
Accrescimento normale Bassa statura Pubertà precoce ... · In “Endocrinologia pediatrica”....
33
- Accrescimento normale - Bassa statura - Pubertà precoce - Pubertà ritardata - Obesità - Diabete mellito - Malattie della tiroide
Transcript of Accrescimento normale Bassa statura Pubertà precoce ... · In “Endocrinologia pediatrica”....

- Accrescimento normale
- Bassa statura
- Pubertà precoce
- Pubertà ritardata
- Obesità
- Diabete mellito
- Malattie della tiroide

L’accrescimento normale
Chirico V, Caruso R, Vicchio P, Moschella E, Malvaso S, Meduri S, Randazzo A, Salpietro V,
Munafò C, Arrigo T
Con il termine generale di accrescimento si intende parlare di processi che riguardano non solo la
crescita staturale, ma anche quella ponderale e la maturazione scheletrica dell’individuo nell’arco
della sua età evolutiva (dallo stadio di zigote alla conclusione della pubertà).
Alla base dei fenomeni di crescita vi sono tre meccanismi fondamentali: l’iperplasia, cioè l’aumento
del numero di cellule, l'ipertrofia o aumento delle dimensioni cellulari e l' osteogenesi, che si
traduce nella maturazione scheletrica.
Questi fenomeni sono strettamente dipendenti da fattori genetici e ambientali, che agiscono
attraverso la mediazione di ormoni o di fattori di crescita: se non interviene nessun impedimento, gli
ormoni consentiranno l' espletamento del pieno potenziale genetico.
I principali fattori fisiologici che influenzano la crescita possono essere distinti in:
- fattori endogeni: genetici ed ormonali;
- fattori esogeni: ambientali, nutrizionali, affettivi e socio-economici.
Durante la vita fetale la crescita, oltre che da fattori genetici, è influenzata da fattori materni, dal
livello di ossigenazione e nutrizione del feto, nonché dal sesso (è lievemente maggiore nei maschi)
e dall’età gestazionale. Un neonato a termine misura in media 50 cm e pesa 3.300 Kg. Dopo il
parto, dalla prima settimana fino ai tre mesi di vita un neonato dovrebbe aumentare di circa 150-200
grammi a settimana. In linea di massima, il peso alla nascita dovrebbe raddoppiare intorno ai cinque
mesi di età, triplicare ad un anno e quadruplicare a 2 anni. Per quel che concerne la statura, il
guadagno dovrebbe essere di circa 24 cm durante il I anno di vita e di 11, 8 cm e 7 cm in media
rispettivamente nel II, III e IV anno di vita. Dal quarto anno di vita e fino alla pubertà la velocità di
crescita si mantiene abbastanza stabile (intorno ai 5-6 cm per anno), con lieve rallentamento nelle
ultime epoche prepuberali. L’inizio della pubertà si accomapagna allo “scatto staturale” (durante il
I° anno guadagno staturale medio di circa 8,5 cm nel maschio e 6,5 cm nelle femmine).
Il calcolo del potenziale genetico (statura bersaglio), rappresentato dall' altezza media dei genitori è
ricavabile dalla seguente formula:
Statura target (bersaglio genetico): Per i maschi: (statura padre + statura madre + 13)/ 2 ®
Per le femmine: (statura padre + statura madre – 13)/ 2 ®
® Con variazioni di circa + 5 cm
dove 13 è un numero fisso che indica approssimativamente la differenza tra le stature medie dei
maschi e delle femmine adulti.

Per quel che riguarda il controllo endocrino della crescita durante la vita fetale esso è influenzato
dalla somatomedina C (SMC) e dall’insulina. Nell’ultimo periodo della vita fetale diviene
prevalente il controllo esercitato dalla SMC, che rappresenta il principale responsabile della crescita
fino ai due anni di vita. Dopo i 2 anni la crescita dipende dal GH. Gli ormoni tiroidei e la SMC
stimolano sia la crescita che la maturazione dei nuclei di ossificazione delle ossa lunghe, grazie al
loro effetto sulle cartilagini di coniugazione; gli steroidi sessuali ed i glucocorticoidi a dosi elevate
hanno un effetto negativo sulla crescita e sulla maturazione ossea.
Tra i fattori nutrizionali, l’ipernutrizione protratta ha un effetto positivo sulla crescita e sulla
maturazione dei nuclei di ossificazione, al contrario della malnutrizione e del malassorbimento che
si accompagnano ad un rallenta,mento della crescita e della maturazione ossea. Anche l’estrazione
socio-economica e l’ambiente familiare esercitano influenze importanti sull’accrescimento del
bambino.
I parametri biometrici e la valutazione della crescita
Il controllo della crescita mediante misurazioni antropometriche inizia già nella vita prenatale,
valendosi principalmente dell’ecografia, e continua dopo la nascita con la rilevazione delle prime
misure essenziali ovvero: il peso, la lunghezza, la circonferenza cranica, misure che
successivamente devono essere prese fino ai tre anni periodicamente (una volta a settimana nel
primo mese di vita; una volta al mese fino a sei mesi e poi ogni tre mesi fino ai tre anni). Dopo i tre
anni si continuerà a misurare la statura eretta ed il peso ogni sei mesi. In caso di statura anormale, al
fine di stabilire l’armonicità o meno dell’eccesso o difetto staturale, è opportuno procedere alla
rilevazione della statura seduta, mediante apposito statimetro. La misura da seduto viene
generalmente rilevata al di sopra dei tre anni di età, in casi particolari (acondroplasia e altri
dimorfismi ossei). La normalità delle misure antropometriche viene giudicata in riferimento a
tabelle (percentili) o grafici standard (deviazioni standard), generalmente sotto forma di curve di
crescita.
Le tavole dei percentili rappresentano l'andamento degli aumenti di peso e di altezza nel tempo, sia
per i maschi sia per le femmine. I grafici vengono realizzati misurando il peso e l'altezza di un
ampio numero di bambini della stessa età, tenendo conto del sesso, dell'etnia e dei diversi fattori
ambientali (Fig.1).

Fig.1 Centili italiani di riferimento
La linea intermedia (mediana) indica il 50 percentile, cioè quel valore al di sopra ed al di sotto del
quale si colloca il 50% della popolazione esaminata. I soggetti che crescono in maniera normale
sono quelli che, a secondo dell'età, si collocano tra i valori del 3° centile e quelli del 95° centile.
Calcolo in DS :la linea intermedia corrisponde alla media matematica dei valori rilevati,
relativamente ad un dato parametro, nella popolazione di controllo. Secondo tale sistema vengono
considerati normali i valori che ricadono entro le +2 e le -2 DS dalla media e patologici per difetto o
per eccesso i valori che si collocano rispettivamente al di sotto delle -2 o al di sopra delle +2 DS.
In entrambi i casi, in condizioni di benessere la curva di crescita ricostruita sarà regolare senza
rallentamenti o accelerazioni, con peso adeguato alla statura e rapporto corretto tra massa magra e
massa grassa. La valutazione seriata (effettuata ogni 6 mesi) dell’altezza e del peso identifica la
velocità di crescita e consente di rilevare tempestivamente la comparsa di un’accelerazione o un
rallentamento dell’accrescimento. La velocità di crescita va confrontata con i percentili di Tanner
della velocità di crescita (Fig.2).

