
Experto Universitario en Genética Médica y Genómica · 2020-01-09 · Diferente contribución de...
69
Experto Universitario en Genética Médica y Genómica Conceptos generales en Genética Humana: Miguel Angel García Pérez Departament de Genética Patrones de transmisión de las enfermedades genéticas I 1
Transcript of Experto Universitario en Genética Médica y Genómica · 2020-01-09 · Diferente contribución de...

Experto Universitario en Genética Médica y Genómica
Conceptos generales en Genética Humana:
Miguel Angel García Pérez
Departament de Genética
Patrones de transmisión de las enfermedades genéticas I
1

ncRNAs, mecanismos epigeneticos, ambiente, interacción gen/ambiente, ….. 2

Diferente contribución de los genes y del ambienteen el desarrollo de las enfermedades humanas
Para cualquier condición, el balance
general de determinantes genéticos y
ambientales puede representarse por un
punto en algún lugar dentro del triángulo3
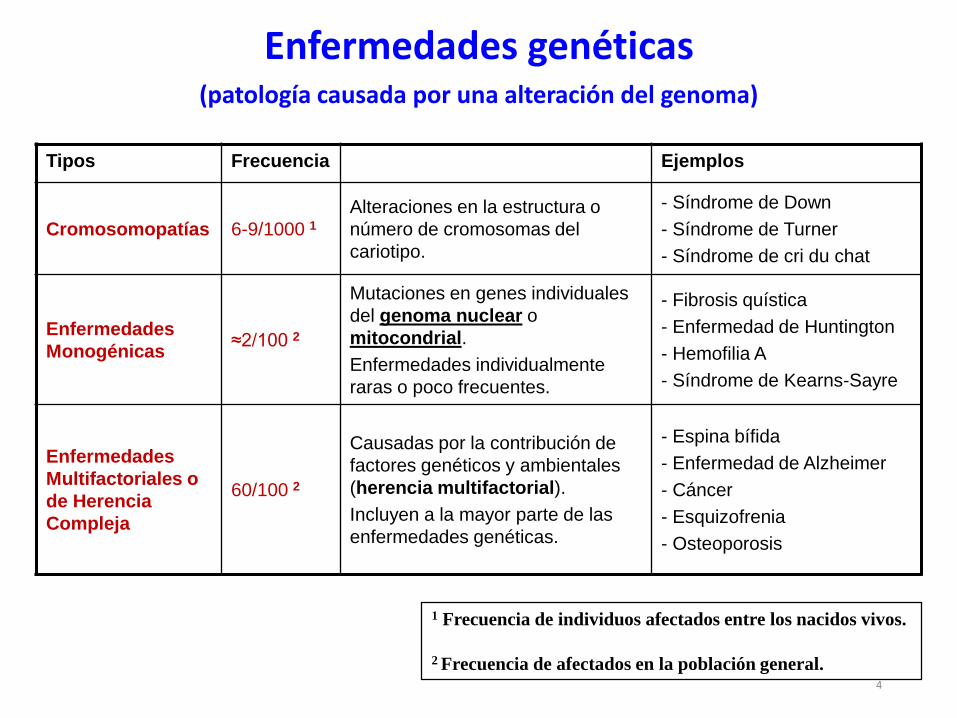
1 Frecuencia de individuos afectados entre los nacidos vivos.
2 Frecuencia de afectados en la población general.
Tipos Frecuencia Ejemplos
Cromosomopatías 6-9/1000 1Alteraciones en la estructura o
número de cromosomas del
cariotipo.
- Síndrome de Down
- Síndrome de Turner
- Síndrome de cri du chat
Enfermedades
Monogénicas≈2/100 2
Mutaciones en genes individuales
del genoma nuclear o
mitocondrial.
Enfermedades individualmente
raras o poco frecuentes.
- Fibrosis quística
- Enfermedad de Huntington
- Hemofilia A
- Síndrome de Kearns-Sayre
Enfermedades
Multifactoriales o
de Herencia
Compleja
60/100 2
Causadas por la contribución de
factores genéticos y ambientales
(herencia multifactorial).
Incluyen a la mayor parte de las
enfermedades genéticas.
- Espina bífida
- Enfermedad de Alzheimer
- Cáncer
- Esquizofrenia
- Osteoporosis
Enfermedades genéticas(patología causada por una alteración del genoma)
4
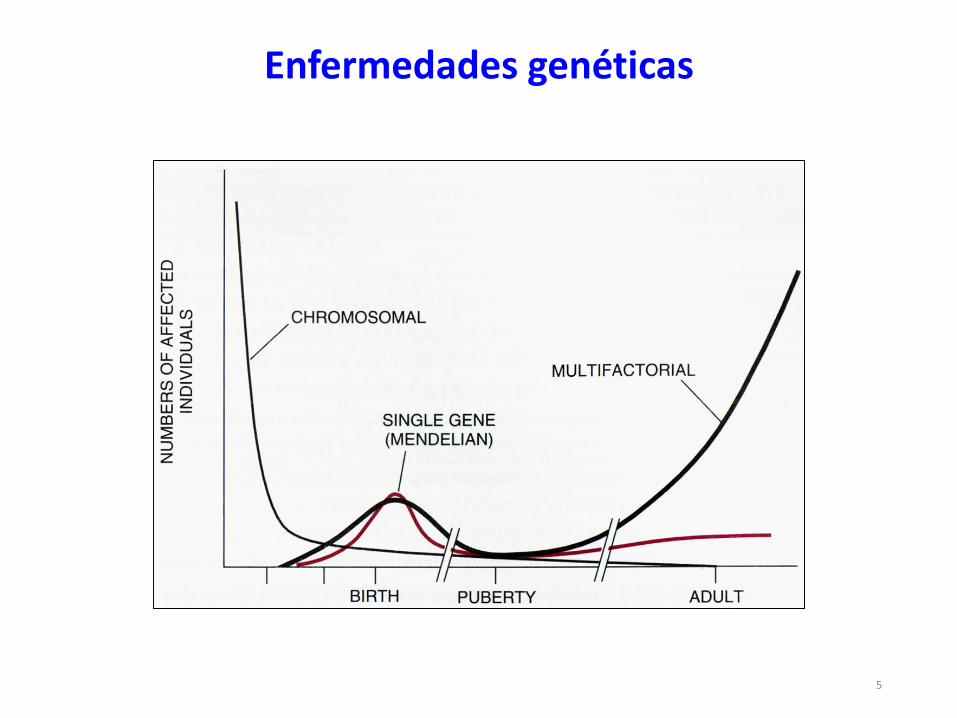
Enfermedades genéticas
5
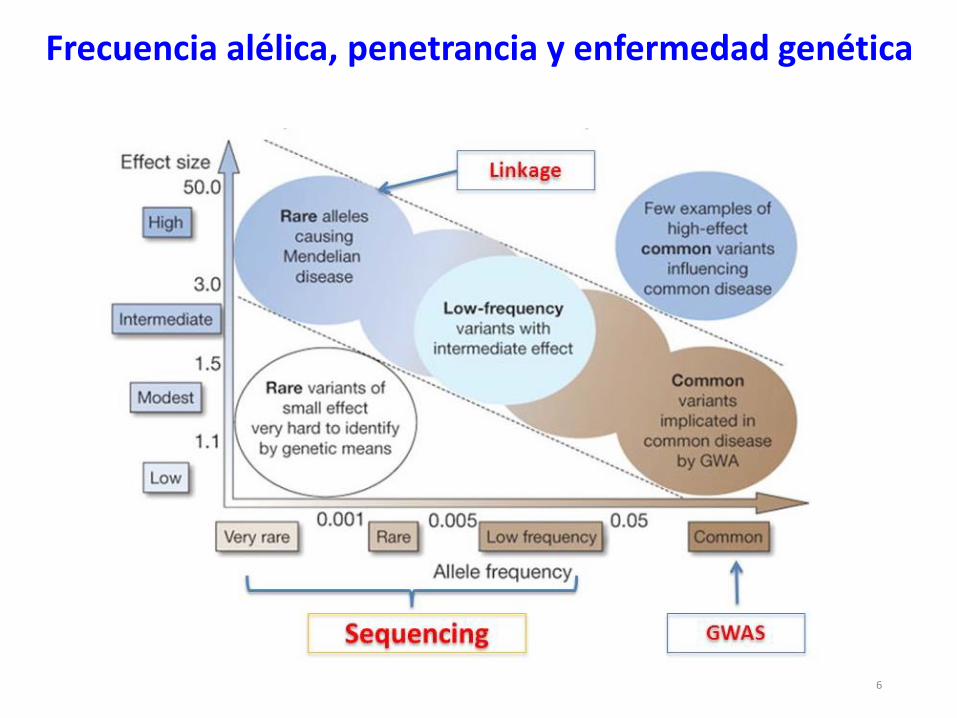
6
Frecuencia alélica, penetrancia y enfermedad genética

Enfermedades monogénicas vs Enfermedades complejas
Las enfermedades monogénicas
suelen mostrar patrones de
herencia obvios y
característicos.
Sin embargo, en las enfermedades
complejas, detectamos una
agregación familiar debido a
factores compartidos (genes y
ambiente) que ocasionan patrones
de herencia complejos.
7

Enfermedades monogénicas
MITOCONDRIAS NÚCLEO
ENFERMEDADES MONOGÉNICAS
Genes núcleo ▶ Herencia mendeliana
Genes mitocondrias ▶ Herencia materna
8

Enfermedades mendelianas(caracteres monogenicos se transmiten siguiendo los principios de Mendel de segregación y de
transmisión independiente)
Se trata de enfermedades individualmente raras o poco comunes.
Enfermedad rara (UE): “Enfermedades, incluidas las de origen genético, que son crónicamente debilitantes o potencialmente mortales y las cuales tienen tan poca prevalencia que se necesitan esfuerzos especiales combinados para combatirlas”.
Prevalencia: menos de 1 caso cada 2000 ciudadanos.
Se estima que existen entre 5.000-7.000 enfermedades raras distintas, que afectan a entre un 3%-8% de la población. La mayoría son de origen genético (80%), donde se incluyen las enfermedades mendelianas.
Para estas enfermedades los recursos terapéuticos son, en general, limitados.
MEDICAMENTOS HUÉRFANOS: Son aquéllos que sirven para diagnosticar, prevenir o tratar las enfermedades raras.
9

http://www.ncbi.nlm.nih.gov/Omim
Bases de datos sobre genes y enfermedades genéticas humanas
10
12 ediciones en papel entre 1966-
1998, iniciada por el Dr. Victor A.
McKusick
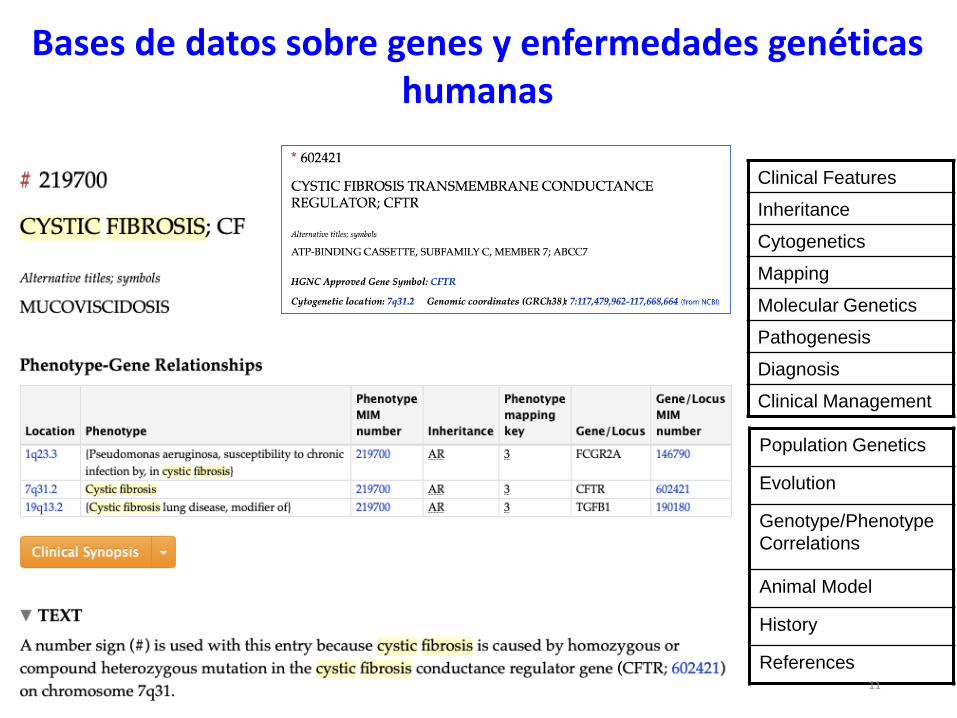
Clinical Features
Inheritance
Cytogenetics
Mapping
Molecular Genetics
Pathogenesis
Diagnosis
Clinical Management
Population Genetics
Evolution
Genotype/Phenotype
Correlations
Animal Model
History
References
Bases de datos sobre genes y enfermedades genéticas humanas
11

http://www.genecards.org/
GeneCards es una base de datos de genes humanos que proporciona
información genómica, proteómica, transcriptómica, genética y
funcional sobre todos los genes humanos conocidos y probables.
http://www.orpha.net/consor/cgi-bin/index.php?lng=ES
El portal sobre enfermedades raras y medicamentos huérfanos
«Ninguna enfermedad es tan rara como
para no merecer nuestra atención»
Bases de datos sobre genes y enfermedades genéticas humanas
12
Visión general de la información biomédicadisponible sobre el gen
buscado

