OUTCOME A LUNGO TERMINE DEI PAZIENTI … dello studio: CSKP-02-2010 OUTCOME A LUNGO TERMINE DEI...
19
Codice dello studio: CSKP-02-2010 OUTCOME A LUNGO TERMINE DEI PAZIENTI PEDIATRICI CON RENE SINGOLO CONGENITO Studio osservazione di coorte prospettico Longitudinale Documento: Protocollo di studio Autore: Dott. Giovanni Montini, Struttura di Nefrologia Pediatrica U. O. di Pediatria Specialistica Cicognani, Azienda Ospedaliero – Universitaria di Bologna Policlinico Sant‟Orsola – Malpighi Versione finale: 27/04/2010 Numero pagine:19
Transcript of OUTCOME A LUNGO TERMINE DEI PAZIENTI … dello studio: CSKP-02-2010 OUTCOME A LUNGO TERMINE DEI...

Codice dello studio: CSKP-02-2010
OUTCOME A LUNGO TERMINE
DEI PAZIENTI PEDIATRICI
CON RENE SINGOLO CONGENITO
Studio osservazione di coorte prospettico
Longitudinale
Documento: Protocollo di studio
Autore: Dott. Giovanni Montini, Struttura di Nefrologia Pediatrica U. O. di Pediatria
Specialistica Cicognani, Azienda Ospedaliero – Universitaria di Bologna Policlinico
Sant‟Orsola – Malpighi
Versione finale: 27/04/2010
Numero pagine:19

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 2
Indice
1. Introduzione 3
1.2 Eziopatogenesi 3
1.3 Anomalie associate 4
1.4 Genetica 5
1.5 Diagnosi 6
2. Progetto di studio 7
2.1 Razionale dello studio 7
2.2 Disegno dello studio 13
2.3 Popolazione 13
2.4 Visite e valutazioni 15
2.5 Obiettivi dello studio 16
2.6 Gestioni dei dati e analisi statistica 16
2.7 Confidenzialità delle informazioni raccolte 16
2.8 Risultati attesi 17
Bibliografia 18
Abbreviazioni
51Cr-EDTA Acido etilene diaminotetracetico marcato con cromo 51
99mTc-DTPA Acido dietilene triaminopentacetico marcato con tecnezio 99m
CAKUT Anomalie congenite e delle vie urinarie
DMSA Acido dimercaptosuccinico
RAG Agenesia renale
RAP Aplasia renale

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 3
1. Introduzione
L‟incidenza del rene singolo congenito nella popolazione varia a seconda degli studi da 1:450
[1] a 1:3200 [2]. Il sesso maschile sembra essere più colpito del sesso femminile con un
rapporto di 1,2-2,3:1. Le forme a carico del rene sinistro sono più frequenti di quelle a carico
del rene destro .
I fattori di rischio per lo sviluppo di tale patologia sembrerebbero:
• Il diabete materno;
• La presenza di arteria ombelicale singola;
• L‟età materna inferiore ai 18 anni;
• L‟esposizione cronica all‟alcol;
• Esposizione ad agenti teratogeni (es. ACE inibitori).
Infine gli Afro-Americani sembrerebbero più colpiti rispetto ai Caucasici [2] .
1.2 Eziopatogenesi
Si distinguono due categorie di rene funzionalmente unico :
APLASIA RENALE (RAP)
Si definisce come l‟alterato sviluppo di un rene dovuto all‟involuzione precoce della gemma
ureterale o all‟alterata differenziazione del blastema metanefrico o all‟alterata connessione tra
la gemma ureterale e il blastema metanefrico. Tutto questo causa quadri di displasia renale o
di rene multicistico che vanno incontro a involuzione o a forme di displasia estrema con
parenchima renale non funzionante [3].

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 4
AGENESIA RENALE (RAG)
Si definisce come il mancato sviluppo del parenchima renale dovuto al mancato inizio della
sequenza pronefro-metanefro o alla mancata formazione della gemma ureterale.
Nell‟agenesia renale si ha la mancata formazione del rene, dell‟uretere e dell‟emitrigono
omolaterale della vescica [4].
Nella pratica clinica, comunque, è difficile nella maggior parte dei casi una diagnosi
differenziale tra le due forme, soprattutto per quei casi di RAP che vanno incontro a
involuzione.
1.3 Anomalie associate
Le anomalie che si associano al rene singolo congenito possono essere urologiche ed
extra-urologiche.
1) Le urologiche sono le più frequenti e si possono riscontrare in circa il 40% dei casi
[5].
Tali anomalie sono rappresentate da:
Reflusso vescico-ureterale nel 15% dei casi;
Ostruzione dalla giunzione pielo-ureterale nel 12% dei casi;
Ostruzione della giunzione uretero-vescicale nel 2% dei casi;
Altre anomalie (duplicazione vescicale, ostruzione del collo vescicale) nel 9%
dei casi.
2) Le anomalie extra-urologiche [6] sono rappresentate da (vedi tabella 1):
Anomalie respiratorie nel 34% dei casi;
Anomalie dei genitali nel 19% dei casi;
Anomalie cardiache nel 15% dei casi;
Anomalie gastrointestinali nel 9% dei casi,
Anomalie ematologiche nel 6% dei casi;
Anomalie neurologiche nel 3% dei casi;
Altre anomalie nel 10% dei casi.

