
16. Struttura elettronica delle molecole e legame chimico · Energia elettrostatica elettrone...
43
16. Struttura elettronica delle molecole e legame chimico Data la notevole differenza di masse, la dinamica elettronica è molto più veloce del moto nucleare ⇒ nuclei immobili nell’analisi della struttura elettronica Approssimazione di Born-Oppenheimer: per fissate coordinate nucleari , , ,⋯ dei nuclei ,,,⋯ , funzione d’onda delle sole coordinate elettroniche 1 2 3 (, , , ) rr r Ψ ⋯ 1 2 3 1 2 3 ˆ (, , , ) (, , , ) H rr r E rr r Ψ = Ψ ⋯ ⋯ dall’equazione di Schroedinger indipendente dal tempo (soluzione stazionaria) con l’Hamiltoniano relativo al solo moto elettronico Coordinate degli elettroni: , , ,⋯ Coordinate dei nuclei ,,,⋯ : , , ,⋯ Funzione d’onda esatta: Ψ( , , ,⋯, , , ,⋯) 1
Transcript of 16. Struttura elettronica delle molecole e legame chimico · Energia elettrostatica elettrone...

16. Struttura elettronica delle molecole e legame chimico
Data la notevole differenza di masse, la dinamica elettronica è molto più veloce del
moto nucleare ⇒ nuclei immobili nell’analisi della struttura elettronica
Approssimazione di Born-Oppenheimer: per fissate coordinate nucleari
��, �� , �� , ⋯ dei nuclei , , �,⋯ , funzione d’onda delle sole coordinate
elettroniche 1 2 3( , , , )r r r� � �
⋯
1 2 3 1 2 3ˆ ( , , , ) ( , , , )H r r r E r r rΨ = Ψ� � � � � �
⋯ ⋯
dall’equazione di Schroedinger indipendente dal tempo (soluzione stazionaria)
con l’Hamiltoniano relativo al solo moto elettronico
Coordinate degli elettroni: � �, � �, � �, ⋯
Coordinate dei nuclei , , �,⋯ : ��, �� , �� , ⋯
Funzione d’onda esatta: Ψ(� �, � �, � �, ⋯, ��, �� , �� , ⋯ )
1

Esempio: molecola di idrogeno ��
HA HB
e1
e2
Hamiltoniano elettronico:
2 2 2 2 2 2 2 2
2 2 2 2 2 21 1 1 2 2 2
ˆ2 2e e
Hm mx y z x y z
∂ ∂ ∂ ∂ ∂ ∂= − + + − + + − ∂ ∂ ∂ ∂ ∂ ∂
ℏ ℏ Energia cinetica
degli elettroni
Energia elettrostatica
elettrone 1 - nuclei
2 2
0 01 1
1 14 4| | | |A B
e er R r Rπε πε
− − −− −� �� �
Energia elettrostatica
elettrone 2 - nuclei
2 2
0 02 2
1 14 4| | | |A B
e er R r Rπε πε
− − +− −� �� �
Energia elettrostatica
elettrone 1 – elettrone 2
2
0 2 1
1+
4 | |e
r rπε+
−� �
Energia elettrostatica
nucleo A – nucleo B
2
0
1+
4 | |B A
eR Rπε −� �
Hamiltoniano totale = Hamiltoniano elettronico + energia cinetica dei nuclei
2

Equazione di Schroedinger indipendente dal tempo per la funzione d’onda
elettronica Ψ(� �, � �)��Ψ � �, � � = �Ψ(� �, � �)
|Ψ � �, � � |�: densità di probabilità con l’elettrone 1 in posizione � � e l’elettrone
2 in posizione � �
L’Hamiltoniano elettronico �� dipende dalle coordinate nucleari!
⇒ L’equazione di Schroedinger è risolta per fissate coordinate nucleari
⇒ La funzione d’onda dipende (parametricamente) dalle coordinate nucleari
⇒ L’energia � dipende (parametricamente) dalla distanza internucleare �
2 2 2, , , , , ,| | ( ) ( ) ( )B A B x A x B y A y B z A zR R R R R R R R R= − = − + − + −
� �
La dipendenza parametrica dalle coordinate nucleari è lasciata implicita.
3

