

Presentazione standard di PowerPoint - chimica.unipd.itmetodi_ibridi.pdf · descrivere ed a...
38
Metodi ibridi [email protected]
-
Upload
phungquynh -
Category
Documents
-
view
217 -
download
0
Transcript of Presentazione standard di PowerPoint - chimica.unipd.itmetodi_ibridi.pdf · descrivere ed a...

Motivazioni (1)
− Gli effetti solvente influenzano significativamente la
struttura e la reattività delle specie in soluzione
− Sono necessari metodi computazionali che riescano a
descrivere ed a prevedere in maniera quantitativa il
comportamento di specie chimiche in soluzione.
− La chiave del successo delle simulazioni computazionali di
sistemi in fasi condensate è la disponibilità di funzioni che
descrivano in modo accurato l’energia potenziale
considerando opportunamente le forze intermolecolari.

Motivazioni (2)
− Teoricamente i metodi quantomeccanici ab-initio (QM) possono essere utilizzati per calcolare sistematicamente le interazioni intermolecolari in soluzione e generare i dati energetici e strutturali necessari.
− In pratica è impossibile trattare un intero sistema in fase condensata mediante calcoli quantistici perché il tempo di calcolo necessario èproibitivo anche per sistemi di piccole dimensioni.
− Approssimazioni In genere vengono utilizzate delle funzioni empiriche per il potenziale / molecular mechanics (MM) force field.

Motivazioni (3)
− I campi di forza MM non sono adeguati e le funzioni empiriche che descrivono il potenziale (e di conseguenza i semplici metodi di dinamica molecolare) non sono in grado di descrivere neppure approssimativamente reazioni chimiche evolvono con rottura o formazione di un legame.
− I sistemi di interesse chimico in biologia computazionale e nella catalisi, per fare un esempio dei campi di maggiore applicazione, sono sistemi in fasi condensate che comportano eventi reattivi e non possono essere descritti da tecniche MM

Motivazioni (4)
− L’approccio alternativo dei metodi ibridi QM/MM
consiste nel suddividere il sistema in due parti trattando
una parte mediante metodi QM e una parte mediante una
metodologia classica MM.
− Lo sviluppo dei metodi ibridi QM/MM che inizia con gli studi
pionieristici di Warshel e Levitt / Singh e Kollman segue
come linea generale l’idea che un sistema chimico composto
da centinaia o migliaia di atomi possa essere partizionato in
una ‘regione’ QM e in una rimanente ‘regione’ classica.
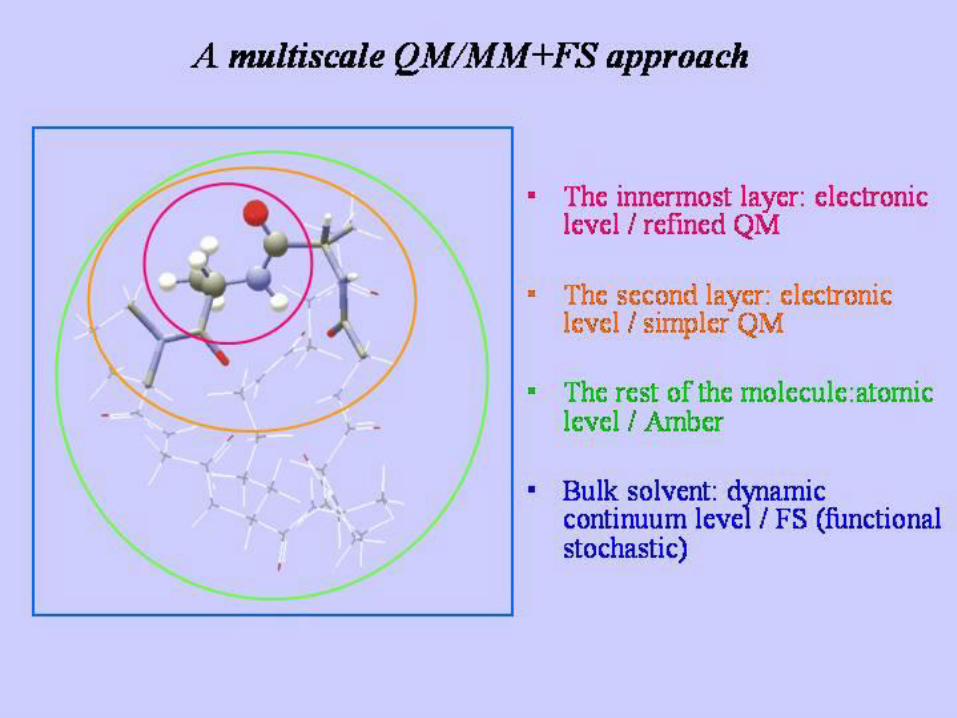

QM/MM (1)
− La parte quantistica permette il calcolo delle proprietà dello stato
fondamentale ed elettronico o dei potenziali di ionizzazione nelle reazioni
chimiche e richiede una limitata (semiempirici) o addirittura assente (ab-
initio) parametrizzazione.
− Gli effetti di polarizzazione del solvente sul soluto sono inclusi nei calcoli
QM mediante un modello che descrive il solvente come un dielettrico
continuo e calcola l’energia di solvatazione usando il campo di reazione
di Onsager.
− I modelli di questo tipo hanno il vantaggio di essere efficienti per i
contributi elettrostatici a lungo raggio nella solvatazione ma non
considerano le specifiche interazioni soluto-solvente. In molti casi è
invece necessario includere esplicitamente queste interazioni.

