
Tricarbonyl−Rhenium Complexes of a Thiol-Functionalized Amphoteric Poly(amidoamine)
10
Tricarbonyl-Rhenium Complexes of a Thiol-Functionalized Amphoteric Poly(amidoamine) Daniela Donghi, † Daniela Maggioni, † Giuseppe D’Alfonso,* ,† Federica Amigoni, † Elisabetta Ranucci, ‡ Paolo Ferruti,* ,‡ Amedea Manfredi, ‡ Fabio Fenili, ‡ Agnese Bisazza, § and Roberta Cavalli § Dipartimento di Chimica Inorganica, Metallorganica e Analitica “L. Malatesta” and Dipartimento di Chimica Organica e Industriale, Universita ` degli Studi di Milano, via Venezian 21, 20133 Milano, and Dipartimento di Scienza e Tecnologia del Farmaco, Universita ` di Torino, Via P. Giuria 9, 1012 Torino, Italy Received July 30, 2009; Revised Manuscript Received October 21, 2009 An amphoteric thiol-functionalized poly(amidoamine) nicknamed ISA23SH 10% was synthesized. Rhenium complexes 1 and 2, containing 0.5 and 0.8 equiv of rhenium, respectively, were easily obtained by reacting ISA23SH 10% with [Re(CO) 3 (H 2 O) 3 ](CF 3 SO 3 ) in aqueous solution at pH 5.5. Both ISA23SH 10% , and its rhenium complexes were soluble in water under physiological conditions. The resultant solutions were stable, even in the presence of cysteine. Rhenium chelation occurred through the S and N atoms of the cysteamine moiety, as demonstrated by 1 H, 13 C, and 15 N NMR spectroscopy. The diffusion coefficients and the hydrodynamic radii of ISA23SH 10% and complex 1 were determined by pulsed gradient spin echo (PGSE) NMR experiments. The radius of the rhenium complexes 1 and 2 was always slightly larger than that of the parent polymer. TEM analysis showed that both complexes form spherical nanoparticles with narrow size distributions. Consistent results were obtained by dynamic light scattering. The observed sizes were in good agreement with those evaluated by PGSE. Preliminary in vitro and in vivo biological studies have been performed on complexes 1 and 2 as well as on the parent ISA23SH 10% . Neither hemolytic activity of the two rhenium complexes and the parent polymer, up to a concentration of 5 mg/mL, nor cytotoxic effects were observed on Hela cell after 48 h at a concentration of 100 ng/mL. In vivo toxicological tests showed that ISA23SH 10% is highly biocompatible, with a maximum tolerated dose (MTD) of 500 mg/kg. No toxic side effects were apparent after the intravenous injection in mice of the two rhenium complexes in doses up to 20 mg/kg. Introduction Poly(amidoamine)s (PAAs) are a family of synthetic polymers obtained by Michael-type polyaddition of amines to bisacryl- amides. Their polymer chain contains tert-amino and amido groups in regular succession. In addition, many reactive functional groups can be introduced as side substituents. 1,2 Several PAAs have been shown to possess a remarkable potential for biomedical applications in that they are biodegrad- able and biocompatible, and owing to their versatility can be tailored to specific applications. In particular, amphoteric PAAs that in extra cellular fluids (pH 7.4) are zwitterionic but predominantly anionic are highly biocompatible and exhibit a “stealth-like” behavior, that is, after intravenous injection in test animals are not captured by the reticulo-endothelial system and have a prolonged permanence in the blood circle. In tumor- bearing animals, they are passively concentrated in the tumor tissues by the so-called EPR (enhanced permeation and reten- tion) effect. 3,4 A typical amphoteric PAA much studied in this respect, nicknamed ISA23, was prepared from 2,2-bis-acryl- amidoacetic acid and 2-methylpiperazine. (See Chart 1.) 4 This PAA can be further functionalized to render it able to bind different metal fragments covalently. Considerable interest has currently been focused on radio- pharmaceuticals incorporating 99m Tc or 186/188 Re. 5-9 The γ emitter 99m Tc is commonly used for diagnostic imaging proce- dures because of its almost ideal physical properties (t 1/2 ) 6.01 h, γ ) 142.7 keV) and easy production from Na 2 99 MoO 4 column generators. The heavier congener rhenium has two radioisotopes of interest for nuclear medicine, both of which have potential for cancer therapy (owing to their - emission) and diagnosis (owing to their γ emission with energies similar to that of 99m Tc): 186 Re (t 1/2 ) 3.7 d, γ ) 137 keV, - ) 1.07 MeV) and 188 Re (t 1/2 ) 16.94 h, γ ) 155 keV, - ) 2.12 MeV). Differently from technetium, however, rhenium occurs in nature as a mixture of two stable isotopes ( 185 Re and 187 Re) in 37.4: 62.6 ratio. Therefore, the preparation and properties of candidate rhenium radiopharmaceuticals can be preliminarily investigated in cold laboratories. Moreover, it may be observed that the results obtained for nonradioactive Re complexes can be extended to the corresponding ones of Tc, owing to the strict chemical similarity existing between the two elements, both actively considered as imaging agents. The conjugation of Re with suitable carriers for targeting cancer cells has attracted of great deal of research. 5-9 In fact, the rhenium complex ion [Re(CO) 3 (H 2 O) 3 ] + is a good candidate for bioconjugation, 10 owing to its fast preparation 11-14 and versatile chemistry. In particular, the three water molecules loosely bound to a stable fac-Re(CO) 3 + core provide convenient conjugation sites. Different chelating agents as well as different coordination routes for obtaining rhenium derivatives with high specificity and stability have been described in the literature. 10,15-17 * To whom correspondence should be addressed. Tel: +39 0250314351. Fax: +39 0250314405. E-mail: [email protected] (G.D.). Tel: +39 0250314128. Fax: +39 0250314129. E-mail: [email protected] (P.F.). † Dipartimento CIMA, Universita ` degli Studi di Milano. ‡ Dipartimento COI, Universita ` degli Studi di Milano. § Universita ` di Torino. Biomacromolecules 2009, 10, 3273–3282 3273 10.1021/bm9008638 CCC: $40.75 2009 American Chemical Society Published on Web 11/16/2009
Transcript of Tricarbonyl−Rhenium Complexes of a Thiol-Functionalized Amphoteric Poly(amidoamine)

Tricarbonyl-Rhenium Complexes of a Thiol-FunctionalizedAmphoteric Poly(amidoamine)
Daniela Donghi,† Daniela Maggioni,† Giuseppe D’Alfonso,*,† Federica Amigoni,†
Elisabetta Ranucci,‡ Paolo Ferruti,*,‡ Amedea Manfredi,‡ Fabio Fenili,‡ Agnese Bisazza,§
and Roberta Cavalli§
Dipartimento di Chimica Inorganica, Metallorganica e Analitica “L. Malatesta” and Dipartimento di ChimicaOrganica e Industriale, Universita degli Studi di Milano, via Venezian 21, 20133 Milano, and Dipartimento
di Scienza e Tecnologia del Farmaco, Universita di Torino, Via P. Giuria 9, 1012 Torino, Italy
Received July 30, 2009; Revised Manuscript Received October 21, 2009
An amphoteric thiol-functionalized poly(amidoamine) nicknamed ISA23SH10% was synthesized. Rheniumcomplexes 1 and 2, containing 0.5 and 0.8 equiv of rhenium, respectively, were easily obtained by reactingISA23SH10% with [Re(CO)3(H2O)3](CF3SO3) in aqueous solution at pH 5.5. Both ISA23SH10%, and its rheniumcomplexes were soluble in water under physiological conditions. The resultant solutions were stable, even in thepresence of cysteine. Rhenium chelation occurred through the S and N atoms of the cysteamine moiety, asdemonstrated by 1H, 13C, and 15N NMR spectroscopy. The diffusion coefficients and the hydrodynamic radii ofISA23SH10% and complex 1 were determined by pulsed gradient spin echo (PGSE) NMR experiments. The radiusof the rhenium complexes 1 and 2 was always slightly larger than that of the parent polymer. TEM analysisshowed that both complexes form spherical nanoparticles with narrow size distributions. Consistent results wereobtained by dynamic light scattering. The observed sizes were in good agreement with those evaluated by PGSE.Preliminary in vitro and in vivo biological studies have been performed on complexes 1 and 2 as well as on theparent ISA23SH10%. Neither hemolytic activity of the two rhenium complexes and the parent polymer, up to aconcentration of 5 mg/mL, nor cytotoxic effects were observed on Hela cell after 48 h at a concentration of 100ng/mL. In vivo toxicological tests showed that ISA23SH10% is highly biocompatible, with a maximum tolerateddose (MTD) of 500 mg/kg. No toxic side effects were apparent after the intravenous injection in mice of the tworhenium complexes in doses up to 20 mg/kg.
