Presentazione di PowerPoint - Piattaforma MOODLE · PDF fileIl trasferimento dipende dal pKa...
50
Farmacocinetica assorbimento, distribuzione Studia l’evoluzione temporale delle concentrazioni di un farmaco e dei suoi metaboliti nei diversi fluidi e tessuti dell’organismo
Transcript of Presentazione di PowerPoint - Piattaforma MOODLE · PDF fileIl trasferimento dipende dal pKa...

Farmacocinetica assorbimento, distribuzione
Studia l’evoluzione temporale delle concentrazioni di un
farmaco e dei suoi metaboliti nei diversi fluidi
e tessuti dell’organismo

OBIETTIVI
Sviluppare nuovi farmaci
Selezionare la via di
somministrazione
Scegliere la forma farmaceutica
Conoscere la capacità di accesso
ad organi e tessuti
Conoscere le vie metaboliche
Caratterizzare i processi di
eliminazione
Stabilire le relazioni con la
risposta farmacologica
Migliorare i risultati dei
trattamenti
L’effetto biologico di un farmaco è
funzione della dose somministrata

Assorbimento
L’assorbimento di un farmaco è il
suo trasferimento dalla sede di
somministrazione al torrente
circolatorio

……..comporta il passaggio di membrane cellulari

Passaggio dei farmaci attraverso le membrane biologiche
in funzione delle loro caratteristiche chimico-fisiche
Caratteristiche del farmaco Passaggio attraverso le membrane
biologiche
Sostanze idrosolubili, non ionizzabili, con
diametro molecolare inferiore a 4 Å (acqua,
urea, alcool)
- Filtrazione attraverso i pori
Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata.
Il trasferimento dipende dal pKa della sostanza
e dal gradiente di pH ai due lati della
membrana
Sostanze idrosolubili non ionizzate con
diametro superiore a 4 Å (glucosio)
- Diffusione facilitata senza dispendio
energetico per mezzo di un trasportatore
Acidi e basi organiche ionizzate -Trasporto attivo con dispendio energetico
mediante un trasportatore
Proteine ed altre grosse molecole - Fagocitosi e pinocitosi (trasporto vescicolare)

Passaggio dei farmaci attraverso le membrane biologiche
in funzione delle loro caratteristiche chimico-fisiche
Caratteristiche del farmaco Passaggio attraverso le membrane
biologiche
PROCESSO PASSIVO
Sostanze idrosolubili, non ionizzabili, con
diametro molecolare inferiore a 4 Å (acqua,
urea, alcool)
- Filtrazione attraverso i pori
Elettroliti deboli (la maggior parte dei farmaci) - Diffusione semplice della forma indissociata.
Il trasferimento dipende dal pKa della sostanza
e dal gradiente di pH ai due lati della
membrana
MECCANISMO DI TRASPORTO
Sostanze idrosolubili non ionizzate con
diametro superiore a 4 Å (glucosio)
- Diffusione facilitata senza dispendio
energetico per mezzo di un trasportatore
Acidi e basi organiche ionizzate -Trasporto attivo con dispendio energetico
mediante un trasportatore
Proteine ed altre grosse molecole - Fagocitosi e pinocitosi (trasporto vescicolare)
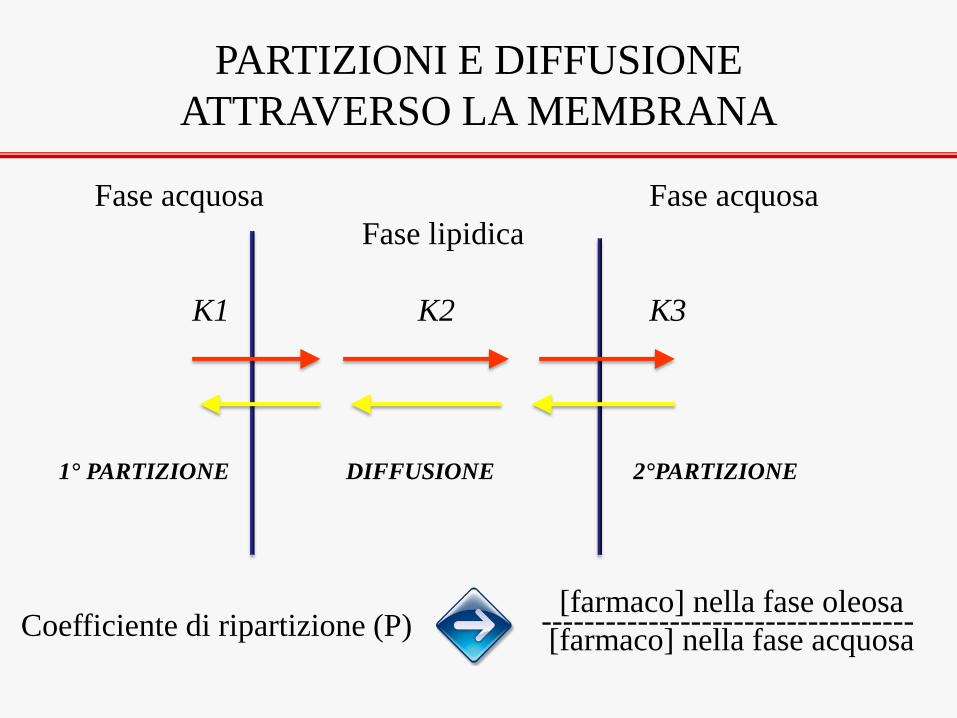
PARTIZIONI E DIFFUSIONE
ATTRAVERSO LA MEMBRANA
Fase acquosa Fase acquosa
Fase lipidica
K1 K2 K3
1° PARTIZIONE DIFFUSIONE 2°PARTIZIONE
[farmaco] nella fase oleosa ----------------------------------- [farmaco] nella fase acquosa Coefficiente di ripartizione (P)