QM/MM (2)
− In teoria la divisione tra le due regioni potrebbe sembrare
piuttosto semplice e netta ma raramente è possibile scrivere
l’energia totale del sistema in termini di due sottosistemi non
interagenti.
− Molto spesso invece le interazioni tra i due sottosistemi sono
piuttosto forti (il caso più ovvio è quello della presenza di un
legame chimico di qualsiasi natura) tanto da rendere
necessari dei trattamenti particolari nella zona tra la regione
QM e la regione MM.

QM/MM (3) / Separazione
− Gli approcci per lo studio delle terminazioni dei metodi QM
possono essere grossolanamente divisi in due gruppi
principali:
1. approcci basati sui link atoms nei quali vengono
aggiunti dei centri addizionali solo nei calcoli QM ma che
non sono presenti nell’intero sistema
2. approcci che presentano una boundary region tra la
regione QM e MM; i centri che si trovano in tale regione
sono soggetti sia a calcoli QM che MM

QM/MM (4) – Condizioni al contorno
− L’energia totale di un sistema QM/MM può essere espressa in modo semplificato come
− EQM rappresenta l’energia della regione QM come se fosse isolata; EMM fornisce l’energia del sistema MM
− EQM/MM è l’energia di interazione tra le regioni QM e MM / tiene conto delle interazioni di van der Waals e delle modificazioni dell’Hamiltoniano QM che possono influenzare la regione MM come ad esempio perturbazioni elettrostatiche
− Infine Econtorno è il termine energetico dovuto ai vincoli imposti nella parte più esterna della regione MM.
/QM MM QM MM contornoE E E E E

Metodi QM
− La scelta del metodo QM da utilizzare nei metodi
ibridi QM/MM segue fondamentalmente gli stessi
criteri da applicare ai puri calcoli QM
− Hartree-Fock (HF)
− Post-HF / Interazione di configurazione (CI),
− Post HF / Metodi perturbativi (MP)
− Metodi Semiempirici: AM1, PM3, l’EVB, ZINDO
− DFT

Metodi MM (1)
− In generale la scelta del modello MM da utilizzare dipende
dal trattamento utilizzato per descrivere la regione di confine
tra QM e MM. La più importante distinzione si ha tra
1. campi di forza di valenza esemplificati da campi di forza
biomolecolari (CHARMM e AMBER) e costruiti da termini
energetici come lo stretch di legame, il bend degli angoli,
etc…
2. campi di forza ionici i cui termini principali sono le forze
elettrostatiche e a corto raggio (van der Waals).

Metodi MM (2)
− Per la scelta del modello MM si devono quindi precedentemente stimare due aspetti:
1. la scelta del campo di forza influenza la carica parziale atomica che a sua volta influisce sulle interazioni QM/MM a lungo raggio. Il campo di forza ionico tende a generare una carica più grande di quella generata dal fitting del potenziale elettrostatico. In molti casi si sviluppano cariche ioniche formali
2. Il trattamento dei legami e i contatti interionici tra le regioni QM e MM seguono generalmente lo stesso approccio utilizzato per le interazioni nella regione MM.
− Nel caso del campo di forza di valenza si identificano facilmente i termini derivati dallo stretch dei legami e dal bend degli angoli e possono quindi essere eliminati i termini che derivano dall’interazione QM.
− Nei campi di forza ionici i termini di interazione attrattiva a corto-raggio QM…MM derivano dalla presenza di cariche MM nell’Hamiltoniano QM e di conseguenza non possono essere separate dalle interazioni a lungo-raggio.

Termini di accoppiamento QM/MM
− I termini a corto raggio o di Lennard-Jones / simile a MM standard
− I termini elettrostatici / varie possibili scelte
− modello meccanico nel quale i calcoli QM sono ottenuti in fase gas
senza accoppiamento elettronico con l’ambiente. Le interazioni
elettrostatiche tra le regioni QM e MM sono omesse od incluse nei
calcoli MM utilizzando un modello classico di carica puntuale per la
distribuzione di carica QM.
− modello elettrostatico nel quale la partizione classica appare come una
distribuzione di cariche esterna nell’Hamiltoniano QM. La polarizzazione
della regione QM da parte della distribuzione di carica della regione MM
avviene nei calcoli della struttura elettronica QM.
− modello di polarizzazione nel quale viene inclusa la polarizzazione della
regione MM dovuta alla distribuzione di carica in QM.
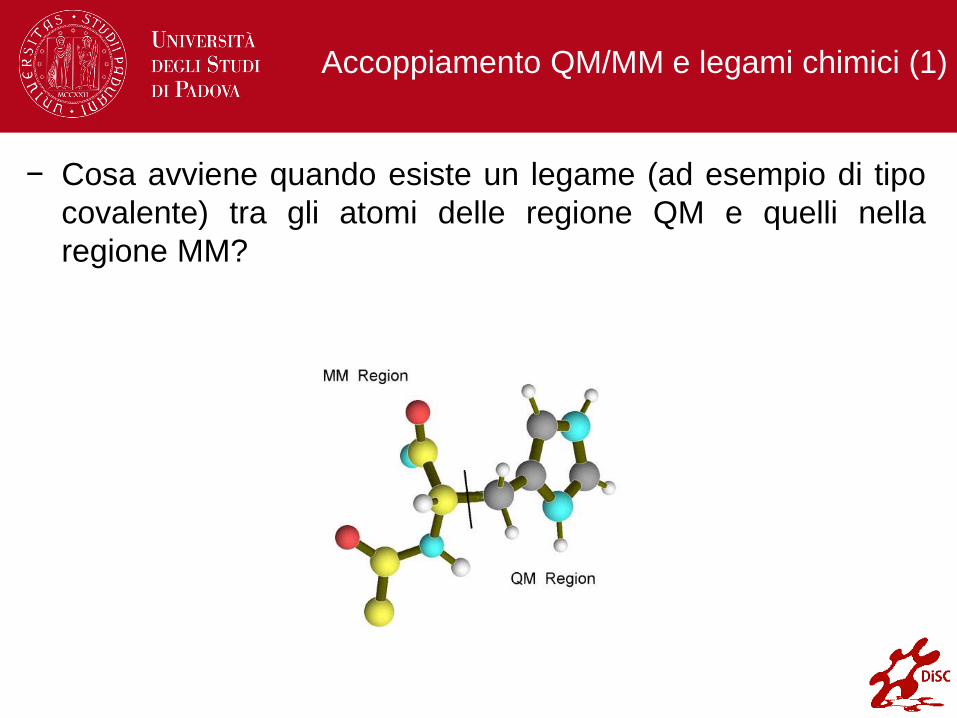
Accoppiamento QM/MM e legami chimici (1)
− Cosa avviene quando esiste un legame (ad esempio di tipo
covalente) tra gli atomi delle regione QM e quelli nella
regione MM?

