
ATTIVITÀ DELL ISTITUTO SUPERIORE DI SANITÀ E LAVORI … · (Domanda di autorizzazione alla...
45
Annarita Meneguz Segretario Commissione per l’Ammissibilità alla Sperimentazione Clinica di Fase I ATTIVITÀ DELL’ISTITUTO SUPERIORE DI SANITÀ E LAVORI DELLA COMMISSIONE PER L’AMMISSIBILITÀ ALLA SPERIMENTAZIONE CLINICA DI FASE I
Transcript of ATTIVITÀ DELL ISTITUTO SUPERIORE DI SANITÀ E LAVORI … · (Domanda di autorizzazione alla...

Annarita Meneguz
Segretario Commissione per l’Ammissibilità alla Sperimentazione Clinica di Fase I
ATTIVITÀ DELL’ISTITUTOSUPERIORE DI SANITÀ E LAVORI
DELLA COMMISSIONEPER L’AMMISSIBILITÀ ALLA
SPERIMENTAZIONE CLINICA DIFASE I


DichiarazioneLe opinioni espresse in questa presentazione sono
attribuibili esclusivamente all’autore

Sperimentazioni di fase 1Sono i primi studi su unnuovo principio attivocondotti sull'uomo, su unpiccolo numero di volontarisani o malati (in generepoche decine).Lo scopo è quello di fornireuna valutazione preliminaresulla farmacocinetica esicurezza della sostanza inesame, e se si confermano leinformazioni ottenute nellafase di ricerca pre clinica.
FIM (First in man) o FTIH (First Time in Human) o FIH (First In Human). Sono i primi studi clinici effettuati come parte di una fase I, in cui il farmaco viene somministrato per la prima volta all'uomo. Gli studi FIM sono tradizionalmente «piccoli» studi di dose-escalation-time in soggetti volontari, e l'obiettivo primario è l'identificazione di un intervallo di dose o la dose adatta per ulteriori studi, basato sulla sicurezza e tollerabilità della sostanza.
4

Massima dose sicura da somministrare
L’obiettivo principale della valutazione dei dati preclinici è
quindi quello individuare la dose per la prima somministrazione clinica
GMP
GLP
GCP

Processo/paradigma dello sviluppo clinico di un farmaco
6
Dagli anni settanta la normativa italiana ha attribuito all’ISS il ruolo di valutare l’ammissibilità dei nuovi farmaci alla
sperimentazione clinica di fase I

Normativa Nazionale
LA NORMATIVA DI RIFERIMENTO ITALIANA
04/04/2016
DPR 439, 21 settembre 2001 Procedure accertamento della composizione e dell’innocuità dei prodotti farmaceutici di nuova istituzione
D.L.vo 211, 24 giugno 2003 Applicazione della buona pratica clinica nella sperimentazione di medicinali per uso clinico. Attuazione Direttiva 2001/20.
D.L.vo 200, 6 nov 2007 Attuazione direttiva 2005/28/CE
D.M. 21 dicembre 2007 Modalità di inoltro della richiesta di autorizzazione all’AC
Legge 8 novembre 2012 n° 189Art.12, comma 9
Centralizzazioni presso l’AIFA delle competenze per tutte le Fasi di sperimentazioni cliniche(in linea con il Regolamento Europeo)
AC

LEGGE 8 NOV. 2012 N.189Le competenze in materia di sperimentazione clinica dei medicinali attribuite daldecreto legislativo 24 giugno 2003, n. 211, all'Istituto superiore di sanità sonotrasferite all'AIFA, la quale si avvale del predetto Istituto, senza nuovi o maggiorioneri a carico della finanza pubblica, ai fini dell'esercizio delle funzioni trasferite,secondo modalità stabilite con decreto del Ministro della salute, sentiti i due entiinteressati. Fino all'adozione del decreto del Ministro della salute, l'Istitutosuperiore di sanità, raccordandosi con l'AIFA, svolge le competenze ad esso giàattribuite, secondo le modalità previste dalle disposizioni previgenti
04/04/2016
Decreto Ministero della Salute 27 aprile 2015

