
Sviluppo di nanoparticelle per imaging di brille amiloidi€¦ · pione, o alla stessa lunghezza...
97
Transcript of Sviluppo di nanoparticelle per imaging di brille amiloidi€¦ · pione, o alla stessa lunghezza...

Sviluppo di nanoparticelle per imaging di
�brille amiloidi
Ruggiero Maria Pesce mat. 739726
Ottobre, 2011

Indice
1 Introduzione 4
1.1 Le malattie neurodegenerative . . . . . . . . . . . . . . . . . . . 4
1.1.1 L'incidenza dell'Alzheimer in Europa e in Italia . . . . . 4
1.1.2 Pro�lo clinico e patologico . . . . . . . . . . . . . . . . . 5
1.1.3 Meccanismo della polimerizzazione delle proteine . . . . 6
1.1.4 Caratteristiche della polimerizzazione dipendente dalla
nucleazione . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.2 Esteri�cazione di Steglich . . . . . . . . . . . . . . . . . . . . . 8
1.3 Polimerizzazione in emulsione . . . . . . . . . . . . . . . . . . . 10
1.3.1 Meccanismo di produzione delle particelle . . . . . . . . 11
1.3.2 Gli intervalli della polimerizzazione in emulsione . . . . . 13
1.3.3 Polimerizzazione in emulsione in condizioni starved . . . 18
1.3.4 E�etto delle limitazioni di�usive sul processo . . . . . . . 20
1.4 ROP : Ring Opening Polymerizazion . . . . . . . . . . . . . . . 23
1.4.1 Polimerizzazione ROP dell'acido polilattico . . . . . . . . 24
1.5 Coloranti azoici . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
1.5.1 Reazioni di copulazione dei sali di diazonio . . . . . . . . 26
2 Materiali e Metodi 29
2.1 Acido polilattico . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.2 2-idrossietil metacrilato(HEMA) . . . . . . . . . . . . . . . . . . 30
i

2.3 Acido ammino benzoico . . . . . . . . . . . . . . . . . . . . . . 30
2.4 Composti per i coloranti azoici . . . . . . . . . . . . . . . . . . . 31
2.5 Iniziatori e emulsionanti per le
polimerizzazioni . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti 35
2.6.1 Light-scattering . . . . . . . . . . . . . . . . . . . . . . . 35
2.6.2 NMR : Risonanza magnetica nucleare . . . . . . . . . . . 38
2.6.3 Analisi alla Fluorescenza . . . . . . . . . . . . . . . . . . 41
2.6.4 Analisi UV . . . . . . . . . . . . . . . . . . . . . . . . . 43
3 Sintesi 44
3.1 PLA route . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.1.1 Sintesi del PLA . . . . . . . . . . . . . . . . . . . . . . . 45
3.1.2 Reazione di esteri�cazione di Steglich . . . . . . . . . . . 46
3.1.3 PLA route : discussione . . . . . . . . . . . . . . . . . . 49
3.2 NPS a partire da HEMA e MMA . . . . . . . . . . . . . . . . . 49
3.3 Sintesi dei macromonomeri . . . . . . . . . . . . . . . . . . . . . 50
3.3.1 Reazione di esteri�cazione di Steglich . . . . . . . . . . . 51
3.3.2 Reazione di copulazione e formazione dei
coloranti azoici . . . . . . . . . . . . . . . . . . . . . . . 56
3.4 Sintesi delle nanoparticelle . . . . . . . . . . . . . . . . . . . . . 60
3.4.1 Analisi al light-scattering . . . . . . . . . . . . . . . . . . 62
4 Analisi prodotti sintetizzati 64
4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici . . . 64
4.1.1 Preparazione degli aggregati . . . . . . . . . . . . . . . . 65
4.1.2 Test dei materiali prodotti . . . . . . . . . . . . . . . . . 66
4.1.3 Test sulla la cinetica di aggregazione delle proteine . . . 72
4.2 Analisi all'UV . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
4.2.1 Analisi all'UV : macromonomeri . . . . . . . . . . . . . . 76
ii

4.2.2 Analisi all'UV : NPs . . . . . . . . . . . . . . . . . . . . 81
5 Conclusioni 87
Bibliogra�a 91
iii

Sommario
Il presente lavoro é incentrato sullo sviluppo e caratterizzazione di nanopar-
ticelle destinate ad essere utilizzate come markers per la determinazione di
�brille amiloidi tramite tecniche di neuro-imaging non invasive.
L'accumulo di tali aggregati proteici nel cervello é un aspetto comune
di molte malattie neurodegenerative, come il morbo di Alzheimer, quello di
Parkinson e la corea di Huntington.
Attualmente come markers di queste �brille vengono utilizzati coloranti
quali la tio�avina T ed una serie di coloranti azoici, come ad esempio il Congo
Red o la Crisamina G. Queste molecole sono in grado di legarsi alle proteine
che formano gli aggregati, sono �uorescenti, e per questo sono largamente
utilizzate nello studio in vitro della cinetica e delle proprietá strutturali delle
�brille amiloidi.
In questo lavoro di tesi l'obiettivo é quello di produrre nanoparticelle fun-
zionalizzate con quattro tipi diversi di coloranti azoici e di utilizzarle per
l'imaging di questi aggregati proteici. I materiali prodotti hanno la carat-
teristica di essere �uorescenti, ovvero se vengono eccitati ad una determinata
lunghezza d'onda emettono ad una lunghezza d'onda diversa da quella di ecc-
itazione e propria del colorante con il quale sono state funzionalizzate. Queste
poi, fatte reagire con gli aggregati proteici ed eccitate alla lunghezza d'onda
propria del materiale, potrobbero presentare un di�erente spettro di emissione
se hanno intergito con le �brille amiloidi. La di�erenza risiede o in una diversa
1

lunghezza d'onda di emissione rispetto a quella registrata per il solo cam-
pione, o alla stessa lunghezza d'onda d'emissione nella presenza di un picco
decisamente piú alto in termini di segnale registrato.
Il grosso vantaggio di utilizzare delle nanoparticelle per l'imaging di �b-
rille amiloidi é che dato la loro maggiore dimensione rispetto ai coloranti
attualmente utilizzati é possibile vederli tramite tecniche di microscopia elet-
tronica(SEM. TEM). Questo potrebbe dare un grosso contributo nel capire i
meccanismi che permettono e regolano il legame tra coloranti e �brille amiloidi
perché sarebbe possibile avere delle immagini chiare di questo atto reattivo,
cosa che adesso non é possibile avere a disposizione.
Le nanoparticelle inoltre sono state sintetizzate a partire da polimeri
biodegradabili e biocompatibili a base di idrossietil-metacrilato (HEMA) e
meta-metilacrilato(MMA). Questo fa si che rispondano alle caratteristiche
necessarie per essere utilizzate in futuro nel campo del drug delivery. Si
avrebbero cosí a disposizione dei materiali che potrebbero essere utilizzati in
tecniche di imaging direttamente in vivo.
Gli step reattivi utilizzati per la produzione sono stati l'esteri�cazione di
Steglich, la polimerizzazione in emulsione e la reazione di copulazione e sintesi
del colorante azoico.
Le molecole utilizzate nella reazione di formazione dei coloranti azoici sono
state il 2-naftolo, l'acido solfanilico e l'acido salicilico. Inoltre, data la sua
capacitá di prestarsi a questo tipo di reazione, si é scelto di utilizzare anche la
tio�avina T, colorante che viene giá utilizzato per l'imaging di �brille amiloidi
con esiti positivi e scienti�camente riconosciuti.
Tutte le reazioni comprendenti il processo di sentesi sono state ottimizzate
sia mediante l'ultilizzo di solventi diversi, sia testando cammini sintetici dif-
ferenti.
2

Il lavoro é stato svolto presso il Dipartimento di Chimica Materiali e Ingeg-
neria Chimica del Politecnico di Milano, per quanto riguarda la produzione e
ottimizzazione dei macromonomeri funzionalizzati. Il seguito del lavoro, quello
riguardante la produzione di nanoparticelle, loro caratterizzazione in termini
di dimensione media, proprietá all'UV e alla �uorescenza, ed il successivo test
su aggregati di �brille-amiloidi é stato condotto presso i laboratori del gruppo
del Professor Massimo Morbidelli all'ETH di Zurigo.
In particolare i macromonomeri e le nanoparticelle sono stati testati su
aggregati proteici di insulina e ne é stata valutata l'e�cienza come markers
tramite analisi alla �uorescenza. Sono state utilizzate queste proteine perché
anche se non sono direttamente collegate alle malattie nuerodegenerative, nella
forma aggregata �nale presentano la stessa conformazione a foglietto β di quelle
che invece lo sono. Inoltre le proteine di insulina sono state largamente studiate
e in letteratura si possono trovare numerose informazioni a riguardo.
3

Capitolo 1
Introduzione
In questo capitolo introduttivo verrá data qualche nozione sulle malattie neu-
rodegenerative in modo tale da chiarire il quadro generale entro cui si in-
serisce questo lavoro. Inoltre saranno spiegate in maniera generale le reazione
utilizzate nella sintesi dei prodotti.
1.1 Le malattie neurodegenerative
1.1.1 L'incidenza dell'Alzheimer in Europa e in Italia
Recenti statistiche indicano che nel mondo 24,3 milioni di persone sono a�ette
da malattie neurodegenerative, con 4,6 milioni di nuovi casi all'anno (un nuo-
vo caso ogni 7 secondi). In Europa i casi demenza sono circa 5 milioni, di cui
oltre 3 milioni dovuti alla malattia di Alzheimer. Questo valore é comunque
destinato ad innalzarsi drasticamente, visto il continuo aumento delle aspetta-
tive di vita; nel 2040 é previsto che il numero di casi raddoppierá nell'Europa
Occidentale e triplicherá nell'Europa dell'Est.
In Italia l'incidenza dell'Alzheimer nella popolazione di etá superiore ai 60
anni è del 3.5% corrispondente, al momento, a circa 528mila malati, con circa
4

1.1 Le malattie neurodegenerative
73mila nuovi casi nell'ultimo anno. La probabilitá di contrarre la malattia sale
al 15-20% nelle persone con etá maggiore agli 80 anni.
1.1.2 Pro�lo clinico e patologico
La malattia esordisce con lieve perdita della memoria, seguita da demenza
lentamente progressiva che ha un decorso di alcuni anni e che porta ad alter-
azioni delle capacitá di linguaggio e delle capacitá visuospaziali. Nella fase
iniziale della malattia i pazienti lamentano incapacitá a trovare le parole e
di�coltá organizzativa e di orientamento. Nella fase intermedia il malato é in-
capace di lavorare, é spesso confuso e so�re di sintomi ansioso-depressivi. Nella
fase terminale della malattia il paziente diventa rigido, muto, incontinente e
molto spesso é costretto a letto.
Dal punto di vista anatomo-patologico tutte queste malattie sono acco-
munate dalla presenza di accumuli di �brille amiloidi nel cervello o in altri
tessuti. Con il termine �brille amiloidi si intendono aggregati allungati di
proteine caratterizzati da una marfologia lunga relativamente dritta, schema
di di�razione crossβ, proprietá di legarsi in maniera selettiva a particolari
coloranti e aventi un nucleo con struttura rigida.
La formazione delle �brille amiloidi coinvolge un riarrangiamento strut-
turale dello stato nativo in una conformazione a fogliettoβ ricca di �brille.
Questa struttura sembra fornire un'impalcatura che favorisce l'assemblamento
delle proteine nella struttura amiloide.
Queste �brille hanno un diametro di circa 10nm e sono tipicamente costitu-
ite da 2-6 proto�lamenti. Indipendentemente dalla natura delle proteine che le
costituiscono presentano tutte la stessa conformazione strutturale : morfologia
crossβ, dove i �li-β sono orientati in maniera perpendicolate tra loro e i fogli-β
sono paralleli all'asse delle �brille.
5

1.1 Le malattie neurodegenerative
1.1.3 Meccanismo della polimerizzazione delle proteine
La maggior parte delle proteine passa dalla fase monomero a quella di polimero
seguendo un modello di polimerizzazione dipendente dalla nucleazione.
In una reazione di questo tipo gli step iniziali sono piú lenti rispetto
agli altri. Questi primi consistono un un numero di equilibri non favorevoli,
�g1.1(2a), che rendono l'inizio della reazione, ovvero la nucleazione, di�cile e cé
una barriera energetica che deve essere superata per far procedere la reazione,
�g1.1(2b). Il picco della curva di energia libera corrisponde alla specie An,in
�g 1.1(2a), che ha un ruolo fondamentale nella polimerizzazione. Dopo la sua
formazione gli step successivi di elongazione diventano termodinamicamente
favorevoli.
La pendenza della curva di energia libera, �g1.1(2b), a ogni valore della di-
mensione degli aggregati é determinata dal prodotto della concentrazione delle
specie e dal rapporto tra la costante cinetica di associazione e dissociazione.
Nella nucleazione la costante di dissociazione é molto maggiore di quella di as-
sociazione. Quando peró si formano i nuclei la pendenza della curva di energia
libera inverte la sua direzione e per tutti gli step successivi la costante di as-
sociazione diventa piú grande di quella di dissociazione. Il nucleo rappresenta
quindi il piú piccolo aggregato di proteine per il quale la costante cinetica di
aggregazione diventa maggiore di quella di dissociazione.
1.1.4 Caratteristiche della polimerizzazione dipendente
dalla nucleazione
La cinetica di questa polimerizzazione mostra un lag phase che rappresenta il
punto debole della fase iniziale della reazione e puó essere descritta con una
funzione t2, �g 1.1(2c) . Questo é causato dal fatto che la costante cinetica di
dissociazione é maggiore di quella di associazione. La durata di questa fase é
6

1.1 Le malattie neurodegenerative
proporzionale alla ripiditá della curva di energia libera e dipende dalla concen-
trazione delle proteine. Minore é la concentrazione di proteine e maggiore é la
lag phase.
La dipendenza dalla concentrazione é controllata sia dal valore delle
costanti cinetiche che dalla dimensione del nucleo (numero di monomeri nel
nucleo).
Questa dipendenza é molto forte per cui esiste un valore critico di concen-
trazione per la formazione del polimero. Questo implica che ad un certo valore
di concentrazione su�ecientemente basso potrebbe esserci la non formazione
di polimero.
La concentraziione critica di proteine é determinata plottando la velocitá
di formazione del polimero contro la concentrazione di proteine, �g1.1(2d).
la lag phase puó essere diminuita o addirittura eliminata utilizzando
all'inizio della reazione una quantitá di nuclei preformati, �g1.1(2e).
Figura 1.1. cinetica di aggregazione dipendente dalla nucleazione
7

1.2 Esteri�cazione di Steglich
1.2 Esteri�cazione di Steglich
L'esteri�cazione di Steglich fu descitta per la prima volta nel 1978 dallo
scienziato Wolfgang Steglich.
Questa reazione permette di ottenere un estere, con rese praticamente uni-
tarie , facendo reagire un acido carbossilico e un alcool con l'ausilio della
DCC (dicicloesilcarbodimmide) in condizioni blande, temperatura e pressione
ambiente e tenue agitazione.
Data la sua versatilitá e la sua facilitá di realizzazione risulta molto comoda
per la preparazione delle ammidi partendo da un acido carbossilico contenente
un gruppo amminico ed un alcool.
La reazione è condotta in solventi organici come diclorometano (DCM),
dimetilformammide (DMF), tetraidrofurano (THF), acetonitrile.
Rispetto all'esteri�cazione di Fischer o�re il vantaggio di non utilizzare un
acido protico come catalizzatore ed evita la formazione di molecole d'acqua,
rendendo piú facili le operazioni di puri�cazione del prodotto �nale.
Lo schema di reazione per un qualsiasi acido carbossilico e un alcool è
schematizzato di seguito.
� L'acido carbossilico reagisce con la DCC a dare l' O-acilisourea, specie
che ha una reattivitá maggiore della corrispondente anidride dell'acido
carbossilico, �gura 1.2.
Figura 1.2. Reazione tra acido carbossilico e DCC
8

