UNIVERSITÀ DEGLI STUDI DI ROMA'LA SAPIENZA' · screening di mutazioni del DNA mitocondriale (50) e...
43
UNIVERSITÀ DEGLI STUDI DI ROMA"LA SAPIENZA" Scuola di Specializzazione in Genetica Medica Direttore Prof. Bruno Dallapiccola Diagnosi molecolare di miopatia miotubulare legata all'X (XLMTM) mediante Denaturing High Performance Liquid Chromatography (DHPLC) Relatore Specializzando Prof. Giuseppe Novelli Dott.ssa Maria Rosaria D'Apice Anno Accademico 1999-2000
Transcript of UNIVERSITÀ DEGLI STUDI DI ROMA'LA SAPIENZA' · screening di mutazioni del DNA mitocondriale (50) e...

UNIVERSITÀ DEGLI STUDI DI ROMA"LA SAPIENZA"
Re
Pr
Scuola di Specializzazione in Genetica MedicaDirettore Prof. Bruno Dallapiccola
Diagnosi molecolare di miopatia miotubulare legata all'X
(XLMTM) mediante Denaturing High Performance Liquid
Chromatography (DHPLC)
latore Specializzando
of. Giuseppe Novelli Dott.ssa Maria Rosaria D'Apice
Anno Accademico1999-2000

INDICE
PREMESSA
INTRODUZIONE
MIOPATIA MIOTUBULARE X-LINKEDClinicaQuadro istopatologicoGene MTM1 e miotubularinaMutazioni e polimorfismi del gene MTM1Diagnosi molecolare di miopatia miotubulare legata all’XDenaturing High Performance liquid Chromatography
MATERIALI E METODICasistica analizzataEstrazione del DNADHPLCSequenziamento direttoStudio dell’inattivazione del cromosoma X
RISULTATI
DISCUSSIONE
BIBLIOGRAFIA

PREMESSA
La miopatia miotubulare X linked (XLMTM; MIM# 310400) è una
grave miopatia congenita, caratterizzata alla nascita da una marcata ipotonia e
debolezza muscolare generalizzata e dalla presenza di piccole cellule muscolari
con nucleo centralizzato sparse tra le fibre muscolari normali. La malattia è
dovuta a mutazioni della miotubularina, una proteina ad attività tirosin-
fosfatasica (PTP) codificata dal gene MTM1, che è composto da 15 esoni e
mappa nella regione Xq28. Sono state attualmente identificate 133 diverse
mutazioni su un totale di 198 pazienti non consanguinei. La mancanza di
mutazioni ricorrenti, unita all’assenza di vere e proprie regioni “hot spot”,
rende la diagnosi molecolare lunga e difficoltosa sia in termini di tempo che
economici. La tecnica attualmente più utilizzata prevede una prima analisi dei
polimorfismi di conformazione (SSCP) dell’intera sequenza codificante del
gene, seguita dal sequenziamento diretto delle varianti identificate.
Allo scopo di definire un protocollo di diagnostica molecolare del gene
MTM1, più rapido e sensibile, abbiamo sviluppato ed elaborato una strategia
molecolare innovativa basata sull'utilizzo della tecnica "Denaturing High
Performance Liquid Chromatography" o DHPLC. Questa tecnica utilizza la
capacità della cromatografia liquida a fase inversa di discriminare, all’interno
di prodotti eterogenei di PCR, molecole di DNA eteroduplici da quelle
omoduplici, in condizioni di parziale denaturazione e sotto diretto controllo
della temperatura. Il vantaggio della DHPLC, rispetto alle tecniche tradizionali,
è essenzialmente dovuto all’elevata sensibilità e riproducibilità, alla facilità di
applicazione, nonché alla rapidità e possibilità di automazione.

INTRODUZIONE
Le miopatie miotubulari, anche dette miopatie centronucleari,
comprendono una serie di miopatie congenite caratterizzate, dal punto di vista
istologico, dalla prevalenza di fibre ipotrofiche con nucleo centralizzato. Il
quadro clinico della miopatia miotubulare è piuttosto variabile in termini di età
di insorgenza, gravità della malattia, quadro istochimico e tipo di eredità (1).
La miopatia miotubulare esiste in una forma X-linked (MIM#310400) (2), una
forma autosomica dominante (MIM# 160150) e una forma autosomica
recessiva (MIM# 255200) (3).
MIOPATIA MIOTUBULARE X-LINKED
Clinica
La miopatia miotubulare X-linked (XLMTM; MIM # 310400) è una
malattia congenita caratterizzata da grave ipotonia e debolezza muscolare
generalizzata, che in particolare interessa i muscoli respiratori, facciali ed
oculari (Figura 1). I movimenti spontanei sono ridotti o assenti ed, alla nascita,
raramente si stabilisce una respirazione normale (4, 5). Spesso si osserva
un’insorgenza prenatale della malattia caratterizzata dalla presenza di
polidramnios e di movimenti fetali ridotti. Nelle portatrici obbligate si
osservano, con una frequenza superiore alla media, aborti spontanei e parti
prematuri. La maggior parte dei pazienti muore per insufficienza respiratoria
tra le prime settimane e i primi 4-5 mesi di vita. Si osserva anche un ridotto
numero di pazienti che sopravvive per molti mesi o anche sino all’età adulta,
specialmente quando sin dalla nascita viene fornito un supporto respiratorio

(3). La ragione di questa diversa progressione clinica rimane ancora
sconosciuta.
Le portatrici generalmente non mostrano manifestazioni cliniche
significative, sebbene in alcuni casi sia stata riportata una leggera debolezza dei
muscoli facciali (3, 6, 7). Sono inoltre descritti in letteratura casi di femmine
affette da miopatia miotubulare legata all’X in seguito a fenomeni di
inattivazione non casuale (eterozigoti estreme) (8, 9).
Quadro istopatologico
Il quadro istopatologico delle biopsie muscolari dei maschi affetti
evidenzia la presenza di molte fibre piccole e rotonde, con nucleo centrale
circondato da aggregati di mitocondri e di elementi contrattili che ricordano i
miotubi fetali (10). La percentuale di fibre contenenti nuclei centralizzati varia,
nella serie di casi riportati, dal 10 al 50% (11-13) oltre che nei diversi campi
microscopici (Figura 2 a, b) (6). Attualmente non è possibile stabilire con
certezza la percentuale minima di questo tipo di fibre necessaria per effettuare
la diagnosi. In circa la metà delle portatrici obbligate, la biopsia muscolare
evidenzia una certa proporzione di fibre con nuclei centralizzati. Questi dati
Figura 1 : Paziente con miopatia miotubulare legata all’X
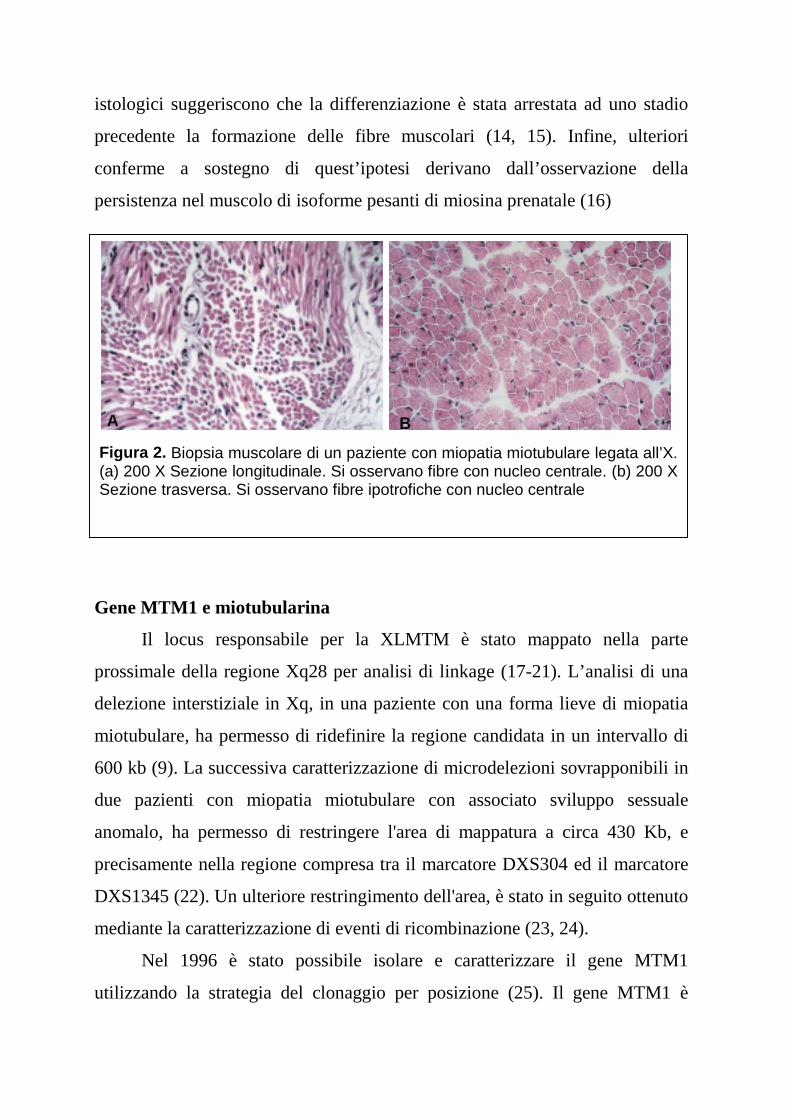
istologici suggeriscono che la differenziazione è stata arrestata ad uno stadio
precedente la formazione delle fibre muscolari (14, 15). Infine, ulteriori
conferme a sostegno di quest’ipotesi derivano dall’osservazione della
persistenza nel muscolo di isoforme pesanti di miosina prenatale (16)
Gene MTM1 e miotubularina
Il locus responsabile per la XLMTM è stato mappato nella parte
prossimale della regione Xq28 per analisi di linkage (17-21). L’analisi di una
delezione interstiziale in Xq, in una paziente con una forma lieve di miopatia
miotubulare, ha permesso di ridefinire la regione candidata in un intervallo di
600 kb (9). La successiva caratterizzazione di microdelezioni sovrapponibili in
due pazienti con miopatia miotubulare con associato sviluppo sessuale
anomalo, ha permesso di restringere l'area di mappatura a circa 430 Kb, e
precisamente nella regione compresa tra il marcatore DXS304 ed il marcatore
DXS1345 (22). Un ulteriore restringimento dell'area, è stato in seguito ottenuto
mediante la caratterizzazione di eventi di ricombinazione (23, 24).
Nel 1996 è stato possibile isolare e caratterizzare il gene MTM1
utilizzando la strategia del clonaggio per posizione (25). Il gene MTM1 è
A B
Figura 2. Biopsia muscolare di un paziente con miopatia miotubulare legata all’X.(a) 200 X Sezione longitudinale. Si osservano fibre con nucleo centrale. (b) 200 XSezione trasversa. Si osservano fibre ipotrofiche con nucleo centrale