Fig.2 Curva della velocità di crescita
La misurazione della circonferenza cranica deve far parte dell’esame clinico di routine del lattante
in quanto fornisce utili informazioni concernenti lo sviluppo della massa cerebrale. Il nastro
millimetrato flessibile, ma non estensibile, deve passare sulla regione frontale al di sopra del bordo
orbitale superiore, in corrispondenza delle bozze frontali, lateralmente in maniera simmetrica e sulla
regione occipitale, in modo da misurare la circonferenza massima. Tale misura va rilevata dalla
nascita ai tre anni.
L’età ossea
La valutazione dell’età ossea è usata per dare informazioni più accurate relativamente allo stato di
sviluppo di un bambino in quanto indice fedele dell’età biologica del soggetto. Viene rilevata
raffrontando la radiografia del polso e della mano sinistra (in particolare nuclei di ossificazione del
carpo e delle falangi) del soggetto in esame con una serie di radiografie standard raffigurate
nell’atlante Greulich and Pyle.
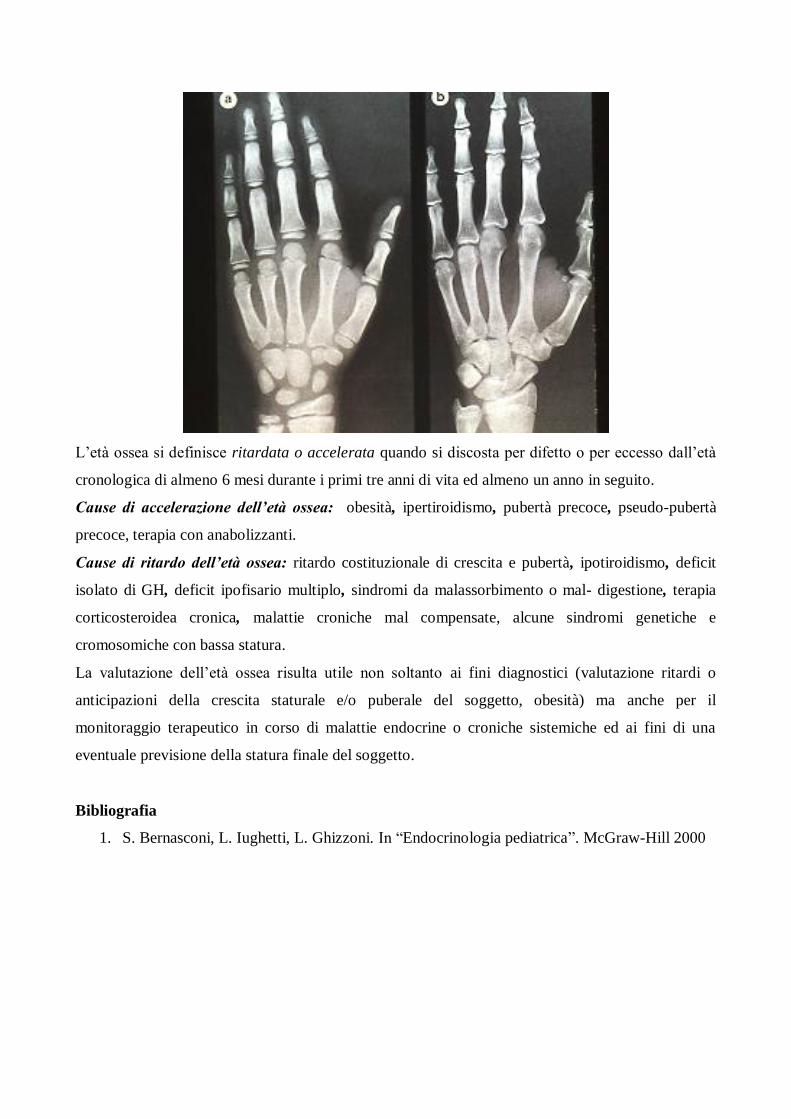
L’età ossea si definisce ritardata o accelerata quando si discosta per difetto o per eccesso dall’età
cronologica di almeno 6 mesi durante i primi tre anni di vita ed almeno un anno in seguito.
Cause di accelerazione dell’età ossea: obesità, ipertiroidismo, pubertà precoce, pseudo-pubertà
precoce, terapia con anabolizzanti.
Cause di ritardo dell’età ossea: ritardo costituzionale di crescita e pubertà, ipotiroidismo, deficit
isolato di GH, deficit ipofisario multiplo, sindromi da malassorbimento o mal- digestione, terapia
corticosteroidea cronica, malattie croniche mal compensate, alcune sindromi genetiche e
cromosomiche con bassa statura.
La valutazione dell’età ossea risulta utile non soltanto ai fini diagnostici (valutazione ritardi o
anticipazioni della crescita staturale e/o puberale del soggetto, obesità) ma anche per il
monitoraggio terapeutico in corso di malattie endocrine o croniche sistemiche ed ai fini di una
eventuale previsione della statura finale del soggetto.
Bibliografia
1. S. Bernasconi, L. Iughetti, L. Ghizzoni. In “Endocrinologia pediatrica”. McGraw-Hill 2000

Bassa Statura
Chirico V, Caruso R, Vicchio P, Moschella E, Malvaso S, Meduri S, Randazzo A, Salpietro V,
Munafò C, Arrigo T
Definizione: Statura inferiore al 3° centile o a -2DS-
Da attenzionare:
a) statura <- 2 DS
b) statura < target genetico
c) velocità di crescita <10°c
d) perdita di classi di centili
Cause di BS e loro frequenza:
1) Ritardo costituzionale di crescita e pubertà
2) Bassa statura familiare circa 80
3) Bassa statura idiopatica
4) Ritardo intrauterino della crescita circa 10
5) Malattie croniche internistiche circa 5
6) Cromosomopatie e sindromi mendeliane circa 1
7) Osteocondrodisplasia circa 1
8) Deficit di GH ed altre endocrinopatie circa 1
Varianti normali di BS sono rappresentate dalla bassa statura familiare, idiopatica e il ritardo
costituzionale di crescita e pubertà.
La BS patologica può essere sproporzionata (sproporzione tronco/arti) dovuta a una displasia
scheletrica o a rachitismo vitamino D resistente.
Le forme patologiche di BS proporzionata (normali segmenti corporei) possono avere origine
prenatale (sindromi, cromosomopatie, ritardo intrauterino della crescita) o postnatale (malattie
croniche internistiche, malnutrizione, farmaci, carenze affettive, disordini endocrini).
Sindrome di Turner (ST): alterazione cromosomica (monosemia o delezione o isocromosoma
della X). Incidenza 1:2000 neonate. Presentano un deficit della lunghezza alla nascita che tende a
peggiorare negli anni e che condiziona negativamente la statura finale . La ST si caratterizza per
alcuni aspetti: ritardo intrauterino della crescita, bassa statura. Amenorrea primaria , infertilità,
ipertransaminasemia, cardiopatia congenita, anomalie renali, malattie autoimmuni (tiroidite,
celiachia, diabete mellito), torace a corazza, cubito valgo, 4° metacarpo corto, pterigio del collo.

Iter diagnostico
Esami di I livello
servono ad escludere patologie croniche internistiche, eventuali alterazioni endocrine
– VES, PCR, AGA,EMA, TGA, Ig, FT4, TSH, SMC, IGFBP3
– Gn, testosterone (m), 17β estradiolo e βHCG (f)
Esami di II e III livello
Vanno eseguiti in base alla patologia sospettata:
- Test da stimolo per la secrezione del GH (almeno due test, sospetto deficit GH)
– PRL (adenoma secernente)
– ritmo circadiano del cortisolo (m. di Cushing)
– Test al GnRH analogo - Test al GnRH (ipogonadismo )
– Test HCG (ipogonadismo nei maschi)
– Ecografia pelvica e/o RMN pelvi ( valutazione organi pelvici nelle femmine)
– Olfattometria (sindrome di Kallmann)
– Cariotipo (s.Turner, s. Down)
– Autoanticorpi anti tiroide, surrene, ovaio (malattie autoimmuni)
– Eco tiroide (Tiroidine cronica autoimmune)
– RMN cerebrale (neoplasia cerebrale)
– Indagine genetica (mirate verso il sospetto diagnostico)
Terapia
Trattare la patologia sottostante (celiachia, MICI, ipotiroidismo, ipercortisolismo…..)
Il trattamento con Ormone della crescita (NOTA 39) viene riservato solo ad alcune categorie:
a) deficit GH ( 2 test da stimolo con risposta di GH <10ng/ml)
b) sindrome di Turner
c) sindrome di Prader Willi
d) insufficienza renale cronica
e) nati piccoli per età gestazionale (previa approvazione della commissione regionale)
Terapia con IGF1 ricombinante (s. Laron)

Bibliografia
1. Hjerrild BE, Mortensen KH, Gravholt CH. Turner syndrome and clinical treatment. Br Med
Bull. 2008;86:77-93
2. Cakan N, Kamat D. Short stature in children: a practical approach for primary care providers.
Clin Pediatr . 2007 ;46:379-85
3. Saenger P, Czernichow P, Hughes I, Reiter EO. Small for gestational age: short stature and
beyond. Endocr Rev. 2007 ;28:219-51