MOLECOLA CON P BASSO
E’ più solubile in acqua che in
olio e quindi sarà catturata
lentamente dalla fase lipidica,
ma sarà ceduta rapidamente
dall’altra parte.
Il processo 1-2 sarà limitante
MOLECOLA CON P ALTO
E’ più solubile in olio che in
acqua e quindi sarà
catturata rapidamente dalla
fase lipidica, ma sarà ceduta
lentamente dall’altra parte.
Il processo 2-3 sarà limitante
1 2 3 1 2 3

MOLECOLA CON P INTERMEDIO
Si verificano le condizioni ottimali di
assorbimento. L’unico fattore limitante
può essere rappresentato dal processo 2
di diffusione all’interno della membrana
che però è molto sottile.
1 2 3
In questo caso la
velocità del
passaggio è regolata
dalla 1° legge di
Fick
A parità di temperatura, di sostanza diffondente e di solvente, la massa
di sale che diffonde attraverso una determinata interfaccia è
direttamente proporzionale al gradiente di concentrazione attraverso la
superficie, all'area della superficie e alla durata del fenomeno
osservato.

Il farmaco è LIPOFILO
Il farmaco non è IONIZZATO
Il farmaco non è LEGATO
•Motilità gastrica
•Presenza di cibo nello stomaco
•pH nel sito di assorbimento
•Area della superficie assorbente
•Flusso ematico
•Eliminazione presistemica
La diffusione passiva di un farmaco
attraverso le membrane è possibile solo se:

AH H+ A- Costante di diss acida = Ka = [H+] [A- ] / [AH]
Per un acido: percentuale ionizzata = 1 + 10(pKa – pH)
100
Per un base: percentuale ionizzata = 1 + 10 (pH – pKa)
100

Il grado di dissociazione dipende dal pH
dell’ambiente in cui si trova
Stomaco: pH = 1,4 Plasma: pH = 7,4
acido acetilsalicilico pKa = 3,4

Succo gastrico Plasma Urina
Anione
A-
Base
libera
B
Base
protonata
BH
Acido
indissociato
AH
gli acidi deboli
tendono ad
accumularsi nei
compartimenti
dove il pH è
più alto, il
contrario fanno
le basi deboli

Influenza del pH sull’assorbimento di
farmaci nel tratto gastrointestinale
Stomaco
pH 1-3.55
digiuno
ileo
pH 8
Piloro pH varia con il riempimento dello stomaco
duodeno
pH 5-6
colon
10
20
30
40
50
60
3.6
4.3 4.7
5.0
7.1
7.2
7.8
8.0
farmaci basici farmaci acidi
chinina aminofenazone
ac. acetilsalicilico
ac. nalidissico
pH intestinale
% f
arm
aco
ass
orb
ito

