
STUDIO C.E.R.P. PROTOCOLLO DI STUDIO - …crc.marionegri.it/cancerpain/docs/protocollo.pdf · 9.1...
122
STUDIO CERP versione 1 del 24.05.2010 1 STUDIO C.E.R.P. PROTOCOLLO DI STUDIO Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone, fentanyl e buprenorfina verso morfina, in pazienti con dolore associato a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica del WHO. EudraCT number: 2010-021017-23 SOTTOPROGETTO Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati. ENTE PROMOTORE E COORDINATORE DELLO STUDIO C.E.R.P. (Center for the Evaluation and Research on Pain) Dipartimento di Oncologia Istituto di Ricerche Farmacologiche MARIO NEGRI Via G. La Masa, 19 20156 Milano
Transcript of STUDIO C.E.R.P. PROTOCOLLO DI STUDIO - …crc.marionegri.it/cancerpain/docs/protocollo.pdf · 9.1...

STUDIO CERP versione 1 del 24.05.2010
1
STUDIO C.E.R.P.
PROTOCOLLO DI STUDIO
Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone, fentanyl e buprenorfina verso morfina, in pazienti con dolore associato
a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica del WHO.
EudraCT number: 2010-021017-23
SOTTOPROGETTO
Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati.
ENTE PROMOTORE E COORDINATORE DELLO STUDIO
C.E.R.P. (Center for the Evaluation and Research on Pain)
Dipartimento di Oncologia
Istituto di Ricerche Farmacologiche MARIO NEGRI
Via G. La Masa, 19
20156 Milano

STUDIO CERP versione 1 del 24.05.2010
2
COMITATI DEL PROGETTO
PRINCIPAL INVESTIGATOR
Oscar Corli (C.E.R.P., Istituto di Ricerche Farmacologiche Mario Negri, Milano)
COMITATO SCIENTIFICO
Giovanni Apolone (C.E.R.P., Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Cinzia Brunelli ( I.R.C.C.S. Istituto Nazionale per la Cura dei Tumori di Milano)
Augusto Caraceni (I.R.C.C.S. Istituto Nazionale per la Cura dei Tumori di Milano)
Silvio Cavuto (ASMN Azienda Ospedaliera, Reggio Emilia)
Tommaso Dragani (I.R.C.C.S. Istituto Nazionale per la Cura dei Tumori di Milano)
Stein Kaasa (Cancer Research and Molecular Medicine, NTNU and St. Olavs University Hospital)
Furio Zucco (A.O. G. Salvini, Garbagnate Milanese, Milano)
WRITING COMMITTEE
Oscar Corli (C.E.R.P., Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Giovanni Apolone (C.E.R.P., Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Augusto Caraceni (I.R.C.C.S. Istituto Nazionale per la Cura dei Tumori di Milano)
Mauro Montanari (C.E.R.P., Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Massimo Pizzuto (SC Cure Palliative, A.O. Istituti Clinici di Perfezionamento, Milano)
Davide Poli (Laboratorio Clinical Trials, Istituto di Ricerche Farmacologiche Mario Negri, Milano)
COORDINAMENTO
Elena Danieli: junior data-management (Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Maria Teresa Greco: referente eventi avversi (Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Simone Mangano: information Technology support (Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Mauro Montanari: senior data-manager & statistician (Istituto di Ricerche Farmacologiche Mario Negri, Milano)
Walter Villani: P.I. & data-management assistant (Istituto di Ricerche Farmacologiche Mario Negri, Milano)
SEGRETERIA SCIENTIFICA E ORGANIZZATIVA
Simona Stupia
Telefono: 02 39014519
Fax: 02 33200231
e-mail: [email protected]

STUDIO CERP versione 1 del 24.05.2010
3
INDICE
SINOSSI DELLO STUDIO 6
ABBREVIAZIONI 11
1. INTRODUZIONE E RAZIONALE 12 1.1 Introduzione 12
1.2 Razionale 13
2. DISEGNO DELLO STUDIO 14
3. OBIETTIVI ED ENDPOINTS 15
3.1 Obiettivi dello studio 15
3.1.1 Obiettivo primario 15
3.1.2 Obiettivi secondari 16
3.1.3 Obiettivo complementare (sottoprogetto) 16
3.2 Endpoints dello studio 16
3.2.1 Endpoint primario di efficacia 16
3.2.2 Endpoint secondari di efficacia 16
3.2.3 Endpoint secondari di sicurezza e tollerabilità 17
3.3 Analisi per sottogruppi 17
3.4 Sottoprogetto 18
4. POPOLAZIONE IN STUDIO 18
4.1 Criteri di inclusione 18
4.2 Criteri di esclusione 18
5. TRATTAMENTI 20
5.1 Regime di trattamento di base/ATC: dosaggi di partenza 20
5.1.1 Situazioni in cui vi è necessità di ridurre il dosaggio suggerito 21
5.1.2 Situazioni in cui vi è necessità di aumentare il dosaggio suggerito 21
5.2 Trattamenti successivi 22
5.2.1 Variazioni di dosaggio del farmaco oppioide ATC 23
5.2.2 Trattamento “aggiuntivo” con oppioidi per ottimizzare la terapia ATC 23
5.2.3 Trattamento “rescue” con oppioidi in presenza di BTP 23
5.2.4 Trattamenti adiuvanti analgesici 24
5.2.5 Cambiamento del farmaco oppioide ATC (Switch) 25
5.2.6 Abbandono del trattamento ATC a base di oppioidi 26

STUDIO CERP versione 1 del 24.05.2010
4
5.3 Altri trattamenti concomitanti ammessi (non rivolti al controllo del dolore) 27
5.4 Uscita dal follow-up 27
5.5 Fornitura dei farmaci 27
6. TIMING E METODOLOGIA DELLO STUDIO 28
6.1 Timing dello studio 28
6.2 Valutazioni 28
6.3 Flow-chart dello studio 28
6.4 Presentazione analitica del timing e delle procedure da svolgere 30
6.4.1 Valutazione della “eleggibilità e randomizzazione” 30
6.4.2 Visita1-valutazione basale 31
6.4.3 Visita2-72 ore 31
6.4.4 Visite 3, 4, 5 e 6 – valutazioni post basali di controllo 32
6.4.5 Completamento dello studio o eventuale interruzione prematura 32
7. EVENTI AVVERSI E REAZIONI AVVERSE 33
7.1 Definizioni 33
7.2 Eventi avversi 33
7.3 Reazioni avverse ai farmaci 33
7.4 Evento avverso serio e reazioni avverse ai farmaci serie 34
7.5 Segnalazioni, documentazione ed ulteriori indagini 34
8. METODI STATISTICI 35
8.1 Gestione dei dati 35
8.2 Popolazioni analizzate 36
8.3 Stima della dimensione del campione 36
8.4 Assegnazione dei soggetti al gruppo di trattamento 36
8.5 Trattamento dei dati mancanti 36
8.6 Descrizione della popolazione 38
8.7 Analisi della variabile primaria di efficacia 38
8.8 Analisi delle variabili secondarie di efficacia 38
8.9 Analisi della tollerabilità 39
8.10 Analisi ad interim 39
8.11 Analisi di sottogruppi 39

STUDIO CERP versione 1 del 24.05.2010
5
9. ORGANIZZAZIONE E RESPONSABILITA’ DELLO STUDIO 40
9.1 Centro di coordinamento 40
9.2 Sperimentatori 40
9.3 Monitor 40
9.4 Conservazione di documenti originali 40
9.5 Pubblicazione dei risultati 40
9.6 Modifiche del protocollo 41
10. BIBLIOGRAFIA 42
11. PROTOCOLLO DEL SOTTOPROGETTO 45
11.1 Introduzione 46
11.2 Disegno dello studio 46
11.3 Obiettivo del sottoprogetto 47
11.4 Timing e metodologia del prelievo biologico 47
11.4.1 Timing 47
11.4.2 Metodologia del prelievo 47
11.5 Analisi genetica e clinica 47
11.6 Aspetti statistici connessi all’analisi genetica 48
11.7 Organizzazione e responsabilità del sottoprogetto 49
11.8 Bibliografia del sottoprogetto 49
12. ASPETTI ETICI ED ASSICURATIVI 50
12.1 Approvazione da parte del comitato etico 50
12.2 Consenso informato allo studio e per la donazione di materiale biologico 50
ALLEGATO I - Definizioni e specifiche 51
ALLEGATO II - Foglio Informativo e Modulo di Consenso Informato 54
ALLEGATO III - Lettera di Informazione per il MMG 63
ALLEGATO IV - Elenco centri partecipanti 65
ALLEGATO V - Schede Raccolta Dati 63
STUDIO CERP versione 1 del 24.05.2010
6
SINOSSI DELLO STUDIO
TITOLO DELLO STUDIO
Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone, fentanyl e buprenorfina verso morfina, in pazienti con dolore associato a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica del WHO.
SOTTOPROGETTO Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati.
EudraCT number 2010-021017-23
VERSIONE Versione 1 del 24 maggio 2010
NAZIONE Italia
N° PREVISTO CENTRI Circa 80
FASE DELLO STUDIO Fase IV, randomizzato, comparativo
INDICAZIONE Trattamento del dolore da cancro da moderato a severo
RAZIONALE
Nella malattia neoplastica il dolore è un sintomo con grave impatto negativo sulla qualità di vita dei malati e ad elevata incidenza, con valori intorno al 70-90% nelle fasi avanzate e metastatiche. Da oltre 20 anni il principale riferimento per il trattamento farmacologico del dolore da cancro sono le linee-guida prodotte dalla Organizzazione mondiale della Sanità (WHO). In tale documento emerge che l’uso dei farmaci oppioidi rappresenta il punto cardine del trattamento, con particolare riferimento agli oppioidi “maggiori” (3° scalino della scala analgesica). I 4 oppioidi maggiori più comunemente prescritti in Italia (morfina e ossicodone orali, fentanyl e buprenorfina transdermici), in base ai dati al momento disponibili, presentano un effetto analgesico parzialmente sovrapponibile ma con percentuali diverse di soggetti non-responders (NR), una diversa necessità di aumento delle dosi nel tempo per mantenere un’adeguata analgesia, un differente ricorso allo switch verso un’altra molecola per inefficacia analgesica. Le osservazioni descritte inducono a pensare che gli oppioidi, pur appartenendo alla stessa famiglia farmacologica, potrebbero esser non pienamente sovrapponibili per quanto riguarda gli effetti clinici prodotti. Importanti differenze sono note sul piano farmacocinetico e farmacodinamico e, più recentemente, anche in termini di farmacogenomica. Alcuni studi pubblicati hanno evidenziato l’esistenza di variazioni geniche responsabili di anomalie riguardanti il trasporto delle molecole attraverso la barriera
STUDIO CERP versione 1 del 24.05.2010
7
ematoencefalica (BEE), la struttura del recettore per gli oppioidi e il sistema enzimatico di metabolismo. Assai limitati, invece, sono gli studi che analizzano i comportamenti degli oppioidi in rapporto all’intero genoma umano (Genome-Wide Association Studies: GWAS). A partire da tutto ciò, si è stabilito di proporre uno studio comparativo tra strategie analgesiche basate sull’impiego dei 4 oppioidi citati, che vada a cercare le possibili differenze in termini di efficacia analgesica, di variazioni di dosi nel tempo, di ricorso a switch, o di abbandono definitivo del trattamento, parallelamente al profilo degli effetti indesiderati. L’associato sottoprogetto metterà in relazione l’assetto genico dei pazienti e i risultati clinici emersi.
OBIETTIVI
Obiettivo primario dello studio è quello di comparare la efficacia analgesica di 4 diverse strategie analgesiche basate su 4 diversi farmaci oppioidi del 3° scalino della scala WHO (la morfina, utilizzata come comparatore, sarà confrontata con buprenorfina, fentanyl e ossicodone) in pazienti con dolore correlato a tumore, di intensità moderata-severa [intensità del dolore medio, riferita alle precedenti 24 ore, ≥ 4, utilizzando una scala numerica di valutazione a 11 punti (0-10 NRS)].
Obiettivi secondari di efficacia consistono nel valutare la performance dei 4 farmaci oppioidi mediante indicatori che descrivono alcune azioni messe in atto dal medico durante il follow-up. Tali azioni saranno descritte attraverso una serie di outcome intermedi, quali le variazioni del dosaggio del farmaco oppioide, il ricorso a cambiamenti del farmaco (switch), l’utilizzo di una terapia aggiuntiva o di salvataggio (rescue therapy), la prescrizione di farmaci adiuvanti analgesici, l’interruzione della terapia (inefficacia/tossicità assoluta)
Obiettivo complementare (sottoprogetto) consiste nell’analizzare l’assetto genico dei pazienti attraverso un’ indagine “genome-wide” al fine di valutare eventuali correlazioni con i risultati di tipo clinico ottenuti, anche ai fini di valutare l’esistenza di situazioni geniche con caratteristiche prognostico-predittivo sugli esiti clinici.
Obiettivi di Safety: Si valuteranno la sicurezza e la tollerabilità inerenti sia le reazioni avverse da farmaci (ADR) in studio, sia gli eventi avversi (AE) e gli Eventi Avversi Seri (SAE) che si verificano durante lo studio.
DISEGNO DELLO STUDIO
Clinical trial randomizzato (RCT), di superiorità, a 4 bracci di trattamento, in aperto, di fase IV, prospettico, multicentrico. La popolazione in studio sarà rappresentata da pazienti con cancro in fase avanzata per presenza accertata di metastasi o per importante progressione locale, che presentano, alla 1° visita, valori di dolore medio provato nelle ultime 24 ore ≥ 4,
STUDIO CERP versione 1 del 24.05.2010
8
misurato utilizzando una NRS. I pazienti eleggibili saranno randomizzati a ricevere uno dei seguenti 4 farmaci oppioidi maggiori: morfina o ossicodone orali e fentanyl o buprenorfina transdermici, come terapia di base, “around-the-clock” (ATC), del dolore. Il follow-up avrà una durata di 28 giorni e prevederà, oltre alla visita basale, altre 5 visite di controllo: dopo 72 ore e ai giorni 7, 14, 21 e 28. Nell’arco dei 28 giorni verranno monitorate sia le variazioni che riguarderanno il dolore sia quelle relative ai trattamenti attuati (variazioni di dose dei farmaci, aggiunta di farmaci rescue e adiuvanti, eventuali switch), sia le reazioni avverse che gli eventi avversi. Il gruppo di pazienti randomizzati a ricevere morfina (trattamento attivo di riferimento) saranno confrontati con quelli randomizzati a ricevere ognuno degli altri 3 farmaci. Saranno quindi realizzati tre confronti (morfina vs fentanyl, morfina vs buprenorfina e morfina vs ossicodone), da cui deriverà la possibile evidenziazione della superiorità di uno o più oppioidi nel confronto con la morfina.
PLANNED SAMPLE SIZE
Obiettivo primario dello studio è testare l'ipotesi nulla che la proporzione di soggetti NR alle terapie analgesiche dopo 28 giorni sia identica tra i trattamenti Buprenorfina, Fentanyl ed Ossicodone verso il trattamento con Morfina contro l’ipotesi alternativa che almeno uno dei tre trattamenti abbia una proporzione di NR inferiore al trattamento con Morfina.
La diminuzione della proporzione di soggetti NR verrà calcolata mettendo a confronto il valore del dolore medio alla visita basale con il dolore medio alla visita finale secondo la relazione [dolorefinale-dolorebasale]/dolorebasale * 100 , dove la misurazione del dolore medio verrà fornita utilizzando una NRS a 11 punti (da 0=nessun dolore a 10= il massimo dolore immaginabile).
Ipotizzando una proporzione di NR nel gruppo Morfina pari al 35% e valutando una diminuzione pari al 40% in uno dei tre trattamenti test, considerando una potenza del test dell’80%, una probabilità di 0.05 per l’errore di tipo I ed infine assumendo un test a due code, otteniamo una dimensione campionaria di 214 pazienti per gruppo per un totale di 856 pazienti. Ipotizzando una perdita del 15% dei pazienti durante lo studio e volendo mantenere quella di cui sopra come la quota minima per poter raggiungere gli obiettivi dello studio, la dimensione finale del campione sarà di 1008 pazienti in totale (252 per braccio). Il calcolo della dimensione iniziale del campione, esclusa l’aggiunta dei drop-out, è stato eseguito con il package nQuery Advisor vers. 7 tramite la procedura che utilizza il test del chi-quadrato per proporzioni per confronti multipli.

STUDIO CERP versione 1 del 24.05.2010
9
PRINCIPALI CRITERI
D’ INCLUSIONE
Lo studio coinvolgerà pazienti:
1. con diagnosi (istologica o citologica) di tumore solido in fase avanzata locale e/o metastatica;
2. con presenza di dolore medio giornaliero ≥4 misurato con NRS e riferito alle ultime 24 ore, attribuibile al tumore, necessitante per la prima volta di un trattamento con oppioidi del 3°scalino/WHO;
3. con aspettativa stimata di vita superiore a un mese;
4. che non hanno precedentemente eseguito, anche saltuariamente, trattamenti con oppioidi del 3° scalino sia ATC sia rescue;
5. in grado di assumere i farmaci in studio, sia per via orale sia transdermica sia sottocutanea;
6. di età pari o superiore a 18 anni.
PRINCIPALI CRITERI
DI ESCLUSIONE
1. partecipazione ad altri progetti di ricerca che sono in conflitto o potrebbero confondere i risultati dello studio;
2. assenza del consenso informato, o ritiro del consenso, per la partecipazione allo studio;
3. presenza di alcune condizioni patologiche mentali o psichiatriche, dovute al tumore o a patologie concomitanti, che interferiscono con lo stato di coscienza o con la capacità di giudizio al punto di compromettere il rispetto del protocollo di studio;
4. controindicazioni di qualsiasi natura all’uso dei farmaci oppioidi;
5. positività di una storia, pregressa o in atto, di abuso di sostanze stupefacenti;
6. diagnosi di neoplasia primitiva cerebrale o di leucemia (acuta o cronica);
7. diagnosi di insufficienza renale cronica conclamata, già in atto;
8. esecuzione di un ciclo di radioterapia o di terapia radiometabolica a scopo antalgico, in corso al momento d’inizio o conclusa da meno di 14 giorni o già programmata entro le 4 settimane dello studio;
9. esecuzione di un ciclo di chemioterapia di 1° linea contemporaneamente all’inizio dello studio, con intervallo di tempo variabile da 7 giorni prima a 7 giorni dopo la vista 1;
10. esecuzione concomitante di trattamenti antalgici con tecniche neurochirurgiche/ablative, o mediante neurostimolazione midollare, o con tecniche anestesiologiche loco-regionali (inclusa la analgesia peridurale o spinale), o mediante ricorso a vertebro/cifoplastica o altre tecniche invasive con rilevanza sul dolore.
STUDIO CERP versione 1 del 24.05.2010
10
SCHEMA TERAPEUTICO
INIZIALE
Dopo randomizzazione i pazienti entreranno nello studio seguendo lo schema posologico di riferimento di seguito descritto.
• Morfina orale SR: 60 mg/24 ore
• Fentanyl TD: 25 mg/h
• Buprenorfina TD: 35 mg/h
• Ossicodone orale SR: 49 mg/24 ore
Sono ammessi scostamenti, in più e in meno, rispetto ai dosaggi di riferimenti anche a partire dal primo giorno.
TRATTAMENTI SUCCESSIVI
Durante il periodo di follow-up, relativamente alla terapia antalgica, è sempre possibile:
• variare il dosaggio giornaliero del farmaco oppioide ATC assegnato
• somministrare un trattamento “aggiuntivo” con oppioidi per ottimizzare la terapia ATC
• aggiungere un trattamento rescue per BTP • aggiungere un trattamento a base di adiuvanti
analgesici • cambiare (switch) l’oppioide • abbandonare del tutto il trattamento con oppioidi, a
favore di altre tecniche analgesiche.
TRATTAMENTI CONCOMITANTI
Sono ammessi eventuali trattamenti specifici antitumorali, di tipo chemioterapico, ormonale e biologico, nel caso in cui lo stadio evolutivo della malattia tumorale ne indichi ancora un possibile utilizzo (si veda, in merito, anche il criterio di esclusione n.13).
Sono inoltre ammessi tutti i trattamenti ritenuti necessari per il controllo di altri sintomi, oltre il dolore, indotti dalla malattia, nonché le terapie atte al controllo di effetti collaterali causati dalla terapia antalgica.
Da ultimo, sono ammesse le terapie relative a eventuali patologie concomitanti.

STUDIO CERP versione 1 del 24.05.2010
11
ABBREVIAZIONI
AE Adverse Event [Eventi Avversi]
ADR AdverseDrugs Reaction (Reazioni Avverse da Farmaci)
ATC Around-The-Clock (terapia nelle 24 ore)
BEE Barriera Emato-Encefalica
BTP Breakthrough Pain
CERP Center for the Evaluation and Research on Pain
CGS Candidates Gene Study
CRF Case Report Form (Scheda Raccolta Dati)
EAPC European Association for Palliative Care
eCRF Electronic Case Report Form (Scheda Raccolta Dati Elettronica)
FANS Farmaci Anti-infiammatori Non Steroidei
FR Full-Responder(s)
GCP Good Clinical Practice
GWAS Genome-Wide Association Study
IK Indice di Karnofsky
IR Immediate Release
ITT Intent-To-Treat [population]
MCAR Missing Completely At Random
NR Non-Responder(s)
NRS Numerical Rating Scale
OEI Opioid Escalation Index
OMEDD Oral Morphine Equivalent Daily Dose
OMS Organizzazione Mondiale della Sanità
PI Pain intensity
PID Pain Intensity Difference
PMI Pain Management Index
PP Per-Protocol [population]
RCT Randomized Clinical Trial
SAE Serious Adverse Event [Evento Avverso Serio]
SR Slow Release
TTS Transdermal Therapeutic System [Fentanyl]
VAS Visual analog scale
WHO World Health Organization

STUDIO CERP versione 1 del 24.05.2010
12
1. INTRODUZIONE E RAZIONALE
1.1 Introduzione
Nei pazienti tumorali in fase avanzata la progressione della malattia, i sintomi associati e gli effetti collaterali delle terapie anti-tumorali influenzano in maniera importante lo stato di salute e la qualità della vita. Il dolore, in particolare, è un sintomo con grave impatto negativo sulla qualità di vita dei malati e ad elevata incidenza, con valori intorno al 70-90% nelle fasi avanzate e metastatiche.
Da oltre 20 anni il principale riferimento per il trattamento farmacologico del dolore da cancro sono le linee-guida (1) prodotte dalla Organizzazione mondiale della Sanità (WHO). In tale documento emerge che l’uso dei farmaci oppioidi rappresenta il punto cardine del trattamento, con particolare riferimento agli oppioidi “maggiori”, inseriti al 3° scalino della scala analgesica. Attualmente alcune molecole di tale scalino sono disponibili anche in Italia (morfina, fentanyl, buprenorfina, ossicodone, metadone e idromorfone), ma il loro impiego rimane ancora complessivamente limitato e spesso circoscritto ai centri specialistici che hanno in cura i malati oncologici.
Nel 2004 è stato lanciato in Italia un progetto promosso dall’Istituto di Ricerche Farmacologiche “Mario Negri” di Milano per migliorare la qualità del trattamento dei pazienti con cancro. Tra le molte attività avviate, nel 2006 è stato intrapreso uno studio di outcome, completato nel 2007, a cui hanno partecipato 110 centri con un reclutamento di 1800 casi, seguiti fino a 3 mesi. Il protocollo dello studio e i principali risultati ottenuti sono stati pubblicati (2, 3, 4). In sintesi, all’inizio della fase osservata circa il 60% dei malati era già in terapia con un farmaco del 3° scalino WHO, mentre del restante 40% circa la metà dei casi (350) passava ad un oppioide maggiore (morfina, fentanyl, buprenorfina, ossicodone) nel follow up successivo. Al tempo basale la maggior parte dei pazienti aveva un dolore, misurato con scale di tipo numerico (0-11), di intensità medio - severa (valore medio=6.8; DS=2.4), spesso non riceveva una terapia al bisogno, non era informato della sua prognosi nel 69% dei casi, ed era in parte ancora in terapia con farmaci anti-tumorali. La valutazione dell’adeguatezza della terapia analgesica, utilizzando un sistema standardizzato (PMI: Pain Management Index), ha identificato una quota compresa tra il 25 ed 50% di casi giudicabili under-treated (5). Nel corso dei 28 giorni di follow-up, nei pazienti che hanno completato lo studio (85%), tutti i parametri analgesici e palliativi considerati (intensità dolore, sollievo dal dolore, soddisfazione nella terapia antalgica, qualità della vita, ecc.) hanno mostrato un significativo miglioramento dal punto di vista clinico. E’ stato, però, possibile identificare un sotto-gruppo di pazienti non-responder ai trattamenti (30%). In tempi successivi sono state eseguite alcune analisi derivate dallo studio osservazionale, che hanno confrontato i diversi farmaci prescritti: in particolare, una valutazione del sottogruppo di pazienti che iniziavano il 3° scalino nel corso dello studio, ha permesso di verificare che i 4 farmaci più comunemente prescritti (morfina e ossicodone orali, fentanyl e buprenorfina transdermici) presentavano un effetto analgesico abbastanza sovrapponibile nel corso del follow-up, ma richiedevano un diverso aumento delle dosi necessarie per mantenere un adeguato livello di analgesia. In più, si osservava una diversa necessità di abbandonare l’oppioide in corso eseguendo uno switch verso un’altra molecola. La natura osservazionale del progetto iniziale non ha permesso di giungere a conclusioni definitive sul valore dei risultati emersi, ma ha dato una forte spinta ad impostare uno studio prospettico randomizzato di tipo pragmatico, che qui viene presentato.

STUDIO CERP versione 1 del 24.05.2010
13
1.2 RAZIONALE
Le osservazioni descritte inducono a pensare che gli oppioidi maggiori, anche se appartenenti alla stessa famiglia farmacologica e anche se inseriti allo stesso livello terapeutico nella scala del WHO, potrebbero non essere del tutto equivalenti e sovrapponibili per quanto riguarda gli effetti prodotti. Importanti differenze sono già state evidenziate sul piano farmacocinetico e farmacodinamico e, più recentemente, in termini di farmacogenomica (6-10).
Gli oppioidi agiscono soprattutto, ma non esclusivamente, sul recettore µ ; l’ampiezza della trasduzione del segnale a partire dal complesso recettore-ligando è influenzata da vari fattori, tra cui l’affinità, la potenza, l’efficacia e l’emivita, che differiscono tra una molecola e l’altra (11-16). Anche la capacità di promuovere la desensibilizzazione e l’endocitosi del complesso ligando-recettore-proteina-G è variabile tra una molecola e l’altra: il recettore non desensibilizzato, che continua a lavorare e a lungo termine, causa modifiche compensatorie nella trasduzione del segnale con alterazioni qualitative del secondo messaggero (17, 18), della trascrizione genica (19,20) e dei neurotrasmettitori (21,22). Questi cambiamenti generalmente provocano una perdita della efficacia analgesica, promuovono l’insorgere della tolleranza e talvolta inducono un’iperalgesia indotta dagli oppioidi stessi. Inoltre, è stato di recente dimostrato che il recettore per gli oppioidi lavora frequentemente come dimero od oligomero in associazione con la proteina-G (23) e questo influisce sull’affinità, potenza ed efficacia della molecola oppioide (24).
Le diversità fin qui descritte sono relative ad aspetti farmacologici e biologici ma possono suggerire anche possibili differenze di comportamento degli oppioidi sul piano clinico. Su questi aspetti sono finora poche le evidenze emerse in ambito della ricerca clinica, anche se non mancano alcuni interessanti rilievi che inducono a intuire diversi comportamenti tra i farmaci. Gli Autori che hanno confrontato la morfina con l’ossicodone (25-28) hanno riscontrato un’analgesia comparabile e, in un caso (28), una minore neurotossicità a carico dell’ossicodone. Dal confronto tra morfina e fentanyl transdermico, invece, emergono un’efficacia comparabile in termini di analgesia (29-31), anche se il fentanyl richiede titolazioni più frequenti con dosaggi più elevati e una minore presenza di stipsi. Questi risultati sono confermati anche da una pooled analysis condotta da Clarks nel 2004 (32) che, però, non prende in considerazione i dosaggi. Uno studio recente (33) confronta tra loro tre oppioidi (morfina, fentanyl e metadone) concludendo che tutti gli oppioidi sono efficaci nel controllo del dolore e richiedono la stessa quantità di farmaci sintomatici e co-analgesici. Però, lo stesso effetto analgesico è ottenuto con aumenti del dosaggio del 18% per il metadone, 38% per la morfina e 96% per il fentanyl.
Inoltre, un’analisi condotta dal C.E.R.P. nel contesto di un ampio studio osservazionale citato precedentemente (4), su una sottopopolazione di 258 pazienti oncologici che iniziavano il 3° scalino con gli stessi 4 oppioidi maggiori proposti in questa ricerca, ha dimostrato che l’effetto analgesico ottenuto dai 4 farmaci dopo 3 settimane di trattamento era molto simile se misurato come valore medio ottenuto nei sottogruppi di pazienti. Questo dato induce a pensare una sostanziale omogeneità di risposta che, in qualche modo, contrasta gli intenti di questo studio. Quando, invece, i dati sono stati rivalutati in termini di soggetti non- responders (senza alcun miglioramento del dolore o, addirittura, con un peggioramento) le differenze sono risultate molto evidenti, variando da un valore del 35% per la morfina al 15% per il fentanyl.
Inoltre, recenti lavori preclinici e clinici (7-9) hanno evidenziato l’esistenza di una serie di geni e di varianti geniche in grado di influenzare, in modo più o meno consistente, l’azione farmacologica e clinica dei farmaci oppioidi.

STUDIO CERP versione 1 del 24.05.2010
14
Sono state evidenziate, ad esempio, alcune variazioni geniche responsabili di atipie riguardanti il trasporto delle molecole attraverso la barriera ematoencefalica (BEE), la struttura del recettore per gli oppioidi e il sistema enzimatico di metabolismo epatico degli stessi. Si tratta, indiscutibilmente, di punti nevralgici dell’attività degli oppioidi; una recente review (34), però, ha ridimensionato l’impatto clinico di alcune di queste varianti.
Assai limitati, invece, sono gli studi che analizzano i comportamenti degli oppioidi (ad esempio, nei soggetti non responders al trattamento) in rapporto all’intero genoma umano (Genome-Wide Association Studies: GWAS) anziché con singoli specifici geni (Candidates Gene Studies). Dai GWAS sono attesi, nei prossimi anni, indicazioni che ineriscono possibili nuovi mediatori del dolore (proteine derivanti da geni rilevati dagli studi) e possibili nuovi target farmacologici, direttamente rilevati da studi sull’uomo (35). In particolare, gli esiti di una ricerca “genome-wide” collegata all’uso degli oppioidi nel dolore da cancro sono al momento poco chiariti.
In sostanza, si ritiene che uno studio comparativo tra strategie analgesiche basate sull’impiego di diversi farmaci oppioidi del 3° scalino, che vada a cercare le possibili differenze di efficacia in termini di pazienti resistenti/refrattari (non-responders: NR) e i comportamenti derivanti in termini di variazioni di dosi necessarie nel tempo, d’incidenza di casi d’insuccesso terapeutico e di ricorso a switch, o di abbandono del trattamento con oppioidi, insieme al profilo degli effetti indesiderati, possa dare un contributo utile ai fini di una scelta mirata nel trattamento del dolore da cancro di entità medio-elevata. L’associato sottoprogetto che metterà in relazione l’assetto genico dei pazienti e i risultati clinici emersi dal confronto tra oppioidi potrà fornire ulteriori conoscenze nella materia.
2. DISEGNO DELLO STUDIO
Lo studio sarà un clinical trial randomizzato (RCT), di superiorità, a 4 bracci di trattamento, in aperto, di fase IV, prospettico, multicentrico, con follow-up di 28 giorni.
La popolazione in studio sarà rappresentata da pazienti con cancro in fase avanzata per presenza accertata di metastasi o per importante progressione locale, che presentano, alla 1° visita, valori di dolore medio provato nelle ultime 24 ore ≥ 4, misurato utilizzando una NRS, Numerical Rating Scale, a 11 punti (0-10). Questi pazienti possono aver precedentemente ricevuto o nessun trattamento antalgico o farmaci del 1° e/o del 2° scalino del WHO (paracetamolo, FANS, codeina, tramadolo). Al momento di inizio dello studio, comunque, necessitano di un trattamento con 3° scalino.
I pazienti eleggibili, informati e consenzienti, aderenti ai criteri di inclusione ed esclusione e privi di contro-indicazioni ai farmaci in studio, saranno inclusi nello studio e randomizzati, secondo modalità standardizzate, a ricevere uno dei seguenti 4 farmaci oppioidi maggiori: morfina o ossicodone orali e fentanyl o buprenorfina transdermici, come terapia di base, “around-the-clock” (ATC), del dolore. Le dosi giornaliere iniziali saranno suggerite in base a criteri standardizzati in base riportati in letteratura (37), ma potranno essere immediatamente modificate, come descritto dettagliatamente nel § 5.1, fino all’ottenimento del dosaggio ottimale per ogni paziente.
Il follow-up avrà una durata di 28 giorni e prevederà, oltre alla visita basale, altre 5 visite di controllo: dopo 72 ore e ai giorni 7, 14, 21 e 28.