Pubertà Precoce
Chirico V, Salpietro A, Barone C, Randazzo A, Malvaso S, Caruso R, Sturiale M, Munafò C,
Arrigo T
L'inizio dello sviluppo puberale (la prima manifestazione clinica è generalmente costituita dallo
sviluppo della mammella nel sesso femminile e dall'aumento di volume dei testicoli nel sesso
maschile), la sua progressione ed il suo completamento presentano un'ampia variabilità nella
popolazione normale.
La pubertà viene definita “precoce” quando i primi segni di sviluppo sessuale compaiono ad una età
inferiore di più di <2.5 S.D. rispetto alla media di riferimento, cioè classicamente prima degli 8 anni
nella femmina e prima dei 9 anni nei maschi, in associazione ad un aumento della velocità di
crescita e della maturazione ossea.
Tab.1 Classificazione delle varie forme di precocità sessuale
Pubertà precoce vera o centrale,
gonadotropino-dipendente
Idiopatica
Secondaria: tumori del SNC, alterazioni congenite
o acquisite del SNC, esposizione prolungata a
steroidi sessuali
Pubertà precoce periferica,
gonadotropino-indipendente
Tumori secernenti HCG, neoplasie secernenti
androgeni nei maschi o estrogeni nelle femmine,
testo tossicosi familiare, sindrome surrenogenitale
congenita (maschi), cisti ovariche, S. di Peutz
Jegers (femmine), S. di Mc Cune-Albright,
ipotiroidismo, S. di Silver Russel
Varianti della maturazione puberale
Telarca prematuro isolato
Menarca prematuro isolato
Adrenarca prematuro isolato
Ginecomastia adolescenziale (maschi)
Macro-orchidia

La pubertà precoce vera (Gn RH-dipendente) è dovuta ad attivazione precoce della secrezione
ipotalamica di GnRH con l'ampiezza e la frequenza della pulsatilità propria della pubertà fisiologica
e conseguente maturazione completa delle gonadi e comparsa dei segni puberali. Si tratta di una
pubertà completa, riguardante sia i caratteri sessuali primari che secondari, isosessuale. E’ molto più
frequente nelle femmine che nei maschi (F:M circa 4:1) e la fascia di età più colpita è quella fra i 6
e gli 8 anni.
Può essere ulteriormente distinta in:
Idiopatica (sporadica, familiare o ereditaria)
Secondaria a patologia endocranica (tumorale e non): tumori intracranici (teratomi, gliomi, cisti,
astrocitoma disgerminoma, amartoma), malformazioni congenite (craniostenosi, idrocefalo),
progressi traumi (perinatali, accidentali), pregresse infezioni (meningite, encefalite) altre cause
(neurofibromatosi, sclerosi tuberosa, SAG trattata tradivamente).
Nelle femmine la causa di gran lunga più frequente è quella idiopatica (oltre il 50%), mentre nei
maschi la causa è organica nei 2/3 dei casi.
La pubertà precoce periferiferica (Gn RH-indipendente) o pseudo-pubertà precoce è dovuta ad
eccessiva produzione periferica di steroidi sessuali (estrogeni o androgeni), non conseguente ad
aumento della secrezione gonadotropinica.
A seconda del sesso del paziente e degli ormoni prodotti, la pseudopubertà precoce può essere
distinta in una forma isosessuale (concordanza tra il sesso e l'ormone prodotto) o in una forma
eterosessuale (aumentati livelli ematici di estrogeni nei maschi o di androgeni nelle femmine).
Da un punto di vista eziopatogenetico possiamo distinguere:
● Forme genetiche: LH, sindrome di McCune-Albright , mutazione DAX1
● Forme neoplastiche: tumori surrenalici, ovarici, testicolari, HCG secernenti
● Forme limitate o reversibili: sindrome adrenogenitale congenita nel maschio, steroidi o
gonadotropine esogene, cisti ovariche.
Tra le forme cosiddette varianti della maturazione puberale, il telarca prematuro, condizione
comunemente osservata nei primi due anni di età, apparirebbe la più frequente forma di precocità
puberale delle femmine. E' una condizione benigna che non pregiudica lo sviluppo e la fertilità delle
bambine affette e può essere indotta o da un'aumentata secrezione endogena di estrogeni o da un
aumentato apporto estrogenico attraverso la dieta o con l'uso di preparati ad uso topico (trattamento
delle sinechie delle piccole labbra, leucorrea da alterata flora vaginale). Non è necessario
intraprendere alcun trattamento se non un attento follow-up, poiché è stato osservato che alcune
forme possono evolvere in un quadro di pubertà precoce vera.

Il pubarca o ircarca prematuro si definisce come la comparsa precoce di pelo pubico o ascellare
(ircarca) prima degli 8 anni nelle femmine e di 9 anni nei maschi in assenza degli altri segni di
maturità sessuale o di virilizzazione, in rapporto ad un'aumentata secrezione di androgeni deboli di
origine surrenalica. E'una condizione benigna che non interferisce con lo sviluppo fisiologico
puberale e non necessita di alcun trattamento.
Il livello sierico delle gonadotropine in condizioni basali o dopo stimolazione con il GnRh test
evidenziano livelli prepuberali. L'età ossea può essere lievemente avanzata per l'età cronologica
senza compromettere la statura finale.
La comparsa di effetti androgenici generalizzati quali l'aumento di volume del pene o del clitoride,
l'aumento staturale, l'irsutismo o l'abbassamento del tono della voce devono indurre il medico ad
escludere la presenza di una sottostante neoplasia androgeno-secernente o un'iperplasia congenita
surrenale da deficit enzimatico mediante un prelievo per il dosaggio del testosterone, del DHEAS e
del 17-idrossiprogesterone.
Il menarca prematuro è una condizione rara caratterizzata dalla comparsa di perdite ematiche
vaginali periodiche senza altri segni di sviluppo sessuale secondario. Il sanguinamento può
manifestarsi già all'età di 1 anno e protrarsi per alcuni anni per poi cessare fino alla comparsa del
menarca fisiologico. Può essere la prima manifestazione della sindrome di McCune-Albright (triade
classica: displasia fibrosa poliostotica, macchie cutanee caffè-latte, pseudopubertà precoce ) o di un
ipotiroidismo giovanile.
Prima di confermare la diagnosi di menarca prematuro è necessario escludere le cause più frequenti
e talora più gravi quali le lesioni traumatiche o infettive della vagina o della cervice, le neoplasie
(rabdomiosarcomi), i corpi estranei, l'esposizione a fonti estrogeniche esogene o l'abuso sessuale. Il
menarca prematuro è una condizione benigna che non necessita di alcun trattamento e non
pregiudica la fertilità futura.
Iter diagnostico
L'anamnesi familiare e l'esame obiettivo associati alla radiografia del carpo ed all'ecografia pelvica
supportati dai risultati degli esami laboratoristici potranno rivelare la causa responsabile.
All’anamnesi valutare: precedenti familiari di pubertà precoce, obesità di lunga durata, assunzione
protratta di steroidi sessuali, precedenti di patologia endocranica, epoca inizio e progressione
manifestazioni puberali, perdite giallastre o ematiche dai genitali.
Alla valutazione auxologia rilevare: statura, peso, velocità di crescita, età ossea, statura dei genitori.

All’esame obiettivo generale valutare: timbro della voce, odore del sudore, sviluppo delle masse
muscolari, untuosità di pelle e capelli, acne, macchie color caffè-latte, esame neurologico, fundus e
campo visivo.
All’esame obiettivo specifico valutare: identificazione stadio puberale, volume e sagoma dei
testicoli, concordanza dei fenomeni puberali, precocità iso-o eterosessuale
Indagini laboratoristiche
● Dosaggio degli steroidi sessuali e delle gonadotropine (nella pubertà precoce vera valori di
gonadotropine più elevati rispetto a quelli attesi per l’età cronologica)
● Test al GnRH (nella pubertà precoce vera il picco LH è maggiore del picco FSH)
Indagini strumentali:
● RX polso e mano sinistra per valutazione età ossea (risulta avanzata e tende a progredire nel
tempo nella pubertà precoce vera)
● Ecografia pelvica (nella femmina aumento di volume dell’ovaio e dell’utero)
● RMN cerebrale (per escludere una causa organica, una volta posta la diagnosi di pubertà precoce
vera)
Trattamento
La terapia medica della pubertà precoce vera si avvale dell’impiego degli analoghi del GnRH che
determinano una ridotta secrezione di FSH ed LH.
La terapia è obbligatoria quando: Età cronologica <6 anni
Età Ossea >2 anni vs Età Cronologica
Previsione statura finale < 2DS vs Target genetico
Il trattamento va sospeso al raggiungimento di un’età ossea di 11-12 nelle femmine e 13 anni nei
maschi.
Bibliografia
1. Wilson JD, Foster DW, Kronenberg HM, Larsenet PR, Williams Textbook of endocrinology.
9th edition. Philadelphia, Saunders Co, 1998.
2. Marie-Christine Lebrethon et al. Management of central isosexual precocity: diagnosis,
treatment, outcome. Current Opinion In Pediatrics 2000; 12:394-399
3 Midyett LK et al, Are pubertal changes in girls before age 8 benign? Pediatrics 2003; 1147-51.
4. Tatò L, Savage MO, Antoniazzi F, Buzi F, Di Maio S, Oostdijk W, Pasquino AM, Raiola G,
Saenger P, Tonini G, Voorhoeve PG; Optimal therapy of pubertal disorders in
precocious/early puberty J Pediatr Endocrinol Metab. 2001;14 Suppl 2: 985-95.

