
MALATTIE RENALI RARE - rtdc.itrtdc.it/Download/Presentazioni_XIII/Egidi.pdf · - Acidosi tubulare...
36
MALATTIE RENALI RARE M. Francesca Egidi Domenico Giannese U.O. Nefrologia Trapianti e Dialisi Azienda Ospedaliero-Universitaria Pisana NEFROLOGIA, DIAGNOSI PRENATALE NON INVASIVA, GENOMICA CARRARA 23 OTTOBRE 2014
Transcript of MALATTIE RENALI RARE - rtdc.itrtdc.it/Download/Presentazioni_XIII/Egidi.pdf · - Acidosi tubulare...

MALATTIE RENALI RARE
M. Francesca Egidi Domenico Giannese
U.O. Nefrologia Trapianti e Dialisi
Azienda Ospedaliero-Universitaria Pisana
NEFROLOGIA, DIAGNOSI PRENATALE NON INVASIVA, GENOMICACARRARA 23 OTTOBRE 2014

Il Parlamento europeo definisce rare le malattie a bassa prevalenza nella popolazione: 5 casi su 10.000 abitanti.
La sterilità di un numero non può definire l’idea dei problemi, numerosi e complessi, connessi a queste patologie.
La scarsa disponibilità di conoscenze scientifiche, che scaturisce dalla rarità, determina spesso lunghi tempi di latenza tra esordio della patologia e diagnosi. Tempi che incidono negativamente sulla prognosi del paziente.
DEFINIZIONE

REGOLAMENTI
Il Ministero della Salute ha individuato con il Decreto ministeriale 279/2001 circa 350 malattie rare, cui assicurare l'assistenza in esenzione dalla quota di partecipazione attraverso una rete di presidi dedicati.
La Regione Toscana con la delibera di Giunta regionale n. 90 del 2009 ha riconosciuto altre 86 patologie e con decreto 1088/2013 ha aggiornato la rete dei presidi regionali dedicati alla diagnosi e cura delle medesime patologie.

REGIONE TOSCANA
Progetto regionale sulle malattie rare si è sviluppato in Toscana fin dal 2001 in collaborazione con le associazioni dei malati raccolte nel forum delle associazioni toscane malattie rare (delibera di giunta regionale n.796/2001).
Il forum è punto di riferimento per i pazienti e dei loro familiari che vivono un'esperienza doppiamente dolorosa rappresentata sia dalla condizione morbosa che dalla condizione di solitudine, legata alla scarsità di conoscenze scientificamente disponibili e professionalmente utilizzabili.

Il modello assistenziale toscano dedicato ai soggetti affetti da malattie rare si fonda sulla tracciabilità dei percorsi diagnostico-terapeutici, la rete dei presidi, lo sviluppo della ricerca ed il registro delle malattie rare.
http://www.malattierare.toscana.it/
REGIONE TOSCANA


Registro malattie rare
Nel 2002 è stato effettuato un censimento preliminare che ha
coinvolto le 4 Aziende ospedeliere e le 12 asl della regione toscana per conoscere il numero di pazienti in carico, l’attività scientifica e l’esperienza diagnostica e terapeutica per ciascuna MR
Nel 2005 è stato attivato il registro toscano delle malattie rare, gestito dalla fondazione G. Monasterio con la finalità di raccogliere i dati inerenti all’epidemiologia delle malattie rare.
Sulla base dei dati inseriti il 30 giugno 2009 sono stati assegnati a ciascun presidio i ruoli specifici (certificazione diagnosi, diagnosi, terapia, follow-up, consulenza genetica etc…)


