
Progettazione e sintesi dei farmaci · micobatteri ha severamente limitato il progredire dei...
60
1 Progettazione e sintesi dei farmaci 1. Il Punto di Partenza (Lead) 1. 1. Introduzione La progettazione di ligandi recettoriali, a parte alcune lievi differenze che verranno tempestivamente indicate, non differisce sostanzialmente dal processo di ricerca e sviluppo di ogni altro farmaco: può essere fatta a partire da punti di partenza molto diversi tra loro. E’ chiaro che l'approccio più immediato sarebbe quello di conoscere la topografia del sito di interazione dei ligandi naturali per disegnare la molecola nuova su di esso. Purtroppo le informazioni che al momento attuale sono disponibili, non permettono di seguire questa via che è invece percorribile per i prodotti destinati ad interagire con gli enzimi. Infatti, mentre per un numero crescente di enzimi è nota la struttura tridimensionale, e quindi la topografia del sito catalitico, al momento attuale non è stato possibile risolvere con la necessaria accuratezza la struttura di alcun recettore ed i modelli recettoriali disponibili sono frutto di elaborazioni teoriche che, per quanto molto utili, non permettono una progettazione sufficientemente accurata. Se non è possibile partire dalla struttura del sito attivo è però generalmente nota la struttura del ligando naturale di un recettore. Questo
-
Upload
truongphuc -
Category
Documents
-
view
216 -
download
0
Transcript of Progettazione e sintesi dei farmaci · micobatteri ha severamente limitato il progredire dei...

1
Progettazione e sintesi dei farmaci
1. Il Punto di Partenza (Lead)
1. 1. Introduzione
La progettazione di ligandi recettoriali, a parte alcune lievi differenze
che verranno tempestivamente indicate, non differisce sostanzialmente dal
processo di ricerca e sviluppo di ogni altro farmaco: può essere fatta a
partire da punti di partenza molto diversi tra loro.
E’ chiaro che l'approccio più immediato sarebbe quello di conoscere
la topografia del sito di interazione dei ligandi naturali per disegnare la
molecola nuova su di esso. Purtroppo le informazioni che al momento
attuale sono disponibili, non permettono di seguire questa via che è invece
percorribile per i prodotti destinati ad interagire con gli enzimi. Infatti,
mentre per un numero crescente di enzimi è nota la struttura
tridimensionale, e quindi la topografia del sito catalitico, al momento
attuale non è stato possibile risolvere con la necessaria accuratezza la
struttura di alcun recettore ed i modelli recettoriali disponibili sono frutto
di elaborazioni teoriche che, per quanto molto utili, non permettono una
progettazione sufficientemente accurata.
Se non è possibile partire dalla struttura del sito attivo è però
generalmente nota la struttura del ligando naturale di un recettore. Questo

2
può essere quindi un adatto prodotto di riferimento per il disegno di nuove
molecole come verrà descritto più ampiamente nella sezione 1.7 (Sintesi
razionale). Da questo punto di vista la progettazione di farmaci che agiscono
sui canali ionici è ovviamente ancora più difficoltosa. In realtà tutti i
farmaci attualmente utilizzati che agiscono con questo meccanismo, sono
stati ottenuti in modo casuale o attraverso modulazione molecolare di
prodotti la cui azione a livello dei canali era stata evidenziata dall'uso
clinico o da un ampio screening farmacologico.
Altri approcci di uso generale nella Chimica Farmaceutica e che si
possono utilizzare per individuare ligandi recettoriali sono: la modifica di
farmaci già noti; lo screening a tappeto o mirato di prodotti di origine
naturale o sintetica; la identificazione di prodotti di origine naturale sia
vegetale che animale; la dissociazione e amplificazione di effetti secondari di
molecole già utilizzate e studiate. A questi si è recentemente aggiunta la
possibilità di realizzare librerie di prodotti di sintesi che offrono la
possibilità di valutare contemporaneamente e in brevissimo tempo migliaia
di prodotti.
Generalmente, quale che sia la via scelta, il prodotto di partenza è
una molecola che necessita di un lavoro più o meno intenso di modulazione
molecolare per ottimizzare la sua interazione con il recettore in termini di
potenza, selettività, efficacia. Se poi lo scopo è quello di ottenere un
farmaco, il cammino sarà ancora più lungo, essendo necessario prendere in

3
considerazione altri fattori di enorme importanza quali la farmacocinetica,
la tossicità, la formulazione.
Perché un processo di ottimizzazione sia possibile e, soprattutto nel
caso di una utilizzazione industriale, rapido ed efficace, è necessario che sia
disponibile un saggio biologico specifico, rapido e possibilmente poco
costoso. Questo ha condizionato da sempre lo sviluppo di farmaci. Mentre
in generale lo sviluppo di antimicrobici è stato rapido per la relativa
semplicità dei saggi microbiologici, la difficoltà di coltivare in vitro i
micobatteri ha severamente limitato il progredire dei farmaci
antitubercolari e antileprotici. Molte delle difficoltà nell'individuare efficaci
farmaci contro le malattie neurodegenerative tipo Alzheimer, dipendono
dalla mancanza di attendibili protocolli di valutazione della attività dei
farmaci, sia a livello preclinico che clinico.
La messa a punto dei metodi di binding, che permettono la
determinazione della affinità recettoriale di molte sostanze in pochissimo
tempo, il perfezionamento dei metodi di analisi enzimatica, l'uso del
computer nel trattamento dei dati, insieme allo sviluppo della robotica,
hanno determinato in questi ultimi anni una svolta, rendendo nuovamente
praticabile lo screening di un gran numero di prodotti.
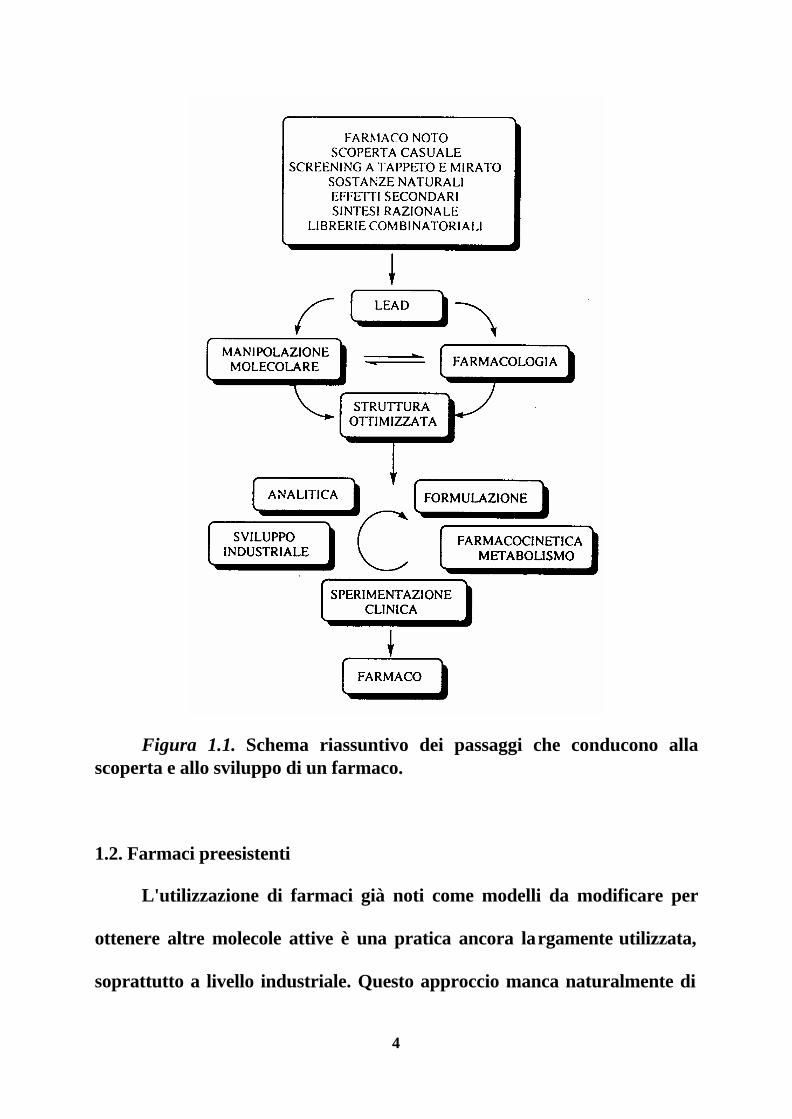
4
Figura 1.1. Schema riassuntivo dei passaggi che conducono alla scoperta e allo sviluppo di un farmaco.
1.2. Farmaci preesistenti
L'utilizzazione di farmaci già noti come modelli da modificare per
ottenere altre molecole attive è una pratica ancora largamente utilizzata,
soprattutto a livello industriale. Questo approccio manca naturalmente di

5
originalità ed ha una giustificazione prevalentemente economica; i prodotti
così ottenuti cadono spesso nella categoria dei cosiddetti “me too
compounds” che permettono a più ditte farmaceutiche di accedere, con
limitati investimenti, a settori di mercato particolarmente remunerativi.
Sebbene molto criticato, questo approccio ha anche una sua validità dal
punto di vista terapeutico in quanto conduce in molti casi a prodotti con
caratteristiche farmacologiche, farmacocinetiche e tossicologiche migliori di
quelle del prodotto di partenza. Inoltre il lavoro fatto permette di stabilire
tutta una serie di relazioni tra la struttura e l'attività che sono spesso
essenziali per la definizione del farmacoforo e del meccanismo di azione. Un
esempio di questo approccio sono i numerosi farmaci anti-H2 derivati dalla
cimetidina, tra i quali ranitidina e famotidina.
Del resto può accadere che durante la ricerca di analoghi di un
farmaco già noto, le modificazioni introdotte producano caratteristiche
farmacologiche del tutto diverse, come è successo nello sviluppo della
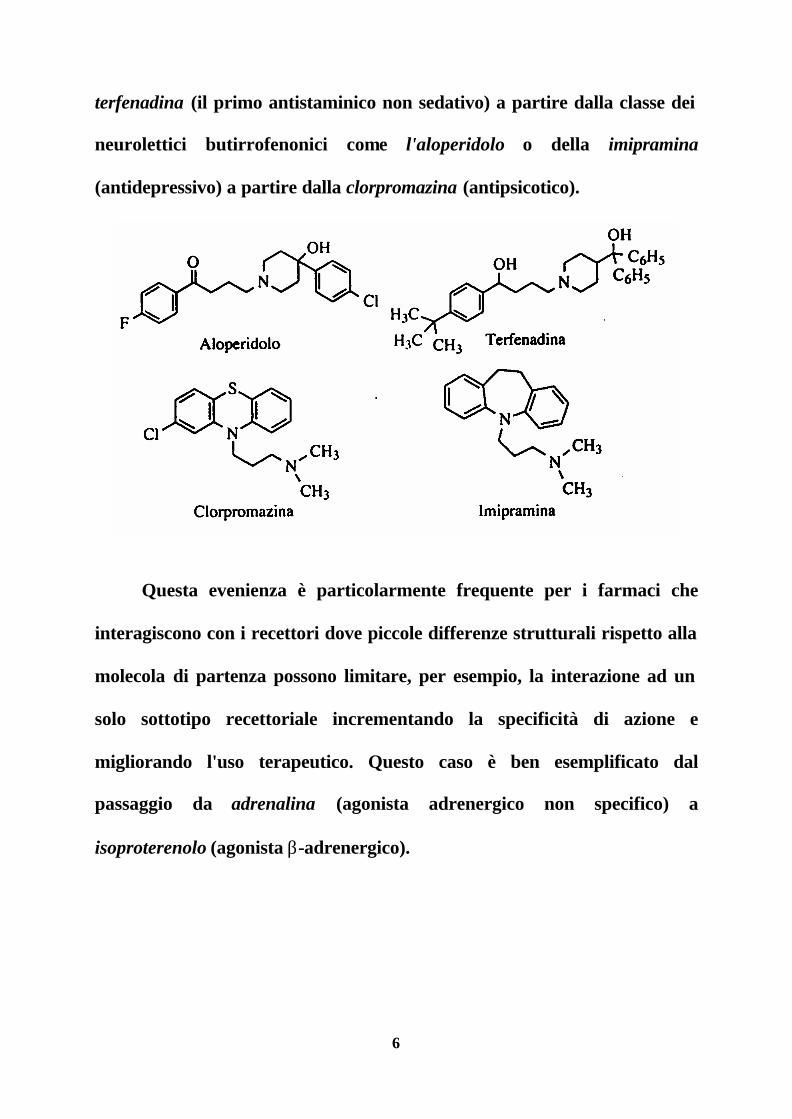
6
terfenadina (il primo antistaminico non sedativo) a partire dalla classe dei
neurolettici butirrofenonici come l'aloperidolo o della imipramina
(antidepressivo) a partire dalla clorpromazina (antipsicotico).
Questa evenienza è particolarmente frequente per i farmaci che
interagiscono con i recettori dove piccole differenze strutturali rispetto alla
molecola di partenza possono limitare, per esempio, la interazione ad un
solo sottotipo recettoriale incrementando la specificità di azione e
migliorando l'uso terapeutico. Questo caso è ben esemplificato dal
passaggio da adrenalina (agonista adrenergico non specifico) a
isoproterenolo (agonista β-adrenergico).