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 5
Anomalie genitali[5] Nel maschio:anomala formazione della
rete testis
Nella femmina: utero unicornato, utero
didelfico.
Anomalie cardiache[6] Difetto di settazione interventricolare
Difetto di settazione interatriale
Pervietà del dotto arterioso
Insufficienza polmonare
Insufficienza mitrale
Disfunzione del ventricolo sinistro
Ipertrofia del ventricolo destro
Associazione dei precedenti
Anomalie gastrointestinali [6] Atresia anale
Fistola retto vescicale
Atresia biliare
Associazione dei precedenti
Labiopalatoschisi
Anomalie ematologiche [6] Anemia di Fanconi
Porpora trombocitopenia idiopatica
Talassemia major
Leucemia mieloide cronica giovanile
Anomalie neurologiche [6] Ipotonia infantile
Epilessia
Ritardo psicomotorio
Altre anomalie [6] Cataratta congenita
Diabete insipido nefritogeno
Anomalie auricolari e delle estremità
Sindrome di Down
Sindrome di Klipper-Feil
Atresia delle coane
Coloboma dell‟iride
Malformazioni vertebrali
Anomalie costali
Tabella 1: Manifeastazioni extrarenali
1.4 Genetica
Il rischio di ricorrenza familiare è elevato per entrambe le forme di RAG e RAP. In
particolare, malformazioni urinarie come RAG, displasia renale e rene multi cistico sono state
frequentemente descritte all‟interno di uno stesso nucleo familiare.

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 6
Diversi sono i geni che sono stati descritti (PAX2, HNF1-beta, TCF2, SALL1, EYA1)
e che si possono associare a tali forme. Nella maggior parte dei casi si tratta di condizioni a
eredità autosomica dominante e penetranza incompleta [7] [8] [9] e in alcuni casi si possono
osservare dei veri e propri quadri sindromici (vedi tabella 1).
1.5 Diagnosi
La diagnosi è prettamente radiologica[5]. Il sospetto diagnostico nasce solitamente
dopo l‟esecuzione di un‟ecografia renale (in epoca prenatale, perinatale o post natale) in cui
non si visualizza il rene nella sua posizione normale. La conferma diagnostica avviene
mediante l‟esecuzione di una scintigrafia renale statica con acido dimercaptosuccinico
(DMSA), marcato con Tecnezio-99m. Al momento della diagnosi è difficile differenziare le
Tabella 2 (modificata da [4]): Sindromi multiorgano associate a RAG o RAP
Sindrome branchio-oto–renale: mutazione autosomica dominante del gene EYA1 (Eyes Absent 1); si manifesta con perdita di udito, appendice pre-auricolare, labiopalatoschisi.
Sindrome di DiGeorge: delezione di 22q11 che si manifesta con anomalie cardiache congenite, ipocalcemia, immunodeficit e disturbi neurocognitivi.
Anemia di Fanconi: causata da mutazione recessiva di molteplici geni, si manifesta soprattutto con pancitopenia.
Sindrome di Fraser: mutazione autosomica recessiva del gene FRAS1 che si manifesta con criptoftalmo, sindattilia cutanea, malformazione della laringe e anomalo sviluppo dei genitali.
Sindrome di Kallmann: mutazione X-linked recessiva del gene Anosmina-1; si manifesta con ipogonadismo ipogonadotropo e anosmia.
Sindrome di Klinefelter: 47,XXY: testicoli piccoli e sclerotici, ginecomastia, azoospermia e ipogonadismo ipergonadotropo.
Sindrome di Rokitansky-Kuster–Hauser: mutazione autosomica dominante del gene WNT4 (wingless-type MMTV integration site family member): assenza dell’utero e della parte superiore della vagina, o presenza di tali strutture in forma rudimentale.
Associazione MURCS: la base genetica è attualmente indefinita, si manifesta con aplasia o ipoplasia del dotto di Muller (MU), malformazioni renali (R) e del somite cervocotoracico (CS).
Sindrome di Poland: la base genetica è attualmente indefinita; si manifesta con ipoplasia unilaterale del muscolo grande pettorale e sindattilia ipsilaterale.
Sindrome delle cisti renali e diabete: mutazione autosomica dominante del gene HNF (hepatocyte nuclear factor): si manifesta con diabete mellito, iperuricemia e malformazioni uterine.
Sindrome di Townes–Brocks: mutazione autosomica dominante del gene SALL1 (sal-like 1/homologue of Drosophila spalt); si manifesta con: ano imperforato, ipoacusia neurosensoriale, ipospadia, pollice bifido.
Sindrome di Williams–Beuren; delezione di 7q11.23, che si manifesta con ritardato sviluppo, anomalie cardiovascolari, ritardo mentale e dismorfia facciale.

