
Parte II. Fisica atomica CAP. 8. La questione dell’atomismo e l ... · sul sistema più semplice,...
70
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita. 1 Parte II. Fisica atomica CAP. 8. La questione dell’atomismo e l’affermazione della teoria cinetica §8.1. La nascita della teoria cinetica §8.2. La stima delle dimensioni delle molecole §8.3. Altre strade per la stima delle dimensioni molecolari. L’esperimento di Rayleigh-Röntgen § 8.4. L’atomismo e le molte vie sperimentali per la determinazione della costante di Avogadro. Cap. 9. Il moto browniano §9.1. Perché trattare la storia del moto browniano §9.2. Cronologia del processo di scoperta del moto browniano § 9.3. Contesto teorico-sperimentale. Evidenza empirica a favore dell’atomismo § 9.4. Le prime ricerche sul moto browniano § 9.5. Analisi della trattazione di Einstein sul moto browniano §9.6. Fluttuazioni e opalescenza vicino al punto critico. Cap. 10. La nascita della spettroscopia §10.1. Cronologia sintetica §10.2. Verifica sperimentale della formula di Balmer e determinazione della costante di Rydberg R Cap. 11. Alle radici della legge del corpo nero di Planck §11.1. Le leggi della radiazione termica §11.2. Tra dati sperimentali e assunzioni teoriche. Verifica della legge di Stefan- Boltzmann e delle leggi di Kirchhoff Cap. 12. Effetto fotoelettrico §12.1. Cronologia essenziale §12.2. Effetto Fotoelettrico: fenomenologia e interpretazioni teoriche §12.3. Agli esperimenti Cap. 13. L’atomo quantizzato di Bohr e l’esperimento di Franck ed Hertz §13.1. L’atomo di Bohr §13.2. Il percorso di Franck ed Hertz §13.3. Che faceva intanto Bohr? §13.4. All’esperimento Cap. 14 Lo spin dell’elettrone Cap. 15. Verso le basse temperature: la scoperta della superconduttività §15.1. In che consiste la superconduttività §15.2. Studio della resistenza elettrica in funzione di T §15.3. Un’altra scoperta importante: l’effetto Meissner §15.4. Le spiegazioni della SC
Transcript of Parte II. Fisica atomica CAP. 8. La questione dell’atomismo e l ... · sul sistema più semplice,...

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
1
Parte II. Fisica atomica CAP. 8. La questione dell’atomismo e l’affermazione della teoria cinetica §8.1. La nascita della teoria cinetica §8.2. La stima delle dimensioni delle molecole §8.3. Altre strade per la stima delle dimensioni molecolari. L’esperimento di Rayleigh-Röntgen § 8.4. L’atomismo e le molte vie sperimentali per la determinazione della costante di Avogadro. Cap. 9. Il moto browniano §9.1. Perché trattare la storia del moto browniano §9.2. Cronologia del processo di scoperta del moto browniano § 9.3. Contesto teorico-sperimentale. Evidenza empirica a favore dell’atomismo § 9.4. Le prime ricerche sul moto browniano § 9.5. Analisi della trattazione di Einstein sul moto browniano §9.6. Fluttuazioni e opalescenza vicino al punto critico. Cap. 10. La nascita della spettroscopia §10.1. Cronologia sintetica §10.2. Verifica sperimentale della formula di Balmer e determinazione della costante di Rydberg R Cap. 11. Alle radici della legge del corpo nero di Planck §11.1. Le leggi della radiazione termica §11.2. Tra dati sperimentali e assunzioni teoriche. Verifica della legge di Stefan-Boltzmann e delle leggi di Kirchhoff Cap. 12. Effetto fotoelettrico §12.1. Cronologia essenziale §12.2. Effetto Fotoelettrico: fenomenologia e interpretazioni teoriche §12.3. Agli esperimenti Cap. 13. L’atomo quantizzato di Bohr e l’esperimento di Franck ed Hertz §13.1. L’atomo di Bohr §13.2. Il percorso di Franck ed Hertz §13.3. Che faceva intanto Bohr? §13.4. All’esperimento Cap. 14 Lo spin dell’elettrone Cap. 15. Verso le basse temperature: la scoperta della superconduttività §15.1. In che consiste la superconduttività §15.2. Studio della resistenza elettrica in funzione di T §15.3. Un’altra scoperta importante: l’effetto Meissner §15.4. Le spiegazioni della SC

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
2
CAP. 8. LA QUESTIONE DELL’ATOMISMO E L’AFFERMAZIONE DELLA TEORIA CINETICA
§8.1. La nascita della teoria cinetica Parleremo nel seguito di atomistica e di teoria cinetica. Che si intende per Atomistica (1850 ca.-1910 ca.): insieme di ipotesi teoriche e di prove sperimentali che contribuirono al processo di legittimazione e di accettazione della teoria cinetica. La teoria cinetica emerge nella seconda metà dell’Ottocento come una concezione radicalmente nuova dei fenomeni del calore (si chiama anche, all’inizio, teoria meccanica del calore). Nasce e cresce in contrapposizione alla vecchia fisica sperimentale e in particolare alla cosiddetta termodinamica fenomenologica (o macroscopica o “generale”). La termodinamica fenomenologica è basata su parametri osservabili (p, V, T, Q, L, U, S, ecc.) e su questi si fondano i suoi principi. La teoria cinetica si basa al contrario su ipotesi microscopiche, in particolare sull’ipotesi che il calore, almeno per i gas, vada identificato con la vis viva o energia cinetica delle molecole (il termine vis viva viene sostituito dalla locuzione moderna ‘energia cinetica’ a partire dagli anni Sessanta dell’Ottocento). Già D. Bernoulli aveva fatto nel Settecento un primo tentativo di fondare la teoria cinetica; un secolo dopo, tra i pionieri troviamo Herapath (1847), Waterstone (1846), Joule (1848), Kronig (1856). I lavori di Kronig stimolano Clausius (1857-59) a occuparsi dello stesso filone di indagine e a loro volta i contributi di Clausius stimolano Maxwell (1859); Boltzmann (dal 1870) fonda la meccanica statistica a partire dalla teoria cinetica dai gas e quindi Gibbs (1902) si occupa dei fenomeni all’equilibrio. Clausius abbraccia una trattazione puramente meccanica del calore mentre con Maxwell e Boltzmann la teoria cinetica prende un indirizzo del tutto nuovo e si coniuga ormai a principi e idee probabilistiche e statistiche. All’inizio la teoria cinetica viene costruita sul sistema più semplice, il gas monoatomico (nei gas le interazioni tra gli atomi sono molto più deboli che nei liquidi e nei solidi e inoltre il gas monoatomico è fatto di singoli atomi). Clausius e il modello di gas perfetto (1857) “Sul tipo di moto che chiamiamo calore”: le particelle hanno volume molto minore del volume totale del gas (v<<V); si muovono di moto rettilineo uniforme fino a che urtano elasticamente con le pareti del recipiente e tra loro; la durata degli urti è infinitesima rispetto al tempo tra due urti successivi e le forze intermolecolari sono trascurabili. Le ipotesi a monte implicite a queste assunzioni sono che: esiste un gran numero N di particelle in moto casuale che obbediscono alle leggi di Newton. Sulla base di queste ipotesi, senza avere in mente un modello molecolare particolare, Clausius costruisce la grandezza pressione (calcolo cinetico della pressione:
p =Nmv 2
3V) e ritrova l’equazione di stato dei gas. Calcola una velocità media per
alcuni gas che trova essere dell’ordine dei 100 m/s (qui bisognerebbe parlare di
velocità quadratiche medie, vqm = v 2 , anche se Clausius in realtà considera i
quadrati della media delle velocità). Sperimentalmente si trova che le velocità quadratiche medie sono dello stesso ordine di grandezza della velocità del suono in quel gas; per l’aria a 0°C e una atmosfera, v 330 m/s. Critica di Buys-Ballot /p. 178 Bellone/: se le particelle sono velocissime come ha sostenuto Clausius perché la diffusione di due gas non è un processo istantaneo? Perché il fumo di sigaretta o i

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
3
vapori di ammoniaca in una stanza sono così lenti a propagarsi in aria? Clausius replica: la velocità di diffusione è rallentata dal gran numero di urti tra le molecole; anche se sono velocissime le molecole possono percorrere spazi piccolissimi in linea retta; è molto improbabile che una molecola si possa muovere di moto rettilineo uniforme per tratti maggiori di quelli definiti dal cammino libero medio). Il concetto di cammino libero medio (1858) Clausius introduce il concetto di clm (distanza media tra due urti successivi) per particelle distribuite a caso. Le molecole sono considerate sfere rigide di diametro d e sezione d’urto d
2. Se due molecole si avvicinano entro d si urtano. Se si considera una molecola di diametro equivalente 2d che urta con nv molecole fisse puntiformi, in t la molecola spazza un cilindro di sezione d
2 e altezza vt contenente nv molecole. Nel tempo t si ha un numero di molecole d
2vt nv che rappresenta anche il numero di
urti subiti dalla molecola in t, da cui l =vt
d2vtnv
=1
d2nv
.
Se le molecole bersaglio si muovono con una velocità relativa rispetto alla molecola
equivalente l =1
2nvd2
(questa formula viene scritta da differenti autori, tra i quali
Maxwell, con lievi modifiche a seconda delle ipotesi sulla cinematica degli urti). Clausius impiega valori medi molecolari; trova anche che per un gas monoatomico,
l’energia cinetica media traslazionale EK trasl =1
2mv 2
=3
2RT , relazione che gli
consente di dare una interpretazione cinetica della temperatura T. Maxwell e la legge di distribuzione delle velocità molecolari (1860) Prima memoria di Maxwell di teoria cinetica; per un gas di sferette elastiche all’equilibrio (cioè a una certa temperatura T) enuncia la legge di distribuzione delle velocità sotto l’ipotesi che le componenti delle velocità di una molecola, vx, vy e vz in direzioni diverse si devono considerare variabili casuali indipendenti che si distribuiscono come gli errori di misura (prima di Maxwell si pensava che le velocità molecolari a una data T fossero tutte uguali). Maxwell introduce elementi probabilistici in teoria cinetica: a livello micro a causa degli urti molecolari un sistema evolve verso uno stato di equilibrio in modo irreversibile nel tempo.
Legge (scritta in forma moderna): N(v) = 4 Nm
2 kT
3
2v 2e
mv 2
2kT con
N = N(v)dv = n°totale di molecole0
; la curva è associata a una data T, per T
crescente le curve si spostano (si allargano e si abbassano) in modo tale da avere area sottesa uguale; la velocità più bassa è 0, per v si ha un asintoto (non esiste un limite superiore per le velocità delle molecole); la curva è asimmetrica rispetto alla velocità più probabile.
N per unità diintervallo di v
v (m/
v v1 2
v più probabile

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
4
Maxwell e il principio di equipartizione dell’energia Nello stato finale di equilibrio (cioè dopo molti urti) l’energia cinetica media traslazionale lungo gli assi coordinati è la stessa per tutti i tipi di particelle; l’energia disponibile, cioè, si distribuisce in parti uguali tra ciascuno dei moti indipendenti in cui le molecole possono assorbire energia. Per un puro moto traslazionale 1
2mvx
2=
1
2mvy
2=
1
2mvz
2 ; se c’è anche una energia cinetica rotazionale (per es. in un
gas biatomico), ciascun contributo è uguale all’ EK rotaz intorno a ciascun asse principale delle particelle. Dal teorema di equipartizione si calcolano i calori specifici (per un gas monoatomico
U =3
2nRT, CV =
dU
ndT=
3
2R, Cp CV = R, Cp =
5
2R ). Ma i valori attesi sono in
disaccordo con i valori sperimentali e inoltre si trova che C=C(T) (Boltzmann risolverà il problema per i gas monoatomici e biatomici, intuendo che nelle molecole ci sono anche moti interni; per una soluzione completa bisogna aspettare la meccanica quantistica, 1929). Il teorema di equipartizione è valido solo in meccanica statistica classica (meccanica newtoniana) e prevede calori specifici indipendenti da T. Maxwell e la viscosità di un gas Esaminiamo la viscosità di un fluido (v. fig.; l’argomento si inquadra nel tema più generale dei fenomeni di trasporto per viscosità, conduzione termica, diffusione le cui leggi generali devono essere ‘ricostruite’ sulla base della teoria cinetica): tavoletta tirata con velocità u; si ha un moto di trascinamento con ‘attrito’ tra i vari strati di fluido; si generano cioè degli sforzi di taglio la cui intensità per unità di superficie è
proporzionale al gradiente di velocità lungo z: =u
z, dove è detto
“coefficiente di viscosità”. z
x
u
Maxwell studia la viscosità di un fluido come trasferimento di quantità di moto da parte di molecole che si urtano tra stati contigui. Prevede che sia indipendente dalla densità del mezzo (e quindi dalla pressione; = nm, con n numero di molecole per unità di volume) contro il senso comune. In effetti con la teoria cinetica risulta
=1
3vl con l
1, =
mv
12 2 r2. La previsione di Maxwell viene confermata
sperimentalmente dallo stesso autore qualche anno dopo. Un punto a favore della teoria cinetica. Uno scoglio durissimo per la teoria cinetica: il secondo principio della termodinamica Ricordare: Antefatti: alle radici della TC o “Teoria meccanica del calore”; la formulazione dei principi della termodinamica fenomenologica (o macroscopica o “generale”). S. Carnot e il II principio (1824), § 4.4.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
5
J. P. Joule e il principio di equivalenza. Verso la formulazione del I principio della termodinamica (Helmholtz, 1847). Q=L+ U (§4.5) Inconciliabilità tra assioma di Carnot (il calorico si conserva) e principio di equivalenza di Joule (il calore si trasforma in lavoro): W. Thomson e R. Clausius (1849-50). (§4.6) Kelvin sulla grandezza calore. Enunciato del II principio e dottrina della dissipazione dell’energia. Enunciato del II principio da parte di Clausius; la grandezza entropia e riformulazione del II principio (1854-1865). S 0 Kelvin riconosce che Q non è del tutto equivalente a L, Q è una forma di energia degradata non integralmente utilizzabile per l’uomo dottrina della dissipazione dell’energia (Kelvin, On a universal tendency in nature to the dissipation of energy, Proc. R. S. Edimburgh, 1852). Gli fa eco Clausius con la wärme Tod: nell’universo c’è una tendenza ad andare verso uno stato di massima entropia S Smax a partire dal quale non c’è più evoluzione (1865, Bellone, p. 165/ pp. 244-45). I guai emergono quando con Maxwell e poi con Boltzmann si tenta di tradurre il II principio della termodinamica nel solo linguaggio della meccanica. Per il I principio (principio di uguaglianza) tutto bene: Q L (espresso nell’ambito della meccanica dei sistemi conservativi, sono validi i criteri di determinismo e di reversibilità di Laplace). Ma in natura i processi L Q sono irreversibili mentre i processi Q L sono irrealizzabili; il II principio stabilisce in natura una tendenza a un cambiamento unidirezionale. Se si vuole tradurre il secondo principio con le leggi della meccanica si va incontro a gravi inconsistenze logiche (crisi profonda del meccanicismo). Maxwell è tra i primi ad accorgersi delle inconsistenze logiche tra reversibilità meccanica e irreversibilità termodinamica (1867, lettera a Tait): il II principio è stato definito su basi macroscopiche con grandezze che definiscono uno stato medio delle molecole ma su scala molecolare chi ne garantisce la validità? Per una singola collisione binaria, scambiando il tempo t con –t l’urto è reversibile ma con molte particelle non posso che avere processi irreversibili. Demone di Maxwell: in grado di conoscere traiettorie e velocità di tutte le molecole, di separare le molecole, creare gradienti di temperature senza lavoro esterno, ecc. Il senso del demone è quello di immaginare un ente in grado di operare a livello microscopico in modo da violare in linea di principio il II principio il quale non può che avere significato statistico. Maxwell afferma così, qualitativamente, che il II principio ha natura probabilistica, ha solo certezza statistica, “ha lo stesso grado di verità contenuto nell’affermazione che se gettate un bicchiere d’acqua in mare, non potete estrarre di nuovo lo stesso bicchiere d’acqua” (lettera a Rayleigh, 1870). Un primo bilancio della teoria cinetica Punti a sfavore: - Se le molecole non sono osservabili come si fa a dar credito alla teoria cinetica? Esistono realmente le molecole? Quanto sono grandi? Quante ce ne sono per unità di volume? - Non spiega la discordanza tra valori sperimentali e attesi per i calori specifici (in particolare, per i gas biatomici Cp/CV =4/3 vs valore sperimentale 1,4). - Paradossi di senso comune: per es. obiezione di Buys-Ballot. - Impossibilità di tradurre il secondo principio della termodinamica con il solo linguaggio della meccanica (§8.4). Punti a favore: - calcolo cinetico della pressione; equazione di stato dei gas; - interpretazione corretta della viscosità; - la teoria cinetica dà supporto all’ipotesi di Avogadro.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
6
Negli ultimi decenni dell’Ottocento i punti a sfavore sembrano prevalere; lo stesso Maxwell si scoraggia e dubita anche della reale esistenza degli atomi. Kelvin nel 1900 parla di nubi che incombono sulla fisica riferendosi proprio alla teoria dinamica del calore. Iniziamo ad analizzare come si dipana la matassa a partire da un filone d’indagine sperimentale che riguarda proprio la determinazione delle dimensioni delle molecole. §8.2. La stima delle dimensioni delle molecole Il primo tentativo di stimare le dimensioni molecolari risale a T. Young1 e si basa sulla teoria dell’attrazione capillare (formulata da Laplace e dallo stesso Young). I fenomeni di capillarità vengono studiati per tutto il Settecento. Come si sa, un liquido ideale ha la proprietà di non avere forma propria e un liquido reale non sempre rispetta questa proprietà: v. gocce di mercurio, di rugiada, menischi, ecc. Il liquido dovrebbe appiattirsi con il baricentro più basso possibile nella configurazione d’equilibrio e invece assume forma sferica (a causa della “tensione superficiale”, concetto introdotto nel 1751 da J. A. Segner).
La cosa si spiega facilmente a livello microscopico: ogni molecola di liquido si trova rispetto alle molecole vicine a una distanza r leggermente superiore alla distanza di equilibrio r0 (v. andamento di U in funzione di r) e quindi è attratta dalle molecole vicine. Così, una molecola tutta interna al liquido, essendo completamente circondata dalle molecole vicine, è soggetta a una forza d’attrazione a risultante nulla. Una molecola sulla superficie esterna del liquido invece è soggetta a una forza risultante diretta verso l’interno del liquido, non controbilanciata, così che la superficie del liquido subisce una forza di compressione. Le leggi della capillarità sono state dedotte su base macroscopica da Laplace e da Young (primi anni dell’Ottocento; equazione di Laplace per la differenza di pressione
interna ed esterna a una goccia sferica di raggio R di liquido: pl pg =lg
R, dove lg
indica la tensione superficiale liquido-gas; equazione di Young: sg = sl + lg cos ,
con angolo di contatto). Il metodo di Young viene riscoperto da Rayleigh nel 18902 e viene ampiamente citato da Kelvin nel 19023 nel suo articolo “On the weights of atoms”. Si tratta di una teoria puramente dinamica (in termini di forze; Young non pensa affatto a una teoria cinetica della materia), rimasta latente per anni a causa del fatto che Young esprime verbalmente e non mediante formule il suo metodo che risulta alla lettura assai oscuro. Tra la tensione T di Young alla superficie libera di un liquido e il termine di Laplace K (dimensionalmente una forza per unità di superficie; K si riferisce alla forza che si esercita tra strati di molecole di liquido) sussiste la relazione a= 3T/K dove a è il range della forza attrattiva di coesione (si ricorda che la tensione superficiale ha le dimensioni di una forza per unità di lunghezza, N/m, o di una energia per unità di superficie). Per valori opportuni di K e di T, Young trova
1T. Young, On the cohesion of fluids, Phil. Trans. 1805, Collected Works, vol. I, p. 461. 2J. Rayleigh, On the theory of surface forces, Phil. Mag., 30 (1890) 478. 3Kelvin, On the weights of atoms, Phil. Mag., 4 (1902) 177.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
7
che la forza di coesione deve avere un range che si estende fino a circa 250 10-6 di inch [10-8 cm]. Young osserva in proposito: “Entro questi limiti di incertezza possiamo ottenere una stima ipotetica della distanza mutua delle particelle di vapore e anche della grandezza reale degli atomi elementari dei liquidi, supponendo che siano approssimativamente in contatto tra di loro; perché se la distanza a cui la forza di coesione inizia è costante alla stessa temperatura e se le particelle di vapore sono condensate quando si avvicinano a
questa distanza, segue che a 60°F la distanza delle particelle di vapor acqueo puro è di circa 250 10-6 inch; e poiché la densità di questo vapore è circa 1/60.000 di quella dell’acqua, la distanza delle particelle deve essere circa 40 volte più grande; per conseguenza, la distanza mutua delle particelle
d’acqua deve essere di circa [0, 025 10-8 cm]. E’ vero che il risultato di questo calcolo differirà considerevolmente in funzione della temperatura delle sostanze confrontate [...]. Questa discordanza tuttavia non invalida del tutto il senso generale della conclusione [...] e sembra abbastanza certo concludere che per quanti errori possano aver influenzato la nostra determinazione, il diametro o
distanza delle particelle d’acqua è tra [0,125 10-8 e 0,025 10-8 cm].” La prima determinazione delle dimensioni delle molecole di J. Loschmidt, 1865 “Zur Grösse der Luftmolecule” (La grandezza delle molecole d’aria), Wien. Ber., 52 (1865) 395-413. “Fino ad ora si era d’accordo sul fatto che nei gas le molecole sono separate l’una dall’altra da distanze così grandi da poter ritenere che i loro diametri siano nella maggior parte dei casi trascurabilmente piccoli. Le molecole stesse si concepivano in moto continuo, e la velocità di questo moto influenzata dalla temperatura. Sul tipo di moto di recente si sono avanzate due concezioni nettamente diverse. L’una, più antica, ritiene che le molecole di gas siano mantenute in posizioni equidistanti tramite forze mutue attrattive e repulsive in una sorta di posizione di equilibrio stabile attorno a cui esse oscillano. Queste forze attrattive e repulsive le si immagina o come originarie e inerenti alla sostanza delle molecole, oppure, e in realtà questa è l’opinione prevalente, condizionate da strati di etere o anche da una sostanza calorica particolare. Questa concezione è riuscita a spiegare la maggior parte dei fenomeni e però alla fine si è constatato che da essa si può dedurre solo ciò che fin dall’inizio si è assunto nelle premesse. E non c’è modo di uscirne. Molto meglio ci riesce la seconda concezione che si rifa a Herapath e Krönig, e che è stata fondata sui lavori di Clausius, Maxwell, Rankine, ecc.. e ha acquistato rapidamente un peso decisivo. Questo secondo punto di vista [teoria cinetica] continua a considerare grandi le distanze tra le molecole di gas, però tralascia il legame che una molecola ha con le sue vicine e la fissa in una posizione, attribuendole però un moto continuo. In virtù di ciò, la molecola si muove in direzione rettilinea senza sapere nulla di un’altra fino a che, esprimendoci in modo approssimativo, non la urta [ipotesi del caos molecolare]. In questo caso segue un urto che avviene completamente secondo le leggi degli urti perfettamente elastici. Rinunciando alle forze mediatrici si era messa da parte una pesante zavorra, si era trovato un calcolo appropriato per ottenere i valori medi nel caos delle molecole che si muovono senza regola e si era pervenuti a certi risultati mediante i quali si poteva sperare, nonostante certe ipotesi ausiliarie come quella sulla forma sferica delle molecole e simili, di comprovare dai fatti stessi una conferma o una smentita convincente. Fino ad ora il decorso della nuova teoria è stato senza dubbio più favorevole. I lavori dei ricercatori sopra citati hanno fornito, da un lato, spiegazioni molto precise e illuminanti degli eventi più importanti nei gas- come per esempio per la pressione [macroscopica], per la conduzione del calore, per la propagazione del suono- mentre dall’altro, con una concezione più approfondita delle relazioni, hanno consentito la determinazione numerica di costanti importanti. Menzioniamo in particolare: la determinazione della velocità media delle molecole, per gas diversi a diverse temperature, quella del rapporto tra la forza viva totale di un gas e la forza viva che corrisponde a quella velocità, entrambe date da Clausius, e inoltre quella del cammino libero medio delle molecole d’aria data da Maxwell e O. E. Meyer. Obiettivo di questo lavoro è quello di ottenere, sulla base di questa teoria, una stima provvisoria per un’altra costante relativa alla grandezza del diametro delle molecole d’aria. A questa grandezza viene in realtà attribuita una piccolezza straordinaria ma il problema se esse

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
8
siano di un milionesimo o di un miliardesimo di mm, o ancora meno, è rimasto finora irrisolto. La nuova teoria è ora in grado di darci informazioni sull’argomento.” [...] [La struttura del ragionamento di Loschmidt: Modello teorico di riferimento: la teoria cinetica di Krönig, Herapath, Clausius, Maxwell (molecole di diametro « cammino libero medio, forze intermolecolari trascurabili, urti elastici) e legge di Avogadro. Obiettivo: determinazione del diametro s delle molecole d’aria. Punto di partenza: concetto di cammino libero medio l stabilito da Clausius (1858) e modificato da Maxwell, dato dalla formula:
l =3
4 N s2 1[ ]
con N numero di molecole per unità di volume (o “densità molecolare”). La [1] viene così modificata:
1 =43
N ls2
1N
=43
44
ls2 1'[ ]
Poiché si sa che nell’ unità di volume (1 cm3) in condizioni normali (legge di Avogadro: volumi uguali di gas diversi a stessa p e T contengono lo stesso numero di molecole) è contenuto lo stesso numero di molecole N, 1/N (che viene definito da Loschmidt “volume di una molecola del gas”) rappresenta la frazione di volume che compete a una molecola; ls2/4 è il “volume dello spostamento molecolare” (pari al volume del cilindro di
diametro di base s e altezza l, spazzato da una molecola tra due urti). La [1’] viene perciò interpretata come volume di una molecola del gas = (16/3) volume dello spostamento di una molecola. Se si suppone che la molecola abbia forma sferica, il volume proprio di una molecola è (4/3) (s/2)3= s3/6; N s3/6 è allora quella frazione del volume unitario occupato dalle N molecole se le si pensasse in quiete e ‘impacchettate’ spazialmente. Dal punto di vista fisico cioè si immagina di comprimere il gas fino allo stato liquido, portando le N molecole a occupare un volume ‘condensato’ di N s3/6. Si definisce allora “coefficiente di condensazione del gas” il rapporto tra la frazione di volume unitario delle molecole del gas condensato e il volume che esse occuperebbero nella fase gassosa: N s3/6: unità di volume = [2]. Da [1] e [2] segue:
s=8 l [3]. Il coefficiente di condensazione del gas si può stimare dividendo il volume di una sostanza allo stato liquido per il suo volume allo stato gassoso in condizioni normali. Naturalmente bisogna considerare il fatto che le molecole, se sono di forma sferica, una volta ‘impacchettate’ nella fase liquida occupano un volume più grande di quello indicato da a causa degli spazi vuoti tra una molecola e l’altra; ma per valutare ordini di grandezza possiamo accontentarci anche di un valore approssimato per , salvo poi apportare correzioni al momento opportuno. Sono stati valutati sperimentalmente valori di per diverse sostanze (per l’acqua 0,00081, per il protossido d’azoto 0,00154, per l’ammoniaca 0,00102, per l’anidride carbonica 0,00204, ecc.) ma sfortunatamente proprio per l’aria, che è la sostanza per la quale al contrario è noto il valore del cammino libero medio l, il dato è assente [al tempo di