Accoppiamento QM/MM e legami chimici (2)
− Metodo dei link atoms: vengono introdotti degli atomi
addizionali che formano un legame covalente con la regione
QM che dovrebbe mimare il legame con la regione MM.
− Questi atomi legati soddisfano la valenza degli atomi QM. Il
più semplice ed ancora più utilizzato atomo legato è l’atomo
di idrogeno che viene posizionato lungo il legame QM/MM
che è stato rotto.
− Possono essere utilizzati anche altri atomi o gruppi funzionali
come gli alogeni o i gruppi metile.
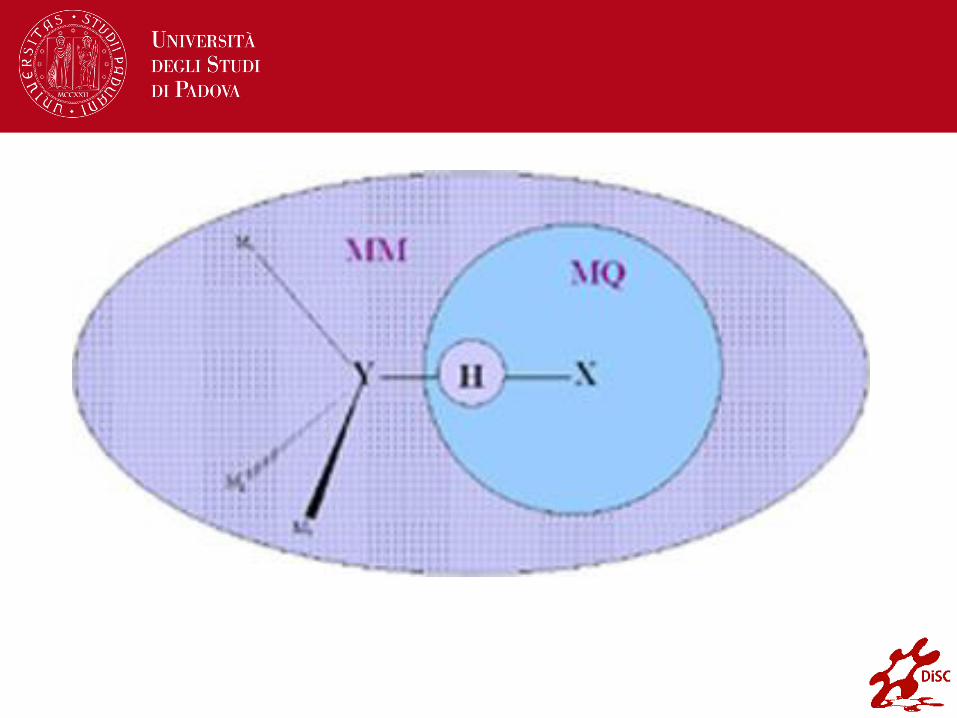

Accoppiamento QM/MM e legami chimici (3)
− I link atoms sono trattati in maniera esatta nei calcoli QM ma non è permesso loro di interagire con gli atomi MM (è come se risultassero invisibili agli atomi MM),
− L’energia e le forze risultano dai termini energetici di legame.
− I termini elettrostatici e di van der Waals che si considerano di non-legame sono trattati separatamente:
1. le interazioni di van der Waals attraverso la regione QM e MM sono determinati in modo esatto come se l’intero sistema fosse classico
2. l’energia di interazione elettrostatica è ottenuta da calcoli quantomeccanici sugli atomi QM insieme ai link atoms in presenza
di tutte le cariche parziali MM escluse quelle prodotte dai link atoms.

Condizioni al contorno
− In tutte le simulazione computazionali di liquidi o proteine si
presenta il problema di come approssimare un sistema
virtualmente infinito mediante una costruzione finita.
− Spesso vengono utilizzate delle condizioni periodiche al
contorno che mimano le condizioni del bulk.
− Un approccio alternativo è lo sviluppo di condizioni
stocastiche al contorno dove si sviluppa nella simulazione
un sistema molecolare finito: non viene replicata una singola
cella ma vengono applicate delle forze al contorno che
interagiscono con gli atomi del sistema

Implementazione (1)
1. Pacchetto di modelling classico nel quale la parte
quantistica QM è integrata come un estensione del campo
di forza classico
2. Pacchetto quantistico QM nel quale l’ambiente classico MM
è incorporato come una perturbazione
3. Schema modulare in cui è presente un programma centrale
di controllo che permette la scelta sia di un metodo QM che
di un metodo MM

Implementazione (2)
− Un buon esempio del primo gruppo è CHARMM, un package che contiene anche l’implementazione QM. Questo programma funziona tradizionalmente bene per sistemi macromolecolari. La capacità di CHARMM può essere estesa con l’inclusione del pacchetto GRACE.
− www.charmm.org