LE MALATTIE RARE IN AMBITO NEFROLOGICO

“EPIDEMIOLOGIA E PROGNOSI DELLA MALATTIA RENALE CRONICA IN ITALIA” GAROFALO C. ET AL G ITAL NEFROL 2012;29 (S58): S3-S11
EPIDEMILOGIA DELLA CKD
STUDIO CARHES (CARDIOVASCULAR RISK IN RENAL PATIENTS OF THE ITALIAN HEALTH EXAMINATION SURVEY) : 7% DELLA POPOLAZIONE ITALIANA
TRA 35 E 79 ANNI, STIMABILE COME 2 MILIONI DI PERSONE, E’ AFFETTA DA INSUFFICIENZA RENALE CRONICA
«EPIDEMIOLOGIA DELLA MALIATTIA RENALE CRONICA IN ITALIA: STATO DELL’ARTE E CONTRIBUTO DELLO STUDIO CARHES.» DE NICOLA ET AL (G ITAL NEFROL 2011; 28 (4) 401-7)
EPIDEMILOGIA DELLA CKD

“The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia” Orphanet J Rare Dis. 2014; 9: 98. Published online Jun 30, 2014. doi: 10.1186/1750-1172-9-98
EPIDEMILOGIA DELLA MALATTIA RENALE RARA

Stima dell’incidenza della malattia renale rara in italia: 196.000 soggetti pari a 0.32% della popolazione compresa tra 35 e 79 anni.
Esiste un rischio concreto che il paziente portatore di una malattia rara renale rimanga isolato se la sua patologia non viene riconosciuta.
EPIDEMILOGIA DELLA MALATTIA RENALE RARA

EPIDEMILOGIA DELLA MALATTIA RENALE RARA
Malattie rare di interesse nefrologico- Acidosi tubulare distale (tipo I, II, III, IV)- Sindrome di Bartter- Sindrome di Gitelman- Sindrome di Dent- Cistinuria- Rene midollare a spugna- Renal-coloboma syndrome- Rene policistico (dominante o recessivo)- SEU- Porpora trombotica trombocitopenica- Sindrome di Alport- Malattia di Fabry- Sindrome di Von Hippel-Lindau- Sindrome di Senior-Loken- Glomerulonefrite membrano-proliferativa tipo II- Sindromi nefrosiche ereditarie- Amiloidosi familiare e senile- ………………..

Difficoltà diagnostiche
Scarsa conoscenza medica.
Alterazioni genetiche non ancora note.
Assenza di biomarcatori specifici di patologia.
Eterogeneità clinica e manifestazioni comuni a diversi quadri patologici.
Assenza di modelli animali.

GESTIONE INTEGRATA
• Medico di medicina generale: spesso primo approccio al paziente non pediatrico
• Specialista (Nefrologo, Pediatra, Genetista…)- inquadramento- certificazione di esenzione (strutture di coordinamento) - terapia specifica- gestione IRC
q terapia conservativaq dialisiq trapianto

GESTIONE CKD NELLA MALATTIA RENALE RARA
Ridurre i fattori di progressione:- Trattare l’ipertensione arteriosa- Interruzione del fumo- Controllo glicometabolico- Ridurre l’obesità- Controllo dell’uricemia
Terapia- Specifica per la malattia (Tiopronina per la cistinuria, Agalsidasi per
la malattia di Fabry…)- Terapia farmacologica/nutrizionale- Trattamento dialitico personalizzato (Iperossaluria primitiva nel
bambino)

TRAPIANTO RENALE
Il trapianto renale nel paziente con patologia rara è una sfida intricata. È necessario valutare:
- Le comorbilità connesse e non connesse alla patologia rara- La possibilità di recidiva della patologia sul rene trapiantato- Se avviare il paziente a un programma di donazione da vivente
(necessaria una valutazione genetica del nucleo familiare o eventualmente escludere i consanguinei)
In quest’ottica è necessario affidare il paziente ad un centro trapianti con esperienza nel campo delle malattie rare

ESPERIENZA DI PISA

Una delle patologie lisosomiali, geneticamente trasmessaattraverso il cromosoma X
L’incidenza stimata è pari a 1:117.000 maschi, colpisce entrambi isessi.
Causata da carenza di Alfa-galattosidasi A (enzima lisosomialeche degrada i glicosfingolipidi) con accumulo patologico di Globotriaosilceramide (Gb3), con conseguente deficit d’organo e morte prematura per disfunzione cellulare
MALATTIA DI FABRY