http://www.ciberer.es
Fundada en 1999, FEDER es una
organización sin ánimo de lucro dirigida
íntegramente por afectados y familiares.
Actúa como una de las principales
plataformas de pacientes de
enfermedades raras en España.
Tiene como misión luchar por los
derechos e intereses de los afectados, con
el fin de mejorar su esperanza y calidad de
vida.
Actúa en nombre de todos los pacientes
(con diagnóstico o en espera de él) que
padecen una de estas patologías.
El CIBER de Enfermedades Raras es uno de
los consorcios públicos establecidos por
iniciativa del Instituto de Salud Carlos III, creado
para servir de referencia, coordinar y potenciar la
investigación sobre las enfermedades raras en
España.
Formado por 62 grupos de investigación,
pertenecientes a diferentes instituciones.
http://web.enfermedades-raras.org
Bases de datos sobre genes y enfermedades genéticas humanas
13
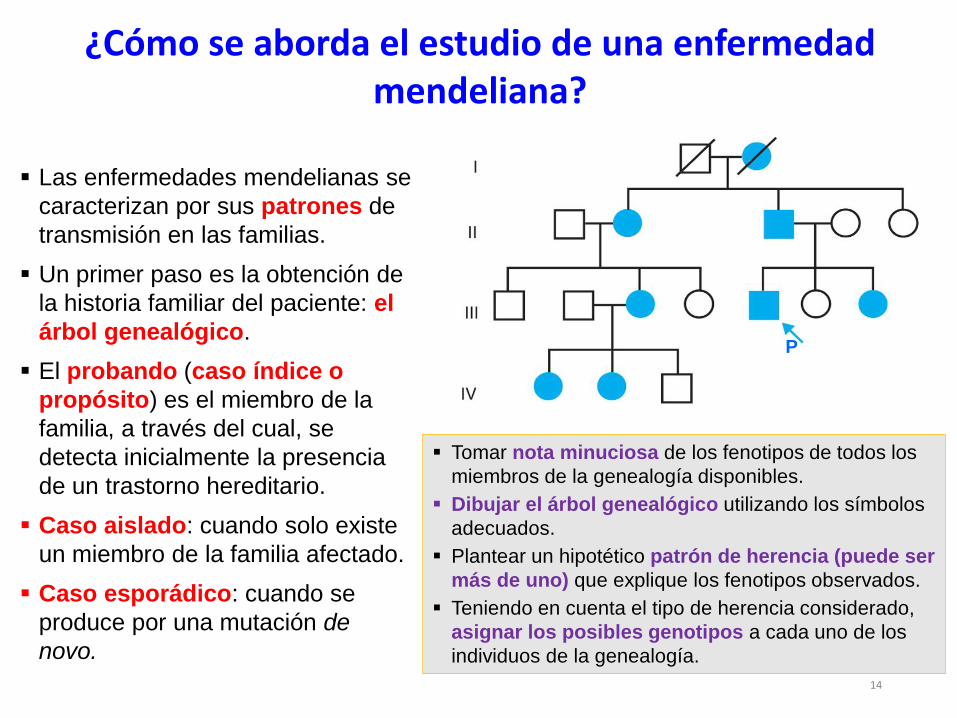
Las enfermedades mendelianas se
caracterizan por sus patrones de
transmisión en las familias.
Un primer paso es la obtención de
la historia familiar del paciente: el
árbol genealógico.
El probando (caso índice o
propósito) es el miembro de la
familia, a través del cual, se
detecta inicialmente la presencia
de un trastorno hereditario.
Caso aislado: cuando solo existe
un miembro de la familia afectado.
Caso esporádico: cuando se
produce por una mutación de
novo.
¿Cómo se aborda el estudio de una enfermedad mendeliana?
Tomar nota minuciosa de los fenotipos de todos los
miembros de la genealogía disponibles.
Dibujar el árbol genealógico utilizando los símbolos
adecuados.
Plantear un hipotético patrón de herencia (puede ser
más de uno) que explique los fenotipos observados.
Teniendo en cuenta el tipo de herencia considerado,
asignar los posibles genotipos a cada uno de los
individuos de la genealogía.
P
14

Símbolos utilizados en la construcción de genealogías
15
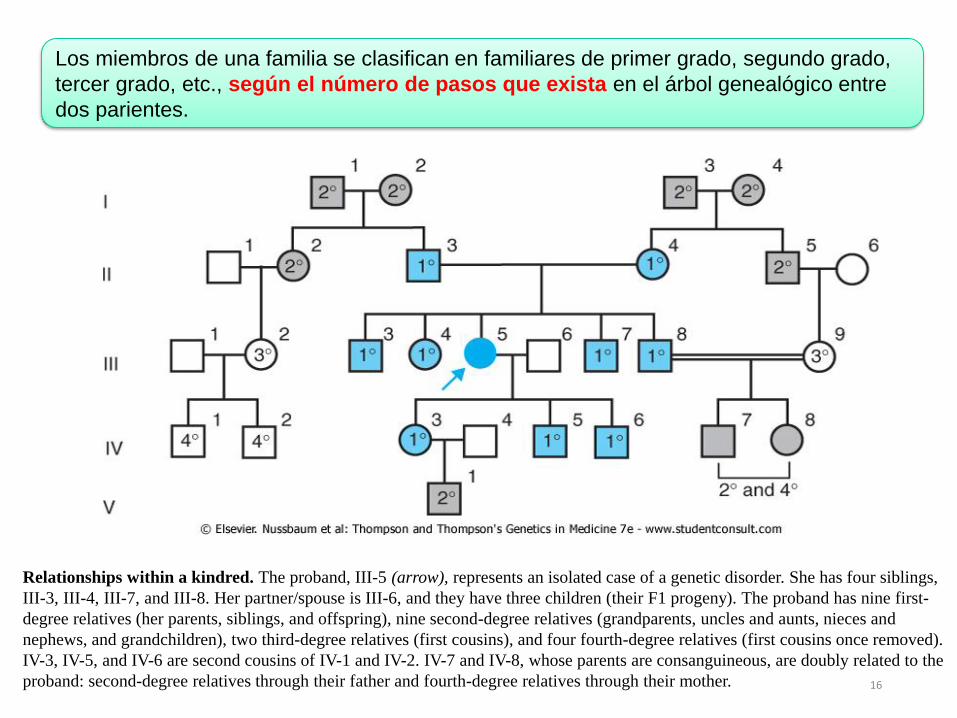
Relationships within a kindred. The proband, III-5 (arrow), represents an isolated case of a genetic disorder. She has four siblings,
III-3, III-4, III-7, and III-8. Her partner/spouse is III-6, and they have three children (their F1 progeny). The proband has nine first-
degree relatives (her parents, siblings, and offspring), nine second-degree relatives (grandparents, uncles and aunts, nieces and
nephews, and grandchildren), two third-degree relatives (first cousins), and four fourth-degree relatives (first cousins once removed).
IV-3, IV-5, and IV-6 are second cousins of IV-1 and IV-2. IV-7 and IV-8, whose parents are consanguineous, are doubly related to the
proband: second-degree relatives through their father and fourth-degree relatives through their mother.
Los miembros de una familia se clasifican en familiares de primer grado, segundo grado,
tercer grado, etc., según el número de pasos que exista en el árbol genealógico entre
dos parientes.
16
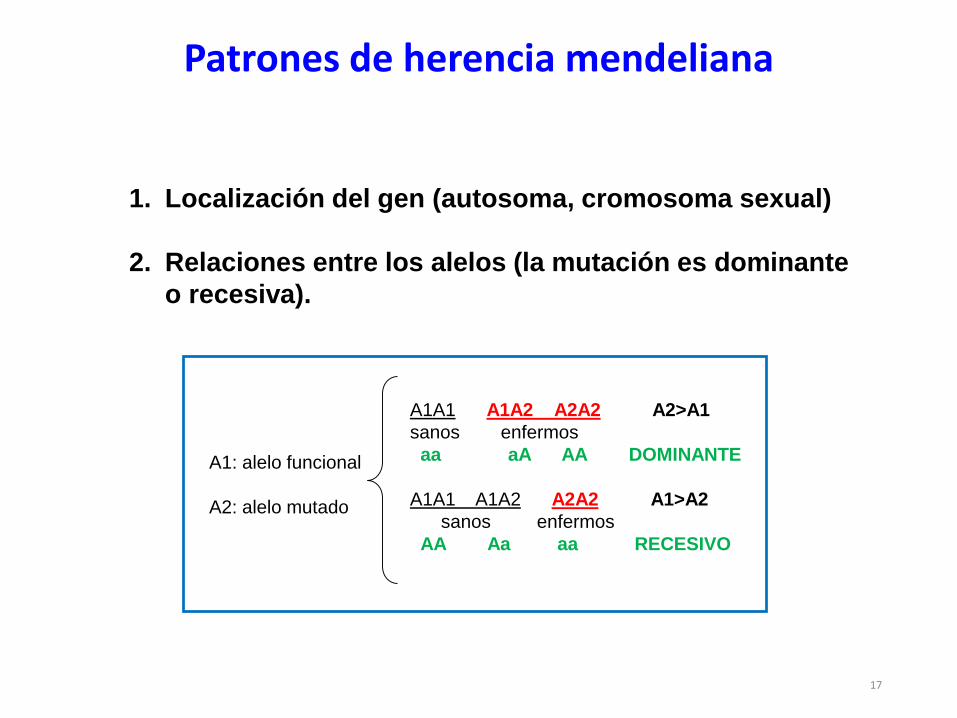
Patrones de herencia mendeliana
1. Localización del gen (autosoma, cromosoma sexual)
2. Relaciones entre los alelos (la mutación es dominante
o recesiva).
A1: alelo funcional
A2: alelo mutado
A1A1 A1A2 A2A2 A2>A1
sanos enfermos
aa aA AA DOMINANTE
A1A1 A1A2 A2A2 A1>A2
sanos enfermos
AA Aa aa RECESIVO
17

Patrones de herencia mendeliana
1. Localización del gen (autosoma, cr. sexual)
2. Relaciones entre los alelos (la mutación es
dominante o recesiva).
PATRONES BÁSICOS
1. Herencia autosómica dominante (A)
2. Herencia autosómica recesiva (B)
3. Herencia ligada al cromosoma X recesiva (C)
4. Herencia ligada al cromosoma X dominante (D)
5. Herencia ligada al cromosoma Y (E)
6. Herencia parcialmente ligada al sexo o
pseudoautosómica.
18
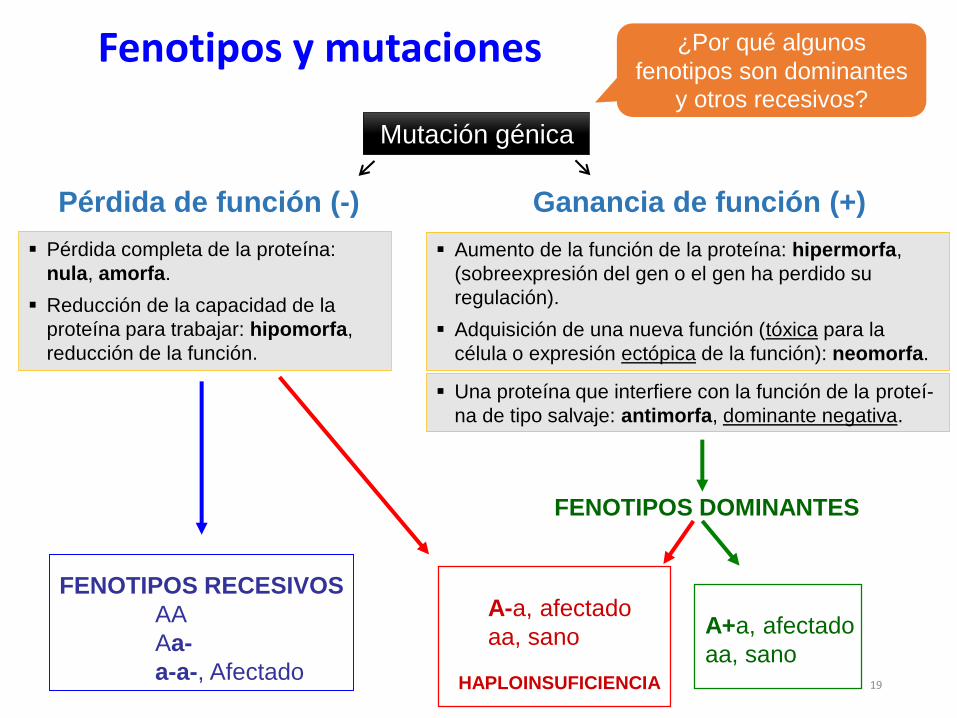
Fenotipos y mutaciones
FENOTIPOS RECESIVOS
AA
Aa-
a-a-, Afectado
FENOTIPOS DOMINANTES
A+a, afectado
aa, sano
A-a, afectado
aa, sano
HAPLOINSUFICIENCIA
¿Por qué algunos
fenotipos son dominantes
y otros recesivos?
Ganancia de función (+)Pérdida de función (-)
Mutación génica
Aumento de la función de la proteína: hipermorfa,
(sobreexpresión del gen o el gen ha perdido su
regulación).
Adquisición de una nueva función (tóxica para la
célula o expresión ectópica de la función): neomorfa.
Pérdida completa de la proteína:
nula, amorfa.
Reducción de la capacidad de la
proteína para trabajar: hipomorfa,
reducción de la función.
Una proteína que interfiere con la función de la proteí-
na de tipo salvaje: antimorfa, dominante negativa.
19
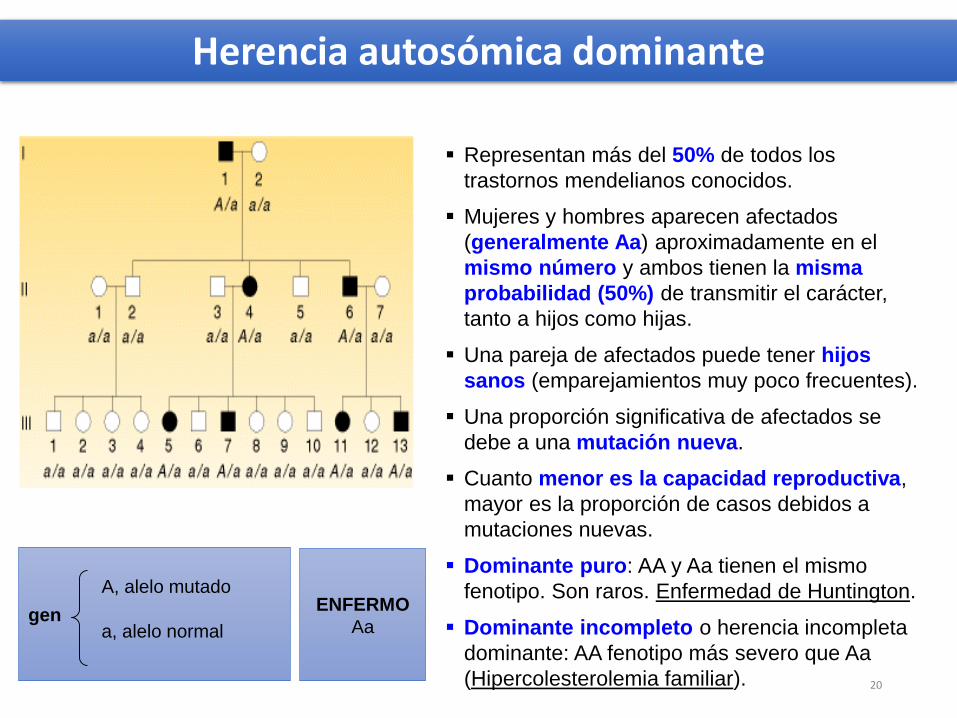
Herencia autosómica dominante
gen
A, alelo mutado
a, alelo normal
ENFERMO
Aa
Representan más del 50% de todos los
trastornos mendelianos conocidos.
Mujeres y hombres aparecen afectados
(generalmente Aa) aproximadamente en el
mismo número y ambos tienen la misma
probabilidad (50%) de transmitir el carácter,
tanto a hijos como hijas.
Una pareja de afectados puede tener hijos
sanos (emparejamientos muy poco frecuentes).
Una proporción significativa de afectados se
debe a una mutación nueva.
Cuanto menor es la capacidad reproductiva,
mayor es la proporción de casos debidos a
mutaciones nuevas.
Dominante puro: AA y Aa tienen el mismo
fenotipo. Son raros. Enfermedad de Huntington.
Dominante incompleto o herencia incompleta
dominante: AA fenotipo más severo que Aa
(Hipercolesterolemia familiar). 20
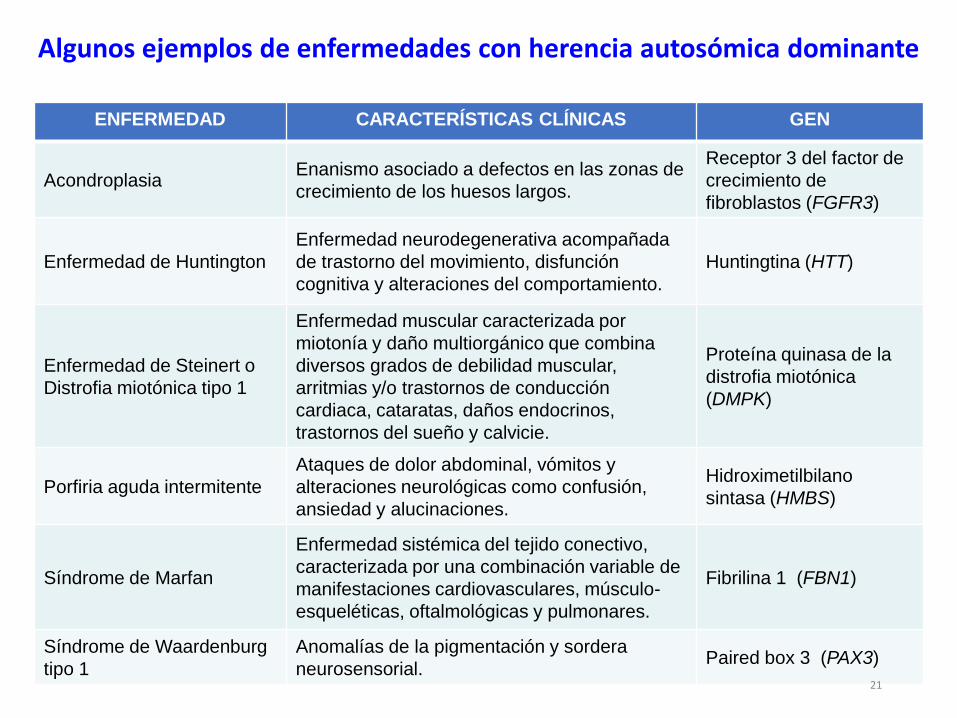
ENFERMEDAD CARACTERÍSTICAS CLÍNICAS GEN
AcondroplasiaEnanismo asociado a defectos en las zonas de
crecimiento de los huesos largos.
Receptor 3 del factor de
crecimiento de
fibroblastos (FGFR3)
Enfermedad de Huntington
Enfermedad neurodegenerativa acompañada
de trastorno del movimiento, disfunción
cognitiva y alteraciones del comportamiento.
Huntingtina (HTT)
Enfermedad de Steinert o
Distrofia miotónica tipo 1
Enfermedad muscular caracterizada por
miotonía y daño multiorgánico que combina
diversos grados de debilidad muscular,
arritmias y/o trastornos de conducción
cardiaca, cataratas, daños endocrinos,
trastornos del sueño y calvicie.
Proteína quinasa de la
distrofia miotónica
(DMPK)
Porfiria aguda intermitente
Ataques de dolor abdominal, vómitos y
alteraciones neurológicas como confusión,
ansiedad y alucinaciones.
Hidroximetilbilano
sintasa (HMBS)
Síndrome de Marfan
Enfermedad sistémica del tejido conectivo,
caracterizada por una combinación variable de
manifestaciones cardiovasculares, músculo-
esqueléticas, oftalmológicas y pulmonares.
Fibrilina 1 (FBN1)
Síndrome de Waardenburg
tipo 1
Anomalías de la pigmentación y sordera
neurosensorial.Paired box 3 (PAX3)
Algunos ejemplos de enfermedades con herencia autosómica dominante
21