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 7
forme derivanti da aplasia renale (soprattutto quelle da involuzione di un rene multi cistico
o di un rene displasico) dalle forme derivanti dall‟agenesia renale.
2 Progetto dello studio
2.1 Razionale dello studio
Negli ultimi anni si è assistito a un aumento notevole degli individui affetti da
patologia renale cronica. Tutto questo ha portato gli studiosi ad approfondire le conoscenze
sui fattori di rischio che potrebbero influenzare l‟evoluzione verso l‟insufficienza renale
cronica, a ricercare marker precoci di danno renale ed elaborare nuove strategie di intervento
per preservare la funzione renale.
Per quanto concerne i fattori di rischio in particolare, l‟attenzione si è focalizzata
maggiormente sull‟obesità, sull‟ipertensione e sul diabete che sono tra le principali cause di
danno renale nella popolazione adulta.
Oltre ai fattori che causano maggiormente l‟insufficienza renale nell‟età adulta, non
bisogna dimenticare quelle condizioni cliniche, quali le anomalie renali congenite, che pur
interessando inizialmente l‟età pediatrica e lo specialista nefrologo pediatra potrebbero
interessare in seguito l‟adulto e il neurologo dell‟adulto.
In età pediatrica infatti la principale causa di insufficienza renale nei bambini è
rappresentata proprio alle anomalie congenite del rene e delle vie urinarie (CAKUT =
Congenital anomalies of the kidney and urinary tract) quali l‟ipodisplasia mono o bilaterale
con o senza uropatia malformativa associata, le valvole dell‟uretra posteriore, il rene a ferro di
cavallo e il rene singolo congenito.
Quest‟ultima condizione clinica è stata per molto tempo considerata a buona prognosi
e raramente annoverato tra i fattori di rischio in grado di ridurre la sopravvivenza del portatore
e/o predisporre il rene residuo a un danno a lungo termine [10].
L‟evoluzione fisiologica del rene singolo nella maggior parte dei casi comporta lo
sviluppo d‟ipertrofia compensatoria [11] con aumento delle dimensioni del rene che possono
arrivare fino a due volte il volume di un rene normale entro i primi 4 anni di vita. [12].
Inoltre, mentre nei soggetti sani la crescita renale avviene fino ai 15 mesi di vita nei
pazienti con rene singolo si prolunga fino ai 22 mesi di vita [13]. Si è visto che l‟ipertrofia

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 8
renale può correlarsi a un meccanismo di iperfiltrazione glomerulare (presente fino al 50% dei
pazienti adulti con monorene) dovuto probabilmente o a un aumento del numero di nefroni o
alla ipertrofia degli stessi [14].
In questi soggetti inoltre i livelli di cistatina C (uno dei marker precoci di danno
renale) sono significativamente più alti rispetto a quelli presenti in una popolazione di
soggetti sani e si nota maggiormente nei soggetti con età superiore ai 12 anni [15].
Questo ha portato i ricercatori a pensare che da una parte il rene singolo potrebbe
presentare un difetto intrinseco e dall‟altra che il processo compensatorio, legato allo sviluppo
dell‟ipertrofia renale e alla conseguente iperfiltrazione, potrebbe rivelarsi nel lungo termine
controproducente e portare allo sviluppo di microalbuminuria, proteinuria, ipertensione
arteriosa [16] e glomerulosclerosi focale come descritto in alcuni studi [4], [17], [18].
Dal punto di vista prognostico infatti accanto ad alcuni studi che hanno riportato la
benignità di tale quadro clinico ve ne sono altri che invece hanno evidenziato un‟evoluzione
più sfavorevole di tale condizione clinica.
In uno studio retrospettivo su 157 adulti con agenesia renale e rene contro laterale
normale, seguiti dal 1960 al 1975 presso la Mayo Clinic (Rochester, Minnesota) [17]:
Il 47% ha sviluppato ipertensione;
Il 19% ha sviluppato proteinuria;
Il 13% ha sviluppato una riduzione della filtrazione glomerulare;
Il 4% è deceduto in seguito alle complicanze dell‟IRC terminale.
Gli Autori dello studio concludevano dicendo che la presenza di RAG poteva
aumentare notevolmente il rischio di sviluppare danno renale.
In un altro studio un gruppo di anatomopatologi guidato da Kiprov si è chiesto se i
pazienti con agenesia renale unilaterale avessero una probabilità maggiore di sviluppare
glomerulosclerosi focale e segmentale [12] rispetto alla popolazione normale. Gli autori
hanno rivisto 586 campioni chirurgici di rene provenienti da 452 biopsie e 134 nefrectomie e
hanno notato che la glomeruolosclerosi focale segmentale era presente in 29 casi (4.9%) e 5 di
questi presentavano agenesia renale unilaterale con correlazione statisticamente significativa
(p < 0.05).

