
perfarescelteconsapevoli - Partecipasalute.it completa.pdf · Ilprogetto Partecipasalute Ilprogetto...
115
LA DISPENSA Orientarsi in salute e sanità per fare scelte consapevoli
Transcript of perfarescelteconsapevoli - Partecipasalute.it completa.pdf · Ilprogetto Partecipasalute Ilprogetto...

LA
DIS
PE
NS
A
Orientarsi in salute e sanità
per fare scelte consapevoli

PROGETTO PARTECIPASALUTE
Realizzato con il sostegno di
Settembre 2008
ISTITUTO DI RICERCHE FARMACOLOGICHE “MARIO NEGRI”, MILANO
Nessuna parte di questo manuale può essere riprodotta o trasmessa sotto ogni
forma e con qualsiasi mezzo – elettronico, meccanico, inclusa la fotocopiatura,
la registrazione e ogni altra forma o sistema d’archiviazione e recupero – senza
l’autorizzazione a utilizzare i contenuti dell’opera originali.
Le richieste d’autorizzazione alla riproduzione o alla citazione del materiale con-
tenuto in questo manuale devono essere inviate a:
Dr.ssa Paola Mosconi
Istituto di Ricerche Farmacologiche “Mario Negri”
Via La Masa 19, 20156 Milano – e-mail: [email protected]
Centro Cochrane italiano

ISBN 978-88-87626-17-9

Come nasce questa dispensaIl corso per rappresentanti di associazioni di cittadini e pazienti organizzatoogni anno da Partecipasalute può contare ora su una dispensa che ne presenta iprincipali contenuti: dalla sperimentazione clinica all’informazione in medicinae sanità; dall’incertezza ai conflitti di interesse, ai comitati etici; da come funzio-nano le agenzie regolatorie dei farmaci al mondo dell’associazionismo in ambitosanitario.La dispensa è cresciuta di anno in anno insieme ai diversi protagonisti dei corsi:scritta per buona parte dai docenti stessi, in una prima versione è stata “speri-mentata” all’interno del corso del 2007 e quindi modificata in base alle indica-zioni e le esigenze emerse tra quei fruitori.Ogni capitolo, inoltre, è stato rivisto da almeno un rappresentante di associa-zione o componente laico di comitato etico.I contenuti della dispensa offrono un quadro generale dei vari temi che verran-no approfonditi durante il corso attraverso incontri con esperti, discussioni e la-vori di gruppo.
AUTORI
l Giovanni Apolone, Istituto di Ricerche Farmacologiche Mario Negri, Milanol Luca Carra, Agenzia di editoria scientifica Zadig, Milanol Sergio Cima, Agenzia di editoria scientifica Zadig, Milanol Cinzia Colombo, Istituto di Ricerche Farmacologiche Mario Negri, Milanol Alessandro Liberati, Centro Cochrane Italiano; Università degli studidi Modena e Reggio Emilia
l Gaia Marsico, Consorzio Mario Negri Sud, Santa Maria Imbaro (Chieti)l Paola Mosconi, Istituto di Ricerche Farmacologiche Mario Negri, Milanol Monica Oldani, Agenzia di editoria scientifica Zadig, Milanol Vanna Pistotti, Istituto di Ricerche Farmacologiche Mario Negri, Milanol Eugenio Santoro, Istituto di Ricerche Farmacologiche Mario Negri, Milanol Roberto Satolli, Agenzia di editoria scientifica Zadig, Milano
REVISORI
l Ines Benedetti, AILS Associazione italiana lotta alla sclerodermia, Milanol Ilaria Carretta, Comitato etico Ospedale San Raffaele, Milanol Maria Gloria De Bernardo, Comitato etico Azienda ospedaliera, Veronal Maria Di Ottavio, Associazione Attivecomeprima, Milanol Alberto Fontana, Unione italiana lotta alla distrofia muscolare (UILDM),Padova
l Carmela Mandas, FDG Federazione diabete giovanile, Cagliaril Annalisa Marzot, Comitato etico ASL 9, Grossetol Marisa Monari, Associazione ADOCM Crisalide, Riminil Silvia Nidasio, MOVI Movimento di volontariato italiano, Fed. Lombardia,Milano
l Rosita Orlandi, Comitato etico indipendente Policlinico di Baril Pierluigi Pennati, ASNPV Associazione Nazionale Psoriasi e Vitiligene Onlus,Milano
l Dafne Rossi, Associazione Serena, Sienal Roberto Trefiletti, Federconsumatori Lombardia, Milanol Luisa Villa, Altroconsumo, Milanol Adele Zuccolini, Comitato etico Arcispedale Santa Maria Nuova,Reggio Emilia
III


IndiceCome nasce questa dispensa . . . . . . . . . . . . . . . . . . . . . III
Prefazione . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . VIISilvio Garattini
Il progetto Partecipasalute . . . . . . . . . . . . . . . . . . . . . . IX
L’ABC della ricerca clinica . . . . . . . . . . . . . . . . . . . . . . . 1Monica OldaniRevisori: Marisa Monari, Adele Zuccolini
Incertezza e conflitti di interesse in medicina . . . . . . . . . . . 29Sergio Cima, Roberto SatolliRevisori: Alberto Fontana, Rosita Orlandi
Meno ricerca ma di migliore qualità . . . . . . . . . . . . . . . . . 49Alessandro LiberatiRevisori: Ines Benedetti, Ilaria Carretta
L’informazione in medicina: come destreggiarsi . . . . . . . . . . 57Luca Carra, Cinzia ColomboRevisore: Maria Di Ottavio
Navigare sulla rete alla ricerca di informazioni di salute . . . . . 71Vanna Pistotti, Eugenio SantoroRevisori: Dafne Rossi, Carmela Mandas
Comitati etici e consenso informato . . . . . . . . . . . . . . . . . 83Alessandro Liberati, Gaia MarsicoRevisori: Maria Gloria De Bernardo, Annalisa Marzot
Le politiche che regolano il mercato dei farmaci: il ruolo
delle agenzie regolatorie . . . . . . . . . . . . . . . . . . . . . . . 93Giovanni Apolone, Paola MosconiRevisori: Luisa Villa, Roberto Trefiletti
L’associazionismo sta cambiando: dall’assistenza alla nascitadi un progetto. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99Cinzia Colombo, Paola MosconiRevisori: Silvia Nidasio, Pierluigi Pennati
V


PrefazioneSilvio Garattini
Il progetto Partecipasalute, reso possibile dal supporto della Compagnia di SanPaolo, nasce con la finalità di rendere il più possibile trasparente il rapporto frail complesso mondo della ricerca e dell’assistenza biomedica e il pubblico conparticolare riferimento ai pazienti. Si parla molto in questi tempi della necessitàdi un maggior coinvolgimento del pubblico nel processo decisionale o di unamaggior partecipazione alla discussione sui grandi problemi della sanità o diuna maggior comprensione dei fattori che permettono la sostenibilità del Servi-zio Sanitario Nazionale, tuttavia si tratta di appelli demagogici a cui non corri-spondono i fatti. E’ infatti difficile trovare iniziative nazionali, regionali, delleASL o delle aziende ospedaliere che si rivolgano a questi problemi; tutta la co-municazione tende ad esaurirsi al livello burocratico o amministrativo chespesso è vissuto dai pazienti solo come limitazione o come complicazione nel-l’acquisire prestazioni che dovrebbero essere invece il frutto di legittimi diritti.D’altra parte, come potrebbe il pubblico essere in grado di esprimere qualsiasiparere sui grandi temi della sanità se solo troppo pochi sono in grado di com-prendere il linguaggio tecnico e spesso inutilmente criptico degli operatori sani-tari? E’ vero che esistono le associazioni dei consumatori e dei pazienti, ma sonogruppi relativamente nuovi per la realtà del nostro Paese. Spesso, pur con tuttala migliore buona volontà, divengono i portavoce di richieste che sono pilotateda specifici interessi economici. Purtroppo il Servizio Sanitario Nazionale è an-cora visto come un corpo estraneo, un’entità che ha il diritto di concedere o dinon concedere determinati interventi. E’ percepito come una fonte di sprechi ocome una fonte di malasanità anche a causa degli interventi massmediatici chetendono a privilegiare pochi eventi negativi rispetto alla stragrande maggioran-za di atti assistenziali positivi. Purtroppo è in grave crisi il rapporto fiducialemedico-paziente ed è in grande aumento il contenzioso sanitario. Pubblicazioni,televisioni e internet hanno aumentato le conoscenze mediche del pubblico,hanno incrementato la fiducia nella ricerca, ma spesso è difficile per il pazien-te fare una ragionevole sintesi fra informazioni spesso contraddittorie. Il medi-co ha per suo conto il problema di orientarsi nella miriade di notizie che ognigiorno escono dal mondo scientifico e inoltre non ha ricevuto nel suo percorsouniversitario la capacità di comunicare in modo efficace e comprensibile.Queste ed altre considerazioni sono alla base della decisione presa da tempo daparte dell’Istituto Mario Negri di essere un interlocutore al servizio del pubblicoe dei pazienti.La dispensa Partecipasalute è uno dei tanti contributi in questo senso ed è il ri-sultato di un corso avanzato tenuto ai cittadini, ai pazienti ed alle loro associa-zioni. E’ dedicata al farmaco e percorre tutto l’itinerario che deve affrontare: dal-la incertezza della ricerca clinica al giudizio degli organismi regolatori, dal ruolodei comitati etici alla difficoltà di reperire informazioni indipendenti. Questa di-spensa è un altro piccolo contributo a rendere i non addetti ai lavori più infor-mati, più critici e più preparati al dialogo con gli operatori sanitari.
VII


Il progetto Partecipasalute
Il progetto Partecipasalute, dal titolo originale Costruire una alleanza strategicatra associazioni di pazienti e cittadini e comunità medico scientifica, è un proget-to di ricerca interdisciplinare tra associazioni di pazienti e cittadini, società me-dico-scientifiche, ricercatori ed esperti di comunicazione e divulgazione scienti-fica. Il progetto è coordinato dall’Istituto di Ricerche Farmacologiche Mario Ne-gri di Milano e si svolge in collaborazione con il Centro Cochrane Italiano el’agenzia di editoria scientifica Zadig. E’ realizzato con il sostegno della Compa-gnia di San Paolo e si avvale di un Comitato tecnico scientifico multidisciplinaredi supporto alle attività. E’ sostenuto anche dal gruppo GRAL, un gruppo dirappresentanti di associazioni di cittadini e pazienti e membri laici di comitatietici che hanno partecipato alle proposte formative del progetto e che collabora-no a molte attività in corso.
Gli obiettivi
Il progetto Partecipasalute è nato per:l orientare i pazienti, i cittadini e le loro associazioni a una partecipazione at-tiva in ambito sanitario e delle scelte in medicina, affiancandoli in un per-corso di formazione e informazione;
l orientare le organizzazioni professionali e scientifiche a un rapporto costrut-tivo con pazienti e cittadini, per accogliere i loro bisogni in particolare ri-guardo alla ricerca clinica e alla diffusione delle informazioni scientifiche;
l creare un tavolo di confronto tra le associazioni di pazienti e cittadini e le or-ganizzazioni scientifiche su tematiche di interesse comune;
l favorire la nascita di una partnership trasparente tra il paziente/cittadino ei servizi sanitari, che aiuti a superare i punti critici di questo rapporto.
Questi infatti sono i presupposti per dare inizio a una collaborazione diffusatra associazioni di pazienti e cittadini e ambiente medico-scientifico, sulla basedi una cultura condivisa della metodologia scientifica e della comunicazioneefficace.
Le attività
Il progetto è attivo dal settembre 2003 e si è sviluppato lungo tre direttive di ri-cerca e attività: conoscenza, empowerment e condivisione.Tutte le attività del progetto sono ampiamente descritte sul sito Partecipasalute(www.partecipasalute.it) nella sezione “Il progetto”:
Conoscenza l indagine in collaborazione con 11 federazioni di pazienti; raccolta dati sulleattività divulgative, sulle attività di ricerca delle associazioni e sulle modalitàdi aggiornamento;
l indagine sul ruolo dei componenti laici nei Comitati etici in collaborazionecon un panel di oncologi coinvolti in progetti di ricerca clinica;
l indagine in collaborazione con la FISM: Federazione italiana società medico-scientifiche su opinioni, attitudine e attività di coinvolgimento delle associa-zioni di cittadini e pazienti;
l indagine sulle aree grigie della ricerca clinica dal punto di vista delle asso-ciazioni di cittadini e pazienti.
Empowerment Sito: messa a punto del sito www.partecipasalute.it rivolto a pazienti, cittadi-ni e loro associazioni sui temi della salute e delle scelte in medicina.L’idea che ha animato i promotori del progetto nella costruzione del sito è tra-smettere attraverso internet fatti, riflessioni e strumenti che spingano versoun’informazione consapevole e scelte ragionate. Ecco quindi, non una replica
IX

delle già tante enciclopedie mediche più o meno chiare e aggiornate disponibilisulla rete, ma alcuni capitoli fondamentali di conoscenza (i tuoi diritti, informatibene, partecipa alla ricerca clinica, attività del progetto), una serie di rubriche(no news usa e getta) su temi non consueti nei siti di salute (miti da sfatare, me-dicina e interesse, l’incertezza della medicina, decisioni condivise, cure utili,inutili non si sa) e, infine, una serie di strumenti attraverso i quali esercitare lospirito critico (il misuratesti, il misurasiti, il misurassociazioni).Corsi: realizzazione di corsi di formazione rivolti a cittadini, pazienti e loro asso-ciazioni e componenti laici di Comitati etici sullametodologia della ricerca clinica.Al momento è in partenza la 3° edizione del percorso formativo.Incontri pubblici di approfondimento: organizzazione di numerosi incontri pub-blici ad hoc di formazione e informazione durante i quali sono stati discussiesempi e ipotesi di collaborazione tra associazioni e comunitàmedico-scientifica.Pubblicazioni: numerosi articoli sul progetto e le tematiche correlate sono statipubblicati, anche su riviste scientifiche internazionali.
Condivisione Consensus conference: realizzazione di iniziative congiunte che si sviluppanoattraverso la stesura di documenti di indirizzo o linee guida di comportamentopratico e nell’organizzazione di conferenze di consenso.l Nel 2005 è stata realizzata la conferenza di consenso su “Bisogni riabilitativie assistenziali delle persone con disabilità da grave cerebrolesione acquisitae delle loro famiglie, nella fase post ospedaliera”.
l Nel 2008, in collaborazione con il Sistema Nazionale Linee Guida dell’Isti-tuto Superiore di Sanità, è stata realizzata la conferenza di consenso daltitolo “Quale informazione per la donna in menopausa sulla terapia ormo-nale sostitutiva?”.
Ricerca: istituzione dello Spazio PARITA “Partecipare alla ricerca insieme alleassociazioni” a cui le associazioni possono rivolgersi per discutere lo sviluppo diun proprio progetto di ricerca indipendente. Il percorso si sviluppa attraversoincontri di discussione e il coinvolgimento delle società medico-scientifiche.
X
Il progetto Partecipasalute

L’ABC della ricerca clinicaMonica OldaniRevisori: Marisa Monari, Adele Zuccolini
L’incertezza della medicina
La conoscenzaprocedeper tentativied errori
«La storia della scienza è lastricata di teorie abbandonate che erano state untempo proclamate assolutamente evidenti» ha affermato il filosofo tedesco KarlPopper, analizzando l’evoluzione dei concetti scientifici. L’immagine non devené stupire né scoraggiare: da sempre la costruzione della conoscenza procedeper acquisizioni graduali, che spesso sono il risultato di una successione di ten-tativi ed errori e che portano alla revisione, a volte parziale a volte totale, di con-vinzioni precedentemente accreditate.Questa, oltre che la storia della scienza in generale, è anche la storia della medi-cina. La medicina moderna ha raggiunto traguardi importanti, contribuendo amigliorare la salute degli individui. I trattamenti sanitari, insieme hanno au-mentato l’aspettativa di vita degli uomini e migliorato la qualità della vita di chiè affetto, per esempio, da malattie croniche. Tuttavia viene spesso negata unasua dimensione imprescindibile e a lungo dissimulata: quella dell’incertezza. Lostesso Ippocrate, che è considerato il padre fondatore della medicina, ben cono-sceva la difficoltà di giudizio, l’ingannevolezza dell’esperienza e la possibilità dierrore nell’esercizio della professione.
Il principiodi autorità
Fino a un’epoca non lontana la scienza medica si è data un gran da fare perconfezionarsi una facciata di infallibilità, sopperendo spesso alla mancanza diconoscenze o di spiegazioni con il principio di autorità. Per secoli, l’ovvio ascen-dente del sapere che guarisce, l’indiscussa autorevolezza di chi lo pratica e la te-nacia delle credenze consolidate hanno lasciato ben poco spazio al dubbio, oquantomeno alla sua espressione.Eppure, la storia stessa della scienza insegna che l’investigazione delle aree diincertezza ha sempre rivestito un ruolo primario nelle conquiste di conoscenza,che spesso hanno avuto come motore primo il sano scetticismo di qualche stu-dioso nei confronti delle nozioni “inconfutabili” del suo tempo e la sua propen-sione a metterle alla prova.
Le dimensioni dell’incertezzaPuò sembrare paradossale che la concezione dell’incertezza come componentelegittima della medicina si sia andata delineando in modo sempre più precisovia via che nella scienza in generale e in quella medica in particolare è cresciutoil ritmo di produzione di nuove scoperte e delle relative applicazioni. Il motivo èche spesso le nuove acquisizioni giungono a rettificare quelle immediatamenteprecedenti e le promesse di soluzioni (diagnosi, cure) definitivamente sicure edefficaci vengono spesso rapidamente ridimensionate. Sono proprio l’accelera-zione del sapere e l’incalzante susseguirsi delle esperienze a mettere in guardiaverso le concezioni date (e pubblicizzate) troppo presto per assodate. Oggi nellascienza lo scenario dell’incertezza è articolato tanto quanto quello della cono-scenza, e nella medicina si ammette che esistono molteplici zone di indetermi-natezza e di imperfezione.
I limitidella conoscenzamedica
A un livello generale vi è l’incertezza che dipende dai limiti propri della cono-scenza medica corrente: si tratta di un’incertezza collettiva, che giustifica l’esi-stenza di quesiti ai quali nessun medico, per quanto esperto e aggiornato, puòdare, in un dato momento, risposta. A livello individuale vi è l’incertezza che de-riva dall’impossibilità per il singolo di padroneggiare tutto il sapere esistente eraggiungibile, sia per i limiti umani nella capacità di accumulare informazionisia per le opportunità non uguali per tutti e dappertutto di accedere ai diversi ti-pi di informazioni. A questa va aggiunta l’incertezza che deriva dalla componen-
1
L’ABC della ricerca clinica

te di soggettività sempre presente nei processi decisionali e nelle scelte operatedi conseguenza.Come se non bastasse, qualsiasi innovazione della medicina, dalla nuova tecni-ca diagnostica al nuovo farmaco, si misura, al momento del suo ingresso nellapratica clinica, con un’ulteriore fonte di incertezza, che ha origine dalla com-plessità dei sistemi ai quali si applica: anche una conoscenza teorica di ottimolivello non può comprendere tutta la variabilità del mondo reale ed è, quindi,inevitabilmente esposta a esiti imprevisti quando venga messa alla prova.
L’influenzadei risvolti socialie culturali
Per finire, nel contesto della cultura medico-scientifica attuale si vanno consoli-dando nuovi tipi di incertezza riguardo alla validità degli interventi curativi. Bu-ona parte dei dubbi posti da alcuni recenti progressi scientifici e tecnici (si pensiper esempio al trapianto di organi, alla diagnosi genetica, alla fecondazione arti-ficiale) non possono essere misurati semplicemente con i parametri, sia teoricisia pratici, della singola disciplina alla quale sono pertinenti, perché richiedonouna serie di valutazioni che vanno al di là del sapere specialistico e riguardano,piuttosto, le implicazioni culturali e i risvolti sociali degli interventi stessi. I con-cetti di benessere, qualità di vita, rischio o prevenzione toccano più da vicino ladimensione etica della salute, davanti alla quale all’incertezza che dipende dallaquantità e dalla qualità delle informazioni tecniche disponibili si aggiungel’incertezza relativa all’impatto degli atti medici sull’esistenza delle persone edelle collettività.D’altra parte, oggi l’incertezza della medicina non tocca più soltanto chi la prati-ca ma anche chi la “subisce”. Il tramonto del modello paternalista-autoritario el’affermarsi di un rapporto medico-paziente basato sulla partecipazione consa-pevole di quest’ultimo alle decisioni che lo riguardano – concretizzatasi formal-mente nella prassi del consenso informato – mette tutti a parte del fatto che lamedicina non è una scienza esatta e che anche in qualità di fruitori si devonooperare delle scelte, assumersene le responsabilità e affrontarne i rischi.
La medicina sperimentale
La scoperta di nuovi trattamenti si accompagna a incertezze relative agli effetti eall’efficacia. Per compensare il più possibile l’indeterminatezza, diminuire ilmargine di imprevedibilità, ridurre al minimo la frequenza degli errori e la gravi-tà dei relativi esiti, è necessario valutare i nuovi trattamenti attraverso speri-mentazioni corrette e rigorose.
Dall’esperienzadel singolomedicoal confrontodelle esperienze
A spingere verso la ricerca di metodi di verifica a cui sottoporre gli interventi me-dici sono stati storicamente, a partire dal XVIII secolo, due fenomeni stretta-mente legati: le scoperte e gli avanzamenti tecnologici della chimica davano av-vio alla produzione di sempre più numerose sostanze dotate di effetti farmacolo-gici e, quindi, di presunti poteri curativi; queste sostanze, utilizzate ancora se-condo criteri del tutto pragmatici, basati il più delle volte sul “sentito dire” o sul“verbo” di qualche notabile dell’arte medica, rivelavano via via di possedere qua-lità diverse da quelle attese e propagandate, oppure di avere, accanto alle pro-prietà terapeutiche, anche il potere di produrre danni più o meno gravi.Del resto, i criteri in base ai quali tradizionalmente si ricorreva a un certo com-posto piuttosto che a un altro derivavano o dalle nozioni accreditate dall’uso po-polare o, nel migliore dei casi, dalle esperienze precedenti del singolo curante.L’idea di far tesoro delle osservazioni fatte sul campo era in linea di principiobuona, e conduceva a unmodo di procedere, empirico e induttivo, che è anche ilfondamento del metodo scientifico moderno. Il problema, allora, era che le espe-rienze del singolo medico passavano al vaglio della sua sola interpretazione eraramente le esperienze personali venivano messe a confronto con quelle di al-tri. L’altra caratteristica della condotta clinica del periodo pre sperimentale èche, in mancanza di nozioni precise sull’efficacia delle diverse cure, esse veniva-no molto spesso utilizzate tutte insieme, in combinazioni che sembravano ispi-rate più al principio melius abundare quam deficere (meglio abbondare chescarseggiare) che non a una conoscenza delle associazioni farmacologiche.
2
L’ABC della ricerca clinica

I pionieri del confronto
L’idea di confrontare in modo sistematico diversi trat-tamenti per avere prove oggettive della loro efficacia ba-lenò con anticipo rispetto allo sviluppo della sperimenta-zione clinica nella mente di un paio di medici particolar-mente illuminati.Il primo, in ordine di tempo, fu James Lind, un medicodella Marina inglese, il quale si mise d’impegno per trova-re il modo di proteggere i suoi marinai dallo scorbuto, unamalattia di origine allora sconosciuta che inevitabilmentecolpiva il personale di bordo durante le traversate di lungadurata sulle rotte oceaniche, annientandone le forze conuna progressione sintomatologica caratterizzata da aste-nia, irritabilità, perdita di peso, dolori muscolari e articola-ri, rigonfiamento delle gengive, alterazioni delle unghie,emorragie sottocutanee. Era il 1747 quando a Lind, im-barcato sulla Salisbury, venne in mente di provare gli ef-fetti di sei diverse integrazioni dietetiche, confrontandolein un gruppo selezionato di dodici marinai colpiti dai clas-sici sintomi, che suddivise in sei coppie.Senza saperlo, Lind attuò per la prima volta nella storiaquello che oggi chiameremmo uno studio clinico controlla-to, pervenendo, oltretutto, a un risultato esemplare: deisei “ricostituenti” che aveva somministrato separatamen-te alle sei diverse coppie di marinai, uno solo, consistentein una porzione giornaliera di agrumi, era stato in grado difar regredire i sintomi dello scorbuto. Lind aveva dimo-strato che nella pratica clinica si poteva smettere di lascia-re tutto al caso e all’improvvisazione e si poteva inveceprocedere secondo criteri razionali. Non aveva la più palli-da idea del perché arance e limoni si fossero distinti perefficacia dai beveroni somministrati in alternativa: nonpoteva immaginare che andassero a compensare una ca-renza di vitamina C legata al suo insufficiente apporto coni cibi disponibili durante le navigazioni, visto che manca-vano ancora più di centocinquant’anni alla scoperta e allacaratterizzazione biochimica delle vitamine.Tuttavia, pur rimanendo all’oscuro dei meccanismi chedavano luogo agli effetti osservati, Lind aveva innanzi-
tutto trovato una cura per una malattia capace di mette-re al tappeto interi equipaggi e, in secondo luogo, avevaper primo praticato il metodo sperimentale, aprendo lastrada alla verifica controllata dei trattamenti medici.Anche se per lungo tempo sia la validità del rimedio da luicollaudato sia il valore scientifico del suo modo di proce-dere non ricevettero l’attenzione che meritavano: primache la Marina di Sua Maestà Britannica decidesse di im-barcare sulle sue navi scorte di agrumi destinate ai mari-nai ci volle un altro mezzo secolo e prima che si riconsi-derasse l’esperimento di Lind alla luce delle sue implica-zioni per la metodologia della ricerca clinica ce ne volleroquasi due.Sorte simile spettò circa cent’anni dopo al medico unghe-rese Ignaz Philipp Semmelweis. Prendendo serviziopresso la prima clinica ostetrica di Vienna, si accorse chenel reparto dove prestavano la loro opera gli studenti dimedicina le puerpere morivano per una grave quanto mi-steriosa setticemia, molto più spesso che in quello fre-quentato solo dalle levatrici; sospettando che a far amma-lare le giovani mamme fossero proprio gli studenti, chesvolgevano anche le esercitazioni necroscopiche e passa-vano dal tavolo anatomico alla sala parto senza lavarsi ac-curatamente le mani, li convinse a disinfettarsele con unasoluzione di cloruro di calcio dopo ogni autopsia, ottenen-do in breve tempo una riduzione della mortalità delle par-torienti dal 12 al 2 per cento. Fatta eccezione per la grati-tudine delle donne che si erano salvate (oltre 400 in solitre anni), Semmelweis ricevette scarsissima considera-zione dai colleghi, i quali gli riservarono, anzi, manifesta-zioni di ostilità e contestazioni tali che finirono per emargi-narlo dalla professione. Anche se li chiamava “particellecadaveriche” e non batteri – in mancanza, nel 1847, di co-noscenze sulle infezioni – Semmelweis aveva scoperto gliagenti causali della funesta febbre puerperale e vi avevatrovato un rimedio, operando, proprio come Lind, un con-fronto controllato tra approcci diversi: l’assenza di precau-zioni igieniche prima, la disinfezione poi.
Quello che non era ancora stato concepito, praticamente, era il modo di proce-dere per confronto delle diverse esperienze, che avrebbe consentito di definire lareale efficacia degli interventi medici mettendoli a paragone gli uni con gli altri.Per la costruzione di un impianto metodologico adatto a fornire verifiche atten-dibili dei diversi approcci terapeutici - principalmente delle ormai numerose so-stanze farmacologiche disponibili - e perché la tutela dei malati in quanto desti-natari degli stessi diventasse una priorità della medicina, si dovette, però, at-tendere fino quasi alla metà del Novecento.
La nascita della sperimentazione clinica
Nel corso del XX secolo si sono realizzati i maggiori sviluppi nella verifica speri-mentale dei farmaci, con la formulazione di metodi che, pur non potendo aspi-rare alla perfezione – obiettivo utopistico se si tiene conto del margine di incer-tezza legato alla complessità con cui si confrontano –, riescono a dare garanziesicuramente molto migliori che in passato.
Il temadella sicurezza
Nei due secoli precedenti erano entrati in circolazione composti terapeutici lacui efficacia e sicurezza, non testate preventivamente in modo sistematico eaffidabile, erano state inevitabilmente messe alla prova sul campo, sostanzial-mente sulla pelle dei pazienti. Spesso con effetti imprevisti e indesiderati an-che gravi.Il tema della sicurezza fu quindi il primo a essere affrontato, negli Stati Uniti al-la fine degli anni Trenta, con un provvedimento che per la prima volta nella sto-
3
L’ABC della ricerca clinica

ria dei medicinali aveva la funzione di tutelare i malati dagli eventuali danniconnessi con il loro impiego (Federal Food, Drug and Cosmetic Act).
Gli studiche verificanol’efficacia
Alla fine degli anni Quaranta, però, si incominciarono a fare le prime mosse an-che sul fronte dell’efficacia, definendo un metodo che consentiva di mettere aconfronto caso per caso trattamenti diversi, in modo sistematico, attendibile eriproducibile.
Assegnarea caso
Nel 1948 il prestigioso British Medical Journal pubblicò i risultati del primostudio controllato e randomizzato - vale a dire uno studio effettuato crismi se-condo i principi della sperimentazione clinica moderna - sugli effetti terapeuti-ci di un farmaco. Il farmaco era la streptomicina, un antibiotico che in base al-le osservazioni sui batteri faceva ben sperare per la cura della tubercolosi. Inquello studio, condotto dal Medical Research Council britannico, fu applicatoper la prima volta il metodo della randomizzazione, che consiste nel suddivide-re i partecipanti in un gruppo sperimentale (sottoposto al trattamento da te-stare) e un gruppo di controllo (sottoposto a un intervento di confronto) secon-do un criterio casuale (il termine random in inglese significa a caso). Fino adallora la comparazione di trattamenti diversi era stata esposta alla possibilitàdi distorsioni che potevano inficiarne i risultati: erano sempre stati gli speri-mentatori, infatti, a decidere a quali soggetti somministrare un certo farmacoe a quali, invece, la sua controparte, con il rischio che la selezione avvenisse,magari anche per scelte fatte in buona fede, secondo criteri non del tutto im-parziali. Con il campionamento randomizzato, invece, si introduceva il casocome arbitro assoluto a garanzia dell’uniformità, e pertanto della confrontabi-lità, dei diversi gruppi di soggetti, partendo dal presupposto che, siccome nonsi può mai ottenere una totale identità di caratteristiche tra i pazienti da stu-diare, solo assegnandoli per sorte al gruppo sperimentale piuttosto che aquello di controllo si ha la possibilità di trovare una quota simile di diversità intutti e due.
Confrontarein cieco
Nella stessa sperimentazione venne introdotto anche un altro principio meto-dologico divenuto poi un requisito essenziale per garantire la qualità degli stu-di clinici: quello della cecità, ovverosia della necessità di tenere i soggetti speri-mentali e in certi casi anche gli sperimentatori all’oscuro dell’opzione terapeu-tica che stavano utilizzando, per evitare che le loro aspettative verso i diversifarmaci messi a confronto possano influenzare la percezione che entrambihanno dei relativi effetti.Nello studio sulla streptomicina, per esempio, neppure i radiologi incaricati divalutare i cambiamenti del quadro polmonare dei malati di tubercolosi parteci-panti furono messi a conoscenza del trattamento abbinato a ciascuna delle la-stre del torace che esaminavano.
Il placebo Negli anni Cinquanta fece la sua comparsa la terza grande innovazione dellaricerca clinica sui farmaci: l’utilizzo di placebo, cioè di sostanze prive di qual-siasi azione farmacologica da mettere a confronto con i veri trattamenti.L’idea nacque dal sospetto che l’efficacia di alcuni trattamenti non derivassetanto dai loro oggettivi effetti farmacologici quanto da fattori soggettivi cheavevano a che fare con le speranze che i pazienti riponevano in una certa cu-ra, con il potere di convincimento che i medici esercitavano nel prescriverlae, in generale, con l’effetto di rassicurazione che l’intervento terapeutico,qualunque esso fosse, esercitava su entrambi – come se l’idea stessa di “farequalcosa” fosse di per sé curativa, indipendentemente dalle proprietà specifi-che della cura scelta. Si tratta, ovviamente, di fattori che entrano in giocosenza la consapevolezza di chi li anima, e che, pertanto, sono difficilmentecontrollabili. A meno che non si decida, al contrario, di sfruttarli ad hoc. Nel-la sperimentazione clinica il placebo viene infatti utilizzato perché faccia datermine di paragone neutro di trattamenti di cui si voglia identificare la realeutilità, in modo da scoprire i casi in cui miglioramenti e guarigioni dipendonosemplicemente dall’atto terapeutico e non dai processi biochimici attivatinell’organismo da uno specifico medicinale.
4
L’ABC della ricerca clinica

Placebo: una bugia per svelare la verità
Il placebo è un trattamento inerte, che viene messo a con-fronto con quello sperimentale per chiarire se gli effetti diquest’ultimo siano davvero dovuti alle sue prerogativespecifiche (se si tratta di un farmaco alla sua azione far-macologica) oppure a qualche altro fattore del tutto indi-pendente.Nel primo caso solo il trattamento sperimentale si riveleràefficace mentre il placebo nonmigliorerà affatto, o al mas-simo di poco, le condizioni dei pazienti; nel secondo casosia il trattamento sperimentale sia il placebo risulterannoefficaci più o meno allo stesso modo.I benefici prodotti da un trattamento inerte sono compresisotto il nome di effetto placebo e si ritiene siano riconduci-bili a fattori di natura psicologica che entrano in gioco nelmomento in cui una persona viene “presa in cura”. Questifattori sono strettamente legati non solo alle caratteristi-che personali del malato ma anche alla qualità del rappor-to che ha con il suo medico.Di questo contributo squisitamente mentale al loro benes-sere gli interessati non sono in genere consapevoli; anzi, il“trucco” funziona proprio perché non è manifesto. Perquesto motivo è necessario che la somministrazione delplacebo nel corso di una sperimentazione avvenga “in cie-
co”, cioè all’insaputa dei soggetti coinvolti: ciascuno di es-si verrà informato del fatto di partecipare a uno studio cheprevede il controllo di un trattamento sperimentale conplacebo, ma non saprà a quale dei due trattamenti (speri-mentale o placebo) è destinato.L’utilizzo di placebo nella ricerca clinica pone, però, que-stioni etiche di rilievo, perché dare come controllo una so-stanza inerte durante la sperimentazione di un nuovotrattamento significa che, se quest’ultimo è davvero effi-cace, per tutta la durata dello studio solo metà dei pazientine avrà tratto beneficio, mentre l’altra metà (quella delgruppo di controllo) avrà assunto pillole finte.Affinché l’utilità scientifica del placebo non si traduca inuna sottrazione di cure, attualmente tutte le normativedi etica della ricerca raccomandano che il ricorso al pla-cebo sia limitato a due solicasi: innanzi tutto quelli in cui,oltre al trattamento da sperimentare, non esista un’al-ternativa terapeutica di comprovata efficacia per la stes-sa malattia; quindi ai casi in cui il farmaco disponibile, alquale bisognerebbe rinunciare a favore del placebo, siasolo un sintomatico per disturbi non gravi (cosicché ri-nunciarvi non comporti conseguenze pesanti per la salu-te dei pazienti).
La scoperta dell’effetto placebo
Negli anni Sessanta l’effetto placebo dimostrò le suepotenzialità nell’ambito della ricerca clinica in uno studioche oggi, alla luce dei principi etici introdotti nei canonidella sperimentazione clinica, verrebbe considerato inop-portuno, ma che all’epoca ebbe l’effetto di una rivelazio-ne; tanto più che l’intervento terapeutico di cui si sma-scherò la totale inutilità era un intervento chirurgico. Cal-deggiato dal suo ideatore, il chirurgo italiano GiuseppeGucci, un intervento di legatura delle arterie mammarie,fatto allo scopo di deviare il sangue in quelle coronarie percompensare l’ischemia cardiaca e prevenire gli attacchi diangina pectoris, suscitò una tale perplessità in un collegastatunitense che questi decise di verificarne l’efficacia ri-spetto a un placebo. Fu così che una ventina di americaniche soffrivano di attacchi anginosi entrò nella sperimenta-zione e venne sottoposta per metà all’intervento di lega-tura dei vasi mammari e per l’altra metà a un finto inter-vento, in cui fu praticata soltanto un’incisione superficialesul torace subito suturata. Alla fine, di identico i malati deidue gruppi avevano solo la cicatrice e la convinzione diavere subìto l’intervento “italiano”; eppure, tutti avverti-rono lo stesso miglioramento dei sintomi.L’obiezione che si può avanzare a un esperimento del ge-nere è scontata: è stato giusto sottoporre le dieci personedel gruppo di controllo a un intervento chirurgico fasullo?Per quanto superficiale, l’intervento placebo non era come
acqua di rose: comportava fare un’anestesia generale,con i rischi connessi; impegnava l’organismo nel processodi riparazione della ferita, con la possibilità di complicanzeinfettive e reazioni infiammatorie; lasciava una cicatrice,con il relativo danno estetico. Tutto questo solo per sbu-giardarne un altro, che si faceva passare per serio e inve-ce era inutile. Il guadagno fu sicuramente considerevoleper la scienza e per i pazienti anginosi a venire - ai qualinessuno più propose la legatura delle arterie mammarie -ma assolutamente nullo per quei dieci che si erano sotto-posti fiduciosi all’esperimento: avevano rischiato di nonottenere la cura migliore nel caso in cui il vero interventosi fosse rivelato valido ed erano stati, per di più, imbro-gliati. E’ vero che dopo si erano sentiti meglio, ma persuggestionarli fino a quel punto forse si sarebbero potutitrovare sistemi meno invasivi!Oggi, il ricorso al placebo nella ricerca clinica è rigidamen-te subordinato a considerazioni che di volta in volta tengo-no conto delle sue implicazioni etiche, vale a dire del dirit-to dei malati a ottenere sempre e comunque la miglioreassistenza che le cognizioni medico-scientifiche consento-no. Ma lunga è stata la strada per arrivare a salvaguarda-re i pazienti, e in particolare quelli che entrano a far partedi studi sperimentali, dalla possibilità di essere “usati” neimodi un po’ troppo disinvolti che, in nome della ricerca, sisono per molto tempo adottati.
Prove di sicurezza dei farmaci
Danni alla saluteprovocatida farmaci
La lista dei rimedi e farmaci resisi responsabili di incidenti isolati o estesi dannialla salute pubblica perché messi in circolazione senza sufficienti verifiche dellaloro efficacia e sicurezza è lunga. Ma tra tutti, almeno un caso può essere qui ri-cordato; non perché sia stato il più importante né l’ultimo della serie, ma perchéaprì la strada sia all’enunciazione rigorosa del metodo sperimentale sia alla co-struzione di un sistema normativo preposto alla tutela dei pazienti. Nel 1937,negli Stati Uniti, un composto a base di sulfamidici fece un centinaio di vittimeperché conteneva un solvente, il glicol-dietilene, di cui non era stata presa inconsiderazione la tossicità.
5
L’ABC della ricerca clinica

A seguito di quell’incidente, negli Stati Uniti fu adottato il Federal Food, Drugand Cosmetic Act (1938) con il quale si stabiliva che per ogni nuovo prodotto ve-nissero presentate prove della sua sicurezza, assegnando alla Food and DrugAdministration, l’autorità nazionale preposta all’autorizzazione della commer-cializzazione dei farmaci, il compito di prendere visione delle suddette prove pri-ma di accordarne l’immissione sul mercato.
Il Codicedi Norimberga
Pochi anni dopo, a breve distanza dalla fine della Seconda guerra mondiale, lascoperta delle atrocità perpetrate a scopo di ricerca, ma senza scrupoli di sorta,dai medici nazisti sui prigionieri dei campi di concentramento (e anche sui ma-lati psichiatrici e sui cosiddetti “ritardati” mentali) ispirò il primo codice deonto-logico della storia che si sia preoccupato di fissare per la sperimentazione sul-l’uomo una serie di norme a difesa dei soggetti che vi si sottopongono. Il Codicedi Norimberga (1946), come fu chiamato dal nome del Tribunale militare incari-cato di giudicare e punire i crimini di guerra, compresi quelli passati sotto l’egi-da della scienza, conteneva anche una prescrizione estremamente innovativa,divenuta poi un principio imprescindibile degli studi clinici: quella di otteneredai partecipanti un esplicito consenso a procedere. Naturalmente, il contenutodi una qualsiasi ricerca non doveva essere accettata dai candidati a partecipar-vi senza che fossero loro fornite informazioni precise, e il loro consenso dovevaessere basato sulla consapevolezza dei vantaggi e dei rischi ai quali potevanoandare incontro: si doveva cioè trattare, secondo la definizione passata in uso,di un “consenso informato”.Veniva così stabilito l’obbligo inderogabile per i ricercatori di rendere consape-voli i soggetti dei loro esperimenti sugli interventi ai quali intendevano sottopor-li, sulle relative implicazioni per la loro salute e sugli scopi degli esperimentistessi, e di espletare questa parte informativa rigorosamente prima di chiederneil consenso. Fu un primo passo, al quale ne fecero seguito diversi altri, andandovia via a perfezionare un sistema di tutela che, a più riprese, dimostrava di nonessere mai abbastanza avveduto. Molti dei provvedimenti relativi alla ricercaclinica, e a quella sui farmaci in particolare, che si susseguirono nella secondametà del secolo scorso nacquero, infatti, in risposta ad avvenimenti molto gravi.
Il Codice di Norimberga
1. Il consenso volontario del soggetto umano è assoluta-mente essenziale. Ciò significa che la persona in questio-ne deve avere capacità legale di dare consenso, deve es-sere in grado di esercitare il libero arbitrio senza l’inter-vento di alcun elemento coercitivo, inganno, costrizione,falsità o altre forme di imposizione o violenza; deve averesufficiente conoscenza e comprensione degli elementi del-la situazione in cui è coinvolto, tali da metterlo in posizio-ne di prendere una decisione cosciente e illuminata.2. L’esperimento dovrà essere tale da fornire risultati utilial bene della società e non altrimenti ricavabili con mezzi ometodi di studio; la natura dell’esperimento non dovrà es-sere né casuale né senza scopo.3. L’esperimento dovrà essere impostato e basato sui risul-tati delle sperimentazioni su animali e sulla conoscenzadella storia naturale del morbo o di altri problemi allo stu-dio, cosicché risultati antecedenti giustifichino lo svolgersidell’esperimento.4. L’esperimento dovrà essere condotto in modo tale daevitare ogni sofferenza o lesione fisica o mentale che nonsia necessaria.5. Non si dovranno condurre esperimenti ove vi sia già apriori ragione di credere che possano sopravvenire la morte
o un’infermità invalidante, eccetto forse quegli esperimentiin cui il medico sperimentatore si presta come soggetto.6. Il grado di rischio da correre non dovrà oltrepassarequello determinato dalla rilevanza umanitaria del proble-ma che l’esperimento dovrebbe risolvere.7. Si dovrà effettuare una preparazione particolare, e par-ticolari attenzioni dovranno essere usate al fine di mettereal riparo il soggetto dell’esperimento da possibilità ancheremote di lesione, invalidità o morte.8. L’esperimento dovrà essere condotto solo da personescientificamente qualificate. Sarà richiesto il più alto gra-do di capacità e attenzione in tutte le fasi dell’esperimentoa coloro che lo conducono o vi sono comunque coinvolti.9. Nel corso dell’esperimento il soggetto umano dovràavere la libera facoltà di porre fine a esso se ha raggiuntouno stato fisico o mentale per cui gli sembra impossibilecontinuarlo.10. Durante l’esperimento lo scienziato responsabile deveessere pronto a interromperlo in qualunque momento se èindotto a credere in buona fede, dopo una ponderata ri-flessione con tutte le sue facoltà, che la continuazione del-l’esperimento comporterebbe probabilmente lesioni, in-validità o morte per il soggetto umano.
La leggeHarris-Kefauver
Dopo il drammatico caso dell’utilizzo di talidomide, la cui sperimentazione pre-liminare non era stata abbastanza accurata da metterne in luce gli effetti tera-togeni (cioè la capacità di provocare malformazioni fetali), nel 1962 gli Stati Uni-ti – che tra l’altro non avevanomai autorizzato al loro interno la commercializza-
6
L’ABC della ricerca clinica

zione di quel farmaco – emanarono un emendamento al Federal Food, Drug andCosmetic Act del 1938, che decretava quale sarebbe stato da quel momento l’itersperimentale che ogni nuovo farmaco doveva percorrere per arrivare all’immis-sione sul mercato. L’emendamento, conosciuto come legge Harris-Kefauver,stabiliva che, per ottenere tale autorizzazione, il prodotto dovesse subire quat-tro livelli successivi di verifica, dalla fase 0 alla fase 3, passando attraverso lostudio delle sue caratteristiche chimiche in laboratorio, la sperimentazione ne-gli animali e infine l’applicazione nell’uomo, che prima sarebbe dovuta avvenirein volontari sani e poi in gruppi di pazienti affetti dalla malattia che era destina-to a curare. L’anno successivo, l’Investigational New Drug decideva che le indu-strie farmaceutiche avevano il dovere di provare la sicurezza e l’efficacia deicomposti per i quali intendevano chiedere l’approvazione alla Food and DrugAdministration e di designare il ricercatore responsabile di quelle prove.Ai vecchi farmaci, ormai in circolazione da anni senza essere passati al vagliodelle nuove norme, pensò il DESI:Drug Efficacy Study Implementation del 1968,un programma avviato sempre dalla Food and Drug Administration statuniten-se, che aveva il compito di definire, per ogni prodotto già in commercio prima del1962, se era efficace o inefficace oppure necessitava di ulteriori studi.
Il protocollo disperimentazionedeve esseresottopostoa un comitatoetico
Un elemento ancora più innovativo fu apportato dalla Dichiarazione di Helsinki,il documento sui principi etici per la ricerca medica concordato dall’Associazio-nemedicamondiale nel 1964, nel quale, oltre a ridefinire in dettaglio il principiodel consenso informato, si sancisce che “il disegno e l’esecuzione di ogni proce-dura sperimentale che coinvolga soggetti umani devono essere chiaramente de-scritti in un protocollo di sperimentazione”; protocollo da sottoporre, prima del-la sua messa in opera, all’esame e all’approvazione di un “comitato etico di revi-sione appositamente istituito, che deve essere indipendente dal ricercatore, dal-lo sponsor e da qualsiasi altro tipo di indebita influenza”. Ai comitati etici indi-pendenti, come furono da allora in poi denominati, sarebbe spettato il compitodi valutare la validità scientifica delle singole ricerche e la conformità alle nor-mative degli Stati in cui si sarebbero svolti, con l’unico interesse di salvaguar-darne i cittadini da eventuali rischi. Nonostante decaloghi e dichiarazioni, con-tinuarono a emergere esempi di ricerche condotte in modo “selvaggio” o per lomeno discutibili sul piano etico, rendendo obbligatorio aggiustare il tiro dellatutela con nuovi strumenti.Gli anni che seguirono la Dichiarazione di Helsinki videro, in effetti, la nascitadi una serie di provvedimenti e di emendamenti alle normative in vigore, cheportarono a perfezionare gli strumenti di difesa dei diritti dei soggetti speri-mentali, e più in generale dei malati, e a precisare le funzioni e le caratteristi-che che i comitati indipendenti di controllo, o Institutional review board, comefurono ribattezzati nel 1971 dalla Food and Drug Administration, dovevanopossedere.
Nascono normecomuni a vari statiper regolamentaregli studi clinici
Nel 1982 vide la luce la prima edizione delle International Ethical Guidelines forBiomedical Research Involving Human Subjects, elaborate dal Council for Inter-national Organizations of Medical Sciences in collaborazione con l’Organizza-zione mondiale della sanità, e nel 1990 si raggiunse, con la International Confe-rence on Harmonization, un accordo tra le principali industrie farmaceutiche ele agenzie governative dei farmaci di Europa, Giappone e Stati Uniti affinché glistudi clinici incominciassero a essere condotti nei diversi Stati secondo normecomuni, onde evitare che farmaci approvati per la vendita in un Paese non po-tessero entrare in commercio in un altro.In conformità con il principio di unificare i criteri per la ricerca clinica, l’annosuccessivo arrivarono le indicazioni di Good Clinical Practice (Norme di buonapratica clinica), via via recepite dalle singole legislazioni nazionali, finalizzate apromuovere la qualità della ricerca clinica, utilizzando al meglio le risorse di-sponibili, e a garantire la sicurezza dei soggetti partecipanti. In esse si ribadivail ruolo dei comitati etici indipendenti, quale principale strumento applicativo dinorme in questione. Ruolo che venne puntualizzato nel 2000 anche dall’Orga-nizzazione mondiale della sanità attraverso l’emanazione delle WHO Operatio-
7
L’ABC della ricerca clinica

nal Guidelines for Ethics Committees That Review Biomedical Research, dove èrichiamata la necessità di stabilire per i comitati etici procedure che “assicurinola coerenza e facilitino la cooperazione”.Anche se le acquisizioni fondamentali sono state raggiunte e formalizzate in do-cumenti che sono tuttora considerati le sue pietre miliari, come la Dichiarazionedi Helsinki o le Norme di buona pratica clinica, la regolamentazione della ricercaclinica continua la sua evoluzione, per esempio attraverso continui aggiorna-menti e revisioni di quei documenti. Né si è interrotta, purtroppo, la tradizionedegli errori e degli imprevisti in cui di tanto in tanto la ricerca incappa. Nellastoria, anche recente, non mancano i casi di trattamenti banditi dai repertoriterapeutici o di cui sono state ridimensionate le indicazioni perché non eranostati valutati abbastanza a fondo prima di essere messi in vendita oppure per-ché hanno rivelato effetti indesiderati che nel contesto un po’ troppo “ideale” de-gli studi sperimentali non erano emersi.Per questo motivo la ricerca clinica deve continuare a fare i conti con quellaquota di incertezza che le è connaturata e non può considerare il proprio compi-to concluso una volta portato un farmaco al traguardo dell’approvazione com-merciale. La sua missione di indagine e di vigilanza deve continuare in quellache oggi è ritenuta l’ultima e decisiva fase della sperimentazione: la sorveglianzapost-marketing, che avviene fuori dai laboratori e dagli ospedali, sul banco diprova molto più complesso e meno controllabile del mondo reale.
Osservare
Descrivere o analizzare
Studiosservazionalie studisperimentali
Generalmente una nuova ricerca è motivata dal fatto che non si possiedono an-cora informazioni esaustive su un dato fenomeno, per esempio l‘evoluzione diuna malattia nel tempo, la sua distribuzione nella popolazione, la precisione diun certo strumento diagnostico nell’identificarla, l’efficacia di uno o di più trat-tamenti nel curarla. Come si può vedere dagli esempi appena citati, gli obiettividi conoscenza che la ricerca si può porre sono di due ordini: teorico e pratico. Inentrambi i casi la priorità è, rispettivamente, sapere di più di una determinatacondizione patologica e trovare una soluzione ai disagi che essa crea ai singoliindividui oppure alla comunità.Il più delle volte il raggiungimento del primo obiettivo rappresenta la via per rea-lizzare successivamente il secondo: quando l’oggetto di studio è una malattia, èovvio che si vada alla ricerca di nozioni teoriche prima di tutto per utilizzarle aifini clinici. Distinguere i due diversi obiettivi di conoscenza ha tuttavia un sensodal punto di vista metodologico, perché a seconda del tipo di informazioni che sidesidera ottenere si effettuano tipi di indagine diversi. A grandi linee, gli obietti-vi teorici si prestano di più a una ricerca di tipo osservazionale, gli obiettivi ap-plicativi di più a una ricerca di tipo sperimentale.La differenza fondamentale tra studi osservazionali e studi sperimentali consi-ste nel fatto che nei primi il ricercatore lascia la realtà che è oggetto del suo stu-dio intatta, mentre nei secondi vi effettua un intervento programmato allo scopodi rilevarne le conseguenze. Ciò significa che osservando si registrano, descrivo-no e misurano gli elementi fenomenologici (le cosiddette variabili) così come so-no naturalmente presenti nella realtà; sperimentando, invece, si corregge la re-altà per rispondere ai propri quesiti di ricerca, e lo si fa escludendo o modifican-do alcune variabili e introducendone altre, per poi registrare, descrivere e misu-rare gli effetti del cambiamento attuato.
Ricercaepidemiologicae ricerca clinica
Il metodo osservazionale è quello utilizzato dalla ricerca epidemiologica, che hala funzione di descrivere la distribuzione delle singole malattie nelle diverse po-polazioni e di individuare i fattori che spiegano tale distribuzione. Gli studi epi-demiologici hanno perciò il compito di scoprire quante persone sono affette dauna data patologia in una certa popolazione in un preciso momento (prevalen-za), quante la sviluppano ex novo nel tempo (incidenza), quali categorie di indi-vidui ne sono maggiormente colpite, quali sono le cause o i fattori di rischio che
8
L’ABC della ricerca clinica