Storia
Nel 1989 descritto interessamento cutaneo (angiocheratoma
corporis diffusum) da William Anderson e Johannes Fabry, dermatologo tedesco
Malattia
xx
xy xx
xxxx
xx xx
xx
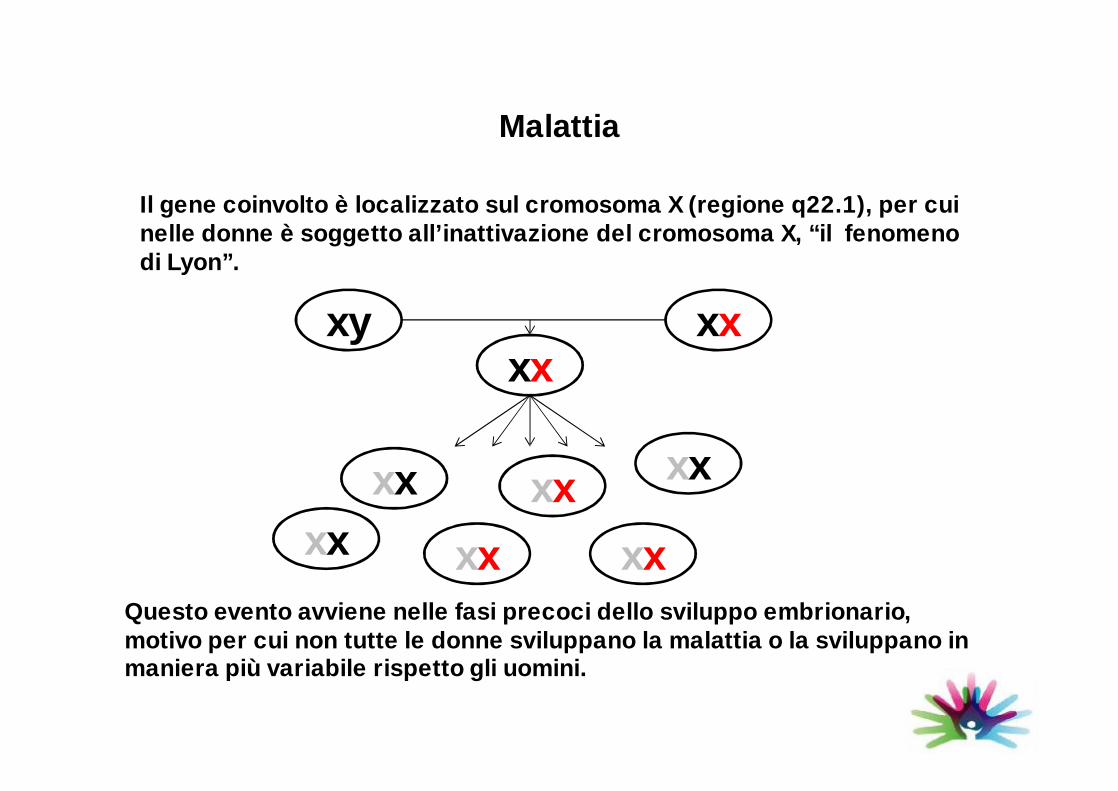
Il gene coinvolto è localizzato sul cromosoma X (regione q22.1), per cui nelle donne è soggetto all’inattivazione del cromosoma X, “il fenomeno di Lyon”.
xx
Questo evento avviene nelle fasi precoci dello sviluppo embrionario, motivo per cui non tutte le donne sviluppano la malattia o la sviluppano in maniera più variabile rispetto gli uomini.
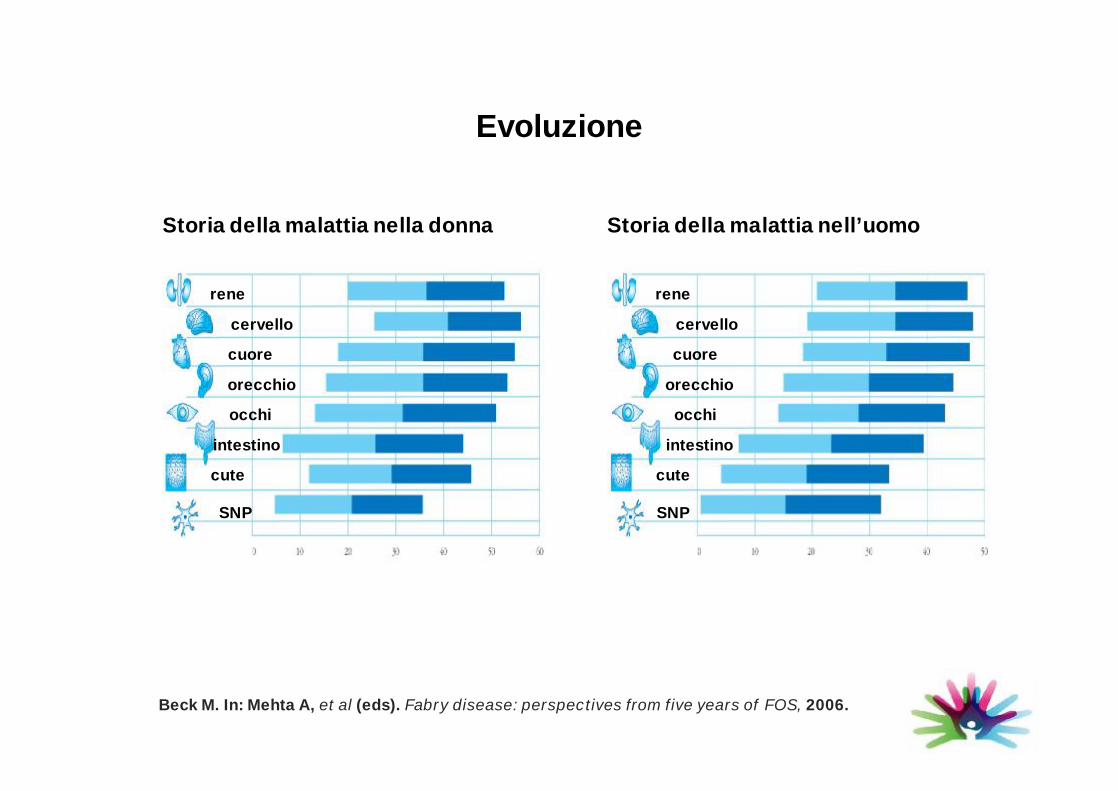
Evoluzione