Introduction
Poly(amidoamine)s (PAAs) are a family of synthetic polymersobtained by Michael-type polyaddition of amines to bisacryl-amides. Their polymer chain contains tert-amino and amidogroups in regular succession. In addition, many reactivefunctional groups can be introduced as side substituents.1,2
Several PAAs have been shown to possess a remarkablepotential for biomedical applications in that they are biodegrad-able and biocompatible, and owing to their versatility can betailored to specific applications. In particular, amphoteric PAAsthat in extra cellular fluids (pH 7.4) are zwitterionic butpredominantly anionic are highly biocompatible and exhibit a“stealth-like” behavior, that is, after intravenous injection in testanimals are not captured by the reticulo-endothelial system andhave a prolonged permanence in the blood circle. In tumor-bearing animals, they are passively concentrated in the tumortissues by the so-called EPR (enhanced permeation and reten-tion) effect.3,4 A typical amphoteric PAA much studied in thisrespect, nicknamed ISA23, was prepared from 2,2-bis-acryl-amidoacetic acid and 2-methylpiperazine. (See Chart 1.)4 ThisPAA can be further functionalized to render it able to binddifferent metal fragments covalently.
Considerable interest has currently been focused on radio-pharmaceuticals incorporating 99mTc or 186/188Re.5-9 The γemitter 99mTc is commonly used for diagnostic imaging proce-dures because of its almost ideal physical properties (t1/2 )6.01 h, γ ) 142.7 keV) and easy production from Na2
99MoO4
column generators. The heavier congener rhenium has tworadioisotopes of interest for nuclear medicine, both of whichhave potential for cancer therapy (owing to their - emission)and diagnosis (owing to their γ emission with energies similarto that of 99mTc): 186Re (t1/2 ) 3.7 d, γ ) 137 keV, - ) 1.07MeV) and 188Re (t1/2 ) 16.94 h, γ ) 155 keV, - ) 2.12 MeV).Differently from technetium, however, rhenium occurs in natureas a mixture of two stable isotopes (185Re and 187Re) in 37.4:62.6 ratio. Therefore, the preparation and properties of candidaterhenium radiopharmaceuticals can be preliminarily investigatedin cold laboratories. Moreover, it may be observed that theresults obtained for nonradioactive Re complexes can beextended to the corresponding ones of Tc, owing to the strictchemical similarity existing between the two elements, bothactively considered as imaging agents.
The conjugation of Re with suitable carriers for targetingcancer cells has attracted of great deal of research.5-9 In fact,the rhenium complex ion [Re(CO)3(H2O)3]+ is a good candidatefor bioconjugation,10 owing to its fast preparation11-14 andversatile chemistry. In particular, the three water moleculesloosely bound to a stable fac-Re(CO)3
+ core provide convenientconjugation sites. Different chelating agents as well as differentcoordination routes for obtaining rhenium derivatives with highspecificity and stability have been described in the literature.10,15-17
* To whom correspondence should be addressed. Tel: +39 0250314351.Fax: +39 0250314405. E-mail: [email protected] (G.D.). Tel:+39 0250314128. Fax: +39 0250314129. E-mail: [email protected](P.F.).
† Dipartimento CIMA, Universita degli Studi di Milano.‡ Dipartimento COI, Universita degli Studi di Milano.§ Universita di Torino.
Biomacromolecules 2009, 10, 3273–3282 3273
10.1021/bm9008638 CCC: $40.75 2009 American Chemical SocietyPublished on Web 11/16/2009

In this article, we propose an amphoteric PAA functionalizedwith a thiol pendant (ISA23SH10%) as polymer carrier andchelating agent (Chart 1). It appeared particularly apt tocoordinate the Re(CO)3
+ fragment because of the presence of2-aminoethanethiol groups, which are known to be good ligandsfor rhenium or technetium complex ions.18-21 Nanosizedcomplexes with different rhenium contents (0.5 or 0.85 metalatoms per thiol-bearing unit) were obtained and characterizedby analytical and spectroscopic techniques. In particular, NMRspectroscopy was used to investigate the coordination site andthe particle size. The complexes were highly stable in waterunder physiological conditions and in preliminary tests showedremarkable biocompatibility both in vitro and in vivo, suggestingthat the metal fragments were tightly coordinated to the PAAbackbone.
Experimental Part
Instruments and Methods. NMR experiments were performed ona DRX400 Bruker spectrometer (operating at 400.13, 100.62, and 40.55MHz for 1H, 13C, and 15N, respectively) equipped with a Bruker 5 mmBBI Z-gradient probe head capable of producing gradients with astrength of 53.5 G/cm. 15N spectra were referred to external CH3NO2.All spectra were acquired at room temperature using standard 1D and2D NMR experiments on samples typically containing 15-30 mg oflyophilized samples dissolved in 500 µL of D2O (or of a D2O/H2O 1/9mixture, to identify the NH protons). The π/2 pulse lengths were 6.9(1H), 9.25 (13C), and 31.0 µs (15N).
2D 1H-13C and 1H-15N HMBC (heteronuclear multiple bondcorrelation) and HSQC (heteronuclear single quantum coherence) NMRexperiments were typically recorded with 2048 data points in 1Hdimension, 128-512 increments in the heteronuclei dimension, and32-512 transients per increment; 1JHC ) 145, nJHC ) 10, 1JHN ) 88,and nJHN ) 8 Hz were used.
1H pulsed field gradient spin echo (PGSE) NMR experiments wereperformed at room temperature on diluted samples (typically 5 mM inrhenium) containing 0.3 M NaCl to prevent polymer aggregation.22
To check the presence of NMR silent particles, 1 µL of anhydrousdioxane was added as internal standard. A number of experiments werealso performed in still more diluted solutions, typically 0.5 mM. Astimulated echo sequence (STE) incorporating bipolar gradient pulseswas used,23 both with and without water suppression, depending onthe concentration of the samples (standard Bruker pulse programsstebpgp1s19 and stebpgp1s, respectively). The advantage of the use ofthe STE sequence is that the observed echo attenuation is coupled tothe spin-lattice relaxation T1 rather than the spin-spin relaxation T2,
and for polymers, T1 is often much longer than T2.24 This allowsmeasurements using longer diffusion times (∆), necessary for measuringlow diffusion coefficients. The gradient strength (G) was linearlyincremented in 16 steps from 20 to 95% of its maximum value.Diffusion time (∆) ) 500 ms and gradient pulse duration (δ) ) 2.4ms were normally used. When water signal was used as the internalstandard for the diffusion, the gradient strength was varied from 2 to95% to have at least six points for the decay of the water signal, andrecovery delay was left longer than 30 s. We determined translationaldiffusion coefficients (Dt) by evaluating the decrease in the intensity Iof at least three 1H resonances (typically H17, H2, H9, and H5resonances were used; see labels in the chart of Table 1). Standarddeviations of the diffusion coefficients were obtained from the linearfitting of eq 1 (where γ is the gyromagnetic ratio of 1H and τ representsthe time between bipolar gradients),25 and the standard deviations ofthe hydrodynamic radii were computed accordingly using the Origindata analysis software package.
From the diffusion coefficients, the hydrodynamic radii, rH, can beobtained through the Stokes-Einstein eq 2, where k is the Boltzmann’sconstant, T is the absolute temperature, η is the viscosity, and c is afactor depending on the size of the diffusing species with values rangingbetween 4 (slip boundary conditions) and 6 (stick boundary conditions).In our case, the value 6 has been used because rH is much higher thanthat of the solvent.26 We have evaluated the viscosity of the solutionsrelative to that of the pure solvent by measuring the diffusion coefficientof the internal standard dioxane in the absence and in the presence ofthe polymer and of NaCl. Actually, dioxane is a suitable standard forthese measures because it has been shown that it does not interact withpolypeptide chains.27 The ratio between the two Dt values of dioxaneresulted in 1.1 in the 5 mM solutions of the free polymer and of complex1, whereas no significant variation was observed for the more dilutesolutions.
IR spectra were acquired on a Bruker Vector 22 FT instrument using0.1 mm CaF2 cells.
Microwave assisted reactions were performed using a MilestoneMicroSYNTH instrument.