STUDIO CERP versione 1 del 24.05.2010
15
Nell’arco dei 28 giorni verranno monitorate sia le variazioni che riguarderanno il dolore provate dai pazienti [intensità, tipologia, presenza e caratteristiche del breakthrough pain (BTP)], sia le variazioni dei trattamenti attuati (variazioni di dose dei farmaci, aggiunta di farmaci rescue e adiuvanti, eventuali switch), sia le reazioni avverse che gli eventi avversi.
Il gruppo di pazienti randomizzati a ricevere morfina (trattamento attivo di riferimento) saranno confrontati con quelli randomizzati a ricevere ognuno degli altri 3 farmaci, considerati sperimentali: saranno quindi realizzati tre confronti (morfina vs fentanyl, morfina vs buprenorfina e morfina vs ossicodone), da cui deriverà la possibile evidenziazione della superiorità di uno o più oppioidi nel confronto con la morfina.
Gli endpoints dello studio saranno diversi, organizzati in accordo agli obiettivi primari, secondari e complementari.
3. OBIETTIVI ED ENDPOINTS
3.1 OBIETTIVI DELLO STUDIO
Lo studio ha un obiettivo primario, due obiettivi secondari e un obiettivo complementare (relativo al sottoprogetto). Obiettivi primari e secondari saranno ottenuti attraverso una serie di analisi condotte nell’intero campione, in accordo ai principi della analisi per intention-to-treat e per-protocol e in sottogruppi pre-identificati.
3.1.1 Obiettivo primario
L’obiettivo primario dello studio è quello di comparare la efficacia analgesica di 4 diverse strategie analgesiche basate su 4 diversi farmaci oppioidi del 3° scalino della scala WHO (la morfina, utilizzata come comparatore, sarà confrontata con buprenorfina, fentanyl e ossicodone) in pazienti con dolore correlato a tumore, di intensità moderata-severa [intensità del dolore medio, riferita alle precedenti 24 ore, uguale o superiore a 4, utilizzando una scala numerica di valutazione a 11 punti (0-10 NRS)]. La valutazione verrà condotta utilizzando una serie di endpoint primari e secondari di efficacia analgesica e di sicurezza.
3.1.2 Obiettivi secondari
a) Valutare la performance dei 4 farmaci analgesici utilizzando indicatori che descrivono alcune azioni messe in atto dal medico, dopo la randomizzazione, durante il follow-up. Tali azioni saranno descritte attraverso una serie di outcome intermedi, quali le variazioni del dosaggio del farmaco oppioide, il ricorso a cambiamenti del farmaco (switch), l’utilizzo di una terapia aggiuntiva e di salvataggio (rescue therapy), la prescrizione di farmaci adiuvanti analgesici, l’interruzione della terapia (inefficacia/tossicità assoluta) e/o l’uscita dallo studio.
b) Utilizzare i dati raccolti nel contesto del RCT al fine di sviluppare nuovi metodi, outcomes ed endpoints per valutare la resa della terapia analgesica in futuri clinical trial e nella pratica clinica. A tal fine il data-base sarà utilizzato per analisi di tipo esplorativo come training sample che richiederanno una seconda valutazione in un campione indipendente (testing sample).

STUDIO CERP versione 1 del 24.05.2010
16
3.1.3 Obiettivo complementare (sottoprogetto)
Analizzare l’assetto genico dei pazienti attraverso un’ indagine “genome-wide” al fine di valutare eventuali correlazioni con i risultati di tipo clinico ottenuti. Valutare l’esistenza di situazioni geniche che abbiano caratteristiche di tipo prognostico-predittivo sugli esiti clinici.
Questa parte dello studio è condotta come un sottoprogetto indipendente nel contesto del RCT che viene descritta a parte in questo protocollo.
3.2 ENDPOINTS DELLO STUDIO
3.2.1 Endpoint primario di efficacia
L’endpoint primario di efficacia è rappresentato dalla proporzione di soggetti che saranno classificati come non rispondenti alla terapia analgesica (non-responders).
Non-Responders (NR) sono i soggetti che avranno soddisfatto il seguente criterio: avere una PID (pain intensity difference) relativa al dolore medio provato nelle 24 ore precedenti, tra visita 6 e visita 1, ≤ 0, a indicare una situazione invariata o peggiorata dell’intensità del dolore medio. Il calcolo dell’intensità del dolore verrà effettuato mediante una scala di tipo NRS ad 11 punti, da 0 (= nessun dolore) a 10 ( il massimo dolore immaginabile).
3.2.2 Endpoint secondari di efficacia
1. Proporzione di soggetti che saranno classificati come non rispondenti alla terapia analgesica (NR), in base al seguente criterio: avere una PID (pain intensity difference) relativa al dolore peggiore provato nelle 24 ore precedenti, tra visita 6 e visita 1, ≤ 0, a indicare una situazione invariata o peggiorata dell’intensità del dolore peggiore. Il calcolo dell’intensità del dolore verrà effettuato mediante una scala di tipo NRS ad 11 punti, da 0 (= nessun dolore) a 10 ( il massimo dolore immaginabile).
2. Proporzione dei soggetti che ottengono una risposta analgesica completa (full-responders).
• Full-Responders (FR) sono i soggetti che avranno soddisfatto il seguente criterio: avere una PID relativa al dolore medio provato nelle 24 ore precedenti, tra visita 6 e visita 1, ≥ 30% del valore basale, ad indicare una risposta ottimale (36) al trattamento analgesico. Anche in questo caso il calcolo dell’intensità del dolore verrà effettuato mediante una scala di tipo NRS ad 11 punti, da 0 (= nessun dolore) a 10 ( il massimo dolore immaginabile).
3. La proporzione di soggetti classificabili come NR e FR dopo le prime 72 ore di trattamento, secondo i criteri descritti nei punti precedenti tra la visita 2 (dopo 72 ore) e visita 1 (basale), al fine di valutare il primo impatto sul dolore dei farmaci assegnati. Anche in questo caso il calcolo dell’intensità del dolore verrà effettuato mediante una scala di tipo NRS ad 11 punti, da 0 (= nessun dolore) a 10 ( il massimo dolore immaginabile).
4. Proporzione di soggetti con un incremento di dose dell’oppioide ≥ al 5% (su base giornaliera, in relazione al dosaggio iniziale, tra fine e inizio del trattamento), misurato attraverso l’OEI % (Opioids Escalation Index-%). Il valore del 5% identifica il cut-off tra gli aumenti di dose considerati normali e quelli di elevata entità (46, 50).

STUDIO CERP versione 1 del 24.05.2010
17
5. Proporzione di soggetti che hanno richiesto il ricorso a un trattamento “aggiuntivo” a base di oppioidi per ottimizzare il trattamento ATC.
6. Proporzione di soggetti che hanno richiesto il ricorso a farmaci analgesici adiuvanti (steroidi, antidepressivi triciclici o di generazione successiva, anticonvulsivanti, bifosfonati, FANS) non finalizzati al trattamento di specifiche tipologie di dolore (neuropatico, osseo, viscerale) ma prescritti con finalità analgesica di rinforzo del trattamento dell’oppioide ATC assegnato per randomizzazione, nel corso del follow-up.
7. Proporzione di soggetti che hanno richiesto il cambio dell’oppioide come terapia ATC (switch), per inadeguatezza dell’analgesia ottenuta, nel corso del follow-up.
8. Proporzione di pazienti che abbandonano lo studio per cause attribuibili al trattamento (inefficacia e/o tossicità).
3.2.3. Endpoint secondari di sicurezza e tollerabilità
Si valuteranno la sicurezza e la tollerabilità inerenti sia le reazioni avverse da farmaci (ADR) in studio, sia gli eventi avversi (AE), così come gli eventi avversi seri (SAE) e le reazioni avverse serie (SADR) che si verificano durante lo studio anche per cause indipendenti dai trattamenti.
A tal fine è prevista una valutazione parallela da parte del paziente stesso e da parte del medico, mediante l’utilizzo di metodi e di scale standardizzate. In particolare si valuterà:
da parte dei pazienti: la frequenza (sì/no) e la severità (mediante scala verbale a 4 punti) delle ADR generalmente riferibili all’uso degli oppioidi, quali stipsi, nausea, vomito, prurito, sonnolenza, confusione, xerostomia, allucinazioni, spasmi/cloni muscolari, gastralgia, disuria, dispnea, prurito.
da parte del medico: la frequenza (sì/no), la severità (lieve, moderata, grave), la correlazione con i trattamenti, e le iniziative intraprese, relative agli eventi avversi (AE, SAE, SADR).
3.3 ANALISI PER SOTTOGRUPPI
Verranno anche condotte analisi in sottogruppi di pazienti che possono presentare aspetti specifici relativi al dolore e ai trattamenti, come ad esempio:
1. Analisi dei soggetti con presenza di BTP, in cui saranno valutate modalità ed esiti e della terapia “rescue” applicata al controllo degli episodi dolorosi.
2. Analisi dei soggetti su cui verrà eseguito uno switch, in cui si valuteranno le modalità di realizzazione e gli esiti derivanti.
3. Analisi dei soggetti che nella fase di titolazione dei 4 oppioidi (prime 72 ore) richiederanno variazioni di dose rispetto alla dose basale. Si considererà anche la relazione tra risposta analgesica/dose necessaria (a 72 ore) e risposta analgesica finale (a 28 giorni), in termini di coerenza tra i due risultati e di predittività della risposta iniziale vs quella finale.

STUDIO CERP versione 1 del 24.05.2010
18
3.4 SOTTOPROGETTO
Ad ogni paziente in studio, informato e consenziente, in qualsiasi momento del follow-up verrà eseguito un prelievo di sangue intero che permetterà l’analisi dell’intero genoma. La sequenza genica ottenuta sarà confrontata, in termini di singoli geni o di cluster di geni, con i risultati clinici esaminati nella ricerca (endpoints primari e secondari). Il protocollo relativo al sottoprogetto è descritto separatamente al § 11.
4. POPOLAZIONE IN STUDIO
In base al calcolo del sample size (§ 8.3) verranno arruolati nello studio 1.008 pazienti (252 per braccio) che rispondono ai criteri di inclusione e ed esclusione qui di seguito descritti.
4.1 CRITERI DI INCLUSIONE
Lo studio coinvolgerà pazienti:
1. con diagnosi (istologica o citologica) di tumore solido in fase avanzata locale e/o metastatica;
2. con presenza di dolore medio giornaliero ≥4 misurato con NRS e riferito alle ultime 24 ore, attribuibile al tumore, necessitante per la prima volta di un trattamento con oppioidi del 3°scalino/WHO;
3. con aspettativa stimata di vita superiore a un mese;
4. che non hanno precedentemente eseguito, anche saltuariamente, trattamenti con oppioidi del 3° scalino sia ATC sia rescue;
5. in grado di assumere i farmaci in studio, sia per via orale sia transdermica sia sottocutanea;
6. di età pari o superiore a 18 anni.
4.2 CRITERI DI ESCLUSIONE
Per essere eleggibile, il paziente non deve possedere alcuno dei seguenti criteri di esclusione:
1. partecipazione ad altri progetti di ricerca che sono in conflitto o potrebbero confondere i risultati dello studio;
2. assenza del consenso informato, o ritiro del consenso, per la partecipazione allo studio;
3. presenza di alcune condizioni patologiche mentali o psichiatriche, dovute al tumore o a patologie concomitanti, che interferiscono con lo stato di coscienza o con la capacità di giudizio al punto di compromettere il rispetto del protocollo di studio;
4. necessità di trattamenti per comorbidità presenti all’inizio dello studio che potrebbero creare interazioni farmacologiche potenzialmente pericolose con gli oppioidi (ad esempio, uso di antifungini conazolici o antibiotici macrolidi);
5. controindicazioni di qualsiasi natura all’uso dei farmaci oppioidi;
6. utilizzo di farmaci oppioidi combinati, nelle loro formulazioni commerciali, con altre molecole (paracetamolo, naloxonici, ecc.)
7. positività di una storia, pregressa o in atto, di abuso di sostanze stupefacenti;

STUDIO CERP versione 1 del 24.05.2010
19
8. impossibilità di garantire la regolarità del follow up;
9. necessità d’inizio del 3° scalino in una situazione di “emergenza” per quanto riguarda l’intensità e la gravità del dolore, che richieda, ad esempio, un intervento di immediato effetto (trattamenti endovenosi) tale da non consentire i tempi necessari per l’ottenimento della randomizzazione e per il successivo inizio della terapia secondo il protocollo;
10. diagnosi di neoplasia primitiva cerebrale o di leucemia (acuta o cronica);
11. diagnosi di insufficienza renale cronica conclamata (49), già in atto;
12. esecuzione di un ciclo di radioterapia o di terapia radiometabolica a scopo antalgico, in corso al momento d’inizio o conclusa da meno di 14 giorni o già programmata entro le 4 settimane dello studio;
13. esecuzione di un ciclo di chemioterapia di 1° linea contemporaneamente all’inizio dello studio, con intervallo di tempo variabile da 7 giorni prima a 7 giorni dopo la vista 1;
14. esecuzione concomitante di trattamenti antalgici con tecniche neurochirurgiche/ablative, o mediante neurostimolazione midollare, o con tecniche anestesiologiche loco-regionali (inclusa la analgesia peridurale o spinale), o mediante ricorso a vertebro/cifoplastica o altre tecniche invasive con rilevanza sul dolore.
NOTA: i criteri di inclusione/esclusione sono tutti requisiti indispensabili per l’ammissione di nuovi
pazienti allo studio. Però, dati gli obiettivi dello studio e le caratteristiche cliniche dei malati, portatori di
tumore in fase avanzata, si sottolinea la necessità di porre particolare attenzione:
al 2° criterio d’inclusione, in quanto si ritiene opportuno che l’inizio del terzo scalino sia da mettere in
correlazione con un’entità del dolore medio-elevata (≥4), altrimenti sarebbe discutibile proprio la scelta
di questo scalino.
al 3° criterio di inclusione (soggetti con aspettativa stimata di vita superiore a un mese) in quanto
un’errata valutazione di questo aspetto, con l’inclusione di numerosi malati che abbiano una
sopravvivenza inferiore a 28 giorni, determinerebbe un elevato numero di “lost”, fattore che potrebbe
diminuire in modo rilevante la potenza dei test statistici e inficiare i risultati.

STUDIO CERP versione 1 del 24.05.2010
20
5. TRATTAMENTI
Insieme all’assegnazione, per randomizzazione, dell’oppioide con cui intraprendere lo studio viene anche specificata l’entità “indicativa” della dose di partenza.
5.1 REGIME DI TRATTAMENTO DI BASE/ATC: DOSAGGI DI PARTENZA
Entrando nel merito dei dosaggi di partenza dei 4 oppioidi in studio, la seguente tabella indica i valori che vengono indicati come standard di riferimento, a cui attenersi salvo le necessarie variazioni.
TABELLA DELLE DOSI MEDIE GIORNALIERE DI PARTENZA PER OGNI OPPIOIDE IN STUDIO:
farmaco oppioide dose MORFINA ORALE SR 60 mg /24 ore FENTANYL TRANSDERMICO 25 µg/h BUPRENORFINA TRANSDERMICA 35 µg/h OSSICODONE ORALE SR 40 mg /24 ore
Tali dosi possono rivelarsi immediatamente adeguate e non richiedere modifiche, oppure risultare inadeguate per eccesso o per difetto. In tal caso si può intervienire subito sui dosaggi fino a raggiungere un buon equilibrio tra efficacia analgesica e tollerabilità.
NOTA: In merito alla dose iniziale giornaliera dell’oppioide, va innanzi tutto precisato che il Comitato Scientifico dello studio ha optato per la soluzione di indicare dei dosaggi standard di riferimento per iniziare il trattamento del 3° scalino. Le ragioni di tale scelta derivano da quanto recentemente emerso dalla Conference di Bristol (febbraio 2010), avente per tema la “Rivalutazione delle raccomandazioni della E.A.P.C. (European Association for Palliative Care) sull’uso degli oppioidi nel dolore da cancro”. In quel contesto è emerso che l’avvio del trattamento di 3° scalino può essere positivamente realizzato utilizzando fin dall’inizio oppioidi a lento rilascio (SR: Slow Release) a dosi standardizzate, dedotte dalla letteratura e dalle comuni abitudini cliniche. Pur partendo da tale presupposto, il curante ha però la facoltà di aggiustare, se necessario, fin dal momento d’inizio o nei giorni successivi, la dose giornaliera di partenza suggerita (37). Tali variazioni di dose vanno intese sia in senso aggiuntivo sia riduttivo, e sono finalizzate ad ottenere il miglior rapporto tra efficacia e tollerabilità dell’oppioide, in ogni singolo malato, nel tempo più breve possibile. Questo metodo viene suggerito dagli Esperti della E.A.P.C. non tanto come alternativa alla titolazione tradizionale (basata sull’impiego di oppioidi a immediato rilascio per poi passare a quelli a lento rilascio), ma come il nuovo metodo di titolazione del farmaco oppioide. Per tale ragione ci si è attenuti a questa proposta.
STUDIO CERP versione 1 del 24.05.2010
21
5.1.1. Situazioni in cui vi è necessità di ridurre il dosaggio suggerito E’ possibile che i pazienti portatori di determinate situazioni cliniche, le principali delle quali sono descritte qui di seguito, necessitino o di una riduzione del dosaggio di standard di partenza o della programmazione, fin dall’inizio di una dose inferiore rispetto a quella suggerita. In particolare questa necessità potrebbe verificarsi in pazienti:
• naÏve per trattamenti continui con oppioidi. In questa categoria rientrano i pazienti che non hanno ricevuto alcun trattamento antalgico o hanno eseguito solo una terapia di 1° scalino/WHO (antinfiammatori). Normalmente, invece, non ne fanno parte i malati che hanno già eseguito un trattamento con 2° scalino (oppioidi “deboli”)
• con età ≥ 80 anni • con altre patologie o deficit funzionali d’organo che normalmente comportano una
riduzione delle dosi dei farmaci. A tali categorie di pazienti, nei cui confronti è necessario porre attenzione nella definizione della dose di un farmaco oppioide, possono aggiungersi altre situazioni particolari identificate di volta in volta dai Ricercatori.
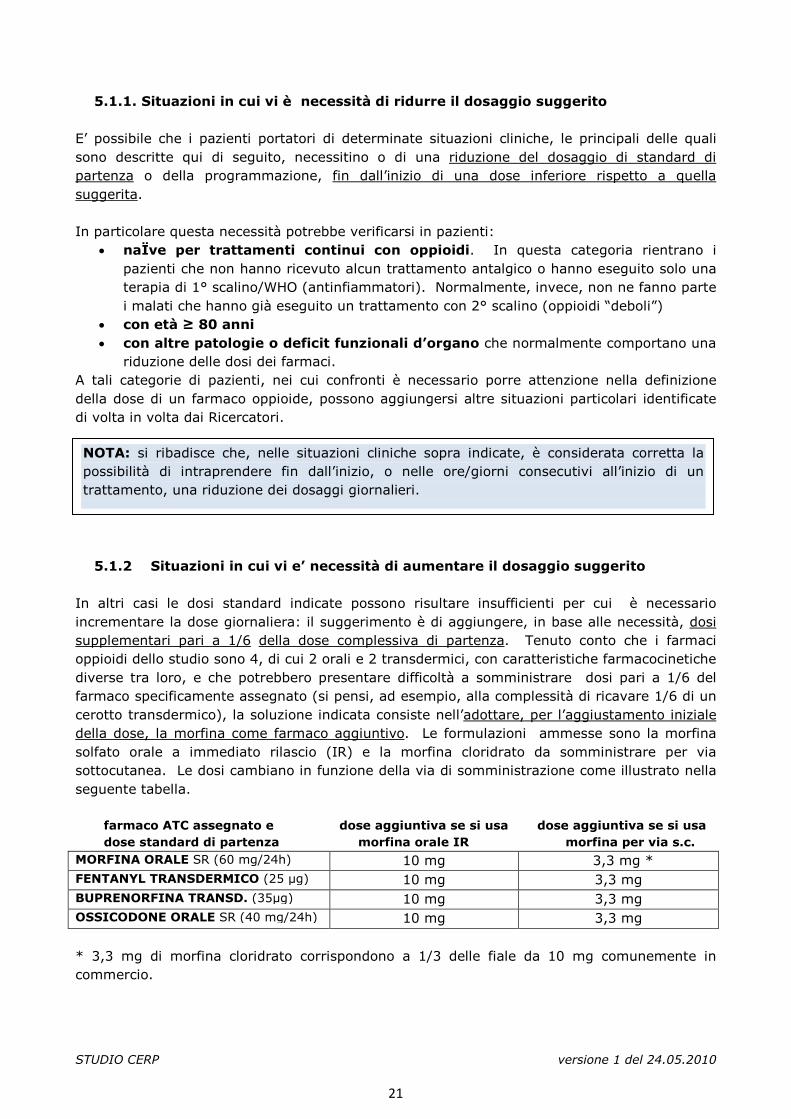
5.1.2 Situazioni in cui vi e’ necessità di aumentare il dosaggio suggerito In altri casi le dosi standard indicate possono risultare insufficienti per cui è necessario incrementare la dose giornaliera: il suggerimento è di aggiungere, in base alle necessità, dosi supplementari pari a 1/6 della dose complessiva di partenza. Tenuto conto che i farmaci oppioidi dello studio sono 4, di cui 2 orali e 2 transdermici, con caratteristiche farmacocinetiche diverse tra loro, e che potrebbero presentare difficoltà a somministrare dosi pari a 1/6 del farmaco specificamente assegnato (si pensi, ad esempio, alla complessità di ricavare 1/6 di un cerotto transdermico), la soluzione indicata consiste nell’adottare, per l’aggiustamento iniziale della dose, la morfina come farmaco aggiuntivo. Le formulazioni ammesse sono la morfina solfato orale a immediato rilascio (IR) e la morfina cloridrato da somministrare per via sottocutanea. Le dosi cambiano in funzione della via di somministrazione come illustrato nella seguente tabella. farmaco ATC assegnato e dose aggiuntiva se si usa dose aggiuntiva se si usa dose standard di partenza morfina orale IR morfina per via s.c. MORFINA ORALE SR (60 mg/24h) 10 mg 3,3 mg * FENTANYL TRANSDERMICO (25 µg) 10 mg 3,3 mg BUPRENORFINA TRANSD. (35µg) 10 mg 3,3 mg OSSICODONE ORALE SR (40 mg/24h) 10 mg 3,3 mg * 3,3 mg di morfina cloridrato corrispondono a 1/3 delle fiale da 10 mg comunemente in commercio.
NOTA: si ribadisce che, nelle situazioni cliniche sopra indicate, è considerata corretta la possibilità di intraprendere fin dall’inizio, o nelle ore/giorni consecutivi all’inizio di un trattamento, una riduzione dei dosaggi giornalieri.

STUDIO CERP versione 1 del 24.05.2010
22
Una situazione clinica a parte riguarda quei malati che, pur non trovandosi in una situazione di emergenza per quanto riguarda il dolore (in tal caso vale il criterio 9 di esclusione), presentano comunque alla prima visita un dolore d’intensità elevata che richiede risposte efficaci e rapide. In tal caso i farmaci dello studio potrebbero, indipendentemente dalla dose, richiedere tempi relativamente lunghi prima di dare sollievo al dolore, date le loro caratteristiche farmacocinetiche. In questi casi è consentito iniziare somministrando simultaneamente l’oppioide assegnato per randomizzazione alla dose suggerita più la dose aggiuntiva di morfina (orale a IR o sottocutanea), secondo la stessa logica descritta sopra per situazioni meno urgenti. L’elemento caratterizzante in questi casi è dato dalla simultaneità dell’impiego del farmaco ATC e della dose aggiuntiva. Tale dose, inoltre, può essere ripetuta più volte fino al raggiungimento del pieno effetto del farmaco ATC assegnato per randomizzazione.
5.2 TRATTAMENTI SUCCESSIVI (DURANTE IL FOLLOW-UP)
Durante il periodo di follow-up, relativamente alla terapia antalgica, è sempre possibile :
• variare il dosaggio giornaliero del farmaco oppioide ATC assegnato
• somministrare un trattamento “aggiuntivo” con oppioidi per ottimizzare la terapia ATC
• aggiungere un trattamento rescue per BTP
• aggiungere un trattamento a base di adiuvanti analgesici
• cambiare (switch) l’oppioide
• abbandonare del tutto il trattamento con oppioidi, a favore di altre tecniche
analgesiche.
IN SINTESI:
• Per ognuno dei 4 oppioidi in studio viene indicata una dose di partenza standard
• Tale dose va in linea di massima rispettata salvo condizioni cliniche che richiedano degli scostamenti
• Il più semplice degli scostamenti consiste in un aumento o in una riduzione di dose da attuare, in genere dopo le prime 24 ore, secondo il parametro indicativo di 1/6 della dose complessiva
• Nei soggetti naÏve per gli oppioidi, negli ultraottantenni o nei pazienti con altre patologie che consigliano una riduzione dei dosaggi degli oppioidi, è possibile partire già con dosi ridotte
• Al contrario nei soggetti molto sofferenti, ma arruolabili allo studio, si può iniziare somministrando contemporaneamente la dose standard insieme alla dose aggiuntiva
• Tutta questa fase iniziale, riconducibile alla titolazione della dose giornaliera del farmaco oppioide, richiede attenzione sotto il profilo clinico e uno scrupoloso monitoraggio per quanto concerne lo studio in cui, a tal fine, è prevista una visita apposita (visita 2) dopo l’inizio del trattamento.

STUDIO CERP versione 1 del 24.05.2010
23
5.2.1 Variazioni di dosaggio del farmaco oppioide ATC Dopo i possibili aggiustamenti iniziali di dose (fase di titolazione), è possibile che anche successivamente le dosi giornaliere del farmaco oppioide vadano modificate per mantenere un adeguato livello di analgesia.
La decisione di variare la dose spetta al medico in base alla valutazione del dolore riscontrato nel paziente (per protocollo, è prevista a ogni visita la valutazione del dolore medio e peggiore delle 24 ore precedenti) e, più in generale, agli esiti della valutazione complessiva medica e delle decisioni contestualmente prese tra medico e malato.
5.2.2 Trattamento “aggiuntivo” con oppioidi per ottimizzare la terapia ATC
Talora i pazienti affetti da dolore correlato al cancro presentano un certo grado di variabilità dell’intensità dolorosa nel corso della giornata: questa situazione non corrisponde al BTP ma consiste solo in moderate oscillazioni del dolore. In questo studio il trattamento ATC del dolore prevede l’uso di 4 oppioidi a lento e costante rilascio del principio attivo, in cui le situazioni appena descritte potrebbero non essere del tutto coperte. In tali casi, come succede di prassi nella clinica, accanto al farmaco ATC, somministrato sistematicamente, può essere utile un farmaco extra, di tipo “aggiuntivo”, da usare al bisogno per ottimizzare il trattamento di base, coprendo i momenti più dolorosi.
A tale fine, il trattamento aggiuntivo previsto in questo studio si avvale (in modo analogo a quanto previsto nella fase di titolazione):
• o dell’uso di morfina orale IR, nella misura indicativa di 1/6 della dose complessiva giornaliera ATC, per ogni singola somministrazione aggiuntiva;
• o della morfina per iniezione sottocutanea, alla dose indicativa di 1/20 del dosaggio complessivo giornaliero.
5.2.3 Trattamento “rescue” con oppioidi in presenza di BTP
per Breakthrough pain (BTP o dolore episodico intenso, secondo una traduzione non letterale in lingua italiana) s’intende “una esacerbazione transitoria del dolore che avviene sia spontaneamente sia in seguito a prevedibili o imprevedibili fattori scatenanti (ad esempio, nel caso del dolore incidente) a fronte di un dolore di base adeguatamente controllato dal trattamento ATC” (39).
E’ stato valutato che la prevalenza media del BTP nei malati con dolore da cancro si attesta intorno al 50% dei casi. L’episodio doloroso è, in genere, di rapida insorgenza e d’intensità molto elevata. Dopo aver fatto diagnosi di BTP (vedere anche allegato 1), si ricorre in genere a trattamenti rescue (di “emergenza”) degli episodi dolorosi, consistenti nella somministrazione di un oppioide potente e a rapido esordio dell’azione.
NOTA: una volta deciso di aumentare la dose dell’oppioide in corso, il medico valuta e decide l’entità di
tale aumento in modo da ottenere il miglior risultato in termini clinici nei confronti del malato. Si ricorda
che l’entità dell’aumento di dose indicato in letteratura mediamente varia tra il 20% e il 50%. In casi
particolari il medico potrà comunque decidere di incrementare la dose in modo diverso dal range
segnalato. Le variazioni potranno essere effettuate in qualsiasi momento nel corso dei 28 giorni di durata
dello studio e per qualsiasi numero di volte, in base alla necessità cliniche.
STUDIO CERP versione 1 del 24.05.2010
24
Per quanto riguarda il farmaco oppioide rescue da utilizzare nello studio, la scelta indicata cadrà su una delle possibilità qui di seguito indicate:
• utilizzo di uno dei farmaci a base di fentanyl transmucosale (pastiglie per mucosa orale con applicatore incorporato/lollipop, compresse orosolubili e sublinguali, spray nasale). Il motivo di questo indirizzo si ricollega alle indicazioni emerse in letteratura per cui vi è “una forte evidenza che le somministrazioni transmucosali di fentanyl sono efficaci, sicure e ben tollerate” (39) e, in particolare, di rapido esordio dell’effetto (superiore a quello ottenibile con oppioidi, anche a immediato rilascio, somministrati per via orale). Tutte queste formulazioni richiedono una titolazione per ottenere l’individuazione della dose corretta da somministrare, che deve attenersi alle indicazioni espresse nel foglio illustrativo di ciascun prodotto.
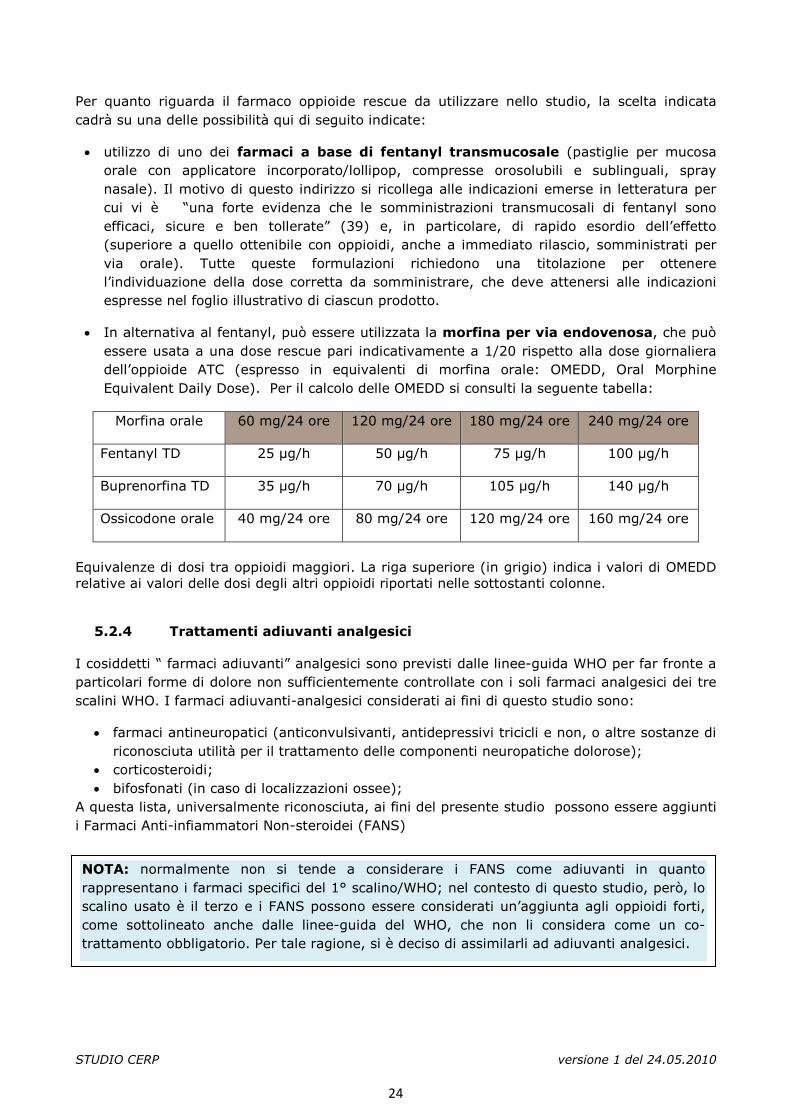
• In alternativa al fentanyl, può essere utilizzata la morfina per via endovenosa, che può essere usata a una dose rescue pari indicativamente a 1/20 rispetto alla dose giornaliera dell’oppioide ATC (espresso in equivalenti di morfina orale: OMEDD, Oral Morphine Equivalent Daily Dose). Per il calcolo delle OMEDD si consulti la seguente tabella:
Morfina orale 60 mg/24 ore 120 mg/24 ore 180 mg/24 ore 240 mg/24 ore
Fentanyl TD 25 µg/h 50 µg/h 75 µg/h 100 µg/h
Buprenorfina TD 35 µg/h 70 µg/h 105 µg/h 140 µg/h
Ossicodone orale 40 mg/24 ore 80 mg/24 ore 120 mg/24 ore 160 mg/24 ore
Equivalenze di dosi tra oppioidi maggiori. La riga superiore (in grigio) indica i valori di OMEDD relative ai valori delle dosi degli altri oppioidi riportati nelle sottostanti colonne.
5.2.4 Trattamenti adiuvanti analgesici
I cosiddetti “ farmaci adiuvanti” analgesici sono previsti dalle linee-guida WHO per far fronte a particolari forme di dolore non sufficientemente controllate con i soli farmaci analgesici dei tre scalini WHO. I farmaci adiuvanti-analgesici considerati ai fini di questo studio sono:
• farmaci antineuropatici (anticonvulsivanti, antidepressivi tricicli e non, o altre sostanze di riconosciuta utilità per il trattamento delle componenti neuropatiche dolorose);
• corticosteroidi; • bifosfonati (in caso di localizzazioni ossee);
A questa lista, universalmente riconosciuta, ai fini del presente studio possono essere aggiunti i Farmaci Anti-infiammatori Non-steroidei (FANS)
NOTA: normalmente non si tende a considerare i FANS come adiuvanti in quanto rappresentano i farmaci specifici del 1° scalino/WHO; nel contesto di questo studio, però, lo scalino usato è il terzo e i FANS possono essere considerati un’aggiunta agli oppioidi forti, come sottolineato anche dalle linee-guida del WHO, che non li considera come un co-trattamento obbligatorio. Per tale ragione, si è deciso di assimilarli ad adiuvanti analgesici.