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
9
Loschmidt non si sa ancora liquefare l’aria]. Ma il chimico H. Kopp (Ann. d. Chemie u. Pharm., 92, 1841, p. 1) è riuscito dalla formula chimica di una sostanza a risalire alla sua densità (o al suo volume specifico) allo stato liquido. Se si considera l’aria una miscela di 4 parti d’azoto e 1 di ossigeno, dai valori delle densità dei due componenti allo stato liquido (1,16 per l’azoto e 1,4545 per l’ossigeno) e al punto di ebollizione Loschmidt risale a un valore della densità dell’aria liquida di 1,224. Questo valore va corretto per tenere conto degli spazi vuoti tra una molecola e l’altra e per compensare gli effetti di temperatura sulla densità, quando si passa dal punto di ebollizione alla temperatura normale. Loschmidt ottiene così per la densità dell’aria liquida L = 1,5. Poiché l’aria è allo stato gassoso 770 volte più leggera dell’acqua, a parità di massa dell’aria allo stato liquido L e gassoso G =VL/VG= G/ L,il valore del coefficiente di condensazione è = 1/(770·1,5) =0,000866. Per quanto riguarda il cammino libero medio per l’aria, esistono diversi valori stimati da Maxwell e da altri ricercatori, che mediati danno l= 0,000140 mm. Dalla [3] s=8 x 0,000866 x 0,000140= 0,000000969 10-6mm = 10 Å. [I valori attuali per il diametro delle molecole di ossigeno e di azoto sono di circa 3Å]. “Questo valore è naturalmente solo una approssimazione ma non è certamente né dieci volte più grande né più piccolo. Se nelle premesse non ci sono errori fondamentali, considerate tutte le correzioni che si possono apportare al cammino libero medio e al coefficiente di condensazione, sussiste la regola che nel campo degli atomi e delle molecole, l’unità di misura appropriata è il milionesimo di mm. Tale lunghezza è circa la settecentesima parte della lunghezza d’onda della luce rossa e come il km è adatto come unità di miglia per le distanze terrestri più grandi, così il milionesimo di mm vale per quelle più piccole. Il
contenuto di 1 mm3 di tali molecole basterebbe a ricoprire 1 m2 di uno strato di materia distribuito con continuità”. [In questa memoria Loschmidt non calcola il “numero di Loschmidt”, cosa che farà in un estratto del suo articolo pubblicato subito dopo (J. Loschmidt, Zeitschr. Math.
u. Physik, 10 (1865)511-512). Nell’estratto l’autore scriverà che in 1 mm3 d’aria sono contenuti 866 bilioni di molecole che salirebbero a un trilione se l’aria fosse liquida. L’estratto è stato scritto probabilmente in gran fretta. Il dato NL=866 1012 molecole/mm3=8,66 1017molecole/cm3 (che corrisponde a un numero di Avogadro NA=4,06 1022 per mole) non è infatti compatibile con i dati riportati nell’articolo completo (8,66 corrisponde se mai a 104). Utilizzando i dati dell’articolo e la [1’] o la [2] si sarebbe ottenuto NL = 6 / s
3= 0,16·1019 molecole/cm3 con NA=3,6 1022 molecole per mole.] Nota: una mole di gas perfetto in condizioni normali occupa sperimentalmente 22,415 l= 22,415 103 cm3; NA= 6 1023 particelle/ (22,415 103 cm3)= 2,7 10 19 particelle/ cm3 . §8.3. Altre strade per la stima delle dimensioni molecolari. L’esperimento di Rayleigh-Röntgen Dopo la stima4 data da Loschmidt nell’ambito della teoria cinetica nel 1865 (d 10-7 cm) altri indizi sperimentali mostravano l’esistenza di un limite di divisibilità della materia; tra questi, la possibilità di ridurre lamine d’oro a uno spessore dell’ordine di 10-5 cm, il comportamento ondulatorio della luce attraverso mezzi trasparenti, il
4Un altro tentativo simile a quello di Loschmidt viene fatto da G. J. Stoney, The internal motions of gases, Phil. Mag., 36 (1868) 132-141.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
10
comportamento di lamine sottili, sia dal punto di vista ottico che rispetto ai fenomeni di tensione superficiale. Il riferimento alla teoria ondulatoria della luce per poter stimare le dimensioni delle molecole viene fatto da A. Cauchy5 intorno al 1835. Su suggerimento di Fresnel, Cauchy osserva che nella dispersione della luce nei mezzi trasparenti ( luce visibile 3,9-7,5 10-5 cm; le della luce si sanno misurare già dai primi decenni dell’Ottocento sfruttando i fenomeni di interferenza), la velocità di propagazione è funzione delle diverse lunghezze d’onda; la luce, cioè, ‘sente’ la struttura discontinua della materia che deve essere molto inferiore alle medie. Kelvin6 affermerà nel 1870 che la scala delle discontinuità non può essere inferiore a 10-4 della lunghezza d’onda della luce (0,5 10-8 cm); e che in base alla teoria cinetica la sfera d’azione molecolare nei liquidi e nei solidi è confrontabile con la della luce. Per quanto riguarda le lamine sottili, un metodo per stimare le dimensioni delle molecole si basa sulle bolle di sapone. Kelvin, in un articolo Sulla dimensione degli atomi del 18837, segue questo ragionamento: l’osservazione qualitativa mostra che la bolla di sapone, man mano che viene gonfiata (cioè al diminuire del suo spessore), assume varie colorazioni da verde a rosso cupo a bianco a nero. In corrispondenza del nero la bolla si rompe e ciò avviene, osserva Kelvin, quando il suo spessore comincia a diventare inferiore a (1/4) giallo bianco = 1/60.000 cm. Per stimare quantitativamente lo spessore della pellicola Kelvin ricorre a considerazioni termodinamiche e a un ragionamento limite. L’assunto da cui parte è che lo spessore della bolla deve avere un limite inferiore di 10-9 cm. In corrispondenza di questo valore, se si tentasse di gonfiare ancora la bolla, il lavoro speso contro la forza di tensione sarebbe infatti sufficiente a trasformare la bolla in vapore. Quindi lo spessore della pellicola deve essere maggiore di tale valore. Kelvin calcola il lavoro L necessario per far aumentare di 10.000 volte la superficie di una pellicola
d’acqua di spessore, poniamo, di d = 1 mm, riducendo così d a 10-4 mm = 10-5 cm. Successivamente calcola che il calore equivalente a questo lavoro farebbe aumentare la temperatura della pellicola di
0,57°C. Volendo ridurre ancora lo spessore a 10-8 cm, il lavoro L’ speso sarebbe 103 L e per conseguenza Q’ diverrebbe 570 volte la quantità di calore che sarebbe necessaria per far crescere di 1°C la temperatura del liquido. La lamina passerebbe così allo stato di vapore. Lo spessore delle macchie nere delle bolle di sapone misurato con metodi diversi (da misure della resistenza elettrica o con metodi ottici) coincide con un valore8 di circa 6 10-7 cm. Approfondimento: lamine e bolle di sapone
Per fare l’acqua saponata ottimale per le bolle di sapone esistono svariate ricette; per es. 160 cm3 di detersivo (Nelsen piatti è particolarmente buono), 35 ml di glicerina; 4 l d’acqua. Oppure: 1 parte di detersivo, 2 parti di glicerina, 3 parti d’acqua. Su questo
5A. Cauchy, Mémoire sur la dispersion, Nouveaux excercises de mathématiques, Praga 1835. 6Kelvin, Nature, 1 (1870) 551. In questo articolo Kelvin propone quattro metodi per risalire alle dimensioni delle molecole: 1) il metodo ottico alla Cauchy basato sulla teoria ondulatoria della luce; 2) un metodo legato a considerazioni sulla “energia di attrazione” tra lastre metalliche a contatto cariche, basato sui fenomeni di elettricità per contatto; 3) un metodo basato sulle forze di tensione superficiale e sull’attrazione capillare (bolle di sapone); 4) un metodo basato sulla teoria cinetica, simile a quello di Loschmidt (di cui all’epoca non conosce i lavori). 7Kelvin, Proc. Roy. Inst., X (1883) 185 in Opere di Kelvin, Utet, p. 680. 8Cfr. J. Perrin, Gli atomi, p. 84.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
11
e i seguenti argomenti, si veda in rete: www.FUNSCI.com (in italiano, sito ottimo); www.exploratorium.edu/ronh/bubbles/bubbles.html. Un testo classico: C. Boys, Soap bubbles and the forces that mould them, 1890. J. Oprea, The mathematics of soap films, Amer. Math. Soc., 2000. Perché in acqua pura non si formano bolle di sapone? L’acqua pura ha una tensione superficiale troppo alta (non forma bolle, si ‘rompe’ a ogni tentativo di formare pellicole; 70 dyn/cm o 7 g/m a 20 °C. diminuisce inoltre con la temperatura). Come si formano allora le bolle o le lamine di sapone? Occorre aggiungere sapone o un altro detensivo (o tensioattivo o surfattante = surface active agents) che abbassi la tensione superficiale del liquido. Il sapone riduce anche l’evaporazione dell’acqua e stabilizza le bolle (effetto Marangoni). La canfora è un esempio di un altro detensivo. Come sono fatte le molecole di sapone? Sono lunghe catene di C e H con una testa idrofila e una coda idrofoba (che ama il grasso e odia l’acqua9). Sulla superficie dell’acqua si forma uno strato monomolecolare.
Nel caso di una pellicola di acqua e sapone, in corrispondenza delle due superfici si formano due strati monomolecolari; nel mezzo le molecole di sapone tendono a separare le molecole d’acqua e abbassano la tensione superficiale dell’acqua.
Perché all’acqua saponata si aggiunge glicerina? La glicerina si attacca alle code delle molecole di sapone, riduce l’evaporazione dell’acqua e quindi stabilizza le bolle o le pellicole. Perché le bolle sono sferiche? A causa della tensione superficiale che opera sempre in modo da rendere minima la superficie della bolla. La forma sferica, come è noto, tra le figure di dato volume (per es. 1 cm3) è quella che ha area minima (tetraedro, 4 facce; cubo, 6 facce; ottaedro, 8 facce, ecc.... sfera, ‘infinite facce’). Che succede quando due o più bolle si incontrano e si fondono (merging)? Quando due bolle uguali si incontrano si elimina parte della superficie esterna e si formano lamine nei punti di contatto (il sistema passa a uno stato di energia minima)
9 Questo spiega per esempio perché il sapone serve a sgrassare i tessuti: all’interno dell’acqua si formano aggregati di molecole di sapone, micelle e membrane; quando tali aggregati incontrano il grasso, vi inseriscono le code mentre le teste attirnao lo sporco verso l’esterno.
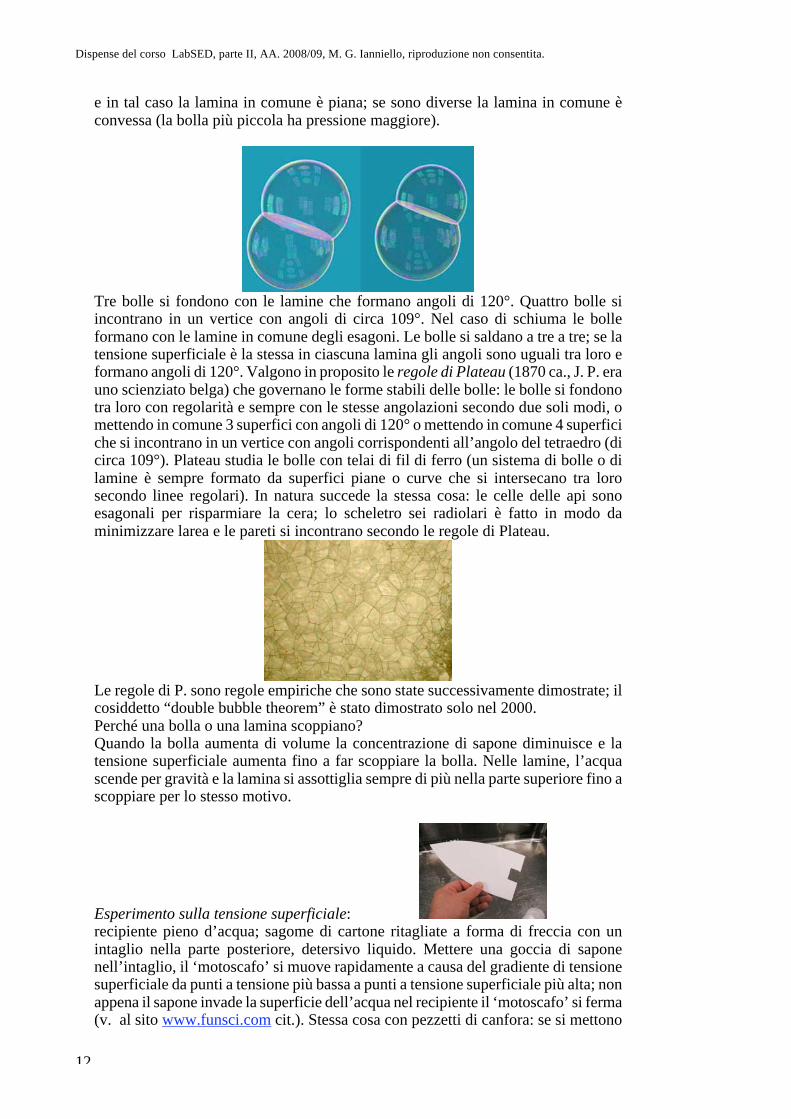
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
12
e in tal caso la lamina in comune è piana; se sono diverse la lamina in comune è convessa (la bolla più piccola ha pressione maggiore).
Tre bolle si fondono con le lamine che formano angoli di 120°. Quattro bolle si incontrano in un vertice con angoli di circa 109°. Nel caso di schiuma le bolle formano con le lamine in comune degli esagoni. Le bolle si saldano a tre a tre; se la tensione superficiale è la stessa in ciascuna lamina gli angoli sono uguali tra loro e formano angoli di 120°. Valgono in proposito le regole di Plateau (1870 ca., J. P. era uno scienziato belga) che governano le forme stabili delle bolle: le bolle si fondono tra loro con regolarità e sempre con le stesse angolazioni secondo due soli modi, o mettendo in comune 3 superfici con angoli di 120° o mettendo in comune 4 superfici che si incontrano in un vertice con angoli corrispondenti all’angolo del tetraedro (di circa 109°). Plateau studia le bolle con telai di fil di ferro (un sistema di bolle o di lamine è sempre formato da superfici piane o curve che si intersecano tra loro secondo linee regolari). In natura succede la stessa cosa: le celle delle api sono esagonali per risparmiare la cera; lo scheletro sei radiolari è fatto in modo da minimizzare larea e le pareti si incontrano secondo le regole di Plateau.
Le regole di P. sono regole empiriche che sono state successivamente dimostrate; il cosiddetto “double bubble theorem” è stato dimostrato solo nel 2000. Perché una bolla o una lamina scoppiano? Quando la bolla aumenta di volume la concentrazione di sapone diminuisce e la tensione superficiale aumenta fino a far scoppiare la bolla. Nelle lamine, l’acqua scende per gravità e la lamina si assottiglia sempre di più nella parte superiore fino a scoppiare per lo stesso motivo.
Esperimento sulla tensione superficiale: recipiente pieno d’acqua; sagome di cartone ritagliate a forma di freccia con un intaglio nella parte posteriore, detersivo liquido. Mettere una goccia di sapone nell’intaglio, il ‘motoscafo’ si muove rapidamente a causa del gradiente di tensione superficiale da punti a tensione più bassa a punti a tensione superficiale più alta; non appena il sapone invade la superficie dell’acqua nel recipiente il ‘motoscafo’ si ferma (v. al sito www.funsci.com cit.). Stessa cosa con pezzetti di canfora: se si mettono

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
13
sulla superficie di acqua ben pulita la canfora si muove da tutte le parti perché sciogliendosi abbassa la tensione superficiale del liquido ed è tirata nei punti dove la tensione superficiale è più alta. Se la superficie dell’acqua è grassa la canfora rallenta e si ferma quando non esistono più differenze di tensione. Forze di coesione e forze di adesione Se si trapassa una bolla o una lamina di sapone con un dito (o un altro oggetto appuntito) ovviamente la bolla o la lamina scoppiano. Non così se si bagna il dito con acqua saponata; nel primo caso entrano in gioco le forze di adesione (agenti tra molecole di tipo diverso), che sono in genere maggiori della forza di coesione (tra molecole di stesso tipo); nel secondo caso invece prevale la forza di coesione. Esperimenti con i telai (strutture 3D)
Reticolo cubico, tetraedrico, conico, ecc. Recipiente con acqua saponata. Immergere i telai nell’acqua saponata e osservare le lamine che si formano. Le forme (triangoli, trapezi, ecc.) si realizzano sempre in modo che le aree delle pellicole siano minori delle aree delle superfici dei telai, con strutture nello stato di minima energia. Le pellicole seguono la legge di “area minima”, in accordo con le regole di Plateau; per uno stesso telaio esistono più modi per minimizzare le aree. E’ possibile fare una bolla cubica? Inserire aria, con una cannuccia preventivamente bagnata in aqua saponata, al centro del telaio cubico (o tetraedrico) estratto dall’acqua saponata.
Con il telaio circolare, fissare un filo ‘lento’ tra un punto e l’altro del telaio; far prevedere come viene teso il filo quando si immerge il telaio nell’acqua saponata e, una volta estratto, si rompe la lamina da un lato del filo. Se si buca per es. la lamina di destra, la pellicola di sinistra viene tirata in modo da avere area minima (a destra, arco di cerchio: massima superficie possibile). Ripetere con un filo chiuso a cappio.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
14
I colori delle bolle o delle lamine di sapone Per un dato spessore l’interferenza può essere costruttiva per certe lunghezze d’onda e distruttiva per altre; i parametri importanti sono: lo spessore d della pellicola, la della luce incidente, eventuali perdite di /2 a seconda di come avviene la riflessione, l’angolo di incidenza della luce. Le condizioni per l’interferenza sono ovviamente le solite.
Perché i colori che si osservano non sono quelli dell’arcobaleno? Dipende dal colore delle onde che interferiscono e da come l’occhio percepisce i colori; se per es. si ha interferenza distruttiva per uno dei colori primari l’occhio percepisce la sintesi dei rimanenti; se si cancella il blu B restanto rosso R e verde V e vedo giallo.
Perché al nero la bolla scoppia? Le più lunghe hanno bisogno di uno spessore d più grande delle più corte (violetto). Man mano che lo spessore si riduce viene cancella to il rosso (vedo B+V), poi il giallo (vedo B), poi il verde (vedo Mag), quindi il B (vedo G); per d molto sottile (d<< visibili, intorno a 25 nm) avviene la cancellazione di tutti i colori per tutte le . Come stimare lo spessore d di una bolla: S= 4 r
2, V=Sd, = m/V 1g/cm3 d=m/S . Per determinare m occorre pesare la bolla (sacchetto di plastica sospeso a dinamometro di opportuna sensibilità; si pesa vuoto e si ripete la pesata dopo aver fatto scoppiare una bolla all’interno del sacchetto; meglio se si sostituisce il sacchetto con una scatola di plastica a pareti trasparenti e rigide per evitare la spinta di Archimede entro cui soffiare la bolla. Un metodo nuovo per la stima delle dimensioni molecolari viene introdotto indipendemente e contemporaneamente da Rayleigh10 e da Röntgen 11 nel 1890 e si basa su misure dello spessore di un film d’olio sull’acqua. Perché l’olio si spande sull’acqua fino a formare uno strato sottilissimo?
10Rayleigh, Proc. Roy. Soc., 47 (1890) 364; Scientific Papers, III, p. 349; vol. IV, p. 530; vol. V, p. 538; vol. VI, p. 534. 11Röntgen, Wied. Annal.,41 (1890) 321.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
15
1, olio
3, aria
2, acqua
Tra la goccia d’olio e l’acqua si formano 3 tensioni superficiali ai limiti dei 3 mezzi (olio 1, acqua 2, aria 3). La goccia starebbe ferma se = 0 mentre, fino a che 12 > 23 + 13 la goccia continua a espandersi fino a che si crea uno strato monomolecolare con molecole d’olio abbastanza distinte l’una dall’altra (in questo caso la della zona occupata dallo strato monomolecolare diventa uguale a quella dell’acqua).] Rayleigh si serve di pezzetti di canfora per indagare che succede quando si fa espandere una goccia di olio sulla superficie dell’acqua. Misura la massa m della minima quantità d’olio sufficiente a impedire i moti della canfora; trova che lo strato d’olio corrispondente a m, di cui misura la superficie, non può essere inferiore a 10-8 cm. Osserva inoltre che la canfora si muove fino a dove il film d’olio è spesso 10,6 10-8 cm mentre si ferma dove lo spessore è di 8,1 10-8 cm. Questo significa che si è raggiunto lo strato monomolecolare per l’olio. Röntgen invece di canfora usa etere e trova analoghe evidenze sperimentali per strati di 5,8 10-8 cm. Per una esecuzione in laboratorio di questa esperienza vedi PSSC. Può essere un buon esempio di ricostruzione di un esperimento storico se contestualizzata nel modo opportuno. § 8.4. Il contributo di Boltzmann (1844-1906) alla teoria cinetica Boltzmann è uno dei fondatori della meccanica statistica; figura tra le più influenti nella fisica degli ultimi tre decenni dell’Ottocento e non solo. L’uomo, il docente, lo scienziato. Programma di ricerca di Boltzmann: dare una interpretazione meccanica, analitica e generale, del secondo principio della termodinamica (nel programma di ricerca di B. si verifica negli anni uno shift da determinismo classico a probabilismo; il riduzionismo meccanicista passa dall’atomismo realista all’atomismo come descrizione mentale). Nasce a Vienna; frequenta l’università di Vienna (allievo di J. Stefan) e si diploma nel 1866; abilitazione nel 1867. Tra il 1869 e il 1876 prof di Fisica matematica a Graz, Wien, Graz. Dopo il suo articolo del 1872 diventa una celebrità. Nel 1875 a Graz sposa Henriette. 1866: “Sul significato meccanico del II principio della teoria del calore” 1872, “Ulteriori studi sull’equilibrio termico delle molecole”; equazione di Boltzmann o equazione di trasporto, teorema H. Per un gas monoatomico non soggetto a forze esterne, un sistema che parte da uno stato iniziale arbitrario non all’equilibrio, per effetto delle collisioni molecolari deve tendere nel tempo a uno stato di equilibrio, caratterizzato da una distribuzione maxwelliana delle velocità. Tale distribuzione è l’unica possibile in quanto rappresenta uno stato stabile per il gas, raggiunto il quale il gas vi permane anche se le molecole continuano a urtarsi. L’equazione comporta l’irreversibilità per l’evoluzione temporale dello stato macroscopico del gas; è la prima equazione che governa l’evoluzione temporale di una probabilità e implica una prova per

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
16
l’irreversibilità dei fenomeni macroscopici (shift di B. da meccanica deterministica a probabilistica) /p. 251 Bellone/
Caso uniforme: sistema isolato con una distribuzione iniziale f=f(v, t)
sistema all’equilibrio in uno stato stazionario caratterizzato da una distribuzione di Maxwell f0(v)
Nel “caso uniforme”, dove f non dipende dalla posizione r (f=f(v,t)) e sono assenti forze esterne (gas rarefatto infinitamente esteso, no pareti), l’equazione di trasporto si semplifica; per t + , allo stato di equilibrio f(v,t) f0(v) con f0 di Maxwell, indipendente dal tempo. Invece di dimostrare questa proprietà per f , B. introduce una funzione H tale che: H t( ) = f v,t( ) log f v, t( )dv e dimostra che H evolve nel tempo
in modo tale che dH/dt 0, con dH/dt=0 nello stato finale. Teorema H: per una distribuzione iniziale arbitraria, H può solo decrescere (a causa degli urti) fino a un valore minimo H0, per t + , H H0, raggiunto il quale il gas si pone in uno stato stazionario f0. B. osserva che -H S, con S entropia di Clausius, sicché il suo teorema H è un modo alternativo per enunciare il II principio della termodinamica. (Nel caso più generale di funzione di distribuzione f(r, v, t), e in presenza di forze esterne, l’equazione di B. descrive l’evoluzione temporale della funzione di distribuzione f come somma di un termine “di streaming” (funzione del numero di molecole in un dato volumetto, e delle eventuali accelerazioni delle molecole) e di un
termine “di collisioni”:f r,v,t( )
t=
f r,v, t( )t
s
+f r,v,t( )
t
c
. Nel caso non uniforme,
f(r,v,t), il minimo per H non corrisponde più a f0 indipendente dal tempo; l’evoluzione verso l’equilibrio avviene in due fasi con tempi di rilassamento diversi). Per ulteriori informazioni v. Baracca, cit. in bibliografia. 1875, Umkehreinwand o paradosso della reversibilità di (Kelvin)-Loschmidt: se il teorema H è basato sulle leggi della meccanica (temporeversibili), per qualunque moto meccanicamente possibile che fa diminuire H ne deve esistere un altro che si ottiene invertendo le velocità delle molecole, incompatibile con la II legge perché ora H aumenta. (Metafisica di fondo di Loschmidt: vuole dimostrare che la morte termica può essere evitata). L’argomentazione di L. era stata già sollevata da Kelvin nel 1874, The kinetic Theory of the dissipation of energy. /Bellone, p. 261/ 1877, replica di B. con un altro fondamentale articolo su “Osservazioni su alcuni problemi di teoria meccanica del calore”: introduce il calcolo delle probabilità (metodo combinatoriale), sottolinea come il II principio sia “intimamente connesso con la teoria delle probabilità mentre il I principio è indipendente da essa”. Questo articolo è un momento fondante della meccanica statistica: indipendentemente da

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
17
come avvenga un processo di trasferimento di energia (per urti, per radiazione, ecc.), un sistema deve tendere verso un macrostato a cui corrisponde probabilità W massima. Per un sistema macroscopico (gas) B. calcola il numero dei modi distinti (“complessioni” diverse dei parametri molecolari, che corrispondono ad altrettanti microstati) in cui un dato macrostato può essere realizzato; quanto più questo numero è elevato tanto più lo stato macro è probabile. La probabilità W è uguale al numero delle possibili realizzazioni di un macrostato attraverso diversi possibili microstati di uguale energia (W misura pertanto la probabilità dello stato); i microstati sono equivalenti (in una cella lo stato microscopico non cambia se si permutano tra loro le molecole in tutti i modi possibili) e hanno a priori la stessa probabilità. Analogia con urna contenente 20 palle nere N e 20 palle bianche B identiche (c’è un solo modo per avere, per es., 20 palle N mentre esistono moltissimi modi distinti per scegliere 20 palle diverse, 20!). Da palle a particelle: B. calcola il numero di modi distinti in cui n particelle possono essere ripartite in k celle in modo che nella i-ma cella ce ne siano ni. Calcola W, logW, manipola, ritrova la forma della funzione H e ‘scrive’ S= klnW+ cost. (v. per es. Baracca, cit.; S=klogW viene in realtà scritto in questa forma per la prima volta da Planck); un gas che parta da uno stato inziale arbitrario evolve verso lo stato che corrisponde al massimo numero di complessioni, cioè verso lo stato più probabile dove W tende a un massimo (S Smax). Il II principio viene reinterpretato come un principio di evoluzione (e quindi di irreversibilità) verso gli stati più probabili in cui S aumenta sempre (stato di massimo disordine; per un dato macrostato esistono molti stati microscopici disordinati equivalenti e la probabilità che uno di essi si verifichi è molto alta). Analogia meccanica: scatole con palline B e N (molecole di gas); agitiamo (collisioni), stato finale grigio. Posso separare B e N? Dal punto di vista meccanico non esiste impossibilità a priori ma la probabilità di avere una miscela grigia è molto maggiore della probabilità di far tornare le palline B e N ordinate. B.: lo stato finale è quello più probabile anche se ci possono essere fluttuazioni intorno a valori medi. B. sottolinea anche di avere fatto uso dell’ipotesi del caos molecolare nel calcolo del numero più probabile di collisioni e questa ipotesi non deriva dalla meccanica. 1884, legge di Stefan-Boltzmann (meccanica statistica all’equilibrio) e altri contributi fondamentali. B. è ormai un’autorità di fama mondiale ma inizia (1888) un periodo di profonda crisi. I prodromi si erano già manifestati quando era stato nominato rettore a Graz; i molti compiti istituzionali lo sfiancano, tumulti studenteschi ecc.; dopo la morte di Kirchhoff nel 1887 viene chiamato a Berlino: B. accetta senza chiedere il permesso alle autorità austriache,e viene pesantemente stigmatizzato per questo. Inizia a manifestarsi la sindrome maniacodepressiva. Nel 1889 muore il figlio Hugo. Vuole lasciare a tutti i costi Graz. Viene chiamato a Monaco nel 1890 su Fisica teorica, torna a Vienna nel 1893 a succedere a Stefan; si trova male: studenti impreparati, situazione politica insoddisfacente, ecc. 1895, Congresso di Lubecca. Riunione dei Naturalisti tedeschi. Meccanicisti (B. e gli studiosi più giovani) vs fenomenologisti (Planck e l’ala radicale degli energetisti Ostwald e Helm; Mach dietro le quinte). 1896, Wiederkehreinwand o paradosso della ricorrenza: E. Zermelo, applica un teorema di Poincaré del 1890 (un sistema puramente meccanico, in accordo con le