Pubertà Ritardata
Chirico V, Salpietro A, Barone C, Randazzo A, Malvaso S, Caruso R, Sturiale M, Munafò C,
Arrigo T
Lo sviluppo puberale si definisce ritardato nella femmina quando all'età di 13 anni non è comparso
ancora il bottone mammario e nel maschio quando a 14 anni non si è osservato ancora un aumento
del volume testicolare (misurabile con l’orchidometro di Prader > 4 cc).
Si parla di ritardo dello sviluppo puberale anche quando la progressione dello sviluppo procede in
maniera troppo lenta ovvero quando il passaggio da uno stadio all'altro - secondo la classificazione
di Tanner- supera i limiti fisiologici: nello specifico nella femmina se più di 12 mesi sono necessari
per il passaggio da uno stadio all'altro di Tanner; nel maschio se più di 9,5 mesi sono necessari per
il passaggio da uno stadio all'altro di Tanner.
Sotto l’aspetto eziopatogenetico si distinguono tre forme principali di pubertà ritardata (Tab.1)
Tab.1
Pubertà tarda costituzionale Variante della norma
Ipogonadismo ipogonadotropo
(difetto a livello ipotalamo-
ipofisario)
Transitorio: malattie croniche o sistemiche (asma,
morbo di Crohn, insufficienza renale); deficit
nutrizionali (malattia celiaca, fibrosi cistica); dispendio
energetico (atleti, ginnasti); endocrinopatie (deficit di
GH, ipotiroidismo, iperprolattinemia)
Permanente: Deficit di gonadotropin (idiopatico, da
malformazioni del SNC); deficit di gonadotropine con
anosmia (S. di Kallmann); deficit isolato di LH;
sindromi polimalformative (S. di Prader-Willi, S. di
Laurence-Moon-Biedl); tumori del SNC; radioterapia
craniale.
Ipogonadismo ipergonadotropo
(difetto a livello gonadico)
Anomalie cromosomi del sesso (S. di Turner, S. di
Klinefelter); danni o agenesie gonadiche.

Iter diagnostico
All’anamnesi valutare: la familiarità per lo stesso problema, nella madre nel caso della femmina,
nel padre nel caso del maschio; l'esistenza di patologie croniche e/o malformative (renale,
cardiaca, intestinale, genitale, respiratoria, flogistica articolare e/o sistemica, diabete, celiachia), la
presenza di particolari manifestazioni cliniche alla nascita (linfedema dei piedi nel sospetto di una
sindrome di Turner), l'eventuale uso cronico di farmaci che possono aver interferito con lo
sviluppo fisico (inteso in maniera globale), un recente significativo calo ponderale, o
l'effettuazione di un attività fisica intensa, solitamente a livello agonistico; lo sviluppo
psicomotorio del soggetto, un eventuale ritardo del linguaggio, un disadattamento scolastico ecc.
che possono indirizzare verso particolari condizioni sindromiche (Klinefelter e varianti ecc.).
All’esame obiettivo definire: la condizione auxologica del paziente (peso, statura, velocità di
crescita secondo i percentili di Tanner-Whitehouse), la presenza di note dismorfiche nell'ipotesi di
una sindrome che si associa a ritardo di sviluppo puberale e/o ipogonadismo (S. di Turner, S. di
Noonan, S. di Prader Willi, S. di Lorence-Moon -Biedl ecc.), la presenza di anomalie associate a
carico di altri organi ed apparati o la presenza di un gozzo di recente comparsa.
Indagini laboratoristiche
● Esami di I livello volti alla valutazione dello stato nutrizionale del soggetto e/o ad escludere
eventuali patologie associate (MICI, celiachia, ipotiroidismo): emocromo, sideremia, ferritina,
VES, PCR, Ig, EMA, TgA , fT4, TSH
● Livelli basali di gonadotropine
● Test dinamici (Test con GnRH)
● Cariotipo (sia in presenza di note dismorfiche e/o di ritardo mentale associati a bassa statura o alta
statura o in caso di importante obesità nel sospetto di una sindrome di Turner, di una sindrome di
Klinefelter o di una sindrome Prader-Willi)
Indagini strumentali
● RX del polso e della mano sinistra, per calcolare l'età scheletrica (secondo Greulich e Pyle):
significativo un ritardo superiore ai 2 anni, rispetto all'età cronologica, in ambedue i sessi.
● Ecografia pelvica
● RMN/TC in caso di sospetto di anomalie o patologie espansive del tratto ipotalamo-ipofisario.
● Olfattometria in caso di sospetto sovraipofisario o in caso di dubbio, effettuare un'olfattometria
per sospetto di S. di Kalmann (è presente di solito alta statura e può essere presente un'importante
ipoacusia o sordità).

Terapia
Il trattamento terapeutico varia in rapporto alla causa sottostante il ritardo puberale. Nelle forme
secondarie a malattie croniche e/o deficit nutrizionali occorre trattare il difetto di base; in caso di
deficit secretivo di gonadotropine o ormoni gonadici si ricorre alla terapia sostitutiva, con strategie
differenti a seconda che si tratti di forma costituzionale o patologica.
Bibliografia
1. Enriette Factor affecting onset of puberty Horm Res 2002;57 (suppl 2) 15-18
2. Karlberg Secular trends in puberal development Horm Res 2002;57 (suppl 2);19-30
3. GE. Faleschini, G. Borotto. La pubertà grave, ritardata. Medico e Bambino pagine elettroniche
2004; 7(1)

Obesità
Munafò C, Chirico V, Caruso R, Salpietro V, Comito D, Malvaso S, Randazzo A, Talenti A,
Arrigo T
Introduzione
Per obesità si intende un accumulo eccessivo e generalizzato di grasso sia nel tessuto sottocutaneo
sia in altri tessuti più profondi, e può essere associata ad alterazione di parametri metabolici con
conseguenze sullo stato di salute fisico e psicologico (WHO, 1998). La misurazione del grasso
corporeo è tecnicamente complessa e le metodiche più accurate non sono adatte per un uso clinico
routinario, ma il loro impiego è giustificato principalmente a scopi di ricerca. Nella comune pratica
clinica si utilizzano stime indirette di grasso corporeo ottenute dalla misura dell’acqua corporea
totale (bioimpedenziometria) o delle pliche di grasso sottocutaneo (plica bicipitale, tricipitale,
sottoscapolare e sovrailiaca). Per convenzione si considera obeso il bambino che abbia uno spessore
della plica tricipitale superiore al 95° percentile e sovrappeso il bambino con un spessore compreso
tra l’85° e il 95° percentile delle tabelle di riferimento. Una modalità alternativa per definire la
condizione di obesità nel bambino è misurare peso e altezza per calcolare il BMI, dato il rapporto
tra peso (Kg) e altezza al quadrato (m²). I vantaggi di questo indice sono dati da vari fattori:
- La semplicità di misura di peso e altezza
- L’ottima correlazione tra BMI e peso corporeo
Il BMI è pertanto un indice attendibile per la diagnosi di obesità nel bambino, per un impiego sia
clinico che epidmiologico. Il Centre for Disease Control (CDC) and Prevention americano definisce
attualmente sovrappeso i bambini e adolescenti con BMI compreso tra l’85° e il 95° percentile e
obesi quelli con BMI superiore al 95° percentile.
Epidemiologia
L’obesità è attualmente considerata il maggior problema di salute pubblica a livello mondiale, causa
di 1,5 milioni di disabilità e di oltre un milione di morti premature all’anno. Si è proposto il
neologismo “globobesity” per indicare la diffusione del fenomeno. L’incremento dell’obesità è stato
infatti documentato non sono nelle nazioni industrializzate ma anche nei Paesi in via di sviluppo. A
partire dagli anni Settanta, ma in particolare nelle ultime due decadi, si è assistito ad un aumento
epidemico in tutte le fasce di età, non risparmiando quella pediatrica. Secondo le ultime stime,
almeno 22 milioni di bambini in età prescolare (WHO) e 155 milioni di bambini e adolescenti di età
compresa tra i 5 e i 17 anni (IOTF) sono sovrappeso; di essi ben 30-41 milioni sono classificabili e
il rischio relativo per un bambino obeso di diventare un adulto obeso,come riportato da una recente