7
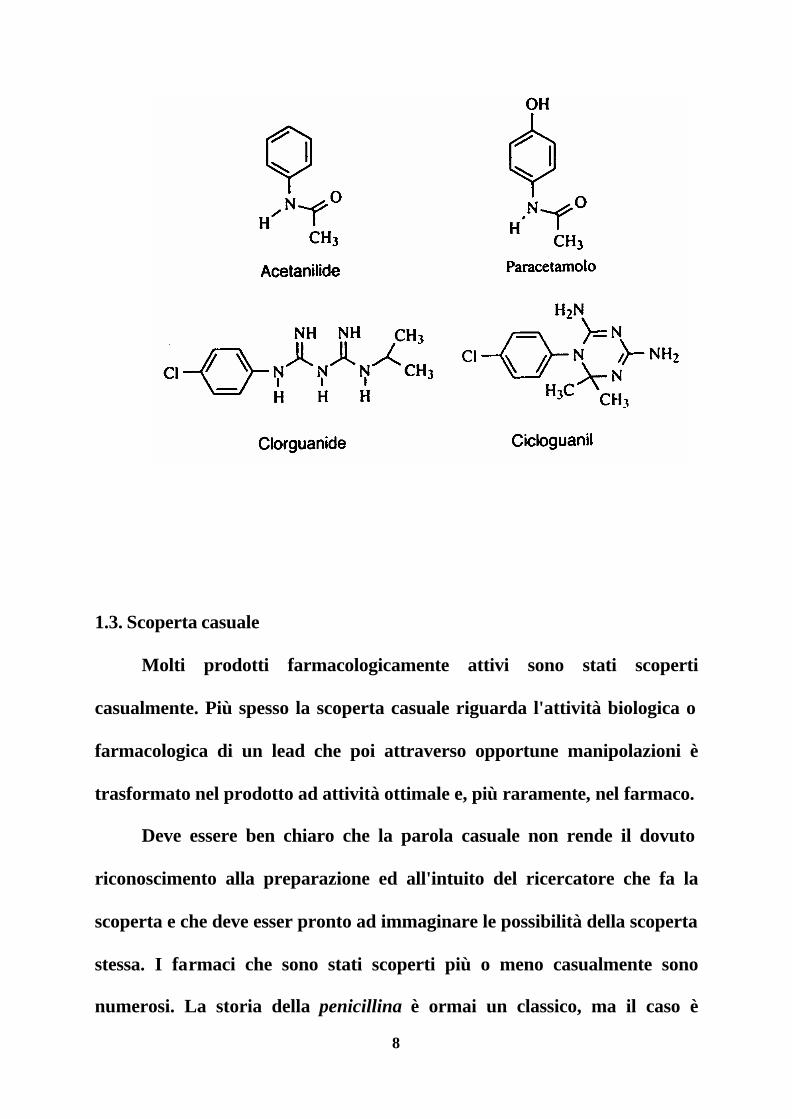
Un caso particolare di questo approccio può essere considerato
l'identificazione e lo studio di metaboliti che è del resto una delle tappe
fondamentali nello sviluppo di un farmaco. Poiché in genere i metaboliti
sono meno tossici, la loro eventuale alta attività farmacologica può portare
all'individuazione di una nuova entità molecolare. Inoltre questo studio può
mettere in evidenza trasformazioni che sono essenziali per l'attività e quindi
aprire la via allo studio e all'utilizzazione di nuove strutture molecolari. Il
paracetamolo, un metabolita della acetanilide, ed il cicloguanil, un
metabolita della clorguanide, sono due farmaci identificati in seguito allo
studio di metaboliti.
8
1.3. Scoperta casuale
Molti prodotti farmacologicamente attivi sono stati scoperti
casualmente. Più spesso la scoperta casuale riguarda l'attività biologica o
farmacologica di un lead che poi attraverso opportune manipolazioni è
trasformato nel prodotto ad attività ottimale e, più raramente, nel farmaco.
Deve essere ben chiaro che la parola casuale non rende il dovuto
riconoscimento alla preparazione ed all'intuito del ricercatore che fa la
scoperta e che deve esser pronto ad immaginare le possibilità della scoperta
stessa. I farmaci che sono stati scoperti più o meno casualmente sono
numerosi. La storia della penicillina è ormai un classico, ma il caso è

9
intervenuto anche nella scoperta di farmaci appartenenti a classi molto
diverse quali isoniazide, iproniazide, clonidina, antabuse, ciclosporina A e
molti altri.
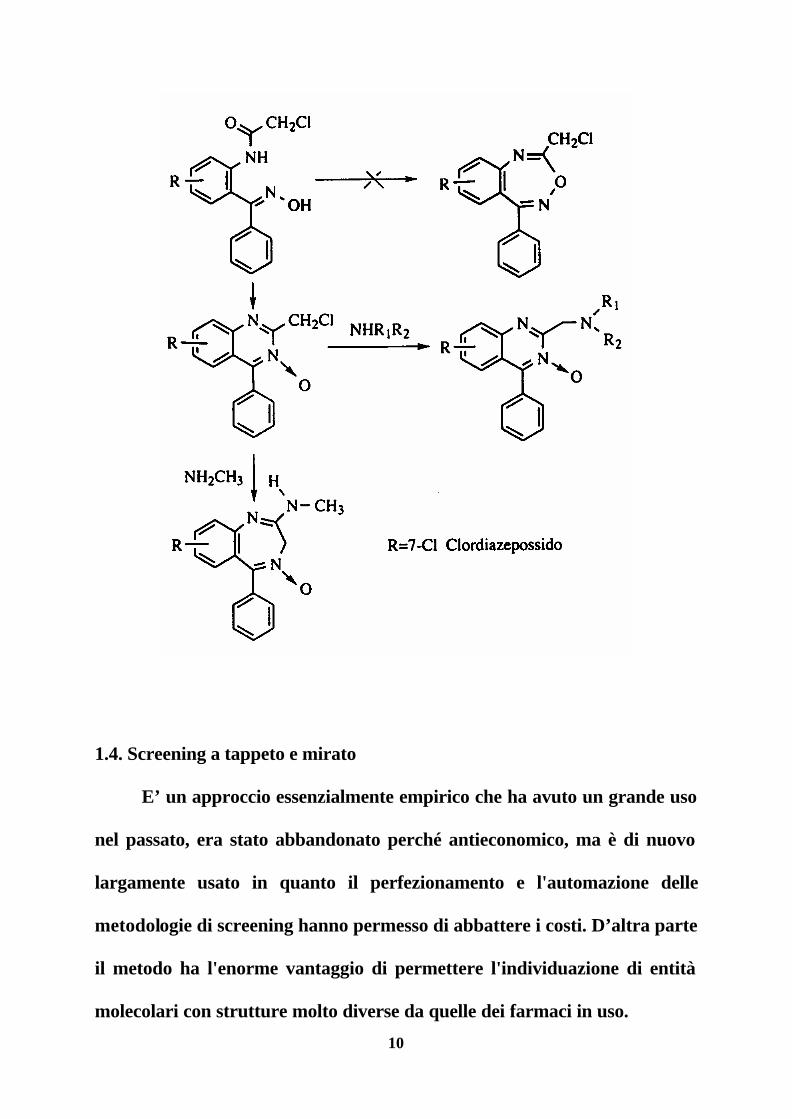
Per fare un esempio che resti nel campo dei recettori basterà
ricordare la storia delle benzodiazepine raccontata dal suo scopritore L.H.
Sternbach. Negli anni cinquanta il suo gruppo di ricerca presso la
Hoffmann-La Roche era impegnato nella ricerca di tranquillanti del tipo
del meprobamato e a questo scopo aveva progettato la sintesi di una serie di
derivati benzoossadiazepinici per poi passare a derivati del tipo
chinazolinico 3-ossido quando si verificò che i prodotti ottenuti non erano
quelli attesi. Vennero sintetizzati molti derivati, tutti inattivi come
tranquillanti. Fu quindi deciso di abbandonare il progetto inviando alla
sperimentazione farmacologica gli ultimi campioni tra i quali uno della cui
struttura non si era completamente sicuri. Si trattava del clordiazepossido,
ottenuto da una reazione inaspettata e con una struttura del tutto originale
rispetto al meprobamato, che avrebbe aperto la strada alla sintesi di una
miriade di congeneri ed alla identificazione di un nuovo meccanismo di
azione e di un nuovo tipo di recettori.
Malgrado i progressi nello screening di massa e nella
razionalizzazione degli approcci, il caso molto probabilmente continuerà ad
avere un ruolo nella scoperta dei farmaci anche per il futuro.
10
1.4. Screening a tappeto e mirato
E’ un approccio essenzialmente empirico che ha avuto un grande uso
nel passato, era stato abbandonato perché antieconomico, ma è di nuovo
largamente usato in quanto il perfezionamento e l'automazione delle
metodologie di screening hanno permesso di abbattere i costi. D’altra parte
il metodo ha l'enorme vantaggio di permettere l'individuazione di entità
molecolari con strutture molto diverse da quelle dei farmaci in uso.

11
Può essere utilizzato fondamentalmente in due modi opposti:
a) provando un prodotto su un elevato numero di saggi biologici.
b) provando molti prodotti su un solo test biologico.
Naturalmente sono possibili soluzioni intermedie a seconda delle
necessità. I prodotti saggiati possono avere le più diverse origini, naturale o
sintetica, possono essere prodotti unici o miscele di prodotti.
Il primo di questi approcci è stato quello sostanzialmente usato e
perfezionato da Paul Jansen il quale ha utilizzato una batteria di recettori
per eseguire prove di binding su molecole di sintesi, ottenute per
combinazione di diversi farmacofori. Da questa ricerca sono derivate molte
molecole, ottimizzate successivamente in farmaci di successo quali
l'aloperidolo, un neurolettico già visto ed il fentanile (analgesico).
Il secondo approccio è quello attualmente più utilizzato, soprattutto
nella ricerca sui farmaci dei recettori. Quando un metodo di binding su un
sistema recettoriale è stato messo a punto, è possibile provare centinaia e

12
migliaia di sostanze in tempi relativamente brevi. Ciò permette, soprattutto
alle industrie, di analizzare le migliaia di prodotti che fanno parte della loro
collezione e tutti gli altri che sarà possibile reperire. Non è improbabile che
al termine della ricerca si siano individuati alcuni prodotti con una capacità
di legame nell'ordine micromolare. Questi prodotti possono essere
ottimizzati con i procedimenti che verranno illustrati in seguito, per
arrivare ad affinità nell'ordine nanomolare che sono ottimali per lo
sviluppo di un farmaco. Un esempio di questo approccio è lo sviluppo di E-
2020 (Donepezil), un prodotto ad azione anticolinesterasica che è in fase di
registrazione come farmaco anti-Alzheimer. Lo screening a tappeto dei
prodotti della collezione della Eisai sull'enzima acetilcolinesterasi, ha
permesso di individuare un derivato piperazinico con una IC50 nel range
micromolare che è stato successivamente ottimizzato al prodotto finale che
ha attività nel range nanomolare.