influenzano la sua insorgenza.Il metodo sperimentale è quello più usato nella ricerca clinica, che ha fonda-mentalmente lo scopo di verificare la validità di qualsiasi intervento di caratteresanitario (trattamenti sia farmacologici sia chirurgici, vaccinazioni, misure igie-niche, restrizioni dietetiche, indagini diagnostiche, provvedimenti ambientali,programmi educativi) che si ipotizza possa servire a prevenire e curare una ma-lattia o a migliorarne la prognosi.La ricerca osservazionale si definisce descrittiva quando si limita a prendere vi-sione di una realtà semplice e a descriverla in dettaglio, e analitica quando siprefigge di capire se esiste una qualche relazione tra diversi fenomeni osservatie di stabilire di che natura essa sia.
Gli studi descrittivi Gli studi descrittivi sono quelli in cui si cerca una risposta alla domanda “checosa succede qui?”, e si descrive, per l’appunto, quello che si vede: i sintomi diuna certa patologia, le alterazioni cellulari di un tipo di tumore, la frequenzacon cui compare un particolare disturbo nei bambini, il numero di decessi inseguito a un incidente, gli intervalli con cui si ripetono le crisi in una specificaforma di epilessia, i danni funzionali dopo dieci anni dall’insorgenza di una ma-lattia cronica, e così via. Le osservazioni di tipo descrittivo possono partire daipotesi da verificare e focalizzarsi su elementi particolari della realtà che si vuolestudiare, oppure possono servire a generare ipotesi, e quindi mantenere inizial-mente un “campo visivo” ampio, per poi restringerlo a quegli aspetti della realtàche si ritenga più utile approfondire.Nonostante la funzione prettamente esplorativa degli studi descrittivi, circoscri-vere il campo di osservazione a un’area di interesse è indispensabile se si vuoleprodurre un corpo ordinato di informazioni pregnanti e non un’accozzaglia ca-suale di dati incoerenti tra loro.
Il caso clinico
Esistono due particolari tipi di studio descrittivo, detti re-soconto di un caso e serie di casi, che si distinguono daglialtri perché consistono nel registrare solo i casi in cui siverificano i fenomeni di interesse. Spesso i casi sono il ri-sultato di osservazioni non programmate e non vengonostudiati in relazione a quanto accade nel resto della popo-lazione, come invece avviene con gli altri metodi della ri-cerca osservazionale.Il resoconto di un caso non è altro che la descrizione diuna singola storia, per esempio un caso clinico, che si pensapossa dare informazioni utili alla comprensione di una datarealtà, per esempio una malattia. La serie di casi è prati-
camente un resoconto di più casi osservati in successione,accomunati da qualche elemento originale, che si conside-ra di alto valore conoscitivo. Serie di casi che hanno granderilevanza ai fini della ricerca sui farmaci sono, per esempio,le registrazioni di effetti collaterali non previsti in fase spe-rimentale, che emergono dopo la commercializzazione deiprodotti, con le stesse caratteristiche in un certo numerodi persone (in serie per l’appunto). Emblematici in questosenso sono stati i due casi già citati dell’associazione trauso della talidomide in gravidanza e malformazioni nei ne-onati e di quella tra uso di dietilstilbestrolo in gravidanza eadenocarcinoma vaginale nelle figlie femmine.
Gli studi analitici Gli studi analitici hanno alla base un quesito del tipo “che relazione c’è tra que-sti due (o più) fenomeni?”, e verificano come tali fenomeni mutano l’uno in rap-porto all’altro. Spesso questo tipo di indagine comporta confrontare l’andamen-to delle variabili da studiare in gruppi diversi di persone oppure in momenti di-versi. Per esempio, se si vuole conoscere il rapporto tra l’esposizione a un com-posto chimico specificatamente presente in alcuni ambienti professionali e uncerto tumore, si possono registrare tutti i casi di diagnosi di quel tumore in ungruppo di lavoratori esposti al composto e, parallelamente, in un gruppo di per-sone che sicuramente non lo sono. Oppure, se si desidera valutare quanto inci-da l’inquinamento da polveri sottili sulla gravità dei sintomi negli asmatici chevivono in una città, si possono annotare i livelli di PM10 registrati dalle centrali-ne di rilevamento e il numero di accessi al Pronto soccorso per crisi d’asma inpiù giorni successivi.Come si può intuire, la funzione più tipica degli studi analitici è quella di andarealla ricerca di associazioni tra diversi fenomeni, di indagarne le eventuali rela-zioni causali e di documentarle con dati quantitativi. In medicina, lo scopo ulti-mo di questo tipo di indagini è, naturalmente, quello di scoprire le cause o i fat-tori di rischio delle malattie, per mettere queste informazioni al servizio della
9
L’ABC della ricerca clinica

prevenzione. Dati gli obiettivi abbastanza specifici che si prefigge, l’osservazionedi tipo analitico ha sempre come presupposto un’ipotesi plausibile inmerito allerelazioni tra gli eventi che vuole esplorare. Vale a dire che, nello sforzo di indivi-duare la causa di un fenomeno, non si sceglie a caso tra le variabili in gioco, maci si concentra su quelle che, in base a conoscenze già acquisite o quanto menoa criteri di verosimiglianza scientifica, hanno una qualche probabilità di influ-enzare il fenomeno stesso. L’ipotesi di partenza è importante – ed è importantesoprattutto che essa sia ragionevole e chiara – perché determina il modo di pro-cedere nell’osservazione.
I metodi per osservare: i disegni di studioIn termini operativi, sia l’osservazione descrittiva sia quella analitica possonoessere effettuate secondo diversi disegni di studio. La scelta di u disegno piutto-sto che un altro dipende soprattutto dal punto di vista che si assume rispetto aifenomeni da osservare: gli eventi possono essere registrati nel presente, facen-done una sorta di fotografia, oppure possono essere esaminati in un periodo ditempo più omeno lungo. Nel primo caso si ricorre a un disegno trasversale e nelsecondo a un disegno longitudinale.
Il disegnotrasversale
Il disegno trasversale consiste nella descrizione di alcune caratteristiche osser-vate in un dato momento in un gruppo di persone. Si può trattare di un gruppodi pazienti affetti da una certa malattia, di cui si desidera delineare nei dettaglile possibili presentazioni cliniche; oppure si può trattare di una grande popola-zione (come quella di un’intera nazione), nella quale si vuole determinare la pre-valenza di una certa condizione patologica o di un certo fattore di rischio.Negli studi di tipo analitico, in cui si vuole indagare il rapporto causa-effetto tradue variabili (una malattia e la sua presunta causa), si parte da una popolazio-ne prescelta di soggetti e all’interno di essa si verifica se alcune caratteristiche sipresentano tra loro associate: poniamo, per esempio, l’abitudine di fumare e ilcancro al polmone. Il principale difetto di questo tipo di studio è che non sempreconsente di dire quale delle due variabili influenzi l’altra. In alcuni casi la dire-zione della relazione causa-effetto è assolutamente inequivocabile, come nel-l’esempio del fumo e del tumore al polmone, in cui è appurato che è l’abitudinedi fumare a determinare il tumore; in altre circostanze, però, la risposta che siottiene può essere semplicemente che in un gruppo di persone due fenomenisono compresenti: può essere che uno dei due sia la causa dell’altro, ma puòanche essere che entrambi siano provocati da un terzo elemento ancora scono-sciuto. Per esempio, in uno studio sul bullismo giovanile si è deciso di indagarecon uno studio trasversale se i ragazzi di alcune scuole che subivano angherieda parte dei compagni avessero un livello di ansia superiore agli altri oppure no.La combinazione delle due condizioni, che in quel caso fu effettivamente trova-ta, può essere interpretata in due modi: è verosimile che subire le minacce deicoetanei generi ansia, ma non si può escludere l’eventualità che i ragazzi ansio-si diventino più facilmente vittime che non persecutori. E’ il dubbio classicodell’uovo e della gallina.Il problema dell’origine dei fatti può essere affrontato con gli studi che copronoun intervallo temporale più ampio. Questo intervallo temporale può svolgersisia nel futuro, per vedere che cosa succede da un certo momento in poi, sia nelpassato, per vedere che cosa è successo prima. Nel primo caso il punto di vistadello studio è prospettico, nel secondo è retrospettivo.
Il disegnolongitudinale
La migliore opportunità per studiare i fenomeni nel tempo è offerta dal disegnolongitudinale, che consiste nell’osservare la popolazione designata per il perio-do di tempo necessario per rilevare le informazioni desiderate. Gli studi longitu-dinali sono gli unici che permettono di rappresentare processi evolutivi: peresempio, di misurare l’incidenza di una malattia, di conoscerne la prognosi o lecomplicanze a lungo termine, di valutare gli esiti a distanza di un interventochirurgico, eccetera.La modalità con cui si svolgono più comunemente gli studi longitudinali è quel-la prospettica, che implica di tenere sotto osservazione la popolazione oggetto distudio (chiamata in questo caso coorte) per un periodo di alcuni giorni o mesi o
10
L’ABC della ricerca clinica

anni, a seconda del tempo necessario alla condizione di cui si valuta la causa oil fattore di rischio per manifestarsi. Generalmente, all’interno della popolazioneprescelta si confrontano due gruppi, che si costituiscono in base alla presenza oassenza della causa o del fattore di rischio sospettati; poi, nel corso del succes-sivo periodo di osservazione (follow up), si va a controllare in quale dei due gruppiil sintomo o la malattia in questione insorgono con maggiore frequenza.Gli studi longitudinali sono il modo più appropriato per indagare le relazionicausali che richiedono sempre un po’ di tempo per esplicitarsi. Anche se sono ipiù complessi e impegnativi da realizzare, da un lato per il vincolo che la lorodurata comporta sia per i ricercatori sia per i soggetti studiati, dall’altro per il ri-schio che nel lungo periodo alcuni di questi ultimi abbandonino, per svariatimotivi, lo studio, alterando così la numerosità e la rappresentatività della coorteprescelta.
Lo studiocaso-controllo
Unmetodo spesso usato in alternativa a quello longitudinale per studiare i rap-porti causa-effetto è il disegno caso-controllo, che in genere viene applicato inun’ottica retrospettiva. Si definisce caso-controllo in quanto consiste nel sele-zionare in partenza due gruppi che si distinguono per la presenza nell’uno(gruppo dei casi) e l’assenza nell’altro (gruppo dei controlli) di un fenomeno (peresempio una malattia) di cui si vuole conoscere la causa, cercando poi nel pas-sato di ciascuno la presenza o l’assenza della variabile che si pensa essere statadeterminante nel provocarlo. Per tornare a un caso già citato, si può indagare sei malati di tumore al polmone (casi) risultino essere stati fumatori più spessodei sani (controlli).Questa soluzione è molto utile quando si vuole risalire a cause o a fattori di ri-schio che non sortiscono i loro effetti in tempi rapidi ma impieganomolti anni senon decenni per tradursi in malattia: è il caso, per esempio, dell’esposizione adagenti cancerogeni e dei tumori. Tuttavia soffre di alcuni limiti che possonocomplicare l’interpretazione dei risultati. Il primo e il più ovvio è legato al fattoche per avere alcune informazioni fondamentali, come quella dell’esposizionepregressa a un fattore di rischio, ci si deve spesso affidare a quanto i singoli sog-getti ricordano del loro passato; e sperare che abbiano buona memoria, perchéa volte il fattore di rischio va ricercato molto lontano, magari nell’infanzia (unamalattia infettiva, una vaccinazione) o ancora più indietro (un farmaco assuntodalla madre durante la gravidanza).Inoltre, selezionando i gruppi da osservare in base alla presenza o assenza diun effetto, non è detto che si facciano sempre le scelte giuste, soprattutto perquanto riguarda il gruppo di controllo: per esempio, nel caso dello studio sullarelazione tra fumo di sigaretta e cancro al polmone, se il gruppo dei casi è ca-ratterizzato dalla presenza di tumori polmonari, il gruppo di controllo più ap-propriato sarà uno in cui non ci sono tumori polmonari o uno in cui non ci so-no tumori di nessun tipo o, ancora, uno in cui non ci sono nemmeno altre ma-lattie dell’apparato respiratorio? La decisione deve essere ben ponderata valu-tando le possibili ripercussioni che tali differenze possono avere sul significatodei risultati. Se, per esempio, nel gruppo di controllo ci sono o tumori diversida quello al polmone o altre malattie respiratorie, è possibile che, a dispettodell’assenza di tumori al polmone, si trovi comunque il dato anamnestico delfumo, perché questo è un fattore di rischio anche per altri tumori e per una se-rie di malattie respiratorie non tumorali. A questo punto si potrebbe conclude-re che, siccome hanno fumato sia gli individui con tumore al polmone sia quel-li che non ce l’hanno, allora non esiste una relazione tra il fumo e il tumore alpolmone. Ma si tratterebbe di un’interpretazione viziata dalla presenza diquelle variabili in più (gli altri tumori, le altre malattie respiratorie), di cui nonsi è considerato il peso.
Evitare o gestire le distorsioni
Distorsione o bias La distorsione, chiamata comunemente con il termine anglosassone bias, è unerrore sistematico presente in uno studio, che si ripercuote sui suoi esiti, deter-minando uno scarto tra risultati trovati e quelli che si sarebbero dovuti ottene-re. In altre parole, è un fattore di disturbo che fa trovare quello che non c’è o non
11
L’ABC della ricerca clinica

fa trovare quello che c’è.Esistono diverse possibilità di bias, ma le tre principali sono:l il bias di selezione, che si verifica se il campione di individui da indagare èstato scelto e assemblato in modo errato;
l il bias di misurazione, che si verifica se i metodi di misurazione non sono va-lidi oppure se si sono usati metodi diversi in pazienti diversi;
l il bias da effetti estranei, che si verifica quando è presente qualche elementonon previsto che è in qualche modo associato alle variabili studiate.
I fattoridi confondimento
La forma di bias più frequente è data dalla presenza di eventuali variabili che siaggiungono a quelle che si è previsto di studiare e che hanno anch’esse a che fa-re con il fenomeno in esame. Per l’effetto che producono, le variabili non previstesono chiamate fattori di confondimento o confondenti. Come visto in preceden-za nell’esempio del fumo e del tumore al polmone, la presenza di fattori interfe-renti può distorcere l’interpretazione dell’associazione tra due eventi al puntoda far sfuggire la relazione che li lega.Analogamente, quando si indaga l’effetto di un certo fattore di rischio sull’insor-genza di una malattia si corre il pericolo di attribuire a esso una responsabilitàche non ha, o che ha solo in parte, se nella storia degli individui studiati esisto-no altri fattori in grado di favorire la stessa malattia.Nelle ricerche osservazionali, che hanno un margine di controllo molto bassosulla realtà che esaminano, ci si trova spesso alle prese con fattori di confondi-mento, e se non è possibile eliminare le variabili aggiuntive in anticipo, quandosi seleziona il campione, bisogna farci i conti dopo, quando è il momento di in-terpretare i dati. E’ anche vero, tuttavia, che spesso nel determinare un fenome-no concorrono più fattori causali (detti in questo caso concause). In questi casila compresenza di diverse variabili connesse al fenomeno oggetto di studio (peresempio diversi fattori di rischio per una stessa malattia) è utile e non va elimi-nata, perché offre l’opportunità di conoscere quanto pesa ciascuno dei fattoricoinvolti e quanto conti la loro combinazione nel determinarlo. L’importante,qualunque tipo di studio si decida di condurre, è cercare di identificare tutte lepossibili fonti di distorsione, o per poterle eliminare in anticipo o per non farsitrarre in inganno da esse.
Aggiustamento Quando non è possibile prevenire i bias nella fase di programmazione dello stu-dio – per esempio facendo molta attenzione a selezionare un campione che siasicuramente rappresentativo oppure selezionando individui che non portinocon sé troppe variabili aggiuntive – vengono in aiuto alcuni metodi di analisistatistica (come la regressione lineare o l’analisi multivariata), che agiscono suidati raccolti nello studio “ripulendoli” dagli effetti delle distorsioni. Tale opera-zione “depurativa” si definisce aggiustamento e serve, per esempio, per tenerein considerazione la differente distribuzione per categorie (età, sesso) dei cam-pioni studiati oppure per eliminare le reciproche interferenze tra più variabiliassociate e misurare il contributo di ciascuna all’effetto osservato.
Scegliere i soggetti da osservare
Il campione La ricerca ideale sarebbe quella che coinvolge tutti gli individui nel cui interesseviene condotta (che nell’insieme formano la cosiddetta popolazione bersaglio otarget), in modo tale da essere sicuri di avere osservato la realtà oggetto di stu-dio in tutta la sua possibile variabilità. Com’è facile immaginare, tale condizioneottimale non è praticabile. Per fare un esempio: volendo effettuare uno studiosui vantaggi di una nuova forma di terapia insulinica, sarà impossibile coinvol-gere tutti i pazienti con diabete di tipo I del paese in cui si vuole registrare il far-maco. Si ricorre pertanto a una soluzione di ripiego, che consiste nel limitare leosservazioni a una sola parte della popolazione bersaglio, detta campione, laquale deve possedere due requisiti ben precisi: essere sufficientemente nume-rosa ed essere rappresentativa della totalità alla quale appartiene.L’ampiezza adeguata del campione permette di evitare che alcuni fenomeni pre-senti nella popolazione di origine non vengano rilevati solo perché si è osservatoun gruppo troppo ristretto di persone, mentre la sua rappresentatività garanti-
12
L’ABC della ricerca clinica

sce che tutta la diversità presente nella popolazione di origine sia presente an-che nel gruppo studiato. Solo con queste premesse è lecito presumere che le in-formazioni ottenute nel campione valgano anche per il resto della popolazione, equindi siano generalizzabili.
Un campionerappresentativo
Le diverse modalità per ottenere un campione a partire da una data popolazio-ne, cioè per selezionare gli individui appropriati che andranno a formarlo, sonodette tecniche di campionamento. C’è la possibilità di comporre semplicementeil campione con i soggetti che è più facile ingaggiare, per esempio con i malatipiù disponibili a partecipare; dato che in questo caso il criterio di selezione è lacomodità si parla di campionamento di comodo. Altrimenti, si può optare per ilcampionamento sistematico, basandosi su principi più oggettivi e riproducibili,come per esempio coinvolgere tra i pazienti di un servizio sanitario solo quelli ilcui cognome inizia con la B oppure solo quelli che sono stati visitati nelle matti-nate di martedì di dieci settimane consecutive. Con entrambe queste tecniche sicorre però il rischio di introdurre elementi di distorsione (bias) che possonoscreditare i risultati dello studio. Nel campione di comodo, per esempio, posso-no finire solo i soggetti che di una data malattia hanno i disturbi più gravi e chesi sono prestati a entrare nello studio proprio per questo motivo, nella speranzadi provare una cura più efficace. Anche nel campione sistematico, che pure dàmigliori garanzie, può capitare di incappare in qualche fattore fuorviante: peresempio, non si può escludere che per qualche motivo contingente i pazientiche accedono a un ambulatorio il mattino appartengano tutti a una categoriaparticolare – quella dei pensionati per dirne una. I due campioni di questi casinon saranno pertanto rappresentativi l’uno della popolazione di soggetti con lamalattia scelta, l’altro della popolazione di pazienti che frequentano il serviziosanitario selezionato.L’unica tecnica che consente di evitare questo tipo di bias è il campionamentocasuale, che consiste nel estrarre a sorte dalla popolazione di origine le personeda includere nel campione. Quando nella popolazione esistono diverse catego-rie di individui (bambini e adulti, maschi e femmine, poveri e ricchi, bianchi eneri, eccetera) e questo è un fatto che può influenzare i risultati (per esempio nelcaso di unamalattia che colpisce più gli uomini che le donne oppure si distribu-isce diversamente nelle diverse etnie), si ricorre a una tecnica che consente dimantenere nel campione la variabilità della popolazione di origine e quindi diavere rappresentate nella stessa proporzione tutte le categorie. Tale tecnica, de-finita campionamento casuale stratificato, consiste nel suddividere la popola-zione in sottogruppi (gli strati) costituiti dalle varie categorie e poi estrarre a sor-
13
L’ABC della ricerca clinica
Nel reparto del dottor Malinverno questa è la situazioneper i 19 pazienti ricoverati tra lunedì e venerdì:
pazienti presenti in repartopazienti presenti in reparto con infezione
Lunedì Martedì Mercoledì Giovedì
P1
P2
P3
P4
P5
P6
P7
P8
P9
Venerdì
Prevalenza: quanti casi di infezione ci sono il lunedì = 5/9Prevalenza: quanti casi di infezione ci sono il venerdì = 3/9Incidenza: quanti nuovi casi di infezione ci sono da lunedì a venerdì = 3/9

te lo stesso numero di individui da ogni strato.
FIGURA 1: Incidenza e prevalenza: l’esempio delle infezioni ospedaliere
Prevalenza e incidenza
La prevalenza è la frequenza con cui una malattia è pre-sente nella popolazione studiata al momento della rileva-zione e quindi corrisponde alla percentuale di persone chein quel preciso momento hanno la malattia. La prevalenzaè un dato statico, che somma tutti i casi di malattia nontenendo conto della loro durata.Al contrario della prevalenza, l’incidenza è un dato dina-mico, in quanto rappresenta la frequenza con cui la malat-tia insorge in un intervallo di tempo definito e corrispondeal numero di nuovi casi che si registrano nel periodo di os-servazione. Per questo motivo l’incidenza è definita anchetasso di insorgenza o di esordio di una malattia.Pur essendo entrambe misure della frequenza con cui unamalattia si manifesta in una popolazione, prevalenza e
incidenza dipingono la situazione secondo due ottiche di-verse, che non sono interdipendenti. Per esempio, unamalattia che è poco frequente ma è caratterizzata daguarigione lenta oppure è cronica e non mortale risulteràavere una prevalenza proporzionalmente maggiore dellasua incidenza, perché il numero degli individui che nesoffrono in ogni dato momento sarà alto, mentre il nu-mero di quelli che si ammalano via via sarà basso. Al con-trario una malattia molto frequente ma con andamentorapido avrà al suo attivo poche vittime nella fotografiascattata dalla prevalenza e molte nel conteggio progres-sivo dell’incidenza.
Frequenze, tassi e rischi
La frequenza è la misura quantitativa più semplice, checorrisponde al numero di eventi di un certo tipo (casi dimalattia o individui malati) contati nella popolazione stu-diata. Di per sé la frequenza dà un’informazione poco rile-vante, perché la sua importanza dipende dalla numerositàdella popolazione in cui gli eventi sono stati contati: trecasi di meningite in una scuola di 500 bambini piuttostoche in una classe di 20 forniscono un’idea ben diversa del-la diffusione della malattia. L’idea del peso relativo del nu-mero degli eventi la dà quindi una proporzione (3/500 o3/20) oppure una percentuale (rispettivamente 0,6 percento e 15 per cento).Un particolare tipo di proporzione dotata anche di una di-mensione temporale è il tasso, che è il numero di eventiche si contano in una data popola-zione con riferimento al periodo diosservazione: per esempio, il nume-ro di suicidi che si verificano in unpaese in un anno è il tasso annualedi suicidi; il numero di persone chemuoiono a causa di una malattia ri-spetto al totale delle persone affetteda quella malattia in un dato inter-vallo di tempo è il tasso di letalitàdella malattia.Il rischio è la frequenza di nuovi casidi malattia in una popolazione su-scettibile di svilupparla nel corso diun certo periodo di tempo, general-mente espressa in percentuale. Que-sto valore, che corrisponde alla pro-babilità che chiunque ha di ammalar-si in quella popolazione, si chiama ri-schio assoluto.Quando, invece, si vuole valutare il peso di un fattore pre-disponente oppure l’effetto di un trattamento sulla proba-bilità di sviluppare una certa malattia si deve fare riferi-
mento a una misura detta rischio relativo. Per calcolare ilrischio relativo si procede in due tappe: innanzitutto sidetermina il rischio assoluto di sviluppare la malattia (cioèla frequenza con cui essa è presente) in due diversi gruppidi individui, gli uni esposti al fattore predisponente oppureal trattamento e gli altri no; poi si mettono a confronto idue valori ottenuti, dividendo il rischio assoluto degli indi-vidui esposti o trattati per quello dei non esposti o trattati.Il risultato della divisione corrisponde al numero di voltein più o in meno che gli individui esposti o trattati hannodi sviluppare la malattia rispetto agli altri: un valore paria 1 significa che non vi è differenza tra i rischi dei duegruppi e che pertanto il fattore predisponente o il tratta-mento sono ininfluenti; un valore >1 indica che il gruppo
degli individui esposti o trattati ha una probabilità più al-ta di ammalarsi; un valore <1 indica che il gruppo degliindividui esposti o trattati ha una probabilità più bassa diammalarsi.
Sperimentare
Come si è già accennato, gli obiettivi della ricerca clinica sono fondamentalmen-te applicativi: trovare un test che consenta di diagnosticare una malattia intempo utile per curarla, individuare il farmaco più idoneo a guarirla o quello piùefficace nel controllarne i sintomi o quello più valido nel prevenirne le compli-canze, escogitare i provvedimenti più utili per migliorare la qualità della vita dei
14
L’ABC della ricerca clinica
TABELLA 1: Tre modi di presentare il beneficio
Numero di deceduti Numero di pazienti trattati
Farmaco 32 1.000
Placebo 41 1.000
Riduzione del rischio assoluto: la proporzione dei pazienti che muoionocon il placebo meno la proporzione dei pazienti che muoiono con il farmaco.
41-32=9 Y 9/1.000=0,9%
Riduzione del rischio relativo: la riduzione del rischio assoluto divisa perla proporzione dei pazienti che muoiono con placebo.
9/41=22%
Numero necessario da trattare (NNT): numero di persone che occorretrattare per salvare una vita.
9:1.000=1:x oppure 1/0,9´100=111Quindi è necessario trattare 111 persone per salvare una vita.

pazienti che ne sono affetti, e così via. Il tutto senza perdere di vista la necessitàche qualsiasi tipo di intervento sia, anche “sostenibile” cioè praticabile a livellodelle comunità alle quali è destinato e accessibile, in termini di costi e di dispo-nibilità, a tutti gli individui che di quelle comunità fanno parte.
Plausibilitàe rilevanza
Uno studio clinico deve, dunque, essere valido su due fronti: essere plausibile,cioè fondato su un’ipotesi ragionevole che valga la pena verificare, ed essere ri-levante, cioè portare a risultati applicabili nella pratica clinica e vantaggiosi perla salute pubblica. Non sarebbe plausibile, per esempio, uno studio che andas-se a valutare gli effetti di un farmaco di cui non si conoscano l’efficacia o la tos-sicità quanto basta almeno per presumere che possa essere effettivamente utilenella cura di una certa malattia. E non sarebbe rilevante uno studio che servasemplicemente a confermare che un nuovo farmaco ha la stessa efficacia e lastessa tollerabilità di uno che è già in commercio, e perciò già provato sul cam-po, da qualche tempo. Plausibilità e rilevanza sono due qualità di natura asso-luta, che dipendono dai contenuti di ricerca in sé. Ma, alla fine, il valore sostan-ziale di uno studio è sempre relativo all’importanza che riveste nella realtà allaquale si applica, nel senso che deve rispondere a esigenze che in quella realtàsono prioritarie.
Opportunitàvalutatada un comitatoetico
Ma non basta. Per quanto possa essere fondato su un’ipotesi interessante,ben disegnato, condotto correttamente e far prevedere ripercussioni utili nellapratica clinica, uno studio può ancora essere inopportuno se è discutibile sulpiano etico. Il valore della conoscenza è commisurato alla probità dei metodiche si utilizzano per ottenerla; ciò significa che un obiettivo scientifico va per-seguito solo se è possibile farlo nel totale rispetto di tutti gli individui che inquesto progetto vengono coinvolti, nel rispetto della loro integrità fisica e psi-chica e dei loro valori.Questo, almeno, è il principio affermato nel Good Clinical Practice (Norme di bu-ona pratica clinica), il documento guida stilato nel 1991 per definire il percorsoche ogni ricercatore deve seguire per condurre una ricerca clinica e le regole chedeve rispettare in ogni fase di essa. La prima di queste regole stabilisce chequalsiasi ricerca clinica sia preceduta dalla formulazione di un dettagliato pro-tocollo di studio e che questo sia sottoposto a un giudizio di opportunità da par-te di un Comitato etico indipendente. Il protocollo deve specificare le premesseteoriche della ricerca e lo scopo che si prefigge e descrivere nei dettagli tutti gliaspetti operativi: la selezione dei pazienti che vi parteciperanno, la scelta deglistrumenti diagnostici che saranno utilizzati, le procedure di trattamento che in-tende instaurare, i benefici che ci si aspetta di osservare, i rischi che si potreb-bero correre, i criteri di valutazione dei risultati.
Il conflittodi interessi
Da diversi anni la buona pratica clinica ha dovuto iniziare a includere tra i suoidoveri morali un’ulteriore forma di tutela, che va ad aggiungersi a quelle neiconfronti dell’errore metodologico e dell’inappropriatezza. Una lunga serie di av-vilenti scoperte ha infatti fatto emergere la necessità di tutelare la ricerca dauna trappola in cui non di rado è incappata e che è la tomba di ogni garanzia dioggettività ed equità: il conflitto di interessi. Tale condizione si genera quando acondurre uno studio sono individui, istituti scientifici o aziende che possono ri-cevere un vantaggio dal fatto che i risultati vadano in una direzione piuttostoche in un’altra. Il classico esempio è la sperimentazione su un farmaco effettua-ta o sponsorizzata dall’azienda che lo produce.
Questione di metodo
Le tappe di unostudio clinico
Una ricerca clinica è un esperimento che deve produrre informazioni scientifi-che, e come tale deve essere organizzato secondo i criteri del metodo sperimen-tale e possederne tutti i requisiti.Le tappe fondamentali dell’organizzazione di uno studio clinico sperimenta-le sono:l partire da un’ipotesi (per esempio: il trattamento A è più efficaceo meno tos-sico del trattamento B; il test diagnostico A individua la malattia a uno sta-
15
L’ABC della ricerca clinica

dio più precoce del test B);l individuare la popolazione alla quale l’ipotesi si applica (i pazienti affetti dal-la malattia per la quale sono indicati i trattamenti A e B; i pazienti che po-trebbero avere la malattia diagnosticabile con i test A e B);
l selezionare dalla popolazione individuata un campione rappresentativo diindividui da candidare allo studio e suddividerlo in un gruppo sperimentale(i pazienti che proveranno il trattamento A; i pazienti che faranno il test A) eun gruppo di controllo (i pazienti che prenderanno il trattamento B; i pa-zienti che faranno il test B);
l stabilire gli esiti (end point) da misurare (l’attenuazione di uno o più sinto-mi o la diminuzione della mortalità con i trattamenti A e B; il miglioramen-to della prognosi della malattia diagnosticata con i test A e B) e i metodi permisurarli.
A questo punto, lo studio clinico può partire con la somministrazione degli in-terventi da confrontare. In questa fase è essenziale che gli individui di entram-bi i gruppi siano trattati esattamente secondo gli stessi criteri, con l’unica va-riante del tipo di intervento assegnato, per evitare che eventuali differenze ne-gli effetti osservati nei due gruppi dipendano non dalla diversa efficacia degliinterventi comparati, ma da variazioni nel modo di effettuarli: un migliora-mento dei sintomi relativamente maggiore nel gruppo sperimentale può esserefallace se, per esempio, il trattamento A è stato somministrato in dosi propor-zionalmente superiori o per più tempo rispetto al trattamento B; risultati dia-gnostici diversi per il test A e per il test B potrebbero dipendere non dalla mag-giore precisione dell’uno in confronto all’altro, ma dalle procedure di esecuzio-ne se queste non sono state equivalenti. Allo stesso modo, gli esiti misurati,nonché i metodi e la prassi per misurarli, devono essere rigorosamente glistessi nei due gruppi.
Passo passo verso il trialAnche se rappresenta il vero e proprio collaudo di ogni nuova terapia – l’unicoche può dire l’ultima parola sull’opportunità di renderla disponibile sul mercatoo di includerla tra gli interventi erogati dai servizi sanitari – lo studio clinico con-trollato e randomizzato (spesso indicato con la sigla RCT, dall’inglese randomi-sed controlled trial) è soltanto il punto di arrivo della sperimentazione. Si pren-da a esempio un farmaco di nuova produzione, di cui per definizione si conosco-no solo le caratteristiche chimiche. Per ovvi motivi precauzionali, la sua sommi-nistrazione ai pazienti, sia pure quelli che si prestano a partecipare a un trial, èsempre preceduta da una serie di indagini che consentono una determinazionepreliminare delle sue proprietà terapeutiche e della sua innocuità. Il suo per-corso dal laboratorio chimico alla farmacia prevede una serie di tappe che ser-vono ad accumulare informazioni diverse.
Lasperimentazionedi un nuovofarmaco
L’idea di suddividere la sperimentazione dei nuovi farmaci in fasi successive èstata introdotta per la prima volta nel 1962, dall’emendamento Harris-Kefauveral Food, Drug and Cosmetic Act statunitense. Nel tempo, poi, le procedure sonostate ridefinite e ottimizzate allo scopo di tutelare meglio i destinatari finali deifarmaci stessi, cioè i pazienti-consumatori, e di preservare i ricercatori coinvoltidalle possibili contestazioni sui metodi utilizzati. Attualmente, l’iter per la “regi-strazione” di un prodotto farmaceutico ai fini commerciali è standardizzato esuddiviso in test pre clinici e test clinici.
I test pre clinici I test pre clinici comprendono lo studio delle caratteristiche biochimiche dellanuova molecola e una prima valutazione della sua efficacia e della sua eventua-le tossicità in laboratorio. Tradizionalmente, per questa valutazione sono sem-pre state utilizzate popolazioni di alcune specie animali allevate allo scopo (topi,ratti, cavie, conigli, gatti, cani, scimmie), ma più di recente si sono sviluppatianche alcuni validi metodi sostitutivi, che consistono nello studio degli effettidei farmaci su batteri e virus o in cellule, tessuti e organi di origine umana isola-ti e mantenuti in coltura (metodi biologici) oppure nella simulazione al compu-ter di processi metabolici e funzionali del corpo umano, anche attraverso mo-delli matematici o meccanici (metodi non biologici).
16
L’ABC della ricerca clinica

La concezione di sistemi di sperimentazione diversi da quelli che utilizzano glianimali è nata, oltre che per motivi etici, dall’osservazione che organismi diffe-renti non reagiscono mai esattamente allo stesso modo a un trattamento eche, quindi, la presenza di un effetto terapeutico o l’assenza di un effetto tossi-co in una o anche in più specie animali non sono una garanzia assoluta che lostesso avvenga nell’uomo. Gli esempi di farmaci passati al vaglio della speri-mentazione animale che hanno riservato sorprese sgradite al momentodell’applicazione nell’uomo non mancano; e, viceversa, anche quelli di farmaciche sono estremamente tossici in alcune specie animali ma non in altre e nonnell’uomo.
I test clinici Il fatto che il modello animale non sia del tutto affidabile è il motivo principaleper cui, superata la parte pre clinica, la sperimentazione prosegue con le provesull’uomo, ovvero i test clinici. Anche questo stadio si svolge con una progres-sione graduale che prevede tre fasi successive.
Fase I Nella fase I il trattamento viene somministrato in un piccolo gruppo di volontarisani, generalmente poche decine, a partire da quantità minime che poi vengonoaumentate gradualmente, per verificare se abbia effetti collaterali e a quali dosiquesti si manifestino. I risultati di questa fase vanno a integrarsi con le indaginifarmacologiche e tossicologiche di quella preclinica, per fornire una prima indi-cazione di come il principio attivo venga assorbito e metabolizzato nell’organi-smo umano e per definirne la tollerabilità, cioè l’assenza di effetti indesideratialle dosi necessarie per ottenere un effetto terapeutico. Sono escluse dalla spe-rimentazione su volontari sani alcune categorie di farmaci, come gli antitumo-rali o gli immunosoppressori, i cui effetti indesiderati sono talmente rilevanti danon essere accettabili se non in vista di un apprezzabile beneficio.
Fase II Nella fase II il trattamento viene provato in un campione un po’ più consistente(un centinaio o poco più) di persone affette dalla malattia target della cura, alloscopo di incominciare a stimarne l’efficacia, di fare un primo bilancio tra rischi ebenefici e di mettere a punto le modalità di utilizzo migliori (dosi, numero e tem-pi di somministrazione) nella condizione per la quale è indicato.
Fase III Nella fase III viene testato in un numero decisamente più alto di malati, variabi-le da qualche centinaio a qualchemigliaio a seconda dell’estensione dello studioa uno o più centri (studi multicentrici), per determinarne la reale efficacia e lavalidità rispetto ad altre opzioni terapeutiche. E’ in quest’ultima fase che di soli-to si ricorre agli studi randomizzati controllati o RCT. Dal punto di vista meto-dologico, il concetto chiave di questo tipo di ricerca è quello del confronto. Ciòche interessa ai fini del giudizio sulla convenienza di introdurre un nuovo trat-tamento è, infatti, la misura della sua efficacia e della sua sicurezza in relazionea quelli già esistenti. L’obiettivo di ogni nuovo farmaco rispetto alla malattia chesi prefigge di curare deve essere quello di superare i suoi “simili”, in termini dicapacità di guarirla o di alleviarne i sintomio in termini di innocuità e tollerabili-tà, in modo da offrire un’alternativa terapeutica valida.Nel corso della sperimentazione clinica, dunque, esso viene confrontato in mo-do sistematico con un altro che abbia le stesse indicazioni oppure, in casi sele-zionati in cui si vuole semplicemente rilevare la sua effettiva capacità di influen-zare il decorso naturale della malattia (quello che essa avrebbe in assenza di cu-re) o in cui un’altra opzione di cura non esiste, con un placebo.Gli studi pre clinici e quelli di fase I, II e III forniscono le informazionisull’efficacia e sulla sicurezza dei farmaci che, alla fine del percorso sperimenta-le, le agenzie e le commissioni di controllo utilizzano per autorizzarne la com-mercializzazione. Complessivamente, la storia pre marketing di ogni nuovo far-maco, dall’inizio della sperimentazione al beneplacito delle autorità sanitarie,dura dai 7 ai 10 anni.
Un registro per i trial
Che la sperimentazione sull’uomo sia essenziale è un fattoincontrovertibile, che debba rispondere a criteri di sicurez-
za oltre che di scientificità è il suo aspetto prioritario. Glistudi pre clinici non rappresentano una garanzia assoluta
17
L’ABC della ricerca clinica

contro sgradite, e a volte francamente brutte, sorprese.Recentissima è la disavventura di sei cittadini britannicisani che hanno partecipato in qualità di volontari alla Spe-rimentazione Di Fase I Del Tgn1412, Un Farmaco BiotechPromosso senza riserve dagli studi pre clinici, compresiquelli su animali da laboratorio. Il TGN1412 è un anticor-po monoclonale che pareva serbare grandi promesse perla cura di alcune malattie autoimmuni, come l’artrite reu-matoide, e di una forma rara di leucemia cronica refratta-ria ai trattamenti convenzionali. La brillante carriera delTGN1412 si è bloccata lo scorso marzo, quando i sei vo-lontari ai quali era stato somministrato per via endoveno-sa sono finiti in Unità di terapia intensiva a causa di unaintensissima reazione infiammatoria generalizzata.Poiché gli studi sperimentali non sono del tutto privi di ri-schi è essenziale che sulla loro qualità e sui loro esiti vi siaun monitoraggio costante. In realtà, ciò è reso difficile dalfatto che l’informazione sulle ricerche cliniche che vengo-no continuamente effettuate in tutto il mondo è decisa-mente parziale, dal momento che fa principalmente affi-damento sulla loro pubblicazione in riviste scientifiche di
larga diffusione. Il che non è affatto la regola: molti studinon arrivano mai alla stampa e, ad aggravare le cose, siaggiunge il fatto che a essere penalizzati maggiormentesono gli studi che hanno portato a risultati sfavorevoli.Con la conseguenza che, consultando le banche dati dellaletteratura medica, si finisce per avere un quadro delle at-tività di sperimentazione clinica e dei loro esiti del tutto in-completo e distorto.Per ovviare a questa situazione, nel maggio del 2005 l’OMSha istituito l’International Clinical Trials RegistryPlatform, con l’obiettivo di garantire la trasparenza el’accessibilità dell’informazione sugli studi clinici. Nel regi-stro internazionale dei trial dovranno essere riportate, se-condo le norme e gli standard stabiliti dall’OMS, tutte lesperimentazioni cliniche effettuate con i relativi risultati.In Italia, inoltre, all’interno dell’Agenzia italiana del far-maco (AIFA) è stato fondato l’Osservatorio nazionalesulla sperimentazione clinica dei medicinali, che attra-verso un registro informatizzato raccoglie i dati riguar-danti tutte le ricerche approvate dai comitati etici locali(https://oss-sper-clin.agenziafarmaco.it/).
Scegliere i candidati a una sperimentazione clinica
La composizione e la tipologia dei partecipanti dipendono innanzitutto da qualisono l’argomento di indagine e l’obiettivo dello studio. Esistono studi che hannoun campo di interesse molto ristretto perché sono finalizzati a stabilire l’utilitàdi un certo trattamento per una categoria particolare di pazienti – l’esigenza puòessere, per esempio, quella di testare la capacità di un farmaco antipertensivodi tenere sotto controllo la pressione dei malati con diabete insulino-dipendente –e studi che hanno un raggio d’azione più ampio perché servono a verificarel’efficacia e la tollerabilità generali di un trattamento. Nei primi la selezione deipartecipanti riguarderà esclusivamente la categoria di pazienti interessata (ipazienti con diabete di tipo I), nei secondi il campione dovrà riprodurre il piùpossibile la variabilità della popolazione generale.
Criteridi eleggibilità
Una volta definito lo scopo dello studio, si stabiliscono i cosiddetti criteri di eleg-gibilità dei soggetti, vale a dire i requisiti che essi devono avere per essere coin-volti. Tali criteri sono tutti quelli che consentono di ottenere un campione cheabbia la composizione desiderata (tipologia di pazienti, età, sesso, eccetera) edia buone garanzie di una corretta partecipazione allo studio (disponibilità afornire le informazioni richieste, a seguire diligentemente le prescrizioni, a pre-sentarsi puntualmente ai controlli, eccetera).Ovviamente, anche se la selezione iniziale viene fatta con criteri abbastanzarestrittivi, la garanzia che tutto vada a buon fine non è assoluta: quello che ac-cade abbastanza spesso è che, nella fase ancora organizzativa dello studio, auna valutazione più approfondita alcuni dei candidati risultino inadatti (peresempio, perché sono già in cura con altri farmaci che creerebbero difficoltà diinterpretazione rispetto al trattamento sperimentale) e non possano quindi es-sere inseriti dal campione, oppure che, a sperimentazione già avviata, alcunidei partecipanti debbano essere esclusi o abbandonino lo studio per una qual-che ragione oggettiva o soggettiva (decesso, sovrapporsi di altre malattie, intol-leranza verso il trattamento, motivi personali che impediscono di continuarela collaborazione).Nel caso di studi su specifiche categorie di pazienti i criteri di inclusione saran-no piuttosto restrittivi; nel caso di studi di più ampio respiro, invece, i criteri diinclusione e di esclusione concerneranno più che altro i requisiti generali (età,sesso) e quelle variabili che possono condizionare l’interpretazione dei risultati(fattori di confondimento).Ma oltre che ad assicurare la corretta delimitazione del campo di indagine e labuona riuscita dello studio, i criteri di inclusione e di esclusione servono a tu-telare i pazienti. Benché tali criteri siano definiti di volta in volta in base allenecessità dei singoli protocolli sperimentali, esistono alcuni principi generali
18
L’ABC della ricerca clinica

riguardanti l’esclusione di soggetti che sarebbero esposti a rischi particolarilegati alla sperimentazione stessa: pazienti con precedenti episodi allergici,pazienti con insufficienza renale grave, pazienti che assumono altri farmacicon i quali quelli sperimentali potrebbero interagire pregiudicandone gli effettiterapeutici.
Controllare e randomizzare
Lo studio clinico controllato randomizzato (RCT), rappresenta lo strumento piùattendibile per verificare la validità di un intervento medico.Come dice il nome, i due requisiti fondamentali di un RCT sono: la presenza diun gruppo di controllo accanto a quello cosiddetto sperimentale (perché speri-menta il trattamento oggetto di studio), vale a dire di un gruppo di individui concaratteristiche analoghe a quelli del gruppo sperimentale che però sono desti-nati a ricevere un trattamento diverso oppure nessun trattamento; la rando-mizzazione, cioè l’attribuzione dei soggetti coinvolti nello studio al gruppo speri-mentale o a quello di controllo secondo un criterio di casualità.Nella storia, anche recente, della ricerca clinica sono state utilizzate altreforme di sperimentazione, prive di uno o di entrambi i requisiti tipici degliRCT: gli studi non controllati, che sperimentano l’intervento oggetto di veri-fica in un unico gruppo di pazienti; gli studi controllati non randomizzatiche prevedono l’esistenza di un gruppo sperimentale e uno di controllo maripartiscono i pazienti nei due gruppi operando delle vere e proprie scelte enon affidandosi al caso. Nella gerarchia di valore degli studi clinici questidue tipi di sperimentazione sono oggi considerati di serie B, perché nonpossono garantire la medesima affidabilità, e quindi validità scientifica, de-gli RCT. A concepire come imperativo il ricorso al controllo e alla randomiz-zazione si è arrivati solo gradualmente, ma i motivi sono ormai chiari e uni-versalmente riconosciuti.
Studionon controllato
Si consideri, per esempio, l’assenza del gruppo di controllo: davanti ai risultatidi uno studio che ha considerato solo il trattamento sperimentale senza con-frontarlo con nient’altro, non si potrà mai essere sicuri fino in fondo che i cam-biamenti osservati siano davvero dovuti al trattamento. Ci si può chiedere, in-fatti, se i malati che sono guariti o che hanno avuto una riduzione dell’intensitàdei sintomi non sarebbero guariti o migliorati comunque, a causa dell’evoluzio-ne naturale della loro malattia. Poniamo il caso che si tratti di una malattia de-stinata a regredire spontaneamente oppure di una malattia caratterizzata dafasi alterne di remissione e riacutizzazione dei sintomi: e se per puro caso il trat-tamento sperimentale fosse stato somministrato proprio in concomitanza conla guarigione naturale o con la fase di miglioramento? Un risultato a favore o asfavore del trattamento sperimentale potrebbe essere solo una questione diconvergenze cronologichementre di fatto esso è del tutto ininfluente sulla storiadella malattia. Ma senza avere provato che cosa succederebbe in sua assenza, èdifficile saperlo.Che ruolo ha, invece, l’assegnazione casuale dei pazienti al gruppo sperimenta-le e al gruppo di controllo? Per essere sicuri che gli effetti di un trattamento nonsiano condizionati da fattori che non c’entrano con le sue proprietà terapeuti-che è necessario fare in modo che le possibili interferenze siano o eliminate opresenti in entrambi i gruppi nella stessa proporzione. Per quanto siano statiarruolati in base ai criteri di inclusione e di esclusione pertinenti agli scopi dellostudio, i soggetti coinvolti in una ricerca sono tutti diversi rispetto una serie dialtre variabili che spesso non è nemmeno tanto facile immaginare e verificare: lemalattie avute nell’infanzia, quelle dei familiari, le abitudini alimentari, e cosivia. Poiché eliminare tutte le fonti di variabilità è impossibile (anche perchémol-te non sono neppure facilmente identificabili) – e per certi versi non è desidera-bile visto che è con l’eterogeneità di tutti i possibili fruitori che un trattamento siconfronterà una volta passato il vaglio scientifico – non resta che cercare di otte-nere lo stesso grado di variabilità all’interno del gruppo sperimentale e all’inter-no del gruppo di controllo.
19
L’ABC della ricerca clinica

Studinon randomizzati
Se l’assegnazione dei soggetti a un gruppo o all’altro fosse fatta da una persona,fosse anche il ricercatore più onesto e rigoroso, nella scelta si insinuerebbe ine-vitabilmente un elemento di soggettività, che potrebbe influenzare i risultatidello studio. L’ostacolo si aggira lasciando fare al caso; il che può significarescegliere i pazienti tirando a testa o croce (randomizzazione semplice) o, ancorameglio, affidarsi a un centro di coordinamento esterno, che dei pazienti conoscesolo i dati anagrafici e li sorteggia usando una tavola di numeri casuali, per poicomunicare la selezione ottenuta ai ricercatori (randomizzazione centralizzata).La randomizzazione serve dunque a equilibrare gli eventuali fattori interferenti,in modo tale che le differenze osservate nel corso dello studio possano, con unalto grado di probabilità, essere ascritte al trattamento.Gli studi controllati non randomizzati utilizzano vari sistemi per formare i grup-pi di controllo: la suddivisione non casuale dei pazienti del campione oppure ilricorso a gruppi di confronto precostituiti. Riguardo al primo caso, si è già vistocome un criterio di selezione non casuale, quale che sia, possa generare una ri-partizione sbilanciata delle differenze individuali. Nel secondo caso, l’impiego diun gruppo di controllo già “pronto” – può essere un gruppo di pazienti ricoveratiin un altro reparto o in un altro ospedale che ricevono una terapia diversa (con-trolli concorrenti) o un gruppo di pazienti che hanno ricevuto una terapia diver-sa in un periodo precedente (controlli storici) – non si avrà mai la certezza che,terapia a parte, i pazienti dei due gruppi siano davvero confrontabili. Può capi-tare che in un gruppo ci sia una quotamaggiore di malati più gravi oppure ce nepossono essere alcuni che hanno contemporaneamente anche altre malattie;inoltre, da reparto a reparto e soprattutto da un momento storico a quello suc-cessivo possono esservi differenze più omeno grandi nei criteri o negli strumen-ti diagnostici utilizzati, negli esami effettuati, nella frequenza dei controlli a di-stanza, eccetera. E non è detto che queste informazioni siano sempre recupera-bili dalle cartelle cliniche esistenti, la cui compilazione non era stata pianificataai fini dello studio.
Tutti all’oscuro per vederci meglio
E’ esperienza comune che le convinzioni e le aspettativepersonali nei confronti di una data realtà hanno il potere diinfluenzare la percezione che si ha di essa. Quelle nei con-fronti delle cure mediche non fanno eccezione, sia sul ver-sante di chi le cure le riceve e le vede come promesse disalute e longevità, sia sul versante di chi le prescrive e levede come occasioni di qualificazione e soddisfazione pro-fessionale.Data la premessa, è lecito sospettare che, in condizioninormali, gli effetti che si rilevano dopo la somministrazio-ne di un nuovo trattamento non siano “tutta farina del suosacco”: i pazienti, che hanno accettato di sperimentarloaspirando a trovare per i loro disturbi una cura più efficacedi quelle già provate, potrebbero inconsciamente essereindotti a sentirsi meglio, e i ricercatori, che nutrono sem-pre la speranza che i loro esperimenti diano risultati posi-tivi, potrebbero vedere più benefici clinici di quanti non cene siano in realtà.Si può anche immaginare che, pur all’interno di un protocol-lo sperimentale prefissato, sia i pazienti sia i medici si com-portino inmodo leggermente diverso a seconda che abbianoa che fare con il trattamento sperimentale, con uno vecchioo con un placebo; per esempio potrebbero, senza renderse-ne conto, essere entrambi un po’ più assidui o attenti nei
confronti del primo: i pazienti nell’autovalutazione dei proprisintomi e i medici nell’esecuzione dei controlli clinici.Dato che è impossibile quantificare il peso di questo tipo diinterferenze, la condizione migliore perché gli effetti di untrattamento sperimentale emergano da uno studio senzacondizionamenti di sorta è la totale ignoranza di pazienti ericercatori rispetto alla sua assegnazione, quella che intermini metodologici si definisce “cecità”.A meno che non sia proprio impossibile mascherare i di-versi trattamenti (come nel caso delle fisioterapie, dellepsicoterapie e spesso degli interventi chirurgici), gli studiclinici vengono quindi effettuati in “cieco”, cioè non dicen-do ai pazienti quale dei trattamenti messi a confrontostanno assumendo, o in “doppio cieco”, cioè tenendo siai pazienti sia i medici all’oscuro della loro distribuzione.Quando il doppio cieco non è attuabile, il rischio di distor-sioni nel giudizio sui cambiamenti clinici può essere supe-rato facendo in modo che vi sia almeno una valutazione incieco dei risultati finali da parte di un osservatore indipen-dente. Ovviamente, l’imparzialità è più facile da ottenerequando i risultati da valutare sono dati oggettivi, comeesami di laboratorio, tracciati elettrocardiografici, imma-gini radiografiche, piuttosto che dati soggettivi, comel’intensità di un sintomo descritta dai pazienti.
Prendere le misure
Gli studi clinici, che vengono dopo quelli più esplorativi della fase preclinica,hanno il compito di fornire risposte pertinenti a quesiti ben precisi: non servonoa capire semplicemente “che cosa succede” dopo la somministrazione del trat-tamento, ma a verificare se un obiettivo terapeutico è raggiunto.
20
L’ABC della ricerca clinica