Beck M. In: Mehta A, et al (eds). Fabry disease: perspectives from five years of FOS, 2006.
Storia della malattia nell’uomoStoria della malattia nella donna
rene
cervello
cuore
orecchio
intestino
occhi
SNP
rene
cute
SNP
cute
intestino
occhi
orecchio
cuore
cervello
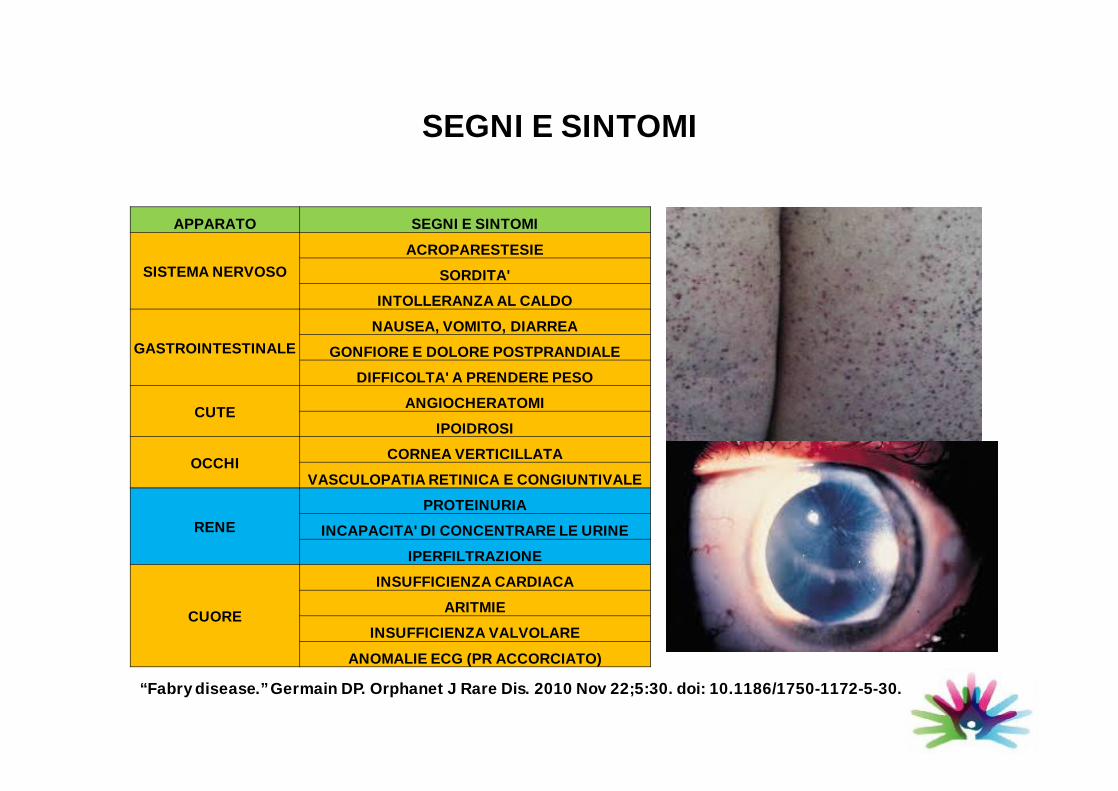
SEGNI E SINTOMI
APPARATO SEGNI E SINTOMI
SISTEMA NERVOSO
ACROPARESTESIE
SORDITA'
INTOLLERANZA AL CALDO
GASTROINTESTINALE
NAUSEA, VOMITO, DIARREA
GONFIORE E DOLORE POSTPRANDIALE
DIFFICOLTA' A PRENDERE PESO
CUTEANGIOCHERATOMI
IPOIDROSI
OCCHICORNEA VERTICILLATA
VASCULOPATIA RETINICA E CONGIUNTIVALE
RENE
PROTEINURIA
INCAPACITA' DI CONCENTRARE LE URINE
IPERFILTRAZIONE
CUORE