trascritto in un mRNA di 4 Kb, ad espressione ubiquitaria ed una forma
alternativa di 2.4 Kb, che utilizza un diverso segnale di poliadenilazione,
selettivamente espresso nel muscolo e nel testicolo. Il gene MTM1 è composto
da 15 esoni (26), che si estendono per una regione genomica di 100 kb ed è
localizzato 20 kb prossimale ad un gene omologo MTMR1. La proteina
codificata, detta miotubularina (Figura 3), contiene una sequenza consensus di
12 aminoacidi per il sito attivo della tirosina fosfatasi (PTP) e la sua attività
fosfatasica è stata confermata su p-nitrofenil fosfato (p-NPP), un substrato
sintetico analogo alla fosfotirosina (27). La miotubularina contiene inoltre un
dominio di legame con le proteine SET (SID), una famiglia di proteine
coinvolte nella regolazione dei meccanismi epigenetici alla base della crescita e
del differenziamento cellulare (28). Diversi omologhi del gene MTM1 sono
stati isolati in topo, nel pesce Danio rerio, in Drosophila, in C. elegans, in S.
pombe e in S. cerevisiae e nel genoma umano, indicando che la miotubularina
appartiene ad un'ampia famiglia genica di tirosin-fosfatasi conservata nel corso
dell'evoluzione. Il possibile ruolo fisiologico di questa proteina viene suggerito
da un dato di recente acquisizione che dimostra che la miotubularina è in
grado, sia in vitro che in vivo, di defosforilare il secondo messaggero lipidico
fosfatidilinositolo 3-fosfato PI(3)P, coinvolto nel processo di trasporto
vescicolare dai compartimenti intracellulari alla membrana plasmatica. È
possibile che nei pazienti con miopatia miotubulare X-linked una regolazione
impropria del PI(3)P interrompa il traffico vescicolare necessario ai processi di
fusione dei mioblasti provocando un arresto nel corretto sviluppo del muscolo.
La miotubularina potrebbe, attraverso il controllo dei livelli cellulari di PI(3)P,
agire come regolatore della localizzazione subcellulare delle proteine con
dominio FYVE intervenendo nei pathway di trasduzione specifici dipendenti
da questi effettori (29). Un'ipotesi alternativa implica l'interazione della
miotubularina con le proteine contenenti i domini SET (Suvar3-9, Enhancer-of-
zeste, Trithorax). La forma mutata della miotubularina mimerebbe l'azione

fisiologica di alcune proteine appartenenti alla famiglia delle miotubularine
mancanti di attività fosfatasica ma capaci di legare i domini SET, come sbf1,
che quando viene superespressa è in grado di indurre in vitro la trasformazione
neoplastica dei miociti e di inibire in coltura la formazione dei miotubi (28).
Mutazioni e polimorfismi del gene MTM1
Il gene MTM1 è mutato nella maggior parte dei pazienti con XLMTM.
Sono state attualmente identificate 133 sono mutazioni differenti in 198
famiglie non imparentate (Figura 3) (30-41). Una lista completa e aggiornata è
presente sul sito web Human Gene Mutation Database (HGMD;
http://www.uwcm.ac.uk).
Le mutazioni del gene MTM1 sono distribuite lungo tutta la sequenza
codificante anche se gli esoni maggiormente interessati sono il 3, 4, 8, 9, 11 e
12. Al momento, la lista delle mutazioni risulta piuttosto eterogenea
comprendendo 55 mutazioni missenso, 40 non senso, 50 microinserzioni e
delezioni, 15 delezioni grandi e 38 mutazioni di splicing (41). La maggior parte
delle mutazioni provoca l’introduzione di un codone di stop e la traduzione di
una proteina tronca, anche se il 26% della mutazioni è costituto da variazioni
missenso che interessano sempre residui aminoacidici ben conservati durante
l'evoluzione. Queste ultime sono localizzate principalmente tra gli esoni 8 e 12
intorno al sito attivo tirosin-fosfatasico (PTP), codificato dall'esone 11, ed al
sito di legame delle proteine SET (SID), codificato dagli esoni 12 e 13. Un
elevato numero di mutazioni missenso mappa comunque negli esoni 8 e 9
senza un'apparente correlazione con i due domini funzionali noti della proteina.
Sono state inoltre descritte varianti polimorfiche non patogenetiche nelle
regioni introniche e nella regione del promotore.

Diagnosi molecolare di miopatia miotubulare legata all’X
La diagnosi clinica ed istologica di XLMTM deve essere confermata
attraverso l’analisi molecolare del gene MTM1. L’importanza della conferma
deriva dalla presenza di fenocopie di cui le più frequenti sono costituite dalle
forme autosomiche dominanti e recessive delle miopatie centronucleari, dalla
forma congenita di distrofia miotonica (DM) e dalla forma più grave di atrofia
muscolare spinale (SMAI). La diagnosi molecolare delle mutazioni del gene
MTM1 si basa sull’analisi delle varianti di conformazione (SSCP) e degli
eteroduplici (HD) dei frammenti di PCR ottenuti dall’amplificazione dei
singoli esoni (32). Le varianti anomale rilevate, vengono quindi isolate e
sequenziate. In genere il protocollo utilizzato prevede una prima analisi degli
esoni 8, 9, 4, 11, 12 e 5, nell’ordine menzionato, in quanto contengono oltre i
due terzi delle mutazioni, e solo in una seconda fase l'analisi degli esoni
rimanenti. Nonostante questo protocollo diagnostico abbia permesso
l’identificazione di un elevato numero di mutazioni si presenta di difficile
applicazione in un laboratorio diagnostico a causa dei lunghi tempi della messa