Gli esiti Il quesito è strettamente legato alle prerogative del trattamento, a quello che daesso ci si può aspettare e a quello che si vuole ottenere: può verificare la riduzio-ne della mortalità nei pazienti cardiopatici o semplicemente la colesterolemiasotto controllo. Mi verrebbe da toglierlo. Lo trovo troppo specifico e lungoPuòanche riguardare il rischio di fratture nelle donne in menopausa con osteoporo-si o semplicemente o l’aumento dei loro valori di densità ossea misurati con lamineralometria ossea computerizzata (MOC). A rigor di logica, le due cose do-vrebbero andare insieme: dal momento che troppo colesterolo nel sangue dan-neggia le coronarie, riducendolo i cardiopatici dovrebbero vivere più a lungo; dalmomento che la fragilità ossea delle donne in menopausa dipende dall’osteo-porosi, un aumento della mineralizzazione del loro scheletro dovrebbe tradursiin una sua maggiore resistenza. Nella pratica, tuttavia, fenomeni pur collegatinon evolvono sempre di pari passo.A dimostrarlo inequivocabilmente sono state le aspettative deluse verso alcunifarmaci che si pensava potessero influenzare in modo significativo la sopravvi-venza dei pazienti: solo negli anni Ottanta furono pubblicati i risultati di dueampi studi clinici in cui si dimostrava che un promettente ipocolesterolemiz-zante (clofibrato), capace di ridurre consistentemente la colesterolemia di pa-zienti cardiopatici, e un efficacissimo antiaritmico (flecainide), capace di elimi-nare quasi completamente le pericolose extrasistoli ventricolari che fanno se-guito all’infarto, aumentavano la mortalità dei pazienti invece di diminuirla.Questo a dispetto dei dati ematochimici e dei segni elettrocardiografici soddisfa-centi. Se i ricercatori si fossero limitati a controllare questi ultimi, consideran-doli indicatori attendibili dell’efficacia clinica dei due farmaci, le conseguenzesarebbero state davvero funeste.Una delle operazioni più delicate implicate nella progettazione degli studi cliniciè quindi la definizione degli esiti, o end point, che dovranno essere misurati perdecidere se un trattamento risponde o meno alle aspettative. Quali sarannoquesti esiti dipende strettamente da quello che si vuole ottenere come risultatofinale. E’ evidente che in qualsiasi studio clinico gli obiettivi ultimi devono esse-re quelli rilevanti per i pazienti e che hanno un’effettiva ricaduta sulla salutepubblica: la guarigione dalla malattia oppure, quando non si possa mettere inconto la guarigione, il prolungamento della sopravvivenza e il miglioramentodella qualità di vita rispetto a quanto la malattia farebbe prevedere. Tra gliobiettivi di alto profilo rientra anche la prevenzione di eventi gravi e invalidanti,come possono essere un infarto del miocardio, un ictus cerebrale, un’emorra-gia, una frattura, un intervento chirurgico urgente, la necessità di dialisi renale.
Esiti primari In generale, per poter vedere l’influenza di un trattamento su questo tipo di esi-ti, che vengono chiamati end point primari, sono necessari tempi molto lunghi.Il che, di solito, rappresenta uno svantaggio: gli studi di lunga durata richiedo-no maggiori investimenti economici e un impegno superiore da parte dei ricer-catori, delle strutture sanitarie e dei pazienti, e sono più esposti ad alcuni in-convenienti, primo fra tutti la perdita di una parte dei partecipanti, che non rie-sce a seguire il programma di cura o non mantiene la stessa assiduità nei con-trolli fino al termine fissato.
Esiti secondari E’ per questo motivo che spesso negli studi clinici vengono fissati obiettivi più “aportata di mano”: di solito sono valori ematochimici (per esempio la colesterole-mia) o parametri fisiologici (per esempio la pressione arteriosa) o modificazionimorfologiche (per esempio le alterazioni della forma del cuore osservabili conl’ecocardiografia, come la dilatazione o l’ipertrofia dei ventricoli). Quelli appenadescritti sono definiti esiti o end point secondari (detti anche surrogati, perchédi fatto “fanno le veci” di quelli primari). Naturalmente sono validi gli end pointsecondari quando è lecito aspettarsi che la loro modificazione si rispecchi fedel-mente nell’effetto complessivo del trattamento su quelli primari (mortalità, fre-quenza di eventi gravi, qualità di vita).
Esiti intermedi Si distinguono infine gli end point intermedi, che pur non essendo paragona-bili a quelli primari, hanno notevole rilevanza sia nella storia naturale di unamalattia sia nell’esperienza dei pazienti; esempi di end point intermedi sono la
21
L’ABC della ricerca clinica

frequenza di crisi di angina pectoris, i sintomi da iperglicemia, la tolleranza al-lo sforzo.Ogni studio può ovviamente porsi più di un obiettivo e prevedere la valutazionedi esiti diversi. Una volta soddisfatta la necessità di individuare degli esiti clini-camente rilevanti, ponendoseli come obiettivi primari, può essere utile rilevareanche alcuni parametri intermedi o secondari. La misurazione di questi ultimipuò rappresentare un obiettivo secondario rispetto al quesito specifico che haispirato lo studio, ma in genere apporta informazioni aggiuntive che possonoessere di supporto alle conclusioni ricavate dall’analisi degli esiti primari o ser-vire a generare nuove ipotesi di ricerca.
Gli esiti in oncologia
Sopravvivenza (overall survival, OS): il tempo che intercor-re tra la diagnosi e il decesso del paziente per qualsiasi causaSopravvivenza specifica (cancer specific survival, CSS):il tempo che intercorre tra la diagnosi e il decesso del pa-ziente a causa del tumoreSopravvivenza senzamalattia (disease-free survival): iltempo che intercorre tra la diagnosi e la ricaduta o il de-cesso del paziente per qualsiasi causaRisposta completa (overall response rate, ORR): la por-
zione di pazienti nella quale si verifica una determinata di-minuzione della massa tumorale per un periodo predefini-to di tempoTempo alla progressione (time-to-progression, TTP): iltempo che intercorre tra la diagnosi e l’obiettiva progressio-ne del tumoreTempo al fallimento della terapia (time-to-treatmentfailure, TTF): il tempo che intercorre tra l’inizio della tera-pia e la sua interruzione per qualsiasi causa
E dopo?Terminati gli studi di fase III, tutti i dati derivati dalle valutazioni pre cliniche ecliniche vanno a formare un dossier che viene sottoposto all’autorità competen-te per la registrazione e l’autorizzazione alla commercializzazione del nuovo far-maco – per l’Italia è l’AIFA, per l’Unione europea l’EMEA, per gli Stati Uniti laFood and Drug Administration (FDA). E’ in questa fase che il prodotto riceve ilnome di fantasia con il quale farà la sua apparizione sul mercato.
Il farmacosul mercato
Una volta arrivato negli scaffali delle farmacie, il farmaco inizia a circolare libe-ramente, previa prescrizionemedica o senza, tra i malati del mondo reale. I qua-li sono consumatori ben più complessi dei soggetti sperimentali. La diffusionedi un nuovo farmaco nella popolazione generale è, pertanto, il banco di provadel suo impatto sulla salute pubblica nelle effettive condizioni d’impiego, equindi rappresenta sia una circostanza molto critica sia una grande opportuni-tà di ricavare ulteriori informazioni su di esso. In particolare, su quegli aspettiche la ricerca clinica, per quanto ben condotta, non può coprire: il manteni-mento dell’efficacia e della tollerabilità nell’uso prolungato, le interazioni con al-tri farmaci, gli effetti sulla sopravvivenza e sulla qualità di vita a lungo termine.Abbandonare un nuovo farmaco al suo destino sarebbe quindi un’occasionemancata, e oltretutto la storia insegna che è anche un bel rischio, un rischioche nel tempo ha già fatto innumerevoli vittime.
Lafarmacovigilanza
Per non perdere la possibilità di acquisire nuove conoscenze sui medicinali viavia messi in commercio e per tutelare i cittadini da eventuali brutte sorprese, èstata istituita l’attività di farmacovigilanza, o sorveglianza post marketing, cheoggi viene considerata la fase IV della ricerca clinica. Obiettivo della farmacovi-gilanza è non solo il rilevamento il più possibile tempestivo di effetti non previ-sti, ma anche la puntualizzazione di quelli previsti. Può infatti capitare chenell’uso generalizzato di un farmaco alcuni effetti collaterali si verifichino piùspesso di quanto era emerso nel corso degli studi sperimentali, oppure che al-cuni tipi di pazienti risultino più sensibili di altri.I risultati dell’attività di sorveglianza possono portare a limitazioni nell’utilizzodel prodotto, a cambiamenti nella posologia raccomandata o all’introduzione dispecifiche avvertenze relative a nuovi effetti collaterali nel foglietto illustrativo,in modo da consentirne un impiego più sicuro ed efficace. In seguito alla segna-lazione di effetti particolarmente gravi, invece, si impongono la revoca dell’auto-rizzazione del farmaco e il suo ritiro dal mercato. Infine, nel caso di osservazionirelative a effetti terapeutici non preventivati può essere presa in considerazione –
22
L’ABC della ricerca clinica

ovviamente previa verifica – l’estensione delle indicazioni del farmaco ad altri di-sturbi o malattie oltre a quelli per i quali era stato ideato.La farmacovigilanza interessa tutti i medicinali in commercio, ma si applica concriteri differenti a diverse categorie di farmaci: per i vaccini e per i farmaci postisotto monitoraggio intensivo, che sono riportati in un apposito elenco redattodal Ministero della salute, vanno segnalate tutte le reazioni avverse, compresequelle già note e quelle non gravi; per tutti gli altri, invece, vanno segnalate sol-tanto le reazioni avverse gravi o quelle inattese, cioè non citate nella scheda tec-nica o nell’autorizzazione rilasciata per la commercializzazione.La sorveglianza sui farmaci è demandata a tutti i medici e gli altri operatori sa-nitari, che hanno l’obbligo di riportare le loro osservazioni su appositi moduli e ditrasmetterle alle Aziende sanitarie o Direzioni sanitarie ospedaliere di riferimen-to; le segnalazioni raccolte a livello locale vengono riportate tutte alla Direzionegenerale per la valutazione dei medicinali del Ministero della salute. Presso l’A-genzia italiana del farmaco, inoltre, è istituito l’Osservatorio nazionale di farma-covigilanza, che attraverso una rete telematica rivolta a tutti i referenti di Regioni,Aziende Sanitarie Locali (ASL), Aziende ospedaliere, Istituti di ricovero e cura acarattere scientifico e delle industrie farmaceutiche, registra segnalazioni e for-nisce informazioni, integrandosi con la banca dati europea EudraVigilance.La forma più comune di segnalazione di effetti avversi e imprevisti di farmaci èquella che viene fatta spontaneamente dai singoli medici caso per caso, ma esi-stono anche forme di monitoraggio programmato per casi selezionati. In ag-giunta, vengono effettuati i cosiddetti studi di esito (o studi di outcome), che so-no ricerche osservazionali condotte in fase post marketing su ampie casistiche,con l’obiettivo di valutare l’effettiva resa di un farmaco di cui si è documentatal’efficacia nel contesto di studi clinici, ma rispetto al quale si hanno dubbi circal’impatto su aspetti importanti della salute nel contesto reale.In ultima analisi, comunque, a svolgere l’attività di farmacovigilanza sono inprima persona i pazienti, i quali riferiscono ai loro curanti di eventuali disturbi odi cambiamenti inattesi verificatisi dopo l’inizio di una nuova cura.
Occhio vigile sui farmaci
Gli ultimi pochi anni sono stati prodighi di cattive notiziesulla sicurezza dei farmaci messi in commercio – o volen-dola vedere dalla parte del mezzo bicchiere pieno, di buo-ne notizie sulla validità della sorveglianza post marketing.Un caso in particolare merita di essere citato, perché, al dilà dei dubbi suscitati circa l’irreprensibilità dell’azienda im-plicata nel gestirlo, riassume in modo esemplare il sensodella farmacovigilanza. La vicenda è quella della ceriva-statina, farmaco molto efficiente nell’abbassare la cole-sterolemia, che però, a breve distanza dal suo ingressonel mercato, rivelò un effetto collaterale tanto raro quantopericoloso. La storia della cerivastatina, che era iniziatanel 1998 con la sua registrazione, finì nel 2001 con il suoritiro dalle farmacie di tutti i paesi nei quali era stata auto-rizzata. Il motivo: nel frattempo, erano morte 31 personeche ne facevano uso, a causa di rabdomiolisi (necrosi dellefibre muscolari). Gli studi iniziati subito dopo le prime se-gnalazioni rivelavano che sulla rarità dell’effetto collatera-le bisognava ricredersi: innanzitutto, con la cerivastatinaesso era molto più frequente che con le altre molecole del-la sua stessa categoria (statine), in secondo luogo la pro-babilità che si manifestasse diventava altissima se la ceri-vastatina veniva assunta in associazione con il gemfibro-
zil, un ipolipemizzante appartenente a una classe farma-cologica diversa (fibrati). Purtroppo, tra i pazienti a rischiodi malattie cardiovascolari la terapia combinata con ceri-vastatina e gemfibrozil non era un’evenienza eccezionale,perché la prima riduce il colesterolo e il secondo i triglice-ridi; e infatti, almeno metà delle vittime della rabdomiolisierano state in cura contemporaneamente con entrambi ifarmaci.Il caso della cerivastatina è un classico esempio dei quesitiche la ricerca lascia irrisolti e ai quali finisce col risponderela farmacovigilanza: come risultò poi, la rabdomiolisi eraun evento raro con le altre statine in commercio (si è sti-mato che ci vorrebbe uno studio su oltre 22.727 pazientiper osservarne un caso), ma con la cerivastatina lo eradieci volte meno, e lo era ancora meno se a prenderla era-no pazienti di oltre 65 anni o pazienti che oltre all’ipercole-sterolemia avevano anche il diabete, e arrivava a esserepersino frequente (capitava a una persona su dieci!) seveniva presa con il gemfibrozil: la fascia di età più avanza-ta, la compresenza di diverse malattie, l’interazione trapiù terapie sono tutti aspetti che tradizionalmente non so-no coperti dalla sperimentazione e finiscono con interes-sare la sorveglianza post marketing.
Interpretare
L’analisi dei datiCome si è detto, uno studio clinico parte con un quesito di ricerca e conun’ipotesi da verificare. Ponendo il caso che in questione vi sia l’efficacia
23
L’ABC della ricerca clinica

dell’ultimo ritrovato contro l’ipertensione (la molecola “XY”) e il suo potere pro-tettivo nei confronti dell’ictus cerebrale rispetto a un trattamento già in com-mercio che serve allo stesso scopo (“A”), l’ipotesi da verificare sarà: nei pazientitrattati con “XY” si verificano meno ictus che nei pazienti trattati con “A”. Conquesto obiettivo si selezionerà un campione di pazienti ipertesi a rischio (conipertensione damoderata a grave) secondo criteri prestabiliti (determinati valoridi pressione arteriosa minima e massima per un certo numero di giorni e di set-timane continuativi); li si assegnerà al trattamento con “XY” o con “A” secondorandomizzazione e in cieco; si proseguiranno i due trattamenti per un certo nu-mero di anni, il tempo necessario a rilevare eventuali eventi cerebrovascolari(end point primario); nel frattempo, si registreranno, prima di somministrare ifarmaci e poi a scadenze prefissate, altri parametri, come la pressione arteriosa,la funzionalità renale, la morfologia cardiaca all’ecocardiografia (end point se-condari), utili a monitorare l’andamento della malattia ipertensiva e delle suecomplicanze.Alla fine, ci si ritroverà con una mole di informazioni, e in particolare, con uncerto numero di ictus avvenuti in ciascuno dei due gruppi di pazienti. Potrebbeessere che il numero degli ictus registrati nel gruppo dei pazienti curati con“XY” sia un po’ inferiore a quello rilevato nell’altro: si può già concludere che“XY” è più efficace di “A” nel prevenire gli eventi cerebrovascolari? Chiaramente,la risposta è negativa. Una differenza tra i due gruppi potrebbe verificarsi ancheper puro caso, soprattutto se la differenza non è, come spesso accade, palese-mente spropositata. Quindi, quanti ictus in meno bisogna registrare per poterassegnare a “XY” la palma dell’antipertensivo più valido con una buona proba-bilità che sia davvero così? Ossia, che davvero “XY” sia in grado di diminuire ilrischio di ictus nei pazienti con ipertensione da moderata a grave?
Test statisticidi significatività
Una volta che si è terminata la raccolta dei dati, cioè si sono annotate tutte lemisurazioni necessarie per rilevare eventuali cambiamenti negli end point scel-ti, il passo successivo è fare in modo che questi dati assumano un significatoapplicabile al contesto clinico, rispondendo a domande come quelle formulatequi sopra. A tale scopo, si sottopongono i dati ad apposite elaborazioni matema-tiche, che hanno il compito di stabilire se le differenze osservate siano significa-tive, vale a dire se siano attribuibili alla diversa efficacia dei diversi trattamentioppure casuali.
Come trarre dai dati le giuste conclusioni
Valore P Esistono procedure statistiche, comprese sottoil nome di test statistici di significatività, che non fornisco-no certezze assolute, ma permettono di calcolare un valo-re, noto come “P”, che rappresenta la probabilità di trarreinvolontariamente dai dati disponibili la conclusione sba-gliata. Com’è ovvio, affinché lo studio abbia una qualcherilevanza, questo valore deve essere molto piccolo. Primadi applicare il test statistico prescelto, è perciò necessariofissare una soglia che P non deve superare perché le diffe-renze osservate possano essere considerate significative;la soglia viene decisa liberamente dai ricercatori, ma disolito si ricorre, per convenzione, a uno di due valori stan-dard di P – 0,05 o 0,01 – che sono definiti livelli di signifi-catività. Adottare un livello di significatività di 0,01 vuoledire avere l’1 per cento di probabilità di considerare veral’ipotesi di partenza mentre in realtà non lo è. In gergostatistico questo tipo di errore è definito errore di I tipo oerrore alfa, e consiste nell’attribuire un significato a risul-tati che sono semplicemente dovuti al caso. Per tornareall’esempio del farmaco “XY”, si avrà una probabilità su100 di sbagliarsi concludendo che esso è più efficace di“A” nel prevenire l’ictus in pazienti con ipertensione. E,specularmente, si avrà il 99 per cento di probabilità diavere ragione.Sempre in termini statistici, si può commettere l’errorecontrario – detto errore di II tipo o errore beta – di consi-derare sbagliata l’ipotesi di partenza quando essa è vera,
e quindi di attribuire al puro caso un risultato che invece èsignificativo. Nell’esempio di “XY” e “A”, fare un errore diII tipo equivale a giudicare la differenza tra gli effetti deidue farmaci del tutto fortuita, mentre in realtà “XY” è dav-vero più efficace di “A”. A questo secondo tipo di errore èstrettamente connesso il concetto di potenza dei test sta-tistici: la potenza di un test corrisponde alla sua capacitàdi non lasciarsi sfuggire una differenza significativa quan-do questa esiste – o, per dirla in termini tecnici, alla pro-babilità che ha di trovarla. La potenza di qualsiasi test sta-tistico dipende molto dalle dimensioni del campione alquale viene applicato: in generale, quanto più numeroso èil campione, tanto maggiore è la potenza del test e, diconseguenza, tanto più alta è la probabilità di interpretarei risultati in modo corretto.
Intervalli di confidenza Un altro metodo per stabilire ilsignificato delle differenze rilevate in uno studio di con-fronto fra trattamenti è quello dei cosiddetti intervalli diconfidenza o limiti fiduciari. Si consideri l’end point se-condario della pressione arteriosa: per parametri comequesto, il dato che si utilizza nel valutare la significativitàdelle differenze tra i gruppi è il valore medio calcolato inciascun gruppo a partire dai valori registrati nei singolisoggetti. Per esempio, nei pazienti trattati con “XY” o con“A”, si saranno registrate modificazioni della pressionearteriosa, presumibilmente abbassamenti, nel corso del
24
L’ABC della ricerca clinica

trattamento antipertensivo: per ognuno dei due gruppi sipuò calcolare che l’abbassamento della pressione è statoin media di un certo numero di mmHg, ma quello che difatto è successo è che ciascun paziente in ciascun gruppoha avuto una sua variazione, che non coincide esatta-mente con quel valore (è un po’ più grande o un po’ piùpiccola). Il valore reale dell’abbassamento della pressio-ne si colloca in qualche punto tra il valore massimo e ilvalore minimo registrati in tutti gli individui. L’intervallodi confidenza tiene conto del range di possibili valori cheil parametro di interesse assume all’interno dei campioni.Quando si confrontano le riduzioni della pressione arte-riosa nei due gruppi dello studio su “XY” e “A” si fa unastima di quanto differiscono.Diversamente dal valore P, che chiarisce se una differenzaè significativa (se “XY” ha un effetto maggiore di “A” sullapressione arteriosa) ma non dice nulla della sua entità (diquanto più grande è l’effetto di “XY” rispetto ad “A”), l’in-tervallo di confidenza fornisce un’indicazione sia dell’enti-tà della differenza riscontrata sia del grado di precisionecon cui il valore stimato la rappresenta. L’intervallo di con-fidenza nel quale si colloca il valore stimato della differen-
za tra le riduzioni della pressione arteriosa dei due gruppi(poniamo, per esempio, 20 mmHg) è espresso da quel va-lore affiancato dall’indicazione della variazione in più o inmeno che si può avere rispetto a esso (20±5 mmHg). Laprobabilità che il valore reale della differenza cada nelrange indicato viene espressa con una percentuale, di so-lito il 95 per cento, a significare non solo che vi è una diffe-renza tra le riduzioni medie della pressione arteriosa con idue trattamenti, ma anche che, con una probabilità del 95per cento, il valore possibile di questa differenza è com-preso tra 15 e 25 mmHg.I test statistici che si possono usare per dimostrare la si-gnificatività delle differenze tra le misurazioni rilevate inuno studio sono molti: di volta in volta si sceglie quale oquali test applicare in base al tipo di confronto da realizza-re (tra differenze o tra variabili) e al tipo di dati da con-frontare (proporzioni, percentuali, medie, eccetera). Iltest migliore è quello con il più piccolo margine di incer-tezza, cioè quello che ha la più bassa probabilità di lascia-re spazio a deduzioni infondate. Come abbiamo detto, ilgrado di affidabilità di un test in questo senso è indicatocome potenza del test.
La garanzia che uno studio clinico deve dare è di essere abbastanza efficientenell’individuare un effetto quando questo di fatto esiste. In generale, il rischio dicadere in errore non vedendo qualcosa che in realtà c’è dipendemolto dalle carat-teristiche del campione, e principalmente dalle sue dimensioni. Soprattuttoquando un evento è poco frequente e le differenze sono piccole, è facile che in uncampione poco numeroso non si rendano visibili. Per esempio, nel casodell’antiaritmico flecainide, ci volle uno studio di grandi dimensioni per dimostra-re che invece di diminuire la mortalità dei pazienti infartuati, come ci si aspettavain base alla sua capacità di controllare le artimie ventricolari, l‘ aumentava di duevolte e mezzo rispetto al placebo: in un campione limitato il numero assoluto didecessi attribuibili al farmaco (che era di 3,7 ogni 100 pazienti trattati) sarebbefacilmente passato inosservato. Come già accennato, la numerosità del campionecondiziona la possibilità di individuare le differeenze significative e di interpretarei risultati in modo corretto. Di solito, quando si avvia uno studio, l’ampiezza delcampione viene determinata a priori, nella fase di progettazione, e viene decisasulla base dei test statistici che si ha intenzione di utilizzare: ogni test, infatti, ne-cessita di un numero minimo di soggetti per dare risultati affidabili.
Dalle prove alle raccomandazioni
Le provedi efficacia
Il “responso” che l’analisi dei dati fornisce sono le cosiddette prove di efficacia.A seconda della qualità dello studio, cioè della sua precisione nell’evidenziaregli effetti dell’intervento sperimentato e nell’evitare le possibili distorsioni in-terpretative, si ottengono prove di efficacia di diverso valore. Quando si devedecidere in merito alla fondatezza scientifica di un determinato atto medico,che sia una terapia farmacologica, un trattamento riabilitativo, un test dia-gnostico o un programma di screening, di solito si prendono in esame tutti glistudi che sono stati realizzati su di esso e si valuta il peso delle prove emerse asuo favore o contro.
I livelli di prova Le prove di efficacia vengono classificate in sei livelli qualitativi, detti livelli diprova, in base alla robustezza metodologica degli studi da cui provengono:l le prove di tipo I sono quelle ottenute da più studi clinici controllati rando-mizzati o da revisioni sistematiche di studi randomizzati;
l le prove di tipo II sono quelle ottenute da un solo studio randomizzato di di-segno adeguato;
l le prove di tipo III sono quelle ottenute da studi di coorte non randomizzaticon controlli concorrenti o storici o loro metanalisi;
l le prove di tipo IV sono quelle ottenute da studi retrospettivi tipo caso-con-trollo o loro metanalisi;
l le prove di tipo V sono quelle ottenute da studi di casistica (come le serie dicasi) senza gruppo di controllo;
25
L’ABC della ricerca clinica

l le prove di tipo VI sono quelle basate sull’opinione di esperti autorevoli o dicomitati di esperti (come le consensus conference).
Il passo successivo consiste nel riassumere le prove di vario livello disponibili inuna sintesi che serva a trasformare i risultati scientifici in indicazioni utili allaloro conversione in atti medici.
Leraccomandazioni
In questo modo, le prove di efficacia su un determinato intervento confluiscononelle raccomandazioni relative alla sua applicazione clinica. Le raccomandazio-ni vengono anch’esse graduate in base alla loro forza, una dimensione che rap-presenta la probabilità che la loro attuazione nella pratica determini un miglio-ramento dello stato di salute dei destinatari.
Il grading Esistono fondamentalmente due sistemi di classificazione (grading) delle rac-comandazioni, che fanno riferimento l’uno al livello qualitativo delle prove asostegno o contrarie all’intervento sanitario implicato e alla concordanza tra idiversi studi su di esso, l’altro al tipo di indicazione che forniscono riguardo al-la realizzazione dell’intervento stesso. Nel primo sistema la forza delle racco-mandazioni decresce dal grado A, che include raccomandazioni basate su pro-ve di qualità superiore, fino al grado D, che comprende raccomandazioni ba-sate su prove di basso livello; nel secondo la forza delle raccomandazioni ri-guarda il parere che esprimono rispetto all’opportunità di attuare l’intervento,e va dal grado A, decisamente favorevole, al grado E, decisamente sfavorevole.La classificazione di prove e raccomandazioni è essenziale dal punto di vistapratico, e infatti rappresenta il sistema di riferimento per la compilazione dellelinee guida, che nella medicina basata sull’evidenza (evidence based medicine)sono i principali strumenti di supporto decisionale all’implementazione degliinterventi sanitari.
Metanalisi e revisioni sistematiche
Nella sua fase di valutazione pre implementazione, e an-che dopo, ogni intervento sanitario viene sottoposto apiù studi sperimentali, e può accadere che non tutti porti-no esattamente alle stesse conclusioni. Le scelte meto-
dologiche, le dimensioni dei campioni, la tipologia dei con-trolli, la durata dei periodi di follow up possono differire dastudio a studio quel tanto che basta per influenzare i risul-tati finali.
26
L’ABC della ricerca clinica
TABELLA 2: Le qualità di uno studio di ricerca
La forza scientifica dei diversi tipi di studi di ricerca: la qualità di una prova scientifica dipende da molti fattori, fra cui iltipo di studio, il numero di osservazioni effettuate nello studio, la selezione dei soggetti, eccetera La gerarchia riportatasotto va dallo studio con forza scientifica maggiore a quello con forza scientifica minore.
Vantaggi Svantaggi
Revisioni sistematichee metanalisi
Sono potenti e minimizzano l’effettodi fattori, chiamati confondenti, che riduconola capacità di attribuire la causa di un eventoal trattamento
Non possono cancellare errorio inappropriatezze che possonoessere presenti nei singoli studiraccolti
Studi randomizzatio semi randomizzati
Sono potenti e minimizzano l’effettodi fattori, chiamati confondenti, che alteranola capacità di attribuire la causa di un eventoal trattamento
Non consentono di evidenziareeventi rari e, al termine dello studio,i soggetti esaminati vengono seguitiper tempi brevi
Studi coorte Potenti e poco costosi Sono fonte di errori e consentonouna generalizzazione limitata
Studi a tempi multipli;studi caso controllo
Potenti e flessibiliPossono verificare un’ipotesi
Sono fonte di errori e consentonouna generalizzazione limitata
Studi cross-over;studi ecologici
Potenti e poco costosi. Non necessitanodi controlli indipendenti, perché tutti i soggettiricevono sia il trattamento in esame sia quellodi controllo
Sono fonte di errori e consentonouna generalizzazione limitata
Casi clinici singolio serie di casi;sorveglianza attivao passiva del paziente
Segnalano l’allarme e sono utili nel casodi eventi rari
Sono fonte di errori e non consentonola generalizzazione

Quando si tratta di decidere qual è il verdetto sull’op-portunità di introdurre un intervento nella pratica clini-ca o di generalizzarne l’uso, ci si può trovare con moltidati a disposizione ma senza un’indicazione definitiva.Se le conclusioni di diverse ricerche su uno stesso ar-gomento non sono perfettamente convergenti si ricor-re alla metanalisi, una tecnica statistica che consentedi combinare i dati di più studi, per generare un datocomplessivo che risponda in modo univoco al quesitoclinico in oggetto.Il vantaggio principale della combinazione dei dati dipiù studi è quello di rendere disponibile un campione
molto ampio – perché il numero finale di soggetti su cuiviene fatta la metanalisi è dato dalla somma dei campionidei singoli studi – che è quindi più affidabile dal punto divista statistico e più rappresentativo dal punto di vistaclinico. In questo modo si riduce l’effetto delle even-tuali distorsioni contenute nei risultati dei singoli studi.La metanalisi è lo strumento operativo delle revisioni si-stematiche.La revisione sistematica consiste nella valutazione cu-mulativa di tutti gli studi, pubblicati e non, su un determi-nato intervento che rispondono al medesimo quesito clini-co e sono comparabili tra loro sul piano metodologico.
Significativo non basta
Dallasignificativitàstatisticaalla significativitàclinica
Nella fase dell’analisi dei dati la statistica è di grande aiuto perché riduce auna frequenza accettabile gli errori di interpretazione; e il fatto è di una certarilevanza visto che, in genere, le informazioni fornite dai dati servono per pren-dere decisioni cliniche. Ciò non toglie che i risultati dei test debbano esserepresi con beneficio di inventario. Il giudizio di significatività emesso da un teststatistico ha, per l’appunto, un valore statistico: si tratta di una descrizioneprobabilistica – anche se, come si è visto, la probabilità che sia corretta puòessere molto alta – e oltretutto deriva dall’osservazione di un campione di di-mensioni necessariamente limitate in un contesto necessariamente artificiale.Anche uno studio guidato da obiettivi plausibili e di ottima qualità metodologi-ca – ben progettato, eseguito con rigore, eticamente ineccepibile e libero daconflitti di interessi – può rivelare dei limiti allorché si prova a trasferirne i risul-tati nella pratica clinica.Il passaggio dalla significatività statistica alla significatività clinica non è sem-pre scontato e diretto. Innanzitutto, nel valutare l’utilità di un intervento sani-tario è necessario tenere conto sia degli effetti positivi sia delle eventuali riper-cussioni negative. E perché possa essere ritenuto valido, bisogna che i beneficiche arreca siano superiori ai danni che può fare. Degli uni e degli altri, dun-que, occorre conoscere l’entità e la probabilità. Per ottenere un beneficio ap-prezzabile si può infatti accettare di esporsi al rischio di subire anche un dan-no, a patto che il rischio sia il più basso possibile e il danno il più lieve possibi-le. E’ ovvio che l’entità del danno che si è disposti a tollerare dipende anchedall’importanza del beneficio che si prevede di ricevere e dalle alternative di-sponibili: un effetto collaterale rilevante può essere ammesso nel caso di untrattamento che serva a contrastare una malattia potenzialmente pericolosa oinvalidante e del quale non esistano equivalenti di pari efficacia (un tipicoesempio sono i trattamenti antitumorali); lo stesso effetto o anche uno menopesante devono essere rifiutati se rischiano di essere più deleteri di quelli dellamalattia e se esistono alternative terapeutiche rispetto alle quali il trattamen-to non offre alcun vantaggio.Perché a un trattamento o a un intervento diagnostico si possa attribuire una si-gnificatività clinica occorre che il rapporto tra i benefici e i rischi che derivano dal-la sua attuazione sia favorevole ai singoli pazienti e alle comunità che lo ricevono.
E allora, ne vale proprio la pena?Come probabilmente si è intuito da quanto descritto finora, l’unica certezza del-la ricerca clinica è che dagli studi sperimentali non emergono risposte definiti-ve. Un errore o una distorsione presente nella progettazione di una ricerca piut-tosto che nella raccolta e nell’interpretazione dei dati è sempre possibile. Sta al-la competenza, all’esperienza e alla serietà professionale dei ricercatori fare inmodo che i loro studi abbiano tutte le carte in regola.Ma se agli errori o agli inconvenienti metodologici più comuni si può pensare inanticipo oppure ovviare in corso d’opera, resta sempre quel margine non con-trollabile di incertezza che accompagna l’ingresso dei risultati scientifici nellapratica clinica.Tuttavia, partire alla ricerca della verità con dei dati obiettivi può essere un van-
27
L’ABC della ricerca clinica

taggio, e per raggiungere questo scopo al momento non si conoscono molte al-ternative agli studi sperimentali. Come afferma Marco Bobbio, cardiologo edesperto di metodologia della ricerca clinica: “Sarebbe un errore passare dalla re-ligione monoteista dei camici bianchi alla monocrazia dei trial, ma non lo è cre-dere che rappresentino un nuovo punto di riferimento della medicina”.In realtà, purché siano razionalmente fondati e mossi da scopi realmente inno-vativi, gli studi clinici possono essere un’opportunità anche per i pazienti, siaperché hanno la possibilità di trovare trattamenti migliori per le loro malattie odi ricevere diagnosi più precise o più precoci, sia perché nel setting sperimenta-le sono sottoposti a controlli più assidui e approfonditi, sono più seguiti nellagestione delle cure, trovano una considerazione più scrupolosa per i benefici o ifastidi che riferiscono.La partecipazione alle sperimentazioni cliniche deve comunque sottostare alprincipio etico fondamentale della libera scelta. Ma affinché sia effettivamentelibera una qualsiasi scelta deve essere consapevole e fondata su una informa-zione completa e trasparente.
I modi per dirlo
I risultati di uno studio sull’efficacia di un trattamentonel prevenire un evento indesiderato (il decesso, un in-farto, una frattura) rispetto a un trattamento di con-trollo (o al placebo) possono essere espressi in terminidi probabilità dei pazienti trattati con l’uno e con l’altrofarmaco di andare incontro all’evento (rischio assolu-to). Dalle percentuali di soggetti colpiti dall’evento pri-ma del termine dello studio in ciascuno dei due gruppisi possono ricavare due tipi di informazioni: il rischiorelativo, cioè il rapporto tra i due rischi assoluti, cheindica quante volte in più o in meno rischiano l’evento ipazienti che assumono il trattamento sperimentale ri-spetto a quelli di controllo; il rischio evitabile, cioè ladifferenza tra il maggiore e il minore dei due rischi as-soluti, che indica quanti eventi si possono preveniretrattando un certo numero di pazienti con il farmacopiù efficace. Questo valore può essere inversamenteespresso come numero di pazienti da trattare perprevenire l’evento (NNT, number needed to treat). Ri-
spetto al rischio relativo quest’ultimo indice dà un’ideapiù precisa dell’entità del beneficio che si trae dall’unoe dall’altro trattamento.Un’altra stima frequentemente usata per rappresentarel’efficacia di un trattamento è il cosiddetto odds ratio,con un termine inglese non traducibile, che corrispondeal rapporto tra la frequenza con cui l’evento da preveni-re si verifica nel gruppo di pazienti assegnati al tratta-mento e la frequenza con cui si verifica nel gruppo dicontrollo. Dal punto di vista numerico, il rischio èespresso come percentuale di casi in cui l’evento si veri-fica (per esempio 20 casi su 100 pazienti o 20 per cen-to), mentre l’odds è il rapporto tra il numero di casi incui l’evento si verifica e il numero di casi in cui non si ve-rifica (quindi 20/80 o 0,25); l’odds ratio di un tratta-mento si calcola dividendo l’odds del gruppo che lo as-sume e l’odds del gruppo di controllo, cosicché un valoredi odds ratio inferiore all’unità significa che l’evento siverifica più raramente nel gruppo trattato.
Prima di dare il consenso
Prima di decidere di partecipare a uno studio è bene leg-gere attentamente la documentazione fornita per il con-senso informato e, nel caso rimanga qualche dubbio, nonesitare a chiedere ulteriori spiegazioni.I punti che è opportuno chiarire prima di prendere una de-cisione sono soprattutto quelli che riguardano l’utilità dellostudio, l’impegno che la partecipazione a esso comporta(in termini di durata totale, frequenza dei controlli, com-plessità della cura, eccetera), gli eventuali rischi.Al medico che raccoglie il consenso si possono per esem-pio fare le seguenti domande:n Di che tipo di trattamento o esame diagnostico sitratta?
n Perché i ricercatori ritengono che il trattamento/esamepossa essere utile?
n E’ già stato sperimentato su altre persone?n Con quali risultati?n Se prima della fine dello studio risultasse che il tratta-mento/esame non è utile o sicuro come si pensava, la
sperimentazione verrà interrotta?n Che effetti collaterali può avere il trattamento?n Se avvertirò degli effetti collaterali riceverò qualche cu-ra per alleviarli, potrò ridurre la dose del trattamento osospenderlo?
n Se, per motivi personali, avrò difficoltà a mantenere lamia partecipazione allo studio potrò uscirne?
n A che cosa mi serviranno le informazioni che dà questoesame diagnostico?
n Se con questo esamemi verrà diagnosticata questa ma-lattia che cure potrò fare? E’ previsto un percorso perfacilitare l’accesso alle cure studiato apposta per le per-sone che partecipano?
n Quanto tempo dura lo studio?n Chi finanzia lo studio?n Sarò rimborsato per le eventuali spese sostenute perpartecipare?
n C’è un’assicurazione che mi tutela in caso di eventualidanni?
Bibliografia di approfondimentol Bobbio M. e Cagliano S. Rischiare di guarire. Roma: Donzelli, 2005.l Straus SE et al. Evidence-basedmedicine. Roma: Il Pensiero Scientifico, 2007.l Dobrilla G. Dottore... mi posso fidare? Roma: Avverbi, 2007
28
L’ABC della ricerca clinica

Incertezza e conflitti di interessein medicinaSergio Cima, Roberto SatolliRevisori: Alberto Fontana, Rosita Orlandi
L’industria della salute
Il settore economico che produce beni e servizi per la salute è stato definito daArnold Relman (ex direttore del New England Journal of Medicine) come “com-plesso medico industriale”. Le sue dimensioni, se si considera anche l’indotto,rappresentano circa il 10 per cento del prodotto interno lordo in Europa, e rag-giungono il 15 per cento negli Stati Uniti. In Italia, secondo Confindustria, l’indu-stria della salute rappresenta la terza impresa del Paese, in termini di prodotto edi occupazione: solo nell’area di Milano, per esempio, contava nel 2003 ben 3.200aziende con 54.000 addetti e un giro d’affari di 10 miliardi di euro l’anno.A livello internazionale il cuore propulsivo del sistema è costituito dalle aziendefarmaceutiche: solo in Europa oggi danno lavoro a oltre mezzo milione di perso-ne. Il mondo dei farmaci, però, rappresenta solo una piccola fetta (attorno al 15per cento, in termini di fatturato) dell’intero settore economico che ruota attor-no alla salute. Accanto ai produttori di farmaci sono cresciuti anche i produttorie i distributori di strumenti e di materiale di consumo (apparecchiature diagno-stiche, attrezzature di intervento, reagenti chimici eccetera) e i fornitori di servi-zi (catene di cliniche, ospedali pubblici e privati, centri diagnostici, eccetera).Come ogni sistema economico, l’industria della salute e le singole imprese chela costituiscono hanno bisogno di crescere per sopravvivere, aumentando il fat-turato e il mercato potenziale. Questa dinamica è speculare alla crescita conti-nua dei costi per i sistemi sanitari e costituisce un problema politico per tutti ipaesi: come favorire una crescita equilibrata del sistema senza mettere a repen-taglio la sostenibilità dei sistemi sanitari e senza imporre una eccessiva e dan-nosa medicalizzazione alla società?
I meccanismiche regolanol’industriadella salute
Per rispondere è importante conoscere i meccanismi del settore. All’interno delsistema, i singoli elementi si muovono in concorrenza tra loro su piani orizzon-tali (tra fornitori di prodotti o servizi analoghi) ma anche con forti sinergie supiani verticali (tra attività che si completano e si favoriscono vicendevolmente).Tutti gli attori in gioco hanno interessi solidali:l centri di diagnosi o di cura, che reclutano un maggior numero di assistiti efatturano un maggior volume di prestazioni;
l produttori di apparecchiature sanitarie;l specialisti, che possono accrescere il numero dei pazienti e di conseguenza ilreddito, la reputazione o il potere;
l ditte farmaceutiche, che spesso agiscono come il veromotore di tutta la catena.L’azione esplicita emirata che ogni singolo elemento del sistemamette in opera perallargare il mercato e promuovere i propri prodotti e servizi specifici è solo l’ultimomiglio di una rete di attività che risulta molto articolata e parte da lontano.Si manifesta attraverso iniziative in tre aree principali: la ricerca clinica, che do-vrebbe produrre innovazione tecnologica da proporre almercato; la definizione del-le malattie, che consente il controllo dei campi di intervento possibili; le campagnedi consapevolezza sulle malattie, per allargare il numero dei cittadini coinvolti.
Ricerca clinica e marketing
Nel quadro sin qui descritto, la scienza ricopre un ruolo ambivalente. Da unaparte, fornendo le prove di efficacia, rappresenta l’unico riferimento certo su cuifondare le scelte appropriate, individuali e di politica sanitaria. Da un’altra par-
29
Incertezza e conflitti di interesse in medicina

te, essendo oggi la ricerca clinica per lo più finanziata direttamente dall’indu-stria della salute, sempre più spesso la si concepisce e la si conduce come ingre-diente del marketing.Occorre prendere atto che, soprattutto in campo biomedico, l’interesse econo-mico ha rimpiazzato la curiosità come forza trainante della ricerca. Si tratta diinteressi ingenti e crescenti: i finanziamenti per la ricerca in campo biomedicosono raddoppiati, arrivando nei soli Stati Uniti alla cifra di 100 miliardi di dolla-ri; su questo totale, quasi il 60 per cento degli investimenti proviene dall’indu-stria, mentre il contributo dei National Institutes of Health è inferiore al 30 percento. Eppure le somme investite oggi dall’industria nella ricerca costituisconomeno di un terzo di quello che le compagnie farmaceutiche spendono per mar-keting, promozione e gestione.
La filiera della ricerca clinicaI costi della ricerca finanziata dall’industria sono raddoppiati in pochi anni,mentre il rendimento in termini di nuovi farmaci è crollato. Si comprende per-ché la filiera della ricerca clinica stia subendo una ristrutturazione che mira amigliorarne l’efficienza. Gli strumenti del processo di ottimizzazione sono quelliclassici della divisione e terziarizzazione del lavoro: vengono affidati a societàspecializzate il disegno e la conduzione dello studio, il reclutamento dei medicisperimentatori e dei pazienti, l’analisi statistica dei dati, la stesura dei rapportie degli articoli, eccetera. Nessun ingranaggio della catena dispone di solide for-me di controllo sull’insieme, che resta in mano allo sponsor.
Perditadi rilevanza
Le distorsioni che questa tendenza produce sono oggetto di una ormai ampialetteratura. L’insieme di questi stravolgimenti si possono però riassumere inuna sola espressione sintetica: perdita di rilevanza. Ci si preoccupa di verificaresolo che le molecole sperimentali siano superiori al placebo nel produrre effettimisurabili ma di dubbio impatto sulla salute, anche per il breve periodo di os-servazione. Un esempio al limite del grottesco, a proposito di uno studio sultrattamento della stitichezza cronica, è stato esaminato in un recente editorialedel Bollettino d’Informazione sui Farmaci. Vi si parla apertamente di vendita del-le malattie “per creare unmercato potenziale sufficientemente ampio ai prodottiche verranno in seguito lanciati”.
L’impotenzadell’etica
I grandi trial multicentrici quasi sempre sono costruiti in modo tale da soddi-sfare le esigenze dei produttori dei farmaci in studio, piuttosto che per ri-spondere alle domande che consentirebbero ai medici di usarli al meglio e aimalati di giovarsene. Tuttavia sono ineccepibili sul piano del metodo. Per que-sto motivo fino a oggi il margine di intervento dei singoli comitati è stato qua-si nullo. Ai comitati etici spesso resta solo la scelta secca tra approvare glistudi così come sono o bocciarli in tronco; due opzioni che lasciano immutatoil corso degli eventi: lo sponsor che riceve un rifiuto in un istituto, ne ha moltialtri a cui rivolgersi. Per uscire da questo stato di cose si può individuare unastrategia che consenta una maggior capacità di influenzare a monte il dise-gno degli studi:l scambiare informazioni e pareri tra i diversi comitati chiamati a decidere suargomenti simili;
l coordinare le proprie decisioni;l ottenere la mediazione dei ricercatori presso l’industria.
La debolezzadelle istituzioni
La statunitense Food and Drug Administration (FDA) e la European MedicinesEvaluation Agency (EMEA) dovrebbero porre un argine alla commercializzazio-ne della ricerca. In effetti l’industria, quando disegna gli studi, si attiene scru-polosamente ai requisiti minimi che sono richiesti da queste autorità:l il rispetto di standard di qualità per gli studi;l la dimostrazione di efficacia e di sicurezza per i farmaci sperimentati.La prima richiesta è quasi sempre soddisfatta. Sul secondo punto invecel’azione delle autorità di controllo è carente: si accontentano che un nuovo far-maco esca bene da un confronto con un termine di paragone irrilevante nel rag-giungere un effetto altrettanto irrilevante.
30
Ricerca clinica e marketing