DECRETO MINISTERIALE 27 APRILE 2015
Decreto del Ministro della salute recante le modalità di esercizio delle funzioni in materia di sperimentazioni cliniche di medicinali trasferite dall’Istituto superiore di sanità all’Agenzia Italiana del Farmaco
Decreta:Articolo 1
(Ambito di applicazione)Il presente decreto disciplina le modalità di esercizio delle funzioni in materia di sperimentazione clinica di fase I che, in base all’art. 12, comma 9, del decreto-legge 13 settembre 2012, n. 158, convertito, con modificazioni, dalla legge 8 novembre 2012, n. 189, sono trasferite dall’Istituto superiore di sanità, di seguito denominato (ISS) all’Agenzia Italiana del Farmaco, di seguito denominata (AIFA), in qualità di autorità competente.
Per lo svolgimento delle funzioni relative alle sperimentazioni di fase I l’AIFA si avvale dell’ISS, secondo le modalità disciplinate dal presente decreto.Ai fini del presente decreto, le sperimentazioni cliniche fase I/II e di fase I/III sono equiparate alle sperimentazioni di fase I.
Articolo 2(Domanda di autorizzazione alla sperimentazione clinica di fase I)
Il promotore della sperimentazione clinica di fase I è tenuto ad acquisire la preventiva autorizzazione dell’AIFA, che si avvale dell’ISS per la valutazione tecnico-scientifica della documentazione presentata a supporto della domanda di autorizzazione. L’ISS svolge tale compito in piena autonomia scientifica ed organizzativa.

DECRETO MINISTERIALE 27 APRILE 2015
Articolo 3(Valutazione della documentazione)
1. L’ISS, esaminata la documentazione e richiedendo, ove necessario,supplementi di documentazione, formula un parere sulla ammissibilità delladomanda di sperimentazione di fase I, avvalendosi della Commissione di cuiall’articolo 7 del decreto del Presidente della Repubblica 21 settembre 2001, n.439, sulla base dei seguenti criteri:valutazione della qualità del prodotto e conformità ai requisiti in materia di fabbricazione e importazione deimedicinali sperimentali e dei medicinali ausiliari;caratteristiche e conoscenze in merito ai medicinali sperimentali e ai medicinali ausiliari;misure di sicurezza previste per ridurre al minimo i rischi;rischio per la salute del soggetto legato alla condizione clinica per la quale il medicinale sperimentale è oggettodi sperimentazione;completezza e adeguatezza della documentazione tecnica a supporto della domanda;valutazione del rapporto fra rischi prevedibili e benefici ipotizzabili in relazione ai risultati dellasperimentazione pre-clinica e sulla base del protocollo clinico proposto.
2. Il parere sull'ammissibilità di cui al comma 1, indica, se del caso, i limiti e le condizioni cui è subordinata la sperimentazione di fase I.
3. Il parere dell’ISS è trasmesso all’AIFA nei termini previsti dal D.P.R. n. 439 del 2001 ai fini dell’adozione del provvedimento da parte del Direttore Generale dell’AIFA.

Osservatorio Nazionale sulla Sperimentazione Clinica dei Medicinali
(OsSC)Operativo da Luglio 2014
Obbligo di inserimento delle richieste diammissibilità alle Sperimentazioni cliniche.
Per poter inserire la richiesta di ammissibilità inOsservatorio è necessario che tutti i Comitati Eticicoinvolti nella sperimentazione siano statiaccreditati dalla Regione

