· di emoglobina fetale” (HPFH) oltre 700 varianti Hb sono state identificate, ma solo 3 ......
79
LE TALASSEMIE www.fisiokinesiterapia.biz
Transcript of · di emoglobina fetale” (HPFH) oltre 700 varianti Hb sono state identificate, ma solo 3 ......

LE TALASSEMIE
www.fisiokinesiterapia.biz

circa il 7% della popolazione mondiale è portatore didiverse malattie ereditarie dell’emoglibina, rendendolele più comuni malattie monogeniche in assoluto
2 gruppi• varianti strutturali dell’emoglobina• talassemie, difetti di sintesi delle catene globiniche
3° gruppo: difetto della normale accensione delladella produzione di emoglobine da fetali adadulte, chiamato “persistenza ereditariadi emoglobina fetale” (HPFH)





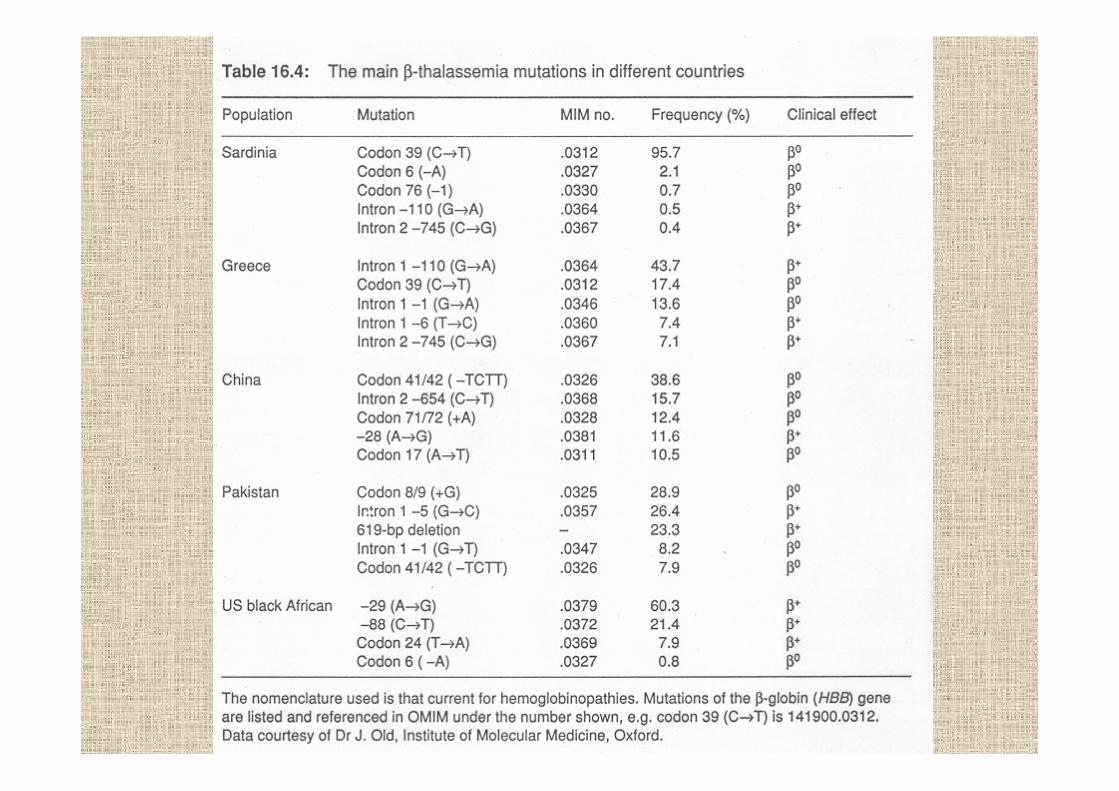
oltre 700 varianti Hb sono state identificate, ma solo 3sono presenti ad alta frequenza in popolazioni differenti
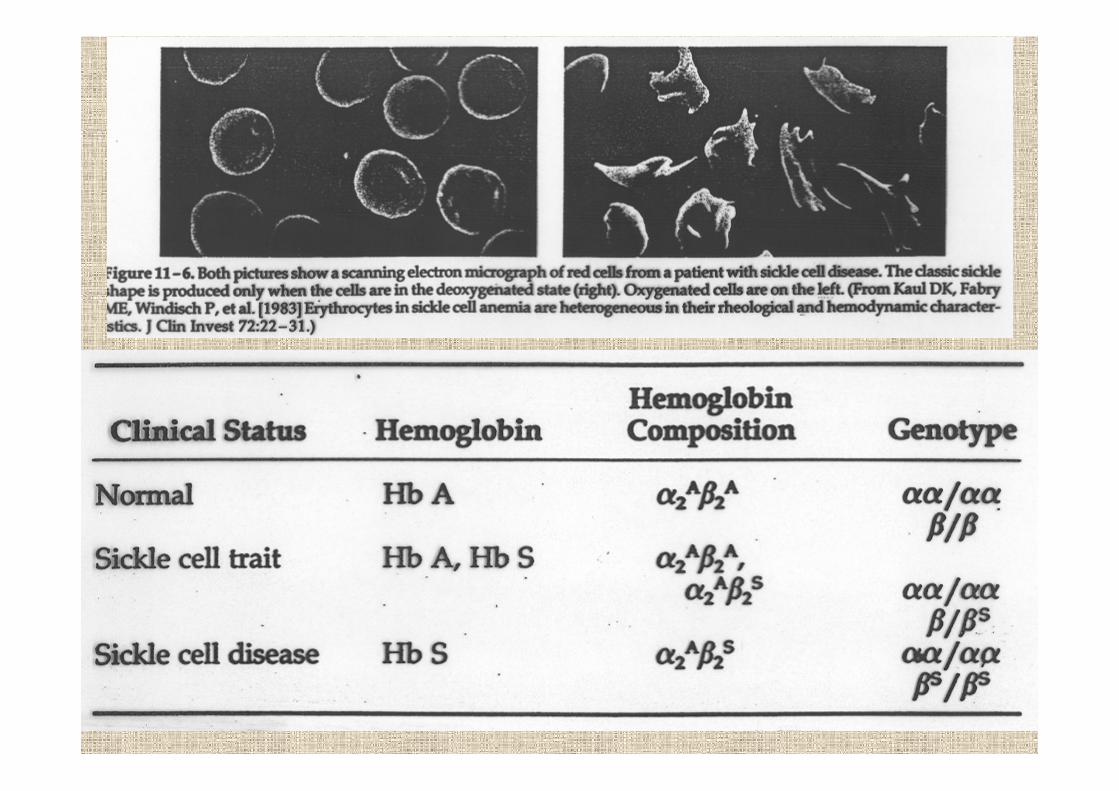
HbS Africa sotto il Saharaparte del Mediterraneomedio-orienteparte dell’India
HbC solo Africa ocidentaleparte del Mediterraneo
HbE IndiaAsia sud-est