Il controllo della ricercaMa di fatto come è possibile rispettare le regole e contemporaneamente piegarleverso il maggior profitto? I meccanismi sono molteplici e non necessariamenteilleciti. Ecco una sintetica carrellata.
Non pubblicarei risultati
Chi finanzia una ricerca e detiene anche la proprietà dei risultati può essere in-dotto a non pubblicarli qualora portino a conclusioni svantaggiose per il farma-co sperimentato. Paradigmatico il caso di un farmaco per la cura dell’ipotiroidi-smo. La ditta produttrice commissionò uno studio per paragonare l’efficaciadella levotiroxamina con quella di tre farmaci generici. La sperimentazione siconcluse con un verdetto di equivalenza: la ditta riuscì a impedire per alcunianni la pubblicazione dei risultati.
Non terminarelo studio
E’ previsto che un trial venga fermato in caso di manifesta superiorità o inferio-rità della nuova cura. Anche la mancanza di fondi è una giustificazione accetta-ta. Spesso è una via di fuga che copre altri guasti della ricerca. La ditta produt-trice del verapamil, un principio attivo che abbassa la pressione, decise di inter-rompere un trial di confronto tra il suo farmaco e le terapie consolidate, addu-cendo difficoltà economiche. Tuttavia in seguito un’analisi indipendente di-mostrò che, se portato a termine, lo studio avrebbe dimostrato l’inferiorità dellanuova terapia rispetto alla vecchia.
Pubblicarein anticipo
Si sta diffondendo la pratica del ‘parto accelerato’. Perché? Quando la terapia insperimentazione si mostra migliore del controllo, la ricerca viene interrotta («perbenefici», come si usa dire). Questi trial hanno l’effetto di indurre la rapida dis-seminazione di nuovi trattamenti. In realtà chi ha esaminato a fondo questi trialha trovato più di un motivo di perplessità:l in genere non vengono illustrate in modo soddisfacente le informazioni chehanno determinato l’interruzione del trial;
l in molti casi viene raggiunto solo uno degli esiti secondari, meno rilevantidal punto di vista clinico.
La manipolazione dei dati
Spesso le ricerche finanziate dall’industria non hanno lo scopo di dirimereun’incertezza e migliorare la pratica clinica. Piuttosto si vuole dimostrare aogni costo l’efficacia del proprio prodotto. Come? Forzando alcuni strumentimetodologici.Ecco le tre strategie che permettono di raggiungere questi due obiettivi:l non sperimentare;l condizionare il protocollo;l controllare la pubblicazione.
Non sperimentareUn’industria che voglia proporre un farmaco alternativo a quelli già in commer-cio può:l dimostrare la superiorità del farmaco rispetto al placebo;l confrontare l’efficacia del nuovo farmaco con quella degli altri.L’industria è più incline a seguire la prima strada, che permette di mostrare conpoca fatica la maggiore efficacia del principio attivo. Il primo studio comparati-vo, chiamato ALLHAT, è stato invece interamente finanziato da un organismostatale statunitense.
Condizionare il protocolloChi finanzia una ricerca può voler intervenire in alcune fasi per trarre dal proprioinvestimento i maggiori benefici possibili. D’altro canto gli autori dello studio ap-pongono le loro firme per assicurare la scientificità di quanto hanno prodotto. Neltempo i finanziatori delle ricerche hanno trovato alcuni modi per addomesticarela ricerca facendo salva la sua scientificità, facendo scelte ben finalizzate.
Scelta dei pazienti All’inizio di uno studio bisogna scegliere quali individui osservare e quanto nu-merosi. Bastano poche persone ad alto rischio di sviluppare un disturbo per
31
Incertezza e conflitti di interesse in medicina

provare l’efficacia preventiva di un farmaco. Questo conduce a utilizzare farma-ci solo in chi può trarne un reale beneficio. L’industria tuttavia è più propensa adilatare il numero delle prescrizioni. La via più facile per ottenere questo risul-tato è includere nello studio molte persone a basso rischio: è noto che le grossecasistiche garantiscono che anche piccole differenze tra i due gruppi a confron-to risultino statisticamente significative. Un trial teso a dimostrare l’efficaciadella statina nel ridurre gli infarti anche in persone non necessariamente concolesterolo alto, ha dovuto arruolare 20.000 persone.
Scelta di un esitosurrogato
L’esito primario è il vero vantaggio per i pazienti: guarigione di una malattia,prolungamento della sopravvivenza, miglioramento della qualità della vita. Tut-tavia spesso gli studi vengono disegnati con traguardi meno ambiziosi: peresempio la semplice riduzione di un fattore di rischio (colesterolo, pressione, gli-cemia). Sono esiti surrogati. E’ dimostrato che alti livelli di colesterolo nel san-gue alzano il rischio di avere un infarto e una morte precoce. Dimostrata la ca-pacità di un farmaco di abbassare il colesterolo si dà per scontato anche il calodella mortalità, che quindi non viene misurato. L’esempio del clofibrate minaquesta certezza: abbassa il colesterolo e aumenta la mortalità per infarto.Esistono almeno tre situazioni in cui l’esito surrogato non può sostituire ilvero esito:l quando non è un fattore coinvolto nel manifestarsi della malattia;l quando ha un ruolo nella malattia ma il modificarlo non determina alcunmiglioramento;
l quando resta estraneo al meccanismo d’azione della terapia sperimentata.
Scelta di un esitocombinato
Per confondere le carte si può mescolare un esito surrogato con uno primario.Ecco cosa è accaduto nel caso del benazepril, un ACE inibitore sperimentato supersone con sofferenza ai reni. L’esito combinato era la somma di un esito sur-rogato, il raddoppio della creatininemia, che è un semplice fattore di rischio diinsufficienza renale, e il ricorso alla dialisi, il vero esito. Il 20 per cento dei pa-zienti trattati con placebo ha avuto il raddoppio della creatinine mia o la neces-sità di ricorrere alla dialisi, contro il 10 per cento di chi assumeva il farmaco. Illettore è indotto a credere che il trattamento dimezzi sia la creatininemia sia ilricorso alla dialisi, in realtà scorporando i dati si scopre che il vantaggio del far-maco sul placebo è dato solo dall’esito surrogato.
Sceltadi un gruppodi controllo
Quando un nuovo principio attivo viene ad aggiungersi ad altri già presenti sulmercato e di provata efficacia, si vorrebbe sapere se la novità dà qualcosa in più.Si dovrebbe quindi condurre una sperimentazione confrontando il principio at-tivo più simile. Spesso invece, l’industria produttrice del farmaco sfidante sce-glie opportunamente il campione. E’ il caso di irbesartan, della classe dei sarta-ni: farmaci contro l’ipertensione che hanno un effetto protettivo sui reni. Al mo-mento della sperimentazione esistevano in commercio altri sartani, ma la capa-cità nefroprotettiva di irbesartan fu confrontata con amlodipina, che notoria-mente non ha alcun effetto sulla funzione renale.In altri casi gli sperimentatori preferiscono agire sul bilancino dei dosaggi: siaumenta o diminuisce il farmaco di confronto a seconda che si voglia dimostra-re la sua minor efficacia o tollerabilità.
Scelta di studiarela non inferiorità
Si può ottenere l’approvazione di un farmaco dimostrando che non è inferiorealle alternative esistenti. Questa scelta potrebbe essere eticamente accettabilese il nuovo farmaco fosse più tollerabile, più facile da somministrare o meno co-stoso. In caso contrario non c’è motivo di abbandonare una cura di provata effi-cacia per una che dichiara in partenza di non essere superiore e non esclude dipoter essere peggiore.
Scelta di comecondurrela ricerca
Esistono due modi per condurre una ricerca: secondo l’analisi dell’efficacia delfarmaco (drug efficacy analysis) oppure secondo l’intenzione di curare (inten-tion to treat), ritenuto più corretto. Questo è un tipo di analisi dei risultati in cuinon vengono valutati solo coloro che hanno effettivamente assunto il tratta-mento, ma tutti coloro che sono stati assegnati a quel tipo di trattamento. I sog-getti possono interrompere precocemente un trattamento o a causa degli effetti
32
Incertezza e conflitti di interesse in medicina

collaterali oppure a causa della malattia che può aver assunto un andamentopiù grave. Non tener conto di questi risultati, potrebbe portare a conclusioni er-rate. Spesso l’arruolamento dei pazienti è escogitato in modo da rendere soloformale l’adozione dell’intention to treat: viene fatto precedere da un periodochiamato run in nel quale tutti i pazienti assumono il nuovo farmaco e alla finedel quale vengono esclusi quelli che non lo hanno tollerato.
Sceltadi svolgere analisiper sottogruppi
Quando però un trattamento non si è rivelato particolarmente efficace i ricerca-tori possono provare a riunire la popolazione osservata in sottogruppi che sa-ranno scomposti e ricomposti secondo determinate caratteristiche cliniche fin-ché non se ne trova uno in cui emerge una differenza statisticamente significati-va a favore del farmaco.Il difetto è duplice:l si individua un gruppo di persone con una caratteristica clinica non rilevan-te (per esempio un farmaco potrà essere indicato solo per persone di etàcompresa tra 50 e 60 anni);
l anche in assenza di un risultato statisticamente significativo sulla globalitàdella popolazione, l’analisi per sottogruppi fa emergere differenze statistica-mente significative che possono essere dovute unicamente al caso.
Sceltadi presentare i dati
Il modo di comunicare i dati può in qualche modo influenzarne la corretta inter-pretazione. In particolare per enfatizzare il successo di una cura si esprimono ivantaggi in termini relativi e gli effetti collaterali in termini assoluti. Se nel grup-po sperimentale si osserva una mortalità dell’1 per cento e nel gruppo di con-trollo del 2 per cento, si dovrebbe affermare che il farmaco ha ridotto la mortali-tà dell’1 per cento. Tuttavia è più vantaggioso dichiarare che il farmaco ha di-mezzato la mortalità. Vale il contrario se invece della mortalità ci fossero gli ef-fetti collaterali.
Controllare la pubblicazioneA ricerca conclusa è possibile modulare l’impatto dell’informazione facilitandola diffusione dei risultati positivi e ostacolando quella dei dati negativi, anche inquesto caso con una scelta oculata.
Non inviarel’articoloa una rivista
Per le riviste scientifiche non vale la stessa regola che detta la scaletta dei quoti-diani: un risultato cattivo non fa notizia, quindi non vale la pena di pubblicarlo.Ma nel momento in cui si valutano i risultati non possono mancare dal comples-so i dati negativi: altrimenti si ottiene un risultato sbilanciato a favore della cura.
Pubblicareun lavoro negativosu una rivistaminore
Le riviste non sono tutte uguali: la loro importanza viene codificata dal numerodi volte che un articolo viene citato da altri autori (impact factor). Si farà di tuttoquindi per pubblicare un articolo positivo su una rivista con alto impact factor,mentre ci si accontenterà di una rivista destinata a rimanere intonsa sugli scaf-fali per articoli negativi. Nel 1988 uno studio che dimostrava l’inefficacia dellanifedipina nel ridurre la mortalità in persone con infarto venne pubblicatasull’EuropeanHeart Journal (che era all’epoca di media importanza – impact fac-tor inferiore a 3). La rivista di punta del settore, Circulation, aveva a quel tempoun’importanza molto maggiore (impact factor superiore a 6). La nifedipina fuprescritta normalmente fino al 1995, quando il National Heart, Lung and BloodInstitute emise un comunicato in cui se ne sconsigliava l’uso. Cosa era succes-so? Pochi giorni prima erano state pubblicate due ricerche su JAMA e su Circu-lation sfavorevoli alla nifedipina.
Ritardarela pubblicazionedi risultatinon favorevoli
Se non si può nascondere il dato negativo di una ricerca se ne può ritardare lapubblicazione. Nel novembre del 1997 vennero pubblicati i dati di uno studioconcluso pochi mesi prima da cui emergeva l’utilità del defibrillatore. La ricercache invece ha dimostrato la sua inefficacia nel ridurre la mortalità si conclusenel marzo del 1998 ma fu pubblicata ad agosto 2000.
Anticiparela diffusionedei risultatifavorevoli
Conclusa una ricerca si possono avere inmano dati positivi che sono destinati agiacere per qualche mese prima di essere pubblicati. Questo periodo di tempoviene percepito come mancato guadagno. Perciò sono sempre più frequenti eraffinate le iniziative che anticipano questi risultati in forma ufficiosa: simposi
33
Incertezza e conflitti di interesse in medicina

in cui vengono presentati i dati in via preliminare; opportune anticipazioni gior-nalistiche. Nessuno si preoccupa di seguire il destino di questi dati: spesso laloro revisione rileva errori o difetti che negano la dignità di pubblicazione.
Gli antidoti contro le deviazioni della ricercaLe strategie per correggere le deviazioni della ricerca sono due: una consiste nelribattere colpo su colpo raffinando la propria capacità di leggere uno studio e fa-cendo emergere i difetti e stilare quindi una lista di possibili trucchi. Tuttavia senon si vuole che la lista si allunghi con le contromosse di ricerca bisogna:l rendere esplicito il conflitto di interessi dei ricercatoril rendere di proprietà pubblica i dati della ricercal rendere obbligatoria la registrazione dei trial in registri pubblicil criticare l’informazionel mutare il panorama culturale
Rendere esplicitoil conflitto
Rendere esplicito il conflitto di interessi dei ricercatori coinvolti invia al lettore ilmessaggio “leggere con particolare attenzione”. La dichiarazione del conflitto diinteressi è ormai pratica diffusa e i dati sembrano confermare che in effetti rag-giunge lo scopo: i lettori sono più critici e attenti e modulano il giudizio anche inbase al tipo di conflitto (per esempio finanziamento dell’intera ricerca o solo bor-se di studio).
Rendere pubblicii dati della ricerca
La proprietà dei dati è questione dibattuta. Chi paga la ricerca è padrone dei da-ti? L’industria che finanzia una ricerca spesso inserisce nel contratto stipulatocon i ricercatori il diritto di veto sulla pubblicazione dei risultati. Un fatto cheassume maggior gravità quando si prende atto che spesso gli sponsor:l raccolgono e analizzano direttamente i dati degli studi;l si arrogano il diritto di sospendere lo studio in qualunque momento dellaconduzione dello stesso;
l sostengono studi nei quali non esiste neppure un comitato indipendenteche decide come condurre e se pubblicare lo studio.
Questa consuetudine riposa su una questione mai risolta: la confusione tra laproprietà dei dati e la loro disponibilità.
I trial devonoessere registrati
Solo metà delle ricerche condotte è rintracciabile. Da un lato dalle banche datidella letteratura scientifica affiora solo una parte delle ricerche pubblicate. Dal-l’altro esistono comunque molte ricerche non pubblicate. Per rendere rintraccia-bile qualsiasi ricerca, e quindi poterne chiedere conto, appare dunque necessariala sua iscrizione in un registro pubblico. Anche il ruolo del comitato etico è fonda-mentale: il suo compito non si esaurisce con l’approvazione di un protocollo macon il controllo del suo rispetto fino alla corretta pubblicazione dei risultati.
L’informazioneva criticata
Anche i giornalisti che divulgano le notizie scientifiche in campo medico sonosoggetti a conflitti di interessi e, qualora non esercitino la loro funzione critica,finiscono per fare semplicemente da cassa di risonanza delle ditte farmaceuti-che (vedi il capitolo L’informazione in medicina: come destreggiarsi).
Mutareil panoramaculturale
C’è chi dubita che le soluzioni sinora proposte cambino davvero un corso dellecose determinato da così potenti interessi economici. Sono quindi indispensa-bili cambiamenti radicali che risolvano a monte i difetti del sistema vigente. E’necessario pensare a:l pubblicare le ricerche on line e lasciare alle riviste cartacee solo la loro anali-si critica;
l aprire nuovi canali di finanziamento per la ricerca indipendente su temi rile-vanti per la salute pubblica;
l imporre per i nuovi farmaci il confronto con i trattamenti già disponibili.
Definizione di malattia
Le malattie non sono entità naturali e sfuggono a ogni tentativo di definizionesu basi puramente empiriche. Sino a nonmolto tempo fa il giudizio clinico veni-va lasciato per lo più al diretto interessato, il quale con il gesto di rivolgersi a un
34
Incertezza e conflitti di interesse in medicina

medico per chiedere aiuto decretava la sua condizione di malato. Oggi invecesono i medici a stilare repertori di condizioni da considerare malattie e a definir-ne i confini e i criteri per riconoscerle prima che l’interessato avverta qualsiasidisturbo. In questo modo il riconoscimento delle malattie è divenuta un’attivitàcompletamente autoreferenziale interna la mondo medico.
Il piano inclinato: ABC della medicalizzazioneNon vi sono soglie identificabili su base oggettiva che segnino il passaggio dalnormale al patologico. Esistono due riferimenti:la sofferenza del diretto interessato, quando compare (criterio clinico);la capacità della medicina di modificare favorevolmente il corso delle cose con isuoi interventi (criterio preclinico).In futuro, qualsiasi condizione umana potrà essere considerata malattia, pur-ché la medicina trovi qualche intervento che dimostri di poterne modificarevantaggiosamente la storia naturale.Questo fenomeno può essere rappresentato graficamente su un piano cartesia-no: un asse è qualitativo, e rappresenta il gradiente tra ciò che è di pertinenzamedica e ciò che non lo è; l’altro è quantitativo e indica la transizione tra ciò cheè normale e ciò che è patologico (figura 1). Nella figura l’ambito di condizioniumane a cui si applica la medicina è rappresentato dall’area in alto a destra,mentre lo spazio in basso a sinistra è quello estraneo agli interventi medici. Perogni aspetto fisico, psichico o sociale (per esempio: densità dei capelli, glicemia,eccetera) si può immaginare un confine che separa i due campi. Nella figura sipropone per la demarcazione un andamento diagonale a forma di S, per tenereconto delle condizioni estreme: anche un aspetto che non è abitualmente di in-teresse per la medicina lo diventa quando si presenta in termini fortemente de-viati dalla norma; viceversa anche un parametro tipicamente pertinente allamedicina, quando è assolutamente normale, non richiede interventi.
FIGURA 1: Il piano della medicalizzazione
La zona di confine è ovviamente sfumata, ma soprattutto è soggetta nel tempoalle già accennate variazioni (indicate dalla freccia: l’area che la racchiude rap-presenta gli ambiti di prossima acquisizione), come risultante delle due compo-nenti (illustrate dalle linee tratteggiate verticale e orizzontale).
Da “non medico” a “medico”Sull’asse qualitativo, si tenderà sempre più ad attribuire l’etichetta di malattia avarianti (anatomiche o fisiologiche) sostanzialmente innocue o di scarsa rile-vanza per la salute. Per queste conversioni, che allargano i confini di ciò che è dipertinenza della medicina (la linea verticale nella figura 1 si sposta a sinistra,verso la posizione tratteggiata), è stato proposto il termine di “non malattie”.
35
Incertezza e conflitti di interesse in medicina
normale
non medico medico
patologico

Il processo attraverso cui una qualsiasi condizione esistenziale, più o meno co-mune, si trasforma in una forma patologica, che giustifichi l’intervento medico,culmina in molti casi nella creazione di un neologismo (come disfunzione eretti-le, fobia sociale, micromastia). Il nuovo termine trova poi una collocazione nellanosologia ufficiale. In tutti i casi è necessaria un’azione coordinata di panel diesperti e di opinion leader del settore.Al proposito, il British Medical Journal ha proposto in tono semiserio una classi-ficazione internazionale delle non malattie (international classification of non-disease), nella quale, in base anche alle segnalazioni dei lettori, sono state in-cluse oltre 200 condizioni. L‘estensione del fenomeno non deve stupire, poichécomprende senz’altro:l gli effetti dell’età;l le peculiarità individuali;l le difficoltà quotidiane;l l’estesa gamma di alterazioni anatomiche o funzionali.
Abbassamento della sogliaPer ogni condizione di pertinenzamedica, si registra una tendenza generale, de-stinata a proseguire nel prossimo futuro, ad allargare sul piano quantitativol’ambito di ciò che viene considerato patologico, a svantaggio della normalità. Lasoglia corrispondente è indicata nella figura 1 dalla linea orizzontale, che sisposta verso il basso, nella posizione indicata dalla linea tratteggiata. I più rile-vanti esempi di questa tendenza riguardano i livelli di glicemia, di pressione ar-teriosa, colesterolo e l’osteoporosi.
Glicemia Alla fine del 1997 una commissione di specialisti, promossa dall’American Dia-betes Association, ha deciso di ridurre il limite di zuccheri nel sangue a digiunooltre il quale si pone la diagnosi di diabete da 140 (soglia fissata nel 1979 da unaltro simile comitato: in precedenza era 160) a 126 milligrammi di glucosio perdecilitro di sangue. Per effetto di questa decisione, in tutto il mondo milioni dipersone che sino ad allora erano considerate sane (essendo prive di disturbi e didanni rilevabili agli organi) hanno cominciato a ricevere l’etichetta di una ma-lattia come il diabete.
Pressionearteriosa
Nella primavera del 2003 il Joint National Committee on Prevention, Detection,Evaluation and Treatment of High Blood Pressure, un comitato governativo sta-tunitense, ha ridotto il limite di normalità a 120 su 80mm di mercurio; i valori dipoco superiori si definiscono col neologismo di pre ipertensione e richiedonoprovvedimenti non farmacologici, ma di stile di vita (esercizio fisico, riduzione delpeso e del sale nei cibi). Si noti che in precedenza si considerava normale unapressione sino a 140 su 90,mentre trenta anni fa il limite arrivava a 160 su 95.
Colesterolo Nell’estate del 2004 un comitato di esperti del National Cholesterol EducationProgram ha emanato la terza edizione degli standard per il tasso di colesterolonel sangue, che aggiornano le linee guida emanate solo tre anni prima.Le novità maggiori sono due. Si riduce a 100 mg/dL la soglia di intervento per ilcolesterolo LDL nei pazienti con rischio ischemico superiore al 20 per cento e siabbassa a 70 mg/dL l’obiettivo da raggiungere. I due valori erano rispettiva-mente 130 e 100 nelle precedenti raccomandazioni. Secondo il coordinatore delcomitato James Cleeman, questa variazione significa che circa 43 milioni diamericani dovrebbero assumere farmaci contro il colesterolo, in particolare sta-tine. Con le linee guida del 2001 si stimava che i potenziali consumatori di stati-ne fossero 36 milioni, con quelle del 1993 circa 12 milioni.
Osteoporosi Gli esperti sanno bene che non esiste alcuna soglia al di sotto della quale il ri-schio di osteoporosi si annulla. E sanno anche che la maggioranza dei soggettiche si ammalano proviene proprio da gruppi con bassi valori del fattore di ri-schio, per la semplice ragione che sono più numerosi in assoluto, pur avendouna minor probabilità di ammalarsi. Puntualmente, anche questa seconda os-servazione viene spesso avanzata come argomento per sostenere la necessità diuna riduzione della soglia, come è avvenuto per esempio recentemente a propo-sito del rischio di fratture in rapporto alla densità dell’osso (vedi figura 2).
36
Incertezza e conflitti di interesse in medicina

FIGURA 2: Rischio di frattura e numero di fratture in funzione della densità minerale ossea(BMD), espressa come deviazione standard dalla media delle donne giovani (Siris ES et al. ArchIntern Med 2004;164:1108)
In fin dei conti quando è utile abbassare una soglia? Per stabilirlo si dovrebberovalutare queste variabili:l anni di vita salvati da un intervento a soglia inferiorel morbilità evitata da un intervento precocel effetti psicologici, sociali e di altra natura provocati da una precoce etichettadi malattia
l morbilità indotta dai trattamenti anticipatil anni di vita persi per effetti iatrogeni.E’ una valutazione nella maggior parte dei casi quasi impossibile, perché richie-derebbe misure accurate dei vantaggi (prove di efficacia, in termini di prolunga-mento della vita e di riduzione della morbilità) e dei danni, misure che non sem-pre sono disponibili o ottenibili per tutti i possibili effetti.In realtà è ben noto che la frequenza e l’entità dei benefici prevedibili si riduconocon l’abbassamento della soglia (in quanto si riduce il rischio di base), mentrequelle dei possibili danni restano costanti: in teoria la soglia ottimale rappre-senta semplicemente il punto di incrocio tra le due linee (vedi figura 3).
FIGURA 3: Benefici ed effetti negativi delle cure in funzione della soglia di intervento (Dirindin N,Vineis P. In buona salute. Torino: Einaudi, 2004.)
37
Incertezza e conflitti di interesse in medicina
Tassodifratture
per1.000persone-anno
Numero
didonneconfr a
tture
Tasso frattureNumero di donne con fratture
Distribuzione BMD
BMD
>1,0 da 1,0a 0,5
da 0,5a 0,0
da 0,0a 0,5-
da -
-
0,5
a 1,0
da -
-
1,0
a 1,5
da -
-
1,5
a 2,0
da -
-
2,0
a 2,5
da -
-
2,5
a 3,0
da 3-
-
,0
a 3,5
<-3,5
50
45
40
35
30
25
20
15
10
5
0
450
400
350
300
250
200
150
100
50
0
Effettisullasalute
NNT
beneficio
danno

In pratica risulterebbe impossibile risolvere la suddetta equazione (e costruire ilgrafico della figura 3), perché la somma algebrica dovrebbe essere fatta tra enti-tà non commensurabili (le due rette del grafico non si incontrano mai, perchésono su due piani diversi: cioè non si può stabilire una soglia al di sotto dellaquale si hanno più danni che benefici e al di sopra della quale accade viceversa).Sul piatto della bilancia infatti vanno messi anche aspetti non direttamentepertinenti la salute, che vengono invece sistematicamente trascurati: spessonon vengono valutati i costi sociali di una diagnosi. Quando un medico dice aqualcuno “lei è malato” o “lei ha bisogno di cure”, cambia la realtà sociale attor-no a quell’individuo. Non è solo una questione di costi sanitari: l’etichetta diunamalattia che dura per tutta la vita cambia in maniera irreversibile ogni rap-porto, anche i più intimi: con i figli, con il coniuge e persino con se stessi.Infine, ma non meno decisiva, vi è l’impossibilità di confrontare i dati su baseoggettiva. La comparazione può essere svolta solo in rapporto a preferenze egiudizi di valore prettamente soggettivi. Infatti gli esiti in questione:l appartengono a domini qualitativamente diversi, per esempio morte rispettoa condizioni di benessere o malessere;
l si distribuiscono su soggetti diversi, rendendo ancora più disparata la valu-tazione: spesso coloro che subiscono effetti negativi non sono neppure sfio-rati dai benefici e viceversa.
Come è possibile stabilire, per esempio, se un anno di vita salvato valga più omeno di 10 anni di sofferenze inutili, soprattutto se i due termini del confrontoriguardano persone diverse?
Anticipazione della diagnosiPer ogni condizione di pertinenza medica, soprattutto in campo oncologico manon solo, si registra una tendenza ad anticipare sull’asse temporale il momentodel riconoscimento, e di conseguenza dell’intervento. E’ un fenomeno stretta-mente connesso a quello dell’abbassamento della soglia, anche se complessiva-mente meglio noto e analizzato, soprattutto nella sua forma più organizzata: loscreening. Esso si fonda sulla convinzione, all’apparenza semplice e convincen-te, che riconoscere una malattia in fase precoce sia sempre un bene, in quantoconsentirebbe maggiori possibilità di cura. In realtà, sulla base delle conoscen-ze disponibili, nella granmole degli interventi di screening (o di controlli periodi-ci) che vengono da varie fonti proposti, è possibile distinguere pochi interventidi provata efficacia, almeno in termini di riduzione della mortalità specifica perla malattia in questione. Anche in questo caso favorevole, manca per lo più unavalutazione degli effetti sulla mortalità generale, quasi impossibile da realizzare(vedi per esempio le polemiche persistenti sullo screeningmammografico ); e so-prattutto manca una stima esauriente degli esiti negativi; anche quando questisono ben noti, non si apprezza la necessità di considerarli, con l’apporto decisi-vo delle preferenze del paziente.
I casi di prostatae polmone
Il ricorso al dosaggio dell’antigene specifico per la prostata (PSA) in soggettiasintomatici si è diffuso estesamente negli ultimi dieci anni nei paesi industria-li, portando con sé un’apparente epidemia di incidenza del tumore, in assenzadi qualsiasi variazione nella mortalità specifica. Per quanto riguarda lo scree-ning del cancro al polmone, dopo una battuta d’arresto seguita all’esito negati-vo di alcuni trial negli anni settanta, si sta assistendo a una ripresa, con la dif-fusione di nuove tecnologie non ancora valutate criticamente (TAC spirale, PET,diagnosi genetica) che rischiano nel prossimo futuro di produrre un impatto si-mile a quello descritto per la prostata.
Aumento del numero necessario da trattare (o da testare)Abbassamento della soglia e anticipazione della diagnosi costituiscono in ulti-ma analisi un incremento della sensibilità diagnostico-terapeutica, intesa insenso lato, a discapito della specificità. Come è noto, questo comporta due per-cezioni illusorie:l un’apparente maggior diffusione della malattia;l un apparente miglioramento della prognosi.
38
Incertezza e conflitti di interesse in medicina

Il risultato finale si traduce in un maggior incentivo a diagnosticare e trattare lamalattia in questione, in un circolo vizioso che è stato definito “ciclo di interven-to crescente” (vedi figura 4).
FIGURA 4: Ciclo di intervento crescente (Black WC et al. New Engl J Med 1993;328:1237)
L’equivalenza tra anticipazione della diagnosi e abbassamento della soglia puòrisultare più chiara se le valutazioni si esprimono in termini di “numero neces-sario da trattare” (NNT). Come è noto, il “numero necessario da trattare” (NNT) èuna misura dell’efficacia di un intervento e rappresenta il numero di pazientiche si devono sottoporre a un certo intervento per ottenere un successo in più(per esempio una morte evitata) rispetto al trattamento di confronto.Il termine NNT può essere applicato a qualsiasi intervento medico, sia esso lasomministrazione di un esame, di un farmaco o un insieme complesso di azioni,che configurino un ciclo di intervento. Quest’ultimo si può definire come l’insie-me di attività che prende le mosse da una indagine e si svolge poi in una catenapiù omeno lunga di interventi diagnostici, terapeutici e di altro genere, concate-nati tra loro (vedi figura 5).
FIGURA 5: La cascata clinica: un esempio
Riferirsi all’intero ciclo di intervento significa scegliere per esempio il nume-ro di morti evitate per cancro alla prostata come parametro per valutarel’NNT della prescrizione di un esame di dosaggio dell’antigene specifico perla prostata (PSA) a un uomo di 50 anni senza sintomi. Oppure il numero didecessi evitati per valutare l’NNT di una diagnosi di diabete formulata allasoglia di glicemia a digiuno di 126 mg/dL. Interpretati in questo modo, ab-bassamento della soglia e anticipazione della diagnosi non rappresentanoaltro che un aumento del numero di soggetti da trattare per ottenere un sin-golo esito positivo.
39
Incertezza e conflitti di interesse in medicina
Più esami
Più diagnosi Più pseudo malattie
Miglioramento prognosiAumento incidenza
L’interventoè efficace
La malattiaè più diffusa!
Antigene prostatico specifico (PSA)in cinquantenne senza disturbi
BiopsiaEcografia
Farmaci, presidi, visite, controlli, esami...
Chirurgia
Impotenza, incontinenza

Nella figura 6, essi sono entrambi rappresentati da un progressivo spostamen-to verso sinistra della soglia di intervento per evitare il verificarsi del singoloevento, raffigurato dal vertice del triangolo sulla destra. Questo spostamentocomporta anche un sicuro incremento assoluto della iatrogenesi.
FIGURA 6: Anticipazione della diagnosi e abbassamento della soglia aumentano entrambe l’NNT
Per inciso, la tendenza verso alti NNT rappresenta un mutamento nel rapportotra curanti e pazienti che non è stato mai sufficientemente messo a fuoco. Persecoli, il “numero da trattare” è stato implicitamente considerato sempre ugualea uno. Il medico dava una cura a un malato per guarire o alleviare i mali di quelmalato; punto e basta. E nella percezione generale (anche per la maggioranzadei medici) le cose sembrano stare ancora così, ma non lo sono. Prendiamo ilcaso del glaucoma. Chi assume farmaci per abbassare la pressione nei bulbidegli occhi ritiene che quell’intervento serva per salvare la sua vista. In realtà, ilsuomedico (a sua volta non del tutto consapevole di ciò) dovrebbe prescrivere lastessa cura a numerose persone nelle stesse condizioni, per ottenere un miglio-ramento di salute in una sola.Ovviamente ancora oggi molti interventi medici hanno un NNT pari o vicino a 1:sono i trattamenti sintomatici e quelli curativi nel senso proprio del termine.Quelli invece con un NNT molto maggiore di 1 si possono genericamente consi-derare come interventi preventivi o preclinici.
I panel che definiscono le malattieUn fiume di denaro alimenta e indirizza le attività degli specialisti (raccolti inpanel, gruppi, società eccetera) che a livello internazionale stabiliscono le defi-nizioni, i criteri diagnostici, le soglie quantitative e qualitative per malattie, sin-dromi, fattori di rischio e condizioni varie.Un esempio di attualità in campo neurologico è la sindrome delle gambe senzariposo (RLS), la cui definizione, adottata internazionalmente nella ricerca, nellapratica e nella valutazione della frequenza del disturbo è frutto di un Internatio-nal Restless Legs Syndrome Study Group, strettamente collegato alla RestlessLegs Syndrome Foundation che annovera come gold sponsor (più di 250 miladollari l’anno) le aziende che producono farmaci per la sindrome.Le strategie per inculcare nella mente di medici e pazienti che una nuova sin-drome è uno “stato di malattia a sé stante, rilevante e frequente" sono ben de-scritte in letteratura:l la prima mossa, a livello locale, consiste nel costituire un “advisory board”,nel quale figurino anche opinion leader riconosciuti;
l poi si passa a sviluppare “linee guida di buona pratica”, a diffondere tra imedici una newsletter, ad avviare un programma di “sostegno per i pazien-ti”, a promuovere associazioni di malati, sino a convincere tutti che la sin-drome in questione è una “malattia seria e credibile”.
All’origine di tutto c’è la concettualizzazione di una condizione umana comemalattia, e la sua definizione in termini che siano compatibili poi con tutte le at-tività necessarie per creare il mercato di uno o più blockbuster (farmaci chevendono almeno per unmiliardo di dollari l’anno), a partire dalla ricerca clinica.
Campagne di consapevolezza sulle malattie“Si possono fare molti soldi dicendo alle persone sane che sono malate”. Conquesto memorabile incipit si apre un articolo pubblicato sul British Medical Jo-
40
Incertezza e conflitti di interesse in medicina
infermitào mortepreclinica: NNT clinica

urnal riguardo l’attività del mercato di malattie (disease mongering), la formapiù spinta e sottile di medicalizzazione promossa dall’industria della salute.L’azione mirata che ogni singolo elemento del sistema mette in opera per pro-muovere i propri prodotti e servizi specifici è solo l’ultima parte di una rete arti-colata di marketing che, partendo da lontano, si propone in generale di amplifi-care l’importanza (per diffusione, gravità, implicazioni economiche e sociali ec-cetera) di una malattia con lo scopo di reclutare pazienti, moltiplicare la presta-zioni, potenziare le strutture, sviluppare l’attività.
Linguaggio e messaggi
Atene, Grecia, 19 Settembre 2005 – Assemblea Annuale del-la European Federation of Neurological Societies (EFNS –Federazione Europea delle Società di Neurologia).“La Sindrome delle Gambe senza Riposo (RLS) è una pa-tologia incredibilmente comunema sottodiagnosticata, cheincide negativamente sulla vita di milioni di persone in tut-to il mondo”, ha spiegato il Professor Dr. Wolfgang, MD,Oertel, Direttore del Dipartimento di Neurologia, Centroper le Malattie Nervose dell’Università Philipps di Marbur-go, Germania.La sindrome delle gambe senza riposo è una delle patolo-
gie neurologiche più comuni al mondo, ma curabili. Si cal-cola che ne sia affetta una persona su 10 di età compresatra i 30 e i 79 anni.Le persone affette da RLS ritengono spesso che la loro pa-tologia possa avere gravi ripercussioni sulle attività quoti-diane e sulla vita nel suo complesso.“I dati presentati oggi sono molto importanti perché raf-forzano le prove a favore dell’effetto benefico di pramipe-xolo, non soltanto per quanto riguarda il sollievo dai sinto-mi principali della patologia, ma anche per quanto riguar-da il miglioramento della qualità della vita dei pazienti”.
Il testo riportato nel box è un comunicato stampa, diffuso nel corso di un recen-te convegno scientifico e ripreso in vario modo da diversi organi d’informazione.Per la sua tipicità si presta bene a essere usato come esempio da analizzare percomprendere il linguaggio delle campagne di sensibilizzazione sulle malattie ecome il messaggio promozionale si articola comunemente.Comunicati stampa di questo tipo si articolano spesso secondo uno schemadettato dalle regole del marketing e adatto a colpire l’attenzione del lettore.
Dare i numeri Il punto di partenza è la quantità di persone colpite da una certa malattia o con-dizione. L’ordine di grandezza è in genere di molti milioni (in Italia, in Europa,nel mondo) e viene citato subito per colpire l’attenzione, ma i dati sono spessoincontrollati e incontrollabili. La maggior parte di questo popolo di infermi igno-ra il suo male.
Suscitare timori Dopo aver gonfiato diffusione e frequenza del male, se ne sottolinea la gravità, intermini di effetti negativi su salute, benessere, economia, lavoro, relazioni so-ciali, eccetera.
Indurre a visiteed esami
Nel 1996 lo slogan di una campagna di sensibilizzazione recitava: «I sintomi abi-tuali dell’epatite sono molto diffusi: aspetto sano, appetito normale, assenza didolore. Se ti senti bene fai le analisi del sangue». Si avvia così la cascata clinica:esami alterati in persone che si ritenevano sane, assegnazione di una etichettadi malattia, interventi medici, complicazioni, effetti collaterali, altri esami, nuo-ve terapie.
Banalizzarela soluzione
Dopo aver ingigantito i rischi che corre chi nasconde la testa sotto la sabbia, siconclude rassicurando: niente paura, c’è giusto una pillola che risolve tutto.
Attori, sponsor e intermediari
Medici e società scientificheI protagonisti delle campagne di sensibilizzazione sono in genere i medici or-ganizzati nelle loro associazioni scientifiche, che svolgono così un ruolo atti-vo di prevenzione e promozione della salute, anziché limitarsi a quello passi-vo di curare i già malati. L’atteggiamento non è privo di implicazioni negative:quando i medici prendono l’iniziativa di intervenire (anche solo con l’infor-mazione) su cittadini sani, o che si ritengono tali, dovrebbero disporre di pro-ve certe circa i benefici attesi e l’assenza di possibili danni, per superarel’inevitabile conflitto d’interessi legato all’espansione della propria attivitàprofessionale.
41
Incertezza e conflitti di interesse in medicina

Cittadini, pazienti e loro associazioniGli strateghi del marketing si sono resi conto che sovvenzionare gli specialisti e irappresentanti dei malati rende, in termini di sviluppo del business e di recluta-mento di nuovi pazienti. Accanto ai medici, in queste campagne vengono spessoschierati anche i pazienti, le cui associazioni assumono la funzione di gruppi dipressione con lo scopo di richiamare una maggiore attenzione sulla specificamalattia di cui soffrono. Anche i rappresentanti dei malati hanno un interesse,personale e d’associazione, a ottenere il riconoscimento della società civile oltrea un impegno anche economico per la patologia di cui sono vittime e paladini. Aciò si aggiungono la loro frequente subalternità culturale ai medici specialisti ela carenza di fonti di informazione indipendenti, col risultato della trasformazio-ne anche di questi soggetti sociali in strumenti della medicalizzazione, più omeno inconsapevoli.Tra i casi segnalati di attività promozionali condotte da istituzioni apparente-mente indipendenti, di cittadini e malati, ma in realtà finanziate dall’industriavi sono, per esempio:l la campagna a favore della diagnosi precoce della broncopneumopatia cro-nica ostruttiva (BPCO) condotta in oltre 80 paesi dal “Progetto mondialeGOLD”;
l la pressione per una più ampia prescrizione di nuovi farmaci per l’Alzheimer(inibitori dell’acetilcolinesterasi ) esercitata dall’Associazione italiana malat-tia di Alzheimer.
Alcune federazioni di associazioni stanno elaborando raccomandazioni su comegestire i rapporti con l’industria farmaceutica. Un esempio sono le linee guidaelaborate dalla britannica Long Term Medical Conditions Alliance, consultabilenel sito dell’organizzazione [www.lmca.org.uk/docs/pharmgds.htm].
Aziende produttrici di farmaci o di altri beni sanitariLe campagne sanitarie sono programmi complessi e molto costosi e quasi sem-pre i fondi provengono dall’industria, in particolare farmaceutica. Ciò non sem-pre è evidente: gli sponsor preferiscono apparire il meno possibile, perché laprovenienza dei finanziamenti non diffonda un’ombra di sospetto sui messaggidella campagna. Spesso lo stesso sponsor finanzia diverse iniziative parziali,messe in opera da varie società scientifiche o associazioni laiche, nel quadro diun più vasto piano promozionale della cui complessità gli organizzatori dei sin-goli eventi non sono consapevoli. In alcune occasioni, soprattutto quando chipaga è una singola azienda, l’investimento si accompagna alla pretesa di rag-giungere il richiamo diretto a un preciso prodotto. Più spesso, però, le campa-gne puntano solo a creare un ambiente culturale genericamente favorevole a di-versi prodotti.In generale le aziende commerciali vedono con tale favore il fatto che i rappre-sentanti dei pazienti (insieme ai medici specialisti) si attivino per chiedere che“si spenda di più” per una certa malattia o condizione, da intervenire semprepiù a monte, spesso favorendo addirittura la nascita di nuove associazioni o laloro fusione in organismi più ampi. Per esempio gli sforzi per costituire un coor-dinamento internazionale delle associazioni di malati, che dal 1999 si sono con-cretizzati nella creazione di International Alliance of Patient’s Organisations[www.patientsorganisations.org], sono sempre stati finanziati dall’industriafarmaceutica, già dalle riunioni preliminari.
Agenzie di pubbliche relazioniLe compagnie di pubbliche relazioni agendo come intermediari tra gli sponsor(che mettono a disposizione i finanziamenti) e le associazioni di medici e malati(che mettono in gioco la loro autorevolezza e credibilità), frappongono uno scher-mo tra i contenuti del messaggio e gli interessi commerciali che ne sono la fonte.
Mass mediaI mezzi di comunicazione di massa rappresentano la cassa di risonanza di tuttele iniziative promozionali in campo medico. Sono indotti a ciò da una naturaleaccondiscendenza nei confronti di chi detiene qualunque forma di potere, sia
42
Incertezza e conflitti di interesse in medicina

esso economico, accademico o politico, e dalla concezione trionfalistica e iper-bolica della medicina e della tecnologia. Queste stigmate “culturali” spiegano iprincipali difetti del giornalismo medico, che assai spesso viene meno al doveredi esercitare una lettura critica delle informazioni di cui si occupa.Ecco i principali vizi correnti dei giornalisti che si occupano di salute:l limitarsi ai pareri di espertil trattare gli specialisti come tuttologil confondere la fantasia con i fattil farsi ingannare dai numeril prendere gli aneddoti come provel leggere acriticamente i risultati degli studil estrapolare i risultati di uno studio clinico: chi legge può avere caratteristi-che ben diverse dai “pazienti medi” analizzati nello studio
l enfatizzare la rilevanza clinical confondere fattori di rischio con malattiel presentare i rischi in modo ingannevole
Strumenti e canali
Poiché le campagne di sensibilizzazione sulle malattie divengono sempre piùnumerose e frequenti, gli organizzatori sono costretti, per così dire, ad alzare lavoce: perché i messaggi raggiungano una vasta platea devono essere trasmessiattraverso una pluralità di mezzi e occasioni, che tendono a coprire spesso tuttala gamma multimediale degli strumenti di comunicazione.
Giornate,settimane, mesi,anni
Secondo le risorse disponibili, lo sforzo promozionale può essere concentrato inuna sola giornata (giornata del morbo di Parkinson, dell’epilessia) o diluito inun arco di tempo maggiore (la settimana del cervello, il mese della salute denta-le, l’anno del cuore). Sostituire nel calendario i morbi ai santi è un espedientenoto agli esperti di teoria della comunicazione: per i media è più facile riportarecome notizie gli eventi puntuali piuttosto che le tendenze di lungo periodo. Unagiornata di sensibilizzazione è quindi un pretesto per parlare di una malattiapiuttosto che di un’altra.
Visite in piazza Sempre più spesso chi organizza campagne di sensibilizzazione sulle malattieprevede un tour nelle principali città in cui si propongono ai passanti materialeinformativo, consulti e controlli. L’iniziativa mira a indurre cittadini che si riten-gono sani a fare visite o esami che altrimenti non avrebbero fatto; si può assimi-lare a un’attività di screening, però in forma spuria, in quanto manca degli ele-menti necessari per farne un programma, cioè un insieme coordinato e control-lato di azioni, e quindi con tutti gli effetti collaterali noti di uno screening nonorganizzato. Inoltre si spezza la relazione triangolare (medico di famiglia, pa-ziente, specialista) il cui delicato equilibrio è indispensabile al buon funziona-mento e alla sopravvivenza del Servizio Sanitario Nazionale.Infine dovrebbe esservi una preoccupazione di immagine. Le iniziative in piaz-za, come molte altre costitutive delle campagne di consapevolezza sulle malat-tie, introducono un linguaggio promozionale nel rapporto tra medico e pazien-te e in quello tra istituzioni e cittadini con tutte le conseguenze consumisticheche ne derivano.
Spot, opuscoli,manifesti e altrimaterialiinformativi
E’ frequente che gli spazi per gli spot o per le inserzioni sui quotidiani, quandoc’è di mezzo un problema di salute, venga concesso gratuitamente, magari at-traverso il meccanismo di Pubblicità Progresso. Altrettanto facile è la disponibi-lità di enti come le Poste o le Ferrovie, che possono fornire formidabili strumenticapillari di distribuzione di materiale.La disponibilità di queste istituzioni si spiega inmolti casi con la convinzione, inperfetta buona fede, che si tratti di propagare il verbo della salute contro la ma-lattia. Le finalità di marketing, neanche tanto nascoste, non vengono percepite.Qui si vuole sottolineare come l’ambiguità sulla reale natura commerciale dellapromozione, camuffata da bene comune, apra spesso le porte a canali di diffu-sione che dovrebbero essere riservati a iniziative realmente indipendenti e dipubblico interesse.
43
Incertezza e conflitti di interesse in medicina

Gli effetti sulla salute e sulla società. Le possibili contromisure
Aumento dell’incertezzaI difetti che si annidano nella ricerca clinica, la crescente medicalizzazione e gliirrisolti conflitti di interesse nel mondo dell’informazione ampliano l’area di in-certezza in cui cadono i giudizi di chi deve valutare il profilo rischio beneficio diuna cura o una notizia di salute. Ecco come.
Risultatidelle ricerchee reale progressodella conoscenza
Se la ricerca clinica si pone obiettivi minimi solo per dare un piccolo vantaggiocommerciale a questo o quel farmaco, lascia senza risposta i quesiti più rilevan-ti per i malati. Un conto è studiare se un farmaco cambia un valore fisiologicocorrelato a una malattia, altro è concludere che una cura piuttosto che un’altraè in grado di aumentare la sopravvivenza o la qualità di vita.Scegliere con cura il campione di uno studio per aumentare la probabilità disuccesso di una cura abbassa l’applicabilità dei risultati della sperimentazione:una volta estesa la cura nella popolazione generale il farmaco ha un profilo ri-schio beneficio meno vantaggioso.Il bagaglio di conoscenze scientifiche aumenta dopo ogni sperimentazione e do-vrebbe permettere di ridurre l’incertezza: ma quale peso dare ai risultati di ri-cerche che si sa che possono essere manipolate per motivi economici?
Diagnosidelle malattie
Una strategia di medicalizzazione aggressiva per rendere malati i sani, attraver-so la proposta di screening di massa senza alcuna prova scientifica di efficacia,determina un aumento di diagnosi di questa o quella malattia, senza però chesiano chiari i vantaggi di avere una diagnosi precoce.Se le soglie si alzano o si abbassano a seconda del vantaggio che può trarnel’industria della salute si perde di vista lo scopo per cui si crea una soglia: ren-dere più vantaggioso possibile il profilo rischio beneficio di un intervento medi-co sulla popolazione.Quando sentirsi malati? Far nascere i criteri diagnostici dall’esigenza di vende-re una cura (un procinetico se si evacua meno di tre volte alla settimana) e nonin funzione di un bisogno di salute, rende incerto il percorso diagnostico e lasuccessiva scelta terapeutica.
Ruolodelle istituzioniscientifiche,associazionie mass media
Le istituzioni scientifiche e le associazioni di pazienti dovrebbero aiutare le per-sone a prendere una decisione piuttosto che un’altra in caso di bisogno. Tutta-via, l’attuale contiguità di questi enti con l’interesse dell’industria, che non ènecessariamente un maggior beneficio per la salute, genera nel pubblico unasfiducia e un rifiuto generalizzato verso il mondo dell’associazionismo e del-l’informazione.
Costi iatrogeniPer ciascuna filiera di cure, tradizionale o nuova, continuerà ad aumentare ladomanda di prestazioni diagnostiche e terapeutiche, nonché il consumo di far-maci e di altri beni e servizi. Inoltre per ogni unità di beneficio non sintomaticoottenibile (una morte o una inabilità o un episodio morboso evitati), continueràa crescere il numero di persone che devono subire un trattamento (NNT), equindi il “mercato potenziale” per la corrispondente filiera di cure. Ne consegueche si moltiplicheranno le fonti e le occasioni di danni iatrogeni, già oggi ingenti.Per esempio si stima che negli Stati Uniti si verifichino 2,2 milioni di eventi av-versi da farmaci solo nei pazienti ricoverati, con oltre 100 mila morti.I tassi iatrogeni sono indipendenti dal fatto che gli interventi siano applicati aindividui con alta o bassa probabilità di ricavarne un beneficio (vedi figura 3),ragion per cui è prevedibile che nel prossimo futuro, con l’aumento del NNTmolte filiere di cure siano destinate ad attraversare il confine oltre al quale idanni iatrogeni superano i vantaggi ottenibili.
Costi socialiE’ facile prevedere che si moltiplicheranno nel prossimo futuro episodi acuti diallarme collettivo come quello suscitato nell’agosto del 2001 dal caso cerivasta-tina, o quello successivo al ritiro del rofecoxib nell’ottobre del 2004. Il susse-
44
Incertezza e conflitti di interesse in medicina
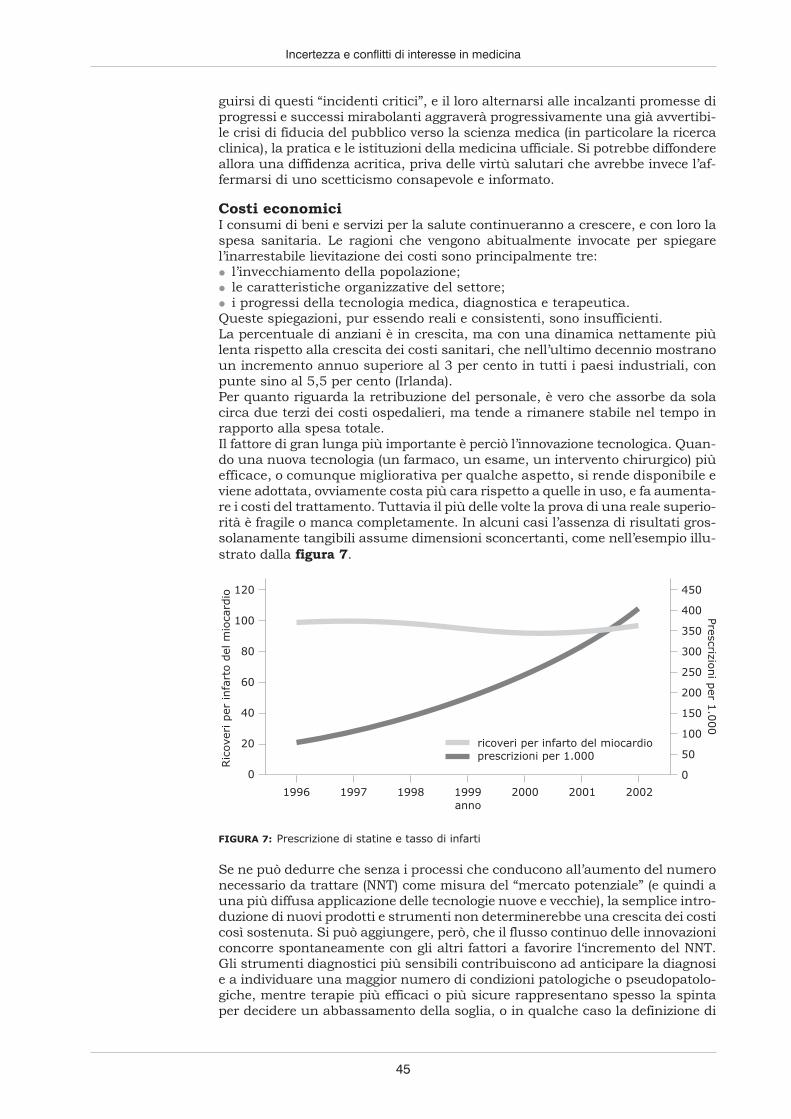
guirsi di questi “incidenti critici”, e il loro alternarsi alle incalzanti promesse diprogressi e successi mirabolanti aggraverà progressivamente una già avvertibi-le crisi di fiducia del pubblico verso la scienza medica (in particolare la ricercaclinica), la pratica e le istituzioni della medicina ufficiale. Si potrebbe diffondereallora una diffidenza acritica, priva delle virtù salutari che avrebbe invece l’af-fermarsi di uno scetticismo consapevole e informato.
Costi economiciI consumi di beni e servizi per la salute continueranno a crescere, e con loro laspesa sanitaria. Le ragioni che vengono abitualmente invocate per spiegarel’inarrestabile lievitazione dei costi sono principalmente tre:l l’invecchiamento della popolazione;l le caratteristiche organizzative del settore;l i progressi della tecnologia medica, diagnostica e terapeutica.Queste spiegazioni, pur essendo reali e consistenti, sono insufficienti.La percentuale di anziani è in crescita, ma con una dinamica nettamente piùlenta rispetto alla crescita dei costi sanitari, che nell’ultimo decennio mostranoun incremento annuo superiore al 3 per cento in tutti i paesi industriali, conpunte sino al 5,5 per cento (Irlanda).Per quanto riguarda la retribuzione del personale, è vero che assorbe da solacirca due terzi dei costi ospedalieri, ma tende a rimanere stabile nel tempo inrapporto alla spesa totale.Il fattore di gran lunga più importante è perciò l’innovazione tecnologica. Quan-do una nuova tecnologia (un farmaco, un esame, un intervento chirurgico) piùefficace, o comunque migliorativa per qualche aspetto, si rende disponibile eviene adottata, ovviamente costa più cara rispetto a quelle in uso, e fa aumenta-re i costi del trattamento. Tuttavia il più delle volte la prova di una reale superio-rità è fragile o manca completamente. In alcuni casi l’assenza di risultati gros-solanamente tangibili assume dimensioni sconcertanti, come nell’esempio illu-strato dalla figura 7.
FIGURA 7: Prescrizione di statine e tasso di infarti
Se ne può dedurre che senza i processi che conducono all’aumento del numeronecessario da trattare (NNT) come misura del “mercato potenziale” (e quindi auna più diffusa applicazione delle tecnologie nuove e vecchie), la semplice intro-duzione di nuovi prodotti e strumenti non determinerebbe una crescita dei costicosì sostenuta. Si può aggiungere, però, che il flusso continuo delle innovazioniconcorre spontaneamente con gli altri fattori a favorire l‘incremento del NNT.Gli strumenti diagnostici più sensibili contribuiscono ad anticipare la diagnosie a individuare una maggior numero di condizioni patologiche o pseudopatolo-giche, mentre terapie più efficaci o più sicure rappresentano spesso la spintaper decidere un abbassamento della soglia, o in qualche caso la definizione di
45
Incertezza e conflitti di interesse in medicina
Ricoveriperinfartodelmiocardio
Prescriz
ioniper1.000
ricoveri per infarto del miocardioprescrizioni per 1.000
anno1996 1997 1998 1999 2000 2001 2002
120
100
80
60
40
20
0
450
400
350
300
250
200
150
100
50
0

nuove entità nosologiche: in entrambi i casi ne consegue un “ciclo di interventocrescente” (vedi figura 4).Infine bisogna considerare che se si riuscisse davvero a contenere i costi sanita-ri, si metterebbe in difficoltà l’industria della salute con tutte le implicite ricadu-te in termini di occupazione e ricchezza. Gli amministratori del sistema sanita-rio sono fortemente contigui al mondo politico, la cui sensibilità alle istanze diun settore economico è direttamente proporzionale alla sua portata. E’ questoprobabilmente il motivo più profondo per cui i tentativi di contenimentodell’inflazione medica (controllo dei prezzi, sistemi di pagamento e di rimborso,ritardi e limiti nella introduzione di novità, liste d’attesa eccetera) sono deboli esolo temporaneamente efficaci.
Sostenibilità socialeLa tesi che una crescita indefinita non sia sostenibile viene spesso giustificatacon l’affermazione che “le risorse sono comunque limitate” e che quindi saràgioco forza, prima o poi, razionarle. In realtà non sembrano esserci limiti teori-ci alla continua dilatazione dei consumi sanitari, o del loro rapporto con il pro-dotto interno lordo, rapporto che ha raggiunto il 15 per cento negli Stati Uniti(figura 8).
FIGURA 8: L’inarrestabile crescita dei costi SSN (Relazioni sulla situazione economica del paese eDpef 2005-2008)
Si pone la questione di come finanziare questa crescita. A parte il pagamento di-retto da parte dei cittadini, vi sono tre fonti principali di finanziamento dellaspesa sanitaria, che nei vari paesi si mescolano in diverse proporzioni:l prelievi fiscali;l contributi previdenziali;l premi assicurativi.Questa formula di finanziamento della sanità si può definire “assicurativa” inquanto si ritiene implicitamente che sia nata per assicurare anche alle fasce piùindigenti della popolazione l’accesso a beni e servizi essenziali. Le condizioni ne-cessarie per la sostenibilità di un sistema “assicurativo” sono le seguenti:l che l’evento assicurato sia relativamente poco frequente;l che sia facilmente determinabile il diritto alla prestazione;l che la prestazione non dia luogo a costi indefiniti.Le transizioni epidemiologica e demografica, con la comparsa di consumatoricronici e prevalenti di cure mediche, mentre da un lato hanno consentito la na-scita dell’industria della salute, dall’altro hanno progressivamente eroso l’equi-librio finanziario del sistema assicurativo, nonostante il suo progressivo allar-gamento a basi contributive sempre più ampie, soprattutto in Europa. Con l’au-mento del numero necessario da trattare (NNT) il rischio assicurato tende ad
46
Incertezza e conflitti di interesse in medicina
Spesa(m
iliardidieuro)
anno
1995 1996 1997 1998 1999 2000 2001 2002 2003 2004 2005
100
90
80
70
60
50
40
30
20
10
0

avvicinarsi a 1 (100 per cento), cioè a trasformare le prestazioni sanitarie, untempo sporadiche e limitate nel tempo, in consumi sempre più generalizzati,anticipati nel tempo e di lunga durata.
Cultura professionale e funzione criticaSi cominciano ad avvertire alcuni segnali di insofferenza al mercato delle ma-lattie. Per quanto riguarda i medici, un risveglio della cultura professionale edetica si manifesta tanto più facilmente quanto minore è il conflitto intrinsecotra l’obbligo di agire nell’interesse del paziente e altri interessi: non a caso, so-no soprattutto i medici di famiglia che si esprimono in maniera critica nei con-fronti degli eccessi e dell’invadenza della medicina. La ragione di questo atteg-giamento è duplice: da una parte c’è l’impostazione centrata sul paziente e suisuoi bisogni complessivi, piuttosto che sulla singola malattia (come avvieneper lo specialista); in secondo luogo, il medico di famiglia non riceve incentivieconomici o di carriera in proporzione alla quantità delle sue prestazioni, anzi,è premiato dal fatto che i suoi assistiti siano sani e si considerino tali. E si tro-va a essere quindi naturalmente estraneo, se non ostile, agli approcci più ag-gressivi e invadenti.
La società civile: resistenza esistenziale e informazioneSi sta manifestando una forma di resistenza che si potrebbe definire “esisten-ziale”, collegata a un generico senso di saturazione da parte dei cittadini, che sisentono chiamati, pur sentendosi bene, a considerarsi bisognosi di cure per unnumero crescente di malattie o rischi. Ogni nuovo abbassamento della soglia oanticipazione della diagnosi, in realtà, produce anche una riduzione della fra-zione di popolazione che accetta di essere reclutata in tutto il ciclo di interventosuccessivo. Per esempio, la recente riduzione dei valori di pressione arteriosache dovrebbero essere raggiunti con il trattamento ha comportato che i pazientiben “controllati” sono oggi solo un terzo dei trattati, anziché la metà come eranoin precedenza.Un ruolo critico ben diverso potrebbe essere svolto dai media e dalle associazio-ni di cittadini e di pazienti, qualora non fossero succubi culturalmente e mate-rialmente rispetto agli interessi dell’industria; basti pensare a organizzazioni diconsumatori come l’americana Public Citizen e alla informazione che produce aproposito di farmaci. Purtroppo, pur essendoci la possibilità che anche in Italiaalcune associazioni indipendenti evolvano verso una maggior incisività anchenel campo della salute, si deve prevedere che questo tipo di informazione sia de-stinata nel prossimo futuro a restare minoritaria rispetto a quella prodotta o in-fluenzata dall’industria. Lo stesso si può dire per i mass media e per l’editoriaspecializzata, anche se un’analisi dettagliata delle pur interessanti esperienzenazionali e internazionali di fonti critiche esula da questo saggio.
Bibliografia di approfondimentol Dirindin N, Vineis P. In buona salute. Torino: Einaudi, 2004.l Bobbio N et al. EBM: conflitti di interessi e trucchi del mestiere. In: LiberatiL. Etica, conoscenza e sanità. Evidence-basedmedicine fra ragione e passio-ne. Roma: Pensiero scientifico, 2005.
l Moynihan R et al., Farmaci che ammalano e case farmaceutiche che ci tra-sformano in pazienti. Bologna: Nuovi Mondi Media, 2005.
l Law J. Big Pharma. Torino: Einaudi, 2006.
47
Incertezza e conflitti di interesse in medicina


