Linfoma a cellule B: Paragone tra due metodiche di ...
52
Linfoma a cellule B: Paragone tra due metodiche di Biologia Molecolare per una diagnosi più efficace Linda Olsson Scuola Superiore Medico Tecnica Locarno Anno 2008/’09 Lavoro di Diploma eseguito presso: Laboratorio di Diagnostica Molecolare dell’Istituto Cantonale di Patologia Locarno Responsabile: Dr. Milo Frattini
Transcript of Linfoma a cellule B: Paragone tra due metodiche di ...

Linfoma a cellule B: Paragone tra due metodiche di Biologia Molecolare per una diagnosi più efficace
Linda Olsson Scuola Superiore Medico Tecnica
Locarno Anno 2008/’09
Lavoro di Diploma eseguito presso:
Laboratorio di Diagnostica Molecolare dell’Istituto Cantonale di Patologia
Locarno
Responsabile: Dr. Milo Frattini

2
Indice
• Riassunto/Abstract………………………………………………. Pag. 3
• Scopo del lavoro…………………………………………………. Pag. 4
• Introduzione………………………………………………………. Pag. 5
• Pazienti materiali e metodi……………………………………… Pag. 8 i. Casistica campioni…………………………………………………………. Pag. 8
ii. Materiale e metodi…………………………………………………………. Pag. 9
a) Estrazione DNA da paraffina………………………………………………. Pag. 9
b) Amplificazione mediante PCR……………………………………………... Pag. 11
c) PCR KRAS…………………………………………………………………... Pag. 12
d) Preparazione gel d’agarosio 1,8% e visualizzazione PCR KRAS……... Pag. 13
e) PCR per linfoma a cellule B metodica LDM-ICP………………………… Pag. 15
f) PCR per linfoma a cellule B metodica BIRD……………………………... Pag. 16
g) Analisi dei frammenti……………………………………………………….. Pag. 17
• Risultati……………………………………………………………. Pag. 20 i. Falsi negativi………………………………………………………………... Pag. 20
ii. Positivi certi…………………………………………………………………. Pag. 20
iii. Negativi certi………………………………………………………………... Pag. 21
iv. Non analizzabili…………………………………………………………….. Pag. 22
• Osservazioni……………………………………………………… Pag. 24
• Conclusioni……………………………………………………….. Pag. 25
• Ringraziamenti……………………………………………………. Pag. 27
• Bibliografia………………………………………………………… Pag. 28
• Allegati…………………………………………………………….. Pag. 29 i. Registro tumori Ticino 2005. Alcuni dati epidemiologici del linfoma.. Pag. 29
ii. Diss et al, J Clin Pathol 1994;47:493-496……………………………... Pag. 31
iii. Protocollo LDM-ICP Fr3-JH…………………………………………….. Pag. 35
iv. Protocollo LDM-ICP Fr2-JH…………………………………………….. Pag. 41
v. Protocollo ditta BIRD…………………………………………………….. Pag. 47

3
Riassunto
Lo scopo di questo lavoro di diploma è stato quello di verificare, tramite tecniche di biologia molecolare, l’efficacia d’individuazione di linfomi a cellule B con una nuova metodica, registrata con marchio CE (Comunità Europea), della ditta BIRD (Biotecnologia Innovativa per Ricerca e Diagnostica, Dia-chem S.r.l. Napoli) per confronto con quella già utilizzata dal Laboratorio di Diagnostica Molecolare dell’Istituto cantonale di Patologia (ICP-LDM), la quale da un 30% di falsi negativi. Durante la differenzazione, e dunque la maturazione dei linfociti a cellule B, avviene un processo chiamato riarrangiamento che permette la creazione di sequenze uniche di DNA per la codificazione delle immunoglobuline. Tali riarrangiamenti sono sfruttati come marker clonali nei linfomi a cellule B. Entrambe sfruttano l’amplificazione di acidi nucleici mediante PCR (Polymerase Chain Reaction) e l’analisi dei frammenti su sequenziatore. La metodica utilizzata ora dal ICP-LDM si basa su due distinte PCR che amplificano due distinte regioni, e che originano due frammenti di lunghezza differente, mentre la metodica del Kit BIRD prevede una semi-nested-PCR, basata comunque sull’amplificazione di due distinte regioni. Le regioni oggetto dell’amplificazione sono uguali tra i due metodi, anche se i primers ibridano in posizioni differenti. L’analisi è stata condotta su un totale di 35 campioni bioptici fissati in formalina ed inclusi in paraffina così riportati: 11 campioni falsi negativi, 12 campioni positivi certi, 10 campioni negativi certi e 3 campioni non analizzabili. Il Kit BIRD è riuscito ad identificare un unico campioni in più rispetto alla metodica LDM-ICP, tuttavia ha anche dato 3 falsi positivi. In campioni negativi certi ha identificato picchi monoclonali; per di più i costi di un kit sono molto maggiori. Ciò ci ha indirizzati sul mantenimento della metodica attualmente in uso presso l’ICP-LDM, e poter affermare che è paragonabile, se non migliore, della metodica della ditta BIRD registrata CE.
Abstract
The Aim of this diploma work was to evaluate, with molecular biology techniques, a new method (European Union registered) from the company BIRD (Biotechnology Innovation for Research and Diagnostics) for the detection of B cell monoclonality, in comparision with the method currently in use at the Laboratory of Molecular Diagnostics of the Institute of Pathology Locarno (LDM-ICP), which currently gives a false negative rate of 30%. Normal lymphoid cells udergo rearrangements causing specifity for the immunoglobilin that they produce. Monoclonal proliferations are presumed to be neoplastic; polyclonal populations are not. Both methods use amplification of nuclear acid with Polymerase chain reaction (PCR) and analysis of fragments with automated DNA sequencing LDM-ICP method use 2 PCR instead of the BIRD method use 2 semi-nested-PCR. Both amplified the same regions, (framework 2 to joining region [Fr2/JH] and framework 3 to joining region [Fr3/JH]) but use different primers. The study was carried out on 36 tissue samples fixed in formalin and wax embedded 6-7 section 3 µm. They were divided into 4 classes: 12 false negative samples, B cell Lymphomas in histology analysis but without monoclonal rearrangements of IgH; 11 true Positive samples, B cell Lymphomas in histology and molecular analysis; 12 true negative samples, T cell Lymphomas or inflammations and 3 not analysable samples, in this sample the DNA was too fragmented for LDM-ICP method. The kit BIRD detected only one sample of true cell B lymphoma more than method LDM- ICP; however it also produced 3 false positives, that is monoclonality in samples with T cell Lymphomas or inflammations. Both significantly higher costs and other increased problems associated with the kit in comparison with the older method led to the decision to maintain the LDM-ICP method and affirm that it is comparable or better than the BIRD method.

4
Scopo del lavoro In questo lavoro è descritto un confronto tra due metodi per individuare la presenza di linfoma a
cellule B in un prelievo bioptico tramite analisi di biologia molecolare. Entrambi prevedono
l’estrazione di acidi nucleici da tessuto fissato in formalina ed incluso in paraffina, la successiva
amplificazione del frammento di interesse con tecniche di PCR (Polymerase Chain Reaction) e
l’analisi dei frammenti su sequenziatore.
Il metodo utilizzato attualmente dal Laboratorio di Diagnostica Molecolare dell’Istituto Cantonale di
Patologia di Locarno (LDM-ICP) è basato sulla letteratura e fornisce un 30% circa di falsi negativi
su campioni classificati come franchi linfomi sulla base delle analisi morfologiche ed
immunoistochimiche eseguite da medici patologi. Lo scopo del presente lavoro è di verificare
l’efficacia d’individuazione di linfomi a cellule B con una nuova metodica registrata con marchio CE
(Comunità Europea), della ditta BIRD (Biotecnologia Innovativa per Ricerca e Diagnostica, Dia-
chem S.r.l. Napoli) per confronto con quella già utilizzata dall’LDM-ICP. Sulla base dei risultati
ottenuti, il responsabile del laboratorio deciderà quale metodica verrà utilizzata in futuro per la
diagnosi di tali neoplasie.
In questo modo avrò l’opportunità di avere una formazione approfondita sulle tecniche di biologia
molecolare applicate nei laboratori di patologia per la diagnosi di malattie neoplastiche; inoltre, i
risultati potranno fornire indicazioni utili al personale del laboratorio nell’ottica di una diagnosi più
efficace del linfoma a cellule B.

5
Introduzione
Il linfoma non Hodgkin a cellule B (NHL) è una proliferazione neoplastica incontrollata di cellule di
origine linfatica, implicate nella difesa immunitaria dell’organismo [3]. Il linfoma a cellule B è una
neoplasia frequente che aumenta con l’avanzare dell’età (solitamente >65 anni) [2,4] con un’
incidenza in Ticino di 20 nuovi casi all’anno per gli uomini e 22 per le donne [4]; la diagnosi viene
posta istopatomorfologicamente su un prelievo bioptico del tessuto interessato (linfonodi, milza,
midollo osseo,..) con l’aiuto della immunofenotipizzazione e della clinica [2]; in caso di dubbio da
parte del medico patologo viene richiesto un supporto molecolare, per confermare o meno la
diagnosi di linfoma a cellule B.
Nel corredo genetico di una cellula B immatura sono presenti tratti di DNA ipervariabili che durante
la loro differenziazione subiscono riarrangiamenti. Questi riarrangiamenti sono determinati da
perdite randomizzate di tratti di DNA con la formazione di un gene, chiamato VDJ, che andrà a
codificare la catena pesante di un’immunoglobulina (IgH), specifica per ogni cellula [1]. Ogni
cellula, duplicandosi darà luogo ad una progenie cellulare, caratterizzata dalla produzione di uno
specifico anticorpo.
In una cellula immatura è presente la regione V dove sono presenti i segmenti VH1, VH2, VH3, VH…,
VHn, una regione D dove sono presenti i segmenti DH1-15 e una regione J dove sono presenti i
segmenti JH1-6 . Durante la differenziazione, e dunque la maturazione, alcuni segmenti delle regioni
sopraccitate vengono eliminati, con conseguente riarrangiamento della zona circostante. I
frammenti mantenuti formano cosi un frammento VDJ specifico per ogni clone [1,5](Vedi figura 1).
Figura 1. Riarrangiamento della catena pesante e variabile delle Immunoglobuline (IgH). Le frecce indicano i due distinti riarrangiamenti; il primo con la formazione della regione DJ, mantenendo solo alcuni tratti del DNA della regione D e della regione J. Il secondo riarrangiamento con la formazione della regione VDJ definitiva per ogni cellula ed è evidenziato con il cerchio.

6
La struttura della regione variabile del gene IgH è composta da una regione VH dove sono presenti
delle zone framework (Fr1, Fr2 e Fr3) che sono conservate per tutti i linfociti B, due zone
determinanti la complementarietà (CDR I e CDR II) ipervariabili e specifiche per ogni clone per la
produzione di un solo anticorpo. Nella regione DH è presente il CDR III, mentre nella zona JH è
presente Fr4. Per unire le diverse regioni (VH, DH e JH) sono presenti delle zone di inserzione “N”
[1] (vedi figura 2).
Nel linfoma a cellule B, in un qualsiasi momento della vita cellulare, un clone di linfocita B può
subire un’alterazione a livello del DNA, solitamente traslocazioni, a seconda del tipo di neoplasia.
Un esempio è il Linfoma diffuso a grandi cellule B (DLBCL) che tipicamente subisce traslocazioni
t(14;18) e t(3;14) [5], mentre il linfoma tipo MALT (Tessuto linfatico mucosa-associato) subisce una
traslocazione t(11;18)(q21;q21) [8].
Queste traslocazioni spesso interessano il promotore del gene. Ciò porta ad un’espressione più
accentuata del gene VDJ di un determinato clone che prende il sopravvento sulle altre cellule
originando un’ espansione neoplastica[9].
La metodica utilizzata ora dal LDM-ICP si basa su due distinte PCR che amplificano due distinte
regioni, originando due frammenti di lunghezza differente. I primers utilizzati per una PCR ibridano
nelle zone Fr2 e JH dando origine a frammenti compresi tra 250 e 350 bp (vedi figura 2), nell’altra
PCR si utilizzano invece primers che si legano nelle zone Fr3 e JH dando origine a frammenti tra
80 e 120 bp di lunghezza [9].
Figura 2.La struttura della regione variabile del gene IgH e posizione dei differenti primers Fr2 FR3 e JH. L’utilizzo di due PCR distinte è importante per ottenere una diagnosi più sensibile.
La metodica del Kit BIRD invece prevede una semi-nested-PCR, basata comunque
sull’amplificazione di due distinte regioni. I primers, nell’amplificazione esterna della PCR Fr2A, si
ibridano nelle zone Fr2 e LJH mentre nella PCR interna si ibridano a livello del Fr2 e VLJH
ottenendo frammenti di grandezza compresa tra 240 e 280 bp. Nell’amplificazione esterna della
PCR Fr3A, i primers, si ibridano nelle zone Fr3 e LJH mentre nella PCR interna si ibridano a livello
delle zone Fr3 e VLJH ottenendo frammenti compresi tra 80 e 120 bp.