1.2 Esteri�cazione di Steglich
� L'alcool attacca questo intermedio formando la DCU (dicicloesilurea),
che precipita solida sul fondo, ed il corrispondente estere, �gura 1.3.
Figura 1.3. Attacco dell'acido sull'O-acilisourea
Utilizzando le ammine la reazione procede senza problemi a dare le cor-
rispondenti ammidi, dato che le ammine sono piú nucleo�le dei rispettivi alcoli.
Nel caso l'esteri�cazione sia lenta c'è la possibilitá che avvenga una reazione
secondaria che diminuisce la resa �nale del prodotto desiderato e complica le
operazioni di puri�cazione del prodotto. Questa reazione consiste nel riarran-
giamento 1,3 dell'O-acilisourea a dare N-acilurea che è una specie stabile che
non reagisce con l'alcool.
Per evitare questa reazione secondaria si aggiungono tracce di DMAP, circa
il 5% in peso rispetto alla DCC. Questo composto, molto piú nucleo�lo dell'al-
cool, si comporta da transfer agent nei confronti del gruppo acile, e la reazione
prosegue come illustrato in �gura 1.4.
Figura 1.4. modalità di reazione della DMAP
9

1.3 Polimerizzazione in emulsione
1.3 Polimerizzazione in emulsione
La polimerizzazione in emulsione é una polimerizzazione con meccanismo
radicalico.
A di�erenza dalla polimerizzazione in bulk o in sospensione con ques-
ta metodologia si riescono ad ottenere particelle di dimensione �nale ridot-
ta, caratteristica principale che devono possedere le NPs �nalizzate al drug
delivery.
Il sistema caratteristico della reazione di polimerizzazione in emulsione
presenta tre distinte fasi:
� una fase acquosa, che contiene l'iniziatore, l'emulsionante, le micelle e
una piccola quantitá di monomero in ragione della sua solubilità;
� le gocce di monomero, disperse in acqua e stabilizzate da un emulsion-
ante;
� le particelle polimeriche stabilizzate dall'emulsionante, le quali con-
tengono, oltre al polimero, una frazione più o meno elevata del monomero
non ancora reagito;
La �gura 1.5, rappresenta l'ambiente di lavoro della polimerizzazione in
emulsione in cui sono visibili le tre fasi distinte.
Figura 1.5. Ambiente della polimerizzazione in emulsione
10

1.3 Polimerizzazione in emulsione
Alla �ne del processo di polimerizzazione si ottengono particelle colloidali
di polimero disperse in fase acquosa e stabilizzate dall'emulsionante. La di-
mensione �nale delle particelle è notevolmente minore di quella iniziale delle
gocce. Questo comportamento é dovuto al fatto che le gocce fungono soltanto
da serbatoio di monomero ed in esse non avvengono reazioni.
Di seguito, �gura 1.6, è riportata un'immagine delle particelle formatesi :
Figura 1.6. nanoparticelle stabilizzate dall'emulsionante
1.3.1 Meccanismo di produzione delle particelle
I meccanismi teorici che spiegano la formazione delle particelle sono i seguenti:
� nucleazione eterogenea in micella : un radicale libero in acqua di�onde
in una micella e la propagazione prosegue all'interno della stessa;
� nucleazione omogenea : la lunghezza della catena radicalica accresciutasi
in acqua supera un valore critico legato al limite di solubilità e determina
la precipitazione di un nucleo di particella;
� nucleazione in goccia : la catena radicalica cresciuta in acqua entra in
una goccia di monomero nella quale prosegue la propagazione;
La successione degli eventi che porta alla nucleazione di una nuova particella
secondo i meccanismi ora discussi è schematicamente riassunta in �gura 1.7:
11

1.3 Polimerizzazione in emulsione
Figura 1.7. meccanismi di produzione delle particelle
In questa trattazione sará escluso il terzo meccanismo di formazione di
nuove particelle. La generazione delle stesse per ingresso in goccia, infatti,
è statisticamente poco favorita rispetto a quello in micella, poiché la super-
�cie speci�ca delle gocce è inferiore a quella delle micelle di molti ordini di
grandezza.
Gli unici meccanismi di nucleazione che verranno considerati sono quello
omogeneo e quella eterogeneo in micella: le particelle formatesi a seguito di
entrambi questi meccanismi sono il vero centro reattivo del sistema e sono
considerate stabili, grazie all'elevato quantitativo di emulsionante caricato nel
sistema.
Una particolaritá della polimerizzazione in emulsione rispetto alle altre
tipologie é la diversa accessibilitá dei radicali. Questi si trovano segregati
in particella per cui, �n tanto che non entra dentro un ulteriore radicale, la
polimerizzazione continua inde�nitamente, la velocitá di terminazione, e quin-
di anche la lunghezza �nale di catena, non sono controllate dalla dimensione
delle particelle ma dall'entrata dei radicali in particella. La terminazione é
12

1.3 Polimerizzazione in emulsione
quindi legata a un fenomeno �sico e non alla reattivitá delle molecole in gioco.
1.3.2 Gli intervalli della polimerizzazione in emulsione
Il meccanismo di formazione delle particelle considerato nel modello è il
seguente: un radicale libero in acqua di�onde in micella e la propagazione
prosegue all'interno della stessa (nucleazione eterogenea in micella). Le parti-
celle formatesi diventano il centro reattivo del sistema e sono considerate stabili
grazie all'elevato quantitativo di emulsionante caricato nel sistema. Nel pro-
cesso di polimerizzazione in emulsione le gocce di monomero, eventualmente
presenti, fungono solo da riserva del monomero stesso; quest'ultimo passa dalle
gocce alla soluzione acquosa man mano che il monomero presente in acqua é
richiamato dalle particelle, all'interno delle quali é consumato dalla reazione.
Esauritesi le gocce la polimerizzazione prosegue �no al consumo del monomero
presente in acqua e in particella.
La velocitá di polimerizzazione in particella é espressa come :
RP = KP ·MP ·NP · n
Nav
÷[mol
cm3s
](1.1)
con
KP = costante cinetica di polimerizzazione
MP = concentrazione di monomero in particella
NP = concentrazione numerale di particelle
n = numero medio di radicali in particella
Nav = numero di Avogadro
Tale velocitá ha andamenti diversi al procedere della reazione, in tal senso
è opportuno dividere il processo in tre intervalli :
� intervallo I : nucleazione delle particelle;
13

1.3 Polimerizzazione in emulsione
� intervallo II : crescita delle particelle;
� intervallo III : esaurimento delle gocce e termine del processo;
Questa suddivisione é valida solo nel caso in cui si assuma come trascur-
abile la nucleazione omogenea rispetto a quella eterogenea: in caso contrario
l'aumento del numero di particelle proseguirebbe anche oltre la �ne
dell'intervallo I, cioè successivamente alla scomparsa delle micelle.
In seguito verranno descritte in dettaglio le tre fasi del processo con relative
equazioni utilizzate nel modello e assunzioni e sempli�cazioni fatte.
line
Intervallo I : nucleazione delle particelle
In questo primo intervallo le riserve di monomero sono sostanzialmente integre
e sono presenti micelle. Quando un radicale prodotto in soluzione acquosa
entra in una micella essa si trasforma in una nuova particella polimerica sec-
ondo la nucleazione eterogenea. Al procedere del tempo si ha una crescita
del numero di particelle polimeriche, le quali, una volta nucleate, aumentano
la loro dimensione richiamando monomero dalla fase acquosa. In seguito a
questo fenomeno di accrescimento si ha un incremento della super�cie interfa-
sica esposta, con conseguente adsorbimento di nuove molecole di tensioattivo in
quantitá tale da garantire la stabilizzazione delle particelle in crescita. Il richi-
amo di emulsionante dalla fase acquosa determina la disgregazione progressiva
di alcune delle micelle che non sono ancora nucleate, mentre la concentrazione
in fase acquosa resta costante e pari al valore di concentrazione critica micel-
lare (CMC). Per questo motivo il numero di particelle formatesi con questo
meccanismo è sicuramente minore del numero di micelle. Quando tutte le mi-
celle inizialmente presenti nel sistema si sono consumate la fase di nucleazione
eterogenea può dirsi terminata.
14

1.3 Polimerizzazione in emulsione
Intervallo II : Crescita delle particelle
In questo intervallo non si ha presenza di micelle in soluzione, il numero di
particelle rimane costante a meno di una lenta crescita dovuta alla nucleazione
omogenea, mentre la loro dimensione aumenta. Essendo caratterizzato dalla
presenza di gocce, questo intervallo non é sempre presente nel processo di
polimerizzazione.
Fin quando sono presenti le gocce monomeriche, si instaura nel sistema il
seguente equilibrio per le attivitá del monomero nelle diverse fasi :
aacquamon = aparticellamon = agocciamon (1.2)
Le gocce di monomero sono pure per cui hanno attivitá unitaria; di con-
seguenza le attivitá e le concentrazioni del monomero in acqua ed in particella
sono costanti e pari al loro valore di saturazione. Essendo costante la concen-
trazione di monomero in particella, lo é anche la sua frazione volumetrica. Per
garantire un rapporto monomero/polimero costante, il monomero migra dalle
gocce alle particelle attraverso la fase acquosa. Questo fenomeno di�usivo fa si
che in tutte le particelle ci sia sempre la stessa frazione di monomero, il quale
non solo compensa quello che via via si converte ma serve anche a mantenere
saturo il nuovo polimero prodotto. Poiché le super�ci speci�che interfasiche
sono molto ampie, dato la dimensione molto piccola delle particelle, le cinetiche
di trasporto sono molto piú rapide della velocitá di polimerizzazione e garan-
tiscono che l'equilibrio delle attivitá sia rispettato. In questa fase sono costanti
sia il numero di particelle polimeriche sia la concentrazione di monomero in
esse presente, pertanto la velocitá di polimerizzazione si mantiene pressoché
costante.
15

1.3 Polimerizzazione in emulsione
Intervallo III : esaurimento delle gocce e
termine del processo
L'ultima fase del processo di polimerizzazione in emulsione paerte quando si
sono consumate sia le micelle che le gocce di monomero, per cui l'equilibrio
che si instaura tra le attivitá del monoro nelle diverse fasi risulta :
aacquamon = aparticellamon (1.3)
Questo equilibrio é stimabile mediante modelli termodinamici e consente
di legare la concentrazione di monomero in acqua e in particella, noti i valori
di saturazione. La maggior parte del monomero é presente nelle particelle, una
certa quantitá, piú o meno ridotta in funzione della solubilitá, é disciolta in
acqua. Nel caso la polimerizzazione sia condotta in modalitá di tipo batch, la
frazione di monomero in particella si consuma progressivamente, richiamando
quello presente in fase acquosa �no al completo esaurimento dello stesso. In
questo caso la velocitá di reazione diminuisce nel tempo.
Le due �gure nella pagina seguente, �gure1.8(a)-(b), rappresentatano l'an-
damento di alcune grandezze tipiche per la polimerizzazione in emulsione nel
tempo.
16

1.3 Polimerizzazione in emulsione
(a) intervalli della polimerizzazione in emulsione.
(b) specie presenti nella polimerizzazione in emulsione.
Figura 1.8. Quadro generale della polimerizzazione in emulsione
17

1.3 Polimerizzazione in emulsione
1.3.3 Polimerizzazione in emulsione in condizioni starved
La polimerizzazione in condizioni starved ovvero a�amate, consta in un pro-
cesso in cui non si ha accumulo di monomero ed é caratterizzato da una lenta
e progressiva addizione di monomero nel tempo in assenza di carico iniziale.
Questa modalitá operativa permette di ottenere delle condizioni diverse da
quelle di un tipico semibatch, inoltre se i tempi di alimentazione sono su�-
cientemente lunghi é veri�cata l'ipotesi di pseudo stazionarietá del monomero,dNmon
dt= 0
In queste condizioni il bilancio sul monomero (Nmon = [mol]) valido in
condizioni semi-batch:
dNmon
dt= Q−Rp · Vw[mol/s] (1.4)
si riduce alla seguente uguaglianza :
Q = Rp · Vw[mol/s] (1.5)
Mediante il controllo della portata di alimentazione si puó regolare la
velocitá di consumo del monomero, e quindi la reattivitá del sistema.
Lavorare in condizioni starved o�re il vantaggio di ottenere un maggiore
controllo dimensionale sulle particelle prodotte. Alimentando lentamente il
monomero i tempi di nucleazione si allungano poiché rallenta la crescita delle
particelle giá formate a causa dell'assenza delle riserve monomeriche costituite
dalle gocce, in questo caso non presenti. L'emulsionante è meno consumato per
adsorbimento sulla super�cie delle particelle e ció permette un allungamento
del periodo di nucleazione. Di conseguenza si ha un aumento della concen-
trazione di particelle, le quali a pari monomero alimentato avranno una dimen-
sione �nale minore. Con questa procedura le diverse grandezze del sistema,
a meno di un transitorio iniziale, si stabilizzano su un valore stazionario che
18

1.3 Polimerizzazione in emulsione
non é piú quello di saturazione proprio del secondo intervallo di una reazione
batch, ma inferiore e dato dalla velocità di reazione.
A titolo di esempio in �gura1.9 è riportato l'andamento sperimentale della
velocità di polimerizzazione ottenuto per una prova completamente semibatch
a velocità di alimentazione di 1 g/min.
Figura 1.9. Andamento della velocitá di polimerizzazione
Questo valore stazionario é legato al bilanciamento fra la velocitá di
alimentazione e la velocitá di reazione.
Gli andamenti discussi nel paragrafo precedente non sono piú propriamente
applicabili a questa modalitá di polimerizzazione. In particolare, mentre l'in-
tervallo I resta invariato, l'intervallo II non é mai presente. La crescita delle
particelle avviene in un contesto analogo a quello presentato nell'intervallo III,
con la di�erenza che la frazione di monomero in particella non decrementa come
nel caso di assenza di alimentazione, ma si mantiene pari al valore stazionario
sino a che prosegue l'addizione. La riduzione della velocitá di polimerizzazione,
conseguenza della diminuzione della concentrazione di monomero in particel-
la, si registra solo quando l'alimentazione viene arrestata. Quanto minore é
la concentrazione pseudo stazionaria, tanto piú breve é il transitorio �nale di
riduzione della velocitá di polimerizzazione
19

1.3 Polimerizzazione in emulsione
In condizioni starved l'addizione di monomero costituisce lo stadio limi-
tante del processo di crescita delle particella. La frazione volumetrica dello
stesso nel luogo di reazione é sempre particolarmente ridotta e, in particolare é
sempre inferiore al valore di saturazione. In queste condizioni possono divenire
particolarmente importanti le limitazioni di�usive ai processi di propagazione
e di terminazione in particella. L'e�etto di tali limitazioni viene discusso nel
seguente paragrafo.
1.3.4 E�etto delle limitazioni di�usive sul processo
In particolare verranno discussi i fenomeni della segregazione delle particelle,
dell'entry e dell'exit dei radicali in particella.
Segregazione delle particelle
Per valutare la concentrazione dei radicali attivi in particella si puó in prima
approssimazione considerare che, in un processo di polimerizzazione volto al-
la produzione di particelle di dimensioni particolarmente ridotte, il numero di
radicali in ogni particella possa essere pari ad uno (radicale che sta propagando)
o a zero (particella polimerica morta). Questo perché nel momento in cui un
nuovo radicale entra in una particella nella quale é in corso la polimerizzazione,
questo reagisce con il radicale presente terminando. In una polimerizzazione
in emulsione i radicali presenti in particella non hanno accesso diretto ad altri
radicali, in questo modo il processo controllante la terminazione non é la ve-
locitá di terminazione stessa, ma i fenomeni di trasferimento di fase tra il bulk
e le particelle.
20

1.3 Polimerizzazione in emulsione
L'andamento del numero di radicali in particella puó essere schematizzato
come in �gura 1.10:
Figura 1.10. andamento del numero di radicali in particella
Entrata dei radicali in particella
L'iniziatore utilizzato in un sistema di polimerizzazione in emulsione é sempre
di carattere idrosolubile. I primi radicali si formano in ambiente acquoso e qui
propagano sino a che la lunghezza di catena da essi raggiunta non sia superiore
ad un valore critico al di lá del quale essi diventano a tal punto insolubili da
non essere piú compatibili con la fase acquosa. La di�erenza di energia libera
della catena radicalica in fase acquosa rispetto a quella in fase organica, cioé
entro la particella rigon�ata di monomero, si rende responsabile dell'ingresso
della catena nella particella stessa: questo fenomeno prende il nome di entrata.
L'entry coinvolge dunque una fase iniziale durante la quale, per
propagazione nel sistema acquoso, si forma un radicale capace di adsorbirsi
nella particella, ed una fase successiva in cui si realizza l'e�ettiva di�usione
della catena attraverso la super�cie di separazione di fase. A�nché l'entry
possa aver luogo é necessario che il raggiungimento della lunghezza critica non
21