Meno ricerca ma di migliore qualitàAlessandro LiberatiRevisori: Ines Benedetti, Ilaria Carretta
Obiettivi delle sperimentazioni cliniche
I pazienti che collaborano a una sperimentazione clinica si sottopongono a disa-gi nella speranza di avere accesso a una terapia migliore di quella esistente, maanche di poter dare un contributo alla collettività e ad altre persone colpite dallastessa malattia.In una situazione ideale lo scopo della ricerca clinica è quello di rispondere aun quesito stimolato dalla pratica clinica, che sulla base delle conoscenze cor-renti non ha una risposta adeguata. Il fine ultimo della ricerca clinica infatti,deve essere quello di produrre conoscenze utili per la cura dei pazienti. Questoobiettivo non è tuttavia sempre perseguito e i protocolli di ricerca suscitanomolti interrogativi sui veri intenti delle sperimentazioni promosse dai ricerca-tori e dalle case farmaceutiche. Perplessità, rabbia, sconforto, sono sentimenticonsueti per chi, abituato alla lettura critica di protocolli di ricerca, si confron-ta quotidianamente con sperimentazioni inutili, o fuorvianti, che perdono divista la nobile funzione della ricerca. In questi casi la ricerca sottrae risorseumane, economiche e strutturali, impegna i pazienti che mettono a disposizio-ne il proprio tempo e il proprio corpo in un progetto che forse non produrràcambiamenti nella cura dei pazienti coinvolti e nemmeno dei pazienti che ver-ranno dopo di loro.
Affidabilità delle sperimentazioni cliniche
Il disegnodello studio
La descrizione dei metodi della ricerca e il disegno dello studio ci permettono dicomprendere in anticipo se una metodologia è affidabile e quali risposte possia-mo o non possiamo ottenere da uno studio clinico. In alcune situazioni la de-scrizione di obiettivi e metodi non è convincente, per esempio se lo studio di-chiara di voler affrontare una questione per la quale sono già disponibili rispo-ste, o affronta un problema che non ha rilevanza clinica, o non dichiara quantipazienti recluterà, o prevede di reclutarne un numero troppo piccolo. Inoltre, ladescrizione dello studio può prevedere una interruzione dello stesso, o la nonpubblicazione a discrezione di chi lo finanzia.
La crisi di credibilità delle sperimentazioni clinicherispetto al paziente
Il mondo della ricerca clinica sta vivendo una profonda crisi di credibilità so-prattutto per quanto riguarda la capacità di tutela degli interessi collettivi e in-dividuali dei pazienti. Per questo molti studiosi hanno promosso iniziative mi-ranti a risvegliare le coscienze assopite dei ricercatori e dei responsabili del fi-nanziamento della ricerca non commerciale. Fra le diverse cause di questa crisidi credibilità sono le seguenti:l scarsa trasparenza nella definizione delle priorità;l mancanza di autonomia dei ricercatori clinici nei confronti dei proprisponsor;
l indebita intrusione degli interessi commerciali nei problemi dei pazienti;l mancanza di fondi per la ricerca indipendente, quella cioè promossa dai ri-cercatori in prima persona a partire dal tentativo di rispondere a reali biso-gni assistenziali;
l scarsa consapevolezza che la ricerca clinica ed epidemiologica è parte in-tegrante dei doveri degli operatori sanitari.
49

Come si vede si toccano questioni molto ampie e complesse e che hanno una ra-dice strutturale negli insufficienti investimenti pubblici nella ricerca. Si tratta al-tresì di problemi che dovrebbero essere affrontati attraverso profonde trasforma-zioni delle politiche sanitarie e degli investimenti sociali, come si vedrà più avanti.Il cittadino informato parteciperebbe attivamente a una sperimentazione clini-ca se fosse preparato all’idea che la sperimentazione è il modo normale dellascienza di progredire e che mancanza di risposte certe da parte della medicinanon è sinonimo di ignoranza. Ma sempre più spesso la medicina si presenta co-me foriera di certezze circa l’efficacia di terapie e interventi personalizzati. La spe-rimentazione appare invece estranea in una società nella quale viene trasmessaun’idea deterministica e non probabilistica della natura della ricerca e dellascienza. Un’idea che finisce per attribuire al termine “sperimentazione” una con-notazione negativa e che determina enormi difficoltà nel far comprendere che lasperimentazione dovrebbe essere parte integrante della normale assistenza ero-gata dai servizi sanitari.1 L’idea che si debba sperimentare suona come unastrana necessità, più suggestiva di pressappochismo e scarsa deontologia pro-fessionale (“vogliono sperimentare sulla mia pelle…”), che di un modo onesto eprofondamente etico di affrontare esplicitamente le situazioni di incertezza.
Ricerca clinica, trasparenza e regoleI ricercatori sono tenuti a documentare in modo completo e accessibile i risulta-ti dei loro studi, e delle sperimentazioni cliniche in particolare. Hanno il dovereetico della trasparenza e devono sentirsi responsabili di tutti i loro atti nel mo-mento in cui partecipano alla produzione di conoscenze verso la comunitàscientifica alla quale appartengono e verso i pazienti cui chiedono di partecipareagli studi (e del pubblico in generale).Questa responsabilità dovrebbe accettare di confrontarsi in modo esplicito etrasparente con almeno tre obblighi:l interpretare con cautela i risultati delle ricerche cliniche e valutarne l’im-portanza e credibilità nel contesto della totalità dei risultati invece che diquella dei singoli studi;
l garantire meccanismi di massima e allargata accessibilità dei risultati dellesperimentazioni cliniche;
l riconoscere l’esistenza dei conflitti di interesse e del loro impatto sui risulta-ti delle sperimentazioni cliniche.
La totalitàdegli studi
Quando si vuole stimare il “valore vero” di una innovazione diagnostico-tera-peutica non si può che fare riferimento alla totalità delle informazioni disponibi-li piuttosto che al risultato di un singolo studio, anche se pubblicato con risaltosu una importante rivista internazionale. Quando i dati sono ancora pochi e nonottenuti indipendentemente da più gruppi di ricerca, è necessario valutare i lororisultati con attenzione. Occorre anche porre attenzione al grande supporto dimarketing e divulgazione che ricevono gli studi quando dimostrano l’efficacia dinuovi prodotti e tecnologie che hanno un potenziale impatto commerciale. Lamancata pubblicazione (publication bias nel gergo tecnico) e la tendenza deglistudi sponsorizzati a dare risultati a favore del trattamento sperimentale (in mi-sura maggiore di quanto atteso su base casuale) sono fattori che devono indur-re alla prudenza. Secondo un recente studio, quasi un terzo degli studi apparsisu riviste ad alto profilo scientifico vengono ridimensionati nel giro di qualcheanno dalla loro prima pubblicazione, non tanto perché le conclusioni degli studifossero sbagliate quanto piuttosto perché l’enfasi data all’interpretazione inizia-le del loro risultato era eccessiva, non teneva conto del grado di imprecisionestatistica dei risultati e della loro non conferma in ambiti differenti.Alcuni studi condotti nel 1998 e 20022,3 hanno valutato criticamente alcuni artico-li scientifici e mostrano come gli autori della maggior parte degli articoli esaminati(77 per cento e 81 per cento nei due campioni rispettivamente) analizzassero i pro-pri risultati senza cercare di interpretarli nel contesto degli altri studi, piuttostodando una lettura parziale e isolata dal contesto. Questi comportamenti sono par-ticolarmente preoccupanti perché portano a una inutile duplicazione di sforzi,3 e aconfusione in chi legge il singolo articolo e vuole farsi un’idea equilibrata.
50
Meno ricerca ma di migliore qualità

Secondo Bradford Hill (uno dei maestri della modernametodologia clinica), ognilavoro scientifico dovrebbe rispondere a quattro quesiti fondamentali:l perché si è intrapreso lo studio?l che cosa hanno fatto esattamente i ricercatori che l’hanno condotto?l quali risultati sono stati ottenuti?l quale significato hanno questi risultati?
La pubblicazionedei risultati
Ormai da un decennio è stato documentato in letteratura quel fenomeno (notocome publication bias) per cui si pubblicano gli studi che hanno risultati positi-vi e non si pubblicano gli studi con risultati nulli o non conclusivi a favore di untrattamento o intervento. Al fenomeno contribuiscono sia i singoli ricercatori,sia le politiche editoriali delle riviste, sia gli sponsor della ricerca. Questo dibat-tito ha occupato, con alterne vicende e variabili ondate di interesse, le paginedelle maggiori riviste scientifiche.
La registrazionedegli studi
La creazione di registri delle sperimentazioni cliniche potrebbe contribuire auna maggiore trasparenza, soprattutto se la registrazione delle sperimentazionicliniche al momento della loro attivazione venisse resa obbligatoria per ricerca-tori e sponsor. Recentemente un importante passo avanti è stato fatto con unapresa di posizione dell’International Committee of Medical Journals Editors(ICMJE) che ha annunciato che, a partire dal luglio 2005, tutte le 11 riviste ade-renti al Comitato avrebbero pubblicato solamente quegli studi registrati al mo-mento del loro inizio in uno dei registri internazionali ad accesso libero esistential mondo. Questa presa di posizione è stata vista come un reale passo avantinella direzione della “uscita dalla clandestinità” di molte sperimentazioni clini-che che vengono iniziate ma che poi, per vari motivi, vengono interrotti e nongiungono alla completa pubblicazione.Oggi esistono a livello internazionale oltre 100 registri nazionali, internazionali,di specialità, di specifiche case farmaceutiche, eccetera, con un grado variabiledi accessibilità e di copertura di informazioni raccolte. In Italia abbiamo peresempio, dal 2002, un sistema obbligatorio – presso l’Osservatorio Sperimenta-zioni Cliniche della Agenzia Italiana del Farmaco – di registrazione di tutte lesperimentazioni cliniche autorizzate dai Comitati Etici, accessibile sia a utentiinterni del sistema (sponsor, regioni e comitati etici) sia al pubblico. Esistonoinoltre altri registri aperti al pubblico, nei quali gli studi registrati non proven-gono da un universo sistematico ma dalla scelta volontaria degli organizzatoridegli studi.Permangono tuttavia ancora molti dubbi sulla reale fattibilità della completaregistrazione di tutte le sperimentazioni cliniche che vengono avviate; sareb-bero in questo senso importanti e necessarie decisioni altrettanto stringentiassunte dalle Autorità che regolano le sperimentazioni cliniche a livello nazio-nale e internazionale.In ogni caso, la registrazione delle sperimentazioni cliniche non può considerar-si la cura per tutti i mali: anche se tutti gli studi fossero completamente regi-strati, e la loro esistenza rintracciabile, questo non garantirebbe la completatrasparenza della loro pubblicazione.
Distorsioni della ricerca clinica
La pubblicazioneselettiva
C’è un altro tipo di distorsione nel processo di pubblicazione (outcome reportingbias o distorsione nel resoconto dei risultati)4,5: essa consiste nel riportare inmodo selettivo e parziale i risultati di uno studio dando enfasi ai risultati che di-mostrano un maggiore effetto dei trattamenti, o comunque non presentandotutto ciò che si era originariamente progettato di studiare. Circa un terzo dei ri-sultati originariamente studiati non viene completamente riportato negli artico-li pubblicati, con particolare censura per quanto riguarda gli effetti collaterali.Fanno sempre parte di questa mancanza di trasparenza le motivazioni che por-tano alla sospensione anticipata delle sperimentazioni cliniche in presenza dirisultati promettenti. La sospensione, in questi casi, non è prevista dal protocol-lo, e questo può portare alla diffusione di risultati erroneamente promettenti.Che la sospensione anticipata di studi in presenza di dati preliminari positivi
51
Meno ricerca ma di migliore qualità

sia comunque una decisione rischiosa è cosa nota da tempo tra gli addetti ai la-vori delle sperimentazioni cliniche. Non è infrequente, peraltro, la possibilità diinterrompere uno studio che sembra dare risultati che non sarebbero graditidallo sponsor che ha un interesse diretto nel trattamento studiato.
I dossierregistratividei farmaci
Resta poi un altro grave limite di omissione apertamente e tranquillamente tolle-rato: quello che permette che i dossier di EMEA e FDA, anche una volta termina-to l’iter registrativo e di approvazione di un farmaco, non siano di libero accesso acausa del segreto industriale e del diritto alla tutela della concorrenza commer-ciale. Questo evidentemente impedisce di valutare in modo trasparente la corret-tezza del processo seguito dalle agenzie regolatorie. E’ inquietante che nessuno siopponga a questa mancanza di trasparenza in un campo nel quale la salute deipazienti dovrebbe essere al primo posto. Eppure, quando si chiede a una pazien-te di partecipare a una sperimentazione clinica, le si chiede proprio di “accettareun potenziale rischio” in nome del principio altruistico del progresso delle cono-scenze e della possibilità di trovare terapie sempre più efficace sicure.
Il medico ècondizionato
Il servizio sanitario nazionale o l’assicurazione privata, finanziatori del sistemasanitario, regolano l’accesso alle prestazioni cliniche per garantire uniformità ditrattamenti, appropriatezza di prestazioni e contenimento dei costi. In questocontesto si inserisce tuttavia l’industria biomedica che produce farmaci, appa-recchiature, materiale di consumo, e che mette in atto interventi di marketingatti a condizionare gli operatori sanitari e a far assumere comportamenti pre-scrittivi per lei vantaggiosi. Di fronte al paziente c’è quindi un professionistadella medicina, esposto a condizionamenti lavorativi, economici, diretti, di pre-stigio e carriera, che deve scegliere tra le opzioni che il finanziatore gli lascia co-me possibilità, ma che è a sua volta bersaglio di una promozione industriale chelo incentiva, con vari mezzi, a consumare risorse.Contrariamente a quello che avviene in molti altri settori industriali, l’informa-zione/pubblicità/propaganda di prodotti sanitari non si rivolge generalmente alconsumatore finale ma al medico, che non consuma e non paga ma prescriveun prodotto a un paziente che spesso non paga direttamente e che, comunque,non sceglie. In base a quali criteri il medico decide quale farmaco o tecnologiaprescrivere? In teoria, sulla base dell’integrazione tra le migliori informazioniscientifiche e la propria personale esperienza.
I conflittidi interesse
Ma la realtà è più complessa: le informazioni disponibili sono asimmetricamen-te prodotte (non tutto quello che dovrebbe essere studiato e ricercato viene fi-nanziato), asimmetricamente distribuite e asimmetricamente recepite nella lorocomplessità e validità. L’informazione commerciale prevale nettamente su quel-la indipendente e il pubblico stesso non è preparato ad avere un sano scettici-smo nei confronti degli avanzamenti veri della medicina. Proprio a partire dallaconsapevolezza di tutto questo ha preso origine, anche in campo medico, la di-scussione sui pericoli del conflitti di interesse.6 In termini generali si dice che siverifica un conflitto di interesse quando ci si trova in una condizione nella qualeil giudizio professionale riguardante un interesse primario (la salute dei pazientio la veridicità dei risultati o di una ricerca o l’oggettività di una informazione)tende a essere indebitamente influenzato da un interesse secondario (guadagnoeconomico, vantaggio personale). Perché si verifichi una condizione di conflittodi interesse è quindi sufficiente che esista un legame in grado di comprometterel’autonomia del professionista nell’ambito di vincoli pure legittimamente impo-sti dal finanziatore. Da questo punto di vista ogni conflitto di interesse è danno-so per l’operato del medico in quanto può minare la sua credibilità di giudizio.Nonostante che il dibattito su questo tema sia molto vivace (anche se spessonon affronta alla radice il problema dei vincoli strutturali che lo determinano)c’è ampio consenso nel ritenere che i conflitti di interesse potenziali od anchesolo ipotizzabili possano essere altrettanto dannosi di quelli reali. Ciò che rendeparticolarmente serio il problema dei conflitti di interesse in campo sanitario èche essi possono esercitare la loro influenza già fin dal momento della produzio-ne delle conoscenze in vario modo: attraverso, per esempio, il disegno “addome-sticato” dei protocolli, la violazione di regole importanti nella conduzione e ana-
52
Meno ricerca ma di migliore qualità

lisi delle sperimentazioni cliniche e attraverso la esagerazione/strumentalizza-zione nell’interpretazione dei risultati.Nell’analisi dell’impatto che il conflitto di interessi può avere sulla produzionedei risultati di sperimentazioni cliniche si è puntata l’attenzione soprattuttosull’esistenza di legami di tipo finanziario tra i ricercatori (o i clinici) e le indu-strie produttrici di farmaci (o di qualsiasi altra tecnologia sanitaria destinata al-la vendita). Questo risultato è frutto non tanto del fatto che questo tipo di inte-ressi sia in assoluto più pericoloso di altri, quanto perché probabilmente è piùfacile da individuare. Molte analisi mostrano che i risultati di ricerche finanziateda organizzazioni aventi fini di lucro, o comunque da chi ha un interesse direttonei risultati della ricerca, tendono a favorire il punto di vista dei finanziatori ri-spetto ai risultati di quelle finanziate da enti non aventi finalità di lucro o da or-ganismi di ricerca indipendenti.I metodi di condizionamento sono molteplici:l la selezione a monte, da parte degli sponsor, dei ricercatori “più adatti” aprodurre certi risultati;
l la selezione del tipo di disegno più favorevole e in particolare quella del“controllo” (placebo al posto di un controllo attivo e controllo attivo usato indosi e modalità subottimali), che è potenzialmente più favorevole al nuovoprodotto;
l la soppressione della pubblicazione dei risultati sfavorevoli o maggior diffi-coltà a pubblicare comunque gli studi negativi (distorsione di pubblicazione).
È su questa sostanziale base empirica che si fonda la preoccupazione che oggicaratterizza chi sostiene la necessità di una vigilanza attiva nei confronti deiconflitti di interesse nel campo della produzione scientifica, con particolare rife-rimento alle sperimentazioni cliniche. Si tratta, ovviamente, della punta di uniceberg, che condiziona oggi l’indipendenza della produzione scientifica e che haa che fare con il modo di concepire l’indipendenza della ricerca e i limiti da porreal mercato e all’interesse privato. Un’ampia discussione su questo tema si trovaanche nel capitolo Incertezza e conflitti di interesse in medicina.
Il finanziamento della ricerca clinica nel SSN
L’assistenza sanitaria è vista sempre di più come costo difficile da controllareinvece che come occasione di sviluppo. I sistemi sanitari peraltro investono pocoin ricerca e, di conseguenza, hanno scarsa capacità di influenzarne natura, per-corso e caratteristiche, finendo per trovarsi nella posizione difensiva dell’acqui-rente disinformato, preoccupato della compatibilità economico-finanziaria dellatecnologia piuttosto che della sua reale necessità, della sua congruità e compati-bilità con l‘attuale configurazione dei servizi e degli spazi di uso appropriato.Un argomento frequentemente utilizzato per giustificare questo stato di cose èche la ricerca, e segnatamente la sperimentazione clinica, non rappresenta uncompito primario del SSN. In realtà, il SSN destina risorse per la ricerca. Il Mini-stero della Salute ha diretta responsabilità di assegnazione e distribuzione delFondo per la Ricerca: responsabilità di gestione attraverso programmi di soste-gno ad hoc della ricerca in campi specifici (trapianti, oncologia, malattie infetti-ve, eccetera) e attraverso il Fondo per la Ricerca Sanitaria rivolto a sostenerel’attività degli Istituti di ricovero e cura a carattere scientifico (IRCCS) e di altreistituzioni del Servizio Sanitario Nazionale che ricevono fondi per la parte cor-rente di ricerca. Una parte minoritaria di questo fondo viene destinato alla co-siddetta ricerca finalizzata che serve a finanziare progetti presentati da vari en-ti. Questa parte del Fondo per la ricerca sanitaria è rivolta al sostegno della ri-cerca sanitaria ma non della sperimentazione clinica in senso proprio, chequindi non ha in Italia (a parte il nuovo meccanismo che sta attualmente atti-vando l’AIFA) un luogo di riferimento preciso per il finanziamento.
I livelli essenzialidi assistenza(LEA)
Attraverso i diversi passaggi legislativi che hanno seguito l’istituzione del Servi-zio Sanitario Nazionale con la legge n. 833 del 1978, si è arrivati nel 2001 al-l’esplicitazione dei livelli essenziali di assistenza (LEA) come: “Le prestazioni [...]garantite dal Servizio Sanitario Nazionale a titolo gratuito o con partecipazione
53
Meno ricerca ma di migliore qualità

alla spesa”.7 Oltre a definire ciò di cui il Servizio sanitario nazionale deve farsicarico nei confronti dei cittadini, il decreto istitutivo dei LEA ha esplicitato an-che che restano al di fuori di essi (parzialmente o interamente, a seconda deglispecifici casi) quelle prestazioni che:l non rispondono a necessità assistenziali tutelate in base ai principi ispirato-ri del Servizio Sanitario Nazionale;
l non soddisfano il principio dell’efficacia, dell’appropriatezza e dell’economi-cità nell’impiego delle risorse in presenza di altre forme di assistenza volte asoddisfare le medesime esigenze.
In questo modo il Servizio Sanitario Nazionale italiano ha scelto la strada di unatteggiamento dinamico rispetto alla possibilità di adeguarsi – all’interno dellalogica dell’appropriatezza e dell’economicità – all’evoluzione delle conoscenzescientifiche. Tuttavia non è chiaro poi come, nel concreto, questa apertura pre-veda una presa in carico attiva e programmata della ricerca, da parte del Servi-zio Sanitario Nazionale, né che cosa esso intenda fare, o far fare, per acquisirequeste conoscenze.I dati regolarmente prodotti dall’Osservatorio per le Sperimentazioni clinichedel Ministero della Salute parlano chiaro su chi fa sperimentazione clinica inItalia. Sulla base del rapporto del 2006 dell’Osservatorio Nazionale delle Speri-mentazioni Cliniche (relativo ai protocolli di ricerca attivati in Italia fino al Di-cembre 2005; disponibile su https://oss-sper-clin.agenziafarmaco.it/) sonostate condotte o sono in corso in Italia 3.527 sperimentazioni cliniche (54 percento circa di fase III, 34 per cento di fase II, 2 per cento di fase I e il restante 10per cento di fase IV o di bioequivalenza). Sul totale, il 74 per cento sono sponso-rizzate dall’industria farmaceutica mentre il 18 per cento sono state promosseda una fonte pubblica del Servizio Sanitario Nazionale, e specificamente 344 daun’Azienda sanitaria locale o Azienda Ospedaliera e 266 da un Istituto di ricove-ro e cura a carattere scientifico.
La ricercaindipendente
Anche in Italia vanno registrati alcuni potenziali segnali positivi nella direzionedello sviluppo di una ricerca indipendente.La prima è rappresentata dal Decreto Ministeriale del Marzo 2005 sulla cosid-detta ricerca non profit8 significativamente intitolato “…Per il miglioramentodella pratica clinica, quale parte integrante dell’assistenza sanitaria…”. Si trat-ta di un provvedimento che tende a facilitare la conduzione e il finanziamento disperimentazioni cliniche non finalizzate alla commercializzazione di un farmacoe all’acquisizione di un marchio di mercato, quanto piuttosto al miglioramentodelle strategie assistenziali.Conseguenza operativa di questo orientamento è il primo bando dell’Agenziaitaliana per il farmaco (AIFA) – attivato nel 2005 – che nasce con lo scopo espli-cito di finanziare le ricerche che non trovano attenzione da parte dell’industriafarmaceutica o di altri enti, e che sono tuttavia vitali per mantenere la capacitàdella ricerca clinico-epidemiologica sul farmaco di rispondere ai bisogni cono-scitivi e operativi del Servizio Sanitario Nazionale. Il bando è articolato per so-stenere tre filoni di ricerca: a) malattie rare; b) grandi trials comparativi; c) studidi farmacovigilanza e di impatto delle strategie di miglioramento della qualitàdell’assistenza. Si tratta di uno sforzo importante, il cui impatto andrà valutatonel tempo in funzione anche della sua capacità di determinare la rinascita diuna rete vitale di gruppi cooperativi di centri presso aziende sanitarie e ospeda-liere, capaci di affrontare questioni di grande rilevanza clinica e assistenziale.Le regioni hanno da questo punto di vista una responsabilità importante di col-laborazione con AIFA per il raggiungimento di questo obiettivo.Va registrato come altro elemento positivo il fatto che anche alcune regioni sistanno muovendo. La Regione Emilia Romagna, per esempio, ha avviato il “Pro-gramma Ricerca e Innovazione della Regione Emilia Romagna” (denominatoPRI-ER),9 volto al potenziamento della ricerca necessaria alla maturazione delleinnovazioni clinico-organizzative e alla verifica della loro effettiva applicazionenella pratica clinica. Il PRI-ER è sostenuto da un fondo indipendente alimentatoda risorse regionali e da contributi di soggetti privati, che non sono tuttavia fi-nalizzati alla sponsorizzazione di specifici progetti ma al sostegno del Fondo nel-
54
Meno ricerca ma di migliore qualità

la sua globalità. Al di là della promozione di specifici progetti che il PRI-ER haavviato in campo oncologico, cerebrovascolare, sulla diagnostica ad alto costo, ealtro ancora, la scommessa del Programma è quella di creare – dentro le aziendesanitarie – una infrastruttura capace di sostenere e promuovere la ricerca comeparte integrante dell’atto assistenziale.
L’esperienzabritannica
Molti sistemi sanitari a livello internazionale hanno da tempo riconosciuto lanecessità di promuovere e sostenere una ricerca capace di rispondere ai quesitiassistenziali e organizzativi più importanti perché la sperimentazione clinicanon commerciale è una componente essenziale dell‘assistenza sanitaria, inquanto occasione per promuovere e acquisire conoscenze relativamente a biso-gni inevasi. L’esempio maggiormente strutturato di intervento del servizio sani-tario pubblico a sostegno e integrazione della ricerca industriale e accademica èdato dal programma Ricerca e Sviluppo britannico che, a partire dal 1995, hamesso a disposizione quote consistenti di risorse per sostenere quella ricercache solitamente non trova finanziamenti commerciali disponibili. Un interes-sante articolo apparso nel 2003 sul British Medical Journal10 spiega come diver-si sistemi sanitari si stiano orientando verso il potenziamento di risorse a favoredella ricerca non commerciale. Si dimostra anche che questo tipo di ricerca è ingrado, laddove sostenuto per un numero sufficiente di anni e con un adeguatoinvestimento infrastrutturale, di incidere su qualità e tipologia delle informazio-ni che vengono rese disponibili.
Prospettive Occorre dunque, sul piano politico e sociale, che:l i temi della ricerca biomedica a livello europeo diventino di pertinenza prio-ritaria degli organismi che si occupano della tutela della sanità pubblica enon siano di pertinenza dell’industria;
l i responsabili delle politiche sanitarie nazionali, regionali e aziendali assu-mano la piena consapevolezza che una capacità di governo della funzione diricerca (e uno sforzo per liberare risorse materiali e organizzative a questoscopo) è un loro compito essenziale;
l le organizzazioni, le società scientifiche e i ricercatori facciano proprio lo slo-gan che un vecchio ma ancora drammaticamente attuale editoriale del Bri-tish Medical Journal di oltre 10 anni fa invocava: “meno ricerca, ma ricercadi migliore qualità e più focalizzata ai bisogni dei pazienti”;11
l i Comitati Etici sfuggano dalla trappola di farsi valutare in base alla capacitàdi “metabolizzare e approvare protocolli” ma si impegnino per regole e moda-lità comuni, richiamando le autorità regionali e nazionali ai propri doveri dicoordinamento, e battendosi per uscire da una situazione di “abbandono alproprio destino” di impotenti esecutori di decisioni prese altrove.
Bibliografia1. Emanuel E, Wendler D, Grady G. What makes clinical Research ethical?
JAMA 2000;283:2701-11. http://www.bioethics.nih.gov/international/readings/framework/clinresethc.pdf
2. Clarke M, Chalmers I. Discussion sections in reports of controlled trialspublished in general medical Journals: islands in search of continents.JAMA 1998;280:280-2
3. Clarke M, Alderson P, Chalmers I. Discussion sections in reports of con-trolled trials published in general medical journals. JAMA 2002;287:2799-801
4. Chan AW, Haart MT, Hrobjiartsson A et al. Empirical evidence for selectivereporting of outcome in randomised comparison of protocols to publishedstudies. JAMA 2004;291:2457-65
5. Chan AW, Krieza-Jeric K, Schmid I, Altman D. Outcome reporting bias inrandomised trials funded by the Canadian Institutes of Health Research.CMAJ 2004;171:735-40
6. Di Pietrantonj C, Demicheli V. I conflitti di interesse nella ricerca finanzia-ta dall’industria farmaceutica. Epidemiol Prev. 2005Mar-Apr;29(2):85-95.
7. Conferenza Stato Regioni. Accordo tra Governo, Regioni e le Province Auto-nome di Trento e Bolzano sui Livelli Essenziali di Assistenza Sanitaria ai
55
Meno ricerca ma di migliore qualità

sensi dell’articolo 1 del decreto legislativo 30 dicembre 1992, n. 502 e suc-cessive modificazioni, 3.1. http://www.ministerosalute.it/alimenti/nutri-zione/nutApprofondimento.jsp?lang=italiano&label=all&id=334
8. Martini N. Documento programmatico sulla sperimentazione clinica deimedicinali. Guida all’adozione dei decreti attuativi. Bollettino Informazio-ne sui Farmaci (BIF) 2004;XI:6-8. http://www.agenziafarmaco.it/wscs_render_attachment_by_id/111.30602.1137602090158e343.pdf?id=111.30608.1137602090224
9. Programma Ricerca & Innovazione Emilia Romagna (PRI ER). DocumentoProgrammatico Novembre 2004. http://asr.regione.emilia-romagna.it/wcm/asr/aree_di_programma/ricercaeinnovazione/pr_prier.htm
10. Chalmers I, Rounding C, Lock K. Descriptive survey of non commercialrandomised control trials in the United Kingdom, 1980-2002. Br Med J2003;327:1017-22
11. Altman D. The scandal of poor medical research. Br Med J 1994;308:283-4
56
Meno ricerca ma di migliore qualità

L’informazione in medicina:come destreggiarsiLuca Carra, Cinzia ColomboRevisore: Maria Di Ottavio
La salute nei mezzi di informazione
La salute, al pari dell’economia e dello sport, ha programmi televisivi dedicati,sezioni di quotidiani, settimanali specializzati. Buona parte della nuova fictiontelevisiva ruota intorno ad argomenti medici: dal “vecchio” E.R. a Doctor House,e di recente anche in teatro si possono vedere rappresentazioni che parlano disalute, test diagnostici, medicina.L’interesse crescente del pubblico per la salute e la medicina si deve in parte al-l’invecchiamento della popolazione – secondo le indagini, sono soprattutto lepersone di mezza età e anziane a essere interessate a questo genere di informa-zione – e in parte alla natura dell’argomento. Pochi temi sono così coinvolgenti evari come questo, che tocca emozioni, sentimenti, paure, speranze, interessi. Lenotizie di salute trattano dalla tecnologia più innovativa, all’azione di solidarie-tà, dal laboratorio di ricerca alla pratica clinica in ospedale, dagli scandali pub-blici alle storie private degli ammalati. Non sorprende dunque che riscuotanocosì tanto successo.
Il medico èla principale fontedi informazione
Ciò detto, per gli italiani la fonte principale di informazioni inmedicina resta an-cora il medico, e solo dopo vengono la televisione, che è comunque in crescitacome fonte, la carta stampata e il passaparola. Da quanto risulta nell’ultimosondaggio eseguito dal Censis sulla domanda di informazione sulla salute inItalia, l’informazione veicolata dai media sembra influenzare soprattutto chi go-de di buona salute e cerca occasioni per migliorare il suo stile di vita, sia attra-verso la prevenzione primaria (alimentazione, esercizio fisico), sia attraverso laprevenzione secondaria (screening, esami). E’ in crescita inoltre la curiosità delpubblico italiano verso le medicine complementari e verso cure volte non tantoa gestire vere e proprie malattie, quanto a migliorare le performance e a “starmeglio”.
Il web,la televisione,i giornali
Internet è una fonte in espansione ed è utilizzato soprattutto dalle fasce più gio-vani, che spesso cercano informazioni pratiche e puntuali per conto di personepiù anziane, che non hanno consuetudine con il web (vedi il capitolo Navigaresulla rete alla ricerca di informazioni di salute).Non è solo il pubblico generale a ricavare informazioni da giornali, programmitelevisivi o internet, anche gli operatori sanitari e i decisori politici utilizzanoqueste fonti.I mass media quindi possono influenzare non solo la consapevolezza, le attitu-dini e le intenzioni di chi li segue, ma anche l’uso delle risorse sanitarie, le prati-che cliniche e le politiche sanitarie. Possono inoltre influenzare quali condizionivengono percepite dal pubblico come malattie che necessitano di un interventomedico.
Diseasemongering
Una considerazione particolare merita il cosiddetto disease mongering, formaaggiornata di medicalizzazione che consiste nel “vendere” come malattia ciò chemalattia non è: dalla calvizie all’iperattività nei bambini, dalla “sintomatologiadepressiva” alla sindrome della stanchezza cronica. Le nuove “malattie alla mo-da”, così come il ben noto fenomeno dell’abbassamento della soglia che stabili-sce il confine fra normalità e malattia (vedi il capitolo L’ABC della ricerca clinica)hanno portato in questi anni a un allargamento del patologico nella vita quoti-diana delle persone, con conseguente aumento da parte dei sani del ricorso aesami e trattamenti.
57
L’informazione in medicina: come destreggiarsi

Nella loro natura di specchio degli interessi, delle ansie, e talvolta delle perversionidell’opinione pubblica, i media sono particolarmente attenti alle notizie in questoambito e le sezioni di scienza e salute di rotocalchi e quotidiani discendono in parteanche dai “Gabinetti di curiosità” (Kunst und wunderkammern) del Seicento, incui si esponevano reperti rari e mostruosi. L’approccio mediatico a certe malattie,quelle rare per esempio, risente di questo tipo di impostazione, così come l’accentoche la stampa pone sulle meraviglie della scienza e i progressi visti come risolutivi(es. terapia genica, clonazione, mappatura del genoma umano, cellule staminali).In buona o cattiva fede, l’informazione dimassa sui temi di salute è una delle formepiù raffinate di alimentazione della credulità popolare, e spesso è priva di qualun-que senso critico. La presenza di rilevanti interessi commerciali fa sì che i mediasiano oggetto privilegiato di sensibilizzazione da parte dell’industria della salute.Questa attenzione, insieme agli altri interessi che ruotano attorno al tema – peresempio dei ricercatori, che vogliono far conoscere i propri studi, o degli editori, chevogliono rendere i propri giornali appetibili per il pubblico e per gli inserzionistipubblicitari – possono influenzare in diversi modi l’informazione, tanto da far sem-brare le notizie di salute dei veri e propri annunci pubblicitari.
Informazionecritica
Negli ultimi anni c’è stato però un risveglio dell’informazione critica in campobiomedico, che ha fatto registrare anche in Italia una maggiore consapevolezzadei conflitti di interesse e dei condizionamenti presenti in questo settore. Que-sto ha portato alcune testate giornalistiche e alcune associazioni di consumato-ri e cittadini a sviluppare strumenti critici di analisi e produzione dell’informa-zione, mirando a standard adeguati di completezza e correttezza. Si tratta tutta-via di fenomeni ancora episodici e isolati, soverchiati da un’industria dell’infor-mazione di salute con una forte componente commerciale.
La filiera dell’editoria medica, tra promozione e informazione
Dalla ricercascientificaalla divulgazionemedica
Fra ricerca scientifica e divulgazione medica non c’è soluzione di continuità.L’indagine epidemiologica, la sperimentazione clinica vengono pensate per es-sere pubblicate su riviste scientifiche, il prestigio e la popolarità delle quali simisura in termini di impact factor (cioè il numero medio di citazioni per artico-lo per ogni rivista). Le ricerche più interessanti vengono accettate per la pub-blicazione dopo essere passate al vaglio dei revisori (cioè alla revisione dei pari,peer-review, che consiste nel sottoporre un articolo scientifico alla lettura cri-tica di uno o più esperti della materia per decidere se pubblicarlo o meno). Almomento della pubblicazione, le ricerche più interessanti vengono commenta-te in un’altra parte della rivista (nell’editoriale) da parte di un opinion leader,esperto della materia. Spesso, i risultati preliminari di una ricerca non ancorapronta per la pubblicazione vengono presentati nell’ambito di convegni e con-gressi e qui si fermano: molti infatti non vengono nemmeno pubblicati sulle ri-viste specialistiche. Il disegno dello studio, così come la sua scelta da parte deiresponsabili editoriali delle riviste, e la scelta dell’opinion leader che la com-menterà possono essere condizionate da interessi commerciali. Dal momentoin cui le ricerche vengono pubblicate in poi, si entra nel regno vero e propriodell’informazione, che viene opportunamente stimolata per trasformare la ri-cerca in “notizia”.
La notiziabilità degli argomenti mediciLa notiziabilità è la qualità di un evento di diventare notizia e in quanto tale essereraccontata dai media. Si tratta di una qualità che conosce tante sfumature quantesono le testate che si occupano di salute. La scelta e il modo di trattare le notizie diun femminile sarà inevitabilmente diverso da un quotidiano o unnewsmagazine.Vi sono, tuttavia, degli elementi fondamentali nella notiziabilità di salute chefanno capire quali sono gli ingredienti basilari della cucina redazionale. Vedia-mo i principali:l una notizia deve essere una novità. In quanto tale, deve avere elementi dioriginalità. L’uomo che morde il cane fa notizia; il cane che morde l’uomo no(a meno che non lo ammazzi);
58
L’informazione in medicina: come destreggiarsi

l una notizia deve essere narrabile. Non bastano dati: devono esserci una sto-ria, delle facce, dei destini;
l una notizia deve contenere una storia con personaggi molto caratterizzati:eroi, vittime, eccetera;
l una notizia importante solitamente è connotata in senso negativo (l’uomoammazzato dal cane, una pandemia in arrivo), o molto positivo (il miracolo,la scoperta di una nuova cura salvavita eccetera);
l una notizia deve riguardaremolte persone, meglio se riguarda (positivamente onegativamente) le categorie cosiddette fragili: donne, bambini, anziani, malati.
Scopo della notizia non è educare il pubblico ma catturarne la curiosità. La se-lezione e la tempestività sono le attività principali delle redazioni. Delle circa100-200 proposte di notizie di salute che arrivano quotidianamente nelle reda-zioni attraverso i comunicati stampa e i lanci di agenzia, ne vengono selezionatealcune secondo queste e altre caratteristiche più pertinenti alla linea editorialedella singola testata.Ciò che salta immediatamente all’occhio è l’estrema uniformità (conformismo)nella scelta delle notizie di medicina fra le diverse testate. La notizia in esclusiva èuna rarità (scoop) che conferma la regola. I motivi di questa relativa uniformitàsono vari: il pool ristretto di riviste internazionali alle quali attingono le redazioni;le pressioni provenienti dagli uffici stampa e di pubbliche relazioni che riferisconodi studi sponsorizzati; infine la ritualità stagionale o scandita da un vero e pro-prio calendario delle giornate dedicate alle diverse malattie che spesso impongo-no un’agenda alla trattazione giornalistica dei diversi temi di salute (il primo di-cembre, giornata mondiale dell’AIDS, l’anno del cuore, la settimana della preven-zione dentale, le campagne di raccolta fondi delle associazioni, eccetera).
La macchina della promozione della notiziaScopo dei media è di dare la notizia importante prima (e meglio) degli altri. Ilgiornalismo scientifico, come le altre forme di giornalismo, è una corsa contro iltempo. Su questomeccanismo si innestano una serie di promotori interessati dinotizie. Le prime sono le stesse riviste scientifiche internazionali, il cui prestigioe la cui ricettività pubblicitaria si accrescono in misura delle citazioni da partedei media. A questo fine è stato messo a punto un sistema di anticipazioni dinotizie, che, opportunamente enfatizzate da brevi comunicati stampa, vengonoinviate via mail alle redazioni dei giornali alcuni giorni prima dall’uscita del nu-mero della rivista scientifica, con un embargo (il divieto di divulgarli entro unacerta data) che teoricamente non dovrebbe essere rotto, pena l’esclusionedall’invio. Istituti scientifici pubblici, privati e aziende, con o senza la mediazio-ne di agenzie stampa, inviano a loro volta comunicati stampa, rinforzati da con-tatti personali con i giornalisti. A questo meccanismo si affianca, per gli eventipiù importanti, l’organizzazione di conferenze stampa, congressi e viaggi chemettono in contatto giornalisti e medici per promuovere con più efficacia le noti-zie e i prodotti di interesse.I viaggi pagati, talvolta di più giorni e in luoghi esotici, sollevano con particolareevidenza il problema dell’esistenza di conflitti di interesse anche per gli operato-ri dei media. Alcune aziende farmaceutiche, di fronte alla graduale presa di co-scienza del problema, hanno elaborato una strategia di coinvolgimento dei gior-nalisti anche e soprattutto su eventi “neutri”, nell’ambito dei quali consolidarecomunque una preziosa conoscenza e consuetudine con gli organi di informa-zione. Un caso classico è l’invito a seguire la consegna dei premi Nobel a Stoc-colma. C’è anche, insomma, chi vola alto. Ma non sempre è così.
Viva la pillola. Dai nostri inviati a Santo Domingo
E’ passato agli annali il caso di una pillola anticoncezionale(Yasmin) di nuova generazione lanciata alcuni mesi fa daiprincipali organi di informazione come “priva di effetti collate-rali”, laddove la letteratura più accreditata riferiva di numero-se segnalazioni di eventi fatali che avevano indotto i medici
olandesi e britannici a sconsigliarla alle pazienti. Alla scorret-tezza informativa si è aggiunta in questo caso il lato farsescorappresentato dal fatto che tutti gli autori degli articoli lauda-tori firmavano i loro articoli da un’isola tropicale dove si eratenuta la conferenza stampa della casa produttrice.
59
L’informazione in medicina: come destreggiarsi

La retorica dei comunicati stampaI comunicati stampa sono tra gli strumenti più comuni attraverso cui giornali,radio e televisione ricevono le informazioni – anche mediche – che poi verrannotrasmesse al pubblico. Sono di solito scritti e diffusi da riviste scientifiche, so-cietà scientifiche o da associazioni di pazienti, in occasione di congressi o di al-tri eventi che richiamano l’attenzione su argomenti specifici. In molti casi peròdietro i firmatari dei comunicati si celano uno o più sponsor che hanno fortiinteressi commerciali collegati alla malattia di cui si tratta. In questi casi le no-tizie sono presentate in termini volutamente allarmistici, in modo che anche isani si sentano malati e aumenti così il numero di potenziali acquirenti del pro-dotto per combattere uno specifico disturbo. Sul sito Partecipasalute è stato ela-borato un “gioco”, chiamato “generatore automatico di comunicati stampa”, cheevidenzia lo schema retorico di base di questo tipo di comunicazione: http://www.partecipasalute.it/informati-bene/generatore-comunicati-001.php.La struttura dei comunicati, insieme ai dati presentati e a qualche frase a ef-fetto, viene poi trasferita spesso in modo acritico all’articolo giornalistico o almateriale informativo che arriva al cittadino. In questo modo il messaggio deicomunicati riceve una risonanza amplificata in direzione dell’effetto volutoda chi li scrive. I comunicati di questo genere si articolano tutti secondo unostesso schema dettato dalle regole del marketing e adatto a colpire l’attenzio-ne del lettore:l ingigantire il problema affermando che tocca milioni di persone per lo più in-consapevoli;
l suscitare timori inducendo a credere che i rischi siano gravi, soprattutto senon si interviene tempestivamente;
l indurre a visite ed esami per creare potenziali malati/clienti;l banalizzare la soluzione, sostenendo che un nuovo prodotto è in grado di ri-solvere facilmente il problema.
Dai congressi ai media: studi preliminari che diventano notizieGli studi presentati a congressi internazionali e nazionali ricevono spesso gran-de risonanza sui giornali, anche quando si tratta di ricerche nelle fasi iniziali,che non hanno risultati definitivi e certi. Solo una parte di questi studi diffusidai media, una volta conclusi, viene pubblicata su riviste mediche, gli altri nonvengono pubblicati. Ricevono quindi grande attenzione di pubblico notizie di ri-sultati parziali e preliminari, che poi possono essere confermati, oppure posso-no essere smentiti o rovesciati dai risultati definitivi degli studi.Lo mostra in modo chiaro un’indagine condotta sulle notizie pubblicate nei prin-cipali giornali statunitensi nei due mesi successivi a cinque congressi svoltisi suAIDS, cardiologia, neuroscienze, oncologia clinica, radiologia (World AIDS Confe-rence e i congressi dell’American Heart Association, della Society for Neuroscien-ce, dell’American Society of Clinical Oncology, della Radiological Society of NorthAmerica). Sui giornali sono state pubblicate in due mesi oltre 250 notizie relativea studi presentati in questi congressi, dei quali la metà ha visto poi la pubblica-zione su riviste mediche autorevoli e prestigiose, un quarto su riviste mediche diprofilo inferiore, e un quarto non è mai stato pubblicato su riviste mediche (neitre anni successivi ai congressi). E’ interessante notare che gli studi delle notizieche hanno ricevuto la prima pagina dei giornali non sono quelli pubblicati poisulle riviste mediche più prestigiose. Insomma gli studi presentati ai congressiscientifici diventano notizie sui mezzi di informazione troppo spesso e troppo pre-sto rispetto ai risultati definitivi, col rischio che vengano divulgate notizie noncorrette. “Per ridurre la diffusione al pubblico di informazioni fuorvianti è neces-sario che chi interviene ai congressi sia molto chiaro nel comunicare ai media lanatura preliminare dei propri studi e i limiti che hanno” commentano gli autoridell’indagine; “da parte loro i giornalisti devono assumere un atteggiamento criti-co verso i dati presentati ai congressi e sottolineare che non sono definitivi, nonsolo per dare un’informazione corretta, ma anche per aiutare i lettori a sviluppareun senso critico verso le notizie che leggono”.1
Tutto questo potrebbe scontrarsi con uno dei principali obiettivi dei congressi
60
L’informazione in medicina: come destreggiarsi

che, se da un lato vogliono permettere e stimolare uno scambio e una discussio-ne tra medici e scienziati, dall’altro vogliono promuovere le attività del singoloricercatore o delle società scientifiche, dello sponsor del convegno o dell’enteche ha finanziato gli studi. Tra questi due interessi, in potenziale conflitto tra lo-ro, si pone la questione di quali informazioni scegliere di divulgare ai media e dicome farlo: entrare nel merito dei limiti degli studi e di risultati parziali ancorada verificare (o smentire), oltre a essere più difficile, non offre la stessa possibili-tà di attirare l’attenzione del pubblico e della comunità scientifica rispetto a da-re risultati come se fossero certi o molto promettenti. Anche per il giornalistadare la notizia di una nuova cura mirabolante è più appealing, anche se quasisempre non corrisponde al vero.
La stampa specializzata per i mediciL’editoria periodica rivolta ai medici costituisce un’industria che conta decine dimigliaia di riviste e siti internet. Tra le diverse testate si distinguono le grandi ri-viste mediche generaliste internazionali (New England Journal of Medicine,JAMA, British Medical Journal, The Lancet) sede del più qualificato aggiorna-mento medico-scientifico disponibile, i cosiddetti throw-away journal (giornaliusa e getta), veicoli ibridi di informazione scientifico-professionale e promozionepiù o meno mascherata, e testate critiche a forte contenuto basato sulle provescientifiche di efficacia, il cui scopo dichiarato è quello di fare informazione me-dica. Nella filiera della promozione di contenuti commerciali si distinguono ithrow-away-journal, che se da un lato fanno informazione sui temi di clinica,professione e politica sanitaria, e in quanto tali sono spesso strumenti utili e avolte insostituibili di aggiornamento, dall’altro possono veicolare informazionipromozionali, oltre che nelle pagine a questo dedicate (pubblicità e redazionali),in articoli giornalistici, di solito legati a eventi congressuali.Questi giornali sono distribuiti gratuitamente ai medici, e in questo modo le-gano strettamente la loro sopravvivenza ai piani pubblicitari delle case farma-ceutiche, le quali peraltro stanno abbandonando il settore per concentrare gliinvestimenti sulla forma più efficace e capillare di propaganda: quella degli in-formatori del farmaco. Benché il medico italiano non sia abituato a pagarsi gliabbonamenti ai giornali professionali, si stanno parimenti diffondendo te-state e siti internet che promuovono una vera e propria controinformazionesulla medicina e i farmaci, che vivono degli abbonamenti o di finanziamentipubblici. Per limitarsi al campo farmaceutico, ricordiamo il francese Prescrire(http://www.prescrire.org/), l’americano Worst Pills (http://www.worstpills.org/) dell’Associazione dei consumatori Public Citizen (http://www.citi-zen.org/), gli italiani Dialogo sui farmaci (http://www.dialogosuifarmaci.it/),il Bollettino italiano dei farmaci (che si raggiunge attraverso il sito http://www.agenziafarmaco.it).
Informazione critica ed errori comuni
Le associazionidi consumatori
Il mondo di internet, ma non solo, diffonde un gran numero di informazioni dimedicina elaborate dalle associazioni di volontariato. Tralasciando le pure in-formazioni di servizio o l’uso sempre più esteso delle rassegne stampa online,merita un’attenzione particolare il giornalismo critico esercitato in particolaredalle associazioni dei consumatori. Oltre all’esempio già ricordato di Public Ci-tizen, che negli Stati Uniti esercita un occhiuto controllo sui processi di appro-vazione e commercializzazione dei farmaci (Worst pills, Best pills), in Italial’Associazione Altroconsumo svolge una funzione di informazione critica suiprezzi ma anche sui temi dell’appropriatezza dei test e delle cure mediche conla testata Salutest. L’Associazione Cittadinanzattiva, a sua volta, con rapportiPIT (Progetto Integrato di Tutela dei diritti del cittadino) salute e altri interven-ti, sviluppa inchieste puntuali sui temi della qualità delle cure, in particolarenegli ospedali.Rivolto anche alle associazioni di pazienti, a cui dedica una sezione, il sito Parte-cipasalute intende fare informazione critica rivolgendosi al grande pubblico.
61
L’informazione in medicina: come destreggiarsi