a punto e della difficoltà di automazione. Inoltre, come è ben noto, il metodo di
ricerca di mutazioni tramite SSCP dipende in larga misura dall’esperienza
dell’operatore e raramente raggiunge una sensibilità superiore al 80%.
Allo scopo di definire un protocollo per la diagnostica molecolare dei
pazienti XLMTM più rapido e sensibile, rispetto a quelli sino ad ora proposti,
abbiamo messo a punto nel nostro laboratorio una metodica alternativa per
l'analisi di mutazioni nel gene MTM1 denominata Denaturing High
Performance Liquid Chromatography (DHPLC), inizialmente descritto da
Underhill e coll. nel 1996 (42) e recentemente applicato con successo alla
ricerca di varianti genomiche di geni di discrete dimensioni, come BRCA1/2
(43-47), e TSC1/2 (48, 49). Questa tecnica è anche applicata con successo allo
screening di mutazioni del DNA mitocondriale (50) e alla ricerca di marcatori
biallelici tipo SNPs (Single Nucleotide Polymorphism) utilizzati per la
mappatura di regioni di suscettibilità a malattie complesse (51) o negli studi
evoluzionistici della genetica di popolazioni (52, 53).
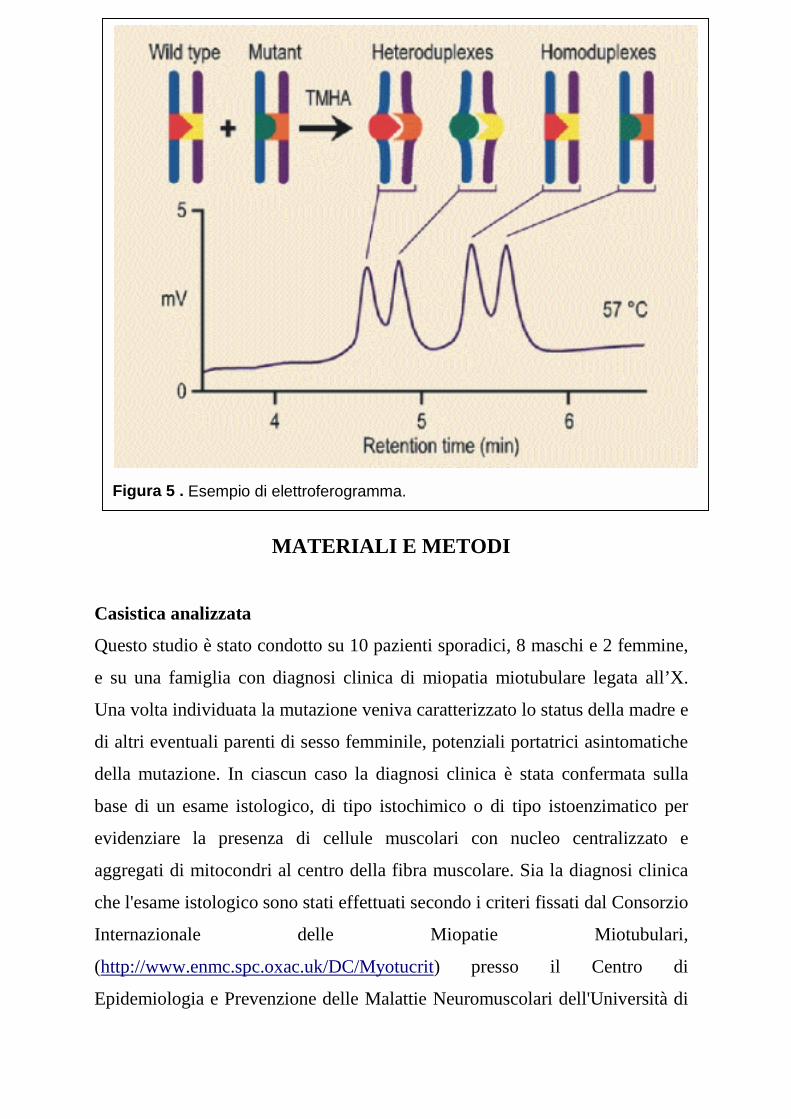
Denaturing High Performance Liquid Chromatography (DHPLC)
La DHPLC (Denaturing High Performance Liquid Chromatography)
(Figura 4) è una metodica di rapido impiego, accurata e automatica per
identificare variazioni nella sequenza di frammenti di DNA amplificati ottenuti
da reazioni di PCR (42). Questa metodica si basa sulla capacità della
cromatografia a fase inversa a coppie ioniche di discriminare tra le molecole di
DNA eteroduplici e omoduplici, sfruttando le differenze dei loro tempi di
ritenzione in una matrice di particelle alchilate, in specifiche condizioni di
parziale denaturazione dipendenti dalla temperatura. L’appaiamento non
corretto che si produce nelle molecole di DNA eteroduplice, a causa della
rinaturazione di due molecole non perfettamente complementari per la
presenza di una mutazione, produce una struttura instabile che destabilizza la
doppia elica. Per questo motivo, alle stesse condizioni di temperatura, la

molecola di DNA eteroduplice si trova in uno stato di parziale denaturazione,
mentre la molecola di DNA omoduplice corrispondente, più stabile, non viene
denaturata (Figura 5). La conseguente riduzione delle cariche negative nelle
porzioni a singolo filamento nel DNA eteroduplice provoca una diminuzione
del numero di legami tra il DNA ed un catione, il trietilammonio acetato, che
funziona come un ponte elettrostatico tra le molecole cariche negativamente
del DNA e la colonna. Una volta sottoposte ad un gradiente di acetonitrile, le
due specie molecolari, mostrando una differente affinità per la colonna,
vengono liberate in tempi differenti. Il campione di DNA, una volta eluito dalla
colonna, viene rilevato da uno spettrofotometro e visualizzato attraverso un
elettroferogramma. I dati relativi ad ogni singolo elettroferogramma vengono
poi immagazzinati automaticamente sotto forma di singoli files. L'amplificato
ottenuto da un eterozigote, essendo costituito sia molecole di DNA omoduplice
che eteroduplice viene eluito in più tempi diversi e produce un
elettroferogramma con più picchi, mentre l'amplificato ottenuto dal DNA di un
omozigote, essendo costituito esclusivamente da DNA omoduplice viene eluito
tutto insieme e visualizzato in un singolo picco. La risoluzione che si riesce ad
ottenere non è uguale per tutte le mutazioni, ma dipende dalla loro posizione
nel frammento di PCR e dalla lunghezza dalla composizione in basi del
frammento stesso.
Figura 4: DHPLC (denaturing high performance liquid chromatography)
MATERIALI E METODI
Casistica analizzata
Questo studio è stato condotto su 10 pazienti sporadici, 8 maschi e 2 femmine,
e su una famiglia con diagnosi clinica di miopatia miotubulare legata all’X.
Una volta individuata la mutazione veniva caratterizzato lo status della madre e
di altri eventuali parenti di sesso femminile, potenziali portatrici asintomatiche
della mutazione. In ciascun caso la diagnosi clinica è stata confermata sulla
base di un esame istologico, di tipo istochimico o di tipo istoenzimatico per
evidenziare la presenza di cellule muscolari con nucleo centralizzato e
aggregati di mitocondri al centro della fibra muscolare. Sia la diagnosi clinica
che l'esame istologico sono stati effettuati secondo i criteri fissati dal Consorzio
Internazionale delle Miopatie Miotubulari,
(http://www.enmc.spc.oxac.uk/DC/Myotucrit) presso il Centro di
Epidemiologia e Prevenzione delle Malattie Neuromuscolari dell'Università di
Figura 5 . Esempio di elettroferogramma.