7
Pertanto, le regioni oggetto dell’amplificazione sono uguali tra i due metodi, anche se i primers
ibridano in posizioni differenti. L’utilizzo di due distinte PCR, con primers che vanno a legarsi in
zone differenti, consente di aumentare la sensibilità di individuare riarrangiamenti patologici: è
sufficiente individuare monoclonalità in un frammento per emettere la diagnosi di linfoma a cellule
B.
Il primer JH, della metodica presente in letteratura [9], e il primer VLJH della metodica BIRD a
livello dell’estremità 5’ hanno coniugato un fluorocromo 6-FAM; in questo modo i prodotti
amplificati possano essere analizzati su sequenziatore automatico a elettroforesi capillare. Lo
strumento possiede un fotometro in grado di rilevare la presenza di molecole coniugate con
fluorocromi eccitate da una sorgente laser, che elabora poi sotto forma di picchi, caratterizzati da
dimensione (proporzionale alla lunghezza del frammento) e altezza (proporzionale alla quantità del
dato riarrangiamento presente nel materiale di partenza) specifiche.
La presenza di uno o due picchi dominanti, di altezza almeno doppia rispetto agli altri, indica una
monoclonalità. Per essere confermata, e dunque poter dare la diagnosi di linfoma a cellule B,
bisogna confermare con altre due ripetizioni dell’analisi ottenendo sempre lo stesso picco con
uguali dimensioni [9].
Nel mio caso, essendo già state eseguite in precedenza le analisi con le conferme relative dal
personale del Laboratorio, per la metodica della letteratura sarà necessario eseguire un’unica volta
le analisi con la metodica LDM-ICP; se i risultati che ottengo in questo lavoro non dovessero
combaciare con quelli dell’analisi eseguita in precedenza, si dovrà procedere a confermare il
risultato con una seconda ripetizione. Queste discrepanze possono accadere in quanto il tessuto
da cui è stato estratto il DNA è differente tra la determinazione precedente (in diagnosi) e quella
attuale. Per la metodica BIRD dovrò invece confermare il risultato come da giusta prassi, non
avendola mai utilizzata in precedenza.
8
Pazienti, materiali e metodi
i) Casistica campioni Le analisi sono state eseguite su:
o 13 campioni positivi certi: Casi microscopicamente classificati come linfomi maligni dai medici
patologi e confermati dall'analisi molecolare con la metodica in uso presso il LDM-ICP
(amplificazione mediante PCR e analisi di frammenti con sequenziatore automatico).
o 9 campioni negativi certi: Casi classificati come negativi per presenza di linfoma, sia sulla base
della valutazione istologica che su quella molecolare con il protocollo attualmente in uso
(questi campioni li abbiamo scelti sulla base di una diagnosi di infiammazione o Linfomi a
cellule T).
o 11 campioni falsi negativi: Casi classificati come Linfomi a cellule B maligni dai medici patologi
ma che non hanno mostrato la presenza di monoclonalità all'analisi molecolare con il protocollo
LDM-ICP.
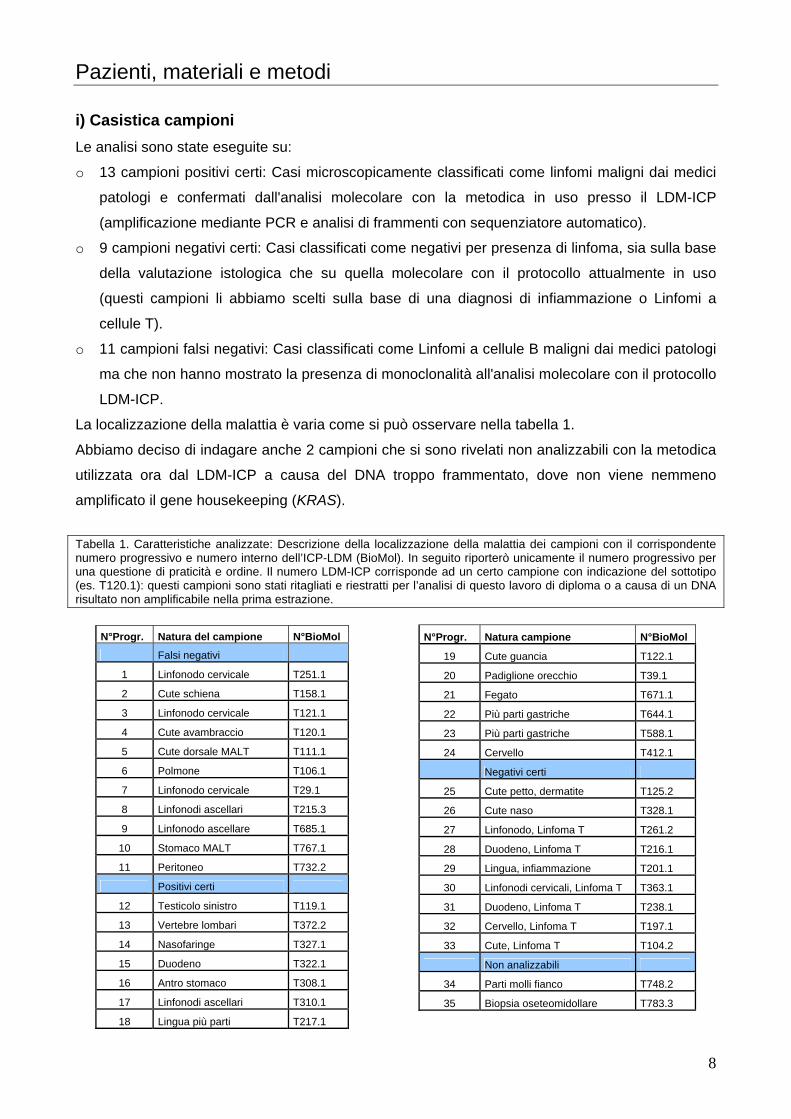
La localizzazione della malattia è varia come si può osservare nella tabella 1.
Abbiamo deciso di indagare anche 2 campioni che si sono rivelati non analizzabili con la metodica
utilizzata ora dal LDM-ICP a causa del DNA troppo frammentato, dove non viene nemmeno
amplificato il gene housekeeping (KRAS).
Tabella 1. Caratteristiche analizzate: Descrizione della localizzazione della malattia dei campioni con il corrispondente numero progressivo e numero interno dell’ICP-LDM (BioMol). In seguito riporterò unicamente il numero progressivo per una questione di praticità e ordine. Il numero LDM-ICP corrisponde ad un certo campione con indicazione del sottotipo (es. T120.1): questi campioni sono stati ritagliati e riestratti per l’analisi di questo lavoro di diploma o a causa di un DNA risultato non amplificabile nella prima estrazione.
N°Progr. Natura del campione N°BioMol
Falsi negativi
1 Linfonodo cervicale T251.1
2 Cute schiena T158.1
3 Linfonodo cervicale T121.1
4 Cute avambraccio T120.1
5 Cute dorsale MALT T111.1
6 Polmone T106.1
7 Linfonodo cervicale T29.1
8 Linfonodi ascellari T215.3
9 Linfonodo ascellare T685.1
10 Stomaco MALT T767.1
11 Peritoneo T732.2
Positivi certi
12 Testicolo sinistro T119.1
13 Vertebre lombari T372.2
14 Nasofaringe T327.1
15 Duodeno T322.1
16 Antro stomaco T308.1
17 Linfonodi ascellari T310.1
18 Lingua più parti T217.1
N°Progr. Natura campione N°BioMol
19 Cute guancia T122.1
20 Padiglione orecchio T39.1
21 Fegato T671.1
22 Più parti gastriche T644.1
23 Più parti gastriche T588.1
24 Cervello T412.1
Negativi certi
25 Cute petto, dermatite T125.2
26 Cute naso T328.1
27 Linfonodo, Linfoma T T261.2
28 Duodeno, Linfoma T T216.1
29 Lingua, infiammazione T201.1
30 Linfonodi cervicali, Linfoma T T363.1
31 Duodeno, Linfoma T T238.1
32 Cervello, Linfoma T T197.1
33 Cute, Linfoma T T104.2
Non analizzabili
34 Parti molli fianco T748.2
35 Biopsia oseteomidollare T783.3

9
ii) Materiale e metodi a) Estrazione DNA da paraffina: Materiale:
o Etanolo 99,8%, Fluka Analytical, Lotto: 1364645
o Xilolo 98,5%, Carlo Erba Reagents, Lotto: 492306
o Kit QIAamp DNA MiniKit Qiagen Tissue, USA, Lotto: 130167744, Cat: NO 51306 contenente i
buffer, Proteinasi K e colonne di purificazione
o Tubi Eppendorf sterili di 2,0 ml, Sarstedt, Nümbrecht, Germania
o Tubi Eppendorf sterili da 1,5 ml, Sarstedt, Nümbrecht, Germania o Eppendorf Centrifuge 5415 R, Basel
o Microtomo, Microm HM 440E, USA
o Vortex, Heidolph REAX 2000, Germania
o Cappa, Heraeus Instrument
o Termoblocco, CH-100 BioLabo, Châtel-St.Denis
o Thermomixer comfort, Eppendorf AG, Hamburg, Germania
o Incubatore, Binden, Tuttlingen
o Ruota, Falc F 205 Rot
o NanoDrop ND-1000 Spectrophotometer, Witec Ag, Littau Protocollo:
A ogni passaggio del protocollo e ogni uscita dalla cappa con le mani cambiare i guanti per evitare
una qualsiasi contaminazione.
Non soffermarsi troppo durante i passaggi con xilolo vista la sua tendenza a frammentare
facilmente il DNA.
Tagliare 6-7 sezioni da 3 µm di tessuto fissato in formalina ed incluso in paraffina con un
microtomo. Inserire i tagli in una provetta da 2,0 ml utilizzando i guanti. Si può utilizzare la stessa
lama per più campioni pulendola dopo ogni taglio con etanolo.
Centrifugare per 15 secondi a 13’200 rpm.
Aggiungere 800 µl di xilolo, vortexare per 15 secondi alla massima velocità, centrifugare per 5
minuti a 13’000 rpm. Eliminare il surnatante.
Aggiungere 500 µl di etanolo, vortexare alla massima velocità per 15 secondi. Centrifugare per 5
minuti a 13’000 rpm, eliminare il surnatante.
Ripetere l’aggiunta di etanolo e la procedura conseguente.
Inserire la provetta con il tappo aperto nel Thermomixer comfort a 45°C e impostare 300 rpm per
circa 5-10 minuti, a seconda della dimensione del pellet.
Una volta evaporato tutto l’etanolo, aggiungere 180 µl di buffer ATL e 20 µl di proteinasi K.
Vortexare e sigillare il tappo con parafilm per evitare che si apra il tappo. Incubare a 56°C per tutta
la notte in un incubatore su una ruota in movimento.

10
Il giorno successivo centrifugare il campione per 15 secondi a 13’200 rpm per togliere i residui
dalle pareti, aggiungere 200 µl di buffer AL, vortexare per 15 secondi alla massima velocità e
incubare in un termoblocco per 10 minuti a 70°C. A fine incubazione centrifugare per 15 secondi a
13’200 rpm. Aggiungere 200 µl di etanolo, vortexare alla massima velocità per 15 secondi.
Centrifugare per 15 secondi a 13’200 rpm.
Pipettare la miscela ottenuta (600 µl circa) in una colonna QIAamp e centrifugare a 8’000 rpm per
1 minuto, staccare la provetta di raccolta e gettare l’eluato, mantenere la colonna con il filtro e
mettere su una provetta di raccolta pulita.
Aggiungere 500 µl di buffer AW1, centrifugare a 8’000 rpm per 1 minuto, staccare la provetta di
raccolta e gettare l’eluato, mantenere la colonna con il filtro e mettere su una provetta di raccolta
pulita.
Aggiungere 500 µl di buffer AW2, centrifugare a 13’200 rpm per 3 minuti, staccare la provetta di
raccolta e gettare l’eluato, mantenere la colonna con il filtro e mettere su una provetta di raccolta
pulita, centrifugare 13’200 rpm per 1 minuto.
Appoggiare la colonna con il filtro su una provetta da 1,5 ml e aggiungere 50 µl di buffer AE sopra
il filtro, incubare a temperatura ambiente per 5 minuti, centrifugare a 8’000 rpm per 1 minuto.
Pipettare l’eluato ottenuto dal passaggio precedente e ricaricare sulla stessa colonna QIAamp,
incubare 5 minuti a temperatura ambiente e centrifugare a 8’000 rpm per 1 minuto.
Gettare colonna con filtro e tenere provetta contenente la soluzione di DNA.
Quantificazione:
Per verificare se si è ottenuto DNA di buona qualità, si utilizza la misurazione allo spettrofotometro.
Per fare il bianco campione, e quindi azzerare la lettura, utilizzare 2 µl di buffer AE. Si passa poi
alla quantificazione degli acidi nucleici presenti nella soluzione utilizzando 2 µl del DNA estratto. Lo
strumento fornisce contemporaneamente letture a 230, 260, 280 e 320 nm. Osservare la
concentrazione di acidi nucleici e in particolare i rapporti 260/280 e 260/230, che devono essere
idealmente compresi tra 1,6 e 2,0. Il DNA assorbe a una lunghezza d’onda di 260 nm, le proteine a
280 nm mentre lo xilolo e l’etanolo a 230 nm. Valori elevati a 230 o 280 denotano pertanto
rispettivamente contaminazione di xilolo/etanolo o proteine (vedi figura 3).

11
Figura 3. Grafici dell’apparecchio nanodrop al momento della quantificazione degli acidi nucleici. Il grafico sulla sinistra indica un campione con una buona concentrazione di acidi nucleici (147,8 ng/µl) e degli ottimi rapporti 260/280 (2,05) e 260/230 (2,05), infatti la curva del grafico prende un andamento a campana spostato sulla sinistra con il picco massimo a 260 nm. Il grafico sulla destra indica un campione contenente una bassa concentrazione di acidi nucleici (2,1 ng/µl); il rapporto 260/280 (1,72) è accettabile mentre il rapporto 260/230 (0,34) non lo è, probabilmente a causa di contaminazione di xilolo o etanolo.
b) Amplificazione mediante PCR Materiale:
o dNTPs (dATP, dTTP, dCTP, dGTP) 100 mM, GE Healthcare, Illustra, UK, Lotto: 3443
11/12/13/14
o dUTP 100 mM, GE Healthcare, Illustra, UK, Lotto: 358155
o Kit AmpliTaq Gold with GeneAmp (Applied Biosystems-Roche), USA, Lotto: K09381;
Scadenza: 31.01.2010 contenente la Taq Gold, PCR buffer e la soluzione di MgCl2 o Acqua deminaralizzata (millipore) autoclavata (Fedegari autoclaven AG) o Tubi Eppendorf da 1,5 ml sterile, Sarstedt, Nümbrecht Germania o Tubi di PCR DNase e RNase free 0,2 ml, Thermo scientific, Axonlab, Baden
o Termociclatore, Bio-Rad USA, DNA Engine, PTC-200
Protocollo:
Per poter eseguire le amplificazioni utilizzare soluzioni di DNA a 25 ng/µl o a 75 ng/µl prendendo
sempre 4 µl di soluzione madre di DNA e aggiungendo una quantità variabile di acqua
demineralizzata autoclavata in base alle concentrazioni ottenute e quantificate con lo
spettrofotometro. Per sapere quanta acqua aggiungere eseguire il calcolo:
((C1.V1)/C2)-4= Volume d’acqua da aggiungere alla soluzione madre di DNA
C1= Concentrazione soluzione madre DNA
V1= Volume costante di soluzione di DNA 4 µl
C2= Concentrazione di DNA che si vuole ottenere 25 ng/ µl o 75 ng/ µl
I campioni diluiti a 25 ng/µl e a 75 ng/µl possono essere conservati a tempo indeterminato a 4°C,
mentre le soluzioni madri sono congelate a -20°C.

12
Tutti i reagenti delle PCR devono essere conservati in congelatore a -20°C.
I reagenti e i campioni devono essere scongelati e centrifugati prima di ogni preparazione, mentre
la Taq Gold polimerasi viene estratta dal congelatore unicamente al momento dell’uso e riposta a
-20°C subito dopo l’aggiunta alla miscela di reazione.
Prima di ogni PCR, preparare i tubi di reazione necessari a seconda del numero dei campioni,
calcolando anche due controlli, uno positivo ed un bianco. Per il controllo positivo utilizzare un
campione di DNA già amplificato precedentemente con successo; per il bianco utilizzare acqua
demineralizzata autoclavata al posto della soluzione di DNA. Il bianco di reazione viene pipettato
sempre per ultimo al fine di verificare che non ci siano state contaminazioni durante l’operazione.
Dopo l’amplificazione pertanto, un esperimento sarà considerato valido se e solo se nel bianco di
reazione non si sarà evidenziata alcuna banda di amplificazione e se nel controllo positivo si sarà
osservata una sola banda di dimensione attesa per quella specifica analisi.
Durante la preparazione della mix aggiungere un volume corrispondente ad un campione in più per
compensare eventuali perdite durante le manipolazioni e i pipettamenti.
Tutti i volumi elencati nelle procedure sono per un unico campione, moltiplicare quindi i volumi dei
reagenti a seconda del numero di campioni da analizzare e dei controlli.
Preparare prima dell’uso le diluizioni di dNTPs e il dUTP. Per i dNTPs, pipettare in una provetta da
1,5 ml 10 µl di ogni nucleotide (da una soluzione madre a concentrazione di 100 mM). Portare a
volume con 60 µl di acqua sterile per ottenere una concentrazione finale 10 mM per ogni
nucleotide. Per il dUTP preparare una provetta da 1,5 ml e pipettare 10 µl di dUTP (da una
soluzione madre a concentrazione di 100 mM), portare a volume con 90 µl di acqua sterile per
ottenere una concentrazione finale 10 mM.
Tutti i passaggi della PCR devono essere eseguiti in ghiaccio con cambio di guanti a ogni
passaggio.
Al momento dell’utilizzo del primer JH per la metodica LDM-ICP e delle mix semi-nested per la
metodica BIRD, lavorare il più possibile al riparo dalla luce (ovvero spegnere la luce della cappa e
usare solo luce ambientale) vista la presenza di un fluorocromo.
Tutti i prodotti d’amplificazione possono essere conservati a 4°C per tempo indeterminato.
c) PCR KRAS
Materiale:
o Primer 17F 100 µM (5’-TGGTGGAGTATTTGATAGTGTA-3’; Invitrogen USA, Lotto: D7328
(A02)964350)
o Primer 18B 100 µM (5’-CATGAAAATGGTCAGAGAA-3’; Invitrogen USA, Lotto: D0328
(A04)964350)
Protocollo:
Il gene houskeeping viene utilizzato come un controllo interno per assicurarsi che il DNA ottenuto
dall’estrazione sia amplificabile e non troppo frammentato. In questo lavoro si è deciso di utilizzare