1.3 Polimerizzazione in emulsione
sia impossibilitato da una precedente terminazione della catena in crescita
con un altro radicale presente in acqua, e che la terminazione non avvenga
neppure nella fase di di�usione della catena dall'acqua alla super�cie della
particella. In tal senso, l'aumento di concentrazione dei radicali da un lato
accelera i fenomeni di iniziazione, mentre dall'altro favorisce le terminazioni
riducendo in tal modo l'e�cienza di entarta.
Uscita dei radicali in particella
Come un radicale libero in soluzione acquosa puó essere catturato da una par-
ticella polimerica, cosí puó anche desorbire da essa: si ha cosí il fenomeno del-
l'exit. Un radicale libero in una catena ad elevato grado di polimerizzazione ha
un'energia libera elevata nell'ambiente acquoso polare non organico, pertanto
non dará luogo a desorbimento. Allo stesso modo una specie attiva super�-
cialmente che sia appena entrata in particella non uscirá dalla stessa, a causa
della propagazione che la rende insolubile in acqua. L'uscita é un fenomeno
proprio dei radicali monomerici che si formano in particella a seguito della
reazione di trasferimento. Il fenomeno dell'uscita diventa rilevante in quanto
i radicali monomerici sono caratterizzati da un elevato coe�ciente di di�u-
sione intraparticellare: in particolare, tenendo conto delle dimensioni ridotte
di una tipica particella colloidale, la velocitá di di�usione risulta molto rapida
se comparata con il tempo necessario alla propagazione del radicale monomeri-
co stesso. La presenza della propagazione farebbe venir meno il fenomeno del
desorbimento, poiché i dimeri o trimeri che si formerebbero sarebbero troppo
idrofobi per dar luogo all'uscita. Il radicale monomerico generato per trasferi-
mento di catena non puó desorbire se il tempo di di�usione é tale da consentire
l'addizione di nuovi radicali in catena. Il radicale R1• deve di�ondere dall'in-
terno della particella verso la super�cie e deve attraversare la stessa prima che
un qualsiasi atto di propagazione possa intervenire, mentre la specie desorbita
deve allontanarsi dalla super�cie esterna della particella di�ondendo verso il
22

1.4 ROP : Ring Opening Polymerizazion
bulk acquoso in tempi su�cientemente rapidi da evitare un riassorbimento nel-
la particella appena abbandonata. Solo se tutto ció ha modo di avvenire prima
di un atto di propagazione, il radicale monomerico da luogo a desorbimento
come illustrato in �gura 1.11:
Figura 1.11. meccanismo dell'exit
Considerare 0.5 come numero medio di radicali in particella é un valore
approssimato perché a causa del fenomeno dell'exit é generalmente piú basso.
1.4 ROP : Ring Opening Polymerizazion
La Ring Opening Polymerization (ROP) é un tipo di polimerizzazione co-
munemente usato per polimerizzare monomeri ciclici, come ammine cicliche e
lattoni. La presenza nella molecola di elementi diversi dal carbonio all'interno
dell'anello fornisce un punto di attacco nucleo�lo o elettro�lo da parte di una
specie iniziatrice agevolando l'apertura dell'anello e quindi la reazione.
La polimerizzazione di monomeri ciclici, come i lattidi, é utilizzata per i
diversi vantaggi che o�re. Rispetto ad una polimerizzazione radicalica o, nel
caso dell'acido lattico a una policondensazione, con questo metodo si riescono a
raggiungere più facilmente pesi molecolari elevati. In particolare questo valore
é controllato dalla conversione e dal rapporto tra monomero e iniziatore. Ad es-
empio nel caso del polilattico si possono raggiungere pesi molecolari dell'ordine
23

1.4 ROP : Ring Opening Polymerizazion
di 100-300000 g/mol al contatrio di una policondensazione dove di�cilmente
si ragguingono pesi molecolari superiori a 10000 Da.
Un altro vantaggio della ROP é la completa assenza di sotto prodotti, cosa
che permette di produrre il polimero direttamente dal monomero ciclico sino
alla sua forma estrusa �nale.
1.4.1 Polimerizzazione ROP dell'acido polilattico
La produzione di acido polilattico puó essere e�ettuata via policondensazione
diretta dell'acido lattico o via ROP.
La prima via ha lo svantaggio che si tratta di una reazione all'equilibrio,
dove é molto di�cile rimuovere le quantitá residue di acqua. Questo limita
il peso molecolare raggiungibile nel prodotto �nale e quindi le sue proprietá
meccaniche.
Nella polimerizzazione ROP si parte dal lattide, ovvero dal diestere ciclico
dell'acido lattico, questo viene aperto con l'ausilio di un catalizzatore e vengono
due molecole di acido lattico.
Tra i catalizzatori usati quello che garantisce la maggior produttivitá é
l'ottanoato di stagno. Questo é anche l'uico ad essere stato accettato dalla
U.S Food and Drug Amministration.
Il meccanismo della reazione si basa su una sorta di �attivazione del
monomero�. Questo, che é il lattide che contiene un gruppo -OH, forma con il
catalizzatore un complesso terziario.
Questa polimerizzazione esibisce un comportamneto �living�; ovvero la
polimerizzazione procede con un numero costante di catene in crescita e dormi-
enti, e le reazioni di terminazione sono assenti. Questo fa sí che il valore di
polidispersitá, de�nito come il rapporto tra il peso molecolare medio ponderale
e medio numerale sia vicino all'unitá.
24

1.4 ROP : Ring Opening Polymerizazion
L'andamento del peso molecolare medio numerale é lineare rispetto alla
conversione e questo é un'ulteriore conferma del fatto che la concentrazione
delle catene attive e dormienti e costante durante la reazione.
Inoltre una conseguenza molto importante del carattere �living� della
reazione é la presenza di gruppi terminali termo e/o redox labili che consentono
di far reagire ulteriormente la catena con un di�erente monomero, ottenendo
dei copolimeri a blocchi o di isolare il prodotto e farlo reagire successivamente
con lo stesso monomero per formare polimeri di peso molecolare maggiore
copolimeri, non ottenibili con le classiche procedure di copolimerizzazione.
25

1.5 Coloranti azoici
1.5 Coloranti azoici
I coloranti azoici derivano formalmente dall'azobenzene e presentano tutti il
gruppo azoico (-N=N-) compreso tra due anelli aromatici, del benzene o naftal-
ene, dell'antracene e di eterocicli aromatici. Si ottengono attraverso la reazione
di copulazione degli azocomposti aromatici con composti che hanno un anel-
lo benzenico attivato, ovvero con un gruppo -OH o −NH2 che favorisce la
sostituzione nucleo�la.
Nella molecola dei coloranti cosí ottenuti il gruppo azoico rappresenta il
gruppo cromoforo, mentre un gruppo amminico, come per esempio il grup-
po dimetilamminico o un ossidrile fenolico costituiscono il gruppo auxocromo.
Oltre ai coloranti del tipo ora descritto che contengono nella loro moleco-
la un solo gruppo azoico (detti monoazoici), ne esistono anche con due, tre
o piú gruppi azoici, ossia coloranti diazoici, triazoici, ecc., che si ottengono
e�ettuando sulla stessa molecola due, tre o più reazioni di copulazione.
Variando opportunamente la natura dei due reagenti della reazione di cop-
ulazione é possibile ottenere una larghissima gamma di coloranti azoici, con
colorazioni che vanno dal giallo al rosso, al verde, all'azzurro e al violetto.
1.5.1 Reazioni di copulazione dei sali di diazonio
I sali di diazonio si preparano per diazotazione delle ammine primarie aro-
matiche. Queste si ottengono per riduzione di nitroderivati che si preparano
facilmente per nitrazione diretta di un substrato aromatico.
I sali di diazonio sono reagenti debolmente elettro�li che reagiscono sola-
mente con anelli aromatici fortemente attivati verso la sostituzione elettro�la.
In �gura1.12 é riportata la reazione generale tra un sale di diazonio e un gener-
ico composto aromatico (a) e a titolo di esempio la reazione tra il cloruro di
benzendiazonio e il fenolo (b).
26

1.5 Coloranti azoici
(a) schema generico della reazione.
(b) reazione tra il cloruro di benzendiazonio e il fenolo.
Figura 1.12. reazioni di copulazione dei sali di diazonio
Le reazioni di copulazione dei sali di diazonio con i fenoli procedono molto
rapidamente in soluzioni leggermente basiche. In queste condizioni una quan-
titá apprezzabile di fenolo é presente come ione fenossido, il quale é piú reat-
tivo del fenolo indissociato nella reazione di sostituzione nucleo�la. Va evitata
peró un'alcalinitá troppo elevata della soluzione ( pH non superiore a 10), in
quanto in queste condizioni il sale di diazonio reagisce con lo ione OH− per
formare un diazenolo oppure la corrispondente base coniugata, come mostrato
in �gura1.13.
Figura 1.13. equilibri a pH maggiore di 10
La copulazione con le ammine é favorita a pH leggermente basico, tra 5 e
7. In queste condizioni la concentrazione del sale di diazonio é massima peró
é limitata la conversione dell'ammina nel suo acido coniugato, il quale non
reagisce con il sale di diazonio. A pH minori di 5 la velocitá di copulazione del
sale con l'ammina é bassa.
Con i derivati del fenolo e dell'anilina la reazione avviene quasi esclusi-
vamente in posizione para se é libera, in caso contrario avviene in posizione
orto.
27

1.5 Coloranti azoici
Gli azocomposti sono intensamente colorati; il gruppo azo (-N=N-) per-
mette ai due anelli benzenici di coniugare reciprocamente, questo porta ad un
esteso sistema di elettroni π delocalizzati con conseguente assorbimento delle
radiazioni nella regione del visibile.
28

Capitolo 2
Materiali e Metodi
Di seguito verranno elencati i materiali utilizzati durante questo lavoro di tesi
assieme ad una loro breve descrizione. Inoltre verranno descritte le metodologie
con le quali sono state condotte le analisi per caratterizzare i prodotti ottenuti.
2.1 Acido polilattico
L'acido polilattico é stato utilizzato come monomero nella polimerizzazione
tramite meccanismo ROP, ottenendo cosí acido polilattico(PLA).
La scelta é caduta su questo monomero per via delle sue caratteristiche in
termini di biocompatibilitá e biodegradabilitá, e per la presenza del gruppo -
OH terminale necessario per la successiva reazione di esteri�cazione di Steglich.
La formula di struttura dell'acido polilattico e del polimero relativo é
illustrata nella pagina seguente in �gura 2.1.
Il composto é stato acquistato dalla SIGMA-ALDRICH, la sua purezza é
del 98%.
29

2.2 2-idrossietil metacrilato(HEMA)
(a) (b)
Figura 2.1. formule di struttura dell'acido lattico (a) e del PLA (b)
2.2 2-idrossietil metacrilato(HEMA)
L'HEMA(ALDRICH, purezza 97%,contenente 220-220ppm di mono-etil etere
idrochinone come inibitore) é il monomero che é stato funzionalizzato con i col-
oranti azoici e poi utilizzato per la produzione di NPs tramite polimerizzazione
radicalica. La sua formula di struttura é illustrata in �gura2.2.
Figura 2.2. 2-idrossietil metacrilato
2.3 Acido ammino benzoico
L'acido amminobenzoico (SIGMA, purezza ≥99%), é un amminoacido. La sua
formula di struttura é riportata in �gura2.3.
Questo viene fatto reagire con L'HEMA secondo la reazione di esteri�-
cazione di Steglich per ottenere l'intermedio con il gruppo amminico aromatico
necessario per la successiva formazione dei coloranti azoici.
30

2.4 Composti per i coloranti azoici
Figura 2.3. acido amminobenzoico
2.4 Composti per i coloranti azoici
I composti con anello aromtico attivato utilizzati nelle reazioni di formazione
dei coloranti azoici sono stati :
� naftolo (ALDRICH, purezza 99%)
� acido salicilico (SIGMA-ALDRICH, purezza 99%)
� acido solfanilico (SIGMA-ALDRICH, purezza 97%)
� tio�avina T (SIGMA,contenuto di colorante 75%)
Nella pagina seguente sono riportate le loro formule di struttura, �gura 2.4
31

2.5 Iniziatori e emulsionanti per le polimerizzazioni
(a) . (b) . (c) .
(d) .
Figura 2.4. composti con anello aromatico attivato, (a) 2-naftolo, (b) acidosalicilico, (c) acido solfonilico, (d) tio�avina T
2.5 Iniziatori e emulsionanti per le
polimerizzazioni
Il catalizzatore a cui si é fatto ricorso per la polimerizzazione ROP é lo stag-
no ottanoato (Sn(C8H15O2)2), scelto perché approvato dalla Food and drug
administration (FDA) come biocompatibile, �gura2.5.
Figura 2.5. ottanoato di stagno
32

2.5 Iniziatori e emulsionanti per le polimerizzazioni
Per la polimerizzazione in emulsione é stato utilizzato il 4,4'-Azobis(acido
cianovalerico), �g2.6.
HOCOCH2CH2C(CH3)(CN)N = NC(CH3)(CN)CH2CH2COOH.
Figura 2.6. 4,4'-Azobis(4-cyanovaleric acid)
Questa scelta é stata fatta perché tale iniziatore é caricato positivamente
data la presenza del gruppo (-COOH) e durante il processo di polimerizzazione
che viene fatto a pH = 10 rimane protonato e non interagisce con l'SDS e con
i macromonomeri.
Per conferire stabilitá alle particelle presenti nel lattice sono stati utilizzati
emulsionanti sia di tipo ionico che di tipo non ionico.
Quelli di tipo ionico, in virtú di una carica positiva o negativa presente su
di loro, generano tra le particelle una repulsione elettrostatica la cui entitá é
correlata alla carica super�ciale delle particelle stesse.
Gli emulsionanti non ionici, o sterici, esplicano il meccanismo stabilizzante
mediante repulsione sterica, dovuta all'impossibilitá di sovrapposizione di due
molecole nello stesso spazio.
La �gura nella pagina seguente, �gura 2.7, illustra i due modi con cui gli
emulsionanti conferiscono stabilitá alle particelle.
33

2.5 Iniziatori e emulsionanti per le polimerizzazioni
Figura 2.7. meccanismi di stabilizzazione delle particelle
Come emulsionante ionico é stato impiegato il sodio-dodecilsolfato
(SDS,NaSO4C12H25 ), �gura 2.8.
Figura 2.8. formula di struttura dell'SDS
Il suo utilizzo é legato all'elevato numero di dati ad esso correlati reperibili
in letteratura, alla sua e�cienza nella generazione di NPs di dimensioni ridotte
e al suo largo uso commerciale.
L'emulsionante di tipo sterico utilizzato é il Tween80 (C64H124O26), formu-
la di struttura nella pagina a seguire, �gura 2.9). La sua scelta é motivata
principalmente alla sua biocompatibilitá, caratteristica fondamentale per ap-
plicazioni di drug delivery. Lo stesso é abitualmente impiegato nell'industria
34

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
alimentare e riconosciuto come sicuro per la salute.
Figura 2.9. formula di struttura del Tween80
2.6 Tecniche utilizzate per la caratterizzazione
dei prodotti ottenuti
I prodotti ottenuti sono stati caratterizzati in termini di dimensioni medie, Z-
potential, proprietá legate alla �uorescenza e all'analisi UV. Sono state anche
condotte delle analisi all'NMR in modo tale da veri�care la struttura �nale de
materiali prodotti.
Nelle pagine seguenti verranno presentate tutte le tecniche utilizzate
assieme ad una breve spiegazione della teoria su cui si basano.
2.6.1 Light-scattering
Per la caratterizzazione delle dimensioni medie e dello Z-potential dei latex
prodotti é stata utilizzata la tecnica d'analisi del light-scattering.
Il light scattering statico(LS) é uno strumento utilizzato per caratterizzare
dal punto di vista dimensionale le NPs prodotte, in termini di diametro medio,
polidispersitá, distribuzione delle dimensioni.
Tale metodo di analisi é una delle piú importanti tecniche sperimentali
nell'investigazione dei sistemi colloidali e consente di ottenere informazioni,
35