13
Lo screening può riguardare, come già accennato, anche miscele di
prodotti. Si può trattare di miscele ottenute attraverso la metodologia della
sintesi combinatoriale (librerie sintetiche), che verrà descritta in seguito,
ma può anche trattarsi di miscele di origine naturale (librerie naturali)
come ad esempio i liquidi di fermentazione di microrganismi. L'asperlicina,
un prodotto della fermentazione dell'Aspergillus alliaceus che ha poi
originato un farmaco non peptidico in grado di bloccare i recettori della
colicistochinina A (CCK-A) è stato identificato in questo modo (vedi sezione
2.3.2).
Il problema principale delle librerie naturali rispetto a quelle
sintetiche è nell'isolamento e nella determinazione della struttura del
prodotto attivo, che malgrado l'enorme progresso fatto dalle relative
metodologie negli ultimi anni è un processo lento, difficile e costoso. Il
vantaggio inestimabile è quello di offrire una enorme varietà di strutture,
difficilmente immaginabili e realizzabili dal chimico sintetico.

14
1.5. Sostanze di origine naturale
I prodotti di origine naturale, in particolare quelli derivati dal regno
vegetale, sono la più antica fonte di sostanze biologicamente attive e di
farmaci. L'evoluzione ha creato una miriade di specie differenti che
debbono sopravvivere nell'ambiente in competizione tra loro. Ciò ha
condotto alla produzione di innumerevoli molecole (metaboliti secondari)
dotate di attività biologica che possono essere utilizzate dall'uomo,
direttamente o dopo opportune manipolazioni, come farmaci.
La scoperta e l'isolamento di prodotti quali atropina, chinina,
morfina, efedrina, d-tubocurarina, reserpina, digitossina, penicillina hanno
rappresentato tappe fondamentali nella medicina ed hanno prodotto una
miriade di derivati di importanza fondamentale in terapia e nello studio dei
meccanismi fisiologici degli organismi viventi.

15
Dopo un periodo di incertezza, dovuto principalmente alle accennate
difficoltà di separazione e identificazione dei principi attivi, questo tipo di
approccio sembra aver riacquistato vitalità e dal regno vegetale la ricerca si
è allargata in altri settori quale quello delle tossine di origine animale. La
ricerca può essere in qualche modo guidata dalle indicazioni della medicina
popolare (folk medicine) anche se spesso questo approccio ha portato a
risultati deludenti.
Tra le più recenti scoperte nel campo delle sostanze di origine
vegetale vanno menzionate la artemisina (antimalarico), il tassolo
(antitumorale); tra i prodotti di brodi di cultura di microrganismi la già
citata asperlicina (antagonista del recettore CCK-A) e la lovastatina
(inibitore della sintesi del colesterolo); tra i veleni e le tossine di origine
animale la tetrodotossina (bloccante dei canali del sodio), la α-
bungarotossina (antagonista dei recettori nicotinici) e la epibatidina
(agonista nicotinico). Questi prodotti sono utilizzati come farmaci o come
mezzi farmacologici di indagine e come modelli per lo sviluppo di nuovi
farmaci.

16
Un caso particolare di prodotti di origine naturale sono le sostanze di
natura peptidica. La scarsa stabilità metabolica e la biodisponibilità
usualmente molto bassa fanno sì che molto raramente essi possano essere
usati come tali. Più frequentemente vengono utilizzati come punti di
partenza per il disegno di analoghi non peptidici, dotati della stessa azione
ma che non presentano problemi farmacocinetici (peptidomimetici). Di
questo approccio si parlerà più avanti.
1.6. Amplificazione di effetti secondari
Durante la sperimentazione preclinica e clinica ma molto più
frequentemente in seguito all'uso su migliaia di pazienti, possono venire
osservati effetti collaterali che, mentre rappresentano un problema per
l'uso del farmaco, possono derivare da meccanismi di azione aventi
interesse terapeutico. Ciò può suggerire di tornare a manipolare la

17
struttura molecolare originale allo scopo di produrre molecole nelle quali
l'effetto sia massimizzato e possibilmente dissociato dall'effetto iniziale.
Da questo punto di vista i sulfamidici sono stati una vera miniera per
lo sviluppo di nuovi farmaci: gli ipoglicemizzanti orali del tipo delle
solfoniluree e i diuretici tiadiazinici rappresentano classi di farmaci di
importanza fondamentale ottenute dalla manipolazione di sulfamidici in
seguito alla osservazione clinica dei loro effetti secondari ipoglicemizzanti e
diuretici.
Nel campo dei farmaci che interagiscono con recettori, un classico
esempio di questo approccio riguarda lo sviluppo del neurolettico
clorpromazina. Questo farmaco, che è usato per il trattamento della
schizofrenia ed ha rivoluzionato il trattamento delle malattie psichiatriche,
è il frutto della osservazione dell'effetto tranquillizzante degli antistaminici
fenotiazinici in particolare della prometazina.
Gli ormoni androgeni come il testosterone hanno, oltre all'effetto
androgeno, quello anabolizzante che determina l'aumento della
utilizzazione dell'azoto proteico con conseguente incremento delle masse

18
muscolari. In alcuni casi patologici questa proprietà è molto utile, mentre
l'azione androgena diventa un effetto collaterale da evitare. Per questa
ragione la molecola del testosterone è stata modificata con lo scopo di
eliminare, o ridurre al minimo, l'azione androgena ed esaltare l'azione
anabolizzante. Ciò ha prodotto farmaci quali lo stanozololo in cui l'azione
androgena è praticamente scomparsa a favore di quella anabolizzante.
Purtroppo questi farmaci hanno anche trovato una utilizzazione non
terapeutica (doping).
Un grande vantaggio di questo approccio è che l'effetto secondario è
quasi sempre osservato direttamente sull'uomo, il che rappresenta una
garanzia per il futuro terapeutico del farmaco eventualmente sviluppato
sulla base di queste osservazioni.
1.7. Sintesi razionale
In questa sede verranno trattati tutti i tentativi di arrivare ad un
farmaco, o ad un lead attendibile, che prendono origine da conoscenze

19
fisiologiche, fisiopatologiche, biochimiche, farmacologiche, chimiche e
quindi hanno un ben preciso punto di partenza dal quale muoversi per
progettare molecole che si ritengono in grado di risolvere il problema
terapeutico o di dare ulteriori informazioni sul funzionamento di un sistema
biologico, nel nostro caso un recettore. Definire ciò un approccio razionale è
probabilmente una esagerazione, visti gli insuccessi che anche in questo
caso sono frequenti. Tuttavia è ragionevole ammettere che questo tentativo
di risolvere il problema poggia su basi più razionali di quelli descritti
precedentemente; il che non vuol assolutamente dire che sia in grado di
dare risultati più certi.
E’ del tutto evidente che il successo di una tale strategia dipende in
modo essenziale dalla conoscenza della fisiologia, della fisiopatologia e dei
processi biochimici che avvengono negli organismi viventi soprattutto a
livello cellulare e molecolare, nonché dalla conoscenza della natura chimica
e della struttura delle molecole coinvolte in questi processi. Di conseguenza,
una volta stabilito lo scopo della ricerca è necessario raccogliere tutti i dati
disponibili per poter progettare con la maggiore razionalità possibile le
molecole da sintetizzare e valutare. Nel caso questi dati siano largamente
insufficienti è probabilmente più opportuno utilizzare uno dei metodi
descritti precedentemente.
Naturalmente anche la modifica di un farmaco già noto,
razionalizzata sulla base di nuove conoscenze acquisite sul suo target,

20
ricade in questo tipo di approccio. Fino a pochi anni fa si riteneva che il sito
di interazione dei farmaci in grado di attivare i recettori muscarinici
dovesse essere di dimensioni ridotte. In effetti la quasi totalità dei
numerosissimi agonisti muscarinici noti, alcuni dei quali sono mostrati di
seguito, era costituita da piccole molecole molto compatte. Questo dogma è
entrato in crisi quando sono stati sintetizzati e trovati, potenti muscarinici
come la xanomelina, per la quale è stato necessario ipotizzare un sito
accessorio di interazione a livello di recettore per accogliere il residuo n-
esilico. E chiaro che sulla base di questi nuovi dati la progettazione di
agonisti muscarinici con strutture più voluminose diventa ragionevole.
Nel caso dei recettori, come già stato detto in precedenza, non si
hanno sufficienti informazioni sulla struttura recettoriale e sul sito di
interazione, a differenza di quanto accade per un numero ogni giorno
crescente di enzimi. Tuttavia, come per gli enzimi è noto il substrato, così
per la maggior parte dei recettori è noto l'agonista fisiologico. Così come si

21
fa per gli enzimi prima che la struttura del sito attivo sia determinata,
anche per i recettori è possibile disegnare agonisti ed antagonisti sulla base
della conoscenza del mediatore naturale.
Questo approccio ha avuto uno straordinario successo ed ha condotto
a molti farmaci e a un numero rilevante di utilissimi mezzi di indagine
farmacologica. La gran parte dei farmaci adrenergici e i loro antagonisti, di
quelli dopaminergici e i loro antagonisti, degli antistaminici, degli
anticolinergici, dei farmaci dei recettori triptaminergici, degli aminoacidi,
della adenosina, degli steroidi è stata ottenuta partendo dalla struttura dei
rispettivi mediatori modificata in modo più o meno razionale. Lo stesso si
può dire per i recettori che hanno come mediatori naturali dei peptidi,
anche se in questo caso la trasformazione è spesso più radicale a causa della
necessità di superare i problemi di stabilità e farmacocinetica cui già si è
fatto cenno. Qui di seguito sono riportati alcuni esempi di farmaci o mezzi
di indagine farmacologica ottenuti con questo approccio.
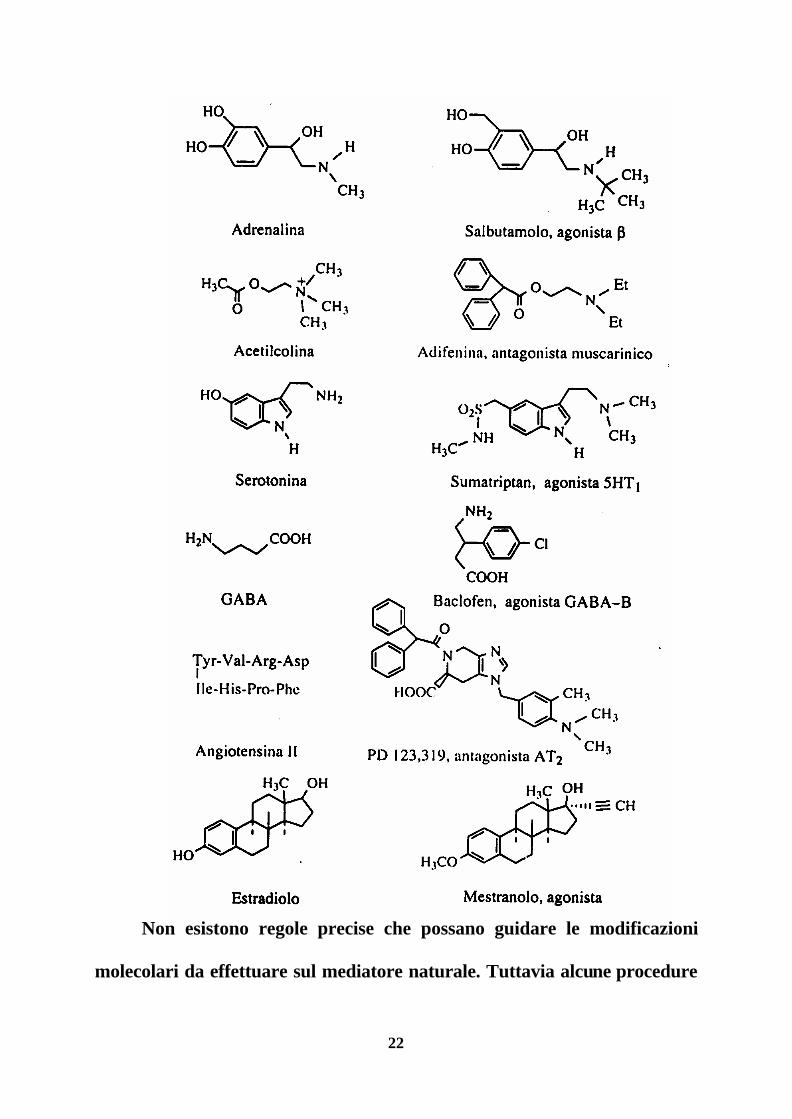
22
Non esistono regole precise che possano guidare le modificazioni
molecolari da effettuare sul mediatore naturale. Tuttavia alcune procedure