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
18
leggi della meccanica deve tornare dopo un tempo sufficientemente lungo, detto tempo di ricorrenza, a una configurazione iniziale entro un certo grado di precisione un numero infinito di volte) a un sistema con un numero finito di gradi di libertà; nell’ambito della TC conclude che H(t) è una funzione quasi periodica del tempo e pertanto non può decrescere sempre (ogni particella di un macrostato nel corso del tempo può assumere attraverso i microstati accessibili anche stati con probabilità più bassa). Il teorema H pertanto non è valido. 1896, Replica di B.: il II principio è estremamente probabile (un macrostato si realizza con grande probabilità) ma non è una certezza assoluta, ci possono essere anche punti dove S diminuisce /fluttuazioni, tanto meno quanto n è grande; B. non riconosce che ci possono anche essere grandi fluttuazioni in sistemi con basso n a differenza di quanto faranno Einstein e Smoluchowski qualche anno dopo, vedi MB e §9.5./; se è pur vero che la probabilità che un sistema torni allo stato iniziale è non nulla è praticamente impossibile e richiede tempi di ricorrenza lunghissimi, non commensurabili con la scala di osservazione umana. B. stima il tempo di ricorrenza per un numero di Loschmidt di molecole di gas con v media nota; perché le molecole ripassino per le stesse posizioni con le stesse velocità ci vogliono 10 alla 10 alla 19 anni. (eccezioni al II principio, estremamente improbabili ma non impossibilità di principio; scimmia davanti a tastiera di computer: la probabilità che la scimmia, digitando sui tasti, scriva la Divina Commedia è estremamente improbabile ma non impossibile) 1899, primo viaggio in America; 1900, a Lipsia su Fisica teorica, primo tentativo di suicidio; 1902, torna a Vienna, malanni vari, lezioni di filosofia della scienza: grande successo iniziale poi si disaffeziona; 1904, secondo viaggio in America, Congresso di S. Louis, scuola estiva a Berkeley; 1906, a Duino (Trieste), si suicida. Com’era B.: da giovane parlava un forte dialetto, anticonformista, oratore vivace e divertente (“notevolissimo polemista, molto temuto nei convegni”, Blaserna), entusiasta (ma altalenante; passava all’improvviso dall’euforia alla malinconia: “sono nato nella notte tra carnevale e mercoledì delle ceneri”), amante della musica e dell’arte (suonava il piano), docente straordinario dallo stile cristallino: “Era un buon conferenziere, nel mio ricordo le sue lezioni sono le più belle e stimolanti che abbia mai seguito. Teneva un corso quadriennale di meccanica, idrodinamica e teoria dell’elasticità, elettricità e magnetismo, teoria cinetica dei gas. Aveva una grande lavagna nel mezzo dove scriveva tutti i calcoli principali, e due altre ai lati dove c’erano i conti secondari. Tutto era scritto in modo molto chiaro e trasparente sicché allora pensai che dalle lavagne si sarebbe potuta ricostruire la lezione intera. Di tutto quello che ci insegnava era così entusiasta che da ogni lezione si usciva con la sensazione che ci fosse stato aperto un mondo nuovo e straordinario” (testimonianza di Lise Meitner (fissione uranio da neutroni lenti), che seguì le sue lezioni dal 1902 al 1905 a Vienna; da E. Broda, p. 19) SPIRITOSO!! Un esempio, dal necrologio per Loschmidt (muore nel 1895): “Ora il corpo di L. si è decomposto nei suoi atomi; in quanti lo possiamo calcolare dai suoi principi e io, affinché in un discorso in onore di un fisico sperimentale non manchi una dimostrazione, ho fatto scrivere questo numero alla lavagna: 10 quadrilioni cioè 1025”. (da E. Broda, p. 29). Posizione filosofica di B.: materialista, realista, combatte gli idealisti (Hegel, Schopenhauer) e i positivisti alla Mach. L’idealismo asserisce che esiste solo l’io e le idee e cerca di spiegare la materia da queste. Il materialismo parte dall’esistenza della materia e cerca di spiegare le sensazioni a partire dalla materia. L’uomo deve le

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
19
proprie idee all’evoluzione (la teoria di Darwin permea tutto il pensiero di B.). “Atomista fino all’impossibile”. Si definisce “reazionario, un sopravvissuto”, ma “non cieco rispetto al nuovo”. Non cita mai né la teoria dei quanti né la RR; cita a volte la radioattività, di continuo i “moti termici incessanti degli atomi”, quasi mai il “cosiddetto MB”. Materialismo vs idealismo: fisico vs psichico, materia vs spirito, oggetto vs soggetto. Mach (su posizioni simili, in Francia, P. Duhem /cfr. Bellone, p. 249 e seg.): empirista radicale (positivista/ empiriocriticista); enfasi posta sull’esperienza, sulle sensazioni pure, veri elementi dell’universo, le sensazioni sono simboli o immagini delle cose, non esiste materia né realta ma solo simboli; qualunque costrutto teorico è bollato come metafisica, no alle ipotesi non comprovabili, in primis alla ipotesi atomica; con l’accento posto sulle sensazioni gli empiristi cercano di superare la divisione tra filosofia idealista e materialista. (Mach e Duhem sono avversari feroci della ‘nuova fisica teorica’; Duhem considera le teorie di campo em di Maxwell e la meccanica statistica di B. “tradimenti della ragione”, la RR di Einstein “una follia contraria alla logica e al buon senso”. Mach non crederà mai agli atomi e alle molecole, “inutili finzioni metafisiche” /cfr. Bellone, p. 273/ B. martire delle sue idee? Si e no. Sul suo suicidio quanto pesarono i dispiaceri per l’atomistica? Forse non moltissimo ma è un fatto che nella Germania di fine Ottocento dominava un clima ostile alla TC dei gas. Nella prefazione al II vol delle sue Lezioni sulla teoria dei gas (1898) scriveva: “Quando la prima parte della “Teoria di gas” era in corso di stampa, avevo già completata questa seconda e ultima parte. Fu proprio allora che cominciarono ad aumentare gli attacchi contro la teoria dei gas. Sono convinto che quegli attacchi siano basati su un malinteso e che il ruolo della teoria dei gas nella scienza non sia ancora esaurito. .....Secondo me sarebbe una grande tragedia per la scienza se la teoria dei gas venisse bandita a causa di un momentaneo atteggiamento ostile verso di essa, come accadde per esempio alla teoria ondulatoria a causa dell’autorità di Newton. Sono consapevole di essere solo un individuo che lotta debolmente contro la corrente del tempo. Posso solo contribuire a fare sì, quando la teoria dei gas verrà rivalutata, che non vi siano troppe cose ancora da scoprire. Perciò in questo libro includerò quelle parti più difficili e soggette a malintesi; e fornirò, almeno come traccia, le trattazioni più facilmente comprensibili di esse”. Bibliografia P. W. Atkins, The second law,1984; ed. it. Il secondo principio, Zanichelli, Bologna,1988. A. Baracca, Manuale critico di meccanica statistica, CULC, Catania,1980. S. G. Brush, The kind of motion we call heat, North Holland pub. Co., Amsterdam,1976. C. Cercignani, Ludwig Boltzmann, the man who trusted atoms, Oxford University Press, 1998. C. Cercignani (a cura di), Ludwig Boltzmann, modelli matematici, fisica e filosofia, Bollati Boringhieri (sottoinsieme dei Populäre Schriften (1905) di L. B.; v. anche E. Broda, Populäre Schriften, Vieweg, 1979). R. Dugas, La théorie physique au sens de Boltzmann, Neuchatel,1956. Y. Elkana, Boltzmann’s scientific research program and its alternatives, in The interaction between science and philosophy, ed. by Y. Elkana, Humanities Press, Jerusalem,1974, pp. 243-279. M. J. Nye, Molecular reality,Macdonald, London,1972.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
20
CAP. 9. IL MOTO BROWNIANO
§9.1. Perché trattare la storia del moto browniano Case study: caso esemplare per conoscere i meccanismi di crescita della fisica Dialettica teoria-esperimento 1. La base empirica è irrinunciabile ma non da sopravvalutare; in particolare, se non esiste una cornice teorica ben strutturata l’esperimento è cieco. (Il moto browniano, osservato nel 1827 viene compreso e inquadrato nella teoria cinetica solo dopo il 1905. Si tratta di una ‘scoperta latente’ su cui si lavora per decenni in assenza di una guida teorica precisa.) 2. Uno stesso risultato o una stessa osservazione sperimentale possono essere interpretati con più teorie. (teoria di Einstein; di Smoluchowski; di Langevin del moto browniano) 3. Influenza e travaso di contesti teorici e/o sperimentali. Programma di ricerca di Einstein di estendere la teoria cinetica dai gas ai liquidi e ai solidi, qui, in particolare, ai liquidi (pressione osmotica; distribuzione della concentrazione con l’altezza, ecc.). La concezione atomica dell’elettricità influenza la ricerca. 4. Tra più teorie rivali quale ha il sopravvento? 5. Le scoperte sono quasi sempre processi e non fenomeni istantanei. 6. Non esistono esperimenti cruciali se non a posteriori. Gli esperimenti di Perrin, considerati dal fisico francese ‘cruciali’, compaiono quando i tempi sono ormai maturi per l’interpretazione del moto browniano in base alla teoria cinetica del calore e i giochi sono già parzialmente conclusi. Ostacoli epistemologici e pregiudizi teorici da superare quando da un vecchio contesto teorico (o paradigma) si passa a un altro (‘rivoluzione’). Passaggio dal continuismo dei fenomenologisti (prevalere del contesto della termodinamica generale) all’atomismo dei sostenitori della teoria cinetica del calore (o termodinamica ristretta). Influenza di elementi ‘metafisici’ Spesso la molla che indirizza la ricerca può essere rappresentata anche da elementi (criteri estetici, fede religiosa, ecc...) del tutto estranei al corpo delle conoscenze scientifiche. Il paradosso della reversibilità e il rifiuto dell’atomismo sono influenzati in parte, anche dal tentativo di evitare la “morte termica”, come conseguenza necessaria del secondo principio della termodinamica (Kelvin,1854: la fine dell’universo è meccanicamente inevitabile; Clausius,1865: l’universo va verso uno stato finale, ecc...) Shift di problemi L’interpretazione del moto browniano evolve e cambia significato nel tempo in funzione della ‘cornice concettuale’ in cui viene inquadrato. Intorno al 1827: particelle organiche autoanimate (contesto biologico) poi, gradualmente, particelle in moto a causa degli urti molecolari (contesto fisico). Già con Perrin, le traiettorie

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
21
browniane sono un esempio di funzioni continue che non ammettono derivata in nessun punto (contesto geometrico-analitico). Sono un esempio di ciò che qualche decennio dopo (Wiener,1920 circa -Mandelbrot,1970 circa) verrà indicato come oggetto frattale, un “mostro” matematico di cui si pensava impossibile dare una trattazione analitica. Secondo questo punto di vista ciò che prima era la norma (funzioni continue che in via eccezionale non ammettono derivate; in fisica, modelli continui e deterministici che in via eccezionale obbediscono alle leggi del caso) viene capovolto (geometria dei frattali che come eccezione ammette funzioni derivabili in ogni punto; fisica del caos, che ammette come casi particolari i fenomeni ordinati). Critica alla ricostruzione razionale dei manuali Sfatare i miti. O, per lo meno, avere la consapevolezza che la ricostruzione sui manuali, operazione spesso necessaria dal punto di vista didattico, è ben diversa dalla storia ‘reale’ della fisica. Il moto browniano viene in generale citato nei manuali come evidenza dell’esistenza degli atomi. La genesi storica è, al contrario, un’altra.
§9.2. Cronologia del processo di scoperta del moto browniano Per tutto l’Ottocento, ricerca fluida; viene studiato il MB e individuate le cause più diverse ( T in seno al liquido, osmosi elettrica, capillarità causata da pori nelle particelle in sospensione, correnti dovute a evaporazione e diffusione nella soluzione, ecc..) Disposizione sperimentale: polveri (per es., pollini o resine) diluite in acqua; microscopio con ingrandimento fino a 500 diametri. 1827, R. Brown, botanico. 1863, L. C. Wiener, fisico: le cause esterne al fluido sono escluse. Il moto diminuisce all’aumentare delle dimensioni delle particelle e della viscosità; rivela la costituzione atomica dei liquidi e l’esistenza di urti tra le molecole e le particelle browniane. Spiegazione in termini di teoria dell’etere + atomi ponderabili. 1867, S. Exner: i moti osservati sono dovuti a microcorrenti nei fluidi, prodotte da gradienti termici (calore e/o luce esterni). G. Cantoni: anche al buio il moto browniano non si indebolisce; è una dimostrazione diretta della teoria meccanica del calore. 1877, Delsaux: il moto browniano è da connettere ai moti del calore molecolare nei fluidi. 1880, J. Thirion: il moto browniano è la dimostrazione visibile della teoria meccanica del calore. Analogia tra le molecole dei gas, i granuli di materia rivelati negli esperimenti ai raggi catodici di Crookes e le molecole nei liquidi. 1888, L. Gouy: il moto browniano rivela l’agitazione interna di un liquido; è la prova visibile dell’ipotesi cineticomolecolare del calore e dell’irregolarità degli urti molecolari. Il moto è più sensibile quanto più le particelle sono piccole, aumenta con la temperatura della soluzione, varia con la natura della soluzione. Il secondo principio a livello microscopico è violato ed ha validità assoluta solo per i “meccanismi grossi”. 1897, Gouy: il moto browniano è un fenomeno governato dal caso. 1905, Il contributo di Einstein (v. oltre). Prime conferme sperimentali dell’ordine di grandezza dei cammini percorsi ottenute da Gouy, Siedentopf, ecc. 1908, Perrin verifica sperimentalmente che la distribuzione verticale delle particelle di un colloide all’equilibrio è esponenziale. 1910, Perrin e collab. misurano gli spostamenti orizzontali di una particella. Varie serie di esperimenti che concordano sul valore per N.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
22
§ 9.3. Contesto teorico-sperimentale. Evidenza empirica a favore dell’atomismo 1860 ca., studio della conduzione elettrica nei gas; scoperta dei raggi catodici: onde o particelle? 1865, Loschmidt: stima del numero di molecole contenute in una certa quantità di materia a partire dalla teoria cinetica. L=N/V 1019 molecole/ cm
3 per un gas in condizioni normali. 1876, Goldstein scopre i raggi canale. 1880 ca., Crookes: scariche elettriche nei gas rarefatti; i rc sono particelle. 1887, Analogia tra la conduzione elettrica nei gas e negli elettroliti.Teoria della dissociazione di soluzioni di Arrhenius (teoria generale degli elettroliti). 1894, J.J. Thomson misura la velocità dei raggi catodici. 1895, Röntgen scopre i raggi X. Scoperta della radioattività naturale. Becquerel: sali di Uranio (“raggi uranici”). Perrin dimostra sperimentalmente che le particelle dei rc sono negative. 1896, Zeeman: un campo magnetico influenza l’emissione delle righe spettrali. 1897, J. J. Thomson: i raggi catodici consistono di cariche elettriche negative (scoperta dell’elettrone). Teoria dell’elettrone di Lorentz; con l’esistenza di elettroni spiega l’effetto Zeeman normale. 1898, Wien dimostra che i raggi canale sono costituiti da particelle . M. Curie, Schmidt: composti del torio. P. e M. Curie: polonio e radio. 1899, Thomson: le particelle emesse nell’effetto fotoelettrico hanno e/m uguale a quello delle particelle dei raggi catodici. Debierre: attinio radioattivo. Rutherford: la radiazione emessa dall’uranio è più complessa della radiazione X (raggi raggi più penetranti). 1900, Villard scopre i raggi nelle emissioni radioattive (componente “ neutra”). 1902, Rutherford e Soddy: teoria delle trasformazioni radioattive (trasmutazioni). N(t) = N0e
t , legge di tipo statistico? 1904, Bragg: i raggi sono atomi (ionizzati di elio), i elettroni. Dei raggi X e non si sa ancora se sono di natura corpuscolare o ondulatoria. Modello atomico a panettone di Thomson. 1905/6, Rutherford stabilisce la natura corpuscolare della radiazione da deviazioni di da radio in campo elettrico e magnetico e stima q/m. Luglio del 1906: teoria cinetica del moto molecolare browniano di Smoluchowski. 1908, Rutherford e Geiger: misura della carica delle da RaC (rivelatore G-R); Geiger: metodo delle scintillazioni. 1909, Geiger e Marsden: scoperta della deviazione a grande angolo delle . Modello atomico planetario di Rutherford.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
23
§ 9.4. Analisi della trattazione di Einstein sul moto browniano (seguiamo l’ordine cronologico con cui i contributi vennero affrontati da Einstein). Articolo I: «Una nuova determinazione delle dimensioni molecolari» (aprile 1905, tesi di dottorato); Articolo II: «Il moto di particelle in sospensione in liquidi in quiete, interpretato secondo la teoria cineticomolecolare del calore» (maggio 1905); Articolo III: «La teoria del moto browniano» (dicembre 1905). A questi primi articoli ne seguiranno altri due nel 1907 e 1908, destinati alla rivista Zeitschrift für Elektrochemie, richiesti all’autore volutamente di taglio divulgativo per diffondere tra i chimici una teoria fondamentale per il loro settore12. Articolo I (tesi di dottorato): La teoria cinetica dei gas ha reso possibili le prime determinazioni della vera grandezza delle molecole mentre finora nei liquidi i fenomeni fisici osservati non sono serviti a determinare le dimensioni delle molecole. Questo stato di cose deriva senza dubbio dalle difficoltà finora insormontabili che hanno impedito lo sviluppo di una teoria cineticomolecolare dettagliata nei liquidi. In questo lavoro dovremo ora dimostrare che è possibile ottenere la grandezza delle molecole di una sostanza disciolta in una soluzione diluita non dissociata, a partire dalla viscosità della soluzione e del solvente puro e dalla diffusione della sostanza soluta nel solvente, a condizione che il volume di una molecola di soluto sia grande rispetto al volume di una molecola di solvente. Una tale molecola di soluto si comporterà con buona approsimazione, con riferimento alla sua mobilità nel solvente e al suo effetto sulla viscosità di questo, come un corpo solido in sospensione nel solvente e sarà possibile applicare le equazioni idrodinamiche al moto del solvente in prossimità di una molecola, considerando ivi il liquido omogeneo e potendo quindi trascurare la sua struttura molecolare. Per la forma dei corpi solidi che devono rappresentare le molecole di soluto, scegliamo la forma sferica.
La tesi di dottorato, facendo riferimento a un liquido anziché a un gas, contiene una novità assoluta. Per la trattazione del problema idrodinamico, in verità non banale, Einstein fa riferimento alle Lezioni di meccanica di G. Kirchhoff. Considerando applicabile l’equazione di Navier-Stokes anche al caso della miscela sferette+solvente, Einstein perviene alla fine alla relazione lineare tra il coefficiente di viscosità k* della miscela e il coefficiente di viscosità k del solvente puro:
k* = k 1+( ) [1]
dove è il volume totale delle sferette nell’unità di volume. D’altra parte, se le sferette hanno raggio P, peso molecolare m e rappresenta la massa di ‘soluto’ per unità di volume, per un numero di Avogadro N di particelle, tale volume è:
=N
m
4
3P 3 [2] .
Per una soluzione di acqua e zucchero all’1% a una data temperatura T e dai dati sperimentali tabulati in letteratura per k*/k, Einstein può così risalire a . Per individuare il coefficiente di diffusione D, Einstein considera, un processo attivo, causato dal gradiente della pressione osmotica in una data direzione ( p/ x) esercitata dal soluto (ritenuta l’unica «forza motrice» presente nel sistema), e un processo resistente dovuto alla forza di Stokes agente su ciascuna particella di soluto. Ottiene così una relazione di proporzionalità diretta tra il flusso di particelle che attraversano
12
A. Einstein, Eine neue Bestimmung, Ann. d. Phys., 19 (1906) 289; Über die von molekularkinetischen Theorie der Wärme geförderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen, Ann. d.
Phys., 17 (1905) 549-560; Theorie der Brownschen Bewegung, Ann. d. Phys., 19 (1906) 371-381; Theoretische Bemerkungen über die Brownsche Bewegung, Zeits. f. Elektroch., 13 (1907) 41-42; Elementare Theorie der Brownschen Bewegung, Zeits. f. Elektroch., 14 (1908) 235-239.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
24
nell’unità di tempo una sezione unitaria e il gradiente di concentrazione / x (con p/ x=(RT/m) / x dalla legge di van’t Hoff), relazione che esprime la legge di
Fick, con un coefficiente di proporzionalità che è proprio il coefficiente di diffusione D:
D =RT
6 k
1
NP [3].
Di qui, noti che siano D e k dai dati sperimentali, è possibile risalire al prodotto NP. Il metodo idrodinamico di Einstein ha consentito di ricavare due equazioni nelle due incognite N e P, ormai completamente determinabili. Dalla [1] e [2] si ottiene infatti la prima equazione
NP 3=
k *
k1
3m
4
mentre dalla [3] si individua la seconda equazione
NP =RT
6 kD .
Sempre per la soluzione di acqua e zucchero, Einstein trova per il raggio P di una molecola di zucchero la stima
P=9,9 10-8 cm e per il numero N di molecole reali per grammomolecola
N=2,1 1022. Questo articolo riporta in calce una breve Appendice, scritta in fase di bozze in data gennaio 1906. Avendo a disposizione dati migliori per il coefficiente di diffusione dello zucchero in acqua e per il coefficiente di viscosità presenti in letteratura nelle tabelle chimicofisiche, Einstein ricalcola:
P=7,8 10-8 cm, N=4,15 1023. Nel 1910 un allievo di J. Perrin, J. Bacelin, troverà in questo articolo degli errori di calcolo, sfuggiti anche al controllo dei severi relatori della tesi di dottorato. Einstein, che nel frattempo aveva conseguito l’abilitazione ed era divenuto dal 1909 professore di fisica all’Università di Zurigo, aveva così incaricato un suo collaboratore, L. Hopf di verificare i calcoli. Trovata l’insidia, dovuta a un errore nel differenziare le componenti della velocità della sferetta nella soluzione, Einstein pubblicherà una
correzione13
al suo lavoro, trovando per la [1]:
k* = k 1+ 2,5( ) [1’]
che porterà a una stima sorprendentemente buona del numero di Avogadro N=6,56 1023.
Nonostante l’errore, l’articolo sulla determinazione delle dimensioni molecolari resta magistrale. Per altro, come ha osservato Pais, tra i contributi dell’anno mirabile di Einstein è quello che «ha avuto più applicazioni pratiche di qualunque altro lavoro
di Einstein»14
e, almeno fino agli anni Settanta, è quello che compare con più frequenza nel quotation index, più dell’articolo sul moto browniano, sulla relatività generale del 1916 e sul quanto di luce del 1905. Articolo II: l’articolo riprende le tematiche principali della tesi di dottorato e inizia con un colpo da maestro di taglio epistemologico. Da una ipotesi (secondo la teoria cinetica in seno ai liquidi esiste un moto di agitazione termica disordinato) segue la previsione sperimentale (possibilità reale di osservare tale moto) e, quindi, la
13A. Einstein, Berichtigung zu meiner Arbeit: «Eine neue Bestimmung der Moleküldimensionen», Ann. d. Phys., 34 (1911) 591-592. 14A. Pais, Sottile è il signore, XXX, p. 104.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
25
conseguenza sul piano della teoria (se l’esperimento conferma l’ipotesi la teoria cinetica ne esce rafforzata): In questo lavoro dovremo mostrare che secondo la teoria cineticomolecolare del calore, particelle di dimensioni visibili al microscopio in sospensione in un liquido, in conseguenza del moto molecolare del calore, devono descrivere moti di ampiezza tale da poter essere facilmente osservati al microscopio. È possibile che i moti che qui discuteremo siano identici al cosiddetto «moto molecolare browniano»; le informazioni che ho su questo tipo di moto sono tuttavia così imprecise che finora non ho potuto formulare alcun giudizio in proposito. Se fosse realmente possibile osservare il movimento in questione insieme alle leggi previste per esso, si dovrebbe ritenere che la termodinamica classica non può essere più considerata esattamente valida già per dimensioni distinguibili al microscopio, e in queste condizioni sarebbe possibile determinare esattamente le dimensioni reali degli atomi. Se viceversa si dovesse scoprire che le previsioni relative a questo moto sono sbagliate, si troverebbe un
serio argomento contro la concezione cineticomolecolare del calore15
. Il leit motiv è dunque quello consueto di dimostrare la realtà di atomi e molecole, come espliciterà Einstein, a distanza di tempo nella sua Autobiografia: Il mio maggior obiettivo [...] è stato quello di trovare fatti che garantissero il più possibile l’esistenza di atomi di dimensione definita. Mentre lo facevo ho scoperto che, in accordo con la teoria atomica, ci sarebbe dovuto essere un moto di particelle microscopiche sospese, aperte all’osservazione senza sapere che le osservazioni relative al moto browniano erano già da tempo familiari. [...] L’accordo di queste considerazioni con l’esperienza, insieme con la determinazione di Planck della vera dimensione molecolare dalla legge di radiazione (per alte T) convinsero gli scettici che erano piuttosto numerosi a quel tempo (Ostwald, Mach) della realtà degli atomi. L’antipatia di questi scienziati nei confronti della realtà atomica deve senza alcun dubbio essere attribuita alla loro inclinazione positivista. Questo è un esempio interessante del fatto che anche gli scienziati di intelligenza ardita e intuizione acuta possono essere bloccati nella interpretazione della realtà a causa di pregiudizi filosofici. Il pregiudizio, che nel frattempo non è affatto morto, consiste nella fede che i fatti da soli possano e debbano fornire la conoscenza scientifica senza il ricorso ad una costruzione concettuale libera. Una tale illusione è possibile solo perché non si ha facilmente dimestichezza con la libera scelta di tali concetti che,
attraverso la verifica e il lungo uso, appaiono essere immediatamente connessi con la base empirica16
. Einstein torna poi a esporre il suo Gedankenexperiment su cui baserà la trattazione teorica: in un recipiente di volume V contenente una soluzione diluita, una membrana semipermeabile divida il volume in un sottovolume V* in cui vengono sciolte z grammomolecole di una sostanza non elettrolita. Se il setto lascia passare il solvente ma non il soluto, sulla parete deve esercitarsi una pressione osmotica p =zRT/V*, data dalla legge di Van’t Hoff.
VV*
parete semipermeabile Se ora, in luogo del soluto sono presenti particelle solide in sospensione impermeabili alla membrana, mentre secondo la termodinamica classica non è previsto che la pressione osmotica agisca perché l’energia libera del sistema non dipende «dalla
15 A. Einstein, Über die von molekularkinetischen Theorie der Wärme, 1905, cit. 16 A. Einstein, Philosopher-scientist,1949, a cura di P. A. Schilpp, New York,1949, pp. 46-49.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
26
posizione della parete e delle particelle in sospensione, ma solo dalla massa totale e dalle proprietà della sostanza sospesa, oltre che dalla pressione e dalla temperatura», secondo la teoria cinetica, anche le particelle sospese devono esercitare la stessa pressione osmotica poiché esse, secondo questa teoria, sono indistinguibili, a meno della taglia, dalle molecole di soluto. Se ci sono n particelle nell’ unità di volume, ce ne saranno =n/V* che esercitano sulla membrana una pressione osmotica p= RT/N, con N numero di Avogadro. Einstein dimostra quindi, e questa è un’altra novità di rilievo, che l’esistenza della pressione osmotica è una conseguenza della teoria cinetica e che particelle in sospensione e molecole di soluto si devono comportare allo stesso modo in una soluzione diluita. Descrive così, in tutta generalità, lo stato istantaneo del sistema mediante le sue variabili di stato p , calcola l’entropia S e successivamente l’energia libera F del sistema da considerazioni probabilistiche sulle posizioni occupate dalle particelle in sospensione a un istante arbitrario e ritrova l’espressione della pressione osmotica:
p =F
V *=
RT
N
Il passo successivo consiste nello studiare lo stato di equilibrio dinamico delle particelle distribuite in modo irregolare nella soluzione sotto l’ipotesi che su ciascuna agisca (per semplicità lungo x) una forza K, funzione della posizione ma non del tempo. All’equilibrio termodinamico, dalla condizione che la variazione dell’energia libera F si annulli per uno spostamento virtuale x della particella sospesa ( F = E-T S =0), deve seguire la condizione che la forza K è equilibrata dalle forze di pressione osmotica
Kp
x= 0
Come ha già fatto nella tesi di dottorato, Einstein determina quindi il coefficiente di diffusione D della sostanza in sospensione presupponendo che nella soluzione dominino due processi inversi, uno di tipo viscoso dovuto alla forza K agente su ciascuna particella e un altro di tipo attivo, di diffusione, conseguenza dei «moti disordinati delle particelle causati dal moto molecolare del calore». All’equilibrio dinamico, il numero di particelle che attraversa una sezione unitaria nell’unità di tempo sollecitate dalla forza K, è direttamente proporzionale al gradiente di concentrazione / x, cioè al numero di particelle che attraversa la stessa sezione nell’unità di tempo per diffusione, dove di nuovo il coefficiente di proporzionalità è dato dalla [3]. Einstein passa così all’analisi statistica del moto disordinato di particelle, generato dal moto termico molecolare. Per questa via ritrova «la nota equazione differenziale della diffusione e si riconosce che D è il coefficiente di diffusione» (nota già da tempo e dedotta su base fenomenologica), mediante una distribuzione di probabilità degli spostamenti delle particelle in funzione del tempo a t+ t a partire da quella al tempo t
f
t= D
2 f
x 2 .
La funzione f esprime il numero di particelle per unità di volume, =f(x, t) e le ipotesi a monte sono che i moti delle particelle siano indipendenti tra loro e che per ogni particella, siano indipendenti i moti descritti in intervalli di tempo t= , molto minori del tempo di osservazione t ma abbastanza grandi da poter ritenere scorrelati i moti eseguiti da una particella in due intervalli di tempo consecutivi. Trovata l’equazione di diffusione, Einstein è immediatamente in grado di trovare la soluzione f(x, t), che