MODULISTICAD.M. 21.12.2007
revisionato con delibera AIFA 28.1.2015
Lettera di accompagnamento
CTA formmodulo di domanda
(App. 5 Appendice_05.doce App. 9 del DM)
Documentazione
AUTORITA’ COMPETENTE-(AIFA)
Istituto Superiore di Sanità (solo fase e I/II e I/IIII)
Comitato Etico
Dossier del prodotto medicinale sperimentale
(Investigational Medicinal Product Dossier-IMPD)
Dossier del prodotto medicinale Non sperimentale (NIMP)
Dossier per lo sperimentatore (Investigator’s Brochure- IB)
Protocollo clinico proposto
In accordo alla CT-1 sez:2.3
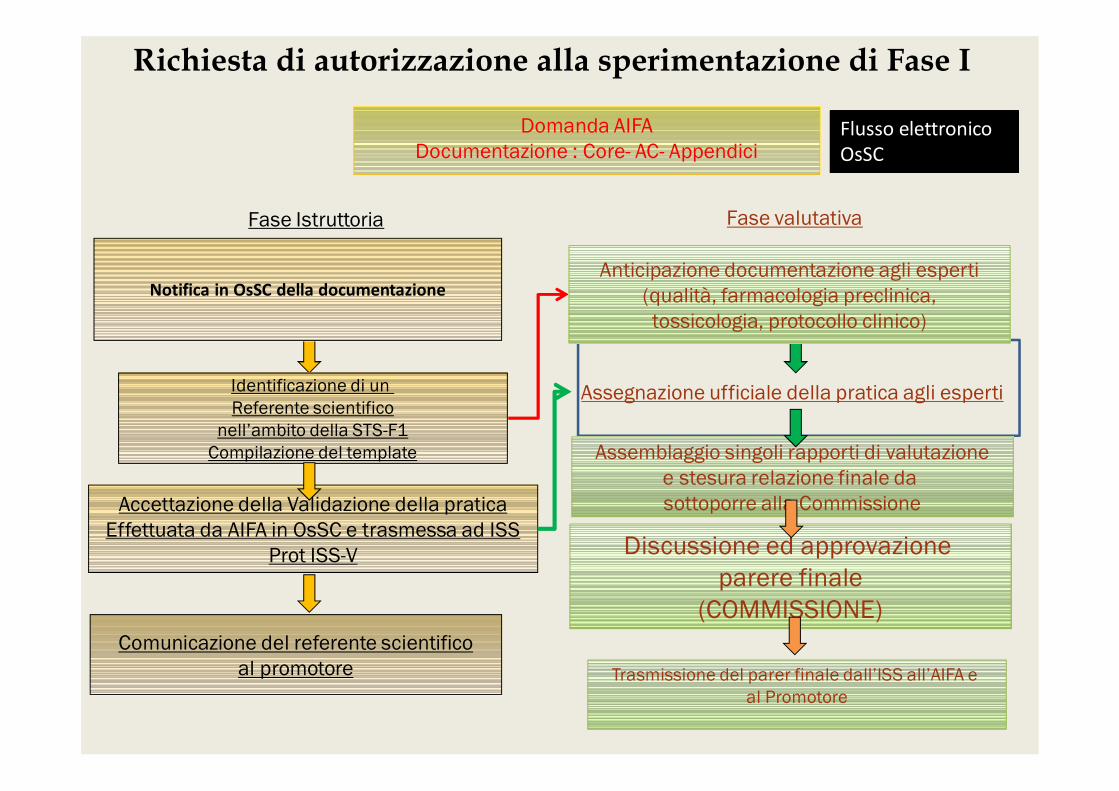
Notifica in OsSC della documentazione
Comunicazione del referente scientificoal promotore
Anticipazione documentazione agli esperti(qualità, farmacologia preclinica,tossicologia, protocollo clinico)
Assemblaggio singoli rapporti di valutazionee stesura relazione finale da sottoporre alla Commissione
Discussione ed approvazione parere finale
(COMMISSIONE)
Trasmissione del parer finale dall’ISS all’AIFA e al Promotore
Identificazione di un Referente scientifico
nell’ambito della STS-F1Compilazione del template
Accettazione della Validazione della praticaEffettuata da AIFA in OsSC e trasmessa ad ISS
Prot ISS-V
Richiesta di autorizzazione alla sperimentazione di Fase I
Domanda AIFADocumentazione : Core- AC- Appendici
Fase Istruttoria Fase valutativa
Flusso elettronicoOsSC
Assegnazione ufficiale della pratica agli esperti