Alcuni esempi di disinformazione in medicina: dalle rassicurazioni all’euristica del terrore
n Un caso recente di rassicurazione di una notizia medi-co-scientifica riguarda lo studio americano denominatoWomen’s Health Initiative che ha mostrato le ricadutenegative di tipo cardiovascolare della terapia ormonalesostitutiva della menopausa. In Italia queste notizie so-no arrivate al grande pubblico quasi sempre attraversoil filtro di autorevoli esperti italiani, che nel darel’informazione hanno sentito il bisogno di spiegare comei risultati statunitensi – di solito presi come oro colato –non fossero estensibili alle “donne italiane”, sia per il di-verso stile di vita, sia per il ricorso molto meno intensoalla terapia ormonale sostitutiva. Le posizioni di medici,specialisti e società scientifiche sull’uso della terapia or-monale non erano comunque concordi tra loro, tantoche le informazioni che sono arrivate alle donne sonostate contrastanti o parziali. Per portare chiarezza in ta-le contesto, è stata organizzata la conferenza di consen-so “Quale informazione per la donna in menopausa sullaterapia ormonale sostitutiva” promossa dal progettoPartecipasalute e dal Sistema nazionale linee guida(Istituto superiore di sanità). La conferenza, che si èsvolta il 16 e 17 maggio 2008 a Torino, ha prodotto unaserie di raccomandazioni sull’uso della terapia ormona-le sostitutiva e su quali informazioni dare alle donne(maggiori informazioni su: http://www.partecipasalu-te.it/cms_2/node/923), da divulgare tra gli esperti e trail pubblico.
n L’imbonimento prevale sul resoconto critico anchequando si parla di screening. Difficilissimo, per esem-pio, riuscire a condurre sulla stampa laica un ragiona-mento corretto sulle nuove offerte di screening di mas-sa: la stampa quotidiana e settimanale, così come latelevisione, si è fatta molto spesso veicolo promozio-nale del controverso screening della prostata, e recen-temente anche del tumore al polmone attraverso laTAC spirale, una nuova tecnologia che consentirebbe diindividuare le formazioni tumorali al loro nascere equindi permettere un intervento precoce e spesso riso-lutivo. A distanza di due anni da quando è stato pubbli-cato su una prestigiosa rivista internazionale lo studio
relativo a questa tecnologia2 un articolo pubblicato sulNew York Times ha denunciato che la fondazione cheaveva dato fondi per lo studio è stata interamente fi-nanziata da una multinazionale del tabacco. Questo hasollevato dubbi sulla possibile influenza della multina-zionale sui risultati dello studio, oltre che sui meccani-smi di trasparenza previsti dalle riviste scientifiche, cherichiedono una dichiarazione di conflitti di interesseagli autori degli articoli (). Nonostante esempi di gior-nalismo di inchiesta e di denuncia come questo, i me-dia in genere hanno difficoltà a entrare nel merito del-l’efficacia di un trattamento o di un intervento diagno-stico, o a distinguere un intervento ancora sperimenta-le da ciò che può essere considerata una tecnologia or-mai matura e appropriata. Nel caso degli screening poi,molto spesso mancano le informazioni chiare e quanti-tative sui benefici e i rischi connessi.
n I media sono molto sensibili alle promesse costituite daproposte futuribili come la terapia genica e le cellulestaminali. Le cellule staminali, di cui si parla da un de-cennio, continuano a essere rilanciate dai mezzi di infor-mazione sotto la pressione di istituti di ricerca come te-rapia salvavita, laddove le ricadute sono di là da venire.3
n La paura della pandemia aviaria è esplosa il 13 settem-bre 2005, a seguito di un congresso organizzato a Maltadalle società scientifiche sponsorizzate dai principaliproduttori di vaccino antinfluenzale e di antivirali. Inquella sede si danno le prime cifre dell’annunciato e“inevitabile” disastro sanitario: 16 milioni di italiani col-piti, 150 mila morti. A pochi giorni di distanza segue larassicurazione: in attesa di un vaccino pandemico chedovrebbe essere pronto entro pochi mesi, conviene vac-cinarsi con il prodotto normale (pensato per l’epidemiastagionale), cosa poi rivelatasi del tutto infondata. In unanno e mezzo di informazione terroristica sulla pande-mia, va registrata almeno una buona notizia, la crescitadel fatturato dei produttori di antivirali, prodotti di fattodestinati a uscire dal commercio per scarso consumo(maggiori informazioni all’indirizzo http://www.parteci-pasalute.it/informati-bene/influenza-001.php).
Le trappole in cui è facile cadereScrivere di salute non è facile, poiché presuppone il dominio del metodo scienti-fico e di quello giornalistico. La difficoltà principale sta da un lato nell’opera didivulgazione corretta della complessità scientifica, psicologica e sociale dei fattimedici e sanitari, e dall’altro nello specifico lavoro giornalistico dell’inchiesta.E’ utile a questo proposito una check list delle potenziali lacune del giornalismomedico, limitandoci agli aspetti più macroscopici. Conoscere queste trappoleconsente da un lato di leggere criticamente l’informazione medico-scientificacorrente, dall’altro di non cadere nelle stesse trappole quando si voglia cimen-tarsi in prima persona nella informazione.
Fidarsidei cosiddettiesperti
Spesso gli articoli e i servizi radiotelevisivi che parlano di salute sono costruiticome passerelle di esperti. Può succedere che ciò oscuri del tutto il disegnodell’articolo, riducendolo a una massa di opinioni. Questo processo è partico-larmente evidente nei servizi televisivi dove l’intervistato, rigorosamente in ca-mice bianco a rinforzo della funzione sciamanica del “dottore”, enumera le sueverità senza possibilità di replica o di contraddittorio. Questo può indebolire lacorrettezza delle informazioni. L’opinione degli esperti, infatti, spesso non è ag-giornata come si crede, o può essere distorta da interessi e pregiudizi.L’esperto, inoltre, può dare informazioni distorte da un conflitto di interessi nondichiarato. Uno studio recente ha mostrato che se una ricerca è finanziata daun’industria, le probabilità che le conclusioni siano pro industria sono quasi ilquadruplo di uno studio indipendente.
62
L’informazione in medicina: come destreggiarsi

Che cosa fare: mantenere un atteggiamento critico sulle affermazioni degliesperti e controllare sempre le fonti delle affermazioni.
Ecco un elenco di fonti da usare per controllare le affermazioni degli esperti:Fonti affidabili l http://www.ceveas.it (in lingua italiana). Centro per la valutazione dell’effi-
cacia dell’assistenza sanitaria, dipartimento dell’Azienda USL di Modena.Ha l’obiettivo di divulgare le migliori prove disponibili su trattamenti e cure,attraverso la valutazione critica dei dati, la loro comprensione e il trasferi-mento delle informazioni a medici, decisori e cittadini.
l http://www.partecipasalute.it/rubrica-8/ (in lingua italiana). Sono dispo-nibili, tra le altre, le rubriche “Pillole sì, pillole no”, curata dalla rivista Dialo-go sui farmaci, che monitora i nuovi farmaci in commercio; “Notizie Cochra-ne”, che presenta le migliori prove di efficacia disponibili su argomenti scelti;“Il sito della settimana” dove vengono recensiti alcuni siti di interesse (vedi ilcapitolo Navigare sulla rete alla ricerca di informazioni di salute).
l http://medlineplus.gov/ (in lingua inglese). E’ il sito per il pubblico dellapiù grande biblioteca medica del mondo, la National Library of Medicine.Fornisce informazioni basate su prove scientifiche su oltre 650malattie e di-sturbi di salute.
l http://www.besttreatments.org/btus/home.jsp (in lingua inglese). Riportai contenuti di Clinical Evidence per pazienti e medici. E’ organizzato per pa-tologie, con informazioni integrative su medicine complementari, esperienzepersonali, prescrizioni di farmaci.
l http://www.prescrire.org/ (in lingua francese)http://www.citizen.org/, http://www.worstpills.org/ (in lingua inglese)
Interrogarelo specialistasbagliato
Per pigrizia, malizia o mancanza di tempo, capita che il giornalista interroghil’esperto sbagliato. I ricercatori in campo biomedico in genere sono molto
specializzati e possono dire cose appropriate e aggiornate solo sul loro am-
bito di studio. Chiedere informazioni sulle nuove cure anticancro a un ricerca-tore che si occupa di studiare la proliferazione cellulare anziché a un oncologoespone al rischio di vere e proprie bufale.Che cosa fare: scegliere la persona da intervistare in base alla pertinenza degliargomenti su cui ha scritto con il tema dell’intervista; integrare, quando possi-bile, con il parere di un medico che abbia una visione ampia, anche se più vaga,del tema, come il medico di famiglia.
Confondere lascienza con lafantascienza
Sia alla televisione sia sui quotidiani e i rotocalchi, la notizia scientifica meritala prima pagina se prefigura una svolta epocale, quando non un miracolo; èmolto comune dare per terapie a portata di mano quelle che sono in realtà spe-rimentazioni allo stadio iniziale, forzando in sede di titolo e sommario, le ricadu-te positive del “progresso medico”. Oltre un terzo degli studi, pubblicati su rivi-ste prestigiose, che riportano risultati molto positivi o annunciano novità ven-gono successivamente smentiti o ridimensionati.4
Che cosa fare: sospettare di articoli o trasmissioni che usano le espressioni:guarigione, miracolo, innovativo, promettente, eccezionale, speranza, vittima.
Farsi ingannaredai numeri
Per mostrare che una notizia è oggettiva e attendibile si usa in genere una preci-sione esasperata nelle cifre che vengono riportate a sostegno dell’informazione.Le strategie usate più spesso in questo campo sono:l l’uso smodato di cifre (con più decimali) che annebbiano la mente del lettore;l l’uso del rischio relativo invece del più utile rischio assoluto. Nel caso delleterapie, esprimere infatti l’efficacia utilizzando una percentuale (riduzionerelativa del rischio) senza dare come riferimento il numero dei casi in cui si èridotto il rischio (assoluto) porta a fraintendere il reale impatto clinico deltrattamento.
Per esempio: un trattamento può ridurre il numero di pazienti che hanno un in-farto da 10 su 100 a 7 su 100. La riduzione in termini relativi è quindi del 30 percento (cioè di 3 persone, su 10 che hanno un infarto). In termini assoluti però lariduzione è pari al 3 per cento, cioè su 100 persone totali che prendono il farma-co, 3 non hanno un infarto.
63
L’informazione in medicina: come destreggiarsi

Che cosa fare:
l sospettare delle fonti informative che usano due decimali: per esempio, il18,63 dei casi…;
l sospendere il giudizio su una ricerca in cui non si riporti il campione su cui èstata condotta;
l privilegiare le informazioni che, insieme alla riduzione relativa del rischio,riportano anche quella assoluta;
l un buon modo per esprimere in modo maggiormente comprensibile l’efficaciadi un intervento è il cosiddetto “numero necessario da trattare” (number nee-ded to treat, NNT), vale a dire il numero di persone che è necessario trattareper ottenere una guarigione/prevenire un esito negativo (infarto, morte, ecce-tera). L’NNT è l’inverso della riduzione assoluta del rischio. Esempio: un certofarmaco ha ridotto il numero di ictus dal 3 per cento al 2 per cento. Il beneficioassoluto è quindi dell’1 per cento (1 caso su 100). L’NNT è uguale a 100: cioèbisogna trattare 100 persone per ottenere l’effetto desiderato in 1 paziente.
Glorie e miserie delle statistiche
I benefici ottenibili con un intervento si possono presenta-re in diversi modi, tutti egualmente corretti da un punto divista tecnico, ma con un effetto profondamente diversosul lettore, sia esso un medico che deve prescrivere, unpaziente che deve seguire una cura o un amministratoreche deve allocare le risorse.Facciamo l’esempio di un farmaco che si propone di pre-venire i malanni di cuore riducendo il tasso di colesterolonel sangue in chi lo ha alto.Per esprimere i risultati si può innanzitutto calcolare la fre-quenza delle morti, dopo un tempo sufficiente, nel gruppodi pazienti trattati rispetto a quello di controllo (costituitoda soggetti identici, ma senza il medicinale).In un esperimento pubblicato qualche anno fa il West ofScotland Coronary Prevention Trial (WOSCOPS), dopo 5anni i morti erano 3 per cento nel gruppo trattato e 4 percento in quello di controllo.
I due dati possono essere confrontati calcolando la diffe-renza: ovvero si può dire che la mortalità in assoluto è ca-lata dell’un per cento (4 meno 3).Questa misura è poco usata nelle pubblicazioni scientifi-che, probabilmente perché fornisce un’immagine dimessadei risultati. Gli autori degli studi preferiscono perciò ingenere calcolare di quanto cala in percentuale il rischio ri-spetto a quello del gruppo di controllo: nel nostro caso, ilcalo dell’un per cento rispetto al 4 di base equivale a unquarto, ovvero il 25 per cento (1 diviso 4).In questo modo, l’apparentemente misero risultato asso-luto si trasforma in una ben più corposa riduzione di mor-talità relativa. In altre parole, quando il rischio di base èbasso (come nel caso in questione, con una mortalità
nel gruppo di controllo del 4 per cento), anche un mode-sto beneficio assoluto rappresenta un cospicuo vantag-gio relativo.Solo la conoscenza di entrambe le espressioni consente diapprezzare la reale entità dell’effetto clinico che ci si puòaspettare. Ma vi sono altri modi di esprimere gli stessi risul-tati, che forniscono punti di vista ancora più interessanti.
Uno dei più utili è il numero di soggetti che si devono trat-tare per evitare un evento, in questo caso una morte.Nell’esempio del farmaco contro il colesterolo si salva, co-me già detto, un paziente ogni 100 trattati per 5 anni. Ilcosiddetto NNT (numero necessario da trattare) saràquindi pari a 100 (100 diviso 1).E’ un numero grande o piccolo? Ci si può fare un’idea, alme-no approssimativa, confrontando il dato con quello di altreterapie di uso corrente: per esempio, si devono trattare confarmaci per abbassare la pressione 70 pazienti anziani percinque anni per salvare una vita; oppure si deve sommini-strare aspirina sempre per cinque anni a 100 adulti maschisenza segni di mal di cuore allo scopo di evitare un infarto.
In generale, il numero di soggetti da trattare è una misurapiù vicina alla pratica clinica e consente anche di fare rapi-damente un calcolo approssimativo del costo per vita sal-vata di una pratica terapeutica. Poniamo che un anno diterapia contro il colesterolo costi circa mille euro per pa-ziente. Poiché è necessario trattare 100 pazienti per 5 an-ni per evitare un decesso, il costo del trattamento per sal-vare una vita è pari a circa 500 mila euro.
da Lettera a un medico sulla cura degli uomini,G. Cosmacini e R. Satolli, Laterza, 2003
Prenderegli aneddoticome prove
La forma di ragionamento non scientifico sorregge le proprie argomentazioni so-prattutto con aneddoti, che hanno una forza suggestiva molto forte, ma nessu-na validità statistica. Si può dare credito a un trattamento solo se è passato at-traverso un processo sperimentale fatto di test di sicurezza ed efficacia a vari li-velli (vedi il capitolo L’ABC della ricerca clinica).Le storie di singole persone sono utilizzate dai giornalisti esperti in modo seletti-vo, come strategia narrativa, senza farle passare per conoscenze scientifiche. Isingoli casi infatti possono illustrare gli effetti di un trattamento, ma non devo-no essere confusi con le prove. Chi legge deve ricordare che dagli aneddoti nonsono possibili generalizzazioni.
Non porrele giuste domandea uno studio clinico
Se il fondare le proprie argomentazioni sulle prove rappresenta un notevolepasso avanti rispetto al giornalismo basato sugli aneddoti o sulle opinioni degliesperti, è anche vero che raramente la cosiddetta “prova” viene esaminata criti-camente. Una singola sperimentazione clinica significa ancora poco perché oc-
64
L’informazione in medicina: come destreggiarsi

corre che il risultato venga replicato. Bisogna poi imparare a distinguere i diver-si tipi di sperimentazione, ognuno dei quali è dotato di una sua maggiore o mi-nore forza scientifica (dagli studi osservazionali a quelli randomizzati e control-lati, dalle revisioni sistematiche alle metanalisi (vedi il capitolo L’ABC della ricer-ca clinica).Anche il cosiddetto processo di peer-review (revisione dei pari), che consiste nelsottoporre un articolo scientifico alla lettura critica di uno o più esperti dellamateria per decidere se pubblicarlo o meno, non garantisce sulla qualità dei ri-sultati. Per mettere alla prova la capacità critiche della peer-review un articolocon 8 gravi errori è stato sottoposto a 420 revisori della rivista JAMA: nessunoha colto più di 5 errori, e la maggior parte non più di due.5
Purtroppo una consapevolezza della natura probabilistica e “retorica” dellascienza è ancora di là da venire.
Estrapolaredalla ricerca puraalla pratica clinica
Si usa spesso estrapolare risultati assolutamente preliminari (condotti su ani-mali o su una selezione molto stretta di pazienti) alla clinica, facendo sembrareormai dietro l’angolo terapie ancora lontane da un’applicazione efficace e gene-ralizzabile.Un’altra forma di estrapolazione scorretta di risultati clinici consiste nel confon-dere efficacia teorica di un trattamento con la sua efficacia sul campo.
Differenza fra efficacia teorica ed efficacia sul campo: il caso del vaccino antinfluenzale
Ogni anno, verso ottobre, i giornali escono con titoli sem-pre uguali, del tipo: “arriva l’influenza, 5 milioni di italiania letto”. In realtà, di questi presunti cinque milioni di per-sone, la quota di individui che si ammalerà di vera e pro-pria influenza varia di anno in anno, ma difficilmente su-pera un terzo del totale.Tutti gli altri malanni sono riconducibili a forme para-influenzali (da altri virus o batteri): i sintomi sono similie difficilmente distinguibili a una visita dal medico.Non c’è da stupirsi, allora, se fra coloro che prendonoil vaccino antinfluenzale solo una parte non si amma-lerà di “influenza”.A questo proposito, gli studi dicono che il vaccino ha
un’efficacia teorica (efficacy) compresa fra il 60-70 percento, ma un’efficacia sul campo (effectiveness) di ap-pena il 10-20 per cento. Che cosa vuole dire questo?L’efficacia teorica misura i casi in cui il vaccino ha evi-tato l’influenza nelle condizioni controllate di uno stu-dio clinico.L’efficacia sul campo, invece, misura quanti reali casi diinfluenza il vaccino riesce a prevenire in quella popolazio-ne “reale” colpita da sintomi influenzali (molti dei qualinon sono stati infettati dai virus influenzali bensì da altrimicrorganismi). Confondere queste due forme di efficaciaporta spesso l’informazione medica a sovrastimare la po-tenza dei farmaci.
La difficoltà di tradurre informazioni che arrivano dalla ricerca pura in cono-scenze applicabili alla ricerca sul campo non riguarda solo l’interpretazione daparte dei giornalisti, ma è intrinseca alla metodologia della ricerca stessa.Quando viene condotto uno studio, si può generare una situazione particolareche non corrisponde alla pratica clinica. Per esempio, la selezione dei pazientida coinvolgere può essere così stringente da non corrispondere alla realtà clini-ca: se uno studio dimostra che un trattamento è efficace in donne senza figlicon più di 50 anni di età, non si può concludere che sia efficace anche in altrigruppi di donne. Questo significa che i nuovi trattamenti devono anche esserestudiati nella pratica clinica prima di sapere come funzionano fuori dal contestodello studio. Alcune estrapolazioni sono comunque necessarie: non si può ri-chiedere di avere prove scientifiche da studi clinici per tutte le indicazioni possi-bili per ogni gruppo di pazienti prima di utilizzare un nuovo trattamento. Occor-re però cautela nelle generalizzazioni.
Enfatizzarele implicazioniclinichedi uno studio
Molti studi giungono a risultati che non cambiano il corso di una malattia;dimostrano semplicemente che, somministrando la tale sostanza, hannomodificato di poco parametri intermedi senza migliorare la qualità di vita delpaziente.Che cosa fare: accertarsi che lo studio prenda in considerazione gli esiticlinici importanti per la vita del paziente (come gli infarti, la mortalità o ledisabilità) e non solo degli esiti intermedi, tipicamente valori di laboratorioo segni fisici (per esempio glicemia, colesterolo, pressione, volume del tu-more) che si ritengono (a ragione o a torto) indicativi dell’evoluzione dellostato di salute.
65
L’informazione in medicina: come destreggiarsi

Scambiareun fattoredi rischioper una malattia
C’è la tendenza, favorita dall’industria della salute (aziende farmaceutiche, me-dici specialisti, televisione e stampa) a trasformare in malattia quelli che sonosemplicemente parametri fisiologici che indicano un rischio aumentato di con-trarre alcuni disturbi: per esempio un aumento di pressione arteriosa, zuccherie colesterolo nel sangue.Questo fenomeno, noto come “medicalizzazione”, si accompagna a un’offertasempre più insistente di screening e di misure di prevenzione primaria e secon-daria alla popolazione sana.“C’è un sacco di soldi da fare se si convincono le persone sane che sono mala-te” così comincia un articolo pubblicato sul British Medical Journal a firma diun giornalista, un medico di famiglia e un professore di farmacologia6, che ri-costruiscono l’influenza delle società farmaceutiche dietro l’invadenza dellamedicina.Il caso della rarefazione delle ossa (osteoporosi) – spiegano Roberto Satolli eGiorgio Cosmacini in Lettera a unmedico sulla cura degli uomini – “è un esempiodi come un fattore di rischio possa essere prima inquadrato come malattia, poidefinito in modo tale da essere applicabile a falangi di persone in buona salute.L’Organizzazione mondiale della sanità ha fissato i criteri di normalità facendoriferimento alla saldezza delle ossa delle donne giovani, in modo tale che il natu-rale processo di invecchiamento sia visto come patologico. Su questa base “leattività promozionali sostenute dall’industria tentano di persuadere milioni didonne sane nel mondo a considerarsi malate”, allo scopo di indurle ad assume-re rimedi costosi che, in barba alla propaganda aggressiva, forniscono nel mi-gliore dei casi solo vantaggi marginali”.
Presentarein modo alteratoi rischi
Spesso i media enfatizzano fonti di rischio con scarso o nullo impatto sanitario,e trascurano di dare un’informazione adeguata su rischi rilevanti. Stampa e te-levisione privilegiano fonti di rischio per le quali si possa facilmente trovare uncolpevole o si possa sospettare un tentativo di censura; che coinvolgano perso-naggi famosi o che possano essere letti come anticamera a guai maggiori; che,pur essendo in realtà poco nocivi, sono molto diffusi.I media influenzano la percezione del rischio che ha il pubblico, come hannomostrato i casi della SARS e dell’influenza aviaria, dove il rischio è stato amplifi-cato al punto tale che in pochi giorni si è registrato una rincorsa alla vaccinazio-ne, anche in assenza di un vaccino efficace,oltre che un calo repentino di con-sumi di carne di pollo (http://www.partecipasalute.it/cms_2/node/271).Nonostante il modo diverso di percepire i rischi da persona a persona, in generepreoccupano di più e sono meno accettabili i rischi:l involontari (es. l’esposizione all’inquinamento) rispetto a quello volontari(es. sport pericolosi o il fumo);
l distribuiti in modo diseguale (alcuni ne beneficiano mentre altri ne soffronole conseguenze);
l ineludibili, anche prendendo precauzioni;l con origini non note o nuove derivanti da cause umane e non da fontinaturali;
l che causano danni nascosti e irreversibili (es. determinano l’insorgere dimalattie molti anni dopo l’esposizione);
l che colpiscono categorie considerate particolarmente meritevoli di attenzio-ne e tutela come bambini, donne e anziani;
l poco compresi dalla scienza;l oggetto di affermazioni contraddittorie da parte delle diverse autorità sani-tarie e ambientali (o, peggio, della stessa fonte).
E’ difficile per chi non lavora con i numeri capire la grandezza di un rischio seviene espressa in percentuali o in termini di probabilità. Ci sono modi differentidi comunicare un rischio, che utilizzano scale o figure, e che rendono più facilela lettura dei dati numerici. In ogni caso, i media dovrebbero sempre specificarequali categorie di persone sono esposte al rischio di cui parlano, cercando diesprimere in modo comprensibile quante probabilità hanno di incorrere nellamalattia o nella condizione in questione. Fare dei paragoni con altri rischi puòaiutare i lettori a decidere quanto quel rischio sia reale per loro.
66
L’informazione in medicina: come destreggiarsi

L’incertezza in medicina, poco raccontata dai mass media
Una delle maggiori difficoltà che i media incontrano è quella di trasmettere alpubblico il carattere intrinsecamente incerto delle decisioni mediche, poichéquesto confligge con il lavoro di estrema semplificazione che viene richiesto algiornalismo. La scarsa consapevolezza dell’incertezza in medicina non si puòcomunque addebitare specificamente ai media, dal momento che essa è un te-ma poco conosciuto dagli stessi medici.Un esempio di questa incertezza riguarda la fascia di età in cui le donne dovreb-bero sottoporsi a mammografia. Nel gennaio 1997 un gruppo di esperti dei Natio-nal Institutes of Health si espresse contro lo screening per le donne tra i 40 e i 50anni di età; la raccomandazione venne ribaltata pochi mesi dopo dal NationalCancer Institute, che indicò l’opportunità di sottoporsi a screening in quella fa-scia di età. Nonostante che dalla maggior parte delle notizie apparse su giornali etelevisioni statunitensi (67 per cento) riguardo alle raccomandazioni espresse daiNational Institutes of Health emergesse un ampio margine di incertezza su que-sto esame diagnostico per la fascia di età in questione, molte suggerivano alla fineche comunque fosse probabilmente utile e opportuno anche per le donne tra i 40e i 50 anni.7 Solo quattro notizie esprimevano la necessità che le donne facesserouna scelta attenta, in mancanza di indicazioni definitive. Il livello di incertezzaespresso, già basso per le notizie derivate dalle raccomandazioni contrariedell’NIH, scompare quasi del tutto nelle notizie che riportano la posizione dell’NCI(favorevole), con un 96 per cento che dichiara l’opportunità dello screening inquella fascia di età.Laddove esiste controversia e incertezza i mezzi di informazione spesso privile-giano una fonte che consente un’informazione più chiara, semplice e priva diincertezza, a sfavore della correttezza e della completezza.
Benefici ed effetti collaterali dei farmaci sui mezzidi informazione: un rapporto non bilanciatoI farmaci vengono trattati dai mezzi di comunicazione di massa spesso in modoparziale e acritico, anche a causa delle fortissime pressioni promozionali pre-senti in questo campo. Un’informazione sbilanciata e incompleta sui farmacipuò essere peraltro pericolosa, inducendo la popolazione a un consumo ecces-sivo e non corretto delle terapie. Per questo motivo, negli ultimi anni si sonomoltiplicati i tentativi da parte di associazioni e istituzioni sanitarie di mettere apunto semplici raccomandazioni da seguire allorquando si parli di farmaci.Ecco le principali domande da porsi per una corretta informazione:
Indicazioni Sono specificate le indicazioni per cui il farmaco è registrato e approvato dalleagenzie regolatorie di riferimento (l’Agenzia europea per i medicinali – EMEA –per l’Europa e la Food and drug administration – FDA – per gli Stati Uniti)?L’approvazione delle agenzie regolatorie fornisce la garanzia che esistano proveche il farmaco ha qualche effetto benefico per le indicazioni per cui è approvato.
Controindicazioni Viene esplicitato quali persone potrebbero avere più danni che benefici dall’as-sunzione del farmaco?
Benefici clinici I benefici descritti hanno un impatto tangibile e importante sulla salute dei pa-zienti? O riguardano benefici non clinici come per esempio l’abbassamento dellivello del colesterolo nel sangue invece della riduzione del rischio di avere infar-ti del miocardio?
Danni clinici Sono descritti gli effetti avversi del farmaco? sono trattati con la stessa precisio-ne e completezza con cui sono descritti i benefici?
Grandezze e cifre Sono presenti numeri che spieghino in modo non ambiguo la grandezza dei be-nefici e dei danni?
Numeri assoluti La differenza tra benefici e danni è espressa in numeri assoluti? I rischi relativipossono generare molti fraintendimenti. In genere ogni numero superiore a 10per cento è un numero relativo. I giornalisti dovrebbero sempre esprime le gran-dezze di benefici e danni in numeri assoluti.
67
L’informazione in medicina: come destreggiarsi

Tempo Per quanto tempo un farmaco o un trattamento devono essere seguiti per avere unbeneficio? come cambia il rapporto benefici/danni durante terapie a lungo termine?
Alternativefarmacologichee non
L’informazione deve riportare le alternative al farmaco o al trattamento in que-stione, siano esse altri farmaci o interventi non farmacologici, indicando benefi-ci e effetti avversi, per dare possibilità di scelta ai pazienti.
Costo Il prezzo del farmaco o della terapia è indicato nell’articolo?
Disegnodello studio
È precisato il tipo di metodo di ricerca usato? Non tutti gli studi sono uguali, eun’indagine di opinione su 100 persone è meno attendibile e sicura di uno studioclinico randomizzato su 1.000 persone. Inoltre, la pubblicazione dello studio suriviste medico scientifiche sottoposte a revisione dei pari, anche se non garanti-sce che i risultati dello studio in merito di sicurezza ed efficacia del farmaco sianodi alta qualità, offre una garanzia maggiore rispetto ai dati presentati ai convegnio ai congressi o pubblicati su riviste non sottoposte a revisione dei pari.
Risultatipreliminari
Viene chiarito se i risultati degli studi riguardano fasi preliminari come quelle inlaboratorio o su animali? È chiaro che esiste una distanza tra l’eventuale scoper-ta e la pratica clinica? È importante anche che siano riportati giudizi fondati e ar-gomentati sulla qualità della ricerca (numerosità, durata, disegno dello studio).
Dichiarare nomie fonti
Per essere trasparenti nei confronti del lettore è necessario che il giornalista indichi inmodo esplicito, indipendentemente dal tono positivo o negativo della notizia, le azien-de chepropongonoprodotti farmaceutici o sponsorizzano studi o ricerche. Le fonti chehanno interessi particolari non sonodi per sé inattendibili,madovrebbero essere cita-te e confrontate sempre con fonti indipendenti o portatrici di interessi contrapposti.
Seguireil percorsodei soldi
Sono dichiarati i conflitti di interesse eventuali degli autori dello studio e glieventuali finanziamenti allo studio da parte dell’industria farmaceutica? Segui-re il percorso dei soldi nella ricerca sui farmaci quando si scrive di medicina esalute è importante come quando si scrive di politica.
Informazionimancanti
Se qualcuno dei punti precedenti non è disponibile, il pubblico è avvisato dellasua mancanza e dell’impatto che può avere sull’interpretazione dei risultati?Dare informazioni incomplete ai lettori può essere fuorviante come dare infor-mazioni non accurate.
Conflittidi interesse
Chi scrive ha interessi in conflitto con la finalità di informare i lettori inmodo cor-retto? Per esempio riguardo ad accettare inviti a convegni organizzati in luoghiesotici con viaggi spesati esistono posizioni contrastanti che vanno dal rifiuto daparte del giornalista di qualsiasi forma di sponsorizzazione e quindi di qualsiasipartecipazione a congressi, considerati fonti non utili di informazione, all’accetta-zione di inviti a congressi a fronte di rapporti chiari e trasparenti con le industriefarmaceutiche che li organizzano. In ogni caso è ritenuto importante stabilireall’interno delle redazioni regole chiare a questo riguardo, che siano rese note.
Farmaci: un’informazione reticente
Abbassare il livello di colesterolo, prevenire l’osteoporosi eprevenire i disturbi cardiovascolari: è sui tre gruppi di farmaciusati per queste condizioni (rispettivamente pravastatina,alendronato e bifosfonato, aspirina) che è stata condottaun’indagine su quattro anni di notizie – dal 1994 al 1998 –apparse su giornali e alle televisioni statunitensi.8 Su oltre200 notizie trovate, il 40 per cento non quantifica i beneficidei farmaci, poco meno della metà ne menziona i potenzialidanni ai pazienti e il 30 per cento ne riporta i costi. Oltre a ciò,tra le notizie che citano un esperto o uno studio scientifico(170 in tutto), la metà ne cita almeno uno con legami finan-ziari con l’industria produttrice del farmaco, dichiarando taleconflitto di interessi in meno del 40 per cento dei casi.Un’indagine canadese recente9 considera le notizie apparsedurante il 2000 sui 24 quotidiani più diffusi nel paese riguardoa cinque farmaci che richiedono prescrizione medica, lanciatisul mercato tra il 1996 e il 2001 (atorvastatina, celecoxib,donepezil, oseltamivir e raloxifene). Su oltre 350 articoli tro-
vati, 190 riportano almeno un beneficio o un danno, e traquesti i benefici sono riportati quasi cinque volte di più deglieffetti avversi; entrambi comunque per la maggior parte del-le volte non vengono quantificati. Negli articoli che li quantifi-cano, un quarto utilizza termini relativi, esponendo chi leggeal rischio di fraintendere la reale portata di tali effetti. Le al-ternative farmaceutiche disponibili sono menzionate in menodella metà degli articoli; il costo del farmaco è dichiarato inmeno di un terzo e solo il 35 per cento indica quanti pazientisono stati coinvolti negli studi. Infine, poco più di un quarto de-gli articoli include informazioni su chi ha finanziato le ricerche.In Italia la situazione non èmigliore: da un’indagine condottanell’ambito delle Conferenza di consenso “Quale informazio-ne per la donna inmenopausa sulla terapia ormonale sostitu-tiva” (vedi box Alcuni esempi di disinformazione in medicina:dalle rassicurazioni all’euristica del terrore) su oltre 220 arti-coli che trattano di terapia ormonale sostitutiva, risulta chepiù del 40 per cento non tratta dei rischi della terapia.
68
L’informazione in medicina: come destreggiarsi

Come valutare la qualità dell’informazione
L’analisi della qualità delle informazioni in medicina è ormai una disciplina svi-luppata, con le sue regole. Tralasciando i dettagli, va notato innanzitutto ciò cheaccomuna i diversi metodi di analisi: per essere giudicato di buona qualità untesto deve essere completo, bilanciato, corretto.La completezza si misura dalla percentuale di argomenti reputati essenziali daun gruppo di lavoro di esperti su un dato argomento; l’equilibro si valuta an-dando a vedere se l’estensore del testo giornalistico ha sentito pareri di diversiesperti, con posizioni diverse, sul tema in questione; la correttezza si valutaconsiderando il tasso di errori fattuali (dati, statistiche, spiegazioni scientifiche)contenuti nell’articolo.Un fattore che condiziona la completezza, e in parte anche le altre due qualità, èla lunghezza dell’articolo (o della durata del servizio radiotelevisivo), ragione percui le valutazioni quali-quantitative degli articoli relative a uno stesso tema ven-gono talvolta normalizzate sulla lunghezza dei testi espressi in parole.
Il programmaHealthNewsReview
Sono numerosi gli studi condotti negli ultimi anni, soprattutto in Canada, Au-stralia e Stati Uniti, rivolti all’analisi della qualità informativa di articoli, comu-nicati e programmi televisivi di salute. L’esperienza più recente e significativa èstata condotta nell’ambito del programma HealthNewsReview, in cui dal 2006al 2008 sono stati analizzati 500 articoli selezionati dai 50 principali quotidiani,dai 3 principali settimanali, agenzie di stampa e televisioni statunitensi sui temidi salute. Presupposto perché l’articolo venisse selezionato era che parlasse diun farmaco, di un prodotto o di una procedura medica.A questi testi è stato applicata una griglia di valutazione, condivisa da analogheesperienze canadesi e australiane, che si compone di dieci punti, ritenuti essen-ziali per una buona informazione medica.Ecco i 10 punti della valutazione di qualità di un articolo:1. La discussione dei costi del prodotto2. La quantificazione dei benefici, in termini assoluti e non solo relativi3. La spiegazione e quantificazione dei potenziali danni4. La comparazione del prodotto con le alternative esistenti5. Il sentire una pluralità di fonti, mai una sola6. Il rifuggire dal disease mongering (trasformando una condizione normale
in una malattia, esagerando l’incidenza di una malattia, eccetera)7. Analizzare il metodo e la qualità delle prove portate dagli studi esaminati8. Verificare la reale novità diagnostica o terapeutica del prodotto rispetto a
quelli esistenti9. Verificare la disponibilità reale del prodotto (esistenza di una approvazione
dell’agenzia regolatoria, entrata nel mercato eccetera)10.Utilizzare i comunicati stampa del produttore (o dell’ente che ha eseguito
la ricerca) solo come elemento di informazione fra gli altri, non limitandosia questa fonte.
Alla luce di questa griglia i revisori di www.Healthnewsreview.org (tre per ogni ar-ticolo) hanno valutato come non soddisfacenti il 62-77 per cento degli articoli,poiché non rispettavano lamaggioranza di questi criteri. 41 articoli su 500 hannocomunque soddisfatto tutti e dieci i requisiti, meritandosi sul sito cinque stelle.10
La logica dell’iniziativa, dell’Università del Minnesota, è che tutti i giudizi ven-gano messi online nonché spediti ai giornalisti, che in linea di massima li han-no apprezzati lamentando la mancanza di specializzazione, di tempo e di spa-zio (lunghezza degli articoli). Questo dovrebbe portare a una presa di coscienzadella cattiva qualità dell’informazione medica e di un suo progressivo migliora-mento. Secondo Ray Moynihan, giornalista scientifico, questo starebbe già av-venendo, come dimostra la diffusione di articoli sul disease mongering regi-strati dal 2006 in poi.
Le domande da porsi sulla qualità degli articoliPer facilitare l’analisi critica delle informazioni in medicina, Partecipasalute hamesso a punto alcune liste di controllo utili a ricordare gli elementi principali di
69
L’informazione in medicina: come destreggiarsi

valutazione della qualità informativa. Essi variano a seconda che il documentoda esaminare sia un sito (“misurasiti”) o un testo (“misuratesti”).Ecco le dieci relative a un articolo:1. cita gli autori degli studi e le relative istituzioni, e dice dove sono stati pub-
blicati o presentati, cioè cita la fonte da cui è stata presa la notizia?2. allarma il lettore enfatizzando la pericolosità e la diffusione della malattia?3. fa riferimento a possibili conflitti di interesse, dice chi ha finanziato lo stu-
dio, il convegno o il farmaco oggetto dell’informazione?4. chiarisce se la cura è una novità?5. informa circa la disponibilità della cura in Italia?6. cita le alternative terapeutiche?7. cita gli studi da cui emergono le prove favorevoli al nuovo farmaco/terapia?8. quantifica i benefici terapeutici utilizzando siamisure relative sia assolute?9. indica gli effetti collaterali del farmaco/terapia?10. indica il costo delle cure?
Bibliografia1. Schwartz LM, Woloshin S, Baczek L. Media coverage of scientific meetings.
Too much, too soon? JAMA 2002;287(21):28592. The International Early Lung Cancer Action Program Investigators.Survi-
val of patients with stage I lung cancer detected on CT screening. N Engl JMed 2006;355:1763-71
3. Milano G, Palmerini C. La rivoluzione delle cellule seminali. Milano: 2005,Feltrinelli
4. Ioannidis JPA. Why most published finaling are false. Plos Med2005;2(8):e124
5. Godlee F, Gale C, Martyn C. Effect on the Quality of Peer Review of Blin-ding Rewiewers. Jama 1998;280:237-240
6. Moynihan R, Heath I, Henry D. Selling sickness: the pharmaceutical in-dustry and disease mongering. BMJ 2002;324(7342):886-91
7. Schwartz LM, Woloshin S. News media coverage of screening mammographyfor women in their 40s and tamoxifen for primary prevention of breast cancer.JAMA 2002;287(23):3136-42
8. Moynihan R, Bero L, Ross-Degnan D et al. Coverage by the news media ofthe benefits and risks of medications. N Engl JMed 2000;342(22):1645- 50
9. Cassels A, HughesME, Cole C et al. Drugs in the news: an analysis of Cana-diannewspaper coverage of newprescription drugs. CMAJ2003;168(9):1133-7
10.Schwitzer G. How do US Journalists cover treatments, tests, products, andprocedures? An evaluation of 550 stories, Plosmedicine 2008:5;e95
Bibliografia di approfondimentol Partecipasalute. http://www.partecipasalute.it/cms_2/informati_benel Cosmacini G, Satolli R. Lettera a un medico sulla cura degli uomini. Ro-ma-Bari: Laterza, 2003
l Lewis R. Medical Journalism. Iowa State University Press, 2001l AAVV. Malati di parole. L’informazione e la comunicazione come terapia: atticonvegno. Azienda per i servizi sanitari medio Friuli
l Vineis P. Nel crepuscolo della probabilità. Torino: 1999, Einaudi
70
L’informazione in medicina: come destreggiarsi

Navigare sulla rete alla ricercadi informazioni di saluteVanna Pistotti, Eugenio SantoroRevisori: Dafne Rossi, Carmela Mandas
Che cosa sono le banche dati
Le riviste scientifiche sono il luogo in cui si rende conto pubblicamente dei reso-conti dei continui aggiornamenti, progressi, scoperte e verifiche in uno specificoambito come quello medico. Per recuperare queste informazioni gli operatori delmondo della sanità possono impiegare strumenti che, in tempi rapidissimi, in-dividuano le fonti (articoli, atti di convegni, pubblicazioni governative, tesi di la-urea, eccetera) riguardanti uno specifico argomento. Questi strumenti sono lebanche dati: archivi informatici tematici relativi a campi disciplinari, che oppor-tunamente interrogati forniscono i dati identificativi del documento/i sull’argo-mento desiderato (titolo, autore/i dell’articolo, volume, anno, pagine della rivi-sta in cui è stato pubblicato, riassunto dell’articolo, descrittori).
La strategia di ricerca nelle banche dati
Formularedomande precise
Sebbene oggi l’accesso alle informazioni sia facile e veloce, onde evitare di rima-nere sommersi da una mole di notizie di scarsa rilevanza, è necessario utilizzareuna metodologia di ricerca che selezioni informazioni davvero utili per la propriaricerca. Il primo passo per un’efficace ricerca è quello di identificare il problemache vogliamo risolvere e formulare una domanda precisa circa l’argomento su cuisi vogliono reperire informazioni, Partendo da una situazione complessa e artico-lata quale può essere quella di un paziente che si vuole documentare su un certoargomento, è necessario tenere in considerazione i seguenti criteri per la formu-lazione delle ricerche e la selezione delle informazioni reperite:l caratteristiche cliniche rilevanti del paziente (età, sesso, quadro clinico, fat-tori prognostici rilevanti, malattie concomitanti);
l tipo di studio al quale si è interessati (eziologia, fattori di rischio, utilità di untest diagnostico, efficacia di un trattamento, superiorità di un trattamentorispetto a un altro, effetti collaterali di un trattamento);
l misura di risultato alla quale si è interessati (mortalità, tasso di incidenza diuna malattia, risoluzione/ miglioramento della sintomatologia, frequenzarecidive, accuratezza di un test).
I descrittori Le principali banche dati indicizzano gli articoli scientifici utilizzando un thesa-urus, ossia un vocabolario di termini controllati (descrittori), standardizzati ecorrelati tra loro che descrivono gli argomenti trattati.
La ricerca a testolibero
La ricerca free text consiste nel cercare liberamente una o più parole all’internodell’intero record, senza limitazione di campi . Questo tipo di ricerca richiede unpo’ di lavoro di messa a punto (di solito la prima esecuzione della ricerca non èla migliore); per ottenere un buon risultato è necessario utilizzare anche sinoni-mi che esprimono lo stesso concetto, singolari/plurali, e considerare che la lin-gua con cui si opera costituisce un criterio di pre-selezione dell’insieme di testisu cui la ricerca potrà dare esito. In questo tipo di ricerca, che generalmente èpiù generoso di risultati ma meno mirato, si ottiene una maggiore selezione fa-cendo uso dei cosiddetti operatori booleani (and, or, not) i quali permettono diassociare (and), sommare (or) o escludere (not) più concetti.Esempio:l Search breast cancer AND tamoxifen
l Search breast cancer OR tamoxifen
l Search breast cancer NOT tamoxifen
71
Navigare sulla rete alla ricerca di informazioni di salute

La prima ricerca (AND) permetterà di recuperare solo quegli articoli in cui il far-maco, il Tamoxifen, viene usato per la cura del tumore della mammella.La seconda ricerca (OR) sommerà tutti gli articoli che trattano qualsiasi proble-ma relativo al tumore della mammella, dalla chirurgia alla prevenzione eccete-ra, e quelli relativi al farmaco, tamoxifen, sia che venga usato per curare il tu-more oppure per altri scopi. Da qui il numero molto elevato degli articoli e lascarsa precisione della ricerca.La terza ricerca (NOT) escluderà il trattamento con tamoxifen nella cura del tu-more della mammella. Vi è da dire che l’operatore NOT èmolto pericoloso, e nor-malmente sconsigliato.Alla luce di quanto si è detto, quindi, è importante, quando vogliamo interrogareuna banca dati, “spendere” un po’ di tempo nel leggere le istruzioni che spiega-no come la banca dati sia stata strutturata in modo da formulare correttamenteil quesito sul quale cerchiamo risposte, recuperando così questo tempo con unbuon livello di pertinenza dei risultati.
Google e PubMedSe si vuole effettuare la ricerca nella letteratura medico scientifica, è più oppor-tuno utilizzare PubMed. Si tratta di una banca dati pubblicata in lingua inglesema che ha un’interfaccia abbastanza semplice (in origine era stata pensata co-me strumento per il cittadino e il paziente). L’accesso è libero e gratuito se ci silimita alla ricerca bibliografica e alla lettura dei riassunti degli articoli scientifici(abstracts) quando disponibili; l’accesso ai testi completi degli articoli scientificiè invece possibile solo se si è abbonati al servizio, piuttosto costoso.Google (anche nella sua componente di Google Scholar) risulta meno efficace diPubMed nella ricerca di studi scientifici. Google Scholar infatti, sebbene ricer-chi tra fonti diverse (PubMed, riviste mediche, tesi, libri, eccetera), non è esau-stivo, non permette di usare i descrittori che PubMed usa per classificare gli ar-ticoli (termini MeSH) e, soprattutto, non è dotato di strumenti per filtrare e sele-zionare la letteratura di interesse offerti invece da PubMed. Google permettel’accesso a documenti di carattere divulgativo, a documenti pubblicati su sitiistituzionali, a documenti di organizzazioni, associazioni, eccetera.Per accedere ai dati di una sperimentazione clinica, contenuti di una linea gui-da o, più in generale, informazioni mediche che risiedono codificate e struttura-te in appositi database è consigliabile l’uso di specifici strumenti disponibili sulweb come illustrato più avanti in questo stesso capitolo.
Le banche dati in medicina
Medline Le banche dati mediche sono più di un centinaio. Medline è certamente la più co-nosciuta. Prodotta dalla National Library of Medicine, la più grande e importantebiblioteca medica nel mondo, ha oggi una versione gratuita su internet chiamataPubMed (http://www.pubmed.gov) che non è solo l’interfaccia web di Medline
72
Navigare sulla rete alla ricerca di informazioni di salute
Figura 1: Elencodi documenti reperiticon PubMed

ma è anche un archivio più ampio che si contraddistingue per la rapidità di ag-giornamento. Ogni giorno sono inserite nella banca dati migliaia di citazioni bi-bliografiche, alcune delle quali si riferiscono addirittura ad articoli non ancorapubblicati. Come detto, attraverso abbonamenti si può avere accesso al testocompleto dell’articolo, e da poco tempo anche a capitoli di libro, creando un pun-to d’incontro importante tra il concetto tradizionale di bibliografia e quello orien-tato al recupero fisico del documento.
Figura 2: Visualizzazione di un documento con PubMed
Cochrane Library Cochrane library (http://www.cochrane.org) è un database elettronico chemettea disposizione le “revisioni sistematiche” della letteratura scientifica, a oggi più di4.500, che rappresentano una sintesi della letteratura sulla efficacia e sicurezzadegli interventi sanitari di tipo preventivo, terapeutico e riabilitativo. Prodottodalla Cochrane Collaboration, Cochrane Library pubblica trimestralmente:l revisioni regolarmente aggiornate sull’efficacia dell‘assistenza sanitaria;l valutazioni e riassunti strutturati di revisioni sistematiche pubblicate sullemaggiori riviste;
l informazioni bibliografiche su oltre 450.000 studi clinici controllati;l informazioni sui Gruppi Collaborativi di Revisione e altre entità della Cochra-ne Collaboration;
l riferimenti a Internet per ulteriori informazioni sull’efficacia degli interventisanitari.
Dal gennaio 2000 sono disponibili anche i riassunti delle singole revisioni scrittein linguaggio più comprensibile e divulgativo rivolte a pazienti e consumatori.Mentre la consultazione della Cochrane Library richiede un abbonamento, le sin-tesi si possono leggere gratuitamente all’indirizzo Cochrane: www.cochrane.org
Embase Embase (http://www.embase.com) è una banca data in lingua inglese con in-formazioni sul farmaco, prodotta da Elsevier, importante editore medico-scien-tifico. L’accesso è a pagamento.
Cinahl Cinahl (http://www.cinahl.com) è una banca dati in lingua inglese su temi col-legati con l’infermieristica, prodotta dall’associazione statunitense degli infer-mieri. L’accesso è a pagamento.
73
Navigare sulla rete alla ricerca di informazioni di salute

I registri di sperimentazioni cliniche
I registri di sperimentazioni cliniche offrono in Internet informazioni sulle speri-mentazioni cliniche, nazionali e internazionali che sono pianificate, in corso oconcluse. Registrare gli studi clinici in questi archivi consente di fornire agli inte-ressati informazioni sullo stato della ricerca clinica condotta a livello internazio-nale, di promuovere la collaborazione tra ricercatori, di favorire il reclutamentodei pazienti nelle sperimentazioni in corso, di prevenire la duplicazione degli stu-di clinici e, non meno importante, di prevenire il finanziamento di ricerche similia quelle già in atto. Al fine di sensibilizzare una maggiore trasparenza nella ricer-ca (e unamaggiore completezza dei registri), negli ultimi anni gli editor delle mag-giori riviste mediche internazionali tra cui NewEngland Journal of Medicine, Lan-cet, Journal of the American Medical Association e British Medical Journal si sonoimpegnati a non pubblicare più i risultati di quelle sperimentazioni cliniche chenon sono state preventivamente registrate in un registro pubblico.Esistono numerosi registri di sperimentazioni cliniche. Alcuni riguardano spe-cifiche patologie, altri, più generali, coprono tutte le aree mediche. La tabella 1
fornisce le indicazioni per accedere ai principali registri disponibili sul web.
TABELLA 1: Esempi di registri di sperimentazioni cliniche accessibili in Internet
ClinicalTrials.gov Tra i registri da segnalare, è importante citare ClinicalTrials.gov (figura 3), il no-to registro delle sperimentazioni cliniche sviluppato alla fine degli anni 90 dallaNational Library of Medicine in collaborazione con la Food and Drug Admini-stration, che oggi presenta circa 60.000 sperimentazioni cliniche condotte in ol-tre 150 paesi.
FIGURA 3: Home page del sito Clinical Trials
OsSC In Italia è invece disponibile l’Osservatorio nazionale sulla sperimentazione cli-nica dei medicinali (OsSC), un registro gestito dall’Agenzia italiana del farmaco(AIFA) che fornisce informazioni sulle sperimentazioni cliniche condotte in Ita-lia, e che ora è aperto al pubblico.
74
Navigare sulla rete alla ricerca di informazioni di salute
Nome registro Indirizzo Area medica
Stroke Trials Registry http://www.stroketrials.org ictus cerebrale
AIDSinfo - Clinical Trials http://www.aidsinfo.nih.gov/clinical_trials HIV/AIDS
Cancer.gov - Clinical Trials http://www.cancer.gov/clinicaltrials oncologia
DEC-NET http://dec-net.marionegri.it pediatria
ClinicalTrials.gov http://www.clinicaltrials.gov tutte
OsSC https://oss-sper-clin.agenziafarmaco.it tutte (nazionali)
ISRCTN register http://www.isrctn.org tutte

Gli indici medici
Guideper individuaresiti affidabili
La ricerca di informazioni mediche sul web non è cosa semplice. A maggior ra-gione non lo è la ricerca di informazioni mediche/sanitarie affidabili. Distri-carsi e saper scegliere tra le numerose offerte presenti sulla rete richiede unapreparazione e una competenza spesso fuori dal comune. C’è chi ha affrontatoquesto problema fornendo gli strumenti per imparare a conoscere i siti affida-bili (per esempio il sito di Partecipasalute, con la sezione “Misurasiti” all’URLhttp://www.partecipasalute.it/cms_2/node/18 e la rubrica “Il sito della set-timana” all’URL http://www.partecipasalute.it/cms_2/buonsito) e chi ha re-alizzato su Internet delle guide ai siti. Tali guide, chiamate anche indici medi-ci, sono aggiornate manualmente attraverso un processo di selezione dei sitiweb che riprendono criteri di qualità e di affidabilità riconosciuti a livello inter-nazionale. I siti web segnalati sono organizzati in base alla specialità medica (oalla malattia), e alla tipologia. Per esempio possono essere raggruppati i link aimigliori siti web che ospitano le riviste mediche del settore, le società scientifi-che, le sperimentazioni cliniche, le linee guida, i portali. Esempi di indici medi-ci sono le aree dedicate alla salute di indici generali come Yahoo! e Google (an-che nella versione italiana). Ask NOAH (tabella 2) è uno strumento dedicatoagli operatori del settore, MedlinePlus (figura 4) è dedicato ai cittadini. Si trat-ta di un portale sviluppato dalla National Library of Medicine per segnalare,attraverso apposite schede, i link ai siti web istituzionali (americani e interna-zionali) che pubblicano informazioni sui trattamenti, sulla prognosi, sulla pre-venzione e sulle sperimentazioni cliniche in corso per numerose malattie.Altro indice meritevole di essere segnalato è quello di Healthfinder, una delleprime esperienze istituzionali di portale informativo sanitario rivolto al cittadi-no (http://www.healthfinder.com).Accanto agli indici medici, Internet offre numerosi indici specialistici, quellicioè specificatamente rivolti a un’area medica o a una malattia ben definita.L’Istituto di Ricerche Farmacologiche “Mario Negri” ha realizzato indici specifi-ci per la cardiologia (tabella 2), l’oncologia, la gastroenterologia, la pneumolo-gia, la neurologia, la dermatologia e la terapia del dolore, dopo un lavoro diraccolta, validazione e classificazione dei siti web. A ogni sito segnalato è asso-ciata anche una descrizione del contenuto che ne illustra i servizi e le possibilicategorie di utenti. Tra le tipologie di siti presenti su questi indici si possonosegnalare quelli delle società scientifiche, delle associazioni di pazienti, dellelinee guida, delle riviste mediche, delle sperimentazioni cliniche, dei congres-si, e dei portali che offrono materiale a scopo formativo o riguardante l’uso deiprincipali farmaci.
75
Navigare sulla rete alla ricerca di informazioni di salute
Nome Indirizzo
Yahoo!/Health/Medicine http://dir.yahoo.com/Health/Medicine
Google/Health/Medicine http://www.google.com/Top/Health/Medicine
Ask NOAH http://www.noah-health.org
MedlinePlus http://www.medlineplus.gov
Healthfinder http://www.healthfinder.gov
CARDIO.CARE http://www.cardiocare.it
DERMA.CARE http://www.dermacare.it
GASTRO.CARE http://www.gastrocare.it
NEURO.CARE http://www.neurocare.it
ONCO.CARE http://www.oncocare.it
PNEUMO.CARE http://www.pneumocare.it
PAIN.CARE http://www.paincare.it
TABELLA 2: Esempi di indici medici su Internet