NB alcune varianti strutturali sono sintetizzate a tassiridotti o sono altamente instabili producendo unfenotipo talassemico
es: beta-codon 26 GAG AAG HbEsi attiva un sito di splicing criptico che causa unprocessamento di mRNA anomalo che produce un fenotipo beta-tal. lieve

le talassemie sono classificate in:
α-tal β-tal δβ-tal εγδβ−tal
sulla base del difetto di sintesi
N.B. ciascuna di queste forme è estremamenteeterogenea
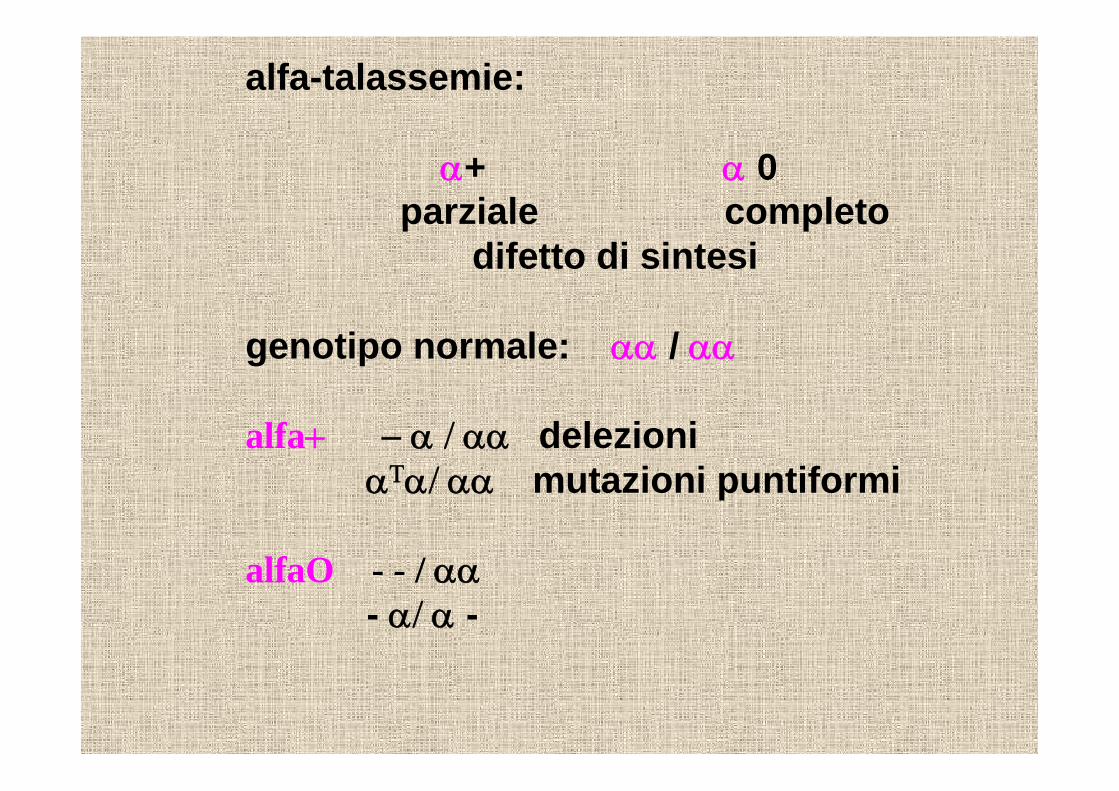
alfa-talassemie:
α+ α 0parziale completo
difetto di sintesi
genotipo normale: αα / αα
alfa+ − α / αα delezioni αΤα/ αα mutazioni puntiformi
alfaO - - / αα- α/ α -

beta talassemie:
beta O no produzione di catene beta
beta + riduzione grave di catene beta
beta ++ riduzione lieve di catene beta
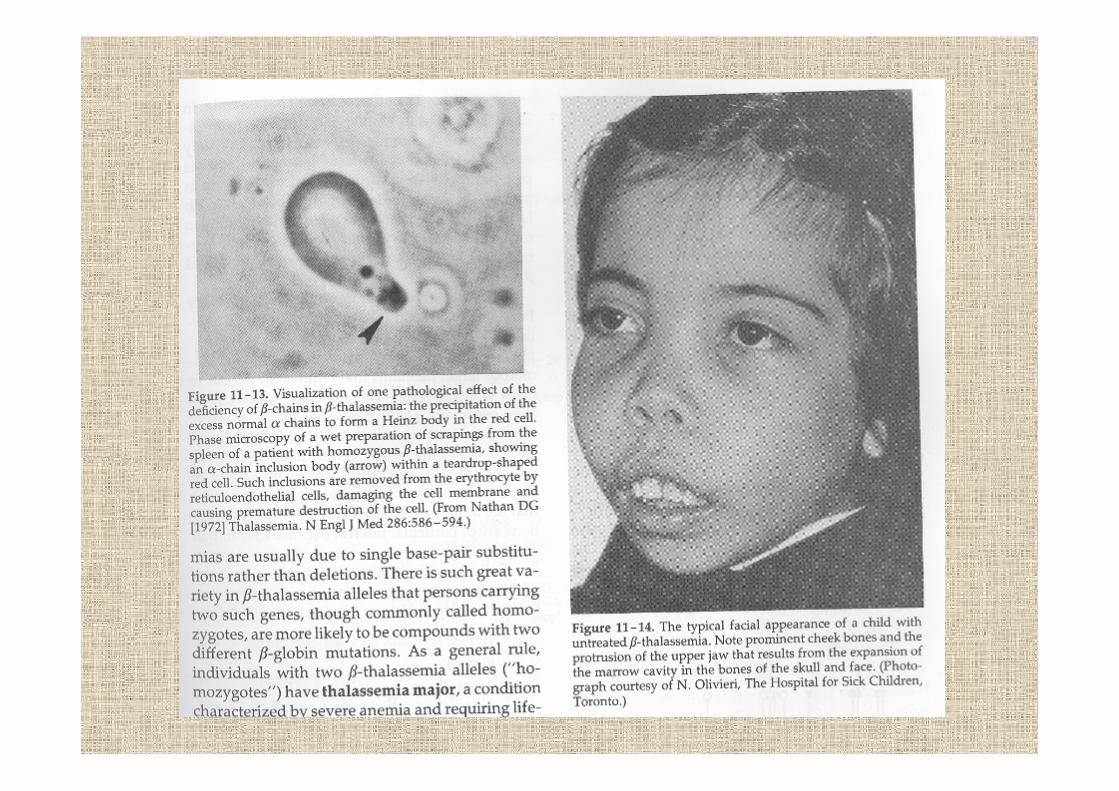