2003-2009: iniziative a sostegno della sperimentazione di fase I
La possibilità di sperimentare i nuovi farmaci nelle fasi precoci disviluppo rappresenta un’ occasione di crescita unica diinnovazione per il Paese.Incontri con responsabili AIFA, rappresentanti degli IRCCS,dell’Accademia e dell’IndustriaAttivazione di progetti formativi (ad es. Master, corsi dispecializzazione) insieme ad AIFA, Università, Industria, IRCCSStesura di linee-guidaPromozione del dibattito culturale
Convegno «Aspetti Scientifici, Etici E Regolatori Delle SperimentazioniCliniche Early Phase” luglio 2010 Organizzato da ISS ed AIFAPubblicazioni

Valutazione preclinica dei vaccini. Biotecnologie e qualità della vita. A. Meneguz A curadi G. Cantelli forti e P. Hrelia . Patron editore 2005Autorità regolatorie e processo registrativo di nuovi farmaci antineoplastici (StefanoVella, Annarita Meneguz). In Sviluppo dei farmaci oncologici con bersaglio molecolare:dalla tradizione all’innovazione (a cura di Dino Amadori). Paletto editore. 2007First-In-Man (FIM) Regulatory Manual (FINAL VERSION). Giovanni Migliaccio, AnnaritaMeneguz, M.G. Galli; F. Belardelli, 2009 ( EATRIS-WP8)Advanced Therapy: rules and experiences of the Phase I Italian Competent Authority.Annarita Meneguz, Dipartimento del Farmaco, Istituto Superiore di Sanità ISS,Pharmaceuticals Policy and Law,12 : 29-33, 2010Non clinical: need of studies on juvenile animals in order to evaluate the toxic potentialbefore any administration to pediatric population, A. Meneguz in Changes in Researchand Development of Medicinal Products since the Paediatric Regulationhttp://cdn.intechopen.com/pdfs/24649/InTech-Changes_in_research_and_development_of_medicinal_products_since_the_paediatric_regulation.pdfVerso una revisione della Direttiva 2001/20 e una maggiore armonizzazioneeuropea dei trial clinici precoci. In .Rapporti ISTISAN: 12/37 A. Meneguz, MF Cometa
Ø http://www.crob.it/crob/files/docs/10/56/73/DOCUMENT_FILE_105673.pdfPopoli P, Cometa MF, Fabi F, Meneguz A. The role of the Istituto Superiore di Sanità as
the competent authority for Phase I trials in the translation of advanced therapies. Annalidell'Istituto Superiore di Sanità 2011;47(01):79-82.
Ø http://www.scielosp.org/pdf/aiss/v47n1/v47n1a16.pdf
Pubblicazioni

2003-2009: iniziative a sostegno della sperimentazione di fase I
Ottimizzazione delle procedure ai fini di fornireil parere qualificato in tempi certi
Potenziamento della segreteria tecnico-scientifica, e amministrativa ed ilcoinvolgimento di un numero crescente diesperti/assessor.

ORGANIZZAZIONE DELL’ATTIVITA’
•COMMISSIONE (DPR 439/01; D.Lvo211/2003 e DM 27 aprile 2015)•Segreteria Tecnico Scientifica (STS-F1)•Segreteria Amministrativa (SA-F1)•ESPERTI Assessor

CommissioneLa Commissione è nominata con Decreto Ministeriale e rimanein carica per 3 anni. Si avvale del supporto della SegreteriaTecnico Scientifica, del contributo degli esperti nominati dalPresidente dell’Istituto Superiore di Sanità e di una Segreteriaamministrativa.
Sedute Ordinarie: la Commissione si riunisce con cadenza mensile(eccetto nel mese di agosto) secondo un calendario stabilito all’iniziodell’anno e pubblicato sul sito della Fase I dell’ISS.Sedute Telematiche: se necessarie al rispetto dei termini per pratiche percui sono pronti tutte le valutazioniSedute Straordinarie: istituite in caso di richieste di pareri allaCommissione che rivestono particolare urgenza in merito a specificheproblematicheAudizioni: ai sensi dell'articolo 4, comma 1, del DPR n.439 del 21settembre 2001, la Commissione può convocare il proponente e/o gliesperti da esso indicati per ottenere chiarimenti sugli aspetti tecnici delladocumentazione.
18