Descrita de forma precisa por George Huntington en 1872.
Trastorno del movimiento (corea).
Disfunción cognitiva (lentitud de pensamiento, dificultad en
recordar, deterioro en las funciones intelectuales).
Alteraciones del comportamiento /psiquiátricas (depresión,
irritabilidad, impulsividad, etc.).
Enfermedad de Huntington – MIM ID #143100
Late onset disease. Los síntomas entre la tercera-
cuarta década de la vida (media en 35-51 años).
La supervivencia media es de 18 años después
del diagnóstico.
Variante Juvenil (5-10% de los casos) aparece en
las dos primeras décadas de vida. Forma más
grave.
Variante senil (25% de los casos) debuta alrededor
de los 50 años. La progresión de la enfermedad es
más lenta.
Las personas que desarrollan la enfermedad, frecuentemente han tenido hijos antes de saber que
llevan el defecto génico. El retraso en la edad de aparición de la enfermedad, reduce el efecto de la
selección natural contra el gen defectivo, explicando su frecuencia en la población, aunque se trate
de un gen con una de las tasas de mutación más baja que se conoce.22

Gen HTT
Alelos normales
(6-35 repeticiones)
Alelos patológicos
Entre 36-39 repeticiones, alto riesgo de desarrollar HD.
A partir de 40 repeticiones, la patología es 100% penetrante.
Las formas juveniles presentan más de 60 repeticiones.
Las formas del adulto tienen entre 36 y 50.
Umbral >35 repeticiones
Enfermedad por expansión de nucleótidos: mutaciones dinámicas
23
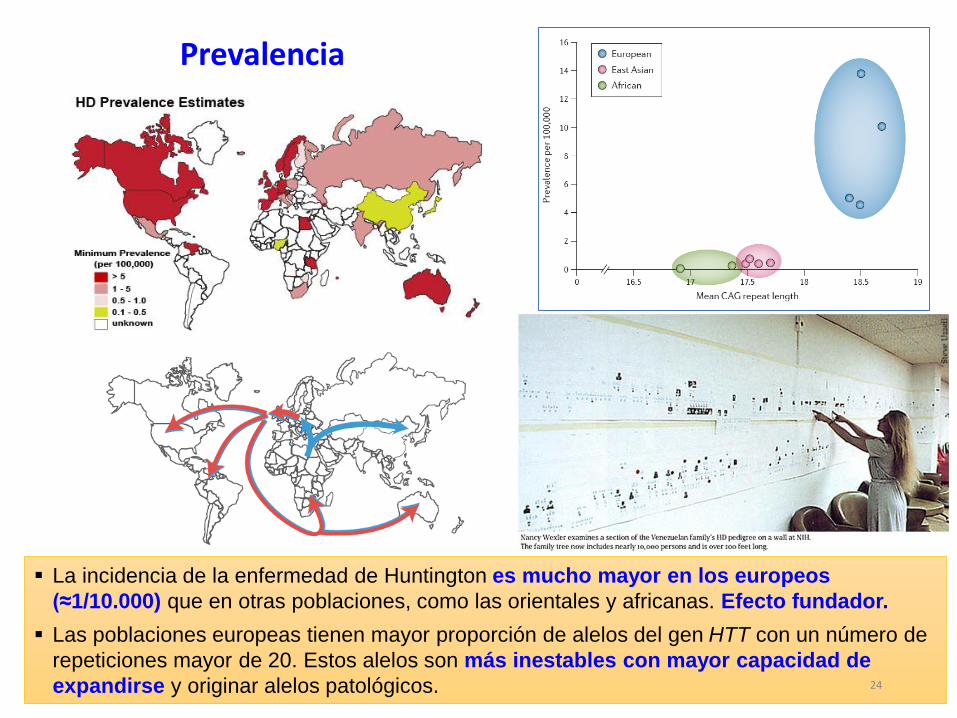
La incidencia de la enfermedad de Huntington es mucho mayor en los europeos
(≈1/10.000) que en otras poblaciones, como las orientales y africanas. Efecto fundador.
Las poblaciones europeas tienen mayor proporción de alelos del gen HTT con un número de
repeticiones mayor de 20. Estos alelos son más inestables con mayor capacidad de
expandirse y originar alelos patológicos.
Prevalencia
24
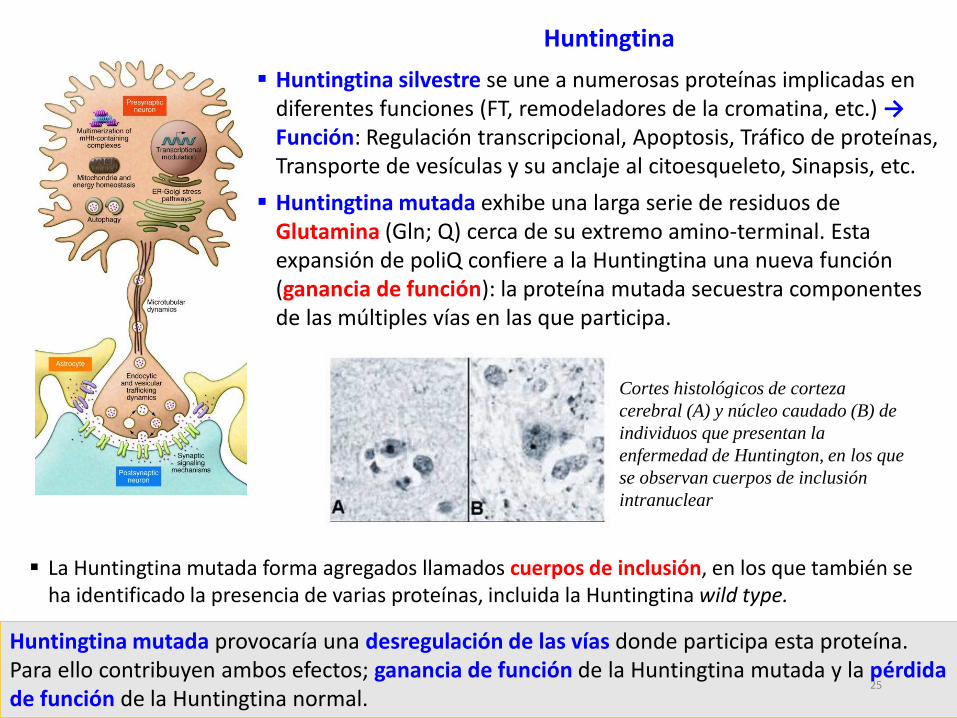
Huntingtina
La Huntingtina mutada forma agregados llamados cuerpos de inclusión, en los que también se ha identificado la presencia de varias proteínas, incluida la Huntingtina wild type.
Cortes histológicos de corteza
cerebral (A) y núcleo caudado (B) de
individuos que presentan la
enfermedad de Huntington, en los que
se observan cuerpos de inclusión
intranuclear
Huntingtina silvestre se une a numerosas proteínas implicadas en diferentes funciones (FT, remodeladores de la cromatina, etc.) →Función: Regulación transcripcional, Apoptosis, Tráfico de proteínas, Transporte de vesículas y su anclaje al citoesqueleto, Sinapsis, etc.
Huntingtina mutada exhibe una larga serie de residuos de Glutamina (Gln; Q) cerca de su extremo amino-terminal. Esta expansión de poliQ confiere a la Huntingtina una nueva función (ganancia de función): la proteína mutada secuestra componentes de las múltiples vías en las que participa.
Huntingtina mutada provocaría una desregulación de las vías donde participa esta proteína. Para ello contribuyen ambos efectos; ganancia de función de la Huntingtina mutada y la pérdida de función de la Huntingtina normal.
25