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 9
Gli stessi ricercatori su 9200 autopsie hanno trovato 7 casi di agenesia renale
unilaterale e in 2 (29%) di questi casi la causa della mote era stata una insufficienza renale
cronica conseguente a sclerosi focale segmentale. Al momento della diagnosi di agenesia
renale e glomeruloslerosi focale segmentale, l‟età media dei pazienti era di 25 anni.
I ricercatori concludevano che la presenza di agenesia renale unilaterale aumentava la
probabilità di sviluppare glomerulo sclerosi focale segmentale rispetto ai soggetti con due reni
normofunzionanti.
Nel 2009, Sanna Cherchi et al. hanno pubblicato uno studio di coorte longitudinale
retrospettivo con lo scopo di osservare l‟outcome a lungo termine dei soggetti con CAKUT
[19]. La popolazione di studio comprendeva 312 pazienti seguiti presso l‟Ospedale Pediatrico
G. Gaslini di Genova tra il 1980 e il 2000 con diagnosi di una delle malformazioni delle vie
urinarie appartenenti alle CAKUT.
I ricercatori hanno identificato sei fenotipi:
(A) Rene singolo con o senza anomaile delle vie urinarie;
(B) Ipodislasia unilaterale con o senza anomalie delle vie urinarie;
(C) Ipodislasia bilaterale con o senza anomalie delle vie urinarie;
(D) Ipodislasia renale associate alla presenza di valvole dell‟uretra posteriore;
(E) Rene multicistico;
(F) Rene a ferro di cavallo.
In particolare, la categoria A comprendeva sia l‟agenesia renale che la „empty renal
fossa‟ riscontrata durante lo studio di imaging (Scintigrafia renale statica ed ecografia renale)
a causa della involuzione di un rene multicistico.
L‟outcome principale dello studio era per le varie categorie l‟insufficienza renale
cronica in terapia dialitica e le variabili prognostiche analizzare erano la presenza di reflusso
vescico-ureterale, i valori di creatinina sierica, la presenza di proteinuria ≥ a 1 g per giorno, se
erano stati seguiti prima o dopo il 1990, l‟ età alla diagnosi, gli interventi chirurgici e l‟
esposizione a farmaci.

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 10
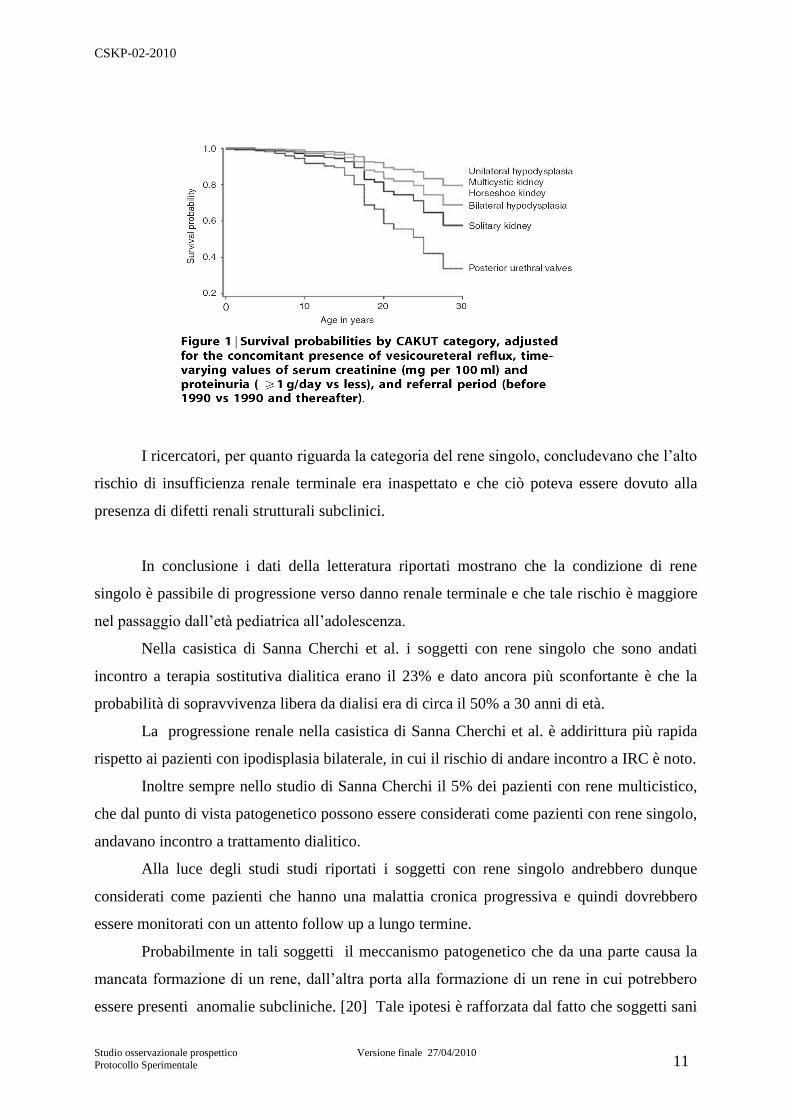
Figura 1 modificata da [19]
Su 312 soggetti studiati 71 (23%) appartenevano alla categoria del rene singolo (vedi
figura 1).
In questa categoria:
I maschi erano 52 (73%);
La diagnosi neonatale è stata posta nel 41% dei casi mentre nel restante
59% è stata più tardiva, in media attorno ai 15 anni d‟età;
all‟età della diagnosi, i livelli sierici di creatinina erano nella norma,
tranne per i pazienti diagnosticati alla nascita, che avevano livelli sierici di creatinina
mediamente più elevati;
alla diagnosi, il 7% presentava proteinuria e il 2,8% ipertensione;
Su 71 soggetti 21 (30%) sono andati incontro a trattamento dialitico
sostitutivo ad un età media di 21± 13.6 anni;
Tra le variabili prognostiche, quelle che hanno influenzato l‟outcome
studiato sono state la presenza di elevati livelli di creatinina sierica, di proteinuria,
soprattutto se > 1g/die e la presenza di reflusso vescicoureterale.
Inoltre gli Autori hanno calcolato la curva di sopravvivenza libera da dialisi a 30 anni
di vita delle varie categorie di CAKUT e questa per il rene singolo era del 50%.
CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 11
I ricercatori, per quanto riguarda la categoria del rene singolo, concludevano che l‟alto
rischio di insufficienza renale terminale era inaspettato e che ciò poteva essere dovuto alla
presenza di difetti renali strutturali subclinici.
In conclusione i dati della letteratura riportati mostrano che la condizione di rene
singolo è passibile di progressione verso danno renale terminale e che tale rischio è maggiore
nel passaggio dall‟età pediatrica all‟adolescenza.
Nella casistica di Sanna Cherchi et al. i soggetti con rene singolo che sono andati
incontro a terapia sostitutiva dialitica erano il 23% e dato ancora più sconfortante è che la
probabilità di sopravvivenza libera da dialisi era di circa il 50% a 30 anni di età.
La progressione renale nella casistica di Sanna Cherchi et al. è addirittura più rapida
rispetto ai pazienti con ipodisplasia bilaterale, in cui il rischio di andare incontro a IRC è noto.
Inoltre sempre nello studio di Sanna Cherchi il 5% dei pazienti con rene multicistico,
che dal punto di vista patogenetico possono essere considerati come pazienti con rene singolo,
andavano incontro a trattamento dialitico.
Alla luce degli studi studi riportati i soggetti con rene singolo andrebbero dunque
considerati come pazienti che hanno una malattia cronica progressiva e quindi dovrebbero
essere monitorati con un attento follow up a lungo termine.
Probabilmente in tali soggetti il meccanismo patogenetico che da una parte causa la
mancata formazione di un rene, dall‟altra porta alla formazione di un rene in cui potrebbero
essere presenti anomalie subcliniche. [20] Tale ipotesi è rafforzata dal fatto che soggetti sani