Ultracentrifugations were performed on a Beckman J2-21 centrifugeequipped with a JA-20 35° fixed angle rotor using Amicon Ultra-4 or
Chart 1. Sketch of the Polymer ISA23 and Copolymer ISA23SH10%
lnII0
) -(γδ)2Dt(∆ - δ3- τ
2)G2 (1)
Dt ) (kT)/cπηrH (2)
3274 Biomacromolecules, Vol. 10, No. 12, 2009 Donghi et al.

Centricon centrifugal filter devices (Millipore) with 3000 nominalmolecular weight limit (NMWL) membranes. The solutions were dilutedup to 1:10, and centrifugation processes were repeated until the IRspectrum of the filtrate did not show any absorption attributable toRe(CO)3 fragments. Because of its high dilution, the filtrate wasnormally evaporated in vacuum and dissolved in a very small volumeof water (100 µL) to detect better the possible presence of carbonyl IRbands. In larger preparations, it has been shown that the centrifugationprocess can be substituted by ultrafiltration using an Amicon apparatusthrough a membrane with a 3000 nominal cut off.
The rhenium content in complexes 1 and 2 was determined directlyon aqueous solutions of the complexes (5 mg in 10 mL of water) withan IRIS Intrepid TJA inductively coupled plasma spectrophotometer(ICP) with TEVA software using the Re atomic emission lines at 221.4,227.5, and 346.0 nm and standard solution at 1000 ppm from Fluka.
Size exclusion chromatography (SEC) traces were obtained withToso-Haas TSK-gel G4000 PW and TSK-gel G3000 PW columnsconnected in series using a Waters model 515 HPLC pump equippedwith a Knauer autosampler 3800, a light scattering (LS) Viscotek 270dual detector, UV detector Waters model 486 operating at 230 nm,and a refractive index detector Waters model 2410. The mobile phasewas a 0.1 M Tris buffer pH 8.00 ( 0.05 with 0.2 M sodium chloride.The flow rate was 1 mL min-1 and, the sample concentration was 1%solutions.
Materials. Analytical grade HPLC solvents were purchased fromFluka and used as received for the synthesis of the polymers. Theorganic solvents used in the syntheses of the organometallic complexeswere purified by standard methods. Ultrapure water (Milli-Q, Millipore,resistivity 18 MΩ cm-2) was used for the preparation of the aqueoussolutions. D2O (99.9%) was purchased from Aldrich and used asreceived. Triethylamine purchased from Fluka was distilled overcalcium hydride. Potassium phosphate monobasic was purchased fromCarlo Erba and used as received. 2-Methylpiperazine was purchasedfrom Fluka and used after sublimation. Its final purity (98%) wasdetermined with acidimetric titration. Thin layer chromatography (TLC)was performed using Macherey-Nagel Alugram SIL G/UV254. N,N′-Bis(acrylamido)acetic acid (BAC) was prepared as reported in theliterature,28 and purity (98%) was determined by NMR spectroscopyand titration. Re2(CO)10 was purchased by Sigma-Aldrich and used asreceived. Re(CO)5Br,29 Re(CO)5(CF3SO3),30 and [Re4(CO)12(µ3-OH)4]31,32 were synthesized according to literature procedures. For the
synthesis of [Re(CO)3(H2O)3](CF3SO3), a literature method33 was used,slightly modified by the addition of a small amount of CF3SO3H. (Seethe Results and Discussion.)
N-tert-Butyloxycarbonyl Cystamine Hydrochloride (Cyst-mBocHCl).34 Triethylamine (2.696 g, 26.65 mmol) was added to a drymethanol solution (100 mL) of cystamine bis-hydrochloride (2.00 g,8.88 mmol), and the solution was stirred for 15 min at roomtemperature. Di-tert-butyldicarbonate (1.941 g, 8.88 mmol) was added,and the stirring was maintained for an additional 2.5 h; the reactionprogress was monitored by TLC (SiO2, eluent: chloroform/isopropylalcohol 1:1, Rf,product ) 0.25). The solvent was then evaporated, and a1 M KH2PO4 aqueous solution (60 mL, pH 4.2) was added. The aqueousphase was extracted with diethyl ether (50 mL) to remove di-N,N′-tert-butyloxycarbonyl cystamine and then brought to pH 9 with 1 MNaOH and extracted with ethyl acetate (6 × 15 mL). The combinedorganic phases were dried (Na2SO4) and evaporated to dryness. Theresidue was dissolved in water at pH g 4 (HCl). The clear solutionobtained was freeze-dried, and N-tert-butyloxycarbonyl-cystamine wasisolated as hydrochloride. Yield: 1.03 g (40%). 1H NMR (D2O, δ):3.31-3.21 (m, 4H, NH3
+-CH2 and CONH-CH2), 2.83 (t, 3J3,2 ) 6.40Hz, 2H, CH2-S), 2.72 (t, 3J2,3 ) 6.40 Hz, 2H, CH2-S), 1.30 (s, 9H,Boc CH3). ESI-MS (m/z): 253 (M + 1), as free base.
Synthesis of the Copolymer ISA23-Cyst-mBoc.35 BAC (2,076 g,0.010 mol) and potassium carbonate (1.402 g, 0.010 mol) were dissolvedin water (3.4 mL). Nitrogen was flushed through the reaction mixturefor 10 min to eliminate all CO2 produced. When the reaction mixturebecame clear, Cyst-mBoc ·HCl (0.295 g, 0.001 mol) and potassiumcarbonate (0.150 g; 0.001 mmol) were added. The reaction mixturewas then continuously stirred for 24 h, and afterward 2-methylpiperazine(0.931 g, 0.009 mol) was added. The solution was maintained for 24 hat 20 °C under nitrogen atmosphere and with occasional stirring. Afterthis period, the crude reaction mixture was diluted to 300 mL withdistilled water, acidified to pH 2.5 with 37% hydrochloric acid andpurified by ultrafiltration through a membrane with a nominal cutoffof 5000. The portion retained was recovered by lyophilization. Yield:1.72 g (45.6%). 1H NMR (D2O, δ): 5.45 (m, 1H, CH-COOH),3.46-3.21 (b, 2H, CH2-N polymer backbone and piperazine ring),3.00 (b, 2H, CH2 piperazine ring), 2.79 (b, 2H, CO-CH2), 2.77 (b,2H, S-CH2), 1.34 (s, 9H, Boc CH3), 1.25 (s, 3H, CH3 piperazine). Mj n
) 19 000, Mj w ) 34000, PD ) 1.8. Content of the disulfide-containingunits: 9.85% on a molar basis, determined by NMR.
Table 1. 1H, 13C, and 15N Chemical Shifts (ppm) of the Free Polymer ISA23SH10% at pH 3.5a
δ 1 (1′) 2 (2′) 3 (3′) 4 (4′) 5 (5′) 6 (6′) 7 (7′) 8 (8′) 9 (9′) 10 (10′)1H 3.50 2.65 8.69 5.48 8.76 2.61 3.10
3.13 (2.80) (2.80) (2.60)(3.46)
13C 48.19 29.90 171.88 58.26 173.05 172.44 30.65 52.36(49.89) (28.72) (171.29) (28.72)
15N -248.9 -248.9
δ 11 12 13 14 15 16 17 18 19 20
1H 3.47 3.31 3.29 3.31 1.28 3.36 2.903.08 2.88 2.71 2.82
13C 48.81 49.58 56.17 55.43 14.00 56.07 18.5015N -333.7 -324.7 -328.5a In parentheses the values of the chemical shifts of the minority part are indicated (primed numbers), when different from those of majority part.
Rhenium-Poly(amidoamine) Complexes Biomacromolecules, Vol. 10, No. 12, 2009 3275

Synthesis of the Copolymer ISA23SH10%.35 ISA23-Cyst-mBoc (1.0g, 0.31 mmol disulfide groups from 1H NMR data) was dissolved indegassed water (30 mL) at pH 8 adjusted by the addition of a fewmicroliters of NaOH (1.0000 N Sigma Aldrich, volumetric standard).1,4-Di-thio-D,L-threitol (0.481 g, 3.09 mmol) was then added. Thereaction mixture was stirred for 15 h at room temperature and thendiluted with water and ultrafiltered through a membrane with amolecular weight cutoff 3000 and eventually freeze dried. Yield: 0.795g (79,5%). The NMR spectrum turned to be superimposable to that ofISA23-Cyst-mBoc, apart from the absence of the Tert-Boc protectinggroup. Therefore, the content of the sulphidryl-containing units wasassumed to be equal to that of the disulfide units present in the parentpolymer. Mj n )10 100, Mj w ) 17 100, PD ) 1.7. Elemental analysis:Found ) C, 46.87; H, 8.05; N, 16.80; S, 0.87% (calcd for(C13H22N4O4)0.9015(C10H17N3O4S)0.0985 ·1.5H2O ) C, 47.23; H, 7.65; N,16.91; S, 0.98); Found ) N/C, 0.358; S/C, 0.019 (calcd ) N/C, 0.358;S/C, 0.021).