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
in modo non invasivo, oltre che sulla dimensione delle particelle anche sulla
struttura degli aggregati e sulle cinetiche di aggregazione.
Il principio di funzionamento si basa sull'interazione tra un raggio di luce
incidente (onda elettromagnetica) e la particella (materia). Quando l'onda
elettromagnetica incontra la molecola, gli elettroni presenti nella molecola
risentono dell'interazione col raggio luminoso ed il centro di massa delle cariche
negative si sposta dalla posizione originale. Si forma un dipolo che oscilla con
la stessa frequenza del raggio incidente ed emette radiazioni elettromagnetiche
in tutte le direzioni. La somma di tutte le radiazioni costituisce la luce di�usa
o �scatterata�. La somma delle radiazioni di�use dai singoli elementi per un
campione investito da un'onda elettromegnetica costituisce la radiazione �s-
catterata�. L'intensitá, la polarizzazione e la distribuzione angolare della luce
�scatterata� dipendono dalla forma e dalla dimensione delle particelle e dal
contrasto ottico delle singole particelle.
Misurando la luce di�usa é possibile ricavare informazioni sulla dimensione
delle particelle, a patto di trascurare lo scattering multiplo, ossia la situazione
in cui un raggio �scatterato� da una particella colpisce un'altra particella e viene
ridi�uso a sua volta; tale fenomeno puó essere giusti�catamente trascurato
a patto di esaminare campioni su�cientemente diluiti e con basso contrasto
ottico, in modo da poter considerare indipendenti tutte le fonti di scattering.
La teoria che trascura lo scattering multiplo é chiamata teoria �Rayleigh-
Debye-Gans�. L'assunzione base é quella che in ogni punto del campione le
particelle siano illuminate da sola radiazione incidente: ció consente di trattare
lo scattering di ogni particella come indipendente dagli altri.
36

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
Questa teoria lega l'intensità della luce di�usa I all'angolo di scattering θ,
ossia l'angolo tra la radiazione incidente e la radiazione di�usa, come mostrato
in �gura2.10.
Figura 2.10. radiazione incidente e angolo di scattering
Nella pratica al posto dell'angolo di scattering θ é piú comodo usare il
modulo del vettore d'onda q :
q =4πn
λ0· senθ
2(2.1)
Dove λ0 é la lunghezza d'onda della radiazione nel vuoto e n é l'indice di
rifrazione del solvente.
Assumendo che tutte le particelle siano uguali e di forma sferica é possibile
ricavare la seguente espressione:
I(q) = I0 ·K1 ·N · V 2p · p(q) · S(q) (2.2)
Dove K1 é una costante di proporzionalitá tipica dello strumento, N é la
concentrazione di particelle, Vp é il volume delle particelle, P(q) é il fattore di
forma e dipende dalla forma delle particelle ed S(q) é il fattore di struttura e
dipende dalla struttura degli aggregati.
37

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
Per geometrie semplici é possibile ricavare i fattori di forma e struttura
e quindi risalire alla dimensione delle particelle. A�ncé sia garantita l'at-
tendibilitá del valore del diametro medio fornito dallo strumento é necessario
avere una distribuzione dimensionale quanto piú possibile unimodale all'in-
terno del campione in analisi. Infatti l'intensitá della luce �scatterata� dalle
particelle dipende dal quadrato del loro volume , quindi dalla sesta potenza del
loro raggio: in tal senso la presenza di particelle grandi oscurerebbe la visibilitá
delle particelle piú piccole, non consentendone l'individuazione e invalidando
la misura.
Da un punto di vista pratico la misura é rapida: il campione diluito viene
messo nella cella e di seguito viene colpito dalla radiazione luminosa. Le in-
formazioni sulla luce �scatterata� consentono di ricavare le dimensioni delle
particelle: tale valore di dimensione ha giá valore statistico, in quanto un gran
numero di particelle ha contribuito allo scattering.
2.6.2 NMR : Risonanza magnetica nucleare
La spettroscopia di risonanza magnetica nucleare (NMR) é la tecnica piú veloce
per poter riconoscere composti chimici ignoti, in particolare in questo lavoro é
stata utilizzata per indagare la formula di struttura del prodotto della reazione
di esteri�cazione cosí da avere un check su quanto ottenuto.
I nuclei degli atomi che possiedono un numero dispari di protoni, di neu-
troni o entrambi, ruotano su se stessi (spin) e quindi possiedono un momento
angolare. I nuclei privi di momento angolare non possiedono spin e non danno
luogo a fenomeni di risonanza magnetica. I nuclei di C12 e O16 cadono in ques-
ta categoria e quindi non vengono rilevati. Poiché i nuclei atomici possiedono
una carica, un nucleo che ruota genera una corrente elettrica e ha un campo
magnetico associato. Il dipolo magnetico µ del nucleo varia per ogni elemento.
Quando un nucleo dotato di spin viene immerso in un campo magnetico, é
sottoposto ad una coppia di forze che lo fanno ruotare per allinearlo col campo
38

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
magnetico esterno. Per un nucleo con spin 1/2, ci sono due
orientamenti permessi del nucleo: parallelo al campo (bassa energia) e contro
il campo (alta energia). Dato che la di�erenza di energia tra i due stati é
piccolissima, dell'ordine di 9E-6 kcal/mol, si raggiunge rapidamente l'equilibrio
tra i due stati e mediamente i nuclei soggetti a campo magnetico si dividono
equamente tra allineati e opposti al campo, �gura2.11.
Figura 2.11. Orientamento dei nuclei in un campo magnetico
Se i nuclei orientati vengono ora colpiti con radiazione elettromagnetica
di opportuna frequenza, gli stati di energia piú bassa (allineati al campo)
assorbono un quanto di energia e ruotano il proprio spin per assumere lo stato
di alta energia (opposti al campo). Quando si veri�ca questa transizione di
spin, si dice che i nuclei sono in risonanza con la radiazione applicata, da qui
il nome risonanza magnetica nucleare.
Siccome il dipolo magnetico µ di un certo nucleo é una costante, si potrebbe
prevedere che tutti i nuclei di un certo tipo debbano risuonare esattamente
alla stessa frequenza in un determinato campo magnetico applicato. Se le
cose stessero cosí, la spettroscopia NMR sarebbe del tutto inutile perché nello
spettro avremmo un unico segnale. Questo vale peró per singoli nuclei nel
vuoto; nella realtá la nube elettronica intorno al nucleo e gli altri atomi vicini
hanno una loro in�uenza per cui il segnale NMR di un certo nucleo risulta
spostato nello spettro a frequenze piú alte o piú basse a seconda degli atomi
circostanti. Questo campo magnetico secondario scherma il nucleo dalla piena
39

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
forza del campo applicato. Il campo magnetico applicato induce, sull'intorno
elettronico di ogni nucleo, un campo magnetico locale opposto (fenomeno di
induzione). La nube elettronica provoca cioé una modulazione locale del campo
applicato, detta schermatura. Tanto pié grande é la densitá elettronica, tanto
maggiore sará questa schermatura, cosí i nuclei che si trovano in un intorno
ricco di elettroni, sentiranno un campo magnetico piú basso e quindi subiranno
la transizione ad una frequenza applicata piú bassa rispetto ai nuclei situati
in intorni poveri di elettroni. Lo spostamento risultante nel segnale NMR per
un dato nucleo é detto spostamento chimico e, in generale, protoni o carboni
adiacenti ad atomi elettronegativi risultano deschermati quindi sentono un
campo magnetico applicato piú intenso e subiscono transizione a frequenze
maggiori e, nello spettro, si trovano ad uno spostamento chimico piú alto,
�gura2.12.
Figura 2.12. Spostamenti per l'Idrogeno
Lo spostamento chimico assoluto risulta tanto maggiore quanto piú grande
é il campo applicato, quindi strumenti diversi fornirebbero spostamenti chimici
assoluti diversi in rapporto alla intensitá del loro campo magnetico. Per questo
si preferisce utilizzare lo spostamento chimico relativo δ de�nito dal rappor-
to tra lo spostamento osservato rispetto al (CH3)4Si in Hz e la frequenza
dell'oscillatore in MHz
In questo modo lo spostamento chimico , misurato in ppm (Hz/MHz), é
indipendente al campo applicato. Al numeratore é stato inserito il tetrametilsi-
lano, (CH3)4Si, nel quale i carboni e i protoni (idrogeni) sono piú fortemente
40

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
schermati di quanto si osserva nella maggior parte delle altre molecole or-
ganiche e che è stato quindi preso come zero nella scala di riferimento. In piú,
ad esempio per l'idrogeno é possibile anche risalire al numero di atomi con
lo stesso spostamento in quanto l'intensitá dell'assorbanza é proporzionale al
numero di protoni che generano il segnale.
2.6.3 Analisi alla Fluorescenza
La �uorescenza é il risultato di un processo in tre fasi che si veri�ca in
determinate molecole (idrocarburi policiclici aromatici in genere o eteroci-
cli) chiamati �uorofori o coloranti �uorescenti Il processo responsabile della
�uorescenza é illustrato dal diagramma di stato elettronico, diagramma di
Jablonski(�gura2.13).
Figura 2.13. diagramma di Jablonsky
La prima fase é quella dell'eccitazione. Un fotone di energia hνEX é fornito
da una sorgente esterna, una lampada a incandescenza o un laser, e assorbita
dal �uoroforo, creando uno stato di singoletto elettronico eccitato (S′1).
Questo stato eccitato si mantiene per un tempo limitato (di solito 1-10
nanosecondi). Durante questo tempo, il �uoroforo subisce cambiamenti con-
formazionali ed é anche soggetto a una moltitudine di possibili interazioni con
l'ambiente molecolare. Questi processi hanno due conseguenze importanti. In
primo luogo, l'energia di S′1 é in parte dissipata, producendo uno stato di sin-
goletto eccitato rilassato (S1), da cui ha origine l'emissione di �uorescenza. In
41

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
secondo luogo, non tutte le molecole eccitate ritornano allo stato fondamentale
dopo l'emissione di �uorescenza.
La terza fase é l'emissione di un fotone di energia hνEM , riportando cosí il
�uoroforo al suo stato fondamentale S0. In seguito a fenomeni di dissipazione
di energia durante il ciclo di vita dello stato eccitato, l'energia di questo fotone
é piú bassa, e quindi sará emessa ad una lunghezza d'onda maggiore. La
di�erenza di energia o di lunghezza d'onda rappresentata da hνEM - hνEX é
chiamato spostamento di Stokes. Lo spostamento di Stokes é fondamentale per
la sensibilitá delle tecniche di �uorescenza perché permette a fotoni d'emissione
di essere rilevati su un basso sfondo, e ben isolati da fotoni di eccitazione. Al
contrario, la spettrofotometria di assorbimento richiede la misurazione della
luce trasmessa a livelli elevati di luce incidente alla lunghezza d'onda stessa.
L'intero processo di �uorescenza é ciclico. A meno che il �uoroforo non
sia irreversibilmente distrutto nello stato eccitato, puó essere ripetutamente
eccitato e analizzato. Il fatto che un �uoroforo puó generare molte migliaia di
fotoni rilevabili é alla base dell' elevata sensibilitá delle tecniche di �uorescenza.
Per molecole poliatomiche in soluzione, le transizioni elettroniche discrete rap-
presentate da hνEM - hνEX nella �gura 2.13 sono sostituiti da spettri di energia
piú ampii chiamati spettro di eccitazione di �uorescenza e spettro di emissione
di �uorescenza. Lo spettro di eccitazione di �uorescenza di una specie �uoro-
fora in una soluzione diluita di solito é identico al suo spettro di assorbimento.
In condizioni diluite lo spettro di emissione di �uorescenza é indipendente dal-
la lunghezza d'onda di eccitazione, questo a causa della parziale dissipazione
di energia di eccitazione durante il tempo in cui si mantiene lo stato eccita-
to. L'intensitá dello spettro di emissione é proporzionale all' ampiezza dello
spettro di eccitazione di �uorescenza alla lunghezza d'onda di eccitazione.
42

2.6 Tecniche utilizzate per la caratterizzazione dei prodotti ottenuti
2.6.4 Analisi UV
Quando un fotone ultravioletto o visibile viene assorbito da una molecola,
questa passa dal suo stato elettronico fondamentale ad uno stato elettronico
eccitato. In un tipico spettro ultravioletto/visibile in ascissa viene riportata
la lunghezza d'onda e in ordinata la percentuale di trasmittanza o di assor-
banza. Se un materiale non è completamente trasparente si veri�cheranno
degli assorbimenti e quindi delle transizioni tra livelli energetici elettronici. In
questo secondo caso lo spettro registrato sará caratterizzato da una serie di
picchi di altezza variabile per ciascuna transizione, in relazione all'intensitá
dell'assorbimento stesso.
Quando un fotone possiede energia hν su�ciente a�nché avvenga una tran-
sizione elettronica, si veri�ca un assorbimento che entro un certo intervallo di
concentrazioni e in presenza di radiazione monocromatica segue la legge di
Lambert-Beer:
Abs = ελ · l · [C] (2.3)
dove ελ rappresenta il coe�ciente di assorbimento molare a una detrminata
lunghezza d'onda λ, [C] é la concentrazione molare della soluzione ed l é il
cammino ottico costituito dallo spessore di campione attraversato dal raggio
luminoso. Il caratteristico assorbimento presentato da diversi tipi di sostanze
dipende dalla presenza di determinati cromofori.
Le tecniche utilizzate in questo lavoro sono dunque molteplici e diverse
tra loro. Per la loro trattazione in dettaglio si fará riferimento alle sezioni
seguenti in cui verranno ricostruiti i passaggi con i quali sono stati sintetizzati i
macromonomeri e le relative NPs, e verranno presentati i risultati delle tecniche
analitiche appena citate.
43

Capitolo 3
Sintesi
Lo scopo del suddetto lavoro di tesi é l'ottenimento di NPs funzionalizzate con
diverse tipologie di coloranti azoici da utilizzare come marker per gli aggregati
di �brille-amiloidi.
Sono state indagate due possibili strade per la produzione di tali NPs; una
che prevede l'utilizzo di LA(acido lattico) come monomero costituente, nell'al-
tra invece le nanoparticelle sono formate da HEMA(2-idrossietil metacrilato)
e MMA(metil-metacrilato).
Per la sintesi delle nanoparticelle si é proceduto con nanoprecipitazione per
il polimero biodegradabile (PLA) sintetizzato via ROP; e polimerizzazione in
emulsione a dare polimeri biocompatibili a partire da monomeri quali MMA e
HEMA.
Mentre la funzionalizzazione delle NPs con il colorante é ottenuta attraverso
esteri�cazione di Steglich per linkare l'acido amminobenzoico al polimero e
ottenere l'intermedio necessario alla formazione del colorante azoico e reazione
di copulazione per formare il colorante azoico.
Nelle pagine che seguiranno verranno descritte in dettaglio le reazioni uti-
lizzate per la sintesi del prodotto �nale. Dopo ogni reazione riportata saranno
44