STUDIO CERP versione 1 del 24.05.2010
25
Date le premesse, nel caso di impiego di adiuvanti dovrà essere specificato se il loro impiego è dovuto a:
• trattamento di particolari tipologie di dolore (neuropatico, osseo, cefalea da metastasi cerebrali,..)
• rinforzo del trattamento ATC
Fatta questa precisazione, i farmaci adiuvanti possono essere liberamente utilizzati (sia in termini di scelta del farmaco, sia di dosaggio) secondo i criteri di necessità e di utilità individuati dal medico.
5.2.5 Cambiamento del farmaco oppioide ATC (switch)
A volte, nella clinica, si constata il non-ottenimento o la perdita dell’effetto analgesico con l’oppioide in uso, nonostante ripetuti aumenti della dose nel tempo.
Questa situazione deve essere considerata con attenzione, sulla base delle seguenti riscontri:
• verificare che la mancata analgesia non sia dovuta a un fattore-dose nel senso che le dosi del farmaco sono di per sé troppo basse e insufficienti a controllare il dolore. Per tale ragione va provato un aumento della dose giornaliera, una o più volte, nei termini espressi nel § 5.2.1, prima di considerare inadeguata la risposta al farmaco oppioide. Si consiglia anche di attenersi a recenti indicazioni (40) in cui la dose pre-switch raggiunta, prima di dichiarare insoddisfacente l’analgesia, dovrebbe essere almeno pari a 120 mg/24 di morfina orale o dosi equivalenti degli altri oppioidi (fentanyl TTS: 50 mg/h; buprenorfina TTS: 70 mg/h; ossicodone orale: 80 mg/24 ore). Questi valori vanno considerati come indicativi, ma sottolineano il fatto che sembra poco ragionevole eseguire uno switch per mancato raggiungimento dell’effetto analgesico a dosi troppo basse degli oppioidi.
• Considerare anche un fattore-tempo, nel senso che tutti gli oppioidi (e in particolare i transdermici) richiedono un certo intervallo per raggiungere uno steady state plasmatico e una stabilizzazione dell’effetto analgesico. Appare quindi opportuno che venga lasciato un periodo di tempo ragionevole dall’inizio dello studio, quantificabile in almeno una settimana, prima di abbandonare il farmaco in atto. Vanno comunque esclusi i casi di evidente mancanza di risposta al farmaco in uso, anche in tempi più brevi.
Se l’attenzione ai due fattori segnalati prima non sortiscono, comunque, un effetto analgesico adeguato o perdono tale effetto nel tempo, è corretto procedere alla sospensione del trattamento con l’oppioide in corso ed eseguire uno switch verso un altro oppioide.
La scelta del nuovo farmaco è affidata all’esperienza personale del medico, e può includere sia un altro dei 4 oppioidi analizzati in questo studio sia una molecola diversa (metadone, idromorfone). Si ricorda, però, che le modalità di esecuzione dello switch e, in particolare, il rapporto di conversione tra il farmaco in uscita e quello in entrata, hanno un loro razionale che può trovare il punto di partenza nella tabella inserita al § 5.2.3.
NOTA: si definisce “switch” di un oppioide il processo per cui si cambia l’oppioide in corso con un altro oppioide in modo da raggiungere un’adeguata efficacia del trattamento e un’adeguata tollerabilità (41).

STUDIO CERP versione 1 del 24.05.2010
26
L’applicazione dei fattori di conversione è comunque un utile riferimento di partenza, ma ogni singolo caso può rispondere in modo variabilmente conforme alle attese, tale da richiedere un attento monitoraggio degli effetti ottenuti nei giorni successivi, e tale da richiedere, talvolta, aggiustamenti, in più o in meno, del dosaggio impostato del nuovo farmaco.
Si ricorda, inoltre, che lo switch rappresenta una scelta terapeutica non solo in caso di constatata inefficacia analgesica del farmaco ma anche in presenza di rilevanti effetti collaterali che non tendono a ridursi nel tempo e che non sono controllabili da opportune terapie mirate su tali effetti.
Da tutto ciò deriva che in caso di switch verrà richiesto di precisare sulla CRF se si tratta di un provvedimento adottato per:
• perdita di efficacia analgesica (dopo tentativi di aumento della dose del farmaco e di adeguato intervallo di tempo intercorso)
• presenza di effetto/i collaterale dì intensità elevata, non controllabile
• entrambe le precedenti cause
• perdita della capacità di assunzione per via orale.
In ogni caso, il malato che esegue uno switch non esce dal follow up ma continua ad essere monitorato settimanalmente fino al termine delle 4 settimane.
5.2.6 Abbandono del trattamento ATC a base di oppioidi
Può infine capitare che nel corso dello studio il trattamento del dolore effettuato con l’oppioide di 3° scalino a un certo momento fallisca completamente, nonostante ogni tentativo di compensazione sia stato provato [aumenti di dose, aggiunta di farmaci rescue e/o adiuvanti per potenziare il trattamento di base (ATC); cambio di oppioide (switch)].
Le ragioni sono probabilmente attribuibili alla comparsa di tolleranza grave, o di iperalgesia da oppioidi, o da altre cause tra cui la progressione di malattia.
In questi casi, descritti in letteratura con un’incidenza intorno al 5-10%, si può abbandonare il trattamento a base di oppioidi per passare ad altre strategie analgesiche rappresentate, in genere, da soluzioni come:
• scelta di trattamenti farmacologici analgesici non oppioidi (FANS, steroidi, adiuvanti, altri analgesici)
• ricorso a tecniche antalgiche non farmacologiche (interventi di neurolesione, tecniche di anestesia loco-regionale, neurostimolazione midollare, interventi di cifo/vertebro plastica, radioterapia, radioterapia metabolica, interventi chirurgici di stabilizzazione ossea o di disostruzione viscerale, applicazione di stent, laserterapia, brachiterapia,…)
• ricorso a sedazione palliativa-terminale, in caso di dolore severo refrattario a qualsiasi trattamento.
Queste opzioni terapeutiche implicano l’uscita del follow-up (vedere § 5.4).

STUDIO CERP versione 1 del 24.05.2010
27
5.3 ALTRI TRATTAMENTI CONCOMITANTI AMMESSI (NON RIVOLTI AL CONTROLLO DEL DOLORE)
Sono ammessi eventuali trattamenti specifici antitumorali, di tipo chemioterapico (ad eccezione di quanto specificato nel criterio di esclusione 13),ormonale e biologico, nel caso in cui lo stadio evolutivo della malattia tumorale ne indichi ancora un possibile utilizzo.
Sono inoltre ammessi tutti i trattamenti ritenuti necessari per il controllo di altri sintomi, oltre il dolore, indotti dalla malattia, nonché le terapie atte al controllo di effetti collaterali causati dalla terapia antalgica.
Da ultimo, sono ammesse le terapie relative a eventuali patologie concomitanti.
5.4 USCITA DAL FOLLOW-UP
Un paziente esce dal follow-up dello studio:
• per ragioni connesse ai farmaci in studio (mancato ottenimento di analgesia, SAE e SADR)
• per ragioni non connesse ai trattamenti (decesso a causa della malattia, trasferimento del paziente ad altra sede di cure)
• per ragioni connesse al paziente (ritiro del consenso, scarsa compliance)
• per ragioni varie (violazioni di protocollo, problemi amministrativi,…)
La CRF prevede la segnalazione dell’uscita dal follow-up, della data e delle relative motivazioni.
5.5 FORNITURA DEI FARMACI
Tutti i predetti farmaci sono stati approvati in Italia per il trattamento del dolore nel paziente con cancro. Essi verranno prescritti dai clinici secondo le abituali procedure in uso nei centri partecipanti. In base a quanto indicato dal comma 1, art. 2 del D.M. del 17/12/2004, trattandosi di farmaci usati per indicazioni specificate nell’autorizzazione all’immissione in commercio, saranno a carico del Servizio Sanitario Nazionale.
STUDIO CERP versione 1 del 24.05.2010
28
6. TIMING E METODOLOGIA DELLO STUDIO
6.1 TIMING DELLO STUDIO
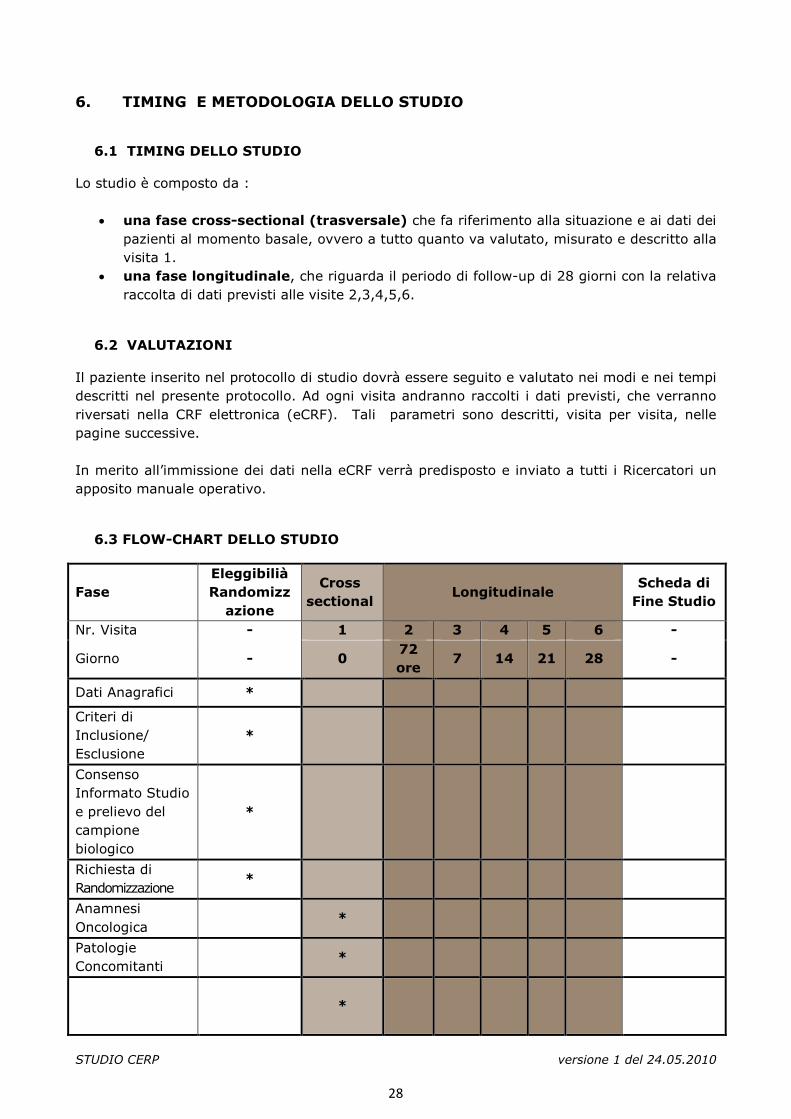
Lo studio è composto da :
• una fase cross-sectional (trasversale) che fa riferimento alla situazione e ai dati dei pazienti al momento basale, ovvero a tutto quanto va valutato, misurato e descritto alla visita 1.
• una fase longitudinale, che riguarda il periodo di follow-up di 28 giorni con la relativa raccolta di dati previsti alle visite 2,3,4,5,6.
6.2 VALUTAZIONI
Il paziente inserito nel protocollo di studio dovrà essere seguito e valutato nei modi e nei tempi descritti nel presente protocollo. Ad ogni visita andranno raccolti i dati previsti, che verranno riversati nella CRF elettronica (eCRF). Tali parametri sono descritti, visita per visita, nelle pagine successive. In merito all’immissione dei dati nella eCRF verrà predisposto e inviato a tutti i Ricercatori un apposito manuale operativo.
6.3 FLOW-CHART DELLO STUDIO
Fase Eleggibilià Randomizz
azione
Cross sectional
Longitudinale Scheda di
Fine Studio
Nr. Visita - 1 2 3 4 5 6 -
Giorno - 0 72 ore
7 14 21 28 -
Dati Anagrafici *
Criteri di Inclusione/ Esclusione
*
Consenso Informato Studio e prelievo del campione biologico
*
Richiesta di Randomizzazione
*
Anamnesi Oncologica
*
Patologie Concomitanti
*
*
STUDIO CERP versione 1 del 24.05.2010
29
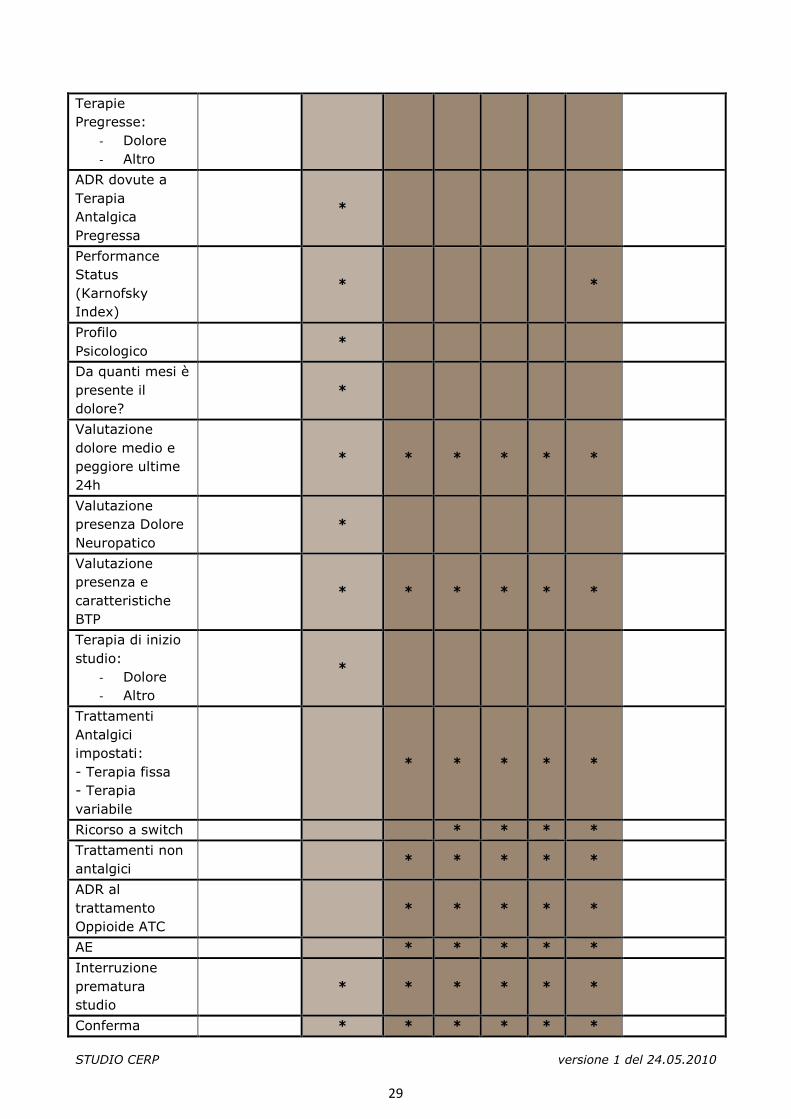
Terapie Pregresse:
- Dolore - Altro
ADR dovute a Terapia Antalgica Pregressa
*
Performance Status (Karnofsky Index)
* *
Profilo Psicologico
*
Da quanti mesi è presente il dolore?
*
Valutazione dolore medio e peggiore ultime 24h
* * * * * *
Valutazione presenza Dolore Neuropatico
*
Valutazione presenza e caratteristiche BTP
* * * * * *
Terapia di inizio studio:
- Dolore - Altro
*
Trattamenti Antalgici impostati: - Terapia fissa - Terapia variabile
* * * * *
Ricorso a switch * * * *
Trattamenti non antalgici
* * * * *
ADR al trattamento Oppioide ATC
* * * * *
AE * * * * *
Interruzione prematura studio
* * * * * *
Conferma * * * * * *
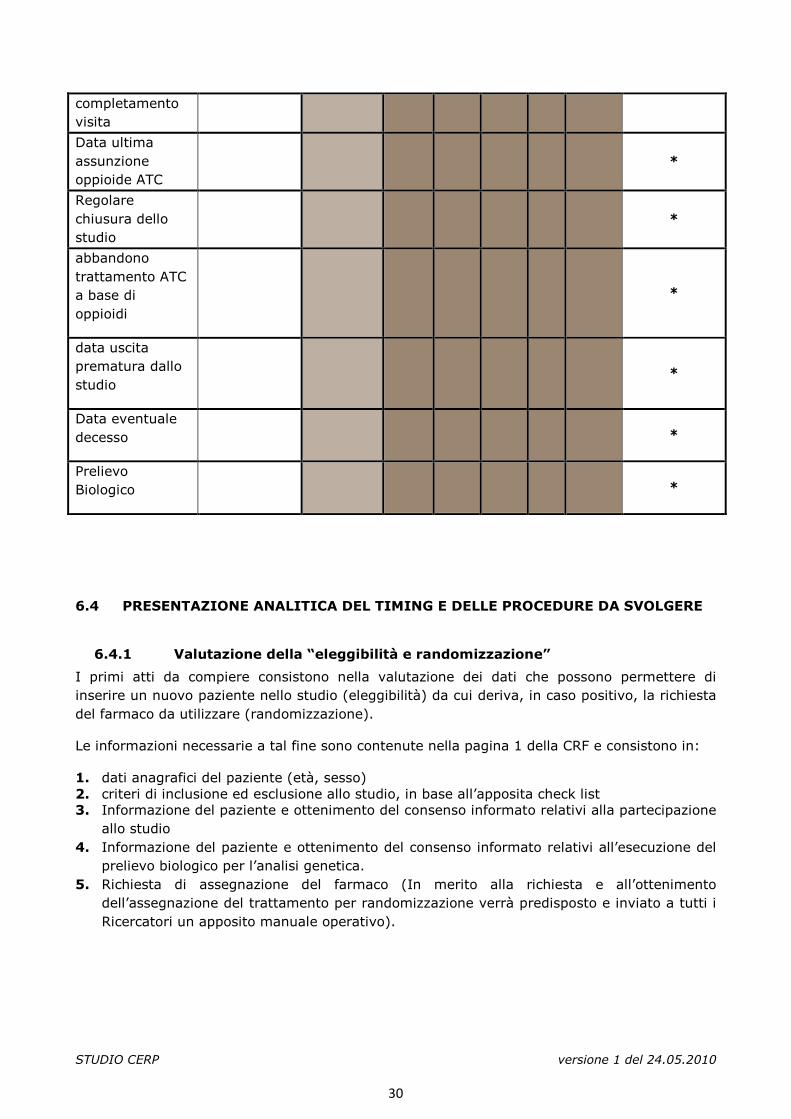
STUDIO CERP versione 1 del 24.05.2010
30
completamento visita
Data ultima assunzione oppioide ATC
*
Regolare chiusura dello studio
*
abbandono trattamento ATC a base di oppioidi
*
data uscita prematura dallo studio
*
Data eventuale decesso
*
Prelievo Biologico
*
6.4 PRESENTAZIONE ANALITICA DEL TIMING E DELLE PROCEDURE DA SVOLGERE
6.4.1 Valutazione della “eleggibilità e randomizzazione”
I primi atti da compiere consistono nella valutazione dei dati che possono permettere di inserire un nuovo paziente nello studio (eleggibilità) da cui deriva, in caso positivo, la richiesta del farmaco da utilizzare (randomizzazione).
Le informazioni necessarie a tal fine sono contenute nella pagina 1 della CRF e consistono in:
1. dati anagrafici del paziente (età, sesso) 2. criteri di inclusione ed esclusione allo studio, in base all’apposita check list 3. Informazione del paziente e ottenimento del consenso informato relativi alla partecipazione
allo studio 4. Informazione del paziente e ottenimento del consenso informato relativi all’esecuzione del
prelievo biologico per l’analisi genetica. 5. Richiesta di assegnazione del farmaco (In merito alla richiesta e all’ottenimento
dell’assegnazione del trattamento per randomizzazione verrà predisposto e inviato a tutti i Ricercatori un apposito manuale operativo).

STUDIO CERP versione 1 del 24.05.2010
31
6.4.2 Visita 1 – valutazione basale
Una volta conclusa la fase descritta nel precedente paragrafo si inseriscono i dati contenuti nella visita1-valutazione basale. Nell’ordine:
1. Anamnesi oncologica sintetica, che comprende l’indicazione del tumore primitivo, la data della sua prima diagnosi, le localizzazioni secondarie presenti in quel momento, i trattamenti antitumorali precedentemente effettuati.
2. Le eventuali patologie concomitanti in atto. 3. Tutti i trattamenti (antalgici e non) in atto prima della visita 1. 4. Le reazioni avverse dovute ai trattamenti antalgici precedenti l’inizio dello studio. 5. Lo stato di performance espresso con l’indice di Karnofsky (IK). 6. il profilo di tipo psicologico (ricavato da EORTC QLQ-C30, versione 3) 7. Una serie di informazioni relative al dolore del malato, includenti:
• da quanti mesi il paziente soffre per dolori dovuti al cancro • l’intensità del dolore medio e peggiore provato nelle ultime 24 ore (misurato con
NRS, su indicazione del paziente). • le caratteristiche e la tipologia del dolore (nocicettivo, neuropatico, misto)
• presenza/assenza di BTP ed eventuali informazioni specifiche
A seguire verranno inserite nella eCRF:
• la nuova terapia oppioide ATC (in base alle indicazioni ricevute dal centro di coordinamento)
• la data esatta in cui tale trattamento inizia (dato basale) • i trattamenti analgesici aggiuntivi, rescue e adiuvanti eventualmente assegnati • tutte le altre terapie, non relative al dolore, prescritte all’inizio dello studio.
6.4.3 Visita 2 - 72 ore
Tenuto conto che in questo studio è stata fatta la scelta di intraprendere il trattamento con oppioidi di 3° scalino iniziando con dosaggi standardizzati di oppioide SR, correggibili immediatamente o nelle ore/giorni successivi come descritto nel § 5.1, si è ritenuto opportuno inserire una visita di controllo a distanza di 72 ore dall’inizio dello studio, per valutare questo periodo iniziale di titolazione e di raggiungimento della dose adeguata. Tale controllo è mirato a capire se e come è cambiato lo schema terapeutico in questa fase d’inizio di terapia e quale tipo di effetti (analgesia ed eventuali ADR) ne sono scaturiti.
Azioni da compiere:
• Misurare l’entità del dolore medio e peggiore relativo alle ultime 24 ore (con NRS, su indicazione del paziente).
• Segnalare la comparsa/persistenza e le caratteristiche del BTP.
• Indicare se si è fatto ricorso a terapia aggiuntiva, di appoggio al trattamento ATC, a partire dalla precedente visita.
La diagnosi di dolore neuropatico si può basare, oltre che sulle conoscenze e all’esperienza del medico, sui criteri suggeriti dal questionario DN4, riportato in allegato 1.
La diagnosi di BTP si può basare, oltre che sulle conoscenze e all’esperienza del medico, sull’algoritmo riportato in allegato 1.

STUDIO CERP versione 1 del 24.05.2010
32
• Inserire lo schema della terapia del dolore fissa (ATC, adiuvante), variabile (terapia aggiuntiva e rescue) e delle altre terapie decise alla visita attuale.
• Indicare lo stato di performance espresso con l’indice di Karnofsky (IK). • Segnalare eventuali ADR, su indicazione del paziente, correlabili alla terapia del dolore
ed eventuali altri AE.
• Indicare l’eventuale interruzione prematura dello studio e relative cause.
6.4.4 Visite 3, 4, 5 e 6 - valutazioni post-basali di controllo
Tali visite corrispondono ai giorni 7, 14, 21 e 28 del follow-up.
Azioni da compiere:
• Misurare l’entità del dolore medio e peggiore relativo alle ultime 24 ore (con NRS, su indicazione del paziente).
• Segnalare la comparsa/persistenza e le caratteristiche del BTP.
• Indicare se si è fatto ricorso a terapia aggiuntiva, di appoggio al trattamento ATC, a partire dalla precedente visita.
• Inserire lo schema della terapia del dolore fissa (ATC, adiuvante), variabile (terapia aggiuntiva e rescue) e delle altre terapie decise alla visita attuale.
• Indicare l’eventuale avvenuto cambio dell’oppioide (switch) e relative cause • Indicare lo stato di performance espresso con l’indice di Karnofsky (IK). • Segnalare eventuali ADR, su indicazione del paziente, correlabili alla terapia del dolore
ed eventuali altri AE. • Indicare l’eventuale interruzione prematura dello studio e relative cause.
6.4.5 Completamento dello studio o eventuale interruzione prematura
Ad ogni visita di controllo, nella CRF, è prevista, come già indicato, la segnalazione di eventuale interruzione prematura dello studio, nel periodo immediatamente precedente o in corso della visita in atto: in tal caso si rimanda alla pagina finale della CRF per le opportune informazioni da inserire. Al termine dell’ultima visita, in ogni caso, verrà richiesto se il paziente ha regolarmente completato lo studio; in caso contrario si richiede di indicarne la causa e la data di interruzione. Ad ogni visita verrà ricordata la necessità di effettuare il prelievo ematico ai fini dell’indagine genetica.

STUDIO CERP versione 1 del 24.05.2010
33
7. EVENTI AVVERSI E REAZIONI AVVERSE
7.1 DEFINIZIONI (G. U. N. 142 DEL 21 GIUGNO 2006)
Per Evento Avverso (AE = Adverse Event) si intende “qualsiasi evento clinico dannoso che si manifesta in un paziente o in un soggetto coinvolto in una sperimentazione clinica cui è stato somministrato un medicinale, e che non ha necessariamente un rapporto causale con questo trattamento”. Per Reazione Avversa ai farmaci (ADR = Adverse Drug Reaction) si intende “qualsiasi reazione dannosa ed indesiderata a un medicinale in fase di sperimentazione, a prescindere dalla dose somministrata”. Per Evento Avverso Serio (SAE = Serious Adverse Event) o Reazione Avversa Seria ai farmaci (SADR = Serious Adverse Drug Reaction) si intende “qualsiasi evento avverso o reazione avversa ai farmaci che, a prescindere dalla dose, ha esito nella morte o mette in pericolo la vita del soggetto, richiede un ricovero ospedaliero o prolunga la degenza in ospedale, o che determina invalidità o incapacità gravi o prolungate, o comporta una anomalia congenita o un difetto alla nascita”.
7.2 EVENTI AVVERSI (AE)
Le informazioni ottenute dal paziente integreranno le osservazioni derivanti dalla visita dello stesso da parte del medico. Per tale descrizione è stata approntata sulla scheda raccolta dati una specifica sezione (presente ad ogni visita) che permette di descrivere l’evento, la gravità, il nesso di causalità ai trattamenti e le eventuali azioni intraprese per affrontare la situazione. Sembra opportuno, inoltre, che il paziente riferisca al medico tutti gli eventi negativi che dovessero insorgere nei periodi di tempo al di fuori delle visite ed eventualmente, in caso di necessità, a contattarlo.
7.3 REAZIONI AVVERSE AI FARMACI (ADR)
Nel presente studio si richiede di porre attenzione agli effetti collaterali legati a tutti i trattamenti dello studio e, in particolare, a quelli riferibili al trattamento con oppioidi, essendo, tra l’altro, l’analisi della tossicità uno degli endpoint secondari di sicurezza e tollerabilità previsti dal protocollo. Al fine del riconoscimento e della valutazione degli effetti collaterali da oppioidi, tenuto anche conto della soggettività di alcuni degli effetti indesiderati di questi farmaci, è stata predisposta un’apposita check-list, inserita nel “Questionario per il paziente” (da riproporre ad ogni visita), che facilita l’individuazione dell’effetto collaterale e ne prevede anche una stima di entità (scala verbale a 4 punti). Le risposte dei pazienti saranno poi riportate sulla CRF.

STUDIO CERP versione 1 del 24.05.2010
34
7.4 EVENTO AVVERSO SERIO (SAE) E REAZIONI AVVERSE AI FARMACI SERIE (SADR)
SAE e SADR sono gli eventi: • ad esito fatale; • che pongano il paziente in pericolo di vita; • che possano determinare disabilità od invalidità al paziente; • che richiedano o prolunghino un'ospedalizzazione; • che possano mettere a rischio il paziente o richiedano un intervento per evitare un esito
riconducibile ad una delle categorie sopra indicate. Nello specifico di questo studio, che include pazienti oncologici con diagnosi di tumore solido in fase avanzata locale e/o metastatica, la progressione di malattia è un evento atteso e la morte, dovuta specificamente a progressione di malattia, non dovrà essere considerata come un SAE e non andrà segnalata. Al contrario, se la morte è dovuta ad altre cause, rientrerà nei SAE o SADR ed entrerà nella obbligatorietà della segnalazione.
7.5 SEGNALAZIONI, DOCUMENTAZIONE ED ULTERIORI INDAGINI
In ottemperanza alla normativa vigente (Dir. Europea 2001/20/E – 04/04/2001; D. Lgs. 211 – 24/06/2003 e successive integrazioni e modifiche), il medico sperimentatore deve segnalare tutti i SAE descritti nel paragrafo precedente. Sulla CRF è predisposta un’apposita scheda per l’identificazione e la valutazione dei SAE: in particolare, su tale scheda è inserita una domanda che interroga sul nesso di causalità tra SAE e trattamenti in corso, che permetterà di individuare, da parte del centro di coordinamento, le SADR dagli altri SAE. Ogni volta che il medico identifica un SAE, connesso o non connesso con i trattamenti, è tenuto ad eseguirne la segnalazione secondo la seguente modalità: Sarà cura dell’Istituto Mario Negri comunicare alle Autorità Sanitarie competenti ogni informazione relativa alla sicurezza dei farmaci in studio, nei tempi e modi compatibili con la normativa vigente.
• compilare il formato elettronico dell’apposita sezione SAE nella CRF • stampare la pagina e inviarla datata e firmata via fax al numero 02-33200231,
all’attenzione della Dr.ssa Maria Teresa Greco (CERP, Istituto Mario Negri), entro 24 ore dall’avvenuta conoscenza.

STUDIO CERP versione 1 del 24.05.2010
35
8. METODI STATISTICI
L'elaborazione statistica dei dati sarà effettuata tramite il package di analisi statistica SAS (Statistical Analysis System) per Windows versione 9.1.
Subito dopo la chiusura e pulizia del database e prima della analisi dei dati verrà stilato il documento chiamato Statistical Analysis Plan (SAP) che sarà composto essenzialmente dai seguenti capitoli:
• Metodi statistici pianificati nel protocollo; • Dimensionamento del campione; • Gestione dei dati mancanti; • Valutazioni di efficacia; • Valutazione di tollerabilità; • Cambiamenti nella strategia di analisi statistica; • Modelli statistici che verranno applicati nell’analisi; • Tabelle, Listati e Figure da inserire in appendice come post-text tables and figures; • Tabelle, Listati e Figure da inserire da inserire nel corpo centrale del report clinico.
8.1 GESTIONE DEI DATI
I dati clinici di questo studio saranno inseriti direttamente dagli sperimentatori attraverso un sistema computerizzato presente all’Istituto Mario Negri attraverso un singolo entry in remoto con verifica elettronica della congruenza dei dati.
Oltre le usuali funzioni di inserimento dati di on-line checks, questo sistema web-based, denominato GCPBASE, prevede un sistema di controllo di autenticità dell’operatore (sperimentatore), un sistema di assegnazione casuale del trattamento (dopo opportuna verifica delle condizioni di eleggibilità e del consenso informato scritto) e un sistema di raccolta dati a riguardo degli eventi avversi seri.
A tempi prefissati e con scadenza regolare, i dati inseriti verranno sistematicamente verificati dallo staff di data-management appositamente costituito all’interno del CERP. Tutti gli errori, incongruenze, omissioni saranno inseriti in moduli di richiesta di verifica (Data Query Form) che il CERP invierà ai rispettivi sperimentatori per le correzioni del caso. Sarà poi cura dello sperimentatore operare le modifiche sui propri dati.
Il singolo soggetto randomizzato sarà identificato da un unico codice univoco generato dal sistema stesso in maniera automatica.
8.2 POPOLAZIONI ANALIZZATE
La popolazione principale per le analisi di efficacia sarà la popolazione Intent-to-treat (ITT) composta da tutti i pazienti eleggibili, randomizzati, con la valutazione della variabile primaria di efficacia al basale e almeno una rilevazione post-basale.
Verrà valutata anche una “Per-protocol population” a supporto della ITT population composta dai pazienti che concluderanno regolarmente lo studio, senza violazioni di protocollo e che avranno assunto lo stesso tipo di trattamento in analisi per tutta la durata dello studio.