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
27
esprime la distribuzione delle variazioni di posizione a t «uguale a quella degli errori casuali, come ci si doveva aspettare»
f x, t( ) =4 D
ex 2
4 Dt
t.
Se dunque particelle, sia che partano da t=0 sia da t, vengono rilasciate nella soluzione, queste, a causa del moto di agitazione termicomolecolare diffondono in modo casuale e i loro spostamenti seguono la legge di Gauss (con = 2Dt ), condizione che gli fa immediatamente ricavare la sua equazione fondamentale del moto browniano per lo spostamento quadratico medio che una particella subisce al tempo t:
x = x 2= 2Dt = t
RT
N
1
3 kP [7].
Einstein è così ormai in grado di tornare al suo tema preferito sulla determinazione del numero di Avogadro. Per una soluzione in acqua con particelle di diametro di 0,001 mm a una data temperatura T (e quindi è noto il coefficiente di viscosità k dell’acqua) per t=1 s, e noto N=6 1023 ottiene infatti la stima dello spostamento medio che una particella browniana subisce (
x=0,8 m). Al contrario, noto
x è
possibile ricavare N: Mi auguro che qualche ricercatore possa presto riuscire a risolvere il problema qui esposto, così
importante per la teoria del calore! Articolo III: confortato dalle prime conferme sperimentali, Einstein completa la sua trattazione: Subito dopo la pubblicazione del mio articolo sul moto di particelle in sospensione in un liquido, interpretato secondo la teoria molecolare del calore, Siedentopf (Jena) mi ha comunicato che insieme ad altri fisici, fra i quali in primo luogo certamente il sig. prof. Gouy (Lione), sono arrivati alla conclusione, attraverso l’ osservazione diretta, che il cosiddetto moto browniano sarebbe causato dal moto termico disordinato delle molecole del liquido. Non solo le proprietà qualitative del moto browniano ma anche l’ordine di grandezza delle traiettorie descritte dalle particelle corrisponde nei
risultati alla teoria17.
Nelle intenzioni di Einstein, la memoria vuole fornire un metodo più generale non solo per dimostrare come il moto browniano dipenda direttamente «dai fondamenti della teoria molecolare del calore» ma anche per ricavare in modo unitario le equazioni fondamentali sia per il moto rettilineo sia rotatorio per una particella browniana in sospensione in un liquido. §9.5. Fluttuazioni e opalescenza vicino al punto critico Nella seconda metà dell’Ottocento iniziano i primi tentativi di spiegare le proprietà ottiche dell’atmosfera, in particolare il blu del cielo, la colorazione rossa del cielo con il Sole all’orizzonte, e il diverso grado di polarizzazione della luce. Clausius 18 tenta per primo di quantificare il fenomeno sulla base di una analogia tra il comportamento ottico delle lamine sottili e quello di particelle di vapore sospese nell’atmosfera. Il risultato a cui perviene si limita a collegare il rapporto delle
17 A. Einstein, Theorie der Brownschen Bewegung, 1906, cit., p. 371. 18R. Clausius, A. d. P., 76 (1849)161.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
28
intensità riflessa e incidente alle dimensioni delle particelle e al colore, e quindi alla lunghezza d’onda della luce. Tyndall19 dedicherà al problema del blu del cielo uno studio sistematico cercando di riprodurre in laboratorio il fenomeno: in un tubo di vetro osserva varie sostanze allo stato di vapore illuminate con sorgenti di diversa intensità. Analizzando con un prisma, a vari angoli, la luce diffusa conferma il fatto che la luce è polarizzata al massimo perpendicolarmente alla direzione di propagazione, che esistono punti neutri, che se si osserva il fascio luminoso a 90° questo mostra una striscia brillante (azzurrina se le particelle sono sufficientemente piccole rispetto alla lunghezza d’onda della luce incidente), mentre l’effetto cessa di essere percepibile se il mezzo è privo di impurità. Il fenomeno da allora sarà noto come “effetto Tyndall”, o fenomeno di opalescenza, e si riferirà sia ai gas che ai liquidi, purché torbidi: per decenni si riterrà infatti che a produrre la “diffusione laterale” della luce sia la presenza di particelle estranee in sospensione nel mezzo (pulviscolo, vapor acqueo, ecc.). I primi tentativi di formulare una teoria coerente della diffusione della luce da parte dell’atmosfera, si devono a Rayleigh che si dedicherà all’argomento a più riprese nell’arco di mezzo secolo. Nella prima memoria sull’argomento, del 187120, Rayleigh trova una legge di attenuazione per la luce primaria del tipo
E = E0 e-hx (1) dove E è l’energia luminosa che attraversa uno strato di spessore x, E0 è l’energia della luce incidente, h, è il coefficiente di trasmissione (o di assorbimento), che dipende linearmente dal numero n di particelle diffondenti per unità di volume e dall’inverso della quarta potenza della lunghezza d’onda della luce. La dipendenza funzionale di h da 4 consente già di giustificare perché il cielo è blu, confrontando i valori di h nei casi di per la luce violetta e rossa: poiché viol è circa la metà di
rosso la luce blu viene diffusa circa 16 volte di più della rossa. Data la legge iniziano i primi tentativi di conferme sperimentali: dal punto di vista delle ipotesi teoriche non c’è tuttavia accordo né sul meccanismo ottico che produce il fenomeno (si parla indifferentemente di riflessione, rifrazione, diffrazione, fluorescenza da sostanze particolari come, per esempio, l’ozono), né sulla densità, forma e natura delle particelle diffondenti. Le risultanze sperimentali, inoltre, non sono direttamente confrontabili perché condotte dai vari sperimentatori con metodi e in condizioni diverse. La situazione non migliora dopo la pubblicazione della seconda memoria di Rayleigh del 188121, dove il fenomeno viene interpretato con la teoria elettromagnetica della luce sotto l’ipotesi che la diffusione sia provocata da diffrazione da particelle sferiche. La terza memoria del 189922 sembra al contrario restringere il campo rispetto ai parametri da controllare. L’inizio dell’articolo contiene già una questione fondamentale: la luce del cielo può essere spiegata per diffrazione da molecole d’aria o bisogna supporre anche la presenza di particelle composte di materia estranea, solida o liquida? Rayleigh ritiene che le sole molecole d’aria possano essere già sufficienti a spiegare il fenomeno. Non si sa per quali associazioni di idee Rayleigh arrivi a questa conclusione: l’idea gli viene sicuramente suggerita da una lettera che Maxwell gli aveva inviato nel 1873 ma non è chiaro perché l’autore arrivi a cambiare opinione dopo tanto tempo. Probabilmente perché nel frattempo ci sono misure di assorbimento che venti anni prima non erano disponibili.
19J. Tyndall, Proc. Roy. Soc., 17 (1868) 223; 37 (1869)384;38 (1869)156. 20Rayleigh, Phil. Mag., 41 (1871) 107, 274, 447. 21Rayleigh, Phil. Mag., 12 (1881) 518. 22Rayleigh , Phil. Mag., 47 (1899) 375.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
29
Nella lettera, che Rayleigh riporta integralmente in una nota, Maxwell si chiede se la teoria della diffusione non possa essere un mezzo per ottenere informazioni sulle dimensioni delle molecole d’aria. Si potrebbe risolvere il problema confrontando misure di assorbimento della luce solare con la formula di Rayleigh ma poiché queste, al momento (cioè al 1873), non sono disponibili si potrebbe preferire di fare misure sull’indice di rifrazione dell’atmosfera. Rayleigh ricalcola allora il coefficiente di assorbimento h in funzione dell’indice di rifrazione μ del mezzo e del numero n di particelle per unità di volume (considera particelle sferiche di diametro molto minore della lunghezza d’onda della luce distribuite in modo casuale nello spazio) e trova:
h = 32 3 μ 1( )2
3n 4 .
Sotto l’ipotesi di assenza di particelle estranee in sospensione studia poi il grado di trasparenza dell’aria scegliendo come parametro significativo misurabile lo spessore x dello strato che la luce attraversa a pressione normale quando subisce una attenuazione di 1/e, dalla legge esponenziale (1). Il valore per x (assumendo = 610-5 cm e μ-1=0,0003) risulta così funzione solo del numero n:
x = 4,4 10-3 n. Per valutare numericamente x avrebbe bisogno di un valore affidabile di n ma “sfortunatamente questo numero, uguale per tutti i gas in accordo con la legge di Avogadro, non può essere ritenuto noto”. Prendendo per buono il valore stimato da Maxwell23 per la costante di Loschmidt, di 19 1018 molecole per cm3 trova x= 83 km. Dal confronto con le misure di assorbimento eseguite da altri ricercatori conclude che, sebbene il suo valore è di circa tre volte inferiore, basta a giustificare il blu del cielo anche in assenza di impurità. L’articolo di Rayleigh del 1899 stabilisce dunque che si può avere diffusione della luce e blu del cielo anche in una atmosfera pura. Questo risultato mentre da un lato darà l’avvio a una serie di misure di assorbimento in alta montagna, eseguite tra gli altri anche da Q. Majorana24, dall’altro, contenendo nella formula per h il numero di molecole di gas per unità di volume ma non le dimensioni delle particelle molecolari (l’unica condizione è che le molecole abbiano dimensioni molto minori di ), offrirà un nuovo procedimento per la determinazione della costante di Avogadro. E in questo nuovo ruolo verrà considerato da Kelvin25 nel 1902. Nel giro di qualche anno si avrà con Smoluchowski ed Einstein una nuova conferma della teoria di Rayleigh ma con una interpretazione radicalmente nuova del fenomeno, visto ormai come diffusione molecolare, che lo renderà un argomento forte a favore della teoria cinetica. Nel 190826 viene pubblicato un contributo di Smoluchowski sulla “Teoria cinetica molecolare dell’opalescenza dei gas allo stato critico” in cui il moto browniano, le variazioni locali di densità nei gas e l’opalescenza vengono interpretati in base allo stesso principio. Il principio riguarda la previsione contenuta in teoria cinetica della possibilità che avvengano fluttuazioni a livello molecolare. In un sistema termodinamico, cioè, accanto a stati di massima probabilità devono esistere stati meno probabili che violano il secondo principio. Questa idea era stata avanzata in particolare sia da Maxwell che da Boltzmann27 qualche anno prima, nel tentativo di difendersi dalle obiezioni di Zermelo (paradosso della ricorrenza) ma l’ipotesi aveva
23J. C. Maxwell, Nature, 8 (1873) 440. 24Q. Majorana, Phil. Mag., 1 (1901) 555. 25Kelvin, Phil. Mag., 4 (1902) 281. 26M. Smoluchowski, A. d. P., 25 (1908) 205. 27L. Boltzmann, A. d. P., 57 (1896) 773-784.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
30
sempre mantenuto un carattere euristico senza necessariamente implicare una possibile conferma sperimentale. Anzi, sia Maxwell che Boltzmann escludevano a priori che le fluttuazioni potessero essere osservabili. Una prima formulazione quantitativa del problema delle fluttuazioni verrà poi data in modo indipendente sia da Gibbs, nel 1902, che da Einstein due anni dopo28 ma con Smoluchowski l’argomento si riveste di un significato del tutto nuovo. Smoluchowski si chiede “quanto sono grandi le deviazioni dallo stato istantaneo, o casuale dallo stato più probabile o medio e se tali deviazioni in qualche caso siano osservabili, questione questa di grande importanza perché tali casi potrebbero fornire un experimentum crucis per decidere l’antica disputa sulla validità della cinetica o della termodinamica”29. Il fisico polacco si era occupato del tema già nel 190430 in occasione della pubblicazione del Boltzmann Festschrift, volume celebrativo per il sessantesimo compleanno del grande fisico viennese, per il quale Smoluchowski aveva presentato un articolo sull’influenza delle disomogeneità di distribuzione delle molecole di un gas sull’entropia e l’equazione di stato. Fin da allora Smoluchowski era interessato a capire se tali fluttuazioni potevano divenire osservabili e costituire così un banco di prova a favore della teoria cinetica. Il nucleo del ragionamento ora si fa più chiaro: a causa dell’agitazione molecolare il numero n di molecole presenti in un dato volume non è costante ma fluttua intorno a un valor medio n0. Nel mezzo si devono così verificare degli “addensamenti casuali
(o locali)” =n n0
n0
, il cui valor medio viene calcolato da Smoluchowski con
considerazioni termodinamiche, a partire dal principio entropia-probabilità di Boltzmann modificato da Einstein31:
=2
n0
;
come si vede dalla formula, se si diminuisce il numero ‘normale’ di molecole n0 presenti, aumentano le fluttuazioni di densità ma sperimentalmente la misura non è più accessibile all’osservazione. Una stima dell’ordine di grandezza delle fluttuazioni in 1 cm3 di gas allo stato normale porta a una deviazione di 10-10 rispetto alla densità normale. Riducendo il volume a un cubo di spigolo pari alla della luce visibile le fluttuazioni aumenterebbero sensibilmente, e di più in un gas che in un liquido, ma sarebbero ugualmente non apprezzabili sperimentalmente. Smoluchowski trova finalmente una via percorribile nel comportamento ottico di un gas vicino al punto critico32 poiché dai calcoli si vede che qui l’addensamento medio33 cresce sensibilmente. Fatti un po’ di conti Smoluchowski trova
=1,13
n04
.
28J.W. Gibbs, Statistical mechanics, New York, 1902; A. Einstein, A. d. P., 14 (1904) 354-362. In questo articolo Einstein calcola la fluttuazione quadratica media dell’energia di un sistema termodinamico; un secondo calcolo delle fluttuazioni si avrà con la trattazione del moto browniano nel 1905. 29Smoluchowski 1908, cit., p. 206. 30M. Smoluchowski, Boltzmann Festschrift, Leipzig, 1904. 31A. Einstein, A. d. P., 19 (1906)373. 32Sperimentalmente si era osservato in laboratorio che la diffusione aumenta vicino al punto critico. M. Avenarius, Ann. Phys. Chem., 151 (1874)306. 33In questo articolo Smoluchowski calcola la fluttuazione media, pari a 2RTrb/Np con b=-(1/v)dv/dp, compressibilità del gas. Il valore quadratico medio delle fluttuazioni, pari a RT/NV(-dp/dv) , verrà calcolato in un lavoro successivo. Cfr. M. Smoluchowski, Phil. Mag., 23 (1912) 165.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
31
La formula (dove = (v-v0)/v0 con v volume specifico) risulta indipendente dal fluido scelto. Se la si applica per esempio a un liquido contenuto in un cubo di spigolo pari a visibile ( =0,6 mm; n0= 109 in condizioni normali) il numero medio di addensamenti salirebbe a 10-2 e il fenomeno dovrebbe essere particolarmente visibile nel caso dell’opalescenza vicino al punto critico. Smoluchowski ricollega così la possibilità di rilevare fluttuazioni di densità nei fenomeni ottici che si presentano nei mezzi torbidi, descritti in particolare dalla teoria di Rayleigh. Se nel mezzo si verificano fluttuazioni di densità queste devono influenzare l’indice di rifrazione34 μ delle particelle diffondenti che nella formula di Rayleigh per il coefficiente di assorbimento figura al quadrato. Vicino al punto critico il valore di h dovrebbe pertanto aumentare enormemente e rendere così ragione del perché un mezzo illuminato appare torbido se lo si osserva perpendicolarmente alla direzione di propagazione. Nell’ottobre 1910 Einstein, dopo aver letto il contributo di Smoluchowski sulla interpretazione cineticomolecolare dell’opalescenza (giudicato da Einstein “un importante lavoro teorico”) elabora a sua volta una “Teoria dell’opalescenza di liquidi omogenei e di miscele vicino al punto critico”35. Dunque, Smoluchowski ha dimostrato che vicino al punto critico si deve verificare opalescenza in misura sensibile. Poiché però non ha calcolato l’intensità della luce diffusa lateralmente, Einstein si accinge a colmare la lacuna. In primo luogo Einstein, come già aveva fatto Smoluchowski, parte dal principio di Boltzmann
S =RN
lg W + cos t.
con N, numero di molecole contenute in una grammomolecola e W probabilità di uno stato di entropia S, che modifica in modo da evidenziare le proprietà statistiche di un sistema termodinamico come
W = cos t.eN
RS S0( )
dove S0 rappresenta l’entropia allo stato di equilibrio termodinamico. Per dare un significato intuitivo a S-S0 Einstein immagina che il sistema, a causa delle fluttuazioni, possa passare spontaneamente dallo stato di equilibrio a uno stato infinitamente vicino, condizione questa che non dovrebbe variare la temperatura del sistema (dU=0). Se si vuole riportare il sistema allo stato di equilibrio occorre fornire dall’esterno un lavoro A con S-S0=A/T0. Si ha così una relazione del tipo
W = cos t .eN
RT 0
A
che consente il calcolo delle fluttuazioni. Poiché inoltre A è pari all’energia utilizzabile dal sistema cambiata di segno, e questa allo stato di equilibrio è minima, si può sostituire ad A, con uno sviluppo in serie di Taylor, una relazione che dipende dal quadrato di parametri osservabili qualunque, sicché il valor medio del lavoro di fluttuazione risulterà pari a (1/2)kT, che è pure uguale a (1/3) dell’energia cinetica media di una molecola di gas monoatomico. Passando al caso particolare di fluttuazioni nella distribuzione spaziale di fluidi (o miscele), a causa dell’agitazione termica molecolare la densità media 0 varia spontaneamente. Einstein calcola di nuovo, per questo caso particolare, il lavoro che bisogna fare per portare l’unità di massa del fluido in modo isotermo dal valore all’equilibro a un valore attuale , ottenendo RT0/N. A questo punto, ricavata dal principio di Boltzmann la legge statistica in base a cui varia la densità di una sostanza omogenea, Einstein è pronto per studiare l’influenza
34Qui viene fatto riferimento alla formula di Lorentz-Lorenz (m2-1)(m2+2) = cost. r, con m indice di rifrazione e r densità del mezzo. 35A. Einstein, A. d. P., 33 (1910) 1275.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
32
che un mezzo praticamente omogeneo e non assorbente esercita su un fascio di luce incidente. Supponendo che il fascio sia monocromatico e polarizzato, a partire dalle equazioni di Maxwell, dopo una lunga serie di calcoli e di considerazioni teoriche, Einstein perviene infine alla equazione [17 b nella memoria originale]
J0
Je
=RT0
N1( )2
+ 2( ) 2
9vp
v
2
4
4 D( )2 cos2
dove J0 è l’intensità della luce diffusa (o di opalescenza), Je è l’intensità della luce incidente, D è la distanza di osservazione tra il volume di gas irradiato e la sorgente, è l’angolo tra il vettore campo elettrico della luce eccitante e il piano normale rispetto alla luce eccitante (o angolo di diffusione), è la costante dielettrica del mezzo ( = μ2), v=1/ è il volume specifico del fluido, dove il quoziente differenziale di p rispetto a v si riferisce a una compressione isoterma. Nel caso di un gas perfetto, essendo +2=3, l’equazione si modifica ancora nella equazione
J0
Je
=RT0
N
1( )2
p
2
4
4 D2 cos2
che coincide con la formula trovata da Rayleigh nel 1899 sommando le onde elementari diffuse dalle singole molecole di gas distribuite in modo casuale nello spazio, fornendo la spiegazione per il blu del cielo. L’ultimo caso trattato da Einstein riguarda le miscele binarie; poiché nell’equazione trovata figurano solo grandezze osservabili la conclusione è che una ricerca quantitativa dei fenomeni qui trattati sarebbe di grande interesse. Perché da una parte sarebbe fondamentale sapere se il principio di Boltzmann dà conto correttamente dei fenomeni trattati, dall’altra grazie a tali ricerche si potrebbe risalire al valore esatto per il numero N [ di Avogadro]. Ma già prima della pubblicazione di questo articolo H. Kamerlingh Onnes e W. H. Keesom36 del laboratorio di fisica di Leida avevano condotto nel 1908 ricerche nel senso che verrà indicato da Einstein tre anni dopo. Con un metodo spettrofotometrico era stato infatti eseguito uno studio sperimentale sull’opalescenza di una sostanza a un componente (etilene) vicino al punto critico. Uno dei due autori, Keesom37, riprende l’argomento e su richiesta esplicita di Einstein riporta in modo più esteso, nel 1911, la derivazione teorica del coefficiente di diffusione s (per coefficiente di diffusione si intende qui il rapporto J0/Je moltiplicato per il fattore (3/16) , dovuto al fatto che Keesom tratta l’intensità della luce diffusa per unità di angolo di apertura del fascio in direzione perpendicolare alla luce incidente), assunta di intensità unitaria. Conducendo misure dell’intensità della luce diffusa iD (prossima alla del sodio) in funzione della temperatura, in un range di qualche grado vicino alla temperatura critica (per l’etilene TK=11,18°C), il valore sperimentale per il coefficiente di diffusione era risultato di sD= 0,0007. Le misure avevano inoltre mostrato una dipendenza di iD inversamente proporzionale a T-TK in accordo con la teoria di Smoluchowski. Si trattava ora di giustificare teoricamente il risultato rispetto alle diverse ipotesi sulla diffusione della luce che si rifacevano principalmente alla teoria di D. Konowalow38, che attribuiva l’opalescenza alla
36Comm. fr. the phys. Lab. of Leiden, n. 104, 1908. 37W. H. Keesom, A. d. P., 35 (1911) 591-598. 38D. Konowalow, A. d. P., 10 (1903) 360.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
33
presenza di polvere; alla teoria di F. G. Donnan39, che si richiamava a complicate spiegazioni in termini di tensione superficiale che si sarebbe formata su piccole gocce di liquido nella transizione liquido-gas; e infine alla teoria di Smoluchowski, rielaborata in modo elegante da Einstein, che interpreta l’opalescenza attribuendola a fluttuazioni di densità. Per quanto riguarda il coefficiente di diffusione Keesom calcola per via teorica prima il contributo dovuto a un elemento di volume della sostanza opalescente a causa delle fluttuazioni dell’indice di rifrazione μ rispetto al valore normale μ0, poi estende la trattazione al numero di elementi di volume contenuti in 1 cm3 e infine a una grammomolecola. La formula trovata coincide con quella di Einstein (17b) a meno di un fattore 2 dovuto al fatto che Keesom ha utilizzato luce non polarizzata
s =2
18N v2 μ0
2 1( )2μ0
2+ 2( )
2 RT
v0
pv
0
dove v è la lunghezza d’onda della luce nel vuoto, v0 è il volume specifico della sostanza. Il valore calcolato di sD per l’etilene vicino alla temperatura critica risulta di 0,00075, dunque in buon accordo con il valore sperimentale. Keesom conclude così di aver trovato “un importante sostegno per l’ipotesi di Smoluchowski”. Dalla formula, inoltre, Keesom dedurrà un valore per il numero di Avogadro N= 75 1022 che converge con i valori allora noti per la costante. Esperimento sulla pressione osmotica Esperimento di simulazione del blu del cielo Esperimento sul moto browniano In aria con la camera a fumo; si osservano corpi chiari su sfondo scuro per diffusione della luce sulle particelle browniane (pb). Esperienza solo qualitativa. MB in una soluzione: si osservano corpi scuri (pb) su sfondo chiaro (soluzione acquosa). Esperienza quantitativa. Materiali: sferette di latex, diametro d=1,1 μm oppure 0,79 μm; microscopio oculare 10x, obiettivo 40x, ingrandimento 400:1 (se si usa l’oculare 16x e l’obiettivo 40x aumenta l’ingrandimento ma si restringe troppo il campo visuale); livella a bolla; righello; acqua distillata; bacchette di vetro per agitare, contagocce; vetrini con uno o più incavi e coprivetrini; videocamera a colori e televisore (se vuoi registrare su cassetta VHS canale 15); carta millimetrata, fogli di acetato, pennarelli, scotch; contasecondi; termometro 0,1 °C; reticolo di diffrazione 100 |||/mm, passo p= 0,01 mm. 1. Preparazione della soluzione browniana: diluire la sospensione delle sferette di lattice in acqua distillata (agitare con bacchetta di vetro) e per tentativi trovare la soluzione migliore (non troppo affollata né troppo diluita, in modo che le pb si muovano casualmente e in modo indipendente l’una dalle altre e siano ben visibili nel campo d’osservazione per un intervallo di tempo di qualche minuto). La soluzione va messa nell’incavo del vetrino e ricoperta con un vetrino coprioggetti per evitare che evapori o che bagni l’obiettivo del microscopio. Collegare la telecamera al televisore e all’oculare del microscopio; ottimizzare messa a fuoco, luminosità e contrasto del televisore; l’ingrandimento della soluzione dipende dalla posizione relativa telecamera-oculare: una volta ottimizzata l’immagine sullo schermo lasciare tutto fisso; evitare la velocità di drift delle pb aggiustando con degli spessori di carta messi sotto il microscopio l’orizzontalità del piano del microscopio stesso (che purtroppo non è dotato di piedini regolabili).
39F.G. Donnan, Chem. News., 90 (1904) 139.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
34
2. Ingrandimento: va determinato il fattore di ingrandimento Itot sullo schermo del televisore Itot= ImicrosxIcamera. Icamera è l’ingrandimento introdotto dalla telecamera + televisore. A tal fine mettere il reticolo di passo noto sul piano del microscopio al posto del vetrino; quando l’immagine del reticolo è a fuoco sullo schermo (aggiustare contrasto e luminosità del televisore) misurare con il righello la distanza xosservata sullo schermo tra due (meglio n e poi dividere) solchi contigui del reticolo: Itot= ImicrosxIcamera = xosservata/p=con p=0,01 mm. Esempio di misura: xosservata = (1,3 ± 0,1) cm = (13 ± 1) mm; Itot= 13/0,01 = 1300 (±100). 3. Determinazione sperimentale del la costante di Avogadro
Con riferimento alla formula di Einstein: NA =t
x2
RT
3 r, t è l’intervallo tra una
osservazione e l’altra (per es. t= (10,0 ± 0,1) s), T (K), temperatura della soluzione browniana (T ambiente) va misurata, x
2 è lo spostamento quadratico medio lungo una direzione e va determinato, R= 8,31 J/molK; a 24°C, coefficiente di viscosità dell’acqua= 0,91 10-3 Ns/m2, r=d/2 è il raggio delle sferette di latex. O si misura NA oppure la costante di Boltzmann k=R/NA. Per determinare x
2 si possono costruire le ‘traiettorie browniane’ (non sono traiettorie reali ma solo la proiezione su un piano orizzontale delle posizioni delle pb, ricordarsi che queste subiscono 1020 urti/s); con la soluzione browniana bene a fuoco, fissare sullo schermo del televisore un foglio di acetato, scegliere una pb ‘buona’ e segnare sul foglio le posizioni occupate dalla particella ogni (per es.) 10 secondi numerandole in sequenza (“traiettoria 1”). Ripetere per altre pb (“traiettoria 2”, 3, ecc.). Rimosso il foglio di acetato riportare la ‘traiettoria’ su carta millimetrata e riprodurre la spezzata rilevando le proiezioni degli spostamenti su un asse, per es. x.
1
2
34
5
6
78
9
10
Traiettoria 1
x
0 Costruire la tabella per ogni traiettoria: indice x
(mm)
x
(mm) x
2
(mm2)
0 1 4 4 16 2 -1 -5 25 3 -2 -1 1 ... ... ... ... ... ... ... Nota: poiché i cammini sono casuali si possono utilizzare anche le proiezioni degli spostamenti lungo y mescolandole alle x (se il moto è browniano le direzioni x e y sono equiprobabili). Per determinare x
2 il modo più corretto sarebbe quello di fare un fit dell’istogramma delle proiezioni degli spostamenti secondo una distribuzione normale stimando la varianza; il metodo è corretto per ‘molti’ dati ma le traiettorie sono tracciabili per pochi punti perché la pb esce dal campo visuale o cambia piano di focalizzazione.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
35
Discutere il modo più sensato per risalire a x2 con Origin (per es. con lo stimatore
della varianza della distribuzione della variabile aleatoria x; con la media campionaria dei quadrati degli spostamenti; si possono inoltre riportare gli spostamenti attorno a 0 e fare il fit con la gaussiana; riportare x
2 vs t e verificare che la relazione è lineare); determinare dai vari x
2 , uno per ogni traiettoria, la media; il valore finale di x
2 va poi diviso per il fattore di ingrandimento Itot al quadrato; valutare e trattare gli errori di misura e alla fine determinare la costante di Avogadro.
Bibliografia R. Maiocchi, La belle epoque dell’atomo, F. Angeli ed., Milano, 1987. B. B. Mandelbrot, Gli oggetti frattali (1975),ed. it. Einaudi,Torino,1987. M. J. Nye, Molecular reality, Macdonald, London,1972. J. Stachel, L’anno memorabile di Einstein, Dedalo, Bari, 2001. A. Einstein, Investigations on the theory of the Brownian Movement, a cura di R. Fürth, Dover Pub.,1956. J. Perrin, Les preuves de la réalité moléculaire, in Les idées modernes sur la constitution de la matière, Gauthier Villars, Paris,1913, pp. 1-53. J. Perrin, Les atomes, Paris, 1913; ed. it. Gli atomi, Editori Riuniti, Roma,1981. J. Perrin, Oeuvres scientifiques de J. Perrin, CNRS, Paris, 1950. Per un suggerimento di schema di lezione sull’argomento, si può vedere per es. A. Carrelli, Le prove sperimentali della teoria cinetica dei gas, Giornale di fisica, 3, 1962, 19-34; p. 24 e seg.
CAP. 10. LA NASCITA DELLA SPETTROSCOPIA
Gli spettri sono come le impronte digitali delle sostanze… ogni elemento chimico ha il suo spettro
§10.1. Cronologia sintetica 1665, classica esperienza di Newton sulla dispersione della luce bianca da prisma. 1752: Lo scienziato scozzese Thomas Melville produce “spettri artificiali”, cioè fiamme colorate. Si riconosce gradualmente che la luce emessa è caratteristica di ciascun elemento. 1800, F. W. Herschel scopre l’infrarosso. Come fa? Invia lo spettro solare sul bulbo di un termometro e osserva le variazioni di temperatura in funzione delle varie componenti. Anche al di là del rosso, dove non si hanno radiazioni visibili, si ha variazione di temperatura. J. W. Ritter scopre l’ultravioletto per via chimica, da riduzione di sali d’argento. La scoperta di Ritter è un caso interessante di influenza del contesto filosofico: Naturphilosophie in Germania, idea delle polarità opposte in natura (elettricità positiva e negativa, polarità magnetiche opposte, luce e ombra, ecc.) e, dunque, se esiste l’IR deve esistere anche l’UV. 1814, J. Fraunhofer scopre oltre 500 righe scure nello spettro visibile del Sole; combina spettroscopio + cannocchiale e osserva la doppia riga scura nello spettro del Sole (che si scoprirà essere identica al doppietto giallo del sodio). Anche la luce delle