FIGURA 4: Home page del sito Medline Plus
FIGURA 5: Home page del sito Cardio.Care
Portali rivolti ai cittadini
Numerosi portali sono stati sviluppati per fornire informazioni ai cittadini. Taliiniziative sono spesso appoggiate dalle società scientifiche, nazionali e interna-zionali, più importanti per far fronte alla continua domanda di “formazione” cheproviene dall’utenza laica. L’affidabilità delle informazioni è garantita dalle so-cietà scientifiche che ne curano direttamente i contenuti. Laddove non sono co-involte società scientifiche ma associazioni di volontariato, gruppi editoriali oorganismi istituzionali, questi portali molto spesso soddisfano criteri di qualitàinternazionali come quelli identificati dalla Health On The Net Foundation (de-nominati HONCode e dai quali discende lo stesso “misurasiti” sviluppato daPartecipasalute) e sono da questa certificati. Quando queste organizzazioni so-no promotrici dello sviluppo di portali rivolti ai pazienti, la qualità dei contenutiè soddisfacente anche in Italia, come dimostra un’indagine svolta dal Censisqualche anno fa.1
76
Navigare sulla rete alla ricerca di informazioni di salute

Cancer.net Le informazioni più aggiornate si trovano nei portali internazionali (tabella 3). Inambito oncologico, per esempio, l’American Society of Clinical Oncology (la princi-pale società scientifica americana – e internazionale – in ambito oncologico) haaperto di recente il portale Cancer.net (fino a qualche mese fa noto con il nome diPLWC, Patients Living With Cancer). Qui i pazienti possono trovare schede detta-gliate su quasi tutte le tipologie di tumore che sintetizzano, per ciascuna di esse,fattori di rischio, sintomi, decorso, diagnosi, terapia e suoi eventuali effetti collate-rali, così come schede dedicate ai trial clinici in corso. Ma trovano anche informa-zioni e suggerimenti su argomenti che spesso sfuggono allemaglie della considera-zione medica, come per esempio il rapporto con il proprio corpo e con la propriaimmagine che cambia, con il sesso, con la fertilità, con la famiglia e con il lavoro.Per tenersi aggiornati, oltre a una newsletter mensile Cancer.net propone agliutenti strumenti tecnologicamente all’avanguardia come i podcast (file audio nelformato MP3 che possono essere scaricati sul proprio computer o lettore MP3) incui vengono spiegati dalla voce di un esperto argomenti di interesse per imalati.
Le iniziativedelle associazionidi pazienti
In un certo senso si tratta di una integrazione di quanto altri portali istituzionali(come per esempio Cancer.gov, il portale del National Cancer Institute) mettono adisposizione del consumatore, che è così libero di consultare più fonti (affidabili) econfrontarne i contenuti. Anche l’iniziativa delle Associazioni di malati porta a ri-sultati significativi come dimostra l’ esperienza italiana dell’Associazione Italianadei Malati di Cancro (AIMaC) sul cui sito gli utenti possono trovare informazionipratiche aggiornate sui vari tipi di tumore e sulle terapie per curarli.
Googlee Microsoft
Altre esperienze da segnalare sono quelle sviluppate da Google e da Microsoft. Idue giganti dell’informatica si sono gettati nel campo della ricerca e distribuzio-ne al pubblico di informazioni sanitarie e attraverso i portali lanciati all’iniziodel 2008 (chiamati rispettivamente Google Health e HealthVault), offrono noti-zie di facile reperimento e fruibilità provenienti da fonti selezionate e “certifica-te” che riguardano qualunque area medica e qualunque patologia.
NHS Choices
Il portalesulla salutedel ServizioSanitario inglese
Un discorso a parte merita il portale della salute che il National Health Servicebritannico ha sviluppato per i cittadini inglesi (figura 6). Il sistema, chiamatoNHS Choices, non solo fornisce informazioni (attraverso qualunque formato,compreso quello audio e video) su qualunque patologia (che può essere selezio-nata anche attraverso un ingegnoso sistema di mappatura costruito sulla im-magine di un corpo umano) o sui servizi offerti dal sistema sanitario inglese(compresa la possibilità di prenotare online visite ospedaliere), ma permette di
77
Navigare sulla rete alla ricerca di informazioni di salute
FIGURA 6: Xxxxx

ottenere informazioni sulle performance “ufficiali” di ospedali e medici che ope-rano nel Regno Unito. Tassi di sopravvivenza, tempi di attesa per un intervento,numero di interventi o esami eseguiti nel corso dell’ultimo anno su una deter-minata area medica, e numerosi altri parametri qualitativi elaborati dal Mini-stero della Salute britannico possono essere confrontati online per permettereal cittadino di scegliere la migliore (oltre che la più vicina) struttura alla qualeaffidarsi per le proprie cure. A ciò si deve aggiungere il fatto che il portale integrastrumenti propri del web 2.0 (vedi il paragrafo più avanti) che permettono al cit-tadino non solo di essere più velocemente e maggiormente informato delle novi-tà, ma soprattutto di “far sentire la sua voce” per esempio esprimendo (e condi-videndo con altri cittadini) giudizi e opinioni sugli ospedali e sui medici.
Un sistemache dialogacon i cittadini
La vera novità di questo genere di portale è proprio l’elevato grado di interazionee condivisione di dati offerto ai cittadini. Il National Health Service, attraverso ilportale NHS Choices offre infatti ai cittadini inglese la possibilità di creare e ac-cedere a un proprio fascicolo sanitario personale basato sul web che si autoali-menta ogni volta che essi usano uno qualunque dei servizi del sistema sanitarioinglese. Gli stessi fascicoli possono essere aggiornati manualmente anche daglistessi pazienti per segnalare altri genere di informazioni sanitarie (stili di vita,diete, ecc) non reperibili attraverso gli usuali canali. Per questioni di riservatez-za, il National Health Service assegna al cittadino il pieno controllo del propriofascicolo che, in ultima istanza, resta l’unico a poter decidere il livello di condivi-sione dei propri dati e i medici con i quali condividerli.
TABELLA 3: Esempi di portali medici su Internet rivolti ai cittadini
Associazioni di malati sul web
Le associazionidi cittadini
In molti paesi l’associazionismo di malati (o di “consumers”) è una realtà che haassunto notevole importanza sia nell’indirizzo delle politiche sanitarie, sia nell’o-rientare parte della ricerca clinica e i fondi necessari a sostenerla. Anche in Italiail fenomeno delle associazioni di malati è in costante crescita negli ultimi anni. Illoro principale obiettivo è di informare chi ne è colpito su come convivere con lamalattia, fornendo non solo suggerimenti clinici e terapeutici, ma offrendo infor-mazioni e servizi che riguardano l’aspetto psicologico e sociale della malattia.In Italia esistono diverse centinaia di associazioni di malati, molte delle quali so-no presenti in Rete con un sito web grazie al quale possono aumentare la pro-pria visibilità ed erogare servizi interattivi. Nonostante l’ampia eterogeneità, talisiti presentano alcune caratteristiche comuni. Innanzitutto sono gestiti diretta-mente da pazienti colpiti dalla malattia di cui si occupa l’associazione, spessocon la collaborazione di medici e ricercatori che garantiscono la qualità delle in-formazioni pubblicate. In secondo luogo offrono informazioni di supporto e aiu-to al paziente e ai suoi familiari (attraverso newsletter, pubblicazioni, accesso adatabase) che sono pensate e prodotte usando forme di comunicazione più vici-ne a loro. Ma soprattutto offrono spazi virtuali interattivi come i forum e i news-group che sono in grado di mettere a confronto le storie, le esperienze e le preoc-cupazione dei malati fornendo così un prezioso sostegno emotivo e psicologico.L’informazione offerta da questa tipologia di siti non è in contrasto con quellaprodotta dai siti delle riviste mediche, dai registri di sperimentazioni cliniche,dai portali delle società medico-scientifiche (che pure incorporano una sezioneper i malati) e dai siti web istituzionali, in generemeno attenti ai reali bisogni dei
78
Navigare sulla rete alla ricerca di informazioni di salute
Nome Indirizzo
Cancer.net http://www.cancer.net
Cancer.gov http://www.cancer.gov
AIMaC http://www.aimac.it
Google HealthMS HealthVaultNHS Choices
http://www.google.com/healthhttp://www.healthvault.comhttp://www.nhs.uk

malati e ai servizi sanitari di cui necessitano per diagnosticare e trattare le pato-logie di cui essi soffrono, ma piuttosto la completa. Il portale Partecipasaluteoffre un’ampia panoramica delle associazioni italiane di malati e di volontaria-to presenti sul web e ne raccoglie i link che sono organizzati per area medica(http://www.partecipasalute.it/cms_2/associazioniitalia).
Le nuove applicazioni del web 2.0
Nel corso dell’ultimo anno hanno iniziato a fare la loro comparsa le applicazioniweb 2.0. Si tratta di sistemi nei quali l’utente assume un ruolo centrale condivi-dendo dati, esperienze giudizi, conoscenze con altri utenti con i quali condivideanaloghi interessi. E’ un nuovo modo di tenersi informati, di discutere, di con-frontarsi, di trovare informazioni e, in definitiva, di creare una massa critica do-tata di una “conoscenza collettiva” che possa aumentare l’empowerment di ognisingolo utente. Lo strumento più conosciuto del web 2.0 è senza dubbio il socialnetwork, un ambiente nel quale i membri che ne fanno parte hanno la possibili-tà di creare contenuti e di condividerli con persone che hanno analoghi interes-si. Questo è possibile grazie al profilo che ogni membro della comunità può pub-blicare sul portale di social networking a cui si è registrato, e a strumenti chepermettono di individuare membri simili della comunità. I contenuti dei socialnetwork possono essere anche commentati e giudicati attivando quel meccani-smo di “selezione naturale” delle informazioni o conoscenze che, secondo gli os-servatori più ottimisti, porta a eliminare le inevitabili distorsioni.Tra i social network più noti si possono citareMySpace e Facebook, che affiancanoapplicazioni di condivisione di contenuti come YouTube (per i video), Flickr (per leimmagini) e SlideShare (per le diapositive). Ognuno di questi strumenti ospita di-verse community (così sono altrimenti detti i gruppi che si formano all’interno deisocial network) di cittadini o pazienti che si confrontano su determinate patologie.
Social networksanitari
Accanto a questi esistono specifiche applicazioni di social network sanitari ri-volti ai cittadini. Una tra le più interessanti è PatientsLikeMe una communitynella quale pazienti che soffrono di disturbi neurologici particolarmente invali-danti (sclerosi laterale amiotrofica, depressione, sclerosi multipla, morbo diParkinson, eccetera) possono condividere informazioni anche sanitarie (come itrattamenti farmacologici assunti e i loro effetti) e storie personali al fine di ge-stire al meglio malattie che, nella vita reale, hanno poche possibilità di aggrega-zione. Un altro genere di esperienza è quella realizzata dal portale CarePagesche negli Sati Uniti è impiegato da centinaia di migliaia di pazienti per tenere undiario clinico con il quale comunicare con i propri conoscenti e i propri cari du-rante la loro degenza in ospedale oppure come strumento di condivisione delleproprie storie al fine di imparare dalle esperienze altrui come gestire la propriamalattia, o per ottenere dalla community un sostegno morale per affrontarla.2
I sistemi di rating Un altro modo che spesso è proposto ai cittadini per partecipare alla formazionedella cosiddetta “intelligenza collettiva” è attraverso l’uso dei sistemi di ratingche permettono di giudicare l’operato di ospedali e medici. Alla base di questi si-stemi c’è l’idea che un’ampia partecipazione di cittadini potrebbe aiutare a iden-tificare i medici e le strutture ospedaliere più meritevoli o, quanto meno, a se-gnalare quelle a cui sarebbe meglio non rivolgersi. Capostipite di questo generedi iniziativa è il portale Revolution Health a cui hanno fatto seguito diverse ini-ziative, anche istituzionali come nel caso di Choices del National Health Servicebritannico. I possibili effetti di tali sistemi sono tuttavia controversi a causadell’ampia arbitrarietà con la quale la qualità dei servizi erogati dagli ospedali ele capacità dei medici sono misurate.3
Cartelle clinichebasate su web
Più concreti sono i sistemi di cartelle cliniche basate sul web (i Personal HealthRecord) che ormai anche i colossi dell’informatica (da Google a Microsoft) hannoiniziato a fornire (gratuitamente) ai cittadini al fine di creare sul web un propriofascicolo sanitario e condividerlo con chi essi desiderano. I termini inseriti in ta-li sistemi sono collegati a dizionari medici che, all’occorrenza, possono spiegar-ne meglio il significato. La propria storia clinica potrebbe così essere disponibile
79
Navigare sulla rete alla ricerca di informazioni di salute

ovunque ci si trovi, rendendo il cittadino, e il medico che ha la possibilità di con-sultarla, più informato (gli inglesi direbbero empowered) e consapevole delle de-cisioni mediche operate.4,5
I wiki Il web 2.0, infine, propone nuovi strumenti per la condivisione e la distribuzionedella conoscenza che sono basati suiwiki. Si tratta di siti web monotematici chesono organizzati come enciclopedie le cui voci sono redatte con la collaborazionedi più persone. Chiunque si sia registrato ha la possibilità di modificare i conte-nuti delle voci mettendo le sue conoscenze a disposizione di altri. Wikipedia èl’applicazionewiki più nota in Internet. La sua notorietà è testimoniata anche dalfatto che qualunque ricerca operata in Google genera come risultato “più pesan-te” un link alla pagina di Wikipedia che illustra la definizione del termine ricerca-to. Il suo uso è diventato così capillare che i portali di alcuni importanti quotidia-ni italiani (per esempio quello di Repubblica) lo usano come principale strumentodi ricerca in Internet, in alternativa allo stesso Google. Wikipedia include anchenumerosi termini medici (molti dei quali presenti nell’edizione italiana) la cuipubblicazione non è ottoposta a verifica da parte di un esperto (la pratica è solosuggerita da Wikipedia ma non obbligatoria). Per queste ragioni occorre cautelaquando, soprattutto in ambito medico, si usaWikipedia come fonte primaria. Nelcaso, meglio sarebbe se i contenuti della voce a cui siamo interessati fossero con-frontati almeno con un’altra fonte presente in Internet (o ancora meglio discussicon il proprio medico) prima di mettere in pratica gli eventuali suggerimenti.
TABELLA 4: Esempi di social network
80
Navigare sulla rete alla ricerca di informazioni di salute
Nome Indirizzo Tipo
MySpace http://www.myspace.com generalista
Facebook http://www.facebook.com generalista
YouTube http://www.youtube.com generalista
Flickr http://www.flickr.com generalista
SlideShare http://www.slideshare.com generalista
PatientsLikeMe http://www.patientslikeme.com social network sanitario
CarePages http://www.carepages.com social network sanitario
Revolution Health http://www.revolutionhealth.com sistema di rating
Google Health http://www.google.com/health sistema di personal health record
MS HealthVault http://www.healthvault.com sistema di personal health record
Wikipedia (inglese) http://www.wikipedia.com wiki
Wikipedia (italiano) http://www.wikipedia.it wiki
FIGURA 7: La home pagedel portale PatientsLikeMe

Bibliografia1. Censis. Il web come consulente globale. Roma 2005. http://www.censis.it/
277/372/5357/5565/cover.asp2. Santoro E. I blog come strumento di condivisione di esperienze tra pazienti.
Partecipasalute 2007. http://www.partecipasalute.it/cms_2/node/6523. Santoro E. Revolution Health e il coinvolgimento dei cittadini: uno strumen-
to di empowerment? Partecipasalute 2007. http://www.partecipasalute.it/cms_2/node/621
4. Santoro E. Le cartelle cliniche personali. Partecipasalute 2008. http://www.partecipasalute.it/cms_2/node/875
5. Santoro E. Google Health e le cartelle sanitarie on line: una nuova sfida perGoogle. Partecipasalute2008, http://www.partecipasalute.it/cms_2/node/919
Bibliografia di approfondimentol Santoro E. Guida alla medicina in rete. Roma: Pensiero Scientifico Editore,2002.
81
Navigare sulla rete alla ricerca di informazioni di salute


Comitati eticie consenso informatoAlessandro Liberati, Gaia MarsicoRevisori: Maria Gloria De Bernardo, Annalisa Marzot
I comitati etici: partecipare alla pari
Un po’ di storia In questo capitolo verrà esaminato il ruolo dei comitati etici, che rappresentanoun’occasione e un’espressione sintetica della continuità tra assistenza e ricerca,globale e locale. Essi iniziarono a svolgere un ruolo significativo intorno al 1960,come organo di consulenza e supporto per casi caratterizzati da complesse impli-cazioni etiche, inizialmente per arginare situazioni limite, e si sono in seguito di-versificati in relazione alle diverse funzioni assunte, distinguendosi in due tipi:l gli Institutional Review Board (IRB), costituiti specificamente per valutarel’eticità dei protocolli di sperimentazione clinica su soggetti umani, rispon-dono anche allo scopo di proteggere le persone dagli abusi perpetrati in no-me della scienza;
l gli Hospital Ethical Committee (HEC), nati verso la metà degli anni settanta inseguito a noti casi giudiziari legati all’interruzione dei trattamenti di sostegno vi-tale, offrono un supporto agli operatori sanitari, incapaci di gestire un carico cosìalto di responsabilità, per scelte non sempre, o quasi mai, tecniche/mediche.
I comitati etici assumono importanza, all’inizio, in particolare negli Stati Uniti:università e ospedali creano comitati per esaminare protocolli di ricerca e modulidi consenso, o per esaminare problemi relativi a interruzione di gravidanza, steri-lizzazione, interruzione di trattamenti in persone in stato vegetativo persistente.
Le violazionidei diritti in nomedella ricerca
Numerose violazioni del diritto alla vita e all’informazione in nome della ricercadi conoscenze mediche furono accertate nel processo sui crimini nazisti, a No-rimberga. Ma anche in molte altre occasioni, in paesi insospettabili, si sono ve-rificati episodi di notevole gravità; un esempio per tutti è il Tuskegee study nelquale dal 1930 al 1972 circa quasi quattrocento uomini di colore affetti da sifili-de vennero lasciati morire senza alcuna cura, anche dopo che questa era statatrovata, per studiare il decorso naturale della malattia. Si può poi ricordare cheHenry Beecher, professore di anestesiologia alla Facoltà di medicina dell’Uni-versità di Harvard, pubblicò nel 1966 un noto articolo nel quale riportava deci-ne di esempi di ricercatori che avevano incluso in ricerche cariche di rischi lepersone senza informarle; e ciò diversi anni dopo la stesura del Codice di No-rimberga. E l’anno dopo Maurice Henry Pappworth denunciò centinaia di speri-mentazioni che coinvolgevano adulti e bambini, condotte in circostanze simili,molte delle quali pubblicate su prestigiose riviste scientifiche. In Gran Bretagnaun grande impulso alla creazione di comitati etici fu dato dalla pubblicazionedel suo libroCavie umane. La sperimentazione sull’uomo (HumanGuinea Pigs).
I comitati etici: quale ruolo
Un importantestrumentodi partecipazione
I comitati etici, che si occupino di sperimentazione dei farmaci o di bioetica cli-nica, riflettono le istanze di una nuova cultura dell’assistenza sanitaria fondatasu informazione, autonomia, democraticità, trasparenza delle strutture sanita-rie, qualità nella gestione dei fondi e della ricerca, collaborazione tra le diverseprofessioni sanitarie. Inoltre assumono un ruolo pedagogico finalizzato a valo-rizzare i soggetti che richiedono l’assistenza sanitaria e a suscitare, nel perso-nale sanitario, una continua riflessione circa la gestione del rapporto con l’assi-stito: in breve sono il luogo per eccellenza della “partecipazione”.Solo i comitati etici che si occupano di sperimentazione dei farmaci esprimonopareri vincolanti per l’avvio di una ricerca, gli altri esprimono pareri di tipo con-sultivo. Per il modo in cui sono costituiti e per i compiti che sono andati ad assu-
83
Comitati etici e consenso informato

mere, i comitati hanno l’opportunità di promuovere lo sviluppo di una pratica euna ricerca libera dalle logiche di mercato e sempre più a servizio dei bisogni disalute. Proprio per questa finalità la loro composizione è eterogenea: infatti dauna parte ci sono i membri cosiddetti tecnici e dall’altra ci sono i membri laicirappresentati da tutte quelle figure che non hanno specifici compiti né sanitariné giuridici né tantomeno amministrativi. Questa composizione “mista”, che è laragione e la scommessa dei comitati etici, favorisce la compenetrazione di diverseprospettive e l’attitudine al confronto, la contaminazione culturale, la democraziadei rapporti e il pluralismo delle visioni, incrina l’aria di autorità e di potere chetradizionalmente ha circondato, e ancora spesso circonda, il mondo medico. Qu-esto tipo di composizione ricorda che le varie questioni affrontate non vanno esa-minate solo da un punto di vista tecnico con l’ ausilio di strumenti e linguaggispecialistici, ma, dal momento che riguardano la vita, la morte, la sofferenza e lepaure delle persone, il ruolo e gli scopi della ricerca e della medicina nel contestosociale, devono essere analizzate in tutte le loro implicazioni.Nei comitati non vi sono (o non dovrebbero esservi) gerarchie, si interviene se-condo la propria esperienza e competenza, si condividono responsabilità e im-pegni, disponibilità all’ascolto. I membri dei comitati etici devono essere indi-pendenti da industrie farmaceutiche, e, almeno alcuni, dall’ente presso cui ope-rano. Il comitato nel suo complesso è (dovrebbe essere) garante nei confrontidella cittadinanza di indipendenza di giudizio e promotore di trasparenza.
Approcciomulticulturale
I comitati etici dovrebbero effettivamente rappresentare gli interessi della colletti-vità e delle persone coinvolte nella sperimentazione clinica, ed essere uno deglistrumenti di garanzia dei diritti in sanità. Nonostante i molti compiti burocraticidi cui nel tempo sono stati caricati, i comitati etici non possono essere consideratisemplicemente come lo strumento amministrativo che garantisce l’adempimentodei requisiti di immissione in commercio dei farmaci. Essi infatti non sono néme-ri organismi di “controllo”, né hanno tra i loro obiettivi ottimizzare la produzionerispetto al mercato: da una parte rappresentano i diritti di cittadini di una comu-nità, dall’altra sono chiamati a essere garanti della corrispondenza della ricerca esperimentazione con obiettivi rilevanti di conoscenza e di cura. Il ruolo di un co-mitato etico appare evidente se si pensa all’importanza che hanno in una realtàsempre più multiculturale i problemi del consenso informato e della consulenzagenetica, in particolare negli screening. Esso infatti diviene luogo di incontro emediazione tra le associazioni, le comunità e tutti coloro che in differenti modi e adiverso titolo operano in ambito sanitario. L’approccio interculturale impone diriflettere su nuove problematiche in cui vengonomessi a confronto diversi model-li di salute e stimola a dare risposte che tengano conto delle differenti sensibilità.
I compiti del comitato etico per la sperimentazione
Composizionee funzionamento
Il comitato etico si occupa degli aspetti scientifici e di quelli relativi alla sicurezza,inoltre può decidere come i soggetti devono essere informati e coinvolti. Esso, fortedelle diverse competenze che ha al suo interno, esamina il protocollo, incontra glisperimentatori che condurranno lo studio, pone domande di vario genere sul valo-re dello studio, sulle aspettative, sui diritti dei pazienti, si informa su studi similigià terminati o in corso. Infine, dopo un’accurata discussione, decide se il progettoha valore scientifico, se introduce novità terapeutiche, se risponde ai bisogni di sa-lute della cittadinanza, se la conduzione dello studio rispetta tutti i diritti delle per-sone coinvolte. Ogni comitato etico deve comparire in un registro istituito presso ilMinistero della salute, così come i pareri che emette devono essere inviati all’Os-servatorio per le Sperimentazioni Cliniche del Ministero della Salute. Solo dopoquesta fase può iniziare la sperimentazione nell’ospedale o sul territorio di compe-tenza del comitato. Il parere del comitato etico è la condizione necessaria per losvolgersi di una sperimentazione. Nel caso degli studi multicentrici, che si svolgo-no in un grande numero di sedi talora anche internazionali, il parere di ogni comi-tato etico vale solo a livello locale. E pur se una direttiva europea prevede l’esisten-za di un parere ‘unico’ emesso dal Centro Coordinatore, ogni comitato è libero di ri-fiutare uno studio, anche fosse l’unico centro a fare questa scelta: per ragioni di ti-po scientifico metodologico, o perché non lo considera rilevante, o perché a livello
84
Comitati etici e consenso informato

locale non si considera opportuno. Il comitato etico non può entrare nel merito deicontenuti del protocollo stesso; ha la possibilità e responsabilità di approvare o re-spingere un protocollo, garantire che si attui in condizioni di “sicurezza”. Il comita-to viene continuamente messo al corrente delle evoluzioni della sperimentazione edi tutti gli eventi indesiderati (dannosi), più omeno gravi, che possono essere colle-gati con i trattamenti in studio. Questa fase, chiamata del monitoraggio, ha in ef-fetti una grande importanza, ma molto spesso non viene mantenuta nel tempo.
I riferimenti normativi dei comitati eticiI riferimenti alti, che delineano il quadro di riferimento in cui devono muoversi icomitati etici sono:l la Dichiarazione di Helsinki;l la Convenzione europea sui diritti umani e la biomedicina (anche detta Con-venzione di Oviedo);
l il Protocollo aggiuntivo alla Convenzione sui diritti dell’uomo e sulla biomedici-na, relativo alla ricerca biomedica.
La Dichiarazione di Helsinki, dell’Associazione Medica Mondiale (AMM), adottatanel giugno 1964, viene periodicamente aggiornata; è il documento internazionaledi riferimento per la ricerca biomedica, in quanto fissa i principi etici che devonoorientare la sperimentazione che coinvolge esseri umani; nasce, in particolare, dal-l’esigenza della medicina di fare chiarezza sui propri ruoli, doveri, responsabilità.La Dichiarazione di Helsinki e la Convenzione europea sui diritti umani e la bio-medicina, hanno l’obiettivo di stabilire i diritti dei cittadini in ambito sanitario,in particolare sanciscono il diritto/dovere della collettività di cercare rispostevalide a bisogni che ancora non hanno trovato una risposta.I documenti citati costituiscono il quadro generale di etica e di diritto nel campodella ricerca, che può essere così sintetizzato:l una ricerca clinica è giustificata solo se paziente e medico sono “incerti” circail trattamento da adottare tra quelli disponibili. La programmazione di unprotocollo di ricerca inizia proprio con il riconoscimento e la definizione delleincertezze sui trattamenti da studiare, a cui segue la traduzione di queste inipotesi di ricerca;
l gli interessi e il bene delle persone coinvolte nella ricerca devono avere prio-rità rispetto agli interessi della società o della scienza;
l la libera scelta dei partecipanti, è una condizione essenziale della ricerca;l la ricerca ha l’obiettivo di contribuire, attraverso un significativo migliora-mento della conoscenza scientifica e delle condizioni della persona, a conse-guire risultati finali di potenziale beneficio per la persona interessata o peraltre persone della stessa età, o che soffrono della stessa malattia;
l la ricerca medica deve essere condotta solo se l’importanza dell’obiettivoprevale sui rischi per il soggetto coinvolto;
l qualsiasi studio volto a dimostrare l’efficacia di un trattamento non può sot-trarre ai partecipanti la miglior terapia disponibile al momento.
Non si possono dimenticare altri due importanti documenti di riferimento:l le Guidelines for Good Clinical Practice (GCP). Le linee guida sviluppate dallaInternational Conference of Harmonization con l’obiettivo di guidare l’attivi-tà di coloro che fanno ricerca clinica riguardano le modalità formali di acqui-sizione del consenso informato dei pazienti, la loro sicurezza, l’integrità deidati. Il documento ha essenzialmente a che fare con le “procedure”, più checon i contenuti della sperimentazione: le linee guida proposte sono vinco-lanti ai fini della registrazione;
l la Direttiva europea del 2001 relativa all’applicazione della buona pratica clini-ca nell’esecuzione delle sperimentazioni cliniche di medicinali per uso umano.Recepita in Italia nel 2003, essa ha sollevato un vivace dibattito, poiché, vistala complessità delle procedure che richiede, è accusata di causare un ostaco-lo grave alla ricerca clinica indipendente, ossia la ricerca che non è promossada industrie farmaceutiche. Scopo della direttiva è regolamentare e “armoniz-zare” il contesto commerciale in cui si trova inserita la medicina e dunque lasperimentazione, creando condizioni di sicurezza (ossia tali per cui le persone
85
Comitati etici e consenso informato
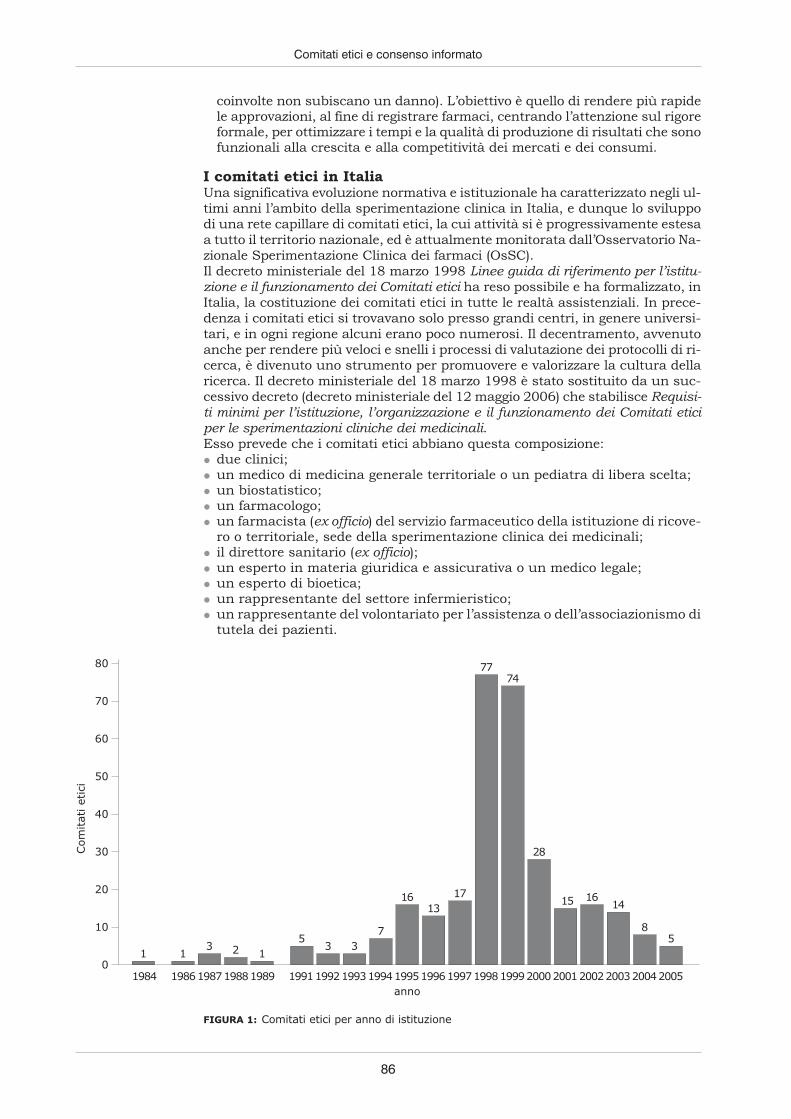
coinvolte non subiscano un danno). L’obiettivo è quello di rendere più rapidele approvazioni, al fine di registrare farmaci, centrando l’attenzione sul rigoreformale, per ottimizzare i tempi e la qualità di produzione di risultati che sonofunzionali alla crescita e alla competitività dei mercati e dei consumi.
I comitati etici in ItaliaUna significativa evoluzione normativa e istituzionale ha caratterizzato negli ul-timi anni l’ambito della sperimentazione clinica in Italia, e dunque lo sviluppodi una rete capillare di comitati etici, la cui attività si è progressivamente estesaa tutto il territorio nazionale, ed è attualmente monitorata dall’Osservatorio Na-zionale Sperimentazione Clinica dei farmaci (OsSC).Il decreto ministeriale del 18 marzo 1998 Linee guida di riferimento per l’istitu-zione e il funzionamento dei Comitati etici ha reso possibile e ha formalizzato, inItalia, la costituzione dei comitati etici in tutte le realtà assistenziali. In prece-denza i comitati etici si trovavano solo presso grandi centri, in genere universi-tari, e in ogni regione alcuni erano poco numerosi. Il decentramento, avvenutoanche per rendere più veloci e snelli i processi di valutazione dei protocolli di ri-cerca, è divenuto uno strumento per promuovere e valorizzare la cultura dellaricerca. Il decreto ministeriale del 18 marzo 1998 è stato sostituito da un suc-cessivo decreto (decreto ministeriale del 12 maggio 2006) che stabilisce Requisi-ti minimi per l’istituzione, l’organizzazione e il funzionamento dei Comitati eticiper le sperimentazioni cliniche dei medicinali.Esso prevede che i comitati etici abbiano questa composizione:l due clinici;l un medico di medicina generale territoriale o un pediatra di libera scelta;l un biostatistico;l un farmacologo;l un farmacista (ex officio) del servizio farmaceutico della istituzione di ricove-ro o territoriale, sede della sperimentazione clinica dei medicinali;
l il direttore sanitario (ex officio);l un esperto in materia giuridica e assicurativa o un medico legale;l un esperto di bioetica;l un rappresentante del settore infermieristico;l un rappresentante del volontariato per l’assistenza o dell’associazionismo ditutela dei pazienti.
FIGURA 1: Comitati etici per anno di istituzione
86
Comitati etici e consenso informato
Comitatietici
anno
1984
1 13 3 3
5 587
16 1614
17
28
15
7774
13
2 1
1986198719881989 199119921993199419951996199719981999200020012002200320042005
80
70
60
50
40
30
20
10
0

“Il comitato etico deve essere istituito, organizzato e funzionante inmodo tale dagarantire l’indipendenza dello stesso”. Per questo non deve esserci “subordina-zione gerarchica nei confronti della struttura ove esso opera”, il comitato eticodeve avere membri “non dipendenti dalla struttura ove opera” e deve essere ga-rantita “la mancanza di conflitti di interesse dei votanti rispetto alla sperimen-tazione proposta”.Nel marzo 2008 i comitati etici sono 270 in Italia; l’Umbria ha 1 comitato etico re-gionale, la Val d’Aosta 1 comitato etico regionale e 1 comitato locale (vedi figura 1).
Tante competenzediverse
Lo scopo della valutazione interdisciplinare, dell’eticità del progetto di ricerca,dovrebbe essere proteggere i diritti, la dignità, la sicurezza, il benessere dei par-tecipanti alla ricerca. La valutazione dell’adeguatezza etica dovrà ricorrere aun’appropriata varietà di competenza ed esperienza, che rifletta, in modo ade-guato, punti di vista professionali e laici. (Protocollo aggiuntivo alla Convenzionesui diritti dell’uomo e sulla biomedicina, relativo alla ricerca biomedica, Art. 9.2)
Chi rappresenta la cittadinanza nei comitati etici?
La disuguaglianzainformativa
Se i comitati etici sono il luogo d’eccellenza dove valutare e promuovere la ricer-ca clinica, probabilmente la partecipazione di rappresentanti della cittadinanzaè ancora troppo limitata. Partecipare ai comitati etici è senza dubbio una formadi grande e importante coinvolgimento, ma soprattutto un esercizio “normale”di democrazia, che è però oggi consentito solo a persone che appartengono almondo del volontariato e dell’associazionismo o a pazienti che ne fanno parte.Una effettiva partecipazione è qualche volta limitata da evidenti differenze cul-turali o di potere, che mettono in soggezione, inibiscono e condizionano la pos-sibilità di esprimersi liberamente o influiscono sulla decisione presa.Il Protocollo aggiuntivo alla Convenzione sui diritti dell’uomo e sulla biomedicina,relativo alla ricerca biomedica, fornisce indicazioni sull’accesso alle informazioniper i cittadini nei comitati etici: nella sua appendice “Informazioni che devonoessere fornite al comitato etico”, è detto chiaramente che del protocollo in di-scussione deve essere fornito un riassunto in lingua corrente, senza ricorso atecnicismi. Né in letteratura, né dalle esperienze raccolte in Italia, risulta che icomitati etici ricevano o richiedano tale documentazione. Nel materiale che per-viene all’attenzione dei membri dei comitati etici le pagine scritte in linguaggiopiù comprensibile sono le informazioni destinate ai pazienti da coinvolgere, chenon rappresentano la traduzione fedele e completa del protocollo. Dunque, èpresente un problema di linguaggi, che non consente ai membri non tecnici deicomitati di capire e intervenire in modo appropriato.Se i membri dei comitati etici fossero nelle condizioni di interagire tra loro allapari, un’area interessante di azione, tutta da sviluppare, potrebbe essere quelladi redigere documenti tesi a informare la cittadinanza sui contenuti concreti, ilsignificato complessivo, i limiti e i contributi delle sperimentazioni oggetto dianalisi. Sicuramente una iniziativa di questo genere andrebbe a riempire unvuoto. La permanenza di questo vuoto corre il rischio di configurarsi come unfallimento radicale del ruolo stesso della presenza di etica nei comitati etici, maforse ancora di più del loro stesso ruolo, proponendo un importante interroga-tivo: i comitati etici sono organismi burocratici di controllo o luogo dove speri-mentare e praticare partecipazione, scuola di diritti condivisi?
Le criticità degli studi
I confini del ruolodi tutela
I comitati etici sono in grado di interpretare il loro ruolo? La mancanza di proveempiriche della reale efficacia dei comitati etici rispetto ai loro obiettivi dichiara-ti è preoccupante. Con la crescita degli interessi commerciali nella sperimenta-zione clinica sui farmaci, e la conseguente possibilità di colossali profitti econo-mici, l’operato dei comitati etici deve essere maggiormente critico e in grado diprendere in considerazione le motivazioni e le implicazioni delle domande chesono alla base dei progetti di ricerca. E’ evidente a tutti che con il diffondersi distudi multicentrici, pianificati con costi elevatissimi dall’industria farmaceuticaper includere migliaia di pazienti e per concludersi nel più breve tempo possibi-
87
Comitati etici e consenso informato

le così da produrre informazioni immediatamente utilizzabili a scopi registrativie di marketing, di fronte ai comitati etici si sono materializzate sfide e compitimolto complessi, resi ancor più difficili dalla mancanza di accordo su quali sia-no i confini del ruolo di tutela che i comitati etici devono esercitare nei confrontidei pazienti. Molti, infatti, considerano ancora che il compito dei comitati eticidebba essere limitato ai problemi di salvaguardia del consenso informato relati-vamente ai protocolli di ricerca e non essere invece più ampio comprendendo lavalutazione degli aspetti scientifici e clinici fondamentali dei progetti di ricerca.Secondo altri invece i comitati etici avrebbero un mandato più ampio che vadalla valutazione degli aspetti essenziali della ricerca (obiettivi, assenza di ri-dondanza, rilevanza clinica e probabilità di realizzare gli scopi prefissati) alla ve-rifica dell‘accettabilità dei rapporti tra sponsor e sperimentatori cosi come ven-gono regolamentati nei contratti, soprattutto a proposito di questioni come lagaranzia che gli sperimentatori abbiano la possibilità di pubblicare e diffondere,senza vincoli o restrizioni, i risultati delle ricerche.1
Le questioni aperteLe sperimentazioni cliniche – come recentemente sottolineato in un articolo divalutazione dell’attività dei comitati etici2 – presentano diversi aspetti critici nel-la progettazione, che rappresentano oggi altrettante questioni aperte circa lacapacità dei comitati etici di svolgere tale compito:l Quando è consentito e appropriato l’uso del placebo?l Come comportarsi con gli studi di equivalenza e di non inferiorità?l Come si forma il gruppo di controllo in una sperimentazione clinica?l Quali i requisiti per la scelta degli esiti?l Quale garanzia che i risultati siano pubblicati?l Come reagire alla frammentazione della partecipazione agli studi?l Come evitare la modifica in corso d’opera dei protocolli di ricerca?
Uso del placebo L’uso del placebo al posto di un farmaco attivo è ovviamente vantaggioso per losponsor di una studio in quanto aumenta la possibilità che il nuovo farmacosperimentale appaia efficace. L’uso del placebo non dovrebbe invece mai esseregiustificato in tutte quelle situazioni terapeutiche nelle quali esiste una alterna-tiva terapeutica efficace disponibile. I comitati etici, e soprattutto i loro compo-nenti non tecnici , dovrebbero essere messi in condizione di valutare la presen-za di alternative terapeutiche.
Studidi equivalenza
Il progetto di uno studio deve avere l’obbiettivo di mostrare la superiorità di unnuovo trattamento rispetto al miglior trattamento disponibile per la stessa indi-cazione terapeutica. Tuttavia negli ultimi anni si è andata invece affermando latendenza a effettuare i cosiddetti studi di equivalenza o di non inferiorità. La ri-cerca commerciale si è astenuta quindi dal cercare farmaci/interventi più attivi,limitandosi a cercare sostanze che abbiano un effetto simile o non peggiore diquelle già disponibili. Tuttavia, stabilire i limiti quantitativi delle differenze en-tro le quali si dichiara che il trattamento è equivalente o comunque non inferio-re è molto complesso.Qual è la differenza che è importante non perdere di vista quando si parla di untrattamento potenzialmente salvavita? E’ del 10 per cento, del 5 per cento o an-che del 2 per cento? Si tratta di domande che dovrebbero suscitare l’attenzionedei comitati etici. I pazienti coinvolti negli studi di equivalenza o di non inferiori-tà non sono infatti quasi mai informati del fatto che il trial non porterà né a loro,né verosimilmente ad altri pazienti, nessun beneficio. I comitati etici dovrebbe-ro essere consapevoli di questo aspetto.
Gruppodi controllo
Anche la scelta del farmaco di controllo e del suo dosaggio è critica per la giusti-ficabilità etico scientifica dello studio. Si tratta evidentemente di un aspettoparticolarmente critico e complesso per il comitato etico da valutare ed è questala ragione principale per la quale ogni proposta di un nuovo studio dovrebbe es-sere preceduta e accompagnata da una revisione sistematica degli studi giacondotti e da una attenta giustificazione dello specifico disegno scelto. La con-troversia sorta a proposito di un importante studio di confronto tra farmaci im-
88
Comitati etici e consenso informato

munosoppressivi è un esempio su cui riflettere e rappresenta molte altre situa-zioni che sono assai più comuni di quanto si creda.3
Scelta degli esiti Gli esiti devono essere misurabili con sufficiente affidabilità e, soprattutto, de-vono avere una difendibile e documentabile rilevanza clinica. L’uso di esiti sur-rogati (tipo la riduzione della colesterolemia), verosimilmente o plausibilmentelegati a esiti clinici forti ed evidenti (morbilità, mortalità), ma spesso senza di-mostrazione, è sempre problematico nel contesto di studi che vogliono dimo-strare l’efficacia clinica di un farmaco o di un intervento. I comitati etici dovreb-bero interrogarsi sulle cautele da usare per studi di questo tipo. I casi recentidella cerivastatina4 e del rofecoxib5 sono importanti campanelli d’allarme.
Pubblicazionedei risultati
Siamo qui di fronte a un aspetto non clinico e tuttavia sostanziale al quale i co-mitati etici dovrebbero prestare attenzione nell’esaminare e approvare i proto-colli. E’ fondamentale assicurarsi che nel protocollo dello studio non esistanocondizioni che permettono allo sponsor dello studio (sia esso privato o pubblico)di esercitare un diritto di veto sulla pubblicazione dei risultati da parte del ricer-catore.6 Chi vuole opporsi al fatto che i comitati etici vigilino su questo aspettofa (volutamente) confusione tra proprietà dei dati e pubblicabilità dei risultati. Ilrecente Decreto ministeriale sulla ricerca indipendente7 fa chiarezza su questopunto e avvia una nuova fase nella quale non sarà più possibile abusare delladiversità di punti di vista sul ruolo dei comitati etici.
Studi multicentrici Un’effettiva criticità con cui sempre più i comitati etici si trovano a confrontarsiè quella degli studi multicentrici multinazionali. Si tratta di studi che – conl’obiettivo dichiarato di reclutare casistiche ampie in un numero molto esteso dicentri – prevedono che tantissimi centri reclutino pochissimi pazienti in un de-terminato intervallo di tempo. Qualcuno li ha definiti gli studi carciofo per indi-carne la complessa funzionalità e non facile maneggiabilità. Con quale logica?Quella di far partecipare molti centri? Quella di aumentare la qualità e rappre-sentatività della ricerca? Difficile pensare siano queste le vere motivazioni.Spiace dover ipotizzare che le ragioni possano essere altre, e che questi studipossano mirare, nella sostanza, a:l far perdere il peso dei singoli comitati etici nella valutazione del protocollo;l rendere impossibile il monitoraggio (ogni centro occupato in contemporaneain tantissime sperimentazioni etero controllate dagli sponsor);
l tener occupati i ricercatori clinici in studi che danno loro riscontri economi-ci (grazie a quote di partecipazione – spesso sostanziose – per ogni pazientearruolato le quali generano imbarazzanti conflitti di interessi nei confrontidei pazienti!) e di prestigio (partecipazioni ai congressi, pubblicazioni inter-nazionali, eccetera).
Inoltre, diminuiscono le garanzie di buona pratica di ricerca nella situazione incui ogni centro coinvolge 3 o 4 pazienti in un trial; questi numeri non consento-no di prendere dimestichezza con un protocollo.
Protocollimodificati in corsa
Altro fenomeno di recente ingresso nel mondo delle sperimentazioni clinichecommerciali è la tendenza a modificare, quando non a stravolgere, in corsod’opera i protocolli alterandone il disegno, interrompendone alcune parti, eccete-ra. Occorre porre attenzione a modifiche che possono sembrare di poca impor-tanza, e che sembrano avere solo un carattere amministrativo , e non consentireche la pubblicazione dello studio sia ottenuta sulla base di un protocollo assai di-verso da quello che i singoli comitati etici avevano originariamente approvato.
Il consenso informato
Informazione e sceltaInformazione e scelta sono il presupposto del “consenso informato”, proceduraformale della comunicazione in cui si fanno partecipi le persone delle diagnosi,delle possibilità, delle attese e delle incertezze relative a un trattamento o a unascelta terapeutica. Una buona informazione è normalmente un diritto, anche sepoco praticato; operare scelte per la propria salute dovrebbe essere una praticanormale e molto semplice, riconducibile al modo con cui dovrebbero essere im-
89
Comitati etici e consenso informato

postate tutte le relazioni. La traduzione formale della scelta in “consenso” haavuto una storia legata, in modo forte, ai contesti assicurativi e medico legali. Inrealtà la firma sul modulo non è solo un atto formale, ma rappresenta simboli-camente un “passaggio culturale” importante: non è più il medico da solo a de-cidere della salute del paziente, ma è il paziente stesso che si informa di ciò cheaccade e sceglie liberamente, consapevolmente e responsabilmente rispetto alleopzioni terapeutiche che gli sono prospettate.
I fondamenticostituzionalidel consensoinformato
L’informazione, e la scelta di essere informati, fanno parte del quadro generaledei diritti costituzionali. La Costituzione Italiana nel secondo comma dell’artico-lo 32 dispone:“Nessuno può essere obbligato a un determinato trattamento sanitario se nonper disposizione di legge. La legge non può in nessun caso violare i limiti impostidal rispetto della persona umana”.L’articolo 32 della Costituzione deve essere letto avendo presente il più generalearticolo 13, “la libertà personale è inviolabile”, che, tradotto nel concreto, signi-fica che le persone hanno il diritto di curarsi e di scegliere la terapia, nessunopuò entrare nel merito delle scelte personali. I trattamenti, in sintesi, non pos-sono mai essere imposti.La Convenzione Europea sui diritti umani e la biomedicina (Convenzione di Ovie-do), che è stata firmata dall’Italia e damolti altri paesi europei, è molto chiara suquesto punto e mentre nell’art. 10 precisa che:“1. Ogni persona ha diritto al rispetto della propria vita privata allorché si trattadi informazioni relative alla propria salute”.“2. Ogni persona ha il diritto di conoscere ogni informazione raccolta sulla pro-pria salute. Tuttavia, la volontà di una persona di non essere informata deve es-sere rispettata”.All’articolo 5 afferma, in modo inequivocabile:“Un intervento nel campo della salute non può essere effettuato se non dopoche la persona interessata abbia dato consenso libero e informato. Questa per-sona riceve innanzitutto una informazione adeguata sullo scopo e sulla naturadell’intervento e sulle sue conseguenze e i suoi rischi. La persona interessatapuò, in qualsiasi momento, liberamente ritirare il proprio consenso”.Anche l’art. 3 della Carta dei diritti fondamentali dell’Unione Europea stabilisceche, “nell’ambito della medicina e della biologia”, deve essere rispettato “il con-senso libero e informato della persona interessata, secondo le modalità definitedalla legge”. Le stesse indicazioni si trovano nella Dichiarazione di Helsinki, ildocumento dell’Associazione Medica Mondiale, e in molti altri
Serveuna comunicazionechiarae trasparente
Naturalmente si può scegliere solo se si è in grado di capire la situazione; ma so-prattutto se si è messi in condizione di capire, e di scegliere. Quindi servonostrumenti intellettuali, tempo e possibilità di confronto. L’autonomia non si ot-tiene perché qualcuno ci offre la possibilità di scegliere, e ovviamente non la ga-rantisce la firma su di un modulo, che mai può sostituire ma solo integrare ilcolloquio. In situazioni di vulnerabilità (la malattia; la poca chiarezza circa dia-gnosi, prognosi, possibilità reali di cura; la paura; la solitudine; l’assoluta e in-superabile asimmetria di potere) l’esercizio dell’autonomia è possibile solo sesostenuto da una comunicazione chiara e trasparente. Purtroppo gli operatorisanitari non sono sempre preparati e disposti a favorire una comunicazione cheabbia le caratteristiche della chiarezza e dell‘esaustività, e il consenso non risul-ta sufficientemente informato. E’ auspicabile che gli operatori sanitari si impe-gnino a garantire tempi e spazi idonei alla comunicazione con il malato e la suafamiglia per affrontare le problematiche connesse con le opzioni terapeuticheprospettate. Si pensi all’importanza e alla delicatezza di questa comunicazionenelle situazioni di fine vita o di opzioni terapeutiche che possono avere conse-guenze per l’autonomia e le capacità del paziente. Nella formazione degli opera-tori sanitari, e in particolare dei medici, gli aspetti relativi alla comunicazionedovrebbero essere maggiormente considerati e affrontati.
Partecipantialla ricerca
Le persone che partecipano agli studi clinici non sono cavie, neppure reclute(termine che deriva dal comune, e universalmente usato, “reclutare pazienti”).
90
Comitati etici e consenso informato

Questi termini che ancora occupano interamente (a livello nazionale, come in-ternazionale) il linguaggio della ricerca clinica – per esempio “in questo studioverranno reclutati o arruolati 1.000 pazienti” – dovrebbero essere banditi e so-stituiti. Il termine cavie è certamente appropriato per descrivere ciò che troppevolte è accaduto, dai crimini nazisti ai casi che si sono moltiplicati nel tempo. E’invece corretto – e auspicabile che venga sempre applicato – l’uso di terminiquali “includere” o “partecipare”.
Bibliografia1. Edwards SRL et al. Research ethics commitees: differences and moral jud-
gement. Bioethics 2004;18:408-272. Bertelè V et al. How can research ethics committee protect patients better?
Br Med J 2003;326:1199-213. Schieppati A et al. Tacrolimus and ciclosporin micremulsion in renal tran-
splantation. The Lancet 2002;360:799-8004. Farmer JA. Learning from the Cerivastatine experience. The Lancet 2001;
358:1383-55. Maxwell SR, Webb DJ. Cox selective inhibitors. The Lancet 2005;365:
449-516. Mann H. Research ethics committes and public dissemination of clinical
trials results. The Lancet 2002;360:406-87. Decreto ministeriale del 17 Dicembre 2004 sulla Sperimentazione no profit
per il miglioramento della pratica clinica quale parte integrante della assi-stenza sanitaria. Gazzetta Ufficiale n. 43 del 22 febbraio 2005
Bibliografia di approfondimentol Liberati A (a cura di). Evidence based medicine tra ragione e passione. Ro-ma: Il Pensiero Scientifico Editore, 2005.
l Marsico G. La sperimentazione umana. Diritti violati/diritti condivisi. Ro-ma: Franco Angeli, 2007.
l Spinsanti S. Chi decide in medicina. Roma: Zadig, 2004.
91
Comitati etici e consenso informato