13
il gene KRAS. KRAS appartiene ad una famiglia genica molto conservata, codifica per una
proteina implicata nella trasduzione del segnale. Questo gene è utilizzato come controllo perché
non subisce variazioni di numero di coppie geniche in tutti i tumori, ovvero non è soggetto a
duplicazioni o delezioni, eventi che potrebbero alterare il risultato [6].
Pipettare in ogni tubo di PCR 2 µl di soluzione di DNA 25 ng/µl; nel caso la concentrazione della
soluzione madre non superi i 25 ng/µl, non diluire il campione ma pipettare direttamente 2 µl di
soluzione madre di DNA.
Preparazione della Mix di reazione:
In un’altra provetta da 1,5 ml pipettare 5 µl di Primer 17F e diluire con 45 µl di acqua sterile
ottenendo cosi una concentrazione 10 µM; lo stesso procedimento viene eseguito con il Primer
18B.
Nella provetta per la mix pipettare 2,5 µl di PCR buffer 10x, 3 µl di MgCl2 25 mM, 0,5 µl di dNTPs
10 mM, 0,1 µl di dUTP 10 mM e 1,25 µl di entrambi i primers 10 µM. Aggiungere 0,4 µl di Taq Gold
polimerasi 5U/µl nella mix. Dopo questa aggiunta terminare velocemente la preparazione della mix
con 14 µl di acqua sterile e pipettare 23 µl di mix in ogni tubo di PCR contenente il DNA.
Amplificazione:
Mettere i tubi di reazione nel termociclatore già impostato precedentemente con il seguente
programma:
Preicubazione per 2 minuti a 50°C (1 ciclo), denaturazione per 10 minuti a 95°C (1 ciclo).
Denaturazione per 15 secondi a 95°C, amplificazione per 30 secondi a 55°C ed estensione per 30
secondi a 72°C; questi passaggi vengono ripetuti per 45 cicli. Estensione finale 3 minuti a 72°C
un’unica volta e poi i campioni vengono mantenuti a 10°C fino a che vengono prelevati dal
termociclatore.
d) Preparazione gel d’agarosio 1,8% e visualizzazione PCR KRAS
Materiale:
o PeqGold Universal Agarose, Axonlab Banen, Lotto: 207324
o Tris Borate EDTA buffer (TBE) 10X, SIGMA USA, Lotto: 038K8410
o Loading buffer (Soluzione di Blu di Bromofenolo), peqlAb Germania
o DNA Marker PeqGold 50 bp DNA-Leiter, Biotechnologie GmbH, peqlab Germania, Lotto:
28112
o GelRed 0.1%, Biotium USA, Lotto: 8G0T19
o ImageMaster VDS, Pharmacia Biotech USA
o Beuta da 100 ml e da 200 ml
o Cilindro da 50 ml e da 100 ml
o Bilancia analitica, Mettler PC 2200

14
o Forno a microonde, Moulinex, symbio
o Camera elettroforetica con supporto, Bio-Rad USA, Wide Mini-Sub Cell CT
o Generatore di corrente, BioRad USA, Power Pac Basic
Protocollo:
Preparazione gel:
Preparare la soluzione TBE 1X in un pallone tarato da 2 l con 200 ml di TBE 10X. Portare a
volume con acqua demineralizzata.
Preparazione del gel da 100 ml: pesare in una beuta da 200 ml 1,8 g di PeqGold Universal
Agarose e aggiungere 100 ml di TBE 1X misurato precedentemente. Sciogliere la soluzione in
microonde a 200 V per circa 10-12 minuti fino a che la soluzione è limpida. Lasciare raffreddare il
gel fino a circa 55°C (non scotta più al tatto) e versare nella camera elettroforetica 15x10 cm dopo
aver posizionato i pettini per creare i pozzetti.
Preparazione del gel 40 ml: pesare in una beuta da 100 ml 0,72 g di PeqGold Universal Agarose e
aggiungere 40 ml di TBE 1X misurato precedentemente. Sciogliere la soluzione in microonde a
200 V per circa 8 minuti fino a che la soluzione è limpida. Lasciare raffreddare il gel fino a circa
55°C (non scotta più al tatto) e versare nella camera elettroforetica 6x10 cm dopo aver posizionato
i pettini per creare i pozzetti.
Lasciare raffreddare il gel almeno un’ora prima dell’utilizzo. Riempire poi la camera elettroforetica
con la soluzione TBE 1X coprendo di circa 5 mm il gel.
Visualizzazione su gel d’agarosio:
Una volta ottenuto l’amplificato visualizzare il risultato su gel d’agarosio 1,8%. Su un foglio di
parafilm pipettare per ogni campione 2 µl di loading buffer e 5 µl di amplificato; per il marker
pipettare sempre 2 µl di loading buffer, 1 µl di DNA Marker e 9 µl di TBE 1x. Pipettare poi 7 µl di
marker e campione in ogni pozzetto del gel. Far migrare a 100 V per circa 30 minuti (fino a che il
colorante blu di bromofenolo non arrivi a circa 1-1,5 cm alla fine del gel).
Per la colorazione delle bande utilizzare una soluzione di GelRed 0.1% diluito con TBE 1x, lasciare
il gel a colorare per almeno 30 minuti. Sciacquare il gel in acqua demineralizzata per eliminare
l’eccesso di soluzione di GelRed 0.1% e osservare le bande ottenute con l’apparecchio
ImageMaster; stampare la fotografia (vedi figura 4). Ci si attende un prodotto d’amplificazione di
250 bp.

15
Figura 4. Visualizzazione, su gel d’agarosio, del prodotto d’amplificazione di 3 campioni e controlli dopo PCR KRAS. Partendo da sinistra ci sono i 3 campioni (1,2 ,3), il bianco di reazione (4) dove non è presente alcuna banda, il controllo positivo (5) che presenta l’unica banda a 250 bp e il marker (M), con tutte le bande di dimensione conosciuta. e) Analisi linfoma cellule B con la metodica in uso presso il ICP-LDM FR2-JH/ FR3-JH Materiale:
o Primer FR2 (5’-TGGRTCCGMCAGSCYYCNGG-3’; Invitrogen USA, Lotto:D4989G11 o Primer FR3 (5’-ACACGGCYSTRTATTACTGT-3’; Invitrogen USA, Lotto:D4989G10 o Primer JH (5’-ACCTGAGGAGACGTGACC-3’; Invitrogen USA, Lotto: O5039C06), il primer è
coniugato al fluorocromo 6-FAM all’estremità 5’ Protocollo:
Per ogni campione in analisi bisogna eseguire due distinte PCR; una PCR amplifica frammenti
dalla regione FR2 alla regione JH, un’altra PCR amplifica frammenti dalla regione FR3 alla regione
JH.
I primers FR2, FR3 e JH vengono preparati sotto cappa prendendo 10 µl di primers soluzione
madre 100 µM e diluendo con 40 µl di acqua demineralizzata autoclavata ottenendo così una
soluzione a concentrazione 20 µM.
Pipettare in ogni tubo 2 µl di soluzione di DNA madre.
Preparazione della Mix di reazione FR2-JH:
Nella provetta della mix pipettare 5 µl di PCR buffer 10x, 3 µl di MgCl2 25 mM, 1 µl di dNTPs 10
mM, 0,2 µl di dUTP 10 mM e 1,5 µl di primer FR2 20 µM e JH 20 µM. Pipettare 0,25 µl di Taq Gold
polimerasi 5U/µl nella mix. Dopo questa aggiunta terminare velocemente la preparazione della mix
con 34,55 µl di acqua sterile e pipettare 48 µl di mix in ogni tubo di PCR contenente il DNA.
Amplificazione FR2-JH:
Mettere i tubi di reazione nel termociclatore impostato precedentemente con il seguente
programma:
Preicubazione per 2 minuti a 50°C (1 ciclo), denaturazione per 10 minuti a 95°C (1 ciclo).
Denaturazione per 1 minuto a 94°C, amplificazione per 1 minuto a 57 °C ed estensione per 2
minuti a 72°C; questi passaggi vengono eseguiti per 45 cicli. Estensione finale 5 minuti a 72°C
3 4 5 M1 2

16
un’unica volta e poi i campioni vengono mantenuti a 10°C fino a che vengono prelevati dal
termociclatore.
Preparazione della Mix di reazione FR3-JH:
Nella provetta della mix pipettare 5 µl di PCR buffer 10x, 3 µl di MgCl2 25 mM, 1 µl di dNTPs 10
mM, 0,2 µl di dUTP 10 mM e 1 µl di primer FR3 20 µM e JH 20 µM. Pipettare 0,25 µl di Taq Gold
polimerasi 5U/µl nella mix. Dopo questa aggiunta terminare velocemente la preparazione della mix
con 35,55 µl di acqua sterile e pipettare 48 µl di mix in ogni tubo di PCR contenente il DNA.
Amplificazione FR3-JH:
Mettere i tubi di reazione nel termociclatore impostato precedentemente con il seguente
programma:
Preicubazione per 2 minuti a 50°C (1 ciclo), denaturazione per 10 minuti a 95°C (1 ciclo).
Denaturazione per 1 minuto a 94°C, amplificazione per 1 minuto a 55 °C ed estensione per 1
minuto e 30 secondi a 72°C; questi passaggi vengono eseguiti per 45 cicli. Estensione finale 5
minuti a 72°C un’unica volta e poi i campioni vengono mantenuti a 10°C fino a che vengono
prelevati dal termociclatore.
f) Analisi Linfoma cellule B Kit Ampli-Lymphoma B (BIRD) Materiale:
o Ampli-Lymphoma B (BIRD) 45 test, Dia-chem Napoli, Lotto:100908/02; Scadenza: 03.2010
o Ampli-Lymphoma B (BIRD) 45 test, Dia-chem Napoli, Lotto:010409/02; Scadenza 10.2010
Protocollo:
Nel Kit BIRD sono contenuti la Mix PCR Fr2A, Mix PCR Fr3A, Mix PCR Fr2A semi-nested, Mix
PCR Fr3A semi-nested, acqua RNase/DNase free, Taq Poymerase (5U/µl) ed un controllo DNA
Fr2A.
Per ogni campione bisogna eseguire due distinte PCR; una che amplifica un frammento dalla zona
Fr2A alla zona LJH ed un’altro che amplifica un frammento dalla zona Fr3A alla zona LJH (PCR
esterna). In seguito vengono fatte altre due PCR distinte più interne utilizzando l’amplificato
ottenuto dalle prime PCR (PCR interna) che vanno ad amplificare un frammento da Fr2A a VLJH
ed un frammento da Fr3A a VLJH rispettivamente (semi nestad PCR).
Per il controllo positivo utilizzare per i primi campioni che si eseguono il controllo dato dal Kit poi, in
seguito, una soluzione di DNA amplificato precedentemente con successo con il Kit, questo
arrangiamento si rende indispensabile in quanto il controllo positivo del Kit è fornito in quantità
limitata.
Pipettare in ogni tubo 2 µl di soluzione di DNA 75 ng/µl; nel caso in cui la concentrazione della
soluzione madre non superi i 75 ng/µl non diluire il campione ma pipettare direttamente 2 µl di
soluzione madre di DNA.

17
Preparazione del tubo di reazione FR2A-JLH e FR3A-LJH (PCR esterna):
Nella provetta della mix pipettare 10 µl della Mix PCR Fr2A e nell’altra provetta 10 µl Mix PCR
Fr3A. Aggiungere in ognuna 0,125 µl di Taq Poymerase (5U/µl) e 13 µl di acqua RNase/DNase
free.
Aggiungere nei tubi di reazione contenente la soluzione di DNA, 23 µl di mix Fr2A dove si vuole
amplificare il frammento Fr2A-LJH, e 23 µl di mix Fr3A dove si vuole amplificare il frammento
Fr3A-LJH.
Amplificazione FR2A-LJH/FR3A-LJH:
Mettere i tubi di reazione nel termociclatore già impostato precedentemente con il seguente
programma:
Denaturazione per 5 minuti a 95°C (1 ciclo). Denaturazione per 1 minuto a 94°C, amplificazione
per 1 minuto a 55 °C ed estensione per 1 minuto a 72°C; questi passaggi vengono eseguiti per 30
cicli. Estensione finale per 10 minuti a 72°C un’unica volta e poi i campioni vengono mantenuti a
10°C fino a che vengono prelevati dal termociclatore.
Preparazione del tubo di reazione FR2A-VJLH e FR3A-VLJH (PCR interna):
Pipettare in ogni tubo di reazione 1,5 µl di amplificato ottenuto dalle PCR FR2A-JLH e FR3A-LJH
(PCR esterne).
Nella provetta della mix preparata precedentemente pipettare 10 µl della Mix PCR Fr2A semi-
nested e nell’altra provetta 10 µl Mix PCR Fr3A semi-nested. Aggiungere in ognuna 0,125 µl di
Taq Poymerase (5U/µl) e 14 µl di acqua RNase/DNase free.
Aggiungere in ogni tubo di reazione contenente il prodotto delle PCR precedenti 24 µl di mix Fr2A
semi-nested per i campioni che si vuole amplificare il frammento Fr2-VLJH, e 24 µl mix Fr3A semi-
nested dove si vuole amplificare il frammento Fr3-VLJH.
Amplificazione FR2A-LJH/FR3A-LJH:
Mettere i tubi di reazione nel termociclatore già impostato precedentemente con il seguente
programma:
Denaturazione per 5 minuti a 95°C (1 ciclo). Denaturazione per 1 minuto a 94°C, amplificazione
per 1 minuto a 55 °C ed estensione per 1 minuto a 72°C; questi passaggi vengono eseguiti per 20
cicli. Estensione finale per 10 minuti a 72°C un’unica volta e poi i campioni vengono mantenuti a
10°C fino a che vengono prelevati dal termociclatore.
g) Analisi dei frammenti Materiale:
o GeneScan-500 LIZ, Applied Biosystems UK, Lotto: 0706072 o Formammide, Applied Biosystems, UK, Lotto: 0809753 o Sequenziatore automatico 3130 Genetic Analyzer della ditta Applied Biosystems USA, Hitachi