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
36
stelle mostra uno spettro con righe scure. Osserva gli spettri delle fiamme di alcune sostanze e dell’arco voltaico. 1832, D. Brewster scopre gli spettri di assorbimento. 1840, Becquerel, prima fotografia dello spettro solare. 1858, Plücker osserva gli spettri nei gas (scarica elettrica nei gas rarefatti). 1859, G. R. Kirchhoff: il doppietto giallo del sodio (da prove alla fiamma) coincide con quello osservato nel Sole. Enuncia la legge secondo cui una fiamma “assorbe” i raggi che essa emette. 1860, Kirchhoff e Bunsen: nascita della spettroscopia. Analisi chimica fondata sull’osservazione dello spettro delle fiamme; come sorgente usano la lampada a gas di Bunsen (fornisce una fiamma poco luminosa e di elevata temperatura al cui interno pongono un filo di platino avvolto ad anello dove prelevano il campione del materiale da esaminare Esperimento); ogni sostanza ha un suo spettro caratteristico di emissione e di assorbimento. L’idrogeno mostra (nel visibile) lo spettro di righe più semplice: poche frequenze distanziate regolarmente. 1885, J.J. Balmer, professore svizzero: la successione delle righe (allora note) dell’idrogeno nel visibile obbedisce a regole precise. 1890, J. Rydberg, spettroscopista svedese: studia lo spettro dell’idrogeno; riassume nella stessa formula empirica la serie di Lyman nell’UV, di Balmer nel visibile, altre serie nell’IR. All’atomo di Bohr: 1913. Nuovo corso alla spettroscopia. Lo spettro dell’idrogeno Lo spettro dell’idrogeno è stato osservato nel corso di anni; la riga più intensa viene rivelata da Ångstrom nel 1853. Nei due decenni successivi vengono rivelate altre tre righe e nel 1891 W. Huggins trova ulteriori righe negli spettri stellari. Balmer nel 1885 scopre che la successione delle righe dell’idrogeno nel visibile obbedisce a regole precise che riassume come:
= Bn2
n2 4, =
1=
1
B
n2 4
n2
=
4
B
1
4
1
n2
= a
1
41
n2
[1] , con B e a
costanti e n=3, 4, 5, 6. Formula di Balmer (formula empirica) in notazione moderna:
=1
= R1
22
1
n2
[1’] con n=3, 4, 5, 6.
Nel 1889 Rydberg generalizza la formula empirica per l’H ad altre serie e ad altri atomi (in particolare alcalini): 1
=R [1/(m +b)2-1/(n+c)2] [2], con R “costante di Rydberg”, b e c costanti,
m=1, 2, 3,…, n=m+1, m+2,…; per b, c=0 si ritrova la formula di Balmer [1]. Formula di Rydberg in notazione moderna:
=1
= RH
1
n f2
1
ni2
[2’] dove si è posto m= nf= 1, 2, 3,.. ; n=ni= nf +1, nf +2,..
nf =1; ni=2, 3, .. serie di Lyman nell’UV; nf =3; ni=4, 5, 6.. serie di Paschen nell’IR; nf =4; ni=5, 6, 7.. serie di Brackett (IR); nf =5; ni=6, 7, 8.. serie di Pfund (IR). R = 3,2899 1015s-1. La costante di Rydberg viene trovata sperimentalmente; il valore è un po’ più piccolo del valore teorico (Rteorico=109735,7 cm-1; Rsper=109677,6 cm-1). Prima del modello

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
37
atomico di Bohr i dati spettroscopici sono impenetrabili; lo spettro dell’atomo di H, il più semplice degli spettri atomici, sarà “la stele di Rosetta della fisica moderna”. La costante di Rydberg si troverà contenere le costanti universali e, m ed h. Rilevanza della formula di Balmer: è evidente quando si introduce il modello quantizzato di Bohr. Prima di Bohr: modello atomico “a panettone” di J.J. Thomson, sfera carica positiva con cariche negative oscillanti intorno a certe posizioni di equilibrio, r 10-10m;
da esperimenti fatti da Geiger e Marsden per Rutherford, con particelle che urtano contro atomi d’oro, si vede che la maggior parte delle attraversa il foglio e viene rivelata mentre qualche viene diffusa a grandi angoli. Gli elettroni presenti negli atomi di Au non influenzano sensibilmente le (mel<<m , con m 7300 mel, q = 2qel, le sono nuclei di 4He): la deflessione è causata da nuclei di Au.
Il modello a panettone non si accorda con questi risultati e viene sostituito dal modello dell’atomo nucleare di Rutherford: la carica positiva è concentrata nel nucleo (r 10-15 m), gli elettroni orbitano intorno, r 10-10m.
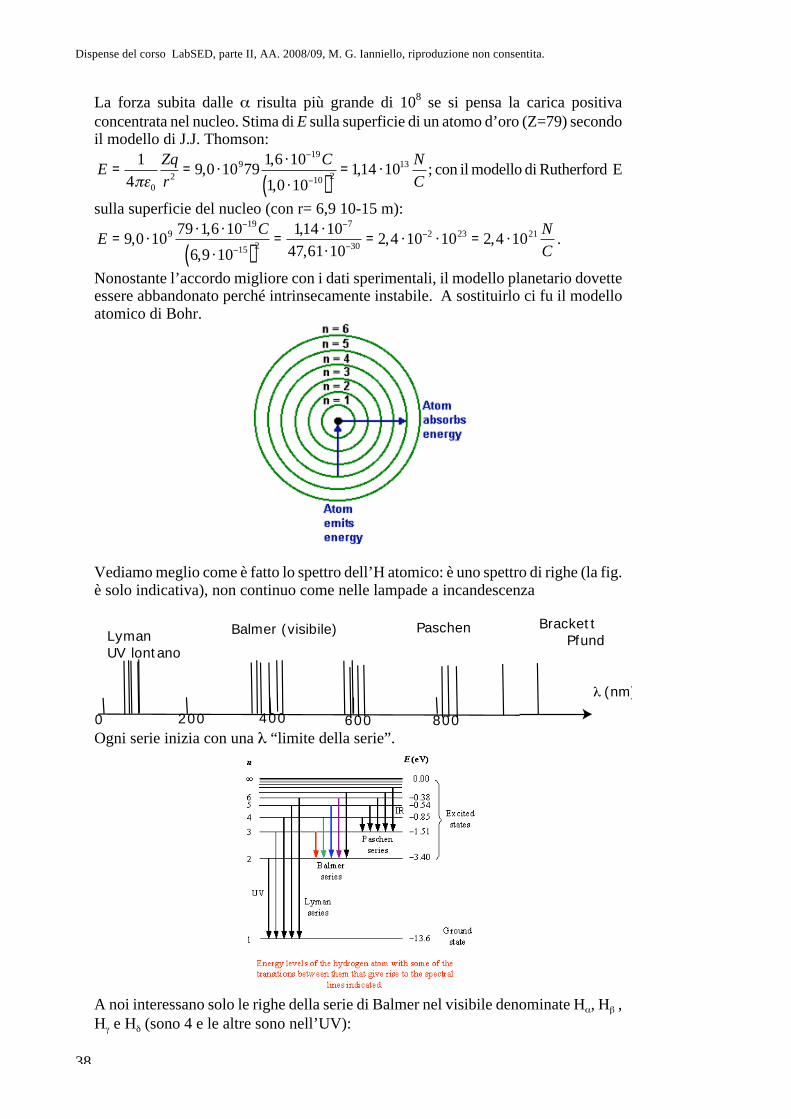
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
38
La forza subita dalle risulta più grande di 108 se si pensa la carica positiva concentrata nel nucleo. Stima di E sulla superficie di un atomo d’oro (Z=79) secondo il modello di J.J. Thomson:
E =1
4 0
Zq
r2 = 9,0 109791,6 10 19C
1,0 10 10( )2 =1,14 1013 N
C; con il modello di Rutherford E
sulla superficie del nucleo (con r= 6,9 10-15 m):
E = 9,0 109 79 1,6 10 19C
6,9 10 15( )2 =
1,14 10 7
47,61 10 30 = 2,4 10 2 1023= 2,4 1021 N
C.
Nonostante l’accordo migliore con i dati sperimentali, il modello planetario dovette essere abbandonato perché intrinsecamente instabile. A sostituirlo ci fu il modello atomico di Bohr.
Vediamo meglio come è fatto lo spettro dell’H atomico: è uno spettro di righe (la fig. è solo indicativa), non continuo come nelle lampade a incandescenza
2000 400 600 800
Lyman
UV lontano
Balmer (visibile) Paschen Brackett
Pfund
(nm)
Ogni serie inizia con una “limite della serie”.
A noi interessano solo le righe della serie di Balmer nel visibile denominate H , H , H e H (sono 4 e le altre sono nell’UV):

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
39
n riga ((nm) colore 3 H 656,28 Rosso 4 H 486,13 Turchese 5 H 434,05 Blu 6 H 410,17 violetto
Come osservare le righe di Balmer nel visibile: ci vuole la lampada di Balmer, o “a idrogeno”; uno strumento per fare osservazioni (spettroscopio a prisma oppure reticolo di Rowland; se si ha uno spettroscopio fisso). Le osservazioni possono essere ‘individuali’ oppure ‘oggettive’. Spettroscopio: che cos’è? Qualsiasi strumento in grado di rilevare lo spettro di assorbimento e di emissione di varie ‘sorgenti’ di ‘luce’ (= radiazione). Ha bisogno di un dispersore (prisma, reticolo, ecc.). Ne esiste una ricca tipologia; il più antico è lo spettroscopio a prisma: è basato sulla dispersione della luce, n=n( ); è buono per il visibile (è di vetro); con opportuni accorgimenti consente di fare osservazioni anche nell’IR (è di cloruro di sodio che non assorbe, come il vetro, l’IR); per es. il satellite IRAS, per le osservazioni astronomiche nell’IR, monta un prisma di NaCl. Altri tipi: spettroscopi interferometrici: le bande di interferenza dipendono dalla lunghezza d’onda della radiazione; vanno bene per spettri da visibile a millimetriche. Spettroscopi con reticoli diffrattivi: come sopra; buoni per visibile, UV e X. Spettrometri di Bragg: basati sull’uso di particolari cristalli come dispersori; buoni per gli X. Come funziona? Gli elementi base sono: sorgente, dispersore e selettore di radiazione. Prisma: disperde per rifrazione la radiazione incidente; l’angolo di rifrazione è funzione di (potere risolutivo cromatico R= , rapporto della lunghezza d’onda
rispetto alla minima differenza di lunghezza d’onda che può essere risolta) Reticolo: disperde per diffrazione la radiazione incidente (vantaggio: la disperde in modo lineare, cioè il potere risolutivo R è costante in tutto il campo spettrale); ricordare: R (= / =mN, minima differenza di lunghezza d’onda che può risolvere un reticolo con N linee e dell’ordine m) è funzione del numero di solchi del reticolo (più è alto il numero di solchi per unità di lunghezza più R è alto; per es. nell’UV e nel visibile 1200-1400 linee/mm con R=104-105). Il nostro reticolo (reticolo di Rowland, introdotto da Rowland intorno al 1875) ha circa 600 linee/mm.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
40
Lampada di Balmer (Leybold, 45113) La individuazione delle righe dell’H è difficile a causa della difficoltà di ottenere idrogeno atomico puro; l’idrogeno gassoso normale è infatti idrogeno molecolare. Se tentiamo di ottenere idrogeno atomico con prove alla fiamma non si ha alcun esito perché le energie in gioco non sono sufficienti a scindere le molecole. Serve pertanto un tubo a scarica dove la tensione applicata accelera gli elettroni che scindono le molecole. La lampada di Balmer è appunto costituita da un tubo a scarica contenente vapor acqueo (molecole di H2O) a bassa pressione alimentato in CA (alimentatore 45114). Quando passa la scarica elettrica la molecola di H2O si scinde in H2O OH+H, con OH gruppo ossidrilico e H idrogeno atomico (non H+ che non dà spettro). La lampada ha un sistema di riciclo dell’acqua grazie alla presenza nel tubo di sostanze ossidanti associate a opportuni catalizzatori. I depositi giallo-bruni sul vetro derivano dagli ossidi metallici; sono brutti a vedersi ma non comportano problemi. Dopo il funzionamento si riottengono molecole di H2O. La parte centrale del tubo si riduce di diametro; il ‘capillare’ che così si ottiene serve a confinare in pratica solo idrogeno atomico e a osservare quindi lo spettro di righe di H senza le bande molecolari di H2 ed eventualmente di O2. Possono essere osservati anche effetti spuri causati dalla presenza di sodio nel vetro, crux dei vecchi spettroscopisti (mettere a bruciare vetro sulla fiamma di un becco Bunsen e osservare la fiamma con lo spettroscopio a prisma; si vede il doppietto giallo del sodio che però con il nostro spettroscopio si risolve a fatica). La lampada va scaldata per 20-30 minuti. Layout sperimentale Materiali: piccolo banco ottico, lampada di Balmer, alimentatore, fenditura40, lente convergente da 5 cm, da 10 cm, reticolo di Rowland, spettroscopio a prisma, schermo semitrasparente. Per l’osservazione individuale si usa lo spettroscopio a prisma, per quella oggettiva il reticolo di Rowland (Leybold, 47123; costante reticolare 6000|||/cm, passo d =1/6000 cm) e lo schermo semitrasparente.
lampada
di Balmer
f=5 cm f= 10 cm
fenditura reticolo
schermo
traslucido
O
In ambiente oscurato: mettere a fuoco la lampada di Balmer sulla fenditura muovendo la lente da 5; mettere a fuoco la luce proveniente dalla fenditura sullo schermo muovendo la lente da 10. Inserire il reticolo fino a che sullo schermo si ottengono ben visibili vari massimi di diffrazione (in partic. del primo ordine).
40 Provare con la fenditura fissa; quella regolabile diffrange troppo sui bordi e provoca una forte riduzione di intensità della luce.
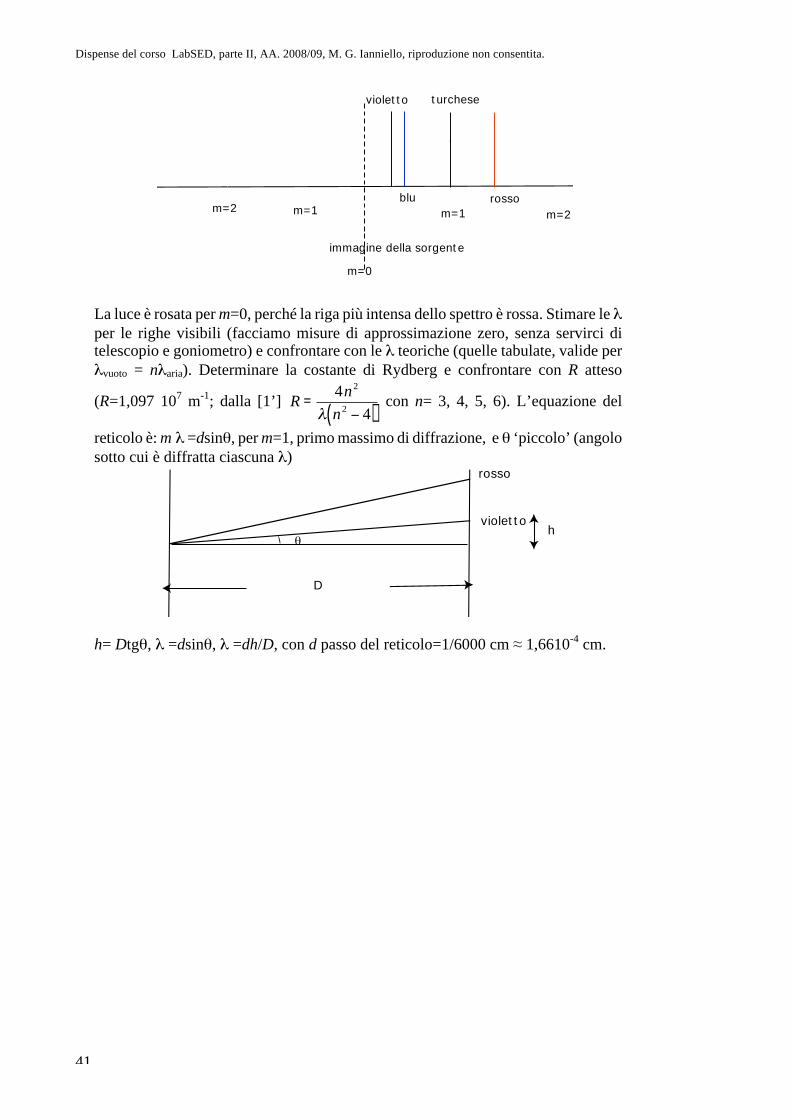
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
41
m=0
m=1 m=2m=1m=2
violetto
blu
turchese
rosso
immagine della sorgente
La luce è rosata per m=0, perché la riga più intensa dello spettro è rossa. Stimare le per le righe visibili (facciamo misure di approssimazione zero, senza servirci di telescopio e goniometro) e confrontare con le teoriche (quelle tabulate, valide per
vuoto = n aria). Determinare la costante di Rydberg e confrontare con R atteso
(R=1,097 107 m-1; dalla [1’] R =4n2
n2 4( ) con n= 3, 4, 5, 6). L’equazione del
reticolo è: m =dsin , per m=1, primo massimo di diffrazione, e ‘piccolo’ (angolo sotto cui è diffratta ciascuna )
D
rosso
violettoh
h= Dtg , =dsin , =dh/D, con d passo del reticolo=1/6000 cm 1,6610-4 cm.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
42
§10.2. Verifica sperimentale della formula di Balmer e determinazione della costante di Rydberg R Come funziona la lampada di Balmer (o “a idrogeno”)? Descrivi lo spettro dell’idrogeno osservato con lo spettroscopio a prisma. Oltre alle righe dell’H, sono osservabili altre righe o bande? Se sì, perché? Nel montaggio dell’esperimento per osservazioni ‘oggettive’, che righe osservi sullo schermo? A partire dalla relazione per i massimi di diffrazione (di ordine m= 0, 1, 2, ):
m = dsin [2] con d passo del reticolo e angolo tra asse ottico e componente monocromatica corrispondente a una riga, misurare le lunghezze d’onda di ciascuna riga dello spettro visibile dell’idrogeno e confrontarla con il valore teorico; determinare R per ciascuna riga e trovarne il valor medio (Ratteso 1,09737 107 m-1). Oppure: riportare su grafico 1/ vs. 1/n2 e dedurre R dalla pendenza della retta). La formula di Balmer predice le per l’idrogeno ma non ne chiarisce l’origine. Come si spiega la formula alla luce della teoria di Bohr del 1913? Che cosa spiega la teoria e quali sono i suoi limiti? Giustificare alla luce della teoria perché la costante di Rydberg contiene le costanti universali e, m e h. Tracciare il diagramma dei livelli di energia per la serie di Balmer nel visibile. Schede Leybold, Fisica atomica e nucleare, La serie di Balmer dell’idrogeno, P6.2.1. J.W. Hänsch, A.L. Schawlow, G.W. Series, Lo spettro dell’idrogeno, in Atomi e nuclei, Le Scienze, Quaderni, 43, 1988. www.fisica.unimo.it/Pdr/Calandra/trattenimento06.pdf www.owlnet.rice.edu/ www.polaris.phys.ualberta.ca/info/Phys29 www.colorado.edu/physics/phys2020 www.pc.chemie.uni-siegen.de/pci/versuche/english/v27-2.html

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
43
CAP. 11. ALLE RADICI DELLA LEGGE DEL CORPO NERO DI PLANCK
§11.1. Le leggi della radiazione termica Quando si studia il comportamento della luce o, più in generale, di un fascio di radiazione elettromagnetica, sappiamo come descriverlo: la ‘luce’ riflette, rifrange, si polarizza, interferisce, ecc. Che si tratti di raggi X, di radiazione UV, di luce visibile, di radiazione IR o di onde radio, varia la lunghezza d’onda della radiazione ma il suo comportamento fisico non cambia. La teoria ondulatoria, nell’ambito dell’elettromagnetismo classico, funziona benissimo e spiega questa classe di fenomeni. Le cose si complicano se siamo in presenza di processi atomici e in particolare di interazione radiazione materia. La spiegazione classica non è più valida e bisogna ricorrere all’ipotesi che la ‘luce’ si comporti come un fascio di particelle; un esempio in merito è rappresentato dal problema della radiazione nera (o in equilibrio termico all’interno di una cavità le cui pareti siano a temperatura T; il problema è detto anche “della radiazione da cavità” o “radiazione di corpo nero”). La spiegazione teorica della radiazione da cavità ha richiesto l’introduzione del quanto d’azione h (h non viene infatti introdotto in connessione con le proprietà dell’atomo ma con la legge di radiazione di corpo nero). La “costante mancante” h, insieme alla quantizzazione dell’energia, viene per la prima volta introdotta da Planck nel dicembre 1900 (ricompare poi con la trattazione di Einstein dell’effetto fotoelettrico del 1905 e dei calori specifici nel 1907 e, ancora, con la trattazione delle proprietà dei gas nel 1912; Bohr introduce h nelle condizioni di quantizzazione dell’atomo nel 1913). Un discorso interessante viene fatto nella Fisica di Berkeley, vol. 4 (“Fisica quantistica”), dove si introduce la costante universale h focalizzando l’attenzione su: 1. problema del corpo nero; 2. effetto fotoelettrico; 3. stabilità e dimensioni degli atomi, che rappresentano altrettanti dilemmi irrisolvibili in fisica classica. I tre problemi vengono affrontati come tre aspetti fondamentali del “Mistero della costante mancante” (cioè h). L’argomento che trattiamo è abbastanza spinoso: la fisica della radiazione non viene quasi mai affrontata, la terminologia che la descrive è fluida (le grandezze in gioco non hanno denominazioni omogenee e cambiano da manuale a manuale: emissività, emittanza, radianza, potere di emissione, ecc.. Più che alla terminologia conviene fare riferimento alle unità di misura indipendentemente dai nomi); gli esperimenti sul tema vengono raramente condotti. Noi affronteremo la verifica della legge di Stefan-Boltzmann e delle leggi di Kirchhoff. La formulazione delle leggi della radiazione termica ha una lunga storia che dura circa cinquant’anni ed è legata strettamente alle tecniche sperimentali di rilevazione delle temperature nell’infrarosso. Le leggi di Kirchhoff, di Stefan, dello spostamento di Wien e i vari tentativi di definire una curva per la distribuzione spettrale dell’energia irradiata si basano in parte su considerazioni teoriche, prevalentemente di natura termodinamica, in parte sui dati sperimentali (sono dunque all’inizio per lo più leggi semiempiriche). La formula di Planck del 1900 viene trovata inizialmente per interpolazione della legge di radiazione di Wien con i dati sperimentali che solo alla fine degli anni Novanta dell’Ottocento si rendono disponibili e in un secondo momento viene reinterpretata teoricamente da Planck sulla base del suo modello a oscillatori. La ‘superformula’ di Planck è in grado comunque di restituire come casi limite tutte le leggi della radiazione termica fino allora trovate. Inseguire con un minimo di fedeltà la storia di queste vicende, e in particolare il ragionamento planckiano, è molto complesso e rimandiamo per approfondimenti al
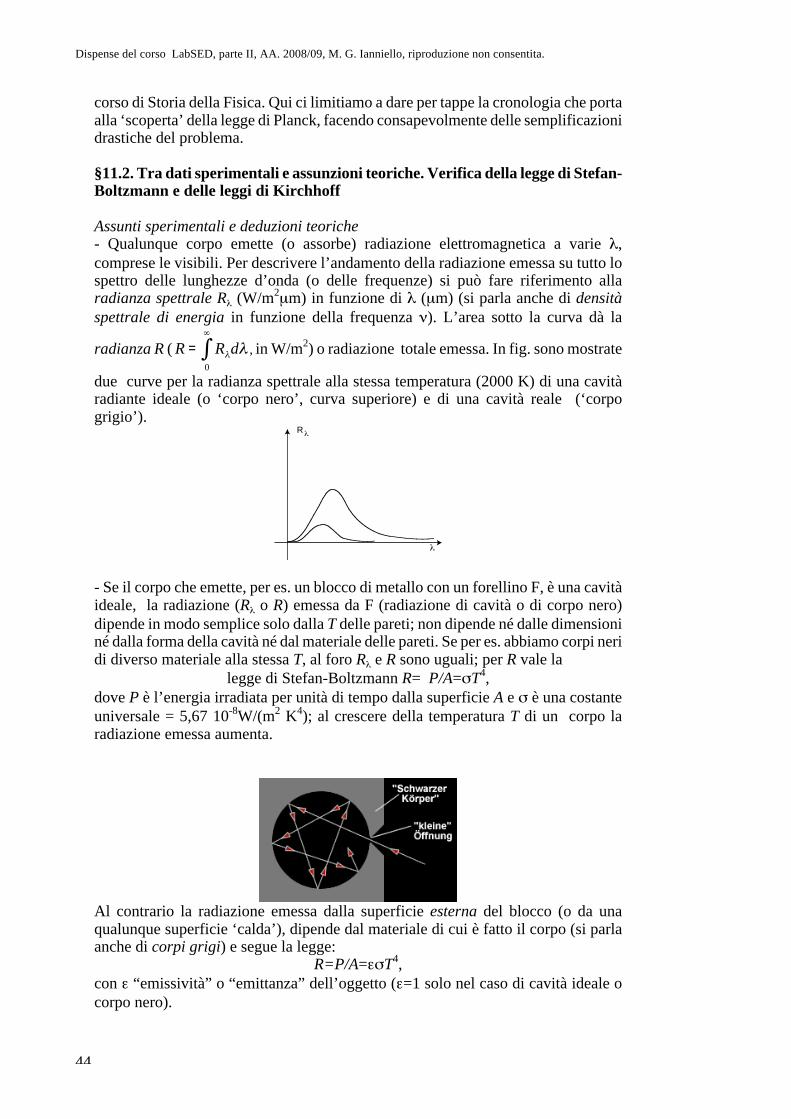
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
44
corso di Storia della Fisica. Qui ci limitiamo a dare per tappe la cronologia che porta alla ‘scoperta’ della legge di Planck, facendo consapevolmente delle semplificazioni drastiche del problema. §11.2. Tra dati sperimentali e assunzioni teoriche. Verifica della legge di Stefan-Boltzmann e delle leggi di Kirchhoff Assunti sperimentali e deduzioni teoriche - Qualunque corpo emette (o assorbe) radiazione elettromagnetica a varie , comprese le visibili. Per descrivere l’andamento della radiazione emessa su tutto lo spettro delle lunghezze d’onda (o delle frequenze) si può fare riferimento alla radianza spettrale R (W/m2μm) in funzione di (μm) (si parla anche di densità
spettrale di energia in funzione della frequenza ). L’area sotto la curva dà la
radianza R ( R = R d0
, in W/m2) o radiazione totale emessa. In fig. sono mostrate
due curve per la radianza spettrale alla stessa temperatura (2000 K) di una cavità radiante ideale (o ‘corpo nero’, curva superiore) e di una cavità reale (‘corpo grigio’).
R
- Se il corpo che emette, per es. un blocco di metallo con un forellino F, è una cavità ideale, la radiazione (R o R) emessa da F (radiazione di cavità o di corpo nero) dipende in modo semplice solo dalla T delle pareti; non dipende né dalle dimensioni né dalla forma della cavità né dal materiale delle pareti. Se per es. abbiamo corpi neri di diverso materiale alla stessa T, al foro R e R sono uguali; per R vale la
legge di Stefan-Boltzmann R= P/A= T4,
dove P è l’energia irradiata per unità di tempo dalla superficie A e è una costante universale = 5,67 10-8W/(m2 K4); al crescere della temperatura T di un corpo la radiazione emessa aumenta.
Al contrario la radiazione emessa dalla superficie esterna del blocco (o da una qualunque superficie ‘calda’), dipende dal materiale di cui è fatto il corpo (si parla anche di corpi grigi) e segue la legge:
R=P/A= T4,
con “emissività” o “emittanza” dell’oggetto ( =1 solo nel caso di cavità ideale o corpo nero).
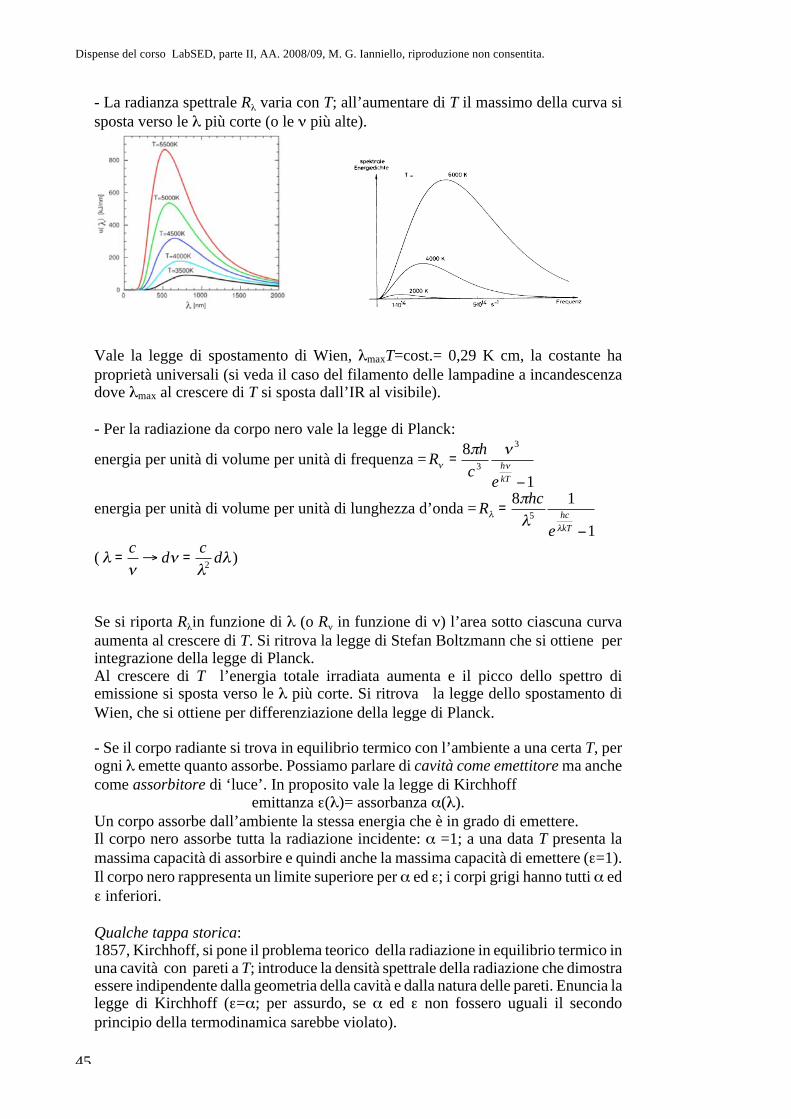
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
45
- La radianza spettrale R varia con T; all’aumentare di T il massimo della curva si sposta verso le più corte (o le più alte).
Vale la legge di spostamento di Wien, maxT=cost.= 0,29 K cm, la costante ha proprietà universali (si veda il caso del filamento delle lampadine a incandescenza dove max al crescere di T si sposta dall’IR al visibile). - Per la radiazione da corpo nero vale la legge di Planck:
energia per unità di volume per unità di frequenza = R =8 h
c 3
3
eh
kT 1
energia per unità di volume per unità di lunghezza d’onda = R =8 hc
5
1
ehc
kT 1
( =c
d =c
2 d )
Se si riporta R in funzione di (o R in funzione di ) l’area sotto ciascuna curva aumenta al crescere di T. Si ritrova la legge di Stefan Boltzmann che si ottiene per integrazione della legge di Planck. Al crescere di T l’energia totale irradiata aumenta e il picco dello spettro di emissione si sposta verso le più corte. Si ritrova la legge dello spostamento di Wien, che si ottiene per differenziazione della legge di Planck. - Se il corpo radiante si trova in equilibrio termico con l’ambiente a una certa T, per ogni emette quanto assorbe. Possiamo parlare di cavità come emettitore ma anche come assorbitore di ‘luce’. In proposito vale la legge di Kirchhoff
emittanza ( )= assorbanza ( ). Un corpo assorbe dall’ambiente la stessa energia che è in grado di emettere. Il corpo nero assorbe tutta la radiazione incidente: =1; a una data T presenta la massima capacità di assorbire e quindi anche la massima capacità di emettere ( =1). Il corpo nero rappresenta un limite superiore per ed ; i corpi grigi hanno tutti ed inferiori.
Qualche tappa storica: 1857, Kirchhoff, si pone il problema teorico della radiazione in equilibrio termico in una cavità con pareti a T; introduce la densità spettrale della radiazione che dimostra essere indipendente dalla geometria della cavità e dalla natura delle pareti. Enuncia la legge di Kirchhoff ( = ; per assurdo, se ed non fossero uguali il secondo principio della termodinamica sarebbe violato).