metanalisi, varia da valori inferiori a 5 a oltre il 40% ed è proporzionale alla durata, al grado di
sovrappeso e all’età. In Italia la prevalenza di sovrappeso ed obesità risulta la più elevata d’Europa,
il 23,9% dei bambini è in sovrappeso, il 13,6% è obeso. La prevalenza di obesità nelle regioni del
Sud Italia è maggiore rispetto al Nord.
Genetica
La regolazione del bilancio energetico è un complicato meccanismo finemente regolato
dall’interazione di numerose sostanze quali neuropeptidi, enzimi, recettori e ormoni. La complessità
di questo sistema non è legata tanto alla quantità delle sostanze implicate nella sua regolazione
quanto alle interazioni tra le varie sostanze e tra queste e l’ambiente. Inoltre l’espressione, l’azione
e talvolta anche la capacità di interazione dei componenti di questo intrigato sistema sono funzione
dell’assetto genetico. Il sistema nervoso centrale gioca sicuramente un ruolo fondamentale nella
regolazione dell’appetito. I sistemi neuronali che regolano la fame e la sazietà, l’accumulo di
energia, la spesa energetica e la produzione endogena di glucosio modulano informazioni
provenienti da vari distretti corporei e veicolati da agenti oressizanti e anoressizanti. Questi
includono segnali ormonali (per esempio la leptina e l’insulina) e segnali provenienti da nutrienti
(per esempio glucosio e acidi grassi liberi), che trasmettono informazioni circa le riserve di energia
dell’organismo. Pertanto il peso corporeo è mantenuto in risposta ad una varietà di stimoli che
consentono, in ultima analisi, l’incremento o la riduzione dell’introito e della spesa energetica. Uno
dei più potenti fattori endogeni anoressizzanti è la leptina. La leptina è un ormone proteico
(codificato dal gene LEP) di 167 amminoacidi; essa è prodotta essenzialmente a livello del tessuto
adiposo ed è proprio l’adipocita, probabilmente sentendo il livello dei trigliceridi o dei suoi
accumulati nella cellula, a mettere in circolo la leptina. Giunta a livello del sistema nervoso centrale
(SNC), la leptina agisce deprimendo l’attività dei neuroni che utilizzano come neurotrasmettitore il
Neuropeptide Y (NPY), che si configurano quindi come i neuroni della “fame”. Di contro, vengono
a essere stimolati quei gruppi di cellule nervose che sintetizzano peptidi come la
proopiomelanocortina (POMP) e i suoi derivati, tra cui l’alfa-melanocyte-stimulating hormon (α-
MSH), nonché i neuroni produttori del cocaine and amphetamine regulated trascript (CART), tra i
più recenti peptidi associati alle funzioni dei neuroni della sazietà. La rete di segnali tra la periferia
e il SNC si completa con altri due neuropeptidi. Il primo di questi, la grelina, è un peptide di 28
amminoacidi che svolge un’azione oressigena. Nell’uomo viene sintetizzata e secreta dalle cellule
gastriche e agisce a livello del nucleo arcuato. L’altro neuropeptide è il peptide YY (PYY) che
viene rilasciato dal tratto gastrointestinale in proporzione del contenuto calorico del pasto; tramite
l’inibizione del NPY determina una riduzione dell’appetito. La scoperta che alcuni soggetti sono
portatori di mutazioni che alterano la funzione o la struttura di un gene ne compromettono l’azione
determinando l’insorgenza di obesità, ci ha aiutato a svelare alcuni meccanismi che sottendono
questo sistema.
Le mutazioni responsabili dell’obesità monogenica
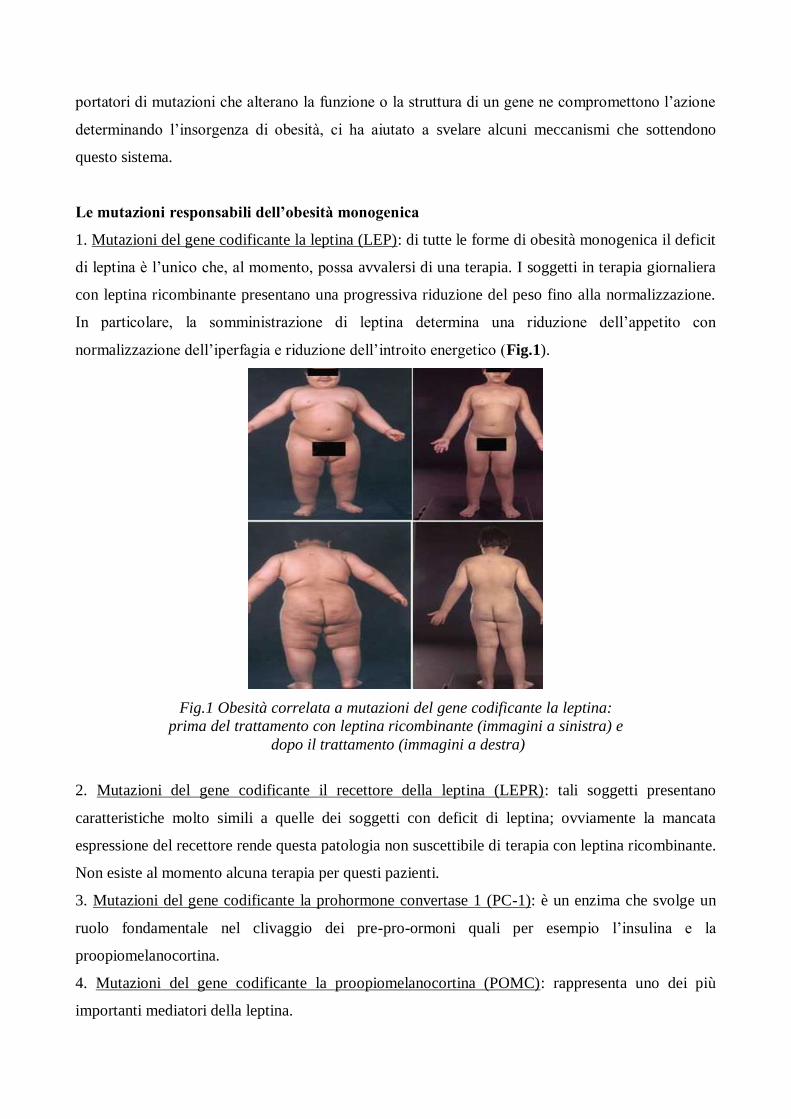
1. Mutazioni del gene codificante la leptina (LEP): di tutte le forme di obesità monogenica il deficit
di leptina è l’unico che, al momento, possa avvalersi di una terapia. I soggetti in terapia giornaliera
con leptina ricombinante presentano una progressiva riduzione del peso fino alla normalizzazione.
In particolare, la somministrazione di leptina determina una riduzione dell’appetito con
normalizzazione dell’iperfagia e riduzione dell’introito energetico (Fig.1).
Fig.1 Obesità correlata a mutazioni del gene codificante la leptina:
prima del trattamento con leptina ricombinante (immagini a sinistra) e
dopo il trattamento (immagini a destra)
2. Mutazioni del gene codificante il recettore della leptina (LEPR): tali soggetti presentano
caratteristiche molto simili a quelle dei soggetti con deficit di leptina; ovviamente la mancata
espressione del recettore rende questa patologia non suscettibile di terapia con leptina ricombinante.
Non esiste al momento alcuna terapia per questi pazienti.
3. Mutazioni del gene codificante la prohormone convertase 1 (PC-1): è un enzima che svolge un
ruolo fondamentale nel clivaggio dei pre-pro-ormoni quali per esempio l’insulina e la
proopiomelanocortina.
4. Mutazioni del gene codificante la proopiomelanocortina (POMC): rappresenta uno dei più
importanti mediatori della leptina.