STUDIO CERP versione 1 del 24.05.2010
36
Per le analisi di tollerabilità verrà presa in considerazione la “Safety population” composta dai pazienti a cui è stato somministrato almeno una dose del trattamento in analisi a loro assegnato.
8.3 STIMA DELLA DIMENSIONE DEL CAMPIONE
Scopo dello studio è testare l'ipotesi nulla che la proporzione di soggetti non-risponder alle terapie analgesiche dopo 28 giorni di trattamento sia identica tra i trattamenti Buprenorfina, Fentanyl ed Ossicodone verso il trattamento con Morfina contro l’ipotesi alternativa che almeno uno dei tre trattamenti di cui sopra abbia una proporzione di non-risponder inferiore al trattamento con Morfina.
La diminuzione della proporzione di soggetti non-responder verrà calcolata mettendo a confronto il valore del dolore medio alla visita basale con il dolore medio alla visita finale secondo la relazione [dolorefinale-dolorebasale]/dolorebasale *100 , dove la misurazione del dolore medio verrà fornita utilizzando una Numerical Rating Scale a 11 punti (da 0=nessun dolore a 10= il massimo dolore immaginabile).
Ipotizzando una proporzione di non-responder nel gruppo Morfina pari al 35% e valutando una diminuzione pari al 40% in uno dei tre trattamenti test, considerando una potenza del test dell’80%, una probabilità di 0.05 per l’errore di tipo I ed infine assumendo un test a due code, otteniamo una dimensione campionaria di 214 pazienti per gruppo per un totale di 856 pazienti. Ipotizzando una perdita del 15% dei pazienti durante lo studio e volendo mantenere quella di cui sopra come la quota minima per poter raggiungere gli obiettivi dello studio, la dimensione finale del campione sarà di 1008 pazienti in totale. Il calcolo della dimensione iniziale del campione, esclusa l’aggiunta dei drop-out, è stato eseguito con il package nQuery Advisor vers. 7 (42) tramite la procedura che utilizza il test del chi-quadrato per proporzioni per confronti multipli (43).
8.4 ASSEGNAZIONE DEI SOGGETTI AL GRUPPO DI TRATTAMENTO
I soggetti che rientrano nei criteri di eleggibilità verranno assegnati casualmente a uno dei quattro bracci di trattamento in accordo al codice di randomizzazione che verrà generato presso l’Istituto Mario Negri mediante opportune procedure. La randomizzazione seguirà un rapporto di 1:1:1:1. I codici di randomizzazione verranno assegnati ai soggetti in ordine strettamente sequenziale. Il farmaco in studio verrà randomizzato all’interno dei centri per garantire il bilanciamento della numerosità dei gruppi di trattamento sia all’interno del singolo centro sia nell’intera popolazione e le cui specifiche verranno esplicitamente dichiarate nel SAP.
E’ previsto che partecipino allo studio circa 80 centri clinici, tra unità di oncologia medica e cure palliative/terapia del dolore.
8.5 TRATTAMENTO DEI DATI MANCANTI
Questo studio richiede che gli investigatori pongano la massima attenzione affinché tutte le informazioni richieste in CRF siano completate in tutte le loro parti ma, essendo questa nel contempo un’indagine approfondita sul dolore dei pazienti affetti da cancro ad uno stadio avanzato, è comunque prevista una significativa percentuale di dati mancanti provenienti da

STUDIO CERP versione 1 del 24.05.2010
37
quei pazienti che terminano prematuramente lo studio per cause dipendenti dalla patologia di cui soffrono o per altre ragioni ad essa connesse.
Si è deciso perciò di porre in atto strumenti di controllo serrato sulla presenza di valori mancanti dovuti alla incompleta compilazione di tutti i campi da parte del medico, richiedendo, per ognuno di essi, le ragioni. Di conseguenza, verranno avviate procedure di sollecitazione per limitare al massimo il loro impatto. In questo modo il meccanismo di produzione dei missing dovrebbe essere mantenuto solo nel caso prima esposto.
Dovessero comunque verificarsi situazioni clinicamente accettabili in cui il dato mancante, a riguardo dell’outcome primario, esula dalle considerazioni di cui sopra, la natura di questa tipologia di missing verrà attentamente vagliata e descritta onde permetterne la classificazione (44) e di conseguenza l’utilizzazione delle procedure più appropriate (multiple imputation, moment method, etc...) per la sua gestione.
La percentuale di dati mancanti sull’outcome primario al di sotto della quale non verrà attuata nessuna procedura di gestione degli stessi verrà definita nel SAP. Verranno effettuate sia le analisi senza dati mancanti sia le analisi basate sulla loro ricostruzione. La decisione di quale di queste analisi sarà utile ai fini della valutazione dei risultati verrà presa dopo la chiusura del database in cui si avrà l’esatta percentuale di dati mancanti. Questa decisione verrà riportata nel SAP.
Entrando nello specifico degli obiettivi di efficacy dello studio, si agirà nel seguente modo:
• analisi principale (valutazione sulla proporzione di non-responder alla fine dello studio): tutti i pazienti che escono prematuramente dallo studio saranno classificati nel seguente modo:
o non-responder se l’uscita è dovuta ad inefficacia analgesica del farmaco o per eventi avversi seri causati dal farmaco o ad entrambe le cause;
o responder o non-responder a seconda delle informazioni cliniche presenti nell’ultima visita valida post-basale.
I risultati di questa analisi saranno poi messi a confronto con due sensitivity analyses in cui applicheremo un Binary Imputation approach con due modalità differenti:
o un’analisi dove tutti i missing finali verranno sostituiti con il valore corrispondente alla classe responder
o un’analisi dove tutti i missing finali verranno sostituiti con il valore corrispondente alla classe non-responder
Il confronto di queste due analisi con quella di base ci permetterà di ottenere un range di possibili risposte con cui circoscrivere le conclusioni.
• analisi secondaria (valutazione longitudinale della proporzione di non-responder): come si vedrà in seguito (§ 8.8) l’analisi di base si avvale della stima dei parametri della regressione con le procedure di Generalized Estimating Equation (42, 45) le quali stimano la matrice di lavoro a partire da tutte le coppie di valori osservati. I risultati di questa analisi saranno messi a confronto con una sensitivity analysis che utilizzerà la stima di massima verosimiglianza solo sui dati osservati.

STUDIO CERP versione 1 del 24.05.2010
38
8.6 DESCRIZIONE DELLA POPOLAZIONE
Tutte le principali variabili, da quelle demografiche fino a quelle di tollerabilità, presenti in questo studio, saranno dettagliatamente descritte con metodiche adatte per ciascuna tipologia. La descrizione avverrà sul totale e separatamente per i quattro trattamenti, dove, in linea generale, verranno riportati:
- per le variabili continue: parametri di tendenza centrale (media aritmetica e mediana) e di dispersione (deviazione standard, min-max, Q1-Q3);
- per le variabili categoriche: frequenza e percentuale; - per le variabili longitudinali di interesse clinico: analisi dei cambiamenti delle visite post-
basali dalla visita basale.
8.7 ANALISI DELLA VARIABILE PRIMARIA DI EFFICACIA
L’analisi della variabile primaria (proporzione di pazienti non-responder al giorno 28) si baserà su un modello di regressione logistica di confronto a coppie tra Morfina e i tre oppioidi: Buprenorfina, Fentanyl ed Ossicodone tramite il Wald test.
Nel caso in cui si dovessero osservare rilevanti differenze tra trattamenti a riguardo di fattori demografici, prognostici o clinici si prevede l’uso di un modello di regressione logistica multivariato in cui verranno presi in considerazione, oltre all’effetto dei trattamenti, tutte quelle variabili che risulteranno essere potenziali fattori di confondimento della risposta clinica. In questo caso la scelta delle covariate da utilizzare nel modello finale verrà fatta preliminarmente con modelli di valutazione della forza di associazione di ogni singola variabile indipendente con la variabile di risposta. In questo contesto verrà escluso dal modello l’effetto dovuto ai trattamenti in studio. L’entità delle differenze per definire un fattore come possibile covariata nell’analisi globale, sarà oggetto di discussione in uno dei paragrafi del SAP.
Come già riferito al paragrafo 8.1, la popolazione principale di analisi è la ITT a cui verrà affiancata un’analisi della popolazione per protocol con identiche caratteristiche di modellazione. Ogni valutazione sarà oggetto di discussione nel rapporto clinico integrato finale.
8.8 ANALISI DELLE VARIABILI SECONDARIE DI EFFICACIA
Per l’andamento della proporzione di soggetti non-responder a tutte le visite post basali si utilizzerà un modello longitudinale per misure binarie aggiustato per le tutte le variabili utilizzate nel modello dell’analisi primaria. Per la stima dei parametri della regressione ci si avvarrà delle procedure di Generalized Estimating Equation (42, 45) le cui specifiche della struttura di covarianza verranno dettagliate nel SAP.
Per tutte le altre analisi, che qui di seguito riportiamo, ci si baserà su un modello analogo a quello descritto per la variabile primaria ed avranno valore puramente esplorativo:
• Proporzione di soggetti full-responder alla fine dello studio
• Proporzione di soggetti full-responder dopo le prime 72 ore
• Proporzione di soggetti che hanno richiesto almeno un cambio di oppioide di 3° scalino (switch) per qualsiasi ragione
• Proporzione di soggetti che hanno richiesto almeno un cambio di oppioide di 3° scalino (switch) causa scarsa efficacia analgesica

STUDIO CERP versione 1 del 24.05.2010
39
• Proporzione di soggetti che hanno richiesto almeno un cambio di oppioide di 3° scalino (switch) causa intolleranza al farmaco
• Proporzione di soggetti che hanno richiesto assunzione di terapia rescue e/o adiuvante
• Proporzione di soggetti che hanno richiesto un aumento del dosaggio dello stesso oppioide di partenza misurato con l’indice OEI ≥ 5%
• Proporzione di soggetti che hanno abbandonato prematuramente lo studio per cause attribuibili al trattamento
8.9 ANALISI DELLA TOLLERABILITÀ
Per le Reazione Avverse la descrizione verrà fatta a tutti i periodi di trattamento ed in base alla loro severità. Per gli Eventi Avversi la descrizione si baserà principalmente sugli eventi seri, su quelli in relazione con il farmaco o che hanno determinato il termine prematuro dello studio o che hanno determinato uno switch.
8.10 ANALISI AD INTERIM
Non sono previste valutazioni ad interim.
8.11 ANALISI DI SOTTOGRUPPI
In queste analisi vengono studiati aspetti specifici del dolore ed il loro trattamento.
Esse sono:
1. Soggetti con presenza di BTP, per una valutazione delle modalità ed esiti e della terapia “rescue” per il controllo degli episodi dolorosi.
2. Soggetti con switch da un oppioide ad un altro oppioide, entrambi di scalino 3, per una valutazione delle modalità di realizzazione e gli esiti derivanti.
3. Soggetti con variazione di dose degli oppioidi assegnati al basale nelle prima 72 ore (visita 2) considerando anche la relazione tra risposta analgesica/dose necessaria a 72 ore e risposta finale.
La modalità con cui verranno condotte queste indagini verterà su analisi di carattere descrittivo limitando l’uso delle inferenza statistica che, in qualsiasi caso, avrà una natura esclusivamente esplorativa.

STUDIO CERP versione 1 del 24.05.2010
40
9. ORGANIZZAZIONE E RESPONSABILITA’ DELLO STUDIO
9.1 CENTRO DI COORDINAMENTO
Lo studio verrà coordinato dal C.E.R.P. (Center for the Evaluation and Research on Pain) in termini di stesura del protocollo, di reclutamento dei centri di ricerca, di gestione del data base relativo alle informazioni inviate per ogni caso arruolato, al data management durante la fase di reclutamento dei pazienti, all’elaborazione statistica dei dati, alla stesura di report finali e alla relativa pubblicazione. Sarà, inoltre, cura del Centro richiedere le necessarie autorizzazioni ai Comitati Etici e definire i rapporti di natura economica con le amministrazioni dei centri clinici che collaboreranno allo studio.
9.2 SPERIMENTATORI
Saranno chiamati a partecipare allo studio, in linea di massima, 80-100 centri clinici nell’ambito sia di reparti/servizi di oncologia medica o di altri ambiti specialistici che normalmente si occupano di trattamenti di pazienti neoplastici, sia di reparti/servizi/hospice di cure palliative o di terapia del dolore.
Ognuno dei centri individuati, che avrà accettato di partecipare al progetto di studio, dovrà arruolare almeno 8 pazienti, in un periodo di tempo compreso tra l’inizio dello studio, previsto in autunno 2010 e la fine dello stesso, prevista orientativamente al termine del 2011.
9.3 MONITOR
E’ prevista un’attività di monitoraggio, gestito dal C.E.R.P., presso i centri che avranno aderito allo studio, finalizzata alla verifica dello stato di avanzamento del reclutamento dei pazienti nello studio, alla corretta realizzazione dello stesso e a un eventuale supporto tecnico ai ricercatori.
9.4 CONSERVAZIONE DEI DOCUMENTI ORIGINALI
L’Istituto Mario Negri avrà cura di conservare e mantenere accessibili alle Autorità regolatorie, per un periodo di dieci anni dopo il termine dello studio, i seguenti documenti:
• copia del protocollo definitivo dello studio (ed eventuali emendamenti) approvato dalle Autorità Amministrative e dai Comitati Etici;
• copie delle autorizzazioni amministrative e delle autorizzazioni dei Comitati Etici con gli elenchi dei loro componenti;
• copie delle schede di raccolta dati; • copie delle relazioni delle visite dei monitor.
9.5 PUBBLICAZIONE DEI RISULTATI
La stesura dei manoscritti connessi alle analisi descritte in questo protocollo sarà a carico del C.E.R.P., i cui componenti, unitamente ai membri del Comitato scientifico, saranno indicati come gli autori principali. Quando possibile, sarà incluso tra i co-autori almeno un nominativo per ciascuno dei centri partecipanti; inoltre tutti i partecipanti saranno citati nelle appendici.

STUDIO CERP versione 1 del 24.05.2010
41
A tutti gli autori sarà richiesto di rivedere e approvare il manoscritto prima della sottomissione per la pubblicazione.
9.6 MODIFICHE DEL PROTOCOLLO
Tutti gli emendamenti al protocollo devono essere effettuati dal promotore dello studio e quindi firmati e datati dallo sperimentatore. Gli emendamenti al protocollo non devono essere implementati senza prima averne ottenuto l’approvazione etica. Fanno eccezione i casi in cui essi sono necessari per eliminare un rischio immediato per il soggetto; in tale evenienza l’emendamento deve essere immediatamente sottoposto al Comitato Etico ma viene implementato senza attenderne l’approvazione. Le modifiche al protocollo che riguardano solo aspetti amministrativi o logistici dello studio, andranno notificate al Comitato Etico.
Per le situazioni che richiedano una violazione del protocollo, lo sperimentatore contatterà il promotore dello studio nelle persone delegate per fax o per telefono. Se possibile tale contatto sarà effettuato prima di implementare qualunque violazione del protocollo. In ogni caso, il contatto con il promotore dello studio dovrà avvenire non appena possibile in modo da discutere la situazione e accordarsi sul corso d’azione più appropriato. I dati riportati nella CRF e nei documenti originali rifletteranno tutte le violazioni di protocollo e i documenti originali descriveranno queste violazioni e le circostanze che le hanno rese necessarie.

STUDIO CERP versione 1 del 24.05.2010
42
10. BIBLIOGRAFIA
1. World Health Organization. Cancer Pain Relief, 2nd edn. Geneva: World Health Organization;1996.
2. Apolone G, Mangano S, Compagnoni A, Negri E, et al. A Multidisciplinary Project to improve the quality of cancer pain management in Italy. Background, methods, and preliminary results. J Ambul Care Manage 2006; 29:332-341
3. Apolone G, Bertetto O, Caraceni A, Corli O, et al. Pain in cancer. An outcome research project to evaluate the epidemiology, the quality and the effects of pain treatment in cancer patients. Health Qol Life Outcomes 2006; 14;4(1):88
4. Apolone G, Corli O, Caraceni A, Negri E, Deandrea S, Montanari M, Greco MT. Pattern and quality of care of cancer pain management. Results from the Cancer Pain Outcome Research Study Group (CPOR SG). Br J Cancer 2009; 100: 1566-74.
5. Deandrea S, Montanari M, Moja L, Apolone G. Prevalence of undertreatment in cancer pain. A review of published literature. Ann Oncol, 2008 Dec;19(12):1985-91.
6. Rhee C, Broadbent AM. Palliation and liver failure: palliative medications dosage guidelines. J Palliat Med 2007;10:677-685.
7. Rakvåg TT, Klepstad P, Baar C, Kvam TM, Dale O, Kaasa S, Krokan HE, Skorpen F.Reyes-Gibby et al., 2007The Val158Met polymorphism of the human catechol-O-methyltransferase (COMT) gene may influence morphine requirements in cancer pain patients. Pain 2005;116:73-78.
8. Campa D, Gioia A, Tomei A, Poli P, Barale R. Association of ABCB1/MDR1 and OPRM1 gene polymorphisms with morphine pain relief. Clin Pharmacol Ther 2008;83:559-566.
9. Ross JR, Riley J, Taegetmeyer AB, Sato H, Gretton S, du Bois RM, Welsh KI. Genetic variation and response to morphine in cancer patients: catechol-O-methyltransferase and multidrug resistance-1 gene polymorphisms are associated with central side effects. Cancer 2008;112:1390-1403.
10. Droney J, Levy J, Quigley C. Prescribing opioids in renal failure. J Opioid Manag 2007;3:309-316.
11. Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol 2007;17:556-564.
12. Arden JR, Segredo V, Wang Z, Lameh J, Sadée W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-targed mu-opioid receptor expressed in HEK 293 cells. J Neurochem 1995;65:1636-1645.
13. Keith E, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem 1996;271:19021–19024.
14. Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. Mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol 1998;53:377–384.
15. Garrido MJ, Troconiz IF. Methadone: a review of its pharmacokinetic/pharmacodynamic properties. J Pharmacol Toxicol Methods 1999;42:61–66.
16. He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell 2002;108:271–282.
17. Bailey CP, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol 2005;5:60–68.
18. Gintzler AR, Chakrabarti S. Post-opioid receptor adaptations to chronic morphine; altered functionality and associations of signaling molecules. Life Sci 2006;79:717–722.
19. Ammon-Treiber S, Hollt V. Morphine-induced changes of gene expression in the brain. Addict Biol 2005;10:81–89.
20. McClung CA, Nestler EJ, Zachariou V. Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J Neurosci 2005;25:6005–6015.
21. Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci 1997;17:796-803.

STUDIO CERP versione 1 del 24.05.2010
43
22. Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology 2003;45:575–584
23. Gomes I, Gupta A, Filipovska J, Szeto H, Pintar J, Devi LA. A role for heterodimerization of m and d opiate receptors in enhancing morphine analgesia. PNAS 2004; 101:, n 14: 5135-5139.
24. Bai M. Dimerization of G-coupled receptors: role in signal transduction. Cellular signaling 2004; 16: 175-186.
25. Bruera E, Belzile M, Pituskin E, Fainsinger R, Darke A, Harsanyi Z, Babul N, Ford I. Randomized, double-blind, cross-over trial comparing safety and efficacy of oral controlled-release oxycodone with controlled-release morphine in patients with cancer pain. J Clin Oncol 1998;16:3222-3229.
26. Mucci LoRusso P, Berman BS, Silberstein PT, et al.Controlled-release oxycodone compared with controlled-release morphine in the treatment of cancer pain: A randomized, double-blind, parallel-groupstudy. European Journal of Pain 1998;2:239–249.
27. Heiskanen TE, Ruismäki PM, Seppälä TA, Kalso EA. Morphine or oxycodone in cancer pain? Acta Oncol 2000;39:941-7D.
28. Lauretti GR, Oliveira GM, Pereira NL. Comparison of sustained-release morphine with sustained-release oxycodone in advanced cancer patients. British Journal of Cancer 2003;89:2027–2030.
29. Ahmedzai S, Brooks D. Transdermal fentanyl versus sustained-release oral morphine in cancer pain: preference, efficacy, and quality of life. The TTS-Fentanyl Comparative Trial Group. J Pain Symptom Manage 1997;13:254-261.
30. Payne R, Mathias SD, Pasta DJ, Wanke LA, Williams R, Mahmoud R. Quality of life and cancer pain: satisfaction and side effects with transdermal fentanyl versus oral morphine. J Clin Oncol 1998;16:1588-1593.
31. van Seventer R, Smit JM, Schipper RM, Wicks MA, Zuurmond WW. Comparison of TTS-fentanyl with sustained-release oral morphine in the treatment of patients not using opioids for mild-to-moderate pain. Current Medical Research and Opinion 2003;19:457–469.
32. Clark AJ, Ahmedzai SH, Allan LG, Camacho F, Horbay GL, Richarz U, Simpson K. Efficacy and safety of transdermal fentanyl and sustained-release oral morphine in patients with cancer and chronic non-cancer pain. Curr Med Res Opin 2004;20:1419-1428.
33. Mercadante S, Porzio G, Ferrera P, Fulfaro F, Aielli F, Verna L, Villari P, Ficorella C, Gebbia V, Riina S, Casuccio A, Mangione S.Sustained-release oral morphine versus transdermal fentanyl and oral methadone in cancer pain management. Eur J Pain 2008;12:1040-1046.
34. Walter C, Lotsch J. Meta-analysis of the relevance of the OPRM1 118A>G genetic variant for pain treatment. Pain 2009; 146: 270-275.
35. Max MB. Moving pain genetics into the Genome-Wide Association era. In “Current Topics in Pain” Castro-Lopes J Editor. IASP Press 2009: 185-197.
36. Kaasa S, Apolone G, Klepstad P, Loge JH, Hjermastad MJ, Corli O, Strasser F et Al. Consensus expert conference on cancer pain assessmente and classification, the need fora n International consensus: working proposal on International standard. In preparation.
37. Klepstad P. Opioid dose titration. Fifth Bristol Opioid conference. Bristol University Press;2010. http://www.ntnu.no/eksternweb/multimedia/archive/00083/5th_Bristol_Opioid_C_83739a.pdf
38. Sittl R, Nuijten M, Nautrup BP. Changes in the prescribed daily doses of transdermal fentanyl and transdermal buprenorphine during treatment of patients with cancer and noncancer pain in Germany: results of a retrospective cohort study. Clin Ther. 2005 Jul;27(7):1022-31.

STUDIO CERP versione 1 del 24.05.2010
44
39. Zeppetella G. Opioids and breakthrough pain. Fifth Bristol Opioid conference. Bristol University Press;2010. http://www.ntnu.no/eksternweb/multimedia/archive/00083/5th_Bristol_Opioid_C_83739a.pdf
40. Dale O, Moksnes K, Kaasa S. Systematic review on switching of strong opioids in cancer patients. An EPCRC opioid guideline project. Fifth Bristol Opioid conference. Bristol University Press; 2010. http://www.ntnu.no/eksternweb/multimedia/archive/00083/5th_Bristol_Opioid_C_83739a.pdf
41. Slatkin NA. Opioid switching and rotation in primary care: implementation and clinical utility. Curr Med Res Opinion 2009; 9: 2133.2150.
42. Clivio L, Tinazzi A, Mangano S, Santoro E. The contribution of information technology: towards a better clinical data management. Drug Devl Res 2006; 67: 145-250.
43. Lachin, J. M. (1977) Sample size determinations for rxc comparative trials. Biometrics, 33: 315-324.
44. Sheu, C.-F. (2000). Regression analysis of correlated binary outcomes. Behavior Research Methods, Instruments, & Computers, 32, 269-273.
45. Little, R.J.A. & Rubin, D.B. (1987) Statistical analysis with missing data. New York, Wiley.
46. O’Brien, R.G., Muller, K.E. Applied Analysis of Variance in Behavioral Science – Marcel Dekker, New York 1993 – Chapter 8 pp. 297-334.
47. Lowe S, Nekolaichuk C, Fainsinger R, Lawlor P. Should the rate of opioid dose escalation be included as a feature in a cancer pain classification system? J Pain and Symptom Manage 2008; 13: 51-57.
48. Treede RD, Jensen TS et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008; 70(18): 1630-5.
49. Niscola P, Scaramucci L, Vischini G et Al. The use of major analgesics in patients with renal disfunction. Curr Drug Targets. 2010 Jun 1;11(6):752-8.
50. Portenoy RK, Hagen NA. Breakthrough pain: definition, prevalence and characteristics.Pain 1990 Jun;41(3):273-81.
51. Davies AN, Dickman A, Reid C, Stevens AM, Zeppetella G; Science Committee of the Association for Palliative Medicine of Great Britain and Ireland. The management of cancer-related breakthrough pain: recommendations of a task group of the Science Committe of the Association for Palliative Medicine of Great Britain and Ireland. Palliative Medicine of Great Britain and Ireland. Eur J Pain. 2009;13:331-8
52. Portenoy RK, Hagen NA. Breakthrough pain: definition, prevalence and characteristics. Pain. 1990;41(3):273-81. e Portenoy (personal communication)”
53. Mercadante S, Dardanoni G, Salvaggio L, Armata MG and Agnello A. Monitoring of opioid therapy in advanced cancer pain patients. J Pain Symptom Manage 1997, 13: 204–212
54. Paice JA, Cohen FL. Validity of a verbally administered numeric rating scale to measure cancer pain intensity. Cancer Nurs. 1997 Apr; 20(2): 88-93.
55. Morita T, Tsuneto S, Shima Y. Proposed definitions for terminal sedation. Lancet. 2001 Jul 28; 358(9278): 335-6.
56. Mercadante S. Opioid titration in cancer pain: a critical review. Eur J Pain. 2007 Nov; 11(8):823-30.

STUDIO CERP versione 1 del 24.05.2010
45
11. PROTOCOLLO DEL SOTTOPROGETTO
Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati
ENTE PROMOTORE E COORDINATORE DELLO STUDIO
C.E.R.P. (Center for the Evaluation and Research on Pain)
Dipartimento di Oncologia
Istituto di Ricerche Farmacologiche MARIO NEGRI
Via G. La Masa, 19
20156 Milano
SEGRETERIA SCIENTIFICA E ORGANIZZATIVA
Simona Stupia
Telefono: 02 39014519
Fax: 02 33200231
e-mail: [email protected]
ENTE RESPONSABILE DEL SOTTOPROGETTO
Unità Operativa “Cure Palliative, Terapia del dolore, Riabilitazione”
I.R.C.C.S. Istituto Nazionale Tumori, Milano
Tel.: 02 23902792
Email: [email protected]

STUDIO CERP versione 1 del 24.05.2010
46
11.1 INTRODUZIONE
Recenti lavori preclinici e clinici (1-10) hanno evidenziato l’esistenza di una serie di geni e di varianti geniche in grado di influenzare, in modo più o meno consistente, l’azione farmacologica e clinica dei farmaci oppioidi.
In particolare, è stata rilevata l’esistenza di alcuni geni responsabili di anomalie strutturali delle proteine coinvolte nel trasporto delle molecole oppioidi attraverso la barriera ematoencefalica (BEE), nella struttura del recettore per gli oppioidi e nel sistema enzimatico inerente il metabolismo epatico degli stessi. Si tratta, indiscutibilmente, di punti nevralgici dell’attività degli oppioidi, ma una recente review (11) ha ridimensionato l’impatto clinico di alcune di queste varianti.
Assai limitati, invece, sono gli studi che analizzano i comportamenti degli oppioidi (ad esempio, nei soggetti non responders al trattamento) con l’intero genoma umano (Genome-Wide Association Studies: GWAS) anziché con singoli specifici geni (Candidates Gene Studies). Dai GWAS sono attesi, nei prossimi anni, indicazioni che ineriscono possibili nuovi mediatori del dolore (proteine derivanti da geni rilevati dagli studi) e possibili nuovi target farmacologici, direttamente rilevati da studi sull’uomo (12 ). In particolare, gli esiti di una ricerca “genome-wide” collegata all’uso degli oppioidi nel dolore da cancro sono al momento del tutto sconosciuti.
L’importanza della farmacogenomica applicata all’impiego degli oppioidi è comunque ancora lungi dall’essere del tutto chiarita e questo studio si indirizza verso la ricerca di nuove conoscenze in questo ambito.
11.2 DISEGNO DELLO STUDIO
L’analisi genetica viene realizzata in parallelo sugli stessi pazienti reclutati per lo “Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone, fentanyl e buprenorfina verso morfina, in pazienti con dolore associato a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica del WHO”, descritto su questo stesso protocollo (si veda § 2 del protocollo generale).
Verranno, però esclusi i pazienti che avranno dato il proprio consenso informato allo studio clinico ma avranno rifiutato il consenso al prelievo di materiale biologico.
Quindi, in termini generali, qualora entrambi i consensi vengano forniti da tutti i pazienti, la numerosità dei soggetti in studio sarà pari a 1008 pazienti con cancro in fase avanzata per presenza accertata di metastasi o per importante progressione locale. E’ comunque prevista la possibilità che determinati pazienti concedano il loro consenso solo allo studio di carattere clinico: in tal caso, la numerosità dei soggetti sottoposti ad analisi genetica sarà inferiore a quella del campione complessivo.

STUDIO CERP versione 1 del 24.05.2010
47
11.3 OBIETTIVI DEL SOTTOPROGETTO
Il sottoprogetto si propone di analizzare l’assetto genetico dei pazienti attraverso un’ indagine “genome-wide”.
La mappatura dell’intero genoma di ogni singolo paziente inserito nel sottoprogetto consentirà di valutare:
• eventuali correlazioni tra risultati clinici e assetto genetico dei pazienti • eventuali indicazioni di tipo prognostico-predittivo sulla possibile risposta ai farmaci
oppioidi in relazione alle caratteristiche genetiche.
11.4 TIMING E METODOLOGIA DEL PRELIEVO BIOLOGICO
11.4.1 Timing
Ai fini di eseguire la mappatura genica dei pazienti è indispensabile che in qualsiasi momento dello studio (dal baseline al giorno 28), possibilmente in concomitanza con altri accertamenti di laboratorio previsti dal medico nella conduzione clinica del caso, venga effettuato un apposito prelievo di sangue, finalizzato alle analisi di tipo genetico. Si ricorda che il prelievo va eseguito solo in quei pazienti che hanno previamente concesso il loro consenso informato al prelievo di materiale biologico.
11.4.2 Metodologia del prelievo
La metodologia della raccolta del prelievo di sangue, della conservazione dello stesso e delle modalità di spedizione all’Unità Operativa “Cure Palliative, Terapia del dolore, Riabilitazione” dell’ I.R.C.C.S. Istituto Nazionale Tumori di Milano, viene descritta in modo dettagliato nel manuale operativo del protocollo generale.
In termini sintetici, si riportano qui di seguito gli aspetti essenziali:
• Prelevare al paziente 3 ml di sangue venoso utilizzando una provetta con EDTA (quelle standard per emocromo).
• Agitare delicatamente la provetta (5-6 volte) e riporla in congelatore ad una temperatura di -20 C°.
• Dopo aver raccolto le provette di tutti i pazienti che hanno aderito allo studio, inviare in un’unica soluzione l’intero materiale all’Unità Operativa “Cure Palliative, Terapia del dolore, Riabilitazione” dell’I.R.C.C.S. Istituto Nazionale Tumori di Milano, tramite corriere (dettagli illustrati nel manuale operativo).
11.5 ANALISI GENETICA E CLINICA
Sarà condotta un’analisi su tutto il genoma per identificare l'associazione tra polimorfismi genetici e la risposta agli oppioidi, utilizzando la piattaforma “Illumina” che consente l'analisi di circa un milione di polimorfismi a singolo nucleotide (SNPs). Per ridurre i costi di ricerca, si impiegherà la strategia del “DNA pooling”, ovvero saranno confrontati due campioni di DNA, ognuno di essi costituito da una miscela equimolecolare di DNA genomico proveniente da un gruppo di pazienti con diverso fenotipo. Tale approccio è stato ampiamente usato per lo studio delle malattie e offre il vantaggio di costi ridotti e di tempi più rapidi rispetto alle analisi dei

STUDIO CERP versione 1 del 24.05.2010
48
singoli campioni (12). L’Istituto Nazionale Tumori, con il gruppo diretto dal Dr. T.A. Dragani, collaboratore allo studio, ha esperienza consolidata nell’uso di tale approccio sperimentale (13,14).
L’analisi GWAS sarà condotta utilizzando due pool di DNA, ognuno dei quali contenenti eguali quantità di DNA proveniente dai pazienti divisi in “non responders” ed in “full responders” sulla base della loro risposta alla terapia con oppioidi. La valutazione della risposta clinica sarà fatta utilizzando le scale presentate dal protocollo principale ed in particolare la definizione dei pazienti responder e non responder sarà formulata utilizzando la Numerical Rating Scale (NRS). Sulla base dell'esperienza passata, si stima che su circa 1000 pazienti che saranno reclutati, circa il 30% sarà costituito da “non responders” ed il resto da “full responders”; i due pool di DNA saranno quindi costituiti da circa 300 e 700 pazienti, rispettivamente. Saranno 16 repliche di ibridizzazione sugli SNP-arrays per ogni pool di DNA, in modo da rendere stabili ed affidabili i risultati che saranno ottenuti. In ogni caso, studi precedenti hanno mostrato che la correlazione delle frequenze alleliche ottenute mediante ibridizzazione su SNP-array in pool di DNA o tramite determinazione del genotipo individuale è statisticamente molto elevata, a sostegno della bontà e dell'affidabilità del metodo del DNA pooling (12).
La successiva genotipizzazione individuale degli SNPs che risulteranno associati con la risposta clinica in esame, sarà condotta utilizzando la piattaforma MassARRAY (Sequenom, Inc., San Diego, CA) con un disegno di PCR multiplex. Tale analisi sarà effettuata da un laboratorio esterno come servizio a pagamento, come è già stato fatto per altri studi diretti dal Dott. Dragani.
Si ritiene che la definizione del profilo genetico associato alla risposta clinica al trattamento con oppiacei del dolore da cancro potrebbe permettere di predire la risposta clinica individuale in futuri studi prospettici, migliorando e personalizzando la qualità della terapia del dolore.
11.6 ASPETTI STATISTICI CONNESSI ALL’ANALISI GENETICA
I dati ottenuti dall’analisi di ibridizzazione su SNP-arrays, in forma di segnali di intensità, saranno utilizzati per determinare le frequenze alleliche di ogni SNP in ognuno dei due pool di DNA e analizzati utilizzando metodi basati sulla varianza casuale delle statistiche di t (15), utilizzando il programma BRB ArrayTools sviluppato dai Dr. Richard Simon e Amy Lam Peng (http://linus.nci. nih.gov / BRB-ArrayTools.html). Per gli SNPs che mostreranno differenze statisticamente significative nella loro frequenza allelica tra i gruppi dei "poor responders" e dei "good responders", saranno stimati i numeri di cromosomi nei due gruppi e sarà quindi creata una tabella di contingenza 2x2 per ogni SNP; tali tabelle saranno analizzate con il test chi-quadrato, ed eventualmente con il test esatto di Fisher. In tal modo, saranno selezionati gli SNPs che mostreranno la migliore associazione statistica con la risposta alla terapia del dolore con oppiacei. Tali SNPs saranno genotipizzati nei singoli individui, come è stato indicato. L’analisi dei genotipi individuali sarà condotta mediante i test statistici standard, quali l’analisi logistica e sarà tenuto conto delle covariate eventualmente associate alla risposta clinica (ad esempio, sesso, età, centro di provenienza).