gen
A, alelo normal
a, alelo mutado
ENFERMO: aa
PORTADOR SANO: Aa
SANO: AA
Hombres y mujeres tienen la misma probabilidad de estar afectados y de transmitir el carácter.
La enfermedad aparece en alguna generación entre la descendencia de padres sanos.
Suele saltar generaciones (pista orientativa).
Los hij@s de un afectad@ son sanos generalmente, a menos que su pareja sea un portador para dicho carácter.
Como los alelos mutantes son, en general, poco frecuentes en la población, la mayoría de los enfermos son heterocigotos compuestos.
La consanguinidad incrementa la probabilidad de tener hij@s afectados.
Herencia autosómica recesiva
Henri de
Toulouse-
Lautrec
26
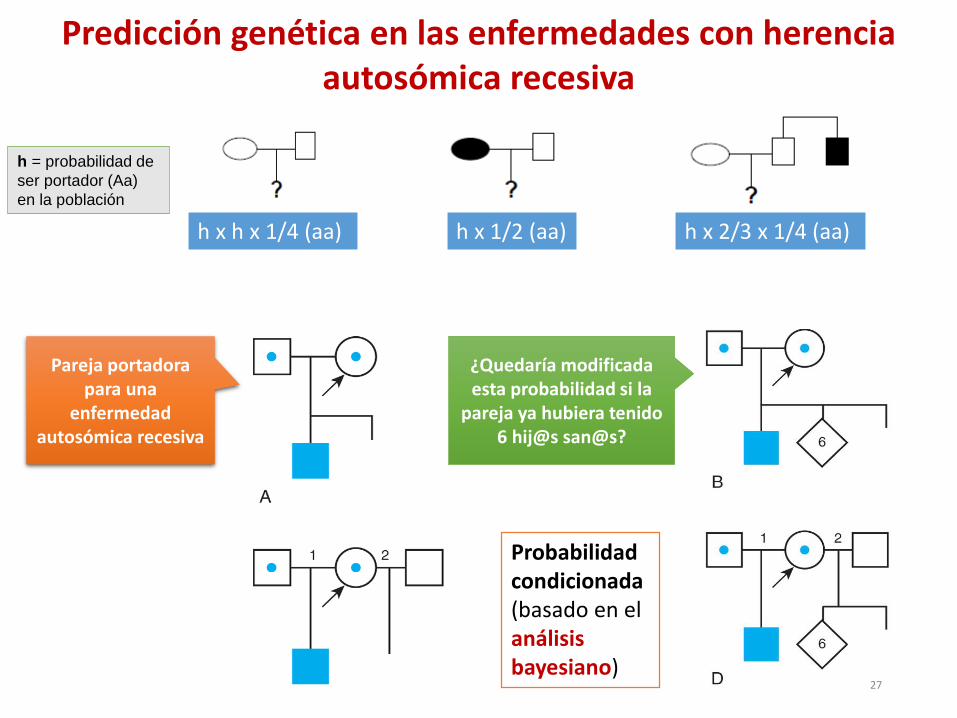
h x h x 1/4 (aa) h x 2/3 x 1/4 (aa) h x 1/2 (aa)
h = 1/22
Pareja portadora para una
enfermedad autosómica recesiva
¿Quedaría modificada esta probabilidad si la
pareja ya hubiera tenido 6 hij@s san@s?
Probabilidad condicionada (basado en el análisis bayesiano)
Predicción genética en las enfermedades con herencia autosómica recesiva
h = probabilidad de
ser portador (Aa)
en la población
27
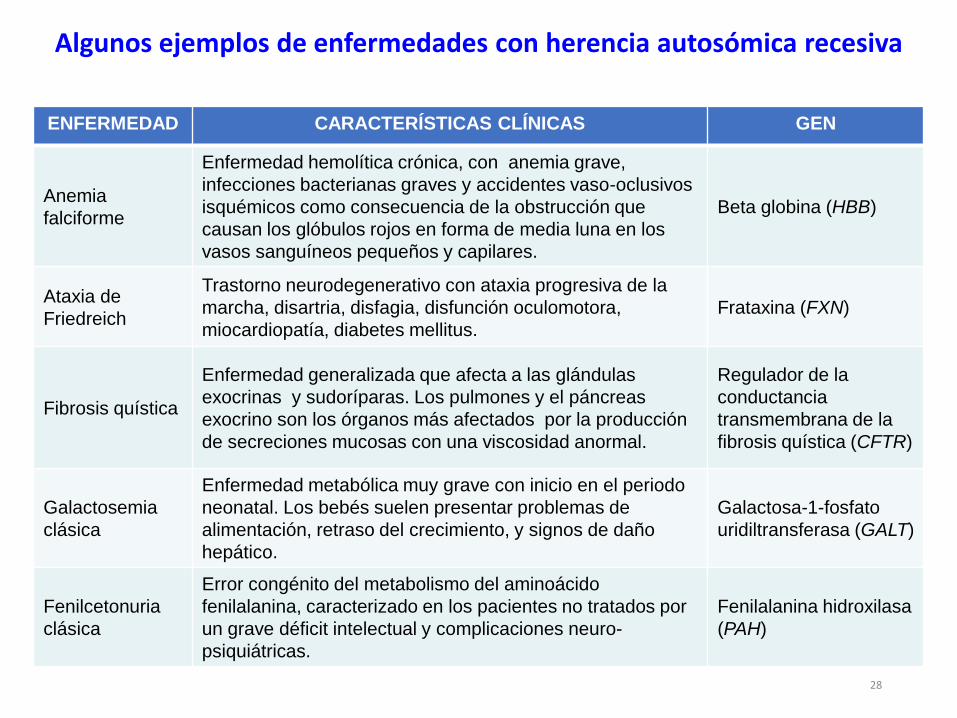
ENFERMEDAD CARACTERÍSTICAS CLÍNICAS GEN
Anemia
falciforme
Enfermedad hemolítica crónica, con anemia grave,
infecciones bacterianas graves y accidentes vaso-oclusivos
isquémicos como consecuencia de la obstrucción que
causan los glóbulos rojos en forma de media luna en los
vasos sanguíneos pequeños y capilares.
Beta globina (HBB)
Ataxia de
Friedreich
Trastorno neurodegenerativo con ataxia progresiva de la
marcha, disartria, disfagia, disfunción oculomotora,
miocardiopatía, diabetes mellitus.
Frataxina (FXN)
Fibrosis quística
Enfermedad generalizada que afecta a las glándulas
exocrinas y sudoríparas. Los pulmones y el páncreas
exocrino son los órganos más afectados por la producción
de secreciones mucosas con una viscosidad anormal.
Regulador de la
conductancia
transmembrana de la
fibrosis quística (CFTR)
Galactosemia
clásica
Enfermedad metabólica muy grave con inicio en el periodo
neonatal. Los bebés suelen presentar problemas de
alimentación, retraso del crecimiento, y signos de daño
hepático.
Galactosa-1-fosfato
uridiltransferasa (GALT)
Fenilcetonuria
clásica
Error congénito del metabolismo del aminoácido
fenilalanina, caracterizado en los pacientes no tratados por
un grave déficit intelectual y complicaciones neuro-
psiquiátricas.
Fenilalanina hidroxilasa
(PAH)
Algunos ejemplos de enfermedades con herencia autosómica recesiva
28
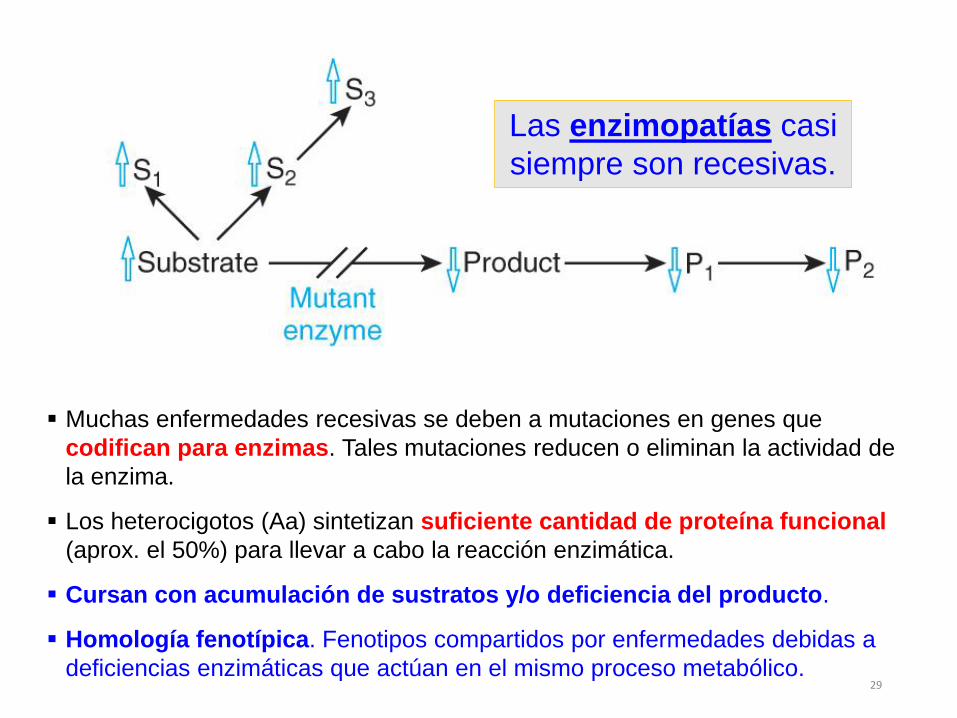
Muchas enfermedades recesivas se deben a mutaciones en genes que
codifican para enzimas. Tales mutaciones reducen o eliminan la actividad de
la enzima.
Los heterocigotos (Aa) sintetizan suficiente cantidad de proteína funcional
(aprox. el 50%) para llevar a cabo la reacción enzimática.
Cursan con acumulación de sustratos y/o deficiencia del producto.
Homología fenotípica. Fenotipos compartidos por enfermedades debidas a
deficiencias enzimáticas que actúan en el mismo proceso metabólico.
Las enzimopatías casi
siempre son recesivas.
29

Enfermedad hereditaria, autosómica
recesiva, con elevada incidencia en la
población de origen europeo (1/2000-2500
recién nacidos). Con una frecuencia de
portador de 1/20-25 personas.
Enfermedad generalizada que afecta a las
glándulas exocrinas y sudoríparas.
Los pulmones y el páncreas exocrino son
los órganos más afectados.
FISIOPATOLOGÍA: Alteración en el
transporte de líquidos y electrolitos a través
de las membranas apicales de los epitelios
glandulares.
Fibrosis quística (MIM ID # 219700): pérdida de función del gen CFTR
(Regulador de la conductancia transmembrana de la fibrosis quística)
30
“Miserable es el niño que al ser besado en la frente tiene gusto a sal. Está embrujado y pronto morirá.” del folklore popular del norte de Europa.
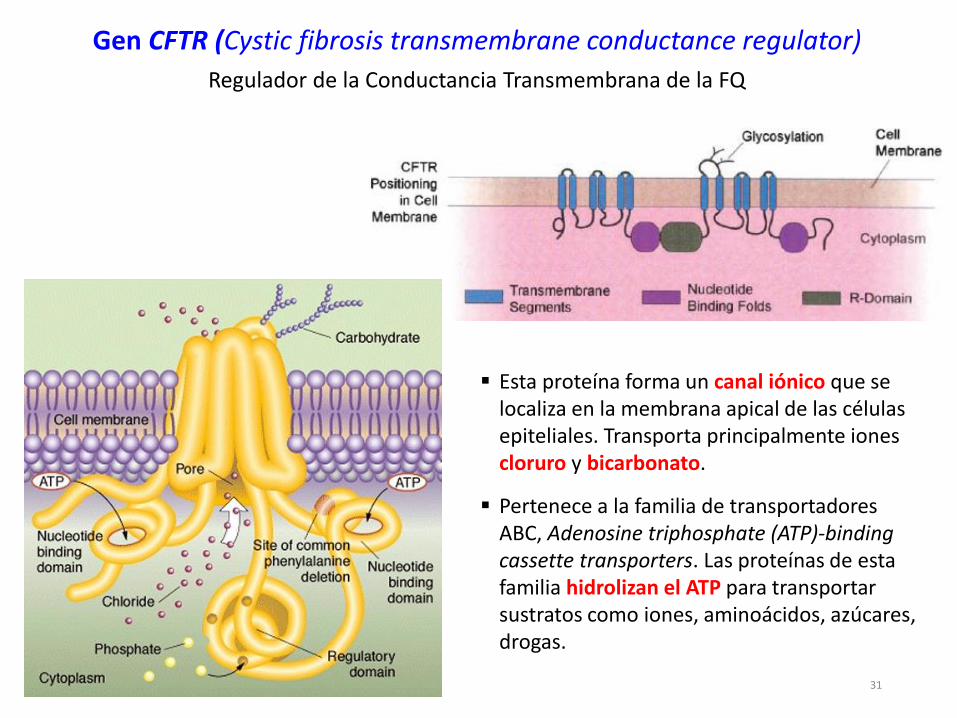
Gen CFTR (Cystic fibrosis transmembrane conductance regulator)
Regulador de la Conductancia Transmembrana de la FQ
Esta proteína forma un canal iónico que se localiza en la membrana apical de las células epiteliales. Transporta principalmente iones cloruro y bicarbonato.
Pertenece a la familia de transportadores ABC, Adenosine triphosphate (ATP)-bindingcassette transporters. Las proteínas de esta familia hidrolizan el ATP para transportar sustratos como iones, aminoácidos, azúcares, drogas.
31

Amplio espectro mutacional en el gen CFTR
Seis clases (I-VI), dependiendo de cómo la mutación afecta a la función del gen
Algunas mutaciones aparecen con mayor frecuencia. Se las considera mutaciones ancestrales, que se
han mantenido por conferir cierta ventaja selectiva (frente a enfermedades como V. cholerae y S. typhi).
La mutación ΔF508 representa, por término medio, el 70% de los alelos mutados en la población de origen
europeo. 32

1. Herencia ligada al cromosoma X (h. ligada al sexo).
2. Herencia ligada al cromosoma Y.
3. Herencia parcialmente ligada al sexo o herencia pseudoautosómica.
Recombinación X - Y
Herencia ligada a los cromosomas sexuales
Cromosoma X
Cromosoma Y
Región pseudoautosómica 1 (PAR1)
Región pseudoautosómica 2 (PAR 2)
Región diferencial de X
Región diferencial de Y
50 genes
1100 genes
2,6 Mb , 24 genes
320 kb, 4 genes
33

Las mujeres pueden ser homocigotas para el alelo funcional, homocigotas
para la mutación, heterocigotas, o heterocigotas compuestas.
Los hombres serán hemicigotos, tanto para el alelo funcional como el
mutado. Expresarán todos los genes situados en el cromosoma X, sean
alelos dominantes o recesivos.
Dominancia / recesividad en genes ligados al X.
La incidencia promedio de desórdenes ligados a X es de 5 casos/10.000 nv.
Los hombres padecerán con mayor frecuencia que las mujeres, las
enfermedades recesivas ligadas a X.
Los hombres padecerán de forma más grave que las mujeres las
enfermedades dominantes ligadas a X.
Herencia ligada al cromosoma X
34

Herencia ligada a X recesiva
La incidencia es mucho más alta en los hombres que en las mujeres.
Las mujeres con hijos afectados son portadoras de la mutación (parientes varones afectados?).
El gen responsable se transmite de un hombre afectado a todas sus hijas.
Las mujeres podrán estar afectadas si el padre lo está y la madre es portadora
(consanguinidad), o a veces como resultado de una inactivación no aleatoria del crom. X.
Una proporción significativa de casos aislados es debida a una nueva mutación.
ENFERME-DAD
CARACTERÍSTICAS CLÍNICAS GEN
Enfermedad de Fabry
Patología multisistémica de almacenamiento lisosómico con manifestaciones neurológicas, cutáneas, renales, cardiovasculares, y cerebrovasculares.
Alfa-galactosidasa A (GALA)
Hemofilia AHemorragias espontáneas o prolongadas, debidas a la deficiencia del factor VIII de la coagulación.
Factor VIII de la coagulación (F8)
Distrofia muscular de Duchenne
Enfermedad neuromuscular caracterizada por atrofia y debilidad musculares progresivas, debido a la degeneración de los músculos esqueléticos, lisos y cardíacos
Distrofina(DMD)
XAXA
XAXa SANOS XAY
XaXa AFECTADOS XaY
35