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
46
1865, Tyndall misura l’intensità totale della radiazione emessa da un filamento di platino annerito, a 5 diverse temperature (corpi anneriti non cavità); 1879, dai dati di Tyndall, Stefan ricava per interpolazione la legge empirica R= T
4. 1884, Boltzmann deduce teoricamente (per via termodinamica) la legge di Stefan. Fine Ottocento: il problema del controllo della T negli altiforni delle industrie siderurgiche tedesche si fa pressante. Nel 1892 H. Rubens realizza un Laboratorio per le radiazioni IR al Politecnico di Berlin-Charlottenburg. Migliorano le tecniche sperimentali per rilevare alte temperature. 1893, Wien ricava teoricamente (per via termodinamica) la legge dello spostamento; tre anni dopo, con ipotesi ad hoc, ricava la legge semiempirica (scritta in notazioni moderne) che per < 5 μm fitta bene i dati sperimentali:
(va bene per corte ma per >5 μm devia sempre più dai dati sperimentali (per altro, all’epoca, gli unici ‘disponibili’); ovviamente Wien non scrive h che è legata alle costanti termodinamiche =h/k=hNA/R, con k costante di Boltzmann e R costante universale dei gas).
1898-1899: Raccolta massiccia di dati sperimentali (Lummer, Pringsheim, Paschen, Rubens, Kurlbaum); si realizzano le prime cavità in materiale ceramico termostatato. La legge di Stefan-Boltzmann viene verificata per la prima volta in modo stringente. Viene studiata la “riflessione selettiva” per alcune frequenze (“metodo dei raggi restanti”). Rayleigh-Jeans (primi del Novecento), da principio di equipartizione dell’energia:
R =8 kT
4
(funziona bene solo per molto lunghe , per 0 la curva diverge, “catastrofe UV” e non ammette un massimo, l’intensità totale irradiata è infinita; la radiazione non può mai essere in equilibrio termico).
R =8 hc
5 ehc
kT
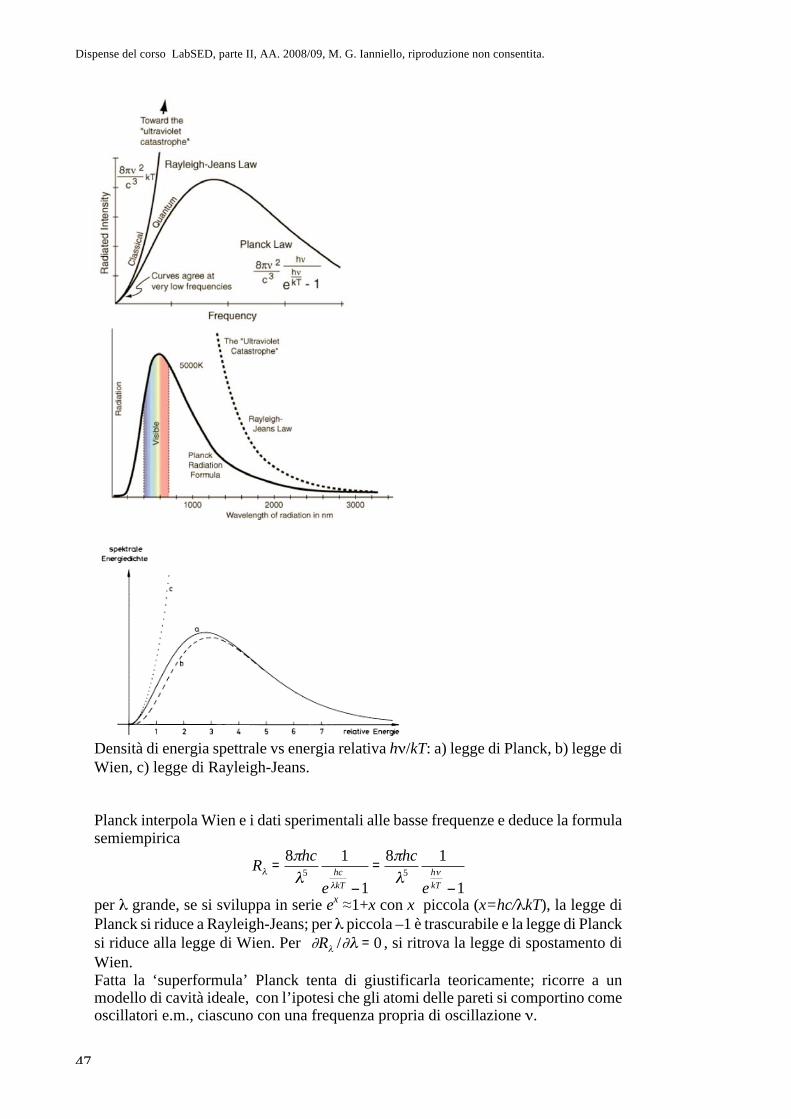
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
47
Densità di energia spettrale vs energia relativa h /kT: a) legge di Planck, b) legge di Wien, c) legge di Rayleigh-Jeans. Planck interpola Wien e i dati sperimentali alle basse frequenze e deduce la formula semiempirica
R =8 hc
5
1
ehc
kT 1
=8 hc
5
1
eh
kT 1
per grande, se si sviluppa in serie ex 1+x con x piccola (x=hc/ kT), la legge di Planck si riduce a Rayleigh-Jeans; per piccola –1 è trascurabile e la legge di Planck si riduce alla legge di Wien. Per R / = 0 , si ritrova la legge di spostamento di Wien. Fatta la ‘superformula’ Planck tenta di giustificarla teoricamente; ricorre a un modello di cavità ideale, con l’ipotesi che gli atomi delle pareti si comportino come oscillatori e.m., ciascuno con una frequenza propria di oscillazione .

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
48
Modello a risonatori di Planck (quello presentato sui manuali corrisponde al modello formulato da Peierls e quindi di circa 20 anni dopo): modello di cavità a pareti perfettamente riflettenti, costituite da risonatori (un risonatore può essere modellizzato con una molla priva di massa con una carica elettrica all’estremo; K molla corrisponde a propria di oscillazione; esiste una infinità di K e quindi una infinità di frequenze proprie di oscillazione. I risonatori sono indipendenti, vale cioè l’ipotesi del caos molecolare, ecc.; per questo Planck può ricorrere a considerazioni statistiche alla Boltzmann). Se arriva calore dall’esterno le molle vibrano, le cariche sono accelerate e irradiano energia nella cavità (=assorbono i colori, cioè le che corrispondono alle loro frequenze di risonanza). All’equilibrio i risonatori di una data assorbono dalla radiazione contenuta nella cavità una quantità pari a quella che emettono. Per far tornare la legge del corpo nero Planck è costretto a fare l’“atto di disperazione” (compromesso e capitolazione) e ammettere che l’energia degli oscillatori è quantizzata; quando un oscillatore passa da uno stato di energia quantizzato a un altro emette un quanto di valore E=h con h = 6,626 10-34 Js. Un oscillatore che permane in uno stato di energia quantizzato non emette né assorbe energia. Agli esperimenti: verifica della legge di Stefan-Boltzmann (Leybold P5.5.2.1) Materiali: forno elettrico 200 W a 220 V, termometro digitale con sensore di temperatura NiCr-Ni, corpo nero e schermo, termopila di Moll e microvoltmetro, banco ottico, supporto, diaframma a iride, cartoncino nero. La radianza (o radiazione totale o potenza totale irradiata dall’unità di superficie, detta anche “intensità di radiazione”) emessa da un corpo nero (CN) segue la legge di
Stefan-Boltzmann: R = T 4 , = 5,67 10 8 W
m2K 4 .
Se il CN si trova in un ambiente a temperatuta T0 (< T del radiatore), assorbe dall’ambiente la radiazione R0= T0
4 con T0 temperatura ambiente. Si considera pertanto la radianza netta R’, pari all’energia per unità di tempo e di superficie sottratta al CN: R'= T 4 T0
4( ) [1].
Montaggio:
Moll
al microvoltmetro
schermo diaframma
forno
al termometro digitale
25 cm
sonda
corpo nero
Il CN è costituito da un cilindretto di ottone, aperto da un lato (foro) e provvisto di un forellino dall’altro per l’inserimento della sonda di temperatura dalla parte posteriore del forno. Inserito il CN nel forno, si fissa a contatto lo schermo; lo schermo è circolare con un foro al centro, uguale al foro del cilindro d’ottone e serve a intercettare solo la radiazione termica del CN e a schermare quella proveniente dalle pareti del forno. Serve inoltre a garantire che la T di riferimento della termopila, T0, resti costante. Disporre la termopila a circa 15 cm dall’apertura del forno oppure disporre il diaframma a metà tra schermo e termopila. Termopila di Moll o pila termoelettrica: serve a convertire la radiazione termica in tensione termoelettrica U. Funziona in base all’effetto Seebeck e dispone di 16

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
49
termoelementi in serie; per i dettagli tecnici vedi Guida. Fino a che la temperatura delle giunzioni dei termoelementi T= T0, T=0 U=0. Non appena T T0, T 0 U 0 con U R’. Microvoltmetro: mettere sotto tensione almeno 10’ prima dell’inizio delle misure; selettore di funzione su V, scegliere il campo di misura adatto (10-4 V). Se il campo di misura non è ‘giusto’ lo strumento emette un fischio e il display non dà indicazioni stabili. V. Guida, in partic. per l’azzeramento della scala mediante il potenziometro di offset. Con l’azzeramento il microvoltmetro considera nulla la radiazione termica ambiente; T0 per convenzione è la temperatuta in corrispondenza della quale la tensione di uscita della termopila è 0. Il forno: dovrebbe scaldare fino a 600 °C ma di fatto non supera 400°C. Il termometro: attenzione che ha due campi di misura, sopra e sotto 200°C (v. Guida). Le misure: fase di riscaldamento; a forno spento misurare T0 con il sensore. Accendere il forno e registrare le misure di U per es. ogni 25°C fino a circa 400°C; riportare in tabella. Spegnere il forno e registrare, nella fase di raffreddamento, U per es. ogni 25°C (cercare di far coincidere i valori di T con quelli registrati nella fase di riscaldamento) fino a una T compresa tra 100 e T0. Alla fine delle misure estrarre il sensore dal CN e misurare T0, schermare la termopila con il cartoncino nero e verificare l’azzeramento del voltmetro. Riportare i valori in tabella incluso T0. Tabella: (°C) T(K) T4-T0
4 (K4) U (μV) U (μV) Graficare U vs T4-T0
4 e verificarne la linearità secondo la [1]. Determinare gli errori di misura, individuare le fonti d’errore e i disturbi ambientali. Si può risalire alla costante dalla pendenza della retta? Perché le U e le U hanno un lieve scarto? Corpi grigi e cubo di Leslie (Leybold P5.5.2.3) Materiali: banco ottico piccolo, cubo di Leslie, supporto, riscaldatore a immersione, recipiente e imbuto, agitatore, termopila di Moll, termometro, microvoltmetro. Verifica della legge di Stefan-Boltzmann per i “corpi grigi” e verifica della legge di Kirchhoff. Nel caso di corpi grigi si ha: R= T
4, con 1, fattore di emissione, varia con la superficie; la radianza netta
è R'= T 4 T04( ) .
Verificare per il cubo di Leslie la linearità tra U e T 4- T40 per ciascuna faccia del
cubo (fare un grafico per faccia bianca, nera, metallica, metallica levigata); il cubo va riempito con acqua calda (bollente), la termopila va posta a circa 10 cm di fronte alla faccia in studio e lasciata nella stessa posizione per tutto il corso delle misure; dopo aver agitato l’acqua misurare T vs U per es. ogni 5°C per ciascuna faccia avendo cura di orientare il sensore del termometro immerso nel cubo, con la punta a contatto con la faccia interna in esame; dopo ogni misura ruotare il cubo di 90° in modo da cambiare faccia; a circa 40°C togliere il sensore, asciugarlo e misurare la temperatura ambiente T0 (schermare Moll con cartone nero, controllare l’azzeramento del voltmetro e riportare V0 in tabella). Tabella: (°C) T(K) T4-T0
4 (K4) U (μV) Discutere i risultati in particolare per le facce nera e bianca. Le pendenze delle rette sono confrontabili con il caso del corpo nero?
CAP. 12. EFFETTO FOTOELETTRICO §12.1. Cronologia essenziale
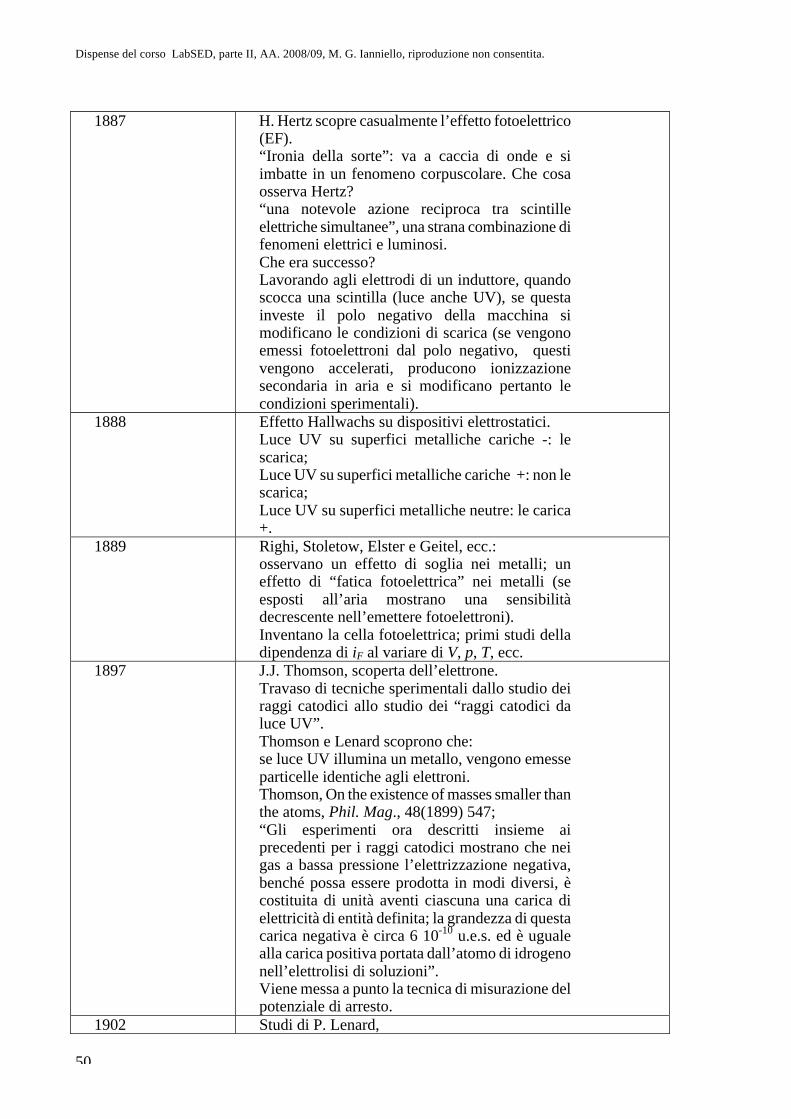
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
50
1887 H. Hertz scopre casualmente l’effetto fotoelettrico
(EF). “Ironia della sorte”: va a caccia di onde e si imbatte in un fenomeno corpuscolare. Che cosa osserva Hertz? “una notevole azione reciproca tra scintille elettriche simultanee”, una strana combinazione di fenomeni elettrici e luminosi. Che era successo? Lavorando agli elettrodi di un induttore, quando scocca una scintilla (luce anche UV), se questa investe il polo negativo della macchina si modificano le condizioni di scarica (se vengono emessi fotoelettroni dal polo negativo, questi vengono accelerati, producono ionizzazione secondaria in aria e si modificano pertanto le condizioni sperimentali).
1888 Effetto Hallwachs su dispositivi elettrostatici. Luce UV su superfici metalliche cariche -: le scarica; Luce UV su superfici metalliche cariche +: non le scarica; Luce UV su superfici metalliche neutre: le carica +.
1889 Righi, Stoletow, Elster e Geitel, ecc.: osservano un effetto di soglia nei metalli; un effetto di “fatica fotoelettrica” nei metalli (se esposti all’aria mostrano una sensibilità decrescente nell’emettere fotoelettroni). Inventano la cella fotoelettrica; primi studi della dipendenza di iF al variare di V, p, T, ecc.
1897 J.J. Thomson, scoperta dell’elettrone. Travaso di tecniche sperimentali dallo studio dei raggi catodici allo studio dei “raggi catodici da luce UV”. Thomson e Lenard scoprono che: se luce UV illumina un metallo, vengono emesse particelle identiche agli elettroni. Thomson, On the existence of masses smaller than the atoms, Phil. Mag., 48(1899) 547; “Gli esperimenti ora descritti insieme ai precedenti per i raggi catodici mostrano che nei gas a bassa pressione l’elettrizzazione negativa, benché possa essere prodotta in modi diversi, è costituita di unità aventi ciascuna una carica di elettricità di entità definita; la grandezza di questa carica negativa è circa 6 10-10 u.e.s. ed è uguale alla carica positiva portata dall’atomo di idrogeno nell’elettrolisi di soluzioni”. Viene messa a punto la tecnica di misurazione del potenziale di arresto.
1902 Studi di P. Lenard,

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
51
Über die lichtelektrische Wirkung, A. d. P., 8 (1902) 149-198. L’intensità I della luce incidente influisce solo sul numero dei fotoelettroni emessi: I ~ ne, ma non sull’energia dei singoli elettroni; il potenziale di arresto dipende solo dalla frequenza della luce incidente V=V( ). Lenard, dalla Nobel lecture (1906): “Ho trovato che la velocità [degli elettroni emessi] è indipendente dalla intensità della luce UV, ed ho pertanto concluso che l’energia all’uscita della placca non proviene affatto dalla luce, ma dall’interno del particolare atomo. La luce ha solo funzione di innesco come quella di una miccia nei confronti di un cannone carico. Trovo che questa conclusione sia importante perché da essa apprendiamo che non solo gli atomi del radio […] contengono riserve di energia, ma anche gli atomi dei diversi elementi”.
1905 Einstein, Über einen die Erzeugung und Verwandlung des Lichtes betreffenden heuristichen Gesichtspunkt, A. d. P., 17(1905) 132. “Di fatto a me sembra che le osservazioni sulla ‘radiazione di corpo nero’, sulla produzione di raggi catodici da parte di luce UV e su altri fenomeni che comportano emissione e conversione di luce, possano essere capiti meglio se si assume che l’energia luminosa sia distribuita discontinuamente nello spazio. Secondo l’ipotesi che qui voglio proporre, quando un raggio di luce si espande partendo da un punto l’energia non si distribuisce su volumi sempre più grandi bensì rimane costituita da un numero finito di quanti di energia localizzati nello spazio e che si muovono senza suddividersi e che possono essere emessi o assorbiti solo come interi”. Einstein, per spiegare la radiazione da corpo nero, ammette che la luce è “come se consistesse di quanti distinti e indipendenti di energia di grandezza (R/N0) [= h ] con = h/k=hN0/R. Einstein estende l’ipotesi anche all’EF studiato da Lenard:
e= (R /N0) -P equazione di Einstein dell’EF; [Ve= h -P, con V potenziale d’arresto, P, lavoro d’estrazione]; “se la formula è corretta P deve essere una funzione lineare della frequenza della luce incidente, la cui pendenza è indipendente dalla natura della sostanza studiata”.
Post 1905
Vari tentativi, da parte di vari autori, di spiegare l’EF in modo “classico” o “semiclassico”.
1916 R.A. Millikan,

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
52
A direct photoelectric determination of Planck’s h, Phys. Rev., 7(1916) 355. Dalla Nobel lecture di Millikan del 1917: “Dopo dieci anni di controlli, cambiamenti, acquisizioni e talvolta cantonate, avendo diretto tutti gli sforzi verso la misura accurata delle energie di emissione dei fotoelettroni _che sembravano a volte funzione della temperatura, a volta delle lunghezze d’onda, ora del materiale (cioè delle f.e.m. di contatto)_ questo lavoro risultò, contrariamente alle aspettative, la prima prova sperimentale del 1914 della esatta validità, entro i limiti di errore sperimentale, della EQUAZIONE DI EINSTEIN e la prima determinazione fotoelettrica diretta della costante di Planck h”.
1925 L’ipotesi dei quanti di luce di Einstein viene finalmente accettata non solo come ipotesi euristica. I quanti di luce vengono denominati “fotoni” nel 1926 da G. Lewis.
§12.2. Effetto Fotoelettrico: fenomenologia e interpretazioni teoriche La fenomenologia Condizioni ottimali Quando luce di opportuna frequenza colpisce una superficie metallica in vuoto spinto vengono emessi elettroni dalla superficie
Luce UV Chimicamente pura, in partic. di metalli alcalini
I fatti sperimentali La teoria ondulatoria non li
spiega! una frequenza di soglia, che
dipende dalla natura del metallo, in corrispondenza della quale si ha EF solo per > 0; per < 0 iF=0 per > 0 iF 0
Se la luce è composta di onde, l’EF si dovrebbe avere per , purché l’intensità della luce incidente sia sufficientemente grande;
v=v( ); (1/2)mv2~ : l’energia
dei fotoelettroni emessi dipende solo da della radiazione incidente ma non dalla sua intensità I.
Secondo la fisica classica, l’energia dei fotoelettroni dipende da I.
L’emissione dei fotoelettroni è istantanea ( t < 10-3s : in realtà è dell’ordine di 10-8) e avviene anche per I debole
Per la TO dovrebbe esserci un tempo di ritardo t tra l’arrivo di una parte del fronte d’onda, l’assorbimento di energia da parte dell’elettrone e la sua

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
53
emissione; per I debole il ritardo dovrebbe essere misurabile.
Il numero dei fotoelettroni emessi dipende da I: ne~I iF~I. Vale l’equazione di Einstein E=h -P
????
Si osservi che l’EF non avviene solo nei metalli: può avvenire anche nei liquidi, nei singoli atomi di un gas, in un semiconduttore, ecc. Inoltre può avvenire anche a frequenze diverse da quelle della luce UV; in generale a grandi non si ha EF; tutte le sostanze hanno comunque una soglia: per es. i metalli alcalini hanno una soglia 0 molto bassa (Li, Na, K, Rb, Cs: l’elettrone di valenza si muove all’esterno di uno strato chiuso ed è poco legato, sicché tali metalli sono più pronti a perdere questo elettrone); altri metalli (come per es. il platino) hanno una soglia 0 alta. La maggior parte dei metalli ha una soglia 0 nella regione UV; altri ce l’hanno nel visibile (come per es. i metalli alcalini). Questo spiega perché nella Cella fotoelettrica di solito il catodo è ricoperto di un metallo alcalino; l’anodo è di platino; la lampada che irradia è al mercurio (emette anche UV) cella con pareti di quarzo. §12.3. Agli esperimenti V. schede esperimenti
CAP. 13. L’ATOMO QUANTIZZATO DI BOHR E L’ESPERIMENTO DI
FRANCK ED HERTZ
§13.1. L’atomo di Bohr Bohr fonda una “nuova meccanica atomica”. Le leggi del moto classiche non valgono più all’interno dell’atomo. Un atomo può esistere solo in determinati stati stazionari discreti, con energie E0, E1, E2, ..; quando l’atomo è eccitato può passare dallo stato fondamentale E0 a uno stato a energia superiore, quindi ‘diseccitarsi’ e tornare allo stato fondamentale. L’atomo può dunque ‘saltare’ (cioè fare transizioni) da uno stato stazionario all’altro. Dati due livelli energetici E2 ed E1 vale la condizione di Bohr: E2-E1=h . Per l’idrogeno Bohr immagina che l’elettrone si muova su orbite circolari con centro nel nucleo e raggio variabile (anch’esso quantizzato). Inoltre postula che il momento angolare dell’elettrone rispetto al nucleo debba essere quantizzato: p= nh/2 , con n intero (detto numero quantico principale). Se l’atomo è a più elettroni servono più numeri quantici; per es. nel modello di Bohr-Sommerfeld le orbite diventano ellittiche con il nucleo in uno dei due fuochi ed è necessario introdurre un secondo numero quantico l (orbitale o azimutale). Per giustificare il fatto che in presenza di un campo magnetico esterno (effetto Zeeman) oppure di un campo elettrico esterno (effetto Stark) i livelli energetici si suddividono occorre introdurre altri 2 numeri quantici (di spin s e il numero quantico magnetico m). La questione dei numeri quantici storicamente è estremamente ostica da capire; vengono proposti vari n. q. che cambiano nel tempo con simboli diversi, che vengono poi ‘rinormalizzati’ più volte fino ad arrivare a 4 numeri quantici.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
54
E
E
E
E
11
0 0
Transizione verso l’alto Transizione verso il basso (assorbimento, viene assorbito un fotone) (emissione, viene emesso un fotone)
Se si hanno più livelli energetici (stati stazionari), per es. n=4, le possibili frequenze di transizione sono in linea teorica n(n-1)/2=6; per es. in emissione:
E
EEE
0
1
2
3
stato fondamentale,n=1
stati eccitati
Per Bohr possono essere assorbite solo quelle righe spettrali che corrispondono a una transizione da uno stato stazionario a uno più elevato; le righe di assorbimento corrispondono pertanto a E1-E0=h 1, E2-E0=h 2, ecc. Analogamente per le righe di emissione vale l’equazione En-Em=h nm. Con la spettroscopia ottica si osservano in linea teorica altrettante righe di emissione. Ma non è detto che si osservino tutte; ci sono infatti “regole di selezione” che non permettono tutte le transizioni possibili. Lo spettro dipende inoltre anche dalla temperatura T: a T ambiente (‘gas freddo’: le energie in gioco sono dell’ordine di kT (1/40) eV) si possono osservare solo certe righe e non altre perché la maggior parte degli atomi si trova allo stato fondamentale E0. Al crescere di T aumenta la probabilità di trovare l’atomo in uno degli stati eccitati secondo la legge esponenziale
Nn
N0
= eEn E0
kT .
Come si eccitano gli atomi di un gas? Aumentando T (eccitazione termica), mediante scarica elettrica, mediante urto con elettroni, ecc. Se gli elettroni-proiettile (di cui si può variare l’ energia E in modo continuo aumentando la tensione anodica) vengono bombardati sugli atomi del gas in modo che
E< E1-E0 urti elastici/ no eccitazione; l’energia cinetica degli elettroni è minore dell’energia di eccitazione al primo stato eccitato;
E> E1-E0 ma E< E2-E0 urti anelastici/ l’atomo passa al primo stato eccitato; E> E2-E0 ma E< E3-E0 l’atomo passa al secondo stato eccitato, ecc.
Gli atomi salgono di livello (si eccitano), poi decadono (ridiscendono di livello in un tempo dell’ordine di 10-8 s) emettendo ‘luce’ di una data frequenza. All’aumentare di E (elettroni-proiettile più energetici), l’atomo può fare transizioni verso livelli sempre più alti fino alla ionizzazione (n ; l’elettrone meno legato dell’atomo viene rimosso dal nucleo e l’atomo diventa, da neutro, uno ione positivo; nel caso dell’elio: He He++e).