STUDIO CERP versione 1 del 24.05.2010
49
11.7 ORGANIZZAZIONE E RESPONSABILITÀ DEL SOTTOPROGETTO
Referente organizzativo e responsabile del sottoprogetto è il Dott. Augusto Caraceni, presso la Fondazione IRCCS Istituto Nazionale Tumori, Via Giacomo Venezian 1, 20133 Milano.
Responsabile per l’analisi del materiale genetico è il Dott. Tommaso A. Dragani, presso la Fondazione IRCCS Istituto Nazionale Tumori, Via Amadeo 42, 20133 Milano.
11.8 BIBLIOGRAFIA DEL SOTTOPROGETTO
1. Rakvåg TT, Klepstad P, Baar C, Kvam TM, Dale O, Kaasa S, Krokan HE, Skorpen F.Reyes-Gibby et al., 2007The Val158Met polymorphism of the human catechol-O-methyltransferase (COMT) gene may influence morphine requirements in cancer pain patients. Pain 2005;116:73-78.
2. Campa D, Gioia A, Tomei A, Poli P, Barale R. Association of ABCB1/MDR1 and OPRM1 gene polymorphisms with morphine pain relief. Clin Pharmacol Ther 2008;83:559-566.
3. Ross JR, Riley J, Taegetmeyer AB, Sato H, Gretton S, du Bois RM, Welsh KI. Genetic variation and response to morphine in cancer patients: catechol-O-methyltransferase and multidrug resistance-1 gene polymorphisms are associated with central side effects. Cancer 2008;112:1390-1403.
4. Lotsch J, Sharke C, Grosch S et Al. The polymorphism A118G of the human mu-opioid receptor gene decrease the pupil constrictor effect of morphine-6 glucoronide but not that of morphine. Pharmakogenetics 2002; 12: 3-9.
5. Bolan EA, Pan YX, Pasternak GW. Functional analysis of MOR.-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse 2004; 51: 11-18.
6. Klepstad P, Dale O, Skorpen F et Al. Genetic variabilità and clinical efficacy of morphine. Acta Anaeshesiol Scand 2005; 49: 902-908.
7. Dugay Y, Baar C, Skorpen F et Al. A novel functional polymorphism in the uridine disphosphateglucoronyltransferase 2B7 promotor with significant impact on promoter activity. Clin Pharmacol Ther 2004; 75: 223-233.
8. Zubieta JK, Heitzeg MM, Smith YR et Al. COMT val met genotype affects mu-opioid neurotransmitter to pain stressor. Science 2003; 299: 1240-1243.
9. Hirschhorn JN. Genomewide association studies – illuminatin biologic pathways. N Engl J Med. 2009 Apr 23, 360 (17): 1699-701.
10. Walter C, Lotsch J. Meta-analysis of the relevance of the OPRM1 118A>G genetic variant for pain treatment. Pain 2009; 146: 270-275.
11. Max MB. Moving pain genetics into the Genome-Wide Association era. In “Current Topics in Pain” Castro-Lopes J Editor. IASP Press 2009: 185-197.
12. Bansal A, van den Boom D, Kammerer S, et al. Association testing by DNA pooling: an effective initial screen. Proc Natl Acad Sci USA. 2002; 99:16871-4.
13. Spinola M, Meyer P, Kammerer S, et al. Association of the PDCD5 locus with lung cancer risk and prognosis in smokers. J Clin Oncol. 2006; 24:1672-8.
14. Spinola M, Leoni VP, Galvan A, et al. Genome-wide single nucleotide polymorphism analysis of lung cancer risk detects the KLF6 gene. Cancer Lett. 2007; 251:311-6.
15. Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics 2003;19:2448-55.

STUDIO CERP versione 1 del 24.05.2010
50
12. ASPETTI ETICI E ASSICURATIVI
12.1 Approvazione da parte del Comitato Etico
Sia lo studio principale che il sottostudio saranno condotti secondo le Good Clinical Practice (ICH/GCP allegato 1 del D.M. del 15.7.1997) nel rispetto dei principi etici in accordo alla Dichiarazione di Helsinki (59° Assemblea dell’Associazione Medica Mondiale, Ottobre 2008 - Seoul).
L’Istituto Mario Negri ha provveduto a stipulare una polizza di assicurazione, in accordo al D.M.
del 17.12.2004 e al D.M. 14/07/2009, con la compagnia LLOYD’S (vedi certificato di assicurazione allegato).
Prima che la sperimentazione abbia inizio, l’Istituto Mario Negri provvederà a ottenere da parte del Comitato Etico di riferimento e da parte dei Comitati Etici dei centri partecipanti l’approvazione del protocollo dello studio, con particolare riguardo al foglio informativo e al modulo di consenso informato.
12.2 Consenso informato allo studio e per la donazione di materiale biologico
Il consenso del paziente a partecipare allo studio sarà richiesto dopo che sia stata fornita al paziente una completa informazione sullo studio e sul sottostudio, prestando particolare attenzione alla spiegazione degli scopi, del modo di gestione e di utilizzo dei dati del paziente. Il diritto del paziente a non concedere il consenso a al ritirarlo in qualsiasi momento dello studio, senza fornire spiegazioni e senza implicazioni al corretto proseguimento dei trattamenti alla sua persona, sarà sempre rispettato. In questo studio si è ritenuto corretto proporre due distinti consensi informati, uno inerente l’adesione al progetto generale dello studio e uno specifico per il prelievo di sangue relativo all’analisi dell’assetto genico. Vedi ALLEGATO 2.

STUDIO CERP versione 1 del 24.05.2010
51
ALLEGATO I
DEFINIZIONI E SPECIFICHE
AUMENTO DI DOSE E’ un metodo per cui si aumenta periodicamente la dose del farmaco in uso in modo da continuare a garantire l’effetto desiderato (nello specifico, l’analgesia). La necessità dell’aumento di dose dipende sia da fattori clinici sia da cambiamenti farmacodinamici che riflettono il potenziale sviluppo dei meccanismi della tolleranza agli oppioidi. Una rapida necessità di aumento di dose è stata identificata come un importante segnale dovuto alla difficoltà di raggiungere un controllo stabile del dolore (47). BREAKTHROUGH PAIN ( BTP) Esacerbazione transitoria del dolore che avviene sia spontaneamente sia in seguito a prevedibili o imprevedibili fattori scatenanti (ad esempio, nel caso del dolore incidente) a fronte di un dolore di base adeguatamente controllato dal trattamento ATC (39, 50).
ALGORITMO PER VALUTARE LA PRESENZA/ASSENZA DI BTP (51,52) 1
2
3
4
5
6
7
8
9
10
11
Dalla stessa fonte bibliografica e dal precedente algoritmo sono state ricavate le domande, inserite nella CRF, per porre diagnosi di presenza di BTP.

STUDIO CERP versione 1 del 24.05.2010
52
DOLORE NEUROPATICO Secondo la definizione della IASP, il dolore neuropatico può essere definito come “un dolore che insorge come conseguenza di una lesione o di una malattia che colpisce il sistema nervoso somato-sensoriale” (48). 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 DOLORE NOCICETTIVO
È IL DOLORE CHE INSORGE PER STIMOLAZIONE DIRETTA DEI NOCICETTORI A CAUSA DI UN TRAUMA TISSUTALE (FUNZIONE DI ALLARME PER UN POTENZIALE DANNO FISICO). SPESSO TALE DOLORE PERSISTE IN RELAZIONE AI PROCESSI INFIAMMATORI/RIPARATIVI CHE CONSEGUONO AL TRAUMA.
QUESTIONARIO DN 4 (criteri di accertamento di una componente neuropatica dolorosa):
Compilare il questionario scegliendo una risposta per ciascuno dei punti proposti nelle domande riportate sotto:
INTERVISTA AL PAZIENTE
Domanda 1: il dolore ha una o più delle seguenti caratteristiche ?
1. BRUCIANTE/URENTE SI NO
2. SENSAZIONE DI FREDDO DOLOROSO SI NO
3. SCOSSE ELETTRICHE SI NO
Domanda 2: il dolore è associato ad uno o piu’ dei seguenti sintomi nell’area del dolore stesso ?
4. FORMICOLIO SI NO
5. PUNTURE DI SPILLO SI NO
6. INTORPIDIMENTO SI NO
7. SENSAZIONE DI PRURITO SI NO
ESAME DEL PAZIENTE
Domanda 3: il dolore è localizzato nella stessa area dove l’esame fisico può rilevare una o piu’ delle seguenti
caratteristiche ?
1. IPOESTESIA AL TATTO SI NO
2. IPOESTESIA ALLA PUNTURA SI NO
Domanda 4: nell’area dolente il dolore può essere causato o peggiorato dallo
3. SFIORAMENTO DELLA PELLE SI NO
PUNTEGGIO COMPLESSIVO DEL PAZIENTE ____/10
NB: la diagnosi di dolore neuropatico viene fatta se il punteggio derivante dal test è > 4

STUDIO CERP
DOLORE NOCICETTIVO E’ il dolore che insorge per stimolazione diretta dei nocicettori a causa di un trauma tissutale (funzione di allarme per un potenziale danno fisico). spesso tale dolore persiste in relazione ai processi infiammatori/riparativi che conseguono al trauma.
DOSE “RESCUE” E DOSE “AGGIUNTIVA” La dose “rescue” o “di salvataggio” consiste in una dose supplementare di oppioide somministrata “al bisogno” insieme al farmaco utilizzato per il dolore di basel’obiettivo di controllare episodi di BTP. Si intendsupplemento di farmaco utilizzato in caso di variazioni di dolore basale nel corso delle 24 ore, senza presentare le caratteristiche di episodio di BTP. EFFICACIA
In medicina per efficacia si intende la (National Cancer Institute website).
NRS (NUMERICAL RATING SCALE) La NRS è uno strumento utilizzato per misurare l’intensità di dolore. Pur esistendo alcune varianti, la più utilizzata è quella a undici punti (0(punteggio) che più rappresenta l’ intensità del loro dolore. Lo zero indica “nessun dolore”; il valore di dieci rappresenta “il massimo dolore immaginabile” (54). OEI % (OPIOID ESCALATION L’Opioid Escalation Index-% è un indicatore della risposta agli oppioidi. Rappresenta l’ entità dell’aumento percentuale medio giornaliero del farmaco, a partire dalla dose iniziale fino alla dose massima raggiunta o, in altri casi, all SEDAZIONE PALLIATIVA/TERMINALE La sedazione palliativa è definita come l’uso di sedativi per alleviare dolori (o altri sintomi) refrattari ad altri trattamenti sintomatici specifici, e consistente nelldel paziente (55). SWITCH DELL’OPPIOIDE Lo “switch” di un oppioide è il processo per cui si cambia l’oppioide in corso con un altro oppioide in modo da raggiungere un’adeguata efficacia del trattamento e/o un’adeguata tollerabilità (56). TERAPIA ADIUVANTE ANALGESICA Secondo il WHO gli adiuvanti sono farmaci che possono contribuire (aumentando l’efficacia dei tre scalini), a ottenere un’attenuazione del dolore nonostante non siano propriamente analgesici (1).
TITOLAZIONE La titolazione ( nella terapia del dolore,) è un metodo per determinare la giusta dose necessaria a ottenere la massima efficacia e i minori effetti collaterali in pazienti che devono cominciare una terapia con oppioidi di 3° scalino (56).
versione 1 del 24.05.2010
53
il dolore che insorge per stimolazione diretta dei nocicettori a causa di un trauma tissutale (funzione di allarme per un potenziale danno fisico). spesso tale dolore persiste in relazione ai processi infiammatori/riparativi che conseguono al trauma.
GGIUNTIVA”
La dose “rescue” o “di salvataggio” consiste in una dose supplementare di oppioide somministrata “al bisogno” insieme al farmaco utilizzato per il dolore di base
di controllare episodi di BTP. Si intende, invece, dose aggiuntiva di oppioidi un supplemento di farmaco utilizzato in caso di variazioni di dolore basale nel corso delle 24 ore, senza presentare le caratteristiche di episodio di BTP.
In medicina per efficacia si intende la capacità di un trattamento di produrre I benefici attesi (National Cancer Institute website).
ING SCALE)
La NRS è uno strumento utilizzato per misurare l’intensità di dolore. Pur esistendo alcune quella a undici punti (0-10), in cui i pazienti scelgono il numero
(punteggio) che più rappresenta l’ intensità del loro dolore. Lo zero indica “nessun dolore”; il valore di dieci rappresenta “il massimo dolore immaginabile” (54).
TION INDEX PERCENTAGE)
% è un indicatore della risposta agli oppioidi. Rappresenta l’ entità dell’aumento percentuale medio giornaliero del farmaco, a partire dalla dose iniziale fino alla dose massima raggiunta o, in altri casi, alla dose finale di un periodo di follow
/TERMINALE
La sedazione palliativa è definita come l’uso di sedativi per alleviare dolori (o altri sintomi) refrattari ad altri trattamenti sintomatici specifici, e consistente nella riduzione della coscienza
Lo “switch” di un oppioide è il processo per cui si cambia l’oppioide in corso con un altro oppioide in modo da raggiungere un’adeguata efficacia del trattamento e/o un’adeguata
ALGESICA
Secondo il WHO gli adiuvanti sono farmaci che possono contribuire (aumentando l’efficacia dei tre scalini), a ottenere un’attenuazione del dolore nonostante non siano propriamente
La titolazione ( nella terapia del dolore,) è un metodo per determinare la giusta dose necessaria a ottenere la massima efficacia e i minori effetti collaterali in pazienti che devono cominciare una terapia con oppioidi di 3° scalino (56).
versione 1 del 24.05.2010
il dolore che insorge per stimolazione diretta dei nocicettori a causa di un trauma tissutale (funzione di allarme per un potenziale danno fisico). spesso tale dolore persiste in relazione ai
La dose “rescue” o “di salvataggio” consiste in una dose supplementare di oppioide somministrata “al bisogno” insieme al farmaco utilizzato per il dolore di base (53) con
e, invece, dose aggiuntiva di oppioidi un supplemento di farmaco utilizzato in caso di variazioni di dolore basale nel corso delle 24 ore,
capacità di un trattamento di produrre I benefici attesi
La NRS è uno strumento utilizzato per misurare l’intensità di dolore. Pur esistendo alcune 10), in cui i pazienti scelgono il numero
(punteggio) che più rappresenta l’ intensità del loro dolore. Lo zero indica “nessun dolore”; il
% è un indicatore della risposta agli oppioidi. Rappresenta l’ entità dell’aumento percentuale medio giornaliero del farmaco, a partire dalla dose iniziale fino alla
a dose finale di un periodo di follow-up ( 53).
La sedazione palliativa è definita come l’uso di sedativi per alleviare dolori (o altri sintomi) a riduzione della coscienza
Lo “switch” di un oppioide è il processo per cui si cambia l’oppioide in corso con un altro oppioide in modo da raggiungere un’adeguata efficacia del trattamento e/o un’adeguata
Secondo il WHO gli adiuvanti sono farmaci che possono contribuire (aumentando l’efficacia dei tre scalini), a ottenere un’attenuazione del dolore nonostante non siano propriamente
La titolazione ( nella terapia del dolore,) è un metodo per determinare la giusta dose necessaria a ottenere la massima efficacia e i minori effetti collaterali in pazienti che devono

STUDIO CERP versione 1 del 24.05.2010
54
ALLEGATO II
FOGLIO INFORMATIVO PER IL PAZIENTE E MODULO DI CONSENSO INFORMATO ALLO STUDIO
Titolo dello studio: Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone, fentanyl e buprenorfina verso morfina, in pazienti con dolore associato a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica del WHO. Sottostudio: Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati.
FOGLIO INFORMATIVO PER IL PAZIENTE
Questo foglio informativo,
che Le è consegnato con congruo anticipo rispetto alla Sua decisione finale di partecipare allo studio, riporta le informazioni essenziali sullo studio clinico a cui Le viene chiesto di partecipare.
E’ importante che Lei legga queste informazioni e che le discuta con il Suo Medico ed i Suoi familiari prima di firmare il consenso alla partecipazione allo studio.
Gentile Signore/a,
le stiamo chiedendo di partecipare ad uno studio clinico. Prima che Lei decida, è importante che abbia tutte le informazioni sul perché questo studio viene fatto e che cosa comporta. Si prenda tutto il tempo necessario per leggere queste informazioni attentamente. Chieda ai medici che l’hanno in cura qualsiasi informazione o spiegazione su aspetti che non le sono chiari. Se non capisce qualche termine guardi la sezione “Cosa vuol dire”.
Innanzi tutto La informiamo che a promuovere e coordinare questo studio è Il Centro per la Ricerca e lo Studio sul Dolore (CERP) dell’Istituto di Ricerche Farmacologiche “Mario Negri” di Milano (www.marionegri.it). L’Istituto “Mario Negri” è una fondazione senza fini di lucro che si occupa di promuovere ricerca in campo sanitario. Lo studio a cui Le chiediamo di partecipare si propone di valutare e comparare l’efficacia di farmaci antidolorifici oppioidi maggiori per il trattamento del dolore. Per svolgere tale studio è necessaria la collaborazione e la disponibilità di persone che, come Lei, sono affette da dolore di entità medio - elevata.
Questo foglio riporta le informazioni sullo studio a cui Le viene chiesto di partecipare. Le è consegnato in anticipo rispetto alla Sua decisione finale, che dovrà essere libera e pienamente informata. E’ importante per Lei ed i Suoi familiari leggere e discutere queste informazioni con il Suo medico; nel caso decidesse di partecipare, Le verrà chiesto di firmare l’allegato Modulo di Consenso Informato che indica la sua accettazione a partecipare allo studio. Quale è lo scopo dello studio? Nel corso della sua malattia ci sono momenti in cui prova dolore, che influisce in modo negativo sulla qualità della sua vita. Esistono molti farmaci contro il dolore (antidolorifici) ma, nel caso il dolore sia di intensità da moderata a severa, le linee guida internazionali (vedi la sezione “Cosa vuol dire”) raccomandano l’utilizzo dei farmaci oppioidi maggiori. Con il termine oppioidi si indicano quelle sostanze che sono in grado di agire su determinati recettori del

STUDIO CERP versione 1 del 24.05.2010
55
sistema nervoso, detti recettori oppioidi (vedi sezione “Cosa vuol dire”), che riescono a ridurre il dolore. Da recenti studi è emerso che in Italia i farmaci oppioidi maggiori più frequentemente utilizzati sono la morfina orale, l’ossicodone orale, il fentanyl transdermico (in cerotto) e la buprenorfina transdermica (pure in cerotto) e, nonostante abbiano la stessa indicazione terapeutica cioè, “togliere il dolore” e abbiano tutti dimostrato di essere efficaci, non esistono studi comparativi che abbiano verificato l’efficacia resa nella pratica corrente. Obiettivo di questo studio è quindi quello di valutare le caratteristiche, e in particolare l’efficacia (capacità di ridurre il dolore) dei tre farmaci oppioidi maggiori rispetto alla morfina, considerato il farmaco di riferimento in questa categoria.
Quali sono le caratteristiche di questo studio?
Questo studio è: � Randomizzato e controllato – cioè prevede la randomizzazione, un metodo di
assegnazione casuale al trattamento con uno dei farmaci oppioidi maggiori. Il centro di coordinamento dello studio determinerà mediante randomizzazione a quale gruppo verrà assegnato;
� di superiorità – cioè si vuole capire se uno dei trattamenti antidolorifici previsti nello studio è migliore degli altri nel diminuire o togliere il dolore al paziente;
� a 4 bracci di trattamento – cioè sono previsti 4 trattamenti alternativi con gli oppioidi maggiori: morfina orale, l’ossicodone orale, il fentanyl transdermico (in cerotto) e la buprenorfina transdermica ( in cerotto);
� in aperto – cioè, tutte le informazioni inerenti alla terapia farmacologica e non, sono note al paziente e al medico;
� prospettico – cioè i pazienti verranno seguiti per un periodo di tempo (28 giorni) e in quel periodo verranno raccolti i dati;
� multicentrico – cioè, condotto in più istituti o ospedali italiani nello stesso momento; � con follow up di 28 giorni – cioè con un periodo di osservazione degli effetti del farmaco
della durata di 28 giorni. La valutazione prevista dallo studio avrà una durata di quattro settimane ma, anche dopo questo periodo, Lei continuerà ad essere trattato dal Suo medico nella maniera più adeguata per alleviare il dolore.
Il protocollo di questo studio contiene tutte le informazioni riguardo le modalità di svolgimento della raccolta dati ed è stato approvato dal Comitato Etico (un organismo indipendente che ha il compito di tutelare l’interesse del paziente che partecipa ad uno studio clinico) della struttura da cui Lei è in cura.
Perché lo studio è stato proposto a me?
Le stiamo proponendo di partecipare a questo studio perché Lei ha una malattia accompagnata da dolore di intensità medio-elevata, che richiede di somministrare farmaci oppioidi maggiori. E’ previsto che partecipino a questo studio 1000 pazienti curati in circa 80-100 Istituti o Ospedali.
Cosa comporta la mia partecipazione?
Se Lei deciderà di partecipare allo studio verrà assegnato in modo casuale ad uno dei 4 gruppi dei farmaci oppioidi maggiori (morfina o ossicodone orali, fentanyl o buprenorfina transdermici). Non sarà né Lei né il Suo medico a scegliere il trattamento. In tal modo sarà reso possibile un confronto equo tra i farmaci usati e alla fine dello studio saremo in grado di dire se esiste una differenza o no tra i trattamenti. Il Suo medico, comunque, può in qualunque momento modificare la Sua terapia o decidere di prescriverLe un altro farmaco in base alle Sue necessità e ai cambiamenti delle Sue condizioni cliniche.
La partecipazione allo studio prevede che il Suo medico trasmetta i dati relativi 1) Alle Sue caratteristiche cliniche, 2) Alla terapia antidolorifica a cui è sottoposto, 3) All’intensità del

STUDIO CERP versione 1 del 24.05.2010
56
dolore - misurato con un questionario in cui Le verrà chiesto di dare un punteggio al Suo dolore da 0 (nessun dolore) a 10 (massimo dolore immaginabile).
Tutti i dati saranno elaborati in forma anonima: solo il Suo medico e le altre persone da cui è in cura saranno a conoscenza del nome del paziente che partecipa allo studio e saranno in grado di legare il nominativo del paziente alla sua situazione clinica.
Le informazioni sulla condizione clinica e sul tipo di trattamento antidolorifico prescritto saranno comunicati a terzi, seppure in forma anonima, che non siano il medico e il paziente stesso: i dati ricevuti saranno infatti elaborati presso l’Istituto di Ricerche Farmacologiche Mario Negri di Milano.
Solo i pazienti che hanno accettato e firmato il consenso informato partecipano allo studio e il paziente che inizialmente accetta di partecipare può comunque ritirare il suo consenso in ogni momento senza dover dare alcuna spiegazione.
Quali sono i vantaggi e gli svantaggi a partecipare allo studio?
In base alle attuali conoscenze i 4 farmaci in studio sono considerati ugualmente efficaci nel controllo del dolore. La partecipazione non comporterà per Lei nessuna nuova conoscenza specifica sul Suo stato di salute né su altri aspetti del trattamento antidolorifico rispetto alle informazioni che Le verranno comunque date dal Suo medico nel contesto del Suo trattamento. La Sua partecipazione a questo studio permetterà di ottenere informazioni scientifiche che potranno in futuro migliorare il trattamento del dolore.
Chi promuove e coordina lo studio? Lo studio è promosso e coordinato dal Centro per la Ricerca e lo Studio del Dolore (C.E.R.P.) dell’Istituto di Ricerche Farmacologiche “Mario Negri” di Milano e fa parte di un progetto più ampio che ha l’obiettivo di aumentare le conoscenze sul trattamento del dolore cronico. E’ l’Istituto di Ricerche Farmacologiche “Mario Negri” che mette a disposizione i fondi per questo studio.
Garanzie a tutela del paziente partecipante allo studio
La partecipazione dei pazienti allo studio è totalmente libera. Questo significa che essi possono liberamente decidere di non partecipare allo studio, e dunque di non firmare il consenso, senza compromettere in nulla l’assistenza che riceveranno in seguito dal proprio medico. Inoltre, i pazienti che inizialmente accettano di partecipare allo studio possono in seguito ritirare in ogni momento il loro consenso senza dover dare alcuna spiegazione. Lei non avrà nessun costo da sostenere per poter partecipare allo studio.
Assicurazione
Tutti i partecipanti allo studio sono coperti da una assicurazione che copre tutti i rischi inerenti alla partecipazione dello studio in accordo alle leggi e alle norme vigenti.
Confidenzialità delle informazioni e tutela della privacy
Quando il paziente firma il proprio consenso a partecipare allo studio, esprime anche il suo consenso al fatto che informazioni sanitarie che lo riguardano vengano utilizzate per gli scopi suddetti e vengano visionate da tutti coloro che sono coinvolti nell’effettuazione dello studio (personale sanitario, personale che elabora i dati, personale ispettivo e quant’altri abilitati dal protocollo di studio e/o dalle normative vigenti).
Tutte le informazioni (personali, cliniche) raccolte durante questo studio sono confidenziali e verranno trattati nel rispetto della normativa vigente, ai sensi del del D.L. n° 196 del 30 giugno 2003 in materia di diritto alla riservatezza dei dati personali e della Deliberazione n° 52 del 24 luglio 2008 in materia delle linee guida per i trattamenti di dati personali nell’ambito delle sperimentazioni cliniche.

STUDIO CERP versione 1 del 24.05.2010
57
Tutte le informazioni saranno eventualmente pubblicate solo in forma anonima e nel loro complesso, evitando accuratamente e rigorosamente qualunque dettaglio che possa in qualche modo consentire a terzi di risalire all’identità del paziente.
Il titolare del trattamento dei dati è l’Istituto di Ricerche Farmacologiche Mario Negri di Milano.
Al titolare del trattamento Lei potrà rivolgersi per far valere i Suoi diritti, così come previsto dall’articolo 7 del Decreto legislativo 196/2003.
Per ogni necessità o domanda, Lei potrà inoltre rivolgersi al Suo medico, che avrà cura di fornirLe ogni ulteriore chiarimento in merito.
Una copia di questo Modulo Informativo e una copia dell’eventuale Consenso Informato restano in possesso del paziente che accetti di partecipare allo studio.
Referente
Referente dello studio è il Professor Silvio Garattini, Istituto di Ricerche Farmacologiche Mario Negri, via Giuseppe La Masa 19, 20156 Milano.
Contatti: Telefono 02 39014519; email: [email protected]
“Cosa vuol dire”:
Linee guida internazionali Le linee guida sono "raccomandazioni di comportamento clinico, elaborate mediante un processo di revisione sistematica della letteratura e delle opinioni di esperti, con lo scopo di aiutare i medici e i pazienti a decidere le modalità assistenziali più appropriate in specifiche situazioni cliniche".
Revisione sistematica Valutazione di tutti gli studi disponibili su un determinato argomento. Il termine sistematica si riferisce al fatto che la revisione deve essere pianificata come un vero e proprio studio preparando un protocollo che esplicita: obiettivi della revisione, modalità di ricerca degli studi, reperimento, valutazione critica e sintesi di tutti gli studi eleggibili per la revisione. Studio clinico controllato e randomizzato Studio in cui i partecipanti sono assegnati in modo casuale (randomizzato) a ricevere il trattamento sperimentale o il trattamento di controllo. Quando gli studi clinici controllati e randomizzati sono condotti in modo appropriato, l'effetto dei trattamenti può essere studiato in gruppi di persone che sono simili all'inizio e trattati allo stesso modo, eccetto che per l'intervento in studio. Quindi qualsiasi differenza vista alla fine nei gruppi può essere attribuita esclusivamente al trattamento e non a errori sistematici o al caso. Randomizzazione Processo di assegnazione casuale dei partecipanti a uno dei bracci di uno studio. La randomizzazione ha lo scopo di rendere simili i gruppi per le loro caratteristiche. Questo permette l'applicabilità dei modelli probabilistici sui quali si regge dal punto di vista metodologico l'intero studio. Recettore per gli oppioidi Proteina della membrana cellulare con grande affinità per i farmaci oppioidi. Il recettore ha un ruolo fondamentale per l’azione del farmaco.

STUDIO CERP versione 1 del 24.05.2010
58
CONSENSO INFORMATO ALLA PARTECIPAZIONE ALLO STUDIO
Questo modulo, deve essere firmato da Lei solo nel caso decida di partecipare allo studio clinico generale.
E’ importante che Lei abbia discusso approfonditamente con il Medico prima di firmare questo consenso, anche sulla base del foglio informativo a cui esso si riferisce
Partecipano allo studio solo i Pazienti che accettano. Il Paziente può ritirare il suo consenso in ogni momento.
Io sottoscritto/a, ________________________________nato/a a ______________________ (prov. ____)
il ___/___/_____ residente in ___________________________________________________ (prov. ____)
nella mia piena capacità di intendere e di volere: � Confermo di essere stato/a dovutamente informato/a circa lo studio e aver avuto tempo
sufficiente per chiedere ulteriori informazioni e per considerare la mia partecipazione ad esso.
� Ho ricevuto una copia del Foglio Informativo per il paziente.
� Tutti i miei diritti mi sono stati spiegati chiaramente.
� Sono consapevole che la partecipazione a questo studio è del tutto volontaria e che sarò libero/a di accettare o rifiutare di prenderne parte, come pure di ritirare il mio consenso in un secondo momento, senza la necessità di motivare la mia decisione e senza influenzare, per questo, la qualità e l’adeguatezza delle successive decisioni terapeutiche sulla mia persona.
� Sono consapevole e acconsento che tutte le informazioni (personali, cliniche) raccolte durante lo studio siano considerate confidenziali e vengano trattate nel rispetto della normativa vigente ai sensi del D.L. n° 196 del 30 giugno 2003 in materia di diritto alla riservatezza dei dati personali e della Deliberazione n° 52 del 24 luglio 2008 in materia delle linee guida per i trattamenti di dati personali nell’ambito delle sperimentazioni cliniche di medicinali
� Acconsento a partecipare allo studio clinico suddetto
Nome del Paziente: ______________________________
Firma del Paziente: _____________________________ Data: _________________
Nome del Medico: ______________________________
Firma del Medico: ______________________________ Data: _________________

STUDIO CERP versione 1 del 24.05.2010
59
FOGLIO INFORMATIVO PER IL PAZIENTE E
MODULO DI CONSENSO INFORMATO AL PRELIEVO BIOLOGICO
Sottostudio: Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati.
FOGLIO INFORMATIVO PER IL PAZIENTE
Gentile Signore/a,
le stiamo chiedendo anche di partecipare ad un sottostudio ovvero uno studio più breve all’interno dello studio clinico precedentemente descritto. In questo sottoprogetto Le chiediamo di donare un Suo campione biologico (sangue). Prima che Lei decida, è importante che abbia tutte le informazioni anche riguardo al sottostudio. Si prenda tutto il tempo necessario per leggere queste informazioni attentamente. Chieda ai medici che l’hanno in cura qualsiasi informazione o spiegazione su aspetti che non le sono chiari. Nel caso Lei acconsentisse a partecipare, Le verrà chiesto di firmare l’allegato Modulo di Consenso Informato per il sottostudio che indica la sua accettazione. La ringraziamo per l’attenzione che vorrà concederci.
Quale è lo scopo di questo sottostudio?
Lo scopo del sottostudio è di valutare possibili correlazioni esistenti tra gli effetti clinici osservati per l’effetto della terapia con gli oppioidi maggiori, e l’assetto genico dei pazienti ovvero la loro struttura genetica. E’ stato infatti ipotizzato da precedenti studi che, tra i vari fattori, un ruolo nella risposta ai farmaci oppioidi sia dovuto alla componente genetica di ogni persona.
Cosa comportala donazione di un Suo campione biologico?
Significa che dovrà sottoporsi ad unico prelievo di sangue per via venosa, che verrà inviato e conservato presso la Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, dove saranno effettuate le analisi. Significa anche che dovrà parallelamente fornire informazioni anagrafiche e cliniche, rientranti nello studio in generale, che saranno usate per valutare e studiare l’eventuale ruolo di particolari fattori genetici nella risposta clinica alla terapia antidolorifica con oppioidi.
Tutti i risultati ottenuti dalle analisi genetiche, le informazioni anagrafiche e cliniche, così come ogni atto medico, sono da considerarsi strettamente confidenziali e sottoposti al vincolo del segreto professionale con la garanzia della massima riservatezza.
Quali rischi comporta il prelievo di sangue?
Il prelievo di sangue potrebbe causare un momentaneo e lieve malessere ed un eventuale livido nella sede del prelievo. Per il resto, se il prelievo è correttamente eseguito, non si prevedono altri effetti.