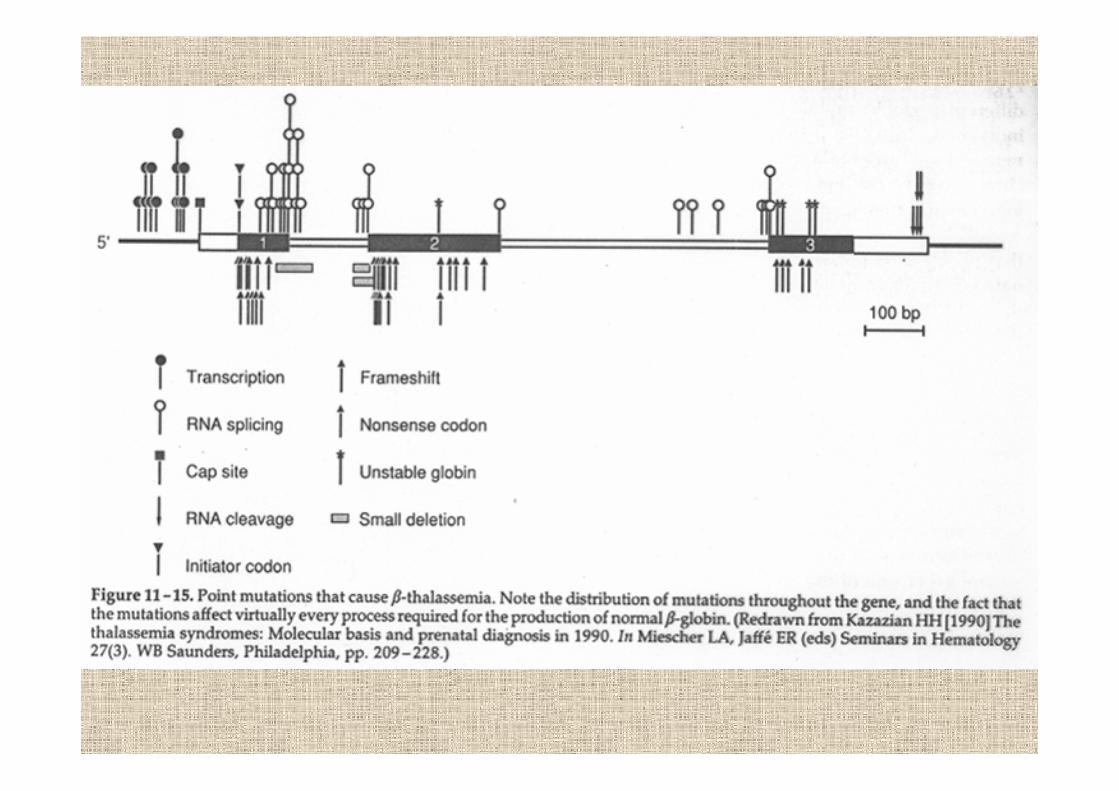
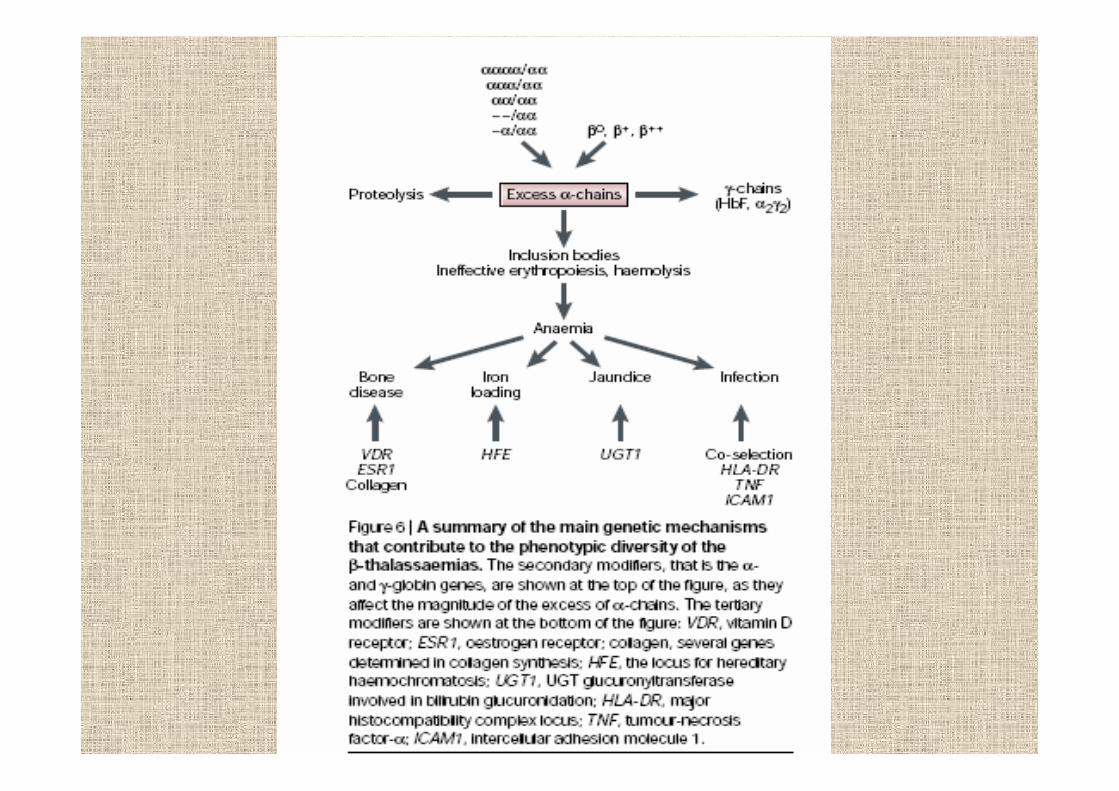
in beta-talassemia mancano catene beta e comeconseguenza aumentano le alfa
aggregazione di alfa nei precursori
maturazione cellulare anomala
prematura distruzione nel midollo osseo
anemia
la gravità delle beta-talassemie dipende dallosbilancio alfa/beta
la gravità dell’anemia influenzerà la gravitàdi splenomegalia,osteoporosi,
sistema endocrino,cuore