INSUFFICIENZA CARDIACA
ARITMIE
INSUFFICIENZA VALVOLARE
ANOMALIE ECG (PR ACCORCIATO)
“Fabry disease.” Germain DP. Orphanet J Rare Dis. 2010 Nov 22;5:30. doi: 10.1186/1750-1172-5-30.
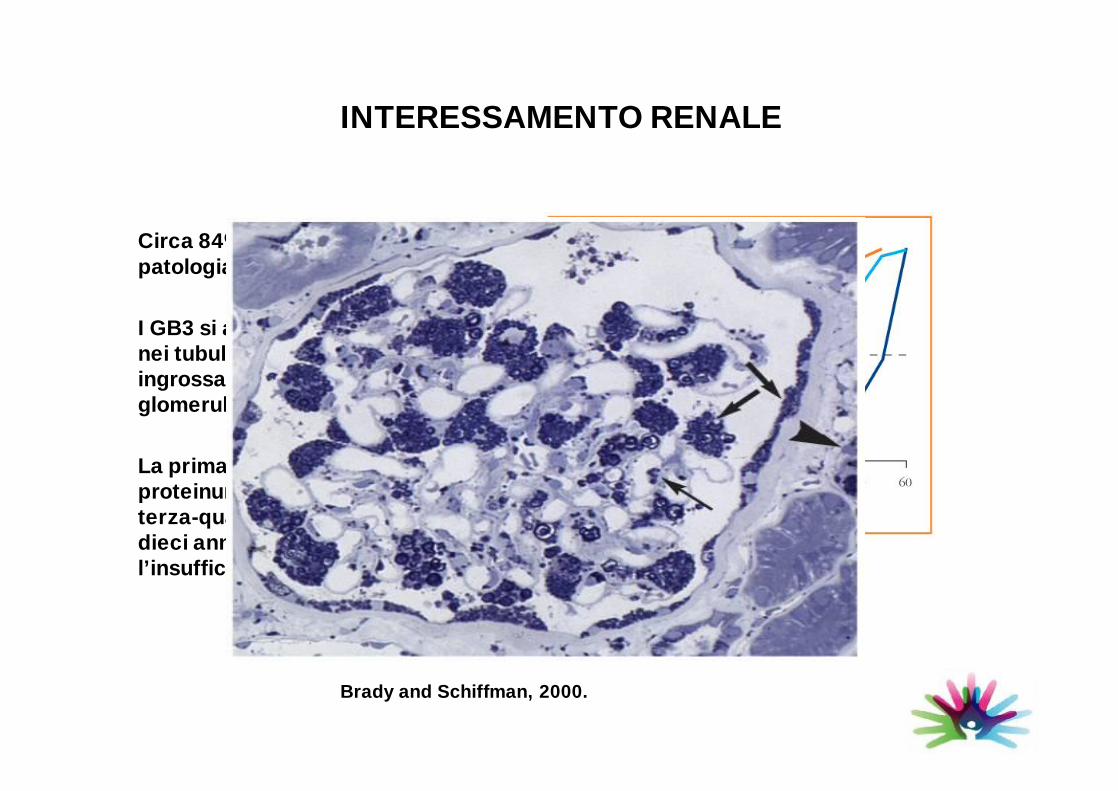
INTERESSAMENTO RENALE
ProteinuriaIRCMorte
Pa
tie
nts
(c
um
ula
tive
%)
Circa 84% dei pazienti presenta unapatologia renale
I GB3 si accumulano nei glomeruli e nei tubuli con conseguenteingrossamento del mesangio e glomerulosclerosi
La prima manifestazione è la proteinuria, che si manifesta nellaterza-quarta decade. A distanza didieci anni circa si manifestal’insufficienza renale.
Branton et al, 2002
Brady and Schiffman, 2000.