15
ENTITA’ DELL’ASSORBIMENTO IN RELAZIONE AI
TEMPI DI CONTATTO CON LA SUPERFICIE
ASSORBENTE GASTROENTERICA
Segmento
dell’intestino
Velocità
di transito
Entità pratica di
assorbimento
Duodeno 5-10
min 15%
Digiuno 2 ore 23%
Ileo 3-6 ore 62%
Motilità gastrica
Presenza di cibo nello
stomaco
pH nel sito di
assorbimento
Area della superficie
assorbente
Flusso ematico
Eliminazione presistemica

Effetto della contemporanea assunzione di cibo
sull’assorbimento di alcuni farmaci
somministrati per via orale
Assorbimento ridotto Assorbimento aumentato
Ampicillina Griseofulvina
Amoxicillina Carbamazepina
Rifampicina Propranololo
Aspirina Metoprololo
Isoniazide Spironolattone
Levodopa Idralazina

Interazioni tra farmaci
Vagolitici Rallentano la
motilità intestinale
Vagomimetici Accelerano la
motilità intestinale
Antiacidi Modificano il pH
gastrico
Cortisonici Effetti lesivi sul
tubo gastroenterico

Variabilità farmacocinetica
di 4 preparazioni di digossina
0 1 2 3 4 50,0
0,5
1,0
1,5
2,0
2,5
3,0 A
B1
B2
C
dig
ossin
a p
lasm
ati
ca (
g/m
l)
Tempo (h)
Le preparazioni farmaceutiche contenevano la stessa dose (0.25 mg/compressa) di
digossina ed erano assunte dagli stessi volontari con le stesse modalità. B1 e B2 erano
preparate dalla stessa ditta
Effetto di primo passaggio
Solubilità del farmaco
Forma farmaceutica

Riassumendo: quali sono i fattori che condizionano
l’assorbimento di un farmaco?
Caratteristiche del farmaco: massa molecolare, stato fisico, carica,
stabilità, solubilità….
Proprietà dell’organismo: morfologia e dimensioni della superficie
assorbente, perfusione dell’area assorbente,
specie, razza, età, stato nutrizionale,
stato di salute…..
Caratteristiche dell’esposizione: dose, via di somministrazione, durata
del contatto con la superficie
assorbente….
Fattori esogeni: formulazione, interazione con altre sostanze,
condizioni fisiche (es. temperatura)…..

Modalità di applicazione e decorso temporale
della concentrazione del farmaco
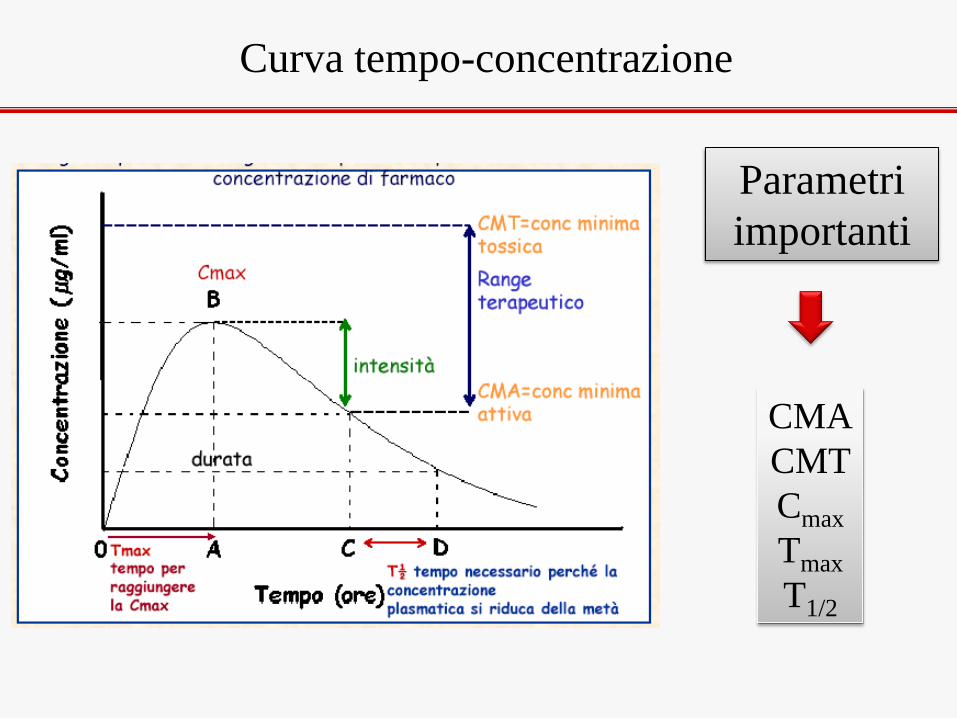
Curva tempo-concentrazione
CMA
CMT
Cmax
Tmax
T1/2
Parametri
importanti