Implementazione (3)
− L’approccio considerato nel secondo gruppo è particolarmente utilizzato quando si ha a che fare con piccole molecole e per la ricerca degli stati di transizione. − accoppiamentro tra GAMESS-UK e AMBER
− MSI Cerius-2 che introduce un modello MM per l’ambiente in un implementazione RI-DFT con ottimizzazione delle coordinate interne per mezzo di TURBOMOLE.
− www.msg.ameslab.gov/GAMESS/GAMESS.html
− http://www.cosmologic.de/turbomole.html

Implementazione (4)
− Vantaggi: impiego flessibile ed aggiornamento facilitato; disponibilità di
vari campi di forza
− Un esempio di sistema modulare include l’accoppiamento di
AMBER con CADPAC e MNDO.
− Altri esempi: QCPE con MOPAC, il MCQUB che combina il MOPAC
con il programma BOSS, ARGUS e HyperChem
http://amber.scripps.edu/
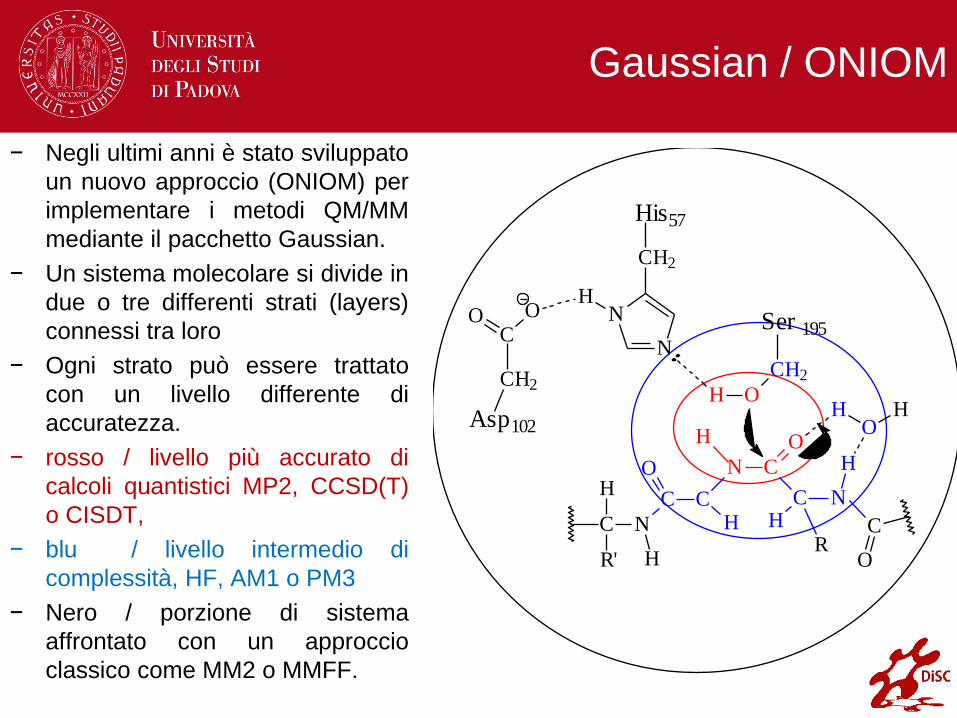
Gaussian / ONIOM
− Negli ultimi anni è stato sviluppato
un nuovo approccio (ONIOM) per
implementare i metodi QM/MM
mediante il pacchetto Gaussian.
− Un sistema molecolare si divide in
due o tre differenti strati (layers)
connessi tra loro
− Ogni strato può essere trattato
con un livello differente di
accuratezza.
− rosso / livello più accurato di
calcoli quantistici MP2, CCSD(T)
o CISDT,
− blu / livello intermedio di
complessità, HF, AM1 o PM3
− Nero / porzione di sistema
affrontato con un approccio
classico come MM2 o MMFF.
HO
H
RN
H
C
H
R'
O
CH
C
O
CN
NH
C
H
CH2
Asp102
COO
H
Ser 195
H O
CH2
His57
N
NH
CH2
C
O

Applicazioni (1)
− Le applicazioni ai metodi ibridi QM/MM sono molteplici
− la predizione della pKa di piccole molecole
− il profilo dell’energia libera per l’associazione di metalli alcalini
− catalisi basata sui metalli di transizione
− reazioni metallorganiche come l’addizione ossidativi di idrogeno molecolare ad un complesso di platino
− sostituenti nel bulk nei processi di catalisi che avvengono nella polimerizzazione delle olefine
− studi di solvatazione
− spettroscopia in fase condensata
− a studi di flessibilità conformazionale
− reazioni chimiche in fasi condensate, agli
− enzimi & DNA

Stato di transizione nell’enzima para-idrossibenzoato
idrossilasi (PHBH) ricavato mediante modelling QM/MM
[L. Ridder, J. N. Harvey, I. M. C. M. Rietjens, J. Vervoort e
A. J. Mulholland, J. Phys. Chem. B, 2003, 107, 2118-2126]

