) 22

3
RISULTATI 24
Espressione delle proteine SHOC2 mutate 24
La traslocazione nel nucleo di SHOC2WT
non è mediata dalla NLS 25
La stabilità in membrana di SHOC2S2G
non dipende dalla palmitolazione 25
Il targeting alla membrana di SHOC2S2G
è mediato dalle LRR N-terminali 26
La traslocazione nel nucleo di SHOC2WT
dipende dalle LRR N-terminali 28
Il ruolo dei motivi KEKE 28
I motivi KEKE sono necessari per l’associazione
di SHOC2S2G ai lipid rafts 31
DISCUSSIONE 33
BIBLIOGRAFIA 36

4
ABBREVIAZIONI
ALL, acute lymphoblastic leukemia
AML, acute myeloid leukemia
BCA, bicinchoninic acid
BSA, bovine serum albumin
CLSM, confocal laser scanning microscopy
DAPI, 4’,6-diamidino-2-phenylindole
EGF, epidermal growth factor
FBS, fetal bovine serum
GH, growth hormone
GPI, glycosylphosphatidylinositol
GTP, guanosine -5’- triphosphate
HRP, horseradish peroxidase
IGF-1, insulin-like growth factor 1
JMML, juvenile myelomonocytic leukemia
LRR, leucine-rich repeat
MAPK, mitogen-activated protein kinase
NF1, neurofibromatosis type 1
NLS, nuclear localization signal
NMT, N-myristoyltransferase
PBS, phosphate buffered saline
PFA, paraformaldehyde
PP1C, protein phosphatase 1 catalytic subunit
RAS, rat sarcoma
SC, Costello syndrome

5
SCFC, cardiofaciocutaneous syndrome
SDS-PAGE, sodium dodecyl sulphate – polyacrylamide gel electrophoresis
SHP-2, Src homology-2 domain-containing phosphatase-2
SL, LEOPARD syndrome
SN, Noonan syndrome
SN/LAH, Noonan-like syndrome with loose anagen hair

6
INTRODUZIONE
LA SINDROME DI NOONAN
La sindrome di Noonan (SN, OMIM 163950) è una malattia dello sviluppo descritta
nel 1963 dalla cardiologa pediatra Jacqueline Noonan che notò, in pazienti giunti
alla sua osservazione per una cardiopatia congenita, la presenza di caratteristiche
facciali simili e di bassa statura (Noonan e Ehmke, 1963). Venne subito riscontrata
una somiglianza con la sindrome di Turner, dalla quale però la SN si differenzia in
quanto colpisce entrambi i sessi, gli individui affetti hanno un assetto cromosomico
normale e in alcune famiglie si osserva una trasmissione autosomica dominante.
L’incidenza della sindrome è compresa tra 1:1000 e 1:2500, anche se fenotipi “mild”
possono presentare una frequenza molto più elevata (1:100) (Mendez e Opitz, 1985).
Caratteristiche cliniche
Le manifestazioni cliniche della SN comprendono bassa statura armonica, facies
dismorfica, difetti cardiaci congeniti, anomalie scheletriche e disabilità intellettive di
grado variabile (Noonan, 1994; Allanson, 2007; van der Burgt, 2007). I principali
dismorfismi facciali sono: fronte alta, ipertelorismo, rima degli occhi rivolta verso il
basso, ptosi palpebrale, epicanto, naso con radice depressa, base larga e punta
bulbosa, orecchie a basso impianto e angolate posteriormente, collo corto con
eccesso di cute sulla nuca e attaccatura bassa dei capelli. Con la crescita, il volto
assume sempre più la forma di un triangolo rovesciato (fronte ampia e mento
appuntito), il collo si allunga e la ridondanza di cute si distende a formare uno
pterigio (Fig. 1). In circa il 50-80% dei casi è presente una cardiopatia congenita, la
più frequente delle quali è la stenosi della valvola polmonare, seguita da un difetto
del setto atrioventricolare, mentre il 20-30% dei pazienti affetti da SN sviluppa una
cardiomiopatia ipertrofica (Burch et al., 1993; Marino et al., 1999). Le anomalie
dell’apparato scheletrico, prevalentemente a carico della colonna vertebrale e del
torace, includono la scoliosi, la fusione delle vertebre cervicali e il petto carenato. Gli
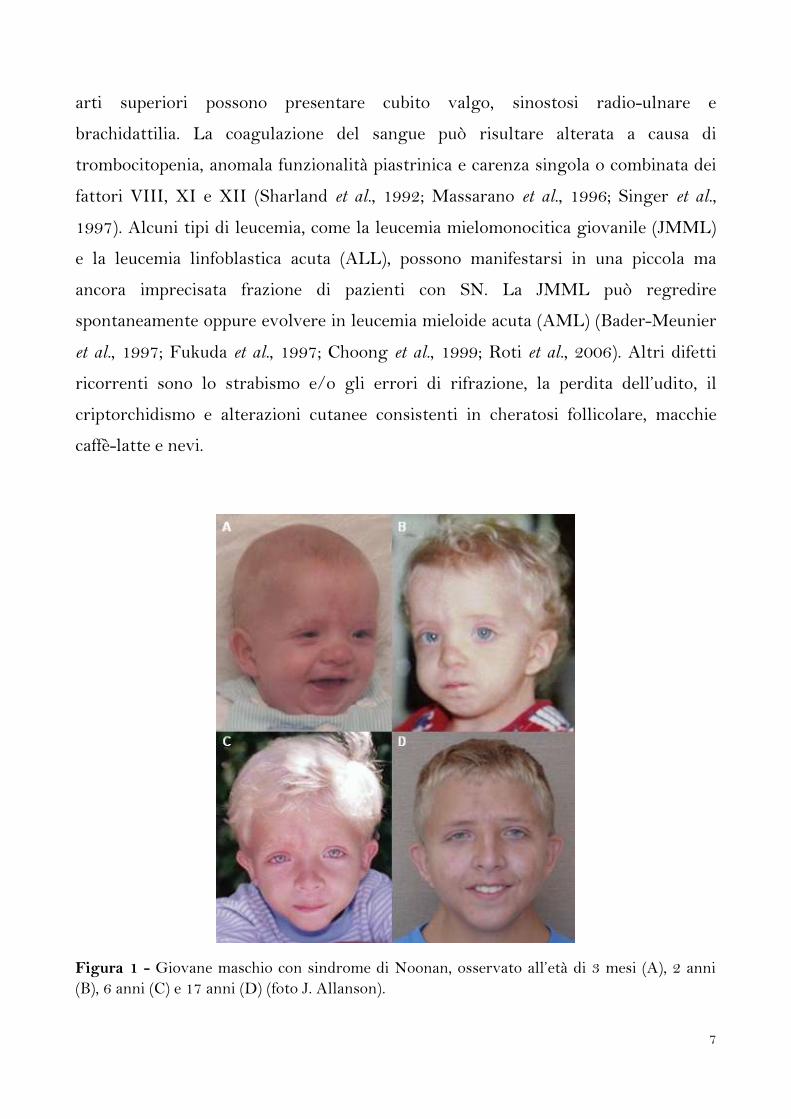
7
arti superiori possono presentare cubito valgo, sinostosi radio-ulnare e
brachidattilia. La coagulazione del sangue può risultare alterata a causa di
trombocitopenia, anomala funzionalità piastrinica e carenza singola o combinata dei
fattori VIII, XI e XII (Sharland et al., 1992; Massarano et al., 1996; Singer et al.,
1997). Alcuni tipi di leucemia, come la leucemia mielomonocitica giovanile (JMML)
e la leucemia linfoblastica acuta (ALL), possono manifestarsi in una piccola ma
ancora imprecisata frazione di pazienti con SN. La JMML può regredire
spontaneamente oppure evolvere in leucemia mieloide acuta (AML) (Bader-Meunier
et al., 1997; Fukuda et al., 1997; Choong et al., 1999; Roti et al., 2006). Altri difetti
ricorrenti sono lo strabismo e/o gli errori di rifrazione, la perdita dell’udito, il
criptorchidismo e alterazioni cutanee consistenti in cheratosi follicolare, macchie
caffè-latte e nevi.
Figura 1 - Giovane maschio con sindrome di Noonan, osservato all’età di 3 mesi (A), 2 anni
(B), 6 anni (C) e 17 anni (D) (foto J. Allanson).