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 12
che donano un rene per il trapianto [21] presentano una prognosi a lungo termine eccellente.
Infatti in uno studio condotto su 3698 donatori, operati tra il 1963 e il 2007 e studiati tra il
2003 e il 2007, il rischio di sviluppo di insufficienza renale era più basso rispetto alla
popolazione normale (con un‟incidenza di 180 casi/milione/anno versus 268
casi/milione/anno nella popolazione generale).
Il fatto dunque che nella popolazione dei monoreni acquisiti il rischio di progressione
verso l‟insufficienza renale sia basso, rinforza l‟ipotesi che nei soggetti con monorene
congenito vi sia un alterazione strutturale inizialmente latente.
Oggi sappiamo che il numero di glomeruli nel singolo rene può variare, normalmente,
da 200mila a 1,8milioni [22]. L‟ipotesi suggerita da Chevalier [20] è che i pazienti con rene
singolo congenito presentino, in alcuni casi, un minor numero di nefroni rispetto alla
popolazione generale.
Il fatto che la presenza di una dotazione ridotta di nefroni possa predisporre
maggiormente allo sviluppo di condizioni patologiche è stato dimostrato per l‟ipertensione
arteriosa e lo sviluppo di danno renale già in soggetti con due reni apparentemente sani [23]
[24],[25].
Un esempio tipico è rappresentato dalla popolazione dei neonati pretermine o con
basso peso per età gestazionale (SGA) che presenta un basso numero di nefroni ed è
maggiormente a rischio di sviluppare insufficienza renale cronica rispetto alla popolazione di
nati a termine (appropriate for gestational age-AGA) [22]. Un altro esempio emerge da uno
studio caso controllo [26] effettuato su soggetti ipertesi deceduti in incidenti stradali che
rispetto a soggetti normotesi presentavano un minor numero di glomeruli (in media 702.379
vs.1.429.200).
E‟ utile quindi seguire nel tempo tali pazienti con un follow-up clinico laboratoristico-
strumentale più mirato a tale condizione. Attualmente, il monitoraggio di questi pazienti
prevede il controllo di:
1. Dieta e stato nutrizionale;
2. Crescita;
3. Pressione arteriosa;
4. Studio del filtrato glomerulare (GFR);