Modello di una superficie di 360 atomi di diamante(100)
[S. Skokov, C. S. Carmer, B. Weiner e M. Frenklach,
Phys. Rev. B., 1994, 49, 5662]
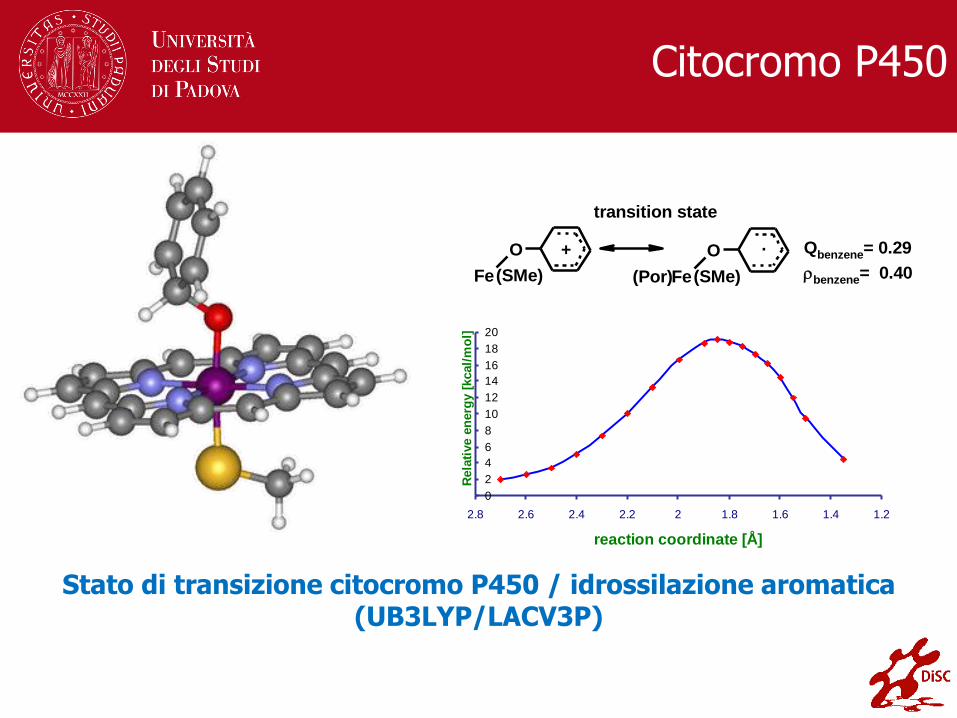
Citocromo P450
0
2
4
6
8
10
12
14
16
18
20
1.21.41.61.822.22.42.62.8
reaction coordinate [Å]
Rela
tive e
nerg
y [
kcal/
mo
l]
O
Fe
O
Fe
+
(SMe)
.
(Por) (SMe)
transition state
Qbenzene= 0.29
benzene= 0.40
Stato di transizione citocromo P450 / idrossilazione aromatica (UB3LYP/LACV3P)