4
Non esistono soluzioni analitiche dell’equazione di Scroedinger per ��
Approssimazione di campo medio (Hartree-Fock) come per gli atomi poli-
elettronici: ciascun elettrone viene descritto da uno spin-orbitale,
ma con orbitali molecolari: si estendono nello spazio attorni ad ambedue i nuclei!
Approssimazione LCAO (Combinazione Lineare di Orbitali Atomici) quale procedura
semplificata per la costruzione degli orbitali atomici.
Per la descrizione degli stati elettronici a bassa energia: orbitali molecolari risultanti
dalla combinazione lineare dei due orbitali atomici 1�: 1 1( ), ( )A Bs sr rϕ ϕ� �
Orbitale di anti-legame 1��∗: combinazione anti-simmetrica
1 * 1 1( ) ( ) ( )A Bs s sr r rσϕ ϕ ϕ∝ −
� � �
Orbitale di legame 1��: combinazione simmetrica
1 1 1( ) ( ) ( )A Bs s sr r rσϕ ϕ ϕ∝ +
� � �

5
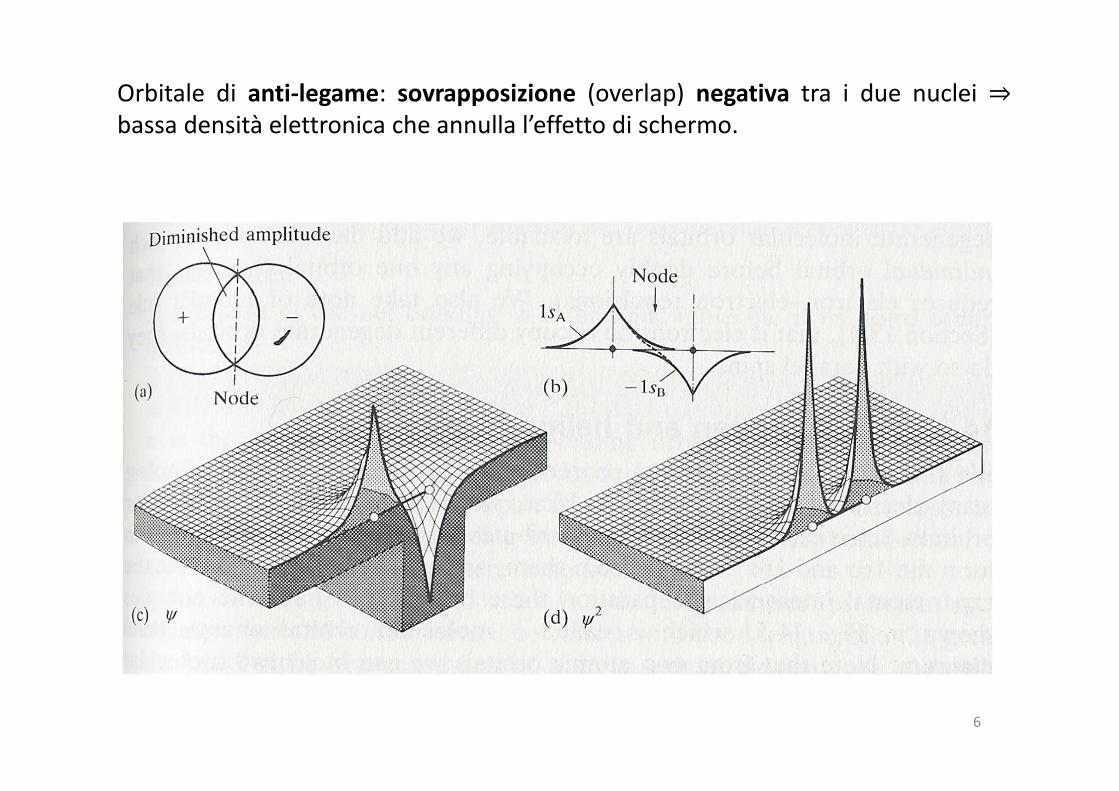
Orbitale di legame: sovrapposizione (overlap) positiva tra i due nuclei ⇒ aumento
della densità elettronica che riduce l’interazione repulsiva tra i due nuclei (effetto
di schermo).
6
Orbitale di anti-legame: sovrapposizione (overlap) negativa tra i due nuclei ⇒bassa densità elettronica che annulla l’effetto di schermo.