18
o Genetic Analyzer, Data Colecction, Software v3.0 o GeneMapper®Software, version 3.7 o MicroAmp Optical 96-Well Reaction Plate, Applied Biosystems USA o Termociclatore, Bio-Rad USA, DNA Engine, PTC-200
o Centrifuge 5415R, Eppendorf, Hamburg, Germania
Protocollo:
La visualizzazione del prodotto ottenuto dalle PCR con le metodiche LDM-ICP e BIRD, si
eseguono con un sequenziatore automatico. I campioni di entrambe le amplificazioni necessitano
di una preparazione prima di essere analizzati con l’apparecchio. Tutte le manipolazioni dei
campioni vengono eseguite sotto cappa e a luce spenta.
La formammide permette di mantenere denaturato il DNA anche a basse temperature. Una fila
della piastra di reazione possiede 8 pozzetti. Dato che l’apparecchio è in grado di pescare 4
campioni contemporaneamente, se non si deve analizzare campioni in un numero multiplo di 4,
aggiungere ai pozzetti vuoti 11 µl di formammide.
Analisi dei frammenti Fr2-JH/Fr3-JH (Metodica LDM-ICP):
Pipettare in un tubo da PCR 1 µl d’amplificato, 0,25 µl di GeneScan-500 LIZ e 10 µl di
formammide.
Inserire i tubi in un termociclatore preimpostato a 90°C per 2 minuti così da denaturare i campioni.
Dopo la denaturazione mettere subito i tubi in una scatola di polistirolo contenete ghiaccio per 15
minuti, coprire i campioni con un foglio d’alluminio. Centrifugare i campioni a 13’200 rpm per 15
secondi, trasferire poi 11 µl di campione nella piastra di reazione nell’ordine predefinito
precedentemente e registrati nel software connesso all’apparecchio. Una volta registrati i campioni
nel software si può precedere con l’analisi.
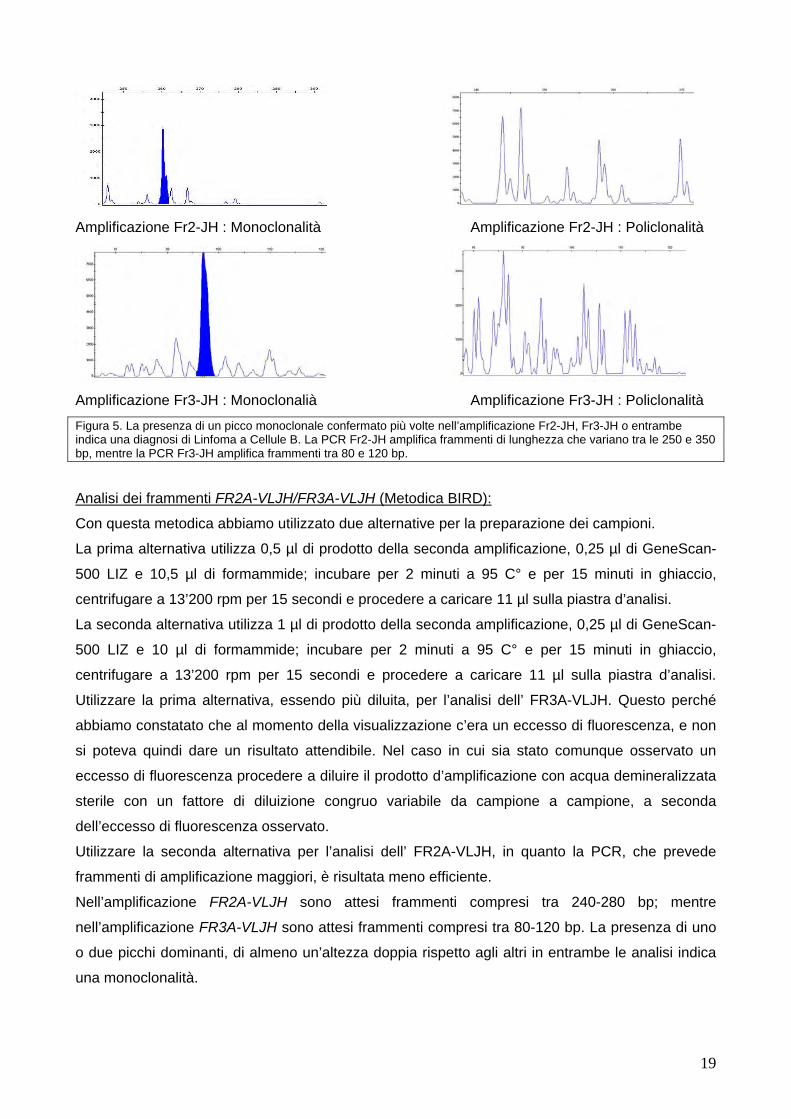
Nell’amplificazione Fr2-JH sono attesi frammenti compresi tra 250-350 bp; mentre
nell’amplificazione Fr3-JH sono attesi frammenti compresi tra 80-120 bp. La presenza di uno o due
picchi dominanti, di almeno un’altezza doppia rispetto agli altri in entrambe le analisi indica una
monoclonalità mentre la presenza di molti picchi indica una policlonalità (vedi figura 5).
19
Amplificazione Fr2-JH : Monoclonalità Amplificazione Fr2-JH : Policlonalità
Amplificazione Fr3-JH : Monoclonalià Amplificazione Fr3-JH : Policlonalità
Figura 5. La presenza di un picco monoclonale confermato più volte nell’amplificazione Fr2-JH, Fr3-JH o entrambe indica una diagnosi di Linfoma a Cellule B. La PCR Fr2-JH amplifica frammenti di lunghezza che variano tra le 250 e 350 bp, mentre la PCR Fr3-JH amplifica frammenti tra 80 e 120 bp. Analisi dei frammenti FR2A-VLJH/FR3A-VLJH (Metodica BIRD): Con questa metodica abbiamo utilizzato due alternative per la preparazione dei campioni.
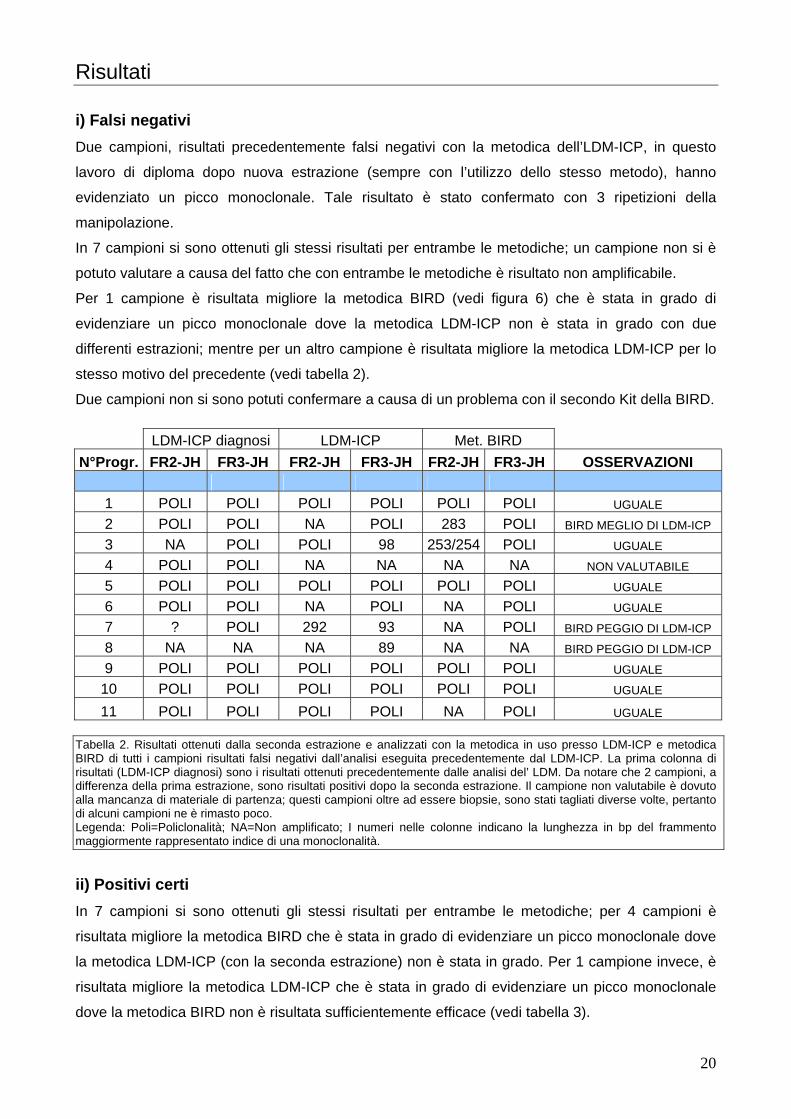
La prima alternativa utilizza 0,5 µl di prodotto della seconda amplificazione, 0,25 µl di GeneScan-
500 LIZ e 10,5 µl di formammide; incubare per 2 minuti a 95 C° e per 15 minuti in ghiaccio,
centrifugare a 13’200 rpm per 15 secondi e procedere a caricare 11 µl sulla piastra d’analisi.
La seconda alternativa utilizza 1 µl di prodotto della seconda amplificazione, 0,25 µl di GeneScan-
500 LIZ e 10 µl di formammide; incubare per 2 minuti a 95 C° e per 15 minuti in ghiaccio,
centrifugare a 13’200 rpm per 15 secondi e procedere a caricare 11 µl sulla piastra d’analisi.
Utilizzare la prima alternativa, essendo più diluita, per l’analisi dell’ FR3A-VLJH. Questo perché
abbiamo constatato che al momento della visualizzazione c’era un eccesso di fluorescenza, e non
si poteva quindi dare un risultato attendibile. Nel caso in cui sia stato comunque osservato un
eccesso di fluorescenza procedere a diluire il prodotto d’amplificazione con acqua demineralizzata
sterile con un fattore di diluizione congruo variabile da campione a campione, a seconda
dell’eccesso di fluorescenza osservato.
Utilizzare la seconda alternativa per l’analisi dell’ FR2A-VLJH, in quanto la PCR, che prevede
frammenti di amplificazione maggiori, è risultata meno efficiente.
Nell’amplificazione FR2A-VLJH sono attesi frammenti compresi tra 240-280 bp; mentre
nell’amplificazione FR3A-VLJH sono attesi frammenti compresi tra 80-120 bp. La presenza di uno
o due picchi dominanti, di almeno un’altezza doppia rispetto agli altri in entrambe le analisi indica
una monoclonalità.
20
Risultati
i) Falsi negativi Due campioni, risultati precedentemente falsi negativi con la metodica dell’LDM-ICP, in questo
lavoro di diploma dopo nuova estrazione (sempre con l’utilizzo dello stesso metodo), hanno
evidenziato un picco monoclonale. Tale risultato è stato confermato con 3 ripetizioni della
manipolazione.
In 7 campioni si sono ottenuti gli stessi risultati per entrambe le metodiche; un campione non si è
potuto valutare a causa del fatto che con entrambe le metodiche è risultato non amplificabile.
Per 1 campione è risultata migliore la metodica BIRD (vedi figura 6) che è stata in grado di
evidenziare un picco monoclonale dove la metodica LDM-ICP non è stata in grado con due
differenti estrazioni; mentre per un altro campione è risultata migliore la metodica LDM-ICP per lo
stesso motivo del precedente (vedi tabella 2).
Due campioni non si sono potuti confermare a causa di un problema con il secondo Kit della BIRD.
LDM-ICP diagnosi LDM-ICP Met. BIRD N°Progr. FR2-JH FR3-JH FR2-JH FR3-JH FR2-JH FR3-JH OSSERVAZIONI
1 POLI POLI POLI POLI POLI POLI UGUALE
2 POLI POLI NA POLI 283 POLI BIRD MEGLIO DI LDM-ICP
3 NA POLI POLI 98 253/254 POLI UGUALE
4 POLI POLI NA NA NA NA NON VALUTABILE
5 POLI POLI POLI POLI POLI POLI UGUALE
6 POLI POLI NA POLI NA POLI UGUALE
7 ? POLI 292 93 NA POLI BIRD PEGGIO DI LDM-ICP
8 NA NA NA 89 NA NA BIRD PEGGIO DI LDM-ICP
9 POLI POLI POLI POLI POLI POLI UGUALE
10 POLI POLI POLI POLI POLI POLI UGUALE
11 POLI POLI POLI POLI NA POLI UGUALE Tabella 2. Risultati ottenuti dalla seconda estrazione e analizzati con la metodica in uso presso LDM-ICP e metodica BIRD di tutti i campioni risultati falsi negativi dall’analisi eseguita precedentemente dal LDM-ICP. La prima colonna di risultati (LDM-ICP diagnosi) sono i risultati ottenuti precedentemente dalle analisi del’ LDM. Da notare che 2 campioni, a differenza della prima estrazione, sono risultati positivi dopo la seconda estrazione. Il campione non valutabile è dovuto alla mancanza di materiale di partenza; questi campioni oltre ad essere biopsie, sono stati tagliati diverse volte, pertanto di alcuni campioni ne è rimasto poco. Legenda: Poli=Policlonalità; NA=Non amplificato; I numeri nelle colonne indicano la lunghezza in bp del frammento maggiormente rappresentato indice di una monoclonalità.
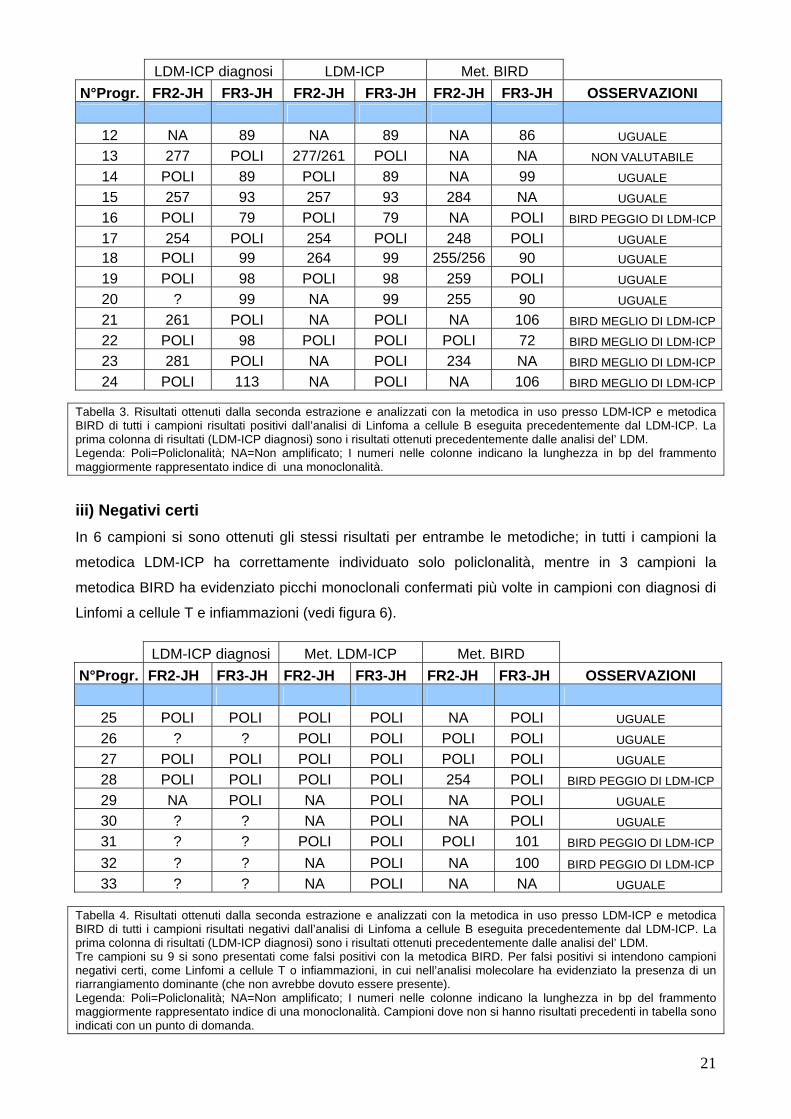
ii) Positivi certi In 7 campioni si sono ottenuti gli stessi risultati per entrambe le metodiche; per 4 campioni è
risultata migliore la metodica BIRD che è stata in grado di evidenziare un picco monoclonale dove
la metodica LDM-ICP (con la seconda estrazione) non è stata in grado. Per 1 campione invece, è
risultata migliore la metodica LDM-ICP che è stata in grado di evidenziare un picco monoclonale
dove la metodica BIRD non è risultata sufficientemente efficace (vedi tabella 3).
21
LDM-ICP diagnosi LDM-ICP Met. BIRD N°Progr. FR2-JH FR3-JH FR2-JH FR3-JH FR2-JH FR3-JH OSSERVAZIONI
12 NA 89 NA 89 NA 86 UGUALE
13 277 POLI 277/261 POLI NA NA NON VALUTABILE
14 POLI 89 POLI 89 NA 99 UGUALE
15 257 93 257 93 284 NA UGUALE
16 POLI 79 POLI 79 NA POLI BIRD PEGGIO DI LDM-ICP
17 254 POLI 254 POLI 248 POLI UGUALE 18 POLI 99 264 99 255/256 90 UGUALE
19 POLI 98 POLI 98 259 POLI UGUALE
20 ? 99 NA 99 255 90 UGUALE
21 261 POLI NA POLI NA 106 BIRD MEGLIO DI LDM-ICP
22 POLI 98 POLI POLI POLI 72 BIRD MEGLIO DI LDM-ICP
23 281 POLI NA POLI 234 NA BIRD MEGLIO DI LDM-ICP
24 POLI 113 NA POLI NA 106 BIRD MEGLIO DI LDM-ICP Tabella 3. Risultati ottenuti dalla seconda estrazione e analizzati con la metodica in uso presso LDM-ICP e metodica BIRD di tutti i campioni risultati positivi dall’analisi di Linfoma a cellule B eseguita precedentemente dal LDM-ICP. La prima colonna di risultati (LDM-ICP diagnosi) sono i risultati ottenuti precedentemente dalle analisi del’ LDM. Legenda: Poli=Policlonalità; NA=Non amplificato; I numeri nelle colonne indicano la lunghezza in bp del frammento maggiormente rappresentato indice di una monoclonalità.
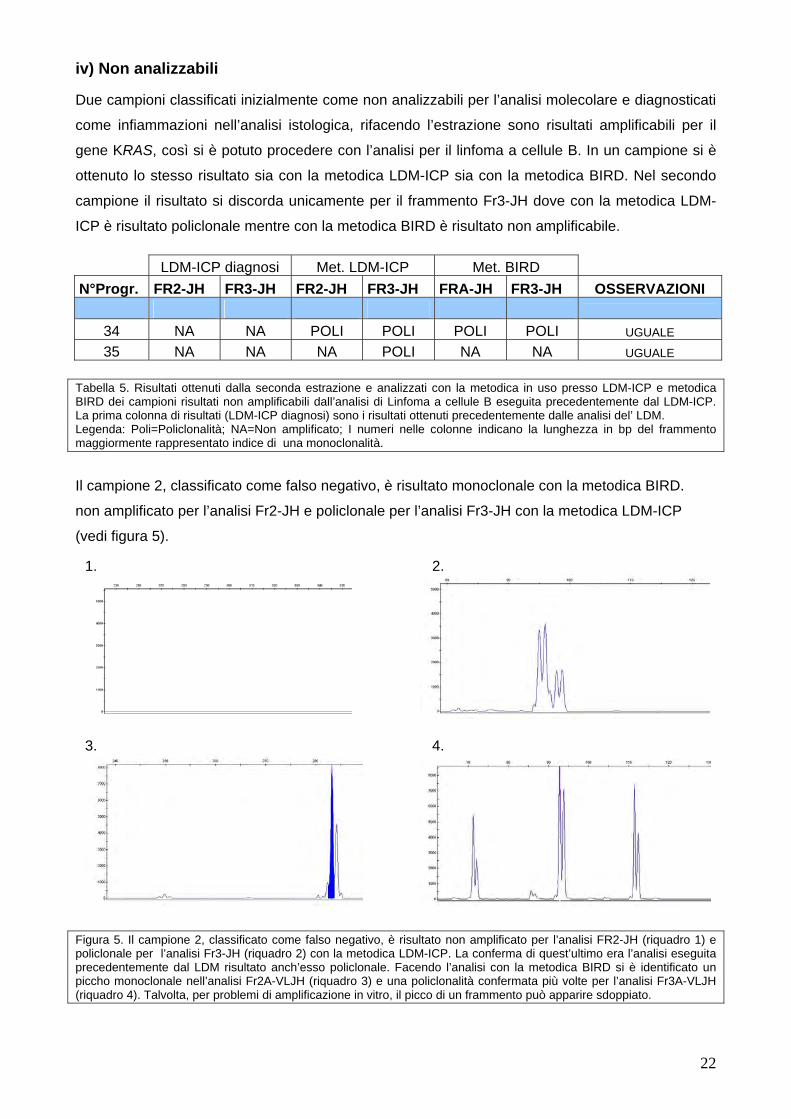
iii) Negativi certi In 6 campioni si sono ottenuti gli stessi risultati per entrambe le metodiche; in tutti i campioni la
metodica LDM-ICP ha correttamente individuato solo policlonalità, mentre in 3 campioni la
metodica BIRD ha evidenziato picchi monoclonali confermati più volte in campioni con diagnosi di
Linfomi a cellule T e infiammazioni (vedi figura 6).
LDM-ICP diagnosi Met. LDM-ICP Met. BIRD N°Progr. FR2-JH FR3-JH FR2-JH FR3-JH FR2-JH FR3-JH OSSERVAZIONI
25 POLI POLI POLI POLI NA POLI UGUALE
26 ? ? POLI POLI POLI POLI UGUALE
27 POLI POLI POLI POLI POLI POLI UGUALE
28 POLI POLI POLI POLI 254 POLI BIRD PEGGIO DI LDM-ICP
29 NA POLI NA POLI NA POLI UGUALE
30 ? ? NA POLI NA POLI UGUALE
31 ? ? POLI POLI POLI 101 BIRD PEGGIO DI LDM-ICP
32 ? ? NA POLI NA 100 BIRD PEGGIO DI LDM-ICP
33 ? ? NA POLI NA NA UGUALE Tabella 4. Risultati ottenuti dalla seconda estrazione e analizzati con la metodica in uso presso LDM-ICP e metodica BIRD di tutti i campioni risultati negativi dall’analisi di Linfoma a cellule B eseguita precedentemente dal LDM-ICP. La prima colonna di risultati (LDM-ICP diagnosi) sono i risultati ottenuti precedentemente dalle analisi del’ LDM. Tre campioni su 9 si sono presentati come falsi positivi con la metodica BIRD. Per falsi positivi si intendono campioni negativi certi, come Linfomi a cellule T o infiammazioni, in cui nell’analisi molecolare ha evidenziato la presenza di un riarrangiamento dominante (che non avrebbe dovuto essere presente). Legenda: Poli=Policlonalità; NA=Non amplificato; I numeri nelle colonne indicano la lunghezza in bp del frammento maggiormente rappresentato indice di una monoclonalità. Campioni dove non si hanno risultati precedenti in tabella sono indicati con un punto di domanda.
22
iv) Non analizzabili Due campioni classificati inizialmente come non analizzabili per l’analisi molecolare e diagnosticati
come infiammazioni nell’analisi istologica, rifacendo l’estrazione sono risultati amplificabili per il
gene KRAS, così si è potuto procedere con l’analisi per il linfoma a cellule B. In un campione si è
ottenuto lo stesso risultato sia con la metodica LDM-ICP sia con la metodica BIRD. Nel secondo
campione il risultato si discorda unicamente per il frammento Fr3-JH dove con la metodica LDM-
ICP è risultato policlonale mentre con la metodica BIRD è risultato non amplificabile.
LDM-ICP diagnosi Met. LDM-ICP Met. BIRD N°Progr. FR2-JH FR3-JH FR2-JH FR3-JH FRA-JH FR3-JH OSSERVAZIONI
34 NA NA POLI POLI POLI POLI UGUALE
35 NA NA NA POLI NA NA UGUALE
Tabella 5. Risultati ottenuti dalla seconda estrazione e analizzati con la metodica in uso presso LDM-ICP e metodica BIRD dei campioni risultati non amplificabili dall’analisi di Linfoma a cellule B eseguita precedentemente dal LDM-ICP. La prima colonna di risultati (LDM-ICP diagnosi) sono i risultati ottenuti precedentemente dalle analisi del’ LDM. Legenda: Poli=Policlonalità; NA=Non amplificato; I numeri nelle colonne indicano la lunghezza in bp del frammento maggiormente rappresentato indice di una monoclonalità.
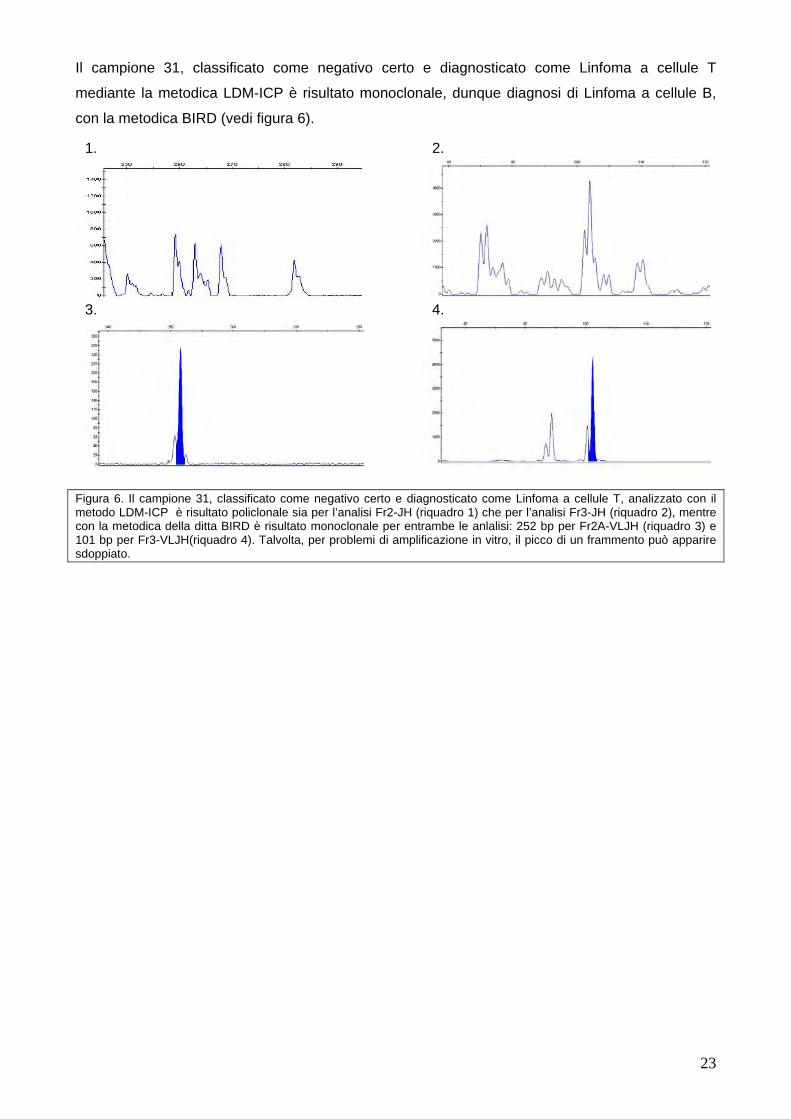
Il campione 2, classificato come falso negativo, è risultato monoclonale con la metodica BIRD.
non amplificato per l’analisi Fr2-JH e policlonale per l’analisi Fr3-JH con la metodica LDM-ICP
(vedi figura 5).
Figura 5. Il campione 2, classificato come falso negativo, è risultato non amplificato per l’analisi FR2-JH (riquadro 1) e policlonale per l’analisi Fr3-JH (riquadro 2) con la metodica LDM-ICP. La conferma di quest’ultimo era l’analisi eseguita precedentemente dal LDM risultato anch’esso policlonale. Facendo l’analisi con la metodica BIRD si è identificato un piccho monoclonale nell’analisi Fr2A-VLJH (riquadro 3) e una policlonalità confermata più volte per l’analisi Fr3A-VLJH (riquadro 4). Talvolta, per problemi di amplificazione in vitro, il picco di un frammento può apparire sdoppiato.
1. 2.
3. 4.
23
Il campione 31, classificato come negativo certo e diagnosticato come Linfoma a cellule T
mediante la metodica LDM-ICP è risultato monoclonale, dunque diagnosi di Linfoma a cellule B,
con la metodica BIRD (vedi figura 6).
Figura 6. Il campione 31, classificato come negativo certo e diagnosticato come Linfoma a cellule T, analizzato con il metodo LDM-ICP è risultato policlonale sia per l’analisi Fr2-JH (riquadro 1) che per l’analisi Fr3-JH (riquadro 2), mentre con la metodica della ditta BIRD è risultato monoclonale per entrambe le anlalisi: 252 bp per Fr2A-VLJH (riquadro 3) e 101 bp per Fr3-VLJH(riquadro 4). Talvolta, per problemi di amplificazione in vitro, il picco di un frammento può apparire sdoppiato.
1. 2.
3. 4.