condizione clinica N°di geni genotipo %prodα funz.




eterozigoti beta-tal possono essere
silentilievi
severi quanto gli omozigoti forma dominante
tal maior più gravetal minor più lieve
fattori ambientali influenzano il fenotipoetà malnutrizione infezioni ricorrenti (popolazioni povere)


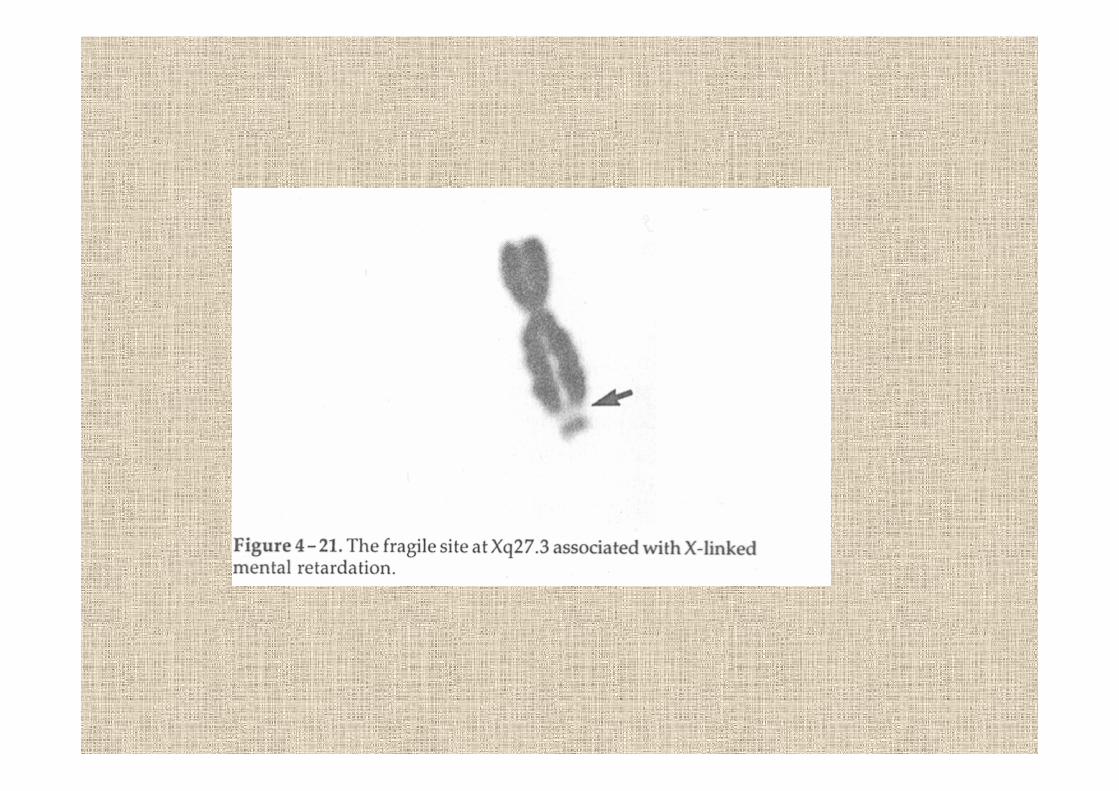
la sindrome del sito fragile del cromosoma XFRAXA
Lubs nel 1969 scopre il sito fragile e lo associaal ritardo mentale1992 identificazione del gene FMR1 responsabile
caratteristiche : faccia lunga triangolare, orecchie a sventolamacroorchidismoRM medio-moderatocapacità del linguaggio gravemente compromessacomportamento autisticosorriso
nelle femmine portatrici gli aspetti sono sfumati