8
Aspetti genetici
La SN può essere trasmessa come carattere autosomico dominante (forma familiare)
oppure può essere causata da mutazioni de novo (forma sporadica). Nel 50% dei casi,
la malattia è dovuta a mutazioni nel gene PTPN11 (Tartaglia et al., 2001), che
codifica per la proteina fosfotirosina fosfatasi SHP-2. In una piccola percentuale di
pazienti sono state identificate mutazioni nei geni SOS1 (Roberts et al., 2007;
Tartaglia et al., 2007), RAF1 (Pandit et al., 2007; Razzaque et al., 2007), KRAS (Carta
et al., 2006; Schubbert et al., 2006), BRAF (Sarkozy et al., 2009), NRAS (Cirstea et al.,
2010) e MEK1 (Nava et al., 2007). Recentemente si è scoperto che anche il gene
RIT1 è coinvolto nella SN (Aoki et al., 2013). Le mutazioni individuate nei diversi
geni-malattia sono in genere missenso e rendono conto del 75% circa dei casi clinici
(Fig. 2). Difetti molecolari nei geni SHOC2 e CBL causano patologie ricollegabili
alla SN, rispettivamente la sindrome di Noonan con “loose anagen hair” o sindrome di
Mazzanti (SN/LAH, OMIM 607721) (Mazzanti et al., 2003; Cordeddu et al., 2009) e
la sindrome “associata a mutazioni in CBL” (Martinelli et al., 2010; Niemeyer et al.,
2010; Perez et al., 2010).
Figura 2 - Geni-malattia noti responsabili della sindrome di Noonan e relativa prevalenza.
PTPN11 50%
?
SOS1 10%
RAF1 5%
5%
KRAS BRAF NRAS MEK1
RIT1 5%

9
Studi di correlazione genotipo-fenotipo hanno dimostrato che:
- mutazioni in PTPN11 sono più frequenti nei pazienti con stenosi della valvola
polmonare (Tartaglia et al., 2002);
- specifiche mutazioni in PTPN11 predispongono allo sviluppo di JMML
(Tartaglia et al., 2003; Kratz et al., 2005);
- mutazioni in RAF1 sono correlate con la cardiomiopatia ipertrofica (Pandit et
al., 2007);
- pazienti con mutazioni in SOS1 presentano una statura media più elevata e
sintomi cutanei significativi (Tartaglia et al., 2007; Zenker et al., 2007);
- mutazioni in KRAS sono associate a craniosinostosi, ritardo mentale e quadro
clinico complessivo generalmente più grave (Kratz et al., 2009).
I geni coinvolti nella SN codificano per proteine che partecipano alla via di
trasduzione del segnale RAS-MAPK implicata in numerosi processi cellulari come la
proliferazione, il differenziamento e il controllo dell’espressione genica (Schubbert et
al., 2007; Shi et al., 2013). La disregolazione di questa via è alla base della patogenesi.
Diagnosi
La diagnosi di SN si basa principalmente sull’osservazione delle caratteristiche
fenotipiche del paziente (Tabella 1) e diversi sistemi di punteggio (Scoring Systems)
sono stati sviluppati per aiutare il medico nella valutazione clinica (Duncan et al.,
1981; van der Burgt et al., 1994). Il sospetto diagnostico di SN può essere
confermato dall’analisi molecolare. Lo studio mutazionale può essere condotto
mediante lo screening consecutivo dei diversi geni oppure attraverso l’utilizzo di un
pannello multigenico. La mancata identificazione di una mutazione non consente
però di escludere la diagnosi, in quanto la causa genetica della malattia rimane
ancora sconosciuta nel 25% circa dei pazienti. La SN va posta in diagnosi
differenziale con la sindrome di Turner (analisi del cariotipo) e con le altre patologie
appartenenti alla famiglia delle RASopatie: la sindrome LEOPARD (SL, OMIM
151100), la sindrome cardiofaciocutanea (SCFC, OMIM 115150), la sindrome di
Costello (SC, OMIM 218040), la neurofibromatosi di tipo I (NF1, OMIM 162200) e

10
la sindrome di Legius (OMIM 611431). Queste malattie dello sviluppo, pur
mostrando un quadro clinico parzialmente sovrapponibile a quello della SN, sono
ben distinte dal punto di vista molecolare. Durante il periodo prenatale, alcuni segni
ecografici come l’igroma cistico, l’idrope fetale e il polidramnios, possono orientare
verso la SN (Zarabi et al., 1983; Benacerraf et al., 1989; Achiron et al., 2000). Nel caso
in cui la mutazione responsabile della patologia sia stata già identificata in un
membro della famiglia, si possono eseguire test genetici su villi coriali o liquido
amniotico.

11
Terapia
Il trattamento della SN richiede un approccio multidisciplinare. La cardiopatia, a
seconda del tipo e della gravità, può essere trattata con terapia farmacologica o
chirurgica. L’ormone della crescita (GH) può essere somministrato a pazienti che ne
sono deficitari o che presentano un difetto dell’asse GH/IGF-1. Il criptorchidismo
non responsivo alla terapia medica e alcune forme gravi di anomalie scheletriche,
strabismo e ptosi palpebrale possono richiedere un intervento chirurgico. Il
trattamento delle patologie della coagulazione viene personalizzato in base allo
specifico difetto diagnosticato e alla sintomatologia. La terapia di stimolazione
cognitiva e la logoterapia sono indicate in caso di ritardo mentale e difficoltà di
linguaggio. La consulenza genetica è utile sia per i pazienti che per le loro famiglie.
12
LA SINDROME DI NOONAN CON “LOOSE ANAGEN HAIR” O
SINDROME DI MAZZANTI
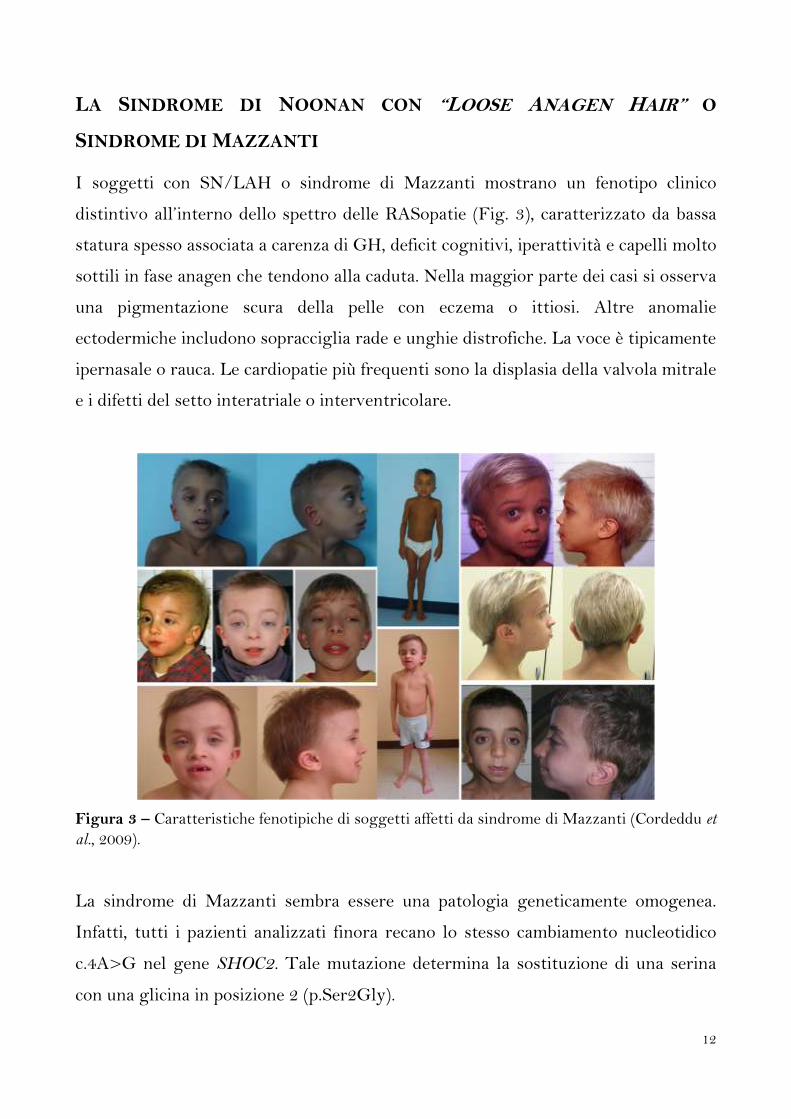
I soggetti con SN/LAH o sindrome di Mazzanti mostrano un fenotipo clinico
distintivo all’interno dello spettro delle RASopatie (Fig. 3), caratterizzato da bassa
statura spesso associata a carenza di GH, deficit cognitivi, iperattività e capelli molto
sottili in fase anagen che tendono alla caduta. Nella maggior parte dei casi si osserva
una pigmentazione scura della pelle con eczema o ittiosi. Altre anomalie
ectodermiche includono sopracciglia rade e unghie distrofiche. La voce è tipicamente
ipernasale o rauca. Le cardiopatie più frequenti sono la displasia della valvola mitrale
e i difetti del setto interatriale o interventricolare.
Figura 3 – Caratteristiche fenotipiche di soggetti affetti da sindrome di Mazzanti (Cordeddu et
al., 2009).
La sindrome di Mazzanti sembra essere una patologia geneticamente omogenea.
Infatti, tutti i pazienti analizzati finora recano lo stesso cambiamento nucleotidico
c.4A>G nel gene SHOC2. Tale mutazione determina la sostituzione di una serina
con una glicina in posizione 2 (p.Ser2Gly).