24
Osservazioni
Nell’ottobre 2008 è stato ordinato il Kit della ditta BIRD con sede a Napoli. Questo Kit contiene
unicamente 45 test, dunque non è bastato per concludere ed ottenere tutti i risultati, soprattutto
conferme e ripetizioni, necessari per il paragone delle due metodiche in esame. A fine marzo 2009
è stato ordinato il secondo Kit della ditta BIRD con arrivo in sede ad inizio aprile 2009;
naturalmente il secondo kit aveva lotto differente e scadenza differente. Durante l’analisi ci siamo
accorti che aveva un problema con l’analisi del frammento Fr3A-VLJH: non è stato amplificato
alcun frammento, nè controlli interni nè campioni amplificati precedentemente con il primo kit dalla
quale si erano ottenuti ottimi risultati.
Per l’analisi del frammento Fr2A-VLJH si sono viceversa ottenuti frammenti di amplificazione
anche se i picchi sono risultati più bassi per confronto con le precedenti analisi ed alcuni campioni
che già in precedenza presentavano picchi bassi con questo kit risultavano non amplificati.
Abbiamo preso subito contatto con la ditta la quale ci ha inviato unicamente la mix per l’analisi
Fr3A-VLJH, senza la Taq polimerasi; fortunatamente nel kit “difettato” c‘era ancora della restante
Taq polimerasi, sufficiente per poter concludere il confronto tra metodiche.

25
Conclusioni
Sulla base dei risultati ottenuti sui pazienti falsi negativi e positivi certi, si può concludere che la
metodica LDM-ICP è paragonabile alla metodica della ditta BIRD: non è stata in grado di
riconoscere 5 campioni su 35 con riarrangiamenti dominanti (individuati invece dal Kit BIRD), ma
ha riconosciuto altri 3 campioni con riarrangiamenti monoclonali (risultati policlonali con il kit BIRD).
Al contrario, nei campioni negativi certi, la metodica LDM-ICP ha correttamente individuato solo
policlonalità, mentre la metodica BIRD ha identificato alcuni picchi monoclonali in campioni risultati
Linfomi a cellule T o infiammazioni, dunque dove non avrebbero dovuto presentarsi riarrangiamenti
dominanti. Questi falsi positivi si sono riscontrati in ben 3 campioni. Quindi la metodica BIRD porta
a classificare come linfoma a cellule B anche pazienti che non sono affetti da tale malattia.
Per una metodica, gli aspetti fondamentali sono due: la sensibilità e la specificità. Il primo
parametro indica quanti pazienti affetti da una data malattia sono individuabili con una data
metodica. Pertanto l’obbiettivo è di avere metodiche altamente sensibili. Da questo punto di vista la
metodica BIRD si fa leggermente preferire rispetto a quella in uso nel LDM-ICP.
La specificità indica invece la possibilità di individuare soltanto i pazienti realmente affetti da una
data malattia. In quest’ottica la metodica BIRD è largamente insufficiente perché porta a
classificare come linfomi a cellule B casi non affetti da tale malattia. Dei due parametri, sensibilità e
specificità, bisogna dare la precedenza al secondo, in quanto una metodica deve essere in grado
di riconoscere con sicurezza unicamente campioni realmente positivi per quella determinata
patologia. Sulla base di queste considerazioni si è deciso di non utilizzare la metodica BIRD per le
future diagnosi, ma di mantenere l’uso della metodica LDM-ICP, che è meno sensibile ma
nettamente più specifica.
Per quanto riguarda le tempistiche, per entrambe le metodiche esse sono comparabili, entrambe
richiedono all’incirca quattro ore e mezza e sono di eguale difficoltà e complessità.
Altra considerazione di cui si deve tener conto è che il costo di un kit è generalmente più elevato
rispetto ad una metodica che utilizza reagenti singoli. Nel caso specifico non è stata fatta una
valutazione precisa perché la metodica BIRD non è risultata specifica, e quindi è stata scartata.
Per di più, nel caso in cui ci sia un problema con un qualsiasi componente del kit (non
funzionamento, contaminazione...), bisogna eliminare tutte le master mix, con considerevole
perdita di tempo (causata dai tempi d’attesa per l’arrivo del nuovo kit e naturalmente quello dovuto
agli accertamenti da parte della ditta, come successo in questo caso) e di costi. Questo problema
non si verifica con la metodica LDM-ICP, che consente di individuare quale sia il reagente alterato
e quindi di procedere alla sostituzione solo di tale reagente in un tempo relativamente breve (dopo
aver eseguito gli opportuni accertamenti) e con sensibili guadagno economico. Queste
considerazioni depongono ulteriormente a favore della metodica LDM-ICP rispetto a quella BIRD.
Un’ultima osservazione interessante è scaturita dalla ri-analisi di pazienti classificati come falsi
negativi: ripetendo l’estrazione, alcuni campioni risultati precedentemente falsi negativi sono
diventati positivi. La ripetizione dell’estrazione ha determinato quindi un aumento della sensibilità

26
della metodica. Di questa importante indicazione il Responsabile del LDM-ICP dovrà tenere conto
nelle analisi diagnostiche future.
In conclusione, il mio lavoro di diploma è servito a confermare che la metodica attualmente in uso
presso l’Istituto Cantonale di Patologia è una buona metodica caratterizzata da elevata specificità
e buona sensibilità, che può essere migliorata provvedendo a rifare l’analisi dopo aver ripetuto
l’estrazione dal DNA genomico.