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 13
5. Studio di marker di danno precoce renale glomerulare (creatininemia, azotemia,
cistatina C, proteinuria e microalbuminuria);
6. Studio di marker di danno precoce tubulare ( B2 microglobulina, Lisozima urinario,
NGAL, NHE3, KIM1, NAG ) [27], [28], [29];
7. Monitoraggio strumentale della morfologia renale (ecografica e scintigrafica).
In particolare per quanto riguarda la determinazione della GFR il gold standard nella
determinazione della clearance renale si basa sul dosaggio radioisotopico di traccianti
glomerulari quali l‟acido dietilene triaminopentacetico marcato con tecnezio 99m (99mTc-
DTPA) o l‟acido etilene diaminotetracetico marcato con cromo 51 (51Cr-EDTA). Tali
tecniche sono infatti più affidabili della valutazione del GFR calcolata tramite la
concetrazione di creatinina plasmatica e meno invasive rispetto alla determinazione della
clearance dell‟inulina che richiede numerosi prelievi [29], [30].
In particolare tra le tecniche basate sul dosaggio radioisotipico, lo “slope-intercept”
method rappresenta quella con maggiore accuratezza diagnostica anche per clearance renali
molto basse (dell‟ordine di 10m/min) [31].
In conclusione i soggetti con rene singolo congenito sembrerebbero presentare un
maggior rischio di sviluppare ipertensione arteriosa, proteinuria e insufficienza renale di vario
grado.
Tali evidenze al momento vengono però alla luce solo da studi retrospettivi condotti
per lo più in soggetti adulti in cui la diagnosi di rene singolo era stata posta per lo più in
adolescenza o nella prima età adulata.
2.2 Disegno dello studio
Si tratta di uno studio osservazionale di coorte prospettico longitudinale della durata di
10 anni.
2.3 Popolazione
Pazienti con diagnosi certa di rene singolo congenito che afferiranno alla struttura di
nefrologia pediatrica dell‟ Unità Operativa di Pediatria Specialistica del Dipartimento Attività
integrata per la salute della donna, del bambino e dell‟adolescente Azienda Ospedaliero –

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 14
Universitaria di Bologna Policlinico Sant‟Orsola – Malpighi diretta da Prof. A. Cicognani dal
1 Aprile 2010 al 1 Aprile 2013.
Per questo studio si prevede l‟arruolamento di circa 100 soggetti.
Criteri di inclusione
Verranno arruolati:
Pazienti con diagnosi di rene singolo congenito già nota effettuata mediante
l‟esecuzione di ecografia renale e confermata con scintigrafia renale statica con
DMSA
Maschi e Femmine
Pazienti con età compresa tra 0-18 anni al momento dell‟entrata nello studio
Pazienti per i quali si sia ottenuto il consenso a partecipare allo studio da parte dei
genitori\esercenti la responsabilità genitoriale e dei ragazzi oltre i 14 anni.
La diagnosi di rene singolo congenito viene definita come presenza alla scintigrafia
renale con DMSA di un singolo parenchima renale funzionante con contributo funzionale
>95% tale condizione corrisponde ad uno dei seguenti casi:
L‟ Agenesia renale vera (RAG);
L‟Aplasia renale congenita con rene funzionalmente unico (RAP)
o Ipodisplasia renale grave;
o Il rene multicistico definito come una massa addominale
multilobulata con cisti a pareti sottili e prive di tessuto renale all‟interno,
uretere atresico, assenza di parenchima renale funzionante alla scintigrafia con
DMSA.
Criteri di esclusione
Presenza al momento dell‟arruolamento nello studio di:
Insufficienza renale definita come filtrato glomerulare calcolato
secondo Schwartz [31] al di sotto dei valori di normalità per età ed età gestazionale
stabiliti dalla National Kidney Foundation's Kidney Disease Outcomes Quality
Initiative clinical practice guidelines for chronic kidney disease in children and
adolescents [32].
Pazienti con rene singolo associato a valvole dell‟uretra posteriore;

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 15
Pazienti diabetici;
Pazienti con cardiopatie o malattie vascolari;
Pazienti con patologie autoimmuni.
2.4 Visite e valutazioni
1) Visite e valutazioni al momento dell‟inclusione nello studio:
Anamnesi dettagliata;
Visita (parametri auxologici + PAO);
Ecografia renale;
Cistografia minzionale (se reflusso > al III grado, profilassi antibiotica);
Esami laboratoristici per funzionalità glomerulare e tubulare (emocromo,
elettroliti, creatininemia, azotemia, cistatina C, esame urine, proteinuria,
microalbuminuria, elettroliti urinari, B2 microglobulina, Lisozima urinario,
NGAL, NHE3, KIM1, NAG, Emogas analisi venosa);
Scintigrafia con DTPA per studio clearance renale al tempo 0 ma non prima
dell‟età di due anni e comunque almeno tre mesi dopo l‟esecuzione di altra
metodica scintigrafica nucleare.
2 ) Visite e valutazioni di follow-up:
Visita, ecografia reni e vie urinarie, prelievo per funzionalità glomerulare e
tubulare ogni sei mesi fino ai due anni di vita poi annualmente (se al momento
dell‟inclusione il bambino ha più di due anni controllo ogni sei mesi per il primo
anno per eventuale completamento diagnostico e poi annuale)
Scintigrafia con DTPA di controllo a tempi stabiliti in base all‟età al momento
dell‟ingresso nello studio:
o Se età ≤ di 2 anni a 5 e 10 anni di età.
o Se età > di 2 anni a 5 e 10 anni di follow-up
Holter pressorio a tempi stabiliti in base all‟età al momento dell‟ingresso nello
studio:
o Se età ≤di 7 anni : all‟età di 7 anni, 10 anni e a fine follow-up;
o Se età >di 7 anni e < di 10 anni: prima determinazione al tempo 0 ed in
seguito a 10 anni ed in seguito a fine follow-up