13
LA PROTEINA SHOC2
SHOC2 è una proteina di 582 aa, costituita da una regione N-terminale ricca in
residui di lisina e acido glutammico (o aspartico) disposti in modo alternato a
formare i motivi KEKE e da 19 ripetizioni di sequenze aminoacidiche ricche in
leucina (dominio LRR), generalmente coinvolte nelle interazioni proteina-proteina
(Fig. 4).
Figura 4 – Struttura schematica della proteina SHOC2. I numeri indicano gli aminoacidi che
delimitano la regione dei motivi KEKE e quella del dominio LRR.
Secondo dati noti in letteratura, SHOC2 è un modulatore positivo della via di
trasduzione del segnale RAS-MAPK e funziona come proteina regolatoria della
subunità catalitica della proteina fosfatasi 1 (PP1C). In seguito all’attivazione di un
recettore transmembrana per fattori di crescita, la proteina SHOC2 lega MRAS-
GTP favorendo la traslocazione in membrana di PP1C. Questa fosfatasi agisce su
RAF1 defosforilando il residuo di serina in posizione 259. RAF1 lega RAS-GTP,
viene attivata e dà inizio alla cascata delle MAP chinasi (Fig. 5) (Rodriguez-Viciana
et al., 2006).
Analizzando in silico la sequenza aminoacidica N-terminale della proteina SHOC2, si
è osservato che la sostituzione Ser2Gly determina la creazione di un sito di
miristilazione. Questa è una forma irreversibile di modificazione proteica che
consiste nel legame dell’acido miristico a un residuo di glicina localizzato
all’estremità N-terminale di un nascente polipeptide. La N-miristilazione è una
modifica co-traduzionale che avviene ad opera dell’N-miristoiltransferasi (NMT) in

14
seguito alla rimozione della metionina iniziale da parte della metionina
aminopeptidasi (Boutin, 1997; Farazi et al., 2001; Resh, 2006).
Figura 5 – Rappresentazione schematica di SHOC2 e dei suoi interattori secondo il modello
proposto da Rodriguez-Viciana et al. (2006).
Esperimenti di incorporazione di acido miristico [3H] hanno confermato la
miristilazione della proteina mutata SHOC2S2G che presenta una localizzazione
cellulare differente da quella di SHOC2 wild type (SHOC2WT). La proteina SHOC2WT
è distribuita nel citoplasma e nel nucleo in cellule starvate ed è localizzata
prevalentemente nel nucleo in cellule starvate e poi stimolate con EGF (Fig. 6A).
Invece, la proteina miristilata SHOC2S2G è costitutivamente localizzata in membrana
plasmatica (Fig. 6B) e ciò comporta un’aumentata attivazione delle MAPK in un
contesto cellulare specifico (Cordeddu et al., 2009). Infatti, nelle cellule di
neuroblastoma (Neuro2A) che esprimono la proteina SHOC2S2G, è stato osservato
un aumento, EGF dipendente, della defosforilazione del residuo di serina in
posizione 259 di RAF1 e un conseguente aumento dell’attivazione di ERK (Fig. 6C).

15
Figura 6 – (A) Localizzazione cellulare della proteina SHOC2WT transientemente espressa in
cellule COS-1. In condizione di deprivazione di siero (a sinistra), SHOC2WT è distribuita nel
citoplasma e nel nucleo mentre, dopo stimolazione con EGF (a destra), la proteina è più
concentrata nel nucleo. (B) Localizzazione cellulare della proteina SHOC2S2G transientemente
espressa in cellule COS-1. SHOC2S2G è localizzata in membrana plasmatica sia in cellule
starvate (a sinistra) che in cellule starvate e poi stimolate con EGF (a destra). Scala: 20 m.
(C) Defosforilazione della Ser259 di RAF1 e attivazione di ERK in cellule Neuro2A co-
trasfettate con costrutti codificanti per RAF1, ERK1 e SHOC2WT o SHOC2S2G, coltivate in
terreno privo di siero per 16h e indotte con EGF per 5, 15 e 30 minuti. Cellule esprimenti
RAF1S259A, una forma mutata della proteina RAF1 con maggiore attività enzimatica (Michaud
et al., 1995), sono state utilizzate come controllo.

16
FINALITÀ DELLA RICERCA
La sostituzione aminoacidica Ser2Gly promuove la N-miristilazione di SHOC2. La
proteina miristilata, al contrario di quella wild type, non è più in grado di traslocare
nel nucleo in seguito a stimolazione con EGF (perdita di funzione) e risulta invece
localizzata costitutivamente in membrana plasmatica (guadagno di funzione).
È noto che l’energia di legame fornita dalla N-miristilazione è relativamente debole
e non sufficiente ad ancorare una proteina alla membrana (Peitzsch e McLaughlin,
1993). Affinché ciò avvenga è necessario che ci sia un secondo segnale costituito da
una sequenza di aminoacidi basici (come nel caso della proteina c-Src), da uno o due
siti di palmitolazione (come nel caso delle proteine Lyn e Fyn) oppure, in
alternativa, la proteina miristilata può essere stabilizzata in membrana da interazioni
con altre proteine. Il secondo segnale sembra essere necessario anche per il targeting
della proteina ad uno specifico tipo di membrana cellulare (Resh, 1999).
Il primo obiettivo di questa tesi è stato quello di comprendere quali fossero i
meccanismi alla base della traslocazione di SHOC2WT nel nucleo e di SHOC2S2G alla
membrana plasmatica. A tal fine ho creato diverse forme mutanti delle due proteine
(mutazioni puntiformi o delezioni) e ne ho analizzato la localizzazione cellulare
tramite microscopia a fluorescenza confocale.
In passato è stato dimostrato che molte proteine leganti catene aciliche sature sono
associate ai lipid rafts (Shenoy-Scaria et al., 1994). Questi sono microdomini
specializzati della membrana plasmatica ricchi di sfingomielina, colesterolo e
gangliosidi, insolubili nei detergenti e presenti in frazioni a bassa densità di un
gradiente di saccarosio. I lipid rafts sono risultati essere coinvolti in numerosi
processi cellulari come l’esocitosi, l’endocitosi e la trasduzione del segnale (Simons e
Toomre, 2000).
Il secondo obiettivo di questa tesi è stato quello di verificare la presenza di una
eventuale associazione di SHOC2S2G miristilata ai lipid rafts. Per tale analisi ho preso
in considerazione anche la proteina SHOC2WT e alcune forme mutanti di SHOC2S2G.

17
MATERIALI E METODI
Analisi in silico
La sequenza aminoacidica di SHOC2 è stata analizzata in silico ed è stato osservato
che la proteina possiede (Fig. 7):
- una regione N-terminale ricca in residui di lisina (evidenziata in giallo);
(http://www.expasv.ch/tools/scanprosite/)
- una predetta sequenza di localizzazione nucleare (NLS) (evidenziata in fucsia);
(http://psort.ims.u-tokio.ac.jp/form2.html)
- un predetto sito di palmitolazione (Cys144) (evidenziato in verde);
(http://csspalm.biocuckoo.org/online.php)
- 19 ripetizioni ricche in leucina (LRR) (18 LRR predette da Pfam, scritte in
blu; 1 LRR predetta da SMART ma non da Pfam, scritta in rosso; LRR
predette con elevata confidenza, evidenziate in celeste).
(http://pfam.sanger.ac.uk/) (http://smart.embl-heidelberg.de/)
Figura 7 – Sequenza aminoacidica e domini funzionali della proteina SHOC2.
Preparazione dei costrutti
Il costrutto codificante la proteina SHOC2WT con tre sostituzioni aminoacidiche
(p.Pro79Ala, p.Arg82Gly, p.Lys83Gly) nella predetta NLS (PGTRKKS, residui 79-
85) (SHOC2NLS) e il costrutto codificante SHOC2S2G con il predetto sito di
palmitolazione mutato (p.Cys144Gly) (SHOC2S2G/C144G) sono stati creati mediante
mutagenesi sito-specifica (QuikChange Site-Directed Mutagenesis Kit, Stratagene).
MSSSLGKEKDSKEKDPKVPSAKEREKEAKASGGFGKESKEKEPKTKGKDAKDGKKDSSAAQP
GVAFSVDNTIKRPNPAPGTRKKSSNAEVIKELNKCREENSMRLDLSKRSIHILPSSIKELTQ
LTELYLYSNKLQSLPAEVGCLVNLMTLALSENSLTSLPDSLDNLKKLRMLDLRHNKLREIPS
VVYRLDSLTTLYLRFNRITTVEKDIKNLSKLSMLSIRENKIKQLPAEIGELCNLITLDVAHN
QLEHLPKEIGNCTQITNLDLQHNELLDLPDTIGNLSSLSRLGLRYNRLSAIPRSLAKCSALE
ELNLENNNISTLPESLLSSLVKLNSLTLARNCFQLYPVGGPSQFSTIYSLNMEHNRINKIPF
GIFSRAKVLSKLNMKDNQLTSLPLDFGTWTSMVELNLATNQLTKIPEDVSGLVSLEVLILSN
NLLKKLPHGLGNLRKLRELDLEENKLESLPNEIAYLKDLQKLVLTNNQLTTLPRGIGHLTNL
THLGLGENLLTHLPEEIGTLENLEELYLNDNPNLHSLPFELALCSKLSIMSIENCPLSHLPP
QIVAGGPSFIIQFLKMQGPYRAMV