27
Ringraziamenti
Vorrei ringraziare innanzitutto la direzione dell’Istituto Cantonale di Patologia, Prof. Dr. Med. Luca
Mazzucchelli, per avermi dato l’opportunità di eseguire il mio lavoro di diploma presso l’ICP.
Un grazie particolare a Dr. Milo Frattini, per la grande disponibilità, il tempo a me dedicato e i
preziosi insegnamenti utili, oltre che al svolgimento di questo lavoro, nel mio prossimo futuro;
Dr.ssa Vittoria Martin per l’aiuto datomi.
Ringrazio Dr. Andrea Boffini, Dr. Giovanni Togni e Professor Claudio Naiaretti per la loro
disponibilità in consigli, correzioni e supporto metodologico; la professoressa Susan Gilbert per il
grande aiuto nella stesura dell’abstract e le presentazioni di inglese, le docenti Daniela Marcacci e
Sonja Marci per la loro pazienza e disponibilità soprattutto nei momenti più stressanti e di
debolezza.
Un sincero grazie anche a tutto il team del Laboratorio di Diagnostica Molecolare, “le ragazze”
della ricerca e della diagnostica, per la loro disponibilità e la loro gentilezza, ai Tecnici del
laboratorio d’istopatologia per il tempo e le spiegazioni a me date.
In ultimo, ma non per questo di minor importanza, vorrei ringraziare le mie 5 compagne di classe:
Cinzia Regazzoni, Damiana Ferrari, Prisca Cima, Samuela Balestra, Sandra Jovic per il sostegno
morale, la professionalità e la grande lealtà dimostrata durante tutto l’anno scolastico, e Dr. Med.
Lorenzo Balestra per la pazienza e i preziosi consigli.

28
Bibliografia
Siti internet: 1. http://www.barbiotech.it/LUCIDI/linfomi_lezione_Dr._Renato_Franco.pdf (ultima apertura
pagina 26.05.2009)
2. http://.lymphoma-net.org (ultima apertura pagina 26.05.2009)
Libri e Pubblicazioni:
3. Scali G.Corso di ematologia I,II,III,2007;237-238
4. Registro Tumori Canton Ticino. Alcuni dati di epidemiologia dei linfomi maligni,2005
5. Swerdlow S H.,Campo E., Lee Harris N., Jaffe E S.,Pileri S A.,Stein H.,Thiele J.,Vardiman J
W.WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.4th
Edition.Lyon,2008;198
6. Bronchud M H.,Foote M.,Giaccone G.,Olopade O.,Workman P..Part I,Genetic Markers in
Sporadic Tumors.Principles of Molecular Oncology.Secon Edition.Totowa,New
Jersey,2004;3:73-150
7. Wu G,Keating A.Biomarkers of Potential Prognostic Significance in diffuse Large B-Cell
Lymphoma.Cancer,2006;106:247-257.
8. Bertoni F.,Zucca E..Delving deeper into MALT lymphoma biology.The Journal of Clinical
Investigation,2006;116:22-26
9. Diss T C.,Pan L.,Peng H.,Wotherspoon A C.,Isaacson P G..Sources of DNA for detecting B
cell monoclonality using PCR.Journal of Clinical Pathology,1994;47:493-496

29
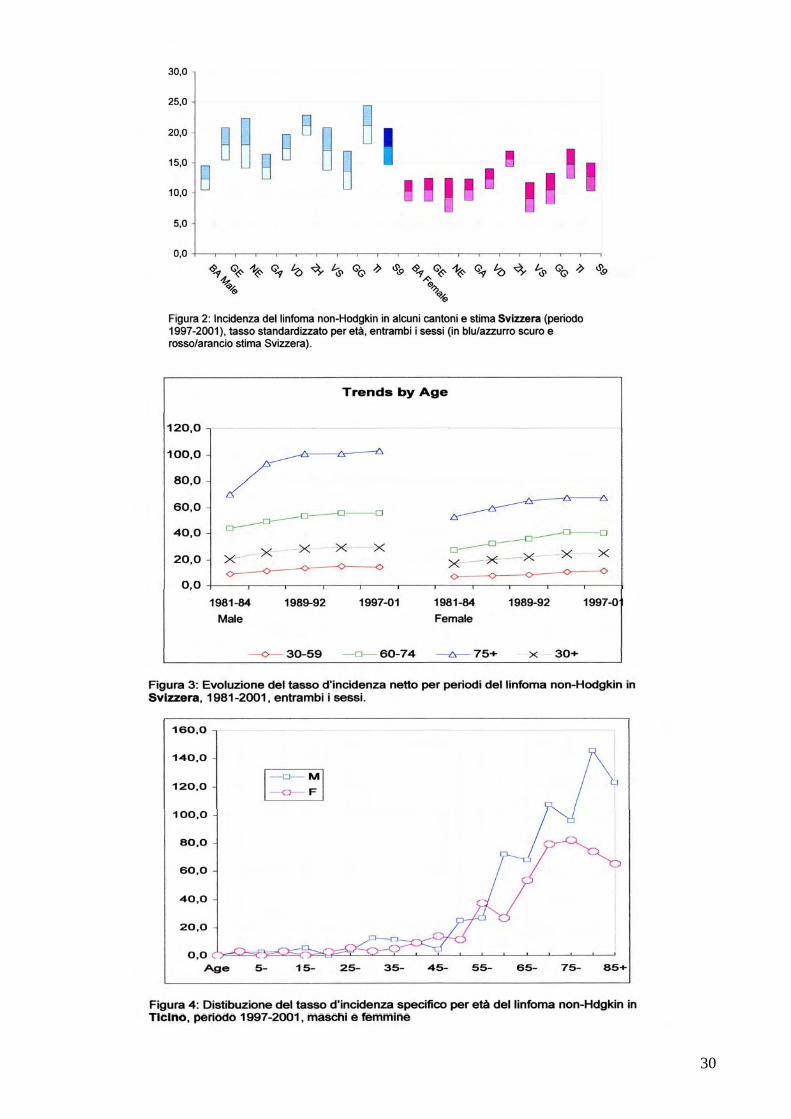
Allegati i) Registro tumori Ticino 2005. Alcuni dati epidemiologici del linfoma Alcuni dati di epidemiologia dei Linfomi maligni Registro Tumori Canton Ticino I linfomi maligni sono tumori generalmente piu’ frequenti nei paesi industrializzati (Figura 1). In Svizzera ogni anno sono 1484 i nuovi casi diagnosticati, mentre in Ticino, secondo i dati del Registro Tumori Ticino, sono annualmente in media 83 le persone colpite da questa malattia. Il linfomi maligni vengono suddivisi in due grandi categorie dette di Hodgkin e non-Hodgkin. I linfomi non-Hodgkin sono i piu’ frequenti. Ogni anno si sviluppano in Ticino 72, dei quali 37 nei maschi e 35 nelle femmine. Il tasso d’incidenza risulta essere piu’ elevato in Ticino rispetto alla media della Svizzera sia per i linfomi di Hodgkin (3.5 per 100000 abitanti in Ticino, rispettivamente 2.6 in Svizzera) sia per i linfomi non-Hodgkin ( 21.1 in Ticino, rispettivamente 17.6 in Svizzera) (Figura 2). Si riscontra una tendenza generalizzata all’aumento di nuovi casi sia negli Stati uniti sia in Svizzera (Figura 3). I Linfomi di Hodgkin sono particolarmente frequenti sia in Svizzera che in Ticino nei giovani e negli anziani, mentre i non-Hodgkin tendono ad aumentare con l’età (Figura 4).. Numerosi i fattori eziologici studiati: tra questi spicca la diretta relazione con alcune infezioni virali come l’HIV/AIDS o batteriche, in particolare l’Helicobacter pylori gastrico. Altre possibili associazioni studiate sono particolari pesticidi utilizzati in agricoltura e fertilizzanti. Malgrado cio’ ancora poco si conosce sugli elementi che scaturiscono la malattia. Un recente studio del Registro tumori Ticino in collaborazione con l’Agenzia Internazionale di Ricerca contro il Cancro dell’OMS pubblicato sul Journal of National Cancer Istitute conferma la relazione del linfoma non-Hodgkin con l’AIDS conclamata ma allo stesso tempo assegna ai medicamenti anti AIDS un potenziale fattore protettivo/preventivo contro l’insorgenza dei linfomi. I progressi in ambito terapeutico hanno permesso di raggiungere globalmente percentuali di sopravvivenza a cinque anni pari a 60-80% delle diagnosi.
30

31
ii) Diss et al, J Clin Pathol 1994;47:493-496

32

33

34

35
iii) Protocollo LDM-ICP Fr2-JH 1. Introduzione I linfomi maligni non Hodgkin (LMNH) a cellule B comprendono numerosi sottogruppi neoplastici con caratteristiche morfologiche, biologiche e genetiche molto eterogenee, tutti generalmente caratterizzati da un riarrangiamento che coinvolge il gene della catena pesante delle immunoglobuline (IgH), con vari partners. Le cellule che portano questi riarrangiamenti acquisiscono capacità proliferative e tendono quindi a riprodursi in modo incontrollato, diventando la sottospecie cellulare maggiormente presente. La dimostrazione di monoclonalità è quindi diagnostica e riveste grande importanza perché permette di distinguere un linfoma da lesioni di linfonodi reattivi, che possiedono caratteristiche morfologiche ed immunofenotipiche difficili da interpretare. L’approccio della PCR consente di amplificare due frammenti (regione framework II e III fino alla regione joining JH), in cui globalmente i riarrangiamenti sono osservati in circa il 70-80% dei linfomi a cellule B. Inoltre, la possibilità di individuare con precisioni le dimensioni del riarrangiamento che si è verificato consentono anche di monitorare il decorso del paziente e verificare la presenza del dato riarrangiamento anche partendo da poche cellule neoplastiche. 2. Materiali clinici e controlli Laboratorio locale 224-Blocco B DNA genomico estratto da: - cellule in RPMI (4.1.IO estrazione DNA da cellule in RPMI - DM) - cellule in Thin Prep (4.1.IO estrazione DNA da cellule in Thin Prep - DM) - cellule in FACSFlow (4.1.IO estrazione DNA da cellule in FACSFlow - DM) - sangue intero (4.1.IO estrazione DNA da sangue intero - DM) - tessuto congelato (4.1.IO estrazione DNA da tessuto congelato - DM) - tessuto fissato in formalina ed incluso in paraffina (4.1.IO estrazione DNA da tessuto fissato -
DM) - sezioni in bianco (4.1.IO estrazione DNA da sezioni in bianco - DM) + bianco di reazione: (Blank) reazione con tutti i reagenti ma senza DNA, serve a verificare che i vari reagenti non siano stati erroneamente contaminati con DNA. + controllo positivo: (C+) reazione con tutti i reagenti e con DNA genomico (di un altro paziente o dello stesso se già stato analizzato) che è stato amplificato con successo in una precedente occasione. 3. Materiali d’uso, apparecchi d’uso e reagenti 3.1 Materiale d'uso • Scatola di polistirolo (sagex) con ghiaccio • Micropipette per PCR sotto cappa DM71 • Micropipette per preparazione Genetic Analyzer sotto cappa DM 59 • Punte sterili con filtro (0.1-10µl / 1-20µl / 1-100µl / 1-200µl / 101-1000µl) • Provette sterili DNase / RNase Free da 1.5 ml • Provette sterili DNase / RNase Free da 0.5 ml • Provette sterili DNase / RNase Free da 0.2 ml • Guanti in lattice senza polvere • Portaprovette in plastica • Vetreria 3.2 Apparecchi d'uso • Mini Centrifuga Eppendorf 5415R DM 69 • Cappa DM 71 • Termociclatori: DM 64, DM 82, DM 83, DM 84, DM 124 e DM 125 • Centrifuga DM 94 • Mini-Cooler • Genetic Analyzer – 3130 DM 68

36
• Cappa DM 59 3.3 Reagenti d’uso Annotare sui moduli relativi il lotto dei reagenti utilizzati, la data di utilizzo e la firma dell’operatore. Reagenti conservati a -20º C (congelatore DM 102- box MILO scorte): • Enzima UNG (Uracyl-DNA Glycosylase-USB o Roche) 1U/µl • dNTPs (dATP, dTTP, dCTP, dGTP) 10mM, precedentemente preparato (4.1.IO aliquote di
dNTPs - DM) • dUTP 10mM, precedentemente preparato (4.1.IO aliquote di dUTP - DM) • AmpliTaq Gold with GeneAmp (Applied Biosystems-Roche)
Il kit contiene l'enzima Taq Gold (5U/µl), il tampone d'incubazione (10x PCR Buffer II) e la soluzione di magnesio cloruro (MgCl2 Solution-25 mM).
Il Kit si conserva a -20°C. • H2O sterile autoclavata (4.1.IO aliquote di H2O sterile autoclavata - DM) Reagenti conservati a -20º C (congelatore DM 108- box MILO): • Hi-Di Formamide (4.1.IO aliquote di Hi-Di Formamide - DM) • H2O sterile autoclavata (4.1.IO aliquote di H2O sterile autoclavata - DM) Reagenti conservati a 4º C (frigorifero DM 67): • Gene Scan TM-500 LIZ Size Standard (Applied Biosystems) 3.4 Primers Sequenze primers:
FR2 5’-TGGRTCCGMCAGSCYYCNGG-3’ JH 5’-ACCTGAGGAGACGGTGACC-3’
Il primer JH è modificato con l’aggiunta in 5’ del fluorocromo 6-FAM. I primers sono aliquotati con procedura a parte (4.1.IO diluizioni primers - DM) 4. Procedimento analitico La preparazione della mix per la PCR si esegue esclusivamente nella cappa DM 71 indossando sempre i guanti, che vengono cambiati ogni volta che si tocca qualcosa fuori dalla cappa e ogni volta che ci si accorge di aver toccato un puntale non più sterile o un bordo di una provetta. 4.1 Preimpostazione del termociclatore Laboratorio locale 326-Blocco B Accendere il termociclatore e impostare il programma “FR2“ (il programma è impostato con lo stesso nome su tutti i termociclatori). 4.2 Aliquotamento del DNA Laboratorio locale 226-Blocco B Preparare e contrassegnare le provette per la reazione a seconda dei campioni da amplificare aggiungendo un Blank (-) e un controllo positivo (C+). Lavorare sotto cappa con i guanti. Centrifugare brevemente in DM 94 le provette e porle in un supporto in ghiaccio. Pipettare: • 2 µl di DNA dei campioni da analizzare della soluzione madre per ogni paziente, oppure • 2 µl di H2O sterile nel Blank (-), oppure • 2 µl DNA di un paziente (o dello stesso se già stato analizzato) che è stato precedentemente amplificato con successo nel C+(+) 4.3 Preparazione della miscela (mix) Laboratorio locale 226-Blocco B Prendere dal congelatore DM 102 i reagenti PCR necessari dalle apposite scatole dal box MILO: • 10x PCR Buffer II • MgCl2 Solution (25 mM)