23
molto empiriche e non sempre estendibili ai diversi tipi di recettore sono
state elaborate durante l'intenso lavoro di ricerca sull'argomento. In
genere, per mantenere l'azione agonista la molecola non deve avere una
dimensione molto diversa da quella del mediatore naturale; al contrario,
l'aggiunta di gruppi ingombranti trasforma molto spesso gli agonisti in
antagonisti. L'introduzione di gruppi che modificano la struttura spaziale
dell'agonista naturale può indurre una specificità verso uno dei sottotipi
recettoriali attivati dallo stesso mediatore.
Seppure ancora largamente imperfetti e molto speculativi, i modelli
recettoriali che sono stati elaborati per quasi tutti i recettori più studiati,
possono essere di aiuto nell'individuare le regioni che permettono
l'inserimento di gruppi che aumentano l'affinità o sono in grado di
discriminare tra sottotipi.
Nel modulare la struttura di un mediatore naturale non va trascurata
la possibilità che i prodotti risultanti possano anche interferire con altri
meccanismi collegati al funzionamento dei sistemi recettoriali quali
l'accoppiamento con le proteine G, i sistemi di uptake, gli enzimi di
degradazione del mediatore. Un'accurata analisi farmacologica del
meccanismo di azione dei prodotti ottenuti è di conseguenza assolutamente
necessaria per definire il loro meccanismo di azione.
Infine va probabilmente inserita in questo capitolo la pratica di
sottoporre a screening gli intermedi di una sintesi razionale. Infatti, poiché

24
tali intermedi hanno spesso una forte somiglianza strutturale con il
prodotto finale, c'è una certa probabilità che anche essi siano attivi con lo
stesso meccanismo. Il farmaco antitubercolare isonicotinoilidrazide
(isoniazide) è stato scoperto in questo modo. Si stava cercando di
sintetizzare l'aldeide isonicotinica dalla quale ottenere il tiosemicabazone,
prodotto finale per il quale era prevedibile attività antitubercolare, e la
isonicotinoilidrazide era un intermedio della sintesi.
1.8. Librerie combinatoriali
Tradizionalmente, come si è visto nei paragrafi precedenti, nuovi lead
sono ottenuti con metodologie che conducono alla individuazione o alla
sintesi di prodotti ognuno dei quali viene sottoposto singolarmente
all'analisi biologica.
Fino a poco tempo fa, il collo di bottiglia di tutto il sistema era
appunto lo screening biologico ed i prodotti chimici si accumulavano in
attesa dei risultati sulla loro attività. Con l'avvento di metodi di screening
che permettono la valutazione di decine e centinaia di prodotti in un solo

25
giorno, lo stadio lento del sistema è diventato la individuazione e la sintesi
chimica dei prodotti da provare i quali, a loro volta, debbono essere il più
possibile diversi tra loro, per poter facilitare la individuazione di nuove
strutture attive. E sorta quindi la necessità di porre la sintesi chimica al
passo con le nuove potenzialità di screening biologico; questo problema è
stato risolto con la sintesi combinatoriale.
1.8.1. Le basi della chimica combinatoriale. L'idea alla base della chimica
combinatoriale è quella di costruire, tutte in una volta e con i principi del
calcolo statistico (combinatorio), vaste collezioni di prodotti (libraries,
librerie) contenenti un alto numero di varianti di una struttura
fondamentale; procedere alla valutazione della loro attività biologica;
isolare ed identificare il/i prodotti eventualmente attivi e quindi procedere,
magari secondo la manipolazione chimica tradizionale, alla ottimizzazione.
Come si vedrà tali librerie possono essere costituite da prodotti in soluzione
o da prodotti ancorati ad una particella solida (bead, perlina). La figura 1.2
illustra il principio della sintesi combinatoriale.
Il numero dei prodotti ottenibili (N) è dipendente dal numero dei
reagenti (building blocks) disponibili in ogni passaggio (n, n', m, m', etc.)
N = n × n' × m × m'
Se in ogni passaggio si usa lo stesso numero di reagenti (B) ed s è il
numero delle volte che essi vengono utilizzati l’espressione diventa

26
N = Bs
Figura 1.2. Illustrazione del principio della chimica combinatoriale a confronto con la sintesi organica classica. A: sintesi organica classica. B: sintesi combinatoriale.
Nella tabella 1.1 è riportato il numero teorico dei peptidi ottenibili
utilizzando tutti i venti aminoacidi naturali (A) in funzione della lunghezza
del peptide. Naturalmente il numero delle possibilità aumenta
enormemente se si utilizzano anche aminoacidi non naturali.
Tabella 1.1. Numero di prodotti ottenibili in funzione della lunghezza del peptide utilizzando i 20 amminoacidi naturali.

27
Se uno considera il numero degli esapeptidi che è possibile ottenere,
facilmente ed in poco tempo, con questa metodologia, usando solo gli
aminoacidi naturali, si rende conto che questo numero supera nettamente il
numero dei prodotti già noti descritti nel Chemical Abstract (circa 1,4 x107)
e può avere un'idea immediata delle possibilità del metodo. Tra l'altro il
metodo si presta molto bene ad essere automatizzato e sono già
commercializzati dei sistemi robotizzati che sono in grado di produrre da
1000 a 2000 prodotti al giorno.
Non esistono ancora in terapia farmaci che siano stati ottenuti con
questo approccio; ciò è comprensibile visto che la metodologia ha avuto
origine (con un lavoro di Mario Geysen che sintetizzò una libreria di alcuni
milioni di esapeptidi) a metà degli anni ottanta; in realtà molte molecole con
questa origine sono in fase, anche avanzata, di sperimentazione e potranno
essere disponibili nel prossimo futuro.
Per utilizzare l'approccio combinatoriale è necessario pianificare,
oltre alla fase ed alla metodologia di sintesi, un adatto metodo di screening

28
delle librerie ottenute e un conveniente, rapido ed efficace metodo di
identificazione dei prodotti rivelatisi attivi. Uno dei vantaggi del metodo è
che la fase di identificazione è necessaria solo se è stata trovata attività e
solo per la libreria, o per la perlina, che hanno mostrato azione biologica.
In ogni caso le varie operazioni sono strettamente connesse, come è
mostrato nella figura 1.3 nella quale sono presi in considerazione i metodi
di sintesi e di analisi più utilizzati al momento attuale e che verranno
descritti più approfonditamente in seguito.

29
Figura 1.3. Schema dei principali metodi di sintesi, analisi e valutazione
della attività biologica utilizzati nella chimica combinatoriale.
Sebbene lo scopo principale della sintesi combinatoriale sia
sicuramente quello di individuare nuovi lead, il metodo si presta anche alla
ottimizzazione, soprattutto in quanto riduce i tempi di sintesi dei prodotti
necessari ad ottenere informazioni sulle relazioni struttura attività, molti
dei quali possono essere ottenuti in una sola sintesi combinatoriale.
1.8.2. Sintesi. Per poter essere utilizzati nella sintesi combinatoriale i
metodi sintetici debbono essere in grado di dare alte rese con una grande
varietà di substrati. Da questo punto di vista la sintesi in fase solida ideata e
messa a punto da Merrifield per i peptidi, è particolarmente utile.
In questa procedura uno dei reagenti è unito covalentemente ad una
adatta fase solida insolubile nei solventi di reazione. Il secondo reagente è
fatto agire in soluzione per produrre il prodotto di reazione che quindi è a
sua volta legato alla fase solida. Ciò permette il facile allontanamento dei
reagenti e catalizzatori (che possono essere usati in eccesso per forzare la
reazione), rende possibile un adeguato lavaggio prima di passare alla
reazione successiva e riduce le perdite che si hanno usualmente in questa
fase, nella sintesi organica in soluzione.
Il problema delle diverse cinetiche di reazione, che si ha quando si
fanno reagire contemporaneamente più prodotti, può essere risolto

30
ricorrendo al metodo della sintesi parcellizzata (split synthesis) di cui si dirà
più avanti. Infatti, nelle sintesi in soluzione nelle quali si fanno reagire molti
substrati contemporaneamente, ognuno con la sua velocità di reazione, c'è
un notevole rischio che la concentrazione dei prodotti ottenuti non sia
omogenea. Questo è un problema quando si va a misurare l'attività
biologica in quanto può determinare la perdita di informazione sull'attività
dei prodotti presenti in concentrazioni minime.
Un problema connesso con la sintesi in fase solida è che, almeno al
momento attuale, delle centinaia di sintesi organiche a disposizione del
chimico sintetico solo una piccola parte sono state adattate alla fase solida.
Ciò rende meno flessibile questo tipo di sintesi. C'è da dire che le cose sono
destinate a cambiare velocemente visto il grande interesse nell'adattare alla
fase solida le sintesi in soluzione più utili.
Nel tentativo di superare questo problema è stata sviluppata una
procedura che combina il metodo di sintesi in fase liquida e quello in fase
solida. Si tratta di attaccare i prodotti da sintetizzare ad un polimero
(polietilenglicol monometiletere) che è solubile in molti comuni solventi
organici, e che quindi permette di operare in soluzione, ma che può essere
precipitato allo stato cristallino per facilitare la fase di purificazione.
I supporti solidi utilizzati sono innanzi tutto il copolimero
polietilene/polistirene che è quello utilizzato da Merrifield e che va
benissimo per la sintesi dei peptidi. La necessità di avere supporti resistenti