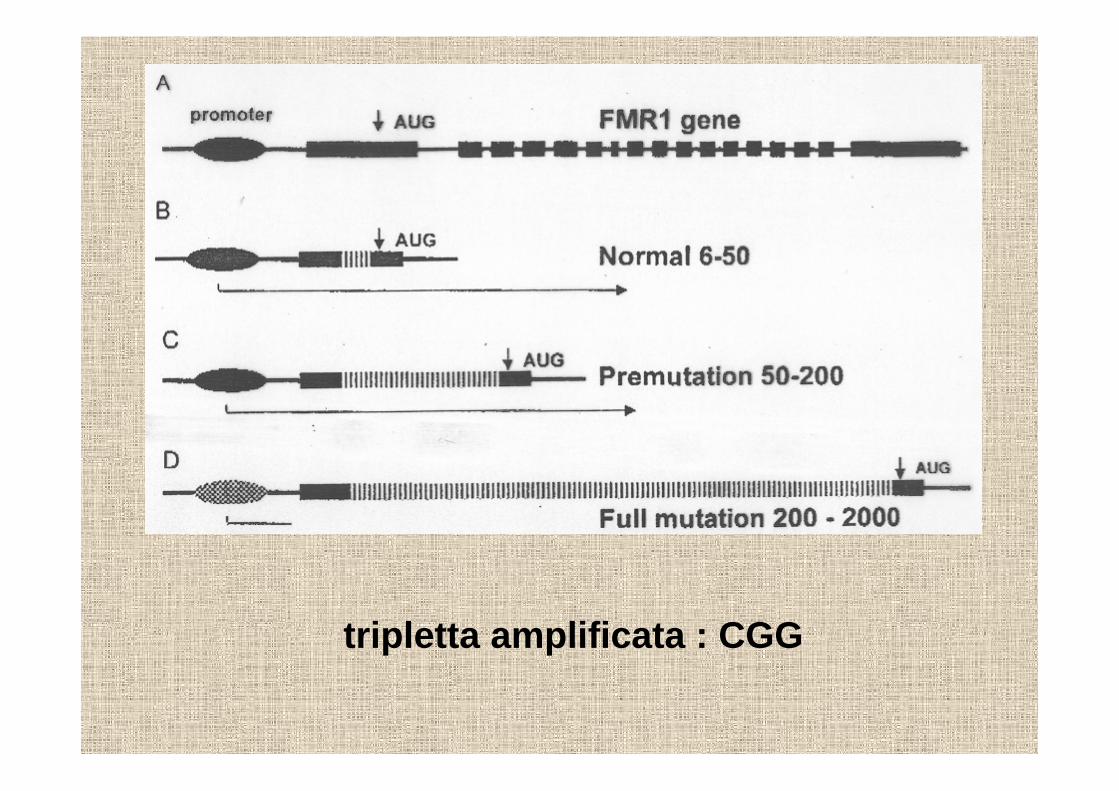
tripletta amplificata : CGG


……
ecc…

ecc…

ecc…


DISTROFIE MUSCOLARI
• le distrofie muscolari sono malattie miogenicheereditarie caratterizzate da “perdita” e debolezza muscolare di gravità e distribuzione variabile
• in numerose forme, il cuore può essere severamentecompromesso, qualche volta anche in assenza di debolezza significativa dal punto di vista clinico
• sulla base della distribuzione della debolezza muscolare più importante, si possono delineare 6 forme più la distrofia muscolare congenita




si osservano spesso variazioni intra- e inter-familiari
LMNA EDMDcardiomiopatia dilatativa con difetti diconduzione ma senza coinvolgimentomuscolarelimb-girdle(AD)1Blipodistrofia parziale di Dunnigan
queste malattie si possono definire alleliche e sono dovutea difetti di assemblaggio della lamina nucleare
si possono osservare variazioni fenotipiche nella stessa famiglia dove segrega la stessa mutazione


la distrofia muscolare di Duchenne (DMD)
è la più frequente e più conosciuta tra le malattie muscolari degenerative dell’infanzia
incidenza 1: 3 500 maschi nati vivi
si trasmette come gene recessivo legato all’Xi maschi sono affetti perché emizigoti e ricevonodalla madre sana il cromosoma X mutato
occasionalmente la malattia si osserva anche nelle donne :45,X s. di Turner o lyonizzazione sfavorevole

DMD:
• esordio tra i 2 e i 5 anni caratterizzato da andaturaanserina, frequenti cadute e difficoltà a salire lescale
• segno di Gowers positivo
• atrofia progressiva prima dei muscoli prossimali degli arti inferiori e poi prossimale degli arti superiori e di seguito sono colpiti i muscoli respiratori
• cardiomiopatia nel 95% dei casi negli ultimi anni divita


DMDdecorso variabile
12 anni circa perdita della deambulazione autonoma20 anni circa decesso
N.B. alto tasso di mutazione de novocirca 1/3 dei casi sporadiciX-linked recessiva
30% dei casi circa hanno QI<75 non progressivo
mosaicismo gonadico


la distrofia muscolare di Becker
è considerata una malattia allelica di DMDè una forma più benigna
l’esordio è più tardivo (12 anni) e il decorso più lentoetà media alla perdita di deamb.:27 annietà media exitus: 42 anni
il cuore è meno frequentemente compromessoQI nella norma