EVOLUZIONE

CASO CLINICO
Maschio, nato nel 1972
Riferiti durante l’infanzia episodi di acroparestesie. Durante l’adolescenza anidrosi e scarsa tolleranza al caldo.
Nel 1995 riferita comparsa di angiocheratomi in corrispondenza del cingolo pelvico e dei fianchi.
Primo riscontro di proteinuria (range non nefrosico) nel 2003 (31 anni) e insufficienza renale (creatininemia 1,3 mg/dl). Nel 2006 a seguito del peggioramento della funzione (2 mg/dl) e per la comparsa di sindrome nefrosica effettuata biopsia renale: Malattia di Fabry, confermata anche dallo studio genetico.
Iniziata terapia con agalsidasi Beta.

CASO CLINICO
Si assiste ad un progressivo peggioramento della funzione renale: Gennaio 2013 creatininemia 7,5 mg/dl.
Si decide di iniziare il trattamento sostitutivo. Il paziente opta per la dialisi peritoneale. In data 16/05/2013 viene posizionato il catetere peritoneale e inizia l’addestramento il 20/05/2013.

CASO CLINICO
Trapianto:- Da cadavere
Soluzione ideale per la sua patologia ma che prospettava lunghi tempi di attesa.
- Da vivente
Figlio unico, non coniugato.
Padre vivente non candidabile per motivi clinici.
Madre vivente di 68 anni. Candidabile?