Quando le dosi sono ripetute?
curva tempo-concentrazione
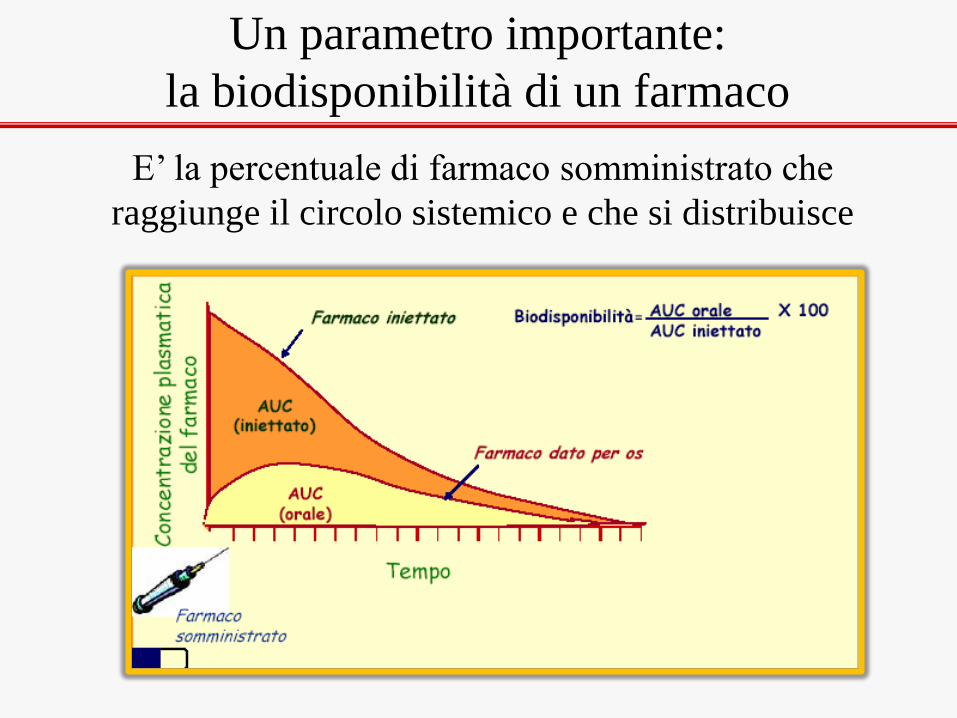
Un parametro importante:
la biodisponibilità di un farmaco
E’ la percentuale di farmaco somministrato che
raggiunge il circolo sistemico e che si distribuisce

Cinetiche di assorbimento
I farmaci che vengono assorbiti per
diffusione passiva seguono una cinetica di
I ordine (esponenziale)
I farmaci che vengono assorbiti per
trasporto attivo seguono una cinetica di
ordine 0 (retta in grafico lineare)

CINETICA DI ASSORBIMENTO DI PRIMO ORDINE
10 mg
90 mg 19 mg
81 mg 27.1 mg
10 mg
9 mg
8.1 mg
100 mg
La quantità di farmaco assorbita è dipendente dalla quantità di farmaco
ancora da assorbire
(10 %)
(10 %)
(10 %)
72.9 mg 7.29 mg (10 %)
34.39 mg
farmaco
da assorbire
farmaco assorbito
farmaco nel plasma

10 mg
90 mg 20 mg
80 mg 30 mg
10 mg
10 mg
10 mg
100 mg
La quantità di farmaco assorbita è indipendente dalla quantità di farmaco
ancora da assorbire
CINETICA DI ASSORBIMENTO DI ORDINE ZERO (trasporto saturato)
farmaco
da assorbire farmaco nel plasma
farmaco assorbito

Attenzione alla scala dei valori riportati sui grafici!!!