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 16
o Se età ≥ di 10 anni: prima determinazione al tempo 0 ed in seguito a 5 e 10
anni di follow-up
CUMS di controllo solo su indicazione clinica ;
In caso di infezione delle vie urinarie con febbre esecuzione di scintigrafia con
DMSA a sei mesi dall‟evento.
2.5 Obiettivi dello studio
Lo studio si pone l‟obiettivo di determinare il rischio per i pazienti con rene singolo
congenito di sviluppare insufficienza renale terminale, insufficienza renale cronica,
ipertensione o proteinuria e gli eventuali fattori prognostici.
Gli outcome dello studio sono definiti nel modo seguente:
1. Insufficienza renale terminale definita secondo i criteri stability dalla National Kidney
Foundation's Kidney Disease Outcomes Quality Initiative clinical practice guidelines
for chronic kidney disease in children and adolescents [33] .
2. Insufficienza renale cronica definita secondo i criteri stability dalla National Kidney
Foundation's Kidney Disease Outcomes Quality Initiative clinical practice guidelines
for chronic kidney disease in children and adolescents [33].
3. Ipertensione arteriosa definita come valori di pressione PAS o PAD ≥ 95° secondo i
valori stabiliti per centile di altezza dal National High Blood Pressure Education
Program Working Group [34].
4. Proteinura su campione di urine definita come rapporto PrU/CrU (mg/mg) stabiliti
dalla National Kidney Foundation conference on proteinuria, albuminuria, risk,
assessment,detection, and elimination (PARADE) [35].
2.6 Gestioni dei dati ed analisi statistica
Le analisi statistiche verranno condotte utilizzando metodi di statistica descrittiva e
inferenziale adeguati alla numerosità del campione ed alle variabili dello studio.
2.7 Confidenzialità delle informazioni raccolte
I dati verranno raccolti in modo anonimo attribuendo ad ogni paziente selezionato
solamente un numero progressivo.

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 17
Il titolare del trattamento dei dati personali è l‟Azienda Ospedaliero-Universitaria di
Bologna, Policlinico S.Orsola-Malpighi e il responsabile è lo Sperimentatore principale dello
studio.
I dati emersi dalle analisi svolte saranno elaborati con metodi statistici per ricavarne le
informazioni che costituiscono lo scopo della ricerca.
Avranno quindi accesso ai dati il Responsabile dello studio e i suoi Collaboratori, che
saranno comunque vincolati all‟obbligo di confidenzialità e di trattamento dei dati stessi in
base alle norme vigenti.
Gli Addetti al monitoraggio e alla verifica, il Comitato Etico e le Autorità Regolatorie
potranno accedere direttamente alla documentazione medica per verificare le procedure dello
studio e/o i dati nella misura prevista dalle norme vigenti.
I dati saranno archiviati in forma anonima e identificati in base al numero di codice e
alle iniziali.
I risultati dello studio costituiranno il materiale per una pubblicazione scientifica, ma
anche in questo lavoro i dati saranno riportati in forma anonima.
2.8 Risultati attesi
Dai dati della letteratura risulta che il rischio di sviluppare l‟ipertensione arteriosa in
questa popolazione è di circa il 47%, quello di sviluppare proteinuria è del 19% e quello di
sviluppare insufficienza renale varia dal 13% al 20% a seconda delle casistiche.
Il nostro studio si pone l‟obiettivo di valutare la prevalenza di insufficienza renale terminale,
insufficienza renale cronica, ipertensione e proteinuria in una popolazione con monorene
seguita in maniera prospettica sin dall‟età pediatrica e di trovare le eventuali variabili
prognostiche che potrebbero influenzarle e quindi di prevenirle.