31
ai più comuni solventi organici, in seguito alla estensione della metodologia
a piccole molecole non peptidiche, ha successivamente spinto alla ricerca di
altri supporti quali varie forme di cellulosa funzionalizzata, vetri a porosità
controllata e copolimeri polietilene/polistirene modificati. Alcune di queste
resine tipo Merrifield, modificate per adattarle a vari substrati, sono
riportate qui di seguito; sono quasi tutte disponibili commercialmente.
Un ruolo importante sulla velocità di reazione, sulla adattabilità ai
diversi ambienti di reazione e sulla valutazione della attività biologica è
giocato dalla struttura che lega la resina alla funzione reattiva (linker,
spaziatore). Esistono una gran varietà di linker e la loro scelta può essere
cruciale per il buon fine della sintesi. Nell'esempio precedente sono mostrati
due semplici linker aromatici; ma per evitare problemi di ingombro sterico,
spesso è necessario avere linker più estesi, quali quelli mostrati qui di
seguito che sono inseriti su resine disponibili commercialmente.
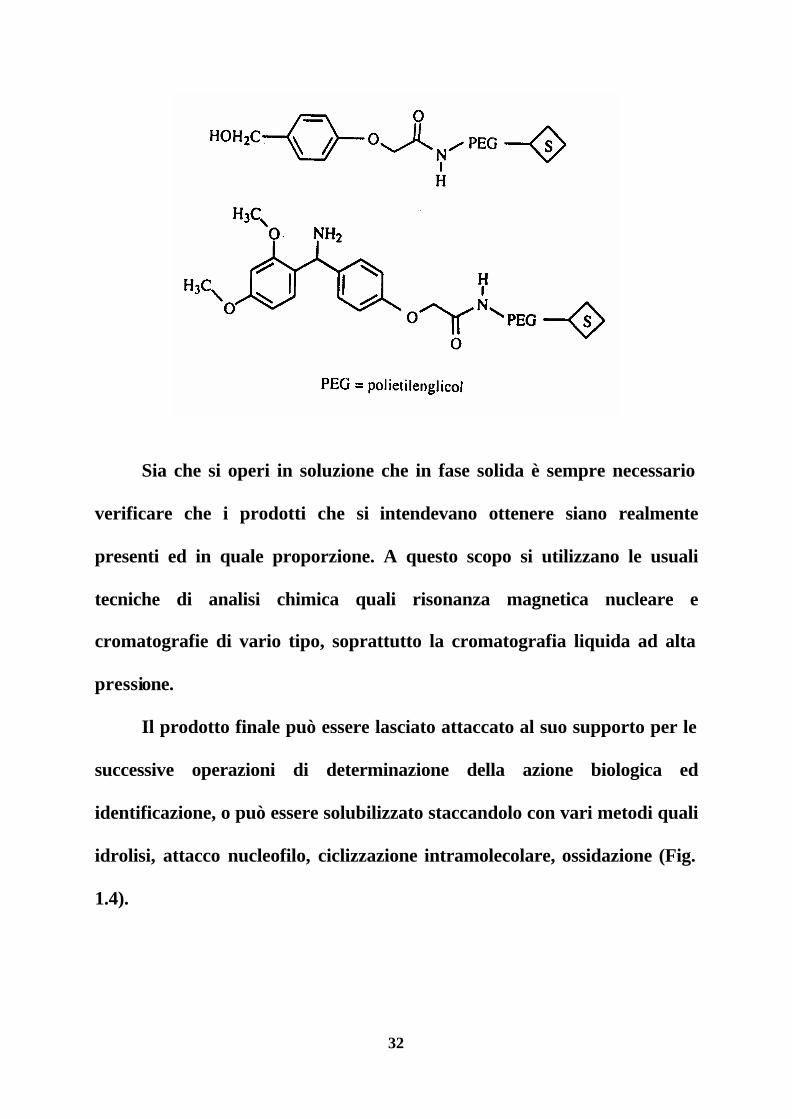
32
Sia che si operi in soluzione che in fase solida è sempre necessario
verificare che i prodotti che si intendevano ottenere siano realmente
presenti ed in quale proporzione. A questo scopo si utilizzano le usuali
tecniche di analisi chimica quali risonanza magnetica nucleare e
cromatografie di vario tipo, soprattutto la cromatografia liquida ad alta
pressione.
Il prodotto finale può essere lasciato attaccato al suo supporto per le
successive operazioni di determinazione della azione biologica ed
identificazione, o può essere solubilizzato staccandolo con vari metodi quali
idrolisi, attacco nucleofilo, ciclizzazione intramolecolare, ossidazione (Fig.
1.4).

33
Figura 1.4. Metodi di scissione di prodotti della sintesi combinatoriale da un supporto solido. A: ciclizzazione molecolare. B: spiazzamento ossidativo, fotolitico, idrolitico. C: spiazzamento nucleofilo.
Nella sintesi combinatoriale sono schematizzabili due procedure
principali:
A) La oligomerizzazione di monomeri. Vengono assemblati
successivamente monomeri bifunzionali diversi mediante un solo tipo di
reazione (Fig. 1.5).
Figura 1.5. Schema di assemblaggio di monomeri su di un supporto solido.
Si tratta naturalmente della stessa sequenza con la quale si
costruiscono singoli polipeptidi, oligonucleotidi, polisaccaridi che però viene
utilizzata secondo le metodologie combinatoriali.

34
In tal modo è possibile costruire un gran numero di librerie di
prodotti estremamente interessanti. Tuttavia, come è stato già accennato, i
peptidi non sono molto indicati come farmaci e una volta identificato un
peptide molto attivo è necessario, il più delle volte, derivare un
peptidomimetico conveniente. Analoghe considerazioni possono essere fatte
per oligonucleotidi e oligosaccaridi. Per questa ragione si sono assemblati
direttamente altri monomeri analoghi a quelli naturali ma in grado di
mostrare caratteristiche di stabilità metabolica e proprietà
farmacocinetiche migliori. Nello schema seguente sono indicate alcune delle
strutture utilizzate per sostituire gli aminoacidi.
Nel caso degli oligonucleotidi una variazione molto utile per
aumentare la stabilità metabolica dei nucleotidi naturali si è rivelata quella
di sostituire l'acido fosforico con l'acido fosfotioico.
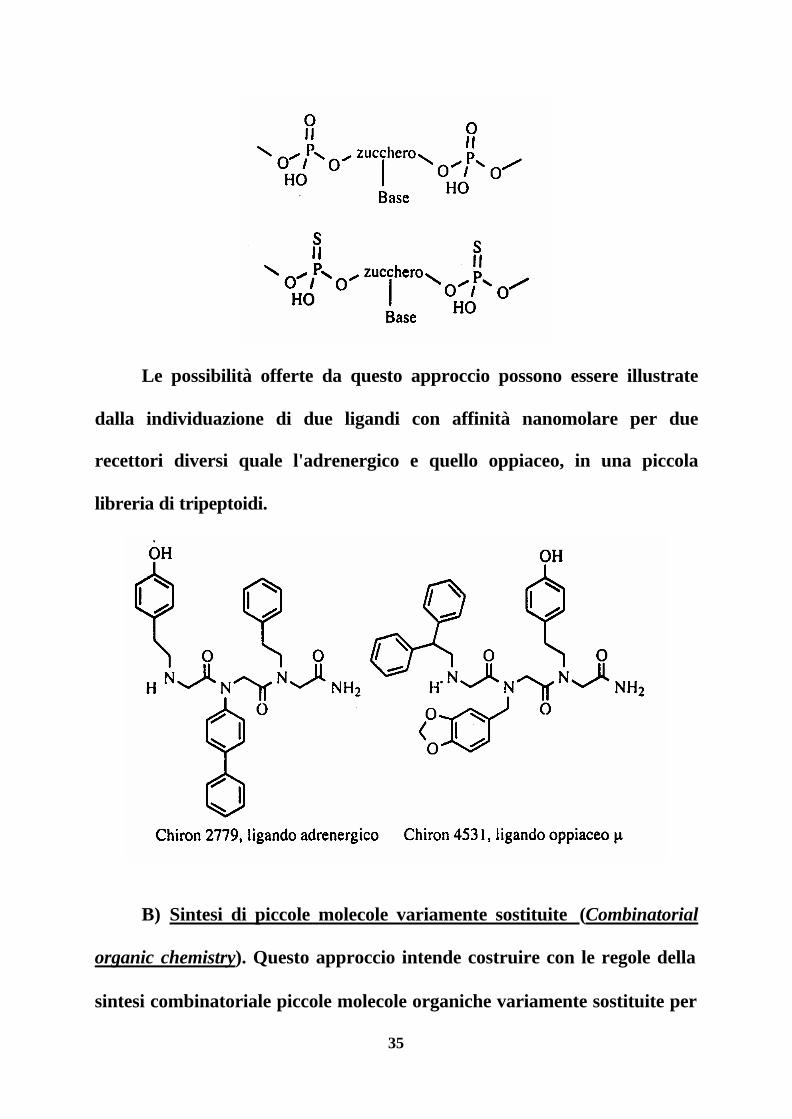
35
Le possibilità offerte da questo approccio possono essere illustrate
dalla individuazione di due ligandi con affinità nanomolare per due
recettori diversi quale l'adrenergico e quello oppiaceo, in una piccola
libreria di tripeptoidi.
B) Sintesi di piccole molecole variamente sostituite (Combinatorial
organic chemistry). Questo approccio intende costruire con le regole della
sintesi combinatoriale piccole molecole organiche variamente sostituite per

36
formare librerie, in genere molto più piccole di quelle ottenibili con
l'oligomerizzazione (Fig. 1.6).
Figura 1.6. Schema di sintesi combinatoriale di piccole molecole organiche.
A questo scopo il chimico deve utilizzare una vasta serie di reazioni
organiche e reagenti molto diversi tra loro. A seconda del tipo di reazioni
coinvolte, al termine della sintesi i prodotti si staccano dal supporto per
passare in soluzione o rimangono attaccati al supporto dal quale, se
necessario, possono essere staccati con uno dei metodi accennati in
precedenza.
Un tipico esempio di questa metodologia che sta assumendo, per ovvie
ragioni sempre più importanza, è la sintesi di una libreria di quaranta
prodotti a nucleo benzodiazepinico effettuata da De Witt che ha chiamato
questo genere di librerie diversomeri .

37
1.8.3. Saggi biologici. Uno dei punti cruciali nella sintesi
combinatoriale è la possibilità di disporre di metodi sensibili, efficaci e
semplici in grado di determinare la attività biologica di miscele contenenti
un grande numero di prodotti in concentrazioni spesso molto ridotte. I
saggi possono essere effettuati sia in soluzione che sui prodotti legati alla
resina di supporto.
Nel primo caso sono utilizzabili i comuni saggi biologici e
farmacologici quali il binding con recettori o la determinazione dell'attività,
generalmente antagonista, su enzimi. Questi saggi sono in genere modificati
allo scopo di renderli adatti alle basse concentrazioni presenti: ad esempio i
recettori possono essere preventivamente immobilizzati. Una trattazione di
queste tecniche va al di la degli scopi di questo libro.
Malgrado ciò, quando la libreria da testare è composta da un grande
numero di prodotti, i risultati non sono sempre attendibili. Per questa
ragione la tendenza attuale è quella di ridurre a numeri più ragionevoli il

38
numero di composti presenti in una libreria da provare tutti insieme (da
centinaia di migliaia ad alcune centinaia). Si ritiene che più è ridotto il
numero di prodotti, più alta sia la attendibilità dei risultati biologici
ottenuti.
Nel secondo caso i metodi debbono tenere conto del fatto che la
presenza del supporto solido può determinare notevoli problemi da
superare per ottenere risultati attendibili. Il principale di questi è che la
presenza del supporto o del sistema di ancoraggio ad esso (linker) può
ostacolare la interazione dei prodotti con le macromolecole biologiche che
costituiscono il bersagli dei prodotti sintetizzati.
Di particolare importanza è la possibilità di individuare i prodotti
attivi nella sintesi in fase solida quando si usa la sintesi parcellizzata o la
toposintesi (vedi di seguito). In questo caso l'individuazione della perlina, lo
spillo o la porzione di chip che portano la molecola attiva è essenziale.
Una delle metodiche più utilizzate in questo caso è quella di ancorare
alla macromolecola biologica che costituisce il bersaglio del prodotto attivo
(anticorpo, recettore) una molecola fluorescente. In tal modo, ad esempio, la
perlina che contiene il prodotto legato, se di dimensioni opportune
(usualmente dal diametro di 50-100 µ), potrà essere riconosciuta e separata
meccanicamente dalle altre per l'analisi della struttura del prodotto.
Alternativamente il recettore (o l'anticorpo) può essere legato ad
enzimi che determinano una risposta visibile (fluorescenza o colorazione) su