18
Nel primo caso sono stati utilizzati il vettore di espressione pcDNA6/V5-SHOC2WT
come stampo e la coppia di primer:
5’-GCCAAACCCAGCAGCTGGGACTGGAGGAAAATCCAGCAATGC-3’ (Forward)
5’-GCATTGCTGGATTTTCCTCCAGTCCCAGCTGCTGGGTTTGGC-3’ (Reverse)
Nel secondo caso sono stati usati il vettore di espressione pcDNA6/V5-SHOC2S2G
come stampo e la coppia di primer:
5’-CCCAGCAGAGGTGGGAGGTTTAGTAAATCTC-3’ (Forward)
5’-GAGATTTACTAAACCTCCCACCTCTGCTGGG-3’ (Reverse)
I costrutti SHOC29-54 e SHOC2S2G/9-54, codificanti le proteine SHOC2WT e
SHOC2S2G prive dei motivi KEKE, sono stati ottenuti tramite PCR e clonaggio del
prodotto di amplificazione nel vettore di espressione eucariotico pcDNA6/V5-HisA
(Invitrogen). Le coppie di primer utilizzate sono riportate nella seguente tabella.
Costrutto Primer Sequenza 5’3’
SHOC29-54 SHOC29-54_NheI_Fw
GCTAGCACCATGAGTAGTAGTTTAGGAAAAGAAAAGGACTCCAGTGCTGCCCAAC
SHOC2_XhoI_Rv CTCGAGGACCATGGCACGATATGGACC
SHOC2S2G/9-54 SHOC2S2G/9-54_NheI_Fw GCTAGCACCATGGGTAGTAGTTTAGGA
AAAGAAAAGGACTCCAGTGCTGCCCAAC
SHOC2_XhoI_Rv CTCGAGGACCATGGCACGATATGGACC
I costrutti codificanti le proteine SHOC2WT con delezioni parziali del dominio LRR
sono stati ottenuti tramite “overlap extension PCR” (Fig. 8). Per ciascun costrutto,
due prodotti di PCR, rappresentanti le regioni che fiancheggiano la sequenza di
DNA da excidere, sono stati preparati utilizzando un primer non chimerico (in Fig.
8, a e d) e un primer chimerico (in Fig. 8, b e c). Quest’ultimo ha una sequenza
nucleotidica che deriva in parte dalla regione fiancheggiante a monte della delezione
e in parte dalla regione fiancheggiante a valle della delezione. Nel passaggio
successivo, i due prodotti di PCR sono stati usati come templato per una PCR di
ligazione che contiene la coppia di primer più esterna (in Fig. 8, a e d).

19
Figura 8 – Rappresentazione schematica del protocollo utilizzato per la “overlap extension
PCR” (modificata da Lee et al., 2004).
L’amplificato è stato infine clonato nel vettore di espressione pcDNA6/V5-HisA. I
costrutti prodotti e i primers utilizzati sono riportati nella seguente tabella.
Costrutto Primer Sequenza 5’3’
SHOC2LRR1-4
SHOC2_NheI_Fw GCTAGCGAGTTCATGTAGTTTTTGTCCAG
SHOC2LRR1-4_Rv CTGAGTTTTGACAACTCTTTGATTG
SHOC2LRR1-4_Fw CAATCAAAGAGTTGTCAAAACTCAG
SHOC2_XhoI_Rv CTCGAGGACCATGGCACGATATGGACC
SHOC2LRR5-9
SHOC2_NheI_Fw GCTAGCGAGTTCATGTAGTTTTTGTCCAG
SHOC2LRR5-9_Rv CTATTCAGTTTCAAGTTTTTGATG
SHOC2LRR5-9_Fw CATCAAAAACTTGAAACTGAATAG
SHOC2_XhoI_Rv CTCGAGGACCATGGCACGATATGGACC
SHOC2LRR10-13
SHOC2_NheI_Fw GCTAGCGAGTTCATGTAGTTTTTGTCCAG
SHOC2LRR10-13_Rv CTCAAGAGAAACCACAAGACTTG
SHOC2LRR10-13_Fw CAAGTCTTGTGGTTTCTCTTGAGG
SHOC2_XhoI_Rv CTCGAGGACCATGGCACGATATGGACC
SHOC2LRR14-18
SHOC2_NheI_Fw GCTAGCGAGTTCATGTAGTTTTTGTCCAG
SHOC2LRR14-18_Rv GATTGAAAGCTTGAGACCAGAC
SHOC2LRR14-18_Fw GTCTGGTCTCAAGCTTTCAATC
SHOC2_XhoI_Rv CTCGAGGACCATGGCACGATATGGACC

20
I costrutti codificanti le proteine SHOC2S2G con le delezioni parziali del dominio
LRR sono stati creati mediante mutagenesi sito-specifica utilizzando il vettore
pcDNA6/V5-SHOC2LRR1-4, pcDNA6/V5-SHOC2LRR5-9, pcDNA6/V5-
SHOC2LRR10-13 o pcDNA6/V5-SHOC2LRR14-18 come stampo e la coppia di primer:
5’-CCAGGCTTGAGTCACCATGGGTAGTAGTTTAGGAAAAG-3’ (Forward)
5’-CTTTTCCTAAACTACTACCCATGGTGACTCAAGCCTGG-3’ (Reverse)
L’introduzione delle mutazioni puntiformi e la creazione delle specifiche delezioni
sono state verificate tramite sequenziamento bidirezionale (ABI BigDye terminator
Sequencing Kit v.3.1 e sequenziatore ABI Prism 3500 Genetic Analyzer, Applied
Biosystems).
Colture cellulari e trasfezione
Per questa ricerca sono state utilizzate cellule COS-1 (American Type Culture
Collection, Manassas, VA, USA) coltivate in terreno DMEM (Dulbecco’s modified
Eagle’s medium, Euroclone, Wetherby, West York, U.K.) supplementato con siero
fetale bovino (FBS) al 10%, L-glutammina 2 mM, 100 unità/ml di penicillina e 100
g/ml di streptomicina (incubazione a 37°C, 5% di CO2, 95% di umidità).
Una volta giunte al 60-70% circa di confluenza, le cellule sono state trasfettate
transientemente con il reagente Fugene6 (Roche) seguendo il protocollo fornito dal
produttore.
Il terreno DMEM complementato solo con gli antibiotici e il fattore di crescita
dell’epidermide (EGF) (Invitrogen) alla concentrazione finale di 30 ng/ml sono stati
usati rispettivamente per starvare e stimolare le cellule.
Omogenati cellulari e dosaggio proteico
Le cellule, trasfettate con i diversi costrutti per 24h, sono state lavate per due volte
con il PBS (soluzione salina tamponata con fosfato) e raccolte mediante l’uso di uno
scraper direttamente nel buffer di lisi RIPA (Tris-HCl 50 mM, pH 8.0, NaCl 150
mM, NP-40 1%, sodio deossicolato 0.5%, SDS 0.1%) addizionato di NaF 20 mM,
Na3VO4 1mM e antiproteasi (una pasticca di CompleteTM, Roche Diagnostic S.p.A.,

21
Applied Science, ogni 10 ml di buffer). I lisati sono stati tenuti in ghiaccio per 30
minuti e poi centrifugati a 16000 g per 20 minuti a 4°C. I supernatanti sono stati
prelevati e la loro concentrazione proteica è stata determinata con il metodo di
dosaggio dell’acido bicinconinico (BCA) (Smith et al., 1985) tramite una opportuna
curva di taratura, ottenuta utilizzando l’albumina di siero bovino (BSA) come
standard.
Preparazione di microdomini di membrana (lipid rafts)
I lipid rafts sono stati isolati secondo la procedura descritta da Parolini et al. (1996).
In breve, 1.5x106 cellule sono state seminate su piastre di 15 cm di diametro e
trasfettate con diversi costrutti per 24h. Poi sono state lavate con PBS e raccolte
mediante l’uso di uno scraper in 750 l di buffer di lisi MBS [2-(N-morpholino)
ethanesulfonic acid 25 mM, pH 6.5, NaCl 150 mM] addizionato di Triton X-100
1%, Na3VO4 1mM e inibitori delle proteasi. Ogni lisato cellulare è stato
omogeneizzato con un pestello di vetro “tight”, unito a una soluzione di saccarosio
all’80% (750 l) e trasferito sul fondo di un tubo da ultracentrifuga. Un gradiente di
saccarosio dal 30% al 5% è stato formato sull’omogenato e il campione è stato
centrifugato a 47000 rpm per circa 18h a 4°C in un rotore SW55Ti (Beckman
Instruments, Palo Alto, CA, USA). Conclusa l’ultracentrifugazione, 12 frazioni da
375 l sono state prelevate a partire dall’alto del gradiente e conservate a 80°C in
attesa di essere analizzate.
Elettroforesi e Western Blot
Un’aliquota (37 l) di ciascuna frazione del gradiente di saccarosio e 30 g di ogni
omogenato cellulare sono stati sottoposti a elettroforesi su gel di poliacrilammide
(7.5%) in condizioni denaturanti (SDS-PAGE) e riducenti (-mercaptoetanolo nel
buffer di preparazione dei campioni). Questo sistema utilizza un tampone Tris base
[tris(idrossimetil)amminometano] 25 mM, glicina 200 mM, SDS 0.1%, pH 7.2. La
corsa elettroforetica è stata effettuata a 180 V per 45 minuti a temperatura ambiente