COMPOSIZIONE DELLA COMMISSIONE(2014-2017)
MEMBRIProf. Gualtiero Walter RICCIARDI (Presidente della Commissione)Prof. Luca PANI (Direttore generale AIFA)Entrambi possono nominare un delegato
ESPERTI ESTERNIProf. Renato Bernardini (Università di Catania)Prof. Paolo Di Bartolomeo (Ospedale Santo Spirito, Pescara)Prof. Armando Santoro (Humanitas Cancer Center, Milano)
ESPERTI INTERNI (AIFA O ISS)Dr. Guido Pantè (AIFA) Dr.ssa Patrizia Popoli (Dip. Farmaco, ISS)Dr. Ugo Testa (Dip. Ematologia, oncologia e medicina molecolare, ISS)
Segretario Dr.ssa Annarita Meneguz (Dipartimento del Farmaco, ISS)
19

Interfaccia regolatoria Esperti-ISS/Commissione/AIFA)
Referente scientifico per singola praticacontact point per l’AC, ed il proponente, segue l’iter di validazione della CTA da parte dei AIFA, istruttoria, individua e contatta gli assessor, elaborazione e discussione della relazione finale per la Commissione
Audizioni “pre-submission”Attività di consulenza e supporto alla preparazione del dossier di fase I e discussione tecnico-scientifica con esperti ISS - Consulenza per i Promotori informale e gratuita
Attività specifiche• Dr.ssa M. Francesca Cometa (Sito web: http://www.iss.it/scf1/, OsSC)• Dr.ssa Fiorella Malchiodi Albedi (Procedure VHP)• Dr.ssa Maria Teresa Tebano (Coordinamento audizioni pre- submission ) • Dr.ssa Annarita Meneguz (Coordinamento formazione esperti , riunioni
Commissione)• Dr.ssa Lucia Palmisano ( interazione con infrastrutture di ricerca ad es ECRIN)• Fulvia Fabi (Banca dati terapia genica e cellulare somatica, D.M. 2.03.2004) TP
STS-F1: Segreteria tecnico-scientifica

ESPERTIEsperti di Qualità/Non Clinica /Clinica (IMPD)
Dipendenti ISS, nominati, sulla base dei loro CV edesperienze specifiche, dal Presidente dell’ISS all’interno diuna lista rinnovata allo scadere del mandato triennale dellaCommissione.Svolgono quotidianamente attività di ricerca scientifica econtrollo nei settori della qualità e della sicurezza efarmacologia pre-clinica e clinicaSono chiamati all’elaborazione di pareri tecnico scientificiFrequentano sedute di aggiornamento interno sulle lineeguida ICH/CHMP e procedure regolatorie, organizzateperiodicamente dalla STS-F1 (Prossima Seduta di formazioneprevista : 15 aprile 2016).
21

Esperti Assessor ISSIMPD qualità (Chimici/Biologici/TA)
B. Barletta, M. Bartolomei, A. Borioni, E.Bossù, B.Brunetto, R. Botta, E. Coccia, K. Cristiano, R. Ferretti, M.C. Galli, G. Gostoli, P. Iacovacci, F. Luciani, F. Micheletti, F.
Nappi, C. Pini (Responsabile Centro per la Ricerca e la Valutazione dei prodotti Immunobiologici), E. Puggioni, R. Tinghino, I. Sestili, L. Valvo, M. Wirz.
IMPD Non clinico C. Ambrosio, M.F. Cometa, G. Diana, G. Maria Rosaria Dominici, Di Felice, F. Fabi, F.
Lozupone, F. Malchiodi Albedi, G. Marano, A. Meneguz, P. Picchieri, P.Popoli, S. Pieretti, R. Potenza, M.T. Tebano
IMPD Clinico R. Arcieri, M. Biffoni, M Boirivant, A. Carè, G. Diana, F. Facchiano, C. Frank, M.
Gabbianelli, M.C. Gagliardi, J. Hassan, G.Marano, L. Palmisano, P. Popoli, F. Pricci, U. Testa 22