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
55
Come visualizzare le transizioni discrete dell’atomo da uno stato stazionario all’altro? Con una curva tensione anodica (=energia degli elettroni urtanti)-corrente IC (di collettore C: l’elettrodo collettore raccoglie gli elettroni che hanno urtato anelasticamente gli atomi del gas e hanno così perso energia; il numero di elettroni raccolti da C è misurato dalla corrente IC, dell’ordine dei pA, rilevata da un nanoamperometro). §13.2. Il percorso di Franck ed Hertz “Sull’eccitazione della riga di risonanza 253,6 mm [2536 Å] del mercurio mediante urti elettronici”, 1914 L’articolo viene considerato come la conferma più brillante e immediata della teoria atomica di Bohr; per questo esperimento e “per la scoperta delle leggi che governano l’urto di un elettrone con un atomo” Franck ed Hertz ricevettero il premio Nobel per la fisica nel 1925. Ma l’articolo in questione non poteva essere la conferma a Bohr? Gli autori all’epoca non conoscono infatti la teoria di Bohr e quando ne vengono a conoscenza non la capiscono almeno fino al 1919; i risultati del 1914 sono sbagliati (i potenziali di eccitazione sono inevitabilmente considerati potenziali di ionizzazione); gli esperimenti del 1914 sono pieni di effetti di disturbo e daranno risultati conclusivi solo intorno al 1924. Vediamo cosa si sapeva all’epoca di Franck ed Hertz. Se elettroni bombardano gli atomi di un gas lo ionizzano (Lenard, Stark, J.J. Thomson); per ionizzare, gli elettroni devono avere una velocità critica, corrispondente a un potenziale critico o tensione di ionizzazione. La teoria di Townsend (1910) spiega inoltre in prima approssimazione i fenomeni di ionizzazione ma a livello microscopico fallisce (per T. gli urti elettrone-atomo sono tutti anelastici). Si tenga presente che all’epoca, qualsiasi fenomeno di luminescenza nel tubo a scarica in cui era contenuto il gas a bassa p veniva considerato ‘ionizzazione’; ancora nulla si sa dello stato eccitato e dei relativi potenziali di eccitazione. F&H partono dal metodo di Lenard “a tre elettrodi”: tra filo caldo F, sorgente degli elettroni, e griglia G veniva applicato il potenziale acceleratore V1 mentre tra elettrodo collettore P e griglia G agiva una tensione di frenamento V2 che impediva agli elettroni primari di raggiungere P. Solo quando l’energia degli elettroni (legata a V1) diventava così grande da ‘ionizzare’ gli atomi del gas, il galvanometro iniziava a rilevare corrente perché in questa situazione V2 spingeva gli ioni positivi verso l’elettrodo collettore e il galvanometro segnava il passaggio di una corrente positiva. O almeno così si credeva all’epoca: in realtà, come si scoprirà in seguito, l’elettrodo collettore poteva caricarsi positivamente anche a causa dei fotoelettroni espulsi dalla sua superficie quando veniva investito dalla radiazione ultravioletta emessa dagli atomi eccitati del gas.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
56
Sulla scia dei lavori di Lenard, Franck ed Hertz modificano la disposizione sperimentale aggiungendo una seconda griglia G2 .
Gli elettroni vengono accelerati da V1 e immessi nello “spazio degli urti” (senza campo), compreso tra le griglie G1 e G2; l’esperimento rappresenta un test per vedere che tipo di urti fanno gli elettroni con gli atomi del gas; se al crescere di V1 si raggiunge un potenziale critico (gli elettroni hanno subito urti anelastici) la griglia G2 fa da filtro e gli elettroni meno energetici vengono ostacolati da V2 e non raggiungono il collettore P. Dalla corrente rilevata, F&H trovano che gli urti prima della ‘ionizzazione’ sono elastici41; migliorando il grado di vuoto nel tubo trovano inoltre che l’assunto di Lenard (potenziale di ionizzazione uguale per tutti i gas) è sbagliato. (Tenere conto che l’elio è l’elemento che ha potenziale di ionizzazione più alto, così come gli altri gas nobili_Ne, A, Kr, Xe, Rn_ hanno potenziali più alti nei vari rispettivi ‘periodi’ della tavola degli elementi). Maggio del 1914: F&H introducono un nuovo metodo di rilevazione dei potenziali di ionizzazione, il metodo dell’urto anelastico tra elettroni e atomi di vapore di mercurio. L’apparato è a simmetria cilindrica, con il filamento di platino F per l’emissione degli elettroni disposto lungo l’asse del cilindro ; il filamento è circondato da una griglia G di platino, attorno alla quale si avvolge una lamina P dello stesso metallo, che funziona da elettrodo collettore, collegata a terra mediante un galvanometro. Tra F e G è applicato un potenziale acceleratore V1, mentre tra G e P può essere prodotto un controcampo. L’apparato sperimentale è inserito in un tubo a vuoto, collegato alla pompa e a un tubo a U che contiene il mercurio da vaporizzare.
41 La massa dell’elettrone collidente è molto più piccola della massa dell’atomo bersaglio; la quantità di moto trasferita dall’elettrone in un urto è pertanto trascurabile.
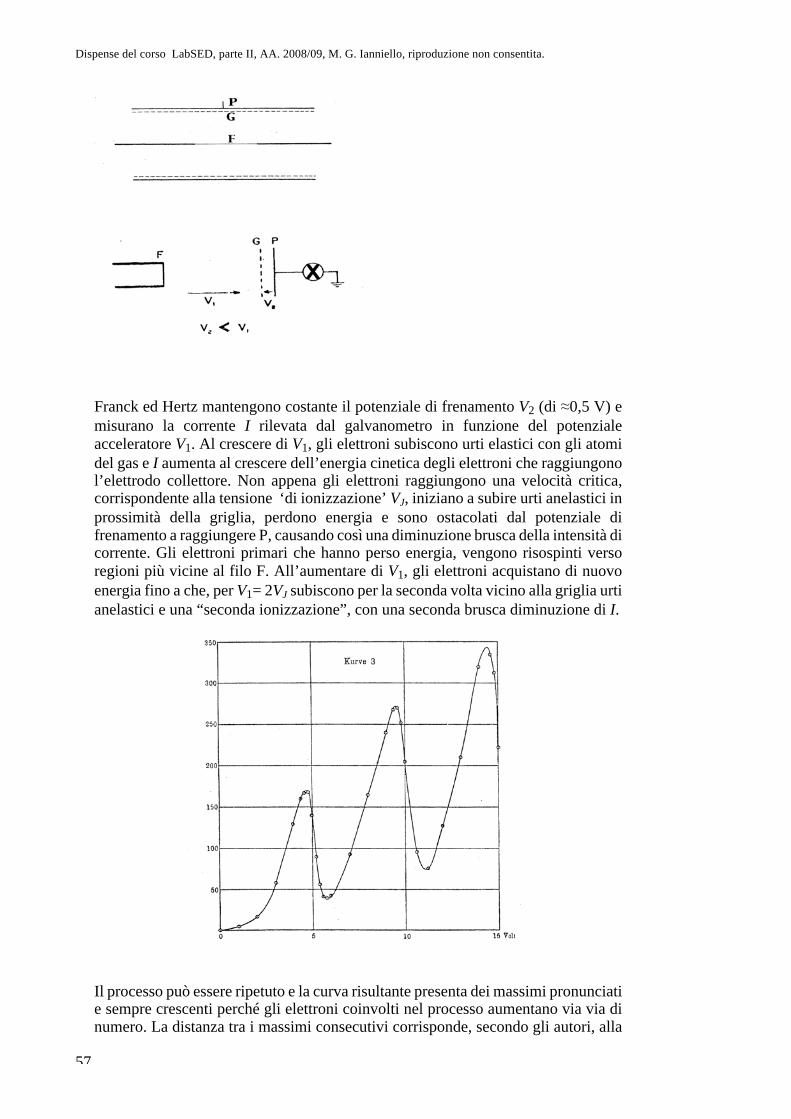
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
57
Franck ed Hertz mantengono costante il potenziale di frenamento V2 (di 0,5 V) e misurano la corrente I rilevata dal galvanometro in funzione del potenziale acceleratore V1. Al crescere di V1, gli elettroni subiscono urti elastici con gli atomi del gas e I aumenta al crescere dell’energia cinetica degli elettroni che raggiungono l’elettrodo collettore. Non appena gli elettroni raggiungono una velocità critica, corrispondente alla tensione ‘di ionizzazione’ VJ, iniziano a subire urti anelastici in prossimità della griglia, perdono energia e sono ostacolati dal potenziale di frenamento a raggiungere P, causando così una diminuzione brusca della intensità di corrente. Gli elettroni primari che hanno perso energia, vengono risospinti verso regioni più vicine al filo F. All’aumentare di V1, gli elettroni acquistano di nuovo energia fino a che, per V1= 2VJ subiscono per la seconda volta vicino alla griglia urti anelastici e una “seconda ionizzazione”, con una seconda brusca diminuzione di I.
Il processo può essere ripetuto e la curva risultante presenta dei massimi pronunciati e sempre crescenti perché gli elettroni coinvolti nel processo aumentano via via di numero. La distanza tra i massimi consecutivi corrisponde, secondo gli autori, alla

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
58
tensione di ‘ionizzazione’ che risulta essere, con le dovute correzioni, di 4,9 V. Franck ed Hertz hanno in realtà individuato il primo potenziale di eccitazione ottica (o “potenziale di risonanza”) del mercurio ma continuano a identificarlo con un “lavoro di ionizzazione”. Nel tentativo di chiarire se il trasferimento di energia alla ‘ionizzazione’ avvenga per quanti, Franck ed Hertz verificano la relazione quantistica eVJ = h tra tensione di ionizzazione e frequenza propria dell’elettrone entrato in oscillazione nell’atomo in seguito a un urto anelastico42. Tale relazione era stata già impiegata dopo il 1909 da Stark nel corso dei suoi studi, nei quali aveva ipotizzato un nesso tra righe spettrali e ionizzazione di atomi soggetti a collisione. E proprio per i vapori di mercurio R.W. Wood aveva misurato la “riga di risonanza” corrispondente a = 2536 Å, e quindi a una frequenza nota. Dal calcolo, il prodotto h corrispondeva esattamente a una energia di 4,84 eV, “un accordo così buono con il valore da noi ottenuto che a stento si può pensare a un caso”. Gli autori concludono così che, quando gli elettroni collidenti hanno raggiunto la velocità critica necessaria a ‘ionizzare’, “l’energia di un raggio di 4,9 V è esattamente uguale a un quanto di energia della riga di risonanza del mercurio a 253,6 mm”. Con il senno di poi, i risultati di Franck ed Hertz (pur essendo fondamentali in quanto rappresentavano la prima misurazione della tensione di eccitazione del mercurio per via puramente elettrica), furono inevitabilmente fraintesi dagli autori. Né l’andamento della curva di Fig. li aiutò: la curva denunciava infatti solo il primo livello di eccitazione del vapore di mercurio (4,9 V), mentre non comparivano potenziali critici più alti; non figurava inoltre alcun indizio di ionizzazione mentre Franck ed Hertz credettero di vederne più d’una. La causa di ciò era da attribuirsi alla pressione del gas (pari a circa 1 mm di Hg), troppo alta. A questa pressione i cammini liberi medi degli elettroni sono troppo corti perché un elettrone possa avere una probabilità sufficiente per guadagnare una energia superiore a 4,9 V necessaria a eccitare il livello più alto (a 6,7 V) e tanto meno i livelli successivi. La bassa sensibilità dell’apparato, al contrario, li mise al riparo da un possibile rischio: quello di osservare a 4,68 V un altro potenziale di eccitazione rilevabile solo per via elettrica perché associato a uno “stato metastabile” del mercurio (scoperto da Franck nel 1919), e quindi a una riga non osservabile otticamente. Lo stesso Hertz osserverà a molti anni di distanza che la mancata rilevazione di questo potenziale critico “fu in effetti una circostanza fortunata poiché a quel tempo non saremmo stati in grado di associare questo quanto di energia allo spettro atomico del mercurio”. La coincidenza numerica tra ciò che Franck ed Hertz ritengono sia il potenziale di ionizzazione e il prodotto h , relativo alla frequenza della riga di risonanza del mercurio, li spinge a pubblicare un articolo che passerà alla storia come l’ “esperimento di Franck ed Hertz” e verrà considerato “la prima osservazione di una riga spettrale in corrispondenza di una certa tensione [di eccitazione]” come richiesto dalla teoria di Bohr. L’idea di Franck ed Hertz è semplice ed elegante: se si bombardano gli atomi di mercurio con elettroni di energia E = 4,9 eV, gli atomi assorbono tale energia e riemettono radiazione di frequenza =E/h osservabile spettroscopicamente. Fatto l’esperimento la previsione è confermata ma qui succede un’altra ‘stranezza’: lo spettro del mercurio contiene la sola riga di risonanza mentre non c’è traccia delle altre righe note, visibili nello spettro d’arco del mercurio. Si noti inoltre che F&H hanno verificato che il trasferimento di energia nell’urto elettrone-
42 Si ricorda che all’epoca prevale ancora un modello di atomo, che contiene elettroni, assimilabile a una carica puntiforme soggetta a una forza di richiamo: una sorta di oscillatore hertziano che assorbe ed emette energia a una certa “frequenza di risonanza”; con l’ipotesi di Planck si assume in più che tale energia sia quantizzata (modelli di questo tipo sono detti “semiclassici”).

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
59
atomo è quantizzato ma questa condizione è compatibile sia con l’ipotesi di Planck sia con il modello di Bohr (che al momento ignorano). Per verificare Bohr bisogna far vedere che nell’atomo avvengono anche transizioni tra stati stazionari di energia più elevati mentre qui si parla solo di una transizione di risonanza. La circostanza di avere osservato una sola riga convince sempre più F&H che 4,9 V sia un potenziale di ionizzazione a cui corrisponde la riga 2536 Å. La confusione tra potenziale di eccitazione, concetto sconosciuto prima di Bohr, e potenziale di ionizzazione era per altro inevitabile a quel tempo. Ma non si trattava solo di una confusione concettuale: l’indistinguibilità sperimentale tra i due potenziali derivava anche dal fatto che, con il metodo di Lenard, le corte lunghezze d’onda emesse nella maggior parte dei casi dagli atomi di gas che si diseccitavano, provocavano effetto fotoelettrico sull’elettrodo collettore. In queste condizioni, il potenziale di frenamento rallentava gli elettroni primari ma accelerava i fotoelettroni e il galvanometro segnava una corrente positiva come se fossero stati rilasciati ioni positivi per ionizzazione. Lo strumento dava cioè la stessa risposta sperimentale in entrambi i casi.
§13.3. Che faceva intanto Bohr? Dopo la pubblicazione della “trilogia” Bohr aveva steso nel 1915 un lungo articolo su “La teoria quantistica della radiazione e la struttura dell’atomo” in difesa del suo modello atomico, sottoposto a critiche nonostante emergessero da più parti evidenze sperimentali favorevoli. Soprattutto nel caso dell’elio Bohr denunciava un disaccordo sensibile tra valori sperimentali della tensione di ionizzazione e valori teorici dedotti dalla sua teoria: A proposito di F&H Bohr dichiara di avere saputo di: “recenti notevoli esperimenti di Franck ed Hertz sulla ionizzazione nel vapore di mercurio. Questi esperimenti mostrano sorprendentemente che un elettrone non perde energia per collisione con un atomo di mercurio se la sua energia è minore di un ben determinato valore, di 4,9 V, ma non appena l’energia è uguale a questo valore l’elettrone ha una grande probabilità di perdere tutta la sua energia per urto con l’atomo. E’ stato inoltre dimostrato che l’atomo, come risultato di un tale urto, emette una radiazione che consiste solo nella riga UV del mercurio di lunghezza d’onda 2536 [Å] e si è sottolineato come, se la frequenza di questa riga è moltiplicata per la costante di Planck, si ottenga un valore che, nel limite degli errori sperimentali, è uguale all’energia acquistata da un elettrone che attraversa una differenza di potenziale di 4,9 V. Franck ed Hertz assumono che 4,9 V corrisponda all’energia necessaria per rimuovere un elettrone dall’atomo di mercurio ma sembra che i loro esperimenti possano forse essere consistenti con il postulato che questa tensione corrisponde solo alla transizione dallo stato normale a qualche altro stato stazionario dell’atomo neutro. Alla luce dell’attuale teoria ci dovremmo aspettare che il valore dell’energia necessaria a rimuovere un elettrone dall’atomo di mercurio possa essere calcolata dal limite delle serie delle righe singole di Paschen 1850, 1403, 1269. Poiché il vapore di mercurio assorbe luce di lunghezza d’onda 1850 le righe di questa serie così come la riga 2536 devono corrispondere a una transizione dallo stato normale dell’atomo ad altri stati stazionari dell’atomo neutro. Un tale calcolo dà 10,5 V per il potenziale di ionizzazione invece di 4,9 V. Se le considerazioni precedenti sono corrette si vedrà che le misure di Franck ed Hertz danno un sostegno molto forte alla teoria considerata in questo articolo. Se, al contrario, il potenziale di ionizzazione del mercurio si dovesse dimostrare così basso come quello assunto da Franck ed Hertz ciò costituirebbe una seria difficoltà per l’interpretazione della costante di Rydberg, comunque per lo spettro del mercurio poiché esso contiene righe di frequenza più grande della riga 2536”. Fu solo nel 1919 che Franck ed Hertz si convertirono finalmente alle idee di Bohr con un articolo dal titolo esplicito su “La conferma della teoria atomica di Bohr nello spettro ottico mediante l’indagine di urti anelastici di elettroni lenti con molecole di gas”. A guerra finita, “nel tempo trascorso, che non ci ha consentito di condurre in proprio esperimenti”, commentano Franck ed Hertz, “il settore è stato affrontato e ampliato soprattutto da parte dei fisici americani, con il risultato che la teoria atomica di Bohr è stata confermata splendidamente qui come negli spettri Röntgen e nelle ricerche sulla struttura fine delle righe dell’elio”.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
60
Tra i fisici americani che costrinsero Franck ed Hertz a staccarsi dal vecchio contesto alla Townsend e a reinterpretare i propri esperimenti alla luce della teoria di Bohr vengono citati J.T. Tate dell’università del Minnesota, H.J. van der Bijl della compagnia Western Electrical di New York, B. Davis e F.S. Goucher della Columbia University, tutti esplicitamente a favore del nuovo modello atomico. Tate, che aveva ripetuto gli esperimenti di Franck ed Hertz ma a pressioni più basse, aveva trovato per la tensione di ionizzazione un valore attorno a 10 V, in ottimo accordo con il valore teorico deducibile dal modello di Bohr. In corrispondenza dello stesso valore, anche Goucher e collaboratori avevano trovato un aumento sensibile della corrente anche se non erano in grado di spiegare quante e quali ionizzazioni avvenissero nel gas. Il motivo della indistinguibilità nella risposta sperimentale era stato individuato da van der Bijl nell’effetto fotoelettrico sugli elettrodi, con la conclusione che il potenziale di ionizzazione doveva essere di circa 10 V e non di 4,9 V, “un risultato che se venisse corroborato sperimentalmente dovrebbe essere in grado di spiegare la teoria di Bohr”. Van der Bijl aveva affrontato l’argomento in un lavoro successivo
dove aveva ribadito che l’eccitazione della riga 2536 forniva una sorgente di luce ultravioletta nel tubo a scarica, in grado di liberare fotoelettroni sull’elettrodo collettore che favorivano così la ionizzazione. Aveva inoltre sollevato la questione dello spettro a una riga trovato da Franck ed Hertz, compatibile con la teoria di Bohr solo in caso di eccitazione e non di ionizzazione, avanzando anche una possibile spiegazione della mancata eccitazione dell’intero spettro all’aumentare della tensione di accelerazione, addebitandola alla presenza di pochi elettroni lenti piuttosto che di un “fascio denso” di elettroni più energetici. A partire dalle conclusioni di van der Bijl, Davis e Goucher, con un abile artificio nei collegamenti del circuito nella disposizione alla Lenard a 4 elettrodi, erano riusciti già nel 1917 a distinguere, sempre nel caso dei vapori di mercurio, la ionizzazione indotta fotoelettricamente dalla ionizzazione per urto vera e propria.
Metodo di Lenard, modificato da Davis e Goucher per distinguere tra corrente fotoelettrica e corrente ionica
Il metodo consisteva nell’applicare tra l’elettrodo collettore P e la griglia G2 una tensione V3 commutabile, in modo da rendere P positivo rispetto a G2 o viceversa. Se la tensione V1 che accelera gli elettroni emessi da F viene aumentata fino a raggiungere il valore del primo potenziale critico (4,9 V), gli atomi diseccitandosi emettono radiazione ultravioletta che investendo gli elettrodi G2 e P può provocare il rilascio di fotoelettroni. Se per esempio V3 è tale che P sia positivo rispetto a G2, i fotoelettroni emessi da P sono frenati dal controcampo mentre quelli emessi da G2 sono attratti da P e il galvanometro segna una corrente Igalv negativa. Se ora, all’aumentare di V1 si raggiunge la tensione di ionizzazione, nello spazio tra G1 e G2
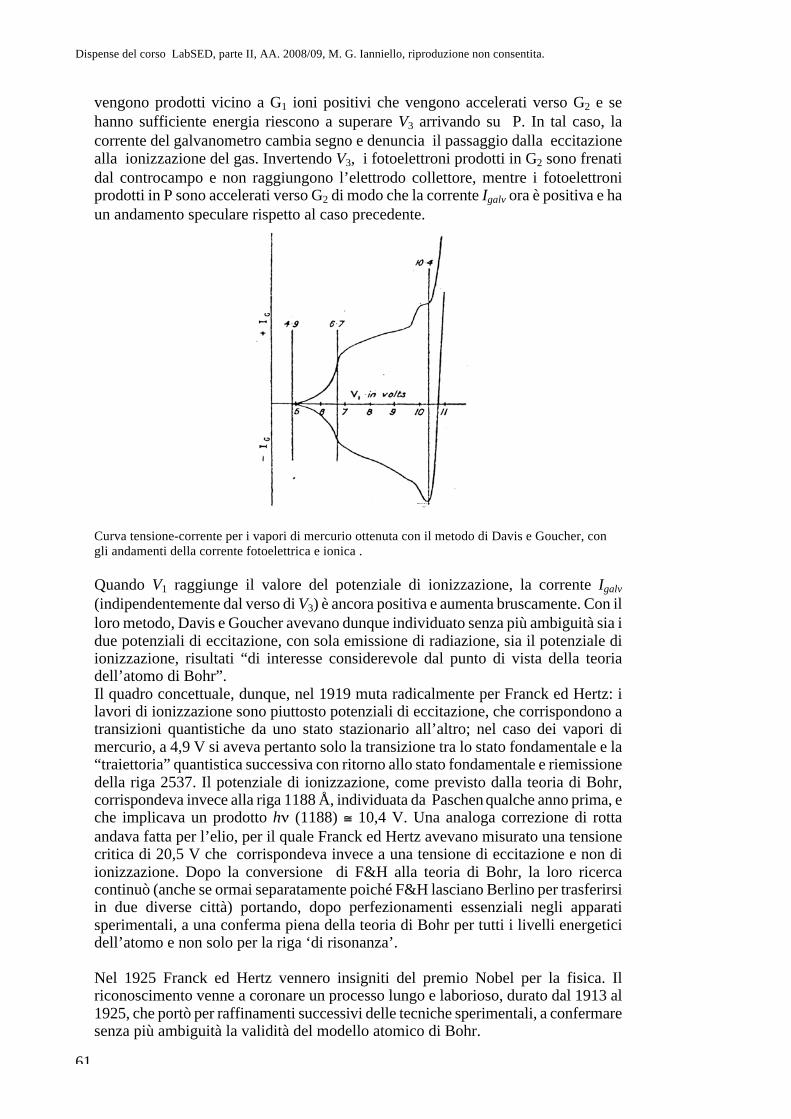
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
61
vengono prodotti vicino a G1 ioni positivi che vengono accelerati verso G2 e se hanno sufficiente energia riescono a superare V3 arrivando su P. In tal caso, la corrente del galvanometro cambia segno e denuncia il passaggio dalla eccitazione alla ionizzazione del gas. Invertendo V3, i fotoelettroni prodotti in G2 sono frenati dal controcampo e non raggiungono l’elettrodo collettore, mentre i fotoelettroni prodotti in P sono accelerati verso G2 di modo che la corrente Igalv ora è positiva e ha un andamento speculare rispetto al caso precedente.
Curva tensione-corrente per i vapori di mercurio ottenuta con il metodo di Davis e Goucher, con gli andamenti della corrente fotoelettrica e ionica .
Quando V1 raggiunge il valore del potenziale di ionizzazione, la corrente Igalv (indipendentemente dal verso di V3) è ancora positiva e aumenta bruscamente. Con il loro metodo, Davis e Goucher avevano dunque individuato senza più ambiguità sia i due potenziali di eccitazione, con sola emissione di radiazione, sia il potenziale di ionizzazione, risultati “di interesse considerevole dal punto di vista della teoria dell’atomo di Bohr”. Il quadro concettuale, dunque, nel 1919 muta radicalmente per Franck ed Hertz: i lavori di ionizzazione sono piuttosto potenziali di eccitazione, che corrispondono a transizioni quantistiche da uno stato stazionario all’altro; nel caso dei vapori di mercurio, a 4,9 V si aveva pertanto solo la transizione tra lo stato fondamentale e la “traiettoria” quantistica successiva con ritorno allo stato fondamentale e riemissione della riga 2537. Il potenziale di ionizzazione, come previsto dalla teoria di Bohr, corrispondeva invece alla riga 1188 Å, individuata da Paschen qualche anno prima, e che implicava un prodotto h (1188) 10,4 V. Una analoga correzione di rotta andava fatta per l’elio, per il quale Franck ed Hertz avevano misurato una tensione critica di 20,5 V che corrispondeva invece a una tensione di eccitazione e non di ionizzazione. Dopo la conversione di F&H alla teoria di Bohr, la loro ricerca continuò (anche se ormai separatamente poiché F&H lasciano Berlino per trasferirsi in due diverse città) portando, dopo perfezionamenti essenziali negli apparati sperimentali, a una conferma piena della teoria di Bohr per tutti i livelli energetici dell’atomo e non solo per la riga ‘di risonanza’.
Nel 1925 Franck ed Hertz vennero insigniti del premio Nobel per la fisica. Il riconoscimento venne a coronare un processo lungo e laborioso, durato dal 1913 al 1925, che portò per raffinamenti successivi delle tecniche sperimentali, a confermare senza più ambiguità la validità del modello atomico di Bohr.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
62
Franck giustificò così, nella sua Nobel Lecture, il percorso di ricerca del 1914:
“Abbiamo preso questo valore per la tensione di ionizzazione (lo stesso abbiamo fatto per l’elio, con la tensione misurata con lo stesso metodo che era di circa 20 V). Cionondimeno, il carattere quantistico del trasferimento di energia non poteva aiutare ma poteva far pensare a noi, che praticamente fin dall’inizio fummo testimoni da vicino degli sviluppi della teoria quantistica di Planck, di impiegare la teoria formulata da Einstein per spiegare l’effetto fotoelettrico! Poiché qui energia luminosa è convertita in energia cinetica degli elettroni, non poteva forse essere nel nostro caso che energia cinetica degli elettroni fosse convertita in energia luminosa? Se questo era il caso sarebbe stato facile
dimostrarlo nel caso del mercurio per l’equazione (1/2) mv2 = h riferita a una riga di 2537 Å che è facilmente accessibile nella regione ultravioletta. Questa riga è la riga di assorbimento di lunghezza d’onda più grande del vapore di mercurio. E’ spesso citata come riga di risonanza-Hg poiché R.W. Wood ha condotto su di essa importanti esperimenti sulla fluorescenza di risonanza. Se la conversione ipotizzata di energia cinetica in luce per urto poteva avere luogo, allora bombardando con elettroni di 4,9 eV doveva apparire la riga 2537 Å e solo questa di tutto lo spettro di righe del mercurio”.
E a proposito del mancato riconoscimento al modello atomico di Bohr aggiunse: “In seguito mi è sembrato totalmente incomprensibile il fatto che mancammo di riconoscere il significato fondamentale della teoria di Bohr al punto che non l’abbiamo mai menzionata neppure una volta nell’articolo in questione [del 1914]. Sfortunatamente non abbiamo potuto rettificare il nostro errore (in parte a causa di circostanze esterne) chiarendo noi stessi le incertezze che ancora sussistevano sperimentalmente”. §13.4. All’esperimento Che dimostra l’esperimento: mediante urto elettronico tra elettroni e atomi di un gas a bassa pressione (nel nostro caso elio) si osserva che quando l’urto è anelastico (trasferimento di energia E dagli elettroni agli atomi) l’atomo fa una transizione a uno stato eccitato in corrispondenza di determinati potenziali critici, in modo discreto e non continuo. L’esperimento ‘visualizza’ il comportamento quantistico degli atomi, rappresentabile, in prima approssimazione, con il modello di Bohr e conferma l’ipotesi di Bohr sull’esistenza nell’atomo di livelli energetici discreti. Si costruisce una curva I di collettore vs. V anodico e si vede che la curva ha dei picchi di corrente (che non ci sarebbero se i livelli energetici fossero continui); si distingue tra picchi di eccitazione e di ionizzazione; si identificano i vari livelli, compresi quelli metastabili (rilevabili elettricamente ma non otticamente). Come è fatto l’apparato (tubo TELTRON 2533/02, contenente ELIO a bassa p, fornito da BCD sistemi)