STUDIO CERP versione 1 del 24.05.2010
60
Quali significati e riscontri deriveranno dal prelievo di sangue e dallo studio in generale?
I risultati di questo studio potrebbero avere ripercussioni per la cura dei pazienti che presentano dolore, sia per capire meglio i meccanismi che determinano l’effetto antidolorifico dei farmaci oppioidi, sia, eventualmente, per la programmazione in futuro di una terapia farmacologica mirata. Per tale ragione si richiede la Sua collaborazione.
Come verrà utilizzato il sangue che mi verrà prelevato?
Il campione di sangue verrà conservato presso la Fondazione IRCCS Istituto Nazionale dei Tumori di Milano fino all’utilizzo per la preparazione del materiale genetico (DNA). Il campione del Suo sangue verrà utilizzato esclusivamente per l’estrazione del DNA. Il DNA ottenuto verrà poi utilizzato per confrontare la risposta ai trattamenti antidolorifici con oppioidi maggiori con l’assetto genetico e, una volta utilizzato, il campione verrà conservato per tutta la durata dello studio fino a due anni successivi alla sua conclusione, in modo da eseguire eventuali analisi di conferma ed eventuali ripetizioni del test, o verrà distrutto, se da Lei richiesto, secondo le procedure standard di smaltimento dei rifiuti biologici in atto nel laboratorio.
Garanzie a tutela del paziente che aderisce al sottostudio
Quando il paziente firma il proprio consenso a partecipare al sottostudio, esprime anche il suo consenso al fatto che informazioni sanitarie che lo riguardano vengano utilizzate per gli scopi suddetti e vengano visionate da tutti coloro che sono coinvolti nell’effettuazione dello studio (personale sanitario, personale che elabora i dati, personale ispettivo e quant’altri abilitati dal protocollo di studio e/o dalle normative vigenti).
Il Suo campione di sangue e le informazioni anagrafiche e cliniche saranno usate solo ed esclusivamente per questo studio.
Tutte le informazioni (personali, cliniche) raccolte durante questo studio sono confidenziali e verranno trattate nel rispetto della normativa vigente ai sensi del del D.L. n° 196 del 30 giugno 2003 in materia di diritto alla riservatezza dei dati personali e della Deliberazione n° 52 del 24 luglio 2008 in materia delle linee guida per i trattamenti di dati personali nell’ambito delle sperimentazioni cliniche.
Tutte le informazioni saranno eventualmente pubblicate solo in forma anonima e nel loro complesso, evitando accuratamente e rigorosamente qualunque dettaglio che possa in qualche modo consentire a terzi di risalire all’identità del paziente
L’adesione alla donazione del proprio campione di sangue è totalmente libera. Questo significa che Lei può liberamente decidere di non fornire il Suo consenso, senza compromettere in nulla l’assistenza che riceverà in seguito da parte del Suo medico. Inoltre, i pazienti che hanno comunque dato il proprio consenso possono ritirarlo in qualsiasi momento senza dover dare nessuna spiegazione.
E’ anche possibile che Lei dia il Suo consenso per partecipare allo studio clinico ma non lo dia per eseguire il prelievo di sangue.
L’adesione alla donazione del campione di sangue non comporta costi aggiuntivi.
Assicurazione
Tutti i partecipanti allo studio sono coperti da una assicurazione che copre tutti i rischi inerenti alla partecipazione dello studio in accordo alle leggi e alle norme vigenti.

STUDIO CERP versione 1 del 24.05.2010
61
Confidenzialità delle informazioni e tutela della privacy
Il titolare del trattamento dei dati è la Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Via Giacomo Venezian 1, 20133 Milano.
Al titolare del trattamento Lei potrà rivolgersi per far valere i Suoi diritti, così come previsto dall’articolo 7 del Decreto legislativo 196/2003.
Per ogni necessità o domanda, Lei potrà inoltre rivolgersi al Suo medico, che avrà cura di fornirLe ogni ulteriore chiarimento in merito.
Una copia di questo Foglio Informativo per il sottostudio e una copia dell’eventuale Consenso Informato a partecipare al sottostudio restano in possesso del paziente che accetti di partecipare al sottostudio.
Referente
Referente per l’analisi del materiale genetico è il Dott. Augusto Caraceni, Fondazione IRCCS Istituto Nazionale dei Tumori di Milano, Via Giacomo Venezian 1, 20133 Milano,
Contatti: tel. 02 23902792; email: [email protected].
Ulteriori informazioni e chiarimenti sullo studio sono disponibili ai siti:
http://crc.marionegri.it/cancerpain/, dove viene presentato il più ampio progetto di ricerca di cui questo studio fa parte e gli ospedali partecipanti.
http://www.partecipasalute.it, dove può trovare una sezione dedicata agli studi clinici e un glossario dove sono spiegati i termini tecnici degli studi.
Dettagli per contattare:
il Suo medico.............……………………………………………… Tel…………………....…....
la Sua infermiera ………………………………………………....... Tel…………………........…
Grazie per aver preso in considerazione questo studio
Data presa visione del paziente …………………………………………………………
Firma per presa visione del paziente …………………………………………………………

STUDIO CERP versione 1 del 24.05.2010
62
CONSENSO INFORMATO AL PRELIEVO DI MATERIALE BIOLOGICO
Io sottoscritto/a, ___________________________________________________________
nato/a a ______________________________ prov. ________il ___/___/_____
residente in ___________________________________________________prov. ____
nella mia piena capacità di intendere e di volere dichiaro:
di essere stato/a messo/a a conoscenza, in modo chiaro e comprensibile, degli scopi del sottostudio ed, in particolare (barrare la voce scelta):
di volermi sottoporre ad un prelievo di sangue venoso per valutare la possibile correlazione tra assetto genetico e risposta clinica al trattamento antidolorifico con farmaci oppioidi.
���� acconsento ���� non acconsento Dichiaro infine di essere a conoscenza che la mia adesione allo studio è volontaria e mi è consentito ritirarla in qualsiasi momento senza dover fornire spiegazioni e che le notizie acquisite saranno assolutamente riservate.
Ho ricevuto copia del foglio informativo per il prelievo di materiale biologico.
Sono consapevole e acconsento che tutte le informazioni (personali, cliniche) raccolte durante lo studio siano considerate confidenziali e vengano trattate nel rispetto della normativa vigente ai sensi del D.L. n° 196 del 30 giugno 2003 in materia di diritto alla riservatezza dei dati personali e della Deliberazione n° 52 del 24 luglio 2008 in materia delle linee guida per i trattamenti di dati personali nell’ambito delle sperimentazioni cliniche di medicinali.
Tutti i dati raccolti (personali e clinici) potranno essere utilizzati nel futuro per la ricerca scientifica sul cancro. La confidenzialità di questi dati sarà assicurata.
Nome del Paziente: ___________________________________
Firma del Paziente: ___________________________________ Data: _________________
Nome del Medico: ___________________________________
Firma del Medico: ___________________________________ Data: _________________

STUDIO CERP versione 1 del 24.05.2010
63
ALLEGATO III
LETTERA DI INFORMAZIONE PER IL MEDICO DI MEDICINA GENERALE
TITOLO DELLO STUDIO
Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone, fentanyl e buprenorfina verso morfina, in
pazienti con dolore associato a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica del WHO.
TITOLO DEL SOTTOPROGETTO
Valutazione, in parallelo, dell’assetto genico dei pazienti e delle possibili correlazioni con gli effetti clinici osservati.
Gentile Collega, con la presente vogliamo informarti che il Sig./la Sig.ra ……………………………………………………………
ha dato il proprio consenso a partecipare allo studio CERP un clinical trial randomizzato (RCT), di superiorità, a 4 bracci di trattamento, in aperto, di fase IV, prospettico, multicentrico e, solamente se ha firmato il Consenso Informato predisposto, anche al sottoprogetto.
DESCRIZIONE DELLO STUDIO Nella malattia neoplastica il dolore è un sintomo con grave impatto negativo sulla qualità di vita dei malati e ad elevata incidenza, con valori intorno al 70-90% nelle fasi avanzate e metastatiche. Da oltre 20 anni il principale riferimento per il trattamento farmacologico del dolore da cancro sono le linee-guida prodotte dalla Organizzazione mondiale della Sanità (WHO). In tale documento emerge che l’uso dei farmaci oppioidi rappresenta il punto cardine del trattamento, con particolare riferimento agli oppioidi “maggiori” (3° scalino della scala analgesica). I 4 oppioidi maggiori più comunemente prescritti in Italia (morfina e ossicodone orali, fentanyl e buprenorfina transdermici), in base ai dati al momento disponibili, presentano un effetto analgesico parzialmente sovrapponibile ma con differenze rispoetto ad alcuni indicatori come la necessità di cambaire il farmaco durante al terapia, la entità di aggiustamenti di dose, le percentuali diverse di soggetti classificabili come non-responders (NR). Le osservazioni descritte inducono a pensare che gli oppioidi, pur appartenendo alla stessa famiglia farmacologica, potrebbero essere non pienamente sovrapponibili per quanto riguarda gli effetti clinici prodotti. Importanti differenze sono note sul piano farmacocinetico e farmacodinamico e, più recentemente, anche in termini di farmacogenomica. Alcuni studi pubblicati hanno evidenziato l’esistenza di variazioni geniche responsabili di anomalie riguardanti il trasporto delle molecole attraverso la barriera ematoencefalica (BEE), la struttura del recettore per gli oppioidi e il sistema enzimatico di metabolismo. Assai limitati, invece, sono gli studi che analizzano i comportamenti degli oppioidi in rapporto all’intero genoma umano (Genome-Wide Association Studies: GWAS). A partire da tutto ciò, sulla abs eanche di numerosi sutid e valutazioni pubblicati dal Fruppo Proponennete nell’arco degli ultimi 5 anni, si è stabilito di proporre uno studio comparativo tra strategie analgesiche basate sull’impiego dei 4 oppioidi citati, che vada a cercare le possibili differenze in termini di efficacia analgesica, di variazioni di dosi nel tempo, di ricorso a switch, o di abbandono definitivo del trattamento, parallelamente al profilo degli effetti indesiderati. L’associato sottoprogetto metterà in relazione l’assetto genico dei pazienti e i risultati clinici emersi. OBIETTIVO DELLO STUDIO L’obiettivo primario dello studio è quello di comparare la efficacia analgesica di 4 diverse strategie analgesiche basate su 4 diversi farmaci oppioidi del 3° scalino della scala WHO (la morfina, utilizzata come comparatore, sarà confrontata con buprenorfina, fentanyl e ossicodone) in pazienti con dolore correlato a tumore, di intensità moderata-severa [intensità del dolore medio, riferita alle precedenti 24 ore, uguale o superiore a 4, utilizzando una scala numerica di valutazione a 11 punti (0-10 NRS)]. La valutazione verrà condotta utilizzando una serie di endpoint primari e secondari di efficacia analgesica e di sicurezza. CARATTERISTICHE DEI PAZIENTI COINVOLTI La popolazione in studio sarà rappresentata da pazienti con cancro in fase avanzata per presenza accertata di metastasi o per importante progressione locale, che presentano, alla 1° visita, valori di

STUDIO CERP versione 1 del 24.05.2010
64
dolore medio provato nelle ultime 24 ore ≥ 4, misurato utilizzando una NRS, Numerical Rating Scale, a 11 punti (0-10). Questi pazienti possono aver precedentemente ricevuto o nessun trattamento antalgico o farmaci del 1° e/o del 2° scalino del WHO (paracetamolo, FANS, codeina, tramadolo). Al momento di inizio dello studio, comunque, necessitano di un trattamento con 3° scalino. Lo studio coinvolgerà pazienti: 1. con diagnosi (istologica o citologica) di tumore solido in fase avanzata locale e/o metastatica; 2. con presenza di dolore medio giornaliero ≥4 misurato con NRS e riferito alle ultime 24 ore,
attribuibile al tumore, necessitante per la prima volta di un trattamento con oppioidi del 3°scalino/WHO;
3. con aspettativa stimata di vita superiore a un mese; 4. che non hanno precedentemente eseguito, anche saltuariamente, trattamenti con oppioidi del 3°
scalino sia ATC sia rescue; 5. in grado di assumere i farmaci in studio, sia per via orale sia transdermica sia sottocutanea; 6. di età pari o superiore a 18 anni.
SCHEMA TERAPEUTICO Dopo la randomizzazione i pazienti entreranno nello studio seguendo lo schema posologico di riferimento di seguito descritto. • Morfina orale SR: 60 mg/24 ore • Fentanyl TD: 25 mg/h • Buprenorfina TD: 35 mg/h • Ossicodone orale SR: 49 mg/24 ore DURATA DELLO STUDIO: Lo studio ha la durata totale di un anno. Questo periodo prevede una fase di reclutamento della durata massima di 11 mesi e un follow-up di 28 giorni dove, oltre alla visita basale, saranno condotte altre 5 visite di controllo: dopo 72 ore e ai giorni 7, 14, 21 e 28.
N° CENTRI E PAZIENTI RICHIESTI: circa 80 centri recluteranno 1008. Il reclutamento medio per centro previsto è di 12 pazienti. Lo studio ha la durata totale di un anno. Il periodo di reclutamento è indicato in 11 mesi e il periodo di follow-up avrà una durata di 28 giorni e prevederà, oltre alla visita basale, altre 5 visite di controllo: dopo 72 ore e ai giorni 7, 14, 21 e 28.
DESCRIZIONE E OBIETTIVI SOTTOPROGETTO Recenti lavori preclinici e clinici hanno evidenziato l’esistenza di una serie di geni e di varianti geniche in grado di influenzare, in modo più o meno consistente, l’azione farmacologica e clinica dei farmaci oppioidi. In particolare, è stata rilevata l’esistenza di alcuni geni responsabili di anomalie strutturali delle proteine coinvolte nel trasporto delle molecole oppioidi attraverso la barriera ematoencefalica (BEE), nella struttura del recettore per gli oppioidi e nel sistema enzimatico inerente il metabolismo epatico degli stessi. Si tratta, indiscutibilmente, di punti nevralgici dell’attività degli oppioidi, ma una recente review ha ridimensionato l’impatto clinico di alcune di queste varianti. Il sottoprogetto si propone di analizzare l’assetto genetico dei pazienti attraverso un’ indagine “genome-wide”. La mappatura dell’intero genoma di ogni singolo paziente inserito nel sottoprogetto consentirà di valutare: • eventuali correlazioni tra risultati clinici e assetto genetico dei pazienti • eventuali indicazioni di tipo prognostico-predittivo sulla possibile risposta ai farmaci oppioidi in
relazione alle caratteristiche genetiche.
Il promotore di questo studio è il CERP, il Centro per la ricerca e Valutazione del Dolore dell’’Istituto di Ricerche Farmacologiche Mario Negri una fondazione eretta a ente morale con sede a Milano, che opera nel campo della ricerca biomedica senza fini di lucro.
RingraziandoLa per l’attenzione, invio cordiali saluti.
Dr. Oscar Corli Responsabile Scientifico del Progetto

STUDIO CERP versione 1 del 24.05.2010
65
ALLEGATO IV
ELENCO DEI CENTRI PARTECIPANTI
OSPEDALE U.O. CITTA' REGIONE
1 ASL Pescara
U.O. Terapia del dolore Cure Palliative Hospice Bouganville P.O. Pescara Pescara Abruzzo
2 ASL CHIETI Hospice D.S.B. Chieti Centro Chieti Abruzzo
3 Ospedale San Carlo U.O. Oncologia Medica Potenza Basilicata
4 A.O. Bianchi-Melacrino-Morelli Oncologia Medica Reggio Calabria Calabria
5 ASL SA1
Assistenza Domiciliare Oncologica ex ASLSA1
Nocera Inferiore (SA) Campania
6 Istituto Nazionale Tumori G. Pascale S.S.D. Terapia Antalgica Napoli Campania
7 A.O. Monaldi
U.O.S. Terapie di Supporto del Cancro del Polmone Malattie Respiratorie SUN Napoli Campania
8 A.O.R.N. Cardarelli Divisione di Oncologia Napoli Campania
9 Ospedale Sacro Cuore - Fatebenefratelli U.O.S. Terapia Antalgica Benevento Campania
10 ASL SALERNO - Distr. EBOLI U.O. Medicina del Dolore e Cure Palliative Eboli Campania
11 A.O. Monaldi Dipartimento Pneumologia Napoli Campania
12 A.O. S.Anna e S.Sebastiano UOC Fisiopatologia del dolore e cure palliative Caserta Campania
13 Medicina Indirizzo Oncologico USL Modena Palliative Care Unit Carpi (MO)
Emilia Romagna
14 A.O. Santa Maria Nuova Oncologia Reggio Emilia Emilia
Romagna
15 AO Santa Croce ASL 3 Terapia del Dolore Fano (PU) Emilia
Romagna
16 AUSL Ravenna - Hospice Lugo Dip. Oncoematologico Hospice Ospedaliero Lugo (RA)
Emilia Romagna
17 Ospedale degli Infermi UO Oncologia Rimini Emilia
Romagna
18 A.O.Universitaria di Bologna U. O. Oncologia Medica Bologna Emilia
Romagna
19 Azienda Ospedaliera Modena Terapie Palliative - Hospice Modena Emilia
Romagna
20 A.O. Universitaria Parma U.O. Oncologia Parma Emilia
Romagna

STUDIO CERP versione 1 del 24.05.2010
66
21 Hospice Casa Madonna Uliveto Hospice Montericcio
di Albinea (RE) Emilia
Romagna
22 Ospedale di Piacenza Oncologia Medica Piacenza Emilia
Romagna
23 Associazione Onlus "Via di Natale" Hospice "Via di Natale" Aviano (PN)
Friuli Venezia Giulia
24 Ospedale Belcolle UO Oncologia Viterbo Lazio
25 Policlinico Umberto I Oncologia B Roma Lazio
26 Policlinico Umberto I UOC Ematologia Roma Lazio
27 Policlinico Tor Vergata U.O.S.A. Medicina del dolore Roma Lazio
28 Policlinico Universitario Umberto I
Scienze Anestesiologiche, Medicina Critica, Terapia del Dolore Roma Lazio
29 Policlinico Umberto I DH Oncologia A Roma Lazio
30 Ospedale S. Eugenio Unità di Cure Domiciliari Roma Lazio
31 Ospedali Galliera S.S.D. Cure Palliative Genova Liguria
32 E.O. Ospedali Galliera Oncologia Medica Genova Liguria
33
ASL 3 Genovese Dip. Cure Primarie Attività Distrettuali S.S. Cure Palliative Genova Liguria
34 A.O.U. San Martino U.O.s. Terapia del dolore e cure palliative Genova Liguria
35 Istituto Nazionale per la Ricerca sul Cancro (IST)
S.C. Terapia Antalgica e Riabilitazione Genova Liguria
36 Hospice Fondazione Castellini Onlus Hospice Melegnano Lombardia
37 A.O. San Gerardo Ambulatorio Cure Palliative e Terapia del Dolore Monza Lombardia
38 Istituto Nazionale dei Tumori Hospice Cure Palliative Milano Lombardia
39 Istituto Scientifico San Raffaele Medicina Oncologica Ambulatorio di Oncologia Milano Lombardia
40 Fondazione Salvatore Maugeri (IRCCS) U.O.C. di Oncologia Medica II Pavia Lombardia
41 Fondazione Salvatore Maugeri (IRCCS) U.O. Cure Palliative Pavia Lombardia
42 Ospedale San Martino - FSM U.O. Cure Palliative Mede (PV) Lombardia
43 Fondazione Don Carlo Gnocchi Onlus
Hospice Santa Maria Delle Grazie Monza Lombardia
44 Azienda Ospedaliera G. Salvini U.O.C. Cure Palliative e Medicina del Dolore
Garbagnate Milanese (MI) Lombardia
45 Hospice "Il Nespolo" ASL Provincia di Lecco Hospice Lecco Lombardia

STUDIO CERP versione 1 del 24.05.2010
67
46 Ospedale Buzzi Unità di Cure Palliative Milano Lombardia
47 Fondazione Salvatore Maugeri (IRCCS) Riabilitazione Oncologica Pavia Lombardia
48 ICP - Ospedale Bassini Unità di Cure Palliative e Terapia del Dolore
Cinisello Balsamo (MI) Lombardia
49 Fondazione Salvatore Maugeri (IRRCS) U.O. Oncologia I Pavia Lombardia
50 Ospedali Riuniti Bergamo Oncologia Medica Bergamo Lombardia
51 Istituto Europeo Oncologia Unità Terapie di Supporto e Cure Palliative Milano Lombardia
52 Ospedale Carlo Poma Struttura Dipartimentale Cure Palliative Mantova Lombardia
53 Multimedica U.O.C. Oncologia Sesto San
Giovanni (MI) Lombardia
54 Ospedale S. Anna U.O.C. Cure Palliative - Hospice Como Lombardia
55 A. O.Valtellina Valchiavenna Unità Cure Palliative Morbegno (SO) Lombardia
56 Policlinico S. Matteo Terapia del dolore Anestesia e Rianimazione I Pavia Lombardia
57 Ospedale di Melegnano Dipartimento di Oncologia Melegnano (MI) Lombardia
58 Ospedale San Paolo S.S. Unità Cure Palliative Milano Lombardia
59 Fondazione Salvatore Maugeri (IRCCS)
Unità Cure Palliative e terapia del dolore Pavia Lombardia
60 Ospedale di Macerata zona terr. 9 ASUR Marche
Terapia del dolore e cure palliative Macerata Marche
61 Osp S.Giovanni Antica Sede Oncologia Medica Torino Piemonte
62 Ospedale S. Andrea - ASL Vercelli Oncologia Medica Vercelli Piemonte
63 AOU San Luigi Gonzaga S.C.D.U. Oncologia Medica Orbassano (TO) Piemonte
64 A.O.U. San Giovanni Battista Oncologia Medica I Torino Piemonte
65 Polo Oncologico di Cuneo S.C. Cure palliative - ASL CN 1 Cuneo Piemonte
66 ASL TO4 Piemonte Servizio Cure Palliative Ivrea Piemonte
67 ASL BA U.O. Cure Palliative Monopoli (BA) Puglia
68 Istituto Nazionale dei Tumori "G.Paolo II"
U.O. Medicina del dolore e cure palliative Bari Puglia
69 Ospedale A. Businco Oncologia Medica Cagliari Sardegna
70 AOU Policlinico G. Martino U.O.C. Oncologia Medica e Hospice Messina Sicilia

STUDIO CERP versione 1 del 24.05.2010
68
71
Ospedale S.Marta A.O.U. Policlinio Vittorio Emanuele Medicina del Dolore Catania Sicilia
72 ASP 9 - Trapani Hospice "Raggio di Sole" Trapani Sicilia
73 CCD Morgagni U.O. Oncologia Medica Catania Sicilia
74 Casa di cura La Maddalena Oncologia Medica Palermo Sicilia
75 AUSL 12 Viareggio Unità di Cure Palliative Viareggio (LU) Toscana
76 Azienda USL 8 Medicina del dolore e Cure Palliative Arezzo Toscana
77 A. O. Universitaria Careggi SC Oncologia Medica Firenze Toscana
78 ASL 1 UMBRIA U.O. Oncologia ASL 1 UMBRIA Città di Castello
(PG) Umbria
79 ULSS 12 VENEZIANA U.O. Oncologia Medica Mestre (VE) Veneto
80 Ospedale di Mirano ASL 13 Veneto
U.O.C. Oncologia e ematologia oncologica Mirano (VE) Veneto
81 Ospedale S. Bortolo ULSS 6 Vicenza
U.O. Terapia del dolore e cure Palliative Vicenza Veneto
82 IOV - Padova Oncologia Medica I Padova Veneto

STUDIO CERP versione 1 del 24.05.2010
69
ALLEGATO V
SCHEDE RACCOLTA DATI

Versione 1.1 (24 mag 2010)
ISTITUTO DI RICERCHE FARMACOLOGICHE MARIO NEGRI
Via Giuseppe La Masa, 19 - 20156 Milano MI - Italy - www.marionegri.it
tel +39 02 39014.1 - fax +39 02 354.6277 - [email protected]
Centro n° Paziente n° Studio CERP1
Studio clinico randomizzato e controllato, in aperto, per comparare l’efficacia analgesica di percorsi terapeutici effettuati con ossicodone,
fentanyl e buprenorfina verso morfina, in pazienti con dolore associato a cancro di intensità moderata-severa, a partire dal momento in cui iniziano il trattamento con 3° scalino della scala analgesica WHO.
Valutazione, in parallelo, dell’assetto genico dei pazienti e delle
possibili correlazioni con gli effetti clinici osservati
SCHEDA RACCOLTA DATI
TITOLO DELLO STUDIO
Studio CERP1

Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Se il soggetto ha aderito anche alla richiesta del prelievo biologico, far
firmare l’apposito modulo del consenso informato scritto e riportare qui
di fianco la data della firma:...........................................................................
1. Partecipazione ad altri progetti di ricerca che sono in conflitto o potrebbero confondere i risultati dello studio............
2. Assenza del consenso informato per la partecipazione allo studio...................................................................................
3. Presenza di condizioni patologiche mentali o psichiatriche, dovute al tumore o a patologie concomitanti, che interfe-riscono con lo stato di coscienza e con la capacità di giudizio al punto da compromettere il rispetto del protocollo di studio.................................................................................................................................................................................
4. Uso di trattamenti per comorbidità presenti all’inizio dello studio che potrebbero creare interazioni potenzialmente pericolose con gli oppioidi (ad esempio: antifungini conazolici o antibiotici macrolidi)................................................
5. Controindicazioni di qualsiasi natura all’uso dei farmaci oppiopidi................................................................................
6. Utilizzo di farmaci oppiodi combinati, nelle loro formulazioni commerciali, con altre molecole (paracetamolo, naloxonici, ...)..........................................................................................................................................
7. Storia pregressa o in atto di abuso di sostanze stupefacenti.............................................................................................
8. Impossibilità a garantire la regolarità del follow-up........................................................................................................
9. Necessità di inizio del 3° scalino in una situazione di “emergenza” per quanto riguarda l’intensità e la gravità del dolore, che richieda, ad esempio, un intervento di immediato effetto (trattamenti endovenosi) tale da non consentire i tempi necessari per l’ottenimento della randomizzazione e per il successivo inizio della terapia secondo il protocollo
10. Diagnosi di neoplasia primitiva cerebrale o di leucemia (acuta o cronica)......................................................................
11. Diagnosi di insufficienza renale cronica già in atto (49)..................................................................................................
12. Esecuzione di un ciclo di radioterapia o di terapia radio metabolica a scopo antalgico, in corso al momento di inizio o concluso da meno di 14 giorni o già programmato entro le 4 settimane dello studio...................................................
13. Esecuzione di un ciclo di chemioterapia di 1° linea contemporaneamente all’inizio dello studio, con intervallo di tempo variabile da 7 giorni prima, a 7 giorni dopo la prima visita..................................................................................
14. Esecuzione concomitante di trattamenti antalgici con tecniche neurochirurgiche/ablative, o mediante neuro stimola-
zione midollare, o con tecniche anestesiologiche loco-regionali (incluse analgesia peridurale o spinale), o mediante
ricorso a vertebro/cifoplastica o altre tecniche invasive con rilevanza sul dolore...........................................................
gg mm aaaa
CONSENSI INFORMATI
Criteri di inclusione 1. Diagnosi istologica o citologica di tumore solido in fase avanzata locale e/o metastatica..............................................
2. Dolore medio giornaliero ≥ 4 misurato con NRS e riferito alle ultime 24 ore, attribuibile al tumore, necessitante per
la prima volta di un trattamento con oppiodi del 3° scalino/WHO..................................................................................
3. Aspettativa di vita superiore ad un mese..........................................................................................................................
4. Il paziente non ha precedentemente eseguito, anche saltuariamente, trattamenti con oppioidi del 3° scalino sia ATC sia rescue.........................................................................................................................................................................
5. Il paziente è in grado di assumere i farmaci in studio, sia per via orale, sia transdermica e sia sottocutanea.................
6. Età pari o superiore ai 18 anni...........................................................................................................................................
Criteri di esclusione
Se il soggetto è eleggibile, si prega di informarlo esaustivamente sulle
finalità dello studio di far firmare l’apposito modulo del consenso infor-
mato scritto e di riportare qui di fianco la data della firma:......................
Eleggibilità e randomizzazione
Data di nascita gg mm aaaa
1. Il paziente è arruolabile solo se ha un’età anagrafica ≥ 18aa
Sesso 2.
No Sì [0] [1]
No Sì [0] [1]
3.
4.
Maschio Femmina [1] [2]
A questo punto richiedere l’assegnazione del farmaco oppioide di partenza e il dosaggio di riferimento, pre-
mendo il pulsante .........
5.
6.
gg mm aaaa
1

2
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
Data rilevazione gg mm aaaa
1.
DATI ANAMNESTICI (1 di 3)
Data della prima diagnosi del tumore primitivo 2 mese anno
Sede del tumore primitivo 4.
Polmone e bronchi..................
Colon e retto............................
Mammella...............................
Prostata....................................
Pancreas..................................
Apparato urinario....................
Stomaco ed esofago.............................
Capo e collo.........................................
Ginecologici.........................................
Fegato e vie biliari...............................
Altra sede/Sede non nota (specificare)
__________________________________________________
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
Presenza di metastasi 6.
Epatica..............................
Polmonare.........................
Ossea.................................
Cerebrale..........................
Addominale......................
Linfonodale......................
se Sì, indicare le sedi:
Altra sede (specificare)....
No Sì [0] [1]
Tumore a prevalente progressione locale 7.
6-1.
6-2.
6-3.
6-4.
6-5.
6-6.
6-7.
Il paziente ha eseguito terapie anti-neoplastiche pregresse ?
Chirurgia..............
Chemioterapia......
Farmaci biologici
Ormonoterapia.....
Radioterapia........
Altre terapie........
No Sì [0] [1] se Sì, indicare quali:
8.
3.
5.
6-8. _____________________________________________
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
8-1.
8-2.
8-3.
8-4.
8-5.
8-6.

3
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
DATI ANAMNESTICI (2 di 3)
Il paziente presenta altre patologie non tumorali concomitanti ?
se Sì, indicare le principali:
No Sì [0] [1] 1.
TERAPIE ANTALGICHE IN CORSO PRIMA DELL’INIZIO DELLO STUDIO
Terapia ATC secondo la scala WHO
2. Nessuna terapia (WHO 0)....
Terapia 1° scalino (WHO 1)
Terapia 2° scalino (WHO 2)
Principio attivo
3.
4.
Altre terapie mirate al controllo del dolore
6. Steroidi.................................
Anticonvulsivanti.................
Antidepressivi.......................
7.
8.
Bifosfonati............................ 9.
Altri adiuvanti analgesici...... 10.
5. Nessuna terapia....................
No Sì [0] [1]
Principio attivo No Sì [0] [1]
1-1.
1-2.
1-3.
3-1.
4-1.
4-2.
6-1.
7-1.
8-1.
9-1.
10-1.

4
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
DATI ANAMNESTICI (3 di 3)
ALTRE TERAPIE NON ANTALGICHE IN CORSO PRIMA DELL’INIZIO DELLO STUDIO
1.
se Sì, indicare quali principi attivi:
Il paziente assume terapie antineoplastiche?
2. Il paziente assume terapie per patologie concomitanti?
3. Il paziente assume terapie per altri sintomi presenti oltre il dolore?
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
se Sì, indicare quali principi attivi:
se Sì, indicare quali principi attivi:
1-1.
1-2.
1-3.
2-1.
2-2.
2-3.
2-4.
3-1.
3-2.
3-3.
3-4.
3-5.
3-6.
3-7.
3-8.
3-9.
3-10.
5
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
REAZIONI AVVERSE DOVUTE ALLA TERAPIA ANTALGICA PREGRESSA
Nel caso in cui abbia assunto una terapia antalgica appena prima di entrare nello studio, il
paziente riferisce di aver sofferto di qualche disturbo?
1.
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
riportare dal questionario del paziente quanto compilato alla visita 1 (rispondere a tutte le domande)
Allucinazioni..........................................
No Un po’ Molto Moltissimo [0] [1] [2] [3]
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito.....................................................
PROMEMORIA: si ricorda la necessità di eseguire il prelievo ematico per la tipizzazione geni-
ca. Vedere le istruzioni negli allegati del protocollo.
Altro 1 _______________________________
Altro 2 _______________________________
No Sì [0] [1]
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
1-9.
1-10.
1-11.
1-12.
1-13.
1-14.

6
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
KARNOFSKY PERFORMANCE STATUS
Indicare l’attuale livello di KPS 1.
Normale attività, non evidenza di malattia..............................................................................
Capace di svolgere una normale attività, segni minori di malattia..........................................
Normale attività con sforzo; qualche segno o sintomo malattia..............................................
Autosufficiente, inabile a svolgere una normale attività lavorativa........................................
Richiede assistenza occasionalmente, ma è in grado di adempiere alla maggior parte delle
proprie necessità.....................................................................................................................
Richiede assistenza particolare e frequente intervento medico...............................................
Inabile; richiede speciali cure ed assistenza............................................................................
Gravemente inabile; l’ospedalizzazione è indicata anche se la morte non è imminente........
Molto malato; richiede ospedalizzazione e terapia di supporto..............................................
Morto.......................................................................................................................................
Moribondo; processi vitali rapidamente ingravescenti...........................................................
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[0]
ASPETTI PSICOLOGICI DURANTE GLI ULTIMI 7 GIORNI
Si è sentito(a) teso(a) ?...........................
Si è preoccupato(a) ?..............................
Si è sentito(a) irritabile ?........................
Si è sentito(a) depresso ?........................
[0] [1]
2.
3.
4.
5.
[2] [3]
No Un pò Molto Moltissimo

7
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
VALUTAZIONE DEL DOLORE NELLE ULTIME 24 ORE
Da quanti mesi il paziente soffre per dolori dovuti al cancro? 1. mesi
Intensità del dolore medio nelle ultime 24 ore 2. NRS: da 0 a 10
Solo nocicettivo (viscerale e/o osseo e/o dei tessuti molli)
Solo neuropatico................................................................
Sia nocicettivo che neuropatico.........................................
[1]
[2]
[3]
Indicare la tipologia del dolore 16.
Insufficienti informazioni per classificare il dolore........... [4]
(vedi protocollo - allegato 1)
Il paziente è arruolabile solo se
l’intensità del suo dolore medio è ≥ 4
Bruciante / urente...................
Sensazione di freddo doloroso
Scosse elettriche......................
4.
5.
6.
Il dolore ha una o più delle seguenti caratteristiche?
No Sì [0] [1]
Formicolio...............................
Punture di spillo.......................
Intorpidimento.........................
7.
8.
9.
Il dolore è associato ad uno o più dei seguenti
sintomi nell’area del dolore stesso?
Sensazione di prurito............... 10.
Il dolore è localizzato nella stessa area dove l’esame
fisico può rilevare una o più delle seguenti
caratteristiche?
Ipoestesia al tatto.....................
Ipoestesia alla puntura.............
11.
12.
Nell’area dolente il dolore può essere causato o peggiorato dallo sfioramento della pelle?
Intervista al paziente
13.
(vedi protocollo - allegato 1)
No Sì [0] [1]
Totale delle risposte affermative 14. (≤ 4 dol. Neuropatico NO, > 4 dol. Neuropatico SI)
No Sì [0] [1]
Esame del paziente
VALUTAZIONE DELLA PRESENZA DI DOLORE NEUROPATICO
Quanti di questi episodi dolorosi intensi il paziente ha provato nelle ultime 24 ore?
Il paziente ha avuto episodi di esacerbazione intensa del dolore rispetto al dolore
medio provato nelle ultime 24 ore ? 15.
In caso affermativo:
Quale farmaco rescue è stato usato: _____________________________________________________
Quante dosi rescue sono state assunte nelle ultime 24 ore?.......................................
Alla dose, per ogni somministrazione, di:
E’ stato usato un farmaco rescue?..............................................................................
(vedi protocollo - allegato 1)
No Sì [0] [1]
No Sì [0] [1]
VALUTAZIONE DELLA PRESENZA DI BREAKTHROUGH PAIN (BTP)
mg µg [1] [2]
Intensità del dolore peggiore nelle ultime 24 ore 3. NRS: da 0 a 10
15-1.
15-2.
15-3.
15-4.
15-6.
15-5.