22
e al suo termine le proteine sono state trasferite su membrana di nitrocellulosa
usando lo strumento Trans-Blot® TurboTM della Bio-Rad (2.5 A per 10 minuti).
La membrana è stata saturata con una soluzione di T-PBS (Na2HPO4 80 mM,
NaH2PO4 25 mM, NaCl 100mM, Tween-20 0.1%) contenente il 5% di latte in
polvere (Bio-Rad), incubata con l’anticorpo primario per 1h a temperatura ambiente,
lavata per 4 volte con T-PBS (1 lavaggio da 15 minuti e 3 da 5 minuti) e infine
incubata con l’anticorpo secondario per 1h a temperatura ambiente. Dopo 4 lavaggi
con T-PBS, la membrana è stata colorata utilizzando un kit per chemiluminescenza
(Super Signal West Pico o Femto, Pierce, Biotechnology Inc., Rockford, USA) e
osservata al Fluorchem (Cell Biosciences, Santa Clara, CA, USA) per la
visualizzazione delle bande immunoreattive.
Gli anticorpi primari e secondari utilizzati e le relative diluizioni sono: anti-V5
monoclonale 1:5000 (Invitrogen), anti--Actina monoclonale 1:5000 (Sigma-
Aldrich), anti-Flotillina1 monoclonale 1:1000 (BD Transduction LaboratoriesTM) e
anti-mouse IgG coniugato alla perossidasi di rafano (HRP) 1:3000 (Sigma-Aldrich).
Microscopia confocale a scansione laser (CLSM)
Le cellule, seminate su vetrini coprioggetto (3x103/vetrino), sono state trasfettate
con i diversi costrutti per 24h, starvate per 16h e stimolate con EGF per 15 minuti.
Poi sono state lavate con PBS e fissate con paraformaldeide (PFA) al 3% per 30
minuti a 4°C. Dopo alcuni lavaggi con il PBS, le cellule sono state permeabilizzate
con Triton X-100 (0.5% in PBS) per 10 minuti a temperatura ambiente.
Al fine di analizzare la localizzazione cellulare delle proteine SHOC2, le cellule sono
state incubate con l’anticorpo primario monoclonale anti-V5 (diluito 1:50 in PBS con
BSA 0.5% e saponina 0.1%) per 1h a temperatura ambiente, lavate due volte con PBS
e incubate con l’anticorpo secondario anti-mouse coniugato al fluoroforo Alexa 594
(Molecular Probes, diluito 1:50 in PBS con BSA 0.5% e saponina 0.1%, colore rosso)
per 1h a temperatura ambiente. Le stesse cellule, lavate con PBS, sono state incubate
con la falloidina coniugata al fluoroforo Alexa 488 (Molecular Probes, diluito 1:100

23
in PBS con BSA 0.5% e saponina 0.1%, colore verde) per 30 minuti a temperatura
ambiente.
I vetrini sono stati montati con il reagente Vectashield antifade contenente il
colorante DAPI per l’identificazione dei nuclei (Vector Laboratories, Burlingame,
CA, USA) e osservati al microscopio confocale a scansione laser Leica TCS SP2
AOBS, usando linee spettrali di eccitazione a 405, 488 e 495 nm, regolate con un
filtro “acousto-optical tunable”. L’acquisizione e l’elaborazione delle immagini sono
state eseguite utilizzando i programmi Leica Confocal Software 2.3 (Leika
Lasertechnik, Heidelberg, Germany) e Adobe Photoshop 7.0 (Adobe Systems
Incorporated, San Jose, CA, USA). I segnali di emissione, provenienti dalle diverse
sonde fluorescenti, sono stati acquisiti in modalità sequenziale.

24
RISULTATI
Espressione delle proteine SHOC2 mutate
L’espressione ectopica di ciascuna forma mutata di SHOC2 è stata verificata tramite
analisi SDS-PAGE e Western Blot (Fig. 9). Tutte le proteine, ad eccezione di quelle
recanti la delezione delle LRR 5-9 e 14-18, sono risultate essere ben espresse.
Figura 9 – Espressione ectopica di SHOC2WT, SHOC2S2G e delle relative forme mutate
(mutanti SHOC2WT, a sinistra e mutanti SHOC2S2G, a destra). In ogni linea dei due gel è stata
caricata una identica quantità di lisato cellulare totale (30 g). Come controllo del contenuto
proteico è stata utilizzata la -Actina. I numeri 45, 50 e 80 kDa si riferiscono ai pesi
molecolare degli standard. La quantità relativa di ciascuna proteina SHOC2 è stata valutata
tramite analisi densitometrica delle bande ed è stata normalizzata come rapporto SHOC2/-
Actina.

25
La traslocazione nel nucleo di SHOC2WT non è mediata dalla NLS
Si è presa in considerazione la possibilità che la predetta NLS (PGTRKKS, residui
79-85) di SHOC2 potesse essere coinvolta nella traslocazione della proteina nel
nucleo. A tal fine, ho creato il mutante SHOC2NLS che possiede una sequenza
consenso modificata per la sostituzione di tre residui aminoacidici (P79A, R82G e
K83G) e ho osservato come questa proteina si distribuiva all’interno della cellula
(Fig. 10). La sua localizzazione cellulare è risultata identica a quella di SHOC2WT.
Infatti, in condizione di deprivazione di siero, SHOC2NLS è distribuita nel citoplasma
e nel nucleo mentre, dopo induzione con EGF, la proteina è più concentrata nel
nucleo. Ciò indica che la NLS non è una reale NLS o che la traslocazione di SHOC2
nel nucleo è mediata da altre specifiche regioni della proteina.
Figura 10 – Rappresentazione schematica della proteina SHOC2NLS e sua localizzazione
cellulare (in condizione di deprivazione di siero, a sinistra e dopo stimolazione con EGF, a
destra). I triangolini neri indicano le posizioni delle sostituzioni aminoacidiche. Scala: 10 m.
La stabilizzazione in membrana di SHOC2S2G non dipende dalla palmitolazione
Un sito putativo di palmitolazione, costituito dalla cisteina in posizione 144, è stato
individuato nella sequenza aminoacidica di SHOC2 tramite analisi in silico. Al fine di
verificare l’esistenza di una correlazione tra stabilizzazione in membrana di
SHOC2S2G e palmitolazione, ho sostituito il residuo di cisteina con uno di glicina e
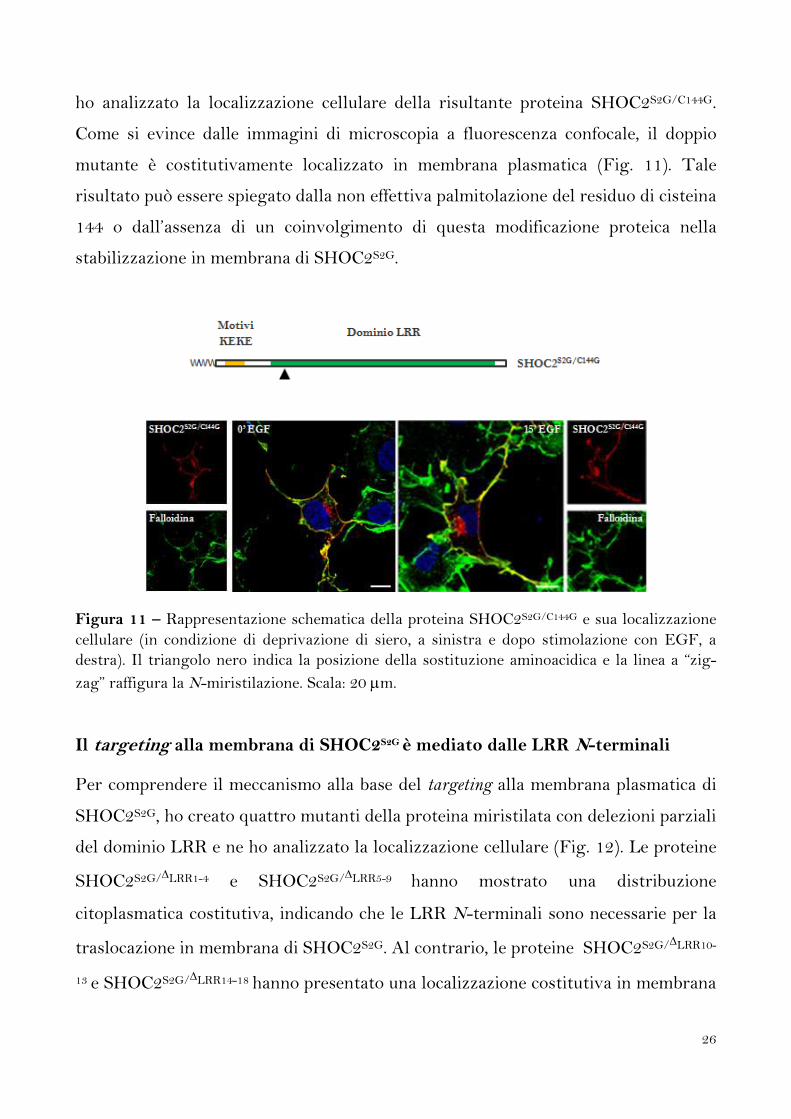
26
ho analizzato la localizzazione cellulare della risultante proteina SHOC2S2G/C144G.
Come si evince dalle immagini di microscopia a fluorescenza confocale, il doppio
mutante è costitutivamente localizzato in membrana plasmatica (Fig. 11). Tale
risultato può essere spiegato dalla non effettiva palmitolazione del residuo di cisteina
144 o dall’assenza di un coinvolgimento di questa modificazione proteica nella
stabilizzazione in membrana di SHOC2S2G.
Figura 11 – Rappresentazione schematica della proteina SHOC2S2G/C144G e sua localizzazione
cellulare (in condizione di deprivazione di siero, a sinistra e dopo stimolazione con EGF, a
destra). Il triangolo nero indica la posizione della sostituzione aminoacidica e la linea a “zig-
zag” raffigura la N-miristilazione. Scala: 20 m.
Il targeting alla membrana di SHOC2S2G è mediato dalle LRR N-terminali
Per comprendere il meccanismo alla base del targeting alla membrana plasmatica di
SHOC2S2G, ho creato quattro mutanti della proteina miristilata con delezioni parziali
del dominio LRR e ne ho analizzato la localizzazione cellulare (Fig. 12). Le proteine
SHOC2S2G/LRR1-4 e SHOC2S2G/LRR5-9 hanno mostrato una distribuzione
citoplasmatica costitutiva, indicando che le LRR N-terminali sono necessarie per la
traslocazione in membrana di SHOC2S2G. Al contrario, le proteine SHOC2S2G/LRR10-
13 e SHOC2S2G/LRR14-18 hanno presentato una localizzazione costitutiva in membrana