Attempted Reaction between ISA23 and [Re(CO)3(H2O)3]-(CF3SO3). A sample of 34.0 mg of ISA23 was dissolved in 2 mL ofwater, and the pH of the solution was adjusted to 5.5 by the additionof a few microliters of NaOH. To this clear solution, 114 µL of a 0.1M solution of [Re(CO)3(H2O)3](CF3SO3) was added, leading to a 5mM rhenium concentration. The pH was readjusted to 5.5 by NaOHaddition. The solution mixture was then heated at 50 °C for 3 h. TheIR spectrum of the mixture showed two CO stretching (νCO 2027s,1917vs cm-1) that can be attributed to [Re4(CO)12(µ3-OH)4],31,32
suggesting that no interaction between [Re(CO)3(H2O)3]+ and ISA23took place. To confirm this, the mixture was centrifuged (3000 cutofffilter, 5000 rpm, 20 °C for 30 min). The IR spectrum of the retainedfraction did not show any CO band, whereas the filtrate showed thebands of [Re4(CO)12(µ3-OH)4].
Reaction between ISA23SH10% and [Re(CO)3(H2O)3](CF3SO3). Asample (17.7 mg) of the copolymer ISA23SH10% (effective cysteamineunit ) 9.85%) was dissolved in 1.2 mL of water and treated with 30µL of a 0.1 M solution of [Re(CO)3(H2O)3]+, leading to a 2.5 mMrhenium concentration. The pH was adjusted to 5.5 by NaOH addition,and the solution was heated at 80 °C for 15 min. The IR spectrumshowed that the ν(CO) bands of the starting material (2037s, 1916vscm-1) were replaced by new ν(CO) bands (2012sh, 2002s, 1908vscm-1), attributable to complex 1. After centrifugation (3000 cutoff filter,7000 rpm, 25 °C for 20 min), the IR spectrum of the retained fractionshowed the ν(CO) bands of 1, whereas no CO bands were present inthe IR spectrum of the filtrate. The retained fraction was lyophilized,affording a white very fine powder used for NMR and ICP analyses.Elemental Analysis: Found ) C, 42.56; H, 7.95; N, 15.17; S, 0.79;Re, 2.8% (calcd for (C13H22N4O4)0.9015 (C10H15N3O4S)0.0985(C3O3Re)0.04925 ·3H2O ) C, 42.50; H, 7.58; N, 15.04; S, 0.87; Re, 2.52); Found )N/C, 0.356; S/C, 0.019 (calcd ) N/C, 0.354; S/C, 0.020).
An analogous procedure was used for the reaction with 1 equiv of[Re(CO)3(H2O)3]+, which afforded complex 2 in 30 min at 80°. Longerreaction times did not cause any spectral changes of the reaction mixture.Elemental analysis indicated the presence of 0.85 equiv of rhenium perthiol group: Found ) C, 41.08; H, 7.31; N, 14.44; S, 0.74; Re, 4.2 (calcdfor (C13H22N4O4)0.9015(C10H15N3O4S)0.0985(C3O3Re)0.0837 ·3H2O ) C, 41.77;H, 7.39; N, 14.67; S, 0.85; Re, 4.18%); Found ) N/C, 0.352; S/C, 0.018(calcd ) N/C, 0.351; S/C, 0.020).
Microwave Synthesis of Complex 1. A sample of ISA23SH10% (30.1mg) dissolved in 2 mL of water was treated with 50 µL of a 0.1 Msolution of [Re(CO)3(H2O)3](CF3SO3), leading to a 2.5 mM rheniumconcentration, in an open glass vessel, under a N2 atmosphere. Afterthe pH value was adjusted to 5.2, the mixture was exposed to microwaveradiation at 500 W for 20 s to reach 80 °C and then at 100 W for 2min while the temperature was maintained constant at 80 °C. The IRspectrum showed the replacement of ν(CO) bands of the startingmaterial with those of complex 1. Centrifugation, as above-described,and IR spectra of retained fraction and filtrate confirmed the occurrenceof the interaction. The reaction was repeated at a lower concentration
using 10.5 mg of ISA23SH10% dissolved in 70 mL of water and 17.5µL of a 0.1 M solution of [Re(CO)3(H2O)3](CF3SO3), leading to a 2.5× 10-5 M rhenium concentration. The pH of the solution was 5.5. Themixture was exposed to microwave radiation at 500 W for 1 min toreach 80 °C and then at 100 W for 30 min, while the temperature wasmaintained constant at 80 °C. The solution was centrifuged, as above,and IR spectra of both the retained fraction and the filtrate wererecorded, showing the formation of complex 1.
Stability of Complexes 1 and 2 under Physiological Con-ditions. A sample of complex 1 (10.0 mg) was dissolved in 10 mL ofa 0.9% w/v NaCl solution at pH 7.4, affording a 0.15 mM solution(referred to the rhenium concentration). The solution was maintainedat 25 °C for 36 h and then centrifuged. No ν(CO) bands were observedin the IR spectrum of the filtrate, whereas the retained fraction showedthe bands of the starting complex, confirming that rhenium coordinationto the polymer had been maintained. The experiment was repeated underthe same conditions with complex 2, obtaining the same results.Moreover, the stability of complex 1 was ascertained under much morediluted conditions (4 × 10-5 M) at 37 °C for 30 h.
Stability of Complexes 1 and 2 in the Presence of Cysteine. Asample of complex 1 (15.0 mg) was dissolved in 0.75 mL of water(affording a 3 mM solution, referred to the rhenium concentration) andtreated with 0.73 mL of a 6.6 mM solution of cysteine (0.0048 mmol).The pH was adjusted to 7.4, and the solution was maintained at roomtemperature for 24 h and then centrifuged. No ν(CO) bands wereobserved in the IR spectrum of the filtrate, whereas the retained fractionshowed the bands of the starting complex. The experiment was repeatedunder the same conditions with complex 2, and the same results wereobtained.
Dynamic Light Scattering Measurements. The average diametersand polydispersity indices of the two rhenium complexes weredetermined by dynamic laser light scattering (DLS) using a 90 Plusinstrument (Brookhaven, NY) at a fixed angle of 90° and a temperatureof 25 °C. Samples were prepared by dispersing the freeze-driedcomplexes in filtered water (1 mg/mL) and diluting before analysis.The polydispersity index represents the size distribution of thenanoparticle population.
Morphological Evaluation. The complex morphology was deter-mined by transmission electron microscopy (TEM). TEM analysis wascarried out using a Philips CM10 instrument (Eindoven, NL). Therhenium complexes 1 and 2 in aqueous solution were spread onto aFormwar-coated copper grid and air-dried before observation.
Hemolytic Activity. The hemolytic activity of the rhenium com-plexes 1 and 2 was evaluated on human blood. Different quantities ofthe two complexes were added to a suspension of erythrocytes (30%v/v) in phosphate buffer pH 7.4 to give the following final concentra-tions: 1.2, 2.0, 3.0, 4.5, and 5.5 mg/mL. A suspension (30% v/v) oferythrocytes in phosphate buffer pH 7.4 was used as the blank. Anothererythrocyte suspension added with an excess of ammonium chloridewas used to obtain complete hemolysis of erythrocytes as 100%hemolytic control. After 90 min of incubation at 37 °C, all sampleswere centrifuged at 2000 rpm for 10 min, and the supernatants wereanalyzed using a Lambda 2 Perkin-Elmer spectrophotometer at awavelength of 543 nm. The percentage of hemolysis was calculatedversus the 100% hemolysis control.