8
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
[1]
TERAPIA ANTALGICA DI INIZIO STUDIO
Morfina orale........
Buprenorfina TD
Ossicodone orale...
Fentanyl TD........
[2]
[3]
[4]
Farmaco oppioide assegnato
per randomizzazione
1.
mg/24h
µg/h
TERAPIA ATC
gg mm aaaa
Dose iniziale
suggerita
Dose iniziale
effettivamente data
60mg/24h
40mg/24h
35µg/h
25µg/h
mg/24h
µg/h
2. 3.
Presenza di una situazione clinica di dolore di elevata intensità con necessità di immediato e
contemporaneo uso di una dose aggiuntiva? (protocollo § 5.1)
4.
se Sì, indicare:
Morfina orale IR (10mg)........
Morfina sottocutanea (3,3mg)
Singola
dose Nr. dosi nelle prime 24h
ALTRE TERAPIE ANTALGICHE OLTRE QUELLA ATC
Al paziente viene impostata una terapia oppioide RESCUE per presenza di BTP?
(non ripetere quanto eventualmente descritto nella sezione precedente) 5.
se Sì, indicare:
Fentanyl transmucosale
Morfina endovena.........
Singola dose
µg/dose
mg/dose
Terapia adiuvante
Al paziente viene data terapia adiuvante analgesica? 6.
se Sì indicare il motivo: 7. Presenza di particolari tipologie di dolore
Appoggio/supplemento alla terapia ATC
[1]
[2]
9. Steroidi.............................
Anticonvulsivanti............
Antidepressivi..................
Principio attivo Via di somm.ne* Dose/die No Sì [0] [1]
10.
11.
Bifosfonati....................... 12.
Altri adiuvanti analgesici 13.
Entrambi [3]
8. FANS...............................
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Data di inizio
del trattamento
Oppioide aggiuntivo
Oppioide rescue
* EV Endovenosa IM Intramuscolare IN Intranasale PO Orale
RT Rettale SC Sottocutanea SU Sublinguale TO Transmucosale orale
TS Transdermico
4-1.
4-4.
4-2.
4-5.
4-3.
4-6.
5-1.
5-3.
5-2.
5-4.
8-1. 8-2. 8-3.
9-1. 9-2. 9-3.
10-1. 10-2. 10-3.
11-1. 11-2. 11-3.
12-1. 12-2. 12-3.
13-1. 13-2. 13-3.

9
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 1 — Valutazione basale — Giorno 0
ALTRE TERAPIE (oltre quella antalgica) AL MOMENTO INIZIALE DELLO STUDIO
1.
se Variata, indicare quali principi attivi:
Terapie antineoplastiche?
2. Terapie per le patologie concomitanti?
3. Terapie sintomatiche-palliative?
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
58. Spuntare qui di fianco quando TUTTE le informazioni per questa visita sono state inserite
Invariata Variata [1] [2]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Invariata Variata [1] [2]
Invariata Variata [1] [2]
se Variata, indicare quali principi attivi:
se Variata, indicare quali principi attivi:
1-1.
1-1-1.
1-1-2.
1-1-3.
2-1.
3-1.
2-1-1.
2-1-2.
2-1-3.
2-1-4.
3-1-1.
3-1-2.
3-1-3.
3-1-4.
3-1-5.
3-1-6.
3-1-7.
3-1-8.
3-1-9.
3-1-10.

10
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 2 —72 ore
Data rilevazione gg mm aaaa
1.
Numero episodi dolorosi...............................................................
Presenza di episodi di BTP dall’ultima visita? 4.
se Sì, indicare, relativamente alle ultime 24h:
Quante dosi rescue sono state assunte nelle ultime 24 ore............
Dosaggio della singola dose rescue: se Fentanyl transmucosale............... µg/dose
se Morfina endovena........................ mg/dose
E’ stato usato un oppioide rescue?................................................
No Sì [0] [1]
No Sì [0] [1]
5.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide aggiuntiva: sono state usate dosi “extra” per dare migliore copertu-
ra al trattamento ATC nelle ultime 24 ore?
VALUTAZIONI RELATIVE ALLE ULTIME 24 ORE
Intensità del dolore medio nelle ultime 24 ore 2. NRS: da 0 a 10
Intensità del dolore peggiore nelle ultime 24 ore 3. NRS: da 0 a 10
4-1.
4-2.
4-3.
4-4.
5-1.
5-3.
5-2.
5-4.

11
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 2 — 72 ore
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE
[1] Morfina orale........
Buprenorfina TD
Ossicodone orale...
Fentanyl TD........
[2]
[3]
[4]
Farmaco oppioide 1.
mg/24h
Terapia ATC Dose
mg/24h
2.
TERAPIA FISSA
Terapia adiuvante analgesica
Al paziente viene data una terapia adiuvante ? 3.
se Sì indicare il motivo 4. Presenza di particolari tipologie di dolore
Appoggio/supplemento alla terapia ATC
[1]
[2]
Indicare la terapia
principale adiuvante
6. Steroidi.............................
Anticonvulsivanti............
Antidepressivi..................
Via di
Principio attivo somm.ne* Dose / die No Sì [0] [1]
7.
8.
Bifosfonati....................... 9.
Altri adiuvanti analgesici 10.
Entrambi [3]
5. FANS...............................
No Sì [0] [1]
* EV Endovenosa IM Intramuscolare IN Intranasale PO Orale
RT Rettale SC Sottocutanea SU Sublinguale TO Transmucosale orale
TS Transdermico
TERAPIA VARIABILE
Terapia aggiuntiva al trattamento attuale ATC
Al paziente viene impostata una terapia aggiuntiva “extra” per dare migliore copertura al
trattamento ATC?
11.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide RESCUE per BTP
Al paziente viene impostata una terapia oppioide RESCUE
per presenza di BTP?
12.
se Sì, indicare:
Fentanyl transmucosale
Morfina endovena.........
Singola dose
µg/dose
mg/dose
Oppioide rescue
No Sì [0] [1]
µg/h
µg/h
5-1. 5-2. 5-3.
6-1. 6-2. 6-3.
7-1. 7-2. 7-3.
8-1. 8-2. 8-3.
9-1. 9-2. 9-3.
10-1. 10-2. 10-3.
11-1.
11-3.
11-2.
11-4.
12-1.
12-3.
12-2.
12-4.

12
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 2 — 72 ore
ALTRE TERAPIE
1.
se Variata, indicare quali principi attivi:
Terapie antineoplastiche?
2. Terapie per le patologie concomitanti?
3. Terapie sintomatiche-palliative?
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
Invariata Variata [1] [2]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Invariata Variata [1] [2]
Invariata Variata [1] [2]
se Variata, indicare quali principi attivi:
se Variata, indicare quali principi attivi:
1-1.
1-1-1.
1-1-2.
1-1-3.
2-1.
3-1.
2-1-1.
2-1-2.
2-1-3.
2-1-4.
3-1-1.
3-1-2.
3-1-3.
3-1-4.
3-1-5.
3-1-6.
3-1-7.
3-1-8.
3-1-9.
3-1-10.
13
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 2 — 72 ore
REAZIONI AVVERSE DOVUTE ALLA TERAPIA ANTALGICA
In relazione alla terapia antalgica data nella visita precedente, il paziente riferisce di aver sof-
ferto di qualche disturbo?
1.
riportare dal questionario del paziente quanto compilato alla visita 2 (rispondere a tutte le domande)
No Sì [0] [1]
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No Un po’ Molto Moltissimo [0] [1] [2] [3]
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito.....................................................
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
1-9.
1-10.
1-11.
1-12.

14
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 2 —72 ore
In caso di interruzione prematura dello studio compilare direttamente la sezione finale della
CRF “Completamento dello studio”.
Si ricorda la necessità di eseguire il prelievo ematico per la tipizzazione genica secondo le
istruzioni presenti negli allegati del protocollo, se non già eseguito.
EVENTI AVVERSI
A suo giudizio, il paziente ha sofferto di qualsiasi evento clinico dannoso, come riportato
in protocollo al capitolo 7 ?
1.
In caso affermativo segnalare gli eventi avversi alla pagina successiva.
3. Spuntare quando TUTTE le informazioni relativa a questa visita sono state inserite
INTERRUZIONE PREMATURA DELLO STUDIO
Il paziente ha interrotto prematuramente lo studio? 2.
No Sì [0] [1]
No Sì [0] [1]
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
15
Visita 2 —72 ore — Eventi avversi
Evento avverso Severità Relazione Data inizio Azione intrapresa
Un Evento avverso è definito serio se è fatale, se pone il paziente in pericolo di vita, se è disabilitante/inabilitante per il paziente, se richiede o prolunga l’ospedalizzazione (a meno che essa
non sia stata pianificata in precedenza) o se richiede interventi per evitare una delle condizioni precedenti.
Ogni evento definito “serio” richiede la notifica entro 24 ore al centro di coordinamento dell’Istituto Mario Negri tramite fax all’attenzione della Dr.ssa Maria Teresa Greco,
gruppo CERP, al nr. di fax 02-33200231
Il medico è pregato di inserire un sintomo per ciascuna riga per: - tutti i nuovi eventi avversi
- tutti gli eventi avversi precedenti che sono peggiorati (colonna “Severità”)
- tutti gli eventi avversi precedenti che hanno modificato la classe di ”Relazione” con il farmaco in studio
- tutte le condizioni mediche precedenti allo studio che sono peggiorate
Severità 1 Lieve
2 Moderata
3 Grave
Relazione con i farmaci in studio 1 Certa
2 Probabile
3 Possibile
4 Dubbia
5 Nessuna
6 Sconosciuta
Azione intrapresa 0 Nessuna azione
1 Aggiustamento della dose/interruzione temporanea
2 Interruzione permanente del farmaco in studio
3 Somm.ne di terapia concomitante
4 Somm.ne di terapia non farmacologica
5 Ospedalizzazione o suo prolungamento
Continua? Data fine
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
5 0 1 2 3 4
gg mm aaaa
Studio CERP1
1-1. 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
L’evento
è “serio” ?
2-1.
3-1.
4-1.
5-1.
6-1.
7-1.
8-1.
9-1.
10-1.
11-1.
12-1.
1-2.
2-2.
3-2.
4-2.
5-2.
6-2.
7-2.
8-2.
9-2.
10-2.
11-2.
12-2.
1-3.
2-3.
3-3.
4-3.
5-3.
6-3.
7-3.
8-3.
9-3.
10-3.
11-3.
12-3.
1-4.
2-4.
3-4.
4-4.
5-4.
6-4.
7-4.
8-4.
9-4.
10-4.
11-4.
12-4.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-5.
2-5.
3-5.
4-5.
5-5.
6-5.
7-5.
8-5.
9-5.
10-5.
11-5.
12-5.
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-6.
2-6.
3-6.
4-6.
5-6.
6-6.
7-6.
8-6.
9-6.
10-6.
11-6.
12-6.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-7.
2-7.
3-7.
4-7.
5-7.
6-7.
7-7.
8-7.
9-7.
10-7.
11-7.
12-7.

16
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 3 — Giorno 7
Data rilevazione gg mm aaaa
1.
Presenza di episodi di BTP dall’ultima visita? 4. No Sì [0] [1]
VALUTAZIONI RELATIVE ALLE ULTIME 24 ORE
Intensità del dolore medio nelle ultime 24 ore 2. NRS: da 0 a 10
Intensità del dolore peggiore nelle ultime 24 ore 3. NRS: da 0 a 10
Numero episodi dolorosi...............................................................
se Sì, indicare, relativamente alle ultime 24h:
Quante dosi rescue sono state assunte nelle ultime 24 ore............
Dosaggio della singola dose rescue: se Fentanyl transmucosale............... µg/dose
se Morfina endovena........................ mg/dose
E’ stato usato un oppioide rescue?................................................ No Sì [0] [1]
5.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide aggiuntiva: sono state usate dosi “extra” per dare migliore copertu-
ra al trattamento ATC nelle ultime 24 ore?
4-1.
4-2.
4-3.
4-4.
5-1.
5-3.
5-2.
5-4.

17
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 3 — Giorno 7
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (1 di 2)
[1] Morfina orale........
Buprenorfina TD
Ossicodone orale...
Fentanyl TD........
[2]
[3]
[4]
Farmaco oppioide
mg/24h
Terapia ATC
Dose
mg/24h
Terapia adiuvante analgesica
Al paziente viene data una terapia adiuvante ? 4.
se Sì indicare il motivo 5. Presenza di particolari tipologie di dolore
Appoggio/supplemento alla terapia ATC
[1]
[2]
Indicare la terapia
principale adiuvante
7. Steroidi.............................
Anticonvulsivanti............
Antidepressivi..................
Via di
Principio attivo somm.ne* Dose / die No Sì [0] [1]
8.
9.
Bifosfonati....................... 10.
Altri adiuvanti analgesici 11.
Entrambi [3]
6. FANS...............................
No Sì [0] [1]
* EV Endovenosa IM Intramuscolare IN Intranasale PO Orale
RT Rettale SC Sottocutanea SU Sublinguale TO Transmucosale orale
TS Transdermico
µg/h
µg/h
6-1. 6-2. 6-3.
7-1. 7-2. 7-3.
8-1. 8-2. 8-3.
9-1. 9-2. 9-3.
10-1. 10-2. 10-3.
11-1. 11-2. 11-3.
TERAPIA FISSA
In caso di avvenuto cambio di oppioide (switch) precisare la causa: 1.
[1] Analgesia inadeguata....................................................
Entrambe......................................................................
Grave Reazione avversa al farmaco, non controllabile
Incapacità alla assunzione orale...................................
[2]
[3]
[4]
Indicare, in ogni caso, la terapia oppioide ATC e il dosaggio:
2. 3.

18
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 3 — Giorno 7
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (2 di 2)
TERAPIA VARIABILE
Terapia aggiuntiva al trattamento attuale ATC
Al paziente viene impostata una terapia aggiuntiva “extra” per dare migliore copertura al
trattamento ATC?
1.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide RESCUE per BTP
Al paziente viene impostata una terapia oppioide RESCUE
per presenza di BTP?
2.
se Sì, indicare:
Fentanyl transmucosale
Morfina endovena.........
Singola dose
µg/dose
mg/dose
Oppioide rescue
No Sì [0] [1]
1-1.
1-3.
1-2.
1-4.
2-1.
2-3.
2-2.
2-4.

19
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 3 — Giorno 7
ALTRE TERAPIE
1.
se Variata, indicare quali principi attivi:
Terapie antineoplastiche?
2. Terapie per le patologie concomitanti?
3. Terapie sintomatiche-palliative?
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
Invariata Variata [1] [2]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Invariata Variata [1] [2]
Invariata Variata [1] [2]
se Variata, indicare quali principi attivi:
se Variata, indicare quali principi attivi:
1-1.
1-1-1.
1-1-2.
1-1-3.
2-1.
3-1.
2-1-1.
2-1-2.
2-1-3.
2-1-4.
3-1-1.
3-1-2.
3-1-3.
3-1-4.
3-1-5.
3-1-6.
3-1-7.
3-1-8.
3-1-9.
3-1-10.
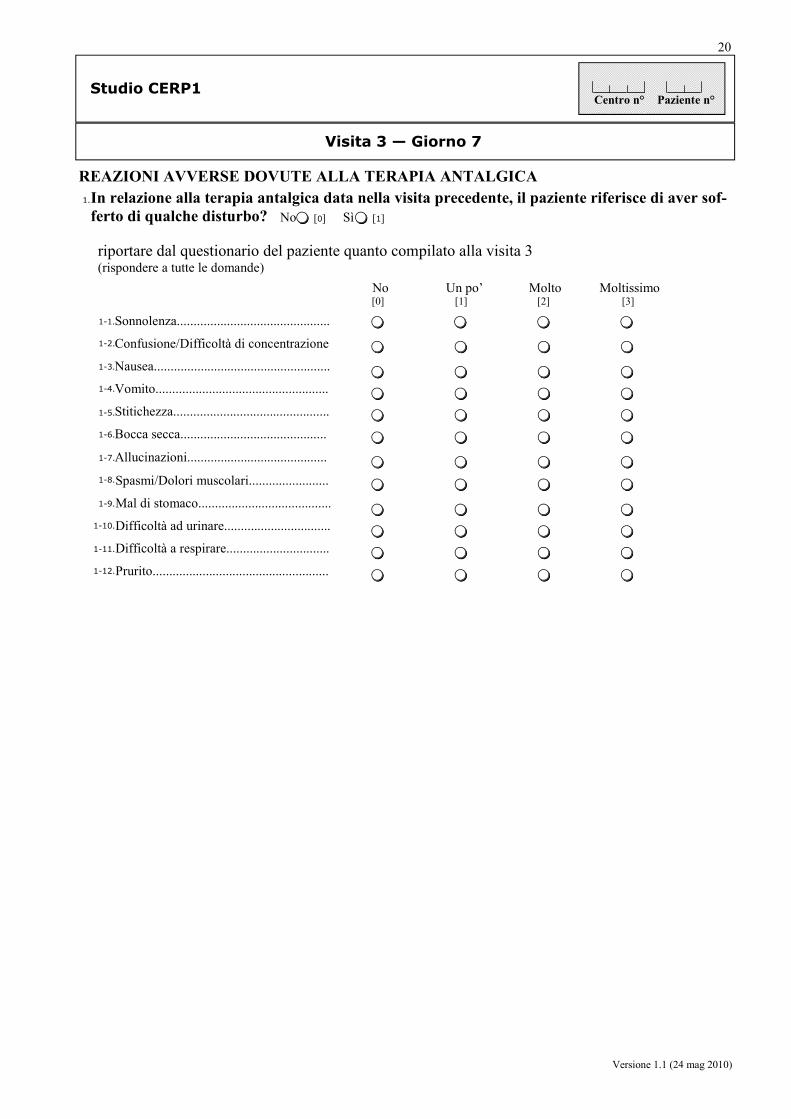
20
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 3 — Giorno 7
REAZIONI AVVERSE DOVUTE ALLA TERAPIA ANTALGICA
In relazione alla terapia antalgica data nella visita precedente, il paziente riferisce di aver sof-
ferto di qualche disturbo?
1.
riportare dal questionario del paziente quanto compilato alla visita 3 (rispondere a tutte le domande)
No Sì [0] [1]
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No Un po’ Molto Moltissimo [0] [1] [2] [3]
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito.....................................................
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
1-9.
1-10.
1-11.
1-12.

21
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 3 — Giorno 7
Si ricorda la necessità di eseguire il prelievo ematico per la tipizzazione genica secondo le
istruzioni presenti negli allegati del protocollo, se non già eseguito.
EVENTI AVVERSI
1.
In caso affermativo segnalare gli eventi avversi alla pagina successiva.
INTERRUZIONE PREMATURA DELLO STUDIO
A suo giudizio, il paziente ha sofferto di qualsiasi evento clinico dannoso, come riportato
in protocollo al capitolo 7 ? No Sì [0] [1]
In caso di interruzione prematura dello studio compilare direttamente la sezione finale della
CRF “Completamento dello studio”.
3. Spuntare quando TUTTE le informazioni relativa a questa visita sono state inserite
Il paziente ha interrotto prematuramente lo studio? 2. No Sì [0] [1]
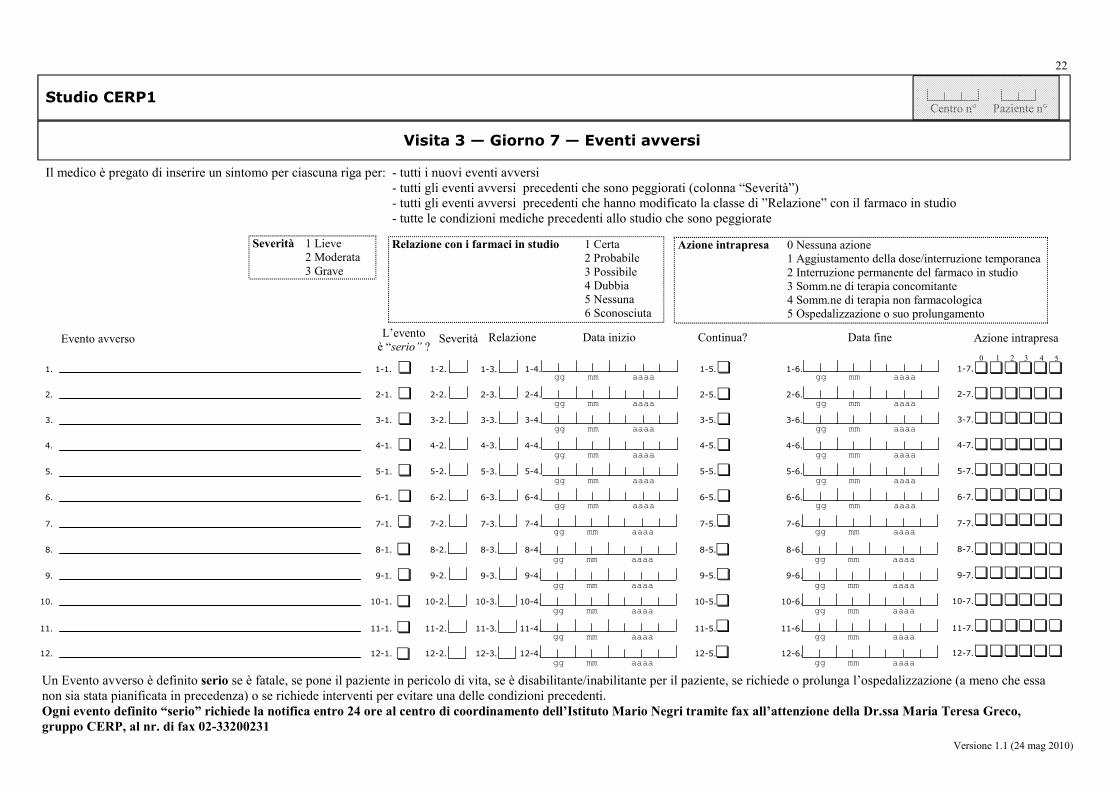
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
22
Un Evento avverso è definito serio se è fatale, se pone il paziente in pericolo di vita, se è disabilitante/inabilitante per il paziente, se richiede o prolunga l’ospedalizzazione (a meno che essa
non sia stata pianificata in precedenza) o se richiede interventi per evitare una delle condizioni precedenti.
Ogni evento definito “serio” richiede la notifica entro 24 ore al centro di coordinamento dell’Istituto Mario Negri tramite fax all’attenzione della Dr.ssa Maria Teresa Greco,
gruppo CERP, al nr. di fax 02-33200231
Il medico è pregato di inserire un sintomo per ciascuna riga per: - tutti i nuovi eventi avversi
- tutti gli eventi avversi precedenti che sono peggiorati (colonna “Severità”)
- tutti gli eventi avversi precedenti che hanno modificato la classe di ”Relazione” con il farmaco in studio
- tutte le condizioni mediche precedenti allo studio che sono peggiorate
Visita 3 — Giorno 7 — Eventi avversi
Severità 1 Lieve
2 Moderata
3 Grave
Relazione con i farmaci in studio 1 Certa
2 Probabile
3 Possibile
4 Dubbia
5 Nessuna
6 Sconosciuta
Azione intrapresa 0 Nessuna azione
1 Aggiustamento della dose/interruzione temporanea
2 Interruzione permanente del farmaco in studio
3 Somm.ne di terapia concomitante
4 Somm.ne di terapia non farmacologica
5 Ospedalizzazione o suo prolungamento
Studio CERP1
Evento avverso Severità Relazione Data inizio Azione intrapresa Continua? Data fine
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-1. 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
L’evento
è “serio” ?
2-1.
3-1.
4-1.
5-1.
6-1.
7-1.
8-1.
9-1.
10-1.
11-1.
12-1.
1-2.
2-2.
3-2.
4-2.
5-2.
6-2.
7-2.
8-2.
9-2.
10-2.
11-2.
12-2.
1-3.
2-3.
3-3.
4-3.
5-3.
6-3.
7-3.
8-3.
9-3.
10-3.
11-3.
12-3.
1-4.
2-4.
3-4.
4-4.
5-4.
6-4.
7-4.
8-4.
9-4.
10-4.
11-4.
12-4.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-5.
2-5.
3-5.
4-5.
5-5.
6-5.
7-5.
8-5.
9-5.
10-5.
11-5.
12-5.
5 0 1 2 3 4
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-6.
2-6.
3-6.
4-6.
5-6.
6-6.
7-6.
8-6.
9-6.
10-6.
11-6.
12-6.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-7.
2-7.
3-7.
4-7.
5-7.
6-7.
7-7.
8-7.
9-7.
10-7.
11-7.
12-7.

23
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 4 — Giorno 14
Data rilevazione gg mm aaaa
1.
Presenza di episodi di BTP dall’ultima visita? 4. No Sì [0] [1]
VALUTAZIONI RELATIVE ALLE ULTIME 24 ORE
Intensità del dolore medio nelle ultime 24 ore 2. NRS: da 0 a 10
Intensità del dolore peggiore nelle ultime 24 ore 3. NRS: da 0 a 10
Numero episodi dolorosi...............................................................
se Sì, indicare, relativamente alle ultime 24h:
Quante dosi rescue sono state assunte nelle ultime 24 ore............
Dosaggio della singola dose rescue: se Fentanyl transmucosale............... µg/dose
se Morfina endovena........................ mg/dose
E’ stato usato un oppioide rescue?................................................ No Sì [0] [1]
5.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide aggiuntiva: sono state usate dosi “extra” per dare migliore copertu-
ra al trattamento ATC nelle ultime 24 ore?
4-1.
4-2.
4-3.
4-4.
5-1.
5-3.
5-2.
5-4.

24
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 4 — Giorno 14
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (1 di 2)
[1] Morfina orale........
Buprenorfina TD
Ossicodone orale...
Fentanyl TD........
[2]
[3]
[4]
Farmaco oppioide
mg/24h
Terapia ATC
Dose
mg/24h
Terapia adiuvante analgesica
Al paziente viene data una terapia adiuvante ? 4.
se Sì indicare il motivo 5. Presenza di particolari tipologie di dolore
Appoggio/supplemento alla terapia ATC
[1]
[2]
Indicare la terapia
principale adiuvante
7. Steroidi.............................
Anticonvulsivanti............
Antidepressivi..................
Via di
Principio attivo somm.ne* Dose / die No Sì [0] [1]
8.
9.
Bifosfonati....................... 10.
Altri adiuvanti analgesici 11.
Entrambi [3]
6. FANS...............................
No Sì [0] [1]
* EV Endovenosa IM Intramuscolare IN Intranasale PO Orale
RT Rettale SC Sottocutanea SU Sublinguale TO Transmucosale orale
TS Transdermico
µg/h
µg/h
6-1. 6-2. 6-3.
7-1. 7-2. 7-3.
8-1. 8-2. 8-3.
9-1. 9-2. 9-3.
10-1. 10-2. 10-3.
11-1. 11-2. 11-3.
TERAPIA FISSA
In caso di avvenuto cambio di oppioide (switch) precisare la causa: 1.
[1] Analgesia inadeguata....................................................
Entrambe......................................................................
Grave Reazione avversa al farmaco, non controllabile
Incapacità alla assunzione orale...................................
[2]
[3]
[4]
Indicare, in ogni caso, la terapia oppioide ATC e il dosaggio:
2. 3.

25
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 4 — Giorno 14
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (2 di 2)
TERAPIA VARIABILE
Terapia aggiuntiva al trattamento attuale ATC
Al paziente viene impostata una terapia aggiuntiva “extra” per dare migliore copertura al
trattamento ATC?
1.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide RESCUE per BTP
Al paziente viene impostata una terapia oppioide RESCUE
per presenza di BTP?
2.
se Sì, indicare:
Fentanyl transmucosale
Morfina endovena.........
Singola dose
µg/dose
mg/dose
Oppioide rescue
No Sì [0] [1]
1-1.
1-3.
1-2.
1-4.
2-1.
2-3.
2-2.
2-4.

26
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 4 — Giorno 14
ALTRE TERAPIE
1.
se Variata, indicare quali principi attivi:
Terapie antineoplastiche?
2. Terapie per le patologie concomitanti?
3. Terapie sintomatiche-palliative?
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
Invariata Variata [1] [2]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Invariata Variata [1] [2]
Invariata Variata [1] [2]
se Variata, indicare quali principi attivi:
se Variata, indicare quali principi attivi:
1-1.
1-1-1.
1-1-2.
1-1-3.
2-1.
3-1.
2-1-1.
2-1-2.
2-1-3.
2-1-4.
3-1-1.
3-1-2.
3-1-3.
3-1-4.
3-1-5.
3-1-6.
3-1-7.
3-1-8.
3-1-9.
3-1-10.
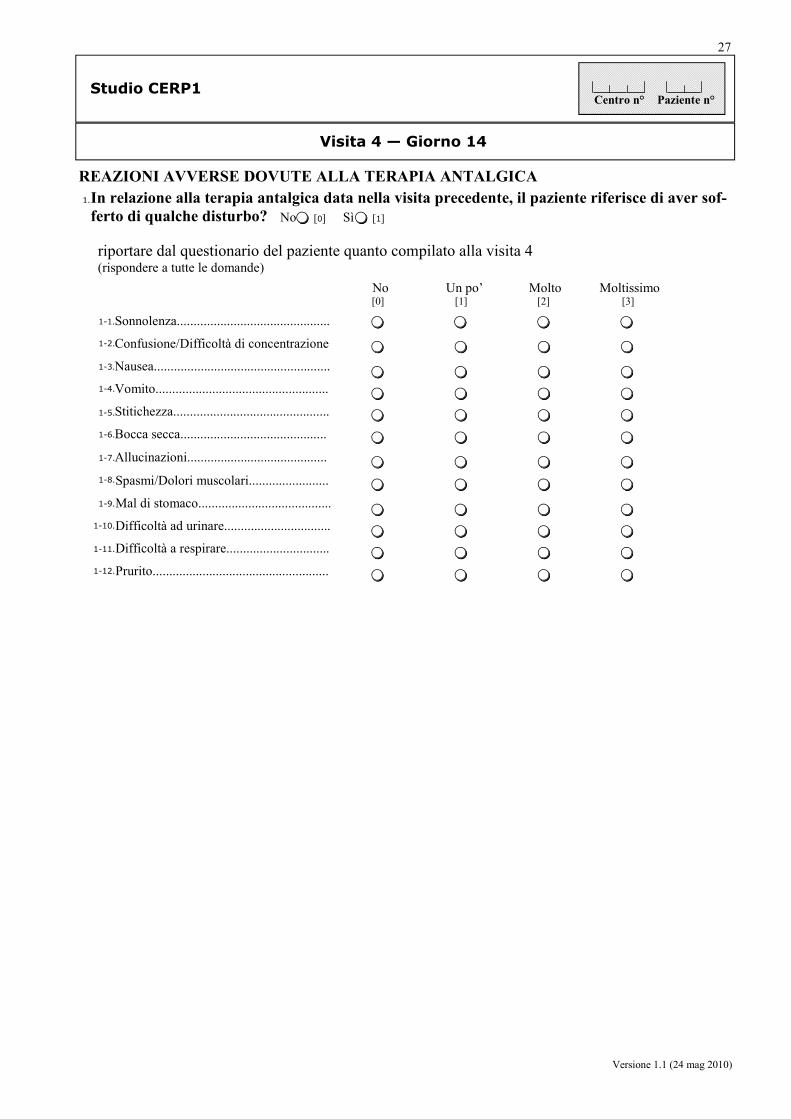
27
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 4 — Giorno 14
REAZIONI AVVERSE DOVUTE ALLA TERAPIA ANTALGICA
In relazione alla terapia antalgica data nella visita precedente, il paziente riferisce di aver sof-
ferto di qualche disturbo?
1.
riportare dal questionario del paziente quanto compilato alla visita 4 (rispondere a tutte le domande)
No Sì [0] [1]
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No Un po’ Molto Moltissimo [0] [1] [2] [3]
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito.....................................................
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
1-9.
1-10.
1-11.
1-12.