No es frecuente encontrar mujeres afectadas de patologías con herencia recesiva ligada a X
Homocigotas para la mutación
La consanguinidad en la familia aumenta la probabilidad de que se forme una pareja entre una portadora y un afectado.
36
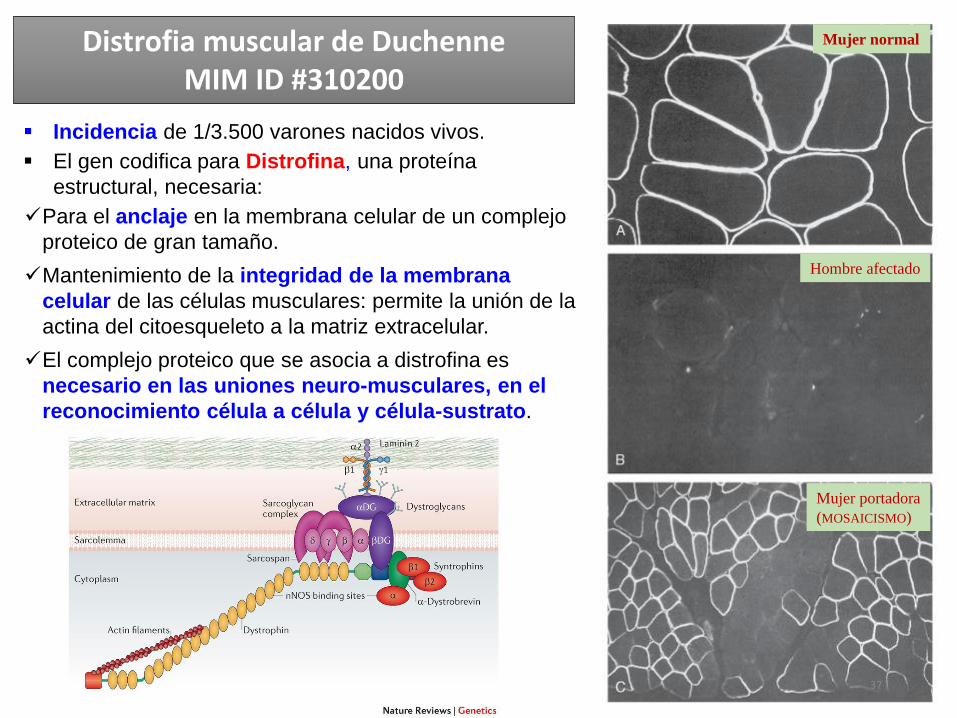
Distrofia muscular de DuchenneMIM ID #310200
Mujer normal
Hombre afectado
Mujer portadora
(MOSAICISMO)
Incidencia de 1/3.500 varones nacidos vivos.
El gen codifica para Distrofina, una proteína
estructural, necesaria:
Para el anclaje en la membrana celular de un complejo
proteico de gran tamaño.
Mantenimiento de la integridad de la membrana
celular de las células musculares: permite la unión de la
actina del citoesqueleto a la matriz extracelular.
El complejo proteico que se asocia a distrofina es
necesario en las uniones neuro-musculares, en el
reconocimiento célula a célula y célula-sustrato.
37

Mujeres afectadas de Distrofia Muscular de Duchenne:
Mujeres heterocigotas afectadas de la enfermedad. Son portadoras de una translocación X-A (mutaciones de novo).
Cada translocación afecta a un autosoma diferente, pero el punto de rotura en el cromosoma X se produce siempre en Xp21.
Una de estas translocaciones en una mujer (Xp;21p) permitió a Worton et al. (Nature 1985), clonar el gen de la distrofina. 38
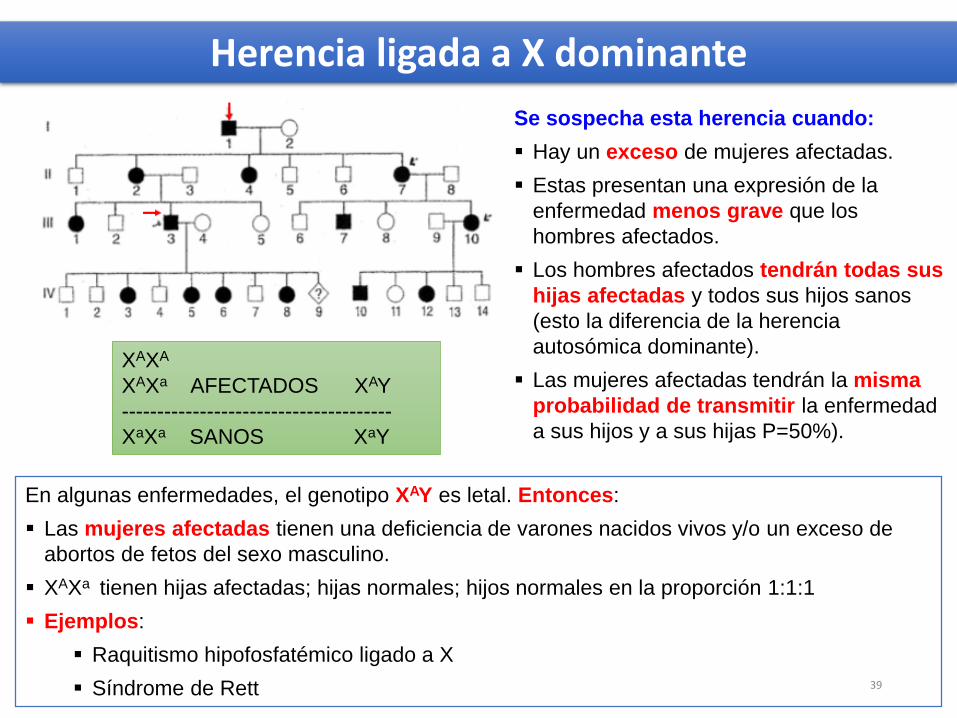
En algunas enfermedades, el genotipo XAY es letal. Entonces:
Las mujeres afectadas tienen una deficiencia de varones nacidos vivos y/o un exceso de
abortos de fetos del sexo masculino.
XAXa tienen hijas afectadas; hijas normales; hijos normales en la proporción 1:1:1
Ejemplos:
Raquitismo hipofosfatémico ligado a X
Síndrome de Rett
Se sospecha esta herencia cuando:
Hay un exceso de mujeres afectadas.
Estas presentan una expresión de la
enfermedad menos grave que los
hombres afectados.
Los hombres afectados tendrán todas sus
hijas afectadas y todos sus hijos sanos
(esto la diferencia de la herencia
autosómica dominante).
Las mujeres afectadas tendrán la misma
probabilidad de transmitir la enfermedad
a sus hijos y a sus hijas P=50%).
Herencia ligada a X dominante
XAXA
XAXa AFECTADOS XAY
--------------------------------------
XaXa SANOS XaY
39

Síndrome de Rett – MIM ID #312750
Frecuencia 1/10.000-15.000 mujeres.
Desarrollo prenatal y neonatal normal seguido de la aparición rápida de sintomatología neurológica a los 6-18 meses.
Ataxia, convulsiones, movimientos característicos de las manos y brazos. Microcefalia, retraso mental grave. Comportamiento irritable, rasgos de autismo.
Alta incidencia de muerte súbita en adultos.
Gen MECP2 (methyl CpG binding protein 2)
El 99% de los casos son esporádicos (70% producidas en la línea germinal paterna).
En raros casos son heredadas de una madre portadorapoco o no afectada (desequilibrio en la inactivación de X).
Codifica una proteína nuclear con cuatro dominios funcionales: Dominio de unión al DNA metilado, Dominio de represión transcripcional, Dominio de unión a RNA, Dominio de unión a factores de procesado del RNA.
40
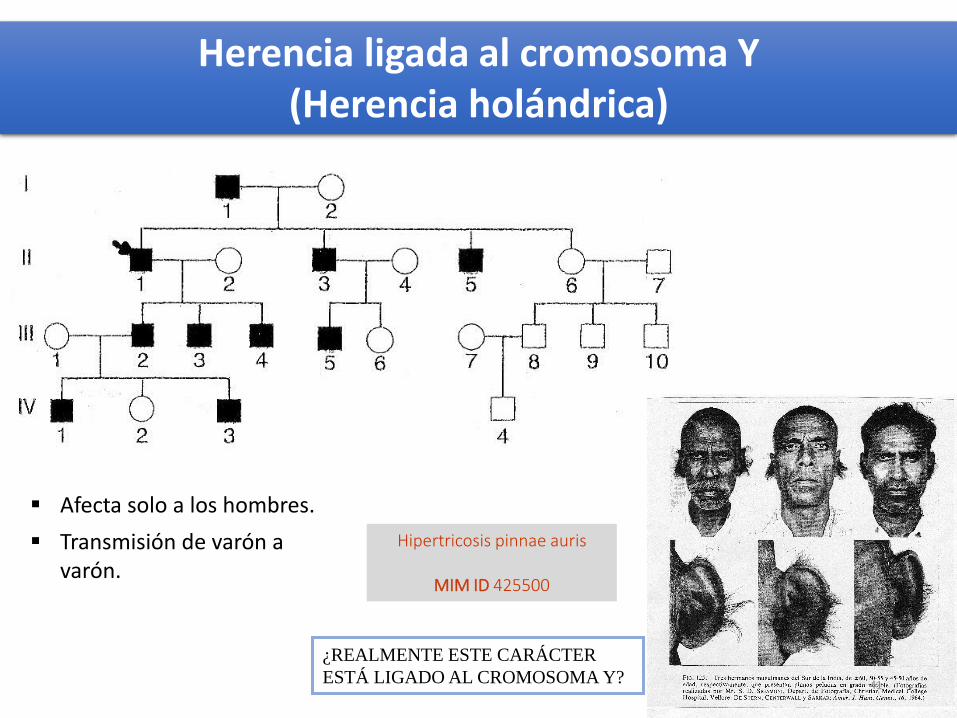
Hipertricosis pinnae auris
MIM ID 425500
¿REALMENTE ESTE CARÁCTER
ESTÁ LIGADO AL CROMOSOMA Y?
Afecta solo a los hombres.
Transmisión de varón a varón.
Herencia ligada al cromosoma Y(Herencia holándrica)
41
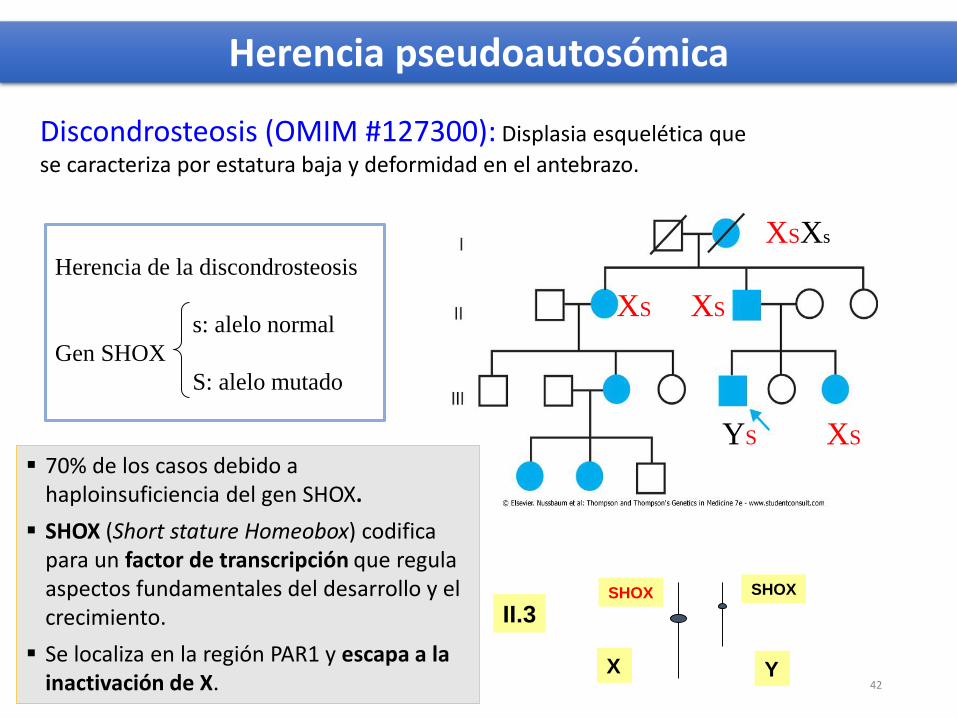
XSXs
XS XS
XSYS
Herencia de la discondrosteosis
s: alelo normal
Gen SHOX
S: alelo mutado
Discondrosteosis (OMIM #127300): Displasia esquelética que
se caracteriza por estatura baja y deformidad en el antebrazo.
Herencia pseudoautosómica
70% de los casos debido a haploinsuficiencia del gen SHOX.
SHOX (Short stature Homeobox) codifica para un factor de transcripción que regulaaspectos fundamentales del desarrollo y el crecimiento.
Se localiza en la región PAR1 y escapa a la inactivación de X.
X Y
SHOX SHOX
II.3
42

Experto Universitario en Genética Médica y Genómica
Conceptos generales en Genética Humana:
Miguel Angel García Pérez
Departament de Genética
Patrones de transmisión de las enfermedades genéticas I
43

Experto Universitario en Genética Médica y Genómica
Conceptos generales en Genética Humana:
Miguel Angel García Pérez
Departament de Genética
Patrones de transmisión de las enfermedades genéticas II
1
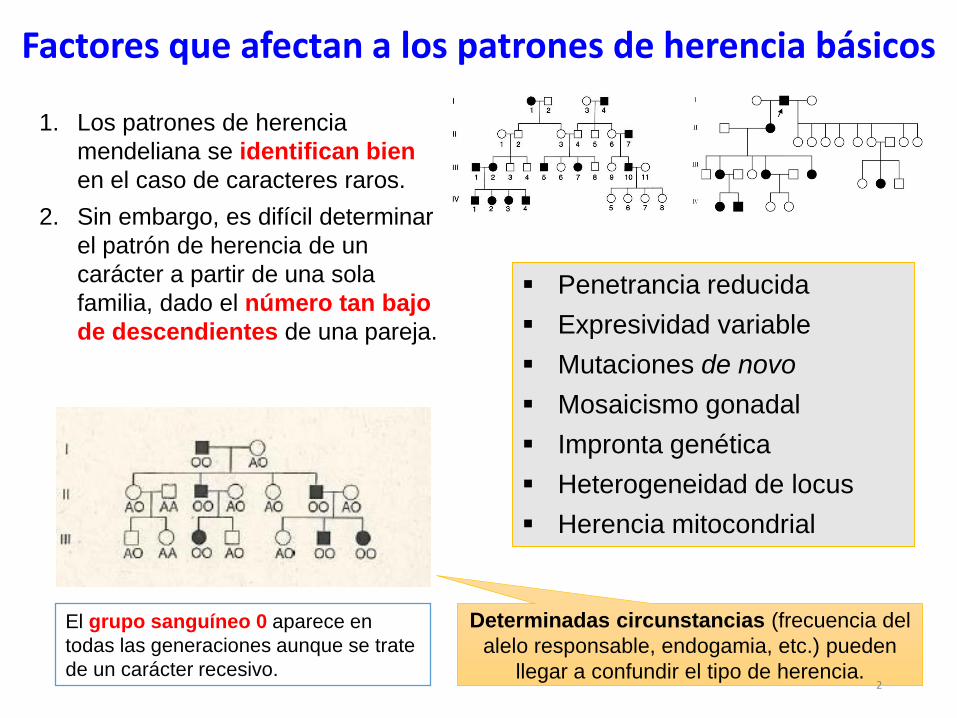
Factores que afectan a los patrones de herencia básicos
1. Los patrones de herencia
mendeliana se identifican bien
en el caso de caracteres raros.
2. Sin embargo, es difícil determinar
el patrón de herencia de un
carácter a partir de una sola
familia, dado el número tan bajo
de descendientes de una pareja.
El grupo sanguíneo 0 aparece en
todas las generaciones aunque se trate
de un carácter recesivo.
Penetrancia reducida
Expresividad variable
Mutaciones de novo
Mosaicismo gonadal
Impronta genética
Heterogeneidad de locus
Herencia mitocondrial
Determinadas circunstancias (frecuencia del
alelo responsable, endogamia, etc.) pueden
llegar a confundir el tipo de herencia.2