Cytotoxicity and Biocompatibility Determination. The in vitrocytotoxicity of the rhenium complexes was evaluated on Hela cellsusing the MTT test (MTT ) (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide). To test the effect of complexes on cell viability,we seeded Hela cells at a density of 5 × 102 in 96-well plates in RPMI-1640 supplemented with 10% of serum (200 µL). The cells wereallowed to attach for 72 h, and the seeding medium was removed andreplaced by experimental medium supplemented with two differentrhenium concentrations (50 and 100 ng/mL). ISA23SH10% and the parenttricarbonyl rhenium complex [Re(CO)3(H2O)3] (CF3SO3) were used asnegative controls at the same concentrations. After 48 h, cell viabilitywas determined by Microplate reader model 450 at 560 nm. The effect
3276 Biomacromolecules, Vol. 10, No. 12, 2009 Donghi et al.

on cell viability of each complex at different concentrations wasexpressed as a percentage by comparing treated cells with cellsincubated with culture medium alone (untreated cells). For the assay,the experiments were performed in triplicate.
Acute toxicity was determined in mice according to standardprotocols by INTOX company (India).
Preliminary in vivo experiments were performed on 6-10 weeksold mice (CD1, Charles River) to evaluate the biocompatibility of thetwo complexes. Volumes (100 or 200 µL) of a 2 mg/mL aqueoussolution of each rhenium complex were administered intravenously inthree mice. The animals were daily controlled for 15 days to observepossible signs of toxicity.
The animal experiments complied with the rules set forth in the NIHGuide for the Care and Use of Laboratory Animals.
Results and Discussion
Synthesis. In this work, [Re(CO)3(H2O)3]+ complex waschosen as the starting reagent for obtaining the PAA-rheniumcomplexes because of its chemical reactivity and ease of thepreparation methods,33,36,37 some of which were suitable forpreparing radioactive derivatives containing the 186,188Reisotopes.11-14 A literature method that involves refluxing for1 h [Re(CO)5OTf] (OTf ) CF3SO3) in water was first at-tempted.33 However, in our experiments, this procedure alwaysafforded [Re(CO)3(H2O)3]OTf contaminated by the “cubane-like” tetranuclear complex [Re4(CO)12(µ3-OH)4]
31,32 (ca. 20%from the IR data). Not surprisingly, this complex does not reactwith the polymer so that when the polymer was treated withsamples of [Re(CO)3(H2O)3]+ contaminated by the “cubane-like” complex the latter was always quantitatively recovered inthe filtrate after ultracentrifugation. The addition to the startingsolution of a few microliters of CF3SO3H (up to pH 2) depressedthe formation of neutral “Re(CO)3(OH)” fragments and gave[Re(CO)3(H2O)3]+ in substantially quantitative yields. Anyway,it is worth mentioning that on increasing the pH to 5.5, the tris-aquo cation [Re(CO)3(H2O)3]+ became thermally unstable andwas completely transformed into [Re4(CO)12(µ3-OH)4] byheating for 30 min at 80 °C.
As polymeric “ligand”, we used an amphoteric PAA nick-named ISA23SH10%, consisting in a PAA of ISA23 structure4
modified by the introduction of ∼10% (on a molar basis) ofcysteamine-deriving units (Chart 1). This was obtained bysubstituting in the ISA23 preparation recipe 10% (on a molarbasis) monoprotected cystamine for an equimolar quantity of2-methylpiperazine and then reductively cleaving the S-Sbonds. The major amine portion of ISA23SH10% is stillconstituted by a 2-methyl piperazine, as in ISA23 (thereforemaintaining its favorable biological properties), with a significantpresence of cysteamine moieties capable of chelating soft metalcenters such as the Re(CO)3
+ core.18-21 The 2-methylpiperazine-deriving moiety of the copolymer is known to have pKa valuesof 3.2 and 7.5.38,39 The pKa value of the 2,2-bis(acrylamido)ace-tic acid deriving moiety is known to be close to 2.3 in all PAAscontaining them.38,39 Considering the pKa values of freecysteamine, that is 10.5 (tert-amine group) and 8.3 (thiolgroup),40,41 the pKa values of the same groups in the cysteamine-deriving moieties may be reasonably expected to be ap-proximately 8.5 and 8.3, respectively. In fact, on the basis ofprevious knowledge,1,2 the former should be lowered by abouttwo pK units as a consequence of the insertion in the PAA chain,whereas the latter should be nearly unchanged. Therefore, atthe pH used in this study (5.5), the copolymer is zwitterionic.
The reactions between ISA23SH10% and [Re(CO)3(H2O)3]+
were monitored by IR spectroscopy, which is a powerful tool
for following the interactions between biological molecules andorganometallic fragments containing CO ligands.42 Indeed, theCO stretching absorptions (hereafter νCO) are strong andsensitive to the coordination environment. The IR spectrum ofa 5 mM (hereafter, the concentrations and the stoichiometricratios are referred to the SH-containing cysteamine part of thepolymer) water solution of ISA23SH10% treated with 0.5 equivof [Re(CO)3(H2O)3]+ (pH 5.5, 80 °C) showed the appearanceof two new broad νCO bands peaking at 2002 and 1908 cm-1
(with a shoulder at 2012 cm-1, see Figure 1), shifted to lowerfrequencies with respect to those of the starting tris-aquocomplex (2037 and 1916 cm-1).
The reaction was complete in 15 min, as ascertained by theIR data and by the lack of νCO bands in the filtrate, obtained byultracentrifugation with a 3000 cutoff filter, to eliminate allrhenium complexes not bound to the polymer. The portionretained from the centrifugation was then freeze-dried, affordinga white powder (complex 1) containing 2.8% w/w of Re, asshown by ICP analysis. The reaction time was further reducedto 2 min by using microwave heating (see the ExperimentalPart), which is in agreement with a recent report on themicrowave syntheses of [Re(CO)3(H2O)3]+ and its bioconju-gates.14 The reaction with 0.5 equiv of rhenium per thiol wasfound to be effective even down to 2.5 × 10-5 M rheniumconcentration. In this case, the reaction time at 80 °C was 3 h,reduced to 30 min by microwave heating.
Interestingly, ISA23SH10% completely inhibits the condensa-tion of the Re(CO)3 units to the “cubane-like” derivative[Re4(CO)12(µ3-OH)4], which occurs upon heating solutions of[Re(CO)3(H2O)3]+ at pH 5.5. The same inhibitory effect doesnot occur with ISA23, indicating that the coordination sites ofrhenium ions are the cysteamine moieties, as confirmed by theNMR data. (See below.)
Figure 1. FTIR spectra in the νCO region in H2O solution of (a) complex1 (s), the starting material [Re(CO)3(H2O)3]+ ( · · · ), and the filtrateafter ultracentrifugation ( · - ·-) and (b) complex 1 (s) and complex2 ( · · · ).
Rhenium-Poly(amidoamine) Complexes Biomacromolecules, Vol. 10, No. 12, 2009 3277

A complex (2) with a higher rhenium content (4.2% w/w)was obtained by an analogous procedure on treating thecopolymer ISA23SH10% with 1 equiv of [Re(CO)3(H2O)3]+ perthiol (pH 5.5, 30 min at 80 °C, ν(CO) at 2009 and 1906 cm-1,Figure 1b). However, the rhenium content of complex 1 isalready adequate for biological applications, and subsequentwork was focused on it.
No reaction occurred at pH >5.5 (7.4 and 10), probablybecause of the fast oligomerization of [Re(CO)3(H2O)3]+
10,43-45
despite the fact that the complexes, once formed, are stable atpH 7.4. In particular, both 1 and 2 were incubated at aconcentration 10-4 M for 36 h (a time fully consistent with thehalf life of the 99mTc and 188Re radioisotopes) under physiologi-cal conditions (pH 7.4 and 0.9% w/v NaCl). The IR spectrumof the filtrate did not show any metal-carbonyl stretching bands,whereas the IR spectrum of the retained portion was identicalto that of the complexes before the incubation. The samecomplexes were also stable for over 18 h in the presence ofcysteine, a potential in vivo ubiquitous competitor for the metalcoordination.