Padova, l'Ospedale Infantile di Alessandria e l'Unità Operativa di Medicina
Molecolare dell'Ospedale Pediatrico Bambino Gesù. Le analisi molecolari sono
state effettuate su DNA estratto da un campione di biopsia muscolare o da un
campione di sangue periferico, prelevato in quantità variabile da 5 ml per i
neonati e 10 ml per i loro genitori e addizionato con EDTA 0.5% per evitarne
la coagulazione.
I soggetti di controllo sono costituiti da pazienti affetti da
patologie non muscolari.
Estrazione del DNA
La preparazione di DNA genomico da sangue periferico è stata ottenuta
mediante le tecniche standard di estrazione con fenolo/cloroformio e
precipitazione etanolica.
Il DNA genomico ottenuto dalle biopsie muscolari è stato preparato
secondo metodiche standard. In breve, i frammenti di muscolo sono stati prima
omogeneizzati e successivamente incubati in una soluzione di lisi contenente
Tris 0.02 mM, NaCl 100mM, EDTA 25mM pH 8.0 SDS 0.2% e proteinasi K
0.1 mg/ml per tutta la notte a 37°C. Il DNA totale è stato estratto con un
tampone saturato fenolo/cloroformio/isoamilico (25:24:1), precipitato in
etanolo e risospeso in H2O bidistillata.
DHPLC
Per lo screening di mutazioni/polimorfismi del gene MTM1 con la metodica
DHPLC è stata eseguita una reazione di PCR per ogni esone utilizzando 50 ng
di DNA genomico, 10 mM Tris HCl pH 8.3, 50 mM KCl, 1.5-2.0 mM MgCl2,
100 µM dNTPs, 0,2 µM primers, 2.5 U AmpliTaq Gold (PE Applied
Biosystems, Foster City, CA). Sono state utilizzate le seguenti condizioni di
amplificazione: un ciclo di denaturazione di 10 min a 94°C, seguito da 30 cicli
di 30 s a 94°C, 30 s a 52°C o 55°C e 30 s a 72°C. Le condizioni specifiche
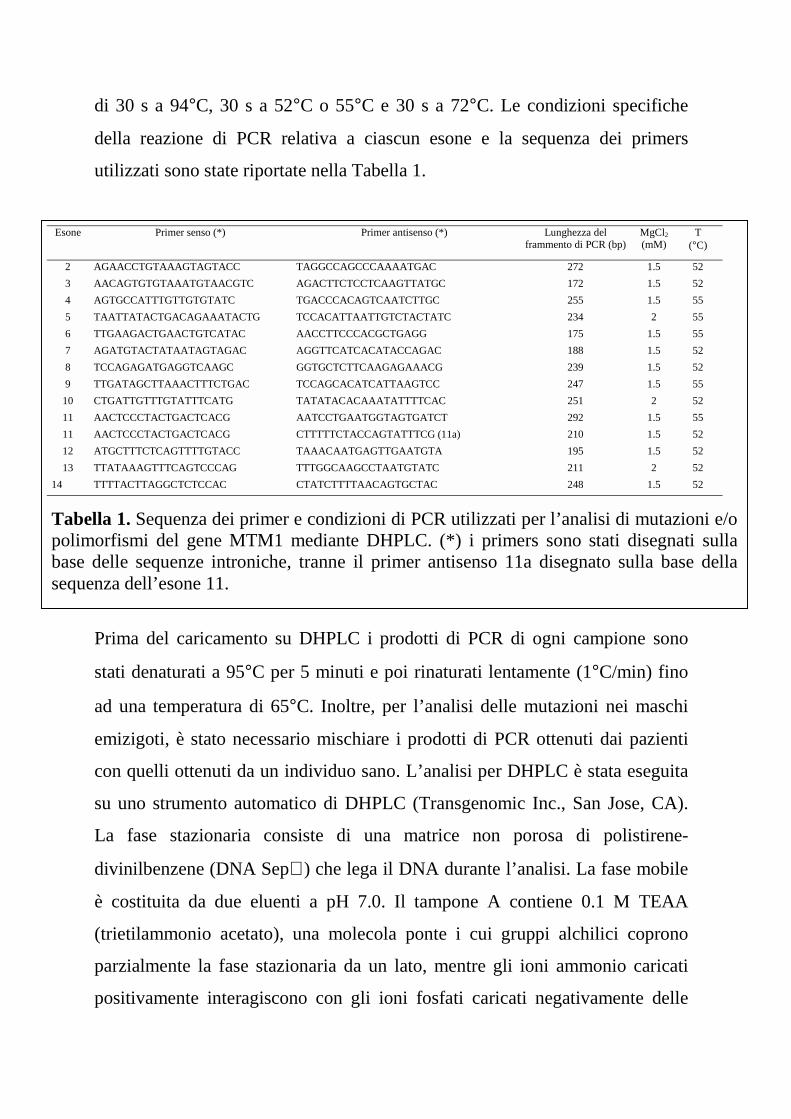
della reazione di PCR relativa a ciascun esone e la sequenza dei primers
utilizzati sono state riportate nella Tabella 1.
Prima del caricamento su DHPLC i prodotti di PCR di ogni campione sono
stati denaturati a 95°C per 5 minuti e poi rinaturati lentamente (1°C/min) fino
ad una temperatura di 65°C. Inoltre, per l’analisi delle mutazioni nei maschi
emizigoti, è stato necessario mischiare i prodotti di PCR ottenuti dai pazienti
con quelli ottenuti da un individuo sano. L’analisi per DHPLC è stata eseguita
su uno strumento automatico di DHPLC (Transgenomic Inc., San Jose, CA).
La fase stazionaria consiste di una matrice non porosa di polistirene-
divinilbenzene (DNA Sep ) che lega il DNA durante l’analisi. La fase mobile
è costituita da due eluenti a pH 7.0. Il tampone A contiene 0.1 M TEAA
(trietilammonio acetato), una molecola ponte i cui gruppi alchilici coprono
parzialmente la fase stazionaria da un lato, mentre gli ioni ammonio caricati
positivamente interagiscono con gli ioni fosfati caricati negativamente delle
Esone Primer senso (*) Primer antisenso (*) Lunghezza delframmento di PCR (bp)
MgCl2
(mM)T
(°C)
2 AGAACCTGTAAAGTAGTACC TAGGCCAGCCCAAAATGAC 272 1.5 52
3 AACAGTGTGTAAATGTAACGTC AGACTTCTCCTCAAGTTATGC 172 1.5 52
4 AGTGCCATTTGTTGTGTATC TGACCCACAGTCAATCTTGC 255 1.5 55
5 TAATTATACTGACAGAAATACTG TCCACATTAATTGTCTACTATC 234 2 55
6 TTGAAGACTGAACTGTCATAC AACCTTCCCACGCTGAGG 175 1.5 55
7 AGATGTACTATAATAGTAGAC AGGTTCATCACATACCAGAC 188 1.5 52
8 TCCAGAGATGAGGTCAAGC GGTGCTCTTCAAGAGAAACG 239 1.5 52
9 TTGATAGCTTAAACTTTCTGAC TCCAGCACATCATTAAGTCC 247 1.5 55
10 CTGATTGTTTGTATTTCATG TATATACACAAATATTTTCAC 251 2 52
11 AACTCCCTACTGACTCACG AATCCTGAATGGTAGTGATCT 292 1.5 55
11 AACTCCCTACTGACTCACG CTTTTTCTACCAGTATTTCG (11a) 210 1.5 52
12 ATGCTTTCTCAGTTTTGTACC TAAACAATGAGTTGAATGTA 195 1.5 52
13 TTATAAAGTTTCAGTCCCAG TTTGGCAAGCCTAATGTATC 211 2 52
14 TTTTACTTAGGCTCTCCAC CTATCTTTTAACAGTGCTAC 248 1.5 52
Tabella 1. Sequenza dei primer e condizioni di PCR utilizzati per l’analisi di mutazioni e/opolimorfismi del gene MTM1 mediante DHPLC. (*) i primers sono stati disegnati sullabase delle sequenze introniche, tranne il primer antisenso 11a disegnato sulla base dellasequenza dell’esone 11.

molecole di DNA interagisce. Il tampone B contiene 0.1 M TEAA e 25%
acetonitrile. L’interazione idrofobica tra la fase stazionaria e le catene
alchiliche delle molecole ponte è ridotta con l’aumento di acetonitrile nella fase
mobile.
I frammenti vengono eluiti con un gradiente lineare di acetonitrile di 2% per
minuto con un flusso di 0.9 ml/min. Il gradiente comprende uno step di
caricamento del DNA, un altro di separazione lineare dei frammenti di PCR,
seguito dal lavaggio e dall’equilibratura della colonna (DNA Sep ). I tempi di
inizio e di fine per ciascun passaggio sono dipendenti dalla sequenza di quel
determinato frammento di PCR .
Le temperature necessarie ad ottenere una risoluzione ottimale delle molecole
eteroduplici sono state calcolate con il programma DHPLC Melt
(http://insertion.stanford.edu/cgi-bin/melt.pl).
Le seguenti tabelle riportano, per ciascun esone analizzato, il gradiente lineare
della fase mobile (percentuali dei tamponi A e B) stabilito con il programma
WAVEMaker e le temperature di denaturazione dell’intero frammento di PCR
consigliate dal programma DHPLC Melt.

MTM1 esone 2
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 50 50 0.9 2.2Gradiente iniziale 0.5 45 55 2.7Gradiente finale 5.0 36 64 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 50 50 7.9Equilibratura finale 7.2 50 50 9.4
T (°C) domini analizzati*53°C 248-280,289-29154°C 166-247,281-28855°C 157-16556°C 1-156La temperatura di denaturazione dell’intero frammento di PCR è 55°CLa temperatura consigliata è 56°C
MTM1 esone 3
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 55 45 0.9 2.2Gradiente iniziale 0.5 50 50 2.7Gradiente finale 5.0 41 59 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 55 45 7.9Equilibratura finale 7.2 55 45 9.4
T (°C) domini analizzati*56°C 18557°C 156-18458°C 1-65,152-15559°C 66-151La temperatura di denaturazione dell’intero frammento di PCR è 58°CLa temperatura consigliata è 59°C