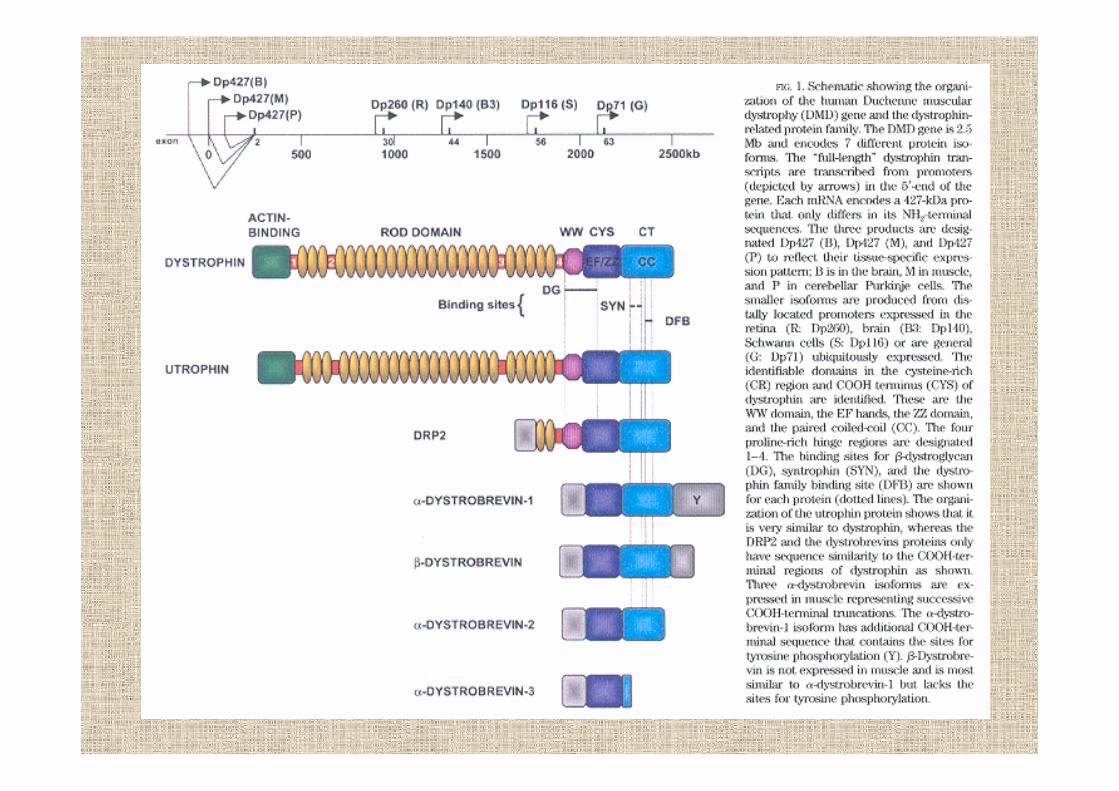
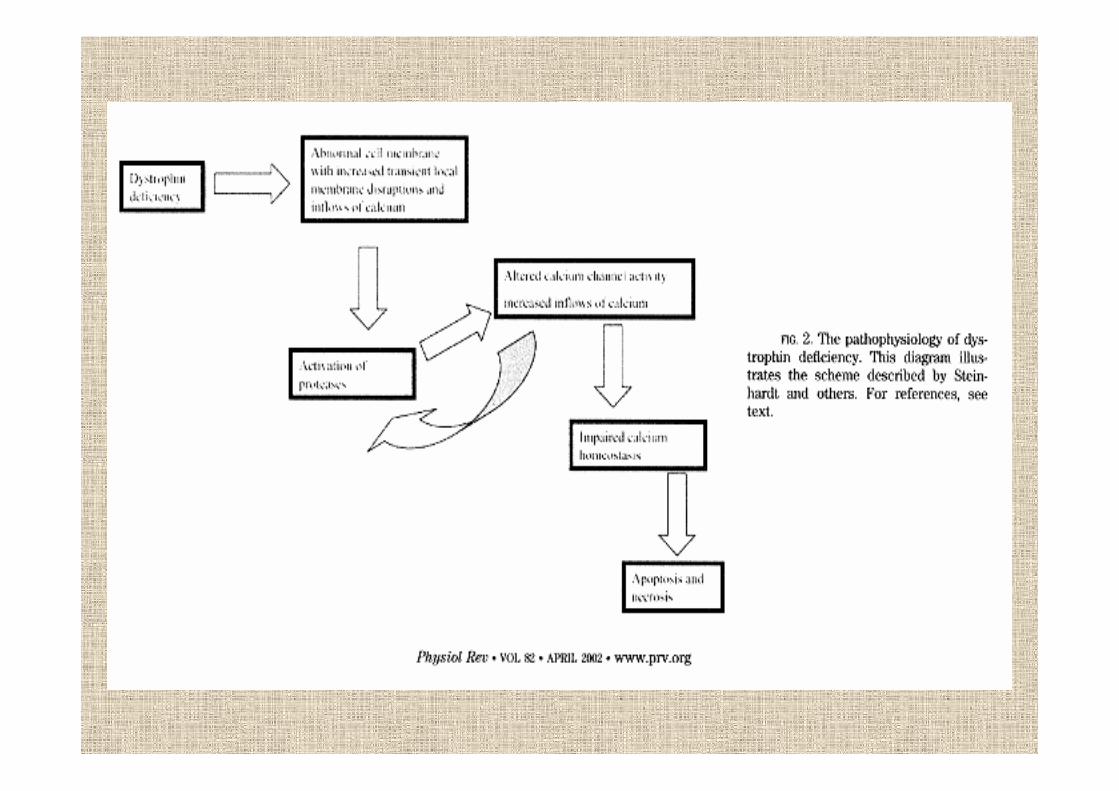
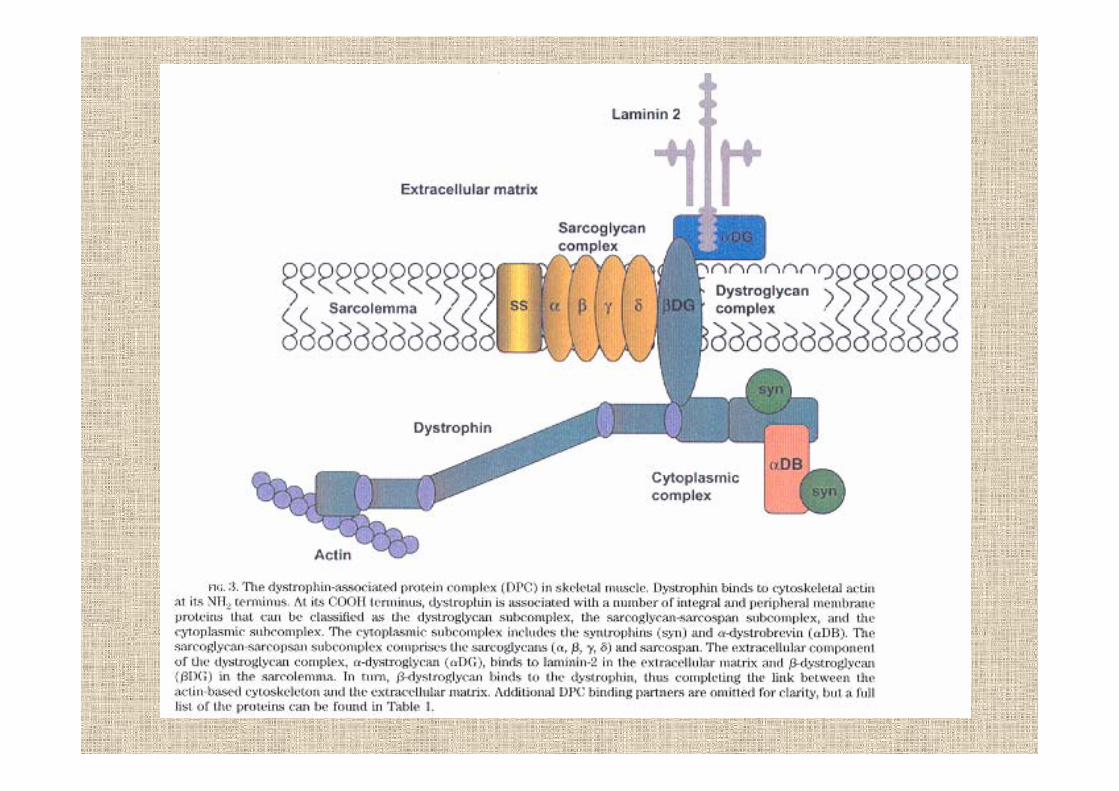
distrofina
espressa soprattutto nel muscolo scheletrico e in piccole quantità nel cervello