37
• aliquota di dNTPs (10 mM each) • aliquota di dUTP (10 mM) • aliquota di H2O sterile • aliquota primer FR2 • aliquota primer JH Scongelare i reagenti, centrifugare brevemente in DM 94 e porli in un supporto in ghiaccio. Prendere il Mini-Cooler dal congelatore DM 102, contenente l’AmpliTaq Gold e l’enzima UNG, e portarlo all’interno della cappa PCR. Per il conteggio del volume della miscela di reazione, in ogni esperimento bisogna calcolare tutti i campioni + il controllo negativo + il controllo positivo e aumentare tale cifra di 1 o più a seconda del numero dei campioni (in generale + 1 ogni 10 campioni circa). Calcolare e pipettare i volumi dei reagenti cominciando dal buffer e proseguendo in ordine, per finire con l’acqua, come da protocollo qui di seguito riportato. conc. iniz. conc.fin. volume X 1 volume per x campioni
H20 34.55 µl . µl PCR Buffer 10 x 1 x 5 µl . µl MgCl2 25 mM 1.5 mM 3 µl . µl
dNTPs 10 mM 0.2 mM 1 µl . µl dUTP 10 mM 0.04 mM 0.2 µl . µl Primer FR2 20 µM 0.6 µM 1.5 µl . µl
Primer JH 20 µM 0.6 µM 1.5 µl . µl UNG 1 U/µl 1 U/s 1 µl . µl Taq Gold 5 U/µl 1.25 U/s 0.25 µl . µl _________ µl Attenzione:
prima di aggiungere il primer JH fluorocromato spegnere la luce bianca della cappa.
dal momento che si aggiunge la UNG e la Taq Gold bisogna portare a termine la preparazione della Mix e l’aggiunta della stessa alle provette contenenti DNA nel minor tempo possibile. In ogni provetta contenente il DNA, aggiungere velocemente 48 µl di miscela precedentemente preparata. 4.4 Amplificazione dei campioni Laboratorio locale 326-Blocco B Aprire il coperchio e alloggiare le provette di PCR nel blocco metallico del termociclatore (DM 64, DM 82, DM 83, DM 84, DM 124 e DM 125), chiudere il coperchio, dare avvio al programma precedentemente impostato qui di seguito riportato:

38
INCUBAZIONE UNG: 50ºC per 2’ DENATURAZIONE INIZIALE: 95ºC per 10’ DENATURAZIONE: 94ºC per 1’ AMPLIFICAZIONE: 57ºC per 1’ per 45 cicli ESTENSIONE: 72ºC per 2’ ESTENSIONE FINALE: 72ºC per 5’ FINE PCR: 10ºC fino a quando i tubi vengono prelevati Terminata la reazione di PCR, togliere le provette dal termociclatore e conservarle a 4 ºC nel frigorifero DM 97. 4.5 Visualizzazione su Genetic Analyzer su 3130 La preparazione si esegue esclusivamente nella cappa DM59 senza guanti con le mani ben pulite (senza l’uso di sapone) e senza luce bianca. 4.5.1 Preimpostazione del termociclatore Laboratorio locale 326-Blocco B Accendere il termoclicatore e impostare il programma “DENATUR” (il programma è impostato con lo stesso nome in tutti i termoclicatori). Dare avvio al programma qui di seguito riportato: DENATURAZIONE : 95ºC per 2h 4.5.2 Aliquotamento dei reagenti Laboratorio locale 325-Blocco B Dato che si utilizza un primer coniugato con un fluorocromo e il marcatore Gene Scan TM-500 LIZ fluorocromato, la preparazione della miscela viene effettuata senza luce sotto cappa (lasciare accesa la luce del locale 325). Togliere le provette di reazione di PCR dal frigorifero DM 97 e centrifugare brevemente in DM 69. Aliquotare 1 µl della reazione di amplificazione di ogni campione, aggiungere 0,25 µl di Gene Scan TM-500 LIZ Size Standard e 10 µl di formamide. 4.5.3 Denaturazione dei campioni Laboratorio locale 326-Blocco B Aprire il coperchio e alloggiare le provette di reazione di PCR nel blocco metallico dei termociclatori (DM 64, DM 82, DM 83, DM 84, DM 124 e DM 125), chiudere il coperchio. Lasciare le provette per 2’. Terminata la denaturazione, togliere le provette dal termociclatore e metterle immediatamente in ghiaccio per 15’ al buio. I campioni denaturati si possono eventualmente conservare a -20ºC nel frigorifero (DM 102-box 3130). 4.5.4 Preparazione della tabella Allestire la tabella seguendo il protocollo dell’apparecchio Genetic Analyzer – 3130 DM 68. 4.5.5 Preparazione della piastra Laboratorio locale 325-Blocco B Centrifugare brevemente in DM 69 i campioni denaturati che si trovano in ghiaccio. Aliquotare i campioni nei pozzetti della piastra secondo la tabella preparata precedentemente. Chiudere la piastra con il coperchio di gomma grigio (“septa”). Inserire la piastra nel supporto nero e assemblarla con il coperchio bianco rigido. 4.5.6 Corsa su 3130 Inserire la piastra nell’apparecchio Genetic Analyzer – 3130 DM 68 come da protocollo.

39
5. Validazione tecnica La validazione della metodica consta di 4 fasi successive.
1. Controllo da parte del patologo di riferimento della qualità del tessuto: il tessuto tumorale deve contenere almeno il 70% di cellule tumorali: qualora venisse ravvisato un quantitativo inferiore, si procede ad una macrodissezione (il patologo segna l’area da cui si deve estrarre il DNA, l’operatore in controluce disegna sulle sezioni in bianco l’area selezionata e quindi procede allo scraping dell’area selezionata con un bisturi sterile).
2. Utilizzo di primers e condizioni di amplificazione ampiamente validati in letteratura in termini di numero di pubblicazioni e che hanno dato (e forniscono) risultati in linea con quanto atteso per frequenza e tipo di alterazione
3. Utilizzo di almeno un bianco di reazione (provetta contenente tutto il necessario e trattata come le altre, ma in cui non è stato aggiunto DNA) e un controllo positivo (provetta contenente DNA di un tessuto che precedentemente è stato amplificato con successo utilizzando le medesime condizioni), il tutto effettuando le manipolazioni in una cappa apposita per l’allestimento delle reazioni di PCR e l’aggiunta di UNG, un enzima che impedisce che eventuali contaminazioni da carry-over (ovvero frutto di precedenti amplificazioni) possano essere amplificati nella reazione in esame. Il controllo negativo deve sempre risultare privo di prodotti di amplificazione, in caso contrario si deve procedere alla ripetizione della PCR di tutti i campioni; occasionalmente, si puo’ osservare una banda, che pero’ è di dimensioni differenti da quelle attese (probabili catene di primer): l’importante è verificare sempre con accuratezza le dimensioni di tale banda, facendo eventualmente correre il gel per un tempo superiore, in modo da ottenere una separazione ottimale delle bande. In questo caso si considera valutabile l’esperimento. Il controllo positivo deve sempre dare un prodotto di amplificazione di dimensioni pari alle attese; qualora cio’ non avvenisse (ed in presenza di prodotti di amplificazioni di dimensioni corrette nei campioni da analizzare), procedere ad eliminare quello specifico controllo positivo e cambiarlo nella prossima reazione di PCR.
4. Ripetizione di tre volte dell’esperimento, partendo da prodotti di amplificazione generati in tempi differenti ed in modo indipendente, per confermare il risultato.
6. Interpretazione ed emissione del risultato 6.1 Allineamento degli elettroferogrammi Aprire il programma GeneMapper utilizzando la username gm e la password 3130User. Cliccare sulla quarta icona da sinistra e importare i file corrispondenti alle corse elettroforetiche dalla cartella Local Disk (D:)\AB Service\Installation\Verification\Fragment Analysis\, dalla cartella con data AAA.MM.GG, selezionare i file che si desiderano e cliccare poi su Add nella maschera di sinistra e poi Add nella maschera di destra. A questo punto i file corrispondenti alle corse elettroforetiche sono stati inseriti in un nuovo progetto. Cliccare su ogni file nel progetto per selezionarlo, e poi cliccare su view (nel menu principale) e quindi su Raw Data, per visualizzare la corsa e vedere se ci sono stati problemi o se la separazione dei frammenti e del marker è avvenuta correttamente. Selezionare sul primo file la casella Analysis Method e poi selezionare Microsatellite Default, quindi cliccare su Analysis Method e digitare sulla tastiera CTRL+D, in questo modo tutti i file verranno analizzati con il metodo Microsatellite Default. Selezionare sul primo file la casella Size Standard e poi selezionare GS500LIZ, quindi cliccare su Size Standard e digitare sulla tastiera CTRL+D, in questo modo tutti i file verranno allineati con il marker GS500LIZ. Selezionare nel menu principale Edit, cliccare su Select All e quindi cliccare sulla 14ma icona (freccia verde orientata a destra): dando il nome al progetto, il software procede all’allineamento dei campioni e alla determinazione delle dimensioni di tutti i frammenti presenti nella corsa elettroforetica, sulla base del marker GS500LIZ. Selezionando il file desiderato e cliccando sulla 6° icona, si apre una maschera che mostra la corsa elettroforetica. Nel banner principale selezionare soltanto il colore blu e quindi cliccare sulla seconda icona da destra, che consente di visualizzare una tabella che riporta i dati salienti della corsa elettroforetica (tra cui le dimensioni dei picchi, l’altezza dei picchi e l’area sottesa i picchi). Cliccando subito sopra il grafico, appare una lente di ingrandimento che permette di selezionare l’area della corsa elettroforetica che corrisponde alle dimensioni attese dell’amplificato. Controllare in questo modo che nel bianco di reazione non ci siano frammenti nell’intervallo desiderato e che il controllo positivo abbia fornito frammenti di dimensioni attese.

40
6.2 Interpretazione dell’elettroferogramma I frammenti attesi sono compresi tra 250 e 350 bp. Ogni picco che si osserva nell’intervallo in questione corrisponde ad un riarrangiamento. La presenza di un picco dominante (che ha un’altezza almeno doppia rispetto agli altri frammenti) indica la possibilità che sia presente un riarrangiamento in quantitativo superiore. Ripetendo l’esperimento per altre due volte, se il picco dominante si conferma tale (controllare che le dimensioni del frammento siano sempre le stesse, un discostamento anche di 1 bp indica la presenza di un riarrangiamento differente) il paziente viene classificato come avente monoclonalità per IgH (che vuol dire che il paziente ha un linfoma), viceversa se il picco in questione non è piu’ la forma maggiormente presente, vuol dire che c’è assenza di monoclonalità (e quindi il paziente è classificato come policlonale): in quest’ultimo caso, comunque, non si puo’ completamente escludere che il paziente abbia un linfoma perché il 20-30% dei pazienti con linfomi non mostrano monoclonalità con la metodica in questione. Bisogna prestare particolare attenzione ai casi in cui il DNA di partenza che viene utilizzato per l’amplificazione è scarso (inferiore a 50 ng totali): in queste condizioni è facile avere false monoclonalità, ma confrontando gli spettri ottenuti da amplificazioni differenti è facile verificare l’effettiva presenza di monoclonalità (il picco dominante ha dimensioni differenti nei vari elettroferogrammi nei casi di falsa monoclonalità, e quindi devono essere classificati come policlonali). Se si osservano 2 picchi dominanti in tutti gli esperimenti, il paziente puo’ essere monoclonale (con un riarrangiamento su ciascuno dei due alleli) o biclonale (due popolazioni cellulari con riarrangiamenti differenti coesistono nell’individuo): in ogni caso il paziente è affetto da un linfoma. Se si osservano 3-5 frammenti dominanti, il paziente è classificato come oligoclonale, ovvero ha poche ma diverse cloni cellulari che sono presenti in quantità abnormi nel proprio organismo. 6.3 Emissione del referto Il referto viene compilato aggiungendo ai dati clinici del paziente il risultato delle analisi molecolari, firmato dall’operatore (o dagli operatori) e dal responsabile dell’analisi, quindi consegnato al patologo di riferimento che procede all’emissione del referto vero e proprio. Una copia cartacea del referto, firmata, viene mantenuta nei raccoglitori nella biblioteca centrale del laboratorio per le analisi dell’anno in corso, o nello studio del responsabile delle analisi per gli anni precedenti, per un periodo massimo di 5 anni. Una copia in formato elettronico del referto viene mantenuta sul server dell’Istituto nella cartella F:\SXIP055\Referti, in cartelle specifiche per le varie analisi molecolari richieste divise per anno. 7. Osservazioni Bibliografia rilevante: Diss et al, J Clin Pathol 1994;47:493-496. 8. Allegati Nessuno.

41
iv) Protocollo LDM-ICP Fr3-JH 1. Introduzione I linfomi maligni non Hodgkin (LMNH) a cellule B comprendono numerosi sottogruppi neoplastici con caratteristiche morfologiche, biologiche e genetiche molto eterogenee, tutti generalmente caratterizzati da un riarrangiamento che coinvolge il gene della catena pesante delle immunoglobuline (IgH), con vari partners. Le cellule che portano questi riarrangiamenti acquisiscono capacità proliferative e tendono quindi a riprodursi in modo incontrollato, diventando la sottospecie cellulare maggiormente presente. La dimostrazione di monoclonalità è quindi diagnostica e riveste grande importanza perché permette di distinguere un linfoma da lesioni di linfonodi reattivi, che possiedono caratteristiche morfologiche ed immunofenotipiche difficili da interpretare. L’approccio della PCR consente di amplificare due frammenti (regione framework II e III fino alla regione joining JH), in cui globalmente i riarrangiamenti sono osservati in circa il 70-80% dei linfomi a cellule B. Inoltre, la possibilità di individuare con precisioni le dimensioni del riarrangiamento che si è verificato consentono anche di monitorare il decorso del paziente e verificare la presenza del dato riarrangiamento anche partendo da poche cellule neoplastiche. 2. Materiali clinici e controlli Laboratorio locale 224-Blocco B DNA genomico estratto da: - cellule in RPMI (4.1.IO estrazione DNA da cellule in RPMI - DM) - cellule in Thin Prep (4.1.IO estrazione DNA da cellule in Thin Prep - DM) - cellule in FACSFlow (4.1.IO estrazione DNA da cellule in FACSFlow - DM) - sangue intero (4.1.IO estrazione DNA da sangue intero - DM) - tessuto congelato (4.1.IO estrazione DNA da tessuto congelato - DM) - tessuto fissato in formalina ed incluso in paraffina (4.1.IO estrazione DNA da tessuto fissato -
DM) - sezioni in bianco (4.1.IO estrazione DNA da sezioni in bianco - DM) + bianco di reazione: (Blank) reazione con tutti i reagenti ma senza DNA, serve a verificare che i vari reagenti non siano stati erroneamente contaminati con DNA.
+ controllo positivo: (C+) reazione con tutti i reagenti e con DNA genomico (di un altro paziente o dello stesso se già stato analizzato) che è stato amplificato con successo in una precedente occasione. 3.Materiali d’uso, apparecchi d’uso e reagenti 3.1 Materiale d'uso • Scatola di polistirolo (sagex) con ghiaccio • Micropipette per PCR sotto cappa DM 71 • Micropipette per preparazione Genetic Analyzer sotto cappa DM 59 • Punte sterili con filtro (0.1-10µl / 1-20µl / 1-100µl / 1-200µl / 101-1000µl) • Provette sterili DNase / RNase Free da 1.5 ml • Provette sterili DNase / RNase Free da 0.5 ml • Provette sterili DNase / RNase Free da 0.2 ml • Guanti in lattice senza polvere • Portaprovette in plastica • Vetreria
3.2 Apparecchi d'uso • Mini Centrifuga Eppendorf 5415R DM 69 • Cappa DM 71 • Termociclatori: DM 64, DM 82, DM 83, DM 84, DM 124 e DM 125 • Centrifuga DM 94 • Mini-Cooler • Genetic Analyzer – 3130 DM 68