39
adatti substrati; un esempio è quello della fosfatasi alcalina ed è mostrato
nella figura 1.7.
Figura 1.7. Visualizzazione di una perlina contenente un prodotto con
affinità per un recettore accoppiato con la fosfatasi alcalina (RE). A: il recettore modificato è mescolato con le perline della libreria. B: il recettore si lega alla perlina che contiene il prodotto affine. C: si aggiunge il substrato dell'enzima; la perlina, alla quale è legato il recettore modificato, si colora (porpora) e può essere individuata ed isolata.
In ogni caso, nella valutazione dei risultati biologici in fase solida va
tenuto conto del fatto che la quantità di prodotto presente in ciascuna
perlina o sito di reazione può non essere omogenea, il che può far rischiare
di perdere informazioni preziose. Infatti la equimolarità dei ligandi è un
criterio fondamentale se si vuole avere una determinazione, per lo meno
semiquantitativa, della affinità.
1.8.4. Analisi. La identificazione dei prodotti attivi in una libreria è un
altro dei passaggi cruciali nella sintesi combinatoriale. Le metodologie

40
utilizzate sono diverse a seconda che la libreria ottenuta sia in soluzione o in
fase solida.
A) Librerie in soluzione. Per l'analisi di librerie in soluzione si
utilizzano i metodi di deconvoluzione e delle sottolibrerie.
I metodi di deconvoluzione, una volta che sia stata riscontrata attività
in una libreria, consistono nel procedere alla sintesi di sottolibrerie dalle
quali sia possibile assegnare ad ogni singolo blocco sintetico la corretta
posizione.
L'esempio riportato di seguito (Fig. 1.8) riguarda una libreria
minima, ma il modo di procedere è identico, anche se più laborioso, per
librerie ampie. Si può verificare che è più vantaggioso, in termini di
rapporto tra numero dei prodotti sintetizzati e numero delle sottolibrerie da
sintetizzare per la deconvoluzione, operare su grosse librerie.

41
Figura 1.8. Schema di deconvoluzione mediante la sintesi di
sottolibrerie. L'ombreggiatura indica le librerie nelle quali si riscontra attività. X rappresenta la miscela BC o c. Nella fase A si fissa la prima posizione e nelle fasi B e C la seconda e la terza posizione.
Nel metodo delle sottolibrerie, queste sono costituite già all'inizio
della sintesi in modo che il risultato delle prove biologiche sia in grado di
dare immediatamente la struttura del prodotto attivo. Applicando
all'esempio precedente questo metodo si opererà come mostrato in fig. 1.9.
Il numero delle sottolibrerie può essere anche molto alto: fino a molte
centinaia.

42
Figura 1.9. Metodo delle sottolibrerie per la identificazione dei prodotti attivi.
Un'altra strategia, che va sotto il nome di metodo delle librerie
ortogonali (ortogonal libraries), è basata su un disegno sperimentale tale
che ogni possibile prodotto appaia solo in due librerie. In una applicazione
di questa strategia 40 cloruri acidi sono stati fatti reagire con un set di 40
nucleofili (alcoli ed ammine) secondo lo schema seguente (Tab. 1.2), per
dare 80 librerie con un totale di 1600 prodotti. In pratica ogni acido è stato
fatto reagire con la miscela di nucleofili e ogni nucleofilo con la miscela
degli acidi; ogni riga e ogni colonna rappresentano librerie diverse.
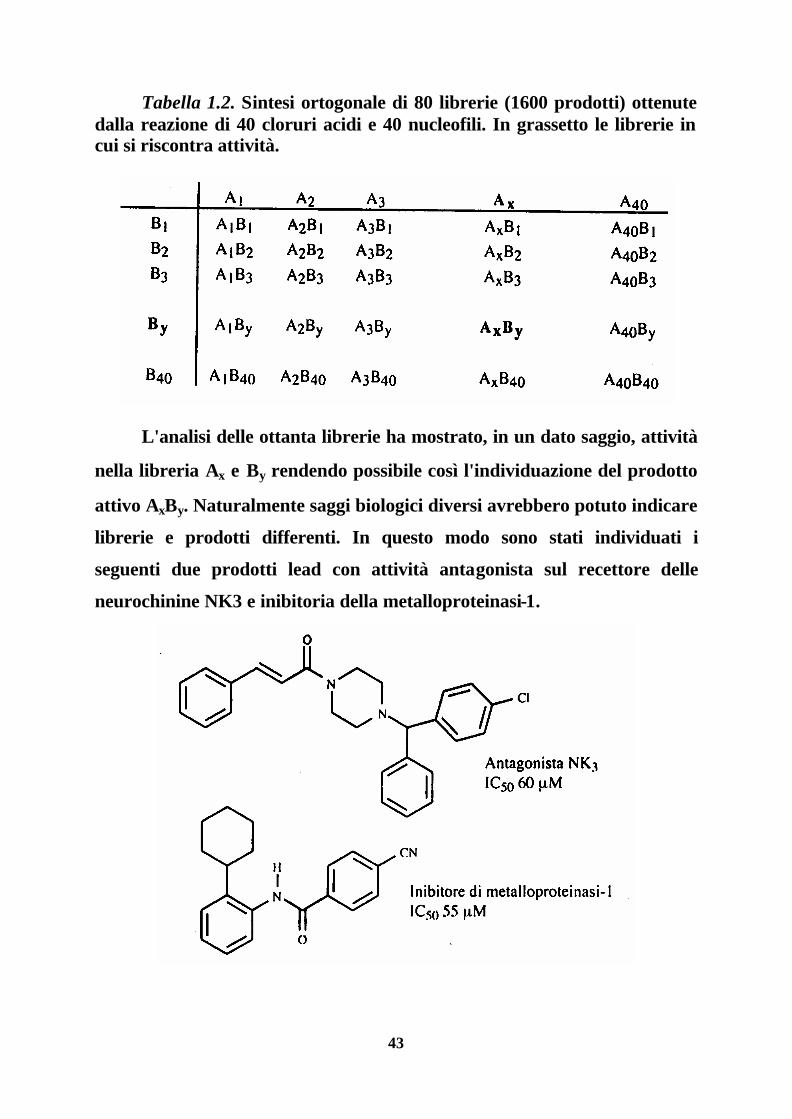
43
Tabella 1.2. Sintesi ortogonale di 80 librerie (1600 prodotti) ottenute dalla reazione di 40 cloruri acidi e 40 nucleofili. In grassetto le librerie in cui si riscontra attività.
L'analisi delle ottanta librerie ha mostrato, in un dato saggio, attività
nella libreria Ax e By rendendo possibile così l'individuazione del prodotto
attivo AxBy. Naturalmente saggi biologici diversi avrebbero potuto indicare
librerie e prodotti differenti. In questo modo sono stati individuati i
seguenti due prodotti lead con attività antagonista sul recettore delle
neurochinine NK3 e inibitoria della metalloproteinasi-1.

44
B) Librerie in fase solida. I metodi per l’analisi dei prodotti legati a
matrici solide sono funzione delle strategie sintetiche utilizzate.
Se si utilizza il metodo della sintesi parcellizzata (split synthesis), che
di regola conduce alla individuazione della perlina che contiene la molecola
attiva, e solo di quella, si può procedere direttamente alla sequenziazione
del prodotto qualora si tratti di oligopeptidi o oligonucleotidi (per esempio
usando il metodo di Edman per i peptidi) oppure alla decodificazione del
codice usato per caratterizzare ogni passaggio della sintesi quando si sia
usato il sistema della sintesi codificata (tag synthesis). Se non si usa la
codificazione è possibile utilizzare anche in questo caso il metodo della
deconvoluzione. A questo scopo vengono spesso conservate parte delle
perline non rimescolate di ogni passaggio della sintesi.
Se invece sono stati usati approcci toposintetici (spatially adressable
libraries), quali il metodo del sacchetto (tea bag synthesis), il metodo degli
spilli (multipin synthesis) o quello fotolitografico, non sono necessarie
ulteriori informazioni in quanto la struttura di ogni prodotto è
perfettamente definita dalla sua collocazione nel sistema di sintesi. I metodi
toposintetici sono anche conosciuti come sintesi parallela (parallel synthesis)
e possono essere adattati anche alla sintesi in soluzione. Maggiori
particolari su questi metodi verranno dati più avanti nella descrizione delle
relative strategie di sintesi.

45
La ricerca in questo campo è molto attiva: recentemente sono stati
riportati metodi nei quali la sequenza sintetica di ogni perlina è codificata
elettronicamente in un microchip (rad iofrequency encoding) inserito nel
cuore della perlina stessa.
1.8.5. Metodo del sacchetto (tea bag synthesis). In questo metodo,
sviluppato per la sintesi di peptidi, la resina di supporto è sigillata in
sacchetti di polipropilene poroso. Gli aminoacidi sono accoppiati mettendo
ogni singolo sacchetto in contatto con la soluzione del singolo aminoacido
attivato. La reazione di accoppiamento per ogni singolo sacchetto è quindi
perfettamente stabilita così come il susseguirsi degli stadi della reazione.
Tutte le altre operazioni (lavaggio, deprotezione) sono invece effettuate
tutte insieme in un singolo recipiente. Al termine della sintesi ogni pacchetto
contiene un singolo oligopeptide di struttura definita che viene usualmente
solubilizzato prima di essere provato biologicamente.
Questa tecnica è sinteticamente molto flessibile, può essere
automatizzata, e permette la sintesi di quantità relativamente molto alte di
oligopeptide (circa 500 µmoli) che ne permettono la purificazione e la
caratterizzazione.
1.8.6. Sintesi parcellizzata (split synthesis). E una delle strategie
sintetiche più utilizzate e si presta alla sintesi di librerie molto grandi. Si