MTM1 esone 4
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 51 49 0.9 2.2Gradiente iniziale 0.5 46 54 2.7Gradiente finale 5.0 37 63 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 51 49 7.9Equilibratura finale 7.2 51 49 9.4
T (°C) domini analizzati*53°C 46-10754°C 1-45,108-274La temperatura di denaturazione dell’intero frammento di PCR è 54°CLa temperatura consigliata è 54°C
MTM1 esone 5
Step minuti %A %B ml/min rilevatoreCaricamento 0.0 52 48 0.9 2.2Gradiente iniziale 0.5 47 53 2.7Gradiente finale 5.0 38 62 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 52 48 7.9Equilibratura finale 7.2 52 48 9.4
T (°C) domini analizzati*54°C 230-25355°C 1-84,148-22956°C 85-147La temperatura di denaturazione dell’intero frammento di PCR è 55°CLa temperatura consigliata è 56°C
MTM1 esone 6
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 54 46 0.9 2.2Gradiente iniziale 0.5 49 51 2.7Gradiente finale 5.0 40 60 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 54 46 7.9Equilibratura finale 7.2 54 46 9.4
T (°C) dominio analizzato*60°C 1-194La temperatura di denaturazione dell’intero frammento di PCR è 60°CLa temperatura consigliata è 60°C
MTM1 esone 7
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 54 46 0.9 2.2Gradiente iniziale 0.5 49 51 2.7Gradiente finale 5.0 40 60 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 54 46 7.9Equilibratura finale 7.2 54 46 9.4
T (°C) domini analizzati*54°C 62-69, 96-10255°C 1-61, 70-95, 103-11856°C 119-207La temperatura di denaturazione dell’intero frammento di PCR è 55°CLa temperatura consigliata è 56°C

MTM1 esone 8
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 51 49 0.9 2.2Gradiente iniziale 0.5 46 54 2.7Gradiente finale 5.0 37 63 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 51 49 7.9Equilibratura finale 7.2 51 49 9.4
T (°C) domini analizzati*55°C 86-8956°C 1-11, 21-22, 34-34, 75-85, 90-9357°C 12-20, 23-33, 35-74, 94-9558°C 96-9959°C 100-259La temperatura di denaturazione dell’intero frammento di PCR è 59°CLa temperatura consigliata è 59°C
MTM1 esone 9
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 50 50 0.9 2.2Gradiente iniziale 0.5 45 55 2.7Gradiente finale 5.0 36 64 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 50 50 7.9Equilibratura finale 7.2 50 50 9.4
T (°C) domini analizzati*53°C 1-654°C 7-1655°C 17-2556°C 26-31, 116-18557°C 32-59, 112-115, 186-18858°C 60-111, 189-247La temperatura di denaturazione dell’intero frammento di PCR è 57°CLe temperature consigliate sono 53°C e 58°C

MTM1 esone 10
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 52 48 0.9 2.2Gradiente iniziale 0.5 47 53 2.7Gradiente finale 5.0 38 62 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 52 48 7.9Equilibratura finale 7.2 52 48 9.4
T (°C) domini analizzati*50°C 236-24551°C 224-23552°C 222-22353°C 1-6, 220-22154°C 7-30, 82-21955°C 31-81La temperatura di denaturazione dell’intero frammento di PCR è 54°CLa temperatura consigliata è 55°C
MTM1 esone 11
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 50 50 0.9 2.2Gradiente iniziale 0.5 45 55 2.7Gradiente finale 5.0 36 64 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 50 50 7.9Equilibratura finale 7.2 50 50 9.4
T (°C) domini analizzati*52°C 284-31153°C 272-28354°C 224-27155°C 218-22356°C 212-21757°C 191-193, 199-21158°C 183-190, 194-19859°C 1-68, 73-78, 173-18260°C 69-72, 79-172La temperatura di denaturazione dell’intero frammento di PCR è 59°CLe temperature consigliate sono 55°C e 60°C

MTM1 esone 12
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 53 47 0.9 2.2Gradiente iniziale 0.5 48 52 2.7Gradiente finale 5.0 39 61 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 53 47 7.9Equilibratura finale 7.2 53 47 9.4
T (°C) domini analizzati*51°C 213-21452°C 207-21253°C 194-20654°C 42-73, 180-19355°C 1-41, 74-77, 164-17956°C 78-163La temperatura di denaturazione dell’intero frammento di PCR è 55°CLa temperatura consigliata è 56°C
MTM1 esone 13
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 52 48 0.9 2.2Gradiente iniziale 0.5 47 53 2.7Gradiente finale 5.0 38 62 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 52 48 7.9Equilibratura finale 7.2 52 48 9.4
T (°C) domini analizzati*53°C 228-23054°C 94-119, 193-22755°C 1-93, 120-192La temperatura di denaturazione dell’intero frammento di PCR è 54°CLa temperatura consigliata è 55°C

MTM1 esone 14
Step tempo %A %B ml/min rilevatoreCaricamento 0.0 52 48 0.9 2.2Gradiente iniziale 0.5 47 53 2.7Gradiente finale 5.0 38 62 7.2Lavaggio iniziale 5.1 0 100 7.3Lavaggio finale 5.6 0 100 7.8Equilibratura iniziale 5.7 52 48 7.9Equilibratura finale 7.2 52 48 9.4
T (°C) domini analizzati*51°C 1-452°C 5-5, 242-24853°C 6-8, 23954°C 9-9, 88-105, 116-11755°C 10-87, 106-115, 118-144, 236-23856°C 145-235La temperatura di denaturazione dell’intero frammento di PCR è 55°CLe temperature consigliate sono 51°C e 56°C
(*) I numeri si riferiscono al nucleotide iniziale e finale del dominio didenaturrazione all’interno del frammento amplificato

Sequenziamento diretto
I prodotti di PCR sono stati purificati con il kit Seq-Prep GD400
(GeneDia) per eliminare primers, dNTPs e dimeri. Per la reazione di sequenza
è stato utilizzato il kit CEQ Dye Terminator Cycle Sequencing (Beckman).
Dopo la rimozione dell’eccesso di ddNTP marcati con il kit Seq-Prep, i
campioni sono stati sottoposti ad elettroforesi capillare su DNA Analysis
Sistem CEQ 2000 (Beckman Coulter).
I primers utilizzati sono gli stessi della reazione di PCR descritti nella tabella 1.
Studio dell'inattivazione del cromosoma X
Il campione costituito da 2�g di DNA genomico è stato digerito con 20
U di HpaII a 37°C per 12 ore. Successivamente un’aliquota della digestione
viene quindi utilizzata come stampo per una reazione di PCR contenente: 10
mM Tris HCl pH 8.3, 50 mM KCl, primer 1�M, dNTPs 250 �M, 0.05 U di
AmpliTaq Gold TM (PE Applied Biosystems, Foster City, CA).
La sequenza dei primers utilizzati, tratta da Tilley et al. (1989), è:
5’-GCTGTGAAGGTTGCTGTTCCTCAT-3’
5’-TCCAGAATCTGTTCCAGAGCGTGC-3’
Sono state utilizzate le seguenti condizioni di amplificazione: un ciclo di
denaturazione di 5 min a 94°C, seguito da 28 cicli di 45 s a 94°C, 30 s a 60°C e
30 s a 72°C.
I prodotti di PCR sono stati successivamente sottoposti ad elettroforesi
capillare su un modello ABI PRISMTM 310 Genetic Analyzer (PE Applied
Biosystems, Foster City, CA). Un microlitro di reazione di PCR è stato unito
con 20 �l di formamide e 0.5 �l del marcatore di peso molecolare fluorescente
(TAMRA GS-500 PE Applied Biosistems, Foster City, CA). Ogni campione è
stato corso per 30 minuti. Durante l’elettroforesi la fluorescenza rilevata nella
regione del laser è stata raccolta e immagazzinata usando il software Genescan

collection (versione 3.1 PE Applied Biosistems, Foster City, CA) alla fine di
ogni corsa.

RISULTATI
In questa tesi vengono riportati i risultati di uno studio effettuato su 10 pazienti
(8 maschi e 2 femmine) con diagnosi clinica di miopatia miotubulare. Inoltre,
lo studio ha riguardato anche una famiglia nella quale la malattia segrega da
più generazioni in maniera chiaramente legata alla X. Per ciascun paziente è
stata amplificata tutta la regione codificante del gene MTM1, tranne gli esoni 1
e 15 per i quali è stato riportato un esiguo numero di mutazioni. Gli amplificati
relativi ai singoli esoni sono stati esaminati mediante analisi di DHPLC. La
tabella 2 riporta, per ogni paziente, l’esone in cui è stato osservato un
elettroferogramma che suggeriva la presenza di una variante. Per discriminare
tra una mutazione ed un polimorfismo, ogni volta venivano analizzati 20
soggetti di controllo. Sono state individuate e caratterizzate 5 nuove mutazioni
e confermati 4 mutazioni precedentemente descritte e un polimorfismo
intronico comune nella popolazione caucasica (Tabella 3) (Figure 8, 11, 12).
Paziente ex 2 ex 3 ex 4 ex 5 ex 6 ex 7 ex 8 ex 9 ex10 ex11 ex 12 ex 13 ex 14
M1 - - - - - - + - - - - - -
F2 - - - - - - + - - - - - -
M3 - - - - - - + - - - - - -
M4 - - - - - - + - - - - - -
M5 - - - - - - - + - - - - -
F6 - - + - - - - - - - - - -
M7 - - - - - - - - - + - - -
M8 - - - - - - - - - + - - -
M9 - - - - - - - - - - - - +
M10 - + - - - - - - - - - - -
Tabella 2. Varianti identificate in ciascun paziente in base all'analisi deglielettroferogrammi dei singoli esoni.