CINETICA DI I° ORDINE
Nell’unità di tempo viene assorbita (attraverso meccanismi
diffusionali o trasporti non saturati) una frazione costante di
farmaco, che è proporzionale alla quantità
che resta da assorbire
CINETICA DI ORDINE ZERO
Nell’unità di tempo viene assorbita una quantità costante di
farmaco, che dipende dalla disponibilità dei siti di trasporto:
caratteristiche dei processi saturabili

Distribuzione
processo per mezzo del quale un farmaco
passa da un distretto corporeo all’altro
fino a raggiungere il sito d’azione,
ovvero dal sangue ai vari compartimenti
dell’organismo

Fattori che influenzano
la distribuzione di un farmaco
Caratteristiche fisico-chimiche del farmaco: grado di ionizzazione
liposolubilità
peso molecolare
Fissazione proteica della molecola
Irrorazione degli organi
cervello, cuore, fegato, reni
Affinità specifica dei tessuti
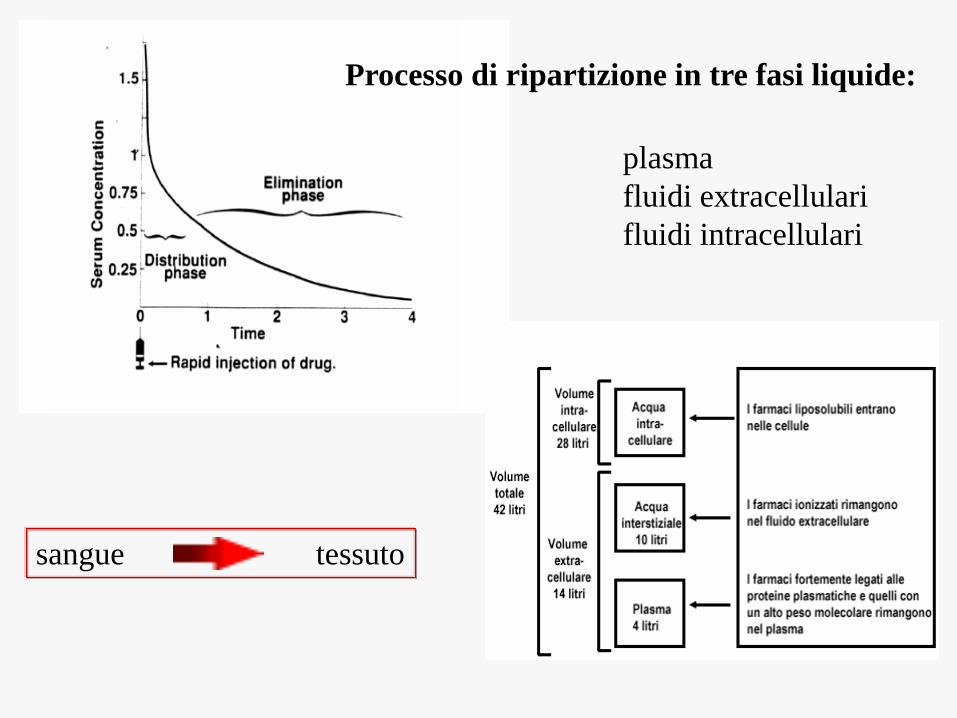
plasma
fluidi extracellulari
fluidi intracellulari
Processo di ripartizione in tre fasi liquide:
sangue tessuto
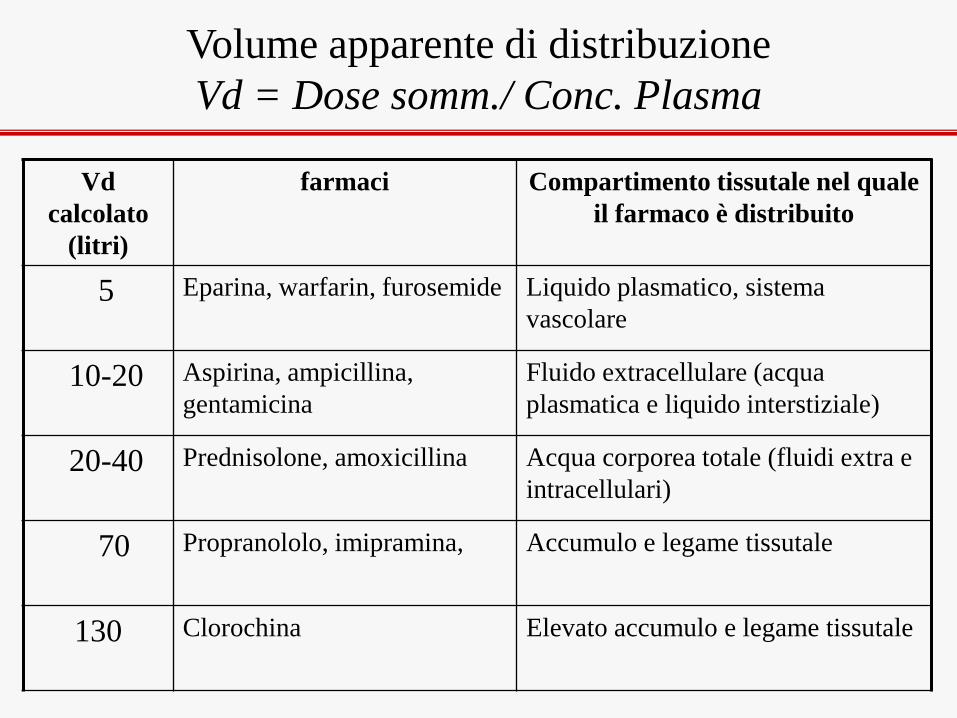
Vd
calcolato
(litri)
farmaci Compartimento tissutale nel quale
il farmaco è distribuito
5 Eparina, warfarin, furosemide Liquido plasmatico, sistema
vascolare
10-20 Aspirina, ampicillina,
gentamicina
Fluido extracellulare (acqua
plasmatica e liquido interstiziale)
20-40 Prednisolone, amoxicillina Acqua corporea totale (fluidi extra e
intracellulari)
70 Propranololo, imipramina, Accumulo e legame tissutale
130 Clorochina
Elevato accumulo e legame tissutale
Volume apparente di distribuzione
Vd = Dose somm./ Conc. Plasma