7
Significato del simbolo �: orbitali molecolari 1�� e 1��∗ sono simmetrici
(invarianti) rispetto a rotazioni attorno all’asse di legame
Orbitale di legame 1��
�
Orbitale di anti-legame 1��∗ � �

8
Energia degli orbitali 1�� (legame) e
1��∗ (anti-legame) dipendente
parametricamente dalla distanza
internucleare �

9
Lo stato fondamentale di ��: 2 elettroni nell’orbitale di legame con spin opposto
(cioé due spin-orbitali con lo stesso orbitale 1�� e spin opposto)
Energia della molecola di �� nello stato
fondamentale come funzione della
distanza internucleare � → energia
potenziale per l’Hamiltoniano vibrazionale
Dal minimo della curva di energia:
distanza di legame Re=0.075 nm e
minimo dell’energia De=436 kJ/mol
Rappresentazione secondo Lewis (doppietti di legame) di ��:
H H H H⇒ −i i

10
Perché non esiste la molecola �!�?
L’energia di �!� è maggiore
degli atomi di �! separati
Orbitali molecolari LCAO per molecole biatomiche omo-nucleari:
Gli orbitali atomici interni (occupati dagli elettroni di core) non sono modificati
poichè non c’è sovrapposizione
Orbitali molecolari di legame (combinazione simmetrica) e di anti-legame
(combinazione antisimmetrica) da orbitali atomici del guscio di valenza
∎ con la stessa energia (2�� con 2��, 2$� con 2$�)
∎ con la stessa simmetria
Simmetria assiale �: invariante rispetto a rotazioni attorno all’asse di legame
Simmetria planare π: invariante rispetto a riflessione attraverso un piano
contenente l’asse di legame

11
Orbitali molecolari dal secondo guscio atomico
La configurazione elettronica dello stato fondamentale secondo il principio di
aufbau usando gli orbitali molecolari

12
Esempio: &�
Ordine di legame≔�
�( � (∗
con ( ((∗) = numero di elettroni in
orbitali di legame (anti-legame)
2 3:[ ]2 2
Lewis: IN NI IN NI
Ordine di legame = 3 (triplo legame)
945 kJ/mole
N He s p
D
⇒ ≡
=
⋮ ⋮
2 4:[ ]2 2
Lewis: : :
Ordine di legame = 2 (doppio legame)
497 kJ/mole
O He s p
O O O O
D
⇒ =
=
2O
2F2 5
:[ ]2 2
Lewis: I I I I
Ordine di legame = 1 (legame singolo)
155 kJ/mole
O He s p
F F F F
D
⋅ ⋅ ⇒ −
=

13
Gli orbitali molecolari LCAO da combinazioni lineari di orbitali atomici con energiesimili, però con pesi diversi
Molecole biatomiche etero-nucleari: gli orbitali atomici dei due atomi hanno
energie diverse!
Esempio di HF: combinazione con pesi diversi dell’orbitale 1s di H e dell’orbitale
2p di F per generare una coppia di orbitali molecolari di legame ed antilegame
antilegameimmetria σNell’orbitale di legame di HF prevale il
contributo dell'orbitale atomico 2p di F.
Il baricentro degli elettroni di legame è spostato verso F ⇒cariche parziali
+δq su H e -δq su F
� � )�*+ �*+
Momento di dipolo molecolare , � �-*+
� � ),

14
Elettronegatività . degli atomi: loro capacità di attrarre gli elettroni di legame.
Esistono diverse parametrizzazioni di elettronegatività: Pauling, Mulliken, etc.
Mulliken: . = 0.187 3 � �-4 �0.17 3, �-4 in eV

15
Compound Bond
Length (Å)
Electronegativity
Difference
Dipole
Moment (D)
HF 0.92 1.9 1.82
HCl 1.27 0.9 1.08
HBr 1.41 0.7 0.82
HI 1.61 0.4 0.44
Esiste una correlazione tra differenza di elettronegatività degli atomi e momento
di dipolo molecolare
D (Debye): unità di misura CGS del momento di dipolo, 1D = 3.33564×10−30 C·m
Importanza dei momenti di dipolo molecolari nel controllare le interazioni
intermolecolari nelle fasi condensate.