gli mRNA a piena lunghezza sono controllati da 3 siti diversinello stesso promotore e sono regolati in modo indipendente
DpB cervello neuroni corticali e ippocampoDpM muscolo e cardiomiocitiDpP cellule di Purkinje e cervelletto
lungo il gene ci sono altri 4 siti-promotore

circa il 65% delle mutazioni DMD e BMD sono grossedelezioni del gene che portano alla totale assenza o aridottissime quantità di distrofina
mutazioni diverse per dimensioni e posizioni
stesso fenotipo
delezioni con scivolamento di lettura del codice
DMD
delezioni con lettura corretta
BMD

circa un terzo delle mutazioni sono piccole delezionio mutazioni puntiformi stop codon distribuite su tutto il gene
la perdita del dominio ROD avvicina i due domini contigui ma non sembra colpire la funzionalità dellaproteina
mutazioni nel DpM aboliscono selettivamente l’espressione nel cuore







sindrome di Steinert o distrofia miotonica
ad insorgenza tardiva (metà della vita)
i sintomi sono evidenti dopo la 3° decade ma alcuni segni possono evidenziarsi già nella 2° decade
DM inizialmente coinvolge i muscoli distalidelle estremità e solo più tardi la muscolatura prossimale
precoce coinvolgimento della testa e del colloptosi e limitazione dei movimenti extraocularifacies caratterizzata da atrofia dei masseteri, degli sterno-cleidomastoidei e dei muscoli temporali
fronte aggrottata assotigliamento dellacalvizie frontale metà inferiore

voce debole, monotona e nasale, difficoltà a parlareper debolezza di laringe e faringe
miotonia: contrazione prolungata di certi muscolidopo percussione o stimolazione elettrica e ritardo dirilascio dopo contrazione volontaria (stretta di mano)
la miotonia può precedere la debolezza muscolare dimolti anni
biopsia muscolare: cambiamenti non specifici
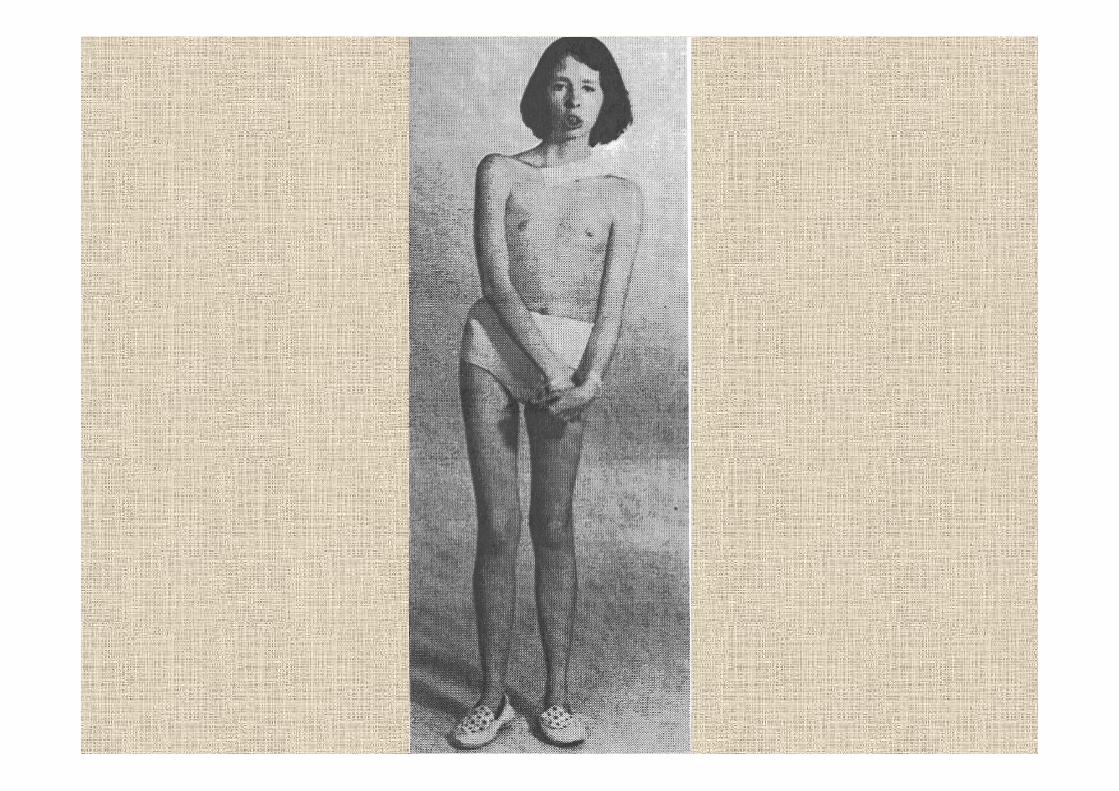

la presenza di cataratta bilaterale è segno clinico distintivo
disfunzione generalizzata del sistema nervoso
con risonanza magnetica si osserva atrofia del cervello
cuore: alterazioni di conduzione (bradicardia) con possibileblocco atrioventricolare e conseguente morte improvvisa
nelle femmine si osserva debolezza uterina che provoca abortività e difficoltà al parto
ecc… ecc… ecc…