− Sitesi aromatica degli amminoacidi / Shikimate pathway
− GAMESS-UK/CHARMM (B3LYP/6-31G*, HF/6-31G*…)
− 25 Å regione QM
OH
O CO2-
CO2-
OH
-O2C
O
CO2-
O
OH
CO2-
H
-O2CChorismate Prephenate
Mutasi Chorismate
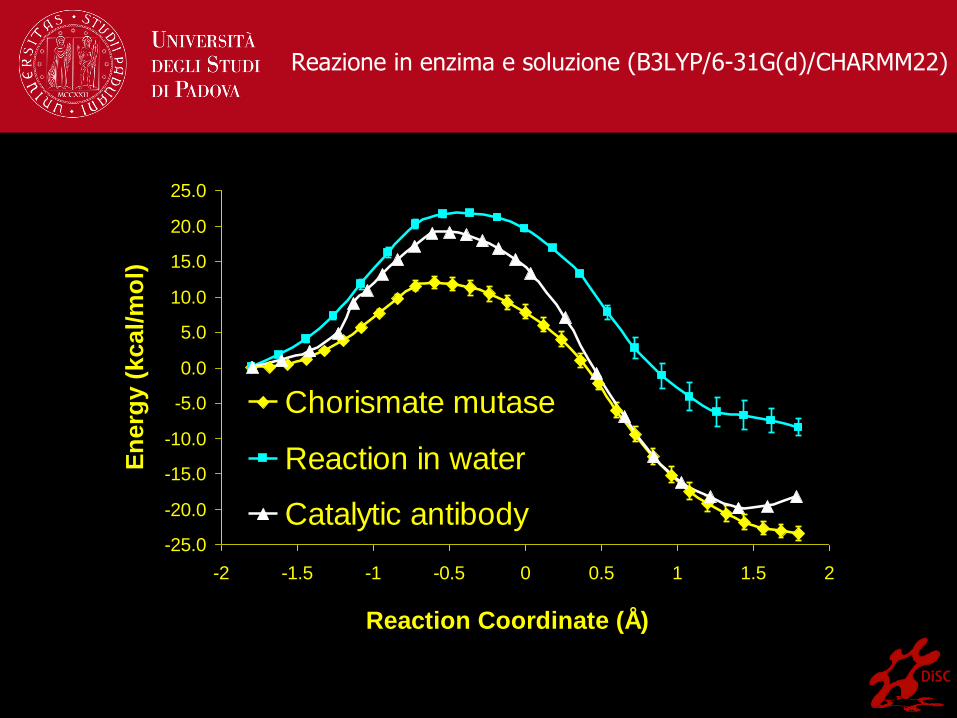
Reazione in enzima e soluzione (B3LYP/6-31G(d)/CHARMM22)
-25.0
-20.0
-15.0
-10.0
-5.0
0.0
5.0
10.0
15.0
20.0
25.0
-2 -1.5 -1 -0.5 0 0.5 1 1.5 2
Reaction Coordinate (Å)
En
erg
y (
kcal/m
ol)
Chorismate mutase
Reaction in water
Catalytic antibody
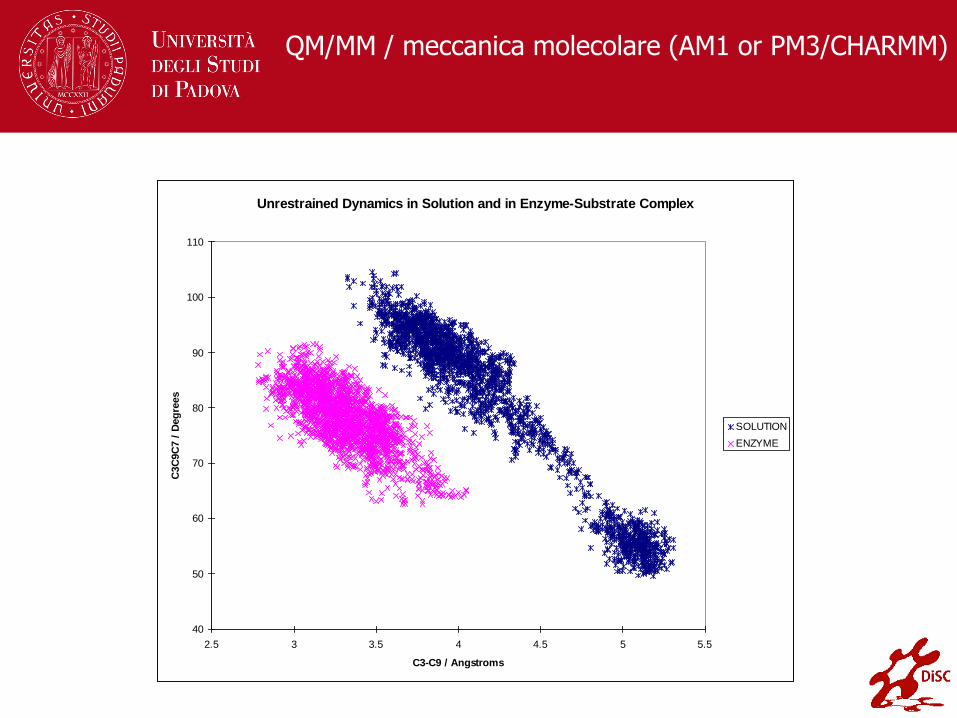
Unrestrained Dynamics in Solution and in Enzyme-Substrate Complex