16
Insieme di distanze di legame ed angoli di legame per la descrizione geometrica di
molecole poliatomiche.
Esempio dell’acqua H2O
2 2:1 :[ ]2 2 2 2
Lewis:
x y zH s O He s p p p
H H
O
i ii i
H H
O
ϑ
1R
2R
1 2Coordinate interne: , ,R R ϑ
Quale angolo di legame?
Molecole poliatomiche

17
• Previsione secondo la procedura VSEPR (Valence Shell Electron Pair Repulsion)
– Con riferimento alla struttura di Lewis, si considerano come
equivalenti le coppie di elettroni di legame e di non legame
– le coppie di elettroni si respingono e tendono a disporsi alla massima
distanza possibile
– i nuclei si dispongono di conseguenza
Per l’acqua (quattro doppietti) si predice
una struttura tetraedrica, con un angolo
di legame di 109.5°, molto vicino al
valore reale, di circa 107°

18
Trattazione quanto-meccanico via costruzione degli orbitali molecolari per combinazione lineare (LCAO) di coppie di orbitali atomici.
Geometria molecolare sbagliata se si utilizzano gli orbitali atomici s e p!
O H
H
1 ( )s H
2 ( )xp O 1 ( )s H
2 ( )yp O
+-+
+
+
-90 !ϑ = °
Per rimediare: utilizzo di orbitali atomici ibridi nella costruzione degli orbitali molecolari.
Orbitali atomici ibridi: combinazioni lineari di orbitali atomici (di uno stesso atomo!) ad energia similare (orbitali s e p dello stesso guscio)

19
Possibili orbitali ibridi del secondo guscio: 1)�$, 2��$�, 3��$�
1) Ibridizzazione �$: combinazione tra 1 orbitale � ed 1 orbitale $ che generano
2 orbitali ibridi 6� e 6� (rimangono 2 orbitali $ puri, cioè non ibridizzati)
1 :h s p= ++
+- - +
2 :h s p= −+
+ -
-+Nota: i piccoli lobi negativi portano un contributo trascurabile all’orbitale molecolare

20
2) Ibridizzazione �$�: combinazione tra 1 orbitale � ed 2 orbitali $ che generano 3orbitali ibridi �$� (rimane 1 orbitale $ puro)
Configurazione trigonale piana con un angolo di 120°

21
3) Ibridizzazione �$�: combinazione tra 1 orbitale � ed 3 orbitali $ che generano 4orbitali ibridi �$� (non rimane nessun orbitale $ puro)
Configurazione tetraedica con
un angolo di 109.5°

22
Esempi di orbitali molecolari derivanti da orbitali atomici ibridi �$�: metano,
ammoniaca e acqua2 2
4Metano :[ ]2 2CH C He s p
H
Hii
iiCLewis: HH iii i
H
H
C HH
Nota: con gli stessi orbitali atomici si formano
anche i 4 corrispondenti orbitali di anti-legame,
che però non hanno alcun ruolo non essendo
occupati (almeno nello stato fondamentale).
4 orbitali molecolari di legame tra i 4 orbitali ibridi
2�$� del Carbonio e gli orbitali 1� dei 4 Idrogeni,
occupati da 4 coppie di elettroni, e che
determinano una struttura geometrica tetraedica

23
2 33Ammoniaca : [ ]2 2NH N He s p
H
Hii
iiNLewis: Hiiii
H
H
N H
3 doppietti di legame dalla combinazione di 1s degli idrogeni
e 2sp3 dell‘azoto, e un doppietto di non legame sull'ultimo
2sp3 dell‘azoto): ha una struttura a piramide trigonale
2 42Acqua: :[ ]2 2
Lewis:
H O O He s p
H H
O
i ii i
H H
O
2 doppietti di legame dalla combinazione di 1s
degli idrogeni e 2sp3 dell‘azoto, e due doppietti di
non legame sui rimanenti 2sp3 dell‘ossigeno:
angolo di legame previsto di 109.5°
Nota: anche con orbitali molecolari derivanti da
orbitali atomici ibridi si genera un momento di dipolo
se gli atomi hanno diversa elettronegatività.