3.1 PLA route
fatte delle considerazioni sugli step utilizzati in modo tale da illustrare il perché
é stata scelta una particolare via sintetica e determinate condizioni operative.
3.1 PLA route
La prima fase del lovoro é stata la produzione dei coloranti utilizzando LA.
La procedura é la seguente: il polimero PLA è stato ottenuto tramite polimer-
izzazione ROP; si é utilizzata la reazione di esteri�cazione di Steglich e si é
proceduto con la reazione di copulazione per formare il colorante azoico.
3.1.1 Sintesi del PLA
Utilizzando L,LA si è ottenuto PLA tramite ring opening polymerization.
Le quantitá di monomero, iniziatore e catalizzatore sono state calco-
late basandosi sulla ricetta pubblicata in letteratura[21-22]. Come catal-
izzatore si é utilizzato lo stagno ottanoato, mentre come iniziatore l'alcool
amilico,(CH3(CH2)4OH), scelto perché ha peso molecolare relativamente alto
e durante il procedere della reazione non evapora facendo rimanere costanti i
rapporti iniziali dei composti caricati.
La sintesi avviene é stata e�ettuata in un pallone di vetro da 50cc com-
pletamente immerso in un bagno ad olio. Per controllare la temperatura del
sistema si é equipaggiato il bagno con un termometro VERTEX. L'intero sis-
tema é sottoposto ad agitazione magnetica. Il pallone é chiuso con un tappo
di gomma, cosa che permette di iniettare altri reagenti senza doverlo aprire.
Si é prodotto PLA a basso peso molecolare, 5000g/mol. 2 grammi di
monomero sono stati pesati e riscaldati �no a fusione (114°C) e poi al raggiung-
imento della temperatura di reazione (130°C). É stato preparato un campione
con 35mg di alcool amilico e e 5.5mg di catalizzatore e lo si é iniettato con una
siringa nel reattore una volta che il lattide ha raggiunto 130°C. La reazione
prosegue per tre ore.
45

3.1 PLA route
3.1.2 Reazione di esteri�cazione di Steglich
Ottenuto il polimero si é passati a legare l'acido amminobenzoico ad esso at-
traverso la reazione di esteri�cazione di Steglich. I reagenti utilizzati sono
PLA, acido amminobenzoico e DCC in quantitá equimolare, perché per ogni
molecola di PLA presenta un -OH che si lega con un gruppo -COOH. Si é
utilizzata DMAP in quantitá catalitica.
Sono stati sciolti 2g di PLA in 30mL di THF in un beker da 250mL e lo si
é collocato in un bagno di ghiaccio. 55mg di acido amminobenzoico sono stati
sciolti a parte in 5mL di THF e aggiunti al reagente nel beker. Il tutto é stato
poi messo sotto tenue agitazione magnetica (circa 150rpm). Si é preparata una
soluzione con 83mg di DCC e 10mg di DMAP in THF e la si é fatta goccialare
nel beker. Per tutto il tempo di questa operazione i reagenti sono stati tenuti
in bagno di ghiaccio. La reazione prosegue quindi per 5 ore a temperatura
ambiente. Al termine della stessa, la soluzione é stata lasciata riposare per
24 ore e si é osservato sul fondo del beker la dicicloesilurea precipitata sotto
forma di cristalli.
Il prodotto ottenuto é stato �ltrato su un �ltro di carta con pori di di-
ametro 2µM; la soluzione é stata poi messa in un rotovapor in modo tale da
far evaporare il THF ed ottenere quindi solo il polimero con legato l'acido
amminobenzoico.
Nella pagine seguenti é illustrato il meccanismo con il quale avviene la
reazione appena descritta, �gure 3.1 , 3.2.
46

3.1 PLA route
Figura 3.1. meccanismo della reazione di esteri�cazione di Steglich
47

3.1 PLA route
Figura 3.2. meccanismo della reazione di esteri�cazione di Steglich
48

3.2 NPS a partire da HEMA e MMA
3.1.3 PLA route : discussione
Con la polimerizzazione ROP e la successiva reazione di esteri�cazione di
Steglich si sono prodotti dei polimeri che hanno il vantaggio di essere
biodegradabili, ma per contro ogni catena é legata ad una sola molecola di acido
amminobenzoico (sull' -OH terminale) e quindi con la reazione di copulazione
si riuscirebbe a funzionalizzare la catena polimerica con una sola molecola di
colorante. Per veri�care al meglio l'e�cacia di questi polimeri funzionalizzati
si é dieciso produrre polimeri con un numero decisamente maggiore di gruppi
funzionali per catena.
Per questo motivo, e per il fatto che le rese dell'esteri�cazione ottenute
non sono mai state piú alte del 60% si é scelto di concentrare il proseguio del
lavoro sullo sviluppo di nanoparticelle a base di MMA e HEMA. Queste sono
costituite per la maggior parte da MMA in quantitá sempre maggiore del 97%,
e aumentando la quantitá di HEMA é possibile aumentare di molto il numero
di gruppi funzionali per ogni molecola di polimero. Utilizzando la polimeriz-
zazione in emulsione si ottiene un prodotto �nale fatto da monomeri contenenti
ognuno un gruppo -OH- e quindi piú possibili punti di attacco per l'acido am-
minobenzoico. Di conseguenza il prodotto �nale é una catena polimerica che
puó avere una ben piú alta concentazione di particelle di colorante azoico.
3.2 NPS a partire da HEMA e MMA
Per questo cammino sintetico, le reazioni coinvolte nell'ottenimento del prodot-
to �nale sono l'esteri�cazione di Steglich, la polimerizzazione in emulsione e
la reazione di copulazione e formazione del colorante azoico. Si é cercato di
individuare la sequenza ottimale con la quale procedere con le reazioni, per
questo sono state e�ettuate delle prove preliminari con quantitá di reagenti
basse.
49

3.3 Sintesi dei macromonomeri
Sono state indagati tre diversi cammini sintetici :
1. formazione del colorante azoico - esteri�cazione di Steglich - polimeriz-
zazione in emulsione
2. esteri�cazione di Steglich - formazione colorante azoico - polimerizzazione
in emulsione
3. esteri�cazione di Steglich - polimerizzazione in emulsione - formazione
colorante azoico
Dopo aver e�ettutato un paio di prove per ogni sequenza si é scelto di
operare seguendo la modalitá indicata al punto numero 2, in quanto o�riva le
migliori garanzie in termini di riproducibilitá delle prove e resa delle reazioni.
La prima via é stata scartata per la di�coltá di sciogliere il colorante azoico nei
solventi da utilizzare per l'esteri�cazione e per le basse rese della stessa, sempre
intorno al 70%. Nel terzo cammino sintetico i problemi incontrati sono legati
al latex di particelle che non era stabile durante il processo di formazione del
colorante azoico. Dunque, il cammino sintetico scelto é quello di sintetizzare
un macromonomero costituito da HEMA funzionalizzato con la molecola di
colorante e poi di copolimerizzarlo con MMA a dare direttamente NPs.
3.3 Sintesi dei macromonomeri
Il primo passo di questa nuova via di sintesi é stato l'ottenimento di un
macromonomero a base di HEMA funzionalizzato con diverse tipologie di
coloranti azoici. Si é dovuto far reagire l'HEMA con l'acido amminoben-
zoico per ottenere l'intermedio necessario per la reazione di formazione del
colorante azoico e poi si é proceduto con la reazione di copulazione ad ottenere
il macromonomero funzionalizzato.
50

3.3 Sintesi dei macromonomeri
3.3.1 Reazione di esteri�cazione di Steglich
Il primo step é stato legare l'acido amminobenzoico all'HEMA attraverso la
reazione di esteri�cazione di Steglich. I reagenti utilizzati sono stati HEMA,
acido amminobenzoico e DCC, sempre in quantitá equimolare per la stessa
ragione espressa in precedenza, e DMAP in quantitá catalitica.
In un beker da 100mL sono stati caricati 1g di acido amminobenzoico e
4.7g di HEMA e sono stati sciolti in 50mL di acetonitrile. Il beker é stato
collocato in un bagno di ghiaccio e messo sotto tenue agitazione magnetica
(circa 150rpm). 1.5g di DCC, precedentemente sciolti in acetonitrile, sono
stati aggiunti goccia a a goccia nel beker. Per il tempo del gocciolamento la
miscela reagente é stata tenuta nel bagno di ghiaccio. La reazione prosegue per
3 ore a temperatura ambiente. Dopo una decina di minuti la soluzione diventa
opaca e sono giá visibili depositi di dicicloesilurea insolubile nel solvente di
reazione. Al termine della reazione si nota la presenza di un corpo di fondo
nel beker, la dicicloesilurea precipita sotto forma di polvere bianca.
La soluzione é stata �ltrata utilizzando una carta da �ltro con pori di
diametro di 110mm. Utilizzando un rotovapor é stato fatto evaporare l'ace-
tonitrile e si é ottenuta una soluzione di colore arancione chiaro di HEMA con
legato l'acido amminobenzoico e HEMA libero.
Le due pagine seguenti mostrano come avviene la reazione appena
descritta,�gure 3.3 , 3.4 .
51

3.3 Sintesi dei macromonomeri
Figura 3.3. meccanismo della reazione di esteri�cazione di Steglich
52

3.3 Sintesi dei macromonomeri
Figura 3.4. meccanismo della reazione di esteri�cazione di Steglich
53

3.3 Sintesi dei macromonomeri
Ester�cazione di Steglich : discussione
L'HEMA é stato utilizzato in quantitá 5:1 molare rispetto all'acido ammi-
nobenzoico per favorire la reazione tra questi due composti ed evitare che
l'acido si leghi con se stesso. La quantitá invece della DCC é equimolare a
quella dell'acido amminobenzoico
In letteratura sono indicati diversi solventi in cui é possibile far avvenire la
reazione, e tutti quanti garantiscono elevate rese. In questo lavoro ne sono stati
testati alcuni al �ne di indivuduare quello piú appropriato per questi reagenti.
Un indicatore del buon esito della reazione é la formazione della dicicloesiurea.
Questo composto é insolubile nel solvente di reazione e precipita; se la reazione
avvenisse con resa unitaria la sua quantitá molare sarebbe pari a quella della
DCC caricata. Per quanti�care l'e�cacia dei diversi solventi si é utilizzato
questo parametro assieme al tempo di risposta del sistema, ovvero il tempo in
cui si é osservato il primo precipitato. La tabella di seguito riporta i risultati
ottenuti per i vari solventi.
solvente resa in dicicloesilurea tempo (ordine di grandezza)THF 55-65% giornoDCM 50-60% ore
acetonitrile 85-95 % minutiDMF 60-70% ore
Tabella 3.1. Resa in DCU per l'esteri�cazione di Steglich con diversi solventi
A fronte dei risultati ottenuti quindi, le reazioni di esteri�cazione sono state
condotte utilizzando l'acetonitrile come solvente.
Analisi all'NMR del prodotto di reazione
Il prodotto della reazione é stato analizzato tramite NMR per avere un check
del buon esito della stessa. La prova é stata condotta sui regenti, HEMA
e acido amminobenzoico, e sul prodotto dell'esteri�cazione. I campioni sono
54

3.3 Sintesi dei macromonomeri
stati disciolti DMSO deuterato in quantitá tale da avere una concentrazione
di 20mg/ML.
(a) acido amminobenzoico. (b) HEMA.
(c) prodotto dell'esteri�cazione.
Figura 3.5. Risultati dell'analisi all'NMR
Dall'NMR del prodotto si puó vedere come il picco dell'-OH proprio dell'aci-
do amminobenzoico sia scomparso, a conferma del fatto che questo composto
ha reagito completamente. I picchi 6 e 7 che corrispondono agli -H dell'anel-
lo benzenico dell'acido ammino benzoico sono spostati verso sinistra dopo la
55

3.3 Sintesi dei macromonomeri
reazione di esteri�cazione. Uno shift verso sinistra rispetto alla posizione che
avevano nel composto singolo lo si nota anche per i picchi 4 e 5 che si riferiscono
all'HEMA.
3.3.2 Reazione di copulazione e formazione dei
coloranti azoici
Ottenuto il monomero di HEMA funzionalizzato con il gruppo amminico
aromatico primario si é proceduto con la reazione per formare il colorante
azoico.
Sono state utilizzate 4 molecole diverse, acido salicilico, 2-naftolo, acido
solfanilico e tio�avina T, per la preparaziome del colorante. Il procedimento
adottato é lo stesso per tutti e tre i composti. Di seguito verranno descritte le
operazioni svolte utilizzando la ti�avina. Si é deciso di illustrare la procedura di
formazione del macromonomero funzionalizzato con questo colorante perché di
esso ne state studiate piú a fondo le caratteristiche ed in modo particolare sono
state condotte delle analisi atte studiarne il comportamento con gli aggregati
proteici confrontandolo con quello della sola molecola di tio�avina T.
Le quantitá delle quattro diverse molecole sono uguali al numero di moli di
acido amminobenzoico utilizzate nel precedente step.
Tutti i passaggi che verranno adesso descritti sono stati condotti a 0°C e
sotto agitazione magnetica (300 rpm).
In un beker da 250 mL sono stati messi 50 mL di acqua distillata e portati
a pH = 1 utilizzando 3.2g acido cloridrico al 30%. Il pH, durante tutte le
operazioni, é stato misurato utilizzando un pHmetro analogico distribuito dalla
�metrohm�.
In seguito é stata aggiunta la miscela recuperata con il rotovapor contenente
HEMA con legato l'acido amminobenzoico e HEMA non reagito. Avere del
56

3.3 Sintesi dei macromonomeri
monomero non reagito non é un problema perché rimane in soluzione nell'acqua
e non partecipa alla reazione di formazione del colorante azoico.
Dopo 10 minuti si é fatta gocciolare lentamente, utilizzando una buretta,
una soluzione di 0.51g di nitrito di sodio (NaNO2) e 21mL di acqua distillata.
La quantitá di nitrito di sodio é equimolare alla quantitá di acido amminoben-
zoico presente nel monomero. Alla �ne dell'operazione il pH misurato risulta
circa 2. In seguito si sono aggiunti 0.8g di carbonato di sodio (Na2CO3),
quantitá anch'essa equimolare con l'acido amminobenzoico, sciolti in 20mL di
acqua distillata �no a che il pH ha raggiunto un valore pari a 3. La miscela é
lasciata reagire per un'ora. A questo punto, la soluzione nel beker é di colore
giallo chiaro. Questa reazione é servita a sintetizzare il sale di diazonio, come
illustrato in �gura 3.6.
In un beker da 500 mL sono stati sciolti 2.32g di thio�avina T in 300mL
di acqua distillata portata a 50°C. Si é fatto ció per favorire la dissoluzione
completa della thio�avina T. Il pH di questa soluzione é stato portato a pH
compreso tra 9 e 10 utilizzando una soluzione di (Na2CO3) e acqua distillata.
Quando si é disciolto tutta la thio�avina T, il beker é stato messo in un bagno
di ghiaccio. A questo punto tramite pipetta é stata aggiunta la miscela di
colore giallo chiaro precedentemente preparata; la soluzione diventa subito di
colore arancione intenso.
Durante questa operazione, che é la vera e propria reazione di copulazione,
é importatnte che il pH si mantenga tra 9 e 10. Per questo si prepara a
parte una soluzione di carbonato di sodio (Na2CO3) e acqua distillata da
utilizzare nel caso il pH scenda al di sotto di tale range di valori. Il controllo
del pH é fondamentale perché se troppo alto si formano dei complessi che non
reagiscono, mentre troppo basso comporta una velocitá di reazione lenta.
La miscela é lasciata reagire per un'ora a 0°C e per un'ora a temperatura
ambiente. Per far precipitare il macromonomero cosí ottenuto si porta il con-
tenuto del beker a pH = 6 utilizzando acido cloridrico al 30% e si lascia riposare
57

3.3 Sintesi dei macromonomeri
il tutto per 30 minuti. Nel beker saranno visibili due fasi separate: le particelle
nella parte alta e la soluzione leggermente colorata sul fondo. Le particelle col-
orate sono state recuperate utilizzando carta da �ltro con pori di diametro
2µM; dopo essiccazione si ottiene una polvere di colore giallo za�erano.
Nelle pagine successive é illustrato il meccanismo della reazione di
copulazione e formazione dei coloranti azoici, �gure 3.6, 3.7.
Figura 3.6. (a) formazione del sale di diazonio; composti ad anello aro-matico attivato: (b) acido salicilico, (c) 2-naftolo, (d) acido solfanilico,(e) tio�avina T
58