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 18
Bibliografia
1. Kass EJ, Bloom D (1992) Anomalies of urinary tract in Edelman CM, Pediatric renal
disease, Little Brown, Boston, pp.2023-2025Rajesh et Al., Congenital renal agenesis:
a rewiew. Saudi J Kidney Dis Transplant 2003; 14(1):336-341]
2. Masahiro et Al., Renal aplasia is the predominant cause of congenital solitary kidneys,
Kidney International(2002), pp.1840-1844
3. Wolff A et Hillman K., Unilateral renal agenesis and the congenital solitary
functioning kidney: developmental, genetic and clinical perspectives, Bju
International(2006); 99, 17-21
4. Zaffanello M. et Al., Are children with congenital solitary kidney at risk for life long
complications? A lack of prediction demands caution Int Urol Nephrol (2009) 41:127–
135.
5. Dursun e al Associated anomalies in children with congenital solitary functioning
kidney..; Pediatr Surg Int (2005) 21:456-459
6. Roodhooft AM, Birnholz JC, Holmes LB., Familial nature of congenital absence and
severe dysgenesis of both kidneys. N Engl J Med1984;310: 1341
7. McPherson E, Carey J, Kramer A et al., Dominantly inherited renal adysplasia. Am J
Med Genet 1987; 26:863–72
8. Arfeen S, Rosborough D, Luger AM, Nolph KD., Familial unilateral renal agenesis
and focal and segmental glomerulosclerosis. Am J Kidney Dis 1993;21: 663
9. Shapiro E, Goldfarb DA, Ritchey ML (2003) The congenital and acquired solitary
kidney. Rev Urol 5:2–8 .
10. Hill LM, Nowak A, Hartle R, Tush B. Fetal compensatory renal hypertrophy with a
unilateral functioning kidney. Ultrasound Obstet Gynecol 2000; 15:191–3
11. Kiprov DD, Colvin RB McCluskey RT, Focal and segmental glomerulosclerosis and
proteinuria associated with unilateral renal agenesis. Lab Invest 1982 Mar¸46(3):275-
81
12. Abidari JM, Park KH, Kennedy WA, Shortliffe LD, Serial follow up of the
contralateral renal size in children with multicystic dysplastic kidney.. J Urol. 2002
Oct;168(4 Pt 2):1821-5; discussion 1825.
13. Douglas-Denton R, Moritz KM, BertramJF, Wintour EM, Compensatory renal growth
after unilateral nephrectomy inthe ovine fetus. J Am Soc Nephrol 2002;13: 406–10
14. Wasilewska A, Zoch-Zwierz W,Jadeszko I et al. Assessment of serum cystatin C in
children with congenital solitary kidney. Pediatr Nephrol 2006; 21:688–93
15. Mei-Zahav M, Korzets Z, Cohen I et al. Ambulatory blood pressure monitoring in
children with a solitary kidney – a comparison between unilateral renal agenesis and
uninephrectomy. Blood Press Monit 2001; 6: 263–7
16. Argueso LR , Ritchey ML , Boyle ETJ et al., Prognosis of patients with unilateral
renal agenesis. Pediatr Nephrol 1992
17. Fotino S., The solitary kidney: a model of chronic hyperfiltration in humans.1989. Am
J Kidney Dis 13:88–98
18. Sanna-Cherchi S. et Al., Renal outcome in patients with congenital anomalies of the
kidney and urinary tract. Kidney International(2009) 76, 528-533

CSKP-02-2010
Studio osservazionale prospettico Versione finale 27/04/2010
Protocollo Sperimentale 19
19. Chevalier et Al., When is one kidney not enough? Kidney International(2009), 76,
475-477
20. Ibrahim HN , Foley R , Tan L et al., Long-term consequences of kidney donation, N
Engl J Med 2009 ; 3 60: 4 59– 4 69.
21. Hughson M, Farris AB 3rd, Douglas-Denton R, Hoy WE, Bertram JF, Glomerular
number and size in autopsy kidneys: the relationship to birth weight.. Kidney Int. 2003
Jun;63(6):2113-22
22. Brenner BM Lawler EV., The hyperfiltration theory: a paradigm shift in nephrology.
(1996) Kidney international 49:1774-1777
23. Brenner BM Garcia DL., Glomeruli and blood pressure. Less of one, more of the
other? (1988) Am J Hypertens 1:335-347
24. Brenner BM MacKenzie HS, Nephron mass as a risk factor for progression of renal
disease (1997) Kidney Int Suppl 63:S124-S127
25. Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with
essential hypertension. N Engl J Med 2003; 348: 101–8
26. Parikh C, Devarajan P. New Biomarkers of acute kidney injury. Crit Care Med
2008;36:S159-S165.
27. Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a urinary biomarker and much
more. Nephrol Dial Transpl 2009.
28. Mitsnefes MM, Kathman TS, Mishra J et al. Serum neutrophil gelatinase-associated
lipocalin as a marker of renal function in children with chronic kidney disease.Pediatr
Nephrol (2007) 22:101–108.
29. Blaufox MD, Aurell M, Bubeck B et al. Report of the Radionuclides in
Nephrourology Committee on renal clearance. J Nucl Med 1996; 37:1883-1890.
30. Piepsz A, Colarinha P, Gordon I et al: Guidelines for glomerular filtration rate
determination in children. EANM guidelines
31. Piciotto G, Cacace G, Cesana P, et al: Estimation of chromium-51 ethylene diamine
tetra-acetic acid plasma clearance: a comparative assessment of simplified techniques.
Eur J Nucl Med 1992; 19:30-35.
32. Schwartz GJ, Brion LP, Spitzer A. The use of plasma creatinine concentration for
estimating glomerular filtration rate in infants, children, and adolescents. Pediatric
Clinics of North America 1987; 34:571-590
33. Hogg RJ, Furth S, Lemley KV, Portman R, Schwartz GJ, Coresh J, Balk E, Lau J,
Levin A, Kausz AT, Eknoyan G, Levey AS; National Kidney Foundation's Kidney
Disease Outcomes Quality Initiative. Pediatrics. 2003 Jun;111(6 Pt 1):1416-21
34. National High Blood Pressure Education Program Working Group. The fourth report
on the diagnosis, evaluation and treatment of high blood pressure in children and
adolescents. Pediatrics 2004;114 (suppl):555-76
35. Hogg RJ, Portman RJ, Milliner D, Lemley KV, Eddy A, Ingelfinger J. Evaluation and
management of proteinuria and nephrotic syndrome in children:recommendations
from a pediatric nephrology panel established at the National Kidney Foundation
conference on proteinuria, albuminuria, risk, assessment,detection, and elimination
(PARADE). Pediatrics. 2000 Jun;105(6):1242-46.