28
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 4 — Giorno 14
Si ricorda la necessità di eseguire il prelievo ematico per la tipizzazione genica secondo le
istruzioni presenti negli allegati del protocollo, se non già eseguito.
EVENTI AVVERSI
1.
In caso affermativo segnalare gli eventi avversi alla pagina successiva.
INTERRUZIONE PREMATURA DELLO STUDIO
A suo giudizio, il paziente ha sofferto di qualsiasi evento clinico dannoso, come riportato
in protocollo al capitolo 7 ? No Sì [0] [1]
In caso di interruzione prematura dello studio compilare direttamente la sezione finale della
CRF “Completamento dello studio”.
3. Spuntare quando TUTTE le informazioni relativa a questa visita sono state inserite
Il paziente ha interrotto prematuramente lo studio? 2. No Sì [0] [1]
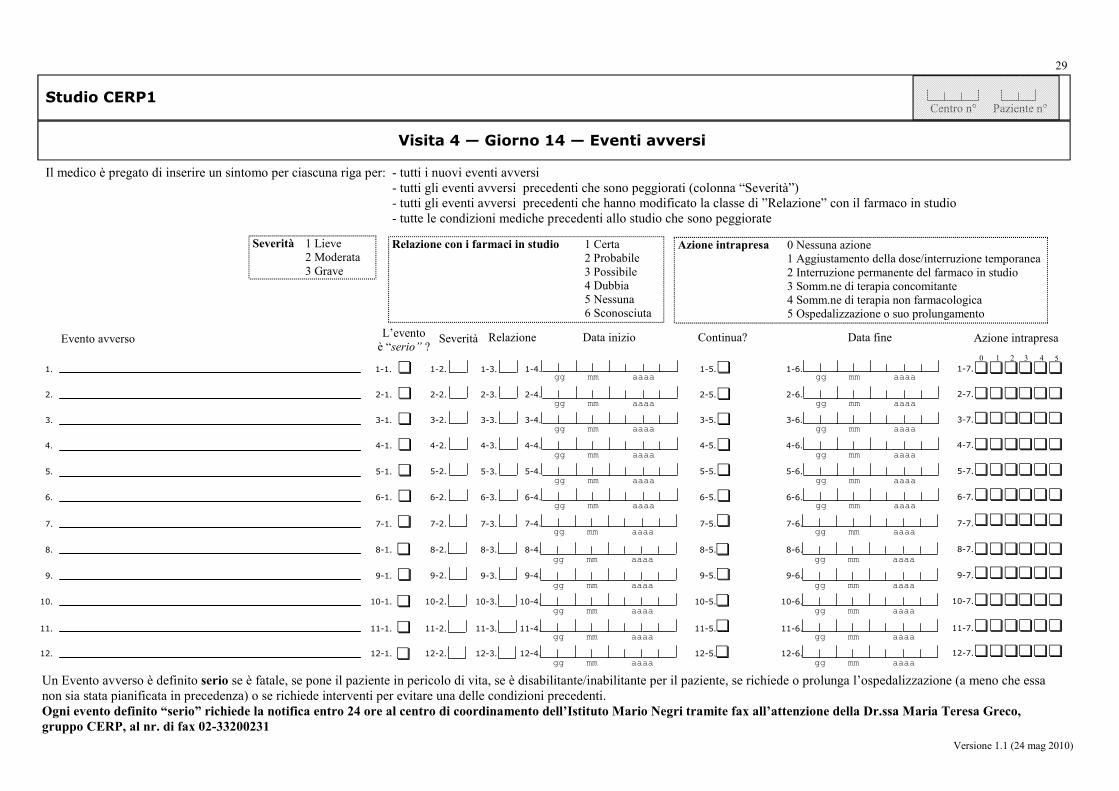
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
29
Un Evento avverso è definito serio se è fatale, se pone il paziente in pericolo di vita, se è disabilitante/inabilitante per il paziente, se richiede o prolunga l’ospedalizzazione (a meno che essa
non sia stata pianificata in precedenza) o se richiede interventi per evitare una delle condizioni precedenti.
Ogni evento definito “serio” richiede la notifica entro 24 ore al centro di coordinamento dell’Istituto Mario Negri tramite fax all’attenzione della Dr.ssa Maria Teresa Greco,
gruppo CERP, al nr. di fax 02-33200231
Il medico è pregato di inserire un sintomo per ciascuna riga per: - tutti i nuovi eventi avversi
- tutti gli eventi avversi precedenti che sono peggiorati (colonna “Severità”)
- tutti gli eventi avversi precedenti che hanno modificato la classe di ”Relazione” con il farmaco in studio
- tutte le condizioni mediche precedenti allo studio che sono peggiorate
Visita 4 — Giorno 14 — Eventi avversi
Severità 1 Lieve
2 Moderata
3 Grave
Relazione con i farmaci in studio 1 Certa
2 Probabile
3 Possibile
4 Dubbia
5 Nessuna
6 Sconosciuta
Azione intrapresa 0 Nessuna azione
1 Aggiustamento della dose/interruzione temporanea
2 Interruzione permanente del farmaco in studio
3 Somm.ne di terapia concomitante
4 Somm.ne di terapia non farmacologica
5 Ospedalizzazione o suo prolungamento
Studio CERP1
Evento avverso Severità Relazione Data inizio Azione intrapresa Continua? Data fine
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-1. 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
L’evento
è “serio” ?
2-1.
3-1.
4-1.
5-1.
6-1.
7-1.
8-1.
9-1.
10-1.
11-1.
12-1.
1-2.
2-2.
3-2.
4-2.
5-2.
6-2.
7-2.
8-2.
9-2.
10-2.
11-2.
12-2.
1-3.
2-3.
3-3.
4-3.
5-3.
6-3.
7-3.
8-3.
9-3.
10-3.
11-3.
12-3.
1-4.
2-4.
3-4.
4-4.
5-4.
6-4.
7-4.
8-4.
9-4.
10-4.
11-4.
12-4.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-5.
2-5.
3-5.
4-5.
5-5.
6-5.
7-5.
8-5.
9-5.
10-5.
11-5.
12-5.
5 0 1 2 3 4
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-6.
2-6.
3-6.
4-6.
5-6.
6-6.
7-6.
8-6.
9-6.
10-6.
11-6.
12-6.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-7.
2-7.
3-7.
4-7.
5-7.
6-7.
7-7.
8-7.
9-7.
10-7.
11-7.
12-7.

30
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 5 — Giorno 21
Data rilevazione gg mm aaaa
1.
Presenza di episodi di BTP dall’ultima visita? 4. No Sì [0] [1]
VALUTAZIONI RELATIVE ALLE ULTIME 24 ORE
Intensità del dolore medio nelle ultime 24 ore 2. NRS: da 0 a 10
Intensità del dolore peggiore nelle ultime 24 ore 3. NRS: da 0 a 10
Numero episodi dolorosi...............................................................
se Sì, indicare, relativamente alle ultime 24h:
Quante dosi rescue sono state assunte nelle ultime 24 ore............
Dosaggio della singola dose rescue: se Fentanyl transmucosale............... µg/dose
se Morfina endovena........................ mg/dose
E’ stato usato un oppioide rescue?................................................ No Sì [0] [1]
5.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide aggiuntiva: sono state usate dosi “extra” per dare migliore copertu-
ra al trattamento ATC nelle ultime 24 ore?
4-1.
4-2.
4-3.
4-4.
5-1.
5-3.
5-2.
5-4.

31
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 5 — Giorno 21
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (1 di 2)
[1] Morfina orale........
Buprenorfina TD
Ossicodone orale...
Fentanyl TD........
[2]
[3]
[4]
Farmaco oppioide
mg/24h
Terapia ATC
Dose
mg/24h
Terapia adiuvante analgesica
Al paziente viene data una terapia adiuvante ? 4.
se Sì indicare il motivo 5. Presenza di particolari tipologie di dolore
Appoggio/supplemento alla terapia ATC
[1]
[2]
Indicare la terapia
principale adiuvante
7. Steroidi.............................
Anticonvulsivanti............
Antidepressivi..................
Via di
Principio attivo somm.ne* Dose / die No Sì [0] [1]
8.
9.
Bifosfonati....................... 10.
Altri adiuvanti analgesici 11.
Entrambi [3]
6. FANS...............................
No Sì [0] [1]
* EV Endovenosa IM Intramuscolare IN Intranasale PO Orale
RT Rettale SC Sottocutanea SU Sublinguale TO Transmucosale orale
TS Transdermico
µg/h
µg/h
6-1. 6-2. 6-3.
7-1. 7-2. 7-3.
8-1. 8-2. 8-3.
9-1. 9-2. 9-3.
10-1. 10-2. 10-3.
11-1. 11-2. 11-3.
TERAPIA FISSA
In caso di avvenuto cambio di oppioide (switch) precisare la causa: 1.
[1] Analgesia inadeguata....................................................
Entrambe......................................................................
Grave Reazione avversa al farmaco, non controllabile
Incapacità alla assunzione orale...................................
[2]
[3]
[4]
Indicare, in ogni caso, la terapia oppioide ATC e il dosaggio:
2. 3.

32
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 5 — Giorno 21
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (2 di 2)
TERAPIA VARIABILE
Terapia aggiuntiva al trattamento attuale ATC
Al paziente viene impostata una terapia aggiuntiva “extra” per dare migliore copertura al
trattamento ATC?
1.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide RESCUE per BTP
Al paziente viene impostata una terapia oppioide RESCUE
per presenza di BTP?
2.
se Sì, indicare:
Fentanyl transmucosale
Morfina endovena.........
Singola dose
µg/dose
mg/dose
Oppioide rescue
No Sì [0] [1]
1-1.
1-3.
1-2.
1-4.
2-1.
2-3.
2-2.
2-4.

33
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 5 — Giorno 21
ALTRE TERAPIE
1.
se Variata, indicare quali principi attivi:
Terapie antineoplastiche?
2. Terapie per le patologie concomitanti?
3. Terapie sintomatiche-palliative?
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
Invariata Variata [1] [2]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Invariata Variata [1] [2]
Invariata Variata [1] [2]
se Variata, indicare quali principi attivi:
se Variata, indicare quali principi attivi:
1-1.
1-1-1.
1-1-2.
1-1-3.
2-1.
3-1.
2-1-1.
2-1-2.
2-1-3.
2-1-4.
3-1-1.
3-1-2.
3-1-3.
3-1-4.
3-1-5.
3-1-6.
3-1-7.
3-1-8.
3-1-9.
3-1-10.
34
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 5 — Giorno 21
REAZIONI AVVERSE DOVUTE ALLA TERAPIA ANTALGICA
In relazione alla terapia antalgica data nella visita precedente, il paziente riferisce di aver sof-
ferto di qualche disturbo?
1.
riportare dal questionario del paziente quanto compilato alla visita 5 (rispondere a tutte le domande)
No Sì [0] [1]
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No Un po’ Molto Moltissimo [0] [1] [2] [3]
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito.....................................................
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
1-9.
1-10.
1-11.
1-12.

35
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 5 — Giorno 21
Si ricorda la necessità di eseguire il prelievo ematico per la tipizzazione genica secondo le
istruzioni presenti negli allegati del protocollo, se non già eseguito.
EVENTI AVVERSI
1.
In caso affermativo segnalare gli eventi avversi alla pagina successiva.
INTERRUZIONE PREMATURA DELLO STUDIO
A suo giudizio, il paziente ha sofferto di qualsiasi evento clinico dannoso, come riportato
in protocollo al capitolo 7 ? No Sì [0] [1]
In caso di interruzione prematura dello studio compilare direttamente la sezione finale della
CRF “Completamento dello studio”.
3. Spuntare quando TUTTE le informazioni relativa a questa visita sono state inserite
Il paziente ha interrotto prematuramente lo studio? 2. No Sì [0] [1]

Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
36
Un Evento avverso è definito serio se è fatale, se pone il paziente in pericolo di vita, se è disabilitante/inabilitante per il paziente, se richiede o prolunga l’ospedalizzazione (a meno che essa
non sia stata pianificata in precedenza) o se richiede interventi per evitare una delle condizioni precedenti.
Ogni evento definito “serio” richiede la notifica entro 24 ore al centro di coordinamento dell’Istituto Mario Negri tramite fax all’attenzione della Dr.ssa Maria Teresa Greco,
gruppo CERP, al nr. di fax 02-33200231
Il medico è pregato di inserire un sintomo per ciascuna riga per: - tutti i nuovi eventi avversi
- tutti gli eventi avversi precedenti che sono peggiorati (colonna “Severità”)
- tutti gli eventi avversi precedenti che hanno modificato la classe di ”Relazione” con il farmaco in studio
- tutte le condizioni mediche precedenti allo studio che sono peggiorate
Visita 5 — Giorno 21 — Eventi avversi
Severità 1 Lieve
2 Moderata
3 Grave
Relazione con i farmaci in studio 1 Certa
2 Probabile
3 Possibile
4 Dubbia
5 Nessuna
6 Sconosciuta
Azione intrapresa 0 Nessuna azione
1 Aggiustamento della dose/interruzione temporanea
2 Interruzione permanente del farmaco in studio
3 Somm.ne di terapia concomitante
4 Somm.ne di terapia non farmacologica
5 Ospedalizzazione o suo prolungamento
Studio CERP1
Evento avverso Severità Relazione Data inizio Azione intrapresa Continua? Data fine
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-1. 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
L’evento
è “serio” ?
2-1.
3-1.
4-1.
5-1.
6-1.
7-1.
8-1.
9-1.
10-1.
11-1.
12-1.
1-2.
2-2.
3-2.
4-2.
5-2.
6-2.
7-2.
8-2.
9-2.
10-2.
11-2.
12-2.
1-3.
2-3.
3-3.
4-3.
5-3.
6-3.
7-3.
8-3.
9-3.
10-3.
11-3.
12-3.
1-4.
2-4.
3-4.
4-4.
5-4.
6-4.
7-4.
8-4.
9-4.
10-4.
11-4.
12-4.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-5.
2-5.
3-5.
4-5.
5-5.
6-5.
7-5.
8-5.
9-5.
10-5.
11-5.
12-5.
5 0 1 2 3 4
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-6.
2-6.
3-6.
4-6.
5-6.
6-6.
7-6.
8-6.
9-6.
10-6.
11-6.
12-6.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-7.
2-7.
3-7.
4-7.
5-7.
6-7.
7-7.
8-7.
9-7.
10-7.
11-7.
12-7.

37
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 6 — Giorno 28
Data rilevazione gg mm aaaa
1.
Presenza di episodi di BTP dall’ultima visita? 4. No Sì [0] [1]
VALUTAZIONI RELATIVE ALLE ULTIME 24 ORE
Intensità del dolore medio nelle ultime 24 ore 2. NRS: da 0 a 10
Intensità del dolore peggiore nelle ultime 24 ore 3. NRS: da 0 a 10
KARNOFSKY PERFORMANCE STATUS
Indicare l’attuale livello di KPS 2.
Normale attività, non evidenza di malattia..............................................................................
Capace di svolgere una normale attività, segni minori di malattia..........................................
Normale attività con sforzo; qualche segno o sintomo malattia..............................................
Autosufficiente, inabile a svolgere una normale attività lavorativa........................................
Richiede assistenza occasionalmente, ma è in grado di adempiere alla maggior parte delle
proprie necessità.....................................................................................................................
Richiede assistenza particolare e frequente intervento medico...............................................
Inabile; richiede speciali cure ed assistenza............................................................................
Gravemente inabile; l’ospedalizzazione è indicata anche se la morte non è imminente........
Molto malato; richiede ospedalizzazione e terapia di supporto..............................................
Morto.......................................................................................................................................
Moribondo; processi vitali rapidamente ingravescenti...........................................................
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[0]
Numero episodi dolorosi...............................................................
se Sì, indicare, relativamente alle ultime 24h:
Quante dosi rescue sono state assunte nelle ultime 24 ore............
Dosaggio della singola dose rescue: se Fentanyl transmucosale............... µg/dose
se Morfina endovena........................ mg/dose
E’ stato usato un oppioide rescue?................................................ No Sì [0] [1]
5.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide aggiuntiva: sono state usate dosi “extra” per dare migliore copertu-
ra al trattamento ATC nelle ultime 24 ore?
4-1.
4-2.
4-3.
4-4.
5-1.
5-3.
5-2.
5-4.

38
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 6 — Giorno 28
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (1 di 2)
[1] Morfina orale........
Buprenorfina TD
Ossicodone orale...
Fentanyl TD........
[2]
[3]
[4]
Farmaco oppioide
mg/24h
Terapia ATC
Dose
mg/24h
Terapia adiuvante analgesica
Al paziente viene data una terapia adiuvante ? 4.
se Sì indicare il motivo 5. Presenza di particolari tipologie di dolore
Appoggio/supplemento alla terapia ATC
[1]
[2]
Indicare la terapia
principale adiuvante
7. Steroidi.............................
Anticonvulsivanti............
Antidepressivi..................
Via di
Principio attivo somm.ne* Dose / die No Sì [0] [1]
8.
9.
Bifosfonati....................... 10.
Altri adiuvanti analgesici 11.
Entrambi [3]
6. FANS...............................
No Sì [0] [1]
* EV Endovenosa IM Intramuscolare IN Intranasale PO Orale
RT Rettale SC Sottocutanea SU Sublinguale TO Transmucosale orale
TS Transdermico
µg/h
µg/h
6-1. 6-2. 6-3.
7-1. 7-2. 7-3.
8-1. 8-2. 8-3.
9-1. 9-2. 9-3.
10-1. 10-2. 10-3.
11-1. 11-2. 11-3.
TERAPIA FISSA
In caso di avvenuto cambio di oppioide (switch) precisare la causa: 1.
[1] Analgesia inadeguata....................................................
Entrambe......................................................................
Grave Reazione avversa al farmaco, non controllabile
Incapacità alla assunzione orale...................................
[2]
[3]
[4]
Indicare, in ogni caso, la terapia oppioide ATC e il dosaggio:
2. 3.

39
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 6 — Giorno 28
TRATTAMENTI ANTALGICI IMPOSTATI ALLA VISITA ATTUALE (2 di 2)
TERAPIA VARIABILE
Terapia aggiuntiva al trattamento attuale ATC
Al paziente viene impostata una terapia aggiuntiva “extra” per dare migliore copertura al
trattamento ATC?
1.
se Sì, indicare:
Morfina orale IR.......
Morfina sottocutanea
Oppioide aggiuntivo
Dose media
giornaliera
mg
mg
No Sì [0] [1]
Terapia oppioide RESCUE per BTP
Al paziente viene impostata una terapia oppioide RESCUE
per presenza di BTP?
2.
se Sì, indicare:
Fentanyl transmucosale
Morfina endovena.........
Singola dose
µg/dose
mg/dose
Oppioide rescue
No Sì [0] [1]
1-1.
1-3.
1-2.
1-4.
2-1.
2-3.
2-2.
2-4.

40
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 6 — Giorno 28
ALTRE TERAPIE
1.
se Variata, indicare quali principi attivi:
Terapie antineoplastiche?
2. Terapie per le patologie concomitanti?
3. Terapie sintomatiche-palliative?
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
se Sì, indicare se variata rispetto alla situazione pregressa:
Invariata Variata [1] [2]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
Invariata Variata [1] [2]
Invariata Variata [1] [2]
se Variata, indicare quali principi attivi:
se Variata, indicare quali principi attivi:
1-1.
1-1-1.
1-1-2.
1-1-3.
2-1.
3-1.
2-1-1.
2-1-2.
2-1-3.
2-1-4.
3-1-1.
3-1-2.
3-1-3.
3-1-4.
3-1-5.
3-1-6.
3-1-7.
3-1-8.
3-1-9.
3-1-10.
41
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 6 — Giorno 28
REAZIONI AVVERSE DOVUTE ALLA TERAPIA ANTALGICA
In relazione alla terapia antalgica data nella visita precedente, il paziente riferisce di aver sof-
ferto di qualche disturbo?
1.
riportare dal questionario del paziente quanto compilato alla visita 6 (rispondere a tutte le domande)
No Sì [0] [1]
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No Un po’ Molto Moltissimo [0] [1] [2] [3]
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito.....................................................
1-1.
1-2.
1-3.
1-4.
1-5.
1-6.
1-7.
1-8.
1-9.
1-10.
1-11.
1-12.

42
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Visita 6 — Giorno 28
Si ricorda la necessità di eseguire il prelievo ematico per la tipizzazione genica secondo le
istruzioni presenti negli allegati del protocollo, se non già eseguito.
EVENTI AVVERSI
A suo giudizio, il paziente ha sofferto di qualsiasi evento clinico dannoso, come riportato
in protocollo alla pagina ...?
1.
In caso affermativo segnalare gli eventi avversi alla pagina successiva.
INTERRUZIONE PREMATURA DELLO STUDIO
No Sì [0] [1]
In caso di interruzione prematura dello studio compilare direttamente la sezione finale della
CRF “Completamento dello studio”.
3. Spuntare quando TUTTE le informazioni relativa a questa visita sono state inserite
Il paziente ha interrotto prematuramente lo studio? 2. No Sì [0] [1]
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
43
Un Evento avverso è definito serio se è fatale, se pone il paziente in pericolo di vita, se è disabilitante/inabilitante per il paziente, se richiede o prolunga l’ospedalizzazione (a meno che essa
non sia stata pianificata in precedenza) o se richiede interventi per evitare una delle condizioni precedenti.
Ogni evento definito “serio” richiede la notifica entro 24 ore al centro di coordinamento dell’Istituto Mario Negri tramite fax all’attenzione della Dr.ssa Maria Teresa Greco,
gruppo CERP, al nr. di fax 02-33200231
Il medico è pregato di inserire un sintomo per ciascuna riga per: - tutti i nuovi eventi avversi
- tutti gli eventi avversi precedenti che sono peggiorati (colonna “Severità”)
- tutti gli eventi avversi precedenti che hanno modificato la classe di ”Relazione” con il farmaco in studio
- tutte le condizioni mediche precedenti allo studio che sono peggiorate
Visita 6 — Giorno 28 — Eventi avversi
Severità 1 Lieve
2 Moderata
3 Grave
Relazione con i farmaci in studio 1 Certa
2 Probabile
3 Possibile
4 Dubbia
5 Nessuna
6 Sconosciuta
Azione intrapresa 0 Nessuna azione
1 Aggiustamento della dose/interruzione temporanea
2 Interruzione permanente del farmaco in studio
3 Somm.ne di terapia concomitante
4 Somm.ne di terapia non farmacologica
5 Ospedalizzazione o suo prolungamento
Studio CERP1
Evento avverso Severità Relazione Data inizio Azione intrapresa Continua? Data fine
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-1. 1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
L’evento
è “serio” ?
2-1.
3-1.
4-1.
5-1.
6-1.
7-1.
8-1.
9-1.
10-1.
11-1.
12-1.
1-2.
2-2.
3-2.
4-2.
5-2.
6-2.
7-2.
8-2.
9-2.
10-2.
11-2.
12-2.
1-3.
2-3.
3-3.
4-3.
5-3.
6-3.
7-3.
8-3.
9-3.
10-3.
11-3.
12-3.
1-4.
2-4.
3-4.
4-4.
5-4.
6-4.
7-4.
8-4.
9-4.
10-4.
11-4.
12-4.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-5.
2-5.
3-5.
4-5.
5-5.
6-5.
7-5.
8-5.
9-5.
10-5.
11-5.
12-5.
5 0 1 2 3 4
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-6.
2-6.
3-6.
4-6.
5-6.
6-6.
7-6.
8-6.
9-6.
10-6.
11-6.
12-6.
gg mm aaaa
gg mm aaaa
gg mm aaaa
1-7.
2-7.
3-7.
4-7.
5-7.
6-7.
7-7.
8-7.
9-7.
10-7.
11-7.
12-7.

44
Versione 1.1 (24 mag 2010)
Centro n° Paziente n° Studio CERP1
Scheda di fine studio
COMPLETAMENTO DELLO STUDIO
Data ultima assunzione del trattamento con
l’oppioide ATC assegnato all’inizio dello studio
1.
se No, indicare il motivo principale:
gg mm aaaa
Il paziente ha concluso regolarmente lo studio ? 2. No Sì [0] [1]
SAE/SADR Insoddisfacente effetto analgesico
Violazioni di protocollo................................................
Il paziente ha ritirato il consenso informato.................
Paziente perso al follow-up..........................................
[5]
Problemi amministrativi...............................................
Decesso.........................................................................
Altro..............................................................................
4.
se Decesso, indicare la data di morte 8. gg mm aaaa
Scarsa compliance / scarsa collaborazione del paziente
[2] [1]
[2]
[3]
[1]
[4]
[7]
Paziente trasferito ad altro centro di cura.....................
[6]
9. E’ stato eseguito il prelievo ematico per l’analisi dell’assetto genico?
[8]
se Altro, specificare ____________________________________________________________ 7.
No Sì [0] [1]
Entrambi [3]
(indicare la data del decesso)
se Sì, indicare quale: Terapia farmacologica con altri tipi di farmaci 5-1-1.
Terapia non farmacologica...............................
Sedazione palliativa-terminale.........................
Altro.................................................................
specificare _____________________________________________________
specificare _____________________________________________________
specificare _____________________________________________________
5-1-2.
5-1-4.
5-1-7.
E’ stata adottata una scelta terapeutica antalgica in sostituzione dell’uso degli oppioidi? No Sì [0] [1]
5-1.
5-1-3.
5-1-5.
5-1-6.
Altre cause................................................................
Abbandono del trattamento ATC a base di oppioidi
[2]
[1]
Se il paziente ha abbandonato il trattamento ATC a base di oppioidi, indicare la causa: 5.
Se il paziente non ha concluso regolarmente lo studio per “Altre cause”, indicare il motivo
principale:
6.
(vedi domanda 6.)
(vedi domanda 5.)
se No, indicare la data di interruzione prematura 3. gg mm aaaa

1
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
Visita 1
Data giorno mese anno
Questionario per il paziente
DA COMPILARE NEL GIORNO DI INGRESSO NELLO STUDIO
VALUTAZIONE DEL DOLORE
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
MEDIO nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispondente
all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
VALUTAZIONE DEI SINTOMI
Se nel corso dell’ultima settimana ha sofferto di qualche sintomo tra quelli di seguito elencati, per favore ne indichi
l’entità:
Studio CERP1
DOLORE MEDIO
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
PEGGIORE nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispon-
dente all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
DOLORE PEGGIORE
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito......................................................
Un po’ Molto Moltissimo Sintomo
Altro........................................................
Altro........................................................
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
VALUTAZIONE DEGLI STATI D’ANIMO
Se nel corso dell’ultima settimana ha sofferto di qualche disturbo tra quelli di seguito elencati, per favore ne indichi
l’entità:
Si è sentito(a) teso(a) ?...........................
Si è preoccupato(a) ?..............................
Si è sentito(a) irritabile ?........................
Si è sentito(a) depresso ?........................
No Un pò Molto Moltissimo Disturbo

2
Versione 1.1 (24 mag 2010)
Visita 2
Data Giorno mese anno
Questionario per il paziente
DA COMPILARE DOPO 72 ORE DALL’ INGRESSO NELLO STUDIO
VALUTAZIONE DEI SINTOMI
Centro n° Paziente n°
Studio CERP1
VALUTAZIONE DEL DOLORE
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
MEDIO nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispondente
all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
DOLORE MEDIO
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
PEGGIORE nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispon-
dente all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
DOLORE PEGGIORE
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
Se dalla visita precedente ha sofferto di qualche sintomo tra quelli di seguito elencati, per favore ne indichi l’entità:
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito......................................................
Un po’ Molto Moltissimo Sintomo

3
Versione 1.1 (24 mag 2010)
Visita 3
Data giorno mese anno
Questionario per il paziente
DA COMPILARE DOPO 7 GIORNI DALL’ INGRESSO NELLO STUDIO
Centro n° Paziente n°
Studio CERP1
VALUTAZIONE DEL DOLORE
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
MEDIO nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispondente
all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
DOLORE MEDIO
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
PEGGIORE nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispon-
dente all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
DOLORE PEGGIORE
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
VALUTAZIONE DEI SINTOMI
Se dalla visita precedente ha sofferto di qualche sintomo tra quelli di seguito elencati, per favore ne indichi l’entità:
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito......................................................
Un po’ Molto Moltissimo Sintomo

4
Versione 1.1 (24 mag 2010)
Visita 4
Data giorno mese anno
Questionario per il paziente
DA COMPILARE DOPO 14 GIORNI DALL’ INGRESSO NELLO STUDIO
Centro n° Paziente n°
Studio CERP1
VALUTAZIONE DEL DOLORE
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
MEDIO nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispondente
all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
DOLORE MEDIO
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
PEGGIORE nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispon-
dente all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
DOLORE PEGGIORE
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
VALUTAZIONE DEI SINTOMI
Se dalla visita precedente ha sofferto di qualche sintomo tra quelli di seguito elencati, per favore ne indichi l’entità:
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito......................................................
Un po’ Molto Moltissimo Sintomo

5
Versione 1.1 (24 mag 2010)
Visita 5
Data Giorno mese anno
Questionario per il paziente
DA COMPILARE DOPO 21 GIORNI DALL’ INGRESSO NELLO STUDIO
Centro n° Paziente n°
Studio CERP1
VALUTAZIONE DEL DOLORE
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
MEDIO nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispondente
all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
DOLORE MEDIO
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
PEGGIORE nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispon-
dente all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
DOLORE PEGGIORE
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
VALUTAZIONE DEI SINTOMI
Se dalla visita precedente ha sofferto di qualche sintomo tra quelli di seguito elencati, per favore ne indichi l’entità:
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito......................................................
Un po’ Molto Moltissimo Sintomo

6
Versione 1.1 (24 mag 2010)
Visita 6
Data giorno mese anno
Questionario per il paziente
DA COMPILARE DOPO 28 GIORNI DALL’ INGRESSO NELLO STUDIO
Centro n° Paziente n°
Studio CERP1
VALUTAZIONE DEL DOLORE
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
MEDIO nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispondente
all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
DOLORE MEDIO
Per favore, facendo un cerchio intorno al numero che meglio ne descrive l’intensità, valuti il suo DOLORE
PEGGIORE nelle ultime 24 ore. La scala descrive l’intensità del dolore partendo dal valore zero, corrispon-
dente all’assenza di dolore, fino al valore 10, corrispondente al dolore più forte che riesce ad immaginare
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
DOLORE PEGGIORE
0 1 2 3 4 5 6 7 8 9 10 nessun
dolore
il dolore più
forte che possa
immaginare
VALUTAZIONE DEI SINTOMI
Se dalla visita precedente ha sofferto di qualche sintomo tra quelli di seguito elencati, per favore ne indichi l’entità:
Sonnolenza..............................................
Confusione/Difficoltà di concentrazione
Nausea.....................................................
Vomito....................................................
Stitichezza...............................................
Bocca secca............................................
Allucinazioni..........................................
No
Spasmi/Dolori muscolari........................
Mal di stomaco........................................
Difficoltà ad urinare................................
Difficoltà a respirare...............................
Prurito......................................................
Un po’ Molto Moltissimo Sintomo

1
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
Scheda rilevamento SAE (Serious Adverse Event) - pag. 1 di 2
Data dell’insorgenza della reazione 3.
Gravità
Decesso.............................................................................. [1]
[2]
gg mm aaaa
Invalidità grave o permanente............................................
Pericolo di vita.................................................................... [4]
[5] Anomalia congenita/deficit alla nascita.............................
[6]
5.
Descrizione della reazione 4.
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
Ospedalizzazione/prolungamento ospedalizzazione..........
[3]
Altre condizioni mediche importanti..................................
Data di nascita gg mm aaaa
1. Sesso 2. Maschio Femmina [1] [2]
INFORMAZIONI DEL PAZIENTE
INFORMAZIONI EVENTO
Azioni intraprese 6.
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
Esito in data
Risoluzione completa.................. [1]
[2]
Miglioramento.............................
Reazione invariata o peggiorata.. [4]
[5] Decesso........................................
[6]
7.
Risoluzione con postumi.............
[3]
Non disponibile...........................
7-1.
Studio CERP1
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
gg mm aaaa
7-2.
7-3.
7-4.
7-5.

2
Versione 1.1 (24 mag 2010)
Centro n° Paziente n°
Scheda rilevamento SAE (Serious Adverse Event) - pag. 2 di 2
INFORMAZIONI SUL FARMACO
A)
B)
C)
Data ultima Via di
1.
Nome farmaco Dosaggio/die somm.ne somm.ne
No Sì [0] [1] Il farmaco è stato sospeso?
Dopo la sospensione, la reazione è migliorata?
Dopo la sospensione, il farmaco è stato ripreso?
Dopo la risomministrazione, i sintomi sono ricomparsi?
NESSO DI CAUSALITA’
A giudizio del medico, qual è il nesso di causalità tra l’evento ed il trattamento in studio? 4.
Certa [1] [2] Possibile Dubbia [4] [5] Nessuna [6] Probabile [3] Sconosciuta
ALTRI PRODOTTI CONCOMITANTI
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
Altre sostanze (piante officinali, omeopatici,integratori, etc..)
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
______________________________________________________________________________
INFORMAZIONI SULLA SEGNALAZIONE
Data della compilazione Status rapporto: Iniziale [1] [2] Follow-up
5.
6.
7.
9. 10.
Farmaci, dosaggi, vie di somm.ne, durata
Inviare via fax copia della scheda all’attenzione della dr.ssa Maria Teresa Greco, CERP,
Istituto Mario Negri, nr. fax 02-33200231
gg mm aaaa
gg mm aaaa
Firma 8.
Studio CERP1
[3] Finale
Dati del segnalatore:
Nome e cognome
1-1. 1-2. 1-3.
1-4.
1-5.
1-6.
1-7.
2.
Il farmaco è stato sospeso?
Dopo la sospensione, la reazione è migliorata?
Dopo la sospensione, il farmaco è stato ripreso?
Dopo la risomministrazione, i sintomi sono ricomparsi?
gg mm aaaa 2-1. 2-2. 2-3.
2-4.
2-5.
2-6.
2-7.
3.
Il farmaco è stato sospeso?
Dopo la sospensione, la reazione è migliorata?
Dopo la sospensione, il farmaco è stato ripreso?
Dopo la risomministrazione, i sintomi sono ricomparsi?
gg mm aaaa 3-1. 3-2. 3-3.
3-4.
3-5.
3-6.
3-7.
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]
No Sì [0] [1]


