¿Qué riesgo tendrían los individuos
de la generación III sanos, de tener
descendencia afectada?
Penetrancia reducida
PENETRANCIA: Capacidad de un
genotipo de expresarse en el fenotipo.
Se mide como el porcentaje de
individuos de un genotipo determinado
que muestra el fenotipo asociado a
dicho genotipo.
La penetrancia reducida se da con
mayor frecuencia en los caracteres
dominantes.
Conocer el grado de penetrancia en
cada patología hereditaria es
determinante para el cálculo del riesgo
genético, un objetivo principal en el
Consejo Genético.
No penetrante
¿Cuál sería la
penetrancia de
este rasgo?
Penetrancia
del 80%
3

Aa
¿Cuál es la probabilidad de que la hija estando sana haya
heredado la mutación A (no-penetrante)?
A (Aa) a (aa)
1/2 1/2
0,2 1
0,1 0,5
0,1/0,6
16,7%
Probabilidad a priori
Probabilidad condicionada
Probabilidad conjunta
Probabilidad a posteriori
Probabilidad Bayesiana
Ejemplo:El carácter muestra una penetrancia del 80%
Por tanto, en este tipo de situación, el 16,7% de los
sanos son portadores de una mutación.
Penetrancia reducida
4
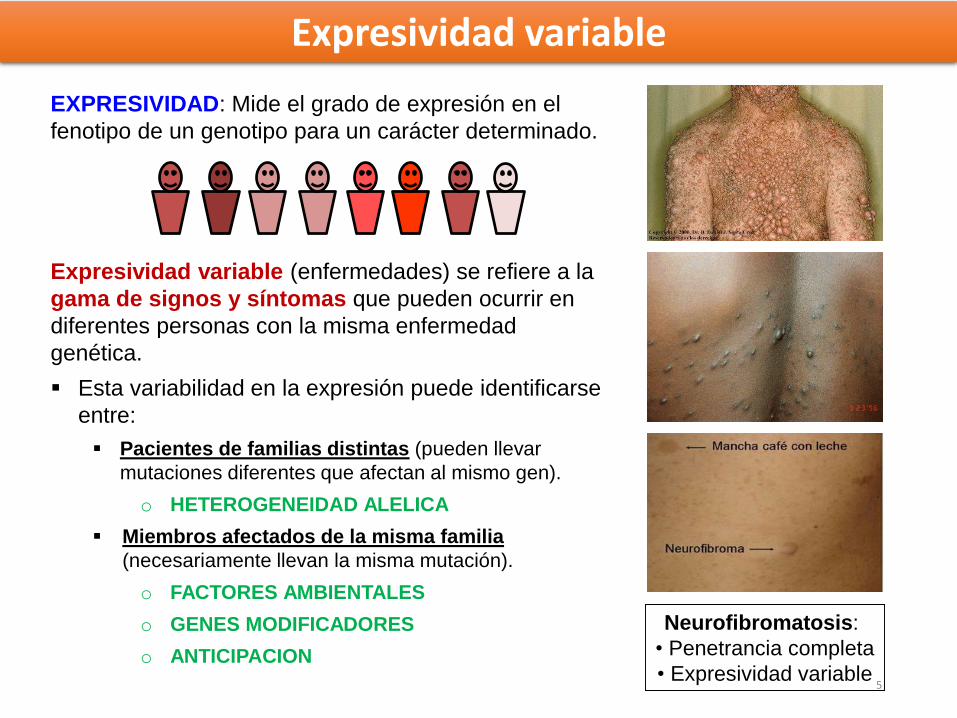
Expresividad variable
Neurofibromatosis:
• Penetrancia completa
• Expresividad variable
EXPRESIVIDAD: Mide el grado de expresión en el
fenotipo de un genotipo para un carácter determinado.
Expresividad variable (enfermedades) se refiere a la
gama de signos y síntomas que pueden ocurrir en
diferentes personas con la misma enfermedad
genética.
Esta variabilidad en la expresión puede identificarse
entre:
Pacientes de familias distintas (pueden llevar
mutaciones diferentes que afectan al mismo gen).
o HETEROGENEIDAD ALELICA
Miembros afectados de la misma familia
(necesariamente llevan la misma mutación).
o FACTORES AMBIENTALES
o GENES MODIFICADORES
o ANTICIPACION
5

Hematoxilina-eosina Inmunofluorescencia
anticuerpo anti-distrofina
Strachan and Read , 4e (Garland Science , Taylor and Francis Group, LLC)
Expresividad variable: heterogeneidad alélica
Distrofia muscular de Duchenne (DMD)
Distrofia muscular de Becker (DMB)
intrón/exón tamaño (pb) cambio pauta deleciones
Heterogeneidad alélica: Situación en la que distintas mutaciones en un mismo gen producen el
mismo o similar fenotipo (variaciones en las manifestaciones clínicas), o incluso dan lugar a cuadros
clínicos diferentes (heterogeneidad clínica o fenotípica; enfermedades alélicas).
6
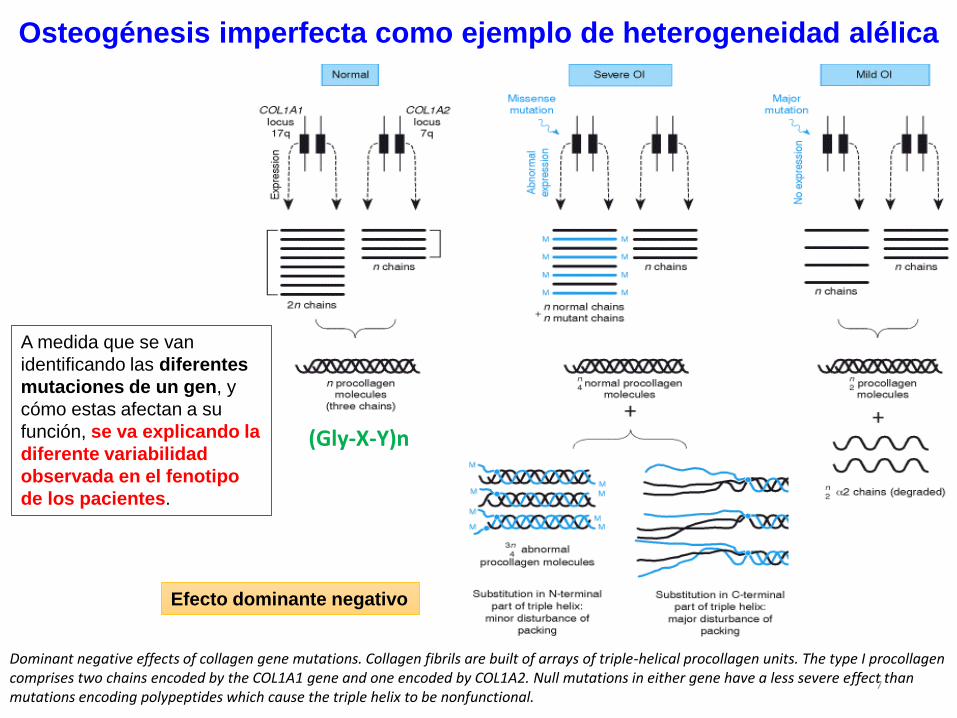
Osteogénesis imperfecta como ejemplo de heterogeneidad alélica
Efecto dominante negativo
Dominant negative effects of collagen gene mutations. Collagen fibrils are built of arrays of triple-helical procollagen units. The type I procollagen comprises two chains encoded by the COL1A1 gene and one encoded by COL1A2. Null mutations in either gene have a less severe effect than mutations encoding polypeptides which cause the triple helix to be nonfunctional.
A medida que se van
identificando las diferentes
mutaciones de un gen, y
cómo estas afectan a su
función, se va explicando la
diferente variabilidad
observada en el fenotipo
de los pacientes.
(Gly-X-Y)n
7
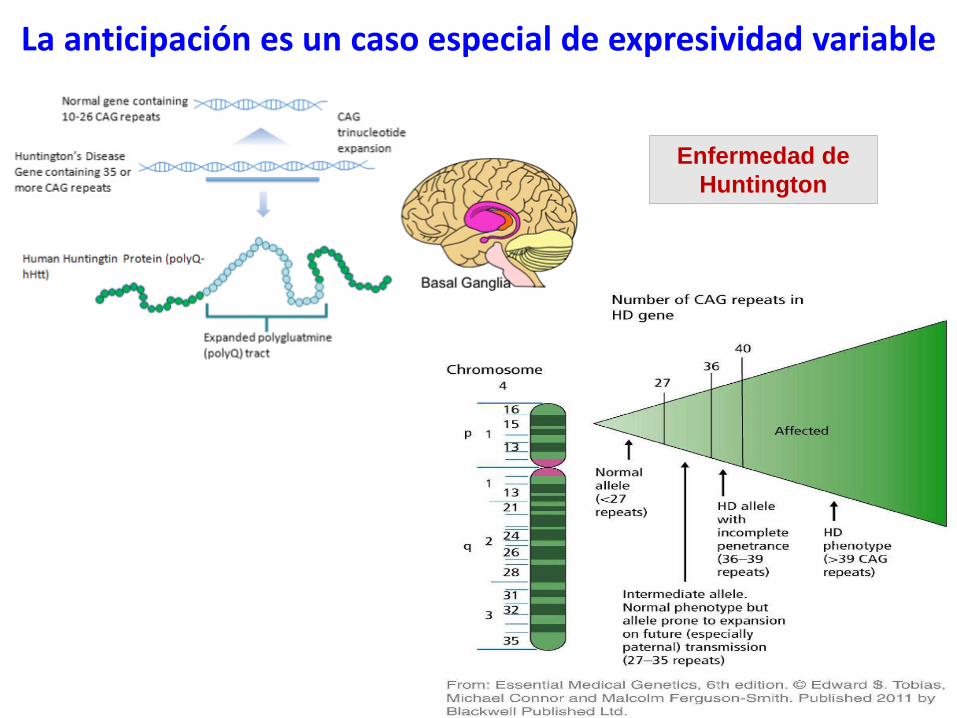
Enfermedad de
Huntington
La anticipación es un caso especial de expresividad variable
8
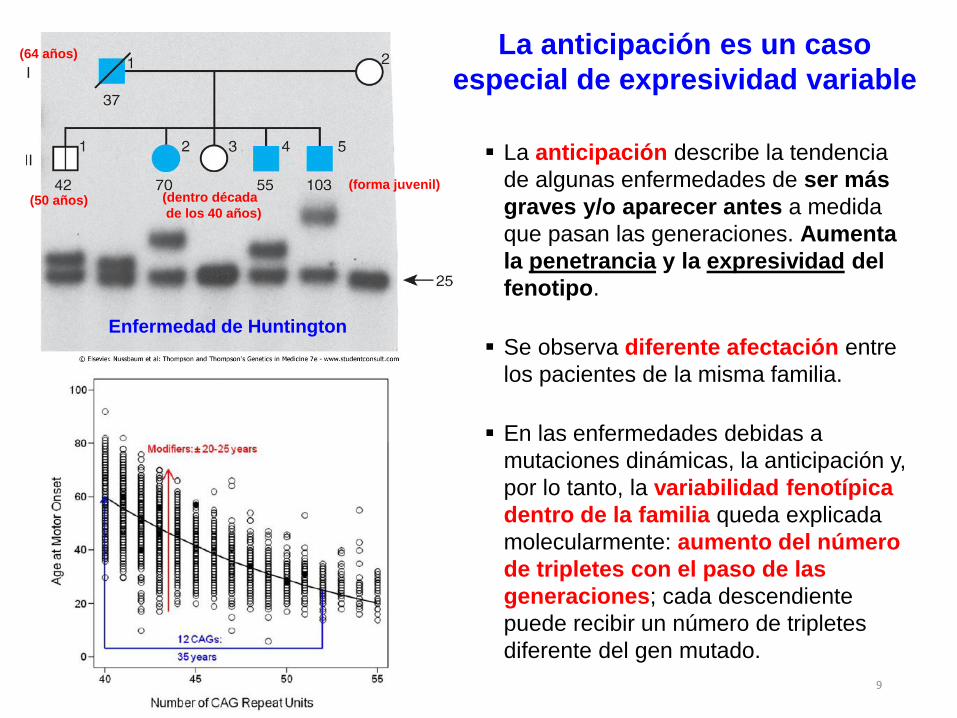
Enfermedad de Huntington
(64 años)
(50 años) (dentro década
de los 40 años)
(forma juvenil)
La anticipación es un caso
especial de expresividad variable
La anticipación describe la tendencia
de algunas enfermedades de ser más
graves y/o aparecer antes a medida
que pasan las generaciones. Aumenta
la penetrancia y la expresividad del
fenotipo.
Se observa diferente afectación entre
los pacientes de la misma familia.
En las enfermedades debidas a
mutaciones dinámicas, la anticipación y,
por lo tanto, la variabilidad fenotípica
dentro de la familia queda explicada
molecularmente: aumento del número
de tripletes con el paso de las
generaciones; cada descendiente
puede recibir un número de tripletes
diferente del gen mutado.
9

Penetrancia versus Expresividad variable
- La penetrancia es una cuestión de todo o nada.
- La expresividad es una cuestión de intensidad.
Causas: combinación de factores genéticos, ambientales y de estilo de vida.
10
Penetrancia reducida y expresividad variable permiten
el mantenimiento de las mutaciones en la población.