Delle mutazioni identificate, 3 sono state evidenziate nell'esone 8, che anche
nel nostro gruppo di pazienti, analogamente a quanto già descritto da altri
ricercatori (41), presenta il maggior numero di mutazioni. La mutazione
T197I (39), evidenziata nel paziente M1, risulta associata con un
quadro clinico grave della malattia che oltre alla classica
sintomatologia muscolare, presentava anche uno sviluppo osseo
anomalo. La mutazione non è stata osservata nei genitori del
paziente, suggerendo pertanto un’origine de novo della stessa.
La seconda mutazione osservata nell'esone 8, la P199S, è stata
identificata allo stato eterozigote in una femmina (F2) affetta con
fenotipo lieve (Figura 6).
Paziente Esone/introne Mutazione Cambio Tipo Referenza
M1 esone 8 644 C→T T197I missenso Novelli G. et al.
F2 esone 8 649 T→C P199S missenso questo studio
M3 esone 8 646 insA (646-647)insA non senso questo studioNovelli G. et al.
M4 esone 8 646 insA (646-647)insA non senso questo studioNovelli G. et al.
M5 esone 9 811 C→T R253X non senso questo studio
F6 introne 3 191-11T→A (191-11)T→A splicing Laporte J. et al.
M7 esone 11 1186 G→A G378R missenso Laporte J. et al.
M8 esone 11 1321 C→G G421R missenso Nishino I. et al.
M8 introne 11 1314+3 A→G (1314+3)A→G polimorfismo Laporte J. et al.
M9 introne 14 1644+2 insG (1644+2)insG splicing questo studio
M10 esone 3 163 C→T R37X non senso Laporte J. et al.
Tabella 3. Mutazioni del gene MTM1 identificate mediante DHPLC
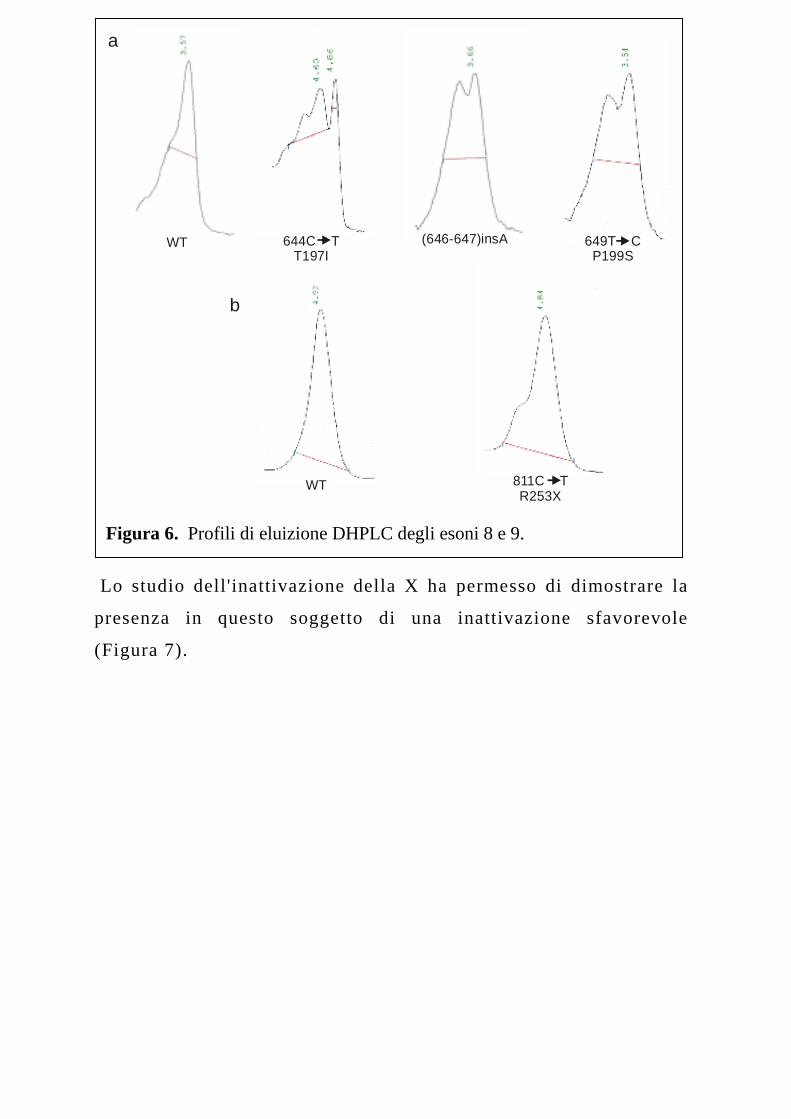
Lo studio dell'inattivazione della X ha permesso di dimostrare la
presenza in questo soggetto di una inattivazione sfavorevole
(Figura 7).
WT (646-647)insA
WT
644C TT197I
649T CP199S
811C TR253X
a
b
Figura 6. Profili di eluizione DHPLC degli esoni 8 e 9.

La mutazione (646-647)insA (Figura 6) (40), che introduce una
singola base tra i nucleotidi 646-647, è stata riscontrata in due
pazienti (M3, M4) non consanguinei provenienti dalla stessa
regione geografica. Questa mutazione determina l'introduzione di
un codone non senso in posizione 198. In entrambi i casi, la
mutazione è risultata associata ad un fenotipo grave della malattia.
Il paziente è deceduto nei primi tre mesi di vita.
La mutazione R253X, individuata nell'esone 9, consiste nella transizione C→T
al nucleotide 811 che introduce un codone non senso in posizione 253 al posto
di un residuo di arginina (Figura 6). Questa mutazione è associata ad
un fenotipo grave della malattia in quanto il paziente (M5) è
deceduto subito dopo la nascita per complicanze respiratorie. La
ricerca della mutazione nella madre e nei nonni materni, ha
1000
500
12823 01219
4000
3000
2000
1000
3525441107
4000
3000
2000
1000
32880 43654
4000
3000
2000
1000
78971 46867
4000
3000
2000
1000
4000
3000
2000
1000
64935
a
b
c
Figura 7. Studio dell’ inattivazione del cromosoma X. (a) Femmina F2affetta da miopatia miotubulare legata all’ X (XLMTM) con pattern diinattivazione sbilanciato. (b) Femmina sana con pattern di inattivazionecasuale. (c)Maschio di controllo.

permesso di stabilire la sua origine nella gametogenesi del nonno
materno (Figura 8).
L’analisi delle varianti evidenziate nell’esone 11 hanno permesso
di identificare il polimorfismo (1314+3)A→G (41) (Figura 9) che
nella nostra popolazione presenta una frequenza di 0.50. Nello
stesso esone sono state identificate due mutazioni missenso, G378R
e G421R, (Figura 9) che sostituiscono due residui aminoacidici
all'interno del sito "consensus" per l'attività tirosina fosfatasica. È
possibile pertanto che queste sostituzioni determinano la perdita di
funzione del sito catalitico della miotubularina.
DXS8377
DXS7423 2
3 2
1
II:2
I:2
II:1
III:1
I:1
DXS8377
DXS7423
3
2 1
1 2
1
DXS8377
DXS7423
3
2
*
Figura 8. Analisi di segregazione della mutazione R253X con i marcatoriDXS8377 e DXS7423, fiancheggianti il gene MTM1. (*) Allele mutato.III:1 probando, II:1 madre portatrice, I:1 nonno materno sano.

La mutazione nonsenso R37X è stata identificata nell’esone 3 e
corrisponde ad una transizione C→T nel sito CpG (Figura 10).
La mutazione (191-11)T→A, individuata nella paziente F6, attiva
un sito di splicing criptico nell'introne 4 (Figura 10). La stessa
mutazione, riportata precedentemente in letteratura, (41) provoca
l’eliminazione dell’esone 4 durante il processo di maturazione
dell'RNA messaggero. Anche in questo caso l'espressione della
malattia era associata all’inattivazione sbilanciata del cromosoma
X.
L'analisi dell'esone 14 ha permesso di identificare una inserzione
(1644+2)insG, (Figura 10) che elimina il sito donatore di splicing
nell'introne 14. L’effetto di questa mutazione sul trascritto maturo
è in corso di studio. Questa mutazione risulta associata ad un
fenotipo lieve della malattia.
WT (1314+3) G A1186G AG378R
1321C GG421R
Figura 9. Profili di eluizione DHPLC dell’ esone 11 .