23
Valutazione scientificaL’elaborazione del parere sulla documentazione sottoposta per l’ammissibilità della sperimentazione si basa sull’esperienza scientifica disponibile e richiede l’applicazione di una metodologia internazionalmente codificata, aderente a norme e linee guida internazionali.Per l’elaborazione dei pareri, gli esperti possono avvalersi, per specificiaspetti regolatori, del supporto dalla STS-F1.
GMPGLP GCP

ØGestione delle pratiche relative alle richieste di ammissibilità di nuove sperimentazioni Cliniche di fase I, trasmissione relazioni della Commissione all’AC e interfaccia amministrativa con il Promotore)
ØGestione delle pratiche relative alle richieste di emendamenti sostanziali, trasmissione relazioni della Commissione all’AC e interfaccia amministrativa con il Promotore)
ØOrganizzazione delle audizioni pre-submission, o ogni altra audizione richiesta dal promotore, o dalla Commissione.
Ø [P. Campagna, A, Tobelli, M.T. Volpe]
Segreteria Amministrativa

0
10
20
30
40
50
60
70
80
90
100
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
Richieste di ammissibilità alla Fase I*
* Richieste in entrata in ISS** Interruzione lavori della Commissione per rinnovo Decreto di Nomina (19 luglio-14 novembre 2011)
1823
44
53
64
51** 51
7568
86
Totali:533 ( 20 su volontario
sano)

Distribuzione Regionale (Sperimentatore responsabile del
coordinamento, G.1 CTA)

04/04/2016
0
10
20
30
40
50
60
70
80
90
100
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
Area Terapeutica oncologica
1823
44
53
64
51 51
7568
13
32 30
4753
44
27
11
34
86
60
351 (65%)

Alcuni degli IMPs oncologici • Inibitori delle chinasi coinvolte nella segnalazione cellulare
(piuttosto aspecifici: sunitinib, sorafenib e il più recenteregorafenib, ma anche quelli più selettivi, come vemurafenib,dabrafenib e crizotinib)
• Combinazioni di inibitori B-RAF (dabrafenib) e inibitori MEK(trametinib).
• Brentuximab vedotin e trastuzumab emtansine (quando, medianteconiugati, li anticorpi monoclonali fanno «rivivere» citotossici maiarrivati in pratica clinica)
• Obinutuzumab anti CD20 MAB• Ibrutinib• Biosimilare rituximab• Ofatumumab• Bevacizumab• Combinazione Obinutuzumab e Polatuzumab Vedotin E
Venetoclax 28

Survey Europea: capire le ragioni per il calo delle SC di Fase II e III e identificare i fattori che determinano la
scelta di un particolare Paese
29

0
10
20
30
40
50
60
70
80
90
100
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
Richieste di ammissibilità alla Fase IProponente non commerciale
1823
44
53
64
51* 51
75
68
412 16
1014
81110 12
86
9
106 (20%)

0
10
20
30
40
50
60
70
80
90
100
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
Richieste di ammissibilità alla Fase ITERAPIA AVANZATA
1823
44
53
64
51 51
75
68
83 6 4 2 4 5 7 7
86
6
52 (10%)

0
10
20
30
40
50
60
70
80
90
100
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015
Richieste di ammissibilità alla Fase IPOPOLAZIONE PEDIATRICA
1823
44
53
64
51 51
7568
2 1 6 7 9 6 8 15 12
86
15
81 (16%)
Regolamento 1901/2006