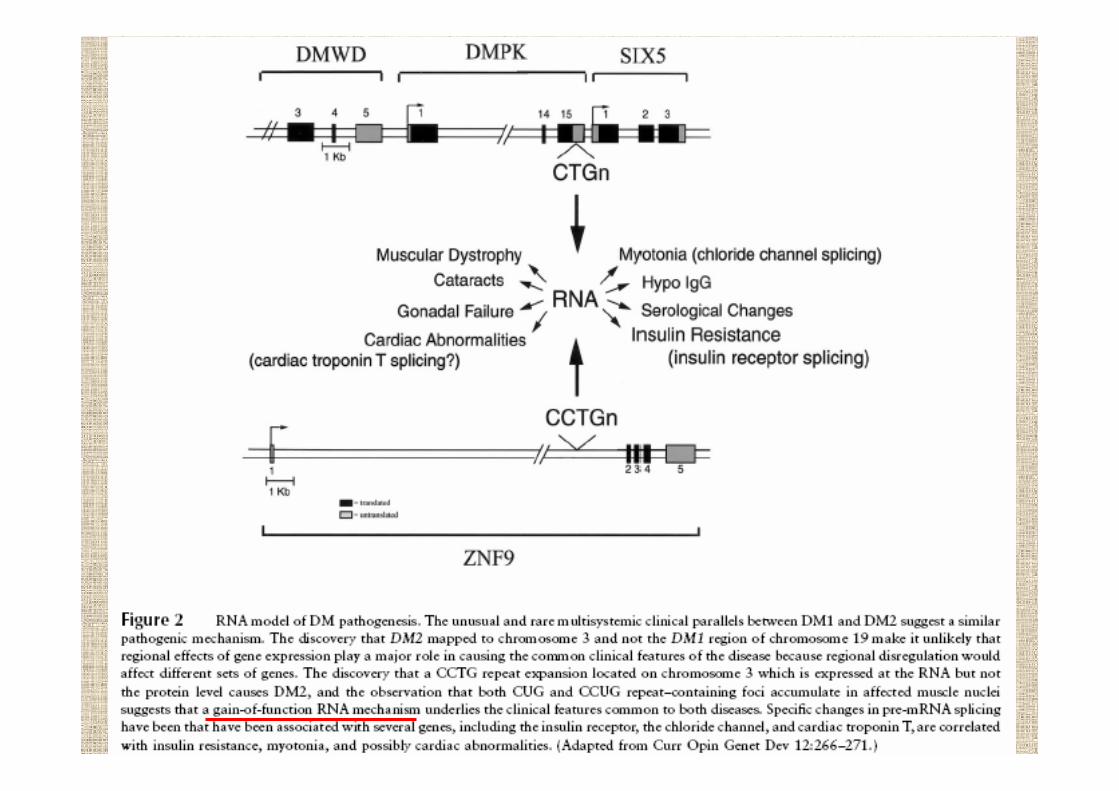
la base molecolare della miotonia di Steinert sta nell’espansione di una tripletta CTG nel gene DMPK(dystrophia-myotonica-protein-kinase)
19q13.2-q13.3
normale 5-27 copie di repeatsaffetto lieve 50-80esordio in età adulta 100-500forma congenita 500-2000
si può osservare anche espansione negativa(reverse anticipation)

l’amplificazione è frequentemente osservata dopo latrasmissione genitore-figlio, ma le amplificazioni estremenon sono mai trasmesse attraverso la linea maschile
fenomeno dell’anticipazione: aumento della gravità ingenerazioni successive

le variazioni sono influenzate da
• n° iniziale di partenza
• n° totale di divisioni cellulari durante la formazionedel tessuto
• specifica selezione in spermatogenesi

individui sani eterozigoti di lunghezza dell’amplificatonel range di normalità passano preferenzialmente aifigli un allele di lunghezza superiore ai 19 CTG (10%)
la malattia viene mantenuta nella popolazione nonostante la ridotta fitness per distorsione della segregazione

trasmissione autosomica dominantecon espressione altamente variabile
molti portatori obbligati sono asintomatici
è quasi sempre la madre che trasmette la formacongenita
è possibile un effetto parentale, infatti i pazienti natida madri affette sono più gravemente colpiti rispetto
a quelli nati con padre affetto

sulla base degli alberi la femmina affetta eterozigote avràun figlio affetto da forma congenita
con probabilità teorica del 50%
e con probabilità osservata pari al 3-9%
dopo la nascita di un figlio affetto il rischio aumentaal 20-37%

lo stato clinico della madre in gravidanza ha importanzasull’esito del nato:
• madre con effetti multisistemici avrà un figlio grave• madre lievemente affetta avrà un figlio non grave
gli affetti DM omozigoti non differiscono dagli eterozigoti
sono state escluse le mutazioni mitocondriali
sono stati esclusi effetti da imprinting

mosaicismo gonosomico
gonadico somatico
sono state osservate differenze in lunghezza dellaripetizione tra spermatozoi e cellule somatiche dellostesso individuo e tra sperma paterno e figlio
avviene amplificazione durante le divisioni mitotichedell’embriogenesi precoce

• non sembra esserci una forte correlazione tralunghezza di CTG e segni clinici
• la lunghezza di CTG nelle cellule del sangue continua a variare lungo l’arco della vita
• il grado di espansione correla con la lunghezza iniziale
• 50% dei pazienti con espansione continua mostra progressione dei sintomi
si ipotizza quindi che la diversità fenotipica in DM possaessere dovuta ad effetti in cis a lungo raggio su altri genidel cromosoma 19 da parte di CTG repeat in funzionedella sua lunghezza risultando in anomalie multisistemiche

il meccanismo attraverso il quale il CTG repeatporta alla manifestazione clinica non è chiaro,forse l’espansione influenza la conformazionedella cromatina vicina provocando disfunzioni
di altre unità di trascrizione
(19 gene rich)
locus nelle vicinanze è DMHP (DM locus associatedhomeodomain protein) espresso nel muscolo scheletrico,
nel cuore e nel cervello)
+ altri geni

DM2 ZNF9 3q
DM non corrispondente il quadro genetico



esiste una forma congenita con ipotonia neonatale,ritardo mentale e motorio e paralisi facciale
la madre trasmette la malattiastoria familiare silente
in utero: movimenti fetali ridotti, piede torto, polidramniosidrope non-immune
NB: la madre ha un allele mediamente più lungo rispettoalla popolazione generale