Influenza della idrofilicità/lipofilicità
del farmaco nella distribuzione

Vd = D/C
D = quantità di farmaco (dose in mg)
C = concentrazione plasmatica (mg/ml)
Per sapere la quantità di farmaco presente nell’organismo:
Vd x C = D
Per sapere il regime terapeutico (dose necessaria ad
ottenere una concentrazione plasmatica desiderata):
D/Vd = C
Utilità pratica del Volume Apparente di Distribuzione

Calcolo di Vd in assenza di eliminazione
Calcolo di Vd in presenza di eliminazione

Diversa permeabilità capillare

PERMEABILITÀ CAPILLARE
glia
Lamina basale
Sinusoidi epatici
Milza
Midollo rosso
Muscoli lisci e
striati
Glomeruli renali
fenestrae
Cervello
Midollo spinale
(barriea emato-encefalica)
giunzione
serrata

Affinità dei farmaci per i diversi tessuti
Lo stesso farmaco può avere affinità diverse
per i diversi tessuti
Il rapporto tra le concentrazioni di farmaco in
un tessuto e nel sangue all’equilibrio di
distribuzione è definito Kp del tessuto
Kp = Ct/Cp

Struttura del farmaco:
diverso coefficiente di ripartizione olio/acqua C
on
cen
trazio
ne n
el S
NC
Tempo (minuti)
15 30 60
Tiopentale
Pentobarbitale
Barbitale
Ac. salicilico
IDR
OF
ILIA
Kp = 1

Co
ncen
trazio
ne t
issu
tale
Tempo (minuti)
Rene
Cervello
Tess. adiposo
Flusso ematico Organo Flusso ematico
ml/min
Polmoni 2500
Rene 650
Fegato 650
Cuore 100
Cervello 350
Tess. adiposo 100
Ossa 125
Muscolo a riposo 375
Kp = 1