33
http://cddf.org/
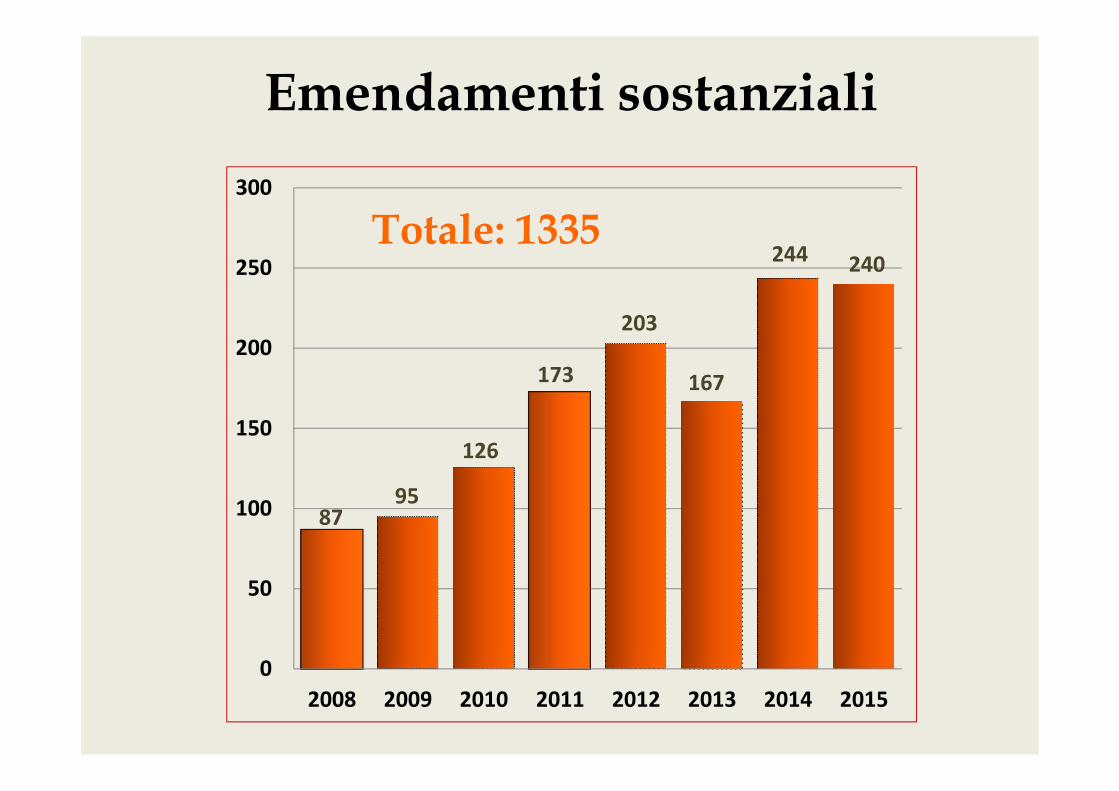
Emendamenti sostanziali
0
50
100
150
200
250
300
2008 2009 2010 2011 2012 2013 2014 2015
8795
126
173
203
167
244 240Totale: 1335

Tempistica Per la sperimentazione di fase I realizzatadirettamente su pazienti oncologici, portatori diAIDS o di altre malattie per le quali i farmacinon possono essere utilizzati su volontari sani,[...] il termine procedurale è ridotto a 30 giorniper la comunicazione dell’esito dell’attivitàistruttoria.
A questa regola fanno eccezione le richieste noncomplete o che, a giudizio degli esperti,necessitino di ulteriore documentazione e per lequali si applica il CLOCK STOP (Pareresospensivo)
DPR 439/2001- D.Lvo 211/2003

Tempistica pubblicata sul sito Anno 2016 - I semestre
18 gennaio19 febbraio18 marzo ( 21 marzo)14 aprile13 maggio21 giugno20 luglio
In accordo al DPR 439/2001 art. 9 (medicinali di particolareimportanza e riduzione dei termini procedimentali), laCommissione valuta le richieste validate da AIFA almeno 30giorni prima della seduta. Tale termine si estende a 60 giorniper la seduta di settembre e 45 giorni per quella di gennaio.
Le date proposte possono subire leggere variazioni.
http://www.iss.it/binary/scf1/cont/Calendario_I_semestre_2016.pdf