È stata condotta un’analisi di linkage sulla famiglia 1 inclusa nella
nostra casistica. I risultati ottenuti erano compatibili con un
coinvolgimento del gene MTM1 (lod score=1,99 per θ=0,
marcatore DXS548) (Figura 11). Tuttavia la ricerca di mutazioni
nella regione codificante del gene non ha permesso di identificare
alcuna variante nucleotidica.
Nessuna mutazione e/o polimorfismi sono stati evidenziati negli
altri esoni esaminati.
WT (1644+2)insG
c
WT (191-11)T A
b
a
WT 163C TR37X
Figura 10. Profili di eluizione DHPLC degli esoni 3, 4 e 14 .

I:1 I:2
II:1DXS548DXS1113DXS8377DXS7423DXS1684
II:2 II:4 II:52 31 14 14 22 2
II:62 31 14 14 22 2
II:7II:8
III:12 41 14 34 12 1
II:32 31 14 14 22 2
III:22 41 14 24 12 1
III:33 41 11 32 12 1
III:43 41 11 32 12 1
III:52 41 14 34 12 1
III:631531
DXS548
DXS548
DXS1113
DXS1113
DXS8377
DXS8377
DXS7423
DXS7423
DXS1684
DXS1684
IV: 1 IV: 2IV: 321442
III:732233
IV: 421442
III:8
IV: 5
III:932641
IV: 63 42 16 34 11 1
IV: 7 IV: 8
II:9
III:10III:112 11 24 34 32 1
III:122 11 24 34 32 1
III:1332731
IV: 921442
IV: 102 31 24 74 32 1
III:14
IV: 11
III:15
IV: 12 IV: 13
II:10
III:16 III:172 31 24 44 32 2
III:182 31 24 44 32 2
III:19
IV: 14
II:11
III:20III:21 III:222 3? ?4 44 42 2
III:23
IV: 15
III:24
IV: 162?442
Figura 11. Analisi della segregazione degli aplotipi della regione Xq28 dellafamiglia 1. In nero è indicato il cromosoma associato alla malattia.

DISCUSSIONE
La miopatia miotubulare legata all’X è una patologia muscolare congenita,
caratterizzata da una grave ipotonia e debolezza muscolare generalizzata nei
maschi affetti. La maggior parte dei pazienti sono casi sporadici
senza una precedente storia familiare e muore nel primo anno di
vita per complicazioni respiratorie. La somiglianza clinica con
altre patologie è spesso il motivo principale della difficoltà a
formulare una diagnosi clinica certa. Il gene responsabile di questa
malattia, MTM1, è costituito da 15 esoni che si estendono per una
regione gnomica di 100 Kb e mutazioni nei due domini noti della
miotubularina sembrano coinvolte nel meccanismo di arresto della
differenziazione dei miociti ad uno stadio “fetale”,
L’individuazione delle mutazioni è importante nella consulenza
genetica e nella diagnosi prenatale, a sostegno della diagnosi
istopatologica e istoenzimatica.
Il protocollo di diagnosi molecolare attualmente utilizzato è basato
sulla combinazione dell’analisi dei polimorfismi di conformazione
(SSCP) e del sequenziamento diretto.
La Denaturing High Performance Liquid Chromatography (DHPLC) è una
metodologia di recente sviluppo, applicata all’analisi di mutazioni, al
rilevamento di SNPs e alla caratterizzazione di alleli negli studi di cancro,
malattie ereditarie ed evoluzione genomica.
Allo scopo di sviluppare un metodo semplice, senza rischi legati all’uso di
sostanze chimiche o radioattive, sensibile e specifico e al tempo stesso rapido
ed economico, in questa tesi abbiamo valutato l’applicabilità della DHPLC per
l’analisi di mutazioni e/o polimorfismi del gene umano MTM1

Abbiamo studiato 10 pazienti con diagnosi clinica di miopatia miotubulare
legata all’X e sono state identificate 9 differenti mutazioni in 10 casi (100%) ed
una variante polimorfa comune. Molte delle mutazioni missenso sono
raggruppate tra gli esoni 8 e 12, intorno al sito attivo PTP
codificato dall’esone 11, mentre altre sostituiscono aminoacidi nel
sito PTP e nel dominio SID. Non abbiamo invece identificato
mutazioni nella famiglia in esame. È attualmente in corso
un’analisi di mutazioni nel promotore o nelle regioni introniche o
negli esoni 1 e 15, in cui sono state riportate solo rare mutazioni.
La sensibilità dell’analisi mediante DHPLC è dipendente dalle
condizioni di corsa del prodotto di PCR nella colonna
cromatografia. Per alcuni esoni è stato necessario provare tre o
quattro temperature in quanto l’amplificato corrispondente
presentava più domini di denaturazione. Tuttavia il caricamento
semiautomatico dei campioni in una micropiastra da 96 pozzetti è
un vantaggio della fase di messa a punto delle condizioni
sperimentali: l’analisi procede anche per un’intera notte senza
l’intervento fisico dell’operatore.
Va sottolineato che nel concetto di sensibilità è compresa l’analisi
dell’elettroferogramma: ogni deviazione dal profilo di eluizione del
campione di DNA di un soggetto sano deve essere considerata una
potenziale mutazione. L’elettroferogramma corrispondente alla
mutazione R253X nell’esone 9 (Figura 8) mostra la presenza di una “spalla”
aggiuntiva rispetto al profilo di eluizione dello stesso prodotto di PCR in un
soggetto sano.
Nella nostra analisi di mutazioni non abbiamo osservato falsi
positivi: la successiva analisi per sequenziamento diretto ha
evidenziato la presenza di una mutazione o di un polimorfismo in
tutti i casi riportati. Riportiamo invece un caso di falso negativo

per l’esone 11: l’elettroferogramma del paziente M8 era simile a
quello corrispondente al polimorfismo (1314+3)A→G nel sito
donatore di splicing dell’introne 11. Il sequenziamento diretto ha
identificato anche la mutazione G421R, oltre alla variante polimorfa. La
mutazione è stata evidenziata in un momento successivo quando la
reazione di PCR è stata eseguita con il primer antisenso 11a
(Tabella 1), disegnato sulla base della sequenza esonica a monte
del sito di splicing.
Un altro vantaggio di questa metodologia è data dalla possibilità di analisi di
mutazioni in soggetti emizigoti semplicemente miscelando i prodotti di PCR di
un maschio malato e di un maschio sano. Il profilo di eluizione è esattamente
sovrapponibile a quello di un soggetto eterozigote per la stessa mutazione.
Tra gli svantaggi dell’analisi di mutazioni mediante DHPLC è da contemplare
il costo iniziale dell’acquisto della strumentazione che, tuttavia, non è superiore
a quello di un sequenziatore automatico standard. I costi di una singola corsa
sono invece sensibilmente bassi (circa 1$): la reazione di PCR non richiede
reagenti speciali come primers biotinilati o fluorescenti o dannosi come
radioisotopi, né enzimi in una fase di manipolazione successiva.
L’analisi di mutazioni mediante DHPLC è applicata di routine per geni
complessi quali BRCA1/2, TSC1/2. È verosimile ritenere che, tra gli sviluppi
futuri dell’applicazione della tecnica, c’è l’analisi di SNPs su grandi segmenti
genomici per la ricerca di polimorfismi di suscettibilità a malattie complesse,
per l’identificazione di marcatori in disequilibrium e per studi evolutivi (52,
53).

BIBLIOGRAFIA
1. Fardeau M, et al. Congenital myopathies. Myology, 2nd edn. New York:
McGra-Hill. 1994:1487-1532.
2. Van Wijngaarden GK, et al. Familial myotubular myopathy.
Neurology 1969 19:901-908.
3. Wallgren-Pettersson C, et al. The myotubular myopathies: differential
diagnosis of the X linked recessive, autosomal dominant, and autosomal
recessive forms and present state of DNA studies. J Med Genet.
1995;32:673-679.
4. Bodensteiner JB. Congenital myopathies. Muscle Nerve. 1994
Feb;17(2):131-144.
5. Wallgren-Pettersson C, et al. Report on the 20th ENMC sponsored
international workshop: myotubular/centronuclear myopathy.
Neuromuscul Disord. 1998;8:521-525.
6. Helliwell TR, et al. Myotubular myopathy: morphological,
immunohistochemical and clinical variation. Neuromuscul Disord. 1998
May;8(3-4):152-61.
7. Heckmatt JZ, et al. Congenital centronuclear (myotubular) myopathy. A
clinical, pathological and genetic study in eight children. Brain. 1985
Dec;108 ( Pt 4):941-64.
8. Dahl N, et al. Miotubular myopathy in a girl with a deletion at Xq27-q28
and unbalanced X inactivation assigns the MTM1 gene to a 600 kb
region. Am J Hum Genet. 1995 May;56(5):1108-1115.
9. Tanner SM, et al. Skewed X-inactivation in a manifesting carrier of X-
linked myotubular myopathy and in her non-manifesting carrier mother.
Hum Genet. 1999 Mar;104(3):249-53.
10. Fardeau M, et al. Congenital myopathies. Skeletal muscle Pathology, eds
Mastaglia. Edinburgh: Churcill Livingstone. 1992.