24
Esempi di orbitali molecolari derivanti da orbitali atomici ibridi �$�: etilene, e
gruppi carbonilici in aldeidi e chetoni
Etilene: C C
H
H H
HPer ciascun C: 3 orbitali 2�$� → 3 orbitali molecolari �
1 orbitale 2$ → 1 orbitale molecolare 7
Implicazione: l’etilene è planare, altrimenti non si avrebbe il legame 7
Aldeide formica: C O
H
H C: 3 orbitali 2�$� → 3 orbitali molecolari �1 orbitale 2$ → 1 orbitale molecolare 7
O: 1 orbitali 2�$� → 1 orbitali molecolari �2 orbitali 2�$� → 2 doppietti di non legame
1 orbitale 2$ → 1 orbitale molecolare 7L’aldeide formica è una
molecola planare!

25
Esempio di orbitali molecolari derivanti da orbitali atomici ibridi �$: acetilene
C C HH
Implicazione: l’acetilene è una molecola lineare!

26
Coniugazione molecolare nel caso di strutture 7 estese: orbitali molecolari
distribuiti su più di due atomi
Caso del butadiene:
geometria planare!
Caso del legame peptidico R-CO-NH-R‘:
ibridizzazione �$� per N e coniugazione tra
l’orbitale $8 di N e l’orbitale molecolare 7di CO → struttura planare di -CO-NH-

27
Spettroscopia elettronica (UV-VIS): transizioni di un elettrone tra due orbitali molecolari. Transizioni a minor energia: HOMO → LUMO
HOMO = Highest Occupied Molecular Orbital
LUMO = Lowest Unoccupied Molecular Orbital
HOMO
LUMOhνννν
Energia
Assorbimento di un
fotone UV-VIS da parte
di una molecola:
promozione di un
elettrone dall’ HOMO al
LUMO.

28
Data una configurazione elettronica con ogni elettrone avente un spin 9: = � 1 2⁄o 9: = 1 2⁄ , risulta definito lo spin elettronico totale <: come somma degli spin
dei singoli elettroni.
Molteplicità di spin (Singoletto, Doppietto, Tripletto, ….): numero di stati degeneri
corrispondenti a valori diversi dello spin totale <:
Esempi: �!:1�� <: =�
�−�
�= 0 Singoletto
=>:1��2� <: =1
2−1
2+1
2=1
2
<: =1
2−1
2−1
2= −
1
2
Doppietto
Stato fondamentale di molecole organiche non radicaliche: Singoletto ?@orbitali occupati da due elettroni con spin opposto →<: = 0
Transizione HOMO → LUMO: Singoletto ?� e Tripletto A� con energia diversa
LUMO
HOMO
LUMO
HOMO
<: = 1
Uno degli stati di Tripletto<: = 0
Stato di Singoletto

292929
Regola di selezione per le transizioni elettroniche :
?@ → ?B
LUMO
HOMO
LUMO
HOMO
hνννν
S0S1 S2
L’assorbimento di radiazione non può provocare la transizione da da Singoletto a
Tripletto, poiché non cambia lo stato di spin degli elettroni.
In ogni stato elettronico, sia fondamentale che eccitato, esistono diversi livelli
vibrazionali. Quindi le transizioni elettroniche sono accompagnate da transizioni
vibrazionali (transizioni vibroniche o vibro-elettroniche)
r0
Energia
Molecola biatomica
0S
1S
Molecola generica
Stato Fondamentale S0
Stato Eccitato S1
νννν0 νννν1 νννν2

30
Spettro di assorbimento come somma di
diverse bande: transizioni dallo stato
fondamentale a diversi livelli vibrazionali
dello stato eccitato.
νννν0νννν1νννν2
Però struttura vibrazionale risolta solo
se le bande vibrazionali sono
sufficientemente strette.
Spettro di assorbimento con una unica banda
per una data transizione elettronica, se le
componenti vibrazionali hanno bande molto
larghe.
Orbitali molecolari localizzati in parte della molecola ⇒ Le transizioni
elettroniche coinvolgono solo parti della molecola, detti gruppi cromofori
� σ σ* (es: composti saturi)
� π π* (es: alcheni, alchini, aromatici)
� n π* (es: composti con N, S, O)
→
→
→