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
63
A
F
K
Celettrodocollettore
al nanoamperometro
Cannone elettronico (filamento+catodo+anodo): filamento F, emette elettroni per effetto termoelettronico; anodo A, a tensione VA accelera gli elettroni emessi da F; eVA è l’energia potenziale acquistata dagli elettroni; C è l’elettrodo collettore ad anello, a piccolo potenziale positivo VC (piccolo rispetto a VA) collegato a un nanoamperometro per rilevare IC: raccoglie gli elettroni più lenti che hanno urtato anelasticamente gli atomi del gas. In condizioni normali gli elettroni emessi da F attraversano l’anello C e non vengono intercettati da questo. La parete interna del tubo è rivestita da uno strato conduttore trasparente, connesso al cilindro anodico e isolato dal collettore. Analogia (fatta da F&H) tra: effetto fotoelettrico: VA (potenziale di arresto)+V0 (potenziale di estrazione) = h (in eV)
V
V
A
0
0
urto elettronico: VA (tensione anodica che accelera gli elettroni) + V0 (potenziale di contatto) = h con h = E2-E1 . E2-E1 = VA+V0; Vion=VA +V0 Veccit=VA’+V0. Come è fatto l’atomo di elio? Ha due elettroni attorno al nucleo (sistema a più elettroni). Il sistema è più complesso rispetto all’atomo di idrogeno, l’unico elemento per il quale il modello di Bohr funzioni. Lo schema dei livelli si presenta con due schemi dei termini, il paraelio e l’ortoelio. All’epoca si pensava che l’elio fosse una mescolanza di due specie; solo dopo l’introduzione dello spin (post 1925) si capì che la separazione dei due termini era dovuta a un momento di spin totale 0 (paraelio: gli spin dei due elettroni sono

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
64
antiparalleli) oppure 1 (ortoelio: spin paralleli; v. per es., M. Born, Fisica atomica, Boringhieri, p. 219). La tensione di ionizzazione dell’elio è di 24,58 eV. Franck ed Hertz, all’epoca delle loro prime esperienze non conoscevano Vion; il diagramma dei livelli dell’elio era parzialmente conosciuto. F&H indagano anche altri gas ‘più semplici’, in particolare i vapori di mercurio. Nelle apparecchiature didattiche si preferisce elio o neon perché emettono nel visibile e quindi sono di più facile accesso all’osservazione. I vapori di mercurio, al contrario, emettono nell’UV, condizione che richiederebbe apparecchiature più costose e metodi di osservazione più sofisticati. Il procedimento di misura Si fa una indagine esplorativa (“scan”) tra 0 e 50 V. In questo scan si vede che ci sono due zone con dei massimi. Per capire se c’è ionizzazione (sappiamo che questa si verifica a circa 24 V), si invertono i poli della batteria che alimenta l’elettrodo collettore (-VC). Se il gas si ionizza C attira ioni positivi e la corrente cambia segno –IC pA ; si indaga meglio la regione al di sotto della ionizzazione, tra 15 e 25 V: si individuano i picchi e con opportune correzioni si risale ai potenziali critici dell’elio. CAP. 14. LO SPIN DELL’ELETTRONE Si veda, in merito, di M.G. Ianniello, M. Ingrassia, S. Petrarca, Lo spin dell’elettrone e una proposta di esperimento ESR, Giornale di Fisica, 47, 3 (2006) 229-248. CAP. 15. VERSO LE BASSE TEMPERATURE: LA SCOPERTA DELLA SUPERCONDUTTIVITÀ
“Corrente elettrica che scorre in eterno”43 §15.1. In che consiste la superconduttività Alcuni materiali, se raffreddati al di sotto di una certa temperatura critica TC (T< TC), hanno resistenza elettrica R nulla; passano cioè dallo stato normale (R 0) allo stato superconduttore (R=0). Nei superconduttori (SC) si instaurano correnti permanenti dette correnti persistenti. Conduzione normale Nei conduttori normali sono gli elettroni a portare corrente; a T ordinaria la conducibilità elettrica è 1/T (più aumenta T più gli elettroni di conduzione che attraversano il reticolo urtano in modo indipendente e perdono energia sotto forma di calore). Il materiale è resistivo. Superconduzione Nei SC sono coppie di elettroni (coppie di Cooper) a supercondurre la corrente (si formano coppie quando l’attrazione elastica del reticolo supera la repulsione coulombiana tra due elettroni). I materiali SC conducono elettricità senza dissipare energia. Gli elettroni a coppie si muovono coerentemente in fase e passano indisturbati attraverso il reticolo come un’ ‘onda di piena’ (non sentono né impurezze né le oscillazioni reticolari). A basse T (vibrazioni del reticolo piccole) il moto degli elettroni è favorito ma la spiegazione non basta per capire la SC (vedi teoria BCS, per es. su www.mfn.unipmn.it/corsi-di L/Fisica1/orientamento/supercond/supercond2.doc).
43 cfr. J. Teichmann, W. Schreier, M. Segre, Experimente die Geschichte machten, pp. 161-168 Bayer. Schulbuch Verlag, München1995.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
65
I parametri fondamentali nella SC Lo stato SC è definito da tre parametri critici, correlati tra loro: la temperatura critica TC (i SC hanno diverse TC a seconda del materiale di cui sono fatti); il campo magnetico esterno critico B0C, la densità massima di corrente JC che un SC può portare. Per T> TC, per B> BC , per J> JC, il campione passa dallo stato SC allo stato normale. Per avere un SC occorre avere la ‘giusta’ combinazione di T, B e J con valori inferiori al valore critico. Quando viene scoperta la SC Nel maggio 1911, da Heike Kamerlingh-Onnes, Leida. La scoperta si deve a 3 prerequisiti:
1860 sviluppo, alla fine dell’Ottocento, della tecnologia delle basse temperature, motivato sia da questioni scientifiche (studio della liquefazione dei gas in termodinamica) sia da esigenze applicative (nella cosiddetta industria del freddo nei birrifici, nei macelli, nelle latterie, ecc.).
1861 Studio, nella seconda metà dell’Ottocento, del comportamento di sostanze vicino allo “zero assoluto”.
1862 Studio della dipendenza della resistenza elettrica dalla temperatura (R=R(T)) in telegrafia (seconda metà dell’Ottocento, posa dei cavi transatlantici). La nascita della ‘industria del freddo’ e la liquefazione dei gas 1852, Joule e W. Thomson (Kelvin) scoprono l’effetto “Joule-Thomson”: un gas reale si raffredda espandendosi anche senza apporto di lavoro esterno. Questo principio è fondamentale per la realizzazione delle macchine frigorifere. Principali protagonisti della “guerra del freddo”: Linde, Dewar, Kamerlingh-Onnes. Carl von Linde, ing., nel 1878 lascia la Scuola Politecnica di Monaco per fondare la “Gesellschaft für Lindes Eismaschinen”, che si impone presto nel mercato con la produzione di ghiacciaie. Nel 1892 Linde torna alla ricerca e tenta di liquefare i gas. Primi tentativi di liquefare i gas: ossigeno Già nel 1877 scienziati francesi erano riusciti a ottenere, mediante espansione di gas soggetti ad alte pressioni, piccole quantità di ossigeno liquido. Qualche anno dopo anche il fisico polacco Z. Florenty von Wrobleski (che morirà nell’esplosione del suo laboratorio), insieme al chimico C. von Olszewski riesce a liquefare qualche cm3 di ossigeno. L’ “aria liquida” 1895, Linde liquefa l’aria (ossigeno e azoto liquido) in grosse quantità da impiegarsi nell’industria dei concimi, degli esplosivi, ecc. Idrogeno ed elio L’inglese J. Dewar, come i colleghi polacchi, era riuscito a liquefare presso i laboratori della Royal Institution di Londra, ossigeno nel modo tradizionale, per espansione. La sfida per i ricercatori nel settore delle basse temperature era tuttavia rappresentata dagli ultimi gas “permanenti”, H ed He. Per l’elio si iniziò a tentare dal 1895, quando questo gas fu scoperto sulla Terra. La ricerca migliorò con gli apparati di Linde che nel frattempo Dewar aveva iniziato a usare. Con queste macchine, Dewar ottenne nel 1898 20 cm3 cubi di idrogeno liquido. Lo scienziato inglese credette di aver liquefatto anche l’elio ma si trattava solo di impurezze. Entra in scena Kamerlingh-Onnes, a Leida e si accende una competizione dura tra Dewar e K-O. K-O, nel 1882 a soli 29 anni viene chiamato sulla prima cattedra di Fisica sperimentale in Olanda. Figlio di un proprietario di fabbrica ha una mentalità imprenditoriale; fonda un laboratorio per le basse temperature, convinto che il laboratorio sia la ‘fabbrica’ della scienza. E’ estremamente attento a scegliere i propri collaboratori. Anche se nel 1901 boccia Einstein che da Milano gli spedisce una domanda di assunzione.
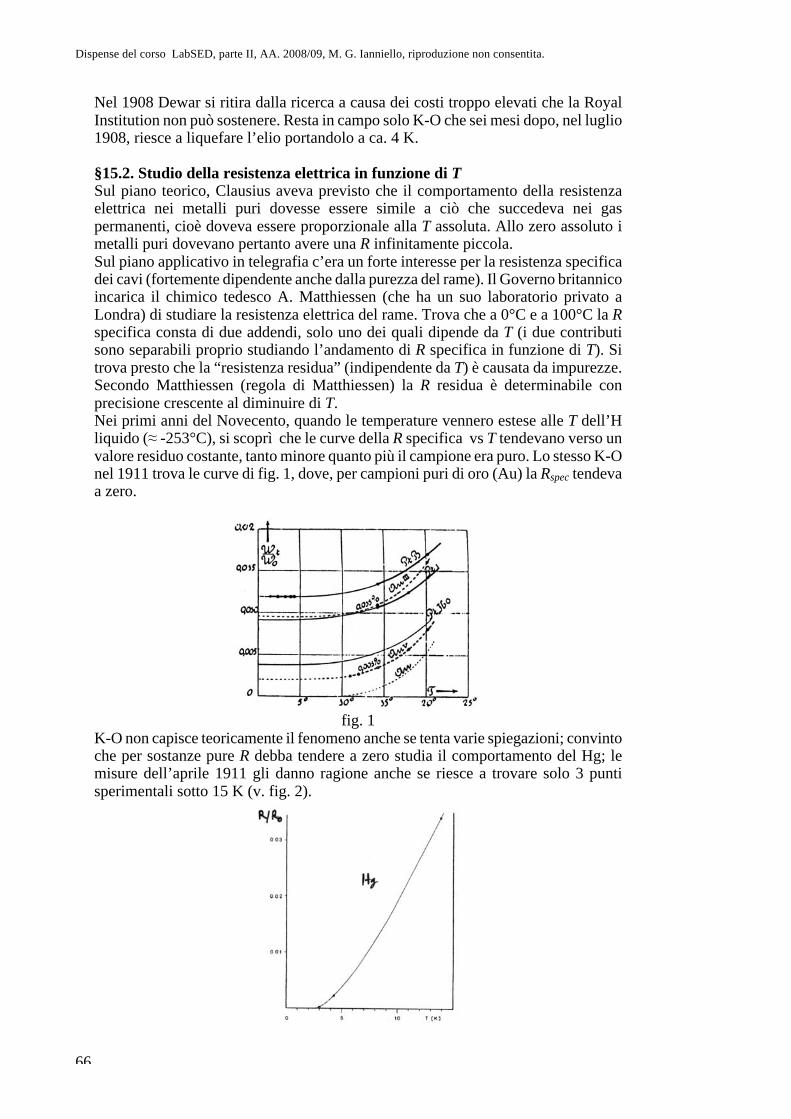
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
66
Nel 1908 Dewar si ritira dalla ricerca a causa dei costi troppo elevati che la Royal Institution non può sostenere. Resta in campo solo K-O che sei mesi dopo, nel luglio 1908, riesce a liquefare l’elio portandolo a ca. 4 K. §15.2. Studio della resistenza elettrica in funzione di T Sul piano teorico, Clausius aveva previsto che il comportamento della resistenza elettrica nei metalli puri dovesse essere simile a ciò che succedeva nei gas permanenti, cioè doveva essere proporzionale alla T assoluta. Allo zero assoluto i metalli puri dovevano pertanto avere una R infinitamente piccola. Sul piano applicativo in telegrafia c’era un forte interesse per la resistenza specifica dei cavi (fortemente dipendente anche dalla purezza del rame). Il Governo britannico incarica il chimico tedesco A. Matthiessen (che ha un suo laboratorio privato a Londra) di studiare la resistenza elettrica del rame. Trova che a 0°C e a 100°C la R specifica consta di due addendi, solo uno dei quali dipende da T (i due contributi sono separabili proprio studiando l’andamento di R specifica in funzione di T). Si trova presto che la “resistenza residua” (indipendente da T) è causata da impurezze. Secondo Matthiessen (regola di Matthiessen) la R residua è determinabile con precisione crescente al diminuire di T. Nei primi anni del Novecento, quando le temperature vennero estese alle T dell’H liquido ( -253°C), si scoprì che le curve della R specifica vs T tendevano verso un valore residuo costante, tanto minore quanto più il campione era puro. Lo stesso K-O nel 1911 trova le curve di fig. 1, dove, per campioni puri di oro (Au) la Rspec tendeva a zero.
fig. 1
K-O non capisce teoricamente il fenomeno anche se tenta varie spiegazioni; convinto che per sostanze pure R debba tendere a zero studia il comportamento del Hg; le misure dell’aprile 1911 gli danno ragione anche se riesce a trovare solo 3 punti sperimentali sotto 15 K (v. fig. 2).
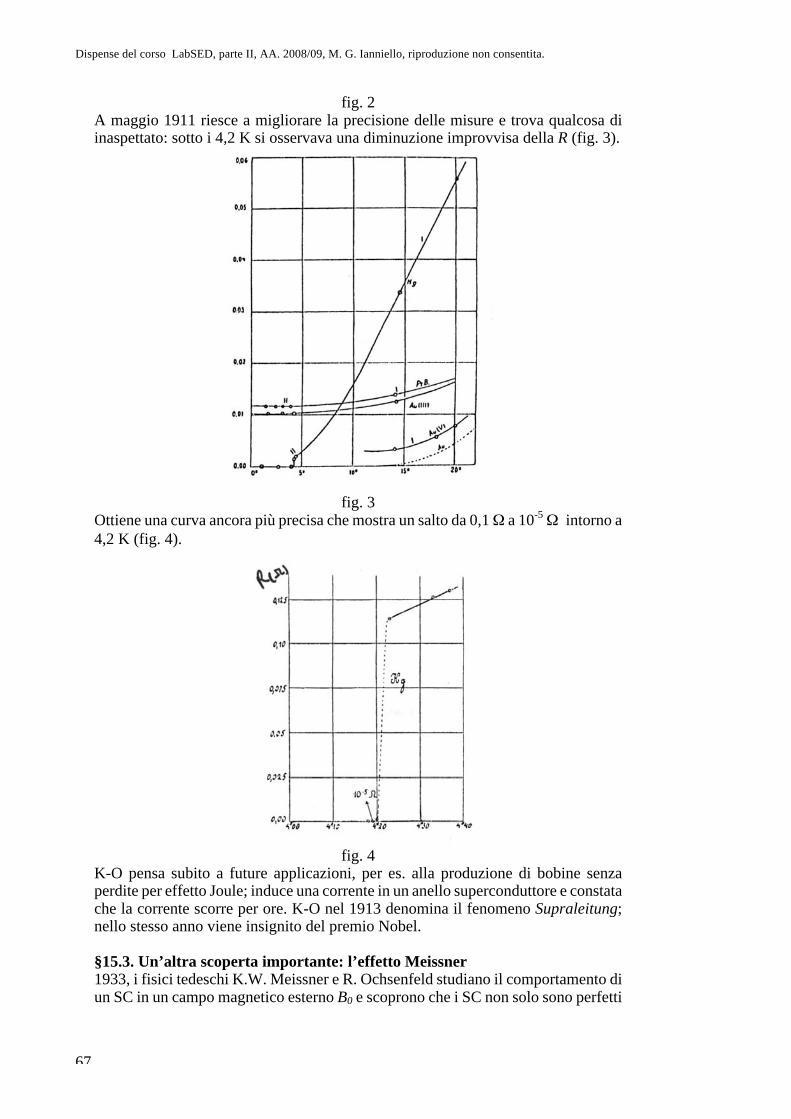
Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
67
fig. 2 A maggio 1911 riesce a migliorare la precisione delle misure e trova qualcosa di inaspettato: sotto i 4,2 K si osservava una diminuzione improvvisa della R (fig. 3).
fig. 3
Ottiene una curva ancora più precisa che mostra un salto da 0,1 a 10-5 intorno a 4,2 K (fig. 4).
fig. 4
K-O pensa subito a future applicazioni, per es. alla produzione di bobine senza perdite per effetto Joule; induce una corrente in un anello superconduttore e constata che la corrente scorre per ore. K-O nel 1913 denomina il fenomeno Supraleitung; nello stesso anno viene insignito del premio Nobel. §15.3. Un’altra scoperta importante: l’effetto Meissner 1933, i fisici tedeschi K.W. Meissner e R. Ochsenfeld studiano il comportamento di un SC in un campo magnetico esterno B0 e scoprono che i SC non solo sono perfetti

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
68
conduttori di elettricità (superconducono) ma anche materiali diamagnetici perfetti44. Nel caso dei SC poiché le correnti che circolano hanno R=0, si comportano come “diamagneti perfetti”. In linea generale, se un SC, raffreddato a T<TC, è immerso in un B0, nel SC circolano correnti indotte che generano un campo BSC opposto a B0. All’interno del SC Btot = B0 + BSC =0 (si dice anche che il SC espelle il campo magnetico interno). Le linee di induzione di B0 non penetrano nel SC che diventa perfettamente diamagnetico. Se B0 esterno è troppo forte, il SC smette di far circolare corrente per non spendere troppa energia e per B0> B0C transisce allo stato normale. Questa spiegazione basata sulla legge (macroscopica) di Faraday-Neumann-Lenz giustifica in prima approssimazione il comportamento diamagnetico dei SC ma ovviamente non basta; la fenomenologia cambia, per es., a seconda del tipo di SC e della sua geometria.
SC di tipo I e II SC di tipo I a basse TC e debole B0: raggiungono lo stato SC a TC < 23 K (per raffreddarli serve elio liquido); espellono tutto il campo magnetico interno (BSC=0); per B0> B0C il campo magnetico esterno entra nel materiale e annulla lo stato SC. SC di tipo II ad alte TC: raggiungono lo stato SC a TC T azoto liquido (77 K); anche con B0 esterni intensi i SC restano tali. 1986, scoperta della SC ad alta T critica (premio Nobel a J.C. Bednorz e K. A. Müller dei lab IBM svizzeri). Nuovo impulso alla tecnologia (sviluppo dei materiali ceramici SC a base di ossidi metallici detti “perovskiti”, del tipo II). Nei SC ad alta TC (HcTS= High critical Temperature Superconductors): “salti di T” intorno a 100 K ( -170°C). Dal 1986, i SC perovskiti lavorano intorno a 35 K;
44 Ricordarsi che il diamagnetismo è una proprietà di tutti i corpi e che una sostanza diamagnatica viene respinta debolmente dal polo di un magnete; gli atomi delle sostanza diamagnetiche non hanno momento di dipolo propri. Se immaginiamo il materiale costituito da atomi contigui, in ciascun atomo gli elettroni ruotano in verso opposto all’atomo contiguo, in modo che i loro momenti magnetici si elidono. In presenza di B0 esterno questo non avviene più e il momento magnetico totale è opposto a B0. Nelle sostanze ferromagnetiche al contrario gli atomi hanno un momento di dipolo proprio e in presenza di B0 i dipoli magnetici si allineano con il campo esterno. v. Manuali.

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
69
nel feb. 1987, si scopre un materiale della classe perovskite che opera a 90 K (YBa2Cu3O7); si può pertanto lavorare con azoto liquido come refrigerante (T azoto liquido 77 K), con ulteriori vantaggi dati dal basso costo dell’azoto liquido e dalla sua grande reperibilità (il 78% dell’aria è azoto). Prima della scoperta dei SC ad alta T si operava intorno a 20 K e perciò l’elio liquido rimaneva il solo refrigerante usato (per l’elio liquido, Tc=4,2 K). §15.4. Le spiegazioni della SC In merito alle spiegazioni del fenomeno della SC, fino agli anni Cinquanta del Novecento si annaspa; alla fine del 1957, J. Bardeen, J. R. Schriefer e L.N. Cooper sviluppano la teoria BCS con l’ipotesi che gli elettroni formino coppie di Cooper (coppie di elettroni con spin opposti si accoppiano e si comportano come bosoni; a T> TC le coppie si rompono, ecc.). Premio Nobel nel 1972. Per spiegazioni, vedi corsi di fisica dello stato solido e di Meccanica Quantistica. La teoria BCS non spiega completamente la SC nei materiali ceramici. In particolare funziona per i SC tradizionali (ante 1986) mentre non tiene per i SC ad alta T critica. Esistono altre teorie in via di sviluppo. Esperimenti Useremo dischi superconduttori del tipo YBCO (a base di ittrio, Y e di bismuto, rame e ossigeno) e BSCCO (a base di bismuto, Bi e di stronzio, calcio, rame e ossigeno). Si tratta di materiali ceramici del tipo II (perovskiti). In genere a scuola si usano questi: ne esistono a base di altri ossidi ma fare attenzione (per esempio, quelli a base di tantalio sono tossici: NO a scuola; comunque meglio usare i guanti). I dischi sono fragili (non farli cadere) e igroscopici (dopo l’uso con azoto liquido asciugare sotto una lampada). Per raffreddare i dischi SC usiamo l’azoto liquido (77 K); per trasportarlo: dewar senza coperchio (per evitare che il thermos esploda); stare in ambiente ventilato; NO sulla pelle; meglio usare guanti e pinze. Ci limitiamo a studiare l’effetto Meissner (disco SC e magnete; confronto tra la levitazione indotta da dischi di tipo YBCO e BSCCO; più dischi SC dello stesso tipo uno sull’altro per osservare che il magnete levita più in alto: l’effetto Meissner è un effetto di volume). Perché il magnete levita Attenzione: nei SC di tipo II il campo magnetico non è espulso completamente ma è costretto in filamenti nel materiale. Sulla superficie del SC circolano supercorrenti in verso tale da avere una polarità speculare a quella del magnete (N-N; S-S). Le forze repulsive fanno levitare il magnete. Il campo magnetico esterno penetra in parte nel volume del SC con “flussoni” (flussi microscopici di campo magnetico quantizzati) che si attaccano (si ‘pinzano’: flux pinning) sulle impurezze del SC che ‘centrano’ il magnete al di sopra del disco SC. Il magnete può essere messo in oscillazione, in rotazione, ecc. Il SC ‘aggiusta’ le correnti in modo da creare sempre un polo speculare a quello del magnete. Il fenomeno dipende anche dalla geometria del SC. A seconda della geometria si possono avere stati misti (mixed-state Meissner effect), cioè regioni in cui circola corrente che si alternano a regioni in cui non circolano correnti (cioè regioni SC che si alternano a regioni NON SC); nelle regioni dove circolano correnti si creano BSC opposti a B0 e B0 non penetra nel SC. Nella geometria cilindrica (grosso modo il nostro caso) i flussoni penetrano nel SC in certe regioni; sul bordo, dove circolano correnti superficiali che espellono BSC, non penetrano. (spiegazione da prendere cum grano salis) Bibliografia

Dispense del corso LabSED, parte II, AA. 2008/09, M. G. Ianniello, riproduzione non consentita.
70
H. Kamerlingh-Onnes, Further experiments with liquid Helium. On the change of electric resistance of pure metals at very low temperatures. The disappearance of the resistance of Mercury, Communications from the Physical laboratory of the Univ. of Leiden, 122 b (1911) 11-15. P. F. Dahl, Superconductivity: its historical roots and development from Mercury to the ceramic oxides, New York, 1992. M. Grilli, Superconduttività, conferenza di orientamento per il Progetto Lauree Scientifiche (16 marzo 2007) Hyperphysics.phy-astr.gsu.edu/hbase/solids/meis.html


