3.3 Sintesi dei macromonomeri
Figura 3.7. meccanismo della reazione di copulazione
59

3.4 Sintesi delle nanoparticelle
Reazione di copulazione e formazione
dei coloranti azoici : discussione
Con la metodologia descritta nel paragrafo precedente sono stati prodotti quat-
tro macromonomeri funzionalizzati ognuno con una diversa molecola. Per og-
nuno dei prodotti ne é stata valutata la resa in termini di percentuali in peso
di particelle colorate recuperate rispetto al valore teorico recuperabile. Nella
seguente tabella sono riportati questi valori, assieme al colore delle particelle
ottenute per i diversi composti aromatici utilizzati per funzionalizzarle.
composto colorante recuperato(% in peso) colore2-naftolo 90-95% rosso vivo
acido solfanilico 80-85% arancione chiaroacido salicilico 80-85% arancione accesotio�avina T 85-90% giallo za�erano
Tabella 3.2. Caratteristiche dei diversi coloranti ottenuti
Con questa procedura, tutto sommato di semplice esecuzione, si sono
ottenuti ottimi risultati in termini di resa per tutte e quattro le molecole
utilizzate, in particolare per il 2-naftolo e la tio�avina T.
É stata dunque messa a punto una via sintetica facilmente riproducibile
per la produzione dei macromonomeri funzionalizzati e che garantisce alte
conversioni dei reagenti ai prodotti desiderati.
La risposta all'analisi all'UV di tali prodotti sará presentata in una
successiva sezione.
3.4 Sintesi delle nanoparticelle
Ottenuti i macromonomeri in forma di polvere, si é passati alla loro succes-
siva polimerizzazione e formazione di nanoparticelle mediante una polimer-
izzazione in emulsione in modalitá batch. In particolare si è proceduto con
60

3.4 Sintesi delle nanoparticelle
una copolimerizzazione utilizzando MMA. Sono stati prodotti dei latex al 5%
di monomero rispetto all'acqua presente. É stata altresí indagata l'in�uen-
za di parametri di processo, quali la quantitá di emulsionante presente, sulle
dimensioni �nale delle NPs.
La ricetta standard per ottenere le NPs é la seguente: su 50g di soluzione,
47.5g sono di acqua distillata e 2,5g dei due monomeri. Per quest'ultimi si é
scelto di utilizzare il macromonomero colorato allo 0.2% rispetto all'MMA, per
cui le quantitá sono 2,495g di MMA e 0,005g di monomero colorato. L'emul-
sionante utilizzato é stato l'SDS. Sono state e�ettuate delle prove con la sua
quantitá pari al 5% e al 10% in peso rispetto ai comonomeri presenti. Come
iniziatore sono stati utilizzati 35mg di 4,4'-Azobis(4-cyanovaleric acid).
La preparazione é identica per tutte e le tipologie di macromonomero col-
orato utilizzato. Come reattore si é usato un pallone di vetro a tre colli da
100mL. In esso sono stati caricati 40mL di acqua distillata portata a pH 10
utilizzando idrossido di sodio (NaOH), per deprotonare i macromonomeri e
renderli solubili in acqua . 25mg di macromonomero colorato sono stati mes-
si nel reattore si é posto il tutto sotto agitazione magnetica �no a completo
scioglimento del macromonomero. Sono stati pesati 2,475g di MMA e poi car-
icati all'interno del reattore. A questo punto é messo caricato l'emulsionante,
la sua quantitá é pari a 125mg, nel caso in cui lo si sia utilizzato al 5%, e 250mg
nel caso al 10%. Il reattore é stato posto in un bagno d'olio per portarlo alla
temperatura di reazione, per questo set di polimerizzazioni si é lavorato a 70°C.
Al bagno d'olio é collegato un termometro che opera un controllo sulla tem-
peratura mantenendola costantemente al valore �ssato (vertex). L'agitazione
magnetica é stata impostata a 300rpm. All'interno del reattore si é realizzata
un'atmosfera inerte facendo gorgogliare gas d'azoto attraverso una cannuccia
di vetro per circa 25 minuti in modo da rimuovere tutto l'ossigeno presente nel
sistema, tale �usso viene in seguito abbassato per garantire un'atmosfera inerte
nel all'interno del reattore. Quando il sistema ha raggiunto la temperatura di
61

3.4 Sintesi delle nanoparticelle
reazione, l'iniziatore, precedentemente disciolto in 5mL d'acqua, é stato iniet-
tato rapidamente nel reattore con una siringa attraverso un tappo di materiale
gommoso che non permette all'ossigeno di raggiungere la zona di reazione. La
stessa prosegue per circa tre ore ed é stata fermata solo dopo aver veri�cato
tramite prelievo e successiva prova gravimetrica di avere conversioni maggiori
del 95%.
Dopo aver fatto delle prove preliminari alla �uorescenza utilizzando queste
NPs e gli aggregati di proteine si é visto che lavorando con questi valori di SDS
gli aggregati proteici venivano destabilizzati. Per questo motivo le polimer-
imerizzazioni del macromonomero con la tio�avina sono state condotte utiliz-
zando SDS allo 0.05%. La polimerizzazione é stata eseguita in modalitá batch
come descritto in precedenza. In questa modo peró alla �ne della polimeriz-
zazione si é notato che il latex non era particolarmente colorato e un pó di
macromonomero rimaneva attaccato alle pareti.
Per risolvere questo problema si é passati ad una polimerizzazione �s-
tarved� utilizzando sempre le stesse quantitá. Nel pallone é stato caricato il
macromonomero disciolto in acqua basica, l'emulsionante e 0.65g di MMA,
quantitá che permette che questo sia completamente solubile in acqua. I
restanti 1.845g di MMA sono stati fatti gocciolare in un'ora e mezzo una vol-
ta che si é creato un ambiente inerte e caricato l'iniziatore. Al termine della
polimerizzazione il latex risulta giallo chiaro e di macromonomero sulle pareti
non se né trovata traccia.
3.4.1 Analisi al light-scattering
Tutti i latex prodotti sono stati caratterizzati in termini di dimensione media
di particella e Z-potential tramite analisi al light-scattering.
La preparazione del campione per le analisi delle dimensioni medie é analo-
ga per tutti i latex prodotti. É stato prelevato 1mL di campione e lo si é
posto all'interno dell'apposita cuvette, �gura 3.8 a, poi si é portato il tutto a
62

3.4 Sintesi delle nanoparticelle
volume utilizzando acqua distillata. Per il calcolo dello Z-potential sono state
utilizzate le couvette in �gura 3.8 b , riempite con 1mL di campione senza
diluirlo.
(a) (b)
Figura 3.8. cuvette utilizzate per le analisi, a : Z-ave, b : Z-pot
I risultati ottenuti in seguito alle analisi al light-scattering sono presentati
nella tabella di seguito.
macromonomero funzionalizzante quantitá sds% z-ave(nm) PDI z-pot(mV)naftolo 10% 36 0,044 -51,7naftolo 5% 77 0,067 -37,7
acido salicilico 10% 41 0,066 -44,4acido salicilico 5% 47 0,058 -76,4acido solfanilico 10% 38 0,2 -45,2acido solfanilico 5% 57 0,042 -41.3tio�avina (batch) 0.05% 236 0,061 -37,4
tio�avina (�starved�) 0.05% 240 0,058 -38,1
Tabella 3.3. Risultati dell'analisi al light-scattering
Si sono dunque prodotte delle NPs aventi tutte delle dimensioni ridotte
che rientrano in pieno nel target richiesto per il loro impiego nel drug delivery
(30-150nm). Il valore della polidispersitá é molto basso e questo indica che si
ha una distrubuzione stretta di dimensioni intorno al valore medio. Inoltre i
valori di Z-potential sono tutti al di sopra del valore di -35mV per cui i sistemi
colloidali risultano avere buona stabilitá.
63

Capitolo 4
Analisi prodotti sintetizzati
La capacitá dei macromonomeri, e delle relative NPs, di legarsi agli aggregati
di proteine β-amiloidi é stata indagata tramite analisi alla �uorescenza. Sono
stati analizzati prima i soli materiali per vedere come rispondevano alla �u-
orescenza, per ognuno é stata individuata la lunghezza d'onda di eccitazione
ottimale a cui corrispondeva un ben de�nito spettro di emissione.
Caratterizzato quindi ogni materiale, si é passato al suo test sugli aggregati
di proteine. Individuati i composti che hanno la capacitá di legarsi agli ag-
gregati, si é utilizzati questi per delle analisi atte a monitorare la loro cinetica
di aggregazione. La modalitá con cui sono state condotte tutte le analisi sará
discussa in dettaglio nelle pagine successive.
Di seguito saranno anche presentati i risultati delle analisi all'UV condotte
per caratterizzare l'assorbanza dei macromonomeri e delle NPs.
4.1 Analisi alla �uorescenza : �binding� degli
aggregati proteici
Per le analisi é stato utilizzato uno spettrometro di �uorescenza prodotto dalla
Perkin Elmer. Nel piatto d'analisi, �gura 4.1, sono stati caricati i campioni di
64

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
macromonomero e Nps, e gli aggregati proteici. Si é lasciato incubare il tutto
per mezz'ora a temperatura ambiente e poi si é proceduto con la prova.
Figura 4.1. piatto utilizzato per l'analisi UV
4.1.1 Preparazione degli aggregati
Gli aggregati proteici di insulina sono stati ottenuti come segue. Insulina in
forma di polvere e' stata disciolta in una soluzione 25µM di HCl con 0.1 M
NaCl a pH 1.6 a una concentrazione di 5.5 g/L. Il tutto é stato �ltralto con
un �ltro per siringhe da 0.2µm e la concentrazione é stata misurata tramite
assorbanza UV a 280 nm. La soluzione é stata caricata in una preovetta
Eppendorf e messa in un termomixer settato a 62°C senza agitazione. Dopo
12 ore la formazione di �brille o é visibile ad occhio nudo.
65

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
4.1.2 Test dei materiali prodotti
Il primo set di prove sono incentrate sulla veri�ca della capacitá dei
nuovi macromonomeri prodotti di legarsi alle �brille amiloidi. I quattro
macromonomeri sono stati sciolti in KPS in quantitá tale da avere una concen-
trazione di 2mg/mL. Le prove sono state condotte con un rapporto tra moli di
colorante e moli di proteina di circa 3. Nel piatto d'analisi sono stati caricati
150µL di soluzione contenente il macromonomero e 60µM di aggregati.
Le analisi sono state condotte e�ettuando uno scan d'emissione; ovvero
si é eccitato il materiale ad una speci�ca lunghezza d'onda, propria di ogni
composto, e si é registrato l'andamento dell'assorbanza a diverse lunghezze
d'onda di emissione.
Nella tabella seguente sono riassunte le lunghezza d'onda a cui sono stati
eccitati i materiali e le relative quantitá utilizzate nel corso di questa analisi.
composto funzionalizzante λex [nm] qtá [µL] qtá proteine(940µM) [µL]2-naftolo 445 150 10.2
ac.solfanilico 460 150 10.2ac.salicilico 350 150 10.2tio�avina 450 150 10.2
Tabella 4.1. quntitá utilizzate durante l'analisi
I risultati delle analisi sono presentati di seguito, �gure 4.2(a)(b)(c).
66

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
(a) linea continua : macromonomero piú proteina,linea tratteggiata : macromonomero.
(b) linea continua : macromonomero piú proteina,linea tratteggiata : macromonomero.
(c) linea continua : macromonomero piú proteina, lineatratteggiata : macromonomero.
Figura 4.2. Emission scan del macromonomero con (a)2-naftolo,(b)acido solfanilico, (c)acido salicilico
.
67

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
Come si puó ben notare dai gra�ci, due dei tre macromonomeri prodotti,
quello funzionalizzato con il 2-naftolo e quello con l'acido solfanilico, subiscono
un aumento dell'assorbanza di emissione dopo l'incubazione con gli aggregati
di insulina e ció indica che questi due macromonomeri sono in grado di legarsi
alle �brille. Il macromonomero con l'acido salicilico invece presenta lo stesso
spettro prima e dopo l'incubazione, che signi�ca che non é in grado di attaccarsi
alle proteine.
I tre macromonomeri hanno strutture simili. La parte indicata con R nelle
�gure precedenti é la stessa, costituita da HEMA e acido ammino benzoico,
e la di�erenza sta nella parte funzionale �nale. Quindi la capacitá di legarsi
o meno agli aggregati proteici é data appunto dal tipo di molecola che si é
utilizzata nella formazione del colorante azoico.
A fronte dei risultati ottenuti, dato l'aumento di un ordine di grandezza che
si ottine dopo l'incubazione con le proteine, si potrebbe pensare ad un'in�uenza
del gruppo con lo zolfo e con l'azoto, contenuti nell'acido solfanilico, nel favorire
il legame con le�brille amiloidi. Inoltre questi due gruppi sono presenti in due
dei coloranti che attualmente vengono utilizzati come markers di tali aggregati,
congo red e tio�avina T.
Anche la presenza di due anelli benzenici vicini potrebbe essere un fattore
che facilita il link con le proteine. Infatti il macromonomero con il 2-naftolo
presenta una crescita nell'assorbanza di emissione dopo la reazione con le �b-
rille, a contrario di quello funzionalizzato con l'acido salicilico. I due composti
hanno in comune un anello benzenico con un gruppo -OH, e di�eriscono perché
il primo é legato ad un altro anello benzoico mentre il secondo ad un gruppo
carbossilico -COOH.
I test sul macromonomero funzionalizzato con la tio�avina sono stati e�et-
tuati in maniera comparata con un campione di sola tio�avina in modo tale
da avere un confronto diretto sull'e�cacia di questo nuovo macromonomero
rispetto a quello comunemente utilizzato.
68

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
Le prime prova e�ettuate sono state uno scan di emissione, con lunghezza
d'onda di eccitazione 450nm e rapporto tra moli di macromonomero e proteina
pari a 2.5 e 0.1, �gure 4.3(a)(b) .
(a) linea continua : macromonomero-proteina, (•) tiofavina-proteina, lineatratteggiata : macromonomero, tio�avina.
(b) linea continua : macromonomero-proteina, • tiofavina-proteina, lineatratteggiata : macromonomero, tio�avina.
Figura 4.3. Emission scan, (a)rapporto moli di colorante-moli proteine =2.5,(b)rapporto moli di colorante-moli proteine = 0.1
.
Il macromonomero funzionalizzato con la tio�avina si lega alle �brille di in-
sulina e di conseguenza l'assorbanza aumenta dopo l'incubazione con le stesse,
4.3(a)(b). Si puó notare come nelle prove condotte in largo eccesso di col-
orante rispetto alle proteine, �g4.3(a), il suo segnale sia addirittura maggiore
di quello della sola tio�avina. Lavorando invece in difetto di colorante rispetto
69

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
alle proteine, �g 4.3(b), la sola tio�avina esibisce un segnale di assorbanza piú
alto. Inoltre confrontando i valori assoluti dell'assorbanza delle due prove si
é notato che il segnale dell'assorbanza di emissione diminuisce passando dal
rapporto moli di colorante-moli di proteine uguale a 2.5 a quello paria 0.1.
Per indagare questo comportamento é stata e�ettuata un'analisi di scan
d'emissione facendo variare questo rapporto tra i due valori considerati nella
prova precedente, �gura 4.4.
Figura 4.4. andamento dell'Abs al variare del rapporto tra moli di colorantee di proteina, (•) tio�avina, (-) macromonomero
Dal gra�co si nota come, nella fase iniziale, aumentando il numero di moli di
colorante rispetto alla proteina si ha una crescita nel valore dell'assorbanza �no
al raggiungimento di un plateau per un valore di questo rapporto pari a 0.1.
In questo range il valore dell'assorbanza della tio�avina é maggiore rispetto
a quello del macromonomero. Superato il valore di 0.5 l'assorbanza inizia
a decrescere �no al raggiungimento di un secondo plateau per un valore del
rapporto tra moli di colorante e proteina pari a 1.7. In queste condizioni invece
é l'assorbanza del macromonomero ad essere piú alta rispetto alla tio�avina.
Questo andamento, che prevede una diminuzione dell'assorbanza del col-
orante all'aumentare del suo numero di moli rispetto alle proteine, potrebbe
essere attribuito ad una sorta di complessazione che lo stesso subisce e in
70