31
Gran parte delle sostanze hanno transizioni elettroniche nell’ultravioletto, e quindi
appaiono trasparenti nel visibile (caso dell’acqua, o dell’acetone).
In caso di doppi legami coniugati o sistemi aromatici l’assorbimento si sposta verso
lunghezze d’onda maggiori. Se si passa da assorbimento dall’UV ad assorbimento nel
visibile si ha una sostanza colorata (es: caroteni, clorofille)
β-carotene
200 300 400 500 600 700-20000
0
20000
40000
60000
80000
100000
120000
140000
160000
ε (l
mol
-1 c
m-1)
Lunghezza d'onda (nm)
Spettro di assorbimento del �-carotene
0 1S S→0 2S S→
323232
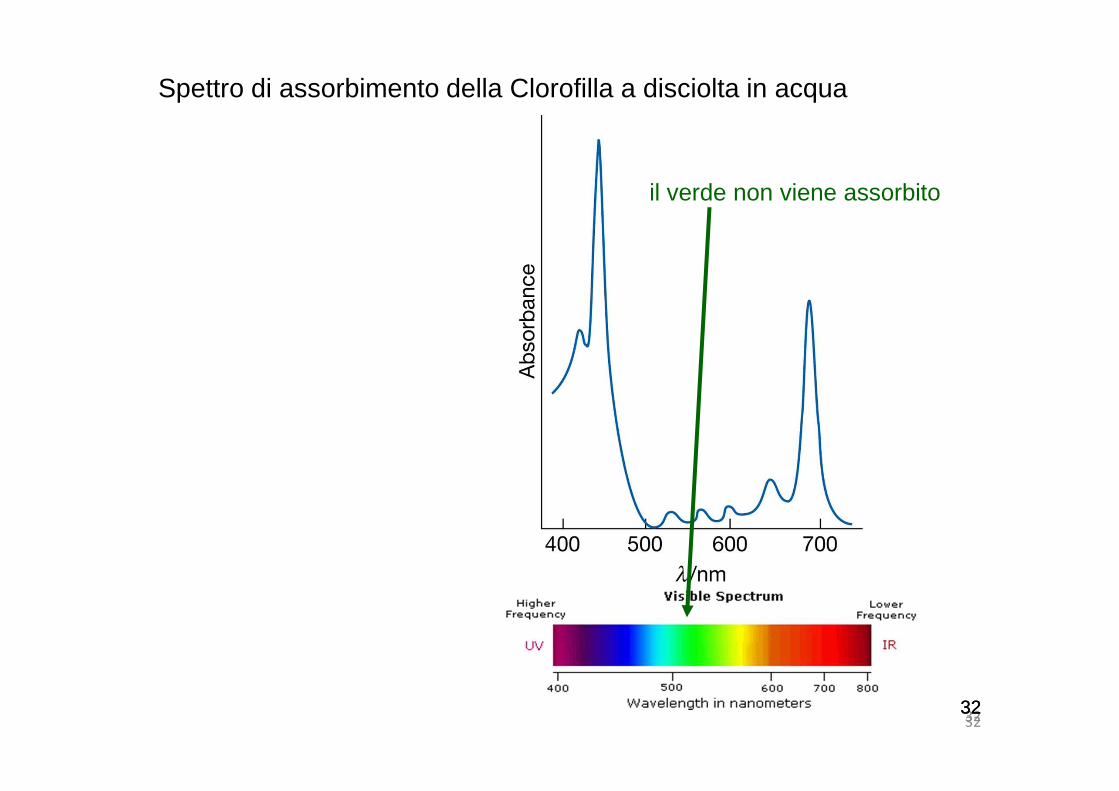
Spettro di assorbimento della Clorofilla a disciolta in acqua
32
il verde non viene assorbito

33
Spettri di assorbimento UV dei principali amminoacidi aromatici (non assorbano
nel visibile!)
Triptofano (Trp)
Fenilalanina (Phe)
Tirosina (Tyr)ε280 (Trp) = 5690 litro / mol cmε280 (Tyr) = 1280 litro / mol cmε280 (Phe) = 160 litro / mol cm