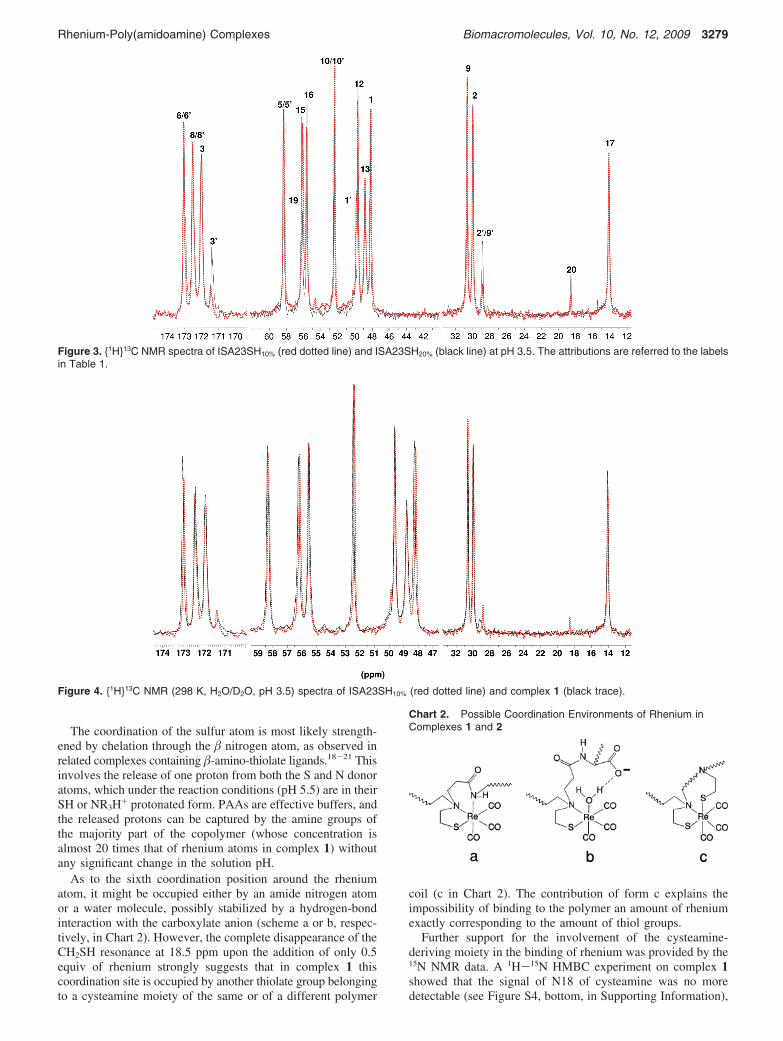
Nuclear Magnetic Resonance Characterization. A detailedmultinuclear (1H, 13C, and 15N) NMR characterization of thecopolymer ISA23SH10% and of its complex with 0.5 equiv ofrhenium per thiol (complex 1) was undertaken, with the aim ofobtaining information on which of the potential binding sitespresent in the polymer (SH, COOH, and amines) was actuallyinvolved in rhenium coordination. The attributions of theresonances of the copolymer have been obtained mainly by 2D1H-13C and 1H-15N HMBC and HSQC experiments and arereported in Table 1. The position of the signals is slightlysensitive to pH: In Table 1, the data acquired at pH 3.5 areshown because of the better spectral resolution at this pH. Thechemical shift values at pH 5.5, that is, the pH value used inmost of the reactivity studies, are slightly different and arereported in Table S1 in the Supporting Information. Theattributions of the signals of the minority part have been assistedby the analysis of the spectra of the copolymer containing adouble percentage of cysteamine (ISA23SH20%). In the Sup-porting Information (Figures S1 and S2), we have reported the1H-13C correlation experiments concerning the latter copolymerfor the sake of clarity.
Typical 1H and 1H13C NMR spectra at pH 3.5 are depictedin Figures 2a and 3, respectively. The positions of the resonancesof the 2-methylpyperazine units are consistent with literaturedata.39,46 The resonances of the thiol-bearing units were hardlydetectable in the spectra, being weak and largely overlappedwith those of the major constituents. However, the proton spectraat pH 3.5 (Figure 2a) showed, in the narrow gap between themultiplets of methylenes 13 and 16, a group of signals that byheterocorrelation experiments were assigned to the protons inthe sites 2′, 9′, and 20. (See the labels of the chart in Table 1.)The corresponding carbon resonances were also detectablein the 13C spectra, together with those of carbons 1′ and 3′(Figure 3).
The attribution of these signals to the thiol-bearing portionis well supported by the comparison with the spectra of theanalogous copolymer with a double percentage of SH(ISA23SH20%), as shown in Figure 3 and Figure S3 in theSupporting Information. The 15N signals of the amide groupsN4′ and N7′ were also overlapped with the corresponding signalsN4 and N7 of the majority part, whereas the signal of thecysteamine N18 was detected at -328.5 ppm (Figure S4 top inthe Supporting Information).
The 1H and 1H13C spectra of the rhenium-containingpolymer (complex 1) showed only minimal variations withrespect to those of the parent polymer. However, these variationshad a diagnostic value because they specifically involved theresolved signals of the thiol-bearing units, as apparent in the13C spectrum reported in Figure 4. The most significant change,besides the broadening of the 13C signals 2′, 3′, and 9′, was thedisappearance of the signal at 18.5 ppm due to carbon atom Rto SH group (C20).
Also, the proton resonance of this site was no more recogniz-able in the 1H spectrum (Figure 2b). This might be due to alow field shift, resulting in the overlap with some of the moreintense signals of the majority part. Indeed, it is known fromliterature data that the 13C resonance of CH2SH fragmentsundergo a downfield shift upon coordination to a metalcenter.41,47,48 A smaller (ca. 3 ppm) downfield shift also occursupon simple deprotonation of the SH group,48 as we verifiedexperimentally on a D2O solution of cysteamine.
Figure 2. 1H NMR spectrum of (a) the free polymer ISA23SH10% and (b) complex 1 (298 K, pH 3.5, H2O/D2O 9:1, water signal suppressed). Theattributions are referred to the labels in Table 1.
3278 Biomacromolecules, Vol. 10, No. 12, 2009 Donghi et al.
The coordination of the sulfur atom is most likely strength-ened by chelation through the nitrogen atom, as observed inrelated complexes containing -amino-thiolate ligands.18-21 Thisinvolves the release of one proton from both the S and N donoratoms, which under the reaction conditions (pH 5.5) are in theirSH or NR3H+ protonated form. PAAs are effective buffers, andthe released protons can be captured by the amine groups ofthe majority part of the copolymer (whose concentration isalmost 20 times that of rhenium atoms in complex 1) withoutany significant change in the solution pH.
As to the sixth coordination position around the rheniumatom, it might be occupied either by an amide nitrogen atomor a water molecule, possibly stabilized by a hydrogen-bondinteraction with the carboxylate anion (scheme a or b, respec-tively, in Chart 2). However, the complete disappearance of theCH2SH resonance at 18.5 ppm upon the addition of only 0.5equiv of rhenium strongly suggests that in complex 1 thiscoordination site is occupied by another thiolate group belongingto a cysteamine moiety of the same or of a different polymer
coil (c in Chart 2). The contribution of form c explains theimpossibility of binding to the polymer an amount of rheniumexactly corresponding to the amount of thiol groups.
Further support for the involvement of the cysteamine-deriving moiety in the binding of rhenium was provided by the15N NMR data. A 1H-15N HMBC experiment on complex 1showed that the signal of N18 of cysteamine was no moredetectable (see Figure S4, bottom, in Supporting Information),
Figure 3. 1H13C NMR spectra of ISA23SH10% (red dotted line) and ISA23SH20% (black line) at pH 3.5. The attributions are referred to the labelsin Table 1.
Figure 4. 1H13C NMR (298 K, H2O/D2O, pH 3.5) spectra of ISA23SH10% (red dotted line) and complex 1 (black trace).
Chart 2. Possible Coordination Environments of Rhenium inComplexes 1 and 2
Rhenium-Poly(amidoamine) Complexes Biomacromolecules, Vol. 10, No. 12, 2009 3279

whereas the signals of the piperazine nitrogen atoms (N11 andN14) were observed at the same positions of the free polymer.A substantial drop in the resolution was also observed withrespect to the 2D map recorded on the free polymer. Thecoordination of nitrogen to a rhenium atom is expected to causenot only a shift in its 15N resonance49-51 but also broadeningof the signal and possible loss of magnetization during the 2Dexperiments for the decrease in the effective T2, resulting fromthe scalar coupling with the quadrupolar rhenium isotopes.Actually, both the 185Re and 187Re natural isotopes of rheniumare quadrupolar (I ) 5/2), and the coupling with these isotopesoften results in broadening or even disappearance of the involvedresonances.49 This well explains the disappearance of the signalof coordinated nitrogen. The additional failure of observing theresonance of the remaining half of the cysteamine-derivingnitrogen atoms (i.e., those not rhenium bonded) can be explainedby taking into account their low concentration (one-half of theinitial one) and their possible spreading in several nonidenticalsites together with the difficulty of achieving a high signal-to-noise ratio in this type of experiment.