27
plasmatica, dimostrando che le LRR C-terminali non sono essenziali per la
traslocazione in membrana di SHOC2S2G.
Figura 12 – Rappresentazione schematica delle proteine SHOC2S2G con delezioni parziali del
dominio LRR e loro localizzazione cellulare (le immagini mostrano la localizzazione delle
proteine mutate in cellule starvate; le stesse localizzazioni si osservano anche in cellule
stimolate con EGF). La linea punteggiata e quella a “zig-zag” raffigurano rispettivamente la
delezione e la N-miristilazione. Scala: 10 m (pannelli in alto) o 20 m (pannelli in basso).

28
La traslocazione nel nucleo di SHOC2WT dipende dalle LRR N-terminali
Anche per SHOC2WT ho costruito i quattro mutanti con le delezioni parziali del
dominio LRR al fine di capire quali regioni della proteina fossero importanti per la
sua traslocazione nel nucleo (Fig. 13). Le proteine SHOC2LRR1-4 e SHOC2LRR5-9
hanno presentato una localizzazione citoplasmatica costitutiva, mentre le proteine
SHOC2LRR10-13 e SHOC2LRR14-18 hanno mostrato una distribuzione intracellulare
identica a quella di SHOC2WT sia in cellule starvate che in cellule starvate e poi
stimolate con EGF. Questi risultati indicano che le LRR localizzate all’estremità N-
terminale del dominio sono essenziali per il trasporto di SHOC2WT dal citoplasma al
nucleo, al contrario quelle localizzate all’estremità C-terminale non sono necessarie.
Il ruolo dei motivi KEKE
Per studiare il ruolo dei motivi KEKE, ho costruito due mutanti con una loro
delezione, uno per SHOC2WT e uno per SHOC2S2G. La proteina SHOC29-54 ha
mostrato una localizzazione nucleare costitutiva (Fig. 14A), dimostrando che la
regione dei KEKE è importante per il trasporto di SHOC2WT dal nucleo al
citoplasma. Invece, la proteina SHOC2S2G/9-54 è risultata essere costitutivamente
distribuita in tutta la cellula (membrana plasmatica, citoplasma e nucleo) (Fig. 14B).
La presenza di questa proteina nel citoplasma indica che è poco stabilizzata in
membrana e che quindi i motivi KEKE contribuiscono all’ancoraggio di SHOC2S2G
alla membrana plasmatica. La proteina osservata nel nucleo, invece, potrebbe essere
una porzione di SHOC2S2G/9-54 non efficientemente miristilata che si comporta come
SHOC29-54 localizzandosi nel compartimento nucleare.

29
Figura 13 – Rappresentazione schematica delle proteine SHOC2WT con delezioni parziali del
dominio LRR e loro localizzazione cellulare (pannelli in alto, le immagini mostrano la
localizzazione delle proteine mutate in cellule starvate; le stesse localizzazioni si osservano
anche in cellule stimolate con EGF; pannelli in basso, localizzazione delle proteine mutate in
cellule starvate, 0’ EGF e in cellule stimolate, 15’ EGF,). La linea punteggiata raffigura la
delezione. Scala: 10 m.

30
Figura 14 – (A) Rappresentazione schematica della proteina SHOC29-54 e sua localizzazione
cellulare (l’immagine mostra la localizzazione della proteina mutata in cellule starvate; la
stessa localizzazione si osserva anche in cellule stimolate con EGF). La linea punteggiata
raffigura la delezione. (B) Rappresentazione schematica della proteina SHOC2S2G/9-54 e sua
localizzazione cellulare (l’immagine mostra la localizzazione della proteina mutata in cellule
starvate; la stessa localizzazione si osserva anche in cellule stimolate con EGF). La linea
punteggiata e quella a “zig-zag” raffigurano rispettivamente la delezione e la N-miristilazione.
Scala: 20m.

31
I motivi KEKE sono necessari per l’associazione di SHOC2S2G ai lipid rafts
L’ultracentrifugazione in gradiente di saccarosio è uno dei metodi che viene
maggiormente utilizzato per lo studio dei lipid rafts, microdomini di membrana
insolubili nei detergenti che flottano nelle frazioni a bassa densità del gradiente.
Dopo averle separate, le frazioni possono essere analizzate tramite Western Blot per
verificare la presenza o meno di una specifica proteina di interesse. Il marcatore
proteico dei lipid rafts è la flotillina.
Esperimenti di questo tipo sono stati condotti per verificare l’esistenza di una
associazione tra SHOC2S2G e i lipid rafts. Come mostrato in Fig. 15, SHOC2S2G è
presente sia nelle frazioni insolubili 5-6 contenenti i lipid rafts che nelle frazioni
solubili 8-12 contenenti le proteine solubilizzate dal detergente e tutte le altre
proteine cellulari. SHOC2WT e il mutante SHOC2S2G/LRR1-4, non localizzati in
membrana, sono presenti solo nelle frazioni solubili del gradiente (10-12) mentre, il
mutante SHOC2S2G/LRR10-13, localizzato in membrana plasmatica come SHOC2S2G, è
presente sia nelle frazioni insolubili (5-6) che in quelle solubili (8-12).
Inaspettatamente, la proteina SHOC2S2G/9-54 priva dei motivi KEKE non si trova
nelle frazioni dei lipid rafts. Ciò può dipendere dalla sua ridotta stabilizzazione in
membrana o da uno specifico ruolo della regione dei KEKE nell’interazione con
proteine localizzate nei lipid rafts. Questa seconda ipotesi è stata confermata
dall’analisi del gradiente di una forma tronca della proteina SHOC2S2G,
SHOC2S2G/171-582 (non caratterizzata in questo studio), che pur essendo poco
stabilizzata in membrana è associata ai rafts.

32
Figura 15 – I lipid rafts, preparati utilizzando cellule COS-1 transientemente trasfettate con i
costrutti mostrati, sono stati separati su un gradiente di densità di saccarosio. Le 12 frazioni
ottenute da ciascun gradiente sono state sottoposte ad analisi immunoblot con gli anticorpi V5
(SHOC2) e Flotillina. Le frazioni 5 e 6 corrispondono alle frazioni insolubili nei detergenti (i
lipid rafts) mentre le frazioni 8-12 corrispondono a quelle solubili.