46
utilizzano il più delle volte perline del diametro di circa 50-100 µ, il che vuol
dire circa 107-108 perline per grammo di resina.
L'idea alla base del metodo, introdotto da Furka nel 1988, è di
suddividere le perline in diverse porzioni uguali (il cui numero dipende
dalla strategia sintetica che si vuol perseguire), sottoporle singolarmente
alla fase di accoppiamento e quindi rimescolare opportunamente il tutto.
Nelle fasi successive si ripeterà l'operazione di separazione e
rimescolamento ad ogni passaggio sintetico.
Questa metodologia, inizialmente concepita per superare le differenti
cinetiche di reazione (prevedibili nella reazione di miscele di reattivi), ha il
grande vantaggio di condurre alla fine a perline che hanno alla superficie
una sola specie molecolare (one bead-one compound).
A seconda della funzionalizzazione del supporto, la quantità di
prodotto su ogni perlina può arrivare a molte nanomoli, una quantità
sufficiente sia alla determinazione della attività biologica che alla
individuazione del prodotto.
Un tipico schema di sintesi di questo tipo è mostrato nella figura 1.10.
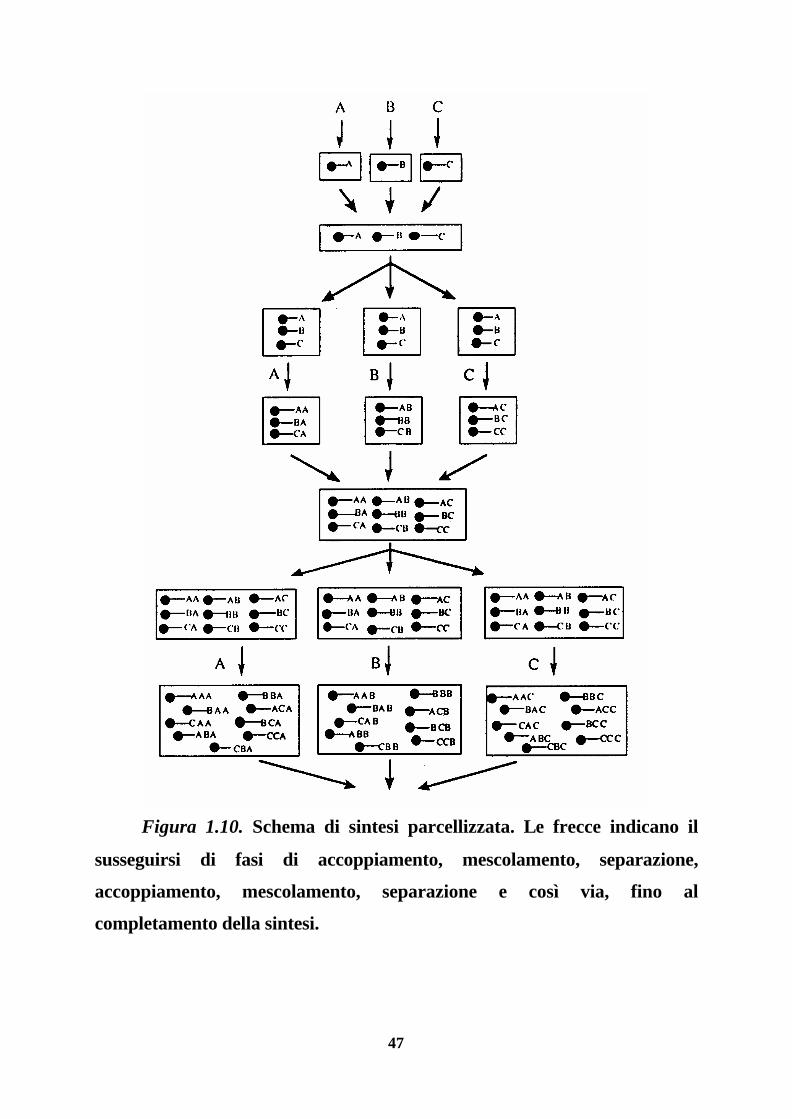
47
Figura 1.10. Schema di sintesi parcellizzata. Le frecce indicano il
susseguirsi di fasi di accoppiamento, mescolamento, separazione,
accoppiamento, mescolamento, separazione e così via, fino al
completamento della sintesi.

48
1.8.7. Sintesi codificata (tag synthesis). Lo schema di sintesi descritto
precedentemente può essere utilizzato come tale solo con quei prodotti che
si prestano ad essere individuati per sequenziazione (quali oligopeptidi e
oligonucleotidi) una volta identificata la perlina contenente il prodotto
attivo, essendo impossibile identificare direttamente la struttura del
prodotto stesso.
Per ovviare a ciò sono stati messi a punto dei metodi di codificazione
che permettono di risalire alla struttura del prodotto attivo. Questi metodi
sono essenzialmente di due tipi.
Nel primo metodo ad ogni passaggio della reazione, utilizzando un
linker bifunzionale, vengono eseguite due tipi di reazioni: una consiste nella
reazione di costruzione della libreria, l'altra consiste nell'attaccare alla
perlina anche un prodotto che permette di riconoscere il reattivo aggiunto
in quel passaggio. Questi prodotti sono in genere aminoacidi o nucleotidi
inseriti in sequenza.
Al termine della sequenza di reazioni ogni perlina conterrà quindi il
prodotto voluto e, ad esempio, un peptide che può essere sequenziato
secondo Edman per raccontare la storia della perlina e quindi definire la
struttura del prodotto attivo (Fig. 1.11).

49
Figura 1.11. Schematizzazione del metodo della sintesi codificata. A-F: blocchi sintetici; a-f: unità codificanti.
Come unità codificanti possono essere usati anche nucleotidi;
l'oligonucleotide risultante può essere amplificato tramite PCR (polimerase
chain reaction) fino a raggiungere la concentrazione adatta ad una facile
sequenziazione.
Nel secondo metodo, messo a punto da Clark Still, i gruppi codificanti
non sono inseriti in sequenza ma si attaccano direttamente alla perlina (Fig.
1.12). E la loro presenza e la loro quantità che codifica con il codice binario
(tipo quello commerciale a barre) il prodotto sintetizzato presente sulla
perlina attiva.
Al termine della sequenza, la perlina trovata attiva viene liberata
dalle molecole codificanti le quali vengono identificate e la loro
composizione stabilisce la struttura del prodotto sulla perlina.
C. Still ha utilizzato una serie di prodotti che si scindono dal supporto
per irradiazione e la cui miscela può essere semplicemente risolta per gas
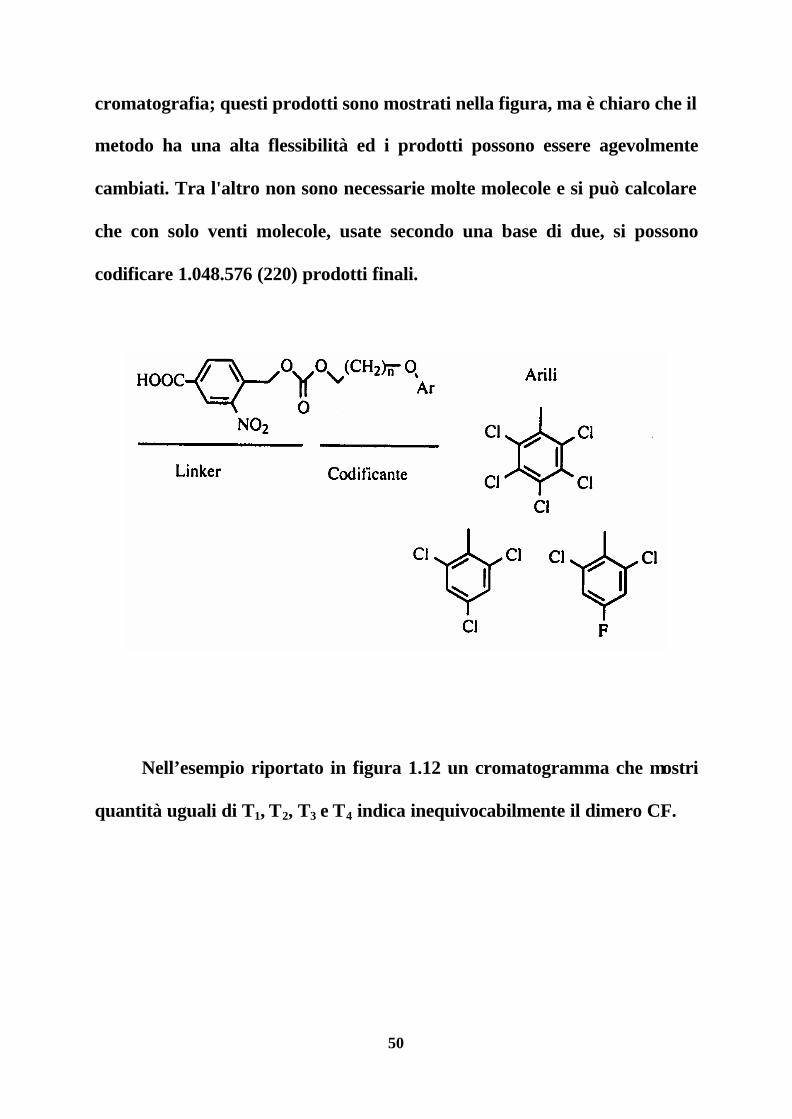
50
cromatografia; questi prodotti sono mostrati nella figura, ma è chiaro che il
metodo ha una alta flessibilità ed i prodotti possono essere agevolmente
cambiati. Tra l'altro non sono necessarie molte molecole e si può calcolare
che con solo venti molecole, usate secondo una base di due, si possono
codificare 1.048.576 (220) prodotti finali.
Nell’esempio riportato in figura 1.12 un cromatogramma che mostri
quantità uguali di T1, T2, T3 e T4 indica inequivocabilmente il dimero CF.

51
Figura 1.12. Schema di sintesi parcellizzata e codificata secondo Still.
Le frecce indicano il susseguirsi delle fasi di mescolamento, separazione e
accoppiamento, secondo la normale sintesi parcellizzata.
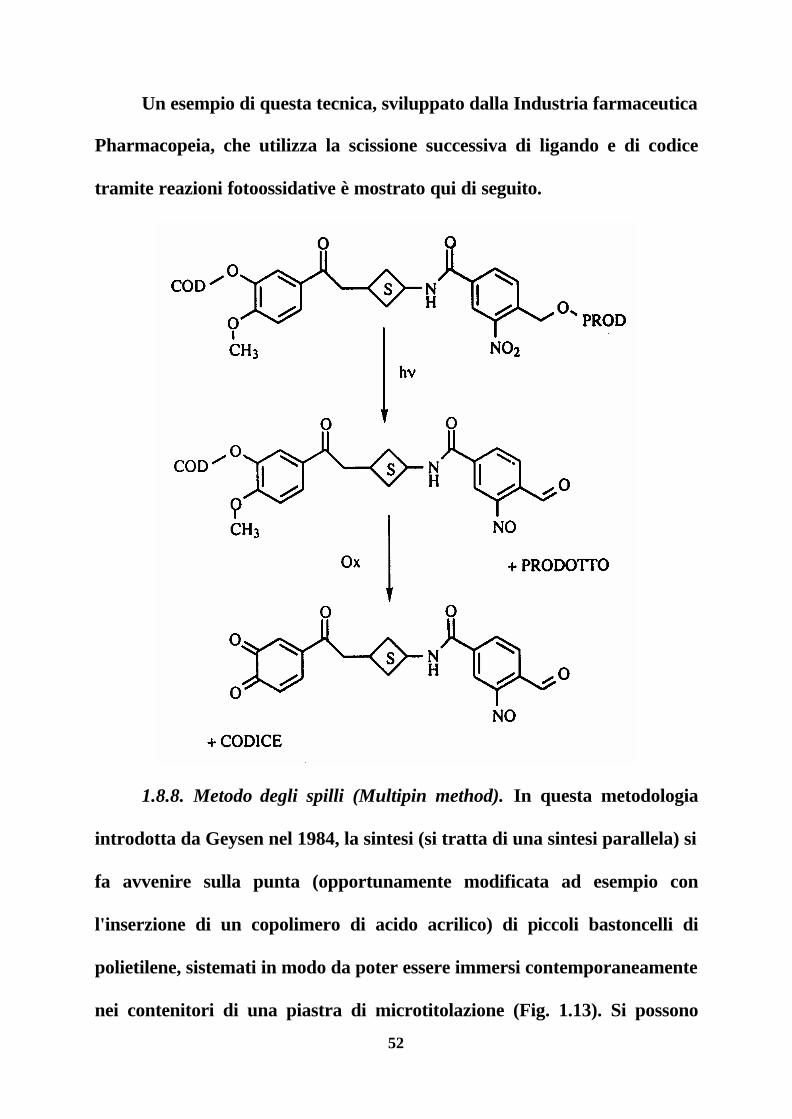
52
Un esempio di questa tecnica, sviluppato dalla Industria farmaceutica
Pharmacopeia, che utilizza la scissione successiva di ligando e di codice
tramite reazioni fotoossidative è mostrato qui di seguito.
1.8.8. Metodo degli spilli (Multipin method). In questa metodologia
introdotta da Geysen nel 1984, la sintesi (si tratta di una sintesi parallela) si
fa avvenire sulla punta (opportunamente modificata ad esempio con
l'inserzione di un copolimero di acido acrilico) di piccoli bastoncelli di
polietilene, sistemati in modo da poter essere immersi contemporaneamente
nei contenitori di una piastra di microtitolazione (Fig. 1.13). Si possono