In related cases in which the disappearance of the 15N signalupon coordination was reported, the presence of dynamicprocesses was invoked as a possible cause of such a finding.52
However, in our case, no significant line width variation wasobserved in 1H13C spectra acquired at higher temperature (343K), allowing us to rule out the hypothesis that the observeddisappearance/broadening of some signals arise from the slowexchange between slightly different coordination environments.
It may be observed that the carboxyl groups do not seem tobe involved in complex formation; in other words, the ampho-teric nature of the carrier is irrelevant in regards to complexation.However, as explained in the Introduction, it imparts to thecarrier a combination of favorable properties from the biologicalstandpoint.
To ascertain whether the formation of complex 1 involvedsome extent of polymer aggregation caused by the coordinationon the same metal center of thiolate groups from differentpolymer coils we have performed two types of NMR checks.First of all, by using a suitable internal standard (see theExperimental Part), we verified that the observed proton signalsaccounted for more than 90% of the amount of weighted sample.This rules out the presence of very large aggregates in solution,which are usually NMR silent.53
Second, we evaluated the polymer size by means of pulsedgradient spin echo (PGSE) NMR experiments. Through thismethod, diffusion coefficients are determined by least-squaresfitting of the signal intensities as a function of the gradientstrength (eq 1),23 and the hydrodynamic radii, rH, of the randompolymers are computed by using the Stokes-Einstein eq 2. Inthis equation, the spherical approximation is used, which is themost convenient model to provide a rough estimation of theparticle size. Moreover, this hypothesis is well supported byTEM analysis. (See below.)
The fittings of the signal attenuations according to eq 1 werewell linear (Figure S5 in the Supporting Information), showingthat under these conditions the samples can be considered tobe substantially monodisperse.54 The values of the diffusioncoefficients and of the hydrodynamic radii so estimated arereported in Table 2. The values for the free copolymer are almostidentical to those previously estimated with the same methodfor the related polymer ISA23.55 The comparison of the size ofthe free polymer and of complex 1 shows that the presence ofrhenium causes a slight increase in the particles size (FigureS5 in the Supporting Information), which is in agreement with
the hypothesis of a certain degree of intercoil interaction aroundthe rhenium centers. On the contrary, the diffusion coefficientsonly slightly vary with pH, at least in the range of 3.5-7.4,and with the concentration, at least in the range of 0.5-5 mM(as thiol groups), and in the presence of 0.3 M NaCl (added toreduce polymer aggregation).22 It has not been possible toanalyze more diluted samples because low intensity signals areaffected by too large errors to afford reliable diffusion coef-ficients.24
Morphological Evaluation. The morphology of complexes1 and 2 was evaluated by TEM analysis. Both complexes (seeFigure 5) form nanoparticles with a quite spherical morphology,average diameter <25 nm, and a narrow size distribution. Inparticular, the size of complex 1 ranged from about 5 to 15nm, whereas the size of complex 2 ranged from about 7 to20 nm. The DLS determinations showed a mean diameter of∼15 nm for complex 1 and of ∼22 nm for complex 2, withpolydispersity indices of 0.1. These values are slightly largerthan those obtained by NMR, as is often found when the resultsof these two techniques are compared.53 These sizes mayrepresent a crucial determinant for the in vivo biodistributionand accumulation of the rhenium complex nanoparticles.Homogeneous distribution and sizes are important propertiesthat can affect the circulation time and intracellular trafficking.Previous studies have shown that small particles have the highercell uptake.56
Preliminary Biological Evaluation. No hemolytic activitywas observed for ISA23SH10% and the two rhenium complexesup to a concentration of 5.5 mg/mL. No significant toxic effectswere observed for the two rhenium complexes on Hela cellsup to 100 ng/mL after 48 h of exposure (Figure 6). Thisobservation time was considered to be amply sufficient for the
Table 2. Translational Diffusion Coefficients (Dt) andHydrodynamic Radii (rH) Estimated through PGSE Experiments(300 K, D2O, NaCl ) 0.3 M)a
sample concentration (mM)b pH Dt × 1011 (m2/s) rH (nm)
ISA23SH10% 0.5 3.5 5.4(1) 3.9(1)ISA23SH10% 5 3.5 4.34(5) 4.4(1)ISA23SH10% 5 5.5 4.41(3) 4.32(3)ISA23SH10% 5 7.4 4.32(3) 4.41(3)complex 1 0.5 3.5 4.1(1) 5.1(1)complex 1 5 5.5 3.57(2) 5.33(4)complex 1 5 3.5 3.7(1) 5.2(2)complex 1 5 5.5 3.22(4) 5.9(1)complex 1 5 7.4 3.38(8) 5.6(2)
a In parentheses is reported the uncertainty on the last digit. The valuesin italics refer to a polymer sample with Mj n ) 20 500 and PD ) 3.8,whereas all other values have been measured on a sample with Mj n )19 000 and PD ) 1.8. b Referred to the thiol groups.
Figure 5. TEM photomicrograph of PAA-rhenium complexes: (a)complex 1 (magnification 105 000×) and (b) complex 2 (magnification145 000×).
3280 Biomacromolecules, Vol. 10, No. 12, 2009 Donghi et al.

envisaged medical application, which requires storage time andin vivo stability consistent with the stability of the radiophar-maceutical involved. (See above.) A slight difference wasobserved between the two concentrations (50 and 100 ng/mL,see Figure 6) showing a dose dependence. The parent polymerISA23SH10% and the free tricarbonyl rhenium complex did notshow any cytotoxic effect at the tested concentrations. Acutetoxicity experiments carried out according to standard protocolsgave a maximum tolerated dose (MTD) value of 500 mg/kgfor ISA23SH10%. Preliminary in vivo studies were performedon the two rhenium complexes, which were intravenouslyinjected in mice in doses up to 20 mg/kg, corresponding torhenium doses up to about 1 mg/kg. All mice survived, and notoxic signs were observed.
Conclusions
This study has shown that a PAA containing cysteamine-deriving moieties carrying thiol pendants is able to tightly bindRe(CO)3
+ fragments, up to a 0.85:1 Re/SH molar ratio. Thecomplexation reaction is fast and occurs upon thermal ormicrowave activation of the reagents. The complexes maintainthe water solubility of the free polymer and survive underphysiological conditions, even in the presence of added excesscysteamine. Moreover, the lack of toxicity has been evidencedby preliminary in vitro and in vivo studies. PGSE, DLS, andTEM measurements indicate that the polymer coils aggregatein solution, giving nanoparticles sufficiently large to avoid rapidrenal excretion. Owing to these favorable properties, coupledto the intrinsic tumor targeting capability of amphoteric PAAs,the rhenium-complexing polymers here reported warrant asignificant diagnostic and therapeutic potential for preparingradiopharmaceutical formulations of rhenium and technetium,owing to the strong similarity of their chemical properties.
Supporting Information Available. NMR data and detailsof the NMR experiments. This material is available free ofcharge via the Internet at http://pubs.acs.org.
References and Notes(1) Ferruti, P.; Marchisio, M. A.; Barbucci, R. Polymer 1985, 26, 1336–
1348.(2) Ferruti, P.; Marchisio, M. A.; Duncan, R. Macromol. Rapid Commun.
2002, 23, 332–355.(3) Maeda, H.; Matsumura, Y. Crit. ReV. Ther. Drug Carrier Syst. 1989,
6, 193–210.(4) Richardson, S.; Ferruti, P.; Duncan, R. J. Drug Targeting 1999, 6,
391–404.(5) Mindt, T.; Struthers, H.; Garcia-Garayoa, E.; Desbouis, D.; Schibli,
R. Chimia 2007, 61, 725–731.
(6) Mazzi, U.; Riondato, M. Radiometal Labelled Complexes for Diag-nostic Imaging and Radiotherapy. In Recent DeVelopment in Bioinor-ganic Chemistry; Saviano, M., Ed.; Transword Research Network:Trivandrum, India; 2006; pp 1-33.
(7) Liu, S. Chem. Soc. ReV. 2004, 33, 445–461.(8) Wynn, A.; Volkert, W. A.; Hoffman, T. J. Chem. ReV. 1999, 99, 2269–
2292.(9) Dilworth, J. R.; Parrott, S. J. Chem. Soc. ReV. 1998, 27, 43–55.
(10) Alberto, R.; Schibli, R.; Waibel, R.; Abram, U.; Schubiger, A. P.Coord. Chem. ReV. 1999, 190-192, 901–919.