42
• Cappa DM 59 3.3 Reagenti d’uso Annotare sui moduli relativi il lotto dei reagenti utilizzati, la data di utilizzo e la firma dell’operatore.
Reagenti conservati a -20º C (congelatore DM 102- box MILO scorte): • Enzima UNG (Uracyl-DNA Glycosylase-USB o Roche) 1U/µl • dNTPs (dATP, dTTP, dCTP, dGTP) 10mM, precedentemente preparato (4.1.IO aliquote di
dNTPs - DM) • dUTP 10mM, precedentemente preparato (4.1.IO aliquote di dUTP - DM) • AmpliTaq Gold with GeneAmp (Applied Biosystems-Roche)
Il kit contiene l'enzima Taq Gold (5U/µl), il tampone d'incubazione (10x PCR Buffer II) e la soluzione di magnesio cloruro (MgCl2 Solution-25 mM). Il Kit si conserva a -20°C.
• H2O sterile autoclavata (4.1.IO aliquote di H2O sterile autoclavata - DM)
Reagenti conservati a -20º C (congelatore DM 108- box MILO): • Hi-Di Formamide (4.1.IO aliquote di Hi-Di Formamide - DM) • H2O sterile autoclavata (4.1.IO aliquote di H2O sterile autoclavata - DM) Reagenti conservati a 4º C (frigorifero DM 67): • Gene Scan TM-500 LIZ Size Standard (Applied Biosystems) 3.4 Primers Sequenze primers:
FR3 5’-ACACGGCYSTRTATTACTGT-3’ JH 5’-ACCTGAGGAGACGGTGACC-3’
Il primer JH è modificato con l’aggiunta in 5’ del fluorocromo 6-FAM. I primers sono aliquotati con procedura a parte (4.1.IO diluizioni primers - DM) 4. Procedimento analitico La preparazione della mix per la PCR si esegue esclusivamente nella cappa DM 71 indossando sempre i guanti, che vengono cambiati ogni volta che si tocca qualcosa fuori dalla cappa e ogni volta che ci si accorge di aver toccato un puntale non più sterile o un bordo di una provetta. 4.1 Preimpostazione del termociclatore Laboratorio locale 326-Blocco B Accendere il termociclatore e impostare il programma “FR3“ (il programma è impostato con lo stesso nome su tutti i termociclatori). 4.2 Aliquotamento del DNA Laboratorio locale 226-Blocco B Preparare e contrassegnare le provette per la reazione a seconda dei campioni da amplificare aggiungendo un Blank (-) e un controllo positivo (C+). Lavorare sotto cappa con i guanti. Centrifugare brevemente in DM 94 le provette e porle in un supporto in ghiaccio. Pipettare: • 2 µl di DNA dei campioni da analizzare della soluzione madre per ogni paziente, oppure • 2 µl di H2O sterile nel Blank (-), oppure • 2 µl DNA di un paziente (o dello stesso se già stato analizzato) che è stato precedentemente
amplificato con successo nel C+(+)
4.3 Preparazione della miscela (mix) Laboratorio locale 226-Blocco B Prendere dal congelatore DM 102 i reagenti PCR necessari dalle apposite scatole dal box MILO: • 10x PCR Buffer II

43
• MgCl2 Solution (25 mM) • aliquota di dNTPs (10 mM each) • aliquota di dUTP (10 mM) • aliquota di H2O sterile • aliquota primer FR3 • aliquota primer JH
Scongelare i reagenti, centrifugare brevemente in DM 94 e porli in un supporto in ghiaccio. Prendere il Mini-Cooler dal congelatore DM 102, contenente l’AmpliTaq Gold e l’enzima UNG, e portarlo all’interno della cappa PCR. Per il conteggio del volume della miscela di reazione, in ogni esperimento bisogna calcolare tutti i campioni + il controllo negativo + il controllo positivo e aumentare tale cifra di 1 o più a seconda del numero dei campioni (in generale + 1 ogni 10 campioni circa). Calcolare e pipettare i volumi dei reagenti cominciando dal buffer e proseguendo in ordine, per finire con l’acqua, come da protocollo qui di seguito riportato. conc. iniz. conc.fin. volume X 1 volume per x campioni
H20 35.55 µl . µl PCR Buffer 10 x 1 x 5 µl . µl MgCl2 25 mM 1.5 mM 3 µl . µl
dNTPs 10 mM 0.2 mM 1 µl . µl dUTP 10 mM 0.04 mM 0.2 µl . µl Primer FR3 20 µM 0.4 µM 1 µl . µl
Primer JH 20 µM 0.4 µM 1 µl . µl UNG 1 U/µl 1 U/s 1 µl . µl Taq Gold 5 U/µl 1.25 U/s 0.25 µl . µl
_________ µl Attenzione:
prima di aggiungere il primer JH fluorocromato spegnere la luce bianca della cappa.
dal momento che si aggiunge la UNG e la Taq Gold bisogna portare a termine la preparazione della Mix e l’aggiunta della stessa alle provette contenenti DNA nel minor tempo possibile.
In ogni provetta contenente il DNA, aggiungere velocemente 48 µl di miscela precedentemente preparata. 4.4 Amplificazione dei campioni Laboratorio locale 326-Blocco B Aprire il coperchio e alloggiare le provette di PCR nel blocco metallico del termociclatore (DM 64, DM 82, DM 83, DM 84, DM 124 e DM 125), chiudere il coperchio, dare avvio al programma precedentemente impostato qui di seguito riportato:

44
INCUBAZIONE UNG: 50ºC per 2’ DENATURAZIONE INIZIALE: 95ºC per 10’ DENATURAZIONE: 94ºC per 1’ AMPLIFICAZIONE: 55ºC per 1’ per 45 cicli ESTENSIONE: 72ºC per 1’30’’ ESTENSIONE FINALE: 72ºC per 5’ FINE PCR: 10ºC fino a quando i tubi vengono prelevati Terminata la reazione di PCR, togliere le provette dal termociclatore e conservarle a 4 ºC nel frigorifero DM 97. 4.5 Visualizzazione su Genetic Analyzer su 3130 La preparazione si esegue esclusivamente nella cappa DM59 senza guanti con le mani ben pulite (senza l’uso di sapone) e senza luce bianca. 4.5.1 Preimpostazione del termociclatore Laboratorio locale 326-Blocco B Accendere il termoclicatore e impostare il programma “DENATUR” (il programma è impostato con lo stesso nome in tutti i termoclicatori). Dare avvio al programma qui di seguito riportato: DENATURAZIONE : 95ºC per 2h 4.5.2 Aliquotamento dei reagenti Laboratorio locale 325-Blocco B Dato che si utilizza un primer coniugato con un fluorocromo e il marcatore Gene Scan TM-500 LIZ fluorocromato, la preparazione della miscela viene effettuata senza luce sotto cappa (lasciare accesa la luce del locale 325). Togliere le provette di reazione di PCR dal frigorifero DM 97 e centrifugare brevemente in DM 69. Aliquotare 1 µl della reazione di amplificazione di ogni campione, aggiungere 0,25 µl di Gene Scan TM-500 LIZ Size Standard e 10 µl di formamide. 4.5.3 Denaturazione dei campioni Laboratorio locale 326-Blocco B Aprire il coperchio e alloggiare le provette di reazione di PCR nel blocco metallico dei termociclatori (DM 64, DM 82, DM 83, DM 84, DM 124 e DM 125), chiudere il coperchio. Lasciare le provette per 2’. Terminata la denaturazione, togliere le provette dal termociclatore e metterle immediatamente in ghiaccio per 15’ al buio. I campioni denaturati si possono eventualmente conservare a -20ºC nel frigorifero (DM 102-box 3130). 4.5.4 Preparazione della tabella Allestire la tabella seguendo il protocollo dell’apparecchio Genetic Analyzer – 3130 DM 68. 4.5.5 Preparazione della piastra Laboratorio locale 325-Blocco B Centrifugare brevemente in DM 69 i campioni denaturati che si trovano in ghiaccio. Aliquotare i campioni nei pozzetti della piastra secondo la tabella preparata precedentemente. Chiudere la piastra con il coperchio di gomma grigio (“septa”). Inserire la piastra nel supporto nero e assemblarla con il coperchio bianco rigido. 4.5.6 Corsa su 3130 Inserire la piastra nell’apparecchio Genetic Analyzer – 3130 DM 68 come da protocollo.

45
5. Validazione tecnica La validazione della metodica consta di 4 fasi successive.
5. Controllo da parte del patologo di riferimento della qualità del tessuto: il tessuto tumorale deve contenere almeno il 70% di cellule tumorali: qualora venisse ravvisato un quantitativo inferiore, si procede ad una macrodissezione (il patologo segna l’area da cui si deve estrarre il DNA, l’operatore in controluce disegna sulle sezioni in bianco l’area selezionata e quindi procede allo scraping dell’area selezionata con un bisturi sterile).
6. Utilizzo di primers e condizioni di amplificazione ampiamente validati in letteratura in termini di numero di pubblicazioni e che hanno dato (e forniscono) risultati in linea con quanto atteso per frequenza e tipo di alterazione
7. Utilizzo di almeno un bianco di reazione (provetta contenente tutto il necessario e trattata come le altre, ma in cui non è stato aggiunto DNA) e un controllo positivo (provetta contenente DNA di un tessuto che precedentemente è stato amplificato con successo utilizzando le medesime condizioni), il tutto effettuando le manipolazioni in una cappa apposita per l’allestimento delle reazioni di PCR e l’aggiunta di UNG, un enzima che impedisce che eventuali contaminazioni da carry-over (ovvero frutto di precedenti amplificazioni) possano essere amplificati nella reazione in esame. Il controllo negativo deve sempre risultare privo di prodotti di amplificazione, in caso contrario si deve procedere alla ripetizione della PCR di tutti i campioni; occasionalmente, si puo’ osservare una banda, che pero’ è di dimensioni differenti da quelle attese (probabili catene di primer): l’importante è verificare sempre con accuratezza le dimensioni di tale banda, facendo eventualmente correre il gel per un tempo superiore, in modo da ottenere una separazione ottimale delle bande. In questo caso si considera valutabile l’esperimento. Il controllo positivo deve sempre dare un prodotto di amplificazione di dimensioni pari alle attese; qualora cio’ non avvenisse (ed in presenza di prodotti di amplificazioni di dimensioni corrette nei campioni da analizzare), procedere ad eliminare quello specifico controllo positivo e cambiarlo nella prossima reazione di PCR.
8. Ripetizione di tre volte dell’esperimento, partendo da prodotti di amplificazione generati in tempi differenti ed in modo indipendente, per confermare il risultato. Nei casi controversi, procedere ad un’ulteriore reazione di PCR indipendente, e fornire il risultato sulla base della maggioranza dei responsi.
6. Interpretazione ed emissione del risultato 6.1 Allineamento degli elettroferogrammi Aprire il programma GeneMapper utilizzando la username gm e la password 3130User. Cliccare sulla quarta icona da sinistra e importare i file corrispondenti alle corse elettroforetiche dalla cartella Local Disk (D:)\AB Service\Installation\Verification\Fragment Analysis\, dalla cartella con data AAA.MM.GG, selezionare i file che si desiderano e cliccare poi su Add nella maschera di sinistra e poi Add nella maschera di destra. A questo punto i file corrispondenti alle corse elettroforetiche sono stati inseriti in un nuovo progetto. Cliccare su ogni file nel progetto per selezionarlo, e poi cliccare su view (nel menu principale) e quindi su Raw Data, per visualizzare la corsa e vedere se ci sono stati problemi o se la separazione dei frammenti e del marker è avvenuta correttamente. Selezionare sul primo file la casella Analysis Method e poi selezionare Microsatellite Default, quindi cliccare su Analysis Method e digitare sulla tastiera CTRL+D, in questo modo tutti i file verranno analizzati con il metodo Microsatellite Default. Selezionare sul primo file la casella Size Standard e poi selezionare GS500LIZ, quindi cliccare su Size Standard e digitare sulla tastiera CTRL+D, in questo modo tutti i file verranno allineati con il marker GS500LIZ. Selezionare nel menu principale Edit, cliccare su Select All e quindi cliccare sulla 14ma icona (freccia verde orientata a destra): dando il nome al progetto, il software procede all’allineamento dei campioni e alla determinazione delle dimensioni di tutti i frammenti presenti nella corsa elettroforetica, sulla base del marker GS500LIZ. Selezionando il file desiderato e cliccando sulla 6° icona, si apre una maschera che mostra la corsa elettroforetica. Nel banner principale selezionare soltanto il colore blu e quindi cliccare sulla seconda icona da destra, che consente di visualizzare una tabella che riporta i dati salienti della corsa elettroforetica (tra cui le dimensioni dei picchi, l’altezza dei picchi e l’area sottesa i picchi). Cliccando subito sopra il grafico, appare una lente di ingrandimento che permette di selezionare l’area della corsa elettroforetica che corrisponde alle dimensioni attese dell’amplificato. Controllare in questo modo che nel bianco di reazione non ci

46
siano frammenti nell’intervallo desiderato e che il controllo positivo abbia fornito frammenti di dimensioni attese. 6.2 Interpretazione dell’elettroferogramma I frammenti attesi sono compresi tra 80 e 120 bp. Ogni picco che si osserva nell’intervallo in questione corrisponde ad un riarrangiamento. La presenza di un picco dominante (che ha un’altezza almeno doppia rispetto agli altri frammenti) indica la possibilità che sia presente un riarrangiamento in quantitativo superiore. Ripetendo l’esperimento per altre due volte, se il picco dominante si conferma tale (controllare che le dimensioni del frammento siano sempre le stesse, un discostamento anche di 1 bp indica la presenza di un riarrangiamento differente) il paziente viene classificato come avente monoclonalità per IgH (che vuol dire che il paziente ha un linfoma), viceversa se il picco in questione non è piu’ la forma maggiormente presente, vuol dire che c’è assenza di monoclonalità (e quindi il paziente è classificato come policlonale): in quest’ultimo caso, comunque, non si puo’ completamente escludere che il paziente abbia un linfoma perché il 20-30% dei pazienti con linfomi non mostrano monoclonalità con la metodica in questione. Bisogna prestare particolare attenzione ai casi in cui il DNA di partenza che viene utilizzato per l’amplificazione è scarso (inferiore a 50 ng totali): in queste condizioni è facile avere false monoclonalità, ma confrontando gli spettri ottenuti da amplificazioni differenti è facile verificare l’effettiva presenza di monoclonalità (il picco dominante ha dimensioni differenti nei vari elettroferogrammi nei casi di falsa monoclonalità, e quindi devono essere classificati come policlonali). Se si osservano 2 picchi dominanti in tutti gli esperimenti, il paziente puo’ essere monoclonale (con un riarrangiamento su ciascuno dei due alleli) o biclonale (due popolazioni cellulari con riarrangiamenti differenti coesistono nell’individuo): in ogni caso il paziente è affetto da un linfoma. Se si osservano 3-5 frammenti dominanti, il paziente è classificato come oligoclonale, ovvero ha poche ma diverse cloni cellulari che sono presenti in quantità abnormi nel proprio organismo. 6.3 Emissione del referto Il referto viene compilato aggiungendo ai dati clinici del paziente il risultato delle analisi molecolari, firmato dall’operatore (o dagli operatori) e dal responsabile dell’analisi, quindi consegnato al patologo di riferimento che procede all’emissione del referto vero e proprio. Una copia cartacea del referto, firmata, viene mantenuta nei raccoglitori nella biblioteca centrale del laboratorio per le analisi dell’anno in corso, o nello studio del responsabile delle analisi per gli anni precedenti, per un periodo massimo di 5 anni. Una copia in formato elettronico del referto viene mantenuta sul server dell’Istituto nella cartella F:\SXIP055\Referti, in cartelle specifiche per le varie analisi molecolari richieste divise per anno. 7. Osservazioni Bibliografia rilevante: Diss et al, J Clin Pathol 1994;47:493-496. 8. Allegati Nessuno.

47
v) Protocollo ditta BIRD

48

49

50

51

52