40
50
60
70
80
90
100
110
2.5 3 3.5 4 4.5 5 5.5
C3-C9 / Angstroms
C3C
9C
7 /
Deg
rees
SOLUTION
ENZYME
QM/MM / meccanica molecolare (AM1 or PM3/CHARMM)
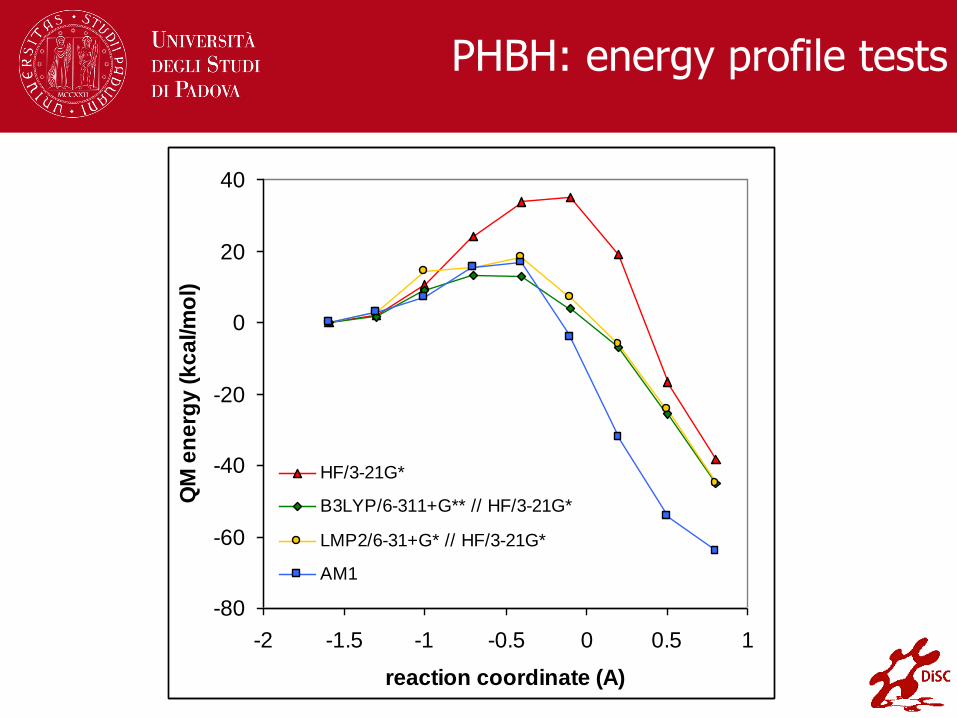
PHBH: energy profile tests
-80
-60
-40
-20
0
20
40
-2 -1.5 -1 -0.5 0 0.5 1
reaction coordinate (A)
QM
en
erg
y (
kc
al/m
ol)
HF/3-21G*
B3LYP/6-311+G** // HF/3-21G*
LMP2/6-31+G* // HF/3-21G*
AM1
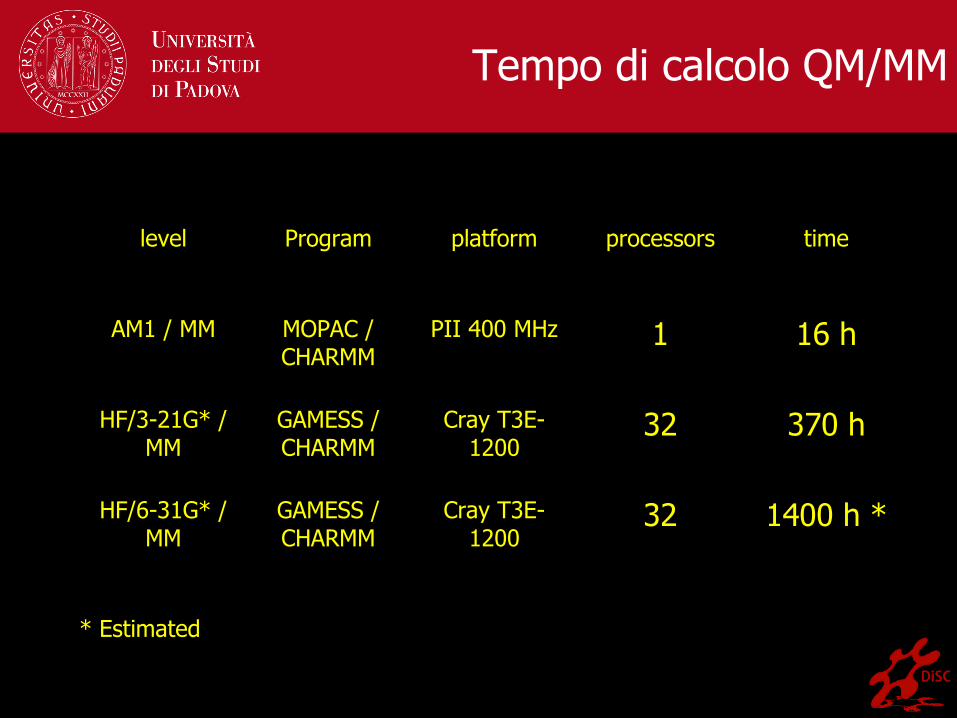
Tempo di calcolo QM/MM
level Program platform processors time
AM1 / MM MOPAC /CHARMM
PII 400 MHz 1 16 h
HF/3-21G* /MM
GAMESS /CHARMM