(11) Schibli, R.; Schwarzbach, R.; Alberto, R.; Ortner, K.; Schmalle, H.;Dumas, C.; Egli, A.; Schubiger, A. Bioconjugate Chem. 2002, 13,750–756.
(12) Alberto, R.; Schibli, R.; Egli, A. (Mallinckrodt Medical Inc.). PatentWO/1998/048848, 1998.
(13) Park, S. H.; Byun, M. W.; Jang, S. H. (Korean Atomic EnergyResearche Inst.). Eur. Pat. Appl. 1797904 A1, 2007.
(14) Causey, P. W.; Besanger, T. R.; Schaffer, P.; Valliant, J. F. Inorg.Chem. 2008, 47, 8213–8221.
(15) Bartholoma, M.; Valliant, J.; Maresca, K. M.; Babich, J.; Zubieta, J.Chem. Commun. 2009, 493–512.
(16) Lim, N. C.; Ewart, C. B.; Bowen, M. L.; Ferreira, C. L.; Barta, C. A.;Adam, M. J.; Orvig, C. Inorg. Chem. 2008, 47, 1337–1345, andreferences therein.
(17) Riondato, M.; Camporese, D.; Martın, D.; Suades, J.; Alvarez-Lorena,A.; Mazzi, U. Eur. J. Inorg. Chem. 2005, 4048–4055.
(18) Lazarova, N.; Babich, J.; Valliant, J.; Schaffer, P.; James, S.; Zubieta,J. Inorg. Chem. 2005, 44, 6763–6770.
(19) Park, S. H.; Gwon, H. J.; Park, K. B. Chem. Lett. 2004, 33, 1278–1279.
(20) Lipowska, M.; Hansen, L.; Xu, X.; Marzilli, P. A.; Taylor, A., Jr.;Marzilli, L. G. Inorg. Chem. 2002, 41, 3032–3041.
(21) Kramer, D. J.; Davidson, A.; Davis, W. M.; Jones, A. G. Inorg. Chem.2002, 41, 6181–6183.
(22) Mendichi, R.; Ferruti, P.; Malgesini, B. Biomed. Chromatogr. 2005,19, 196–201.
(23) Stilbs, P. Prog. Nucl. Magn. Reson. Spectrosc. 1987, 19, 1–45.(24) Grythe, K. F.; Hansen, F. K.; Walderhaug, H. J. Phys. Chem. B 2004,
108, 12404–12412.(25) Wu, D.; Chen, A.; Johnson, C. S., Jr. J. Magn. Reson., Ser. A 1995,
115, 123–126.(26) Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Chem.
Soc. ReV. 2008, 37, 479–489.(27) Wilkins, D. K.; Grimshaw, S. B.; Receveur, V.; Dobson, C. M.; Jones,
J. A.; Smith, L. J. Biochemistry 1999, 38, 16424–16431.(28) Ferruti, P.; Ranucci, E.; Trotta, F.; Gianasi, E.; Evagorou, G. E.; Wasil,
M.; Wilson, G.; Duncan, R. Macromol. Chem. Phys. 1999, 200, 1644–1654.
(29) Schmidt, S.; Trogler, W.; Basolo, F. Inorg. Synth. 1985, 23, 41–46.(30) Schmidt, S.; Nitschke, J.; Trogler, W. Inorg. Synth. 1989, 26, 113–
117.(31) Herberhold, M.; Suess, G. Angew. Chem., Int. Ed. Engl. 1975, 14,
700.(32) Herberhold, M.; Suess, G.; Ellerman, J.; Gaebelein, H. Chem. Ber.
1978, 111, 2931–2941.(33) He, H.; Lipowska, M.; Xu, X.; Taylor, A. T.; Carlone, M.; Marzilli,
L. Inorg. Chem. 2005, 44, 5437–5446.(34) Jacobson, K. A.; Fischer, B.; Ji, X. Bioconjugate Chem. 1995, 6, 255–
263.(35) Garnett, M.; Ferruti, P.; Ranucci, E. (University of Nottingham,
Universita degli Studi di Milano). Patent WO/2008/038038, 2008.(36) Alberto, R.; Egli, A.; Abram, U.; Hegetschweiler, K.; Gramlich, V.;
Schubiger, A. J. Chem. Soc., Dalton Trans. 1994, 2815–2820.(37) Lazarova, N.; James, S.; Babich, J.; Zubieta, J. Inorg. Chem. Commun.
2004, 7, 1023–1026.(38) Ferruti, P.; Manzoni, S.; Richardson, S. C. W.; Duncan, R.; Pattrick,
N. G.; Mendichi, R.; Casolaro, M. Macromolecules 2000, 33, 7793–7800.
(39) Franchini, J.; Ranucci, E.; Ferruti, P.; Rossi, M.; Cavalli, R. Biom-acromolecules 2006, 7, 1215–1222.
(40) Sugiura, Y.; Hojo, Y.; Tamai, Y.; Tanaka, H. J. Am. Chem. Soc. 1976,98, 2339–2341.
(41) Fleischer, H.; Dienes, Y.; Mathiasch, B.; Schmitt, V.; Schollmeyer,D. Inorg. Chem. 2005, 44, 8087–8096.
(42) Bioorganometallics: Biomolecules, Labeling, Medicine; Jaouen, G.,Ed.; Wiley-VCH: Weinheim, Germany, 2006.
(43) Salignac, B.; Grundler, P.; Cayemittes, S.; Frey, U.; Scopelliti, R.;Merbach, A.; Hedinger, R.; Hegetschweiler, K.; Alberto, R.; Prinz,U.; Raabe, G.; Kolle, U.; Hall, S. Inorg. Chem. 2003, 42, 3516–3526.
Figure 6. Viability of Hela cells after 48 h of incubation with thecomplexes 1 and 2 with respect to untreated cells as positive control(100%). The parent polymer ISA23SH10% and the free tricarbonylrhenium complex did not show any cytotoxic effect at the testedconcentrations.
Rhenium-Poly(amidoamine) Complexes Biomacromolecules, Vol. 10, No. 12, 2009 3281

(44) Egli, A.; Hegetschweiler, K.; Alberto, R.; Abram, U.; Schibli, R.;Hedinger, R.; Gramlich, V.; Kissner, R.; Schubiger, P. A. Organo-metallics 1997, 16, 1833–1840.
(45) Alberto, R.; Egli, A.; Abram, U.; Hegetschweiler, K.; Gramlich, V.;Schubiger, P. J. Chem. Soc., Dalton Trans. 1994, 2815–2820.
(46) Ranucci, E.; Suardi, M. A.; Annunziata, R.; Ferruti, P.; Chiellini, F.;Bartoli, C. Biomacromolecules 2008, 9, 2693–2704.
(47) Briand, G. G.; Cooper, B. F. T.; MacDonald, D. B. S.; Martin, C. D.;Schatte, G. Inorg. Chem. 2006, 45, 8423–8429.
(48) Isab, A.; Wazeer, M. Spectrochim. Acta, Part A 2007, 66, 364–370.(49) Multinuclear NMR; Mason, J., Ed.; Plenum Press, New York, 1987.(50) Pazderski, L.; Szlyk, E.; Sitkowski, J.; Kamienski, B.; Kozerski, L.;
Tousek, J.; Marek, R. Magn. Reson. Chem. 2006, 44, 163–170.(51) Marek, R.; Lycka, A. Curr. Org. Chem. 2002, 6, 35–66.
(52) Jantti, A.; Wagner, M.; Suontamo, R.; Kolhemainen, E.; Rissanen,K. Eur. J. Inorg. Chem. 1998, 1555–1562.
(53) Sanna, C.; La Mesa, C.; Mannina, L.; Stano, P.; Viel, S.; Segre, A.Langmuir 2006, 22, 6031–6041.
(54) Walderhaug, H.; Hansen, F. K.; Abrahmsen, S.; Persson, K.; Stilbs,P. J. Phys. Chem. B 1993, 97, 8336–8342.
(55) Griffiths, P. C.; Paul, A.; Khayat, Z.; Wan, K.-W.; King, S. M.; Grillo,I.; Schweins, R.; Ferruti, P.; Franchini, J.; Duncan, R. Biomacromol-ecules 2004, 5, 1422–1427.
(56) Desai, M. P.; Labhasetawar, V.; Walter, E.; Levy, R. J.; Amidon, G. L.Pharm. Res. 1997, 14, 1568–1573.
BM9008638
3282 Biomacromolecules, Vol. 10, No. 12, 2009 Donghi et al.


