Caso clinico
Madre:Non comorbilità significative, non ipertesa, non diabetica.
Funzione renale normale, confermata anche da filtrato separato renale con metodica scintigrafica.
Esame urine con sedimento negativo e proteinuria assente.
Portatrice di malattia di Fabry?
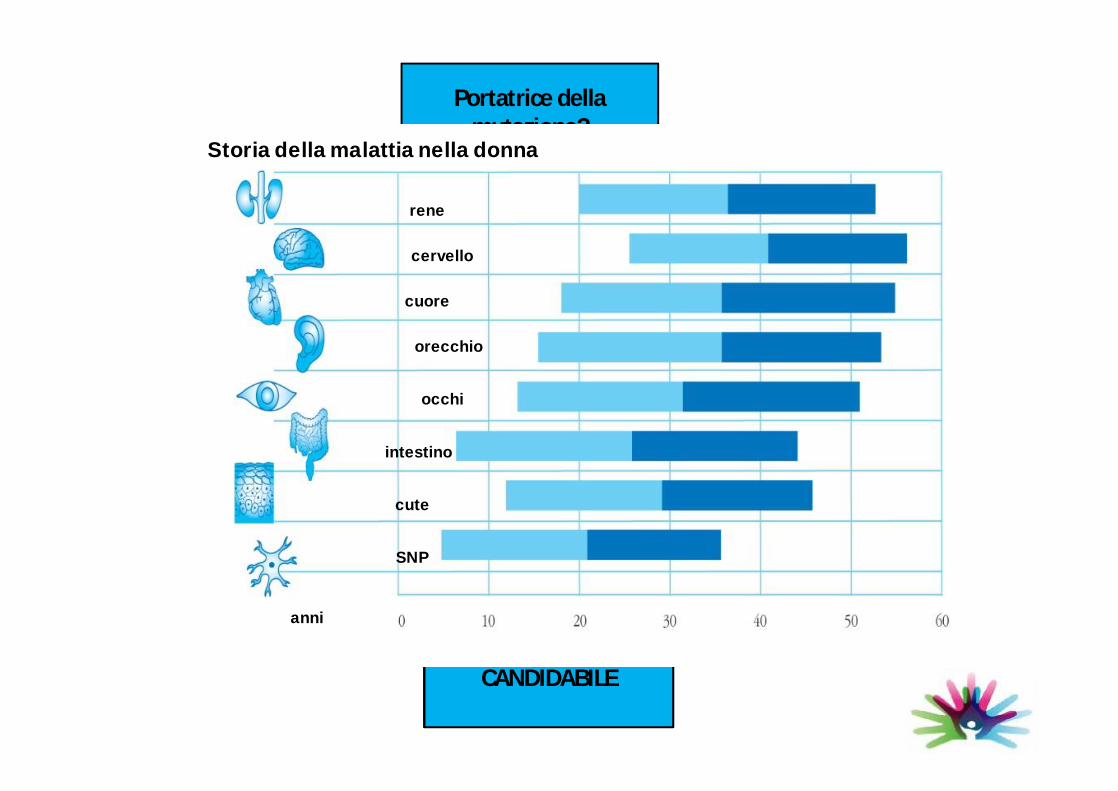
Portatrice della mutazione?
CANDIDABILE
Manifestazioni cliniche di malattia
Inattivazione precoce del cromosoma con
mutazione
Mutazione ex novo
NO
SI
NO
CANDIDABILE
Storia della malattia nella donna
cervello
cuore
orecchio
intestino
occhi
SNP
rene
cute
anni

Caso clinico
In data 21 Maggio 2014 effettuato il trapianto da donatrice (madre)
vivente
Il paziente viene dimesso in data 29 Maggio 2014: creatininemia 1.9 mg/dl
01 Ottobre 2014: creatininemia 1.8 mg/dl. Proteinuria assente
Il paziente continua con le somministrazioni di agalsidasi Beta.

Conclusioni
E’ necessaria una sempre maggior informazione ed aggiornamento del
personale medico in merito alle malattie rare.
E’ importante concentrare i pazienti verso centri di riferimento specializzati per patologia, al fine di ottimizzare la gestione del malato.
L’individuazione dei centri trapianto in grado di gestire la patologia renale rara dovrebbe essere estesa in ambito nazionale.

Conclusioni
Rimane di fondamentale importanza potenziare la rete di gestione delle malattie rare coinvolgendo la medicina del territorio, presidi ospedalieri periferici, i centri di alta specializzazione e le strutture di genetica.
E’ necessario in tal senso anche il coinvolgimento delle associazione dei malati per migliorare l’assistenza di pazienti e dei familiari che combattono con una patologia che rimane emotivamente impegnativa da affrontare.

“Prima le buone notizie: è stato dato il suo nome a una nuova malattia.”
Wiet van Broeckhoven

Grazie dell’attenzione


