53
produrre fino a 2 µmoli per spillo. Si possono utilizzare formati diversi, ma
il più comune è costituito da 96 bastoncelli supportati su una piastra di
polietilene che si adattano ad una piastra con altrettante vaschette. Le
vaschette sono riempite con il reattivo opportuno, secondo la strategia
combinatoriale scelta, e l'operazione viene ripetuta quante volte è
necessario. Ogni prodotto finale è univocamente determinato dalla
posizione e dalla storia chimica della vaschetta corrispondente. Questo
metodo che è naturalmente adatto per costruire un numero limitato di
prodotti, si presta ad essere automatizzato. L'analisi biologica può essere
fatta sul prodotto legato o su prodotti solubilizzati quando si sia usato un
linker opportuno.
Figura 1.13. Attrezzatura per sintesi combinatoriale con il metodo
degli spilli.
Nella figura 1.14 è esemplificata una semplicissima strategia per
questo tipo di approccio. Il disegno mostra la disposizione dei reagenti nelle

54
vaschette e il tipo di molecole che si trovano sulla testa dello spillo dopo la
reazione.
Come già accennato, questo metodo può essere facilmente adattato
alla sintesi in fase liquida facendo avvenire le reazioni in batterie di
contenitori opportunamente disegnati.
Figura 1.14. Esempio di sintesi con il metodo degli spilli. Strategia a
strisce ortogonali .
1.8.9. Metodo fotolitografico. Questo metodo, nel quale vengono
combinate la tecnica fotolitografica e quella fotochimica, è particolarmente
adatto per la sintesi di polipeptidi, oligonucleotidi e prodotti analoghi.
Si possono utilizzare vetrini da microscopio di vetro borosilicato sui
quali vengono ancorati i monomeri protetti con un proteggente che si può
eliminare per fotolisi. L'uso di maschere protettive permette la attivazione e
quindi la successiva reazione di zone ben definite del supporto. Il numero
dei prodotti che si possono sintetizzare è limitato solo dalla risoluzione, che
deve permettere di identificare chiaramente due siti contigui. Al termine il

55
vetrino è esposto alla interazione con il recettore usualmente accoppiato ad
un prodotto fluorescente. Dopo accurato lavaggio, la scansione con un
microscopio a fluorescenza permette di identificare il prodotto attivo la cui
struttura è definita dalla sua posizione sulla lastrina. Il metodo è stato
miniaturizzato: in questo caso è essenziale raggiungere una buona
risoluzione tra siti di sintesi contigui; si possono avere 40.000 siti di sintesi
distinguibili in uno spazio di un centimetro quadro. Questa tecnica
sviluppata dalla Affimax è anche nota sotto il nome di VLSIPS (very large-
scale immobilized polimer synthesis).
Seguendo la schematizzazione della figura 1.15: 1) un adatto gruppo
reattivo (per esempio un aminoacido) è innestato su di un supporto solido;
2) il gruppo amminico, su cui si effettueranno le successive reazioni, viene
opportunamente protetto con un proteggente fotolabile (X); 3, 4) usando
una maschera opportunamente disegnata, una parte del supporto viene
esposta ed irradiata per eliminare il gruppo proteggente dai gruppi ivi
localizzati; 5) i gruppi deprotetti vengono fatti reagire con il reattivo
opportuno (A); 6) i gruppi introdotti vengono di nuovo protetti; 7) una
nuova maschera viene usata per selezionare i gruppi da deproteggere; 8, 9)
i gruppi deprotetti vengono fatti reagire con un nuovo reagente (B); 10) si
procede ad una nuova protezione e così via fino al completamento del
disegno sperimentale.

56
Figura 1.15. Procedimento per la sintesi fotolitografica. X = gruppo proteggente fotosensibile. A, B = reagenti.
Utilizzando questa metodologia su un semplicissimo sistema quale
quello dell'esempio precedente si può operare secondo la figura 1.16.

57
Figura 1.16. Una semplice strategia di sintesi fotolitografica. a: illumina; b: accoppia; c: proteggi.
1.8.10. Esempi di applicazioni. Utilizzando alcune delle strategie
descritte in precedenza sono state costruite in questi ultimi tempi un gran
numero di librerie. I due esempi che seguono vogliono semplicemente dare
una idea della utilizzazione di questa metodologia. Gordon e collaboratori
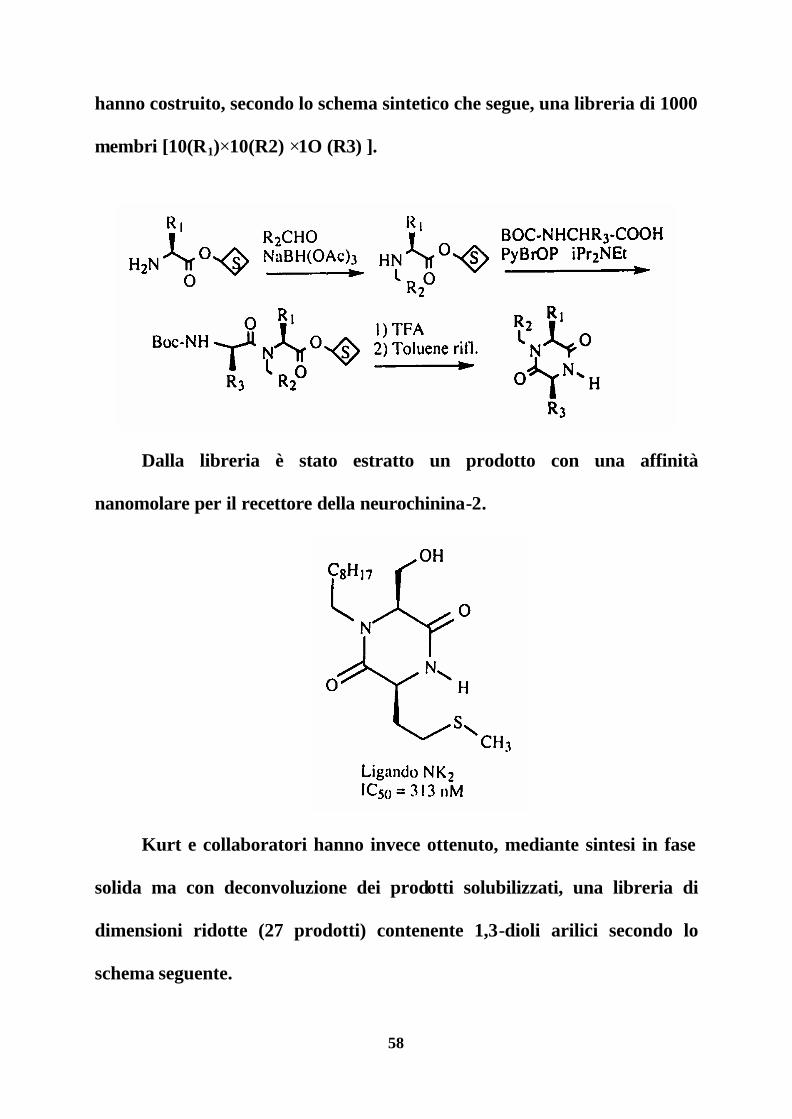
58
hanno costruito, secondo lo schema sintetico che segue, una libreria di 1000
membri [10(R1)×10(R2) ×1O (R3) ].
Dalla libreria è stato estratto un prodotto con una affinità
nanomolare per il recettore della neurochinina-2.
Kurt e collaboratori hanno invece ottenuto, mediante sintesi in fase
solida ma con deconvoluzione dei prodotti solubilizzati, una libreria di
dimensioni ridotte (27 prodotti) contenente 1,3-dioli arilici secondo lo
schema seguente.
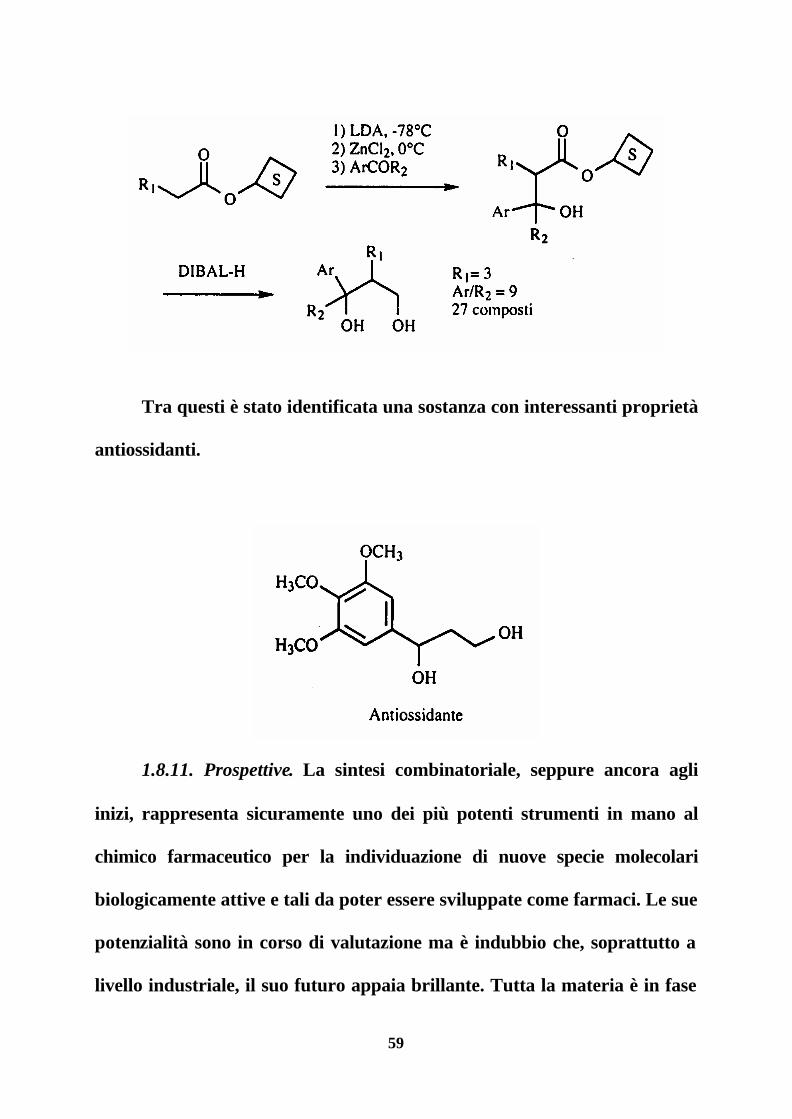
59
Tra questi è stato identificata una sostanza con interessanti proprietà
antiossidanti.
1.8.11. Prospettive. La sintesi combinatoriale, seppure ancora agli
inizi, rappresenta sicuramente uno dei più potenti strumenti in mano al
chimico farmaceutico per la individuazione di nuove specie molecolari
biologicamente attive e tali da poter essere sviluppate come farmaci. Le sue
potenzialità sono in corso di valutazione ma è indubbio che, soprattutto a
livello industriale, il suo futuro appaia brillante. Tutta la materia è in fase

60
di rapida crescita ed è molto probabile che alcune delle metodologie
attualmente usate, e che sono state descritte in questo capitolo, diventino
rapidamente obsolete in favore di altre più semplici, affidabili ed
economiche.
1. 9. Conclusioni
La ricerca di una molecola di riferimento (lead) rimane uno dei punti
fondamentali nella progettazione e nello sviluppo di nuovi farmaci e mezzi
farmacologici di indagine.
Alle vecchie strategie, che per lo più rimangono valide, si sono
aggiunti nuovi strumenti quali la sintesi combinatoriale, già vista in questo
capitolo, e l'uso del computer nella analisi delle relazioni struttura attività e
nella progettazione di nuove molecole a cui sarà dedicato un capitolo a
parte.
In ogni caso è assolutamente importante ricordare che
l'individuazione di un nuovo lead non è che il principio di un cammino, che
può essere inaspettatamente lungo ed impervio, prima di condurre, se mai
lo farà, all'agognato farmaco, o all'utile strumento di indagine.


