5. Mutazioni del gene codificante il recettore 4 delle melanocortine (MC4R): è il modello di obesità
monogenica in assoluto più interessante; le mutazioni su questo gene determinano obesità
essenziale; il fenotipo di questi soggetti è caratterizzato da obesità severa ad esordio precoce e
iperfagia.
6. Mutazioni del gene codificante il cocaine and amphetamine regulated transcript (CART): altro
modulatore dell’azione della leptina, insieme al POMC e MC4R.
L’obesità come modello di patologia multigenica
Circa il 5% dei casi di obesità, soprattutto se ad esordio precoce, è conseguenza dell’alterazione di
un singolo gene. Quindi la stragrande quantità di casi di obesità (circa il 95%) è il frutto
dell’interazione di numerosi geni predisponenti all’obesità con un ambiente obesogenico. Il
bambino obeso, nella maggior parte dei casi non mangia cibi particolari, ma egli manga troppo di
tutto. Il bambino obeso spende energie, come ogni altro bambino, ma le sue spese sono equivalenti
a quelle di un bambino non obeso e non sono affatto rapportate al suo peso, perché altrimenti non
sarebbe grasso.
Esame obiettivo
L’esame clinico di un bambino o adolescente in sovrappeso/obeso in genere consente di orientarsi
tra le varie forme di obesità: secondaria a patologia endocrina o sindromica oppure idiopatica.
Il bambino affetto da obesità essenziale presenta caratteristiche molto precise:
- L’obesità si instaura gradualmente nel tempo
- La distribuzione del tessuto adiposo è uniforme a livello del viso, tronco e arti
- La statura è medio-alta
- Non sono presenti stigmate malformtive
- È spesso presente pseudoipogenitalismo
- Normale è lo sviluppo psicomotorio e la performance scolastica
- La maturazione puberale è tendenzialmente accelerata, così come la maturazione ossea è in
genere avanzata rispetto all’età cronologica
- Spesso è presente valgismo delle ginocchia, piede piatto
- Si rilevano, soprattutto in epoca adolescenziale, strie a livello di cosce, fianchi e glutei
- Può essere presente acanthosis nigricans (iperpigmentazione a livello del collo e delle
ascelle)

Diagnosi differenziale dell’obesità in età pediatrica
Cause endocrine
Sindrome di Cushing, Ipotiroidismo, Iperinsulinismo, Deficit di ormone della crescita,
Disfunzione ipotalamica, Sindrome di Prader-Willi, Sindrome dell’ovaio policistico,
Pseudoipoparatiroidismo tipo I.
Sindromi generiche
Sindrome di Turner, Sindrome di Laurence-Moon-Biedl, Sindrome di Alstrom-Hallgren.
Altre sindromi
Sindrome di Cohen, Sindrome di Carpenter.
Indagini di laboratorio
Devono essere effettuate in relazione al grado di obesità e alla familiarità per fattori di rischio
cardiovascolare (diebete mellito, ipertensione, dislipidemia, malattia cardiovascolare), per cercare
di individuare precocemente i marker di una sindrome metabolica secondaria alla stato di
sovrappeso del bambino.
Lo screening comprende: glicemia a digiuno, insulinemia a digiuno, profilo lipidico, transaminasi,
misurazione della pressione arteriosa.
Nei pazienti sovrappeso, nei quali si evidenzi ipertensione, va eseguito un approfondimento
diagnostico mediante: visita specialistica cardiologica, ECG ed ecocardiografia; esame delle urine
standard e dosaggio della microlbuminuria; dosaggio di creatininemia e potassiemia.
Complicanze
L’obesità infantile non è solo un problema estetico come spesso tuttora si crede: essa può infatti
condurre a molte complicanze nel breve e nel lungo periodo, sia di natura metabolica, che di
pertinenza cardiovascolare, gastroenterologica, respiratoria, ortopedica e psicologica. Pressoché
tutte, peraltro, possono regredire con il recupero di un peso normale.
Complicanze psicologiche. I bambini obesi o sovrappeso vanno incontro a notevoli stress e
difficoltà a livello sociale e psicologico. E’ spesso frequente una stigmatizzazione sociale nella
scuola, sul posto di lavoro e nelle varie occasioni di tipo sociale. I bambini in età scolare sono
frequentemente presi in giro, intimiditi ed esclusi dalle varie attività. I disturbi psicologici sono
comuni nel bambino obeso. Anche nei bambini apparentemente in salute, una adeguata valutazione
psicologica spesso maschera problemi emozionali significativi.

Complicanze metaboliche. Le complicanze metaboliche, le più temute, possono riguardare il
controllo degli zuccheri (insulino-resistenza e iperinsulinismo, ridotta tolleranza al glucosio, diabete
mellito di tipo 2) e dei lipidi (aumento del colesterolo e/o dei trigliceridi, riduzione del colesterolo
HDL, aumento del colesterolo LDL) nel sangue, fattori questi che insieme all’aumento della
pressione arteriosa, tipica dell’obesità, predispongono alla comparsa di malattie cardiovascolari in
età adulta. I pazienti hanno un incremento della secrezione basale di insulina, della stimolazione alla
secrezione di insulina e della resistenza all’insulina.
Complicanze cardiovascolari. A queste età precoci è però già possibile ed anzi frequente
l’ipertensione (e in generale, circa il 60% degli obesi sono ipertesi), così come sono possibili forme
più o meno precoci di aterosclerosi, con aumentato rischio futuro di infarto e ictus cerebrale.
L’obesità infantile determina certamente un aumentato rischio di contrarre – una volta adulto –
malattie coronariche gravi come l’angina pectoris, l’insufficienza coronarica, l'infarto.
Complicanze gastroenterologiche. La steatosi epatica non alcolica (un accumulo di grasso a
livello del fegato determinato dall’obesità e non dal consumo di alcolici), problema che, se non
risolto, può evolvere, anche già in età pediatrica, fino alla cirrosi.
Complicanze respiratorie. Si può avere riduzione della capacità respiratoria, dispnea da sforzo,
ridotta ventilazione con riduzione dei livelli d’ossigeno, fino alla cosiddetta sindrome di Pickwick,
nella quale si hanno vere e proprie brevi perdite di coscienza durante le normali attività di vita.
Sono anche possibili ostruzioni catarrali delle vie respiratorie, bronchiti ricorrenti, asma, aumento
dei livelli di anidride carbonica in circolo. I bambini obesi sono più predisposti ad avere disturbi
respiratori soprattutto nel sonno, in particolare le apnee, con frequenti risvegli ed un sonno
disturbato che si riflette poi sul resto della giornata, quando i bambini tenderanno ad essere
sonnolenti, o al contrario nervosi e iperattivi, a soffrire frequentemente di cefalea o a rendere meno
bene a scuola.
Complicanze ortopediche. L’eccesso di peso ha lo stesso effetto sul corpo della costruzione di
troppi piani in un palazzo: le fondamenta e le strutture di sostegno tenderanno a cedere. Per questo
le complicanze ortopediche sono così frequenti, soprattutto il ginocchio valgo (le gambe ad X), i
piedi piatti e la scoliosi. Molto di frequente la prima alterazione che si manifesta è il piede piatto,
successivamente sono coinvolte le ginocchia, e alla fine la somma di posture errate può determinare
un atteggiamento scoliotico della schiena. Da non sottovalutare, inoltre, l’accrescimento eccessivo
della metafisi prossimale-mediale della tibia e la lussazione dell’epifisi della testa del femore.
Complicanze endocrinologiche. I bambini in sovrappeso hanno un’età ossea avanzata, sono più
alti in relazione all’età e presentano uno sviluppo puberale più precoce rispetto ai bambini non in
sovrappeso. Nelle bambine con obesità è poi frequente la presenza di cicli mestruali alterati.

Più avanti con l’età, inoltre, non è raro riscontrare in queste ragazze la sindrome dell’ovaio
policistico, causa di non pochi disturbi ginecologici ed endocrini.
Complicanze cutanee. Una complicanza cutanea che spesso indica un problema nel controllo degli
zuccheri, è la cosiddetta acanthosis nigricans (pelle ispessita, vellutata e grigiastra a livello delle
pieghe cutanee e del collo), ma ci sono anche infezioni cutanee croniche, smagliature, intertrigine
(una dermatite dovuta a sfregamento soprattutto a livello delle ascelle, delle pieghe inguinali e
sottomammarie), acne, irsutismo.
Prevenzione e trattamento
Il mantenimento del peso ideale è auspicabile non solo per fattori estetici, ma anche per prevenire le
possibili complicanze dell’obesità. I bambini sovrappeso, ch non vengono trattati possono rimanere
tali anche da adulti. Durante l’età pediatrica, una volta instaurata un’obesità, è molto difficile
mettere in pratica un programma efficace per ridurre e mantenere il peso a livelli accettabili senza
una partecipazione attiva e una motivazione da parte del bambino e della sua famiglia. Le tecniche
che vengono utilizzate per ridurre il peso negli adulti, come gli interventi chirurgici, la
farmacoterapia e l’introduzione di palloncini nello stomaco, sono controindicate nei bambini.
Anche le diete nettamente ipocaloriche sono inadeguate al momento che possono ritardare la
crescita e lo sviluppo in momenti critici dell’età pediatrica.
Per trattare con successo l’obesità bisogna prendere in considerazione almeno i seguenti aspetti:
1. Modificazione della dieta e del suo contenuto calorico
2. Definizione e applicazione pratica di programmi di attività fisica appropriati
3. Modifiche del comportamento del bambino
4. Coinvolgimento della famiglia nella terapia.
Per quanto riguarda la modificazione della dieta e del suo contenuto calorico si consiglia una dieta
ipocalorica bilanciata, ben condotta e finalizzata ad acquisire un corretto comportamento
alimentare, caratterizzata da una ripartizione bilanciata dell’apporto energetico rappresentato per il
50-60% dai carboidrati, per il 28-30% dai grassi e per il 10-15 % dalle proteine. Le modificazioni
più rilevanti sono: la ripartizione degli alimenti in 5 pasti al giorno, la valorizzazione della prima
colazione, la riduzione dell’apporto dei grassi e proteine di origine animale, la limitazione degli
zuccheri a rapido assorbimento, l’aumento delle fibre alimentari. Per ciò che concerne l’attività
fisica, bisognerebbe abituare il bambino ai giochi all’aperto, ridurre il tempo dedicato alla
televisione, videogiochi o computer a favore di attività più dinamiche. Spronare il bambino a fare le
scale, piuttosto che prendere l’ascensore, fare lunghe passeggiate, andare a scuola a piedi.
I risultati che si ottengono sono comunque limitati nel tempo, i controlli nel tempo mostrano