Herencia autosómica recesiva.
Herencia ligada al X recesiva.
Herencia dominante (mutación de novo); mayor
complicación si hay mosaicismo gonadal.
Alteración en el cariotipo.
Enfermedad congénita no genética.
?
Familia sin antecedentes de la
enfermedad presente en el miembro
afectado de la generación III.
Esta situación (casos esporádicos) es
común en enfermedades que conllevan
graves malformaciones y/o son
genéticamente letales.
Las mutaciones de novo complican la interpretación del árbol genealógico
11

Mosaicismo gonadal
Las mutaciones postcigóticas originan
mosaicos con dos o más líneas celulares
genéticamente diferentes.
Si las mutaciones afectan a la línea
germinal darán origen a un individuo en el
que se formarán gametos normales y
gametos portadores de la mutación
(mosaicismo germinal o gonadal).
Si afecta tanto a células germinales como
a células somáticas: mosaicismo
gonosómico.
Como consecuencia una pareja sin
antecedentes familiares podrá tener más
de un hijo afectado de una enfermedad
dominante.
Aunque se conozca el modo de herencia
de la enfermedad, es muy complicado
determinar el riesgo de recurrencia.
Si el gen responsable se ha clonado, el análisis
molecular es de gran utilidad en estos casos. 12
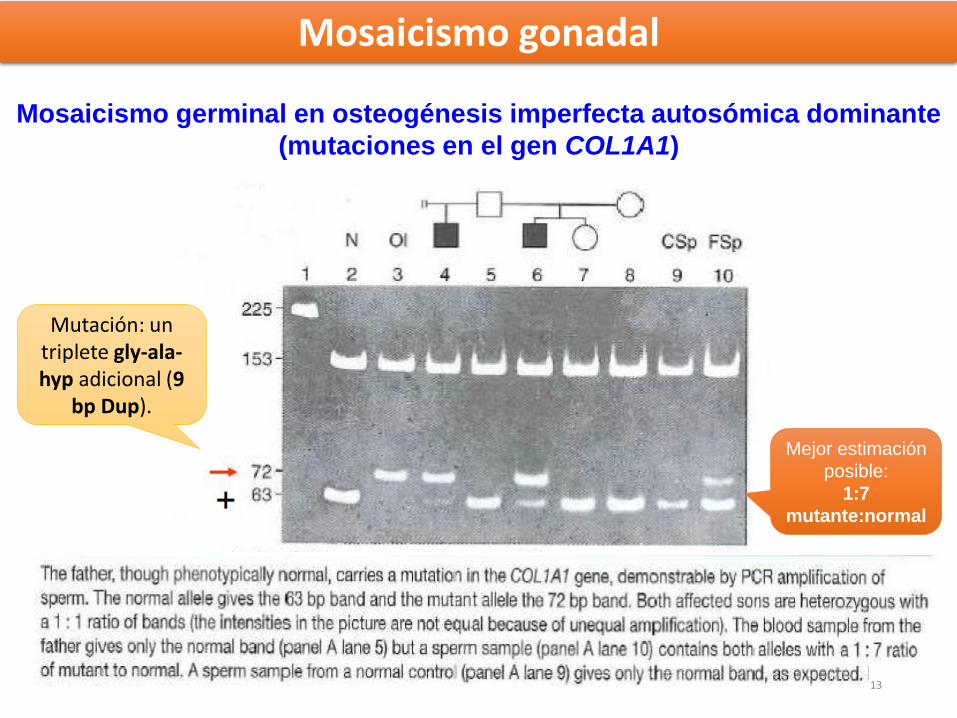
Mosaicismo germinal en osteogénesis imperfecta autosómica dominante
(mutaciones en el gen COL1A1)
Mutación: un triplete gly-ala-hyp adicional (9
bp Dup).
Mosaicismo gonadal
Mejor estimación
posible:
1:7
mutante:normal
13
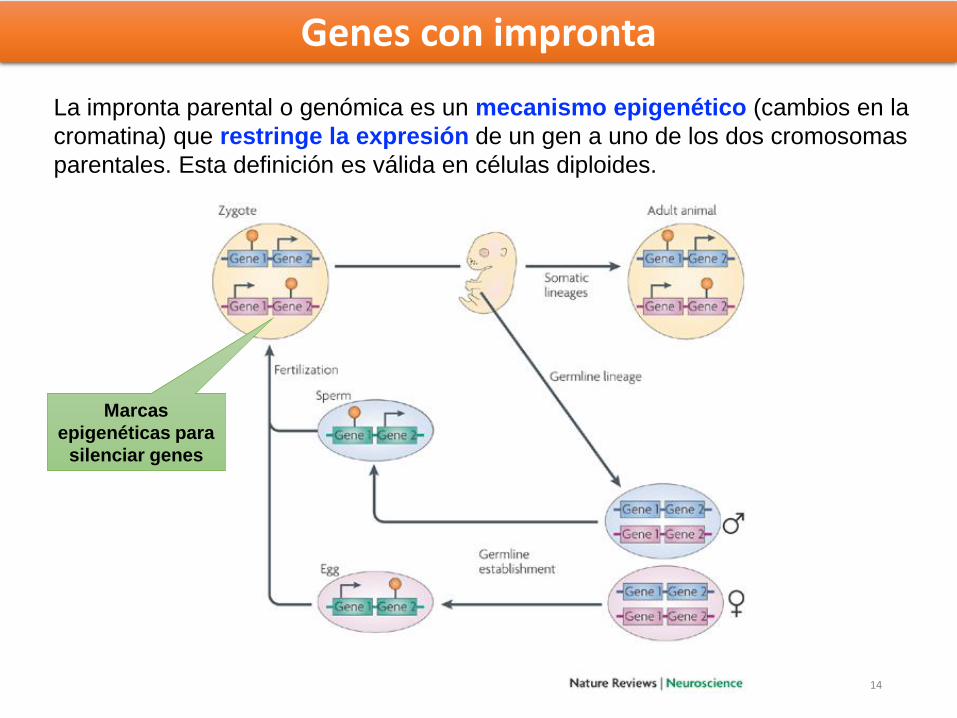
La impronta parental o genómica es un mecanismo epigenético (cambios en la
cromatina) que restringe la expresión de un gen a uno de los dos cromosomas
parentales. Esta definición es válida en células diploides.
Marcas
epigenéticas para
silenciar genes
Genes con impronta
14
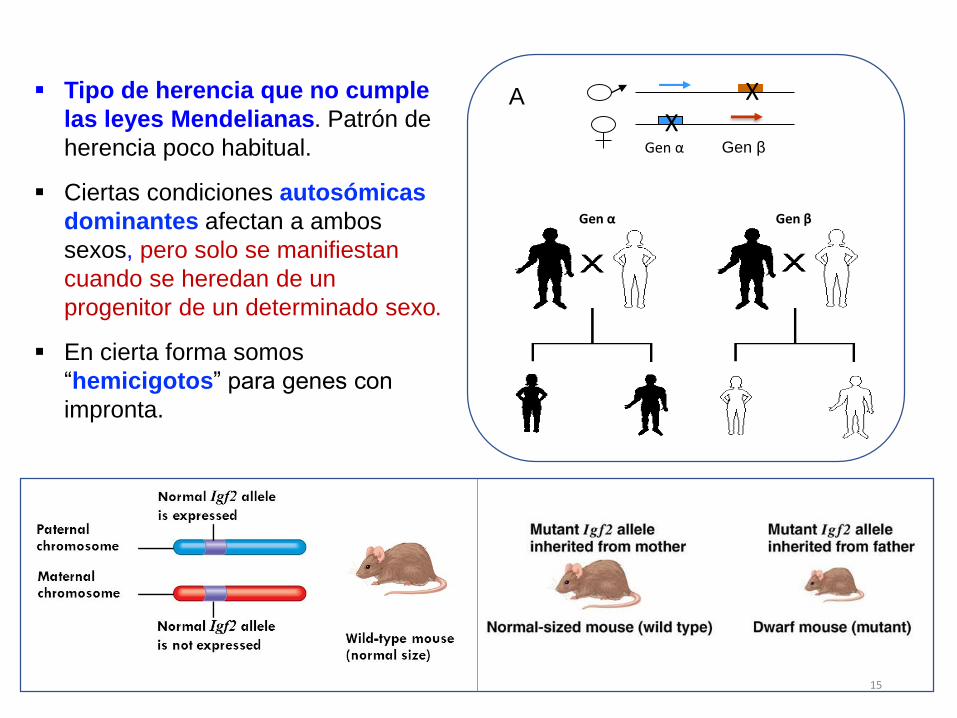
Gen α Gen β
Gen α Gen β
XX
A Tipo de herencia que no cumple
las leyes Mendelianas. Patrón de
herencia poco habitual.
Ciertas condiciones autosómicas
dominantes afectan a ambos
sexos, pero solo se manifiestan
cuando se heredan de un
progenitor de un determinado sexo.
En cierta forma somos
“hemicigotos” para genes con
impronta.
15
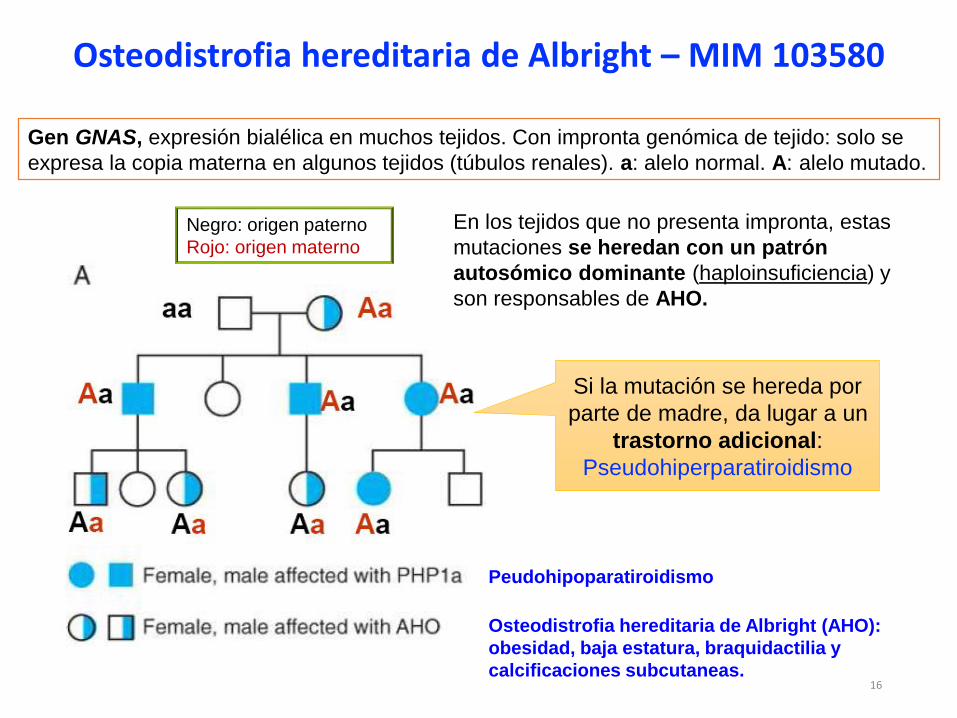
Negro: origen paterno
Rojo: origen materno
Osteodistrofia hereditaria de Albright (AHO):
obesidad, baja estatura, braquidactilia y
calcificaciones subcutaneas.
Peudohipoparatiroidismo
Gen GNAS, expresión bialélica en muchos tejidos. Con impronta genómica de tejido: solo se
expresa la copia materna en algunos tejidos (túbulos renales). a: alelo normal. A: alelo mutado.
En los tejidos que no presenta impronta, estas
mutaciones se heredan con un patrón
autosómico dominante (haploinsuficiencia) y
son responsables de AHO.
Si la mutación se hereda por
parte de madre, da lugar a un
trastorno adicional:
Pseudohiperparatiroidismo
Osteodistrofia hereditaria de Albright – MIM 103580
16
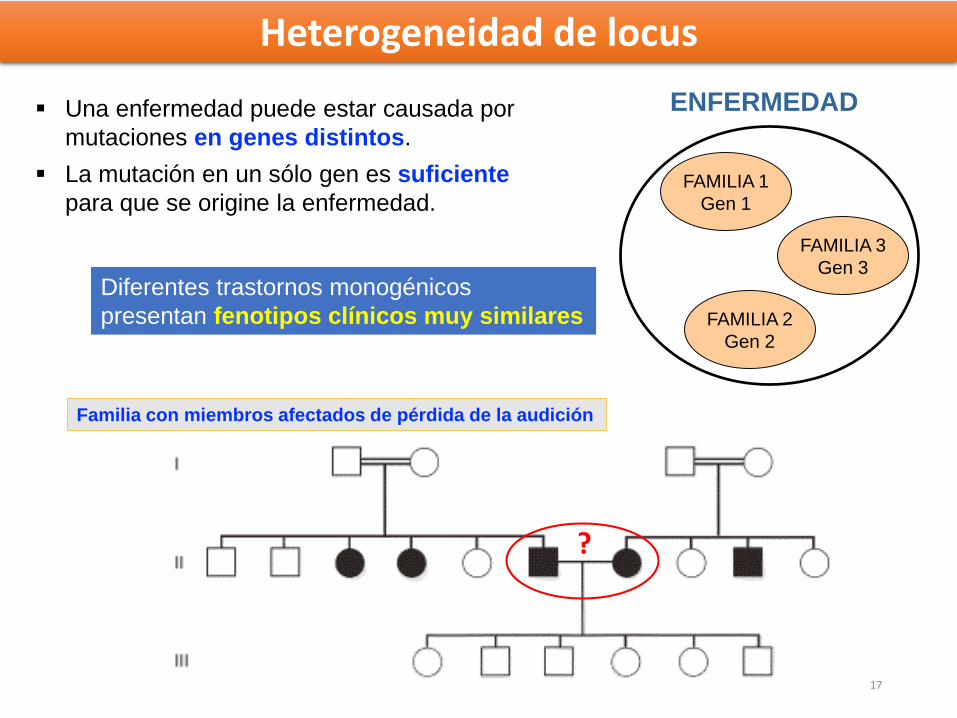
Heterogeneidad de locus
Una enfermedad puede estar causada por
mutaciones en genes distintos.
La mutación en un sólo gen es suficiente
para que se origine la enfermedad.
ENFERMEDAD
FAMILIA 1
Gen 1
FAMILIA 2
Gen 2
FAMILIA 3
Gen 3
Familia con miembros afectados de pérdida de la audición
aa bb
Diferentes trastornos monogénicos
presentan fenotipos clínicos muy similares
?
17

Search Albinism
Results 95
1 #203200. ALBINISM, OCULOCUTANEOUS, TYPE
II; OCA2
Gene map locus 15q11.2-q12
2 #606952. ALBINISM, OCULOCUTANEOUS, TYPE
IB; OCA1B
Gene map locus 11q14-q21
3 #203290. ALBINISM, OCULOCUTANEOUS, TYPE
III; OCA3
Gene map locus 9p23
15q11.2-q12:
Melanosomal
membrane
protein
11q14-q21:
Tyrosinase
9p23: Tyrosinase-
related protein 1
18