33
DISCUSSIONE
SHOC2 è una proteina ubiquitaria composta quasi interamente da ripetizioni ricche
in leucina (dominio LRR), con una sequenza N-terminale ricca in lisina (motivi
KEKE) (Fig. 4). Questa proteina agisce da modulatore positivo della via di
trasduzione del segnale RAS-MAPK favorendo l’interazione di RAF1 con RAS
(Rodriguez-Viciana et al., 2006).
La mutazione missenso c.4A>G (p.Ser2Gly) nel gene SHOC2 è responsabile di una
malattia dello sviluppo nota come SN/LAH o sindrome di Mazzanti. Esperimenti in
vitro hanno dimostrato che la sostituzione aminoacidica Ser2Gly promuove la N-
miristilazione della proteina e causa l’aberrante targeting di SHOC2 alla membrana
plasmatica (Fig. 6B) (Cordeddu et al., 2009).
La N-miristilazione non è sufficiente ad ancorare una proteina alla membrana.
Affinché ciò avvenga è necessario che ci sia un secondo segnale costituito da una
sequenza di aminoacidi basici oppure da uno o due siti di palmitolazione (Resh,
1999). La proteina SHOC2 non possiede una sequenza aminoacidica polibasica né
cisteine all’estremità N-terminale che consentano il legame dell’acido palmitico.
Nonostante la Cys144 fosse stata predetta essere un sito di palmitolazione, il suo
coinvolgimento nella stabilizzazione in membrana di SHOC2S2G è stato escluso dal
momento che il doppio mutante SHOC2S2G/C144G ha mostrato una localizzazione
costitutiva in membrana plasmatica (Fig. 11).
Alla luce di questi risultati, si è presa in considerazione la possibilità che la
traslocazione in membrana di SHOC2S2G potesse dipendere da interazioni proteina-
proteina. L’analisi della localizzazione cellulare dei mutanti SHOC2S2G con delezioni
parziali del dominio LRR ha dimostrato che le LRR N-terminali sono coinvolte nel
targeting di SHOC2S2G alla membrana plasmatica (Fig. 12). La delezione dei motivi
KEKE, invece, comporta una minore stabilizzazione in membrana della proteina
SHOC2S2G/9-54 indicando che la regione dei KEKE contribuisce all’ancoraggio in
membrana di SHOC2S2G (Fig. 14B).

34
In condizione di deprivazione di siero, la proteina SHOC2WT mostra una
distribuzione citoplasmatica e nucleare mentre, dopo stimolazione con EGF,
SHOC2WT è localizzata per lo più nel nucleo. Tale osservazione suggerisce un ruolo
inatteso per questa proteina nella trasduzione del segnale (controllo dell’espressione
genica) (Fig. 6A).
L’ipotesi che la traslocazione nel nucleo di SHOC2WT potesse dipendere dalla
predetta NLS è stata esclusa in quanto la proteina mutata SHOC2NLS presenta una
distribuzione identica a quella di SHOC2WT, sia in cellule starvate che in cellule
stimolate con EGF (Fig. 10).
Dall’analisi della localizzazione cellulare dei mutanti SHOC2WT con delezioni
parziali del dominio LRR si è dedotto che le LRR N-terminali sono implicate nella
traslocazione di SHOC2WT nel compartimento nucleare (Fig. 13). Invece, i motivi
KEKE sono risultati necessari per il trasporto di SHOC2WT dal nucleo al citoplasma
(Fig. 14A).
È ormai noto da diversi anni che nella membrana plasmatica esistono microdomini,
definiti lipid rafts, con una particolare composizione lipidica e proteica. Essi
contengono elevate concentrazioni di sfingomielina, colesterolo e gangliosidi. Le
proteine che generalmente sono associate ai rafts hanno lunghi domini
transmembrana, una o due catene aciliche sature oppure un’ancora di
glicosilfosfatidilinositolo (GPI). Si ritiene che i rafts organizzino tali proteine per il
loro trasporto in piccole vescicole o per favorirne l’attivazione in maniera integrata,
come nel caso della trasduzione del segnale. La proteina HRAS, per esempio, utilizza
il ricircolo dei lipid rafts per ottenere l’amplificazione del segnale proliferativo (Roy
et al., 1999).
Esperimenti di ultracentrifugazione in gradiente di saccarosio hanno indicato che la
proteina SHOC2S2G miristilata è associata ai rafts e che tale localizzazione è mediata
dalla regione dei KEKE (Fig. 15). Resta da comprendere quale sia l’effetto di stimoli
extracellulari sull’associazione di SHOC2S2G ai lipid rafts e quale sia il ruolo di questi
microdomini nella modulazione dell’attivazione del pathway RAS–MAPK mediata da
SHOC2S2G.

35
In conclusione, i risultati della presente ricerca dimostrano che:
la localizzazione cellulare della proteina SHOC2 è mediata da interazioni
proteina-proteina;
le ripetizioni ricche in leucina N-terminali sono necessarie per il targeting di
SHOC2S2G alla membrana plasmatica e per la traslocazione nel nucleo di
SHOC2WT;
i motivi KEKE contribuiscono all’ancoraggio in membrana di SHOC2S2G,
mediano l’associazione di SHOC2S2G con i lipid rafts e sono necessari per il
trasporto di SHOC2WT dal nucleo al citoplasma.
Ulteriori studi saranno necessari per identificare gli interattori delle proteine
SHOC2WT e SHOC2S2G (esperimenti di co-immunoprecipitazione, spettrometria di
massa e sistema del doppio ibrido in lievito) e per analizzare come la diversa
localizzazione cellulare dei mutanti per delezione di SHOC2 influisce sulla via di
trasduzione del segnale RAS-MAPK (saggi di luciferasi).

36
BIBLIOGRAFIA
Achiron R, Heggesh J, Grisaru D, Goldman B, Lipitz S, Yagel S, Frydman M. Noonan
syndrome: a cryptic condition in early gestation. Am J Med Genet 92:159-165 (2000)
Allanson JE. Noonan syndrome. Am J Med Genet Part C Semin Med Genet 145C:274-279
(2007)
Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, Ogata T, Takada F, Yano M,
Ando T, Hoshika T, Barnett C, Ohashi H, Kawame H, Hasegawa T, Okutani, T,
Nagashima T, Hasegawa S, Funayama R, Nagashima T, Nakayama K, Inoue S, Watanabe
Y, Ogura T, Matsubara Y. Gain-of-function mutations in RIT1 cause Noonan syndrome,
a RAS/MAPK pathway syndrome. Am J Hum Genet 93:173-180 (2013)
Bader-Meunier B, Tchernia G, Miélot F, Fontaine JL, Thomas C, Lyonnet S, Lavergne JM,
Dommergues JP. Occurrence of myeloproliferative disorder in patients with Noonan
syndrome. J Pediatr 130:885-889 (1997)
Benacerraf BR, Greene MF, Holmes LB. The prenatal sonographic features of Noonan’s
syndrome. J Ultrasound Med 8:59-63 (1989)
Boutin JA. Myristoylation. Cell Signal 9:15-35 (1997)
Burch M, Sharland M, Shinebourne E, Smith G, Patton M, McKenna W. Cardiologic
abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic
assessment of 118 patients. J Am Coll Cardiol 22:1189-1192 (1993)
Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A,
Pizzuti A, Grammatico P, Zampino G, Dallapiccola B, Gelb BD, Tartaglia M. Germline
missense mutations affecting KRAS Isoform B are associated with a severe Noonan
syndrome phenotype. Am J Hum Genet 79:129-135 (2006)
Choong K, Freedman MH, Chitayat D, Kelly EN, Taylor G, Zipursky A. Juvenile
myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol 21:523-527
(1999)
Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F,
Merbitz-Zahradnik T, König R, Kratz CP, Pantaleoni F, Dentici ML, Joshi VA,
Kucherlapati RS, Mazzanti L, Mundlos S, Patton MA, Silengo MC, Rossi C, Zampino G,
Digilio C, Stuppia L, Seemanova E, Pennacchio LA, Gelb BD, Dallapiccola B,