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
questa nuova struttura risulta piú di�coltosa l'interazione con le proteine. Il
fatto che per alti valori di rapporto tra moli di colorante e moli di proteine il
macromonomero presenti un segnale piú alto potrebbe signi�care che risente
meno di questo fenomeno.
Indagate le capacitá dei macromonomeri di legarsi o meno agli aggre-
gati proteici si é passati al test delle ralative nanoparticelle. Per ragioni di
tempo si é riusciti a testare solo quelle fatte con il macromonomero conte-
nente la tio�avina. La scelta di partire da quest'ultimo sta nel fatto che il
rispettivo macromonomero ha o�erto i migliori risultati in termini di segnale
d'assorbanza.
Sono stati testati i due latex prodotti l'uno in modalitá batch e l'altro in
modalitá �starved�. La modalitá di analisi é la stessa delle prove precedenti.
Nel piatto sono stati caricati 150 µL di NPs e 10,2µL di aggregati proteici,
dopo un tempo di incubazione pari a mezz'ora si é passati alla prova. La
lunghezza d'onda di eccitazione é quella propria della tio�avina, 450nm.
Nella pagine che segue sono presenati i risultati, �g4.5(a)(b).
71

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
(a) linea continua : NPs-proteina, lineatratteggiata : NPs.
(b) linea continua : NPs-proteina, lineatratteggiata : NPs.
Figura 4.5. Emission scan, (a)NPs prodotte in modalitá �starved�,(b)NPs prodotte in modalitá batch
.
Anche le NPs formate dal macromonomero con la tio�avina, dunque, sono
in grado di legarsi agli aggregati di proteine. Come si puó vedere dai gra�ci
l'aumento di assorbanza dopo l'incubazione con le �brille é netto. In termi-
ni di segnale le NPs formate in modalitá �starved� presentano un valore di
assorbanza piú elevato rispetto a quelle formate in modalitá batch.
4.1.3 Test sulla la cinetica di aggregazione delle proteine
Nelle prove e�ettuate in precedenza si sono utilizzati direttamente gli aggregati
di proteine; in questo set di analisi invece si parte da �monomeri� di insulina e si
72

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
monitora con l'utilizzo del colorante tutta la cinetica che porta alla formazione
degli aggregati.
Nello speci�co in queste prove é stato utilizzato il macromonomero con la
tio�avina e le relative NPs prodotte in modalitá �starved�.
A seconda del rapporto tra moli di colorante e moli di proteine si é
proceduto con una prova on-line o una prova o�-line.
Nel caso in cui si é lavorato con rapporto un rapporto colorante/proteina
pari a 3 si é eseguita una prova o�-line. Questa consiste nel far avvenire
l'aggregazione delle proteine nel termomixer, nella modalitá descritta in prece-
denza, prelevare ogni ora 10.2µL di campione ed iniettarlo nel piatto dove sono
giá stati caricati 150µL di colorante. Dopo aver aspettato mezz'ora per l'in-
cubazione si é proceduto alla misura di �uorescenza di emissione, si é eccitato il
campione alla lunghezza propria di eccitazione e ne si é registrata l'assorbanza
di emissione. Si é proceduto con questa modalitá perché per basse concen-
trazioni di proteina la cinetica di aggregazione risulta molto piú lenta e puó
durare anche settimane.
Utilizzando invece un rapporto tra moli di colorante e proteine pari a 0.1
si é potuti procedere con una prova on-line. Ovvero é stata preparata una
soluzione di insulina monomerica di 5.5g/L, il piatto d'analisi é stato caricato
con 150µL di questa soluzione e sono stati aggiunti 10µL di colorante. Prima
di caricare il piatto nel �uorimetro si é portato questo alla temperatura di
aggregazione, 60°C, e poi si é proceduto con la prova e�ettuando un'analisi di
assorbanza di emissione ogni ora.
73

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
I primi risultati che saranno presentati di seguito,�g4.6, riguardano le prove
fatte utilizzanzo il macromonomero con la tio�avina a due diversi rapporti di
moli tra colorante e proteine. Le analisi sono state fatte anche su un campione
di sola tio�avina alle stesse condizioni in modo tale da avere un confronto
diretto con essa.
(a) (•) tio�avina, linea tratteggiata :macromonomero.
(b) (•) tio�avina, linea tratteggiata :macromonomero.
Figura 4.6. cinetica di aggregazione, (a)modalitá on-line,(b)in modalitá o�-line
.
Il risultato della prova on-line é che la cinetica di aggregazione delle proteine
in presenza del macromononomero é analoga a quella con la tio�avina, con solo
un e�etto trascurabile sulla durata della lag-phase. La di�erenza del segnale
di assorbanza �nale dipende dalla capacitá del colornate di emettere dopo
74

4.1 Analisi alla �uorescenza : �binding� degli aggregati proteici
essere stato eccitato, é da notare comunque come questi numeri siano gli stessi
ottenuti nelle prove con gli aggregati.
La prova o�-line esibisce lo stesso andamento cinetico visto nella pro-
va on-line e questo é un ulteriore importante conferma del fatto che il
macromonomero non in�uenza il processo di aggregazione delle proteine.
Il comportamento delle Nps per monitorare la cinetica di aggregazione é
stato indagato con un'analisi in modalitá on-line, �gura4.7 .
Figura 4.7. cinetica di aggregazione monitorata con le NPS, modalitá on-line
La cinetica di aggregazione presenta una lag-phase di circa 5 ore, analoga a
quella osservata in precedenza. La diminuzione di assorbanza nella fase iniziale
é indipendente dalla cinetica di aggregazione e dipende dalle sole nanoparticelle
che si trovano a contatto con una soluzione a pH = 1.6. Infatti lo stesso
andamento lo si é ritrovato facendo un'analisi di �uorescenza delle sole NPs in
presenza del bu�er a pH = 1.6 nel quale vengono disciolte le proteine, �g4.8.
75

4.2 Analisi all'UV
Figura 4.8. NPs in bu�er a pH = 1.6
4.2 Analisi all'UV
I macromonomeri e le NPs prodotte sono state analizzate tramite spettoscopia
all'UV per analizzare gli assorbimenti e le transizioni tra livelli energetici elet-
tronici. Nelle pagine che seguiranno verranno presentati i risultati ottenuti
tramite questa analisi.
4.2.1 Analisi all'UV : macromonomeri
Gli andamenti dell'assorbanza in funzione della lunghezza d'onda per i vari
campioni al variare della loro concentrazione sono presentati nelle pagine a
seguire, �gure 4.9, 4.10, 4.11, 4.12. Per ogni campione é stata poi formulata
la legge costitutiva che lega la concentrazione dello stesso al suo assorbimento
attraverso il coe�ciente di assorbimento molare ed il cammino ottico.
76

4.2 Analisi all'UV
(a) andamento dell'assorbanza al variare di λ.
(b) (•) punti sperimentali, (-) regressione lineare .
Figura 4.9. macromonomero funzionalizzato con il 2-naftolo
coe�cente di assorbimento molecolare ελ = 49645 mol cm L-1composto [C] mg/mL λmax Abs2-naftolo 0.1 491 1.1992-naftolo 0.08 491 1.022-naftolo 0.05 491 0.6472-naftolo 0.025 491 0.3342-naftolo 0.02 491 0.3182-naftolo 0.01 491 0.164
Tabella 4.2. Abs del macromonomero con il 2-naftolo a diverse concentrazioni
77
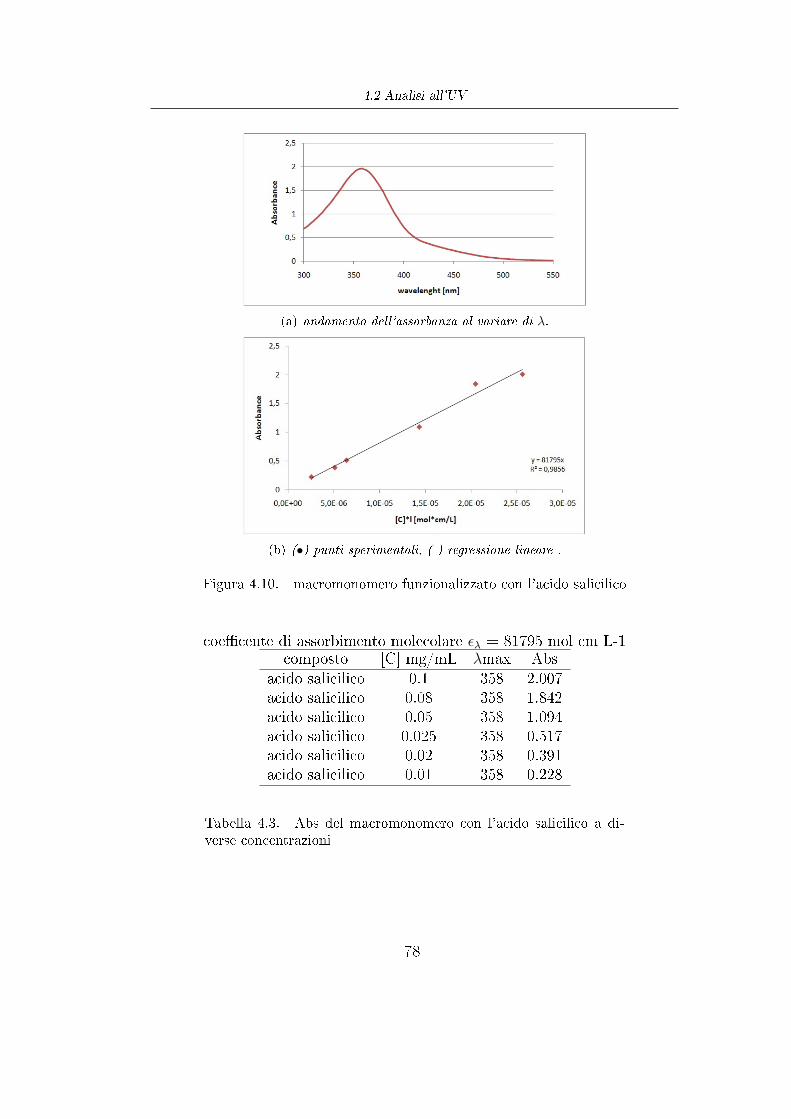
4.2 Analisi all'UV
(a) andamento dell'assorbanza al variare di λ.
(b) (•) punti sperimentali, (-) regressione lineare .
Figura 4.10. macromonomero funzionalizzato con l'acido salicilico
coe�cente di assorbimento molecolare ελ = 81795 mol cm L-1composto [C] mg/mL λmax Abs
acido salicilico 0.1 358 2.007acido salicilico 0.08 358 1.842acido salicilico 0.05 358 1.094acido salicilico 0.025 358 0.517acido salicilico 0.02 358 0.391acido salicilico 0.01 358 0.228
Tabella 4.3. Abs del macromonomero con l'acido salicilico a di-verse concentrazioni
78

4.2 Analisi all'UV
(a) andamento dell'assorbanza al variare di λ.
(b) (•) punti sperimentali, (-) regressione lineare .
Figura 4.11. macromonomero funzionalizzato con l'acido solfanilico
coe�cente di assorbimento molecolare ελ = 11867 mol cm L-1composto [C] mg/mL λmax Abs
acido solfanilico 0.1 291 0.294acido solfanilico 0.08 291 0.191acido solfanilico 0.05 291 0.168acido solfanilico 0.025 291 0.065acido solfanilico 0.02 291 0.031acido solfanilico 0.01 291 0.017
Tabella 4.4. Abs del macromonomero con l'acido solfanilico a di-verse concentrazioni
79
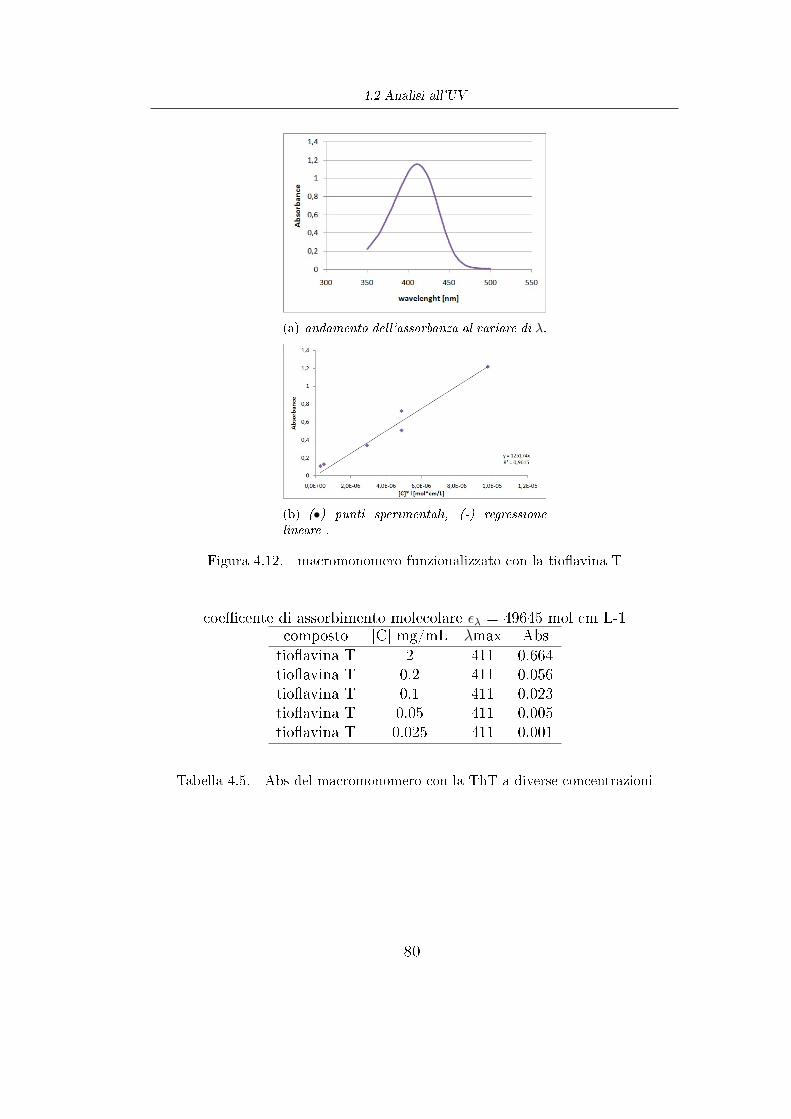
4.2 Analisi all'UV
(a) andamento dell'assorbanza al variare di λ.
(b) (•) punti sperimentali, (-) regressionelineare .
Figura 4.12. macromonomero funzionalizzato con la tio�avina T
coe�cente di assorbimento molecolare ελ = 49645 mol cm L-1composto [C] mg/mL λmax Abstio�avina T 2 411 0.664tio�avina T 0.2 411 0.056tio�avina T 0.1 411 0.023tio�avina T 0.05 411 0.005tio�avina T 0.025 411 0.001
Tabella 4.5. Abs del macromonomero con la ThT a diverse concentrazioni
80

4.2 Analisi all'UV
4.2.2 Analisi all'UV : NPs
Prodotte le nanoparticelle tramite copolimerizzazione tra MMA e
macromonomero funzionalizzato le si é analizzate all'UV. Si sono confrontati
i risultati con quelli riguardarnti i soli macromonomeri per osservare l'e�etto
che la formazione di Nps ha sull'assorbanza del macromonomero. Nel piatto
d'analisi sono stati caricati 150 µL di campione, per e�ettuare le diluzioni
si é utilizzata acqua a pH = 10. Per ogni serie di campioni su uno é stato
e�ettuato uno scan tra 270nm e 550nm per individuare la lunghezza d'onda
massima di assorbimento e poi per gli altri si é misurata l'assorbanza a quella
lunghezza d'onda.
I risultati sono presentati nelle pagine seguenti, �gure 4.13, 4.14, 4.15, 4.16.
81

4.2 Analisi all'UV
(a) andamento dell'assorbanzaal variare di λ.
(b) (•) punti sperimentali,(-) regressione lineare .
(c) andamento dell'assorbanzaal variare di λ.
(d) (•) punti sperimentali,(-) regressione lineare .
Figura 4.13. NPs : MMA - macromonomero con 2-naftolo: (a),(b)sds 10%; (c),(d) sds 5%
macromonomero con [C] mg/mL λmax Abs (10%sds) Abs (5%sds)2-naftolo 0.01 491 0.172 0.2392-naftolo 0.008 491 0.145 0.2012-naftolo 0.005 491 0.102 0.1372-naftolo 0.001 491 0.033 0.0372-naftolo 0.0005 491 0.018 0.029
Tabella 4.6. Abs delle NPs con 2-naftolo a diverse concentrazionie quantitá di sds
82