un’elevata frequenza di recidive a 4-10 anni di distanza. Solo il 50 % dei pazienti riesce a
conservare un peso inferiore rispetto a prima. In ogni caso appare consigliabile che il medico,
nell’interesse del singolo paziente, dia inizio ad un prudente trattamento dietetico e fisico,
combinato con la modificazione del comportamento e con la terapia della famiglia. Lo scopo ultimo
dovrebbe essere quello di favorire l’accrescimento e fornire un deciso sostegno sociale e
psicologico.
Bibliografia
1. Pediatria di Nelson. 18° Edizione

Diabete Mellito
Chirico V, Salpietro A, Vicchio P, Deak A, Loddo I, Malvaso S, Caruso R, Munafò C, Arrigo T
Diabete Tipo 1: da distruzione della β cellula pancreatica e deficit insulinico assoluto
Diabete Tipo 2: da insulino resistenza con deficit relativo di secrezione o da prevalente deficit di
secrezione insulinica
Altri tipi di Diabete:
a) Difetti genetici della β-cellula b) Difetti genetici dell’azione insulinica
c) Malattie del pancreas esocrino d) Endocrinopatie
e) Farmaci f) Infezioni
g) Da forme rare di malattie immunitarie h) Da malattie genetiche associate al diabete
i) Diabete gestazionale
Epidemiologia ed eziopatogenesi
- Prevalenza: 1% , età 0/15 anni - Incidenza annuale: 5-20/100.000, età 0-15 anni
- Nazione più colpita: Finlandia - Regione Italiana più colpita: Sardegna
- Rischio per i fratelli: 4-10 % - Rischio per i figli: 1-2 %
- Fattori genetici: Predisponesti - Fattori virali: Scatenanti
Il Diabete tipo 1 può essere suddiviso nei seguenti quattro stadi:
1) Prediabete
2) Insorgenza clinica del diabete
3) Parziale remissione o Luna di miele
4) Insulino-dipendenza permanente
PREDIABETE
Può essere suddiviso in fasi:
La prima fase è rappresentata dalla predisposizione genetica, condizione necessaria ma non
sufficiente per lo sviluppo del diabete. Alleli genetici correlati ad aumentato rischio: HLA DR3-
DR4 – DQA1*0501*0301 – DQB1*0201*302;
Aplotipi, quali ad esempio HLA DR2 – DQA1*0102 DQB1*0602, determinano invece una
resistenza alla comparsa del diabete stesso.
È una specifica combinazione allelica, più che la presenza o l'assenza di un residuo sulla molecola
HLA, a conferire suscettibilità alla malattia o protezione da questa.

La seconda fase comporta eventi scatenanti (agenti virali, alimenti), che danno origine all’attacco
autoimmune (ICA) nei confronti delle beta-cellule (insulite).
ICA sono una classe eterogenea di immunoglobuline, dirette contro determinanti antigenici insulari.
Tre diverse molecole sono state dimostrate, quali antigeni bersaglio degli ICA : l’enzima
glutammico decarbossilasi,GAD (identificato nel 1990, più comune adulti), la tirosin-fosfatasi
insulare IA-2 e il ganglioside GM2-1. L’insulina è una molecola autoantigenica e gli IAA (Ab anti-
insulina) sono marker aggiuntivi di rischio e i primi ad apparire.
Nella terza fase non si osservano ancora manifestazioni cliniche, ma si verifica un progressivo
declino della secrezione insulinica in risposta alla somministrazione di glucosio endovena (IVGTT).
Marker metabolici
Il test più usato per valutare la funzione ß-cellulare nella predizione di DMT1 è quello della risposta
precoce insulinemica al carico endovena di glucosio (IVGTT). Si calcola la First Phase Insulin
Response (FPIR), data dalla somma dei valori di insulinemia al tempo +1 e +3 minuti dalla fine
dell'infusione endovenosa di glucosio. Viene considerata patologica una FPIR che sia inferiore al 1°
percentile di normalità secondo gli standard di riferimento della SIEDP, suddivisi per stadio
puberale.
è stata costruita una life table analysis in base alla risposta insulinemica all'IVGTT nei parenti di I
grado con ICA positivi: se la risposta è inferiore al 1° percentile aumenta il rischio di DM1 (entro 4
anni circa)
Il rilascio dell’insulina così danneggiato nella “First Phase Insulin Response (FPIR)” associato alla
presenza di marcatori immunologici (GAD – IA-2) conferisce pressappoco il 100% di Rischio di
diabete tipo 1 nei successivi 5 anni nei soggetti con familiarità di I grado per diabete tipo 1.
Nella quarta fase compare la ridotta tolleranza alla somministrazione orale di glucosio (OGTT); è la
fase in cui la malattia diventa clinicamente evidente, e vi è una residua produzione di insulina, ma
dopo un periodo di remissione più o meno lungo, fa seguito la distruzione di tutte le cellule beta,
con deficit di insulina.
Definizione tolleranza glucidica
Con i nuovi criteri si definisce diabetico un soggetto che dopo 2 controlli glicemici, a digiuno,
presenta un valore della glicemia compreso tra 126 e 139 mg/dl e che dopo prova da carico orale
con glucosio abbia a 2 ore un valore di glicemia > di 200 mg/dl.
Intolleranza al glucosio o IGT (Imparied Glucose Tolerance) se il soggetto presenta dopo carico
orale di glucosio a 2 ore valori glicemici compresi tra 140 ma <199.

Alterata glicemia a digiuno o IFG (Imparied Fasting Glucose) se il soggetto avrà valori tra 110-
125 a digiuno . NORMALE, se la glicemia è < 110 mg/dl
IN SINTESI
È la malattia metabolica più frequente in età pediatrica
È determinata da distruzione autoimmune delle cellule beta
L’incidenza è più alta nei paesi sviluppati (Italia 7-36/100.000)
Predisposizione preesistente (HLA DR3, DR4, DQ2)
Fattori ambientali (enterovirus, rosolia, alimenti, etc..)
Associazioni con altre malattie autoimmuni (celiachia, tiroidite…)
Esordio classico nel 70-80% dei casi (poliuria-polidipsia)
Esordio subdolo più frequente a 10-20 anni (LADA)
SEGNI E SINTOMI PRECOCI
Legati all’iperglicemia: poliuria, polidipsia, polifagia, disturbi visivi
“ alla disidratazione e diseletrolitemia: astenia, dimagramento
SEGNI E SINTOMI TARDIVI
Legati alla chetonemia: anoressia, nausea, vomito, alito acetonemico
“ all’iperosmolarità: alterazioni stato mentale
“ all’acidosi: dolori addominali, respiro di Kussmaul
“ all’ipopotassemia: ileo paralitico, crampi muscolari, disritmie
INSORGENZE ATIPICHE
Enuresi persistente
Dolori addominali con o senza vomito
Candidosi vaginali recidivanti
Guadagno inconsistente di peso o perdita
Fatica,irritabilità, diminuzione del rendimento scolastico
Ricorrenti infezioni della pelle