37
Wittinghofer A, Ahmadian MR, Tartaglia M, Zenker M. A restricted spectrum of NRAS
mutations causes Noonan syndrome. Nat Genet 42:27-29 (2010)
Cordeddu V, Di schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, Cecchetti S,
Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Digilio MC,
Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche Kerstin, Ferrero GB, Anichini C,
Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R,
Bzzicalupo P, Gelb BD, Tartaglia M. Mutation of SHOC2 promotes aberrant protein N-
myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet
41:1022-1026 (2009)
Duncan WJ, Fowler RS, Farkas LG, Ross RB, Wright AW, Bloom KR, Huot DJ, Sondheimer
HM, Rowe RD. A comprehensive scoring system for evaluating Noonan syndrome. Am J
Med Genet 10:37-50 (1981)
Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-
myristoylation. J Biol Chem 276:39501-39504 (2001)
Fukuda M, Horibe K, Miyajima Y, Matsumoto K, Nagashima M. Spontaneus remission of
juvenile chronic myelomonocytic leukemia in an infant with Noonan syndrome. J Pediatr
Hematol Oncol 19:177-179 (1997)
Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergsträsser E, Emanuel PD, Hasle H,
Kardos G, Klein C, Kojima S, Stary J, Trebo M, Zecca M, Gelb BD, Tartaglia M, Loh ML.
The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan
syndrome/myeloproliferative disease. Blood 106:2183-2185 (2005)
Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F, Oudesluys-Murphy AM, Di
Rocco C, Kloska SP, Tartaglia M, Zenker M. Craniosynostosis in patients with Noonan
syndrome caused by germline KRAS mutations. Am J Med Genet A 149A:1036-1040
(2009)
Lee J, Lee HJ, Shin MK, Ryu WS. Versatile PCR-mediated insertion or deletion mutagenesis.
Biotechniques 36:398-400 (2004)
Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in
children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of
atrioventricular canal. J Pediatr 135:703-706 (1999)

38
Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F, Caputo V, Silvano M,
Buscherini F, Ferrara G, Digilio MC, Cavaliere ML, van Hagen JM, Zampino G, van der
Burgt I, Ferrero GB, Mazzanti L, Screpanti I, Yntema HG, Nillesen WM, Savarirayan R,
Zenker M, Dallapiccola B, Gelb BD, Tartaglia M. Heterozygous germline mutations in
the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum
Genet 87:250-257 (2010)
Massarano AA, Wood A, Tait RC, Stevens R, Super M. Noonan syndrome: Coagulation and
clinical aspects. Acta Paediatr 85:1181-1185 (1996)
Mazzanti L, Cacciari E, Cicognani A, Bergamaschi R, Scarano E, Forabosco A. Noonan-like
syndrome with loose anagen hair: a new syndrome? Am J Med Genet A 118A:279-286
(2003)
Mendez HM, Opitz JM. Noonan syndrome: A review. Am J Med Genet 21:493-506 (1985)
Michaud NR, Fabian JR, Mathes KD, Morrison DK. 14-3-3 is not essential for RAF1 function:
identification of Raf-1 proteins that are biologically activated in a 14-3-3- and Ras-
indipendent manner. Mol Cell Biol 15:3390-3397 (1995)
Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, Aoki Y, Matsubara Y, Arveiler
B, Lacombe D, Pasmant E, Parfait B, Baumann C, Héron D, Sigaudy S, Toutain A, Rio M,
Goldenberg A, Leheup B, Verloes A, Cavé H. Cardio-facio-cutaneous and Noonan
syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype
relationships and overlap with Costello syndrome. J Med Genet 44:763-771 (2007)
Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ,
Sakamoto KM, Gorr TA, Mehta P, Schmid I, Kropshofer G, Corbacioglu S, Lang PJ,
Klein C, Schlegel PG, Heinzmann A, Schneider M, Starý J, van den Heuvel-Eibrink MM,
Hasle H, Locatelli F, Sakai D, Archambeault S, Chen L, Russell RC, Sybingco SS, Ohh M,
Braun BS, Flotho C, Loh ML. Germline CBL mutations cause developmental
abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42:794-800
(2010)
Noonan JA, Ehmke DA. Associated noncardiac malformations in children with congenital
heart disease. J Pediatr 63:468-470 (1963)
Noonan JA. Noonan syndrome. An update and review for the primary pediatrician. Clin
Pediatr (Phila) 33:548-555 (1994)

39
Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz
W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C,
Mundel P, López Siquero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I,
Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD.
Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with
hypertrophic cardiomyopathy. Nat Genet 39:1007-1012 (2007)
Parolini I, Sargiacomo M, Lisanti MP, Peschle C. Signal transduction and
glycophosphatidylinositol-linked proteins (lyn, lck, CD4, CD45, G proteins, and CD55)
selectively localize in Triton-insoluble plasma membrane domains of human leukemic cell
lines and normal granulocytes. Blood 87:3783-3794 (1996)
Peitzsch RM, McLaughlin S. Binding of acylated peptides and fatty acids to phospholipid
vesicles: pertinence to myristoylated proteins. Biochemistry 32:10436-10443 (1993)
Pérez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, Philip N, Derain-Court J,
Cassinat B, Lachenaud J, Kaltenbach S, Salmon A, Désirée C, Pereira S, Menot ML, Royer
N, Fenneteau O, Baruchel A, Chomienne C, Verloes A, Cavé H. Germline mutations of the
CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic
leukaemia. J Med Genet 47:686-691 (2010)
Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma
K, Katayama H, Nakagawa M, Fujiwara Y, Matsushima M, Mizuno K, Tokuyama M,
Hirota H, Muneuchi J, Higashinakagawa T, Matsuoka R. Germline gain-of-function
mutations in RAF1 cause Noonan syndrome. Nat Genet 39:1013-1017 (2007)
Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated
and palmitoylated proteins. Biochim Biophys Acta 1451:1-16 (1999)
Resh MD. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat Chem Biol
2:584-590 (2006)
Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y,
Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations
in SOS1 cause Noonan syndrome. Nat Genet 39:70-74 (2007)
Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. A phosphatase
holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-
Ras effector to modulate Raf activity. Mol Cell 22:217-230 (2006)

40
Roti G, La Starza R, Ballanti S, Crescenzi B, Romoli S, Foà R, Tartaglia M, Aversa F, Fabrizio
Martelli M, Mecucci C. Acute lymphoblastic leukaemia in Noonan syndrome. Br J
Haematol 133:448-450 (2006)
Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge M, Stang E, Rolls B, Hancock JF,
Parton RG. Dominant-negative caveolin inhibits H-Ras function by disrupting
cholesterol-rich plasma membrane domains. Nat Cell Biol 1:98-105 (1999)
Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, Scioletti AP, Esposito
G, Cordeddu V, Lepri F, Petrangeli V, Dentici ML, Mancini GM, Selicorni A, Rossi C,
Mazzanti L, Marino B, Ferrero GB, Silengo MC, Memo L, Stanzial F, Faravelli F, Stuppia
L, Puxeddu E, Gelb BD, Dallapiccola B, Tartaglia M. Germline BRAF mutations in
Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and
associated phenotypic spectrum. Hum Mutat 30:695-702 (2009)
Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, van der Burgt I, Musante L,
Kalscheuer V, Wehner LE, Nguyenn H, West B, Zhang KY, Sistermans E, Rauch A,
Niemeyer CM, Shannon K, Kratz CP. Germline KRAS mutations cause Noonan
syndrome. Nat Genet 38:331-336 (2006)
Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer.
Nat Rev Cancer 7:295-308 (2007)
Sharland M, Patton MA, Talbot S, Chitolie A, Bevan DH. Coagulation-factor deficiencies and
abnormal bleeding in Noonan’s syndrome. Lancet 339:19-21 (1992)
Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine3 of Src family
protein tyrosine kinase determines palmitoylation and localization in caveolae. J Cell Biol
126:353-363 (1994)
Shi GX, Cai W, Andres DA. Rit subfamily small GTPase: regulators in neuronal
differentiation and survival. Cell Signal 25:2060-2068 (2013)
Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31-39 (2000)
Singer ST, Hurst D, Addiego JE Jr. Bleeding disorders in Noonan syndrome: three case
reports and review of the literature. J Pediatr Hematol Oncol 19:130-134 (1997)
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto
EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid.
Anal Biochem 150:76-85 (1985)

41
Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I,
Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Mutations
in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome.
Nat Genet 29:465-468 (2001)
Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, Brunner HG, Bertola
DR, Crosby A, Ion A, Kucherlapati RS, Jeffery S, Patton MA, Gelb BD. PTPN11
mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation,
and phenotypic heterogeneity. Am J Hum Genet 70:1555–1563 (2002)
Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hählen K, Hasle H, Licht
JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia,
myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148-150 (2003)
Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K,
Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C,
Vasta I, Gibson K, Curry CJ, Siquero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-
Sagi D, Gelb BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan
syndrome. Nat Genet 39:75-79 (2007)
van der Burgt I, Berends E, Lommen E, van Beersum S, Hamel B, Mariman E. Clinical and
molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet
53:187-191 (1994)
van der Burgt I. Noonan syndrome. Orphanet J Rare Dis 14:2-4 (2007)
Zarabi M, Mieckowski GC, Mazer J. Cystic hygroma associated with Noonan’s syndrome. J
Clin Ultrasound 11:398-400 (1983)
Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I, Doerr HG, Gaspar H,
Hofbeck M, Gillessen-Kaesbach G, Koch A, Meinecke P, Munddlos S, Nowka A, Rauch A,
Reif S, von Schnakenburg C, Seidel H, Wehner LE, Zweier C, Bauhuber S, Matejas V,
Kratz CP, Thomas C, Kutsche K. SOS1 is the second most common Noonan gene but
plays no major role in cardio-facio-cutaneous syndrome. J Med Genet 44:651-656 (2007)


