Interazione con il bersaglio farmacologico
Dispersione in siti di legame casuali
Siti di deposito e accumulo
Dove va a finire il farmaco?
Interazione con le proteine plasmatiche
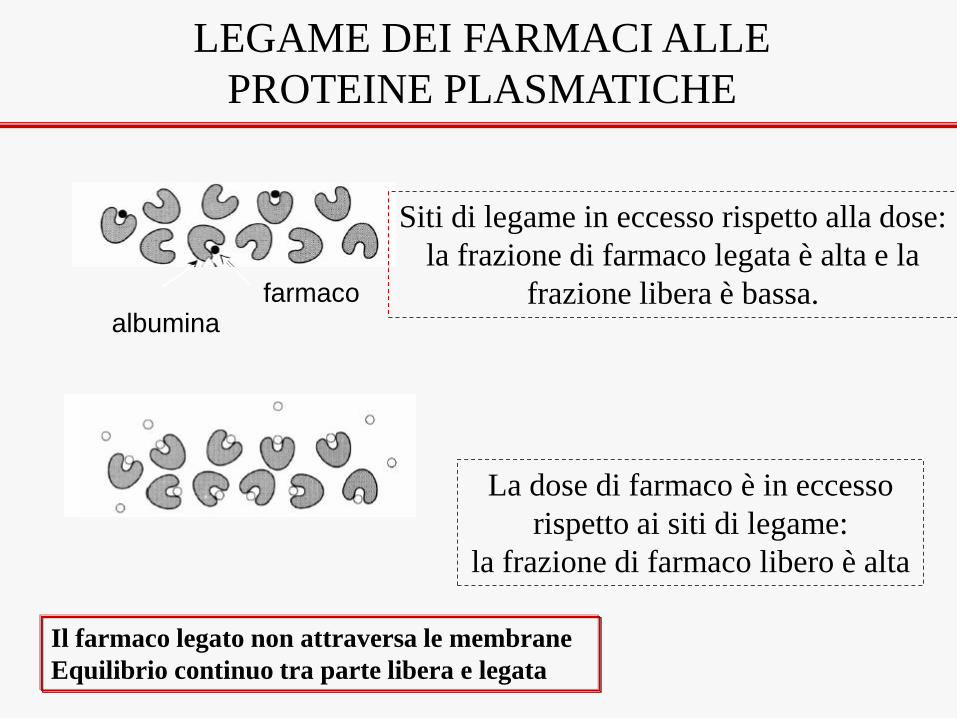
farmaco albumina
Siti di legame in eccesso rispetto alla dose:
la frazione di farmaco legata è alta e la
frazione libera è bassa.
La dose di farmaco è in eccesso
rispetto ai siti di legame:
la frazione di farmaco libero è alta
LEGAME DEI FARMACI ALLE
PROTEINE PLASMATICHE
Il farmaco legato non attraversa le membrane
Equilibrio continuo tra parte libera e legata

Perché è importante?
Warfarin
anticoagulante (INR=2-3)
legato alla albumina per il 97-99%
Un paziente in trattamento con warfarin colpito da
attacco epilettico: somministrazione di fenitoina:
INR sale a 10.
INR=International Normalised Ratio, indice del tempo di coagulazione.
Circa 1 in condizioni fisiologiche

Perché è importante?
•Se un farmaco si lega molto alle proteine plasmatiche
è necessario dare una dose di carico per avere un
effetto terapeutico
•Se un farmaco si lega alle proteine plasmatiche e un
altro agente compete per tale legame e lo spiazza:
se era legato per il 97% e viene spiazzato per il 3%
la sua concentrazione libera raddoppia!
se era legato per il 70% e viene spiazzato per il 3%
la sua concentrazione libera varia ma non drasticamente!
EFFICACE TOSSICO

Farmaci molto legati...
• Legati alle albumine o alle glicoproteine alfa:
– FANS
– warfarin
– ceftiofur
– doxiciclina
– furosemide
– chinidina
– diazepam
– propranololo
– …

effetto albumine a1-glicoproteine lipoproteine
- - ins. epatica contracc.orali ipertiroidismo
cirrosi sindr. nefrosica traumi
polmonite
traumi
ustioni
età
+ + esercizio fisico artrite reumatoide diabete
insuff.renale età (anziano) ipotiroidismo
ipotiroidismo infarto cardiaco sindr. nefrosica
malattia celiaca
Alterazioni della concentrazione delle proteine plasmatiche
in relazione ad alcune condizioni fisiopatologiche

La barriera placentare
eparina
insulina
benzodiazepine
barbiturici

I farmaci non penetrano uniformemente nell’organismo
Particolari tessuti possono mostrare concentrazioni di farmaco superiori o inferiori a quello del plasma
Alcuni tessuti possono comportarsi
in modo analogo al plasma (si possono considerare un compartimento comune)
Compartimenti

Modello a un compartimento

Modello a due compartimenti