DIFFICOLTA’ NELLA DIAGNOSI
Neonati con sintomi nascosti
Enuresi e/o poliuria diagnosticate come infezione delle vie urinarie
Dolori addominali o vomito diagnosticati come coliche addominali-appendiciti
Iperventilazione diagnosticata e trattata come patologia respiratoria
Polidipsia erroneamente diagnosticate come abitudine o necessità psicogena di bere
Diagnosi erronee (13-7%)
Sepsi urinaria (45%)
Addome acuto (30%)
Disturbi dell’adolescenza (13%)
Anoressia nervosa (6%)
Asma bronchiale-Polmonite (3%)
Ipertiroidismo (3%)
CHETOACIDOSI
• Circa il 15-30% delle diagnosi avvengono per KAD
• Causa di mortalità nel 6-10% dei casi
• La causa principale di morte è l’edema cerebrale
• KAD: glicemia >300 mg/dl
pH <7.2 (<7.1 KAD severa)
HCO3 < 12 mEq/l
Terapia della chetoacidosi
• Sol.fisiologica nelle prime due ore e poi idratazione + insulina + K, eventuale sol glucosata
se glicemia <250mg/dl
Cardini della terapia del diabete
Insulinoterapia (rapida, analogo rapida, intermedia, analogo intermedia, premiscelate,
tramite siringhe, penne o microinfusore)
Piano alimentare (carboidrati 50-60%, lipidi 25-30%, protidi 15%)
Autocontrollo (determinazioni pluriquotidiane di glicemia e glicosuria/chetonuria)
Esercizio fisico (regolare e programmato)

Valore percentuale Hba1c Livello di rischio
< 6 % Rientra nella media
< 7,5 % Appena superiore alla media
< 8,5 % Controllo da tenere sott’occhio
< 10 % Alto
> 10 % Molto alto
CARATTERISTICHE DIFFERENZIALI DEL DIABETE
Tipo 1 Tipo 2
Livelli di insulina assenti o ridotti Normali o aumentati
Sintomatologia importante spesso assente
Chetosi presente assente
Peso normopeso obesità o sovrappeso
Età esordio (anni) infanzia-adolescenza >35
Comparsa complicanze parecchi anni spesso presenti
croniche dopo l’esordio alla diagnosi
Prevalenza 0.6% 3-7
Familiarità modesta importante
Sistema HLA correlato non correlato
Autoimmunità presente assente
Terapia insulina dieta,
ipoglicemizzanti orali
Segni e sintomi da iperisulinismo sono: acanthosis nigricans, skin tags, dislipidemia, ipertensione,
obesità truncale. La resistenza periferica all’insulina rappresenta il primum movens e caratterizza
il T2DM.
L’insulino resitenza (IR) si definisce come una ridotta capacità dell’insulina a concentrazioni usuali
di promuovere l’utilizzo periferico del glucosio, sopprimere la produzione epatica di glucosio e
inibire la liberazione delle VLDL.

L’IR si caratterizza per valori di insulina a digiuno >15, durante l’OGTT oltre 150 e dopo due ore
all’OGTT di 75 μU/ml. L’HOMA è un indice affidabile per valutare l’IR secondo la formula:
glicemia basale (mmol/l) x Insulina basale (μU/ml)/22.5. Valori patologi sono considerati >2.5 nei
bambini e >4 negli adolescenti.
Bibliografia
1. Couper J, Donaghue K; International Society for Pediatric and Adolescent Diabetes (ISPAD).
Phases of diabetes. Pediatr Diabetes. 2007 ;8:44-7.
2. Vanelli M, Chiarelli F. Treatment of diabetic ketoacidosis in children and adolescents . Acta
Biomed 2003 ; 74:59-68.
3. Lorini R, Vanelli M. Normal values of first-phase insulin response to intravenous glucose in
healthy Italian children and adolescents. The Prediabetes Study Group of the Italian Society for
Pediatric Endocrinology and Diabetology (SIEDP). J Pediatr Endocrinol Metab. 1996 ;9 :163-7
4. Ten S, Maclaren N. Insulin resistance sindrome in children .JCEM 89; 2526-2539; 2004
5. Calcaterra V, Klersy C, Muratori T, Telli S, Caramagna C, Scaglia F, Cisternino M, Larizza D
Prevalence of metabolic syndrome (MS) in children and adolescents with varying degrees of
obesity. Clin Endocrinol. 2008 ;68:868-72.

Malattie della tiroide
Chirico V, Piraino B, Moschella E, Meduri S, Malvaso Barone C, Sturiale M, Munafò C,
Arrigo T
IPOTIROIDISMO
È una condizione clinica legata alla carenza degli ormoni della tiroide in relazione alle necessità
metaboliche dell'organismo.
La carenza di ormoni tiroidei può essere dovuta alla:
• insufficiente produzione ormonale
• assente liberazione ormonale
• ridotta o assente sensibilità periferica agli ormoni tiroidei
Ipotiroidismo congenito: segni e sintomi precoci
Neonato: Peso alla nascita aumentato; Postmaturità; Ritardata emissione del meconio e/o stipsi;
Ittero neonatale protratto; Distensione addominale e/o ernia ombelicale; Torpore e difficoltà alla
suzione; Pianto rauco; Fontanella posteriore > 0.5 cm; Cute marezzata.
Lattante: Mixedema; Cute marezzata e secca; Sella nasale appiattita; Macroglossia; Addome
batraciano; Psuedoipertrofia muscolare; Ampia fontanella anteriore; Ritardo psicomotorio;
suzione torpida; Letargia; Stipsi; Rallentamento della crescita.
Ipotiroidismo Congenito
Frequenza 1- 4000; Rapporto F:M 2.5 : 1;
Cause
• Agenesia ( 30-40 %)
• Ectopia ( 40-50 %)
• Ipoplasia ( 5 %)
• Difetti dell’ormonogenesi ( 10%)
• Insufficienza ipotalamo-ipofisaria ( 5 %)
Si può associare a : Malformazioni cardiache, multiple, a carico SNC (5.5 %)
Prima della dimissione dai reparti di maternità viene eseguito obbligatoriamente a tutti i neonati lo
screening neonatale. Il valore di TSH > 10μU/ml è patologico, al richiamo il neonato viene
sottoposto a prelievo anche degli ormoni tiroidei.
Prima dell’avvio del trattamento eseguire: ecografia tiroidea o scintigrafia tiroidea, RX nucleo
distale del femore, test di livello, inquadramento auxologico.

La terapia va avviata quanto prima possibile, alla posologia iniziale di L-T4 10-12 µ/Kg/die, dopo 1
mese dosare FT4 e TSH. Successivi controlli clinico-laboratoristici ed eventuali adattamenti
posologici andrebbero previsti ogni 3 mesi per tutto il 1° anno ed ogni 6 mesi successivamente con
l’obiettivo di mantenere la FT4 ai limiti alti della normalità ed il TSH ai limiti della dosabilità.
Tiroidite Cronica Autoimmune
È un’affezione cronica della tiroide a patogenesi autoimmunitaria, catterizzata da:
a) infiltrazione linfocitaria diffusa della tiroide
b) vario grado di gozzo o atrofia tiroidea
c) stato funzionale di eu-ipotiroidismo (raro iper)
La TC rappresenta la più frequente causa di gozzo e di ipotiroidismo in aree iodio-sufficienti. E’
rara prima dei 5 anni. Ha una prevalenza generale di 1.3 – 3 %, con picco di incidenza fra 10 e 18
anni, colpisce maggiormente il sesso femminile. Vi sono precedenti familiari di tireopatia nel 30 %
dei casi. Frequentemente vi è associazione con altre malattie autoimmuni e/o cromosomopatie.
La diagnosi si basa su storia familiare e personale, quadro clinico, dosaggio anticorpi antitiroide
(perossidasi, microsomi, TRAb), e ecografia tiroidea.
Criteri per la diagnosi
Almeno 2 dei seguenti 3 criteri:
1. Alterazioni ecografiche specifiche
2. Aumento degli anticorpi anti-perossidasi
3. Aumento di volume della tiroide
N.B. La funzione tiroidea può essere normale o variamente alterata (ipotiroidismo conclamato o
subclinico, perfino ipertiroidismo transitorio)
La terapia con LT4 a dosaggio di 1-2 μg/kg viene avviata in caso di gozzo e/o ipotiroidismo.
La terapia con LT4 determina una riduzione delle dimensioni del gozzo, ma non modifica né il
titolo anticorpale nè l’ecostruttura tiroidea.
Prognosi
50% guarigione completa
25% diminuita riserva tiroidea
25% danno permanente con ipotiroidismo
Il trattamento con LT4 non sembra aver influenzato il decorso dell’HT

Bibliografia
1. Dias VM, Campos AP, Chagas AJ, Silva RM. Congenital hypothyroidism: etiology. Pediatr
Endocrinol Metab. 2010 ;23:815-26.
2. Rastogi MV, LaFranchi SH. Congenital hypothyroidism.Orphanet J Rare Dis. 2010 10;5:17.
3. Brown RS. Autoimmune thyroid disease: unlocking a complex puzzle. Curr Opin Pediatr.
2009; 21:523-8.


