Emissione del parere della Commissione entro i tempi previsti dalla norma e in linea con la media
di altre AC europee
2008 2009 2010 2011 2012 2013 2014
Valutazioni di sperimentazioni cliniche di Fase I su pazienti*Dati agenzie europee
*UK: 25-28 giorni*GE: 30 giorni*FR: 30 giorni

Richieste di ammissibilità alla Fase I(FIM)
ANNO TOTALE FIM 2013 75 15 (20%) 12 Protocolli Fase I
(80%)
3 Protocolli Fase I/II
(20%)2014 68 14 (21%) 7 Protocolli Fase I
(50%) 7 Protocolli Fase
I/II(50%)
2015 84 18 (21%) 9 Protocolli Fase I (50%)
9 Protocolli Fase I/II
(50%)38

Tema caldo
Riguarda gli studi l’adattabilità applicata agli studi didose ranging di Fase II e perfino gli studi di Fase I per lasicurezza.Il dibattito sull’etica, i vantaggi, le opportunità e le sfidepresentate da questi disegni è in corso.
http://www.medelis.com/clinical-cancer-research-abstracts/adaptive-trial-design/
39

EU e USA RP e linea guida
40
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM201790.pdf
http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003616.pdf
2010
• La FDA ed EMEA sembrano essere «più ricettive» ai trial adattativi di Fase III o di Fase II/III di quanto non fossero a pochi anni fa.

Iniziative future
Stimolare il dibattito per un confronto fra tutti gli addettiai lavori sull’incrementata complessità dei disegni deglistudi oncologici anche di fase precoce (adaptive trials),anche tramite un secondo Convegno congiuntoISS/AIFA/ Commissione.
Rapporto ISTISAN: Obiettivi e interpretazione dei datidi qualità e degli studi pre-clinici e dei protocolli asupporto delle sperimentazioni clinica di fase 1, inclusigli studi esploratoriReclutamento interno di ulteriori AssessorFormazione e tutoraggio dei nuovi Assessor
41

REGOLAMENTO DEL PARLAMENTO EUROPEO E DEL CONSIGLIOsulla sperimentazione clinica di
medicinali per uso umano, e che abroga la direttiva 2001/20/CE

L’ISS può essere una risorsa • Gli SM dovranno stabilire l'organismo o gli organismi
appropriati ai fini della valutazione• Gli SM garantiscono che le persone incaricate di convalidare e
valutare la domanda non abbiano conflitti di interesse, sianoindipendenti dallo sponsor, dall'istituzione cui fa capo il sitodi sperimentazione e dagli sperimentatori coinvolti, e sianoesenti da qualsiasi indebito condizionamento.
• Gli SM garantiscono che la valutazione sia effettuatacongiuntamente da un numero ragionevole di persone cheposseggono collettivamente le qualifiche e l'esperienzanecessarie.

ConclusioniLa sperimentazione clinica di fase I costituisce un momentoparticolarmente importante nello sviluppo di un nuovofarmaco.Tale attività richiede competenze specifiche da parte deivalutatori, una preparazione adeguata da parte deglisperimentatori, la disponibilità di strutture dedicate.La possibilità di sperimentare i nuovi farmaci nelle fasiprecoci di sviluppo rappresenta un importante occasione dicrescita ed innovazione per il Paese.L’Istituto Superiore di Sanità ha avviato negli anni una seriedi iniziative volte a facilitare/promuovere la sperimentazionedi fase I in Italia.La stretta collaborazione con l’Autorità Compente, AIFA, e iltrasparente confronto con l’Industria, l’Accademia e leStrutture cliniche, rappresenta un requisito indispensabile peril raggiungimento di obiettivi concreti e soddisfacenti
44

Buon Lavoro a Tutti
True genius resides in the capacity for evaluation uncertainty , hazardous,
and conflicting information
(W. Churchill)
45


