4.2 Analisi all'UV
(a) andamento dell'assorbanzaal variare di λ.
(b) (•) punti sperimentali,(-) regressione lineare .
(c) andamento dell'assorbanzaal variare di λ.
(d) (•) punti sperimentali,(-) regressione lineare .
Figura 4.14. NPs : MMA - macromonomero con ac.salicilico : (a),(b) sds10%; (c),(d) sds 5% sds 10%
macromonomero con [C] mg/mL λmax Abs (10%sds) Abs (5%sds)acido salicilico 0.01 360 0.144 0.155acido salicilico 0.008 360 0.102 0.125acido salicilico 0.005 360 0.078 0.104acido salicilico 0.001 360 0.028 0.016acido salicilico 0.0005 360 0.013 0.011
Tabella 4.7. Abs delle NPs con ac.salicilico a diverse concentrazionie quantitá di sds
83

4.2 Analisi all'UV
(a) andamento dell'assorbanzaal variare di λ.
(b) (•) punti sperimentali,(-) regressione lineare .
(c) andamento dell'assorbanzaal variare di λ.
(d) (•) punti sperimentali,(-) regressione lineare .
Figura 4.15. NPs : MMA - macromonomero con ac.solfanilico : (a),(b) sds10%; (c),(d) sds 5% sds 10%
macromonomero con [C] mg/mL λmax Abs (10%sds) Abs (5%sds)acido solfanilico 0.01 360 0.149 0.167acido solfanilico 0.008 360 0.124 0.104acido solfanilico 0.005 360 0.69 0.076acido solfanilico 0.001 360 0.019 0.032acido solfanilico 0.0005 360 0.010 0.011
Tabella 4.8. Abs delle NPs con ac.salicilico a diverse concentrazionie quantitá di sds
84

4.2 Analisi all'UV
Figura 4.16. NPs : MMA - macromonomero con tio�avina : sds 0.15%;(•) punti sperimentali, (-) regressione lineare
macromonomero con [C] mg/mL λmax AbsTio�avina 0.008 411 2.62Tio�avina 0.005 411 1.59Tio�avina 0.004 411 1.02Tio�avina 0.002 411 0.89Tio�avina 0.0001 411 0.51
Tabella 4.9. Abs delle NPs con ac.salicilico a diverse concentrazionie quantitá di sds
85
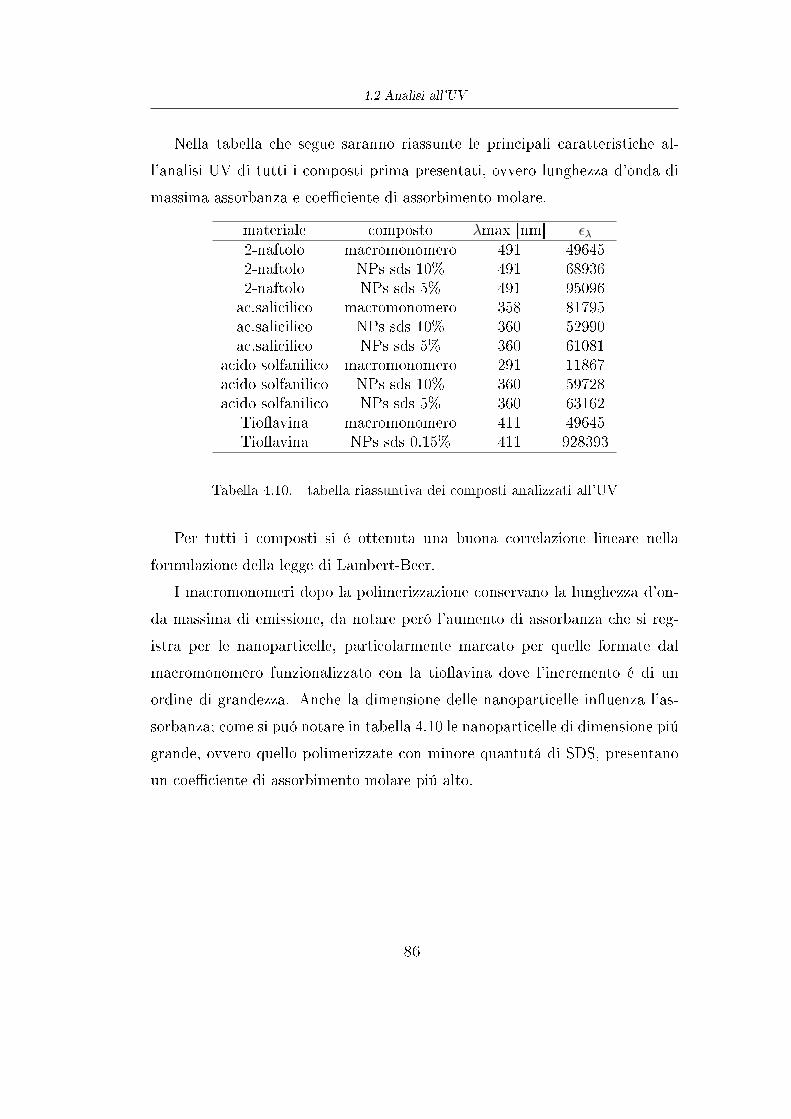
4.2 Analisi all'UV
Nella tabella che segue saranno riassunte le principali caratteristiche al-
l'analisi UV di tutti i composti prima presentati, ovvero lunghezza d'onda di
massima assorbanza e coe�ciente di assorbimento molare.
materiale composto λmax [nm] ελ2-naftolo macromonomero 491 496452-naftolo NPs sds 10% 491 689362-naftolo NPs sds 5% 491 95096ac.salicilico macromonomero 358 81795ac.salicilico NPs sds 10% 360 52990ac.salicilico NPs sds 5% 360 61081
acido solfanilico macromonomero 291 11867acido solfanilico NPs sds 10% 360 59728acido solfanilico NPs sds 5% 360 63162
Tio�avina macromonomero 411 49645Tio�avina NPs sds 0.15% 411 928393
Tabella 4.10. tabella riassuntiva dei composti analizzati all'UV
Per tutti i composti si é ottenuta una buona correlazione lineare nella
formulazione della legge di Lambert-Beer.
I macromonomeri dopo la polimerizzazione conservano la lunghezza d'on-
da massima di emissione, da notare peró l'aumento di assorbanza che si reg-
istra per le nanoparticelle, particolarmente marcato per quelle formate dal
macromonomero funzionalizzato con la tio�avina dove l'incremento é di un
ordine di grandezza. Anche la dimensione delle nanoparticelle in�uenza l'as-
sorbanza; come si puó notare in tabella 4.10 le nanoparticelle di dimensione piú
grande, ovvero quello polimerizzate con minore quantutá di SDS, presentano
un coe�ciente di assorbimento molare piú alto.
86

Capitolo 5
Conclusioni
In questo lavoro di tesi sono stati prodotti nuovi materiali da utilizzare per
l'imaging di �brille amiloidi.
Utilizzando la reazione di esteri�cazione di Steglich, la reazione di cop-
ulazione e la successiva formazione di coloranti azoici tramite formazione dei
sali di diazonio, sono stati formulati quattro di�erenti macromonomeri formati
da HEMA-acido ammibenzoico funzionalizzati con quattro molecole ad anello
aromatico attivato: 2-naftolo, acido solfanilico, acido salicilico e tio�avina T.
Il loro successivo test sulle �brille amiloidi tramite analisi alla �uorescenza
ha portato come risultato che tre su quattro sono in grado di legarsi con le pro-
teine. Data la loro struttura simile a meno della molecola diversa con la quale
é stato formulato il colorante diazoico, si sono potute fare delle considerazioni
sui gruppi funzionali che favoriscono l'interazione con le proteine.
Il macromonomero con la tio�avina T é quello che ha o�erto i migliori
risultati in termine di segnale di assorbanza di emissione. Per questo com-
posto sono state condotte delle analisi per confrontare il suo comportamento
rispetto alla sola tio�avina, colorante che viene attualmente e largamente uti-
lizzato per gli studi delle �brille amiloidi. Si é riscontrato che lavorando con
un basso rapporto tra moli di colorante e moli di proteine il segnale della solo
87

5 � Conclusioni
tio�avina é maggiore; spostandosi verso rapporti piú alti invece il segnale del
macromonomero risulta piú grande. Inoltre confrontando i segnali di assor-
banza ottenuti a diversi valori di rapporto tra moli di colorante e proteine si
é visto che all'aumentare di questo numero il segnale diventava piú basso. Si
é ipotizzato quindi un possibile meccanismo di complessazione del colorante
che entra in gioco quando questo é presente in grandi quantitá e che sfavorisce
l'interazione con le �brille amiloidi. Tuttavia questa ipotesi va confermata e
approfondita con ulteriori analisi.
Per quanto riguarda gli altri macromonomeri si é notato che quello con
l'acido solfanilico e quello con il 2-naftolo sono in grado di legare le �brille
amiloidi, al contrario di quello con l'acido salicilico.
Questi risultati dunque o�rono un interessante spunto di lavoro per il
futuro. Utilizzando queste reazioni di facile esecuzione é possibile produrre in
maniera mirata una serie di nuovi materiali con gruppi funzionali diversi per
monitorare piú a fondo quali sono e�ettivamente le molecole che favoriscono
il legame con le �brille amiloidi e ottenere nuove importanti informazioni a
riguardo.
La fase successiva del lavoro é stata quella di copolimerizzare i
macromonomeri con MMA tramite modalitá batch. Sono stati prodotte NPs
a due diverse quantitá di SDS, 10% e 5% in peso rispetto ai due monomeri.
Dall'analisi al light-scattering si é notato come questi latex siano di buona
qualitá, dimensione tra 30-70nm, bassa polidispersitá e z-potential nel range
che assicura dei lattici di buona stabilitá. Per queste caratteristiche unite al
fatto che per produrle si sono utilizzati materiali biocompatibili e biodegrad-
abili rende le nanoparticelle prodotte buoni candidati per applicazioni nel
campo del drug delivery.
Per il test delle NPs sugli aggregati proteici si é dovuta condurre una
88

5 � Conclusioni
polimerizzazione con quantitá di SDS piú bassa per evitare la destabiliz-
zazione degli aggregati proteici. Si é scelto di concentrarsi a questo punto sul
macromonomero con la tio�avina in modo tale da poter avere un riscontro
con i risultati della sola tio�avina largamente riportati in letteratura. Si é
condotta una polimerizzazione �starved' ' con quantitá di SDS pari allo 0.05%.
Il successivo test del latex ottenuto con le �brille amiloidi ha dimostrato che
anche le NPs con la tio�avina sono in grado di legarsi agli aggregati proteici.
Il lavoro é poi proseguito studiando la cinetica di aggregazione dei
�monomeri� di insulina. Come agenti per l'imaging delle proteine in questo
esperimento sono stati utilizzati il macromonomero con la tio�avina e le relative
NPs. I risultati dell'analisi sono che entrambi i composti sono in grado di
monitorare la cinetica di aggregazione della proteina modello: seppure con
segnali di assorbanza di diversa intensitá; l'andamento qualitativo é analogo
a quello ottenuto con la sola tio�avina e indicando come questi composti non
in�uenzino la modalitá di aggregazione.
Le NPs utilizzate per questa analisi hanno dimensione di 240nm, tuttavia
questa é una variabile che si puó facilmente modi�care variando il quantitativo
di emulsionante utilizzato nella polimerizzazione. Un possibile lavoro futuro
potrebbe essere studiare l'e�etto che le dimensioni delle NPs hanno sulla
cinetica di aggregazione.
In conclusione con questo lavoro di tesi sono stati prodotti nuovi materiali
per lo studio dell'aggregazione delle �brille amiloidi. La loro diversa capacitá di
legarsi agli aggregati proteici ha fornito delle importatnti indicazioni su quali
sono i gruppi funzionali che la favoriscono e questo é un aspetto che andrá
sicuramente sviluppato in futuro.
Anche le NPs con il macromonomero con la tio�avina hanno dato buoni
risultati in seguito alle analisi con gli aggregati proteici. Inoltre date le loro
89

5 � Conclusioni
dimensioni, maggiori di quelle dei coloranti attualmente utilizzati, potrebbero
essere analizzate al microscopio di trasmissioe elettronica(TEM-SEM). Questo
tipo di analisi potrebbe sicuramente dare un contributo nel capire i meccanismi
di interazione tra colorante e �brille amiloidi.
90

Bibliogra�a
[1] William E.Klunk, Manik L.Debnath, Jay W.Pettergrew : Development of
Small Molecule Probes For Beta-Amyloid Protein of Alzheimer's Disease
, ELSEVIER Science Ltd, Neurobiology Of ging, 1994.
[2] G.Blessed, B.E.Tomlison,M.Roth : The Associations Between Quanti-
tative Measures of Dementia and Senile Changes in the Cerebral Grey
Matter of Elderly Subject), Br.J.Psychiatry, 1968.
[3] R.N. Kalaria : The Blood Brain Barrier and Cerebral Microcirculation
in Alzheimer Disease , Cerebrovascular and Brain Metabolism Rewiews,
1992.
[4] William E.Klunk, J.W.Abraham, Jay W.Pettergrew : Quantitative Eval-
uation of Congo Red Binding to Amiloyd-like Proteins with Beta-Pleated
Sheet Conformation, J.Histochemistry and Cytochemistry, 1989.
[5] J.Hardy, G.A.Higgins: Alzheimer's Disease: The Amyloid Cascade
Hypotesis, science, 1992
[6] M.Lindgren, P.Hammasrstrom : Amyloid oligomers : spettroscopic
characterization of amyloidogenic proteins states, The FEBS Journal,
2010
[7] R.Khurana, C.Coleman, S.Singh : Mechanism of Thio�avin T binding to
amyloid �brils, Journal of Structural Biology, 2005
91

Bibliogra�a
[8] M.Krebs, E.Bromley, A.Donald : The binding of Thio�avi T to amyloid
�brils : localisation and implications, Journal of Structural Biology, 2005
[9] M.Lindgren, K.Sorgjerd, P.Hammarstrom : Detection ann Characteriza-
tion of Aggregate, Pre�brillar Amyloidogenic Oligomers,and Proto�brils
Using Fluorescence Spectroscopy, biophysical Journal, 2005
[10] M.Biancalana, K.Makabe, A.Koide : Molecular Mecanism of Thio�avin
T Binding to the Surface of beta-Rich peptide Self Assemblies, Journal
Molecular Biology, 2009
[11] P.Hammstrom, R.Simon, P.Konradson: A Fluorescent Pentametric Thio-
phene Derivative Detecs in Vitro-Formed Pre�brillar Protein Aggregates,
Biochemistry, 2010
[12] M.Biancalana, S.Kloide: Molecular mechanism of Thio�avin T binding to
amyloid �brils , Biochimica and BioPhysica Acta, 2010
[13] C.Kitts, D.Vanden Bout: A spectroscopic Study of 2-[4'-
(Dimethylamino)phenyl]-benzothiazole Binding to InsulinAmyloid Fibrils,
2001
[14] D.B.Lund: Physical properties of foods, AVI, Journal of Physics, 1983
[15] M.Dumoulin, C.Dobson: probing the origins, diagnosis and treatment of
amyloid disease using antibodies, Biochimie, 2004
[16] G.Gorbenko, V.Trusova, E.Kirilova: New �uorescent probes for detection
and characterization of amyloid �brils , Chemical Physics Letters, 2010
[17] S.Kumar, J.Udgaonkur: Mechanism of amyloid �bril formation by
proteins, Current Science, 2010
[18] H.Naiki, K.Higouchi: Fluorometric determination of amyloid �brils in
vitro using the �uorescence dye Thi�avin T, Anal.Biochemistry, 2008
Chemistry, 2004
[19] M.Groenning, L.Olsen : Study on the binding of Thio�avin T to beta-
sheet-rich and non beta-sheet cavities, Journal Structural Biology, 2007
92

Bibliogra�a
[20] G.Westermark, K.Johnson : Staining methods for identi�cation of amyloid
in tissue, Methods Enzimology, 2006
[21] Y.Yu, G.Storti, M.Morbidelli : Kinetics of Ring-Opening Polymerization
of L,L-Lactide, I&EC Research, 2011
[22] Y.Yu, G.Storti, M.Morbidelli : Ring-Opening Polymerization of
L,L-Lactide: Kinetic and Modeling Study, Macromolecules, 2009
93


