Le politiche che regolanoil mercato dei farmaci: il ruolodelle agenzie regolatorieGiovanni Apolone, Paola MosconiRevisori: Luisa Villa, Roberto Trefiletti
Da molecole promettenti a buoni farmaci
I progressidella medicina
Negli ultimi anni la medicina ha registrato importanti progressi nella compren-sione dei fattori, molecolari e non, legati allo sviluppo delle principali malattie.Nonostante i miglioramenti ottenuti nella cura e nel controllo della progressionedelle malattie grazie ai nuovi farmaci, c’è ancora molto da fare in termini di risul-tati a lungo termine per la maggior parte delle più comuni malattie di tipo croni-co. Se si prende come esempio la patologia tumorale maligna – a tutt’oggi tra leprime cause di morte, di sofferenza e di consumo di risorse nei paesi occidentali –si osserva che c’è stato un enorme incremento delle conoscenze che derivano dal-la cosiddetta scienza di base, però i miglioramenti osservati negli ultimi 10 anniin termini di percentuale di pazienti guaribili e di sopravvivenza riguardano po-che forme tumorali e sono in parte attribuibili alla prevenzione primaria (riduzio-ne dei rischi) e secondaria (diagnosi anticipata), alla tempestività delle cure chi-rurgiche più che alla successiva terapia con farmaci. Inoltre, il ruolo dei farmaci ècertamente importante nelle fasi di terapia adiuvante quando i pazienti ricevonodeterminate terapie per ridurre il rischio di ricadute dopo l’asportazione del tu-more, mentre resta marginale in occasione di progressione e metastasi (cioè, cel-lule tumorali che colpiscono altri tessuti). Tuttavia, nonostante il contrasto esi-stente tra promesse e risultati, le aspettative dei pazienti e dei medici sulla ricer-ca e sui nuovi farmaci sono certamente ben riposte e nei prossimi anni si assiste-rà a un incremento nella disponibilità di nuovi farmaci basati su meccanismi diazione innovativi, o più mirati a specifici obiettivi molecolari o clinici.
Molti interessigravitano intornoai farmaci
Nulla si può dare per scontato soprattutto in un contesto sempre più dominatoda unmercato globale e quindi governato da interessi prevalentemente economi-ci. Al fine di aumentare la probabilità che promettenti molecole diventino buonifarmaci, non basta attendere che i risultati dalla ricerca si trasformino in cam-biamenti di salute, ma è necessario creare e sostenere una interazione coordina-ta tra tecnologia, biologia, ricerca clinica, pratica clinica e politica sanitaria, inquanto il processo basato sul passaggio passivo e automatico dalla teoria alla ri-cerca clinica ed eventualmente alla pratica corrente non può essere dato perscontato. I momenti critici di questo modello sono molti, ma essenzialmente lacriticitàmaggiore risiede nella qualità scientifica dei dati e nell’etica della ricerca.
Le basi razionali delle sperimentazioni
Dalla sempliceosservazione allasperimentazione
C’è naturalmente accordo sul fatto che nuovi interventi medico sanitari – com-presi i nuovi farmaci – debbano essere provati in maniera completa prima di es-sere offerti a tutta la popolazione, al fine di garantire il cittadino. E’ meno intuibi-le, e quindi talvolta meno accettabile, che per documentare la qualità di un far-maco in termini di sicurezza e efficacia sia necessario utilizzare complesse e lun-ghe procedure che talvolta durano anni e coinvolgono gruppi di pazienti, più omeno consapevoli di essere parte di una sperimentazione scientifica. Perché nonbasta basarsi sulle osservazioni, ma è invece necessario sperimentare? Nel pas-sato, in effetti bastava applicare i nuovi farmaci su pochi topolini e su pochi casiper osservare grandi e inequivocabili effetti. Oggi invece, in un contesto dove pre-valgono le malattie croniche e non curabili – dove cioè è molto difficile guarire e
93
Le politiche che regolano il mercato dei farmaci: il ruolo delle agenzie regolatorie

l’obiettivo è ridurre i rischi, controllare il decorso della malattia e migliorare il be-nessere del paziente – è più difficile identificare i miglioramenti attribuibili con si-curezza al farmaco. In assenza di eclatanti effetti e di adeguati controlli, la sem-plice osservazione non è un approccio abbastanza valido e affidabile per docu-mentare il valore di un nuovo trattamento medico. E’ pertanto necessario accu-mulare una serie di prove descritte in termini di fasi di sperimentazione, sia preclinica (essenzialmente su animali) sia clinica (in soggetti umani: fasi I, II, III e IV),che permettano di dare un giudizio scientifico, cioè motivabile e basato su espe-rienze ripetibili, sul valore di un farmaco.
Necessità di studiclinici controllatie randomizzati
Il perno di tutte queste prove è lo studio clinico che valuta l’efficacia e la sicurezzadi un farmaco inmodo controllato. Si tratta di uno studio in cui vi sono due grup-pi di pazienti, uno dei quali è trattato con il nuovo farmaco in valutazione e l’altrono, e i pazienti sono assegnati ai due gruppi in modo randomizzato, come si dicein gergomedico, cioè assegnati dal caso e non su scelta del medico o del paziente.Inoltre, non basta dimostrare che un farmaco sia efficace, in termini assoluti orelativi, ma è necessario anche documentarne la sicurezza (presenza e quantitàdi effetti indesiderati), sia con valutazioni durante lo sviluppo (pre clinico e clini-co) sia con una attenta vigilanza durante la commercializzazione. Accanto, quin-di, alla necessità di documentare l’efficacia del farmaco, cioè il fatto che esso svol-ge l’azione terapeutica che ci si aspettava e globalmente fa bene, diventa semprepiù importante avere dati sulla sua sicurezza in modo da poter decidere sia a li-vello di popolazione, sia a livello di singolo caso, se i benefici siano superiori aiproblemi, cioè ai cosiddetti effetti collaterali indesiderati: è il rapporto rischio/be-neficio. Il più delle volte gli studi clinici condotti durante lo sviluppo del farmaconon sono in grado di quantificare con certezza la sicurezza di un farmaco: bastapensare alla possibilità che un importante effetto collaterale sia presente con unaprobabilità di 1 caso su 1.000 o su 10.000 casi per capire che, se si sono studiatiin tutto poche migliaia di casi, dal punto di vista statistico quell’importante effet-to collaterale potrebbe non essersi manifestato, ma si svelerà inevitabilmente du-rante la commercializzazione quando il prodotto sarà utilizzato da centinaia dimigliaia di soggetti. Inoltre anche la valutazione di ampie casistiche può non es-sere sufficiente se gli studi pre clinici, non sono stati condotti con attenzione. Ilcaso del talidomide è solo uno dei tanti storici esempi che sottolineano l’impor-tanza della farmacovigilanza dopo la commercializzazione.
Il caso Talidomide
La Talidomide è un farmaco commercializzato in tutto ilmondo, tranne che negli USA, dalla fine degli anni cinquan-ta, dapprima come anticonvulsivante e poi come sedativo.Veniva considerato particolarmente sicuro in quanto ancheeventuali sovra dosaggi non risultavano mai essere letali, alcontrario dei barbiturici, unici sedativi allora disponibili.Erano stati condotti studi su animali soprattutto permettere in evidenza la tossicità acuta, ma era stata datascarsa attenzione alla possibile tossicità cronica. Dato ilteorico profilo di sicurezza, fu molto spinto il suo utilizzoin gravidanza.Una lettera di 15 righe, scritta nel 1961 da un medico au-
straliano alla rivista The Lancet, che segnalava un eccessodi malformazioni in bambini nati da donne che avevanoassunto farmaci a base di talidomide, scatenò l’attenzionesulla capacità del farmaco di provocare malformazionicongenite, anche incompatibili con la vita.In breve si stimò in migliaia il numero di casi coinvolti, siscoprì la natura del problema conducendo studi appro-priati su cavie animali gravide e si tolse il farmaco dalmercato. Il caso della Talidomide insegnò che nessun far-maco dovrebbe essere fino a che il suo effetto sull’uomonon sia stato ben studiato, con modelli e test appropriatianche rispetto a specifiche condizioni (età, gravidanza).
La prima segnalazione degli effetti della talidomide, pubblicata sulla rivista The Lancet
Gentile Signore,le malformazioni congenite sono presenti in circa l’1,5 percento dei neonati. Negli ultimi mesi ho osservato chel’incidenza di malformazioni severe multiple in neonatipartoriti da donne che avevano fatto uso di Talidomidedurante la gravidanza, come antivomito o sedativo, è dialmeno il 20 per cento.Queste anomalie sono presenti nelle strutture che si svi-luppano nel mesenchima, cioè le ossa e la muscolatura in-
testinale. Lo sviluppo delle ossa sembra essere influenza-to in modo rilevante, dando come esito polidattilia [nume-ro di dita superiore alla norma], sindattilia [fusione di dueo più dita della mano/piede] e alterazioni nello sviluppodelle ossa lunghe [femore e radio anormalmente corti].Qualcuno dei lettori della rivista ha visto simili malforma-zioni in neonati partoriti da donne che avevano preso que-sto farmaco durante la gravidanza?
W.G. McBride
94
Le politiche che regolano il mercato dei farmaci: il ruolo delle agenzie regolatorie

Registrare nuovi farmaci: i sistemi europeo e americano
EMEA (European Medicines Agency)L’Agenzia Europea per i Medicinali (http://www.emea.europa.eu) è un organodecentrato dell’Unione Europea con sede a Londra. Il suo compito principale èdi tutelare e promuovere la sanità pubblica e la salute degli animali mediante lavalutazione e il controllo dei medicinali per uso umano e veterinario. Attraversouna rete di oltre 4.000 esperti europei essa coordina le risorse scientifiche di piùdi 40 autorità nazionali competenti di 30 paesi della vasta area UE-SEE. Essacontribuisce alle attività internazionali dell’Unione Europea mediante i suoi la-vori nell’ambito della farmacopea europea, dell’organizzazione mondiale dellasanità, delle conferenze trilaterali (UE, Giappone e Stati Uniti) sull’armonizza-zione e di altre organizzazioni e iniziative internazionali.La valutazione scientifica dei nuovi farmaci si basa su tre principi: qualità, sicu-rezza ed efficacia. L’EMEA dipende nell’Unione Europea dal Direttorato genera-le dell’industria, dunque considera il farmaco più un bene di consumo che unbene per la salute. Il sistema europeo di autorizzazione dei medicinali prevededue distinte procedure: una centralizzata e l’altra decentralizzata (o di mutuoriconoscimento).
Proceduracentralizzata
Le aziende farmaceutiche presentano all’EMEA un’unica richiesta di introdu-zione in commercio valida per tutti i paesi dell’Unione Europea. Il comitato per imedicinali per uso umano (CHMP, Committee for Medicinal Products for Hu-man use) o il comitato per i medicinali veterinari (CVMP, Committee for Medici-nal Products for Veterinary use) effettuano una valutazione unica e sono re-sponsabili dell’intero iter. Il CHMP è inoltre responsabile delle attività post regi-strazione compresa l’estensione o la variazione delle indicazioni d’utilizzo. Se ilcomitato, dopo aver esaminato la pratica, giunge alla conclusione che la quali-tà, la sicurezza e l’efficacia del medicinale sono state sufficientemente dimostra-te, esprime un parere favorevole, che è trasmesso alla Commissione. Essa allo-ra concede l’autorizzazione all’immissione in commercio valida per tutto il terri-torio dell’Unione Europea. La procedura centralizzata è obbligatoria per tutti iprodotti biotecnologici e per i farmaci orfani, cioè i farmaci per malattie rare.
Proceduradecentralizzatao di mutuoriconoscimento
Questa procedura non riguarda farmaci innovativi, ma prodotti per cui si ha unprevalente interesse commerciale. La procedura prevede che l’autorizzazioneall’immissione in commercio rilasciata da uno stato membro dell’Unione Euro-pea (che agisce da paese di riferimento e viene denominato Stato membro di ri-ferimento o Reference Member State-RMS) venga estesa a uno o più stati mem-bri (denominati Stati membri interessati o Concerned Member States-CMS) in-dicati dal richiedente.E’ la corsia preferenziale per raggiungere nuovi mercati quando un farmaco siagià commerciabile all’interno di uno stato dell’Unione Europea. Condizione ne-cessaria per l’applicazione della procedura di mutuo riconoscimento è che i do-cumenti e il dossier di registrazione del medicinale siano gli stessi in tutti glistati membri coinvolti nella procedura.
La procedura centralizzata resta quella maggiormente utilizzata. Il CHMP, inol-tre, risolve le dispute nei casi in cui vi è un disaccordo fra gli stati membri relati-vamente all’autorizzazione di vendita di un prodotto medicinale particolare (ar-bitration procedure). Il CHMP si occupa anche dei casi di rinvio (Community re-ferral procedure) che si avviano quando ci sono delle preoccupazioni concer-nenti farmaci e salute pubblica o dove sono in gioco altri interessi della UnioneEuropea.Nel 2001 è stato costituito il Comitato per i medicinali orfani (COMP, Committeefor Orphan Medicinal Products), la cui funzione consiste nell’esaminare do-mande di assegnazione di persone od aziende che intendono sviluppare i cosid-detti medicinali. Nel 2004 infine è stato costituito un nuovo comitato, HMPC(Committee on Herbal Medicinal Products), per i farmaci di origine vegetale.A dieci anni dalla sua inaugurazione, l’organizzazione dell’EMEA e i suoi compi-
95
Le politiche che regolano il mercato dei farmaci: il ruolo delle agenzie regolatorie

ti sono stati rivisti (insieme alle normative europee che regolano l’autorizzazioneall’immissione in commercio dei farmaci) allargando significativamente il man-dato dell’Agenzia. Oltre a una serie di cambiamenti organizzativi, verrà conferi-ta all’agenzia maggiore responsabilità, in particolare per accelerare l’accesso deipazienti a nuovi farmaci e migliorare la fornitura di informazioni a tutti coloroche fanno uso di prodotti farmaceutici. Al momento ci sono due aspetti moltocritici del lavoro dell’EMEA: l’agenzia non richiede studi di tipo comparativo pervalutare qual è il reale valore aggiunto di un nuovo principio attivo rispetto auno già in commercio; e il lavoro è per gran parte riservato e quindi non c’è tra-sparenza sui dati disponibili e sulle procedure.
FDA (Food and Drug Administration)La FDA (http://www.fda.gov) è l’ente americano che regolamenta da più di unsecolo l’approvazione di nuovi farmaci; è considerata una delle più vecchie isti-tuzioni americane a difesa del consumatore. Il mandato della FDA recita:“La FDA è responsabile della protezione della sanità pubblica assicurando la si-curezza, l’efficacia e la sicurezza degli farmaci umani, dei prodotti biologici, deidispositivi medici, dell’approvvigionamento di generi alimentari, dei cosmetici edei prodotti che emettono radiazioni. La FDA è inoltre responsabile per i pro-gressi in sanità pubblica favorendo lo sviluppo di innovazioni che rendono lemedicine e gli alimenti più efficaci, più sicuri e accessibili e aiutando il pubblicoa ottenere informazioni corrette e scientificamente valide per usare al megliomedicine e alimenti con la finalità di migliorare la loro salute”.La FDA si avvale di uno staff di più di 9.000 persone, organizzate in diversi di-partimenti, di cui almeno due si occupano dei farmaci, il CDER (Center for DrugEvaluation and Research) e il CBER (Center for Biologicals Evaluation and Re-search) a loro volta organizzati in sezioni specializzate, dotate di propri espertiche dipendono in maniera diretta dall’Agenzia. Il meccanismo attraverso il qualela FDA approva l’immissione in commercio di nuovi farmaci è stato rivisto com-pletamente durante gli anni Novanta al fine di garantire sia una attenta valuta-zione del rapporto rischio/beneficio del farmaco, sia una rapida valutazionedello stesso in modo da garantire quanto più possibile una veloce e pronta di-sponibilità del prodotto al cittadino. Tali obiettivi sono ottenuti grazie all‘azionecombinata di 3 meccanismi:l fast track (letteralmente percorso veloce) è una procedura che prevedeun’interazione diretta, attraverso riunioni di lavoro, tra la FDA e il gruppoche sviluppa un nuovo farmaco, al fine di concordare durante il cammino(dalla pre clinica alle fasi cliniche I-IV) il miglior modo per studiare il farma-co in valutazione; è riservata a farmaci cosiddetti salva vita;
l priority review (revisione prioritaria) è una procedura che viene richiestadopo che il nuovo farmaco è stato presentato per l’approvazione alla FDA,implica una accelerazione dei tempi di approvazione di un nuovo farmaco(da 10 a 6 mesi); anche in questo caso la richiesta viene fatta solo in casiparticolari;
l accelerated approval (approvazione accelerata e anticipata) è una procedurache può essere richiesta per farmaci che hanno dimostrato di essere poten-zialmente utili per condizioni cliniche che mettono a rischio la vita, il cuipercorso di valutazione della reale efficacia non è ancora stato concluso, madi cui – tuttavia – sono disponibili risultati così importanti e promettenti dasuggerire una precoce introduzione nel mercato.
La FDA ha inoltre formali programmi di collaborazione sia con esperti esterni eindipendenti, di cui si avvale per ricevere consigli, sia con agenzie e istituti pub-blici di ricerca, come l’NCI (National Cancer Institute) e l’NIH (National Instituteof Health).In questi anni FDA è stata oggetto di critica dopo che alcuni farmaci approvatio in fase di approvazione hanno prodotto gravi effetti per la popolazione (peresempio il caso del Vioxx), e la discussione che ha occupato sia la stampa me-dico scientifica sia la stampa laica è stata molto vivace; sul sito Partecipasalu-te, nella rubrica “Medicina e interesse” sono presenti numerosi articoli su que-sto tema.
96
Le politiche che regolano il mercato dei farmaci: il ruolo delle agenzie regolatorie

Le principali differenze tra EMEA e FDA
E’ chiaro dalla descrizioni sopra riportate che le due agen-zie hanno modalità organizzative, di finanziamento e di la-voro differenti. Alcuni aspetti vanno sottolineati:n i tempi di approvazione sono in generale più lunghi perEMEA rispetto a FDA, soprattutto per la disponibilità difast track e priority review;
n la FDA ha maggiori competenze interne per valutare i sin-goli prodotti e quindi non deve dipendere dalla collabo-razione di esperti provenienti dai singoli stati membri;
n la FDA si può avvalere di gruppi esterni di esperti, tecni-ci e laici, con cui confrontarsi in caso di dubbi sulle deci-sioni da prendere;
n EMEA si deve confrontare con un mercato formalmentesimile in termini di dimensioni, ma estremamente varie-gato in termini di lingue, culture, economie e modelli as-sistenziali;
n infine, EMEA esiste solo da poco più di dieci anni, men-tre la FDA da più di cento.
AIFA, Agenzia Italiana del FarmacoL’AIFA (http://www.agenziafarmaco.it), agenzia regolatoria italiana, è un orga-nismo di diritto pubblico che opera sulla base degli indirizzi e della vigilanza delMinistero della salute, in autonomia, trasparenza ed economicità, in raccordocon le regioni, l’Istituto superiore di sanità, gli Istituti di ricovero e cura a carat-tere scientifico, le associazioni dei pazienti, i medici e le società Scientifiche, ilmondo produttivo e distributivo.Le principali attività dell’AIFA sono le seguenti:l registrazione e farmacovigilanza;l prezzi rimborso e mercato;l valutazione europea;l rapporti con EMEA;l sperimentazione e ricerca;l produzione e controllo;l affari amministrativi.Le prime quattro sono strettamente correlate alla registrazione e uso dei farmaci.L’AIFA assicura inoltre l’unitarietà dell’assistenza farmaceutica sul territorionazionale nonché l’accesso ai farmaci innovativi e ai farmaci per le malattie ra-re, attraverso un processo di registrazione conforme alle procedure previste dal-la normativa vigente nell’Unione Europea.I requisiti di qualità, sicurezza ed efficacia di tutti i medicinali – ivi compresiemoderivati, vaccini, radiofarmaci, medicinali a base di erbe e medicinali omeo-patici – sono assicurati (sia nel caso di nuove autorizzazioni all’immissione incommercio, sia nel caso di variazioni) attraverso le valutazioni chimico farmace-utiche, biologiche, farmaco tossicologiche e cliniche che l’AIFA effettua avvalen-dosi di esperti interni ed esterni, degli esperti dell’Istituto Superiore di Sanità edella Commissione Tecnico Scientifica. Tutte queste valutazioni vengono effet-tuate in conformità alle normative e protocolli europei e alle Linee Guida redattedall’EMEA e dall’ICH (International Conference on Harmonization). Per garanti-re che i farmaci immessi sul mercato siano sicuri l’AIFA gestisce un servizio difarmacovigilanza che effettua il monitoraggio continuo delle segnalazioni di rea-zioni avverse al fine di identificare tempestivamente eventuali segnali di rischiolegati all’uso dei farmaci e di assicurare un rapporto rischio/beneficio favorevo-le per la popolazione. Una volta che i farmaci sono stati approvati dall’EMEA,l’AIFA si occupa di classificare i farmaci nei rispettivi livelli diversi di rimborsa-bilità e, quando opportuno, discute anche del prezzo al pubblico.
I nuovi farmaci anti tumorali e il debito di informazione
I pazienti oncologici in particolare ripongono grandi spe-ranze nelle novità, con il presupposto che il nuovo sia me-glio. In realtà, benché ci siano stati molti annunci miraco-listici, pochi sono stati i farmaci veramente innovativi nelsettore oncologico, soprattutto se si chiede al farmaco unreale beneficio clinico, cioè un aumento della sopravvi-venza dei pazienti rispetto alle terapie standard, oppureun beneficio in termini di qualità della vita.L’EMEA e l’FDA hanno un ruolo molto importante in questoprocesso perché dettano le regole e i tempi della speri-mentazione e perché sono le prime a valutare il valore
delle nuove medicine e sono responsabili della loro immis-sione in commercio.Fino a circa 10 anni fa non esistevano molte differenze trale due Agenzie, ora formalmente FDA appare più sensibilealle opinioni dei cittadini, mentre EMEA appare un po’ piùrigida e conservativa. In realtà, a un’analisi del comporta-mento delle due Agenzie nel giudicare i prodotti anti tu-morali valutati e approvati negli ultimi anni, non emergeuna grande differenza: entrambe le Agenzie hanno appro-vato i nuovi farmaci permettendo l’utilizzo di piccoli studi,non comparativi e non randomizzati, dove l’efficacia dei
97
Le politiche che regolano il mercato dei farmaci: il ruolo delle agenzie regolatorie

farmaci è stata valutata non utilizzando la sopravvivenzama semplici indicatori di attività del farmaco.In particolare uno studio pubblicato nel 2005 da Apolone ecollaboratori sul British Journal of Cancer1 documenta che:n sono stati approvati dall’EMEA, nei 10 anni considerati,14 farmaci anti tumorali per 27 differenti indicazioni, 14nuove e 13 estensioni di indicazioni precedenti;
nmolti farmaci non sono supportati da studi clinici con-trollati randomizzati e l’obiettivo più utilizzato dagli stu-di è la riduzione della massa tumorale in risposta al far-maco e non la sopravvivenza;
n più in particolare, su 48 studi presenti nei dossier deifarmaci approvati, solo 25 sono studi clinici randomizza-ti, e riguardano la metà delle relative indicazioni. Gli al-
tri sono studi clinici non randomizzati e studi clinici ran-domizzati non comparativi, cioè in assenza di altri far-maci di confronto;
n per quanto riguarda gli obiettivi utilizzati, solo per dueindicazioni è stato considerato come obiettivo primariola sopravvivenza, negli altri casi è stata valutata la ri-sposta del tumore al farmaco (cioè la riduzione dellamassa tumorale), o la sopravvivenza libera da malattia,cioè l’intervallo tra l’intervento terapeutico e la ricom-parsa della malattia;
n infine, dalla valutazione della sopravvivenza, è emersoche l’aumento medio attribuibile ai nuovi farmaci eramolto piccolo (in media intorno a un mese e mezzo) emai superiore a 3 mesi.
Enti regolatori e partecipazione dei cittadiniIl processo di approvazione di un nuovo farmaco necessita che molte e diversecompetenze siano presenti: tutte le parti interessate devono trovare una giustacollocazione nel processo e tra queste anche, naturalmente, le rappresentanzedi pazienti e cittadini. La FDA prima, e l’EMEA più recentemente, stanno met-tendo in pratica differenti modelli di coinvolgimento diretto di cittadini, di pa-zienti e di loro associazioni.La FDA ha istituito, già da diversi anni, un sistema per integrare il punto di vi-sta dei pazienti e delle sue rappresentanze nella valutazione clinica e scientificadel dossier sui nuovi farmaci. I rappresentanti dei cittadini e dei pazienti sonostati coinvolti per avere un collegamento tra commissioni medico scientifiche eutilizzatori tramite diversi programmi e approcci.2 Nell’intento di fornire questotipo di formazione, la FDA ha predisposto un documento per descrivere, attra-verso una dozzina di domande, le modalità di coinvolgimento;3 inoltre, sono sta-ti sviluppati programmi specifici per i rappresentanti dei pazienti con cancro.4
Il modello FDA potrebbe essere importato a livello europeo per dare maggiorepossibilità ai rappresentanti dei pazienti di partecipare attivamente e per fare inmodo che il processo di coinvolgimento incida concretamente sul percorso diapprovazione del farmaco.L’EMEA da marzo 2005 ha pubblicato diversi documenti (disponibili in linguainglese sul sito dell’Agenzia, www.emea.europa.eu) finalizzati, che mirano a darvita a un gruppo di lavoro con rappresentanti di pazienti. Secondo questi docu-menti, per poter collaborare con l’EMEA le associazioni devono avere alcune ca-ratteristiche tra cui – oltre alla presenza di uno statuto registrato, all’interesseper i farmaci, alla rappresentatività a livello europeo – la trasparenza sulle fontidi finanziamento e sul loro utilizzo. Infatti, come è stato sottolineato ancora re-centemente (http://www.partecipasalute.it/associazioni/european-patients-forum.pdf) la trasparenza e la mancanza di conflitti di interessi da parte dellestesse associazioni di cittadini e pazienti rimane un nodo ancora difficile da ri-solvere. La consultazione dei cittadini e delle organizzazioni avviene solo su ri-chiesta di una delle parti coinvolte nel processo di approvazione (comitatoscientifico, working parties/scientific advisory groups o rapporteur). L’EMEA haanche affermato la necessità di una riorganizzazione del suo sito in modo darenderlo più fruibile alla navigazione da parte di cittadini e pazienti. Si proponeinoltre la diffusione capillare delle informazioni rilevanti sui farmaci, collabora-re alle indagini di farmacovigilanza, assicurare trasparenza delle informazioni.
Bibliografia1. Apolone G et al. Ten years of marketing approvals of anti-cancer drugs in
Europe. Regulatory policy and guidance documents need to find a balancebetween different pressures. BJC 2005;93:504-9
2. FDA: Cfr. http://www.fda.gov/oashi/patrep/patientrep.html#overview.3. When a Patient Speaks… Patient Representatives to FDA Advisory Commit-
tees, http://www.fda.gov/oashi/patrep/patbroc.html4. FDA: http://www.fda.gov/oashi/cancer/cpat.html
98
Le politiche che regolano il mercato dei farmaci: il ruolo delle agenzie regolatorie

L’associazionismo sta cambiando:dall’assistenza alla nascita di un progettoCinzia Colombo, Paola MosconiRevisori: Silvia Nidasio, Pierluigi Pennati
La partecipazione dei cittadini
I pazienti hanno sempre avuto il ruolo passivo di fruitori dei servizi sanitari edelle terapie, all’interno di un paradigma paternalistico consuetudinario nellasanità e nella salute. Tale paradigma sta lasciando il posto, in alcune situazioni,a un modello alternativo che vede il cittadino come consapevole e collaborativoprotagonista che partecipa alle scelte che riguardano la sua salute, l’organizza-zione dei servizi sanitari, e le priorità di ricerca. La partecipazione dei cittadini,come dimostrato in alcuni progetti sperimentali, potrebbe contribuire a miglio-rare e rendere più accessibili i servizi e a migliorare la salute e la qualità della vi-ta dei pazienti, e potrebbe migliorare l’appropriatezza dei trattamenti. I pazientie le organizzazioni che sono coinvolti e partecipano alle scelte hanno maggioreconsapevolezza dei propri diritti, acquisiscono conoscenze di carattere organiz-zativo, clinico e politico, hanno un maggiore controllo sulla loro situazione disalute e sui trattamenti cui sono sottoposti. Le organizzazioni, inoltre, possonocontribuire a migliorare situazioni problematiche, sostenendo cambiamenti or-ganizzativi o politici, influenzando linguaggi, stili, contenuti. Questo capitolodescrive il ruolo delle organizzazioni di pazienti in Italia nella condivisione delleresponsabilità con i diversi protagonisti del settore sanitario.
Le organizzazioni non profit che operano nel settore sanitario
In Italia sono numerose le organizzazioni non profit che operano nel settore sanita-rio: oltre 21.000 nel 2003, secondo la rilevazione sulle organizzazioni di volontaria-to effettuata dall’Istat nel biennio 2004-2005.1 Queste organizzazioni compongonouna realtà in continua trasformazione in cui stanno nascendo sempre più spessofederazioni di singole organizzazioni locali. Le organizzazioni di pazienti rivolgonospesso la loro attenzione ad aspetti legati all’assistenza sanitaria e sono ancora po-co orientate a richiedere per i cittadini che rappresentano uno spazio nella discus-sione sullo sviluppo, la pianificazione e l’organizzazione dei servizi sanitari o nell’i-dentificazione delle priorità di ricerca.2 La corresponsabilità, cioè la condivisionedelle responsabilità con i protagonisti del settore sanitario, pur nei distinti ambitidi competenza, in molte realtà è ancora un progetto da avviare. I cambiamenti diruolo e responsabilità delle organizzazioni sono piuttosto lenti e, a seconda dellepatologie e delle zone del territorio italiano, tuttavia si assiste a una lenta matura-zione, favorita da significativi mutamenti del panorama sanitario di riferimento.
Le premesse del cambiamentoDiverse condizioni rendono necessario, in questo momento storico, un cambia-mento del rapporto fra i cittadini e il sistema sanitario:l anche in Italia – come in altri paesi sviluppati – condizioni di vita migliori,cure di qualità più elevata e capacità diagnostiche avanzate hanno trasfor-mato molte patologie da acute in croniche, creando un costante contatto deipazienti e dei loro familiari con i servizi sanitari. Si pensi a esempio al pa-ziente oncologico, che resta in contatto con l’ospedale o il centro presso cui èstato curato almeno cinque anni dopo aver concluso le terapie primarie (pe-riodo di follow up), al paziente diabetico, al paziente sieropositivo, che dopola diagnosi sono seguiti e assistiti per tutto l’arco della vita. Vivendo a lungocon una malattia e imparando a gestirne le conseguenze, molte persone svi-luppano un alto grado di capacità e competenza;
99
L’associazionismo sta cambiando: dall’assistenza alla nascita di un progetto

l critiche sonomosse dall’opinione pubblica al sistema sanitario e alle sue po-litiche. La comunicazione sui problemi, sulle prospettive, sulle collocazionidei servizi, sulle buone pratiche è carente. Poco si sta facendo concretamen-te per quanto riguarda il miglioramento dell’informazione sui risultatidell’assistenza sanitaria nei confronti dei cittadini e dei pazienti;
l la sostenibilità economica del sistema sanitario è sempre più problematica e alcittadino/utente è richiesta maggior responsabilità nell’uso delle risorse; il co-involgimento permette ai governi di condividere decisioni difficili e promuovecomportamenti virtuosi. D’altra parte, i cittadini e le loro organizzazioni hannobisogno di rielaborare attraverso il dialogo e la discussione valori conflittuali escelte difficili riguardanti questioni importanti, se devono poi essere in grado dianalizzarne tutte le implicazioni e valutare la scelta delle priorità;
l il rapporto medico paziente sta cambiando, e ha subito l’influenza della di-scussione in corso da tempo nei paesi anglosassoni sul passaggio dal pater-nalismo medico a un rapporto di collaborazione. Si sta sviluppando di con-seguenza la ricerca di modelli di partecipazione e condivisione dei ruoli;
l si allarga e cresce la medicina basata su evidenze: molte delle risposte alledomande sugli effetti delle terapie possono essere ottenute consultando re-visioni sistematiche e prove di efficacia disseminate da enti e progetti pub-blici, che si occupano anche della traduzione di revisioni internazionali;
l nuove patologie, alcune create ad hoc per interessi economici (secondo il fe-nomeno del disease mongering – commercio di malattie – già ricordato an-che in capitoli precedenti) si affacciano sul panorama sanitario e spingonoverso una forte e inutile medicalizzazione.
Una strada verso coinvolgimento e partecipazione
Il ruolo delle organizzazioni di pazientiIn questo panorama i cittadini e le organizzazioni con sempre maggior frequenzasono chiamati a partecipare al dibattito sulla salute e vengono coinvolti nellescelte sanitarie e nella valutazione dei servizi. Alcune associazioni hanno assuntoun carattere transnazionale: sono molto attente agli aspetti riguardanti l’accessoe l’organizzazione di servizi e l’accesso ai trattamenti e fanno sentire la loro voceproprio su questi aspetti. Altre sono passate da un ruolo di spinta verso il miglio-ramento delle politiche sanitarie alla gestione di progetti di informazione e ricer-ca: infatti partecipano alla ricerca clinica e sociologica che le riguarda e su moltifronti riprovazione sociale (verso talune malattie, trattamenti, prevenzione) sonoautorevoli consulenti delle autorità sanitarie per quanto riguarda ogni aspettodella loro patologia. In altri casi hanno organizzato autonomamente o in collabo-razione con il sistema sanitario servizi informativi e di consulenza.
L’esempio di Europa Donna: un ruolo riconosciuto a livello medico e istituzionale
1992: sei paesi europei lanciano – sull’esempio di un’ana-loga iniziativa americana – una coalizione per la lotta con-tro il tumore al seno.1996: in Italia nasce il Forum Italiano, che unisce a sua vol-ta più di un centinaio di associazioni sparse sul territorionazionale. Il mondo medico e politico vengono attivamentecoinvolti, vengono raccolti dati, attivati tavoli di discussionecomuni, avviati progetti di collaborazione. Gruppi trasver-sali di parlamentari, in diversi Paesi, appoggiano la lottacontro il tumore al seno sostenendo iniziative per la diffu-sione di informazioni, attivazione di programmi di scree-ning, miglioramento dell’assistenza sanitaria. In Italia è at-tivo il gruppo Europa Donna Parlamento, costituito da par-lamentari dei diversi schieramenti politici che annualmente
discute i risultati ottenuti e programma le attività future.2006: i paesi aderenti sono 39.
In concreto Europa Donna:l è partner alla pari del maggior congresso europeo sultumore al seno (EBCC European Breast Cancer Confe-rence);
l ha contribuito a istituire un gruppo di sostegno al Parla-mento Europeo che si è fatto promotore della stesura diuna Risoluzione Europea per il tumore al seno. La Riso-luzione è stata recepita dai paesi aderenti e sta portan-do cambiamenti, nella prevenzione e assistenza;
l ha creato un forte movimento di aggregazione di gran-de traino per tutte le realtà periferiche.
L’indipendenza dal mondo medico e dalle industrie farmaceutichePer molte realtà associative la questione che rimane da affrontare e superare èquella dell’indipendenza di ruolo, ovvero dell’affrancamento dal legame cultura-le e sanitario che spesso hanno con il mondo medico; in molti comitati direttivi
100
L’associazionismo sta cambiando: dall’assistenza alla nascita di un progetto

di organizzazioni la sproporzione tra componente medico tecnica e rappresen-tanza di cittadini/pazienti è notevolmente a favore della prima. Per acquisireautonomia e indipendenza in questo senso è fondamentale per le organizzazioniaccrescere la capacità di orientarsi in modo corretto nel campo dell’informazio-ne sulla salute e distinguere il ruolo che deve avere il rappresentante dei cittadi-ni/pazienti rispetto agli altri decisori. Le associazioni dei pazienti devono poteraccedere a informazioni comprensibili e di buona qualità, altrimenti non po-tranno partecipare alle scelte che li riguardano personalmente o collaborare almiglioramento del sistema sanitario.E’ inoltre necessario che le organizzazioni aumentino la consapevolezza del ruo-lo strategico che rivestono e degli interessi di cui sono oggetto, in particolare daparte dell’industria: si pensi alla richiesta di modifica della direttiva europeache regolamenta la pubblicità sui farmaci, per ottenere una pubblicità diretta alpubblico dei farmaci di prescrizione, o alla creazione di sezioni o siti ad hoc pernon tecnici da parte di aziende farmaceutiche o istituti o enti di ricerca; oppurealla creazione all’interno delle aziende farmaceutiche di uffici specificamentedestinati a gestire i rapporti con il mondo del volontariato, o alle donazioni cheprovengono da parte delle case farmaceutiche.
Una strada verso coinvolgimento e partecipazione
Verso la nascita di un progettoLe trasformazioni in corso nel mondo dell’associazionismo stanno spingendoverso la nascita di nuovi progetti che coinvolgono il pubblico e i pazienti. In Ita-lia si è ancora agli inizi, mentre in paesi come Stati Uniti, Regno Unito, Austra-lia, per motivi storici e culturali, le associazioni di pazienti sono da tempo attivee pretendono di essere riconosciute come un interlocutore indispensabile per ledecisioni medico sanitarie. Negli ultimi decenni questa consapevolezza si starafforzando nel nostro paese – “Niente su di noi senza di noi” è lo slogan condivi-so da molte associazioni – e le organizzazioni o federazioni di pazienti semprepiù spesso rifiutano di delegare le proprie decisioni all’esperto, per definire eportare avanti in prima persona le proprie richieste ed esigenze.
Le organizzazioni dei pazienti promuovono iniziative...Già da tempo in Italia molte organizzazioni di pazienti organizzano corsi di for-mazione al proprio interno e collaborano a campagne di sensibilizzazione, masolo di recente alcune di esse hanno iniziato a promuovere o sostenere progettidi ricerca e azioni di lobby. Esempi in questo senso arrivano dal bando per la ri-cerca indipendente dell’Agenzia italiana del farmaco (AIFA), a cui hanno parte-cipato nel 2005 alcuni progetti di associazioni di pazienti, promossi insieme asocietà scientifiche o istituti di ricerca, così come dalle richieste al ParlamentoEuropeo presentate dal Forum Europa Donna contro il tumore al seno, chehanno portato a una risoluzione parlamentare. Infine, attraverso la creazione diuna rete di organizzazioni a livello europeo, un gruppo di attivisti per la lottacontro l’AIDS – l’Italian community advisory board (I-CAB) membro dell’E-CAB(corrispettivo europeo) – ha rapporti con industrie farmaceutiche di cui legge ediscute i protocolli clinici, occupandosi anche di rileggere e proporre eventualimodifiche ai moduli di consenso informato.Queste esperienze testimoniano il passaggio di interesse e di azione da un am-bito assistenziale a interventi di lobby su aspetti legislativi e sulla produzione diricerca scientifica, passaggio che si può compiere grazie anche al rapporto privi-legiato delle associazioni con i pazienti e i cittadini che rappresentano, ricono-sciuto sempre più spesso dagli altri decisori che si occupano di salute.
…e sono chiamate a partecipareSono oramai frequenti le iniziative organizzate da enti istituzionali, istituti di ri-cerca, industrie farmaceutiche, che coinvolgono cittadini, pazienti e loro orga-nizzazioni. Le finalità sono diverse e possono andare dal miglioramento dei ser-vizi sanitari, alla riduzione di spese sanitarie non appropriate, alla scelta degliambiti di ricerca, oppure, al contrario, alla spinta verso il consumo di prestazio-
101
L’associazionismo sta cambiando: dall’assistenza alla nascita di un progetto

ni e prodotti sanitari. In questo panorama variegato alcune esperienze locali eregionali sono condotte insieme ai cittadini e ai pazienti per aumentare la loropartecipazione alle decisioni di salute e la loro consapevolezza sui diritti di salu-te o sulla comunicazione del rischio nella pratica clinica. Nei riquadri sono sin-teticamente riportati, a titolo di esempio, alcune iniziative significative in corsoin Emilia Romagna, una tra le regioni più sensibili e attive su queste tematiche.Si tratta di progetti nati da un coinvolgimento istituzionale, che hanno una rica-duta pratica sul territorio, e sono composti da gruppi multidisciplinari di medi-ci, operatori sanitari, cittadini e pazienti.
Il laboratorio per il cittadino competente di Modena
L’AUSL di Modena ha promosso nel 2000 il Laboratorio peril cittadino competente. Si tratta di un gruppo di lavoromultidisciplinare che comprende medici, operatori sanita-ri, cittadini e pazienti nato grazie alla collaborazione tra ilCentro per la valutazione dell‘ efficacia dell‘assistenza sa-nitaria (CeVeas) e il comitato consultivo misto di Sassuo-lo, che si pone gli obiettivi di:l aumentare la possibilità e capacità di cittadini e pazienti dicomprendere i linguaggi utilizzati per i temi di la salute;
l sviluppare un linguaggio comune tra medici, infermieri,farmacisti, cittadini e rappresentanti di associazioni;
l produrre strumenti di comunicazione scientificamentecorretti e comprensibili.
Finora si è occupato di:l organizzare corsi di formazione rivolti a operatori sani-tari e rappresentanti di organizzazioni di volontariato;
l produrre materiale informativo su: alimentazione, or-ganizzazioni di volontariato a sostegno di persone larin-gotomizzate, comportamenti a rischio per persone conpacemaker, diabete, allattamento al seno, osteoporosi.
Per maggiori informazioni si può vedere il sito: http://www.ausl.mo.it/comunicazione/lab.php
Il laboratorio dei cittadini per la salute di Bologna
La tua disdetta aiuta chi aspettaL’invito rivolto ai cittadini nella campagna organizzata dalLaboratorio dei cittadini per la salute di Bologna è di disdi-re le visite prenotate presso le strutture pubbliche a cui sirinuncia, alleggerendo le liste di attesa.Il Laboratorio aveva stimato che, nelle strutture dell’AUSLdella sola città di Bologna durante l’anno 2003, ben18.000 tra visite ed esami non erano stati effettuati per-ché chi li aveva prenotati non si era presentato all’appun-tamento e non ne aveva dato comunicazione tempestiva:queste prestazioni non avevano potuto così essere rimes-se a disposizione di altri cittadini.Con l’aiuto delle associazioni e organizzazioni presenti nelLaboratorio, radicate nel territorio e nel tessuto sociale,che hanno contribuito alla diffusione del messaggio attra-verso le proprie pubblicazioni, i siti web e la distribuzione
di materiale informativo nei propri locali, si sono ottenutiimportanti risultati: sono 17.400 in più dell’anno prece-dente le prenotazioni di visite ed esami disdetti dai cittadi-ni bolognesi nei dodici mesi (novembre 2004/ novembre2005) in cui si è svolta la campagna di comunicazione.Nato nel 2002 su progetto dell’AUSL di Bologna, il Labora-torio di Bologna prende le mosse dall’esperienza del Labo-ratorio per il cittadino competente di Modena, e finora, si èoccupato di:l screening: migliorare la comunicazione, aumentare l’a-desione ai programmi di screening regionali promuo-vendo incontri e iniziative informativi e indagini quanti-tative e qualitative;
l liste di attesa.Per maggiori informazioni si può vedere il sito: http://www.ausl.bologna.it/labcitsal/
Il Programma ricerca e innovazione dell’Emilia Romagna (PRI E-R)Il programma è nato come una iniziativa istituzionale mirataa creare programmi di collaborazione con le Aziende sanita-rie della regione allo scopo di sviluppare attività di ricerca einnovazione in modo sistematico (LR n. 29/2004). Il proget-to mira quindi a integrare la ricerca e l’innovazione nel nor-male funzionamento del sistema trasformando gli assetti or-ganizzativi e gestionali. Con finanziamenti pubblici e privatisono stati avviati progetti nell’area cardiologia, oncologica enell’ambito della continuità assistenziale e integrazione so-ciosanitaria. All’interno dei progetti oncologici orientati a:l uso appropriato dei nuovi farmaci oncologici;l uso della tomografia a emissione di positroni (PET);l benefici e rischi dei nuove modalità di trattamento ra-dioterapico del tumore al seno all’interno delle strutturedel Servizio sanitario regionale;
l efficacia e impatto sulla qualità della vita di programmiche valutano la situazione del paziente dopo la terapiaprimaria; grande enfasi è stata data alla collaborazionecon le organizzazioni di pazienti presenti sul territorio(una quindicina) che sono state coinvolte sia nella fasedi programmazione delle ricerche sia nella fase operati-va e contribuiranno alla fase di divulgazione dei risultati.
Lisa Beveridge, dell’associazione Crisalide di Rimini così rife-
risce l’esperienza di una rappresentante di associazione cheha partecipato al gruppo di lavoro sui nuovi farmaci oncolo-gici: “Partecipare a questi incontri è stato stimolante ma an-che molto faticoso, soprattutto all’inizio: il primo ostacolo èstato il linguaggio. Abbiamo dovuto leggere documenti tuttiin inglese, con termini medici e tecnici, abbiamo dovuto leg-gere molto, e, nonostante la mezza giornata di corso di for-mazione che abbiamo seguito, affrontare questioni e aspettitecnici non è stato semplice”. A questo si è aggiunta la neces-sità di valutare la letteratura medica e le questioni affrontatenegli incontri seguendo un metodo di giudizio rigoroso, a cuinon tutti i rappresentanti di organizzazioni erano abituati.“Chi partecipa a un tavolo di ricerca deve sapere astrarsi dal-l’esperienza personale e portare un punto di vista che sia utileai pazienti che rappresenta”. Nonostante le difficoltà ad ac-cettare la presenza di pazienti da parte dei medici, la volontàdi lavorare insieme alla fine è prevalsa: “a una prima reazio-ne di chiusura da parte dei clinici e dopo un picco di tensione,si è creato un clima di collaborazione per creare un nucleo diinformazioni da condividere e divulgare” conclude Lisa.Per maggiori informazioni sul progetto PRI E-R: http://asr.regione.emilia-romagna.it/wcm/asr/aree_di_programma/ricercaeinnovazione/pr_prier.htm
102
L’associazionismo sta cambiando: dall’assistenza alla nascita di un progetto

Panorama internazionale: un’alleanza all’insegnadell’incertezza della medicina
La James Lind AllianceStoricamente le esperienze di condivisione delle responsabilità con i diversi at-tori del mondo sanitario sono più radicate nel mondo anglosassone.3 Una diqueste iniziative è la James Lind Alliance (http://www.lindalliance.org). Natanel Regno Unito alla fine del 2004 in seguito alla decisione del Medical ResearchCouncil inglese di coinvolgere i pazienti e i consumatori nelle diverse fasi dellaconduzione degli studi clinici, è stata fondata da INVOLVE, dalla Royal Societyof Medicine e dalla James Lind Library.4 Questa organizzazione, che basa lapropria filosofia sul riconoscimento della medicina come disciplina caratteriz-zata da ambiti in incertezza, si pone gli obiettivi di:l indirizzare la ricerca verso priorità stabilite da pazienti e clinici;l definire, in modo condiviso da pazienti e clinici, i quesiti di ricerca sull’effi-cacia di trattamenti ancora senza prove;
l aumentare la conoscenza del pubblico e degli operatori sanitari sull’incer-tezza riguardo agli effetti dei trattamenti.
La James Lind Alliance funziona come una coalizione composta da ricercatori,medici, operatori sanitari, pazienti e consumatori e si avvale di gruppi di lavoromisti che fino a ora si sono occupati in particolare di identificare aree di incer-tezza nei disturbi d’asma, e più in generale di rendere noto il concetto di incer-tezza che accompagna l’effettiva utilità dei trattamenti e di sviluppare un data-base sulle incertezze degli effetti dei trattamenti (DUETs, http://www.duets.nhs.uk/). Gruppi di cittadini e pazienti sono invitati a porre quesiti sui tratta-menti, che segnalano aree di incertezza tra cui identificare aree di ricerca dasviluppare.
Il progetto Partecipasalute
Primo bilancio di un’alleanza strategica fra associazionie comunità medico scientificaIl progetto Partecipasalute (attivo dal 2003) è stato una palestra importante perraccogliere dati ed esperienze intorno all’idea di una partecipazione attiva deiconsumatori e delle loro rappresentanze.
Un percorsoa ostacoli
Attraverso le attività di Partecipasalute è possibile individuare oggi, in Italia, i li-miti più rilevanti per passare dall’assistenza alla nascita di un progetto, e pro-porre alcuni spunti per superarli:l esistono ancora notevoli difficoltà da parte di medici, ricercatori e decisori dipolitica sanitaria ad accettare le organizzazioni di pazienti come possibili in-terlocutori nel dibattito della salute; a ciò si affianca il fatto che l’esperienzariportata dalle organizzazioni ha ancora un carattere prevalentemente emo-zionale o rivendicativo, piuttosto che costruttivo e complementare all’espe-rienza medica e gestionale;
l le organizzazioni sono ancora lontane dall’idea di tutela dei diritti che si fon-di sulle conoscenze mediche basate su prove di efficacia; tale idea intendeapplicare principi scientifici alle iniziative di promozione e di prevenzione disalute in modo indipendente ma coordinato con gli sforzi dei clinici, degliepidemiologi, dei funzionari di sanità pubblica e dei decisori politici;
l una barriera importante all’acquisizione di informazioni basate su prove di ef-ficacia è legata alla difficoltà di accedere alle informazioni, sia per un motivolinguistico – la lingua del mondo medico sanitario è l’inglese – sia per scarsitàdi fonti ad accesso libero e gratuito, sia per mancanza di esperienza e metododa parte delle organizzazioni stesse nel raccogliere in modo autonomo e legge-re in modo critico informazioni di salute scientificamente provate.
Spunti per andareoltre
Un tentativo di offrire accesso a informazioni di qualità basate sulle prove scien-tifiche è rappresentato dalla rubrica pubblicata sul sito Partecipasalute “Cureutili, inutili, non si sa”, che traduce i riassunti strutturati delle revisioni prodot-
103
L’associazionismo sta cambiando: dall’assistenza alla nascita di un progetto

te dalla Cochrane collaboration e le versioni delle revisioni destinate ai non tec-nici, pubblicandoli insieme a capitoli di un testo medico (Clinical Evidence) cheaiutano e definire il contesto degli argomenti trattati. L’esperienza è nelle fasiiniziali e le modalità di divulgazione delle informazioni, così come il loro effetto,sono motivo di discussione, oltre che oggetto di valutazione tramite un questio-nario. Il web si sta dimostrando una risorsa importante per divulgare informa-zioni: esistono siti di qualità (spesso in inglese) che possono aiutare a trovare in-formazioni rilevanti e provate.
Strategie per promuovere la partecipazione
Per leorganizzazioni
Organizzare e partecipare a programmi di empowerment con percorsi di forma-zione può essere utile per poi partecipare in modo più consapevole e attivo a ta-voli di confronto, progetti o discussioni e per acquisire professionalità maggiorenello svolgimento del proprio ruolo.
Per gli operatoriche voglionocoinvolgerele organizzazioni
Per evitare che la presenza e la partecipazione di organizzazioni, cittadini o pa-zienti in gruppi di lavoro o progetti siano solo formali, è necessario che vengasvolta una continua verifica sull’efficacia delle iniziative di coinvolgimento, eche vengano promosse e divulgate le iniziative rivolte ai cittadini e ai pazienti.L’esempio della Carta dei servizi è indicativo: le Carte dei servizi sono una op-portunità per la partecipazione, perché sono contemporaneamente guida aiservizi e patto con i cittadini. La Carta dei servizi fu istituita per la prima volta inItalia dal DPCM 27 aprile 1994, ma la legge numero 328 del 2000 prevede lasua adozione come requisito necessario all’accreditamento, fornendo un im-pulso alla sua diffusione. Si tratta di uno strumento obbligatorio per chi erogaservizi, scritto dalle Aziende Sanitarie Locali per cittadini, organizzazioni dipazienti ed enti locali, ma spesso la sua formulazione non tiene conto delle ne-cessità e richieste dei suoi destinatari, così come dimostrato da una indaginesu 19 documenti.5
Per concludere E’ importante che le organizzazioni possano partecipare alla pari ai lavori dicommissioni, gruppi di lavoro, progetti di ricerca, per portare la voce dei pazien-ti che rappresentano. Perché ciò si realizzi pienamente è necessario da un latoche le organizzazioni si affermino e si affianchino nel ruolo di esperti, svilup-pando le proprie conoscenze, capacità critiche e i propri strumenti operativi,dall’altro che medici, ricercatori e politici superino quelle barriere culturali e so-ciali che li portano a relegare le organizzazioni nel ruolo di passivi ascoltatori divoci che si pretendono autorevoli.
Bibliografia1. Le organizzazioni di volontariato in Italia http://www.istat.it/salastam-
pa/comunicati/non_calendario/20051014_002. Indagine sulle associazioni/federazioni dei pazienti, http://www.parteci-
pasalute.it/attivita/indagini-001.php3. Mosconi P et al. Come, dove, quando vengono coinvolti i cittadini in sanità.
Clinical Governance, agosto 2005:24-30; http://www.partecipasalute.it/partecipa/clinical-governance.pdf
4. Il gruppo INVOLVE, nato nel 2003, promuove iniziative di coinvolgimentodi pazienti e cittadini (http://www.invo.org.uk).
5. Carta dei servizi: uno strumento utile per il Cittadino?, http://www.parte-cipasalute.it/diritti/carta-servizi-001.php
104
L’associazionismo sta cambiando: dall’assistenza alla nascita di un progetto


