34
Misure di assorbanza su un soluto disciolto in un solvente che non assorbe radiazione: quale dipendenza dalla concentrazione C del soluto?
D: percorso ottico = distanza percorsa dalla radiazione nel campione
(normalmente misurato in cm)
Legge di Lambert-Beer: E = F E DC
C: concentrazione (moli/litro)
F(E): coefficiente di estinzione molare (litro/moli cm), dipende
dalla lunghezza d’onda, ma non dalla concentrazione (o da D)
Nota: se le concentrazioni sono espresse come 9GD C9�⁄ , allora F(E)ha dimensioni C9� 9GD⁄

353535
campione0I
x0 l
x
)(xI
0
0Idx
xdI )(dx
xdI )(− : tasso di diminuzione (nello spazio)del numero di fotoni
)()(
xKcIdx
xdI =− K: costante di prop.
dx
xId
dx
xdI
xIKc
)(ln)(
)(
1 ==−
KcxIbxaxI −=+= 0ln)(ln
Per :lx = 00 0
0 010
ln ( ) ln ln ln ( ) ln( )
1log ln
( ) 2.3 ( ) 2.3
II l I Kcl Kcl I I l
I l
I I KA cl
I l I l
= − = − =
= = =
ε

36
Evoluzione degli stati eccitati dopo l’assorbimento di radiazione
Dagli stati S2,S3,.. si ha un rapido
decadimento non radioattivo allo stato S1
in tempi di circa 10H�� s
IC = Internal Conversion: energia in
eccesso dissipata come energia
termica del solvente circostante.S0
S1
S2
S3
hν
IC
energia

37
Lo stato S1 ha tempi di vita variabili ma tipicamente di nanosecondi, e può
decadere secondo diversi processi:
1) Decadimento IC allo stato fondamentale S0,
senza emissione di radiazione (“decadimento non radiativo”).
S0
S1
S2
S3
energia
IC

38
2) Il decadimento ad S0 con emissione di
luce: Fluorescenza.
S0
S1
S2
S3
energia
F

39
S0
S1
S2
S3
energia
3) La conversione ISC (Inter System
Crossing) ad uno stato di tripletto T1. Lo
stato T1 a sua volta può decadere non
radiativamente ad S0 o con emissione di
un fotone, processo detto:
Fosforescenza (P).
I tempi di vita di T1 sono molto lunghi (da
microsecondi a secondi o minuti) a causa
della transizione proibita
T1
ISC
P

404040
Rappresentazione schematica degli stati e dei processi coinvolti nei processi di
assorbimento / emissione / decadimento
Diagramma di Jablonski
A FP
IC
IC
S0
S1
S2
S3
T1
ISC

414141
Fluorescenza
A
S0
S1
S2
S3
La fluorescenza avviene sempre a lunghezze
d’onda uguali o maggiori dell’assorbimento.Si
può osservare struttura vibronica anche nella
fluorescenza

42
Si definisce la resa quantica di fluorescenza come il rapporto tra il numero di
fotoni emessi ed il numero di fotoni assorbiti
è un numero compreso tra 0 e 1assorbiti fotoni .
emessi fotoni .
nr
nrfl =Φ
H2O, pH = 7
I principali fluorofori biologici intrinseci sono gli amminoacidi aromatici, in
particolare tirosina e triptofano.
Le basi degli acidi nucleici sono pochissimo fluorescenti. Sono fluorescenti
alcuni coenzimi come NADH, FAD e FMN.

434343
Sorgente
Campione
Schema di una spettrometro di fluorescenza UV-VIS (fluorimetro)
Monocromatore di eccitazione (prisma, reticolo)
Rivelatore di intensità luminosa (tubo fotomoltiplicatore)
Monocromatore di emissione(prisma, reticolo)






![PROBLEMA Gruppo 1 Elettrostatica...PROBLEMA Gruppo 1 Elettrostatica 1. Data la famiglia di funzioni = − ln 1− definite nell’intervallo [-20;20], determina i valori dei due parametri](https://static.fdocumenti.com/doc/165x107/60aaa586efbbe27dcf39d2de/problema-gruppo-1-problema-gruppo-1-elettrostatica-1-data-la-famiglia-di-funzioni.jpg)