11. Ambler MW, et al. X-linked myotubular recessive myopathy: I. Clinical
and pathologic findings in a family. Hum Pathol 1984 Jun;15(6):566-
574.
12. Ambler MW, et al. X-linked myotubular recessive myopathy: II. Muscle
morphology and human myogenesis Hum Pathol 1984 Dec;15(12):1107-
1120.
13. Sasaki T, et al. Muscle histochemistry in myotubular (centronuclear)
myopathy. Brain Dev. 1989;11(1):26-32.
14. Sarnat HB. Myotubular myopathy: arrest of morphogenesis of myofibres
associated with persistence of fetal vimentin and desmin. Four cases
compared with fetal and neonatal muscle. Can J Neurol Sci. 1990
May;17(2):109-23.
15. Sawchak JA, et al. Centronuclear myopaty eterogeneity:distinction of
clinical types by myosin isoform patterns. Neurol. 1991 41:135:140.
16. Van der Ven PF, et al. Abnormal expression of intermediate filament
proteins in X-linked myotubular myopathy is not reproduced in vitro.
Neuromuscul Disord. 1995 Jul;5(4):267-75.
17. Thomas NS, et al. X linked neonatal centronuclear/myotubular
myopathy: evidence for linkage to Xq28 DNA marker loci. J Med Genet.
1990 May 27(5):284-7.
18. Darnfors C, et al. X-linked myotubular myopathy: a linkage study. Clin
Genet. 1990 May;37(5):335-40.
19. Lehesjoki AE, et al. X linked neonatal myotubular myopathy: one
recombination detected with four polymorphic DNA markers from
Xq28. J Med Genet. 1990 May;27(5):288-291.
20. Liechti-Gallati S, et al. X-linked centronuclear myopathy: mapping the
gene to Xq28. Neuromuscul Disord. 1991;1(4):239-45.

21. Janssen EA, et al. The gene for X-linked myotubulr myopathy is located
in an 8 Mb region at the border of Xq27.3 and Xq28. Neuromuscul
Disord. 1994 Sep-Nov;4(5-6):455-461.
22. Hu LJ, et al. Deletions in Xq28 in two boys with myotubular myopathy
and abnormal genital development define a new contiguous gene
syndrome in a 430 kb region. Hum Mol Genet. 1996 Jan;5(1):139-43.
23. Dahl N, et al. X linked myotubular myopathy (MTM1) maps between
DXS304 and DXS305, closely linked to the DXS455 VNTR and a new,
highly informative microsatellite marker (DXS1684). J Med Genet. 1994
Dec;31(12):922-924.
24. Hu LJ, et al. X-linked myotubular myopathy: refinement of the gene to a
280 Kb region with new and highly informative microsatellite markers.
Human Genet. 1996; 98:178-181.
25. Laporte J, et al. A gene mutated in X-linked myotubular myopathy
defines a new putative tyrosine phosphatase family conserved in yeast.
Nat Genet. 1996 Jun;13(2):175-82.
26. Laporte J, et al. Genomic organization of the MTM1 gene implicated in
X-linked myotubular myopathy. Eur J Hum Genet. 1998 Jul-
Aug;6(4):325-30.
27. Laporte J, et al. Characterization of the myotubularin dual specifity
phosphatase gene family from yeast to human. Hum Mol Genet 1998
Oct;7(11):1703-1712.
28. Cui X, et al. Association of SET domain and myotubularin-related
proteins modulates growth control. Nat Genet. 1998;18:331-337.
29. Taylor GS, et al. Myotubularin, a protein phosphatase mutated in
myotubular myopathy, dephosphorilates the lipid second messenger,
phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA 2000 Aug
1;97(16):8910-8915.

30. Guiraud-Chaumeil C, et al. A mutation in the MTM1 gene invalidates a
previous suggestion of nonallelic heterogeneity in X-linked myotubular
myopathy. Am J Hum Genet. 1997 Jun;60(6):1542-1544.
31. de Gouyon BM, et al. Characterization of mutations in the myotubularin
gene in twenty six patients with X-linked myotubular myopathy. Hum
Mol Genet. 1997;6:1499-1504.
32. Laporte J, et al. Mutations in the MTM1 gene implicated in X-linked
myotubular myopathy. ENMC International Consortium on Myotubular
Myopathy. European Neuro-Muscular Center. Hum Mol Genet.
1997;6:1505-1511.
33. Donnelly A, et al. A novel mutation in exon b (R259C) of the MTM1
gene is associated with a mild myotubular myopathy. Mutations in brief
no. 125. Online. Hum Mutat. 1998;11(4):334.
34. Watanabe T, et al. X-linked recessive myotubular myopathy with a
splice-site mutation in the myotubularin gene. No To Hattatsu. 1998
Nov;30(6):523-7.
35. Nishino I, et al. MTM1 gene mutations in Japanese patients with the
severe infantile form of myotubular myopathy. Neuromuscul Disord.
1998 Oct;8(7):453-8.
36. Tanner SM, et al. Confirmation of prenatal diagnosis results of X-linked
recessive myotubular myopathy by mutational screening, and description
of three new mutations in the MTM1 gene. Hum Mutat. 1998;11(1):62-
8.
37. Buj-Bello A, et al. Identification of novel mutations in the MTM1 gene
causing severe and mild forms of X-linked myotubular myopathy. Hum
Mutat. 1999 Oct;14(4):320-325.
38. Tanner SM, et al. Characterization of 34 novel and six known MTM1
gene mutations in 47 unrelated X-linked myotubular myopathy patients.
Neuromuscul Disord. 1999;9:41-49.

39. Novelli G, et al. 1999 Mutation Acc H971495 Hum Genet 105:373.
40. Novelli G, et al. 1999 Mutation Acc H971496 Hum Genet 105:374.
41. Laporte J, et al. MTM1 mutations in X-linked myotubular myopathy.
Hum Mutat 2000;15(5):593-409 Review.
42. Underhill PA, et al. A pre-Columbian human Y chromosome-specific
transition and its implications for human evolution. Proc. Natl. Acad.
Sci. USA 1996;93 :196-200.
43. Wagner T.M.U. et al. BRCA1-related breast cancer in austrian breast
and ovarian cancer families: specific BRCA1 mutations and pathological
characteristics. Int. J. Cancer 1998 77:354-360
44. Wagner T.M.U. et al. Global sequence diversity of BRCA2: analisys of
71 breast cancer families and 95 control individuals of worldwide
populations. Hum. Mol. Gen. 1999, 8:413-423.
45. Gross E. et al. A comparison of BRCA1 mutation analysis by direct
sequencing, SSCP and DHPLC. Hum. Genet. 1999, 105:72-78
46. Wagner T. et al. Denaturing High Performance Liquid Chromatography
detects reliably BRCA1 and BRCA2 mutations. Genomics 1999,
62:369-376.
47. Arnold N. et al. A highly sensitive, fast, and economical technique for
mutation analysis in hereditary breast and ovarian cancers. Hum Mutat
1999, 14:333-339.
48. Choy YS, et al. Superiority of denaturing high performance liquid
chromatography over single-stranded conformation-sensitive gel
electrophoresis for mutation detection in TSC2. Ann. Hum. Genet. 1999,
63:383-391.
49. Jones A. C. et al. Application and evaluation of denaturing HPLC for
molecular genetic analysis in tuberous sclerosis. Hum. Genet 2000,
106:663-668.

50. Van den Bosch B. J. C. et al. Mutation analysis of the entire
mitochondrial genome using denaturing high performance liquid
chromatography. Nucleic Acid Res. 2000, 28:e89
51. Giordano M. et al. Identification by Denaturing High Performance
Liquid Chromatography of numerous polymorphisms in a candidate
region for multiple sclerosis susceptibility. Genomics 1999, 56:247-253
52. Underhill P.A. et al. Detection of numerous Y chromosome biallelic
polymorphisms by Denaturing High Performance Liquid
Chromatography. Genome research 1997, 7:996-1005
53. Underhill, P.A. Y chromosome sequence variation and the history of
human populations. Nat Genet 2000 26(3):358-361

INDICE
PREMESSA 1
INTRODUZIONE 2
MIOPATIA MIOTUBULARE X-LINKED 2Clinica 2Quadro istopatologico 3Gene MTM1 e miotubularina 3Mutazioni e polimorfismi del gene MTM1 5Diagnosi molecolare di miopatia miotubulare legata all’X 6Denaturing High Performance liquid Chromatography 7
MATERIALI E METODI 9Casistica analizzata 9Estrazione del DNA 9DHPLC 10Sequenziamento diretto 20Studio dell’inattivazione del cromosoma X 20
RISULTATI 22
DISCUSSIONE 25
BIBLIOGRAFIA 28