Herencia mitocondrial
Globalmente, las enfermedades causadas por mutaciones
en el mtDNA tienen una prevalencia poblacional
aproximada de 1/10.000 individuos con afectación
fenotípica.
Se estima que 1/200 individuos tiene mutaciones
detectables.
Proyecto 1000 genomas: “∼90% of the individuals carry at
least one heteroplasmy. At least 20% of individuals harbor
heteroplasmies reported to be implicated in disease”
El mtDNA representa el 0,5%
del DNA celular.
La mayor parte de las células
tiene unas 1000 moléculas de
mtDNA distribuidas entre
cientos de mitocondrias por
célula.19
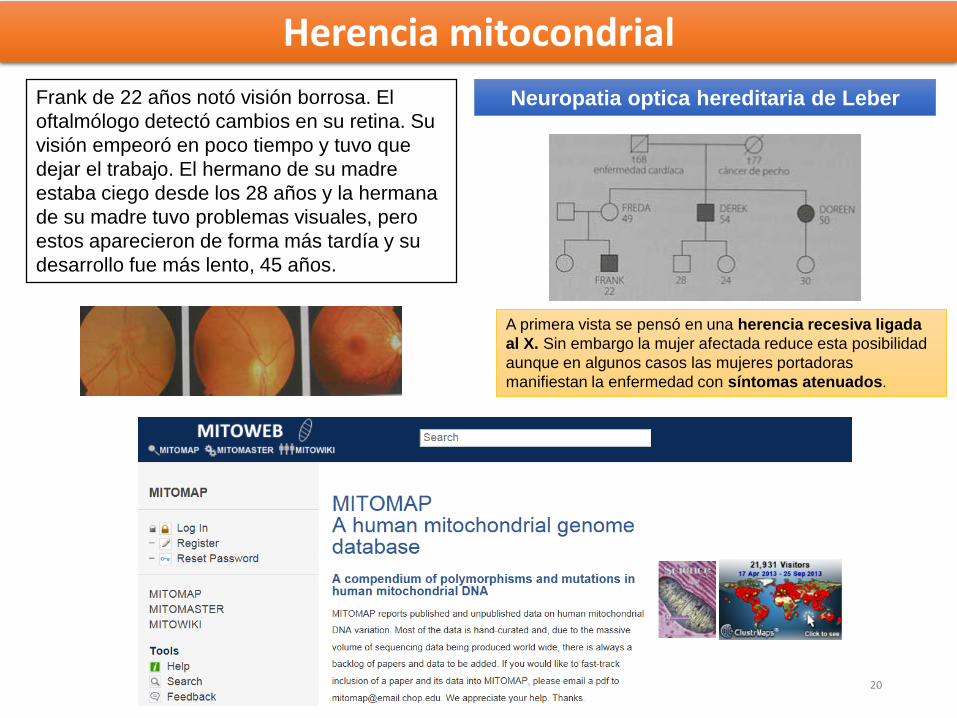
Frank de 22 años notó visión borrosa. El
oftalmólogo detectó cambios en su retina. Su
visión empeoró en poco tiempo y tuvo que
dejar el trabajo. El hermano de su madre
estaba ciego desde los 28 años y la hermana
de su madre tuvo problemas visuales, pero
estos aparecieron de forma más tardía y su
desarrollo fue más lento, 45 años.
Neuropatia optica hereditaria de Leber
A primera vista se pensó en una herencia recesiva ligada
al X. Sin embargo la mujer afectada reduce esta posibilidad
aunque en algunos casos las mujeres portadoras
manifiestan la enfermedad con síntomas atenuados.
Herencia mitocondrial
20

Características de la herencia mitocondrial
1. Herencia materna
2. Segregación replicativa
3. Homoplasmia/heteroplasmia
4. Efecto umbral fenotípico asociado a heteroplasmia
Cuando el número de copias de mtDNA mutado sobrepase un nivel determinado, diferente
para cada tejido, la producción de ATP se verá afectada y, por debajo de un nivel umbral,
aparecerán las manifestaciones de la enfermedad.
Para que la enfermedad afecte
clínicamente es necesaria la superación
de un umbral crítico de la proporción de
moléculas de mtDNA portadoras de la
mutación perjudicial en las células del
tejido afectado. A consecuencia
de ello, las enfermedades mitocondriales
se caracterizan por penetrancia
reducida y expresión variable.
21
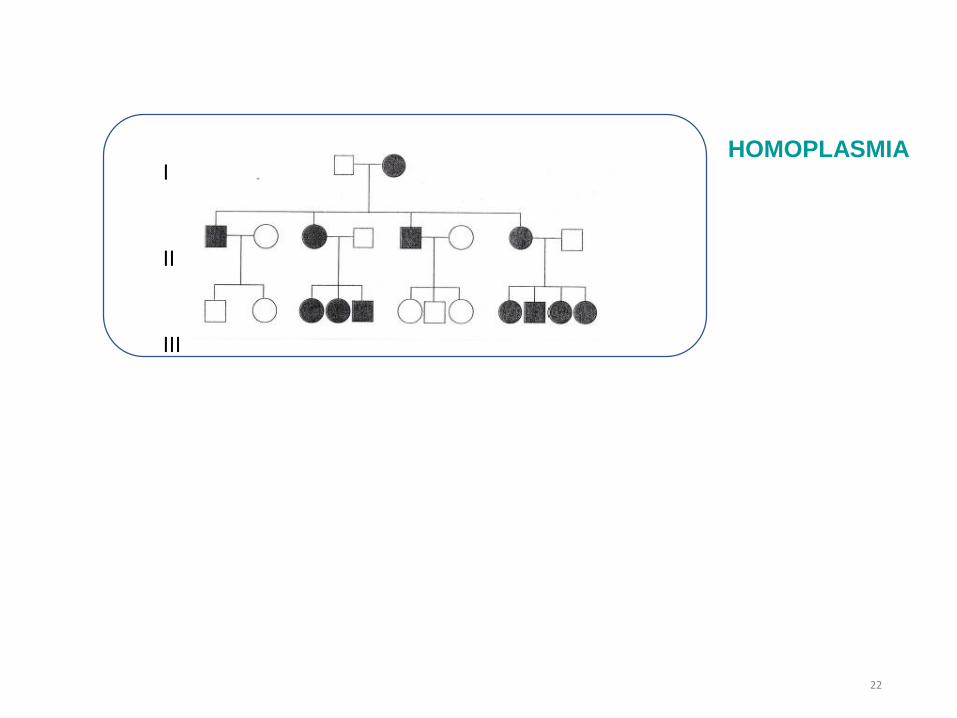
I
II
III
HOMOPLASMIA
HETEROPLASMIA
Se producen los típicos
saltos generacionales
en la expresión de la enfermedad
22
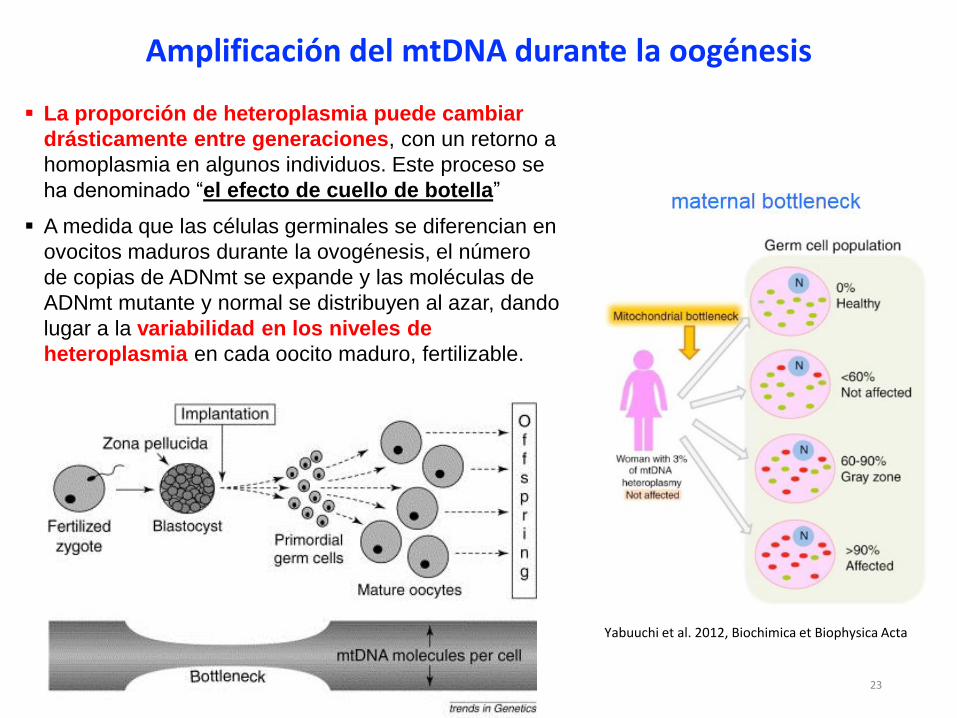
Amplificación del mtDNA durante la oogénesis
La proporción de heteroplasmia puede cambiar
drásticamente entre generaciones, con un retorno a
homoplasmia en algunos individuos. Este proceso se
ha denominado “el efecto de cuello de botella”
A medida que las células germinales se diferencian en
ovocitos maduros durante la ovogénesis, el número
de copias de ADNmt se expande y las moléculas de
ADNmt mutante y normal se distribuyen al azar, dando
lugar a la variabilidad en los niveles de
heteroplasmia en cada oocito maduro, fertilizable.
Yabuuchi et al. 2012, Biochimica et Biophysica Acta
23
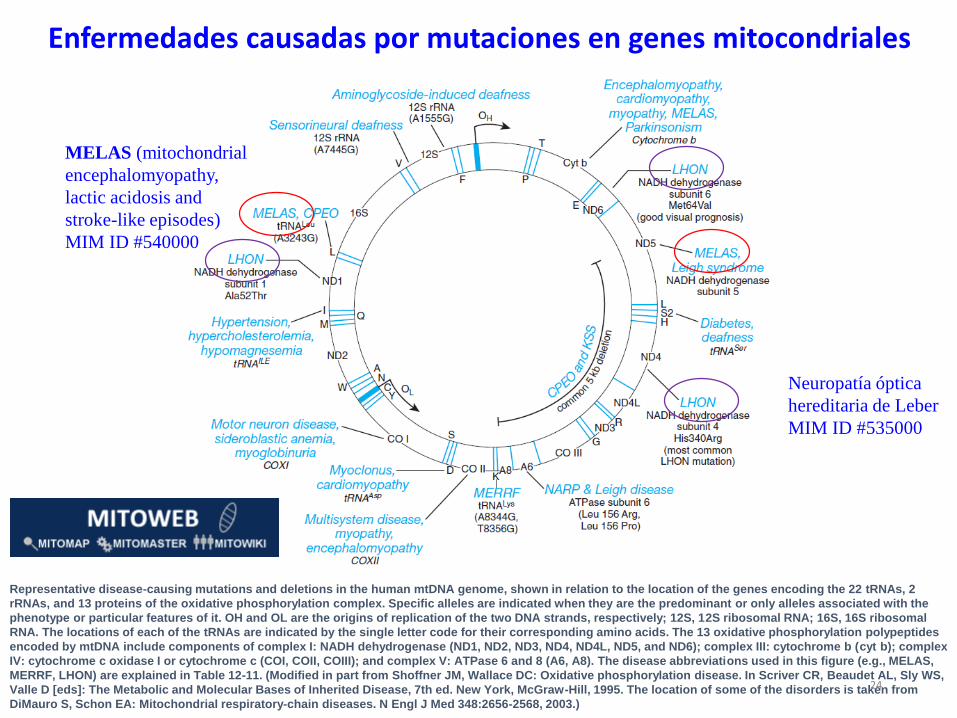
Representative disease-causing mutations and deletions in the human mtDNA genome, shown in relation to the location of the genes encoding the 22 tRNAs, 2
rRNAs, and 13 proteins of the oxidative phosphorylation complex. Specific alleles are indicated when they are the predominant or only alleles associated with the
phenotype or particular features of it. OH and OL are the origins of replication of the two DNA strands, respectively; 12S, 12S ribosomal RNA; 16S, 16S ribosomal
RNA. The locations of each of the tRNAs are indicated by the single letter code for their corresponding amino acids. The 13 oxidative phosphorylation polypeptides
encoded by mtDNA include components of complex I: NADH dehydrogenase (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6); complex III: cytochrome b (cyt b); complex
IV: cytochrome c oxidase I or cytochrome c (COI, COII, COIII); and complex V: ATPase 6 and 8 (A6, A8). The disease abbreviations used in this figure (e.g., MELAS,
MERRF, LHON) are explained in Table 12-11. (Modified in part from Shoffner JM, Wallace DC: Oxidative phosphorylation disease. In Scriver CR, Beaudet AL, Sly WS,
Valle D [eds]: The Metabolic and Molecular Bases of Inherited Disease, 7th ed. New York, McGraw-Hill, 1995. The location of some of the disorders is taken from
DiMauro S, Schon EA: Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656-2568, 2003.)
Neuropatía óptica
hereditaria de Leber
MIM ID #535000
MELAS (mitochondrial
encephalomyopathy,
lactic acidosis and
stroke-like episodes)
MIM ID #540000
Enfermedades causadas por mutaciones en genes mitocondriales
24
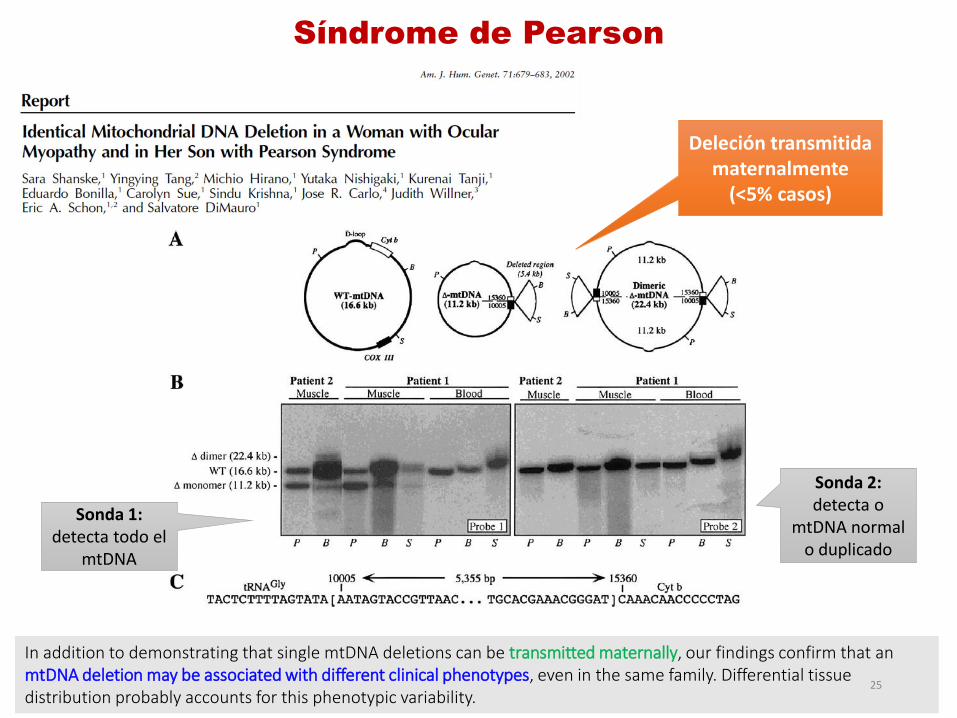
Síndrome de Pearson
In addition to demonstrating that single mtDNA deletions can be transmitted maternally, our findings confirm that an mtDNA deletion may be associated with different clinical phenotypes, even in the same family. Differential tissue distribution probably accounts for this phenotypic variability.
Sonda 1: detecta todo el
mtDNA
Sonda 2: detecta o
mtDNA normal o duplicado
Deleción transmitida maternalmente
(<5% casos)
25

Experto Universitario en Genética Médica y Genómica
Conceptos generales en Genética Humana:
Miguel Angel García Pérez
Departament de Genética
Patrones de transmisión de las enfermedades genéticas II
26


