Cray T3E-1200
32 370 h
HF/6-31G* /MM
GAMESS /CHARMM
Cray T3E-1200
32 1400 h *
* Estimated
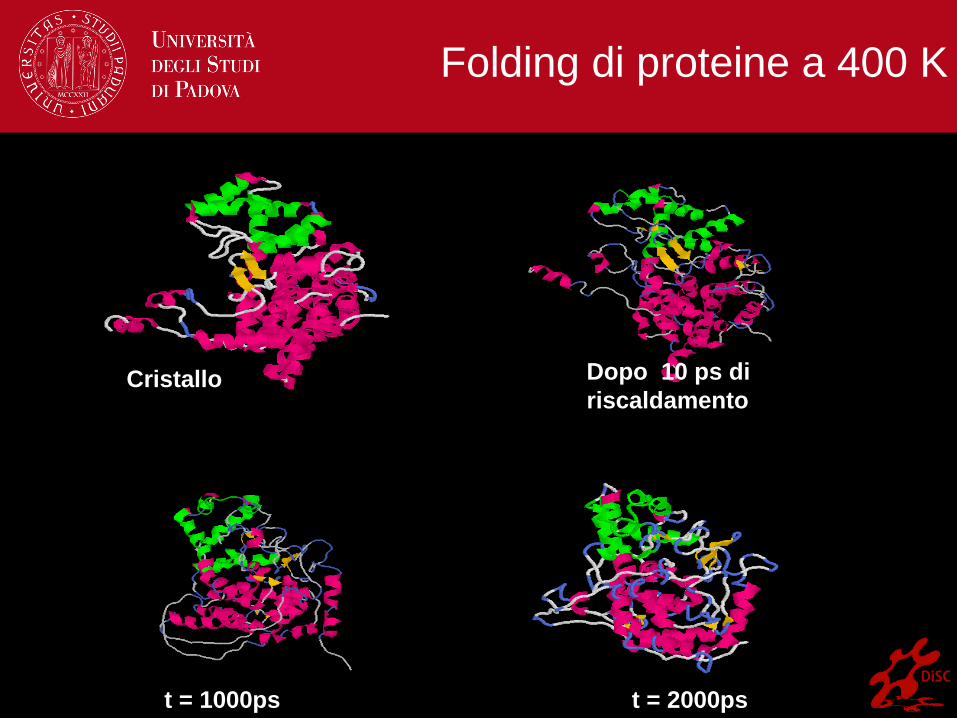
Cristallo Dopo 10 ps di
riscaldamento
t = 1000ps t = 2000ps
Folding di proteine a 400 K
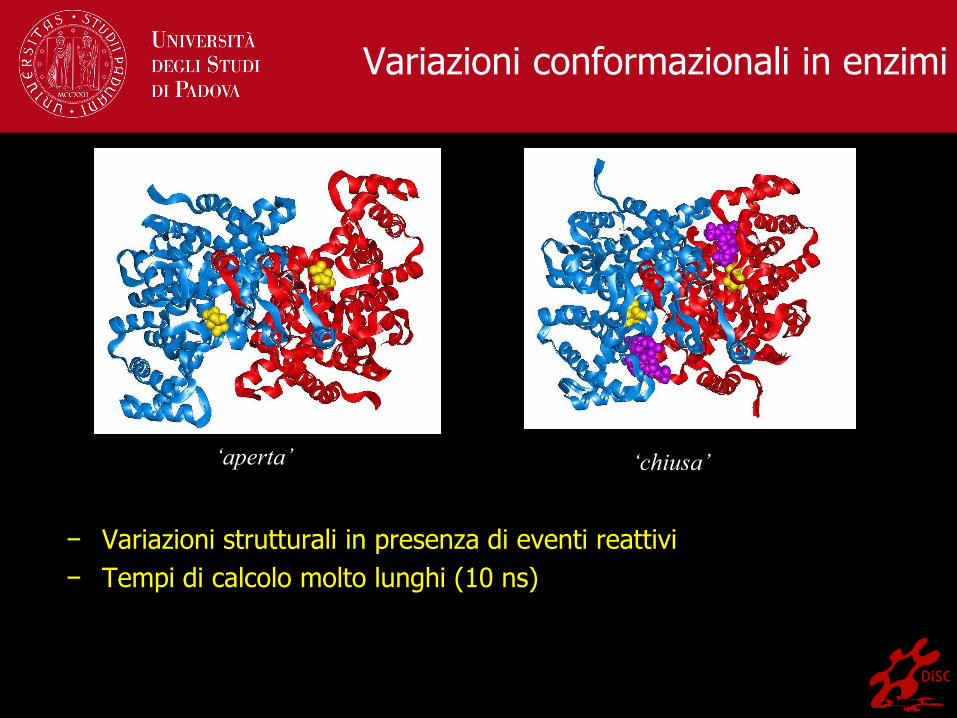
Variazioni conformazionali in enzimi
− Variazioni strutturali in presenza di eventi reattivi
− Tempi di calcolo molto lunghi (10 ns)
‘aperta’ ‘chiusa’
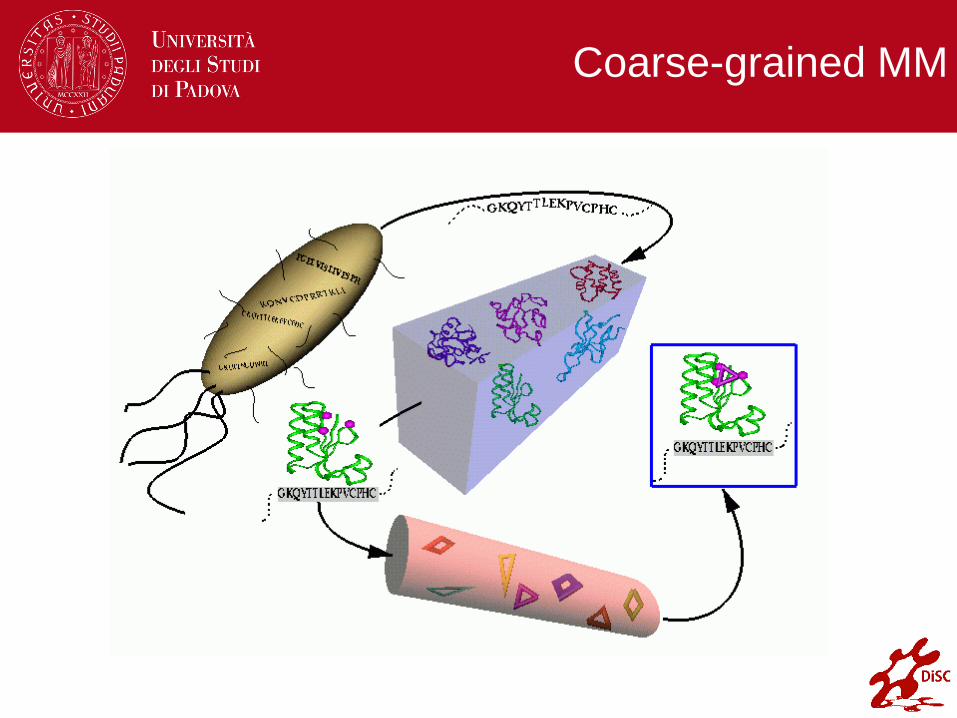
Coarse-grained MM
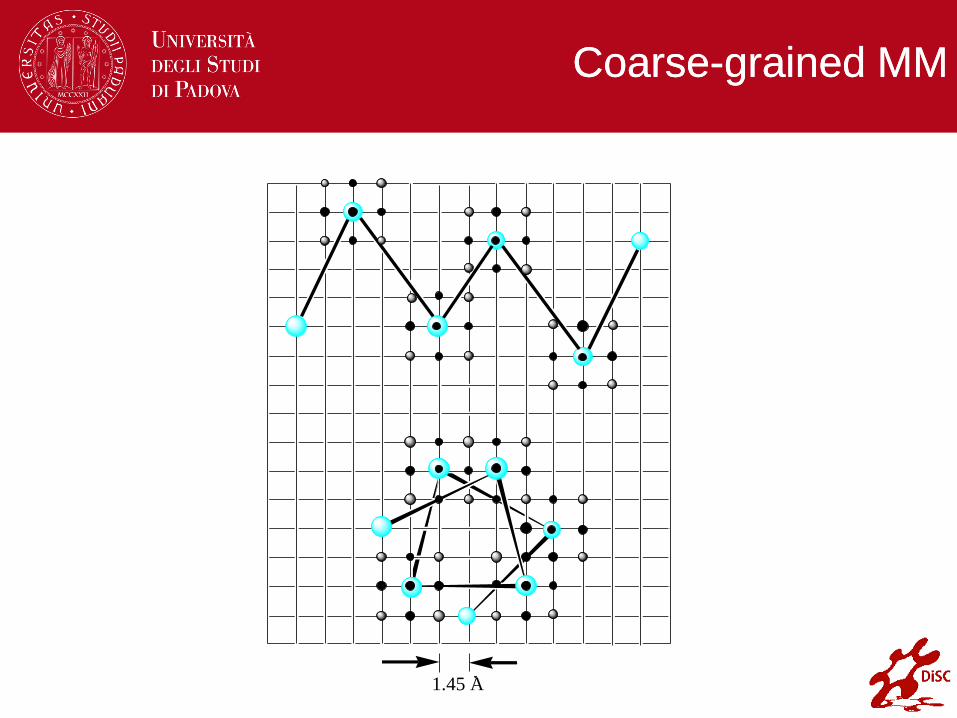
Coarse-grained MM
1.45 A
Coarse-grained MM



















